Healthcare analytics, AI solutions for biological big data, providing an AI platform for the biotech, life sciences, medical and pharmaceutical industries, as well as for related technological approaches, i.e., curation and text analysis with machine learning and other activities related to AI applications to these industries.
Chemotherapy Benefit in Early Breast Cancer Patients
Larry H Bernstein, MD, FCAP, Curator
LPBI
Agendia’s MammaPrint® First and Only Genomic Assay to Receive Level 1A Clinical Utility Evidence for Chemotherapy Benefit in Early Breast Cancer Patients
Clinical high-risk patients with a low-risk MammaPrint® result, including 48 percent node-positive, had five-year distant metastasis-free survival rate in excess of 94 percent, whether randomized to receive adjuvant chemotherapy or not
MammaPrint could change clinical practice by substantially de-escalating the use of adjuvant chemotherapy and sparing many patients an aggressive treatment they will not benefit from
Forty-six percent overall reduction in chemotherapy prescription among clinically high-risk patients
April 19, 2016 / B3C newswire / —Agendia, Inc., together with the European Organisation for Research and Treatment of Cancer (EORTC) and Breast International Group (BIG), announced results from the initial analysis of the primary objective of the Microarray In Node-negative (and 1 to 3 positive lymph node) Disease may Avoid ChemoTherapy (MINDACT) study at the American Association for Cancer Research Annual Meeting 2016 in New Orleans, LA.
Using the company’s MammaPrint® assay, patients with early-stage breast cancer who were considered at high risk for disease recurrence based on clinical and biological criteria had a distant metastasis-free survival at five years in excess of 94 percent.The MammaPrint test—the first and only genomic assay with FDA 510(k) clearance for use in risk assessment for women of all ages with early stage breast cancer—identified a large group of patients for whom five-year distant metastasis–free survival was equally good whether or not they received adjuvant chemotherapy (chemotherapy given post-surgery).
“The MINDACT trial design is the optimal way to prove clinical utility of a genomic assay,” said Prof. Laura van ’t Veer, CRO at Agendia, Leader, Breast Oncology Program, and Director, Applied Genomics at UCSF Helen Diller Family Comprehensive Cancer Center. “It gives the level 1A clinical evidence (prospective, randomized and controlled) that empowers physicians to clearly and confidently know when chemotherapy is part of optimal early-stage breast cancer therapy. In this trial, MammaPrint (70-gene assay) was compared to the standard of care physicians use today, to decide what is the best treatment option for an early-stage breast cancer patient.”
The MINDACT trial is the first prospective randomized controlled clinical trial of a breast cancer recurrence genomic assay with level 1A clinical evidence and the first prospective translational research study of this magnitude in breast cancer to report the results of its primary objective.
Among the 3,356 patients enrolled in the MINDACT trial, who were categorized as having a high risk of breast cancer recurrence based on common clinical and pathological criteria (C-high), the MammaPrint assay reduced the chemotherapy treatment prescription by 46 percent.Using the 70-gene assay, MammaPrint, 48 percent of lymph-node positive breast cancer patients considered clinically high-risk (Clinical-high) and genomic low-risk (MammaPrint-low) had an excellent distant metastasis-free survival at five years in excess of 94 percent.
“Traditionally, physicians have relied on clinical-pathological factors such as age, tumor size, tumor grade, lymph node involvement, and hormone receptor status to make breast cancer treatment decisions,” said Massimo Cristofanilli, MD, Associate Director of Translational Research and Precision Medicine at the Robert H. Lurie Comprehensive Cancer Center, Northwestern University in Chicago. “These findings provide level 1A clinical utility evidence by demonstrating that the detection of low-risk of distant recurrence reported by the MammaPrint test can be safely used in the management of thousands of women by identifying those who can be spared from a toxic and unnecessary treatment.”
MINDACT is a randomized phase III trial that investigates the clinical utility of MammaPrint, when compared (or – “used in conjunction with”) to the standard clinical pathological criteria, for the selection of patients unlikely to benefit from adjuvant chemotherapy. From 2007 to 2011, 6,693 women who had undergone surgery for early-stage breast cancer enrolled in the trial (111 centers in nine countries). Participants were categorized as low or high risk for tumor recurrence in two ways: first, through analysis of tumor tissue using MammaPrint at a central location in Amsterdam; and second, using Adjuvant! Online, a tool that calculates risk of breast cancer recurrence based on common clinical and biological criteria.
Patients characterized in both clinical and genomic assessments as “low- risk” are spared chemotherapy, while patients characterized as “high- risk” are advised chemotherapy. Those with conflicting results are randomized to use either clinical or genomic risk (MammaPrint) evaluation to decide on chemotherapy treatment.
The MINDACT trial is managed and sponsored by the EORTC as part of an extensive and complex partnership in collaboration with Agendia and BIG, and many other academic and commercial partners, as well as patient advocates.
“These MINDACT trial results are a testament that the science of the MammaPrint test is the most robust in the genomic breast recurrence assay market. Agendia will continue to collaborate with pharmaceutical companies, leading cancer centers and academic groups on additional clinical research and in the pursuit of bringing more effective, individualized treatments within reach of cancer patients,” said Mark Straley, Chief Executive Officer at Agendia. “We value the partnership with the EORTC and BIG and it’s a great honor to share this critical milestone.”
Breast cancer is the most frequently diagnosed cancer in women worldwide(1). In 2012, there were nearly 1.7 million new breast cancer cases among women worldwide, accounting for 25 percent of all new cancer cases in women(2).
CRISPR/Cas9, Familial Amyloid Polyneuropathy (FAP) and Neurodegenerative Disease, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 2: CRISPR for Gene Editing and DNA Repair
CRISPR/Cas9, Familial Amyloid Polyneuropathy ( FAP) and Neurodegenerative Disease
Curator: Larry H. Bernstein, MD, FCAP
CRISPR/Cas9 and Targeted Genome Editing: A New Era in Molecular Biology
The development of efficient and reliable ways to make precise, targeted changes to the genome of living cells is a long-standing goal for biomedical researchers. Recently, a new tool based on a bacterial CRISPR-associated protein-9 nuclease (Cas9) from Streptococcus pyogenes has generated considerable excitement (1). This follows several attempts over the years to manipulate gene function, including homologous recombination (2) and RNA interference (RNAi) (3). RNAi, in particular, became a laboratory staple enabling inexpensive and high-throughput interrogation of gene function (4, 5), but it is hampered by providing only temporary inhibition of gene function and unpredictable off-target effects (6). Other recent approaches to targeted genome modification – zinc-finger nucleases [ZFNs, (7)] and transcription-activator like effector nucleases [TALENs (8)]– enable researchers to generate permanent mutations by introducing doublestranded breaks to activate repair pathways. These approaches are costly and time-consuming to engineer, limiting their widespread use, particularly for large scale, high-throughput studies.
The Biology of Cas9
The functions of CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) and CRISPR-associated (Cas) genes are essential in adaptive immunity in select bacteria and archaea, enabling the organisms to respond to and eliminate invading genetic material. These repeats were initially discovered in the 1980s in E. coli (9), but their function wasn’t confirmed until 2007 by Barrangou and colleagues, who demonstrated that S. thermophilus can acquire resistance against a bacteriophage by integrating a genome fragment of an infectious virus into its CRISPR locus (10).
Three types of CRISPR mechanisms have been identified, of which type II is the most studied. In this case, invading DNA from viruses or plasmids is cut into small fragments and incorporated into a CRISPR locus amidst a series of short repeats (around 20 bps). The loci are transcribed, and transcripts are then processed to generate small RNAs (crRNA – CRISPR RNA), which are used to guide effector endonucleases that target invading DNA based on sequence complementarity (Figure 1) (11).
Figure 1. Cas9 in vivo: Bacterial Adaptive Immunity
In the acquisition phase, foreign DNA is incorporated into the bacterial genome at the CRISPR loci. CRISPR loci is then transcribed and processed into crRNA during crRNA biogenesis. During interference, Cas9 endonuclease complexed with a crRNA and separate tracrRNA cleaves foreign DNA containing a 20-nucleotide crRNA complementary sequence adjacent to the PAM sequence. (Figure not drawn to scale.)
One Cas protein, Cas9 (also known as Csn1), has been shown, through knockdown and rescue experiments to be a key player in certain CRISPR mechanisms (specifically type II CRISPR systems). The type II CRISPR mechanism is unique compared to other CRISPR systems, as only one Cas protein (Cas9) is required for gene silencing (12). In type II systems, Cas9 participates in the processing of crRNAs (12), and is responsible for the destruction of the target DNA (11). Cas9’s function in both of these steps relies on the presence of two nuclease domains, a RuvC-like nuclease domain located at the amino terminus and a HNH-like nuclease domain that resides in the mid-region of the protein (13).
To achieve site-specific DNA recognition and cleavage, Cas9 must be complexed with both a crRNA and a separate trans-activating crRNA (tracrRNA or trRNA), that is partially complementary to the crRNA (11). The tracrRNA is required for crRNA maturation from a primary transcript encoding multiple pre-crRNAs. This occurs in the presence of RNase III and Cas9 (12).
During the destruction of target DNA, the HNH and RuvC-like nuclease domains cut both DNA strands, generating double-stranded breaks (DSBs) at sites defined by a 20-nucleotide target sequence within an associated crRNA transcript (11, 14). The HNH domain cleaves the complementary strand, while the RuvC domain cleaves the noncomplementary strand.
The double-stranded endonuclease activity of Cas9 also requires that a short conserved sequence, (2–5 nts) known as protospacer-associated motif (PAM), follows immediately 3´- of the crRNA complementary sequence (15). In fact, even fully complementary sequences are ignored by Cas9-RNA in the absence of a PAM sequence (16).
Cas9 and CRISPR as a New Tool in Molecular Biology
The simplicity of the type II CRISPR nuclease, with only three required components (Cas9 along with the crRNA and trRNA) makes this system amenable to adaptation for genome editing. This potential was realized in 2012 by the Doudna and Charpentier labs (11). Based on the type II CRISPR system described previously, the authors developed a simplified two-component system by combining trRNA and crRNA into a single synthetic single guide RNA (sgRNA). sgRNAprogrammed Cas9 was shown to be as effective as Cas9 programmed with separate trRNA and crRNA in guiding targeted gene alterations (Figure 2A).
To date, three different variants of the Cas9 nuclease have been adopted in genome-editing protocols. The first is wild-type Cas9, which can site-specifically cleave double-stranded DNA, resulting in the activation of the doublestrand break (DSB) repair machinery. DSBs can be repaired by the cellular Non-Homologous End Joining (NHEJ) pathway (17), resulting in insertions and/or deletions (indels) which disrupt the targeted locus. Alternatively, if a donor template with homology to the targeted locus is supplied, the DSB may be repaired by the homology-directed repair (HDR) pathway allowing for precise replacement mutations to be made (Figure 2A) (17, 18).
Cong and colleagues (1) took the Cas9 system a step further towards increased precision by developing a mutant form, known as Cas9D10A, with only nickase activity. This means it cleaves only one DNA strand, and does not activate NHEJ. Instead, when provided with a homologous repair template, DNA repairs are conducted via the high-fidelity HDR pathway only, resulting in reduced indel mutations (1, 11, 19). Cas9D10A is even more appealing in terms of target specificity when loci are targeted by paired Cas9 complexes designed to generate adjacent DNA nicks (20) (see further details about “paired nickases” in Figure 2B).
The third variant is a nuclease-deficient Cas9 (dCas9, Figure 2C) (21). Mutations H840A in the HNH domain and D10A in the RuvC domain inactivate cleavage activity, but do not prevent DNA binding (11, 22). Therefore, this variant can be used to sequence-specifically target any region of the genome without cleavage. Instead, by fusing with various effector domains, dCas9 can be used either as a gene silencing or activation tool (21, 23–26). Furthermore, it can be used as a visualization tool. For instance, Chen and colleagues used dCas9 fused to Enhanced Green Fluorescent Protein (EGFP) to visualize repetitive DNA sequences with a single sgRNA or nonrepetitive loci using multiple sgRNAs (27).
Wild-type Cas9 nuclease site specifically cleaves double-stranded DNA activating double-strand break repair machinery. In the absence of a homologous repair template non-homologous end joining can result in indels disrupting the target sequence. Alternatively, precise mutations and knock-ins can be made by providing a homologous repair template and exploiting the homology directed repair pathway.
B. Mutated Cas9 makes a site specific single-strand nick. Two sgRNA can be used to introduce a staggered double-stranded break which can then undergo homology directed repair.
C. Nuclease-deficient Cas9 can be fused with various effector domains allowing specific localization. For example, transcriptional activators, repressors, and fluorescent proteins.
Targeting Efficiency and Off-target Mutations
Targeting efficiency, or the percentage of desired mutation achieved, is one of the most important parameters by which to assess a genome-editing tool. The targeting efficiency of Cas9 compares favorably with more established methods, such as TALENs or ZFNs (8). For example, in human cells, custom-designed ZFNs and TALENs could only achieve efficiencies ranging from 1% to 50% (29–31). In contrast, the Cas9 system has been reported to have efficiencies up to >70% in zebrafish (32) and plants (33), and ranging from 2–5% in induced pluripotent stem cells (34). In addition, Zhou and colleagues were able to improve genome targeting up to 78% in one-cell mouse embryos, and achieved effective germline transmission through the use of dual sgRNAs to simultaneously target an individual gene (35).
A widely used method to identify mutations is the T7 Endonuclease I mutation detection assay (36, 37) (Figure 3). This assay detects heteroduplex DNA that results from the annealing of a DNA strand, including desired mutations, with a wildtype DNA strand (37).
Figure 3. T7 Endonuclease I Targeting Efficiency Assay
Genomic DNA is amplified with primers bracketing the modified locus. PCR products are then denatured and re-annealed yielding 3 possible structures. Duplexes containing a mismatch are digested by T7 Endonuclease I. The DNA is then electrophoretically separated and fragment analysis is used to calculate targeting efficiency.
Another important parameter is the incidence of off-target mutations. Such mutations are likely to appear in sites that have differences of only a few nucleotides compared to the original sequence, as long as they are adjacent to a PAM sequence. This occurs as Cas9 can tolerate up to 5 base mismatches within the protospacer region (36) or a single base difference in the PAM sequence (38). Off-target mutations are generally more difficult to detect, requiring whole-genome sequencing to rule them out completely.
Recent improvements to the CRISPR system for reducing off-target mutations have been made through the use of truncated gRNA (truncated within the crRNA-derived sequence) or by adding two extra guanine (G) nucleotides to the 5´ end (28, 37). Another way researchers have attempted to minimize off-target effects is with the use of “paired nickases” (20). This strategy uses D10A Cas9 and two sgRNAs complementary to the adjacent area on opposite strands of the target site (Figure 2B). While this induces DSBs in the target DNA, it is expected to create only single nicks in off-target locations and, therefore, result in minimal off-target mutations.
By leveraging computation to reduce off-target mutations, several groups have developed webbased tools to facilitate the identification of potential CRISPR target sites and assess their potential for off-target cleavage. Examples include the CRISPR Design Tool (38) and the ZiFiT Targeter, Version 4.2 (39, 40).
Applications as a Genome-editing and Genome Targeting Tool
Following its initial demonstration in 2012 (9), the CRISPR/Cas9 system has been widely adopted. This has already been successfully used to target important genes in many cell lines and organisms, including human (34), bacteria (41), zebrafish (32), C. elegans (42), plants (34), Xenopus tropicalis (43), yeast (44), Drosophila (45), monkeys (46), rabbits (47), pigs (42), rats (48) and mice (49). Several groups have now taken advantage of this method to introduce single point mutations (deletions or insertions) in a particular target gene, via a single gRNA (14, 21, 29). Using a pair of gRNA-directed Cas9 nucleases instead, it is also possible to induce large deletions or genomic rearrangements, such as inversions or translocations (50). A recent exciting development is the use of the dCas9 version of the CRISPR/Cas9 system to target protein domains for transcriptional regulation (26, 51, 52), epigenetic modification (25), and microscopic visualization of specific genome loci (27).
The CRISPR/Cas9 system requires only the redesign of the crRNA to change target specificity. This contrasts with other genome editing tools, including zinc finger and TALENs, where redesign of the protein-DNA interface is required. Furthermore, CRISPR/Cas9 enables rapid genome-wide interrogation of gene function by generating large gRNA libraries (51, 53) for genomic screening.
The Future of CRISPR/Cas9
The rapid progress in developing Cas9 into a set of tools for cell and molecular biology research has been remarkable, likely due to the simplicity, high efficiency and versatility of the system. Of the designer nuclease systems currently available for precision genome engineering, the CRISPR/Cas system is by far the most user friendly. It is now also clear that Cas9’s potential reaches beyond DNA cleavage, and its usefulness for genome locus-specific recruitment of proteins will likely only be limited by our imagination.
Scientists urge caution in using new CRISPR technology to treat human genetic disease
The bacterial enzyme Cas9 is the engine of RNA-programmed genome engineering in human cells. (Graphic by Jennifer Doudna/UC Berkeley)
A group of 18 scientists and ethicists today warned that a revolutionary new tool to cut and splice DNA should be used cautiously when attempting to fix human genetic disease, and strongly discouraged any attempts at making changes to the human genome that could be passed on to offspring.
Among the authors of this warning is Jennifer Doudna, the co-inventor of the technology, called CRISPR-Cas9, which is driving a new interest in gene therapy, or “genome engineering.” She and colleagues co-authored a perspective piece that appears in the March 20 issue of Science, based on discussions at a meeting that took place in Napa on Jan. 24. The same issue of Science features a collection of recent research papers, commentary and news articles on CRISPR and its implications. …..
A prudent path forward for genomic engineering and germline gene modification
Scientists today are changing DNA sequences to correct genetic defects in animals as well as cultured tissues generated from stem cells, strategies that could eventually be used to treat human disease. The technology can also be used to engineer animals with genetic diseases mimicking human disease, which could lead to new insights into previously enigmatic disorders.
The CRISPR-Cas9 tool is still being refined to ensure that genetic changes are precisely targeted, Doudna said. Nevertheless, the authors met “… to initiate an informed discussion of the uses of genome engineering technology, and to identify proactively those areas where current action is essential to prepare for future developments. We recommend taking immediate steps toward ensuring that the application of genome engineering technology is performed safely and ethically.”
Amyloid CRISPR Plasmids and si/shRNA Gene Silencers
Santa Cruz Biotechnology, Inc. offers a broad range of gene silencers in the form of siRNAs, shRNA Plasmids and shRNA Lentiviral Particles as well as CRISPR/Cas9 Knockout and CRISPR Double Nickase plasmids. Amyloid gene silencers are available as Amyloid siRNA, Amyloid shRNA Plasmid, Amyloid shRNA Lentiviral Particles and Amyloid CRISPR/Cas9 Knockout plasmids. Amyloid CRISPR/dCas9 Activation Plasmids and CRISPR Lenti Activation Systems for gene activation are also available. Gene silencers and activators are useful for gene studies in combination with antibodies used for protein detection. Amyloid CRISPR Knockout, HDR and Nickase Knockout Plasmids
CRISPR-Cas9-Based Knockout of the Prion Protein and Its Effect on the Proteome
The molecular function of the cellular prion protein (PrPC) and the mechanism by which it may contribute to neurotoxicity in prion diseases and Alzheimer’s disease are only partially understood. Mouse neuroblastoma Neuro2a cells and, more recently, C2C12 myocytes and myotubes have emerged as popular models for investigating the cellular biology of PrP. Mouse epithelial NMuMG cells might become attractive models for studying the possible involvement of PrP in a morphogenetic program underlying epithelial-to-mesenchymal transitions. Here we describe the generation of PrP knockout clones from these cell lines using CRISPR-Cas9 knockout technology. More specifically, knockout clones were generated with two separate guide RNAs targeting recognition sites on opposite strands within the first hundred nucleotides of the Prnp coding sequence. Several PrP knockout clones were isolated and genomic insertions and deletions near the CRISPR-target sites were characterized. Subsequently, deep quantitative global proteome analyses that recorded the relative abundance of>3000 proteins (data deposited to ProteomeXchange Consortium) were undertaken to begin to characterize the molecular consequences of PrP deficiency. The levels of ∼120 proteins were shown to reproducibly correlate with the presence or absence of PrP, with most of these proteins belonging to extracellular components, cell junctions or the cytoskeleton.
Recent advances in genome engineering technologies based on the CRISPR-associated RNA-guided endonuclease Cas9 are enabling the systematic interrogation of mammalian genome function. Analogous to the search function in modern word processors, Cas9 can be guided to specific locations within complex genomes by a short RNA search string. Using this system, DNA sequences within the endogenous genome and their functional outputs are now easily edited or modulated in virtually any organism of choice. Cas9-mediated genetic perturbation is simple and scalable, empowering researchers to elucidate the functional organization of the genome at the systems level and establish causal linkages between genetic variations and biological phenotypes. In this Review, we describe the development and applications of Cas9 for a variety of research or translational applications while highlighting challenges as well as future directions. Derived from a remarkable microbial defense system, Cas9 is driving innovative applications from basic biology to biotechnology and medicine.
The development of recombinant DNA technology in the 1970s marked the beginning of a new era for biology. For the first time, molecular biologists gained the ability to manipulate DNA molecules, making it possible to study genes and harness them to develop novel medicine and biotechnology. Recent advances in genome engineering technologies are sparking a new revolution in biological research. Rather than studying DNA taken out of the context of the genome, researchers can now directly edit or modulate the function of DNA sequences in their endogenous context in virtually any organism of choice, enabling them to elucidate the functional organization of the genome at the systems level, as well as identify causal genetic variations.
Broadly speaking, genome engineering refers to the process of making targeted modifications to the genome, its contexts (e.g., epigenetic marks), or its outputs (e.g., transcripts). The ability to do so easily and efficiently in eukaryotic and especially mammalian cells holds immense promise to transform basic science, biotechnology, and medicine (Figure 1).
For life sciences research, technologies that can delete, insert, and modify the DNA sequences of cells or organisms enable dissecting the function of specific genes and regulatory elements. Multiplexed editing could further allow the interrogation of gene or protein networks at a larger scale. Similarly, manipulating transcriptional regulation or chromatin states at particular loci can reveal how genetic material is organized and utilized within a cell, illuminating relationships between the architecture of the genome and its functions. In biotechnology, precise manipulation of genetic building blocks and regulatory machinery also facilitates the reverse engineering or reconstruction of useful biological systems, for example, by enhancing biofuel production pathways in industrially relevant organisms or by creating infection-resistant crops. Additionally, genome engineering is stimulating a new generation of drug development processes and medical therapeutics. Perturbation of multiple genes simultaneously could model the additive effects that underlie complex polygenic disorders, leading to new drug targets, while genome editing could directly correct harmful mutations in the context of human gene therapy (Tebas et al., 2014).
Eukaryotic genomes contain billions of DNA bases and are difficult to manipulate. One of the breakthroughs in genome manipulation has been the development of gene targeting by homologous recombination (HR), which integrates exogenous repair templates that contain sequence homology to the donor site (Figure 2A) (Capecchi, 1989). HR-mediated targeting has facilitated the generation of knockin and knockout animal models via manipulation of germline competent stem cells, dramatically advancing many areas of biological research. However, although HR-mediated gene targeting produces highly precise alterations, the desired recombination events occur extremely infrequently (1 in 106–109 cells) (Capecchi, 1989), presenting enormous challenges for large-scale applications of gene-targeting experiments.
Genome Editing Technologies Exploit Endogenous DNA Repair Machinery
To overcome these challenges, a series of programmable nuclease-based genome editing technologies have been developed in recent years, enabling targeted and efficient modification of a variety of eukaryotic and particularly mammalian species. Of the current generation of genome editing technologies, the most rapidly developing is the class of RNA-guided endonucleases known as Cas9 from the microbial adaptive immune system CRISPR (clustered regularly interspaced short palindromic repeats), which can be easily targeted to virtually any genomic location of choice by a short RNA guide. Here, we review the development and applications of the CRISPR-associated endonuclease Cas9 as a platform technology for achieving targeted perturbation of endogenous genomic elements and also discuss challenges and future avenues for innovation. ……
Figure 4Natural Mechanisms of Microbial CRISPR Systems in Adaptive Immunity
…… A key turning point came in 2005, when systematic analysis of the spacer sequences separating the individual direct repeats suggested their extrachromosomal and phage-associated origins (Mojica et al., 2005; Pourcel et al., 2005; Bolotin et al., 2005). This insight was tremendously exciting, especially given previous studies showing that CRISPR loci are transcribed (Tang et al., 2002) and that viruses are unable to infect archaeal cells carrying spacers corresponding to their own genomes (Mojica et al., 2005). Together, these findings led to the speculation that CRISPR arrays serve as an immune memory and defense mechanism, and individual spacers facilitate defense against bacteriophage infection by exploiting Watson-Crick base-pairing between nucleic acids (Mojica et al., 2005; Pourcel et al., 2005). Despite these compelling realizations that CRISPR loci might be involved in microbial immunity, the specific mechanism of how the spacers act to mediate viral defense remained a challenging puzzle. Several hypotheses were raised, including thoughts that CRISPR spacers act as small RNA guides to degrade viral transcripts in a RNAi-like mechanism (Makarova et al., 2006) or that CRISPR spacers direct Cas enzymes to cleave viral DNA at spacer-matching regions (Bolotin et al., 2005). …..
As the pace of CRISPR research accelerated, researchers quickly unraveled many details of each type of CRISPR system (Figure 4). Building on an earlier speculation that protospacer adjacent motifs (PAMs) may direct the type II Cas9 nuclease to cleave DNA (Bolotin et al., 2005), Moineau and colleagues highlighted the importance of PAM sequences by demonstrating that PAM mutations in phage genomes circumvented CRISPR interference (Deveau et al., 2008). Additionally, for types I and II, the lack of PAM within the direct repeat sequence within the CRISPR array prevents self-targeting by the CRISPR system. In type III systems, however, mismatches between the 5′ end of the crRNA and the DNA target are required for plasmid interference (Marraffini and Sontheimer, 2010). …..
In 2013, a pair of studies simultaneously showed how to successfully engineer type II CRISPR systems from Streptococcus thermophilus (Cong et al., 2013) andStreptococcus pyogenes (Cong et al., 2013; Mali et al., 2013a) to accomplish genome editing in mammalian cells. Heterologous expression of mature crRNA-tracrRNA hybrids (Cong et al., 2013) as well as sgRNAs (Cong et al., 2013; Mali et al., 2013a) directs Cas9 cleavage within the mammalian cellular genome to stimulate NHEJ or HDR-mediated genome editing. Multiple guide RNAs can also be used to target several genes at once. Since these initial studies, Cas9 has been used by thousands of laboratories for genome editing applications in a variety of experimental model systems (Sander and Joung, 2014). ……
The majority of CRISPR-based technology development has focused on the signature Cas9 nuclease from type II CRISPR systems. However, there remains a wide diversity of CRISPR types and functions. Cas RAMP module (Cmr) proteins identified in Pyrococcus furiosus and Sulfolobus solfataricus (Hale et al., 2012) constitute an RNA-targeting CRISPR immune system, forming a complex guided by small CRISPR RNAs that target and cleave complementary RNA instead of DNA. Cmr protein homologs can be found throughout bacteria and archaea, typically relying on a 5′ site tag sequence on the target-matching crRNA for Cmr-directed cleavage.
Unlike RNAi, which is targeted largely by a 6 nt seed region and to a lesser extent 13 other bases, Cmr crRNAs contain 30–40 nt of target complementarity. Cmr-CRISPR technologies for RNA targeting are thus a promising target for orthogonal engineering and minimal off-target modification. Although the modularity of Cmr systems for RNA-targeting in mammalian cells remains to be investigated, Cmr complexes native to P. furiosus have already been engineered to target novel RNA substrates (Hale et al., 2009, 2012). ……
Although Cas9 has already been widely used as a research tool, a particularly exciting future direction is the development of Cas9 as a therapeutic technology for treating genetic disorders. For a monogenic recessive disorder due to loss-of-function mutations (such as cystic fibrosis, sickle-cell anemia, or Duchenne muscular dystrophy), Cas9 may be used to correct the causative mutation. This has many advantages over traditional methods of gene augmentation that deliver functional genetic copies via viral vector-mediated overexpression—particularly that the newly functional gene is expressed in its natural context. For dominant-negative disorders in which the affected gene is haplosufficient (such as transthyretin-related hereditary amyloidosis or dominant forms of retinitis pigmentosum), it may also be possible to use NHEJ to inactivate the mutated allele to achieve therapeutic benefit. For allele-specific targeting, one could design guide RNAs capable of distinguishing between single-nucleotide polymorphism (SNP) variations in the target gene, such as when the SNP falls within the PAM sequence.
CRISPR/Cas9: a powerful genetic engineering tool for establishing large animal models of neurodegenerative diseases
Zhuchi Tu, Weili Yang, Sen Yan, Xiangyu Guo and Xiao-Jiang Li
Animal models are extremely valuable to help us understand the pathogenesis of neurodegenerative disorders and to find treatments for them. Since large animals are more like humans than rodents, they make good models to identify the important pathological events that may be seen in humans but not in small animals; large animals are also very important for validating effective treatments or confirming therapeutic targets. Due to the lack of embryonic stem cell lines from large animals, it has been difficult to use traditional gene targeting technology to establish large animal models of neurodegenerative diseases. Recently, CRISPR/Cas9 was used successfully to genetically modify genomes in various species. Here we discuss the use of CRISPR/Cas9 technology to establish large animal models that can more faithfully mimic human neurodegenerative diseases.
Neurodegenerative diseases — Alzheimer’s disease(AD),Parkinson’s disease(PD), amyotrophic lateral sclerosis (ALS), Huntington’s disease (HD), and frontotemporal dementia (FTD) — are characterized by age-dependent and selective neurodegeneration. As the life expectancy of humans lengthens, there is a greater prevalence of these neurodegenerative diseases; however, the pathogenesis of most of these neurodegenerative diseases remain unclear, and we lack effective treatments for these important brain disorders.
CRISPR/Cas9, Non-human primates, Neurodegenerative diseases, Animal model
There are a number of excellent reviews covering different types of neurodegenerative diseases and their genetic mouse models [8–12]. Investigations of different mouse models of neurodegenerative diseases have revealed a common pathology shared by these diseases. First, the development of neuropathology and neurological symptoms in genetic mouse models of neurodegenerative diseases is age dependent and progressive. Second, all the mouse models show an accumulation of misfolded or aggregated proteins resulting from the expression of mutant genes. Third, despite the widespread expression of mutant proteins throughout the body and brain, neuronal function appears to be selectively or preferentially affected. All these facts indicate that mouse models of neurodegenerative diseases recapitulate important pathologic features also seen in patients with neurodegenerative diseases.
However, it seems that mouse models can not recapitulate the full range of neuropathology seen in patients with neurodegenerative diseases. Overt neurodegeneration, which is the most important pathological feature in patient brains, is absent in genetic rodent models of AD, PD, and HD. Many rodent models that express transgenic mutant proteins under the control of different promoters do not replicate overt neurodegeneration, which is likely due to their short life spans and the different aging processes of small animals. Also important are the remarkable differences in brain development between rodents and primates. For example, the mouse brain takes 21 days to fully develop, whereas the formation of primate brains requires more than 150 days [13]. The rapid development of the brain in rodents may render neuronal cells resistant to misfolded protein-mediated neurodegeneration. Another difficulty in using rodent models is how to analyze cognitive and emotional abnormalities, which are the early symptoms of most neurodegenerative diseases in humans. Differences in neuronal circuitry, anatomy, and physiology between rodent and primate brains may also account for the behavioral differences between rodent and primate models.
Mitochondrial dynamics–fusion, fission, movement, and mitophagy–in neurodegenerative diseases
Neurons are metabolically active cells with high energy demands at locations distant from the cell body. As a result, these cells are particularly dependent on mitochondrial function, as reflected by the observation that diseases of mitochondrial dysfunction often have a neurodegenerative component. Recent discoveries have highlighted that neurons are reliant particularly on the dynamic properties of mitochondria. Mitochondria are dynamic organelles by several criteria. They engage in repeated cycles of fusion and fission, which serve to intermix the lipids and contents of a population of mitochondria. In addition, mitochondria are actively recruited to subcellular sites, such as the axonal and dendritic processes of neurons. Finally, the quality of a mitochondrial population is maintained through mitophagy, a form of autophagy in which defective mitochondria are selectively degraded. We review the general features of mitochondrial dynamics, incorporating recent findings on mitochondrial fusion, fission, transport and mitophagy. Defects in these key features are associated with neurodegenerative disease. Charcot-Marie-Tooth type 2A, a peripheral neuropathy, and dominant optic atrophy, an inherited optic neuropathy, result from a primary deficiency of mitochondrial fusion. Moreover, several major neurodegenerative diseases—including Parkinson’s, Alzheimer’s and Huntington’s disease—involve disruption of mitochondrial dynamics. Remarkably, in several disease models, the manipulation of mitochondrial fusion or fission can partially rescue disease phenotypes. We review how mitochondrial dynamics is altered in these neurodegenerative diseases and discuss the reciprocal interactions between mitochondrial fusion, fission, transport and mitophagy.
Applications of CRISPR–Cas systems in Neuroscience
Genome-editing tools, and in particular those based on CRISPR–Cas (clustered regularly interspaced short palindromic repeat (CRISPR)–CRISPR-associated protein) systems, are accelerating the pace of biological research and enabling targeted genetic interrogation in almost any organism and cell type. These tools have opened the door to the development of new model systems for studying the complexity of the nervous system, including animal models and stem cell-derived in vitro models. Precise and efficient gene editing using CRISPR–Cas systems has the potential to advance both basic and translational neuroscience research.
Cellular neuroscience, DNA recombination, Genetic engineering, Molecular neuroscience
Figure 3: In vitro applications of Cas9 in human iPSCs.close
a | Evaluation of disease candidate genes from large-population genome-wide association studies (GWASs). Human primary cells, such as neurons, are not easily available and are difficult to expand in culture. By contrast, induced pluripo…
The development of the CRISPR/Cas9 system has made gene editing a relatively simple task. While CRISPR and other gene editing technologies stand to revolutionize biomedical research and offers many promising therapeutic avenues (such as in the treatment of HIV), a great deal of debate exists over whether CRISPR should be used to modify human embryos. As I discussed in my previous Insight article, we lack enough fundamental biological knowledge to enhance many traits like height or intelligence, so we are not near a future with genetically-enhanced super babies. However, scientists have identified a few rare genetic variants that protect against disease. One such protective variant is a mutation in the APP gene that protects against Alzheimer’s disease and cognitive decline in old age. If we can perfect gene editing technologies, is this mutation one that we should be regularly introducing into embryos? In this article, I explore the potential for using gene editing as a way to prevent Alzheimer’s disease in future generations. Alzheimer’s Disease: Medicine’s Greatest Challenge in the 21st Century Can gene editing be the missing piece in the battle against Alzheimer’s? (Source: bostonbiotech.org) I chose to assess the benefit of germline gene editing in the context of Alzheimer’s disease because this disease is one of the biggest challenges medicine faces in the 21st century. Alzheimer’s disease is a chronic neurodegenerative disease responsible for the majority of the cases of dementia in the elderly. The disease symptoms begins with short term memory loss and causes more severe symptoms – problems with language, disorientation, mood swings, behavioral issues – as it progresses, eventually leading to the loss of bodily functions and death. Because of the dementia the disease causes, Alzheimer’s patients require a great deal of care, and the world spends ~1% of its total GDP on caring for those with Alzheimer’s and related disorders. Because the prevalence of the disease increases with age, the situation will worsen as life expectancies around the globe increase: worldwide cases of Alzheimer’s are expected to grow from 35 million today to over 115 million by 2050.
Despite much research, the exact causes of Alzheimer’s disease remains poorly understood. The disease seems to be related to the accumulation of plaques made of amyloid-β peptides that form on the outside of neurons, as well as the formation of tangles of the protein tau inside of neurons. Although many efforts have been made to target amyloid-β or the enzymes involved in its formation, we have so far been unsuccessful at finding any treatment that stops the disease or reverses its progress. Some researchers believe that most attempts at treating Alzheimer’s have failed because, by the time a patient shows symptoms, the disease has already progressed past the point of no return.
While research towards a cure continues, researchers have sought effective ways to prevent Alzheimer’s disease. Although some studies show that mental and physical exercise may lower ones risk of Alzheimer’s disease, approximately 60-80% of the risk for Alzheimer’s disease appears to be genetic. Thus, if we’re serious about prevention, we may have to act at the genetic level. And because the brain is difficult to access surgically for gene therapy in adults, this means using gene editing on embryos.
With the latest CRISPR/Cas9 advance, the exhortation “turn on, tune in, drop out” comes to mind. The CRISPR/Cas9 gene-editing system was already a well-known means of “tuning in” (inserting new genes) and “dropping out” (knocking out genes). But when it came to “turning on” genes, CRISPR/Cas9 had little potency. That is, it had demonstrated only limited success as a way to activate specific genes.
A new CRISPR/Cas9 approach, however, appears capable of activating genes more effectively than older approaches. The new approach may allow scientists to more easily determine the function of individual genes, according to Feng Zhang, Ph.D., a researcher at MIT and the Broad Institute. Dr. Zhang and colleagues report that the new approach permits multiplexed gene activation and rapid, large-scale studies of gene function.
The new technique was introduced in the December 10 online edition of Nature, in an article entitled, “Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex.” The article describes how Dr. Zhang, along with the University of Tokyo’s Osamu Nureki, Ph.D., and Hiroshi Nishimasu, Ph.D., overhauled the CRISPR/Cas9 system. The research team based their work on their analysis (published earlier this year) of the structure formed when Cas9 binds to the guide RNA and its target DNA. Specifically, the team used the structure’s 3D shape to rationally improve the system.
In previous efforts to revamp CRISPR/Cas9 for gene activation purposes, scientists had tried to attach the activation domains to either end of the Cas9 protein, with limited success. From their structural studies, the MIT team realized that two small loops of the RNA guide poke out from the Cas9 complex and could be better points of attachment because they allow the activation domains to have more flexibility in recruiting transcription machinery.
Using their revamped system, the researchers activated about a dozen genes that had proven difficult or impossible to turn on using the previous generation of Cas9 activators. Each gene showed at least a twofold boost in transcription, and for many genes, the researchers found multiple orders of magnitude increase in activation.
After investigating single-guide RNA targeting rules for effective transcriptional activation, demonstrating multiplexed activation of 10 genes simultaneously, and upregulating long intergenic noncoding RNA transcripts, the research team decided to undertake a large-scale screen. This screen was designed to identify genes that confer resistance to a melanoma drug called PLX-4720.
“We … synthesized a library consisting of 70,290 guides targeting all human RefSeq coding isoforms to screen for genes that, upon activation, confer resistance to a BRAF inhibitor,” wrote the authors of the Nature paper. “The top hits included genes previously shown to be able to confer resistance, and novel candidates were validated using individual [single-guide RNA] and complementary DNA overexpression.”
A gene signature based on the top screening hits, the authors added, correlated with a gene expression signature of BRAF inhibitor resistance in cell lines and patient-derived samples. It was also suggested that large-scale screens such as the one demonstrated in the current study could help researchers discover new cancer drugs that prevent tumors from becoming resistant.
Familial amyloid polyneuropathy type I is an autosomal dominant disorder caused by mutations in the transthyretin (TTR ) gene; however, carriers of the same mutation exhibit variability in penetrance and clinical expression. We analyzed alleles of candidate genes encoding non-fibrillar components of TTR amyloid deposits and a molecule metabolically interacting with TTR [retinol-binding protein (RBP)], for possible associations with age of disease onset and/or susceptibility in a Portuguese population sample with the TTR V30M mutation and unrelated controls. We show that the V30M carriers represent a distinct subset of the Portuguese population. Estimates of genetic distance indicated that the controls and the classical onset group were furthest apart, whereas the late-onset group appeared to differ from both. Importantly, the data also indicate that genetic interactions among the multiple loci evaluated, rather than single-locus effects, are more likely to determine differences in the age of disease onset. Multifactor dimensionality reduction indicated that the best genetic model for classical onset group versus controls involved the APCS gene, whereas for late-onset cases, one APCS variant (APCSv1) and two RBP variants (RBPv1 and RBPv2) are involved. Thus, although the TTR V30M mutation is required for the disease in Portuguese patients, different genetic factors may govern the age of onset, as well as the occurrence of anticipation.
Autosomal dominant disorders may vary in expression even within a given kindred. The basis of this variability is uncertain and can be attributed to epigenetic factors, environment or epistasis. We have studied familial amyloid polyneuropathy (FAP), an autosomal dominant disorder characterized by peripheral sensorimotor and autonomic neuropathy. It exhibits variation in cardiac, renal, gastrointestinal and ocular involvement, as well as age of onset. Over 80 missense mutations in the transthyretin gene (TTR ) result in autosomal dominant disease http://www.ibmc.up.pt/~mjsaraiv/ttrmut.html). The presence of deposits consisting entirely of wild-type TTR molecules in the hearts of 10– 25% of individuals over age 80 reveals its inherent in vivo amyloidogenic potential (1).
FAP was initially described in Portuguese (2) where, until recently, the TTR V30M has been the only pathogenic mutation associated with the disease (3,4). Later reports identified the same mutation in Swedish and Japanese families (5,6). The disorder has since been recognized in other European countries and in North American kindreds in association with V30M, as well as other mutations (7).
TTR V30M produces disease in only 5–10% of Swedish carriers of the allele (8), a much lower degree of penetrance than that seen in Portuguese (80%) (9) or in Japanese with the same mutation. The actual penetrance in Japanese carriers has not been formally established, but appears to resemble that seen in Portuguese. Portuguese and Japanese carriers show considerable variation in the age of clinical onset (10,11). In both populations, the first symptoms had originally been described as typically occurring before age 40 (so-called ‘classical’ or early-onset); however, in recent years, more individuals developing symptoms late in life have been identified (11,12). Hence, present data indicate that the distribution of the age of onset in Portuguese is continuous, but asymmetric with a mean around age 35 and a long tail into the older age group (Fig. 1) (9,13). Further, DNA testing in Portugal has identified asymptomatic carriers over age 70 belonging to a subset of very late-onset kindreds in whose descendants genetic anticipation is frequent. The molecular basis of anticipation in FAP, which is not mediated by trinucleotide repeat expansions in the TTR or any other gene (14), remains elusive.
Variation in penetrance, age of onset and clinical features are hallmarks of many autosomal dominant disorders including the human TTR amyloidoses (7). Some of these clearly reflect specific biological effects of a particular mutation or a class of mutants. However, when such phenotypic variability is seen with a single mutation in the gene encoding the same protein, it suggests an effect of modifying genetic loci and/or environmental factors contributing differentially to the course of disease. We have chosen to examine age of onset as an example of a discrete phenotypic variation in the presence of the particular autosomal dominant disease-associated mutation TTR V30M. Although the role of environmental factors cannot be excluded, the existence of modifier genes involved in TTR amyloidogenesis is an attractive hypothesis to explain the phenotypic variability in FAP. ….
ATTR (TTR amyloid), like all amyloid deposits, contains several molecular components, in addition to the quantitatively dominant fibril-forming amyloid protein, including heparan sulfate proteoglycan 2 (HSPG2 or perlecan), SAP, a plasma glycoprotein of the pentraxin family (encoded by the APCS gene) that undergoes specific calcium-dependent binding to all types of amyloid fibrils, and apolipoprotein E (ApoE), also found in all amyloid deposits (15). The ApoE4 isoform is associated with an increased frequency and earlier onset of Alzheimer’s disease (Ab), the most common form of brain amyloid, whereas the ApoE2 isoform appears to be protective (16). ApoE variants could exert a similar modulatory effect in the onset of FAP, although early studies on a limited number of patients suggested this was not the case (17).
In at least one instance of senile systemic amyloidosis, small amounts of AA-related material were found in TTR deposits (18). These could reflect either a passive co-aggregation or a contributory involvement of protein AA, encoded by the serum amyloid A (SAA ) genes and the main component of secondary (reactive) amyloid fibrils, in the formation of ATTR.
Retinol-binding protein (RBP), the serum carrier of vitamin A, circulates in plasma bound to TTR. Vitamin A-loaded RBP and L-thyroxine, the two natural ligands of TTR, can act alone or synergistically to inhibit the rate and extent of TTR fibrillogenesis in vitro, suggesting that RBP may influence the course of FAP pathology in vivo (19). We have analyzed coding and non-coding sequence polymorphisms in the RBP4 (serum RBP, 10q24), HSPG2 (1p36.1), APCS (1q22), APOE (19q13.2), SAA1 and SAA2 (11p15.1) genes with the goal of identifying chromosomes carrying common and functionally significant variants. At the time these studies were performed, the full human genome sequence was not completed and systematic singlenucleotide polymorphism (SNP) analyses were not available for any of the suspected candidate genes. We identified new SNPs in APCS and RBP4 and utilized polymorphisms in SAA, HSPG2 and APOE that had already been characterized and shown to have potential pathophysiologic significance in other disorders (16,20–22). The genotyping data were analyzed for association with the presence of the V30M amyloidogenic allele (FAP patients versus controls) and with the age of onset (classical- versus late-onset patients). Multilocus analyses were also performed to examine the effects of simultaneous contributions of the six loci for determining the onset of the first symptoms. …..
The potential for different underlying models for classical and late onset is supported by the MDR analysis, which produces two distinct models when comparing each class with the controls. One could view the two onset classes as unique diseases. If this is the case, then the failure to detect a single predictive genetic model is consistent with two related, but different, diseases. This is exactly what would be expected in such a case of genetic heterogeneity (28). Using this approach, a major gene effect can be viewed as a necessary, but not sufficient, condition to explain the course of the disease. Analyzing the cases but omitting from the analysis of phenotype the necessary allele, in this case TTR V30M, can then reveal a variety of important modifiers that are distinct between the phenotypes.
The significant comparisons obtained in our study cohort indicate that the combined effects mainly result from two and three-locus interactions involving all loci except SAA1 and SAA2 for susceptibility to disease. A considerable number of four-site combinations modulate the age of onset with SAA1 appearing in a majority of significant combinations in late-onset disease, perhaps indicating a greater role of the SAA variants in the age of onset of FAP.
The correlation between genotype and phenotype in socalled simple Mendelian disorders is often incomplete, as only a subset of all mutations can reliably predict specific phenotypes (34). This is because non-allelic genetic variations and/or environmental influences underlie these disorders whose phenotypes behave as complex traits. A few examples include the identification of the role of homozygozity for the SAA1.1 allele in conferring the genetic susceptibility to renal amyloidosis in FMF (20) and the association of an insertion/deletion polymorphism in the ACE gene with disease severity in familial hypertrophic cardiomyopathy (35). In these disorders, the phenotypes arise from mutations in MEFV and b-MHC, but are modulated by independently inherited genetic variation. In this report, we show that interactions among multiple genes, whose products are confirmed or putative constituents of ATTR deposits, or metabolically interact with TTR, modulate the onset of the first symptoms and predispose individuals to disease in the presence of the V30M mutation in TTR. The exact nature of the effects identified here requires further study with potential application in the development of genetic screening with prognostic value pertaining to the onset of disease in the TTR V30M carriers.
If the effects of additional single or interacting genes dictate the heterogeneity of phenotype, as reflected in variability of onset and clinical expression (with the same TTR mutation), the products encoded by alleles at such loci could contribute to the process of wild-type TTR deposition in elderly individuals without a mutation (senile systemic amyloidosis), a phenomenon not readily recognized as having a genetic basis because of the insensitivity of family history in the elderly.
Safety and Efficacy of RNAi Therapy for Transthyretin Amyloidosis
Transthyretin amyloidosis is caused by the deposition of hepatocyte-derived transthyretin amyloid in peripheral nerves and the heart. A therapeutic approach mediated by RNA interference (RNAi) could reduce the production of transthyretin.
Methods We identified a potent antitransthyretin small interfering RNA, which was encapsulated in two distinct first- and second-generation formulations of lipid nanoparticles, generating ALN-TTR01 and ALN-TTR02, respectively. Each formulation was studied in a single-dose, placebo-controlled phase 1 trial to assess safety and effect on transthyretin levels. We first evaluated ALN-TTR01 (at doses of 0.01 to 1.0 mg per kilogram of body weight) in 32 patients with transthyretin amyloidosis and then evaluated ALN-TTR02 (at doses of 0.01 to 0.5 mg per kilogram) in 17 healthy volunteers.
Results Rapid, dose-dependent, and durable lowering of transthyretin levels was observed in the two trials. At a dose of 1.0 mg per kilogram, ALN-TTR01 suppressed transthyretin, with a mean reduction at day 7 of 38%, as compared with placebo (P=0.01); levels of mutant and nonmutant forms of transthyretin were lowered to a similar extent. For ALN-TTR02, the mean reductions in transthyretin levels at doses of 0.15 to 0.3 mg per kilogram ranged from 82.3 to 86.8%, with reductions of 56.6 to 67.1% at 28 days (P<0.001 for all comparisons). These reductions were shown to be RNAi mediated. Mild-to-moderate infusion-related reactions occurred in 20.8% and 7.7% of participants receiving ALN-TTR01 and ALN-TTR02, respectively.
ALN-TTR01 and ALN-TTR02 suppressed the production of both mutant and nonmutant forms of transthyretin, establishing proof of concept for RNAi therapy targeting messenger RNA transcribed from a disease-causing gene.
Alnylam May Seek Approval for TTR Amyloidosis Rx in 2017 as Other Programs Advance
Officials from Alnylam Pharmaceuticals last week provided updates on the two drug candidates from the company’s flagship transthyretin-mediated amyloidosis program, stating that the intravenously delivered agent patisiran is proceeding toward a possible market approval in three years, while a subcutaneously administered version called ALN-TTRsc is poised to enter Phase III testing before the end of the year.
Meanwhile, Alnylam is set to advance a handful of preclinical therapies into human studies in short order, including ones for complement-mediated diseases, hypercholesterolemia, and porphyria.
The officials made their comments during a conference call held to discuss Alnylam’s second-quarter financial results.
ATTR is caused by a mutation in the TTR gene, which normally produces a protein that acts as a carrier for retinol binding protein and is characterized by the accumulation of amyloid deposits in various tissues. Alnylam’s drugs are designed to silence both the mutant and wild-type forms of TTR.
Patisiran, which is delivered using lipid nanoparticles developed by Tekmira Pharmaceuticals, is currently in a Phase III study in patients with a form of ATTR called familial amyloid polyneuropathy (FAP) affecting the peripheral nervous system. Running at over 20 sites in nine countries, that study is set to enroll up to 200 patients and compare treatment to placebo based on improvements in neuropathy symptoms.
According to Alnylam Chief Medical Officer Akshay Vaishnaw, Alnylam expects to have final data from the study in two to three years, which would put patisiran on track for a new drug application filing in 2017.
Meanwhile, ALN-TTRsc, which is under development for a version of ATTR that affects cardiac tissue called familial amyloidotic cardiomyopathy (FAC) and uses Alnylam’s proprietary GalNAc conjugate delivery technology, is set to enter Phase III by year-end as Alnylam holds “active discussions” with US and European regulators on the design of that study, CEO John Maraganore noted during the call.
In the interim, Alnylam continues to enroll patients in a pilot Phase II study of ALN-TTRsc, which is designed to test the drug’s efficacy for FAC or senile systemic amyloidosis (SSA), a condition caused by the idiopathic accumulation of wild-type TTR protein in the heart.
Based on “encouraging” data thus far, Vaishnaw said that Alnylam has upped the expected enrollment in this study to 25 patients from 15. Available data from the trial is slated for release in November, he noted, stressing that “any clinical endpoint result needs to be considered exploratory given the small sample size and the very limited duration of treatment of only six weeks” in the trial.
Vaishnaw added that an open-label extension (OLE) study for patients in the ALN-TTRsc study will kick off in the coming weeks, allowing the company to gather long-term dosing tolerability and clinical activity data on the drug.
Enrollment in an OLE study of patisiran has been completed with 27 patients, he said, and, “as of today, with up to nine months of therapy … there have been no study drug discontinuations.” Clinical endpoint data from approximately 20 patients in this study will be presented at the American Neurological Association meeting in October.
As part of its ATTR efforts, Alnylam has also been conducting natural history of disease studies in both FAP and FAC patients. Data from the 283-patient FAP study was presented earlier this year and showed a rapid progression in neuropathy impairment scores and a high correlation of this measurement with disease severity.
During last week’s conference call, Vaishnaw said that clinical endpoint and biomarker data on about 400 patients with either FAC or SSA have already been collected in a nature history study on cardiac ATTR. Maraganore said that these findings would likely be released sometime next year.
The first medication for a rare and often fatal protein misfolding disorder has been approved in Europe. On November 16, the E gave a green light to Pfizer’s Vyndaqel (tafamidis) for treating transthyretin amyloidosis in adult patients with stage 1 polyneuropathy symptoms. [Jeffery Kelly, La Jolla]
The most clinically advanced RNA interference (RNAi) therapeutic achieved a milestone in April when Alnylam Pharmaceuticals in Cambridge, Massachusetts, reported positive results for patisiran, a small interfering RNA (siRNA) oligonucleotide targeting transthyretin for treating familial amyloidotic polyneuropathy (FAP). …
FAP is characterized by the systemic deposition of amyloidogenic variants of the transthyretin protein, especially in the peripheral nervous system, causing a progressive sensory and motor polyneuropathy.
FAP is caused by a mutation of the TTR gene, located on human chromosome 18q12.1-11.2.[5] A replacement of valine by methionine at position 30 (TTR V30M) is the mutation most commonly found in FAP.[1] The variant TTR is mostly produced by the liver.[citation needed] The transthyretin protein is a tetramer. ….
Proteome Sciences plc has strongly encouraged by two recent reports that emphasise the importance of protein profiling to improve outcomes in cancer treatment. These highlight the growing need for more detailed, personal assessment of protein profiles to improve the management of cancer treatment.
In the first study two groups from University College London and Cancer Research UK demonstrated that genetic mutations in cancer can lead to changes in the proteins on the cell surface1. These are new sequences which are seen as foreign by the body’s immune system and, with appropriate immunotherapy, the level of response in lung cancer was greatly enhanced.
However many of the patients with these types of mutations unfortunately still did not respond which highlighted the need for deeper analysis of the protein expression in tumours in order to better appreciate the mechanisms that contribute to treatment failure.
The second study, led by Professor Nigel Bundred of Manchester University, reported that use of two drugs that act on the same breast cancer target, an over-expressing protein called Her-2, were able to eradicate detectable tumours in around 10% of those treated in just 11 days, with 87% of those treated having a proteomic change indicating cells had stopped growing and/or cell death had increased2.
Whilst these results appear very promising it is worth noting that the over-expressing Her-2 target is only present in about 20% of breast tumours meaning this combination therapy was successful in clearing tumours in just 2% of the total breast cancer population.
Dr. Ian Pike, Chief Operating Officer of Proteome Sciences commented, “Both these recent studies should rightly be recognised as important steps forward towards better cancer treatment. However, in order to overcome the limitations of current drug therapy programs, a much deeper and more comprehensive analysis of the complex protein networks that regulate tumour growth and survival is required and will be essential to achieve a major advance in the battle to treat cancer.
“Our SysQuant® workflows provide that solution. As an example, in pancreatic cancer3 we have successfully mapped the complex network of regulatory processes and demonstrate the ability to devise personalised treatment combinations on an individual basis for each patient. A retrospective study with SysQuant® to predict response to the targeted drug Sorafenib in liver cancer is in process and we are planning further prospective trials to guide personalised treatment selection in liver cancer.
“We are already delivering systems-wide biology solutions through SysQuant® and TMTcalibrator™ programs to our clients that are generating novel biological data and results using more sensitive profiling that are helping them to better understand their drug development programs and to provide new biomarkers for tracking patient response in clinical trials.
“We are strongly positioned to deliver more comprehensive analysis of proteins and cellular pathways across other areas of disease and in particular to extend the use of SysQuant® with other leading cancer research groups in liver and other cancers.”
Proteome Sciences has also expanded its offering in personalised medicine through the use of its TMTcalibrator™ technology to uniquely identify protein biomarkers that reveal active cancer and other disease processes in body fluid samples. The importance of these ‘mechanistic’ biomarkers is that they are essential to monitor that drugs are being effective and that they can be used as early biomarkers of disease recurrence.
Using SysQuant® and TMTcalibrator™, Proteome Sciences can deliver more comprehensive analysis and provide unparalleled levels of sensitivity and breadth of coverage of the proteome, enabling faster, more efficient drug development and more accurate disease diagnosis.
Discovering ‘Outlier’ Enzymes
Researchers at TSRI and Salk Institute have discovered ‘Outlier’ enzymes that could offer new targets to treat type 2 diabetes and inflammatory disorders.
A team led by scientists at The Scripps Research Institute (TSRI) and the Salk Institute for Biological Studies have discovered two enzymes that appear to play a role in metabolism and inflammation—and might someday be targeted with drugs to treat type 2 diabetes and inflammatory disorders. The discovery is unusual because the enzymes do not bear a resemblance—in their structures or amino-acid sequences—to any known class of enzymes.
The team of scientists nevertheless identified them as “outlier” members of the serine/threonine hydrolase class, using newer techniques that detect biochemical activity. “A huge fraction of the human ‘proteome’ remains uncharacterized, and this paper shows how chemical approaches can be used to uncover proteins of a given functionality that have eluded classification based on sequence or predicted structure,” said co-senior author Benjamin F. Cravatt, chair of TSRI’s Department of Chemical Physiology.
“In this study, we found two genes that control levels of lipids with anti-diabetic and anti-inflammatory activity, suggesting exciting targets for diabetes and inflammatory diseases,” said co-senior author Alan Saghatelian, who holds the Dr. Frederik Paulsen Chair at the Salk Institute. The study, which appeared as a Nature Chemical Biology Advance Online Publication on March 28, 2016, began as an effort in the Cravatt laboratory to discover and characterize new serine/threonine hydrolases using fluorophosphonate (FP) probes—molecules that selectively bind and, in effect, label the active sites of these enzymes.
Pulling FP-binding proteins out of the entire proteome of test cells and identifying them using mass spectrometry techniques, the team matched nearly all to known hydrolases. The major outlier was a protein called androgen-induced gene 1 protein (AIG1). The only other one was a distant cousin in terms of sequence, a protein called ADTRP. “Neither of these proteins had been characterized as an enzyme; in fact, there had been little functional characterization of them at all,” said William H. Parsons, a research associate in the Cravatt laboratory who was co-first author of the study.
Experiments on AIG1 and ADTRP revealed that they do their enzymatic work in a unique way. “It looks like they have an active site that is novel—it had never been described in the literature,” said Parsons. Initial tests with panels of different enzyme inhibitors showed that AIG1 and ADTRP are moderately inhibited by inhibitors of lipases—enzymes that break down fats and other lipids. But on what specific lipids do these newly discovered outlier enzymes normally work?
At the Salk Institute, the Saghatelian laboratory was investigating a class of lipids it had discovered in 2014. Known as fatty acid esters of hydroxy fatty acids (FAHFAs), these molecules showed strong therapeutic potential. Saghatelian and his colleagues had found that boosting the levels of one key FAHFA lipid normalizes glucose levels in diabetic mice and also reduces inflammation.
“[Ben Cravatt’s] lab was screening panels of lipids to find the ones that their new enzymes work on,” said Saghatelian, who is a former research associate in the Cravatt laboratory. “We suggested they throw FAHFAs in there—and these turned out to be very good substrates.” The Cravatt laboratory soon developed powerful inhibitors of the newly discovered enzymes, and the two labs began working together, using the inhibitors and genetic techniques to explore the enzymes’ functions in vitro and in cultured cells.
Co-first author Matthew J. Kolar, an MD-PhD student, performed most of the experiments in the Saghatelian lab. The team concluded that AIG1 and ADTRP, at least in the cell types tested, appear to work mainly to break down FAHFAs and not any other major class of lipid. In principle, inhibitors of AIG1 and ADTRP could be developed into FAHFA-boosting therapies.
“Our prediction,” said Saghatelian, “is that if FAHFAs do what we think they’re doing, then using an enzyme inhibitor to block their degradation would make FAHFA levels go up and should thus reduce inflammation as well as improve glucose levels and insulin sensitivity.” The two labs are now collaborating on further studies of the new enzymes—and the potential benefits of inhibiting them—in mouse models of diabetes, inflammation and autoimmune disease.
“One of the neat things this study shows,” said Cravatt, “is that even for enzyme classes as well studied as the hydrolases, there may still be hidden members that, presumably by convergent evolution, arrived at that basic enzyme mechanism despite sharing no sequence or structural homology.”
Other co-authors of the study, “AIG1 and ADTRP are atypical integral membrane hydrolases that degrade bioactive FAHFAs,” were Siddhesh S. Kamat, Armand B. Cognetta III, Jonathan J. Hulce and Enrique Saez, of TSRI; and co-senior author Barbara B. Kahn of Beth Israel Deaconess Medical Center and Harvard Medical School
New Weapon Against Breast Cancer
Molecular marker in healthy tissue can predict a woman’s risk of getting the disease, research says.
Harvard Stem Cell Institute (HSCI) researchers at Dana-Farber Cancer Institute (DFCI) and collaborators at Brigham and Women’s Hospital (BWH) have identified a molecular marker in normal breast tissue that can predict a woman’s risk for developing breast cancer, the leading cause of death in women with cancer worldwide.
The work, led by HSCI principal faculty member Kornelia Polyak and Rulla Tamimi of BWH, was published in an early online release and in the April 1 issue of Cancer Research.
The study builds on Polyak’s earlier research finding that women already identified as having a high risk of developing cancer — namely those with a mutation called BRCA1 or BRCA2 — or women who did not give birth before their 30s had a higher number of mammary gland progenitor cells.
In the latest study, Polyak, Tamimi, and their colleagues examined biopsies, some taken as many as four decades ago, from 302 participants in the Nurses’ Health Study and the Nurses’ Health Study II who had been diagnosed with benign breast disease. The researchers compared tissue from the 69 women who later developed cancer to the tissue from the 233 women who did not. They found that women were five times as likely to develop cancer if they had a higher percentage of Ki67, a molecular marker that identifies proliferating cells, in the cells that line the mammary ducts and milk-producing lobules. These cells, called the mammary epithelium, undergo drastic changes throughout a woman’s life, and the majority of breast cancers originate in these tissues.
Doctors already test breast tumors for Ki67 levels, which can inform decisions about treatment, but this is the first time scientists have been able to link Ki67 to precancerous tissue and use it as a predictive tool.
“Instead of only telling women that they don’t have cancer, we could test the biopsies and tell women if they were at high risk or low risk for developing breast cancer in the future,” said Polyak, a breast cancer researcher at Dana-Farber and co-senior author of the paper.
“Currently, we are not able to do a very good job at distinguishing women at high and low risk of breast cancer,” added co-senior author Tamimi, an associate professor at the Harvard T.H. Chan School of Public Health and Harvard Medical School. “By identifying women at high risk of breast cancer, we can better develop individualized screening and also target risk reducing strategies.”
To date, mammograms are the best tool for the early detection, but there are risks associated with screening. False positive and negative results and over-diagnosis could cause psychological distress, delay treatment, or lead to overtreatment, according to the National Cancer Institute (NCI).
Mammography machines also use low doses of radiation. While a single mammogram is unlikely to cause harm, repeated screening can potentially cause cancer, though the NCI writes that the benefits “nearly always outweigh the risks.”
“If we can minimize unnecessary radiation for women at low risk, that would be good,” said Tamimi.
Screening for Ki67 levels would “be easy to apply in the current setting,” said Polyak, though the researchers first want to reproduce the results in an independent cohort of women.
AIG1 and ADTRP are atypical integral membrane hydrolases that degrade bioactive FAHFAs
Enzyme classes may contain outlier members that share mechanistic, but not sequence or structural, relatedness with more common representatives. The functional annotation of such exceptional proteins can be challenging. Here, we use activity-based profiling to discover that the poorly characterized multipass transmembrane proteins AIG1 and ADTRP are atypical hydrolytic enzymes that depend on conserved threonine and histidine residues for catalysis. Both AIG1 and ADTRP hydrolyze bioactive fatty acid esters of hydroxy fatty acids (FAHFAs) but not other major classes of lipids. We identify multiple cell-active, covalent inhibitors of AIG1 and show that these agents block FAHFA hydrolysis in mammalian cells. These results indicate that AIG1 and ADTRP are founding members of an evolutionarily conserved class of transmembrane threonine hydrolases involved in bioactive lipid metabolism. More generally, our findings demonstrate how chemical proteomics can excavate potential cases of convergent or parallel protein evolution that defy conventional sequence- and structure-based predictions.
Figure 1: Discovery and characterization of AIG1 and ADTRP as FP-reactive proteins in the human proteome.
(a) Competitive ABPP-SILAC analysis to identify FP-alkyne-inhibited proteins, in which protein enrichment and inhibition were measured in proteomic lysates from SKOV3 cells treated with FP-alkyne (20 μM, 1 h) or DMSO using the FP-biotin…
Willems, L.I., Overkleeft, H.S. & van Kasteren, S.I.Current developments in activity-based protein profiling. Bioconjug. Chem.25, 1181–1191 (2014).
Berger, A.B., Vitorino, P.M. & Bogyo, M.Activity-based protein profiling: applications to biomarker discovery, in vivo imaging and drug discovery. Am. J. Pharmacogenomics4,371–381 (2004).
Simon, G.M. & Cravatt, B.F.Activity-based proteomics of enzyme superfamilies: serine hydrolases as a case study. J. Biol. Chem.285, 11051–11055 (2010).
National Institutes of Health (NIH)-sponsored screening centers provide academic researchers with a special opportunity to pursue small-molecule probes for protein targets that are outside the current interest of, or beyond the standard technologies employed by, the pharmaceutical industry. Here, we describe the outcome of an inhibitor screen for one such target, the enzyme protein phosphatase methylesterase-1 (PME-1), which regulates the methylesterification state of protein phosphatase 2A (PP2A) and is implicated in cancer and neurodegeneration. Inhibitors of PME-1 have not yet been described, which we attribute, at least in part, to a dearth of substrate assays compatible with high-throughput screening. We show that PME-1 is assayable by fluorescence polarization-activity-based protein profiling (fluopol-ABPP) and use this platform to screen the 300,000+ member NIH small-molecule library. This screen identified an unusual class of compounds, the aza-β-lactams (ABLs), as potent (IC50 values of approximately 10 nM), covalent PME-1 inhibitors. Interestingly, ABLs did not derive from a commercial vendor but rather an academic contribution to the public library. We show using competitive-ABPP that ABLs are exquisitely selective for PME-1 in living cells and mice, where enzyme inactivation leads to substantial reductions in demethylated PP2A. In summary, we have combined advanced synthetic and chemoproteomic methods to discover a class of ABL inhibitors that can be used to selectively perturb PME-1 activity in diverse biological systems. More generally, these results illustrate how public screening centers can serve as hubs to create spontaneous collaborative opportunities between synthetic chemistry and chemical biology labs interested in creating first-in-class pharmacological probes for challenging protein targets.
Protein phosphorylation is a pervasive and dynamic posttranslational protein modification in eukaryotic cells. In mammals, more than 500 protein kinases catalyze the phosphorylation of serine, threonine, and tyrosine residues on proteins (1). A much more limited number of phosphatases are responsible for reversing these phosphorylation events (2). For instance, protein phosphatase 2A (PP2A) and PP1 are thought to be responsible together for > 90% of the total serine/threonine phosphatase activity in mammalian cells (3). Specificity is imparted on PP2A activity by multiple mechanisms, including dynamic interactions between the catalytic subunit (C) and different protein-binding partners (B subunits), as well as a variety of posttranslational chemical modifications (2, 4). Within the latter category is an unusual methylesterification event found at the C terminus of the catalytic subunit of PP2A that is introduced and removed by a specific methyltransferase (leucine carbxoylmethyltransferase-1 or LCMT1) (5, 6) and methylesterase (protein phosphatase methylesterase-1 or PME-1) (7), respectively (Fig. 1A). PP2A carboxymethylation (hereafter referred to as “methylation”) has been proposed to regulate PP2A activity, at least in part, by modulating the binding interaction of the C subunit with various regulatory B subunits (8–10). A predicted outcome of these shifts in subunit association is the targeting of PP2A to different protein substrates in cells. PME-1 has also been hypothesized to stabilize inactive forms of nuclear PP2A (11), and recent structural studies have shed light on the physical interactions between PME-1 and the PP2A holoenzyme (12).
There were several keys to the success of our probe development effort. First, screening for inhibitors of PME-1 benefited from the fluopol-ABPP technology, which circumvented the limited throughput of previously described substrate assays for this enzyme. Second, we were fortunate that the NIH compound library contained several members of the ABL class of small molecules. These chiral compounds, which represent an academic contribution to the NIH library, occupy an unusual portion of structural space that is poorly accessed by commercial compound collections. Although at the time of their original synthesis (23) it may not have been possible to predict whether these ABLs would show specific biological activity, their incorporation into the NIH library provided a forum for screening against many proteins and cellular targets, culminating in their identification as PME-1 inhibitors. We then used advanced chemoproteomic assays to confirm the remarkable selectivity displayed by ABLs for PME-1 across (and beyond) the serine hydrolase superfamily. That the mechanism for PME-1 inhibition involves acylation of the enzyme’s conserved serine nucleophile (Fig. 3) suggests that exploration of a more structurally diverse set of ABLs might uncover inhibitors for other serine hydrolases. In this way, the chemical information gained from a single high-throughput screen may be leveraged to initiate probe development programs for additional enzyme targets.
Projecting forward, this research provides an example of how public small-molecule screening centers can serve as a portal for spawning academic collaborations between chemical biology and synthetic chemistry labs. By continuing to develop versatile high-throughput screens and combining them with a small-molecule library of expanding structural diversity conferred by advanced synthetic methodologies, academic biologists and chemists are well-positioned to collaboratively deliver pharmacological probes for a wide range of proteins and pathways in cell biology.
The frequency and proliferative activity of tissue-specific stem and progenitor cells are suggested to correlate with cancer risk. In this study, we investigated the association between breast cancer risk and the frequency of mammary epithelial cells expressing p27, estrogen receptor (ER), and Ki67 in normal breast tissue. We performed a nested case-control study of 302 women (69 breast cancer cases, 233 controls) who had been initially diagnosed with benign breast disease according to the Nurses’ Health Studies. Immunofluorescence for p27, ER, and Ki67 was performed on tissue microarrays constructed from benign biopsies containing normal mammary epithelium and scored by computational image analysis. We found that the frequency of Ki67+ cells was positively associated with breast cancer risk among premenopausal women (odds ratio [OR]=10.1, 95% confidence interval [CI]=2.12-48.0). Conversely, the frequency of ER+ or p27+ cells was inversely, but not significantly, associated with subsequent breast cancer risk (ER+: OR=0.70, 95% CI=0.33-1.50; p27+: OR=0.89, 95% CI=0.45-1.75). Notably, high Ki67+/low p27+ and high Ki67+/low ER+ cell frequencies were significantly associated with a 5-fold higher risk of breast cancer compared to low Ki67+/low p27+ and low Ki67+/low ER+ cell frequencies, respectively, among premenopausal women (Ki67hi/p27lo: OR=5.08, 95% CI=1.43-18.1; Ki67hi/ERlo: OR=4.68, 95% CI=1.63-13.5). Taken together, our data suggest that the fraction of actively cycling cells in normal breast tissue may represent a marker for breast cancer risk assessment, which may therefore impact the frequency of screening procedures in at-risk women.
Scientists have discovered a group of six proteins that may help to divulge secrets of how we age, potentially unlocking new insights into diabetes, Alzheimer’s, cancer, and other aging-related diseases.
The proteins appear to play several roles in our bodies’ cells, from decreasing the amount of damaging free radicals and controlling the rate at which cells die to boosting metabolism and helping tissues throughout the body respond better to insulin. The naturally occurring amounts of each protein decrease with age, leading investigators to believe that they play an important role in the aging process and the onset of diseases linked to older age.
The research team led by Pinchas Cohen, M.D., dean and professor of the University of Southern California Leonard Davis School of Gerontology, identified the proteins and observed their origin from mitochondria and their game-changing roles in metabolism and cell survival. This latest finding builds upon prior research by Dr. Cohen and his team that uncovered two significant proteins, humanin and MOTS-c, hormones that appear to have significant roles in metabolism and diseases of aging.
Unlike most other proteins, humanin and MOTS-c are encoded in mitochondria. Dr. Cohen’s team used computer analysis to see if the part of the mitochondrial genome that provides the code for humanin was coding for other proteins as well. The analysis uncovered the genes for six new proteins, which were dubbed small humanin-like peptides, or SHLPs, 1 through 6 (pronounced “schlep”).
After identifying the six SHLPs and successfully developing antibodies to test for several of them, the team examined both mouse tissues and human cells to determine their abundance in different organs as well as their functions. The proteins were distributed quite differently among organs, which suggests that the proteins have varying functions based on where they are in the body. Of particular interest is SHLP 2, according to Dr. Cohen. The protein appears to have insulin-sensitizing, antidiabetic effects as well as neuroprotective activity that may emerge as a strategy to combat Alzheimer’s disease. He added that SHLP 6 is also intriguing, with a unique ability to promote cancer cell death and thus potentially target malignant diseases.
Proteins That May Protect Against Age Related Illnesses Discovered
Tested in both mice and human cells, the proteins may lead to greater understanding of aging-related diseases, from diabetes to Alzheimer’s to cancer, according to researchers at USC Davis School of Gerontology who led the study.
Researchers have identified a group of six proteins that they believe may divulge secrets of how we age and unlock new insights into diabetes, Alzheimer’s, cancer, and other aging-related diseases.
Nicknamed “Schlep” or “SHLP” for “small humanin-like peptides,” these proteins appear to play several big roles in our bodies’ cells, from decreasing the amount of damaging free radicals and controlling the rate at which cells die, to boosting metabolism and helping tissues throughout the body respond better to insulin. The naturally occurring amounts of each protein decrease with age, leading researchers to believe that they play an important role in the aging process and the onset of age-related diseases.
The research team led by Pinchas Cohen, dean and professor of the USC Leonard Davis School of Gerontology, identified the tiny proteins for the first time and observed their surprising origin: organelles in the cell called mitochondria and their game-changing roles in metabolism and cell survival.
This latest finding builds upon prior research by Cohen and his team that uncovered two significant proteins, humanin and MOTS-c, hormones that appear to have significant roles in metabolism and diseases of aging.
The cell proliferation antigen Ki-67 organises heterochromatin
MichalSobecki,
KarimMrouj
Montpellier Institute of Molecular Genetics (IGMM) CNRS UMR 5535, Centre National de la Recherche Scientifique (CNRS), Montpellier, France; Faculty of Sciences, University of Montpellier, Montpellier, France
Contribution: Acquisition of data, Analysis and interpretation of data, Drafting or revising the article
No competing interests declared
</div>”>KarimMrouj,
AlainCamasses
Montpellier Institute of Molecular Genetics (IGMM) CNRS UMR 5535, Centre National de la Recherche Scientifique (CNRS), Montpellier, France; Faculty of Sciences, University of Montpellier, Montpellier, France
Contribution: Conception and design, Acquisition of data, Analysis and interpretation of data
No competing interests declared
</div>”>AlainCamasses,
NikolaosParisis
Montpellier Institute of Molecular Genetics (IGMM) CNRS UMR 5535, Centre National de la Recherche Scientifique (CNRS), Montpellier, France; Faculty of Sciences, University of Montpellier, Montpellier, France
Contribution: Conception and design, Acquisition of data, Analysis and interpretation of data, Drafting or revising the article
RNA Molecular Biology, Center for Microscopy and Molecular Imaging, Fonds de la Recherche Nationale, Université Libre de Bruxelles, Charleroi-Gosselies, Belgium
Contribution: Conception and design, Acquisition of data, Analysis and interpretation of data
Antigen Ki-67 is a nuclear protein expressed in proliferating mammalian cells. It is widely used in cancer histopathology but its functions remain unclear. Here, we show that Ki-67 controls heterochromatin organisation. Altering Ki-67 expression levels did not significantly affect cell proliferation in vivo. Ki-67 mutant mice developed normally and cells lacking Ki-67 proliferated efficiently. Conversely, upregulation of Ki-67 expression in differentiated tissues did not prevent cell cycle arrest. Ki-67 interactors included proteins involved in nucleolar processes and chromatin regulators. Ki-67 depletion disrupted nucleologenesis but did not inhibit pre-rRNA processing. In contrast, it altered gene expression. Ki-67 silencing also had wide-ranging effects on chromatin organisation, disrupting heterochromatin compaction and long-range genomic interactions. Trimethylation of histone H3K9 and H4K20 was relocalised within the nucleus. Finally, overexpression of human or Xenopus Ki-67 induced ectopic heterochromatin formation. Altogether, our results suggest that Ki-67 expression in proliferating cells spatially organises heterochromatin, thereby controlling gene expression.
A protein called Ki-67 is only produced in actively dividing cells, where it is located in the nucleus – the structure that contains most of the cell’s DNA. Researchers often use Ki-67 as a marker to identify which cells are actively dividing in tissue samples from cancer patients, and previous studies indicated that Ki-67 is needed for cells to divide. However, the exact role of this protein was not clear. Before cells can divide they need to make large amounts of new proteins using molecular machines called ribosomes and it has been suggested that Ki-67 helps to produce ribosomes.
Now, Sobecki et al. used genetic techniques to study the role of Ki-67 in mice. The experiments show that Ki-67 is not required for cells to divide in the laboratory or to make ribosomes. Instead, Ki-67 alters the way that DNA is packaged in the nucleus. Loss of Ki-67 from mice cells resulted in DNA becoming less compact, which in turn altered the activity of genes in those cells.
Genomics and epigenetics link to DNA structure, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 1: Next Generation Sequencing (NGS)
Genomics and epigenetics link to DNA structure
Larry H. Bernstein, MD, FCAP, Curator
LPBI
Sequence and Epigenetic Factors Determine Overall DNA Structure
Atomic-level simulations show electrostatic forces between each atom. [Alek Aksimentiev, University of Illinois at Urbana-Champaign]
The traditionally held hypothesis about the highly ordered organization of DNA describes the interaction of various proteins with DNA sequences to mediate the dynamic structure of the molecule. However, recent evidence has emerged that stretches of homologous DNA sequences can associate preferentially with one another, even in the absence of proteins.
Researchers at the University of Illinois Center for the Physics of Living Cells, Johns Hopkins University, and Ulsan National Institute of Science and Technology (UNIST) in South Korea found that DNA molecules interact directly with one another in ways that are dependent on the sequence of the DNA and epigenetic factors, such as methylation.
The researchers described evidence they found for sequence-dependent attractive interactions between double-stranded DNA molecules that neither involve intermolecular strand exchange nor are mediated by DNA-binding proteins.
“DNA molecules tend to repel each other in water, but in the presence of special types of cations, they can attract each other just like nuclei pulling each other by sharing electrons in between,” explained lead study author Hajin Kim, Ph.D., assistant professor of biophysics at UNIST. “Our study suggests that the attractive force strongly depends on the nucleic acid sequence and also the epigenetic modifications.”
The investigators used atomic-level supercomputer simulations to measure the forces between a pair of double-stranded DNA helices and proposed that the distribution of methyl groups on the DNA was the key to regulating this sequence-dependent attraction. To verify their findings experimentally, the scientists were able to observe a single pair of DNA molecules within nanoscale bubbles.
“Here we combine molecular dynamics simulations with single-molecule fluorescence resonance energy transfer experiments to examine the interactions between duplex DNA in the presence of spermine, a biological polycation,” the authors wrote. “We find that AT-rich DNA duplexes associate more strongly than GC-rich duplexes, regardless of the sequence homology. Methyl groups of thymine act as a steric block, relocating spermine from major grooves to interhelical regions, thereby increasing DNA–DNA attraction.”
The findings from this study were published recently in Nature Communications in an article entitled “Direct Evidence for Sequence-Dependent Attraction Between Double-Stranded DNA Controlled by Methylation.”
After conducting numerous further simulations, the research team concluded that direct DNA–DNA interactions could play a central role in how chromosomes are organized in the cell and which ones are expanded or folded up compactly, ultimately determining functions of different cell types or regulating the cell cycle.
“Biophysics is a fascinating subject that explores the fundamental principles behind a variety of biological processes and life phenomena,” Dr. Kim noted. “Our study requires cross-disciplinary efforts from physicists, biologists, chemists, and engineering scientists and we pursue the diversity of scientific disciplines within the group.”
Dr. Kim concluded by stating that “in our lab, we try to unravel the mysteries within human cells based on the principles of physics and the mechanisms of biology. In the long run, we are seeking for ways to prevent chronic illnesses and diseases associated with aging.”
Direct evidence for sequence-dependent attraction between double-stranded DNA controlled by methylation
Jejoong Yoo, Hajin Kim, Aleksei Aksimentiev, and Taekjip Ha Nature Communications 7 11045 (2016) DOI:10.1038/ncomms11045BibTex
Although proteins mediate highly ordered DNA organization in vivo, theoretical studies suggest that homologous DNA duplexes can preferentially associate with one another even in the absence of proteins. Here we combine molecular dynamics simulations with single-molecule fluorescence resonance energy transfer experiments to examine the interactions between duplex DNA in the presence of spermine, a biological polycation. We find that AT-rich DNA duplexes associate more strongly than GC-rich duplexes, regardless of the sequence homology. Methyl groups of thymine acts as a steric block, relocating spermine from major grooves to interhelical regions, thereby increasing DNA–DNA attraction. Indeed, methylation of cytosines makes attraction between GC-rich DNA as strong as that between AT-rich DNA. Recent genome-wide chromosome organization studies showed that remote contact frequencies are higher for AT-rich and methylated DNA, suggesting that direct DNA–DNA interactions that we report here may play a role in the chromosome organization and gene regulation.
Formation of a DNA double helix occurs through Watson–Crick pairing mediated by the complementary hydrogen bond patterns of the two DNA strands and base stacking. Interactions between double-stranded (ds)DNA molecules in typical experimental conditions containing mono- and divalent cations are repulsive1, but can turn attractive in the presence of high-valence cations2. Theoretical studies have identified the ion–ion correlation effect as a possible microscopic mechanism of the DNA condensation phenomena3, 4, 5. Theoretical investigations have also suggested that sequence-specific attractive forces might exist between two homologous fragments of dsDNA6, and this ‘homology recognition’ hypothesis was supported by in vitro atomic force microscopy7 and in vivo point mutation assays8. However, the systems used in these measurements were too complex to rule out other possible causes such as Watson–Crick strand exchange between partially melted DNA or protein-mediated association of DNA.
Here we present direct evidence for sequence-dependent attractive interactions between dsDNA molecules that neither involve intermolecular strand exchange nor are mediated by proteins. Further, we find that the sequence-dependent attraction is controlled not by homology—contradictory to the ‘homology recognition’ hypothesis6—but by a methylation pattern. Unlike the previous in vitro study that used monovalent (Na+) or divalent (Mg2+) cations7, we presumed that for the sequence-dependent attractive interactions to operate polyamines would have to be present. Polyamine is a biological polycation present at a millimolar concentration in most eukaryotic cells and essential for cell growth and proliferation9, 10. Polyamines are also known to condense DNA in a concentration-dependent manner2, 11. In this study, we use spermine4+(Sm4+) that contains four positively charged amine groups per molecule.
Sequence dependence of DNA–DNA forces
To characterize the molecular mechanisms of DNA–DNA attraction mediated by polyamines, we performed molecular dynamics (MD) simulations where two effectively infinite parallel dsDNA molecules, 20 base pairs (bp) each in a periodic unit cell, were restrained to maintain a prescribed inter-DNA distance; the DNA molecules were free to rotate about their axes. The two DNA molecules were submerged in 100mM aqueous solution of NaCl that also contained 20 Sm4+molecules; thus, the total charge of Sm4+, 80 e, was equal in magnitude to the total charge of DNA (2 × 2 × 20 e, two unit charges per base pair; Fig. 1a). Repeating such simulations at various inter-DNA distances and applying weighted histogram analysis12 yielded the change in the interaction free energy (ΔG) as a function of the DNA–DNA distance (Fig. 1b,c). In a broad agreement with previous experimental findings13, ΔG had a minimum, ΔGmin, at the inter-DNA distance of 25−30Å for all sequences examined, indeed showing that two duplex DNA molecules can attract each other. The free energy of inter-duplex attraction was at least an order of magnitude smaller than the Watson–Crick interaction free energy of the same length DNA duplex. A minimum of ΔG was not observed in the absence of polyamines, for example, when divalent or monovalent ions were used instead14, 15.
Figure 1: Polyamine-mediated DNA sequence recognition observed in MD simulations and smFRET experiments.
(a) Set-up of MD simulations. A pair of parallel 20-bp dsDNA duplexes is surrounded by aqueous solution (semi-transparent surface) containing 20 Sm4+ molecules (which compensates exactly the charge of DNA) and 100mM NaCl. Under periodic boundary conditions, the DNA molecules are effectively infinite. A harmonic potential (not shown) is applied to maintain the prescribed distance between the dsDNA molecules. (b,c) Interaction free energy of the two DNA helices as a function of the DNA–DNA distance for repeat-sequence DNA fragments (b) and DNA homopolymers (c). (d) Schematic of experimental design. A pair of 120-bp dsDNA labelled with a Cy3/Cy5 FRET pair was encapsulated in a ~200-nm diameter lipid vesicle; the vesicles were immobilized on a quartz slide through biotin–neutravidin binding. Sm4+ molecules added after immobilization penetrated into the porous vesicles. The fluorescence signals were measured using a total internal reflection microscope. (e) Typical fluorescence signals indicative of DNA–DNA binding. Brief jumps in the FRET signal indicate binding events. (f) The fraction of traces exhibiting binding events at different Sm4+ concentrations for AT-rich, GC-rich, AT nonhomologous and CpG-methylated DNA pairs. The sequence of the CpG-methylated DNA specifies the methylation sites (CG sequence, orange), restriction sites (BstUI, triangle) and primer region (underlined). The degree of attractive interaction for the AT nonhomologous and CpG-methylated DNA pairs was similar to that of the AT-rich pair. All measurements were done at [NaCl]=50mM and T=25°C. (g) Design of the hybrid DNA constructs: 40-bp AT-rich and 40-bp GC-rich regions were flanked by 20-bp common primers. The two labelling configurations permit distinguishing parallel from anti-parallel orientation of the DNA. (h) The fraction of traces exhibiting binding events as a function of NaCl concentration at fixed concentration of Sm4+ (1mM). The fraction is significantly higher for parallel orientation of the DNA fragments.
Unexpectedly, we found that DNA sequence has a profound impact on the strength of attractive interaction. The absolute value of ΔG at minimum relative to the value at maximum separation, |ΔGmin|, showed a clearly rank-ordered dependence on the DNA sequence: |ΔGmin| of (A)20>|ΔGmin| of (AT)10>|ΔGmin| of (GC)10>|ΔGmin| of (G)20. Two trends can be noted. First, AT-rich sequences attract each other more strongly than GC-rich sequences16. For example, |ΔGmin| of (AT)10 (1.5kcalmol−1 per turn) is about twice |ΔGmin| of (GC)10 (0.8kcalmol−1 per turn) (Fig. 1b). Second, duplexes having identical AT content but different partitioning of the nucleotides between the strands (that is, (A)20 versus (AT)10 or (G)20 versus (GC)10) exhibit statistically significant differences (~0.3kcalmol−1 per turn) in the value of |ΔGmin|.
To validate the findings of MD simulations, we performed single-molecule fluorescence resonance energy transfer (smFRET)17 experiments of vesicle-encapsulated DNA molecules. Equimolar mixture of donor- and acceptor-labelled 120-bp dsDNA molecules was encapsulated in sub-micron size, porous lipid vesicles18 so that we could observe and quantitate rare binding events between a pair of dsDNA molecules without triggering large-scale DNA condensation2. Our DNA constructs were long enough to ensure dsDNA–dsDNA binding that is stable on the timescale of an smFRET measurement, but shorter than the DNA’s persistence length (~150bp (ref. 19)) to avoid intramolecular condensation20. The vesicles were immobilized on a polymer-passivated surface, and fluorescence signals from individual vesicles containing one donor and one acceptor were selectively analysed (Fig. 1d). Binding of two dsDNA molecules brings their fluorescent labels in close proximity, increasing the FRET efficiency (Fig. 1e).
FRET signals from individual vesicles were diverse. Sporadic binding events were observed in some vesicles, while others exhibited stable binding; traces indicative of frequent conformational transitions were also observed (Supplementary Fig. 1A). Such diverse behaviours could be expected from non-specific interactions of two large biomolecules having structural degrees of freedom. No binding events were observed in the absence of Sm4+ (Supplementary Fig. 1B) or when no DNA molecules were present. To quantitatively assess the propensity of forming a bound state, we chose to use the fraction of single-molecule traces that showed any binding events within the observation time of 2min (Methods). This binding fraction for the pair of AT-rich dsDNAs (AT1, 100% AT in the middle 80-bp section of the 120-bp construct) reached a maximum at ~2mM Sm4+(Fig. 1f), which is consistent with the results of previous experimental studies2, 3. In accordance with the prediction of our MD simulations, GC-rich dsDNAs (GC1, 75% GC in the middle 80bp) showed much lower binding fraction at all Sm4+ concentrations (Fig. 1b,c). Regardless of the DNA sequence, the binding fraction reduced back to zero at high Sm4+ concentrations, likely due to the resolubilization of now positively charged DNA–Sm4+ complexes2, 3, 13.
Because the donor and acceptor fluorophores were attached to the same sequence of DNA, it remained possible that the sequence homology between the donor-labelled DNA and the acceptor-labelled DNA was necessary for their interaction6. To test this possibility, we designed another AT-rich DNA construct AT2 by scrambling the central 80-bp section of AT1 to remove the sequence homology (Supplementary Table 1). The fraction of binding traces for this nonhomologous pair of donor-labelled AT1 and acceptor-labelled AT2 was comparable to that for the homologous AT-rich pair (donor-labelled AT1 and acceptor-labelled AT1) at all Sm4+ concentrations tested (Fig. 1f). Furthermore, this data set rules out the possibility that the higher binding fraction observed experimentally for the AT-rich constructs was caused by inter-duplex Watson–Crick base pairing of the partially melted constructs.
Next, we designed a DNA construct named ATGC, containing, in its middle section, a 40-bp AT-rich segment followed by a 40-bp GC-rich segment (Fig. 1g). By attaching the acceptor to the end of either the AT-rich or GC-rich segments, we could compare the likelihood of observing the parallel binding mode that brings the two AT-rich segments together and the anti-parallel binding mode. Measurements at 1mM Sm4+ and 25 or 50mM NaCl indicated a preference for the parallel binding mode by ~30% (Fig. 1h). Therefore, AT content can modulate DNA–DNA interactions even in a complex sequence context. Note that increasing the concentration of NaCl while keeping the concentration of Sm4+ constant enhances competition between Na+ and Sm4+ counterions, which reduces the concentration of Sm4+ near DNA and hence the frequency of dsDNA–dsDNA binding events (Supplementary Fig. 2).
Methylation determines the strength of DNA–DNA attraction
Analysis of the MD simulations revealed the molecular mechanism of the polyamine-mediated sequence-dependent attraction (Fig. 2). In the case of the AT-rich fragments, the bulky methyl group of thymine base blocks Sm4+ binding to the N7 nitrogen atom of adenine, which is the cation-binding hotspot21, 22. As a result, Sm4+ is not found in the major grooves of the AT-rich duplexes and resides mostly near the DNA backbone (Fig. 2a,d). Such relocated Sm4+ molecules bridge the two DNA duplexes better, accounting for the stronger attraction16, 23, 24, 25. In contrast, significant amount of Sm4+ is adsorbed to the major groove of the GC-rich helices that lacks cation-blocking methyl group (Fig. 2b,e).
Figure 2: Molecular mechanism of polyamine-mediated DNA sequence recognition.
(a–c) Representative configurations of Sm4+ molecules at the DNA–DNA distance of 28Å for the (AT)10–(AT)10 (a), (GC)10–(GC)10 (b) and (GmC)10–(GmC)10 (c) DNA pairs. The backbone and bases of DNA are shown as ribbon and molecular bond, respectively; Sm4+ molecules are shown as molecular bonds. Spheres indicate the location of the N7 atoms and the methyl groups. (d–f) The average distributions of cations for the three sequence pairs featured in a–c. Top: density of Sm4+ nitrogen atoms (d=28Å) averaged over the corresponding MD trajectory and the z axis. White circles (20Å in diameter) indicate the location of the DNA helices. Bottom: the average density of Sm4+ nitrogen (blue), DNA phosphate (black) and sodium (red) atoms projected onto the DNA–DNA distance axis (x axis). The plot was obtained by averaging the corresponding heat map data over y=[−10, 10] Å. See Supplementary Figs 4 and 5 for the cation distributions at d=30, 32, 34 and 36Å.
If indeed the extra methyl group in thymine, which is not found in cytosine, is responsible for stronger DNA–DNA interactions, we can predict that cytosine methylation, which occurs naturally in many eukaryotic organisms and is an essential epigenetic regulation mechanism26, would also increase the strength of DNA–DNA attraction. MD simulations showed that the GC-rich helices containing methylated cytosines (mC) lose the adsorbed Sm4+ (Fig. 2c,f) and that |ΔGmin| of (GC)10 increases on methylation of cytosines to become similar to |ΔGmin| of (AT)10 (Fig. 1b).
To experimentally assess the effect of cytosine methylation, we designed another GC-rich construct GC2 that had the same GC content as GC1 but a higher density of CpG sites (Supplementary Table 1). The CpG sites were then fully methylated using M. SssI methyltransferase (Supplementary Fig. 3; Methods). As predicted from the MD simulations, methylation of the GC-rich constructs increased the binding fraction to the level of the AT-rich constructs (Fig. 1f).
The sequence dependence of |ΔGmin| and its relation to the Sm4+ adsorption patterns can be rationalized by examining the number of Sm4+ molecules shared by the dsDNA molecules (Fig. 3a). An Sm4+ cation adsorbed to the major groove of one dsDNA is separated from the other dsDNA by at least 10Å, contributing much less to the effective DNA–DNA attractive force than a cation positioned between the helices, that is, the ‘bridging’ Sm4+ (ref. 23). An adsorbed Sm4+ also repels other Sm4+ molecules due to like-charge repulsion, lowering the concentration of bridging Sm4+. To demonstrate that the concentration of bridging Sm4+ controls the strength of DNA–DNA attraction, we computed the number of bridging Sm4+ molecules, Nspm (Fig. 3b). Indeed, the number of bridging Sm4+ molecules ranks in the same order as |ΔGmin|: Nspm of (A)20>Nspm of (AT)10≈Nspm of (GmC)10>Nspm of (GC)10>Nspm of (G)20. Thus, the number density of nucleotides carrying a methyl group (T and mC) is the primary determinant of the strength of attractive interaction between two dsDNA molecules. At the same time, the spatial arrangement of the methyl group carrying nucleotides can affect the interaction strength as well (Fig. 3c). The number of methyl groups and their distribution in the (AT)10 and (GmC)10 duplex DNA are identical, and so are their interaction free energies, |ΔGmin| of (AT)10≈|ΔGmin| of (GmC)10. For AT-rich DNA sequences, clustering of the methyl groups repels Sm4+ from the major groove more efficiently than when the same number of methyl groups is distributed along the DNA (Fig. 3b). Hence, |ΔGmin| of (A)20>|ΔGmin| of (AT)10. For GC-rich DNA sequences, clustering of the cation-binding sites (N7 nitrogen) attracts more Sm4+ than when such sites are distributed along the DNA (Fig. 3b), hence |ΔGmin| is larger for (GC)10 than for (G)20.
Figure 3: Methylation modulates the interaction free energy of two dsDNA molecules by altering the number of bridging Sm4+.
(a) Typical spatial arrangement of Sm4+ molecules around a pair of DNA helices. The phosphates groups of DNA and the amine groups of Sm4+ are shown as red and blue spheres, respectively. ‘Bridging’ Sm4+molecules reside between the DNA helices. Orange rectangles illustrate the volume used for counting the number of bridging Sm4+ molecules. (b) The number of bridging amine groups as a function of the inter-DNA distance. The total number of Sm4+ nitrogen atoms was computed by averaging over the corresponding MD trajectory and the 10Å (x axis) by 20Å (y axis) rectangle prism volume (a) centred between the DNA molecules. (c) Schematic representation of the dependence of the interaction free energy of two DNA molecules on their nucleotide sequence. The number and spatial arrangement of nucleotides carrying a methyl group (T or mC) determine the interaction free energy of two dsDNA molecules.
Genome-wide investigations of chromosome conformations using the Hi–C technique revealed that AT-rich loci form tight clusters in human nucleus27, 28. Gene or chromosome inactivation is often accompanied by increased methylation of DNA29 and compaction of facultative heterochromatin regions30. The consistency between those phenomena and our findings suggest the possibility that the polyamine-mediated sequence-dependent DNA–DNA interaction might play a role in chromosome folding and epigenetic regulation of gene expression.
Rau, D. C., Lee, B. & Parsegian, V. A.Measurement of the repulsive force between polyelectrolyte molecules in ionic solution: hydration forces between parallel DNA double helices. Proc. Natl Acad. Sci. USA81, 2621–2625 (1984).
Raspaud, E., Olvera de la Cruz, M., Sikorav, J. L. & Livolant, F.Precipitation of DNA by polyamines: a polyelectrolyte behavior. Biophys. J.74, 381–393 (1998).
Grosberg, A. Y., Nguyen, T. T. & Shklovskii, B. I.The physics of charge inversion in chemical and biological systems. Rev. Mod. Phys.74, 329–345 (2002).
Thomas, T. & Thomas, T. J.Polyamines in cell growth and cell death: molecular mechanisms and therapeutic applications. Cell. Mol. Life Sci.58, 244–258 (2001).
The author and his father have seen several relatives succumb to mental illness.CREDIT PHOTOGRAPH BY DAYANITA SINGH FOR THE NEW YORKER
In the winter of 2012, I travelled from New Delhi, where I grew up, to Calcutta to visit my cousin Moni. My father accompanied me as a guide and companion, but he was a sullen and brooding presence, lost in a private anguish. He is the youngest of five brothers, and Moni is his firstborn nephew—the eldest brother’s son. Since 2004, Moni, now fifty-two, has been confined to an institution for the mentally ill (a “lunatic home,” as my father calls it), with a diagnosis of schizophrenia. He is kept awash in antipsychotics and sedatives, and an attendant watches, bathes, and feeds him through the day.
My father has never accepted Moni’s diagnosis. Over the years, he has waged a lonely campaign against the psychiatrists charged with his nephew’s care, hoping to convince them that their diagnosis was a colossal error, or that Moni’s broken psyche would somehow mend itself. He has visited the institution in Calcutta twice—once without warning, hoping to see a transformed Moni, living a secretly normal life behind the barred gates. But there was more than just avuncular love at stake for him in these visits. Moni is not the only member of the family with mental illness. Two of my father’s four brothers suffered from various unravellings of the mind. Madness has been among the Mukherjees for generations, and at least part of my father’s reluctance to accept Moni’s diagnosis lies in a grim suspicion that something of the illness may be buried, like toxic waste, in himself.
Rajesh, my father’s third-born brother, had once been the most promising of the Mukherjee boys—the nimblest, the most charismatic, the most admired. But in the summer of 1946, at the age of twenty-two, he began to behave oddly, as if a wire had been tripped in his brain. The most obvious change in his personality was a volatility: good news triggered uncontained outbursts of joy; bad news plunged him into inconsolable desolation. By that winter, the sine curve of Rajesh’s psyche had tightened in its frequency and gained in its amplitude. My father recalls an altered brother: fearful at times, reckless at others, descending and ascending steep slopes of mood, irritable one morning and overjoyed the next. When Rajesh received news of a successful performance on his college exams, he vanished, elated, on a two-night excursion, supposedly “exercising” at a wrestling camp. He was feverish and hallucinating when he returned, and died of pneumonia soon afterward. Only years later, in medical school, did I realize that Rajesh was likely in the throes of an acute manic phase. His mental breakdown was the result of a near-textbook case of bipolar disorder.
Jagu, the fourth-born of my father’s siblings, came to live with us in Delhi in 1975, when I was five years old and he was forty-five. His mind, too, was failing. Tall and rail thin, with a slightly feral look in his eyes and a shock of matted, overgrown hair, he resembled a Bengali Jim Morrison. Unlike Rajesh, whose illness had surfaced in his twenties, Jagu had been troubled from his adolescence. Socially awkward, withdrawn from everyone except my grandmother, he was unable to hold a job or live by himself. By 1975, he had visions, phantasms, and voices in his head that told him what to do. He was still capable of extraordinary bursts of tenderness—when I accidentally smashed a beloved Venetian vase at home, he hid me in his bedclothes and informed my mother that he had “mounds of cash” stashed away, enough to buy “a thousand” replacement vases. But this episode was symptomatic: even his love for me extended the fabric of his psychosis and confabulation.
Unlike Rajesh, Jagu was formally diagnosed. In the late nineteen-seventies, a physician in Delhi examined him and determined that he had schizophrenia. But no medicines were prescribed. Instead, Jagu continued to live at home, half hidden away in my grandmother’s room. (As in many families in India, my grandmother lived with us.) For nearly a decade, she and my father maintained a fragile truce, with Jagu living under her care, eating meals in her room and wearing clothes that she stitched for him. At night, when Jagu was consumed by his fears and fantasies, she put him to bed like a child, with her hand on his forehead. She was his nurse, his housekeeper, his only friend, and, more important, his public defender. When my grandmother died, in 1985, Jagu joined a religious sect in Delhi and disappeared, until his death, a dozen years later.
……
at schizophrenia runs in families was evident even to the person who first defined the illness. In 1911, Eugen Bleuler, a Swiss-German psychiatrist, published a book describing a series of cases of men and women, typically in their teens and early twenties, whose thoughts had begun to tangle and degenerate. “In this malady, the associations lose their continuity,” Bleuler wrote. “The threads between thoughts are torn.” Psychotic visions and paranoid thoughts flashed out of nowhere. Some patients “feel themselves weak, their spirit escapes, they will never survive the day. There is a growth in their heads. Their bones have turned liquid; their hearts have turned into stone. . . . The patient’s wife must not use eggs in cooking, otherwise he will grow feathers.” His patients were often trapped between flickering emotional states, unable to choose between two radically opposed visions, Bleuler noted. “You devil, you angel, you devil, you angel,” one woman said to her lover.
Bleuler tried to find an explanation for the mysterious symptoms, but there was only one seemingly common element: schizophrenic patients tended to have first-degree relatives who were also schizophrenic. He had no tools to understand the mechanism behind the heredity. The word “gene” had been coined just two years before Bleuler published his book. The notion that a mental illness could be carried across generations by unitary, indivisible factors—corpuscles of information threading through families—would have struck most of Bleuler’s contemporaries as mad in its own right. Still, Bleuler was astonishingly prescient about the complex nature of inheritance. “If one is looking for ‘theheredity,’ one can nearly always find it,” he wrote. “We will not be able to do anything about it even later on, unless the single factor of heredity can be broken down into many hereditary factors along specific lines.”
In the nineteen-sixties, Bleuler’s hunch was confirmed by twin studies. Psychiatrists determined that if an identical twin was schizophrenic the other twin had a forty-to-fifty-per-cent chance of developing the disease—fiftyfold higher than the risk in the general population. By the early two-thousands, large population studies had revealed a strong genetic link between schizophrenia and bipolar disorder. Some of the families described in these studies had a crisscrossing history that was achingly similar to my own: one sibling affected with schizophrenia, another with bipolar disorder, and a nephew or niece also schizophrenic.
“The twin studies clarified two important features of schizophrenia and bipolar disorder,” Jeffrey Lieberman, a Columbia University psychiatrist who has studied schizophrenia for thirty years, told me. “First, it was clear that there wasn’t a single gene, but dozens of genes involved in causing schizophrenia—each perhaps exerting a small effect. And, second, even if you inherited the entire set of risk genes, as identical twins do, you still might not develop the disease. Obviously, there were other triggers or instigators involved in releasing the illness.” But while these studies established that schizophrenia had a genetic basis, they revealed nothing about the nature of the genes involved. “For doctors, patients, and families in the schizophrenia community, genetics became the ultimate mystery,” Lieberman said. “If we knew the identity of the genes, we would find the causes, and if we found the causes we could find medicines.”
In 2006, an international consortium of psychiatric geneticists launched a genomic survey of schizophrenia, hoping to advance the search for the implicated genes. With 3,322 patients and 3,587 controls, this was one of the largest and most rigorous such studies in the history of the disease. Researchers scanned through the nearly seven thousand genomes to find variations in gene segments that were correlated with schizophrenia. This strategy, termed an “association study,” does not pinpoint a gene, but it provides a general location where a disease-linked gene may be found, like a treasure map with a large “X” scratched in a corner of the genome.
The results, reported in 2009 (and updated in 2014) in the journal Nature, were a dispiriting validation of Bleuler’s hunch about multiple hereditary factors: more than a hundred independent segments of the genome were associated with schizophrenia. “There are lots of small, common genetic effects, scattered across the genome,” one researcher said. “There are many different biological processes involved.” Some of the putative culprits made biological sense—if dimly. There were genes linked to transmitters that relay messages between neurons, and genes for molecular channels that move electrical signals up and down nerve cells. But by far the most surprising association involved a gene segment on chromosome 6. This region of the genome—termed the MHC region—carries hundreds of genes typically associated with the immune system.
“The MHC-segment finding was so strange and striking that you had to sit up and take notice,” Lieberman told me. “Here was the most definitive evidence that something in the immune system might have something to do with schizophrenia. There had been hints about an immunological association before, but this was impossible to argue with. It raised an endlessly fascinating question: what was the link between immune-response genes and schizophrenia?”
In the first years of her career in brain research, Beth Stevens thought of microglia with annoyance if she thought of them at all. When she gazed into a microscope and saw these ubiquitous cells with their spidery tentacles, she did what most neuroscientists had been doing for generations: she looked right past them and focused on the rest of the brain tissue, just as you might look through specks of dirt on a windshield.
“What are they doing there?” she thought. “They’re in the way.’”
Stevens never would have guessed that just a few years later, she would be running a laboratory at Harvard and Boston’s Children’s Hospital devoted to the study of these obscure little clumps. Or that she would be arguing in the world’s top scientific journals that microglia might hold the key to understanding not just normal brain development but also what causes Alzheimer’s, Huntington’s, autism, schizophrenia, and other intractable brain disorders.
Microglia are part of a larger class of cells—known collectively as glia—that carry out an array of functions in the brain, guiding its development and serving as its immune system by gobbling up diseased or damaged cells and carting away debris. Along with her frequent collaborator and mentor, Stanford biologist Ben Barres, and a growing cadre of other scientists, Stevens, 45, is showing that these long-overlooked cells are more than mere support workers for the neurons they surround. Her work has raised a provocative suggestion: that brain disorders could somehow be triggered by our own bodily defenses gone bad.
A type of glial cell known as an oligodendrocyte
In one groundbreaking paper, in January, Stevens and researchers at the Broad Institute of MIT and Harvard showed that aberrant microglia might play a role in schizophrenia—causing or at least contributing to the massive cell loss that can leave people with devastating cognitive defects. Crucially, the researchers pointed to a chemical pathway that might be targeted to slow or stop the disease. Last week, Stevens and other researchers published a similar finding for Alzheimer’s.
This might be just the beginning. Stevens is also exploring the connection between these tiny structures and other neurological diseases—work that earned her a $625,000 MacArthur Foundation “genius” grant last September.
All of this raises intriguing questions. Is it possible that many common brain disorders, despite their wide-ranging symptoms, are caused or at least worsened by the same culprit, a component of the immune system? If so, could many of these disorders be treated in a similar way—by stopping these rogue cells?
Schizophrenia is a heritable brain illness with unknown pathogenic mechanisms. Schizophrenia’s strongest genetic association at a population level involves variation in the major histocompatibility complex (MHC) locus, but the genes and molecular mechanisms accounting for this have been challenging to identify. Here we show that this association arises in part from many structurally diverse alleles of the complement component 4 (C4) genes. We found that these alleles generated widely varying levels of C4A and C4B expression in the brain, with each common C4 allele associating with schizophrenia in proportion to its tendency to generate greater expression of C4A. Human C4 protein localized to neuronal synapses, dendrites, axons, and cell bodies. In mice, C4 mediated synapse elimination during postnatal development. These results implicate excessive complement activity in the development of schizophrenia and may help explain the reduced numbers of synapses in the brains of individuals with schizophrenia.
Science 31 Mar 2016; http://dx.doi.org:/10.1126/science.aad8373Complement and microglia mediate early synapse loss in Alzheimer mouse models. Soyon Hong1, Victoria F. Beja-Glasser1,*, Bianca M. Nfonoyim1,*,…., Ben A. Barres6, Cynthia A. Lemere,2, Dennis J. Selkoe2,7, Beth Stevens1,8,†
Synapse loss in Alzheimer’s disease (AD) correlates with cognitive decline. Involvement of microglia and complement in AD has been attributed to neuroinflammation, prominent late in disease. Here we show in mouse models that complement and microglia mediate synaptic loss early in AD. C1q, the initiating protein of the classical complement cascade, is increased and associated with synapses before overt plaque deposition. Inhibition of C1q, C3 or the microglial complement receptor CR3, reduces the number of phagocytic microglia as well as the extent of early synapse loss. C1q is necessary for the toxic effects of soluble β-amyloid (Aβ) oligomers on synapses and hippocampal long-term potentiation (LTP). Finally, microglia in adult brains engulf synaptic material in a CR3-dependent process when exposed to soluble Aβ oligomers. Together, these findings suggest that the complement-dependent pathway and microglia that prune excess synapses in development are inappropriately activated and mediate synapse loss in AD.
Genome-wide association studies (GWAS) implicate microglia and complement-related pathways in AD (1). Previous research has demonstrated both beneficial and detrimental roles of complement and microglia in plaque-related neuropathology (2, 3); however, their roles in synapse loss, a major pathological correlate of cognitive decline in AD (4), remain to be identified. Emerging research implicates microglia and immune-related mechanisms in brain wiring in the healthy brain (1). During development, C1q and C3 localize to synapses and mediate synapse elimination by phagocytic microglia (5–7). We hypothesized that this normal developmental synaptic pruning pathway is activated early in the AD brain and mediates synapse loss.
Complex machinery
It’s not surprising that scientists for years have ignored microglia and other glial cells in favor of neurons. Neurons that fire together allow us to think, breathe, and move. We see, hear, and feel using neurons, and we form memories and associations when the connections between different neurons strengthen at the junctions between them, known as synapses. Many neuroscientists argue that neurons create our very consciousness.
Glia, on the other hand, have always been considered less important and interesting. They have pedestrian duties such as supplying nutrients and oxygen to neurons, as well as mopping up stray chemicals and carting away the garbage.
Scientists have known about glia for some time. In the 1800s, the pathologist Rudolf Virchow noted the presence of small round cells packing the spaces between neurons and named them “nervenkitt” or “neuroglia,” which can be translated as nerve putty or glue. One variety of these cells, known as astrocytes, was defined in 1893. And then in the 1920s, the Spanish scientist Pio del Río Hortega developed novel ways of staining cells taken from the brain. This led him to identify and name two more types of glial cells, including microglia, which are far smaller than the others and are characterized by their spidery shape and multiple branches. It is only when the brain is damaged in adulthood, he suggested, that microglia spring to life—rushing to the injury, where it was thought they helped clean up the area by eating damaged and dead cells. Astrocytes often appeared on the scene as well; it was thought that they created scar tissue.
This emergency convergence of microglia and astrocytes was dubbed “gliosis,” and by the time Ben Barres entered medical school in the late 1970s, it was well established as a hallmark of neurodegenerative diseases, infection, and a wide array of other medical conditions. But no one seemed to understand why it occurred. That intrigued Barres, then a neurologist in training, who saw it every time he looked under a microscope at neural tissue in distress. “It was just really fascinating,” he says. “The great mystery was: what is the point of this gliosis? Is it good? Is it bad? Is it driving the disease process, or is it trying to repair the injured brain?”
Barres began looking for the answer. He learned how to grow glial cells in a dish and apply a new recording technique to them. He could measure their electrical qualities, which determine the biochemical signaling that all brain cells use to communicate and coördinate activity.
“From the second I started recording the glial cells, I thought ‘Oh, my God!’” Barres recalls. The electrical activity was more dynamic and complex than anyone had thought. These strange electrical properties could be explained only if the glial cells were attuned to the conditions around them, and to the signals released from nearby neurons. Barres’s glial cells, in other words, had all the machinery necessary to engage in a complex dialogue with neurons, and presumably to respond to different kinds of conditions in the brain.
Why would they need this machinery, though, if they were simply involved in cleaning up dead cells? What could they possibly be doing? It turns out that in the absence of chemicals released by glia, the neurons committed the biochemical version of suicide. Barres also showed that the astrocytes appeared to play a crucial role in forming synapses, the microscopic connections between neurons that encode memory. In isolation, neurons were capable of forming the spiny appendages necessary to reach the synapses. But without astrocytes, they were incapable of connecting to one another.
Hardly anyone believed him. When he was a young faculty member at Stanford in the 1990s, one of his grant applications to the National Institutes of Health was rejected seven times. “Reviewers kept saying, ‘Nah, there’s no way glia could be doing this,’” Barres recalls. “And even after we published two papers in Science showing that [astrocytes] had profound, almost all-or-nothing effects in controlling synapses’ formation or synapse activity, I still couldn’t get funded! I think it’s still hard to get people to think about glia as doing anything active in the nervous system.”
Marked for elimination
Beth Stevens came to study glia by accident. After graduating from Northeastern University in 1993, she followed her future husband to Washington, D.C., where he had gotten work in the U.S. Senate. Stevens had been pre-med in college and hoped to work in a lab at the National Institutes of Health. But with no previous research experience, she was soundly rebuffed. So she took a job waiting tables at a Chili’s restaurant in nearby Rockville, Maryland, and showed up at NIH with her résumé every week.
After a few months, Stevens received a call from a researcher named Doug Fields, who needed help in his lab. Fields was studying the intricacies of the process in which neurons become insulated in a coating called myelin. That insulation is essential for the transmission of electrical impulses.
As Stevens spent the following years pursuing a PhD at the University of Maryland, she was intrigued by the role that glial cells played in insulating neurons. Along the way, she became familiar with other insights into glial cells that were beginning to emerge, especially from the lab of Ben Barres. Which is why, soon after completing her PhD in 2003, Stevens found herself a postdoc in Barres’s lab at Stanford, about to make a crucial discovery.
Barres’s group had begun to identify the specific compounds astrocytes secreted that seemed to cause neurons to grow synapses. And eventually, they noticed that these compounds also stimulated production of a protein called C1q.
Conventional wisdom held that C1q was activated only in sick cells—the protein marked them to be eaten up by immune cells—and only outside the brain. But Barres had found it in the brain. And it was in healthy neurons that were arguably at their most robust stage: in early development. What was the C1q protein doing there?
The answer lies in the fact that marking cells for elimination is not something that happens only in diseased brains; it is also essential for development. As brains develop, their neurons form far more synaptic connections than they will eventually need. Only the ones that are used are allowed to remain. This pruning allows for the most efficient flow of neural transmissions in the brain, removing noise that might muddy the signal.
But it was unknown how exactly the process worked. Was it possible that C1q helped signal the brain to prune unused synapses? Stevens focused her postdoctoral research on finding out. “We could have been completely wrong,” she recalls. “But we went for it.”
It paid off. In a 2007 paper, Barres and Stevens showed that C1q indeed plays a role in eliminating unneeded neurons in the developing brain. And they found that the protein is virtually absent in healthy adult neurons.
Now the scientists faced a new puzzle. Does C1q show up in brain diseases because the same mechanism involved in pruning a developing brain later goes awry? Indeed, evidence was already growing that one of the earliest events in neurodegenerative diseases such as Alzheimer’s, Parkinson’s, and Huntington’s was significant loss of synapses.
When Stevens and Barres examined mice bred to develop glaucoma, a neurodegenerative disease that kills neurons in the optic system, they found that C1q appeared long before any other detectable sign that the disease was taking hold. It cropped up even before the cells started dying.
This suggested the immune cells might in fact cause the disease, or at the very least accelerate it. And that offered an intriguing possibility: that something could be made to halt the process. Barres founded a company, Annexon Biosciences, to develop drugs that could block C1q. Last week’s paper published by Barres, Stevens, and other researchers shows that a compound being tested by Annexon appears to be able to prevent the onset of Alzheimer’s in mice bred to develop the disease. Now the company hopes to test it in humans in the next two years.
Paths to treatments
To better understand the process that C1q helps trigger, Stevens and Barres wanted to figure out what actually plays the role of Pac-Man, eating up the synapses marked for death. It was well known that white blood cells known as macrophages gobbled up diseased cells and foreign invaders in the rest of the body. But macrophages are not usually present in the brain. For their theory to work, there had to be some other mechanism. And further research has shown that the cells doing the eating even in healthy brains are those mysterious clusters of material that Beth Stevens, for years, had been gazing right past in the microscope—the microglia that Río Hortega identified almost 100 years ago.
Now Stevens’s lab at Harvard, which she opened in 2008, devotes half its efforts to figuring out what microglia are doing and what causes them to do it. These cells, it turns out, appear in the mouse embryo at day eight, before any other brain cell, which suggests they might help guide the rest of brain development—and could contribute to any number of neurodevelopmental diseases when they go wrong.
Meanwhile, she is also expanding her study of the way different substances determine what happens in the brain. C1q is actually just the first in a series of proteins that accumulate on synapses marked for elimination. Stevens has begun to uncover evidence that there is a wide array of protective “don’t eat me” molecules too. It’s the balance between all these cues that regulates whether microglia are summoned to destroy synapses. Problems in any one could, conceivably, mess up the system.
Evidence is now growing that microglia are involved in several neurodevelopmental and psychiatric problems. The potential link to schizophrenia that was revealed in January emerged after researchers at the Broad Institute, led by Steven McCarroll and a graduate student named Aswin Sekar, followed a trail of genetic clues that led them directly to Stevens’s work. In 2009, three consortia from around the globe had published papers comparing DNA in people with and without schizophrenia. It was Sekar who identified a possible pattern: the more a specific type of protein was present in synapses, the higher the risk of developing the disease. The protein, C4, was closely related to C1q, the one first identified in the brain by Stevens and Barres.
McCarroll knew that schizophrenia strikes in late adolescence and early adulthood, a time when brain circuits in the prefrontal cortex undergo extensive pruning. Others had found that areas of the prefrontal cortex are among those most ravaged by the disease, which leads to massive synapse loss. Could it be that over-pruning by rogue microglia is part of what causes schizophrenia?
To find out, Sekar and McCarroll got in touch with Stevens, and the two labs began to hold joint weekly meetings. They soon demonstrated that C4 also had a role in pruning synapses in the brains of young mice, suggesting that excessive levels of the protein could indeed lead to over-pruning—and to the thinning out of brain tissue that appears to occur as symptoms such as psychotic episodes grow worse.
If the brain damage seen in Parkinson’s and Alzheimer’s stems from over-pruning that might begin early in life, why don’t symptoms of those diseases show up until later? Barres thinks he knows. He notes that the brain can normally compensate for injury by rewiring itself and generating new synapses. It also contains a lot of redundancy. That would explain why patients with Parkinson’s disease don’t show discernible symptoms until they have lost 90 percent of the neurons that produce dopamine.
It also might mean that subtle symptoms could in fact be detected much earlier. Barres points to a study of nuns published in 2000. When researchers analyzed essays the nuns had written upon entering their convents decades before, they found that women who went on to develop Alzheimer’s had shown less “idea density” even in their 20s. “I think the implication of that is they could be lifelong diseases,” Barres says. “The disease process could be going on for decades and the brain is just compensating, rewiring, making new synapses.” At some point, the microglia are triggered to remove too many cells, Barres argues, and the symptoms of the disease begin to manifest fully.
Turning this insight into a treatment is far from straightforward, because much remains unclear. Perhaps an overly aggressive response from microglia is determined by some combination of genetic variants not shared by everyone. Stevens also notes that diseases like schizophrenia are not caused by one mutation; rather, a wide array of mutations with small effects cause problems when they act in concert. The genes that control the production of C4 and other immune-system proteins may be only part of the story. That may explain why not everyone who has a C4 mutation will go on to develop schizophrenia.
Nonetheless, if Barres and Stevens are right that the immune system is a common mechanism behind devastating brain disorders, that in itself is a fundamental breakthrough. Because we have not known the mechanisms that trigger such diseases, medical researchers have been able only to alleviate the symptoms rather than attack the causes. There are no drugs available to halt or even slow neurodegeneration in diseases like Alzheimer’s. Some drugs elevate neurotransmitters in ways that briefly make it easier for individuals with dementia to form new synaptic connections, but they don’t reduce the rate at which existing synapses are destroyed. Similarly, there are no treatments that tackle the causes of autism or schizophrenia. Even slowing the progress of these disorders would be a major advance. We might finally go after diseases that have run unchecked for generations.
“We’re a ways away from a cure,” Stevens says. “But we definitely have a path forward.”
The complement protein C4 is a non-enzymatic component of the C3 and C5 convertases and thus essential for the propagation of the classical complement pathway. The covalent binding of C4 to immunoglobulins and immune complexes (IC) also enhances the solubilization of immune aggregates, and the clearance of IC through complement receptor one (CR1) on erythrocytes. Human C4 is the most polymorphic protein of the complement system. In this review, we summarize the current concepts on the 1-2-3 loci model of C4A and C4B genes in the population, factors affecting the expression levels of C4 transcripts and proteins, and the structural, functional and serological diversities of the C4A and C4B proteins. The diversities and polymorphisms of the mouse homologues Slp and C4 proteins are described and contrasted with their human homologues. The human C4 genes are located in the MHC class III region on chromosome 6. Each human C4 gene consists of 41 exons coding for a 5.4-kb transcript. The long gene is 20.6 kb and the short gene is 14.2 kb. In the Caucasian population 55% of the MHC haplotypes have the 2-locus, C4A-C4B configurations and 45% have an unequal number of C4A and C4B genes. Moreover, three-quarters of C4 genes harbor the 6.4 kb endogenous retrovirus HERV-K(C4) in the intron 9 of the long genes. Duplication of a C4 gene always concurs with its adjacent genes RP, CYP21 and TNX, which together form a genetic unit termed an RCCX module. Monomodular, bimodular and trimodular RCCX structures with 1, 2 and 3 complement C4 genes have frequencies of 17%, 69% and 14%, respectively. Partial deficiencies of C4A and C4B, primarily due to the presence of monomodular haplotypes and homo-expression of C4A proteins from bimodular structures, have a combined frequency of 31.6%. Multiple structural isoforms of each C4A and C4B allotype exist in the circulation because of the imperfect and incomplete proteolytic processing of the precursor protein to form the beta-alpha-gamma structures. Immunofixation experiments of C4A and C4B demonstrate > 41 allotypes in the two classes of proteins. A compilation of polymorphic sites from limited C4 sequences revealed the presence of 24 polymophic residues, mostly clustered C-terminal to the thioester bond within the C4d region of the alpha-chain. The covalent binding affinities of the thioester carbonyl group of C4A and C4B appear to be modulated by four isotypic residues at positions 1101, 1102, 1105 and 1106. Site directed mutagenesis experiments revealed that D1106 is responsible for the effective binding of C4A to form amide bonds with immune aggregates or protein antigens, and H1106 of C4B catalyzes the transacylation of the thioester carbonyl group to form ester bonds with carbohydrate antigens. The expression of C4 is inducible or enhanced by gamma-interferon. The liver is the main organ that synthesizes and secretes C4A and C4B to the circulation but there are many extra-hepatic sites producing moderate quantities of C4 for local defense. The plasma protein levels of C4A and C4B are mainly determined by the corresponding gene dosage. However, C4B proteins encoded by monomodular short genes may have relatively higher concentrations than those from long C4A genes. The 5′ regulatory sequence of a C4 gene contains a Spl site, three E-boxes but no TATA box. The sequences beyond–1524 nt may be completely different as the C4 genes at RCCX module I have RPI-specific sequences, while those at Modules II, III and IV have TNXA-specific sequences. The remarkable genetic diversity of human C4A and C4B probably promotes the exchange of genetic information to create and maintain the quantitative and qualitative variations of C4A and C4B proteins in the population, as driven by the selection pressure against a great variety of microbes. An undesirable accompanying byproduct of this phenomenon is the inherent deleterious recombinations among the RCCX constituents leading to autoimmune and genetic disorders.
C4A isotype is responsible for effective binding to form amide bonds with immune aggregates or protein antigens, while C4B isotype catalyzes the transacylation of the thioester carbonyl group to form ester bonds with carbohydrate antigens.
Derived from proteolytic degradation of complement C4, C4a anaphylatoxin is a mediator of local inflammatory process.
Schizophrenia and the Synapse
Genetic evidence suggests that overactive synaptic pruning drives development of schizophrenia.
Compared to the brains of healthy individuals, those of people with schizophrenia have higher expression of a gene called C4, according to a paper published inNature today (January 27). The gene encodes an immune protein that moonlights in the brain as an eradicator of unwanted neural connections (synapses). The findings, which suggest increased synaptic pruning is a feature of the disease, are a direct extension of genome-wide association studies (GWASs) that pointed to the major histocompatibility (MHC) locus as a key region associated with schizophrenia risk.
“The MHC [locus] is the first and the strongest genetic association for schizophrenia, but many people have said this finding is not useful,” said psychiatric geneticist Patrick Sullivan of the University of North Carolina School of Medicine who was not involved in the study. “The value of [the present study is] to show that not only is it useful, but it opens up new and extremely interesting ideas about the biology and therapeutics of schizophrenia.”
Schizophrenia has a strong genetic component—it runs in families—yet, because of the complex nature of the condition, no specific genes or mutations have been identified. The pathological processes driving the disease remain a mystery.
Researchers have turned to GWASs in the hope of finding specific genetic variations associated with schizophrenia, but even these have not provided clear candidates.
“There are some instances where genome-wide association will literally hit one base [in the DNA],” explained Sullivan. While a 2014 schizophrenia GWAS highlighted the MHC locus on chromosome 6 as a strong risk area, the association spanned hundreds of possible genes and did not reveal specific nucleotide changes. In short, any hope of pinpointing the MHC association was going to be “really challenging,” said geneticist Steve McCarroll of Harvard who led the new study.
Nevertheless, McCarroll and colleagues zeroed in on the particular region of the MHC with the highest GWAS score—the C4 gene—and set about examining how the area’s structural architecture varied in patients and healthy people.
The C4 gene can exist in multiple copies (from one to four) on each copy of chromosome 6, and has four different forms: C4A-short, C4B-short, C4A-long, and C4B-long. The researchers first examined the “structural alleles” of the C4 locus—that is, the combinations and copy numbers of the different C4 forms—in healthy individuals. They then examined how these structural alleles related to expression of both C4Aand C4B messenger RNAs (mRNAs) in postmortem brain tissues.
…………..
Schizophrenia risk from complex variation of complement component 4
Schizophrenia is a heritable brain illness with unknown pathogenic mechanisms. Schizophrenia’s strongest genetic association at a population level involves variation in the major histocompatibility complex (MHC) locus, but the genes and molecular mechanisms accounting for this have been challenging to identify. Here we show that this association arises in part from many structurally diverse alleles of the complement component 4 (C4) genes. We found that these alleles generated widely varying levels of C4A and C4B expression in the brain, with each common C4 allele associating with schizophrenia in proportion to its tendency to generate greater expression of C4A. Human C4 protein localized to neuronal synapses, dendrites, axons, and cell bodies. In mice, C4 mediated synapse elimination during postnatal development. These results implicate excessive complement activity in the development of schizophrenia and may help explain the reduced numbers of synapses in the brains of individuals with schizophrenia.
Cannon, T. D. et al. Cortex mapping reveals regionally specific patterns of genetic and disease-specific gray-matter deficits in twins discordant for schizophrenia. Proc. Natl Acad. Sci. USA99, 3228–3233 (2002)
Cannon, T. D. et al. Progressive reduction in cortical thickness as psychosis develops: a multisite longitudinal neuroimaging study of youth at elevated clinical risk. Biol. Psychiatry77,147–157 (2015)
Garey, L. J. et al. Reduced dendritic spine density on cerebral cortical pyramidal neurons in schizophrenia. J. Neurol. Neurosurg. Psychiatry65, 446–453 (1998)
Glantz, L. A. & Lewis, D. A. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch. Gen. Psychiatry57, 65–73 (2000)
Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature511, 421–427 (2014)
International Schizophrenia Consortium et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature460, 748–752 (2009)
Schizophrenia Psychiatric Genome-Wide Association Study Consortium. Genome-wide association study identifies five new schizophrenia loci. Nature Genet . 43, 969–976 (2011)
The strongest genetic association found in schizophrenia is its association to genetic markers across the major histocompatibility complex (MHC) locus, first described in three Nature papers in 2009. …
Schizophrenia: From genetics to physiology at last
Jianxin Shi1, et al. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature 460, 753-757 (6 August 2009) | doi:10.1038/nature08192; Received 29 May 2009; Accepted 10 June 2009; Published online 1 July 2009; Corrected 6 August 2009
Schizophrenia, a devastating psychiatric disorder, has a prevalence of 0.5–1%, with high heritability (80–85%) and complex transmission1. Recent studies implicate rare, large, high-penetrance copy number variants in some cases2, but the genes or biological mechanisms that underlie susceptibility are not known. Here we show that schizophrenia is significantly associated with single nucleotide polymorphisms (SNPs) in the extended major histocompatibility complex region on chromosome 6. We carried out a genome-wide association study of common SNPs in the Molecular Genetics of Schizophrenia (MGS) case-control sample, and then a meta-analysis of data from the MGS, International Schizophrenia Consortium and SGENE data sets. No MGS finding achieved genome-wide statistical significance. In the meta-analysis of European-ancestry subjects (8,008 cases, 19,077 controls), significant association with schizophrenia was observed in a region of linkage disequilibrium on chromosome 6p22.1 (P = 9.54 × 10-9). This region includes a histone gene cluster and several immunity-related genes—possibly implicating aetiological mechanisms involving chromatin modification, transcriptional regulation, autoimmunity and/or infection. These results demonstrate that common schizophrenia susceptibility alleles can be detected. The characterization of these signals will suggest important directions for research on susceptibility mechanisms.
Editor’s Summary 6 August 2009
Schizophrenia risk: link to chromosome 6p22.1
A genome-wide association study using the Molecular Genetics of Schizophrenia case-control data set, followed by a meta-analysis that included over 8,000 cases and 19,000 controls, revealed that while common genetic variation that underlies risk to schizophrenia can be identified, there probably are few or no single common loci with large effects. The common variants identified here lie on chromosome 6p22.1 in a region that includes a histone gene cluster and several genes implicated in immunity.
Letter
Hreinn Stefansson1,48, et al. Common variants conferring risk of schizophrenia. Nature460, 744-747 (6 August 2009) | doi:10.1038/nature08186; Received 16 March 2009; Accepted 5 June 2009; Published online 1 July 2009
Schizophrenia is a complex disorder, caused by both genetic and environmental factors and their interactions. Research on pathogenesis has traditionally focused on neurotransmitter systems in the brain, particularly those involving dopamine. Schizophrenia has been considered a separate disease for over a century, but in the absence of clear biological markers, diagnosis has historically been based on signs and symptoms. A fundamental message emerging from genome-wide association studies of copy number variations (CNVs) associated with the disease is that its genetic basis does not necessarily conform to classical nosological disease boundaries. Certain CNVs confer not only high relative risk of schizophrenia but also of other psychiatric disorders1, 2, 3. The structural variations associated with schizophrenia can involve several genes and the phenotypic syndromes, or the ‘genomic disorders’, have not yet been characterized4. Single nucleotide polymorphism (SNP)-based genome-wide association studies with the potential to implicate individual genes in complex diseases may reveal underlying biological pathways. Here we combined SNP data from several large genome-wide scans and followed up the most significant association signals. We found significant association with several markers spanning the major histocompatibility complex (MHC) region on chromosome 6p21.3-22.1, a marker located upstream of the neurogranin gene (NRGN) on 11q24.2 and a marker in intron four of transcription factor 4 (TCF4) on 18q21.2. Our findings implicating the MHC region are consistent with an immune component to schizophrenia risk, whereas the association with NRGN and TCF4 points to perturbation of pathways involved in brain development, memory and cognition.
Letter
The International Schizophrenia Consortium. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature460, 748-752 (6 August 2009) | doi:10.1038/nature08185; Received 11 February 2009; Accepted 8 June 2009; Published online 1 July 2009; Corrected 6 August 2009
Schizophrenia is a severe mental disorder with a lifetime risk of about 1%, characterized by hallucinations, delusions and cognitive deficits, with heritability estimated at up to 80%1, 2. We performed a genome-wide association study of 3,322 European individuals with schizophrenia and 3,587 controls. Here we show, using two analytic approaches, the extent to which common genetic variation underlies the risk of schizophrenia. First, we implicate the major histocompatibility complex. Second, we provide molecular genetic evidence for a substantial polygenic component to the risk of schizophrenia involving thousands of common alleles of very small effect. We show that this component also contributes to the risk of bipolar disorder, but not to several non-psychiatric diseases.
The Psychiatric GWAS Consortium Steering Committee. A framework for interpreting genome-wide association studies of psychiatric disorders. Molecular Psychiatry (2009) 14, 10–17; doi:10.1038/mp.2008.126; published online 11 November 2008
Genome-wide association studies (GWAS) have yielded a plethora of new findings in the past 3 years. By early 2009, GWAS on 47 samples of subjects with attention-deficit hyperactivity disorder, autism, bipolar disorder, major depressive disorder and schizophrenia will be completed. Taken together, these GWAS constitute the largest biological experiment ever conducted in psychiatry (59 000 independent cases and controls, 7700 family trios and >40 billion genotypes). We know that GWAS can work, and the question now is whether it will work for psychiatric disorders. In this review, we describe these studies, the Psychiatric GWAS Consortium for meta-analyses of these data, and provide a logical framework for interpretation of some of the conceivable outcomes.
The purpose of this article is to consider the ‘big picture’ and to provide a logical framework for the possible outcomes of these studies. This is not a review of GWAS per se as many excellent reviews of this technically and statistically intricate methodological approach are available.7, 8, 9, 10, 11, 12 This is also not a review of the advantages and disadvantages of different study designs and sampling strategies for the dissection of complex psychiatric traits. We would like to consider how the dozens of GWAS papers that will soon be in the literature can be synthesized: what can integrated mega-analyses (meta-analysis is based on summary data (for example, odds ratios) from all available studies whereas ‘mega-analysis’ uses individual-level genotype and phenotype data) of all available GWAS data tell us about the etiology of these psychiatric disorders? This is an exceptional opportunity as positive or negative results will enable us to learn hard facts about these critically important psychiatric disorders. We suggest that it is not a matter of ‘success versus failure’ or ‘optimism versus pessimism’ but rather an opportunity for systematic and logical approaches to empirical data whereby both positive and appropriately qualified negative findings are informative.
The studies that comprise the Psychiatric GWAS Consortium (PGC; http://pgc.unc.edu) are shown in Table 1. GWAS data for ADHD, autism, bipolar disorder, major depressive disorder and schizophrenia from 42 samples of European subjects should be available for mega-analyses by early 2009 (>59 000 independent cases and controls and >7700 family trios). To our knowledge, the PGC will have access to the largest set of GWAS data available.
A major change in human genetics in the past 5 years has been in the growth of controlled-access data repositories, and individual phenotype and genotype data are now available for many of the studies in Table 1. When the PGC mega-analyses are completed, most data will be available to researchers via the NIMH Human Genetics Initiative (http://nimhgenetics.org). Although the ready availability of GWAS data is a benefit to the field by allowing rapid application of a wide range of analytic strategies to GWAS data, there are potential disadvantages. GWAS mega-analysis is complex and requires considerable care and expertise to be done validly. For psychiatric phenotypes, there is the additional challenge of working with disease entities based largely on clinical description, with unknown biological validity and having both substantial clinical variation within diagnostic categories as well as overlaps across categories.13 Given the urgent need to know if there are replicable genotype–phenotype associations, a new type of collaboration was required.
The purpose of the PGC is to conduct rigorous and comprehensive within- and cross-disorder GWAS mega-analyses. The PGC began in early 2007 with the principal investigators of the four GAIN GWAS,14 and within six months had grown to 110 participating scientists from 54 institutions in 11 countries. The PGC has a coordinating committee, five disease-working groups, a cross-disorder group, a statistical analysis and computational group, and a cluster computer for statistical analysis. It is remarkable that almost all investigators approached agreed to participate and that no one has left the PGC. Most effort is donated but we have obtained funding from the NIMH, the Netherlands Scientific Organization, Hersenstichting Nederland and NARSAD.
The PGC has two major specific aims. (1) Within-disorder mega-analyses: conduct separate mega-analyses of all available GWAS data for ADHD, autism, bipolar disorder, major depressive disorder, and schizophrenia to attempt to identify genetic variation convincingly associated with any one of these five disorders. (2) Cross-disorder mega-analyses: the clinically-derived DSM-IV and ICD-10 definitions may not directly reflect the fundamental genetic architecture.15 There are two subaims. (2a) Conduct mega-analysis to identify genetic variation convincingly associated with conventional definitions of two or more disorders. This nosological aim could assist in delineating the boundaries of this set of disorders. (2b) An expert working group will convert epidemiological and genetic epidemiological evidence into explicit hypotheses about overlap among these disorders, and then conduct mega-analyses based on these definitions (for example, to examine the lifetime presence of idiopathic psychotic features without regard to diagnostic context).
The goal of the PGC is to identify convincing genetic variation-disease associations. A convincing association would be extremely unlikely to result from chance, show consistent effect sizes across all or almost all samples and be impervious to vigorous attempts to disprove the finding (for example, by investigating sources of bias, confirmatory genotyping, and so on). Careful attention will be paid to the impact of potential sources of heterogeneity17 with the goal of assessing its impact without minimizing its presence.
Biological plausibility is not an initial requirement for a convincing statistical association, as there are many examples in human genetics of previously unsuspected candidate genes nonetheless showing highly compelling associations. For example, multiple SNPs in intron 1 of the FTO gene were associated with body mass index in 13 cohorts with 38 759 participants18 and yet ‘FTO’ does not appear in an exhaustive 116 page compilation of genetic studies of obesity.19 Some strong associations are in gene deserts: multiple studies have found convincing association between prostate cancer and a region on 8q24 that is ~250 kb from the nearest annotated gene.20 Both of these examples are being intensively investigated and we suspect that a compelling mechanistic ‘story’ will emerge in the near future. The presence of a compelling association without an obvious biological mechanism establishes a priority research area for molecular biology and neuroscience of a psychiatric disorder.
The PGC will use mega-analysis as the main analytic tool as individual-level data will be available from almost all samples. To wield this tool appropriately, a number of preconditions must be met. First, genotype data from different GWAS platforms must be made comparable as the direct overlap between platforms is often modest. This requires meticulous quality control for the inclusion of both SNPs and subjects and attention to the factors that can cause bias (for example, population stratification, cryptic relatedness or genotyping batch effects). Genotype harmonization can be accomplished using imputation (21, 22, for example) so that the same set of ~2 million23, 24 directly or imputed SNP genotypes are available for all subjects. Second, phenotypes need to be harmonized across studies. This is one of the most crucial components of the PGC and we are fortunate to have world experts directing the work. Third, the mega-analyses will assess potential heterogeneity of associations across samples.
A decision-tree schematic of the potential outcomes of the PGC mega-analyses is shown in Figure 1. Note that many of the possibilities in Figure 1 are not mutually exclusive and different disorders may take different paths through this framework. It is possible that there eventually will be dozens or hundreds of sequence variants strictly associated with these disorders with frequencies ranging from very rare to common.
………
GWAS has the potential to yield considerable insights but it is no panacea and may well perform differently for psychiatric disorders. Even if these psychiatric GWAS efforts are successful, the outcomes will be complex. GWAS may help us learn that clinical syndromes are actually many different things—for example, proportions of individuals with schizophrenia might evidence associations with rare CNVs of major effect,5, 6 with more common genetic variation in dozens (perhaps hundreds) of genomic regions, between genetic variation strongly modified by environmental risk factors, and some proportion may be genetically indistinguishable from the general population. Moreover, as fuel to long-standing ‘lumper versus splitter’ debates in psychiatric nosology, empirical data might show that some clinical disorders or identifiable subsets of subjects might overlap considerably.
The critical advantage of GWAS is the search of a ‘closed’ hypothesis space. If the large amount of GWAS data being generated are analyzed within a strict and coherent framework, it should be possible to establish hard facts about the fundamental genetic architecture of a set of important psychiatric disorders—which might include positive evidence of what these disorders are or exclusionary evidence of what they are not. Whatever the results, these historically large efforts should yield hard facts about ADHD, autism, bipolar disorder, major depressive disorder and schizophrenia that may help guide the next era of psychiatric research.
Pe’er I, Yelensky R, Altshuler D, Daly MJ. Estimation of the multiple testing burden for genomewide association studies of nearly all common variants. Genet Epidemiol 2008; 32: 381–385. | Article | PubMed |
Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R et al. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med 2008; 358: 667–675. | Article | PubMed | ChemPort |
Letter
Hreinn Stefansson1,36, et al. Large recurrent microdeletions associated with schizophrenia. Nature455, 232-236 (11 September 2008) | doi:10.1038/nature07229; Received 17 April 2008; Accepted 8 July 2008; Corrected 11 September 2008
Reduced fecundity, associated with severe mental disorders1, places negative selection pressure on risk alleles and may explain, in part, why common variants have not been found that confer risk of disorders such as autism2, schizophrenia3 and mental retardation4. Thus, rare variants may account for a larger fraction of the overall genetic risk than previously assumed. In contrast to rare single nucleotide mutations, rare copy number variations (CNVs) can be detected using genome-wide single nucleotide polymorphism arrays. This has led to the identification of CNVs associated with mental retardation4, 5 and autism2. In a genome-wide search for CNVs associating with schizophrenia, we used a population-based sample to identify de novoCNVs by analysing 9,878 transmissions from parents to offspring. The 66 de novo CNVs identified were tested for association in a sample of 1,433 schizophrenia cases and 33,250 controls. Three deletions at 1q21.1, 15q11.2 and 15q13.3 showing nominal association with schizophrenia in the first sample (phase I) were followed up in a second sample of 3,285 cases and 7,951 controls (phase II). All three deletions significantly associate with schizophrenia and related psychoses in the combined sample. The identification of these rare, recurrent risk variants, having occurred independently in multiple founders and being subject to negative selection, is important in itself. CNV analysis may also point the way to the identification of additional and more prevalent risk variants in genes and pathways involved in schizophrenia.
The C4 gene can exist in multiple copies (from one to four) on each copy of chromosome 6, and has four different forms: C4A-short, C4B-short, C4A-long, and C4B-long. The researchers first examined the “structural alleles” of the C4 locus—that is, the combinations and copy numbers of the different C4 forms—in healthy individuals. They then examined how these structural alleles related to expression of both C4Aand C4B messenger RNAs (mRNAs) in postmortem brain tissues.
From this the researchers had a clear picture of how the architecture of the C4 locus affected expression ofC4A and C4B. Next, they compared DNA from roughly 30,000 schizophrenia patients with that from 35,000 healthy controls, and a correlation emerged: the alleles most strongly associated with schizophrenia were also those that were associated with the highest C4A expression. Measuring C4A mRNA levels in the brains of 35 schizophrenia patients and 70 controls then revealed that, on average, C4A levels in the patients’ brains were 1.4-fold higher.
C4 is an immune system “complement” factor—a small secreted protein that assists immune cells in the targeting and removal of pathogens. The discovery of C4’s association to schizophrenia, said McCarroll, “would have seemed random and puzzling if it wasn’t for work . . . showing that other complement components regulate brain wiring.” Indeed, complement protein C3 locates at synapses that are going to be eliminated in the brain, explained McCarroll, “and C4 was known to interact with C3 . . . so we thought well, actually, this might make sense.”
McCarroll’s team went on to perform studies in mice that revealed C4 is necessary for C3 to be deposited at synapses. They also showed that the more copies of the C4 gene present in a mouse, the more the animal’s neurons were pruned.
Synaptic pruning is a normal part of development and is thought to reflect the process of learning, where the brain strengthens some connections and eradicates others. Interestingly, the brains of deceased schizophrenia patients exhibit reduced neuron density. The new results, therefore, “make a lot of sense,” said Cardiff University’s Andrew Pocklington who did not participate in the work. They also make sense “in terms of the time period when synaptic pruning is occurring, which sort of overlaps with the period of onset for schizophrenia: around adolescence and early adulthood,” he added.
“[C4] has not been on anybody’s radar for having anything to do with schizophrenia, and now it is and there’s a whole bunch of really neat stuff that could happen,” said Sullivan. For one, he suggested, “this molecule could be something that is amenable to therapeutics.”
UniProtKB
Derived from proteolytic degradation of complement C4, C4a anaphylatoxin is a mediator of local inflammatory process. It induces the contraction of smooth muscle, increases vascular permeability and causes histamine release from mast cells and basophilic leukocytes.
Non-enzymatic component of C3 and C5 convertases and thus essential for the propagation of the classical complement pathway. Covalently binds to immunoglobulins and immune complexes and enhances the solubilization of immune aggregates and the clearance of IC through CR1 on erythrocytes. C4A isotype is responsible for effective binding to form amide bonds with immune aggregates or protein antigens, while C4B isotype catalyzes the transacylation of the thioester carbonyl group to form ester bonds with carbohydrate antigens.
In 1956, Francis Crick—co-discoverer of DNA’s helical structure—postulated what is now considered to be a central doctrine of the biological sciences stating that “The central dogma of molecular biology deals with the detailed residue-by-residue transfer of sequential information. It states that such information cannot be transferred back from protein to either protein or nucleic acid.” What Crick was suggesting was that DNA makes RNA and, in turn, RNA makes protein.
In the time since the initial proposal of the central dogma, scientists have come to understand that there are not only instances of reverse information flow from RNA to DNA, but chemical alterations to RNA structures that can have a profound effect on gene regulation. The discovery of these alterations has added a critical dimension to how scientists view the genetic code and recently spawned an entirely new field of study within molecular biology: the epitranscriptome.
Now, a recent study by scientists at the University of Chicago and Tel Aviv University has revealed evidence that provides a promising new lever in the control of gene expression. The researchers describe a small chemical modification to RNA that can significantly boost the conversion of genes to proteins.
The findings from this study were published recently in Nature through an article entitled “The dynamic N1-methyladenosine methylome in eukaryotic messenger RNA.”
“Epigenetics, the regulation of gene expression beyond the primary information encoded by DNA, was thought until recently to be mediated by modifications of proteins and DNA,” explained co-senior study author Gidi Rechavi, Ph.D., chair in oncology at Tel Aviv University’s Sackler Faculty of Medicine and head of the Cancer Research Center at Sheba Medical Center. “The new findings bring RNA to a central position in epigenetics.”
“This discovery further opens the window on a whole new world of biology for us to explore,” added co-senior study author Chuan He, Ph.D., professor in the department of chemistry and investigator within the Howard Hughes Medical Institute at the University of Chicago. “These modifications have a major impact on almost every biological process.”
Previously, Dr. He’s laboratory discovered the first RNA demethylase that reverses the most prevalent mRNA methylation N6-methyladenosine (m6A), implying that the addition and removal of the methyl group could dramatically affect these messengers and the outcome of gene expression—as also seen for DNA and histones—which subsequent research found to be true.
In the current study, the investigators described a second functional mRNA methylation, N1-methyladenosine (m1A). Like m6A, the small modification is evolutionarily conserved and common, present in humans, rodents, and yeast. However, its location and effect on gene expression reflect a new form of epitranscriptome control.
“The discovery of m1A is extremely important, not only because of its own potential in affecting biological processes but also because it validates the hypothesis that there is not just one functional modification,” Dr. He stated. “There could be multiple modifications at different sites where each may carry a distinct message to control the fate and function of mRNA.”
From their findings, the research team estimates that that m1A may be present on transcripts of more than one out of three expressed human genes—suggesting that m1A, like m6A, may be a mechanism by which cells rapidly boost the expression of hundreds or thousands of specific genes.
“mRNA is the perfect place to regulate gene expression because they can code information from transcription and directly impact translation—you can add a consensus sequence to a group of genes and use a modification of the sequence to readily control several hundred transcripts simultaneously,” Dr. He said. “If you want to rapidly change the expression of several hundred or a thousand genes, this offers the best way.”
The scientists were excited by their findings and have plans for future studies that will examine the role of m1A methylation in human development, for diseases such as diabetes and cancer, and its potential as a target for therapeutic uses.
“This study represents a breakthrough discovery in the exciting, nascent field of the ‘epitranscriptome,’ which is how RNAs are regulated, akin to the genome and the epigenome,” commented Christopher Mason, Ph.D., associate professor at Weill Cornell Medicine, who was not affiliated with the study. “What is important about this work is that m6A was recently found to enrich at the ends of genes, and now we know that m1A is what is helping regulate the beginning of genes, and this opens up many questions about revealing the ‘epitranscriptome code’ just like the histone code or the genetic code.”
The dynamic N1-methyladenosine methylome in eukaryotic messenger RNA
Gene expression can be regulated post-transcriptionally through dynamic and reversible RNA modifications. A recent noteworthy example is N6-methyladenosine (m6A), which affects messenger RNA (mRNA) localization, stability, translation and splicing. Here we report on a new mRNA modification, N1-methyladenosine (m1A), that occurs on thousands of different gene transcripts in eukaryotic cells, from yeast to mammals, at an estimated average transcript stoichiometry of 20% in humans. Employing newly developed sequencing approaches, we show that m1A is enriched around the start codon upstream of the first splice site: it preferentially decorates more structured regions around canonical and alternative translation initiation sites, is dynamic in response to physiological conditions, and correlates positively with protein production. These unique features are highly conserved in mouse and human cells, strongly indicating a functional role for m1A in promoting translation of methylated mRNA.
Figure 1: Development of m1A-seq to map a newly identified constituent of mammalian mRNA.
a, Chemical structures of m1A and m6A. Methyl groups (-CH3) are in red and the positive charge (+) on m1A is in blue. b, LC-MS/MS quantitation of m1A, m6A and Ψ in human and mouse mRNA isolated from the indicated cell types. …
Figure 3: m1A occurs in GC-rich sequence contexts and in genes with structured 5′ UTRs.
a, Sequence frequency logo for a set of 192 adenosines in peak areas that have a higher mismatch rate in immunoprecipitation relative to input (FC ≥ 6) in HepG2 demonstrates the GC-rich context of m1A. b, Length-adjusted minimum free energy…
Figure 5: m1A in mRNA is a dynamic modification that responds to changing physiological and stress conditions, and varies between tissues.
a, LC-MS/MS quantification of m1A (left, grey) and m6A (right, black) in mRNA of untreated and glucose-starved (upper panels) or heat shock-treated (lower panels) HepG2 cells, presented as percentage of unmodified A. Mean values ± s.e.m…
RNA modification discovery suggests new code for control of gene expression
A new cellular signal discovered by a team of scientists at the University of Chicago and Tel Aviv University provides a promising new lever in the control of gene expression. Gene expression study
The study, published online Feb. 10 in the journal Nature, describes a small chemical modification that can significantly boost the conversion of genes to proteins. Together with other recent findings, the discovery enriches a critical new dimension to the “Central Dogma” of molecular biology: the epitranscriptome.
“This discovery further opens the window on a whole new world of biology for us to explore,” said Chuan He, the John T. Wilson Distinguished Service Professor in Chemistry, Howard Hughes Medical Institute investigator and senior author of the study. “These modifications have a major impact on almost every biological process.”
The central dogma of molecular biology describes the cellular pathway where genetic information from DNA is copied into temporary RNA “transcripts,” which provide the recipe for the production of proteins. Since Francis Crick first postulated the theory in 1956, scientists have discovered a multitude of modifications to DNA and proteins that regulate this process.
Only recently, however, have scientists focused on investigating dynamic modifications that specifically target the RNA step. In 2011, He’s group discovered the first RNA demethylase that reverses the most prevalent mRNA methylation N6-methyladenosine, or m6A, implying that the addition and removal of the methyl group could dramatically affect these messengers and impact the outcome of gene expression, as also seen for DNA and histones. Subsequently, scientists discovered that the dynamic and reversible methylation of m6A dramatically controlled the metabolism and function of most cellular messenger RNA, and thus, the production of proteins.
In the new Nature study, researchers from UChicago and Tel Aviv University describe a second functional mRNA methylation, N1-methyladenosine, or m1A. Like m6A, the small modification is evolutionarily conserved and common, and present in humans, rodents and yeast, the authors found. But its location and effect on gene expression reflect a new form of epitranscriptome control and suggest an even larger cellular “control panel.”
“The discovery of m1A is extremely important, not only because of its own potential in affecting biological processes, but also because it validates the hypothesis that there is not just one functional modification,” He said. “There could be multiple modifications at different sites where each may carry a distinct message to control the fate and function of mRNA.”
The researchers estimated that m1A was present on transcripts of more than one out of three expressed human genes. Methylated genes exhibited enhanced translation compared to unmethlyated genes, producing protein levels nearly twice as high in all cell types. This increase suggests that m1A, like m6A, may be a mechanism by which cells rapidly boost the expression of hundreds or thousands of specific genes, perhaps during important processes such as cell division, differentiation or under stress.
“mRNA is the perfect place to regulate gene expression, because they can code information from transcription and directly impact translation; you can add a consensus sequence to a group of genes and use a modification of the sequence to readily control several hundred transcripts simultaneously,” He said. “If you want to rapidly change the expression of several hundred or a thousand genes, this offers the best way.”
However, despite their complementary effects, m1A and m6A exert their influence on mRNA through different pathways. While studies have found that m6A localizes predominantly to the tail of messenger RNA molecules, increasing their translation and turnover rate, m1A was found largely near the start codon of mRNA transcripts, where protein translation begins. The different mechanisms could allow for finer tuning of post-transcriptional gene expression, or the selective activation of particular genes in different physiological situations.
“This study represents a breakthrough discovery in the exciting, nascent field of the ‘epitranscriptome,’ which is how RNAs are regulated, akin to the genome and the epigenome,” said Christopher Mason, associate professor at Weill Cornell Medicine, who was not affiliated with the study. “What is important about this work is that m6A was recently found to enrich at the ends of genes, and now we know that m1A is what is helping regulate the beginning of genes, and this opens up many questions about revealing the ‘epitranscriptome code’ just like the histone code or the genetic code.”
Future studies will examine the role of m1A methylation in human development, diseases such as diabetes and cancer, and its potential as a target for therapeutic uses.
Citation: “The dynamic N1-methyladenosine methylome in eukaryotic messenger RNA,” Nature, Feb. 10, 2016, by Chuan He, Dan Dominissini, Sigrid Nachtergaele, Qing Dai, Dali Han, Wesley Clark, Guanqun Zheng, Tao Pan and Louis Dore from the University of Chicago, and Sharon Moshitch-Moshkovitz, Eyal Peer, Nitkan Kol, Moshe Shay Ben-Haim, Ayelet Di Segni, Mali Salmon-Divon, Oz Solomon, Eran Eyal, Vera Hershkovitz, Ninette Amariglio and Gideon Rechavi from Tel Aviv University. DOI: 10.1038/nature16998
Funding: National Institutes of Health, Howard Hughes Medical Institute, Flight Attendant Medical Research Institute, Israel Science Foundation, Israeli Centers of Excellence Program, Ernest and Bonnie Beutler Research Program, Chicago Biomedical Consortium, Damon Runyon Cancer Research Foundation and Kahn Family Foundation.
The meeting is aimed at bringing in students and postdocs as well as faculty involved in RNA modification and epitranscriptome research. In addition to talks, there will be a poster session and reception.
I. Cancer Causing Genes?
I have read an article recently that explained cancer as just plain “bad luck.” I do not believe this to be the case. There are harmful initiators of cancer and it can be associated with unfortunate timing, exposures, poor protections, faulty adaptation and circumstances beyond control. There are approximately 2,619 known genes that are associated with cancer expression and I am confident that this number will change as more research continues. These types of genes are also called oncogenes. These genes can be directly or indirectly associated with cancer initiation, progression, expression, survival and demise.
II. Epigenetics?
Epigenetics plays an important role in the turn on or off signaling of these genes by influencing DNA protection mechanisms without altering the DNA sequencing. Epigenetics refers to high-level information residing above the genetic code. While each cell in the body is equipped with the same genetic manual, epigenetic instructions tell cells how to make a difference. These instructions determine the access to pages with genetic information by directing the way the DNA is packaged into chromatin. DNA organized in loose chromatin is readily available for gene expression. Conversely, DNA tightly packed into dense chromatin has the letters of genetic code effectively buried and unavailable for reading and transcription. Distinct epigenetic marks decide which sets of genes may be expressed and which genes are kept silent.
III. DNA Methylation Hypomethylation and Hypermethylation?
In previous articles (blogs), we discussed the methylation process of DNA. Simply, methylation is the mechanism by which DNA is in homeostasis (healthy sequencing) or in harmful dysfunctional sequencing. Hypomethylation (decreased or low) and Hypermethylation (increased or high) are the two extremes that initiate harmful consequences upon DNA and ultimately lead to and result in unfavorable conditions and change in DNA sequencing performance. Ultimately, this leads to unfavorable function and potentially bad signals altering cell health and survival, predisposing new cell lines to “bad” mutations.
DNA methylation patterns undergo complex changes in cancer. The total amount of methylated cytosine is usually decreased resulting in global (extensive) hypomethylation. Decreased cytosine methylation typically affects satellite DNA, repetitive sequences, and CpG sites. In genetics, CpG is a site where cytosine (C) lies next to guanine (G) in the DNA sequence. (The p indicates that C and G are connected by a phosphodiester bond.) Methylation of DNA occurs at any CpG site. The cause of reduced amount of methylcytosine observed in human tumors has not been determined and remains under investigation. Despite global hypomethylation, high activity of DNA methyltransferases has been detected in multiple human tumor types. This increase may be related to higher proliferation rate of malignant cells.
Besides global (extensive) hypomethylation, most cancers also show focal hypermethylation in distinct subsets of promoter-associated CpG islands as well. Affected genes are permanently silenced, since methylation marks are propagated through mitosis and are maintained in the malignant clone. Aberrant (diverging from normal) hypermethylation occurring in transformed cells serves as an alternative mechanism for inactivation of tumor suppressor genes. Hundreds to thousands of genes can be epigenetically silenced by CpG island hypermethylation in human cancer suggesting a general disturbance of epigenetic memory. Methylation affects individual cancer patients with varying extent. While some patients have minimal changes, others show concordant hypermethylation of multiple genes. This phenomenon was first described as CpG island methylator phenotype (CIMP) in colorectal cancer and confirmed in many other types of cancer and leukemia. Epigenetic DNA methylation changes in cancer appear to be considerably more frequent events than genetic mutations. Mass sequencing of more than 20,000 transcripts in breast and colorectal cancers revealed about 80 harmless and less than 15 potentially oncogenic mutations per tumor. Balanced methylation and protective epigenetics are essential in minimizing cancer-causing mutations.
IV. Not Just Bad Luck!
Cancer is, in fact, a scary diagnosis and it is unfortunate when it occurs. However, I would not term it “bad luck,” as this implies a powerless and hopeless end. The gift of life is filled with miraculous movements that defy the laws of nature. Protective epigenetics can be considered miraculous and spontaneous and is what becomes necessary to move toward favorable adaptations that increase health, longevity and survival. Moreover, it is the mechanism by which succeeding familial generations can benefit in protections against cancer. It’s not just about our own protective actions on gene expression but also for the protective adaptability of future generations. There is hope and record of individuals having spontaneous regression of cancer. I have treated many in my clinical career! Encourage the expression of your healthy genes by protective epigenetics!
This structure (bottom left) of the malaria parasite’s proteasome, obtained using the revolutionary Cryo-Electron Microscopy technique, enabled the design of a specific inhibitor (front) against the mosquito-borne malaria parasite (pictured at back). [University of Melbourne]
With media attention recently focused on the spread of the Zika virus, it’s easy to forget about the mosquito-borne disease that has been credited with killing one out of every two people who have ever lived—malaria. Currently, close to 50 percent of the world’s population live in malaria-endemic areas, leading to between 200–500 million new cases and close to 500,000 deaths annually (mostly children under the age of five).
Adding to the complexities of trying to control this disease is that resistance to the most effective antimalarial drug, artemisinin, has developed in Southeast Asia, with fears it will soon reach Africa. Artemisinin-resistant species have spread to six countries in five years.
A collaborative team of scientists from Stanford University, University of California, San Francisco, University of Melbourne, and the MRC in Cambridge have used cutting-edge technology to design a smarter drug to combat the resistant strain.
“Artemisinin causes damage to the proteins in the malaria parasite that kill the human cell, but the parasite has developed a way to deal with that damage. So new drugs that work against resistant parasites are desperately needed,” explained coauthor Leann Tilley, Ph.D., professor and deputy head of biochemistry and molecular biology in the Bio21 Molecular Science and Biotechnology Institute at The University of Melbourne.
Malaria is caused by the protozoan parasite from the genus Plasmodium. Five different species are known to cause malaria in humans, with P. falciparum infection leading to the most deaths. The parasite is transmitted through the bite of the female mosquito and ultimately ends up residing within the host’s red blood cells (RBCs)—replicating and then bursting forth to invade more RBCs in a recurrently timed cycle.
“This penetration/replication/breakout cycle is rapid—every 48 hours—providing the opportunity for large numbers of mutations that can produce drug resistance,” said senior study author Matthew Bogyo, Ph.D., professor in the department of pathology at Stanford Medical School. “Consequently, several generations of antimalarial drugs have long since been rendered useless.”
The compound that investigators developed targets the parasites proteasome—a protein degradation pathway that removes surplus or damaged proteins through a cascade of proteolytic reactions.
“The parasite’s proteasome is like a shredder that chews up damaged or used-up proteins. Malaria parasites generate a lot of damaged proteins as they switch from one life stage to another and are very reliant on their proteasome, making it an excellent drug target,” Dr. Tilley noted.
The scientists purified the proteasome from the malaria parasite and examined its activity against hundreds of different peptide sequences. From this, they were able to design inhibitors that selectively targeted the parasite proteasome while sparing the human host enzymes.
The findings from this study were published recently in Nature through an article titled “Structure- and function-based design of Plasmodium-selective proteasome inhibitors.”
Additionally, scientists at the MRC used a new technique called Single-Particle Cryo-Electron Microscopy to generate a three-dimensional, high-resolution structure of a protein, based on thousands composite images.
The researchers tested the new drug in red blood cells infected with parasites and found that it was as effective at killing the artemisinin resistant parasites as it was for the sensitive parasites.
“The compounds we’ve derived can kill artemisinin-resistant parasites because those parasites have an increased need for highly efficient proteasomes,” Dr. Bogyo commented. “So, combining the proteasome inhibitor with artemisinin should make it possible to block the onset of resistance. That will, in turn, allow the continued use of that front-line malaria treatment, which has been so effective up until now.”
“The new proteasome inhibitors actually complement artemisinin drugs,” Dr. Tilley added. “Artemisinins cause protein damage and proteasome inhibitors prevent the repair of protein damage. A combination of the two provides a double whammy and could rescue the artemisinins as antimalarials, restoring their activity against resistant parasites.”
The scientists were excited by their results, as they may provide a much-needed strategy to combat the growing levels of resistance for this deadly pathogen. However, the researchers tempered their exuberance by noting that many more drug libraries needed to be screened before clinical trials can begin.
“The current drug is a good start, but it’s not yet suitable for humans. It needs to be able to be administered orally and needs to last a long time in the blood stream,” Dr. Tilley concluded.
Regulatory DNA Engineered, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 2: CRISPR for Gene Editing and DNA Repair
Regulatory DNA engineered
Larry H. Bernstein, MD, FCAP, Curator
LPBI
New Type of CRISPR Screen Probes the Regulatory Genome
February 8, 2016 | When a geneticist stares down the 3 billion DNA base pairs of the human genome, searching for a clue to what’s gone awry in a single patient, it helps to narrow the field. One of the most popular places to look is the exome, the tiny fraction of our DNA―less than 2%―that actually codes for proteins. For patients with rare genetic diseases, which might be fully explained by one key mutation, many studies sequence the whole exome and leave all the noncoding DNA out. Similarly, personalized cancer tests, which can help bring to light unexpected treatment options, often sequence the tumor exome, or a smaller panel of protein-coding genes.
Unfortunately, we know that’s not the whole picture. “There are a substantial number of noncoding regions that are just as effective at turning off a gene as a mutation in the gene itself,” says Richard Sherwood, a geneticist at Brigham and Women’s Hospital in Boston. “Exome sequencing is not going to be a good proxy for what genes are working.”
Sherwood studies regulatory DNA, the vast segment of the genome that governs which genes are turned on or off in any cell at a given time. It’s a confounding area of genetics; we don’t even know how much of the genome is made up of these regulatory elements. While genes can be recognized by the presence of “start” and “stop” codons―sequences of three DNA letters that tell the cell’s molecular machinery which stretches of DNA to transcribe into RNA, and eventually into protein―there are no definite signs like this for regulatory DNA.
Instead, studies to discover new regulatory elements have been somewhat trial-and-error. If you suspect a gene’s activity might be regulated by a nearby DNA element, you can inhibit that element in a living cell, and see if your gene shuts down with it.
With these painstaking experiments, scientists can slowly work their way through potential regulatory regions―but they can’t sweep across the genome with the kind of high-throughput testing that other areas of genetics thrive on. “Previously, you couldn’t do these sorts of tests in a large form, like 4,000 of them at once,” says David Gifford, a computational biologist at MIT. “You would really need to have a more hypothesis-directed methodology.”
Recently, Gifford and Sherwood collaborated on a paper, published in Nature Biotechnology, which presents a new method for testing thousands of DNA loci for regulatory activity at once. Their assay, called MERA (multiplexed editing regulatory assay), is built on the recent technology boom in CRISPR-Cas9 gene editing, which lets scientists quickly and easily cut specific sequences of DNA out of the genome.
So far, their team, including lead author Nisha Rajagopal from Gifford’s lab, has used MERA to study the regulation of four genes involved in the development of embryonic stem cells. Already, the results have defied the accepted wisdom about regulatory DNA. Many areas of the genome flagged by MERA as important factors in gene expression do not fall into any known categories of regulatory elements, and would likely never have been tested with previous-generation methods.
“Our approach allows you to look away from the lampposts,” says Sherwood. “The more unbiased you can be, the more we’ll actually know.”
A New Kind of CRISPR Screen
In the past three years, CRISPR-Cas9 experiments have taken all areas of molecular biology by storm, and Sherwood and Gifford are far from the first to use the technology to run large numbers of tests in parallel. CRISPR screens are an excellent way to learn which genes are involved in a cellular process, like tumor growth or drug resistance. In these assays, scientists knock out entire genes, one by one, and see what happens to cells without them.
This kind of CRISPR screen, however, operates on too small a scale to study the regulatory genome. For each gene knocked out in a CRISPR screen, you have to engineer a strain of virus to deliver a “guide RNA” into the cellular genome, showing the vicelike Cas9 molecule which DNA region to cut. That works well if you know exactly where a gene lies and only need to cut it once—but in a high-throughput regulatory test, you would want to blanket vast stretches of DNA with cuts, not knowing which areas will turn out to contain regulatory elements. Creating a new virus for each of these cuts is hugely impractical.
The insight behind MERA is that, with the right preparation, most of the genetic engineering can be done in advance. Gifford and Sherwood’s team used a standard viral vector to put a “dummy” guide RNA sequence, one that wouldn’t tell Cas9 to cut anything, into an embryonic stem cell’s genome. Then they grew plenty of cells with this prebuilt CRISPR system inside, and attacked each one with a Cas9 molecule targeted to the dummy sequence, chopping out the fake guide.
Normally, the result would just be a gap in the CRISPR system where the guide once was. But along with Cas9, the researchers also exposed the cells to new, “real” guide RNA sequences. Through a DNA repair mechanism called homologous recombination, the cells dutifully patched over the gaps with new guides, whose sequences were very similar to the missing dummy code. At the end of the process, each cell had a unique guide sequence ready to make cuts at a specific DNA locus—just like in a standard CRISPR screen, but with much less hands-on engineering.
By using a large enough library of guide RNA molecules, a MERA screen can include thousands of cuts that completely tile a broad region of the genome, providing an agnostic look at anywhere regulatory elements might be hiding. “It’s a lot easier [than a typical CRISPR screen],” says Sherwood. “The day the library comes in, you just perform one PCR reaction, and the cells do the rest of the work.”
In the team’s first batch of MERA screens, they created almost 4,000 guide RNAs for each gene they studied, covering roughly 40,000 DNA bases of the “cis-regulatory region,” or the area surrounding the gene where most regulatory elements are thought to lie. It’s unclear just how large any gene’s cis-regulatory region is, but 40,000 bases is a big leap from the highly targeted assays that have come before.
“We’re now starting to do follow-up studies where we increase the number of guide RNAs,” Sherwood adds. “Eventually, what you’d like is to be able to tile an entire chromosome.”
Far From the Lampposts
Sherwood and Gifford tried to focus their assays on regions that would be rich in regulatory elements. To that end, they made sure their guide RNAs covered parts of the genome with well-known signs of regulatory activity, like histone markers and transcription factor binding sites. For many of these areas, Cas9 cuts did, in fact, shut down gene expression in the MERA screens.
But the study also targeted regions around each gene that were empty of any known regulatory features. “We tiled some other regions that we thought might serve as negative controls,” explains Gifford. “But they turned out not to be negative at all.”
The study’s most surprising finding was that several cuts to seemingly random areas of the genome caused genes to become nonfunctional. The authors named these DNA regions “unmarked regulatory elements,” or UREs. They were especially prevalent around the genes Tdgf1 and Zfp42, and in many cases, seemed to be every bit as necessary to gene activity as more predictable hits on the MERA screen.
These results caught the researchers so off guard that it was natural to wonder if MERA screens are prone to false positives. Yet follow-up experiments strongly supported the existence of UREs. Switching the guide RNAs from aTdgf1 MERA screen and a Zfp42 screen, for example, produced almost no positive results: the UREs’ regulatory effects were indeed specific to the genes near them.
In a more specific test, the researchers chose a particular URE connected to Tdgf1, and cut it out of a brand new population of cells for a closer look. “We showed that, if we deleted that region from the genome, the cells lost expression of the gene,” says Sherwood. “And then when we put it back in, the gene became expressed again. Which was good proof to us that the URE itself was responsible.”
From these results, it seems likely that follow-up MERA screens will find even more unknown stretches of regulatory DNA. Gifford and Sherwood’s experiments didn’t try to cover as much ground around their target genes as they might have, because the researchers assumed that MERA would mostly confirm what was already known. At best, they hoped MERA would rule out some suspected regulatory regions, and help show which regulatory elements have the biggest effect on gene expression.
“We tended to prioritize regions that had been known before,” Sherwood says. “Unfortunately, in the end, our datasets weren’t ideally suited to discovering these UREs.”
Getting to Basic Principles
MERA could open up huge swaths of the regulatory genome to investigation. Compared to an ordinary CRISPR screen, says Sherwood, “there’s only upside,” as MERA is cheaper, easier, and faster to run.
Still, interpreting the results is not trivial. Like other CRISPR screens, MERA makes cuts at precise points in the genome, but does not tell cells to repair those cuts in any particular way. As a result, a population of cells all carrying the same guide RNA can have a huge variety of different gaps and scars in their genomes, typically deletions in the range of 10 to 100 bases long. Gifford and Sherwood created up to 100 cells for each of their guides, and sometimes found that gene expression was affected in some but not all of them; only sequencing the genomes of their mutated cells could reveal exactly what changes had been made.
By repeating these experiments many times, and learning which mutations affect gene expression, it will eventually be possible to pin down the exact DNA bases that make up each regulatory element. Future studies might even be able to distinguish between regulatory elements with small and large effects on gene expression. In Gifford and Sherwood’s MERA screens, the target genes were altered to produce a green fluorescent protein, so the results were read in terms of whether cells gave off fluorescent light. But a more precise, though expensive, approach would be to perform RNA sequencing, to learn which cuts reduced the cell’s ability to transcribe a gene into RNA, and by how much.
A MERA screen offers a rich volume of data on the behavior of the regulatory genome. Yet, as with so much else in genetics, there are few robust principles to let scientists know where they should be focusing their efforts. Histone markers provide only a very rough sketch of regulatory elements, often proving to be red herrings on closer examination. And the existence of UREs, if confirmed by future experiments, shows that we don’t yet even know which areas of the genome to rule out in the hunt for regulatory regions.
“Every dataset we get comes closer and closer to computational principles that let us predict these regions,” says Sherwood. As more studies are conducted, patterns may emerge in the DNA sequences of regulatory elements that link UREs together, or reveal which histone markers truly point toward regulatory effects. There might also be functional clues hidden in these sequences, hinting at what is happening on a molecular level as regulatory elements turn genes on and off in the course of a cell’s development.
For now, however, the data is still rough and disorganized. For better and for worse, high-throughput tools like MERA are becoming the foundation for most discoveries in genetics—and that means there is a lot more work to do before the regulatory genome begins to come into focus.
CORRECTED 2/9/16: Originally, this story incorrectly stated that only certain cell types could be assayed with MERA for reasons related to homologous recombination. In fact, the authors see no reason MERA could not be applied to any in vitro cell line, and hope to perform screens in a wide range of cell types. The text has been edited to correct the error.


















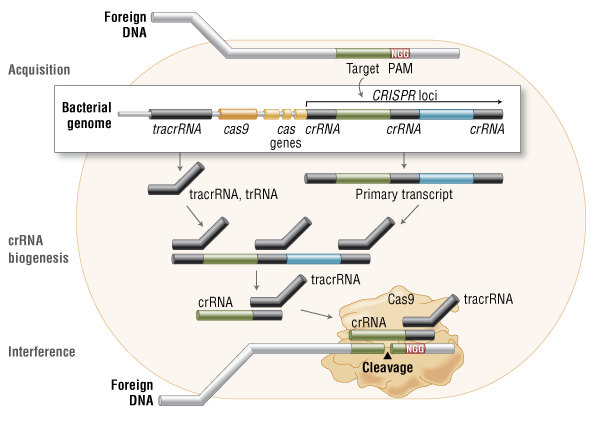{kind=link}
{kind=link}
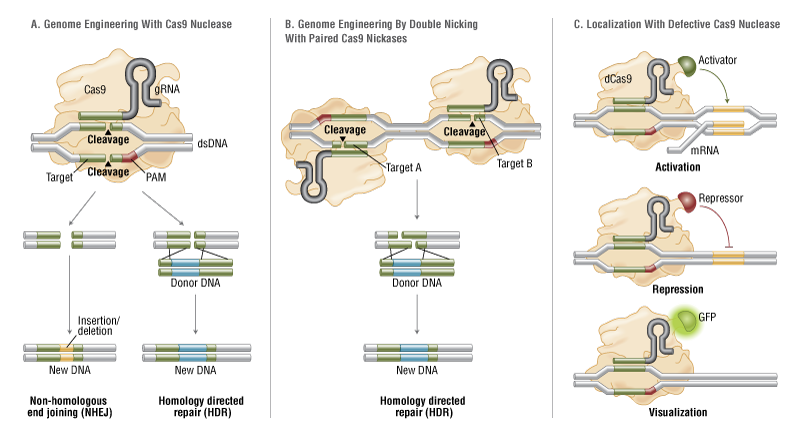{kind=link}
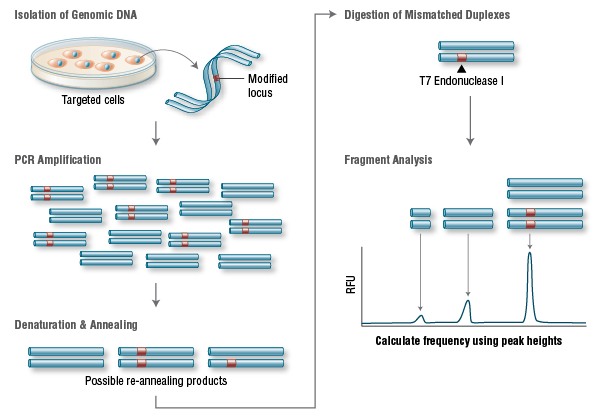{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}