Pros and Cons of Drug Stabilizers for Arterial Elasticity as an Alternative or Adjunct to Diuretics and Vasodilators in the Management of Hypertension.
Author, and Content Consultant to e-SERIES A: Cardiovascular Diseases: Justin Pearlman, MD, PhD, FACC
and
Article Curator: Aviva Lev-Ari, PhD, RN
This article presents the 2013 Thought Frontier on Hypertension and Vascular Compliance.
Conceptual development of the subject is presented in the following nine parts:
1. Physiology of Circulation and Role of Arterial Elasticity
2. Isolated Systolic Hypertension caused by Arterial Stiffening may be inadequately treated by Diuretics or Vasodilatation Antihypertensive Medications
3. Physiology of Circulation and Compensatory Mechanism of Arterial Elasticity
4. Vascular Compliance – The Potential for Novel Therapies
- Novel Mechanism for Disease Etiology: Modulation of Nuclear and Cytoskeletal Actin Polymerization.
- Genetic Therapy targeting Vascular Conductivity
- Regenerative Medicine for Vasculature Function Protection
5. In addition to curtailing high pressures, stabilizing BP variability is a potential target for management of hypertension
6. Mathematical Modeling: Arterial stiffening explains much of primary hypertension
7. Classification of Blood Pressure and Hypertensive Treatment Best Practice of Care in the US
8. Genetic Risk for High Blood Pressure
9. Is it Hypertension or Physical Inactivity: Cardiovascular Risk and Mortality – New results in 3/2013.
Summary By Justin D. Pearlman MD ME PhD MA FACC
1. Physiology of Circulation and Role of Arterial Elasticity
- Simplistically, high blood pressure stems from too much volume (salt water) for the vascular space, or conversely, too little space for the volume. Biological signals, such as endothelin, hypoxia, acidosis, nitric oxide, can modify vascular volume by constricting muscles in blood vessel walls. Less simplistically the physics of circulation are governed by numerous factors, with essentials detailed below.
- The vascular space has two major circuits: pulmonary (lungs) and systemic (body).
- Compliance (C) relates change in volume (ΔV) to change in pressure (ΔP) as a measure of the strength of elasticity, where elasticity summarizes the intrinsic forces that return to original shape after deformation: C = ΔV/ΔP . Those values can be estimated by ultrasound imaging with Doppler blood velocity estimation, by MRI, or invasively. Related properties can also be measured, such as wave propagation time or fractional flow reserve.
- The vascular system is dynamic, with frequency components and reactive elements. The fundamental frequency is governed by the heart rate delivering a stroke volume forward into the vasculature; a heart rate of 60/minute corresponds to the frequency of 1 Hertz (1 cycle/second). The pressure rise due to the ejection of stroke volume is called the pulse pressure.
- Numerous factors affect blood flow, including blood composition (affected by anemia or blood dilution), leakiness of vessels, elasticity, wave propagation, streamlines, viscosity, osmotic pressure (affected by protein deficiency and other factors),
- In a static system, the driving force relates linearly flow by way of resistance (R in units of dyn·s·cm−5): V=IR (Ohm’s law).
- Pulmonary:

- Systemic:
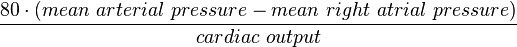
- Pulmonary:
- In a dynamic, reactive system, the relation between the driving potential (pressure gradient), and current (blood flow) is governed by a differential equation. However, use of complex numbers and exponentials recovers simplicity similar to Ohm’s law:
- Variables take the form
 , where t is time, s is a complex parameter, and A is a complex scalar. Complex values simply mean two dimensional, e.g., magnitude (as in resistance) plus phase shift (to account for reactive components).
, where t is time, s is a complex parameter, and A is a complex scalar. Complex values simply mean two dimensional, e.g., magnitude (as in resistance) plus phase shift (to account for reactive components). - Complex version of Ohm’s law:
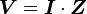 where V and I are the complex scalars in the voltage and current respectively and Z is the complex impedance.
where V and I are the complex scalars in the voltage and current respectively and Z is the complex impedance. - Frequency dependent “resistance” is captured by the term impedance.
- Variables take the form
- Breathing in increases the return of blood to the heart, adding to pulse variation.
- Dynamic elastance (Eadyn) relates volume variation (VVS) to pressure variation (PPV): Eadyn=PPV/SVV
- PPV(%) = 100% × (PPmax − PPmin)/[(PPmax + PPmin)/2)]
- where PPmax and PPmin are the maximum and minimum pulse pressures determined during a single respiratory cycle
- SVV(%) = 100% × [(SVmax − SVmin)/SVmean]
- where SVmax and SVmin are the maximum and minimum standard deviation of arterial pressure about the mean arterial pressure during a single respiratory cycle
- PPV(%) = 100% × (PPmax − PPmin)/[(PPmax + PPmin)/2)]
- The nervous system provides both stimulants and inhibitors (sympathetic and vagal nerves) to regulate blood vessel wall muscle tone and also heart rate. Many medications, and anesthetic agents in particular, reduce those responses to stimuli, so the vessels dilate, vascular impedance lowers, pressures drop, and autoregulation is impaired.
- Diuretics aim to decrease volume of circulating fluid, vasodilators aim to increase the vascular space, and elasticity treatments will aim to preserve or improve the ability to accommodate changes in volume of fluid.
- Vessel dilation near the skin promotes heat loss.
- Vascular elasticity is impaired by atherosclerosis, menopause, and endothelial dysfunction (impaired nitric oxide signals response, impaired endothelin response).
- Elastance in a cyclic pressure system of systole-diastole (contraction-dilation) presents impedance as a pulsatile load on the heart. Inotropy describes the generation of pressure by cardiac contraction, lusiotropy the compliance of the heart to accept filling with minimal back pressure to the lungs. Chronic exposure to elevated vascular impedance leads to impairment of lusiotropy (diastolic failure, stiff heart) and inotropy (systolic failure, weak heart).
2. Isolated Systolic Hypertension caused by Arterial Stiffening may be inadequately treated by Diuretics or Vasodilatation Antihypertensive Medications
- Stiff or “lead pipe” blood vessels drop pressure precipitously to dangerously low levels in response to diuretics.
- Stiff walls due to fibrosis or scar tissue have limited ability to dilate
- Red clover isoflavones improve vascular compliance loss from menopause.
- Fish oils may improve loss of vascular compliance from diabetes.
3. Physiology of Circulation and Compensatory Mechanism of Arterial Elasticity
Antihypertensive agents have focused on the following approaches:
-
Reduced volume (diuretics)
- Diuretics enhance renal excretion of sodium. Sodium is the major determinant of circulating volume. Too much blood volume for the amount of vascular space elevates blood pressure. Clinical trials show that use of diuretics to lower blood pressure can prevent strokes, non-inferior to vasodilators and recommended as first line agents.
- The most common prescriptions, a mild diuretic, hydrochlorothiazide (HCTZ), is known to improve blood vessel compliance by reducing cell turgor, which explains why its full onset of benefit as well as its slow offset when stopped can take more than one month.
- Chlorthalidone – Some evidence suggests that chlorthalidone may be superior to hydrochlorothiazide for the treatment of hypertension. However, a recent study concluded: chlorthalidone in older adults was not associated with fewer adverse cardiovascular events or deaths than hydrochlorothiazide. However, it was associated with a greater incidence of electrolyte abnormalities, particularly hypokalemia.
-
Increased vascular space (vasodilation)
- Alternatively, the pressure can be lowered by increasing the vascular space for a given vascular volume. Examples of mediators for arterial tone (degree of dilation) include nitric oxide, prostacyclin and endothelin.
|
Class |
Description |
| Hyperpolarization mediated (Calcium channel blocker) | Changes in the resting membrane potential of thecell affects the level of intracellular calciumthrough modulation of voltage sensitive calcium channelsin the plasma membrane. |
| cAMP mediated | Adrenergic stimulation results in elevated levelsof cAMP and protein kinase A, which results inincreasing calcium removal from the cytoplasm. |
| cGMP mediated (Nitrovasodilator) | Through stimulation of protein kinase G.Until 2002, the enzyme for this conversion wasdiscovered to be mitochondrial aldehyde dehydrogenase.Proc. Natl. Acad. Sci. USA 102 (34): 12159–12164. doi:10.1073/pnas.0503723102http://www.pnas.org/content/102/34/12159.long |
|
Class |
Example |
| Hyperpolarization mediated (Calcium channel blocker) | adenosineamlodipine (Norvasc),diltiazem (Cardizem,Dilacor XR) andnifedipine (Adalat, Procardia). |
| cAMP mediated | prostacyclin |
| cGMP mediated (Nitrovasodilator) | nitric oxide |
-
Reduced pulsatile force (beta blockers)
These work by blocking certain nerve and hormonal signals to the heart and blood vessels, thus lowering blood pressure. Frequently prescribed beta blockers include
- metoprolol (Lopressor, Toprol XL)
- carvedilol (Coreg)
- nadolol (Corgard)
- penbutolol (Levatol).
- Metabolized nebivolol increases vascular NO production, involves endothelial ß2-adrenergic receptor ligation, with a subsequent rise in endothelial free [Ca2+]i and endothelial NO synthase–dependent NO production
-
Angiotensin-converting enzyme (ACE) inhibitors
These allow blood vessels to widen by preventing the hormone angiotensin from affecting blood vessels. Frequently prescribed ACE inhibitors include captopril (Capoten), lisinopril (Prinivil, Zestril) and ramipril (Altace).
-
Angiotensin II receptor blockers
These help blood vessels relax by blocking the action of angiotensin. Frequently prescribed angiotensin II receptor blockers include losartan (Cozaar), olmesartan (Benicar) and valsartan (Diovan).
Another very commonly prescribed drug class of medication counteracts hardening of arteries.
Atheroma lipids have enzyme systems that explicitly disassemble cholesterol esters and reconstruct them inside blood vessel walls,e.g., Anacetrapib, Genetic variants that improve cholesterol levels are stimulating development of additional medications.
We can propose that atheroma build up in arterial blood vessel walls constitutes a maladaptive defense against aneurysm and risk of vessel rupture from hypertension.
Arguably, HMG-CoA reductase inhibitors, statin therapy is a second example of a medication that helps protect vascular elasticity, both by its lipid effects and its anti-inflammatory effects.
The best-selling statin is atorvastatin, marketed as Lipitor (manufactured by Pfizer) and Torvast. By 2003, atorvastatin became the best-selling pharmaceutical in history,[4] with Pfizer reporting sales of US$12.4 billion in 2008.[5] As of 2010, a number of statinsare on the market: atorvastatin (Lipitor and Torvast), fluvastatin (Lescol), lovastatin (Mevacor, Altocor, Altoprev), pitavastatin(Livalo, Pitava), pravastatin (Pravachol, Selektine, Lipostat), rosuvastatin (Crestor) and simvastatin (Zocor, Lipex).[6] Several combination preparations of a statin and another agent, such as ezetimibe/simvastatin, are also available.
References for Statins from:
http://en.wikipedia.org/wiki/Statin
Clinical Considerations of Statin Therapy’s manifold effects, in
Compensatory Effects in the Physiology of Circulation
Before declaring vessel elasticity a new and highly desirable treatment target, consider that it is not firmly established that hardening of arteries (loss of elasticity) is entirely maladaptive.
In parallel with any focus on increasing vascular elasticity or compliance, each of the issues discussed, below merits scrutiny and investigation.
Cardiac Circulation Dynamics
Endothelium morphology, rheological properties of intra vasculature fluid dynamics and blood viscosity provided explanation for shear stress of vessels under arterial pressure
and
Aging and Vasculature Diminished Elasticity
While among other reasons for Hypertension increasing prevalence with aging, arterial stiffening is one.
Yet, stiffer vessels are more efficient at transmitting pressure to distal targets. With aging, muscle mass diminishes markedly and the contribution to circulation from skeletal muscle tissue compressions combined with competent venous valves fades.
and
and
Aging and Myocardial Diminished Contractility and Ejection Fraction
With aging heart contractility diminishes. These issues can cause under perfusion of tissues, inadequate nutrient blood delivery (ischemia), lactic acidosis, tissue dysfunction and multi-organ failure. Hardened arteries may compensate. Thus, pharmacotherapy to increase Arterial Elasticity may be counterindicated for patients with mild to progressive CHF.
http://pharmaceuticalintelligence.com/2013/05/05/bioengineering-of-vascular-and-tissue-models/
and
and
http://pharmaceuticalintelligence.com/2012/10/17/chronic-heart-failure-personalized-medicine-two-gene-test-predicts-response-to-beta-blocker-bucindolol/
Our biosystems are highly interdependent, and we cannot leap to conclusions without careful thorough evidence. Increasing arterial elastance will lower vascular impedance and change the frequency components of our pulsatile perfusion system.
MOST comprehensive review of the Human Cardiac Conduction System presented to date:
Diminished contractility will increase the amount of energy needed to maintain circulation. It will change efficiency dramatically – consider the difference between periodically pushing someone sitting on a swing at the resonance frequency if the pendulum versus significantly off resonance.
http://pharmaceuticalintelligence.com/2013/04/14/mitochondrial-metabolism-and-cardiac-function/
and
Increased Arterial Elasticity – Potential Risk to Myocardium
The hypothesis that we should focus on cellular therapies to increase vascular compliance may decrease the circulation efficiency and result in worsening of cardiac right ventricular morphology and development of Dilated cardiomyopathy and hypertrophic cardiomyopathy (muscle thickening and diastolic failure), an undesirable outcome resulting from an attempt to treat the hypertension.
4. Vascular Compliance – The Potential of Noval Therapies
-
Novel Mechanism for Disease Etiology for the Cardiac Phenotype: Modulation of Nuclear and Cytoskeletal Actin Polymerization.
Lamin A/C and emerin regulate MKL1–SRF activity by modulating actin dynamics
Nature (2013) doi:10.1038/nature12105
Published online 05 May 2013
Affiliations
Cornell University, Weill Institute for Cell and Molecular Biology/Department of Biomedical Engineering, Ithaca, New York 14853, USA
Chin Yee Ho &
Jan Lammerding
Brigham and Women’s Hospital/Harvard Medical School, Department of Medicine, Boston 02115, Massachusetts, USA
Chin Yee Ho,
Diana E. Jaalouk &
Jan Lammerding
Institute of Biotechnology, University of Helsinki, 00014 Helsinki, Finland
Maria K. Vartiainen
Present address: American University of Beirut, Department of Biology, Beirut 1107 2020, Lebanon.
Diana E. Jaalouk
Contributions
C.Y.H., D.E.J. and J.L. conceived and designed the overall project, with valuable help from M.K.V. C.Y.H. and D.E.J. performed the experiments. C.Y.H., D.E.J. and J.L. analysed data. C.Y.H. and J.L. wrote the paper.
Corresponding author Jan Lammerding
Laminopathies, caused by mutations in the LMNA gene encoding the nuclear envelope proteins lamins A and C, represent a diverse group of diseases that include Emery–Dreifuss muscular dystrophy (EDMD), dilated cardiomyopathy (DCM), limb-girdle muscular dystrophy, and Hutchison–Gilford progeria syndrome1. Most LMNA mutations affect skeletal and cardiac muscle by mechanisms that remain incompletely understood. Loss of structural function and altered interaction of mutant lamins with (tissue-specific) transcription factors have been proposed to explain the tissue-specific phenotypes1. Here we report in mice that lamin-A/C-deficient (Lmna−/−) and LmnaN195K/N195K mutant cells have impaired nuclear translocation and downstream signalling of the mechanosensitive transcription factor megakaryoblastic leukaemia 1 (MKL1), a myocardin family member that is pivotal in cardiac development and function2. Altered nucleo-cytoplasmic shuttling of MKL1 was caused by altered actin dynamics in Lmna−/− and LmnaN195K/N195K mutant cells. Ectopic expression of the nuclear envelope protein emerin, which is mislocalized in Lmnamutant cells and also linked to EDMD and DCM, restored MKL1 nuclear translocation and rescued actin dynamics in mutant cells. These findings present a novel mechanism that could provide insight into the disease aetiology for the cardiac phenotype in many laminopathies, whereby lamin A/C and emerin regulate gene expression through modulation of nuclear and cytoskeletal actin polymerization.
http://www.nature.com/nature/journal/vaop/ncurrent/full/nature12105.html
-
Genetic Therapy to Conductivity Disease
-
Regenerative Medicine for Vasculature Function Protection
and
and
5. Stabilizing BP Variability is the next Big Target in Hypertension Management
Hypertension caused by Arterial Stiffening is Ineffectively Treated by Diuretics and Vasodilatation Antihypertensives
Barcelona, Spain – An aging population grappling with rising rates of hypertension and other cardiometabolic risk factors should prompt an overhaul of how hypertension is diagnosed and monitored and should spur development of drugs with entirely new mechanisms of action, one expert says. Speaking here at the 2013 International Conference on Prehypertension and Cardiometabolic Syndrome, meeting cochair Dr Reuven Zimlichman (Tel Aviv University, Israel) argued that the definitions of hypertension, as well as the risk-factor tables used to guide treatment, are no longer appropriate for a growing number of patients.
Most antihypertensives today work by producing vasodilation or decreasing blood volume and so are ineffective treatments in ISH patients. In the future, he predicts, “we will have to start looking for a totally different medication that will aim to improve or at least to stabilize arterial elasticity: medication that might affect factors that determine the stiffness of the arteries, like collagen, like fibroblasts. Those are not the aim of any group of antihypertensive medications today.”
Zimlichman believes existing databases could be used to develop algorithms that take this progression of disease into account, in order to better guide hypertension management. He also points out that new ambulatory blood-pressure-monitoring devices also measure arterial elasticity. “Unquestionably, these will improve our ability to diagnose both the status of the arteries and the changes of the arteries with time as a result of our treatment. So if we treat the patient and we see no improvement in arterial elasticity, or the patient is worse, something is wrong, something is not working—either the patient is not taking the medication, or our choice of medication is not appropriate, or the dose is insufficient, etc.”
http://www.theheart.org/article/1502067.do
Oslo, Norway – New research that is only just starting to be digested by the hypertension community indicates that visit-to-visit variability in blood-pressure readings will likely become another way of looking for “at-risk” hypertensive patients and in fact is likely to be more reliable as an indicator of cardiovascular risk than the currently used mean BP.
The Goal of Stabilizing BP variability
June 29, 2010 Lisa Nainggolan
Discussing the importance of this issue for guidelines and clinical practice, Dr Tony Heagerty (University of Manchester, UK) told the recent European Society of Hypertension (ESH) European Meeting on Hypertension 2010: “We are poking around in the dark, offering treatment blankly across a large community, and probably treating a lot of people who don’t need to be treated, while not necessarily treating the highest-risk patients. We should stop being reassured by ‘occasional’ normal BPs. The whole game now is, can we improve the identification of our ‘at-risk’ individuals?”
Heagerty was speaking at a special plenary session on late-breaking research discussing BP variability as a risk factor. This issue has emerged following new analyses reported at the ACC meeting and published in a number of papers in the Lancet and Lancet Neurology earlier this year, which showed that variability in blood pressure is a much stronger determinant of both stroke and coronary disease outcome than average blood pressure.
http://www.theheart.org/article/1093553.do
Three years later, 2/1/2013, Zimlichman also argued that definitions of essential and secondary hypertension have changed very little over the past few decades and have typically only been tweaked up or down related to other CV risk factors. Diastolic hypertension has been the primary goal of treatment, and treatment goals have not adequately taken patient age into account (in whom arterial stiffening plays a larger role), and they have typically relied too heavily on threshold cutoffs, rather than the “linear progression” of risk factors and their impact on organ damage.
6. Mathematical Modeling: Arterial stiffening provides sufficient explanation for primary hypertension
Klas H. Pettersen, Scott M. Bugenhagen, Javaid Nauman, Daniel A. Beard, Stig W. Omholt
(Submitted on 3 May 2013 (v1), last revised 6 May 2013 (this version, v2))
Hypertension is one of the most common age-related chronic diseases and by predisposing individuals for heart failure, stroke and kidney disease, it is a major source of morbidity and mortality. Its etiology remains enigmatic despite intense research efforts over many decades. By use of empirically well-constrained computer models describing the coupled function of the baroreceptor reflex and mechanics of the circulatory system, we demonstrate quantitatively that arterial stiffening seems sufficient to explain age-related emergence of hypertension. Specifically, the empirically observed chronic changes in pulse pressure with age, and the impaired capacity of hypertensive individuals to regulate short-term changes in blood pressure, arise as emergent properties of the integrated system. Results are consistent with available experimental data from chemical and surgical manipulation of the cardio-vascular system. In contrast to widely held opinions, the results suggest that primary hypertension can be attributed to a mechanogenic etiology without challenging current conceptions of renal and sympathetic nervous system function. The results support the view that a major target for treating chronic hypertension in the elderly is the reestablishment of a proper baroreflex response.
Klas H. Pettersen1, Scott M. Bugenhagen2, Javaid Nauman3, Daniel A. Beard2 & Stig W. Omholt3
1Department of Mathematical and Technological Sciences, Norwegian University of Life Science, Norway
2Department of Physiology, Medical College of Wisconsin, Milwaukee, Wisconsin, USA
3NTNU Norwegian University of Science and Technology, Department of Circulation and Medical Imaging, Cardiac Exercise Research Group, Trondheim, Norway
Correspondence should be addressed to: KHP (klas.pettersen@gmail.com)
Keywords: hypertension, mechanogenic, baroreceptor signaling, cardiovascular model, arterial stiffening
Author contributions: K.H.P. and S.W.O. designed the study. K.H.P. constructed the
integrated model and performed the numerical experiments with contributions from
D.A.B. and S.M.B.. J.N. extracted and compiled empirical test data from the HUNT2
Survey. S.W.O, K.H.P. and D.A.B. wrote the paper.
7. Classification of Blood Pressure and Hypertensive Treatment:
Best Practice of Care in the US
8. Genetic Risk for High Blood Pressure
Hypertension.2013; 61: 931doi: 10.1161/HYP.0b013e31829399b2
Blood Pressure Single-Nucleotide Polymorphisms and Coronary Artery Sisease (page 995)
Blood pressure (BP) is considered a major cardiovascular risk factor that is influenced by multiple genetic and environmental factors. However, the precise genetic underpinning influencing interindividual BP variation is not well characterized; and it is unclear whether BP-associated genetic variants also predispose to clinically apparent cardiovascular disease. Such an association of BP-related variants with cardiovascular disease would strengthen the concept of BP as a causal risk factor for cardiovascular disease. In this issue of Hypertension, analyses within the Coronary ARtery DIsease Genome-Wide Replication And Meta-Analysis consortium indicate that common genetic variants associated with BP in the population, indeed, contribute to the susceptibility for coronary artery disease (CAD). Lieb et al tested 30 single-nucleotide polymorphisms—that based on prior studies were known to affect BP—for their association with CAD. In total, data from 22 233 CAD cases and 64 762 controls were analyzed. The vast majority (88%) of BP-related single-nucleotide polymorphisms were also shown to increase the risk of CAD (as defined by an odds ratio for CAD >1; Figure). On average, each of the multiple BP-raising alleles was associated with a 3% (95% confidence interval, 1.8%–4.3%) risk increase for CAD.
Masked Hypertension in Diabetes Mellitus (page 964)
The first important finding in the IDACO study of masked hypertension (MH) in the population with diabetes mellitus and non–diabetes mellitus was that antihypertensive treatment converted some sustained hypertensives into sustained normotensives; this resulted in an increased cardiovascular disease risk in the treated versus untreated normotensive comparator group (Figure). Not surprisingly, normalization of blood pressure (BP) with treatment did not eliminate the lifetime cardiovascular disease burden associated with prior elevated BP nor did it correct other cardiometabolic risk factors that clustered with the hypertensive state.
The second important IDACO finding was that treatment increased the prevalence of MH by decreasing conventional BP versus daytime ambulatory BP (ABP) by a ratio of ≈3 to 2. The clinical implication of increased prevalence of MH with therapy in the population of both diabetes mellitus and non–diabetes mellitus was that these subjects did not receive sufficient antihypertensive therapy to convert MH into normalized ABP (ie, treated, normalized ABP being the gold standard for minimizing cardiovascular disease risk). Indeed, there is a transformation-continuum from sustained hypertension to MH and finally to sustained normotension with increasing antihypertensive therapy. These IDACO findings strongly suggest that many physicians mistakenly have their primary focus on normalizing in-office rather than out-of-office home BP and/or 24-hour ABP values and this results in an increased prevalence of MH. However, what constitutes optimal normalized ABP will remain empirical until established in randomized controlled trials.
Genetic Risk Score for Blood Pressure (page 987)
Elevated blood pressure (BP) is a strong, independent, and modifiable risk factor for stroke and heart disease. BP is a heritable trait, and genome-wide association studies have identified several genetic loci that are associated with systolic BP, diastolic BP, or both. Although the variants have modest effects on BP, typically 0.5 to 1.0 mm Hg, their presence may act over the entire life course and, therefore, lead to substantial increase in risk of cardiovascular disease (CVD). However, the independent impact of these variants on CVD risk has not been established in a prospective setting. Havulinna et al genotyped 32 common single-nucleotide polymorphisms in several Finnish cohorts, with up to 32 669 individuals after exclusion of prevalent CVD cases. The median follow-up was 9.8 years, during which 2295 incident CVD events occurred. Genetic risk scores were created for systolic BP and diastolic BP by multiplying the risk allele count of each single-nucleotide polymorphism by the effect size estimated in published genome-wide association studies on BP traits. The GRSs were strongly associated with baseline systolic BP, diastolic BP, and hypertension (all P<10–62). Hazard ratios for incident CVD increased roughly linearly by quintile of systolic BP or diastolic BP GRS (Figure). GRSs remained significant predictors of CVD risk after adjustment for traditional risk factors, even including BP and use of antihypertensive medication. These findings are consistent with a lifelong effect of these variants on BP and CVD risk.
Related Articles on Genetics and Blood Pressure
- Wolfgang Lieb,
- Henning Jansen,
- Christina Loley,
- Michael J. Pencina,
- Christopher P. Nelson,
- Christopher Newton-Cheh,
- Sekar Kathiresan,
- Muredach P. Reilly,
- Themistocles L. Assimes,
- Eric Boerwinkle,
- Alistair S. Hall,
- Christian Hengstenberg,
- Reijo Laaksonen,
- Ruth McPherson,
- Unnur Thorsteinsdottir,
- Andreas Ziegler,
- Annette Peters,
- John R. Thompson,
- Inke R. König,
- Jeanette Erdmann,
- Nilesh J. Samani,
- Ramachandran S. Vasan,
- andHeribert Schunkert
- , on behalf of CARDIoGRAM
Hypertension. 2013;61:995-1001, published online before print March 11 2013,doi:10.1161/HYPERTENSIONAHA.111.00275
- Stanley S. Franklin,
- Lutgarde Thijs,
- Yan Li,
- Tine W. Hansen,
- José Boggia,
- Yanping Liu,
- Kei Asayama,
- Kristina Björklund-Bodegård,
- Takayoshi Ohkubo,
- Jørgen Jeppesen,
- Christian Torp-Pedersen,
- Eamon Dolan,
- Tatiana Kuznetsova,
- Katarzyna Stolarz-Skrzypek,
- Valérie Tikhonoff,
- Sofia Malyutina,
- Edoardo Casiglia,
- Yuri Nikitin,
- Lars Lind,
- Edgardo Sandoya,
- Kalina Kawecka-Jaszcz,
- Jan Filipovský,
- Yutaka Imai,
- Jiguang Wang,
- Hans Ibsen,
- Eoin O’Brien,
- and Jan A. Staessen
- , on behalf of the International Database on Ambulatory blood pressure in relation to Cardiovascular Outcomes (IDACO) Investigators
Hypertension. 2013;61:964-971, published online before print March 11 2013,doi:10.1161/HYPERTENSIONAHA.111.00289
- Aki S. Havulinna,
- Johannes Kettunen,
- Olavi Ukkola,
- Clive Osmond,
- Johan G. Eriksson,
- Y. Antero Kesäniemi,
- Antti Jula,
- Leena Peltonen,
- Kimmo Kontula,
- Veikko Salomaa,
- and Christopher Newton-Cheh
Hypertension. 2013;61:987-994, published online before print March 18 2013,doi:10.1161/HYPERTENSIONAHA.111.00649
9. Is it Hypertension or Physical Inactivity: Cardiovascular Risk and Mortality – New results in 3/2013.
Heart doi:10.1136/heartjnl-2012-303461
- Epidemiology
- Original article
Estimating the effect of long-term physical activity on cardiovascular disease and mortality: evidence from the Framingham Heart Study
+Author Affiliations
1Department of Epidemiology and Preventive Medicine, Monash University, Melbourne, Australia
2Biostatistics Unit, Group Health Research Institute, Seattle, Washington, USA
3Obesity and Population Health Unit, Baker IDI Heart and Diabetes Institute, Melbourne, Australia
Correspondence toDr Susan M Shortreed, Biostatistics Unit, Group Health Research Institute, 1730 Minor Avenue, Suite 1600, Seattle, WA 98101, USA; shortreed.s@ghc.org
- Published Online First 8 March 2013
Abstract
Objective In the majority of studies, the effect of physical activity (PA) on cardiovascular disease (CVD) and mortality is estimated at a single time point. The impact of long-term PA is likely to differ. Our study objective was to estimate the effect of long-term adult-life PA compared with long-term inactivity on the risk of incident CVD, all-cause mortality and CVD-attributable mortality.
Design Observational cohort study.
Setting Framingham, MA, USA.
Patients 4729 Framingham Heart Study participants who were alive and CVD-free in 1956.
Exposures PA was measured at three visits over 30 years along with a variety of risk factors for CVD. Cumulative PA was defined as long-term active versus long-term inactive.
Main outcome measures Incident CVD, all-cause mortality and CVD-attributable mortality.
Results During 40 years of follow-up there were 2594 cases of incident CVD, 1313 CVD-attributable deaths and 3521 deaths. Compared with long-term physical inactivity, the rate ratio of long-term PA was 0.95 (95% CI 0.84 to 1.07) for CVD, 0.81 (0.71 to 0.93) for all-cause mortality and 0.83 (0.72 to 0.97) for CVD-attributable mortality. Assessment of effect modification by sex suggests greater protective effect of long-term PA on CVD incidence (p value for interaction=0.004) in men (0.79 (0.66 to 0.93)) than in women (1.15 (0.97 to 1.37)).
Conclusions
- Cumulative long-term PA has a protective effect on incidence of all-cause and CVD-attributable mortality compared with long-term physical inactivity.
- In men, but not women, long-term PA also appears to have a protective effect on incidence of CVD.
Summary – PENDING
1. Kannel WB, Gordan T (1978) Evaluation of cardiovascular risk in the elderly: the Framingham study. Bull N Y Acad Med 54:573–591.
40. Kirkwood TBL (1977) Evolution of ageing. Nature 270:301–304.
41. Nakayama Y et al. (2001) Heart Rate-Independent Vagal Effect on End-Systolic Elastance of the Canine Left Ventricle Under Various Levels of Sympathetic Tone. Circulation 104:2277–2279.
42. Cohen A (1991) A Padé approximant to the inverse Langevin function. Rheologic Acta 30:270–273.
43. Brown AM, Saum WR, Tuley FH (1976) A comparison of aortic baroreceptor discharge in normotensive and spontaneously hypertensive rats. Circ Res 39:488–496.
44. Smith H (2011) in Texts in Applied Mathematics, Texts in Applied Mathematics. (Springer New York, New York, NY), pp 119–130.
Other related articles were published on this Open Access Online Scientific Journal including the following:
Pearlman, JD and A. Lev-Ari 5/24/2013 Imaging Biomarker for Arterial Stiffness: Pathways in Pharmacotherapy for Hypertension and Hypercholesterolemia Management
Lev-Ari, A. 5/17/2013 Synthetic Biology: On Advanced Genome Interpretation for Gene Variants and Pathways: What is the Genetic Base of Atherosclerosis and Loss of Arterial Elasticity with Aging
Bernstein, HL and A. Lev-Ari 5/15/2013 Diagnosis of Cardiovascular Disease, Treatment and Prevention: Current & Predicted Cost of Care and the Promise of Individualized Medicine Using Clinical Decision Support Systems
Pearlman, JD and A. Lev-Ari 5/7/2013 On Devices and On Algorithms: Arrhythmia after Cardiac Surgery Prediction and ECG Prediction of Paroxysmal Atrial Fibrillation Onset
Pearlman, JD and A. Lev-Ari 5/4/2013 Cardiovascular Diseases: Decision Support Systems for Disease Management Decision Making
Aviva Lev-Ari, PhD, RN and Larry H. Bernstein, MD, FACP, 3/7/2013
Aviva Lev-Ari, PhD, RN, 4/28/2013
Aviva Lev-Ari, PhD, RN, 5/29/2012
Mitochondrial Dysfunction and Cardiac Disorders
Curator: Larry H Bernstein, MD, FACP


















. Image:Cardiac a...")
. Image:Cardiac a...")

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}