Voluntary and Involuntary S- Insufficiency
Writer and Curator: Larry H Bernstein, MD, FCAP
Transthyretin and the Stressful Condition
Introduction
This article is written among a series of articles concerned with stress, obesity, diet and exercise, as well as altitude and deep water diving for extended periods, and their effects. There is a reason that I focus on transthyretin (TTR), although much can be said about micronutients and vitamins, and fat soluble vitamins in particular, and iron intake during pregnancy. While the importance of vitamins and iron are well accepted, the metabolic basis for their activities is not fully understood. In the case of a single amino acid, methionine, it is hugely important because of the role it plays in sulfur metabolism, the sulfhydryl group being essential for coenzyme A, cytochrome c, and for disulfide bonds. The distribution of sulfur, like the distribution of iodine, is not uniform across geographic regions. In addition, the content of sulfur found in plant sources is not comparable to that in animal protein. There have been previous articles at this site on TTR, amyloid and sepsis.
Transthyretin and Lean Body Mass in Stable and Stressed State
A Second Look at the Transthyretin Nutrition Inflammatory Conundrum
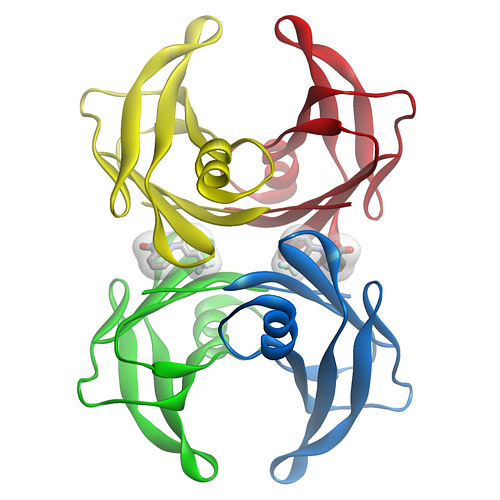
Stabilizers that prevent transthyretin-mediated cardiomyocyte amyloidotic toxicity
Thyroid Function and Disorders
http://pharmaceuticalintelligence.com/2015/02/05/thyroid-function-and-disorders/
Proteomics, Metabolomics, Signaling Pathways, and Cell Regulation: a Compilation of Articles in the Journal http://pharmaceuticalintelligence.com
Malnutrition in India, high newborn death rate and stunting of children age under five years
Vegan Diet is Sulfur Deficient and Heart Unhealthy
http://pharmaceuticalintelligence.com/2013/11/17/vegan-diet-is-sulfur-deficient-and-heart-unhealthy/
How Methionine Imbalance with Sulfur-Insufficiency Leads to Hyperhomocysteinemia
http://pharmaceuticalintelligence.com/2013/04/04/sulfur-deficiency-leads_to_hyperhomocysteinemia/
Amyloidosis with Cardiomyopathy
http://pharmaceuticalintelligence.com/2013/03/31/amyloidosis-with-cardiomyopathy/
Advances in Separations Technology for the “OMICs” and Clarification of Therapeutic Targets
Sepsis, Multi-organ Dysfunction Syndrome, and Septic Shock: A Conundrum of Signaling Pathways Cascading Out of Control
Automated Inferential Diagnosis of SIRS, sepsis, septic shock
Transthyretin and the Systemic Inflammatory Response
Transthyretin has been widely used as a biomarker for identifying protein-energy malnutrition (PEM) and for monitoring the improvement of nutritional status after implementing a nutritional intervention by enteral feeding or by parenteral infusion. This has occurred because transthyretin (TTR) has a rapid removal from the circulation in 48 hours and it is readily measured by immunometric assay. Nevertheless, concerns have been raised about the use of TTR in the ICU setting, which prompts a review of the actual benefit of using this test in a number of settings. TTR is easily followed in the underweight and the high risk populations in an ambulatory setting, which has a significant background risk of chronic diseases. It is sensitive to the systemic inflammatory response syndrom (SIRS), and needs to be understood in the context of acute illness to be used effectively. There are a number of physiologic changes associated with SIRS and the injury/repair process that will affect TTR and will be put in context in this review. The most important point is that in the context of an ICU setting, the contribution of TTR is significant in a complex milieu. copyright @ Bentham Publishers Ltd. 2009.
Transthyretin as a marker to predict outcome in critically ill patients.
Arun Devakonda, Liziamma George, Suhail Raoof, Adebayo Esan, Anthony Saleh, Larry H. Bernstein.
Clin Biochem Oct 2008; 41(14-15): 1126-1130
A determination of TTR level is an objective method od measuring protein catabolic loss of severly ill patients and numerous studies show that TTR levels correlate with patient outcomes of non-critically ill patients. We evaluated whether TTR level correlates with the prevalence of PEM in the ICUand evaluated serum TTR level as an indicator of the effectiveness of nutrition support and the prognosis in critically ill patients.
TTR showed excellent concordance with patients classified with PEM or at high malnutrition risk, and followed for 7 days, it is a measure of the metabolic burden. TTR levels did not respond early to nutrition support because of the delayed return to anabolic status. It is particularly helpful in removing interpretation bias, and it is an excellent measure of the systemic inflammatory response concurrent with a preexisting state of chronic inanition.
The Stressful Condition as a Nutritionally Dependent Adaptive Dichotomy
Yves Ingenbleek and Larry Bernstein
Nutrition 1999;15(4):305-320 PII S0899-9007(99)00009-X
The injured body manifests a cascade of cytokine-induced metabolic events aimed at developing defense mechanisms and tissue repair. Rising concentrations of counterregulatory hormones work in concert with cytokines to generate overall insulin and insulin-like growth factor 1 (IGF-1), postreceptor resistance and energy requirements grounded on lipid dependency. Dalient features are self-sustained hypercortisolemia persisting as long as cytokines are oversecreted and down-regulation of the hypothalamo-pituitary-thyroid axis stabilized at low basal levels. Inhibition of thyroxine 5’deiodinating activity (5’DA) accounts for the depressed T3 values associated with the sparing of both N and energy-consuming processes. Both the liver and damaged territories adapt to stressful signals along up-regulated pathways disconnected from the central and peripheral control systems. Cytokines stimulate 5’DA and suppress the synthesis of TTR, causing the drop of retinol-binding protein (RBP) and the leakage of increased amounts of T4 and retinol in free form. TTR and RBP thus work as prohormonal reservoirs of precursor molecules which need to be converted into bioactive derivatives (T3 and retinoic acids) to reach transcriptional efficiency. The converting steps (5’DA and cellular retinol-binding protein-1) are activated to T4 and retinol, themselves operating as limiting factors to positive feedback loops. …The suicidal behavior of TBG, CBG, and IGFBP-3 allows the occurrence of peak endocrine and mitogenic influences at the site of inflammation. The production rate of TTR by the liver is the main determinant of both the hepatic release and blood transport of holoRBP, which explains why poor nutritional status concomitantly impairs thyroid- and retinoid-dependent acute phase responses, hindering the stressed body to appropriately face the survival crisis. …
abbreviations: TBG, thyroxine-binding globulain; CBG, cortisol-binding globulin; IGFBP-3, insulin growth factor binding protein-3; TTR, transthyretin; RBP, retionol-binding protein.
Why Should Plasma Transthyretin Become a Routine Screening Tool in Elderly Persons?
Yves Ingenbleek.
J Nutrition, Health & Aging 2009.
The homotetrameric TTR molecule (55 kDa as MM) was first identified in cerebrospinal fluid (CSF). The initial name of prealbumin (PA) was assigned based on the electrophoretic migration anodal to albumin. PA was soon recognized as a specific binding protein for thyroid hormone. and also of plasma retinol through the mediation of the small retinol-binding protein (RBP, 21 kDa as MM), which has a circulating half-life half that of TTR (24 h vs 48 h).
There exist at least 3 goos reasons why TTR should become a routine medical screening test in elderly persons. The first id grounded on the assessment of protein nutritional status that is frequently compromized and may become a life threatening condition. TTR was proposed as a marker of protein-energy malnutrition (PEM) in 1972. As a result of protein and energy deprivation, TTR hepatic synthesis is suppressed whereas all plasma indispensable amino acids (IAAs) manifest declining trends with the sole exception of methionine (Met) whose concentration usually remains unmodified. By comparison with ALB and transferrin (TF) plasma values, TTR did reveal a much higher degree of reactivity to changes in protein status that has been attributed to its shorter biological half-life and to its unusual tryptophan richness. The predictive ability of outcome offered by TTR is independent of that provided by ALB and TF. Uncomplicated PEM primarily affects the size of body nitrogen (N) pools, allowing reduced protein syntheses to levels compatible with survival. These adaptiver changes are faithfully identified by the serial measurement of TTR whose reliability has never been disputed in protein-depleted states. On the contrary, the nutritional relevance of TTR has been controverted in acute and chronic inflammatory conditions due to the cytokine-induced transcriptional blockade of liver synthesis which is an obligatory step occurring independently from the prevailing nutritional status. Although PEM and stress ful disorders refer to distinct pathogenic mechanisms, their combined inhibitory effects on TTR liber production fueled a long-lasting strife regarding a poor specificity. Recent body compositional studies have contributed to disentagling these intermingled morbidities, showing that evolutionary patterns displayed by plasma TTR are closely correlated with the fluctuations of lean body mass (LBM).
The second reason follows from advances describing the unexpected relationship established between TTR and homocysteine (Hcy), a S-containing AA not found in customary diets but resulting from the endogenous transmethylation of dietary methionine. Hcy may be recycled to Met along a remethylation pathway (RM) or irreversibly degraded throughout the transsulfuration (TS) cascade to relase sulfaturia as end-product. Hcy is thus situated at the crossrad of RM and TS pathways which are in equilibrium keeping plasma Met values unaltered. Three dietary water soluble B viatamins are implicated in the regulation of the Hcy-Met cycle. Folates (vit B9) are the most powerful agent, working as a supplier of the methyl group required for the RM process whereas cobalamines (vit B12) and pyridoxine (vit B6) operate as cofactors of Met-synthase and cystathionine-β-synthase. Met synthase promotes the RM pathway whereas the rate-limiting CβS governs the TS degradative cascade. Dietary deficiency in any of the 3 vitamins may upregulate Hcy plasma values, an acquied biochemiucal anomaly increasingly encountered in aged populations.
The third reason refers to recent and fascinating data recorded in neurobiology and emphasizing the specific properties of TTR in the prevention of brain deterioration. TTR participates directly in the maintenance of memory and normal cognitive processes during the aging process by acting on the retinoid signaling pathway. Moreover, TTR may bind amyloid β peptide in vitro, preventing its transformation into toxic amyloid fibrils and amyloid plaques. TTR works as a limiting factor for the plasma transport of retinoid, which in turn operates as a limiting determinant of both physiologically active retinoic acid (RA) derivatives, implying that any fluctuation in protein status might well entail corresponding alterations in cellular bioavailability of retinoid compounds. Under normal aging circumstances, the concentration of retinoid compounds declines in cerebral tissues together with the downregulation of RA receptor expression. In animal models, depletion of RAs causes the deposition of amyloid-β peptides, favoring the formation of amyloid plaques.
Prealbumin and Nutritional Evaluation
Larry Bernstein, Walter Pleban
Nutrition Apr 1996; 12(4):255-259.
http://nutritionjrnl.com/article/S0899-9007(96)90852-7
We compressed 16-test-pattern classes of albumin (ALB), cholesterol (CHOL), and total protein (TPR) in 545 chemistry profiles to 4 classes by conveerting decision values to a number code to separate malnourished (1 or 2) from nonmalnourished (NM)(0) patients using as cutoff values for NM (0), mild (1), and moderate (2): ALB 35, 27 g/L; TPR 63, 53 g/L; CHOL 3.9, 2.8 mmol/L; and BUN 9.3, 3.6 mmol/L. The BUN was found to have to have too low an S-value to make a contribution to the compressed classification. The cutoff values for classifying the data were assigned prior to statistical analysis, after examining information in the structured data. The data was obtained by a natural experiment in which the test profiles routinely done by the laboratory were randomly extracted. The analysis identifies the values used that best classify the data and are not dependent on distributional assumptions. The data were converted to 0, 1, or 2 as outcomes, to create a ternary truth table (eaxch row in nnn, the n value is 0 to 2). This allows for 34 (81) possible patterns, without the inclusion of prealbumin (TTR). The emerging system has much fewer patterns in the information-rich truth table formed (a purposeful, far from random event). We added TTR, coded, and examined the data from 129 patients. The classes are a compressed truth table of n-coded patterns with outcomes of 0, 1, or 2 with protein-energy malnutrition (PEM) increasing from an all-0 to all-2 pattern. Pattern class (F=154), PAB (F=35), ALB (F=56), and CHOL (F=18) were different across PEM class and predicted PEM class (R-sq. = 0.7864, F=119, p < E-5). Kruskall-Wallis analysis of class by ranks was significant for pattern class E-18), TTR (6.1E-15) ALB (E-16), CHOL (9E-10), and TPR (5E-13). The medians and standard error (SEM) for TTR, ALB, and CHOL of four TTR classes (NM, mild, mod, severe) are: TTR = 209, 8.7; 159, 9.3; 137, 10.4; 72, 11.1 mg/L. ALB – 36, 0.7; 30.5, 0.8; 25.0, 0.8; 24.5, 0.8 g/L. CHOL = 4.43, 0.17; 4.04, 0.20; 3.11, 0.21; 2.54, 0.22 mmol/L. TTR and CHOL values show the effect of nutrition support on TTR and CHOL in PEM. Moderately malnourished patients receiving nutrition support have TTR values in the normal range at 137 mg/L and at 159 mg/L when the ALB is at 25 g/L or at 30.5 g/L.
An Informational Approach to Likelihood of Malnutrition
Larry Bernstein, Thomas Shaw-Stiffel, Lisa Zarney, Walter Pleban.
Nutrition Nov 1996;12(11):772-776. PII: S0899-9007(96)00222-5.
http://dx.doi.org:/nutritionjrnl.com/article/S0899-9007(96)00222-5
Unidentified protein-energy malnutrition (PEM) is associated with comorbidities and increased hospital length of stay. We developed a model for identifying severe metabolic stress and likelihood of malnutrition using test patterns of albumin (ALB), cholesterol (CHOL), and total protein (TP) in 545 chemistry profiles…They were compressed to four pattern classes. ALB (F=170), CHOL (F = 21), and TP (F = 5.6) predicted PEM class (R-SQ = 0.806, F= 214; p < E^-6), but pattern class was the best predictor (R-SQ = 0.900, F= 1200, p< E^-10). Ktuskal-Wallis analysis of class by ranks was significant for pattern class (E^18), ALB (E^-18), CHOL (E^-14), TP (@E^-16). The means and SEM for tests in the three PEM classes (mild, mod, severe) were; ALB – 35.7, 0.8; 30.9, 0.5; 24.2, 0.5 g/L. CHOL – 3.93, 0.26; 3.98, 0.16; 3.03, 0.18 µmol/L, and TP – 68.8, 1.7; 60.0, 1.0; 50.6, 1.1 g/L. We classified patients at risk of malnutrition using truth table comprehension.
Downsizing of Lean Body Mass is a Key Determinant of Alzheimer’s Disease
Yves Ingenbleek, Larry Bernstein
J Alzheimer’s Dis 2015; 44: 745-754.
http://dx.doi.org:/10.3233/JAD-141950
Lean body mass (LBM) encompasses all metabolically active organs distributed into visceral and structural tissue compartments and collecting the bulk of N and K stores of the human body. Transthyretin (TTR) is a plasma protein mainly secreted by the liver within a trimolecular TTR-RBP-retinol complex revealing from birth to old age strikingly similar evolutionary patterns with LBM in health and disease. TTR is also synthesized by the choroid plexus along distinct regulatory pathways. Chronic dietary methionine (Met) deprivation or cytokine-induced inflammatory disorders generates LBM downsizing following differentiated physiopathological processes. Met-restricted regimens downregulate the transsulfuration cascade causing upstream elevation of homocysteine (Hcy) safeguarding Met homeostasis and downstream drop of hydrogen sulfide (H2S) impairing anti-oxidative capacities. Elderly persons constitute a vulnerable population group exposed to increasing Hcy burden and declining H2S protection, notably in plant-eating communities or in the course of inflammatory illnesses. Appropriate correction of defective protein status and eradication of inflammatory processes may restore an appropriate LBM size allowing the hepatic production of the retinol circulating complex to resume, in contrast with the refractory choroidal TTR secretory process. As a result of improved health status, augmented concentrations of plasma-derived TTR and retinol may reach the cerebrospinal fluid and dismantle senile amyloid plaques, contributing to the prevention or the delay of the onset of neurodegenerative events in elderly subjects at risk of Alzheimer’s disease.
Amyloidogenic and non-amyloidogenic transthyretin variants interact differently with human cardiomyocytes: insights into early events of non-fibrillar tissue damage
Pallavi Manral and Natalia Reixach
Biosci.Rep.(2015)/35/art:e00172 http://dx.doi.org:/10.1042/BSR20140155
TTR (transthyretin) amyloidosis are diseases characterized by the aggregation and extracellular deposition of the normally soluble plasma protein TTR. Ex vivo and tissue culture studies suggest that tissue damage precedes TTR fibril deposition, indicating that early events in the amyloidogenic cascade have an impact on disease development. We used a human cardiomyocyte tissue culture model system to define these events. We previously described that the amyloidogenic V122I TTR variant is cytotoxic to human cardiac cells, whereas the naturally occurring, stable and non-amyloidogenic T119M TTR variant is not. We show that most of the V122I TTR interacting with the cells is extracellular and this interaction is mediated by a membraneprotein(s). In contrast, most of the non-amyloidogenic T119M TTR associated with the cells is intracellular where it undergoes lysosomal degradation. The TTR internalization process is highly dependent on membrane cholesterol content. Using a fluorescent labelled V122I TTR variant that has the same aggregation and cytotoxic potential as the native V122I TTR, we determined that its association with human cardiomyocytes is saturable with a KD near 650nM. Only amyloidogenic V122I TTR compete with fluorescent V122I force ll-binding sites. Finally, incubation of the human cardiomyocytes with V122I TTR but not with T119M TTR, generates superoxide species and activates caspase3/7. In summary, our results show that the interaction of the amyloidogenic V122I TTR is distinct from that of a non-amyloidogenic TTR variant and is characterized by its retention at the cell membrane, where it initiates the cytotoxic cascade.
Emerging roles for retinoids in regeneration and differentiation in normal and disease states
Lorraine J. Gudas
Biochimica et Biophysica Acta 1821 (2012) 213–221
http://dx.doi.org:/10.1016/j.bbalip.2011.08.002
The vitamin (retinol) metabolite, all-transretinoic acid (RA), is a signaling molecule that plays key roles in the development of the body plan and induces the differentiation of many types of cells. In this review the physiological and pathophysiological roles of retinoids (retinol and related metabolites) in mature animals are discussed. Both in the developing embryo and in the adult, RA signaling via combinatorial Hoxgene expression is important for cell positional memory. The genes that require RA for the maturation/differentiation of T cells are only beginning to be cataloged, but it is clear that retinoids play a major role in expression of key genes in the immune system. An exciting, recent publication in regeneration research shows that ALDH1a2(RALDH2), which is the rate-limiting enzyme in the production of RA from retinaldehyde, is highly induced shortly after amputation in the regenerating heart, adult fin, and larval fin in zebrafish. Thus, local generation of RA presumably plays a key role in fin formation during both embryogenesis and in fin regeneration. HIV transgenic mice and human patients with HIV-associated kidney disease exhibit a profound reduction in the level of RARβ protein in the glomeruli, and HIV transgenic mice show reduced retinol dehydrogenase levels, concomitant with a greater than 3-fold reduction in endogenous RA levels in the glomeruli. Levels of endogenous retinoids (those synthesized from retinol within cells) are altered in many different diseases in the lung, kidney, and central nervous system, contributing to pathophysiology.
The Membrane Receptor for Plasma Retinol-Binding Protein, A New Type of Cell-Surface Receptor
Hui Sun and Riki Kawaguchi
Intl Review Cell and Molec Biol, 2011; 288:Chap 1. Pp 1:34
http://dx.doi.org:/10.1016/B978-0-12-386041-5.00001-7
Vitamin A is essential for diverse aspects of life ranging from embryogenesis to the proper functioning of most adul torgans. Its derivatives (retinoids) have potent biological activities such as regulating cell growth and differentiation. Plasma retinol-binding protein (RBP) is the specific vitamin A carrier protein in the blood that binds to vitamin A with high affinity and delivers it to target organs. A large amount of evidence has accumulated over the past decades supporting the existence of a cell-surface receptor for RBP that mediates cellular vitamin A uptake. Using an unbiased strategy, this specific cell-surface RBP receptor has been identified as STRA6, a multi-transmembrane domain protein with previously unknown function. STRA6 is not homologous to any protein of known function and represents a new type of cell-surface receptor. Consistent with the diverse functions of vitamin A, STRA6 is widely expressed in embryonic development and in adult organ systems. Mutations in human STRA6 are associated with severe pathological phenotypes in many organs
such as the eye, brain, heart, and lung. STRA6 binds to RBP with high affinity and mediates vitamin A uptake into cells. This review summarizes the history of the RBP receptor research, its expression in the context of known functions of vitamin A in distinct human organs, structure/function analysis of this new type of membrane receptor, pertinent questions regarding its very existence, and its potential implication in treating human diseases.
Choroid plexus dysfunction impairs beta-amyloid clearance in a triple transgenic mouse model of Alzheimer’s disease
Ibrahim González-Marrero, Lydia Giménez-Llort, Conrad E. Johanson, et al.
Front Cell Neurosc Feb2015; 9(17): 1-10
http://dx.doi.org:/10.3389/fncel.2015.00017
Compromised secretory function of choroid plexus (CP) and defective cerebrospinal fluid (CSF) production, along with accumulation of beta-amyloid (Aβ) peptides at the blood-CSF barrier (BCSFB), contribute to complications of Alzheimer’s disease (AD). The AD triple transgenic mouse model (3xTg-AD) at 16 month-old mimics critical hallmarks of the human disease: β-amyloid (Aβ) plaques and neurofibrillary tangles (NFT) with a temporal-and regional-specific profile. Currently, little is known about transport and metabolic responses by CP to the disrupted homeostasis of CNS Aβ in AD. This study analyzed the effects of highly-expressed AD-linked human transgenes (APP, PS1 and tau) on lateral ventricle CP function. Confocal imaging and immunohistochemistry revealed an increase only of Aβ42 isoform in epithelial cytosol and in stroma surrounding choroidal capillaries; this buildup may reflect insufficient clearance transport from CSF to blood. Still, there was increased expression, presumably compensatory, of the choroidal Aβ transporters: the low density lipoprotein receptor-related protein1 (LRP1) and the receptor for advanced glycation end product (RAGE). A thickening of the epithelial basal membrane and greater collagen-IV deposition occurred around capillaries in CP, probably curtailing solute exchanges. Moreover, there was attenuated expression of epithelial aquaporin-1 and transthyretin(TTR) protein compared to Non-Tg mice. Collectively these findings indicate CP dysfunction hypothetically linked to increasing Aβ burden resulting in less efficient ion transport, concurrently with reduced production of CSF (less sink action on brain Aβ) and diminished secretion of TTR (less neuroprotection against cortical Aβ toxicity). The putative effects of a disabled CP-CSF system on CNS functions are discussed in the context of AD.
Endoplasmic reticulum: The unfolded protein response is tangled In neurodegeneration
Jeroen J.M. Hoozemans, Wiep Scheper
Intl J Biochem & Cell Biology 44 (2012) 1295–1298
http://dx.doi.org/10.1016/j.biocel.2012.04.023
Organelle facts•The ER is involved in the folding and maturation ofmembrane-bound and secreted proteins.•The ER exerts protein quality control to ensure correct folding and to detect and remove misfolded proteins.•Disturbance of ER homeostasis leads to protein misfolding and induces the UPR.•Activation of the UPR is aimed to restore proteostasis via an intricate transcriptional and (post)translational signaling network.•In neurodegenerative diseases classified as tauopathies the activation of the UPR coincides with the pathogenic accumulation of the microtubule associated protein tau.•The involvement of the UPR in tauopathies makes it a potential therapeutic target.
The endoplasmic reticulum (ER) is involved in the folding and maturation of membrane-bound and secreted proteins. Disturbed homeostasis in the ER can lead to accumulation of misfolded proteins, which trigger a stress response called the unfolded protein response (UPR). In neurodegenerative diseases that are classified as tauopathies, activation of the UPR coincides with the pathogenic accumulation of the microtubule associated protein tau. Several lines of evidence indicate that UPR activation contributes to increased levels of phosphorylated tau, a prerequisite for the formation of tau aggregates. Increased understanding of the crosstalk between signaling pathways involved in protein quality control in the ERand tau phosphorylation will support the development of new therapeutic targets that promote neuronal survival.
Chemical and/or biological therapeutic strategies to ameliorate protein misfolding diseases
Derrick Sek Tong Ong and Jeffery W Kelly
Current Opin Cell Biol 2011; 23:231–238
http://dx.doi.org:/10.1016/j.ceb.2010.11.002
Inheriting a mutant misfolding-prone protein that cannot be efficiently folded in a given cell type(s) results in a spectrum of human loss-of-function misfolding diseases. The inability of the biological protein maturation pathways to adapt to a specific misfolding-prone protein also contributes to pathology. Chemical and biological therapeutic strategies are presented that restore protein homeostasis, or proteostasis, either by enhancing the biological capacity of the proteostasis network or through small molecule stabilization of a specific misfolding-prone protein. Herein, we review the recent literature on therapeutic strategies to ameliorate protein misfolding diseases that function through either of these mechanisms, or a combination thereof, and provide our perspective on the promise of alleviating protein misfolding diseases by taking advantage of proteostasis adaptation.


. Image:Cardiac a...")
. Image:Cardiac a...")
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}