Amyloidosis describes the various clinical syndromes that occur as a result of damage by amyloid deposits in tissues and organs throughout the body. Systemic amyloidosis is a relatively rare multisystem disease caused by the deposition of misfolded protein in various tissues and organs. The term amyloid describes the deposition in the extracellular space of certain proteins in a highly characteristic, insoluble fibrillar form. The disease entity is a disorder of misfolded or misassembled proteins. There is extracellular amyloid fiber laid down as cross β-sheets disrupting organ function, which may affect the pancreas, kidney, autonomic nervous system, the heart, and in one form causes carpal tunnel syndrome.
It may present to almost any specialty, and diagnosis is frequently delayed. Cardiac involvement is a leading cause of morbidity and mortality, especially in primary light chain (AL) amyloidosis and in both wild-type and hereditary transthyretin amyloidosis. The heart is also occasionally involved in acquired serum amyloid A type (AA) amyloidosis and other rare hereditary types. Clinical phenotype varies greatly between different types of amyloidosis, and even the cardiac presentation has a great spectrum. The incidence of amyloidosis is uncertain, but it is thought that the most frequently diagnosed
AL amyloidosis has an annual incidence of 6 to 10 cases per million population in
the United Kingdom and United States.
The molecular basis for this particular phenomenon came with the extensive work done on multiple myeloma, antibody structure, and light chains. In 1950, the discovery of a familial amyloid polyneuropathy was described in Portugal, and there were similar diseases in Sweden and Japan. There were 72 known variants of transthyretin (
TTR) in 1995, and now there are 100. In addition, the occurance of different TTR associated variants with and without (amyloid) is found is Brazil, UK, US, Israel, Spain, France, Germany, Denmark, and Africa. The table of variants, organ damage, and geographic location is too large to place on this document. If we refer to amyloid cardiomyopathy, it is exclusively a primary amyloidopathy, not secondary to light chain disorders or an inflammatory disease. If we consider amyloidosis, we also have to consider family history, organ dysfunction, and we have to make a distinction between primary cardiac involvement, autonomic nervous system instability, and the two coexisting. Familial amyloid polyneuropathy (FAP) is an extremely debilitating and progressive disease that is only treatable by liver transplantation. Primary amyloid cardiomyopathy has been treated by heart transplant. The qualifying statement here is, it depends.
Primary and Secondary Amyloidoses
Amyloid was originally described by pathologists based on microscopy. Amyloidoses are a systemic primary or secondary disease. There are distinctions to be made based on location and type. The clinical significance of amyloid disease varies enormously, ranging from incidental asymptomatic deposits to localized disease through to rapidly fatal systemic forms that can affect multiple vital organs.
Common causes of secondary amyloidosis are – light chain production (AL) as in plasma cell dyscrasia, amyloid A (AA), senile systemic amyloidosis (diagnosed rarely in life). The systemic amyloidoses are designated by a capital A (for amyloid) followed by the abbreviation for the chemical identity of the fibril protein. Thus,
TTR amyloidosis is abbreviated
ATTR, and
immunoglobulin light chain type amyloidosis is abbreviated AL. Both normal-sequence TTR and variant-sequence TTR form amyloidosis. Normal-sequence TTR forms cardiac amyloidosis in elderly people, termed senile cardiac amyloidosis (SCA). When it was recognized that SCA is often accompanied by microscopic deposits in many other organs, the alternative name senile systemic amyloidosis (SSA) was proposed. Both terms are now used.
Currently available therapy is focused on reducing the supply of the respective amyloid fibril precursor protein and supportive medical care, which together have greatly improved survival. Chemotherapy and anti-inflammatory treatment for the disorders that underlie AL and
AA amyloidosis are guided by serial measurements of the respective circulating amyloid precursor proteins, i.e. serial serum free light chains in AL and
serum amyloid A protein in AA type.
Quality of life and prognosis of some forms of hereditary systemic amyloidosis can be improved by liver and other organ transplants. Various new therapies, ranging from silencing RNA, protein stabilizers to monoclonal antibodies, aimed at inhibiting fibril precursor supply, fibril formation or the persistence of amyloid deposits, are in development; some are already in clinical phase.
What is transthyretin (TTR)?
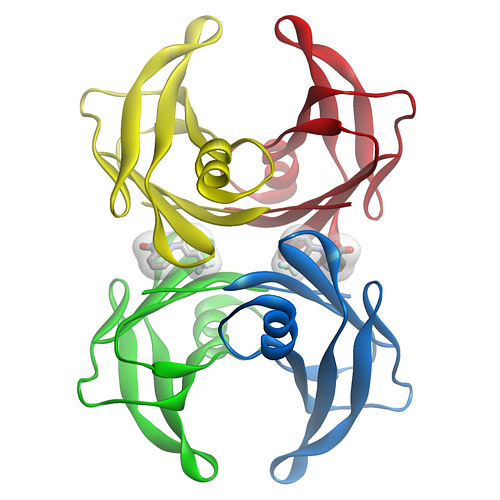
TTR is a tetramer of 4 127 amino acid subunits synthesized by the liver that circulates as a transporter of thyroxin, and with retinol-binding protein, transports vitamin A. It was originally defined by the migration in electrophoresis more anodal to albumin, hence, prealbumin. It is present in cerebrospinal fluid, secreted by the choroid plexus. The TTR monomer contains 8 antiparallel
beta pleated sheet domains. TTR can be found in plasma and in cerebrospinal fluid and is synthesized by the choroid plexus of the brain and, to a lesser degree, by the retina. Its gene is located on the long arm of chromosome 18 and contains 4 exons and 3 introns.
The concentration in serum can be expected to be above 20 mg/dL in a health adult, but the protein decreases by 1 mg/dL/day postoperatively, and it decreases with acute or chronic renal failure, pneumonia or sepsis, rising again with the onset of anabolism. Patients in the pulmonary intensive care unit have TTR levels that remain low for 7-10 days, but followup data for the remainder of the hospital stay or in relationship to readmission in the six months after release from hospital care was not part of the study.
A decrease in TTR is associated with the systemic inflammatory response, whereby, the liver reprioritizes the synthesis of proteins with an increase in acute phase reactants (APRs), namely,
C-reactive protein (CRP) and a-1 acid glycoprotein, and decreased albumin and TTR. The inflammatory condition maintains a euthyroid status with decreased TTR because of the availability of free thyroxine in equilibrium with the lower binding protein. This has been referred to sick euthyroid status. The role in thyroxine transport is not insignificant, as chronic protein malnutrition is associated with hypothyroidism, as originally described by Prof. Yves Ingenbleek, Univ. Louis Pateur, Starsbourg, Fr. in Senegalese children with Kwashiorkor. However, the importance of TTR as a unique biomarker is not to be downgraded because of what is often refered to as “an inverted APR”.
Transthyretin was discovered to be a good reflection of the “lean body mass”, by Vernon Young, MIT, and Ingenbleek, as a result of 3 decades of study. The ratio of S:N being 1:20 in plant proteins and 1:12.5 in animal sources, is closely related to methylation reactions and sustained deficiency of S intake results in elevated homocysteine level.
What is FAP?
Familial amyloid polyneuropathy (FAP), also called transthyretin-related hereditary amyloidosis, transthyretin amyloidosis or Corino de Andrade’s disease, is an autosomal dominant neurodegenerative disease. It is a form of amyloidosis, and was first identified and described by Portuguese neurologist Mário Corino da Costa Andrade, in the 1950s.FAP is distinct from senile systemic amyloidosis (SAS), which is not inherited, and which was determined to be the primary cause of death for 70% of supercentenarians who have been autopsied.
Familial amyloid polyneuropathy (FAP) is an extremely debilitating and progressive disease that is only treatable by liver transplantation. Primary amyloid cardiomyopathy has been treated by heart transplant. The qualifying statement here is, it depends. Those patients with TTR-amyloidopathy have a specific gene substitution in the TTR gene. Consequently, there is circulation TTR, but it is not effectively involved in thyroxine transport.
Characteristics.
Usually manifesting itself between 20 and 40 years of age, it is characterized by pain, paresthesia, muscular weakness and autonomic dysfunction. In its terminal state, the kidneys and the heart are affected. FAP is characterized by the systemic deposition of amyloidogenic variants of the transthyretin protein, especially in the peripheral nervous system, causing a progressive sensory and motor polyneuropathy. The age at symptom onset, pattern of organ involvement, and disease course vary, but most mutations are associated with cardiac and/or nerve involvement. The gastrointestinal tract, vitreous, lungs, and carpal ligament are also frequently affected. When the peripheral nerves are prominently affected, the disease is termed familial amyloidotic polyneuropathy (FAP). When the heart is involved heavily but the nerves are not, the disease is called familial amyloid cardiomyopathy (FAC). Regardless of which organ is primarily targeted, the general term is simply amyloidosis-transthyretin type, abbreviated ATTR.
Genetics.
- TTR mutations accelerate the process of TTR amyloid formation and are the most important risk factor for the development of clinically significant ATTR. More than 85 amyloidogenic TTR variants cause systemic familial amyloidosis. The variant TTR is mostly produced by the liver. Amyloidogenic TTR mutations destabilize TTR monomers or tetramers, allowing the molecule to more easily attain an amyloidogenic intermediate conformation. The tetramer has to dissociate into misfolded monomers to aggregate into a variety of structures including amyloid fibrils. Because most patients are heterozygotes, they deposit both mutant and wild type TTR subnits.
- Familial amyloid polyneuropathy has an autosomal dominant pattern of inheritance. FAP is caused by a mutation of the TTR gene, located on human chromosome 18q12.1-11.2. A replacement of valine by methionine at position 30 (TTR V30M) is the mutation most commonly found in FAP.
- The disease in the TTR V30M kindreds was termed FAP because early symptoms arose from peripheral neuropathy, but these patients actually have systemic amyloidosis, with widespread deposits often involving the heart, gastrointestinal tract, eye, and other organs.
- TTR V122I: This variant, carried by 3.9% of African Americans and over 5% of the population in some areas of West Africa, increases the risk of late-onset (after age 60 years) cardiac amyloidosis. It appears to be the most common amyloid-associated TTR variant worldwide. Affected patients usually do not have peripheral neuropathy.
- TTR T60A: This variant causes late-onset systemic amyloidosis with cardiac, and sometimes neuropathic, involvement. This variant originated in northwest Ireland and is found in Irish and Irish American patients.
- TTR L58H: Typically affecting the carpal ligament and nerves of the upper extremities, this variant originated in Germany. It has spread throughout the United States but is most common in the mid-Atlantic region.
- TTR G6S: This is the most common TTR variant, but it appears to be a neutral polymorphism not associated with amyloidosis. It is carried by about 10% of people of white European descent.
Cardiac transthyretin (TTR) amyloidosis
Cardiac amyloidosis of transthyretin fibril protein (ATTR) type is an infiltrative cardiomyopathy characterised by ventricular wall thickening and diastolic heart failure. More than 27 different precursor proteins have the propensity to form amyloid fibrils. The particular precursor protein that misfolds to form amyloid fibrils defines the amyloid type and predicts the patient’s clinical course. Several types of amyloid can infiltrate the heart, resulting in progressive diastolic and systolic dysfunction, congestive heart failure, and death. Increased access to cardiovascular magnetic resonance imaging has led to a marked increase in referrals to St George’s University of London, London (Dr. Jason Dungu) of Caucasian patients with wild-type ATTR (senile systemic) amyloidosis and Afro-Caribbean patients with the hereditary ATTR V122I type. Both subtypes present predominantly as isolated cardiomyopathy. The differential diagnosis includes cardiac amyloid light-chain (AL) amyloidosis, which has a poorer prognosis and can be amenable to chemotherapy.
Clinical Presentation
Cardiac amyloidosis, irrespective of type, presents as a restrictive cardiomyopathy characterized by progressive diastolic and subsequently systolic biventricular dysfunction and arrhythmia.1 Key “red flags” to possible systemic amyloidosis include nephrotic syndrome, autonomic neuropathy (eg, postural hypotension, diarrhea), soft-tissue infiltrations (eg, macroglossia, carpal tunnel syndrome, respiratory disease), bleeding (eg, cutaneous, such as periorbital, gastrointestinal), malnutrition/cachexia and genetic predisposition (eg, family history, ethnicity). Initial presentations may be cardiac, with progressive exercise intolerance and heart failure. Other organ involvement, particularly in AL amyloidosis, may cloud the cardiac presentation (eg, nephrotic syndrome, autonomic neuropathy, pulmonary or bronchial involvement). Pulmonary edema is not common early in the disease process, but pleural and pericardial effusions and atrial arrhythmias are often seen. Syncope is common and a poor prognostic sign. It is typically exertional or postprandial as part of restrictive cardiomyopathy, sensitivity to intravascular fluid depletion from loop diuretics combined with autonomic neuropathy, or conduction tissue involvement (atrioventricular or sinoatrial nodes) or ventricular arrhythmia. The latter may rarely cause recurrent syncope. Disproportionate septal amyloid accumulation mimicking hypertrophic cardiomyopathy with dynamic left ventricular (LV) outflow tract obstruction is rare but well documented. Myocardial ischemia can result from amyloid deposits within the microvasculature. Atrial thrombus is common, particularly in AL amyloidosis
Diagnosis and Treatment
imaging – Cardiovascular Magnetic Resonance in Cardiac Amyloidosis*.
Cardiac amyloidosis can be diagnostically challenging. Cardiovascular magnetic resonance (CMR) can assess abnormal myocardial interstitium. In cardiac amyloidosis, CMR shows a characteristic pattern of global subendocardial late enhancement coupled with abnormal myocardial and blood-pool gadolinium kinetics. The findings agree with the transmural histological distribution of amyloid protein and the cardiac amyloid load.
*AM Maceira; J Joshi; SK Prasad; J Charles Moon, et al. Royal Brompton Hospital, London;
The diagnosis of amyloidosis requires histological identification of amyloid deposits. Congo Red staining renders amyloid deposits salmon pink by light microscopy, with a characteristic apple green birefringence under polarized light conditions. Additional immunohistochemical staining for precursor proteins identifies the type of amyloidosis. Ultimately, immunogold electron microscopy and mass spectrometry confer the greatest sensitivity and specificity for amyloid typing.
Treatment of cardiac amyloidosis is dictated by the amyloid type and degree of involvement. Consequently, early recognition and accurate classification are essential.
Novel diagnostic and surveillance approaches using imaging (echocardiography, cardiovascular magnetic resonance), biomarkers (brain natriuretic peptide [BNP], high-sensitivity troponin), new histological typing techniques, and current and future treatments, including approaches directly targeting the amyloid deposits.
Etiology
Amyloidosis is caused by the extracellular deposition of autologous protein in an abnormal insoluble β-pleated sheet fibrillary conformation—that is, as amyloid fibrils. More than 30 proteins are known to be able to form amyloid fibrils in vivo, which cause disease by progressively damaging the structure and function of affected tissues. Amyloid deposits also contain minor nonfibrillary constituents, including serum amyloid P component (SAP), apolipoprotein E, connective tissue components (glycosaminoglycans, collagen), and basement membrane components (fibronectin, laminin). Amyloid deposits can be massive, and cardiac or other tissues may become substantially replaced. Amyloid fibrils bind Congo red stain, yielding the pathognomonic apple-green birefringence under cross-polarized light microscopy that remains the gold standard for identifying amyloid deposits.
AL Amyloidosis
AL amyloidosis is caused by deposition of fibrils composed of monoclonal immunoglobulin light chains and is associated with clonal plasma cell or other B-cell dyscrasias. The spectrum and pattern of organ involvement is very wide, but cardiac involvement occurs in half of cases and is sometimes the only presenting feature. Cardiac AL amyloidosis may be rapidly progressive. Low QRS voltages, particularly in the limb leads, are common. Thickening of the LV wall is typically mild to moderate and is rarely >18 mm even in advanced disease. Cardiac AL amyloid deposition is accompanied by marked elevation of the biomarkers BNP and cardiac troponin, even at an early stage. Involvement of the heart is the commonest cause of death in AL amyloidosis and is a major determinant of prognosis; without cardiac involvement, patients with AL amyloidosis have a median survival of around 4 years, but the prognosis among affected patients with markedly elevated BNP and cardiac troponin (Mayo stage III disease) is on the order of 8 months.
Hereditary Amyloidoses
Mutations in several genes, such as transthyretin, fibrinogen, apolipoprotein A1, and apolipoprotein A2 can be responsible for hereditary amyloidosis, but by far the most common cause is variant ATTR amyloidosis (variant ATTR) caused by mutations in the transthyretin gene causing neuropathy and, often, cardiac involvement.
TTR gene mutation
The most common is the Val122Ile mutation. In a large autopsy study that included individuals with cardiac amyloidosis, the TTR Val122Ile allele was present in 3.9% of all African Americans and 23% of African Americans with cardiac amyloidosis. Penetrance of the mutation is not truly known and is associated with a late-onset cardiomyopathy that is indistinguishable from senile cardiac amyloidosis.
Pathology, Presentation, and Management of Amyloidoses
More than 100 genetic variants of TTR are associated with amyloidosis. Most present as the clinical syndrome of progressive peripheral and autonomic neuropathy. Unlike wild-type ATTR or variant ATTR Val122Ile, the features of other variant ATTR include vitreous amyloid deposits or, rarely, deposits in other organs. Cardiac involvement in variant ATTR varies by mutations and can be the presenting or indeed the only clinical feature. For example, cardiac involvement is rare in variant ATTR associated with Val30Met (a common variant in Portugal or Sweden), but it is almost universal and develops early in individuals with variant ATTR due to Thr60Ala mutation (a mutation common in Ireland).
Senile Systemic Amyloidosis (Wild-Type ATTR)
Wild-type TTR amyloid deposits are found at autopsy in about 25% of individuals >80 years of age. The prevalence of wild-type TTR deposits leading to the clinical syndrome of wild-type ATTR cardiac amyloidosis is unknown. Wild-type ATTR is a predominantly cardiac disease, and the only other significant extracardiac feature is a history of carpal tunnel syndrome, often preceding heart failure by 3 to 5 years. Extracardiac involvement is most unusual.
Both wild-type ATTR and ATTR due to Val122Ile are diseases of the >60-year age group and are often misdiagnosed as hypertensive heart disease. Wild-type ATTR has a strong male predominance, and the natural history remains poorly understood, but studies suggest a median survival of about 7 years from presentation. Recent developments in cardiac magnetic resonance (CMR), which have greatly improved detection of cardiac amyloid during life, suggest that wild-type ATTR is more common than previously thought: It accounted for 0.5% of all patients seen at the UK amyloidosis center until 2001 but now accounts for 7% of 1100 cases with amyloidosis seen since the end of 2009. There appears to be an association between wild-type ATTR and history of myocardial infarctions, G/G (Val/Val) exon 24 polymorphism in the alpha2-macroglobulin (alpha2M), and the H2 haplotype of the tau gene36; the association of tau with Alzheimer’s disease raises interesting questions as both are amyloid-associated diseases of aging.
ECG of a patient with cardiac AL amyloidosis showing small QRS voltages (defined as ≤6 mm height), predominantly in the limb leads and pseudoinfarction pattern in the anterior leads.
Echocardiography is characteristic. Typical findings include concentric ventricular thickening with right ventricular involvement, poor biventricular long-axis function with normal/near-normal ejection fraction and valvular thickening (particularly in wild-type or variant ATTR). Diastolic dysfunction is the earliest echocardiographic abnormality and may occur before cardiac symptoms develop. Biatrial dilatation in presence of biventricular, valvular, and interatrial septal thickening 53 is a useful clue to the diagnosis.
Transthoracic echocardiogram with speckle tracking. The red and yellow lines represent longitudinal motion in the basal segments, whereas the purple and green lines represent apical motion. This shows loss of longitudinal ventricular contraction at the base compared to apex.
Biomarkers.
High-sensitivity troponin is abnormal in >90% of cardiac AL patients, and the combination of BNP/NT-proBNP plus troponin measurements is used to stage and risk-stratify patients with AL amyloidosis at diagnosis. Very interestingly, the concentration of BNP/NT-proBNP in AL amyloidosis may fall dramatically within weeks after chemotherapy that substantially reduces the production of amyloidogenic light chains. The basis for this very rapid phenomenon, which is not mirrored by changes on echocardiography or CMR, remains uncertain, but a substantial fall is associated with improved outcomes.
Cardiac Magnetic Resonance.
CMR provides functional and morphological information on cardiac amyloid in a similar way to echocardiography, though the latter is superior for evaluating and quantifying diastolic abnormalities. An advantage of CMR is in myocardial tissue characterization. Amyloidotic myocardium reveals subtle precontrast abnormalities (T1, T2), but extravascular contrast agents based on chelated gadolinium provide the key information.
CMR with the classic amyloid global, subendocardial late gadolinium enhancement pattern in the left ventricle with blood and mid-/epimyocardium nulling together.
Recently, the technique of equilibrium contrast CMR has demonstrated much higher extracellular myocardial volume in cardiac amyloid than any other measured disease. It is anticipated that accurate measurements of the expanded interstitium in amyloidosis will prove useful in serial quantification of cardiac amyloid burden.
Sequential static images from a CMR TI scout sequence. As the inversion time (TI) increases, myocardium nulls first (arrow in image 3), followed by blood afterwards (arrow in image 6), implying that there is more gadolinium contrast in the myocardium than blood—a degree of interstitial expansion such that the “myocrit” is smaller than the hematocrit.
Tissue biopsy.
To confirm amyloidosis, including familial TTR amyloidosis, the demonstration of amyloid deposition on biopsied tissues is essential. With Congo red staining, amyloid deposits show a characteristic yellow-green birefringence under polarized light. Tissues suitable for biopsy include: subcutaneous fatty tissue of the abdominal wall, skin, gastric or rectal mucosa, sural nerve, and peritendinous fat from specimens obtained at carpal tunnel surgery. Sensitivity of endoscopic biopsy of gastrointestinal mucosa is around 85%; biopsy of the sural nerve is less sensitive. It is ideal to show that these amyloid deposits are specifically immunolabeled by anti-TTR antibodies.
Serum variant TTR protein.
TTR protein normally circulates in serum or plasma as a soluble protein having a tetrameric structure [Kelly 1998, Rochet & Lansbury 2000]. Normal plasma TTR concentration is 20-40 mg/dL (0.20-0.40 mg/mL). Pathogenic mutations in TTR cause conformational change in the TTR protein molecule, disrupting the stability of the TTR tetramer, which is then more easily dissociated into pro-amyloidogenic monomers.
After immunoprecipitation with anti-TTR antibody, serum variant TTR protein can be detected by mass spectrometry. Approximately 90% of TTR variants so far identified are confirmed by this method. Mass shift associated with each variant TTR protein is indicated.
Molecular genetic testing.
- TTR is the only gene in which mutations are known to cause familial TTR amyloidosis.
- Identified in many individuals of different ethnic backgrounds; found in large clusters in Portugal, Sweden, and Japan.
- The gene has four exons; and all the hitherto-identified mutations are in exons 2, 3, or 4.
GeneReviews designates a molecular genetic test as clinically available only if the test is listed in the GeneTests Laboratory Directory by either a US CLIA-licensed laboratory or a non-US clinical laboratory.
- Molecular genetic testing of TTR by sequence analysis (may be preceded by targeted mutation analysis)
- Although deletion/duplication testing is available clinically, no exonic or whole-gene deletions or duplications involving TTR have been reported to cause familial transthyretin amyloidosis.
- However, with newly available deletion/duplication testing methods, it is theoretically possible that such mutations may be identified in affected individuals in whom prior testing by sequence analysis of the entire coding region was negative.
- Predictive testing for at-risk asymptomatic adult family members requires prior identification of the disease-causing mutation in the family.
- Prenatal diagnosis and preimplantation genetic diagnosis (PGD) for at-risk pregnancies require prior identification of the disease-causing mutation in the family.
Genetically Related (Allelic) Disorders
Familial euthyroid hyperthyroxinemia is caused by normal allelic variants in TTR, including Gly6Ser, Ala109Thr, Ala109Val, and Thr119Met (see Table 5) [Nakazato 1998, Benson 2001, Saraiva 2001]. The TTR protein binds approximately 15% of serum thyroxine. These mutations increase total serum thyroxine concentration because of their increased affinity for thyroxine, however, they increase neither free thyroxine nor free triiodothyronine. Therefore, individuals with these sequence variants develop no clinical symptoms (i.e., they are euthyroid).
Senile systemic amyloidosis (SSA; previously called senile cardiac amyloidosis) results from the pathologic deposition of wild-type TTR, predominantly in the heart. Pathologic deposits are also seen in the lungs, blood vessels, and the renal medulla of the kidneys [Westermark et al 2003]. SSA affects mainly the elderly but is rarely diagnosed during life.
Sensorimotor neuropathy and autonomic neuropathy progress over ten to 20 years. Various types of cardiac conduction block frequently appear. Cachexia is a common feature at the late stage of the disease. Affected individuals usually die of cardiac failure, renal failure, or infection.
Cardiac amyloidosis.
Cardiac amyloidosis, mainly characterized by progressive cardiomyopathy, has been reported with more than two thirds of TTR mutations. In some families with specific TTR mutations, such as Asp18Asn, Val20Ile, Pro24Ser, Ala45Thr, Ala45Ser, His56Arg, Gly57Arg, Ile68Leu, Ala81Thr, Ala81Val, His88Arg, Glu92Lys, Arg103Ser, Leu111Met, or Val122Ile, cardiomyopathy without peripheral neuropathy is a main feature of the disease.
Cardiac amyloidosis is usually late onset. Most individuals develop cardiac symptoms after age 50 years; cardiac amyloidosis generally presents with restrictive cardiomyopathy. The typical electrocardiogram shows a pseudoinfarction pattern with prominent Q wave in leads II, III, aVF, and V1-V3, presumably resulting from dense amyloid deposition in the anterobasal or anteroseptal wall of the left ventricle. The echocardiogram reveals left ventricular hypertrophy with preserved systolic function. The thickened walls present “a granular sparkling appearance.”
Among the mutations responsible for cardiac amyloidosis, Val122Ile is notable for its prevalence in African Americans. Approximately 3.0%-3.9% of African Americans are heterozygous for Val122Ile . The high frequency of Val122Ile partly explains the observation that in individuals in the US older than age 60 years, cardiac amyloidosis is four times more common among blacks than whites.
Leptomeningeal (oculoleptomeningeal) amyloidosis.
Amyloid deposition is seen in the pial and arachnoid membrane, as well as in the walls of vessels in the subarachnoid space associated with TTR mutations including Leu12Pro, Asp18Gly, Ala25Thr, Val30Gly, Ala36Pro, Gly53Glu, Gly53Ala, Phe64Ser, Tyr69His, or Tyr114Cys. Individuals with leptomeningeal amyloidosis show CNS signs and symptoms including: dementia, psychosis, visual impairment, headache, seizures, motor paresis, ataxia, myelopathy, hydrocephalus, or intracranial hemorrhage. When associated with vitreous amyloid deposits, leptomeningeal amyloidosis is known as familial oculolepto-meningeal amyloidosis (FOLMA). In leptomeningeal amyloidosis protein concentration in the cerebrospinal fluid is usually high, and gadolinium-enhanced MRI typically shows extensive enhancement of the surface of the brain, ventricles, and spinal cord.
Genotype-Phenotype Correlations.
In subsets of families with the Val30Met mutation, considerable variation in phenotypic manifestations and age of onset is observed. It is hypothesized that genetic modifiers and non-genetic factors contribute to the pathogenesis and progression of familial TTR amyloidosis. The vast majority of individuals with familial TTR amyloidosis are heterozygous for a TTR mutation. It has been clinically and experimentally demonstrated that the normal allelic variant c.416C>T (Thr119Met) has a protective effect on amyloidogenesis in individuals who have the Val30Met mutation.
Cardiac amyloidosis is caused by Asp18Asn, Val20Ile, Pro24Ser, Ala45Thr, Ala45Ser, His56Arg, Gly57Arg, Ile68Leu, Ala81Thr, Ala81Val, His88Arg, Glu92Lys, Arg103Ser, Leu111Met, or Val122Ile. Peripheral and autonomic neuropathy are absent or less evident in persons with these mutations.
Leptomeningeal amyloidosis is associated with Leu12Pro, Asp18Gly, Ala25Thr, Val30Gly, Ala36Pro, Gly53Glu, Gly53Ala, Phe64Ser, Tyr69His, or Tyr114Cys.
Penetrance.
It is generally accepted that the penetrance is much higher in individuals in endemic foci than outside of endemic foci. In Portugal, cumulative disease risk in individuals with the Val30Met mutation is estimated at 80% by age 50 and 91% by age 70 years, whereas the risk in French heterozygotes is 14% by age 50 and 50% by age 70 years. In Sweden, the penetrance is much lower: 1.7% by age 30, 5% by age 40, 11% by age 50, 22% by age 60, 36% by age 70, 52% by age 80, and 69% by age 90, respectively.
Nomenclature
The neuropathy associated with TTR mutations, now called familial TTR amyloidosis, was formerly referred to as one of the following:
- Familial amyloid polyneuropathy type I (or the Portuguese-Swedish-Japanese type)
- Familial amyloid polyneuropathy type II (or the Indiana/Swiss or Maryland/German type)
Prevalence
The Val30Met mutation, found worldwide, is the most widely studied TTR variant and is responsible for the well-known large foci of individuals with TTR amyloid polyneuropathy in Portugal, Sweden, and Japan. Numerous families with various non-Val30Met mutations have also been identified worldwide.
Small transthyretin (TTR) ligands as possible therapeutic agents in TTR amyloidoses.
Almeida MR, Gales L, Damas AM, Cardoso I, Saraiva MJ. Porto, Portugal.
Curr Drug Targets CNS Neurol Disord. 2005 Oct;4(5):587-96.
In transthyretin (TTR) amyloidosis TTR variants deposit as amyloid fibrils giving origin, in most cases, to peripheral polyneuropathy, cardiomyopathy, carpal tunnel syndrome and/or amyloid deposition in the eye. The amino acid substitutions in the TTR variants destabilize the tetramer, which may dissociate into non native monomeric intermediates that aggregate and polymerize in amyloid fibrils that further elongate. Since this is a multi-step process there is the possibility to impair TTR amyloid fibril formation at different stages of the process namely by tetramer stabilization, inhibition of fibril formation or fibril disruption. Based on the proposed mechanism for TTR amyloid fibril formation we discuss the action of some of the proposed TTR stabilizers such as derivatives of some NSAIDs (diflunisal, diclofenac, flufenamic acid, and derivatives) and the action of amyloid disrupters such as 4′-iodo-4′-deoxydoxorubicin (I-DOX) and tetracyclines. Among all these compounds, TTR stabilizers seem to be the most interesting since they would impair very early the process of amyloid formation and could also have a prophylactic effect.
Clusterin regulates transthyretin amyloidosis.
Lee KW, Lee DH, Son H, Kim YS, Park JY, et al. Gyeongnam National University, South Korea
Clusterin has recently been proposed to play a role as an extracellular molecular chaperone, affecting the fibril formation of amyloidogenic proteins. The ability of clusterin to influence amyloid fibril formation prompted us to investigate whether clusterin is capable of inhibiting TTR amyloidosis. Here, we report that clusterin strongly interacts with wild-type TTR and TTR variants V30M and L55P under acidic conditions, and blocks the amyloid fibril formation of TTR variants. In particular, the amyloid fibril formation of V30M TTR in the presence of clusterin is reduced to level similar to wild-type TTR. We also demonstrated that clusterin is an effective inhibitor of L55P TTR amyloidosis, the most aggressive form of TTR diseases. The mechanism by which clusterin inhibits TTR amyloidosis appears to be through stabilization of TTR tetrameric structure.
Prognosis.
Cardiac amyloidosis in general has a poor prognosis, but this differs according to amyloid type and availability and response to therapy. Treatment may be classified as follows: supportive therapy (ie, modified heart-failure treatment including device therapy); therapies that suppress production of the respective amyloid fibril precursor protein (eg, chemotherapy in AL amyloidosis); and novel strategies to inhibit amyloid fibril formation or to directly target the amyloid deposits or stabilize the precursor protein (especially in ATTR with drugs such as tafamidis or diflunisal). Cardiac transplantation, although rarely feasible, can be very successful in carefully selected patients.
Reducing Amyloid Fibril Precursor Protein Production
Treatment of amyloidosis is currently based on the concept of reducing the supply of the respective amyloid fibril precursor protein. In AL amyloidosis, therapy is directed toward the clonal plasma cells using either cyclical combination chemotherapy or high-dose therapy with autologous stem cell transplantation.
The newer treatment options include bortezomib (a proteosome inhibitor)105 and the newer immunomodulatory drugs lenalidomide and pomalidomide. Bortezomib combinations appear to be especially efficient in amyloidosis with high rates of near-complete clonal responses, which appear to translate into early cardiac responses.106–108 Phase II (bortezomib in combination with cyclophosphamide or doxorubicin) and phase III (bortezomib, melphalan, and dexamethasone compared to melphalan and dexamethasone as front-line treatment) trials are underway.
AA amyloidosis is the only other type of amyloidosis in which production of the fibril precursor protein can be effectively suppressed by currently available therapies. Anti-inflammatory therapies, such as anti-tumor necrosis factor agents in rheumatoid arthritis, can substantially suppress serum amyloid A protein production, but very little experience has been obtained regarding cardiac involvement, which is very rare in this particular type of amyloidosis.
TTR is produced almost exclusively in the liver, and TTR amyloidosis has lately become a focus for novel drug developments aimed at reducing production of TTR through silencing RNA and antisense oligonucleotide therapies. ALN-TTR01, a systemically delivered silencing RNA therapeutic, is already in phase I clinical trial. Liver transplantation has been used as a treatment for variant ATTR for 20 years, to remove genetically variant TTR from the plasma. Although this is a successful approach in ATTR Val30Met, it has had disappointing results in patients with other ATTR variants, which often involve the heart. The procedure commonly results in progressive cardiac amyloidosis through ongoing accumulation of wild-type TTR on the existing template of variant TTR amyloid. The role of liver transplantation in non-Val30Met–associated hereditary TTR amyloidosis thus remains very uncertain.
Inhibition of Amyloid Formation
Amyloid fibril formation involves massive conformational transformation of the respective precursor protein into a completely different form with predominant β-sheet structure. The hypothesis that this conversion might be inhibited by stabilizing the fibril precursor protein through specific binding to a pharmaceutical has lately been explored in TTR amyloidosis. A key step in TTR amyloid fibril formation is the dissociation of the normal TTR tetramer into monomeric species that can autoaggregate in a misfolded form. In vitro studies identified that diflunisal, a now little used nonsteroidal anti-inflammatory analgesic, is bound by TTR in plasma, and that this enhances the stability of the normal soluble structure of the protein. Studies of diflunisal in ATTR are in progress. Tafamidis is a new compound without anti-inflammatory analgesic properties that has a similar mechanism of action. Tafamidis has just been licensed for neuropathic ATTR, but its role in cardiac amyloidosis remains uncertain, and clinical trial results are eagerly awaited. Higher-affinity “superstabilizers” are also in development.
Conclusion
Cardiac amyloidosis remains challenging to diagnose and to treat. Key “red flags” that should raise suspicion include clinical features indicating multisystem disease and concentric LV thickening on echocardiography in the absence of increased voltage on ECG; the pattern of gadolinium enhancement on CMR appears to be very characteristic. Confirmation of amyloid type is now possible in most cases through a combination of immunohistochemistry, DNA analysis, and proteomics. A variety of novel specific therapies are on the near horizon, with potential to both inhibit new amyloid formation and enhance clearance of existing deposits.
Future Prospects
Jeffery W. Kelly, the former Dean of Graduate Studies (2000-2008) and Vice President of Academic Affairs (2000-2006), currently is the Chairman of Molecular and Experimental Medicine and the Lita Annenberg Hazen Professor of Chemistry within the Skaggs Institute of Chemical Biology at The Scripps Research Institute in La Jolla, California.
The work on folding proteins by the Kelly Group focuses on
[1] understanding protein misfolding and aggregation and on developing both chemical
[2] and biological strategies
[3] to ameliorate diseases caused by protein misfolding and/or aggregation.
Besides studying the structural and energetic basis behind protein folding, his laboratory also studies the etiology of neurodegenerative diseases linked to protein aggregation, including Alzheimer’s disease, Parkinson’s Disease, and the familial gelsolin and transthyretin-based amyloidoses–publishing over 250 peer-reviewed papers in this area to date. He has also provided insight into genetic diseases associated with loss of protein function, such as lysosomal storage diseases.
Kelly has cofounded three biotechnology companies, FoldRx Pharmaceuticals (with Susan Lindquist), now owned by Pfizer, Proteostasis Therapeutics, Inc. (with Andrew Dillin and Richard Morimoto) (a private corporation) and Misfolding Diagnostics (with Xin Jiang and Justin Chapman; a private corporation). The Kelly laboratory discovered the first regulatory agency-approved drug that slows the progression of a human amyloid disease using a structure-based design approach. This drug, now called Tafamidis or Vyndaqel, slowed the progression of familial amyloid polyneuropathy in an 18 month placebo controlled trial and in an 18 month extension study sponsored by FoldRx Pharmaceuticals (acquired by Pfizer in 2010). Vyndaqel or Tafamidis was approved for the treatment of Familial amyloid Polyneuropathy by the European Medicines Agency in late 2011. Kelly also discovered that diflunisal kinetically stabilizes transthyretin, enabling a placebo controlled clinical trial with it to ameliorate familial amyloid polyneuropathy–the results of which will be announced in 2013. Proteostasis Therapeutics, Inc. is developing first-in-class drugs that adapt the proteostasis network to ameliorate both loss-of-function misfolding diseases and gain-of-toxic function diseases linked to protein aggregation.
In addition to discovering the first drug that slows the progression of a human amyloid disease, the Kelly Laboratory is credited with demonstrating that transthyretin conformational changes alone are sufficient for amyloidogenesis, discovering the first example of functional amyloid in mammals, making major contributions toward understanding β-sheet folding, discovering the “enhanced aromatic sequon”–sequences that are more efficiently glycosylated by cells and sequences which stabilize the proteins that they are incorporated into as a consequence of N-glycosylation and was corresponding author on and contributed some of the key experimental data demonstrating that altering cellular proteostasis capacity has the potential to alleviate protein misfolding and aggregation diseases.
Native state kinetic stabilization as a strategy to ameliorate protein misfolding diseases: a focus on the transthyretin amyloidoses. Johnson SM, Wiseman RL, Sekijima Y, Green NS, Adamski-Werner SL, Kelly JW.
http://www.ncbi.nlm.nih.gov/pubmed/16359163Small molecule-mediated protein stabilization inside or outside of the cell is a promising strategy to treat protein misfolding/misassembly diseases. Herein we focus on the transthyretin (TTR) amyloidoses and demonstrate that preferential ligand binding to and stabilization of the native state over the dissociative transition state raises the kinetic barrier of dissociation (rate-limiting for amyloidogenesis), slowing and in many cases preventing TTR amyloid fibril formation. Since T119M-TTR subunit incorporation into tetramers otherwise composed of disease-associated subunits also imparts kinetic stability on the tetramer and ameliorates amyloidosis in humans, it is likely that small molecule-mediated native state kinetic stabilization will also alleviate TTR amyloidoses.
Energetic characteristics of the new transthyretin variant A25T may explain its atypical central nervous system pathology.
Sekijima Y, Hammarström P, Matsumura M, Shimizu Y, Iwata M, Tokuda T, Ikeda S, Kelly JW.
Transthyretin (TTR) is a tetrameric protein that must misfold to form amyloid fibrils. Misfolding includes rate-limiting tetramer dissociation, followed by fast tertiary structural changes that enable aggregation. Amyloidogenesis of wild-type (WT) TTR causes a late-onset cardiac disease called senile systemic amyloidosis. The aggregation of one of > 80 TTR variants leads to familial amyloidosis encompassing a collection of disorders characterized by peripheral neuropathy and/or cardiomyopathy. Prominent central nervous system (CNS) impairment is rare in TTR amyloidosis. Herein, we identify a new A25T TTR variant in a Japanese patient who presented with CNS amyloidosis at age 42 and peripheral neuropathy at age 44. The A25T variant is the most destabilized and fastest dissociating TTR tetramer published to date, yet, surprising, disease onset is in the fifth decade. Quantification of A25T TTR in the serum of this heterozygote reveals low levels relative to WT, suggesting that protein concentration influences disease phenotype. Another recently characterized TTR CNS variant (D18G TTR) exhibits strictly analogous characteristics, suggesting that instability coupled with low serum concentrations is the signature of CNS pathology and protects against early-onset systemic amyloidosis. The low A25T serum concentration may be explained either by impaired secretion from the liver or by increased clearance, both scenarios consistent with A25T’s low kinetic and thermodynamic stability. Liver transplantation is the only known treatment for familial amyloid polyneuropathy. This is a form of gene therapy that removes the variant protein from serum preventing systemic amyloidosis. Unfortunately, the choroid plexus would have to be resected to remove A25T from the CSF-the source of the CNS TTR amyloid. Herein we demonstrate that small-molecule tetramer stabilizers represent an attractive therapeutic strategy to inhibit A25T misfolding and CNS amyloidosis. Specifically, 2-[(3,5-dichlorophenyl)amino]benzoic acid is an excellent inhibitor of A25T TTR amyloidosis in vitro.
Prevention of Transthyretin Amyloid Disease by Changing Protein Misfolding Energetics
Per Hammarström*, R. Luke Wiseman*, Evan T. Powers, Jeffery W. Kelly†
Genetic evidence suggests that inhibition of amyloid fibril formation by small molecules should be effective against amyloid diseases. Known amyloid inhibitors appear to function by shifting the aggregation equilibrium away from the amyloid state. Here, we describe a series of transthyretin amyloidosis inhibitors that functioned by increasing the kinetic barrier associated with misfolding, preventing amyloidogenesis by stabilizing the native state. The trans-suppressor mutation, threonine 119 → methionine 119, which is known to ameliorate familial amyloid disease, also functioned through kinetic stabilization, implying that this small-molecule strategy should be effective in treating amyloid diseases.
R104H may suppress transthyretin amyloidogenesis by thermodynamic stabilization, but not by the kinetic mechanism characterizing T119 interallelic trans-suppression.
Sekijima Y, Dendle MT, Wiseman RL, White JT, D’Haeze W, Kelly JW.
The tetrameric protein transthyretin (TTR) forms amyloid fibrils upon dissociation and subsequent monomer misfolding, enabling misassembly. Remarkably, the aggregation of one of over 100 destabilized TTR variants leads to familial amyloid disease. It is known that trans-suppression mediated by the incorporation of T119M subunits into tetramers otherwise composed of the most common familial variant V30M, ameliorates disease by substantially slowing the rate of tetramer dissociation, a mechanism referred to as kinetic stabilization of the native state. R104H TTR has been reported to be non-pathogenic, and recently, this variant has been invoked as a trans-suppressor of amyloid fibril formation. Here, we demonstrate that the trans-suppression mechanism of R104H does not involve kinetic stabilization of the tetrameric structure, instead its modest trans-suppression most likely results from the thermodynamic stabilization of the tetrameric TTR structure. Thermodynamic stabilization increases the fraction of tetramer at the expense of the misfolding competent monomer decreasing the ability of TTR to aggregate into amyloid fibrils. As a consequence of this stabilization mechanism, R104H may be capable of protecting patients with modestly destabilizing mutations against amyloidosis by slightly lowering the overall population of monomeric protein that can misfold and form amyloid.


. Image:Cardiac a...")
. Image:Cardiac a...")

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}