Is CRISPR a Solution to Familial Amyloid Polyneuropathy?
Author and Curator: Larry H. Bernstein, MD, FCAP
What FAP is
Familial amyloid polyneuropathy (FAP), also called transthyretin-related hereditary amyloidosis, transthyretin amyloidosis abbreviated also as ATTR ( hereditary form), or Corino de Andrade’s disease,[1] is an autosomal dominant[2] neurodegenerative disease. It is a form of amyloidosis, and was first identified and described by Portugueseneurologist Mário Corino da Costa Andrade, in 1952.[3] FAP is distinct from senile systemic amyloidosis (SSA), which is not inherited, and which was determined to be the primary cause of death for 70% of supercentenarians who have been autopsied.[4]
Usually manifesting itself between 20 and 40 years of age, it is characterized by pain, paresthesia, muscular weakness and autonomic dysfunction. In its terminal state, the kidneys and the heart are affected. FAP is characterized by the systemic deposition of amyloidogenic variants of the transthyretin protein, especially in the peripheral nervous system, causing a progressive sensory and motor polyneuropathy.
FAP is caused by a mutation of the TTR gene, located on human chromosome 18q12.1-11.2.[5] A replacement of valine by methionine at position 30 (TTR V30M) is the mutation most commonly found in FAP.[1] The variant TTR is mostly produced by the liver.[citation needed] The transthyretin protein is a tetramer. The tetramer has to dissociate into misfolded monomers to aggregate into a variety of structures including amyloid fibrils. Because most patients are heterozygotes, they deposit both mutant and wild type TTR subnits.
FAP is inherited in an autosomal dominant manner.[2] This means that the defective gene responsible for the disorder is located on anautosome (chromosome 18 is an autosome), and only one copy of the defective gene is sufficient to cause the disorder, when inherited from a parent who has the disorder.
FAP can be ameliorated by liver transplantation. Wikipedia https://en.wikipedia.org/wiki/Transthyretin-related_hereditary_amyloidosis
ATTRV30M Amyloidosis
|
- Familial amyloid polyneuropathy (FAP) or transthyretin (TTR) amyloid polyneuropathy is a progressive sensorimotor and autonomic neuropathy of adulthood onset. Weight loss and cardiac involvement are frequent; ocular or renal complications may also occur. The prevalence worldwide is unknown, but the prevalence in the general population in Japan has recently been estimated at around 1 per million.
- FAP is clinically heterogeneous, with the clinical presentation depending on the genotype and geographic origin. FAP usually presents as a length-dependent sensory polyneuropathy with autonomic disturbances. Inaugural manifestations are paresthesiae, pain or trophic lesions of the feet, gastrointestinal disorders or weight loss. The most pronounced sensory loss involves pain and temperature sensation. Motor loss occurs later. Autonomic features include postural hypotension, and gastrointestinal and genitourinary disorders.
- FAP is transmitted as an autosomal dominant trait and is caused by mutations in the TTR gene (18q12.1). 100 TTR mutations have been identified so far and are associated with varying patterns of organ involvement, age of onset and disease progression. The most common variant is the TTR Val30Met substitution for which several endemic foci have been identified most notably from Portugal, Japan and Sweden. However, the Val30Met phenotype varies between these countries.
- Detection of amyloid-associated TTR mutations is required for diagnosis. However, identification of a disease-causing mutation is not considered as diagnostic because penetrance is variable. Clinical observation and tissue biopsy (from the nerve or kidney, labial salivary glands, subcutaneous fat tissue or rectal mucosa) are required for a definitive diagnosis: amyloid deposits are characterized by Congo red staining on light microscopy and green birefringence on polarized light microscopy.
- The differential diagnosis should include diabetic neuropathy, chronic inflammatory demyelinating polyneuropathy (see this term), and light chain (AL), gelsolin and apolipoprotein A1 amyloidosis (see these terms). Antenatal diagnosis through chorionic villus sampling should be proposed to patients with early-onset (< 40 years) forms of FAP.
- Genetic counseling should be offered to affected families and presymptomatic detection of relatives of an index case is important to allow early diagnosis.
- Management of FAP should be multidisciplinary, involving a neurologist, geneticist, cardiologist and liver surgeon. Liver transplantation (LT) is currently the only treatment for preventing synthesis of the amyloidogenic variants of TTR. LT can stop progression of the disease during its early stages. Symptomatic treatments are essential for sensorimotor and autonomic neuropathy and visceral complications.
- FAP is a severe and disabling disease. Severe cardiac, renal and ocular manifestations may develop. Death occurs within a mean interval of 10.8 years after onset of the inaugural symptoms and may occur suddenly or may be secondary to infections or cachexia.
Lancet Neurol. 2011 Dec;10(12):1086-97. doi: 10.1016/S1474-4422(11)70246-0.
Familial amyloid polyneuropathy
Planté-Bordeneuve V1, Said G. Author information
Familial amyloid polyneuropathies (FAPs) are a group of life-threatening multisystem disorders transmitted as an autosomal dominant trait. Nerve lesions are induced by deposits of amyloid fibrils, most commonly due to mutated transthyretin (TTR). Less often the precursor of amyloidosis is mutant apolipoprotein A-1 or gelsolin. The first identified cause of FAP-the TTR Val30Met mutation-is still the most common of more than 100 amyloidogenic point mutations identified worldwide. The penetrance and age at onset of FAP among people carrying the same mutation vary between countries. The symptomatology and clinical course of FAP can be highly variable. TTR FAP typically causes a nerve length-dependent polyneuropathy that starts in the feet with loss of temperature and pain sensations, along with life-threatening autonomic dysfunction leading to cachexia and death within 10 years on average. TTR is synthesised mainly in the liver, and liver transplantation seems to have a favourable effect on the course of neuropathy, but not on cardiac or eye lesions. Oral administration of tafamidis meglumine, which prevents misfolding and deposition of mutated TTR, is under evaluation in patients with TTR FAP. In future, patients with FAP might benefit from gene therapy; however, genetic counselling is recommended for the prevention of all types of FAP.
The course and prognostic factors of familial amyloid polyneuropathy after liver transplantation
David Adams, Didier Samuel, Catherine Goulon-Goeau, …, Henri Bismuth, Gérard Said
DOI: http://dx.doi.org/10.1093/brain/123.7.1495 1495-1504
Familial amyloid polyneuropathy (FAP) associated with mutations of the transthyretin (TTR) gene is the most common type of FAP, a devastating disease causing death within 10 years after the first symptoms. Because most of the amyloidogenic mutated TTR is secreted by the liver, transplantation is widely used to treat these patients, but long-term quantitative evaluation of the effects of liver transplantation on the progression of the neuropathy are not available. We have treated 45 patients with symptomatic TTR-FAP, including 43 with the Met30 TTR gene mutation, and report on the results of periodic evaluation of markers of neuropathy in 25 of them, who have been followed for more than 2 years after liver transplantation (mean follow-up 4 years). The overall survival rates at 1 and 5 years were 82 and 60%, respectively. Urinary incontinence and a low Norris score at liver transplantation were associated with poorer outcome. The motor score stabilized in seven of 11 patients (64%) with mild sensorimotor neuropathy (walking unaided) and in two of the eight patients (25%) with severe sensorimotor deficit (walking with aid) at liver transplantation. In five other patients, deterioration of motor deficit occurred only within the first year after liver transplantation, but was progressive after this interval in two patients. None of the six patients with pure sensory neuropathy developed motor loss and superficial sensory loss remained unchanged. Two years after liver transplantation, the rate of myelinated axon loss in nerve biopsy specimens was markedly lower in seven transplanted patients (0.9/mm2 of endoneurial area/month) than in non-transplanted patients (70/mm2 of endoneurial area/month). Symptoms of dysautonomia and quantitated cardiocirculatory autonomic tests remained unchanged. In all patients, serum mutated TTR decreased to 2.5% of pre-liver transplantation values and remained at this level during follow-up. We presently recommend liver transplantation in FAP patients at onset of first symptoms and exclusion of those with a Norris score below 55 and/or with urinary incontinence.
Familial Amyloid Polyneuropathy (Amyloidotic transthyretin related neuropathy [ATTR]
Transthyretin (TTR) amyloidosis is an autosomal dominant disorder caused by the deposition of insoluble amyloid fibrils around peripheral nerves and in various tissues, including the heart muscle. Based on the predominant organ involvement, several distinct subtypes have been reported.
Familial amyloid polyneuropathy (FAP) aka TTR amyloid neuropathy is characterized by slowly progressive, peripheral sensorimotor polyneuropathy and autonomic dysfunction. Disease onset is usually in the third to fourth decade of life. Sensory neuropathy starts in the lower extremities with paresthesia, impaired pain and temperature sensation, followed by loss of motor function. Autonomic neuropathy usually manifests with orthostatic hypotension, constipation alternating with diarrhea, vomiting, impotence or hypohidrosis. Unrelated to neuropathy, other organs manifestations may include cardiomyopathy, vitreous opacities and CNS amyloidosis.
Leptomeningeal amyloidosis aka oculoleptomeningeal amyloidosis affects predominantly the central nervous system, sometimes combined with visual impairment.
Cardiac amyloidosis usually manifests in the sixth decade of life with progressive left ventricular hypertrophy and restrictive cardiomyopathy. In a subset of families with cardiac amyloidosis, peripheral neuropathy may be completely absent or very mild.
Treatment: Currently, the only effective treatment for FAP is an orthotopic liver transplant to stop production of misfolded amyloid protein. In patients with severe amyloid cardiomyopathy, a heart transplant may be necessary. Different drugs designed to prevent or alleviate accumulation of TTR amyloid protein (transthyretin amyloidois inhibitors) are currently under investigation.
Contemporary Reviews in Cardiovascular Medicine
- Transthyretin (TTR) Cardiac Amyloidosis
Frederick L. Ruberg, John L. Berk
Circulation.2012; 126: 1286-1300 doi: 10.1161/CIRCULATIONAHA.111.078915
The systemic amyloidoses are a family of diseases induced by misfolded or misassembled proteins. Extracellular deposition of these proteins as soluble or insoluble cross β-sheets disrupts vital organ function.1 More than 27 different precursor proteins have the propensity to form amyloid fibrils.2 The particular precursor protein that misfolds to form amyloid fibrils defines the amyloid type and predicts the patient’s clinical course. Several types of amyloid can infiltrate the heart, resulting in progressive diastolic and systolic dysfunction, congestive heart failure, and death. Treatment of cardiac amyloidosis is dictated by the amyloid type and degree of involvement. Consequently, early recognition and accurate classification are essential.3
The diagnosis of amyloidosis requires histological identification of amyloid deposits. Congo Red staining renders amyloid deposits salmon pink by light microscopy, with a characteristic apple green birefringence under polarized light conditions (Figure 1). Additional immunohistochemical staining for precursor proteins identifies the type of amyloidosis (Figure 2).4 Ultimately, immunogold electron microscopy and mass spectrometry confer the greatest sensitivity and specificity for amyloid typing.5,6
Transthyretin participates in beta-amyloid transport from the brain to the liver- involvement of the low-density lipoprotein receptor-related protein 1?
Mobina Alemi, Cristiana Gaiteiro, Carlos Alexandre Ribeiro, Luís Miguel Santos,João Rodrigues Gomes, Sandra Marisa Oliveira, ….., Maria João Saraiva & Isabel Cardoso
Scientific Reports 6, Article number: 20164 (2016) doi:10.1038/srep20164
Transthyretin (TTR) binds Aβ peptide, preventing its deposition and toxicity. TTR is decreased in Alzheimer’s disease (AD) patients. Additionally, AD transgenic mice with only one copy of the TTR gene show increased brain and plasma Aβ levels when compared to AD mice with both copies of the gene, suggesting TTR involvement in brain Aβ efflux and/or peripheral clearance. Here we showed that TTR promotes Aβ internalization and efflux in a human cerebral microvascular endothelial cell line, hCMEC/D3. TTR also stimulated brain-to-blood but not blood-to-brain Aβ permeability in hCMEC/D3, suggesting that TTR interacts directly with Aβ at the blood-brain-barrier. We also observed that TTR crosses the monolayer of cells only in the brain-to-blood direction, as confirmed by in vivo studies, suggesting that TTR can transport Aβ from, but not into the brain. Furthermore, TTR increased Aβ internalization by SAHep cells and by primary hepatocytes from TTR+/+ mice when compared to TTR−/− animals. We propose that TTR-mediated Aβ clearance is through LRP1, as lower receptor expression was found in brains and livers of TTR−/− mice and in cells incubated without TTR. Our results suggest that TTR acts as a carrier of Aβ at the blood-brain-barrier and liver, using LRP1.
Transthyretin (Prealbumin) in Health and Disease: Nutritional Implications
Annual Review of Nutrition
Vol. 14: 495-533 (Volume publication date July 1994)
DOI: 10.1146/annurev.nu.14.070194.002431
Y Ingenbleek, and V Young
Plasma Transthyretin as a Biomarker of Lean Body Mass and Catabolic States1,2
Yves Ingenbleek3,* and Larry H Bernstein4
Adv Nutr Sep 2015; 6:572-580, 2015 doi: 10.3945/an.115.008508
Plasma transthyretin (TTR) is a plasma protein secreted by the liver that circulates bound to retinol-binding protein 4 (RBP4) and its retinol ligand. TTR is the sole plasma protein that reveals from birth to old age evolutionary patterns that are closely superimposable to those of lean body mass (LBM) and thus works as the best surrogate analyte of LBM. Any alteration in energy-to-protein balance impairs the accretion of LBM reserves and causes early depression of TTR production. In acute inflammatory states, cytokines induce urinary leakage of nitrogenous catabolites, deplete LBM stores, and cause an abrupt decrease in TTR and RBP4 concentrations. As a result, thyroxine and retinol ligands are released in free form, creating a second frontline that strengthens that primarily initiated by cytokines. Malnutrition and inflammation thus keep in check TTR and RBP4 secretion by using distinct and unrelated physiologic pathways, but they operate in concert to downregulate LBM stores. The biomarker complex integrates these opposite mechanisms at any time and thereby constitutes an ideally suited tool to determine residual LBM resources still available for metabolic responses, hence predicting outcomes of the most interwoven disease conditions.
Evaluating the binding selectivity of transthyretin amyloid fibril inhibitors in blood plasma
Hans E. Purkey, Michael I. Dorrell, and Jeffery W. Kelly*
Department of Chemistry and The Skaggs Institute of Chemical Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, MB12, La Jolla, CA 92037
http://www.pnas.org/content/98/10/5566.full.pdf
Transthyretin (TTR) tetramer dissociation and misfolding facilitate assembly into amyloid fibrils that putatively cause senile systemic amyloidosis and familial amyloid polyneuropathy. We have previously discovered more than 50 small molecules that bind to and stabilize tetrameric TTR, inhibiting amyloid fibril formation in vitro. A method is presented here to evaluate the binding selectivity of these inhibitors to TTR in human plasma, a complex biological fluid composed of more than 60 proteins and numerous small molecules. Our immunoprecipitation approach isolates TTR and bound small molecules from a biological fluid such as plasma, and quantifies the amount of small molecules bound to the protein by HPLC analysis. This approach demonstrates that only a small subset of the inhibitors that saturate the TTR binding sites in vitro do so in plasma. These selective inhibitors can now be tested in animal models of TTR amyloid disease to probe the validity of the amyloid hypothesis. This method could be easily extended to evaluate small molecule binding selectivity to any protein in a given biological fluid without the necessity of determining or guessing which other protein components may be competitors. This is a central issue to understanding the distribution, metabolism, activity, and toxicity of potential drugs.
Amyloid diseases are characterized by the conversion of soluble proteins or peptides into insoluble b-sheet-rich amyloid fibrils. There are currently 17 different human proteins known to form amyloid fibrils in vivo (1–4). These fibrils, or their oligomeric precursors, are thought to cause pathology either through disruption of normal cellular function or by direct toxicity (5–8). X-ray fibril diffraction and electron microscopy reconstruction of amyloid fibrils reveal filaments that have a lamellar cross b-sheet structure wrapped around one another (9–13). Folded proteins form amyloid fibrils through partial unfolding triggered by a change of local environment, a mutation in the protein, or both (8, 14–20).
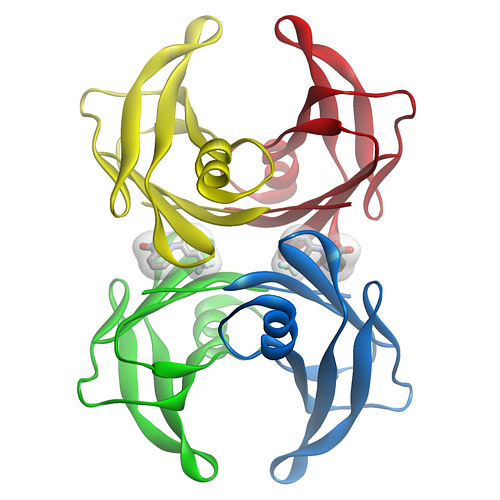
Transthyretin (TTR) is a tetrameric protein composed of identical 127-aa subunits that fold into a b-sandwich tertiary structure. It is found in both the plasma (3.6 mM) and cerebrospinal fluid (CSF) (0.36 mM) of humans. The TTR tetramer has two negatively cooperative C2 symmetric thyroxine (T4)-binding sites (21–23). In the CSF, it binds and transports the thyroid hormone T4 and the retinol-binding protein (RBP), which in turn transports vitamin A. In the plasma, only 10–15% of TTR has T4 bound to it, as thyroid-binding globulin has an order of magnitude higher affinity for T4 than does TTR (24). TTR fibril formation is linked to two amyloid diseases in which the fibrils are composed of full-length protein. Deposition of wild-type TTR is associated with cardiac dysfunction in the disease senile systemic amyloidosis (SSA) (25, 26). More than 70 different single-site mutants have been linked to early-onset amyloid deposition in diseases with a spectrum of clinical manifestations, collectively referred to as familial amyloid polyneuropathy (FAP) (27–35).
We have discovered compounds, through both screening and structure-based design, that dramatically inhibit TTR amyloid fibril formation in vitro (36–42). To stabilize the TTR tetramer and thus prevent amyloid fibril formation in SSA and FAP, these small molecules must be able to selectively bind to TTR in human blood plasma over all other plasma proteins. Possible competitors include thyroid-binding globulin (TBG), which has an order of magnitude higher affinity for TTR’s natural ligand, T4; and albumin, which binds numerous hydrophobic small molecules and is present at a concentration two orders of magnitude higher than TTR, as well as the other plasma proteins. Historically, one was forced to choose two or three of the most likely protein competitors and evaluate their relative affinities for the small molecule in comparison to the protein of interest. The advantage of the approach outlined within this article is that the binding selectivity of TTR amyloid inhibitors in human plasma is determined without having to make assumptions as to which proteins may competitively bind the TTR ligand. Compounds that bind to TTR selectively in plasma are the best candidates for further evaluation in animal models and, ultimately, in human clinical trials.
Analysis of Nonsteroidal Antiinflammatory Drugs (NSAIDs)
The first compounds evaluated for selective binding to TTR in plasma were the NSAIDs previously identified to be potent TTR amyloid inhibitors in vitro (37, 42). Because these compounds are already approved by the Food and Drug Administration, they could easily be evaluated in human clinical trials for another indication if they proved to be selective TTR binders in human plasma. However, none of the NSAIDs exhibited significant selectivity for binding TTR in human plasma at a concentration of 10.8 mM (Table 1), although most exhibit a submicromolar Kd (37, 50). The most selective NSAIDs, flufenamic acid (1) and mefenamic acid (2), only had 0.2 eq of a maximum of 2 molar eq bound to TTR. However, fenoprofen (7), flurbiprofen (6), flufenamic acid (1), mefenamic acid (2), and diflunisal (4) all have maximal therapeutic plasma concentrations exceeding 20 mM (51, 52). When these compounds were incubated with plasma at their maximal therapeutic concentrations, flufenamic acid and diflunisal showed increased binding selectivity (stoichiometry) to TTR (Table 1). Diflunisal (4) is notable in that its 224 mM maximal therapeutic concentration leads to 0.85 eq of drug bound to TTR. Increasing the concentration of all other NSAIDs to their maximal therapeutic dose did not result in dramatic increases in binding selectivity, likely because of binding to other plasma proteins.
Analysis of the Remaining Lead Compounds. Approximately 40 additional small molecule amyloid inhibitors were evaluated for their ability to bind to TTR in plasma by using our immunoprecipitationyHPLC approach (Fig. 2). These compounds were derived from screening or structure-based design and identified as promising by an in vitro fibril formation assay (refs. 40–42; V. H. Oza, H. M. Petrassi, and J.W.K., unpublished data). Lead compounds having diverse structures including biaryls, biarylamines, stilbenes, and dibenzofurans showed promising selectivity (Table 2). Three compounds from this group (9-11) possess excellent TTR-binding selectivity in plasma. At a concentration of 10.8 mM, they exhibit saturation of .1 of the two possible binding sites in the TTR tetramer. Determination of the TTR-binding affinities of these three compounds in buffer shows that the Kd values do not correlate with the plasma binding selectivity (Table 2). For example, inhibitors 9 and 10 have greater than an order of magnitude difference in Kd but nearly identical binding selectivity (stoichiometry) to TTR in plasma. Moreover, compounds with modest binding selectivity, such as flufenamic acid (1), have been previously determined to have Kd values very similar to those exhibiting excellent selectivities (37). Mass spectrometry confirmed the identity of inhibitors 9-12 isolated by immunoprecipitation HPLC as the compounds that were initially incubated with plasma.
Binding to plasma proteins is an important factor in determining the overall distribution, metabolism, activity, and toxicity of a drug (55). In this particular case, we desire small molecules that bind to the plasma protein TTR over protein competitors whose identities are not known and likely change with the small molecule under evaluation. This binding is known to stabilize TTR’s normally folded state, thus preventing the conformational changes required for amyloidogenicity (14, 36). The immunoprecipitationyHPLCbased binding selectivity method outlined above allows quantification of the stoichiometry of small molecule binding to TTR in human plasma in the presence of all other plasma proteins and numerous competing small molecules without the use of tags, such as radiolabels, on the small molecule. Immunoprecipitation has been used previously to determine the stoichiometry of metal ion binding to specific plasma proteins (56, 57). However, to our knowledge, this is the first use of the technique to determine the binding selectivity of a small molecule to a single protein in human plasma. In principle, this approach is applicable to evaluate the binding selectivity of small molecules to any plasma protein, provided that a highly selective antibody for the protein can be generated, binding does not block the epitope or destroy it by conformational change, and appropriate wash steps can be introduced to avoid nonspecific binding of the small molecule and other proteins to the resin.
The Kelly Group Research
Transthyretin amyloid diseases: understanding the mechanism of proteotoxicity and inhibition of amyloid fibril formation
Figure 1: Three dimensional structure of the flufenamic acid-transthyretin tetramer complex. The small organic molecule, flufenamic acid, inhibits the conformational changes of transthyretin associated with amyloid fibril formation. Courtesy of Steven Johnson.
Transthyretin is a 55 kDa homotetrameric protein (Figure 1) that transports L-thyroxine and holo-retinol binding protein in the serum and cerebrospinal fluid of humans. We discovered that conformational changes alone enable transthyretin aggregation. Transthyretin amyloid formation in vivo is initiated by dissociation of the native tetramer under a denaturation stress of unknown origin. Subsequently, the resulting monomers can partially denature and misassemble into amyloid fibrils and other amorphous aggregates. Aggregation by transthyretin monomers under acidic conditions (conditions that render transthyretin amyloidogenesis fast on a laboratory time scale) occurs via a downhill polymerization mechanism, which means that every step along the amyloid formation pathway is energetically favorable and fast relative to tetramer dissociation. Thus, tetramer dissociation is rate limiting for transthyretin amyloid formation in the case of the wild type protein and for the vast majority of mutants.
Stabilizing the native tetrameric state of transthyretin should ameliorate transthyretin amyloid diseases. This hypothesis is supported by the observation of trans-suppression, in which compound heterozygotes expressing both a disease-associated mutation (e.g., V30M) and a trans-suppressor mutation (e.g., T119M) do not develop transthyretin amyloid disease. We have shown that the trans-suppressor mutation T119M inhibits amyloid formation by kinetically stabilizing (i.e., dramatically slowing the dissociation of) mixed transthyretin tetramers.
We have designed numerous small molecules based on the crystal structures of transthyretin that are now established to avidly bind to the unoccupied L-thyroxine binding sites within transthyretin. We have shown that small molecule binding to these sites inhibits amyloid formation in vitro by selectively stabilizing the native tetrameric state over the dissociative transition state, thus raising the energetic barrier for tetramer dissociation, dramatically slowing the rate-limiting step in the aggregation pathway. Over the past decade, we have identified and synthesized over 1000 small molecule inhibitors of transthyretin amyloid formation that group into half a dozen distinct families. The inhibitors are typically composed of two differentially-substituted aromatic rings connected by linkers of variable chemical composition.
These small molecule kinetic stabilizers either just bind to transthyretin or bind and then react chemoselectively with only one of eight lysine ε-amino groups within transthyretin. In collaboration with Dr. Ian Wilson (The Scripps Research Institute, Department of Molecular Biology), we systematically ranked a myriad of possibilities for the three substructures composing a typical transthyretin kinetic stabilizer. We used these data in a substructure combination strategy to develop very potent and selective transthyretin kinetic stabilizers. In collaboration with Dr. Joel Buxbaum (The Scripps Research Institute, Department of Molecular and Experimental Medicine), these and other compounds are being tested in cell and mouse models.
These data should allow us to be able to predict the structures of potent and selective transthyretin amyloidogenesis inhibitors that are largely devoid of characteristics undesirable for a clinical candidate.
In a recently completed placebo-controlled, double-blind clinical trial carried out by FoldRx Pharmaceuticals (a company that Kelly cofounded), benzoxazoles discovered by the Kelly Laboratory and developed by FoldRx Pharmaceuticals proved safe and effective at halting the progression of familial amyloid polyneuropathy by a myriad of metrics. This transthyretin kinetic stabilizer, now named Tafamidis, provides the first pharmacological evidence that the process of amyloid fibril formation causes the transthyretin amyloid diseases—reinvigorating efforts of other investigators and companies to do the same in other amyloid diseases. In collaboration with Dr. Martha Skinner and Dr. John Berk at Boston University, another kinetic stabilizer discovered by the Kelly Laboratory, diflunisal, is being tested in a second placebo-controlled human clinical trial for familial amyloid polyneuropathy that is currently enrolling patients.
In collaboration with Dr. Bill Balch (The Scripps Research Institute, Department of Cell Biology) and Dr. Luke Wiseman (The Scripps Research Institute, Department of Molecular and Experimental Medicine), we investigated the relationship between the secretion of destabilized transthyretin variants and the pathology of the disease they cause. The earliest onset (onset at 20-30 years of age) and most severe transthyretin amyloid diseases are generally associated with mutations that strongly destabilize the transthyretin tetramer, resulting in facile transthyretin dissociation and misfolded monomer misassembly into aggregates in the peripheral nerves. However, the most destabilized variants characterized to date, A25T and D18G transthyretin, do not cause an early onset systemic amyloid disease because they are intercepted by the degradation component of the proteostasis network within liver cells—the liver is where most of the transthyretin in the blood plasma is produced. Because of endoplasmic reticulum-associated degradation of these highly destabilized transthyretin variants within the secretory pathway of liver cells, the concentration of the destabilized mutant transthyretin in blood plasma would not be high enough to enable the amyloidogenesis responsible for pathology. Interestingly, these mutants lead to a very rare brain disease, with an intermediate age of onset (40-50 years old), because the choroid plexus is more permissive in its ability to secrete highly destabilized transthyretin variants for reasons that we are seeking to understand. These results suggest that endoplasmic reticulum-assisted folding mediated by the proteostasis network determines protein secretion in a tissue-specific manner, and we propose that its competition with endoplasmic reticulum-associated degradation may explain the appearance of tissue-selective amyloid diseases.
Is there a CRISPR alternative?
CRISPR biotech Intellia strikes licensing deal with Regeneron, readies IPO
Dive Brief:
- Intellia Therapeutics, one of several companies working to develop CRISPR/Cas 9 technology commercially, on Monday filed to go public in an initially priced $120 million IPO. Concurrently, the biotech announced a collaboration and licensing deal with Regeneron Pharmaceuticals aimed at advancing up to 10 CRISPR-based programs, focusing primarily on liver diseases.
- Regeneron will pay Intellia $75 million upfront, as well as investing $50 million in Intellia’s forthcoming IPO. Additionally, the agreement includes as much as $320 million in milestone payments, according to SEC filing documents.
- The first program to be co-developed with Regeneron will target a rare genetic disorder known as ATTR, which can causes severely impaired nerve or cardiac function.
Intellia hopes CRISPR gene-editing can cure the genetic disorder ATTR, which is caused by a buildup of the transthyretin (TTR) protein in tissue. By “knocking out” TTR expression in the liver, Intellia could reduce or eliminate the disease-causing buildup.
A view by Larry H. Bernstein, MD, FCAP
I can only wish them success in a project that may well be a search for the Trojan Horse. TTR is also synthesized in the choroid plexus. In addition, knocking out TTR expression in the liver is not likely to fit a mechanism leading to TTR buildup in tissue. The TTR is a plasma tetrameric transport protein released into the circulation for transport of TH with a bound retinol-binding protein carrying one retinol per tetrameric protein. Other considerations are the nuclear release and the cell binding that have been extensively studied. The buildup in tissues is a misfolded protein – amyloid, which has extensively studied fibrils. Another issue is the actual functional transport of hormone and delivery of retinoid to cross the blood brain barrier.
Transthyretin participates in betaamyloid transport from the brain to the liver- involvement of the lowdensity lipoprotein receptor-related protein 1?
Mobina Alemi1,2,3, Cristiana Gaiteiro1,2, Carlos Alexandre Ribeiro1,2, Luís Miguel Santos1,2, João Rodrigues Gomes1,2, Sandra Marisa Oliveira1,2,3, Pierre-Olivier Couraud4, Babette Weksler5, Ignacio Romero6, Maria João Saraiva1,2 & Isabel Cardoso1
file:///C:/Users/Larry/Documents/Transthyretin%20participates%20in%20beta-amyloid%20transport%20from%20the%20brain%20to%20the%20liver%20srep20164.pdf
Distinctive binding and structural properties of piscine transthyretin
Claudia Follia, Nicola Pasquatob, Ileana Ramazzinaa, Roberto Battistuttab;c, Giuseppe Zanottib;c, Rodolfo Bernia;
FEBS Letters 555 (2003) 279-284
The thyroid hormone binding protein transthyretin (TTR) forms a macromolecular complex with the retinol-specific carrier retinol binding protein (RBP) in the blood of higher vertebrates. Piscine TTR is shown here to exhibit high binding affnity for L-thyroxine and negligible affnity for RBP. The 1.56 A % resolution X-ray structure of sea bream TTR, compared with that of human TTR, reveals a high degree of conservation of the thyroid hormone binding sites. In contrast, some amino acid differences in discrete regions of sea bream TTR appear to be responsible for the lack of protein-protein recognition, providing evidence for the crucial role played by a limited number of residues in the interaction between RBP and TTR. Overall, this study makes it possible to draw conclusions on evolutionary relationships for RBPs and TTRs of phylogenetically distant vertebrates.
Jeffery Kelly, Ph.D., Lita Annenberg Hazen Professor of Chemistry
Chairman, Department of Molecular and Experimental Medicine
California Campus, Scripps Institute
The Skaggs Institute for Chemical Biology
San Diego, CA
Studies directed at understanding the principles of b-sheet folding utilizing rapid kinetic and thermodynamic measurements are ongoing and focused on the WW domain, a 34-residue 3-stranded b-sheet. Chemical synthesis of the WW domain allows us to incorporate unique amino acids into the fold to probe structural determinants of transition state and ground state structure.
Awards & Professional Activities
- Searle Scholar Award in Biomedical Sciences, 1991-1994
- Camille and Henry Dreyfus Teacher Scholar Award, 1994
- Texas A&M University Teacher Scholar Award, 1994-1995
- The Biophysical Society National Lecturer, 1999
- The Protein Society – Dupont Young Investigator Award, 1999
- Alumni Distinguished Achievement Award, State University of New York at Fredonia, 2000
- Arthur C. Cope Scholar Award, American Chemical Society, 2001
Selected References
Deechongkit, S.; Nguyen, H.; Dawson, P.E.; Gruebele, M.; Kelly, J.W. “Context Dependent Contributions of Backbone H-Bonding to b-Sheet Folding Energetics” Nature 2004, 430, 101-105.
Cohen,F.: Kelly, J.W. “Therapeutic Approaches to Protein Folding Diseases” Nature, 2003, 426, 905-910.
Hammarstrom, P.; Wiseman, R.L.; Powers, E.T.; Kelly, J.W., “Prevention of Transthyretin Amyloid Disease by Changing Protein Misfolding Energetics” Science 2003, 299, 713-716.
Sawkar, A.; Cheng, W-C.; Beutler, E.; Wong, C.-H.; Balch, W.E.; Kelly, J.W. “Chemical Chaperones Increase the Cellular Activity of N370S β glucosidase; A Therapeutic Strategy for Gaucher’s Disease” Proc.Natl.Acad.Sci., 2002, 99, 15428-15433.
Links
The Skaggs Institute for Chemical Biology
The Skaggs Institute Scientific Report
Other Related articles articles published in this Open Access Online Scientific Journal, include the following:
Alzheimer’s disease, snake venome, amyloid and transthyretin
Stabilizers that prevent transthyretin-mediated cardiomyocyte amyloidotic toxicity
Transthyretin and Lean Body Mass in Stable and Stressed State
A Second Look at the Transthyretin Nutrition Inflammatory Conundrum
Licensing deal with Regeneron to accelerate CRISPR biotech Intellia (Jennifer Doudna’s Start Up) for an IPO
The relationship of stress hypermetabolism to essential protein needsNutrition and Aging
The relationship of S amino acids to marasmic and kwashiorkor PEM
The matter of stunting in the Ganges Plains
Adenosine Receptor Agonist Increases Plasma Homocysteine
Voluntary and Involuntary S-Insufficiency
Thyroid Function and Disorders
More Complexity in Protein Evolution
Proteins: An evolutionary record of diversity and adaptation
Malnutrition in India, High Newborn Death Rate and Stunting of Children Age Under Five Years
Vegan Diet is Sulfur Deficient and Heart Unhealthy
Metabolomics: its Applications in Food and Nutrition Research
Late Onset of Alzheimer’s Disease and One-carbon Metabolism
How Methionine Imbalance with Sulfur-Insufficiency Leads to Hyperhomocysteinemia
Amyloidosis with Cardiomyopathy
Expanding the Genetic Alphabet and Linking the Genome to the Metabolome
Proteomics and Biomarker Discovery
The role of biomarkers in the diagnosis of sepsis and patient management
How to deal with the most common form of inherited amyloidoses?


 5 to >1,000 polyps) observed in mutation carriers within a single family
5 to >1,000 polyps) observed in mutation carriers within a single family


. Image:Cardiac a...")
. Image:Cardiac a...")
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}