1. Brown M.A., Lindheimer M.D., de Swiet M. The classification and diagnosis of the hypertensive disorders of pregnancy: statement from the International Society for the Study of Hypertension in Pregnancy (ISSHP) Hypertens Pregnancy. 2001;20:IX–XIV. [PubMed: 12044323]
2. Chesley L.C., Annitto J.E., Cosgrove R.A. The familial factor in toxemia of pregnancy. Obstet Gynecol. 1968;32:303–311. [PubMed: 5742111]
3. Thornton J.G., Macdonald A.M. Twin mothers, pregnancy hypertension and pre-eclampsia. Br J Obstet Gynaecol. 1999;106:570–575.[PubMed: 10426615]
4. O’Shaughnessy K.M., Ferraro F., Fu B. Identification of monozygotic twins that are concordant for preeclampsia. Am J Obstet Gynecol. 2000;182:1156–1157. [PubMed: 10819852]
5. Chappell S., Morgan L. Searching for genetic clues to the causes of pre-eclampsia. Clin Sci (Lond) 2006;110:443–458. [PubMed: 16526948]
6. Cnattingius S. The epidemiology of smoking during pregnancy: smoking prevalence, maternal characteristics, and pregnancy outcomes. Nicotine Tob Res.2004;6(Suppl. 2):S125–140. [PubMed: 15203816]
7. Salonen Ros H., Lichtenstein P., Lipworth L. Genetic effects on the liability of developing pre-eclampsia and gestational hypertension. Am J Med Genet.2000;91:256–260. [PubMed: 10766979]
8. Redman C.W., Sargent I.L. Latest advances in understanding preeclampsia. Science. 2005;308:1592–1594. [PubMed: 15947178]
9. Cooper D.W., Brennecke S.P., Wilton A.N. Genetics of pre-eclampsia. Hypertens Pregnancy. 1993;12:1–23.
10. Esplin M.S., Fausett M.B., Fraser A. Paternal and maternal components of the predisposition to preeclampsia. N Engl J Med. 2001;344:867–872.[PubMed: 11259719]
11. Skjaerven R., Vatten L.J., Wilcox A.J. Recurrence of pre-eclampsia across generations: exploring fetal and maternal genetic components in a population based cohort. BMJ. 2005;331:877. [PMCID: PMC1255793] [PubMed: 16169871]
12. Haig D. Genetic conflicts in human pregnancy. Q Rev Biol. 1993;68:495–532. [PubMed: 8115596]
13. GOPEC Disentangling fetal and maternal susceptibility for pre-eclampsia: a British multicenter candidate-gene study. Am J Hum Genet. 2005;77:127–131. [PMCID: PMC1226184] [PubMed: 15889386]
14. Mutze S., Rudnik-Schoneborn S., Zerres K. Genes and the preeclampsia syndrome. J Perinat Med. 2008;36:38–58. [PubMed: 18184097]
15. Colhoun H., McKeigue P., Davey Smith G. Problems of reporting genetic associations with comlex outcomes. Lancet. 2003;361:865–872.[PubMed: 12642066]
16. Wacholder S., Chanock S., Garcia-Closas M. Assessing the probability that a positive report is false: an approach for molecular epidemiology studies. J Natl Cancer Inst. 2004;96:434–442. [PubMed: 15026468]
17. Isermann B., Sood R., Pawlinski R. The thrombomodulin-protein C system is essential for the maintenance of pregnancy. Nat Med. 2003;9:331–337.[PubMed: 12579195]
18. Brenner B. Thrombophilia and pregnancy loss. Thromb Res. 2002;108:197–202. [PubMed: 12617981]
19. Lin J., August P. Genetic thrombophilias and preeclampsia: a meta-analysis. Obstet Gynecol. 2005;105:182–192. [PubMed: 15625161]
20. Dalmaz C.A., Santos K.G., Botton M.R. Relationship between polymorphisms in thrombophilic genes and preeclampsia in a Brazilian population. Blood Cells Mol Dis. 2006;37:107–110. [PubMed: 16963292]
21. Fabbro D., D’Elia A.V., Spizzo R. Association between plasminogen activator inhibitor 1 gene polymorphisms and preeclampsia. Gynecol Obstet Invest.2003;56:17–22. [PubMed: 12867763]
22. Gerhardt A., Goecke T.W., Beckmann M.W. The G20210A prothrombin-gene mutation and the plasminogen activator inhibitor (PAI-1) 5G/5G genotype are associated with early onset of severe preeclampsia. J Thromb Haemost. 2005;3:686–691. [PubMed: 15842353]
23. Shah N.C., Pringle S., Struthers A. Aldosterone blockade over and above ACE-inhibitors in patients with coronary artery disease but without heart failure.J Renin Angiotensin Aldosterone Syst. 2006;7:20–30. [PubMed: 17083070]
24. Medica I., Kastrin A., Peterlin B. Genetic polymorphisms in vasoactive genes and preeclampsia: a meta-analysis. Eur J Obstet Gynecol Reprod Biol.2007;131:115–126. [PubMed: 17112651]
25. Inoue I., Rohrwasser A., Helin C. A mutation of angiotensinogen in a patient with preeclampsia leads to altered kinetics of the renin-angiotensin system.J Biol Chem. 1995;270:11430–11436. [PubMed: 7744780]
26. Brennecke S.P., Gude N.M., Di Iulio J.L. Reduction of placental nitric oxide synthase activity in pre-eclampsia. Clin Sci (Lond) 1997;93:51–55.[PubMed: 9279203]
27. Banyasz I., Bokodi G., Vannay A. Genetic polymorphisms of vascular endothelial growth factor and angiopoietin 2 in retinopathy of prematurity. Curr Eye Res. 2006;31:685–690. [PubMed: 16877277]
28. Papazoglou D., Galazios G., Koukourakis M.I. Vascular endothelial growth factor gene polymorphisms and pre-eclampsia. Mol Hum Reprod.2004;10:321–324. [PubMed: 14997002]
29. Foidart J., Hustin J., Dubois M. The human placenta becomes haemochorial at the 13th week of pregnancy. Int J Dev Biol. 1992;36:451–453.[PubMed: 1445791]
30. Jauniaux E., Watson A., Hempstock J. Onset of maternal arterial blood flow and placental oxidative stress. A possible factor in human early pregnancy failure. Am J Pathol. 2000;157:2111–2122. [PMCID: PMC1885754] [PubMed: 11106583]
31. Perkins A.V. Endogenous anti-oxidants in pregnancy and preeclampsia. Aust N Z J Obstet Gynaecol. 2006;46:77–83. [PubMed: 16638026]
32. Wickens D., Wilkins M.H., Lunec J. Free radical oxidation (peroxidation)products in plasma in normal and abnormal pregnancy. Ann Clin Biochem.1981;18:158–162. [PubMed: 7283366]
33. Canto P., Canto-Cetina T., Juarez-Velazquez R. Methylenetetrahydrofolate reductase C677T and glutathione S-transferase P1 A313G are associated with a reduced risk of preeclampsia in Maya-Mestizo women. Hypertens Res. 2008;31:1015–1019. [PubMed: 18712057]
34. Gebhardt G.S., Peters W.H., Hillermann R. Maternal and fetal single nucleotide polymorphisms in the epoxide hydrolase and gluthatione S-transferase P1 genes are not associated with pre-eclampsia in the Coloured population of the Western Cape, South Africa. J Obstet Gynaecol. 2004;24:866–872.[PubMed: 16147638]
35. Laasanen J., Romppanen E.L., Hiltunen M. Two exonic single nucleotide polymorphisms in the microsomal epoxide hydrolase gene are jointly associated with preeclampsia. Eur J Hum Genet. 2002;10:569–573. [PubMed: 12173035]
36. Ohta K., Kobashi G., Hata A. Association between a variant of the glutathione S-transferase P1 gene (GSTP1) and hypertension in pregnancy in Japanese: interaction with parity, age, and genetic factors. Semin Thromb Hemost. 2003;29:653–659. [PubMed: 14719182]
37. Descamps O.S., Bruniaux M., Guilmot P.F. Lipoprotein metabolism of pregnant women is associated with both their genetic polymorphisms and those of their newborn children. J Lipid Res. 2005;46:2405–2414. [PubMed: 16106048]
38. Kim Y.J., Williamson R.A., Chen K. Lipoprotein lipase gene mutations and the genetic susceptibility of preeclampsia. Hypertension. 2001;38:992–996.[PubMed: 11711487]
39. Atkinson K.R., Blumenstein M., Black M.A. An altered pattern of circulating apolipoprotein E3 isoforms is implicated in preeclampsia. J Lipid Res.2009;50:71–80. [PubMed: 18725658]
40. Hubel C.A., Roberts J.M., Ferrell R.E. Association of pre-eclampsia with common coding sequence variations in the lipoprotein lipase gene. Clin Genet.1999;56:289–296. [PubMed: 10636447]
41. Zhang C., Austin M.A., Edwards K.L. Functional variants of the lipoprotein lipase gene and the risk of preeclampsia among non-Hispanic Caucasian women. Clin Genet. 2006;69:33–39. [PubMed: 16451134]
42. Roberts J.M., Pearson G., Cutler J. Summary of the NHLBI Working Group on Research on Hypertension During Pregnancy. Hypertension.2003;41:437–445. [PubMed: 12623940]
43. Wang J.X., Knottnerus A.M., Schuit G. Surgically obtained sperm, and risk of gestational hypertension and pre-eclampsia. Lancet. 2002;359:673–674.[PubMed: 11879865]
44. Hiby S.E., Walker J.J., O’Shaughnessy K.M. Combinations of maternal KIR and fetal HLA-C genes influence the risk of preeclampsia and reproductive success. J Exp Med. 2004;200:957–965. [PMCID: PMC2211839] [PubMed: 15477349]
45. Parham P. MHC class I molecules and KIRs in human history, health and survival. Nat Rev Immunol. 2005;5:201–214. [PubMed: 15719024]
46. Moreau P., Contu L., Alba F. HLA-G gene polymorphism in human placentas: possible association of G*0106 allele with preeclampsia and miscarriage.Biol Reprod. 2008;79:459–467. [PubMed: 18509163]
47. Tan C.Y., Ho J.F., Chong Y.S. Paternal contribution of HLA-G*0106 significantly increases risk for pre-eclampsia in multigravid pregnancies. Mol Hum Reprod. 2008;14:317–324. [PubMed: 18353802]
48. LaMarca B.D., Ryan M.J., Gilbert J.S. Inflammatory cytokines in the pathophysiology of hypertension during preeclampsia. Curr Hypertens Rep.2007;9:480–485. [PubMed: 18367011]
49. Alexander B.T., Cockrell K.L., Massey M.B. Tumor necrosis factor-alpha-induced hypertension in pregnant rats results in decreased renal neuronal nitric oxide synthase expression. Am J Hypertens. 2002;15:170–175. [PubMed: 11863253]
50. Sharma A., Satyam A., Sharma J.B. Leptin, IL-10 and inflammatory markers (TNF-alpha, IL-6 and IL-8) in pre-eclamptic, normotensive pregnant and healthy non-pregnant women. Am J Reprod Immunol. 2007;58:21–30. [PubMed: 17565544]
51. Elahi M.M., Asotra K., Matata B.M. Tumor necrosis factor alpha -308 gene locus promoter polymorphism: an analysis of association with health and disease. Biochim Biophys Acta. 2009;1792:163–172. [PubMed: 19708125]
52. Saarela T., Hiltunen M., Helisalmi S. Tumour necrosis factor-alpha gene haplotype is associated with pre-eclampsia. Mol Hum Reprod. 2005;11:437–440. [PubMed: 15901845]
53. Bombell S., McGuire W. Tumour necrosis factor (-308A) polymorphism in pre-eclampsia: meta-analysis of 16 case-control studies. Aust N Z J Obstet Gynaecol. 2008;48:547–551. [PubMed: 19133041]
54. Renaud S.J., Macdonald-Goodfellow S.K., Graham C.H. Coordinated regulation of human trophoblast invasiveness by macrophages and interleukin 10.Biol Reprod. 2007;76:448–454. [PubMed: 17151353]
55. Makris A., Xu B., Yu B. Placental deficiency of interleukin-10 (IL-10) in preeclampsia and its relationship to an IL10 promoter polymorphism. Placenta.2006;27:445–451. [PubMed: 16026832]
56. Daher S., Sass N., Oliveira L.G. Cytokine genotyping in preeclampsia. Am J Reprod Immunol. 2006;55:130–135. [PubMed: 16433832]
57. Goddard K.A., Tromp G., Romero R. Candidate-gene association study of mothers with pre-eclampsia, and their infants, analyzing 775 SNPs in 190 genes. Hum Hered. 2007;63:1–16. [PubMed: 17179726]
58. Kamali-Sarvestani E., Kiany S., Gharesi-Fard B. Association study of IL-10 and IFN-gamma gene polymorphisms in Iranian women with preeclampsia.J Reprod Immunol. 2006;72:118–126. [PubMed: 16863661]
59. Faisel F., Romppanen E.L., Hiltunen M. Polymorphism in the interleukin 1 receptor antagonist gene in women with preeclampsia. J Reprod Immunol.2003;60:61–70. [PubMed: 14568678]
60. Haggerty C.L., Ferrell R.E., Hubel C.A. Association between allelic variants in cytokine genes and preeclampsia. Am J Obstet Gynecol. 2005;193:209–215.[PubMed: 16021081]
61. Zusterzeel P.L., Peters W.H., Burton G.J. Susceptibility to pre-eclampsia is associated with multiple genetic polymorphisms in maternal biotransformation enzymes. Gynecol Obstet Invest. 2007;63:209–213. [PubMed: 17167268]
62. Buimer M., Keijser R., Jebbink J.M. Seven placental transcripts characterize HELLP-syndrome. Placenta. 2008;29:444–453. [PubMed: 18374411]
63. Raijmakers M.T., Roes E.M., Steegers E.A. The C242T-polymorphism of the NADPH/NADH oxidase gene p22phox subunit is not associated with pre-eclampsia. J Hum Hypertens. 2002;16:423–425. [PubMed: 12037698]
64. Rosta K., Molvarec A., Enzsoly A. Association of extracellular superoxide dismutase (SOD3) Ala40Thr gene polymorphism with pre-eclampsia complicated by severe fetal growth restriction. Eur J Obstet Gynecol Reprod Biol. 2009;142:134–138. [PubMed: 19108943]
65. Arngrimsson R., Sigurardo-ttir S., Frigge M.L. A genome-wide scan reveals a maternal susceptibility locus for pre-eclampsia on chromosome 2p13. Hum Mol Genet. 1999;8:1799–1805. [PubMed: 10441346]
66. Laivuori H., Lahermo P., Ollikainen V. Susceptibility loci for preeclampsia on chromosomes 2p25 and 9p13 in Finnish families. Am J Hum Genet.2003;72:168–177. [PMCID: PMC378622] [PubMed: 12474145]
67. Moses E.K., Lade J.A., Guo G. A genome scan in families from Australia and New Zealand confirms the presence of a maternal susceptibility locus for pre-eclampsia, on chromosome 2. Am J Hum Genet. 2000;67:1581–1585. [PMCID: PMC1287935] [PubMed: 11035632]
68. Lachmeijer A.M., Arngrimsson R., Bastiaans E.J. A genome-wide scan for preeclampsia in the Netherlands. Eur J Hum Genet. 2001;9:758–764.[PubMed: 11781687]
69. Lander E., Kruglyak L. Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet. 1995;11:241–247.[PubMed: 7581446]
70. Zintzaras E., Kitsios G., Harrison G.A. Heterogeneity-based genome search meta-analysis for preeclampsia. Hum Genet. 2006;120:360–370.[PubMed: 16868762]
71. Akolekar R., Etchegaray A., Zhou Y. Maternal serum activin a at 11-13 weeks of gestation in hypertensive disorders of pregnancy. Fetal Diagn Ther.2009;25:320–327. [PubMed: 19776595]
72. Roten L.T., Johnson M.P., Forsmo S. Association between the candidate susceptibility gene ACVR2A on chromosome 2q22 and pre-eclampsia in a large Norwegian population-based study (the HUNT study) Eur J Hum Genet. 2009;17:250–257. [PMCID: PMC2696227] [PubMed: 18781190]
73. Fitzpatrick E., Johnson M.P., Dyer T.D. Genetic association of the activin A receptor gene (ACVR2A) and pre-eclampsia. Mol Hum Reprod.2009;15:195–204. [PMCID: PMC2647107] [PubMed: 19126782]
74. Riento K., Ridley A.J. Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol. 2003;4:446–456. [PubMed: 12778124]
75. Kandabashi T., Shimokawa H., Miyata K. Inhibition of myosin phosphatase by upregulated rho-kinase plays a key role for coronary artery spasm in a porcine model with interleukin-1beta. Circulation. 2000;101:1319–1323. [PubMed: 10725293]
76. Ark M., Yilmaz N., Yazici G. Rho-associated protein kinase II (rock II) expression in normal and preeclamptic human placentas. Placenta. 2005;26:81–84. [PubMed: 15664415]
77. Johnson M.P., Roten L.T., Dyer T.D. The ERAP2 gene is associated with preeclampsia in Australian and Norwegian populations. Human Genetics.2009;126(5):655–666. PMCID: PMC2783187. [PMCID: PMC2783187] [PubMed: 19578876]
78. WTCCC Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678.[PMCID: PMC2719288] [PubMed: 17554300]
79. Bezerra P.C., Leao M.D., Queiroz J.W. Family history of hypertension as an important risk factor for the development of severe preeclampsia. Acta Obstet Gynecol Scand. 2010;89:612–617. [PubMed: 20423274]
80. Hatada I., Mukai T. Genomic imprinting of p57KIP2, a cyclin-dependent kinase inhibitor, in mouse. Nat Genet. 1995;11:204–206. [PubMed: 7550351]
81. Oudejans C.B., Mulders J., Lachmeijer A.M. The parent-of-origin effect of 10q22 in pre-eclamptic females coincides with two regions clustered for genes with down-regulated expression in androgenetic placentas. Mol Hum Reprod. 2004;10:589–598. [PubMed: 15208369]
82. Rigourd V., Chauvet C., Chelbi S.T. STOX1 overexpression in choriocarcinoma cells mimics transcriptional alterations observed in preeclamptic placentas. PLoS One. 2008;3:e3905. [PMCID: PMC2592700] [PubMed: 19079545]
83. Berends A.L., Bertoli-Avella A.M., de Groot C.J. STOX1 gene in pre-eclampsia and intrauterine growth restriction. BJOG. 2007;114:1163–1167.[PubMed: 17617193]
84. Iglesias-Platas I., Monk D., Jebbink J. STOX1 is not imprinted and is not likely to be involved in preeclampsia. Nat Genet. 2007;39:279–280. author reply 280–271. [PubMed: 17325670]
85. Kivinen K., Peterson H., Hiltunen L. Evaluation of STOX1 as a preeclampsia candidate gene in a population-wide sample. Eur J Hum Genet.2007;15:494–497. [PubMed: 17290274]
86. Yu L., Chen M., Zhao D. The H19 gene imprinting in normal pregnancy and pre-eclampsia. Placenta. 2009;30:443–447. [PubMed: 19342096]
87. Nussbaum R.L., McInnes R.R., Willard H.F. In: Genetics in medicine. 6th ed. Thompson and Thompson, editor. Saunders; Philadelphia: 2004. pp. 289–309.
88. Treloar S.A., Cooper D.W., Brennecke S.P. An Australian twin study of the genetic basis of preeclampsia and eclampsia. Am J Obstet Gynecol.2001;184:374–381. [PubMed: 11228490]
89. Ronningen K.S., Paltiel L., Meltzer H.M. The biobank of the Norwegian mother and child cohort study: a resource for the next 100 years. Eur J Epidemiol. 2006;21:619–625. [PMCID: PMC1820840] [PubMed: 17031521]
90. Kho E.M., McCowan L.M., North R.A. Duration of sexual relationship and its effect on preeclampsia and small for gestational age perinatal outcome. J Reprod Immunol. 2009;82:66–73. [PubMed: 19679359]
 and
and 
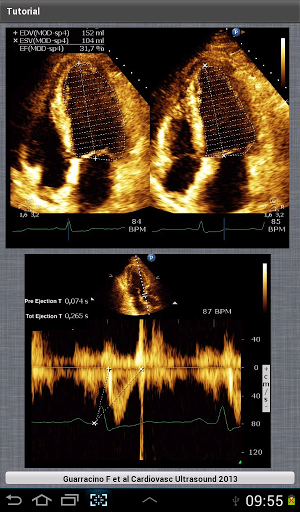



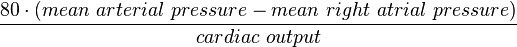
 , where t is time, s is a complex parameter, and A is a complex scalar. Complex values simply mean two dimensional, e.g., magnitude (as in resistance) plus phase shift (to account for reactive components).
, where t is time, s is a complex parameter, and A is a complex scalar. Complex values simply mean two dimensional, e.g., magnitude (as in resistance) plus phase shift (to account for reactive components).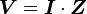 where V and I are the complex scalars in the voltage and current respectively and Z is the complex impedance.
where V and I are the complex scalars in the voltage and current respectively and Z is the complex impedance.