Precision Medicine for Future of Genomics Medicine is The New Era
Demet Sag, PhD, CRA, GCP
Are we there yet? Life is a journey so the science.
Governor Brown announced Precision Medicine initiative for California on April 14, 2015. UC San Francisco is hosting the two-year initiative, through UC Health, which includes UC’s five medical centers, with $3 million in startup funds from the state. The public-private initiative aims to leverage these funds with contributions from other academic and industry partners.
With so many campuses spread throughout the state and so much scientific, clinical and computational expertise, the UC system has the potential to bring it all together, said Atul Butte, MD, PhD, who is leading the initiative.
At the beginning of 2015 President Obama signed this initiative and assigned people to work on this project.
Previously NCI Director Harold Varmus, MD said that “Precision medicine is really about re-engineering the diagnostic categories for cancer to be consistent with its genomic underpinnings, so we can make better choices about therapy,” and “In that sense, many of the things we’re proposing to do are already under way.”
The proposed initiative has two main components:
- a near-term focus on cancers and
- a longer-term aim to generate knowledge applicable to the whole range of health and disease.
Both components are now within our reach because of advances in basic research, including molecular biology, genomics, and bioinformatics. Furthermore, the initiative taps into converging trends of increased connectivity, through social media and mobile devices, and Americans’ growing desire to be active partners in medical research.
Since the human genome is sequenced it became clear that actually there are few genes than expected and shared among organisms to accomplish same or similar core biological functions. As a result, knowledge of the biological role of such shared proteins in one organism can be transferred to another organism.
I remember when I was screening the X-chromosome by using deletion/duplication mapping and using P elements and bar balancers as a tool to keep the genome stable to identify transregulating elements of ovo gene, female germline specific Drosophila melanogaster germline sex determination gene. At the time for my dissertation, I screened X-chromosome using 45 deficiency strains, I found that these trans-regulating regions were grouped into 12 loci based on overlapping cytology. Five regions were trans-regulating activators, and seven were trans-regulating repressors; extrapolating to the entire genome, this result predicted nearly 85 loci. This one gene may expressed three proteins at different time of development and activate/downregulate various regions to accommadate proper system development in addition to auto-regulate and gene dose responses. Drosophila has only four chromosomes but the cellular interactions and signaling mechanisms are still complicated yet as not complicated as human. I do appreciate the new applications and upcoming changes.
Now, the technology is much better and precision is the key to establish to use in clinics. However, we have new issues to overcome like computing such a big data, align properly, analyze effectively, compare and contrast the outcomes to identify the variations that may function in on population, or two etc. At the end of the day collaboration, standardization, and data sharing are few of the key factors.
It is necessary to generate a dynamic yet controlled standardized collection of information with ever changing and accumulating data so Gene Ontology Consortium is created. Three independent ontologies can be reached at (http://www.geneontology.org) developed based on :
- biological process,
- molecular function and
- cellular component.

We need a common language for annotation for a functional conservation. Genesis of the grand biological unification made it possible to complete the genomic sequences of not only human but also the main model organisms and more. some examples include:
- the budding yeast, Saccharomyces cerevisiae,
- the nematode worm Caenorhabditis elegans
- the fruitfly Drosophila melanogaster,
- the flowering plant Arabidopsis thaliana
- fission yeast Schizosaccharomyces pombe
- the mouse , Mus musculus
On the other hand, as we know there are allelic variations that underlie common diseases and complete genome sequencing for many individuals with and without disease is required. However, there are advantages and disadvantages as we can carry out partial surveys of the genome by genotyping large numbers of common SNPs in genome-wide association studies but there are problems such as computing the data efficiently and sharing the information without tempering privacy. Therefore we should be mindful about few main conditions including:
- models of the allelic architecture of common diseases,
- sample size,
- map density and
- sample-collection biases.
This will lead into the cost control and efficiency while identifying genuine disease-susceptibility loci. The genome-wide association studies (GWAS) have progressed from assaying fewer than 100,000 SNPs to more than one million, and sample sizes have increased dramatically as the search for variants that explain more of the disease/trait heritability has intensified.
In addition, we must translate this sequence information from genomics locus of the genes to function with related polymorphism of these genes so that possible patterns of the gene expression and disease traits can be matched. Then, we may develop precision technologies for:
- Diagnostics
- Targeted Drugs and Treatments
- Biomarkers to modulate cells for correct functions
With the knowledge of:
- gene expression variations
- insight in the genetic contribution to clinical endpoints ofcomplex disease and
- their biological risk factors,
- share etiologic pathways
therefore, requires an understanding of both:
- the structure and
- the biology of the genome.
These studies demonstrated hundreds of associations of common genetic variants with over 80 diseases and traits collected under a controlled online resource. However, identifying published GWAS can be challenging as a simple PubMed search using the words “genome wide association studies” may be easily populated with unrelevant GWAS.
National Human Genome Research Institute (NHGRI) Catalog of Published Genome-Wide Association Studies (http://www.genome.gov/gwastudies), an online, regularly updated database of SNP-trait associations extracted from published GWAS was developed.
Therefore, sequencing of a human genome is a quite undertake and requires tools to make it possible:
- to explore the genetic component in complex diseases and
- to fully understand the genetic pathways contributing to complex disease
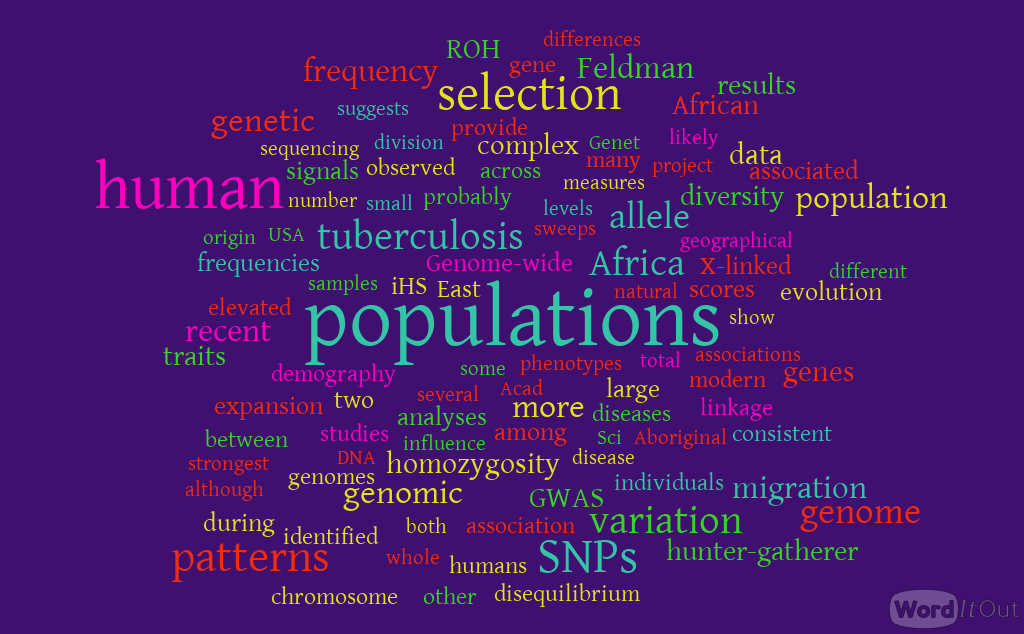
The rapid increase in the number of GWAS provides an unprecedented opportunity to examine the potential impact of common genetic variants on complex diseases by systematically cataloging and summarizing key characteristics of the observed associations and the trait/disease associated SNPs (TASs) underlying them.
With this in mind, many forms can be established:
- to describe the features of this resource and the methods we have used to produce it,
- to provide and examine key descriptive characteristics of reported TASs such as estimated risk allele frequencies and odds ratios,
- to examine the underlying functionality of reported risk loci by mapping them to genomic annotation sets and assessing overrepresentation via Monte Carlo simulations and
- to investigate the relationship between recent human evolution and human disease phenotypes.
This procedure has no clear path so there are several obstacles in the actual functional variant that is often unknown. This may be due to:
- trait/disease associated SNPs (TASs),
- a well known SNP+ strong linkage disequilibrium (LD) with the TAS,
- an unknown common SNP tagged by a haplotype
- rare single nucleotide variant tagged by a haplotype on which the TAS occurs, or
- Copy Number variation (CNV), a linked copy number variant.
There can be other factors such as
- Evolution,
- Natural Selection
- Environment
- Pedigree
- Epigenetics
Even though heritage is another big factor, the concept of heritability and its definition as an estimable, dimensionless population parameter as introduced by Sewall Wright and Ronald Fisher almost a century ago.
As a result, heritability gain interest since it allows us to compare of the relative importance of genes and environment to the variation of traits within and across populations. The heritability is an ongoing mechanism and remains as a main factor:
- to selection in evolutionary biology and agriculture, and
- to the prediction of disease risk in medicine.
Reported TASs associated with two or more distinct traits
| Chromosomal region |
Rs number(s) |
Attributed genes |
Associated traits reported in catalog |
| 1p13.2 |
rs2476601, rs6679677 |
PTPN22 |
Crohn’s disease, type 1 diabetes, rheumatoid arthritis |
| 1q23.2 |
rs2251746, rs2494250 |
FCER1A |
Serum IgE levels, select biomarker traits (MCP1) |
| 2p15 |
rs1186868, rs1427407 |
BCL11A |
Fetal hemoglobin, F-cell distribution |
| 2p23.3 |
rs780094 |
GCKR |
CRP, lipids, waist circumference |
| 6p21.33 |
rs3131379, rs3117582 |
HLA / MHC region |
Systemic lupus erythematosus, lung cancer, psoriasis, inflammatory bowel disease, ulcerative colitis, celiac disease, rheumatoid arthritis, juvenile idiopathic arthritis, multiple sclerosis, type 1 diabetes |
| 6p22.3 |
rs6908425, rs7756992, rs7754840, rs10946398, rs6931514 |
CDKAL1 |
Crohn’s disease, type 2 diabetes |
| 6p25.3 |
rs1540771, rs12203592, rs872071 |
IRF4 |
Freckles, hair color, chronic lymphocytic leukemia |
| 6q23.3 |
rs5029939, rs10499194 |
TNFAIP3 |
Systemic lupus erythematosus, rheumatoid arthritis |
| 7p15.1 |
rs1635852, rs864745 |
JAZF1 |
Height, type 2 diabetes* |
| 8q24.21 |
rs6983267 |
Intergenic |
Prostate or colorectal cancer, breast cancer |
| 9p21.3 |
rs10811661, rs1333040, rs10811661, rs10757278, rs1333049 |
CDKN2A, CDKN2B |
Type 2 diabetes, intracranial aneurysm, myocardial infarction |
| 9q34.2 |
rs505922, rs507666, rs657152 |
ABO |
Protein quantitative trait loci (TNF-α), soluble ICAM-1, plasma levels of liver enzymes (alkaline phosphatase) |
| 12q24 |
rs1169313, rs7310409, rs1169310, rs2650000 |
HNF1A |
Plasma levels of liver enzyme (GGT), C-reactive protein, LDL cholesterol |
| 16q12.2 |
rs8050136, rs9930506, rs6499640, rs9939609, rs1121980 |
FTO |
Type 2 diabetes, body mass index or weight |
| 17q12 |
rs7216389, rs2872507 |
ORMDL3 |
Asthma, Crohn’s disease |
| 17q12 |
rs4430796 |
TCF2 |
Prostate cancer, type 2 diabetes |
| 18p11.21 |
rs2542151 |
PTPN2 |
Type 1 diabetes, Crohn’s disease |
| 19q13.32 |
rs4420638 |
APOE, APOC1, APOC4 |
Alzheimer’s disease, lipids |
* The well known association of JAZF1 with prostate cancer was reported with a p value of 2 × 10−6, which did not meet the threshold of 5 × 10−8 for this analysis.
.
Allele-Frequency Data for Nine Reproducible Associations
|
|
|
|
frequency |
|
|
| gene |
diseasea |
SNP |
associated alleleb |
Europeand |
Africane |
δf |
FST |
reference(s)c |
| CTLA4 |
T1DM |
Thr17Ala |
Ala |
.38 (1,670) |
.209 (402) |
.171 |
.06 |
Osei-Hyiaman et al. 2001; Lohmueller et al. 2003 |
| DRD3 |
Schizophrenia |
Ser9Gly |
Ser/Ser |
.67 (202) |
.116 (112) |
.554 |
.458 |
Crocq et al. 1996; Lohmueller et al.2003 |
| AGT |
Hypertension |
Thr235Met |
Thr |
.42 (3,034) |
.91 (658) |
.49 |
.358 |
Rotimi et al. 1996; Nakajima et al.2002 |
| PRNP |
CJD |
Met129Val |
Met |
.72 (138) |
.556 (72) |
.164 |
.049 |
Hirschhorn et al. 2002; Soldevila et al. 2003 |
| F5 |
DVT |
Arg506Gln |
Gln |
.044 (1,236) |
.00 (251) |
.044 |
.03 |
Rees et al. 1995; Hirschhorn et al.2002 |
| HFE |
HFE |
Cys382Tyr |
Tyr |
.038 (2,900) |
.00 (806) |
.038 |
.024 |
Feder et al. 1996; Merryweather-Clarke et al. 1997 |
| MTHFR |
DVT |
C677T |
T |
.3 (188) |
.066 (468) |
.234 |
.205 |
Schneider et al. 1998; Ray et al.2002 |
| PPARG |
T2DM |
Pro12Ala |
Pro |
.925 (120) |
1.0 (120) |
.075 |
.067 |
Altshuler et al. 2000; HapMap Project |
| KCNJ11 |
T2DM |
Asp23Lys |
Lys |
.36 (96) |
.09 (98) |
.27 |
.182 |
Florez et al. 2004 |
aCJD = Creutzfeldt-Jacob disease; DVT = deep venous thrombosis; HFE = hemochromatosis; T1DM = type I diabetes; T2DM = type II diabetes.
bThe associated allele is the SNP associated with disease, regardless of whether it is the derived or the ancestral allele. The frequencies for this allele are given.
cThe reference that claims this to be a reproducible association, as well as the reference from which the allele frequencies were taken. For allele frequencies obtained from a meta-analysis, only the reference claiming reproducible association is given.
dAllele frequency obtained from the literature involving a European population. Either the general population frequency or the frequency in control groups in an association study was used. To reduce bias, when a control frequency was used for Europeans, a control frequency was also used for Africans. The total number of chromosomes surveyed is given in parentheses after each frequency.
eAllele frequency obtained from the literature involving a West African population. The total number of chromosomes surveyed is given in parentheses after each frequency.
fδ = The difference in the allele frequency between Europeans and Africans.
Allele-Frequency Data for 39 Reported Associations
|
|
|
|
frequency |
|
|
| gene |
disease/phenotypea |
SNP |
associated alleleb |
Europeand |
Africane |
δf |
FST |
referencec |
| ADRB1 |
MI |
Arg389Gly |
Arg |
.717 (46) |
.467 (30) |
.251 |
.1 |
Iwai et al. 2003 |
| ALOX5AP |
MI, stroke |
rs10507391 |
T |
.682 (44) |
.159 (44) |
.523 |
.425 |
Helgadottir et al. 2004 |
| CAT |
Hypertension |
−844 (C/T) |
Tg |
.714 (42) |
.659 (44) |
.055 |
0 |
Jiang et al. 2001 |
| CCR2 |
AIDS susceptibility |
Ile64Val |
Val |
.87 (46) |
.813 (48) |
.057 |
0 |
Smith et al. 1997 |
| CD36 |
Malaria |
Y to stop |
Stop |
0 (46) |
.083 (48) |
.083 |
.062 |
Aitman et al. 2000 |
| F13 |
MI |
Val34Leu |
Val |
.762 (42) |
.795 (44) |
.033 |
0 |
Kohler et al. 1999 |
| FGA |
Pulmonary embolism |
Thr312Ala |
Ala |
.2 (40) |
.5 (42) |
.3 |
.159 |
Carter et al. 2000 |
| GP1BA |
CAD |
Thr145Met |
Met |
.022 (46) |
.167 (48) |
.145 |
.095 |
Gonzalez-Conejero et al.1998 |
| ICAM1 |
MS |
Lys469Glu |
Lys |
.643 (42) |
.875 (48) |
.232 |
.12 |
Nejentsev et al. 2003 |
| ICAM1 |
Malaria |
Lys29Met |
Met |
0 (46) |
.354 (48) |
.354 |
.335 |
Fernandez-Reyes et al.1997 |
| IFNGR1 |
Hp infection |
−56 (C/T) |
T |
.455 (44) |
.604 (48) |
.15 |
.023 |
Thye et al. 2003 |
| IL13 |
Asthma |
−1055 (C/T) |
T |
.196 (46) |
.25 (44) |
.054 |
0 |
van der Pouw Kraan et al. 1999 |
| IL13 |
Bronchial asthma |
Arg110Gln |
Gln |
.273 (44) |
.119 (42) |
.154 |
.05 |
Heinzmann et al. 2003 |
| IL1A |
AD |
−889 (C/T) |
T |
.295 (44) |
.391 (46) |
.096 |
0 |
Nicoll et al. 2000 |
| IL1B |
Gastric cancer |
−31 (C/T) |
T |
.826 (46) |
.375 (48) |
.451 |
.335 |
El-Omar et al. 2000 |
| IL3 |
RA |
−16 (C/T) |
C |
.739 (46) |
.875 (48) |
.136 |
.037 |
Yamada et al. 2001 |
| IL4 |
Asthma |
−590 (T/C) |
T |
.174 (46) |
.708 (48) |
.534 |
.436 |
Noguchi et al. 1998 |
| IL4R |
Asthma |
Gln576Arg |
Arg |
.295 (44) |
.565 (46) |
.27 |
.118 |
Hershey et al. 1997 |
| IL6 |
Juvenile arthritis |
−174 (C/G) |
G |
.5 (44) |
1 (46) |
.5 |
.494 |
Fishman et al. 1998 |
| IL8 |
RSV bronchiolitis |
−251 (T/A) |
Th |
.659 (44) |
.229 (48) |
.43 |
.301 |
Hull et al. 2000 |
| ITGA2 |
MI |
807 (C/T) |
T |
.316 (38) |
.25 (48) |
.066 |
0 |
Moshfegh et al. 1999 |
| LTA |
MI |
Thr26Asn |
Asn |
.357 (42) |
.5 (44) |
.143 |
.018 |
Ozaki et al. 2002 |
| MC1R |
Fair skin |
Val92Met |
Met |
.068 (44) |
0 (44) |
.068 |
.047 |
Valverde et al. 1995 |
| NOS3 |
MI |
Glu298Asp |
Asp |
.5 (44) |
.136 (44) |
.364 |
.247 |
Shimasaki et al. 1998 |
| PLAU |
AD |
Pro141Leu |
Pro |
.659 (44) |
.979 (48) |
.32 |
.287 |
Finckh et al. 2003 |
| PON1 |
CAD |
Arg192Gln |
Arg |
.174 (46) |
.727 (44) |
.553 |
.461 |
Serrato and Marian 1995 |
| PON2 |
CAD |
Cys311Ser |
Ser |
.826 (46) |
.762 (42) |
.064 |
0 |
Sanghera et al. 1998 |
| PTGS2 |
Colon cancer |
−765 (G/C) |
C |
.238 (42) |
.292 (48) |
.054 |
0 |
Koh et al. 2004 |
| PTPN22i |
RA |
Arg620Trp |
Trp |
.084 (1,120) |
.024 (818) |
.059 |
.03 |
Begovich et al. 2004 |
| SELE |
CAD |
Ser128Arg |
Arg |
.091 (44) |
.021 (48) |
.07 |
.025 |
Wenzel et al. 1994 |
| SELL |
IgA nephropathy |
Pro238Ser |
Ser |
.065 (46) |
.333 (48) |
.268 |
.183 |
Takei et al. 2002 |
| SELP |
MI |
Thr715Pro |
Thr |
.864 (44) |
.977 (44) |
.114 |
.063 |
Herrmann et al. 1998 |
| SFTPB |
ARDS |
Ile131Thr |
Thr |
.5 (44) |
.348 (46) |
.152 |
.025 |
Lin et al. 2000 |
| SPD |
RSV infection |
Met11Thr |
Met |
.568 (44) |
.478 (46) |
.09 |
0 |
Lahti et al. 2002 |
| TF |
AD |
Pro570Ser |
Pro |
.957 (46) |
.935 (46) |
.022 |
0 |
Zhang et al. 2003 |
| THBD |
MI |
Ala455Val |
Ala |
.87 (46) |
.848 (46) |
.022 |
0 |
Norlund et al. 1997 |
| THBS4 |
MI |
Ala387Pro |
Pro |
.341 (44) |
.083 (48) |
.258 |
.166 |
Topol et al. 2001 |
| TNFA |
Infectious disease |
−308 (A/G) |
A |
.182 (44) |
.205 (44) |
.023 |
0 |
Bayley et al. 2004 |
| VCAM1 |
Stroke in SCD |
Gly413Ala |
Gly |
1 (46) |
.938 (48) |
.063 |
.041 |
Taylor et al. 2002 |
aAD = Alzheimer disease; AIDS = acquired immunodeficiency syndrome; ARDS = acute respiratory distress syndrome; CAD = coronary artery disease; Hp = Helicobacter pylori; MI = myocardial infarction; MS = multiple sclerosis; RA = rheumatoid arthritis; RSV = respiratory syncytial virus; SCD = sickle cell disease.
bThe associated allele is the SNP associated with disease, regardless of whether it is the derived or the ancestral allele. The frequencies for this allele are given.
cThe reference that reported association with the listed disease/phenotype.
dFrequency obtained from the Seattle SNPs database for the European sample. The total number of chromosomes surveyed is given in parentheses after each frequency.
eFrequency obtained from the Seattle SNPs database for the African American sample. The total number of chromosomes surveyed is given in parentheses after each frequency.
fδ = The difference in the allele frequency between African Americans and Europeans.
gAssociated allele in database is A.
hAssociated allele in reference is A.
iThis SNP was not from the Seattle SNPs database; instead, allele frequencies from Begovich et al. (2004) were used.
They reported that “The SNPs associated with common disease that we investigated do not show much higher levels of differentiation than those of random SNPs. Thus, in these cases, ethnicity is a poor predictor of an individual’s genotype, which is also the pattern for random variants in the genome. This lends support to the hypothesis that many population differences in disease risk are environmental, rather than genetic, in origin. However, some exceptional SNPs associated with common disease are highly differentiated in frequency across populations, because of either a history of random drift or natural selection. The exceptional SNPs given are located in AGT, DRD3, ALOX5AP, ICAM1, IL1B, IL4, IL6, IL8, and PON1.
Of note, evidence of selection has been observed for AGT (Nakajima et al. 2004), IL4(Rockman et al. 2003), IL8 (Hull et al. 2001), and PON1 (Allebrandt et al. 2002). Yet, for the vast majority of the common-disease–associated polymorphisms we examined, ethnicity is likely to be a poor predictor of an individual’s genotype.”
In 2002 the International HapMap Project was launched:
- to provide a public resource
- to accelerate medical genetic research.
Two Hapmap projects were completed.
In phase I the objective was to genotype at least one common SNP every 5 kilobases (kb) across the euchromatic portion of the genome in 270 individuals from four geographically diverse population.
In Phase II of the HapMap Project, a further 2.1 million SNPs were successfully genotyped on the same individuals.
The re-mapping of SNPs from Phase I of the project identified 21,177 SNPs that had an ambiguous position or some other feature indicative of low reliability. These are not included in the filtered Phase II data release. All genotype data are available from the HapMap Data Coordination Center located at (http://www.hapmap.org) and dbSNP (http://www.ncbi.nlm.nih.gov/SNP).
In the Phase II HapMap we identified 32,996 recombination hotspots (an increase of over 50% from Phase I) of which 68% localized to a region of≤5 kb. The median map distance induced by a hotspot is 0.043 cM (or one crossover per 2,300 meioses) and the hottest identified, on chromosome 20, is 1.2 cM (one crossover per 80 meioses). Hotspots account for approximately 60% of recombination in the human genome and about 6% of sequence.
In addition to many previously identified regions in HapMap Phase I including LARGE, SYT1 andSULT1C2 (previously called SULT1C1), about 200 regions identified from the Phase II HapMap that include many established cases of selection, such as the genes HBB andLCT, the HLA region, and an inversion on chromosome 17. Finally, in the future, whole-genome sequencing will provide a natural convergence of technologies to type both SNP and structural variation. Nevertheless, until that point, and even after, the HapMap Project data will provide an invaluable resource for understanding the structure of human genetic variation and its link to phenotype.
HMM libraries, such as PANTHER, Pfam, and SMART, are used primarily
- to recognize and
- to annotate conserved motifs in protein sequences.
In the genomic era, one of the fundamental goals is to characterize the function of proteins on a large scale.
PANTHER, for relating protein sequence relationships to function relationships in a robust and accurate way under two main parts:
- the PANTHER library (PANTHER/LIB)- collection of “books,” each representing a protein family as a multiple sequence alignment, a Hidden Markov Model (HMM), and a family tree.
- the PANTHER index (PANTHER/X)- ontology for summarizing and navigating molecular functions and biological processes associated with the families and subfamilies.
PANTHER can be applied on three areas of active research:
- to report the size and sequence diversity of the families and subfamilies, characterizing the relationship between sequence divergence and functional divergence across a wide range of protein families.
- use the PANTHER/X ontology to give a high-level representation of gene function across the human and mouse genomes.
- to rank missense single nucleotide polymorphisms (SNPs), on a database-wide scale, according to their likelihood of affecting protein function.
PRINTS is ” a compendium of protein motif ‘fingerprints’. A fingerprint is defined as a group of motifs excised from conserved regions of a sequence alignment, whose diagnostic power or potency is refined by iterative databasescanning (in this case the OWL composite sequence database)”.
The information contained within PRINTS is distinct from, but complementary to the consensus expressions stored in the widely-used PROSITE dictionary of patterns.
However, the position-specific amino acid probabilities in an HMM can also be used to annotate individual positions in a protein as being conserved (or conserving a property such as hydrophobicity) and therefore likely to be required for molecular function. For example, a mutation (or variant) at a conserved position is more likely to impact the function of that protein.
In addition, HMMs from different subfamilies of the same family can be compared with each other, to provide hypotheses about which residues may mediate the differences in function or specificity between the subfamilies.
Several computational algorithms and databases for comparing protein sequences developed and matured profile methods (Gribskov et al. 1987;Henikoff and Henikoff 1991; Attwood et al. 1994):
- particularly Hidden Markov Models (HMM;Krogh et al. 1994; Eddy 1996) and
- PSI-BLAST (Altschul et al. 1997),
The profile has a different amino acid substitution vector at each position in the profile, based on the pattern of amino acids observed in a multiple alignment of related sequences.
Profile methods combine algorithms with databases:
A group of related sequences is used to build a statistical representation of corresponding positions in the related proteins. The power of these methods therefore increases as new sequences are added to the database of known proteins.
Multiple sequence alignments (Dayhoff et al. 1974) and profiles have allowed a systematic study of related sequences. One of the key observations is that some positions are “conserved,” that is, the amino acid is invariant or restricted to a particular property (such as hydrophobicity), across an entire group of related sequences.
The dependence of profile and pattern-matching approaches (Jongeneel et al. 1989) on sequence databases led to the development of databases of profiles
- BLOCKS,Henikoff and Henikoff 1991;
- PRINTS,Attwood et al. 1994) and
- patterns (Prosite,Bairoch 1991) that could be searched in much the same way as sequence databases.
Among the most widely used protein family databases are
- Pfam (Sonnhammer et al. 1997;Bateman et al. 2002) and
- SMART (Schultz et al. 1998;Letunic et al. 2002), which combine expert analysis with the well-developed HMM formalism for statistical modeling of protein families (mostly families of related protein domains).
Either knowing its family membership to predict its function, or subfamily within that family is enough (Hannenhalli and Russell 2000).
- Phylogenetic trees (representing the evolutionary relationships between sequences) and
- dendrograms (tree structures representing the similarity between sequences) (e.g.,Chiu et al. 1985; Rollins et al. 1991).
The PANTHER/LIB HMMs can be viewed as a statistical method for scoring the “functional likelihood” of different amino acid substitutions on a wide variety of proteins. Because it uses evolutionarily related sequences to estimate the probability of a given amino acid at a particular position in a protein, the method can be referred to as generating “position-specific evolutionary conservation” (PSEC) scores.
The process for building PANTHER families include:
- Family clustering.
- Multiple sequence alignment (MSA), family HMM, and family tree building.
- Family/subfamily definition and naming.
- Subfamily HMM building.
- Molecular function and biological process association.
Of these, steps 1, 2, and 4 are computational, and steps 3 and 5 are human-curated (with the extensive aid of software tools).
Conclusion:
Precision medicine effort is the beginning of a new journey to provide better health solutions.
Further Reading and References:
Human Phenome Project:
Freimer N., Sabatti C. The human phenome project. Nat. Genet. 2003;34:15–21.
Jones R., Pembrey M., Golding J., Herrick D. The search for genenotype/phenotype associations and the phenome scan. Paediatr. Perinat. Epidemiol. 2005;19:264–275.
Stearns F.W. One hundred years of pleiotropy: A retrospective. Genetics.2010;186:767–773.
Welch J.J., Waxman D. Modularity and the cost of complexity. Evolution.2003;57:1723–1734.
Albert A.Y., Sawaya S., Vines T.H., Knecht A.K., Miller C.T., Summers B.R., Balabhadra S., Kingsley D.M., Schluter D. The genetics of adaptive shape shift in stickleback: Pleiotropy and effect size. Evolution. 2008;62:76–85.
Brem R.B., Yvert G., Clinton R., Kruglyak L. Genetic dissection of transcriptional regulation in budding yeast. Science. 2002;296:752–755.
Morley M., Molony C.M., Weber T.M., Devlin J.L., Ewens K.G., Spielman R.S., Cheung V.G. Genetic analysis of genome-wide variation in human gene expression. Nature. 2004;430:743–747. [PMC free article] [PubMed]
Wagner G.P., Zhang J. The pleiotropic structure of the genotype-phenotype map: The evolvability of complex organisms. Nat. Rev. Genet. 2011;12:204–213.
Cooper Z.N., Nelson R.M., Ross L.F. Informed consent for genetic research involving pleiotropic genes: An empirical study of ApoE research. IRB. 2006;28:1–11.
Model Organisms:
Worm Sequencing Consortium. The C. elegans Sequencing Consortium Genome sequence of the nematode C. elegans: a platform for investigating biology. Science.1998;282:2012–2018.
Adams MD, et al. The genome sequence of Drosophila melanogaster. Science.2000;287:2185–2195.
Meinke DW, et al. Arabidopsis thaliana: a model plant for genome analysis. Science. 1998;282:662–682. [PubMed]
Chervitz SA, et al. Using the Saccharomyces Genome Database (SGD) for analysis of protein similarities and structure. Nucleic Acids Res. 1999;27:74–78.
The FlyBase Consortium The FlyBase database of the Drosophila Genome Projects and community literature. Nucleic Acids Res. 1999;27:85–88.
Blake JA, et al. The Mouse Genome Database (MGD): expanding genetic and genomic resources for the laboratory mouse. Nucleic Acids Res. 2000;28:108–111.
Ball CA, et al. Integrating functional genomic information into the Saccharomyces Genome Database. Nucleic Acids Res. 2000;28:77–80.
Venter, J.C., Adams, M.D., Myers, E.W., Li, P.W., Mural, R.J., Sutton, G.G., Smith, H.O., Yandell, M., Evans, C.A., Holt, R.A., et al. 2001. The sequence of the human genome. Science 291: 1304–1351.
Lander, E.S., Linton, L.M., Birren, B., Nusbaum, C., Zody, M.C., Baldwin, J., Devon, K., Dewar, K., Doyle, M., FitzHugh, W., et al. 2001. Initial sequencing and analysis of the human genome. Nature 409: 860–921.
Mi, H., Vandergriff, J., Campbell, M., Narechania, A., Lewis, S., Thomas, P.D., and Ashburner, M. 2003. Assessment of genome-wide protein function classification for Drosophila melanogaster. Genome Res.
Ashburner, M., Ball, C.A., Blake, J.A., Botstein, D., Butler, H., Cherry, J.M., Davis, A.P., Dolinski, K., Dwight, S.S., Eppig, J.T., et al. The Gene Ontology Consortium. 2000. Gene ontology: Tool for the unification of biology. Nat. Genet. 25: 25–29.
Computational Biology
Attwood TK, Beck ME, Bleasby AJ, Parry-Smith DJ. PRINTS–a database of protein motif fingerprints. Nucleic Acids Res. 1994 Sep;22(17):3590-6.
Obenauer JC, Yaffe MB. Computational prediction of protein-protein interactions.
Methods Mol Biol. 2004;261:445-68. Review.
Aitken A. Protein consensus sequence motifs. Mol Biotechnol. 1999 Oct;12(3):241-53. Review.
Bork P, Koonin EV. Protein sequence motifs. Curr Opin Struct Biol. 1996 Jun;6(3):366-76. Review.
Hodgman TC. The elucidation of protein function by sequence motif analysis. Comput Appl Biosci. 1989 Feb;5(1):1-13. Review.
Altschul, S.F., Madden, T.L., Schäffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25: 3389–3402.
Spencer CC, et al. The influence of recombination on human genetic diversity.PLoS Genet. 2006;2:e148.
Petes TD. Meiotic recombination hot spots and cold spots. Nature Rev. Genet.2001;2:360–369.
Griffiths RC, Tavaré S. The age of a mutation in a general coalescent tree. Stoch Models. 1998;14:273–295. doi: 10.1080/15326349808807471.
Gauderman WJ. Sample size requirements for matched case-control studies of gene-environment interaction. Stat Med. 2002;21(1):35–50. doi: 10.1002/sim.973.
Attwood, T.K., Beck, M.E., Bleasby, A.J., and Parry-Smith, D.J. 1994. PRINTS—A database of protein motif fingerprints. Nucleic Acids Res. 22: 3590–3596.
Bairoch, A. 1991. PROSITE: A dictionary of sites and patterns in proteins. Nucleic Acids Res. 19 Suppl: 2241–2245.
Bairoch, A. and Apweiler, R. 2000. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28: 45–48.
Bateman, A., Birney, E., Cerruti, L., Durbin, R., Etwiller, L., Eddy, S.R., Griffiths-Jones, S., Howe, K.L., Marshall, M., and Sonnhammer, E.L. 2002. The Pfam protein families database. Nucleic Acids Res. 30: 276–280.
Sonnhammer, E.L., Eddy, S.R., and Durbin, R. 1997. Pfam: A comprehensive database of protein domain families based on seed alignments. Proteins 28:405–420.
Swets, J.A. 1988. Measuring the accuracy of diagnostic systems. Science 240:1285–1293. [PubMed]
Thomas, P.D., Kejariwal, A., Campbell, M.J., Mi, H., Diemer, K., Guo, N., Ladunga, I., Ulitsky-Lazareva, B., Muruganujan, A., Rabkin, S., et al. 2003. PANTHER: A browsable database of gene products organized by biological function, using curated protein family and subfamily classification. Nucleic Acids Res. 31: 334–341.
HUGO Gene Nomenclature Committee (2011). HGNC Database.http://www.genenames.org/.
Population Genomics, GWAS, Inheritance, Heritability, Migration, Selection an Evolution:
Dayhoff, M.O., Barker, W.C., and McLaughlin, P.J. 1974. Inferences from protein and nucleic acid sequences: Early molecular evolution, divergence of kingdoms and rates of change. Orig. Life 5: 311–330.
Joseph Lachance Disease-associated alleles in genome-wide association studies are enriched for derived low frequency alleles relative to HapMap and neutral expectations BMC Med Genomics. 2010; 3: 57.
Joseph Lachance, Sarah A. Tishkoff Biased Gene Conversion Skews Allele Frequencies in Human Populations, Increasing the Disease Burden of Recessive Alleles Am J Hum Genet. 2014 October 2; 95(4): 408-420.
Hemalatha Kuppusamy, Helga M. Ogmundsdottir, Eva Baigorri, Amanda Warkentin, Hlif Steingrimsdottir, Vilhelmina Haraldsdottir, Michael J. Mant, John Mackey, James B. Johnston, Sophia Adamia, Andrew R. Belch, Linda M. Pilarski Inherited Polymorphisms in Hyaluronan Synthase 1 Predict Risk of Systemic B-Cell Malignancies but Not of Breast Cancer PLoS One. 2014; 9(6): e100691.
Joseph Lachance, Sarah A. Tishkoff Population Genomics of Human Adaptation
Annu Rev Ecol Evol Syst. Author manuscript; available in PMC 2014 November 5.
Published in final edited form as: Annu Rev Ecol Evol Syst. 2013 November; 44: 123–143
Joseph Lachance, Sarah A. Tishkoff SNP ascertainment bias in population genetic analyses: Why it is important, and how to correct it Bioessays.
Erik Corona, Rong Chen, Martin Sikora, Alexander A. Morgan, Chirag J. Patel, Aditya Ramesh, Carlos D. Bustamante, Atul J. Butte Analysis of the Genetic Basis of Disease in the Context of Worldwide Human Relationships and Migration PLoS Genet. 2013 May; 9(5): e1003447.
Olga Y. Gorlova, Jun Ying, Christopher I. Amos, Margaret R. Spitz, Bo Peng, Ivan P. Gorlov J Derived SNP Alleles Are Used More Frequently Than Ancestral Alleles As Risk-Associated Variants In Common Human Diseases Bioinform Comput Biol.
Ani Manichaikul, Wei-Min Chen, Kayleen Williams, Quenna Wong, Michèle M. Sale, James S. Pankow, Michael Y. Tsai, Jerome I. Rotter, Stephen S. Rich, Josyf C. Mychaleckyj Analysis of Family- and Population-Based Samples in Cohort Genome-Wide Association Studies Hum Genet.
Altshuler D, Daly MJ, Lander ES. Genetic mapping in human disease. Science. 2008; 322(5903):881–888. doi: 10.1126/science.1156409.
Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447(7145):661–678. doi: 10.1038/nature05911.
Kotowski IK, Pertsemlidis A, Luke A, Cooper RS, Vega GL, Cohen JC, Hobbs HH. A spectrum of PCSK9 Alleles contributes to plasma levels of low-density lipoprotein cholesterol. American Journal of Human Genetics.2006;78(3):410–422. doi: 10.1086/500615.
Tomlinson I, Webb E, Carvajal-Carmona L, Broderick P, Kemp Z, Spain S, Penegar S, Chandler I, Gorman M, Wood W. et al. A genome-wide association scan of tag SNPs identifies a susceptibility variant for colorectal cancer at 8q24.21. Nature Genetics. 2007;39(8):984–988. doi: 10.1038/ng2085.
Todd JA, Walker NM, Cooper JD, Smyth DJ, Downes K, Plagnol V, Bailey R, Nejentsev S, Field SF, Payne F. et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nature Genetics. 2007;39(7):857–864. doi: 10.1038/ng2068.
Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A. et al. Finding the missing heritability of complex diseases. Nature. 2009;461(7265):747–753. doi: 10.1038/nature08494.
Maher B. Personal genomes: The case of the missing heritability. Nature.2008;456(7218):18–21. doi: 10.1038/456018a.
Clark AG, Boerwinkle E, Hixson J, Sing CF. Determinants of the success of whole-genome association testing. Genome Res. 2005;15(11):1463–1467. doi: 10.1101/gr.4244005.
Clarke AJ, Cooper DN. GWAS: heritability missing in action? European Journal of Human Genetics. 2010;18:859–861. doi: 10.1038/ejhg.2010.35.
Moore JH, Williams SM. Epistasis and its implications for personal genetics. Am J Hum Genet. 2009;85(3):309–320. doi: 10.1016/j.ajhg.2009.08.006.
Goldstein DB. Common genetic variation and human traits. N Engl J Med.2009;360(17):1696–1698. doi: 10.1056/NEJMp0806284.
Hirschhorn JN. Genomewide association studies–illuminating biologic pathways. N Engl J Med. 2009;360(17):1699–1701. doi: 10.1056/NEJMp0808934.
Iles MM. What can genome-wide association studies tell us about the genetics of common disease? PLoS Genet. 2008;4(2):e33. doi: 10.1371/journal.pgen.0040033.
Myles S, Davison D, Barrett J, Stoneking M, Timpson N. Worldwide population differentiation at disease-associated SNPs. BMC Med Genomics.2008;1:22. doi: 10.1186/1755-8794-1-22.
Lohmueller KE, Mauney MM, Reich D, Braverman JM. Variants associated with common disease are not unusually differentiated in frequency across populations. Am J Hum Genet. 2006;78(1):130–136. doi: 10.1086/499287.
Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA.2009;106(23):9362–9367. doi: 10.1073/pnas.0903103106.
Wang WYS, Barratt BJ, Clayton DG, Todd JA. Genome-wide association studies: Theoretical and practical concerns. Nature Reviews Genetics.2005;6(2):109–118. doi: 10.1038/nrg1522.
Hacia JG, Fan JB, Ryder O, Jin L, Edgemon K, Ghandour G, Mayer RA, Sun B, Hsie L, Robbins C. et al. Determination of ancestral alleles for human single-nucleotide polymorphisms using high-density oligonucleotide arrays. Nat Genet. 1999;22(2):164–167. doi: 10.1038/9674.
Lohmueller KE, Pearce CL, Pike M, Lander ES, Hirschhorn JN. Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nat Genet. 2003;33(2):177–182. doi: 10.1038/ng1071.
Wang WY, Pike N. The allelic spectra of common diseases may resemble the allelic spectrum of the full genome. Med Hypotheses. 2004;63(4):748–751. doi: 10.1016/j.mehy.2003.12.057.
HapMart. http://hapmart.hapmap.org/BioMart/martview/
Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, Gibbs RA, Belmont JW, Boudreau A, Hardenbol P, Leal S. et al. A second generation human haplotype map of over 3.1 million SNPs. Nature.2007;449(7164):851–861. doi: 10.1038/nature06258.
Rotimi CN, Jorde LB. Ancestry and disease in the age of genomic medicine. N Engl J Med. 2010;363(16):1551–1558. doi: 10.1056/NEJMra0911564.
Ganapathy G, Uyenoyama MK. Site frequency spectra from genomic SNP surveys. Theor Popul Biol. 2009;75(4):346–354. doi: 10.1016/j.tpb.2009.04.003.
Nielsen R, Hubisz MJ, Clark AG. Reconstituting the frequency spectrum of ascertained single-nucleotide polymorphism data. Genetics.2004;168(4):2373–2382. doi: 10.1534/genetics.104.031039.
Watterson GA, Guess HA. Is the most frequent allele the oldest? Theor Popul Biol. 1977;11(2):141–160. doi: 10.1016/0040-5809(77)90023-5.
Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29(1):308–311. doi: 10.1093/nar/29.1.308.
Spencer CC, Deloukas P, Hunt S, Mullikin J, Myers S, Silverman B, Donnelly P, Bentley D, McVean G. The influence of recombination on human genetic diversity. PLoS Genet. 2006;2(9):e148. doi: 10.1371/journal.pgen.0020148.
Kimura M. The number of heterozygous nucleotide sites maintained in a finite population due to steady flux of mutations. Genetics. 1969;61(4):893–903.
Johnson AD, O’Donnell CJ. An open access database of genome-wide association results. BMC Med Genet. 2009;10:6. doi: 10.1186/1471-2350-10-6.
Kimura M, Ohta T. The age of a neutral mutant persisting in a finite population. Genetics. 1973;75(1):199–212.
McVean GA, et al. The fine-scale structure of recombination rate variation in the human genome. Science. 2004;304:581–584.
Slatkin M, Rannala B. Estimating the age of alleles by use of intraallelic variability. Am J Hum Genet. 1997;60(2):447–458.
Green R, Krause J, Briggs A, Maricic T, Stenzel U, Kircher M, Patterson N, Li H, Zhai W, Fritz M. et al. A Draft Sequence of the Neanderthal Genome. Science. 2010;328:710–722. doi: 10.1126/science.1188021.
Bamshad M, Wooding SP. Signatures of natural selection in the human genome. Nat Rev Genet. 2003;4(2):99–111. doi: 10.1038/nrg999.
Hernandez RD, Williamson SH, Bustamante CD. Context dependence, ancestral misidentification, and spurious signatures of natural selection. Mol Biol Evol. 2007;24(8):1792–1800. doi: 10.1093/molbev/msm108.
Bustamante CD, et al. Natural selection on protein-coding genes in the human genome. Nature. 2005;437:1153–1157.
Personalized Medicine in Cancer [3]
| Personalized Medicine in Cancer [3] |
larryhbern |
| Advances in Gene Editing Technology: New Gene Therapy Options in Personalized Medicine |
2012pharmaceutical |
| Big Data for Personalized Medicine and Biomarker Discovery, May 5-6, 2015 | Philadelphia, PA |
2012pharmaceutical |
| Tweets by @pharma_BI and by @AVIVA1950 for @PMWCIntl, #PMWC15, #PMWC2015 LIVE @Silicon Valley 2015 Personalized Medicine World Conference |
2012pharmaceutical |
| Presentations Content for Track One @Silicon Valley 2015 Personalized Medicine World Conference, Mountain View, CA, January 26 to January 28, 2015 |
2012pharmaceutical |
| Views of Content Presentations – Track One @Silicon Valley 2015 Personalized Medicine World Conference, Mountain View, CA, January 26 to January 28, 2015 |
2012pharmaceutical |
| Word Associations of Twitter Discussions for 10th Annual Personalized Medicine Conference at the Harvard Medical School, November 12-13, 2014 |
2012pharmaceutical |
| 8:30AM–12:00PM, January 28, 2015 – Morality, Ethics & Public Law in PM, LIVE @Silicon Valley 2015 Personalized Medicine World Conference, Mountain View, CA |
2012pharmaceutical |
| 2:00PM–5:00PM, January 27, 2015 – Personalizing Evidence in the Learning Healthcare System & Biomarker Discovery Technologies, LIVE @Silicon Valley 2015 Personalized Medicine World Conference, Mountain View, CA |
2012pharmaceutical |
| 9:15AM–2:00PM, January 27, 2015 – Regulatory & Reimbursement Frameworks for Molecular Testing, LIVE @Silicon Valley 2015 Personalized Medicine World Conference, Mountain View, CA |
2012pharmaceutical |
| 7:45AM–9:15AM, January 27, 2015 – Risk, Reward & Innovation, LIVE @Silicon Valley 2015 Personalized Medicine World Conference, Mountain View, CA |
2012pharmaceutical |
| 3:30PM –5:15PM, January 26, 2015 – NGS Applications: Impact of Genomics on Cancer Care @Silicon Valley 2015 Personalized Medicine World Conference, Mountain View, CA |
2012pharmaceutical |
| 2:15PM – 3:00PM, January 26, 2015 – Impact of Genomics on Cancer Care @Silicon Valley 2015 Personalized Medicine World Conference, Mountain View, CA |
2012pharmaceutical |
| 1:00PM – 1:15PM, January 26, 2015 – Clinical Methodologies of NGS – LIVE @Silicon Valley 2015 Personalized Medicine World Conference, Mountain View, CA |
2012pharmaceutical |
| 10:30AM-12PM, January 26, 2015 – NGS Applications: Impact of Genomics on Cancer Care – LIVE @Silicon Valley 2015 Personalized Medicine World Conference, Mountain View, CA |
2012pharmaceutical |
| 9AM-10AM, January 26, 2015 – Newborn & Prenatal Diagnosis – LIVE @Silicon Valley 2015 Personalized Medicine World Conference, Mountain View, CA |
2012pharmaceutical |
| 7:55AM – 9AM, January 26, 2015 – Introduction and Overview – LIVE @Silicon Valley 2015 Personalized Medicine World Conference, Mountain View, CA |
2012pharmaceutical |
| Hamburg, Snyderman to Address Timely Issues in Personalized Medicine at 2015 Personalized Medicine World Conference in Silicon Valley |
2012pharmaceutical |
| The Personalized Medicine Coalition welcomes the Administration’s focus on Personalized Medicine |
2012pharmaceutical |
| Silicon Valley 2015 Personalized Medicine World Conference, Mountain View, CA, January 26, 2015, 8:00AM to January 28, 2015, 3:30PM PST |
2012pharmaceutical |
|
TOTAL Views of Presentation Content per Presentation: 10th Annual Personalized Medicine Conference at the Harvard Medical School, November 12-13, 2014 |
2012pharmaceutical |
|
|
Silicon Valley 2015 Personalized Medicine World Conference, Mountain View, CA, January 26, 2015, 8:00AM to January 28, 2015, 3:30PM PST |
2012pharmaceutical |
|
|
FDA Commissioner, Dr. Margaret A. Hamburg on HealthCare for 310Million Americans and the Role of Personalized Medicine |
2012pharmaceutical |
|
|
Tweeting on the 10th Annual Personalized Medicine Conference at the Harvard Medical School, November 12-13, 2014 |
2012pharmaceutical |
|
|
Content of the Presentations at the 10th Annual Personalized Medicine Conference at the Harvard Medical School, November 12-13, 2014 |
2012pharmaceutical |
|
|
2:15PM 11/13/2014 – Panel Discussion Reimbursement/Regulation @10th Annual Personalized Medicine Conference at the Harvard Medical School, Boston |
2012pharmaceutical |
|
|
1:00PM 11/13/2014 – Panel Discussion Genomics in Prenatal and Childhood Disorders @10th Annual Personalized Medicine Conference at the Harvard Medical School, Boston |
2012pharmaceutical |
|
|
11:30AM 11/13/2014 – Role of Genetics and Genomics in Pharmaceutical Development @10th Annual Personalized Medicine Conference at the Harvard Medical School, Boston |
2012pharmaceutical |
|
|
10:15AM 11/13/2014 – Panel Discussion — IT/Big Data @10th Annual Personalized Medicine Conference at the Harvard Medical School, Boston |
2012pharmaceutical |
|
|
8:30AM 11/13/2014 – Harvard Business School Case Study: 23andMe @10th Annual Personalized Medicine Conference at the Harvard Medical School, Boston |
2012pharmaceutical |
|
|
8:00AM 11/13/2014 – Welcome from Gary Gottlieb, M.D., Partners HealthCare @10th Annual Personalized Medicine Conference at the Harvard Medical School, Boston |
2012pharmaceutical |
|
|
4:00PM 11/12/2014 – Panel Discussion Novel Approaches to Personalized Medicine @10th Annual Personalized Medicine Conference at the Harvard Medical School, Boston |
2012pharmaceutical |
|
|
3:15PM 11/12/2014 – Discussion Complex Disorders @10th Annual Personalized Medicine Conference at the Harvard Medical School, Boston |
2012pharmaceutical |
|
|
1:45PM 11/12/2014 – Panel Discussion – Oncology @10th Annual Personalized Medicine Conference at the Harvard Medical School, Boston |
2012pharmaceutical |
|
|
1:15PM 11/12/2014 – Keynote Speaker – International Genetics Health and Disease @10th Annual Personalized Medicine Conference at the Harvard Medical School, Boston |
2012pharmaceutical |
|
|
11:30AM 11/12/2014 – Personalized Medicine Coalition Award & Award Recipient Speech @10th Annual Personalized Medicine Conference at the Harvard Medical School, Boston |
2012pharmaceutical |
|
|
11:00AM 11/12/2014 – Keynote Speaker – Past, Present and Future of Personalized Medicine @10th Annual Personalized Medicine Conference at the Harvard Medical School, Boston |
2012pharmaceutical |
|
|
9:20AM 11/12/2014 – Panel Discussion – Genomic Technologies @10th Annual Personalized Medicine Conference at the Harvard Medical School, Boston |
2012pharmaceutical |
|
|
8:50AM 11/12/2014 – Keynote Speaker – CEO, American Medical Association @10th Annual Personalized Medicine Conference at the Harvard Medical School, Boston |
2012pharmaceutical |
|
|
8:20AM 11/12/2014 – Special Guest Keynote Speaker – The Future of Personalized Medicine @10th Annual Personalized Medicine Conference at the Harvard Medical School, Boston |
2012pharmaceutical |
|
| 8:00AM 11/12/2014 – Welcome & Opening Remarks @10th Annual Personalized Medicine Conference at the Harvard Medical School, Boston |
2012pharmaceutical |
|
| Hashtags and Twitter Handles for 10th Annual Personalized Medicine at Harvard Medical School, 11/12 – 11/13/2014 |
2012pharmaceutical |
|
| Personalized Medicine Coalition (PMC) – Upcoming Events |
2012pharmaceutical |
|
| 10th Annual Personalized Medicine Conference at the Harvard Medical School, November 12-13, 2014, The Joseph B. Martin Conference Center at Harvard Medical School, 77 Avenue Louis Pasteur, Boston, MA |
2012pharmaceutical |
|
| Personalized Medicine Coalition Recognizes Mark Levin with Award for Leadership |
2012pharmaceutical |
|
| Research and Markets: Global Personalized Medicine Report 2014 – Scientific … – Rock Hill Herald (press release) |
2012pharmaceutical |
|
| The Role of Medical Imaging in Personalized Medicine |
Dror Nir |
|
| CardioPredict™ Personalized Medicine Molecular Diagnostic Test |
2012pharmaceutical |
|
| Life Sciences Circle Event: Next omics – Personalized Medicine beyond Genomics, December 11, 2013 5:30-8:30PM, The Broad Institute, Cambridge |
2012pharmaceutical |
|
| Issues in Personalized Medicine: Discussions of Intratumor Heterogeneity from the Oncology Pharma forum on LinkedIn |
sjwilliamspa |
|
| Personalized medicine-based diagnostic test for NSCLC |
ritusaxena |
|
| Personalized Medicine and Colon Cancer |
tildabarliya |
|
| Systems Diagnostics – Real Personalized Medicine: David de Graaf, PhD, CEO, Selventa Inc. |
2012pharmaceutical |
|
| Helping Physicians identify Gene-Drug Interactions for Treatment Decisions: New ‘CLIPMERGE’ program – Personalized Medicine @ The Mount Sinai Medical Center |
2012pharmaceutical |
|
| Issues in Personalized Medicine in Cancer: Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing |
sjwilliamspa |
|
| Ethical Concerns in Personalized Medicine: BRCA1/2 Testing in Minors and Communication of Breast Cancer Risk |
sjwilliamspa |
|
| Personalized Medicine: Clinical Aspiration of Microarrays |
sjwilliamspa |
|
| The Promise of Personalized Medicine |
larryhbern |
|
| Personalized Medicine in NSCLC |
larryhbern |
|
| Attitudes of Patients about Personalized Medicine |
larryhbern |
|
| Understanding the Role of Personalized Medicine |
larryhbern |
|
| Directions for Genomics in Personalized Medicine |
larryhbern |
|
| Personalized Medicine: An Institute Profile – Coriell Institute for Medical Research: Part 3 |
2012pharmaceutical |
|
| Paradigm Shift in Human Genomics – Predictive Biomarkers and Personalized Medicine – Part 1 |
2012pharmaceutical |
|
| Harnessing Personalized Medicine for Cancer Management, Prospects of Prevention and Cure: Opinions of Cancer Scientific Leaders @ http://pharmaceuticalintelligence.com |
2012pharmaceutical |
|
| Nanotechnology, personalized medicine and DNA sequencing |
tildabarliya |
|
| Personalized medicine gearing up to tackle cancer |
ritusaxena |
|
| Personalized Medicine Company Genection launched |
ritusaxena |
|
| Personalized Medicine: Cancer Cell Biology and Minimally Invasive Surgery (MIS) |
2012pharmaceutical |
|
| The Way With Personalized Medicine: Reporters’ Voice at the 8th Annual Personalized Medicine Conference,11/28-29, 2012, Harvard Medical School, Boston, MA |
2012pharmaceutical |
|
| Personalized Medicine Coalition: Upcoming Events |
2012pharmaceutical |
|
| Highlights from 8th Annual Personalized Medicine Conference, November 28-29, 2012, Harvard Medical School, Boston, MA |
2012pharmaceutical |
|
| Personalized medicine-based cure for cancer might not be far away |
ritusaxena |
|
| GSK for Personalized Medicine using Cancer Drugs needs Alacris systems biology model to determine the in silico effect of the inhibitor in its “virtual clinical trial” |
2012pharmaceutical |
|
| Congestive Heart Failure & Personalized Medicine: Two-gene Test predicts response to Beta Blocker Bucindolol |
2012pharmaceutical |
|
| Personalized Medicine as Key Area for Future Pharmaceutical Growth |
2012pharmaceutical |
|
| Clinical Genetics, Personalized Medicine, Molecular Diagnostics, Consumer-targeted DNA – Consumer Genetics Conference (CGC) – October 3-5, 2012, Seaport Hotel, Boston, MA |
2012pharmaceutical |
|
| AGENDA – Personalized Diagnostics, February 16-18, 2015 | Moscone North Convention Center | San Francisco, CA Part of the 22nd Annual Molecular Medicine Tri-Conference |
2012pharmaceutical |
|
| Arrowhead’s 6th Annual Personalized & Precision Medicine Conference is coming to San Francisco, October 29-30, 2014 |
2012pharmaceutical |
| Personalized Cardiovascular Genetic Medicine at Partners HealthCare and Harvard Medical School |
2012pharmaceutical |
|
| Precision Medicine for Future of Genomics Medicine is The New Era |
Demet Sag, Ph.D., CRA, GCP |
|
| Precision Medicine Initiative: Now is a State Initiative in California |
2012pharmaceutical |
|
| 1:30 pm – 2:20 pm 3/26/2015, LIVE Precision Medicine: Who’s Paying? @ MassBio Annual Meeting 2015, Cambridge, MA, Sonesta Hotel, 3/26 – 3/27, 2015 |
2012pharmaceutical |
|
| We Celebrate >600,000 Views for our 2,830 Scientific Articles in Life Sciences and Medicine |
2012pharmaceutical |
|
| attn #3: Investors in HealthCare — Platforms in the Ecosystem of Regulatory & Reimbursement – Integrated Informational Platforms in Orthopedic Medical Devices, and Global Peer-Reviewed Scientific Curations: Bone Disease and Orthopedic Medicine – Draft |
2012pharmaceutical |
|
| Foundation Medicine: Roche has Taken Over at $1.2B and 52.4 percent to 56.3 percent of Foundation Medicine on a fully diluted basis |
2012pharmaceutical |
|
| Bridging the Gap in Precision Medicine @UCSF |
2012pharmaceutical |
|
| Germline Genes and Drug Targets: Medicine more Proactive and Disease Prevention more Effective. |
2012pharmaceutical |
|
| Proteomics – The Pathway to Understanding and Decision-making in Medicine |
larryhbern |
|
| Multi-drug, Multi-arm, Biomarker-driven Clinical Trial for patients with Squamous Cell Carcinoma called the Lung Cancer Master Protocol, or Lung-MAP launched by NCI, Foundation Medicine, and Five Pharma Firms |
2012pharmaceutical |
|
| Preventive Care: Anticipated Changes caused by Genomics in the Clinic and Personalised Medicine |
2012pharmaceutical |
|
| Cancer Labs at School of Medicine @ Technion: Janet and David Polak Cancer and Vascular Biology Research Center |
2012pharmaceutical |
|
| Reprogramming Adult Patient Cells into Stem Cells: the Promise of Personalized Genetic Therapy |
2012pharmaceutical |
|
| US Personalized Cancer Genome Sequencing Market Outlook 2018 – |
2012pharmaceutical |
|
| Summary of Translational Medicine – e-Series A: Cardiovascular Diseases, Volume Four – Part 1 |
larryhbern |
|
| Introduction to Translational Medicine (TM) – Part 1: Translational Medicine |
larryhbern |
|
| Cancer Diagnosis at the Crossroads: Precision Medicine Driving Change, 9/14 – 9/17/2014, Sheraton Seattle Hotel, Seattle WA |
2012pharmaceutical |
|
| Genomic Medicine and the Bioeconomy: Innovation for a Better World May 12–16, 2014 • Boston, MA |
2012pharmaceutical |
|
| Institute of Medicine (IOM) Report on Genome-based Therapeutics and Companion Diagnostics |
2012pharmaceutical |
|
| “Medicine Meets Virtual Reality” – NextMed-MMVR21 Conference 2/19 – 2/22/2014, Manhattan Beach Marriott, Manhattan Beach, CAView |
2012pharmaceutical |
|
|
|
|
|
|
|
|
Read Full Post »



 and
and