Healthcare analytics, AI solutions for biological big data, providing an AI platform for the biotech, life sciences, medical and pharmaceutical industries, as well as for related technological approaches, i.e., curation and text analysis with machine learning and other activities related to AI applications to these industries.
DISCUSSION – Genomics-driven personalized medicine for Pancreatic Cancer, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 1: Next Generation Sequencing (NGS)
DISCUSSION – Genomics-driven personalized medicine for Pancreatic Cancer
Reporter: Aviva Lev-Ari, PhD, RN
[bold face added, ALA]
Integrated Patient-Derived Models Delineate Individualized Therapeutic Vulnerabilities of Pancreatic Cancer –>>> Personalized Tumor Models Could Help Identify Combination Therapies for Hard-to-Treat Cancers
PDAC has a particularly poor prognosis, and even with new targeted therapies and chemotherapy, the survival is poor. Here, we show that patient-derived models can be developed and used to investigate therapeutic sensitivities determined by genetic features of the disease and to identify empirical therapeutic vulnerabilities. These data reveal several key points that are of prime relevance to pancreatic cancer and tumor biology in general.
The Challenges of Using Genetic Analysis to Inform Treatment in PDAC
Precision oncology is dependent on the existence of known vulnerabilities encoded by high-potency genetic events and drugs capable of exploiting these vulnerabilities. At present, the repertoire of actionable genetic events in PDAC is limited.
Rare BRAF V600E mutations are identified in PDAC and could represent the basis for targeted inhibition, as our group and others have previously published (Collisson et al., 2012; Witkiewicz et al., 2015).
Similarly,
germline BRCA deficiency is the basis for ongoing poly(ADP-ribose) polymerase (PARP) inhibitor clinical trials (Lowery et al., 2011).
As shown here, out of 28 cases, only one genetic event was identified that yielded sensitivity to a therapeutic strategy. In this case, existence of the matched model allowed us to confirm the biological relevance of the
STAG2 mutation by showing sensitivity of the model to a DNA cross-linking agent.
Therefore, annotated patient-derived models provide a substrate upon which to functionally dissect the significance of novel and potentially actionable genetic events that occur within a tumor.
Another challenge of genomics-driven personalized medicine is
assessing the effect of specific molecular aberrations on therapeutic response in the context of complex genetic changes present in individual tumors.
KRAS has been proposed to modify therapeutic dependency to EZH2 inhibitors (Kim et al., 2015), and in the models tested, responses to this class of drugs were not uniformly present in cases harboring mutations in chromatin-remodeling genes.
This finding suggests that, although tumors acquire genetic alterations in specific genes, the implicated pathway may not be functionally inactive or therapeutically actionable. Therefore, annotated patient derived models provide a unique test bed for interrogating specific therapeutic dependencies in a genetically tractable system.
Empirical Definition of Therapeutic Sensitivities and Clinical Relevance
Cell lines offer the advantage of the ability to conduct high throughput approaches to interrogate many therapeutic agents. A large number of failed clinical trials have demonstrated the difficulty in treating PDAC. Based on the data herein, the paucity of clinical success is, most probably, due to the diverse therapeutic sensitivity of individual PDAC cases, suggesting that, with an unselected patient population, it will be veritably impossible to demonstrate clinical benefit. Additionally,
very few models exhibited an exceptional response to single agents across the breadth of a library encompassing 305 agents.
We could identify only one tumor that was particularly sensitive to MEK inhibition and another model that was sensitive to
EGFR and
tyrosine kinase inhibitors.
In contrast to the limited activity of single agents, combination screens yielded responses at low-dose concentrations in the majority of models. Specific combinations were effective across several models, indicating that, by potentially screening more models, therapeutic sensitivity clades of PDAC will emerge. In the pharmacological screens performed in this study,
MEK inhibition, coupled with MTOR, docetaxel, or tyrosine kinase inhibitors, was effective in _30% of models tested.
Resistance to MEK inhibitors occurs through several mechanisms, including
Upregulation of oncogenic bypass signaling pathways such as AKT, tyrosine kinase, or MTOR (mammalian target of rapamycin) signaling.
In the clinic, the MEK and MTOR inhibitors (e.g., NCT02583542) are being tested. An intriguing finding from the drug screen was
sensitivity of a subset of models to combined MEK and docetaxel inhibition. This combination has been observed to synergistically enhance apoptosis and inhibit tumor growth in human xenograft tumor models (Balko et al., 2012; McDaid et al., 2005) and is currently being tested in a phase III study in patients with KRAS-mutated, advanced non-small-cell lung adenocarcinoma (Ja¨ nne et al., 2016).
Interestingly, in the models tested herein, there was limited sensitivity imparted through
the combination of gemcitabine and MEK inhibition.
This potentially explains why the combination of MEK inhibitor and gemcitabine tested in the clinic did not show improved efficacy over gemcitabine alone (Infante et al., 2014).
Another promising strategy that emerged from this study involves using
CHK or BCL2 inhibitors as agents that drive enhanced sensitivity to chemotherapy.
Together, the data suggest that the majority of PDAC tumors have intrinsic therapeutic sensitivities, but the challenge is to prospectively identify effective treatment.
Patient-Derived Model-Based Approach to Precision Medicine
This study supports a path for guiding patient treatment based on the integration of genetic and empirically determined sensitivities of the patient’s tumor (Figure S7). In reference to defined genetic susceptibilities, the models provide a means to interrogate the voracity of specific drug targets. Parallel unbiased screening enables the discovery of sensitivities that could be exploited in the clinic. The model-guided treatment must be optimized, allowing for the generation of data in a time frame compatible with clinical decision making and appropriate validation.
In the present study, the majority of models were developed, cell lines were drug screened, and select hits were validated in PDX models within a 10- to 12-month window (Figure S7). This chronology would allow time to inform frontline therapy for recurrent disease for most patients who were surgically resected and treated with a standard of care where the median time to recurrence is approximately 14 months (Saif, 2013).
Although most models were generated from surgically resected specimens, two of the models (EMC3226 and EMC62) were established from primary tumor biopsies, indicating that this approach could be used with only a limited amount of tumor tissue available.
In the context of inoperable pancreatic cancer, application of data from a cell-line screen without in vivo validation in PDX would permit the generation of sensitivity data in the time frame compatible with treatment.
[We] acknowledge that model-guided treatment is also not without significant logistical hurdles, including the availability of drugs for patient treatment, clinically relevant time frames, patient-performance status, toxicity of combination regiments, and quality metrics related to model development and therapeutic response evaluation.
Additionally, it will be very important to monitor ex vivo genetic and phenotypic divergence with passage and try to understand the features of tumor heterogeneity that could undermine the efficacy of using models to direct treatment. As shown here, drug sensitivities remained stable with passage in cell culture and, importantly, were confirmed in PDX models, suggesting that the dominant genetic drivers and related therapeutic sensitivities are conserved.
In spite of these challenges, progressively more effort is going into the development of patient-derived models for guidance of disease treatment (Aparicio et al., 2015; Boj et al., 2015; Crystal et al., 2014; van de Wetering et al., 2015).
Several ongoing trials use PDX models to direct a limited repertoire of agents (e.g., NCT02312245, NCT02720796, and ERCAVATAR2015). Given the experience here, PDAC cell lines would provide the opportunity to rapidly interrogate a larger portfolio of combinations that could be used to guide patient care and provide a novel approach to precision medicine.
Validation of this approach would require the establishment of challenging multi-arm or N-of-1 clinical trials. However, considering the dire outcome for PDAC patients and the long-lasting difficulty in developing effective treatments, this non-canonical approach might be particularly impactful in pancreatic cancer.
Using Online Mendelian Inheritance in Man (OMIM) database and the Human Genome Mutation Database (HGMD) Pro 2015.2 for Quantification of the growth in gene-disease and variant-disease associations, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 1: Next Generation Sequencing (NGS)
Using Online Mendelian Inheritance in Man (OMIM) database and the Human Genome Mutation Database (HGMD) Pro 2015.2 for Quantification of the growth in gene-disease and variant-disease associations
Reporter: Aviva Lev-Ari, PhD, RN
Reanalysis of Clinical Exome Data Over Time Could Yield New Diagnoses
NEW YORK (GenomeWeb) – Clinical exomes that are re-evaluated in a systematic way could yield new diagnoses and prove useful to clinicians, according to a study published yesterday in Genetics in Medicine.
A team of researchers from Stanford University set out to examine whether nondiagnostic clinical exomes could provide new information for patients if they were re-examined with current bioinformatics software and knowledge of disease-related variants as presented in the literature.
Clinical exome sequencing yields no diagnosis for about 75 percent of patients evaluated for possible Mendelian disorders, wrote senior author Gill Bejerano and his colleagues. But a reanalysis of exome and phenotypic data from 40 such individuals using current methods identified a definitive diagnosis for four of them — 10 percent — the team said.
In these cases, the causative variant was de novo and found in a relevant autosomal-dominant disease gene. At the time these exomes were first sequenced, the researchers wrote, the existing literature on these causative genes was either “weak, nonexistent, or not readily located.” When the exomes were re-examined by his team, Bejerano noted, the supporting literature was more robust.
In addition to re-analyzing exome data, the researchers have been working on establishing causality for novel candidate disease genes through patient matches. For this, the team has been using the GeneMatcher website, which allows them to find other clinicians and researchers around the world who have patients, or animal models, with mutations in the same genes as their own patients. Through an API developed by the Matchmaker Exchange project, GeneMatcher submitters can also query the PhenomeCentral and Decipher databases. As of March, more than 4,000 genes had been submitted to GeneMatcher from more than 1,300 submitters in 48 countries, and 1,900 matches had been made, Sobreira reported.
Her team has so far submitted data from 104 families, involving 280 genes, and has had 314 matches so far, involving 113 genes. Several cases have been successes, meaning the researchers could establish that a candidate gene is indeed disease causing, and several others are pending, both from Hopkins and from other groups. The total number of solved cases tracing their success to GeneMatcher is currently unknown, Sobreira said, but the organizers are planning to survey submitters about their success rate in the near future.
Disease related changes in proteomics, protein folding, protein-protein interaction, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 1: Next Generation Sequencing (NGS)
Disease related changes in proteomics, protein folding, protein-protein interaction
Curator: Larry H. Bernstein, MD, FCAP
LPBI
Frankenstein Proteins Stitched Together by Scientists
The Frankenstein monster, stitched together from disparate body parts, proved to be an abomination, but stitched together proteins may fare better. They may, for example, serve specific purposes in medicine, research, and industry. At least, that’s the ambition of scientists based at the University of North Carolina. They have developed a computational protocol called SEWING that builds new proteins from connected or disconnected pieces of existing structures. [Wikipedia]
Unlike Victor Frankenstein, who betrayed Promethean ambition when he sewed together his infamous creature, today’s biochemists are relatively modest. Rather than defy nature, they emulate it. For example, at the University of North Carolina (UNC), researchers have taken inspiration from natural evolutionary mechanisms to develop a technique called SEWING—Structure Extension With Native-substructure Graphs. SEWING is a computational protocol that describes how to stitch together new proteins from connected or disconnected pieces of existing structures.
“We can now begin to think about engineering proteins to do things that nothing else is capable of doing,” said UNC’s Brian Kuhlman, Ph.D. “The structure of a protein determines its function, so if we are going to learn how to design new functions, we have to learn how to design new structures. Our study is a critical step in that direction and provides tools for creating proteins that haven’t been seen before in nature.”
Traditionally, researchers have used computational protein design to recreate in the laboratory what already exists in the natural world. In recent years, their focus has shifted toward inventing novel proteins with new functionality. These design projects all start with a specific structural “blueprint” in mind, and as a result are limited. Dr. Kuhlman and his colleagues, however, believe that by removing the limitations of a predetermined blueprint and taking cues from evolution they can more easily create functional proteins.
Dr. Kuhlman’s UNC team developed a protein design approach that emulates natural mechanisms for shuffling tertiary structures such as pleats, coils, and furrows. Putting the approach into action, the UNC team mapped 50,000 stitched together proteins on the computer, and then it produced 21 promising structures in the laboratory. Details of this work appeared May 6 in the journal Science, in an article entitled, “Design of Structurally Distinct Proteins Using Strategies Inspired by Evolution.”
“Helical proteins designed with SEWING contain structural features absent from other de novo designed proteins and, in some cases, remain folded at more than 100°C,” wrote the authors. “High-resolution structures of the designed proteins CA01 and DA05R1 were solved by x-ray crystallography (2.2 angstrom resolution) and nuclear magnetic resonance, respectively, and there was excellent agreement with the design models.”
Essentially, the UNC scientists confirmed that the proteins they had synthesized contained the unique structural varieties that had been designed on the computer. The UNC scientists also determined that the structures they had created had new surface and pocket features. Such features, they noted, provide potential binding sites for ligands or macromolecules.
“We were excited that some had clefts or grooves on the surface, regions that naturally occurring proteins use for binding other proteins,” said the Science article’s first author, Tim M. Jacobs, Ph.D., a former graduate student in Dr. Kuhlman’s laboratory. “That’s important because if we wanted to create a protein that can act as a biosensor to detect a certain metabolite in the body, either for diagnostic or research purposes, it would need to have these grooves. Likewise, if we wanted to develop novel therapeutics, they would also need to attach to specific proteins.”
Currently, the UNC researchers are using SEWING to create proteins that can bind to several other proteins at a time. Many of the most important proteins are such multitaskers, including the blood protein hemoglobin.
Histone Mutation Deranges DNA Methylation to Cause Cancer
In some cancers, including chondroblastoma and a rare form of childhood sarcoma, a mutation in histone H3 reduces global levels of methylation (dark areas) in tumor cells but not in normal cells (arrowhead). The mutation locks the cells in a proliferative state to promote tumor development. [Laboratory of Chromatin Biology and Epigenetics at The Rockefeller University]
They have been called oncohistones, the mutated histones that are known to accompany certain pediatric cancers. Despite their suggestive moniker, oncohistones have kept their oncogenic secrets. For example, it has been unclear whether oncohistones are able to cause cancer on their own, or whether they need to act in concert with additional DNA mutations, that is, mutations other than those affecting histone structures.
While oncohistone mechanisms remain poorly understood, this particular question—the oncogenicity of lone oncohistones—has been resolved, at least in part. According to researchers based at The Rockefeller University, a change to the structure of a histone can trigger a tumor on its own.
This finding appeared May 13 in the journal Science, in an article entitled, “Histone H3K36 Mutations Promote Sarcomagenesis Through Altered Histone Methylation Landscape.” The article describes the Rockefeller team’s study of a histone protein called H3, which has been found in about 95% of samples of chondoblastoma, a benign tumor that arises in cartilage, typically during adolescence.
The Rockefeller scientists found that the H3 lysine 36–to–methionine (H3K36M) mutation impairs the differentiation of mesenchymal progenitor cells and generates undifferentiated sarcoma in vivo.
After the scientists inserted the H3 histone mutation into mouse mesenchymal progenitor cells (MPCs)—which generate cartilage, bone, and fat—they watched these cells lose the ability to differentiate in the lab. Next, the scientists injected the mutant cells into living mice, and the animals developed the tumors rich in MPCs, known as an undifferentiated sarcoma. Finally, the researchers tried to understand how the mutation causes the tumors to develop.
The scientists determined that H3K36M mutant nucleosomes inhibit the enzymatic activities of several H3K36 methyltransferases.
“Depleting H3K36 methyltransferases, or expressing an H3K36I mutant that similarly inhibits H3K36 methylation, is sufficient to phenocopy the H3K36M mutation,” the authors of the Science study wrote. “After the loss of H3K36 methylation, a genome-wide gain in H3K27 methylation leads to a redistribution of polycomb repressive complex 1 and de-repression of its target genes known to block mesenchymal differentiation.”
Essentially, when the H3K36M mutation occurs, the cell becomes locked in a proliferative state—meaning it divides constantly, leading to tumors. Specifically, the mutation inhibits enzymes that normally tag the histone with chemical groups known as methyls, allowing genes to be expressed normally.
In response to this lack of modification, another part of the histone becomes overmodified, or tagged with too many methyl groups. “This leads to an overall resetting of the landscape of chromatin, the complex of DNA and its associated factors, including histones,” explained co-author Peter Lewis, Ph.D., a professor at the University of Wisconsin-Madison and a former postdoctoral fellow in laboratory of C. David Allis, Ph.D., a professor at Rockefeller.
The finding—that a “resetting” of the chromatin landscape can lock the cell into a proliferative state—suggests that researchers should be on the hunt for more mutations in histones that might be driving tumors. For their part, the Rockefeller researchers are trying to learn more about how this specific mutation in histone H3 causes tumors to develop.
“We want to know which pathways cause the mesenchymal progenitor cells that carry the mutation to continue to divide, and not differentiate into the bone, fat, and cartilage cells they are destined to become,” said co-author Chao Lu, Ph.D., a postdoctoral fellow in the Allis lab.
Once researchers understand more about these pathways, added Dr. Lewis, they can consider ways of blocking them with drugs, particularly in tumors such as MPC-rich sarcomas—which, unlike chondroblastoma, can be deadly. In fact, drugs that block these pathways may already exist and may even be in use for other types of cancers.
“One long-term goal of our collaborative team is to better understand fundamental mechanisms that drive these processes, with the hope of providing new therapeutic approaches,” concluded Dr. Allis.
Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape
Missense mutations (that change one amino acid for another) in histone H3 can produce a so-called oncohistone and are found in a number of pediatric cancers. For example, the lysine-36–to-methionine (K36M) mutation is seen in almost all chondroblastomas. Lu et al. show that K36M mutant histones are oncogenic, and they inhibit the normal methylation of this same residue in wild-type H3 histones. The mutant histones also interfere with the normal development of bone-related cells and the deposition of inhibitory chromatin marks.
Several types of pediatric cancers reportedly contain high-frequency missense mutations in histone H3, yet the underlying oncogenic mechanism remains poorly characterized. Here we report that the H3 lysine 36–to–methionine (H3K36M) mutation impairs the differentiation of mesenchymal progenitor cells and generates undifferentiated sarcoma in vivo. H3K36M mutant nucleosomes inhibit the enzymatic activities of several H3K36 methyltransferases. Depleting H3K36 methyltransferases, or expressing an H3K36I mutant that similarly inhibits H3K36 methylation, is sufficient to phenocopy the H3K36M mutation. After the loss of H3K36 methylation, a genome-wide gain in H3K27 methylation leads to a redistribution of polycomb repressive complex 1 and de-repression of its target genes known to block mesenchymal differentiation. Our findings are mirrored in human undifferentiated sarcomas in which novel K36M/I mutations in H3.1 are identified.
Mitochondria? We Don’t Need No Stinking Mitochondria!
Diagram comparing typical eukaryotic cell to the newly discovered mitochondria-free organism. [Karnkowska et al., 2016, Current Biology 26, 1–11]
The organelle that produces a significant portion of energy for eukaryotic cells would seemingly be indispensable, yet over the years, a number of organisms have been discovered that challenge that biological pretense. However, these so-called amitochondrial species may lack a defined organelle, but they still retain some residual functions of their mitochondria-containing brethren. Even the intestinal eukaryotic parasite Giardia intestinalis, which was for many years considered to be mitochondria-free, was proven recently to contain a considerably shriveled version of the organelle.
Now, an international group of scientists has released results from a new study that challenges the notion that mitochondria are essential for eukaryotes—discovering an organism that resides in the gut of chinchillas that contains absolutely no trace of mitochondria at all.
“In low-oxygen environments, eukaryotes often possess a reduced form of the mitochondrion, but it was believed that some of the mitochondrial functions are so essential that these organelles are indispensable for their life,” explained lead study author Anna Karnkowska, Ph.D., visiting scientist at the University of British Columbia in Vancouver. “We have characterized a eukaryotic microbe which indeed possesses no mitochondrion at all.”
Mysterious Eukaryote Missing Mitochondria
Researchers uncover the first example of a eukaryotic organism that lacks the organelles.
Monocercomonoides sp. PA203VLADIMIR HAMPL, CHARLES UNIVERSITY, PRAGUE, CZECH REPUBLIC
Scientists have long thought that mitochondria—organelles responsible for energy generation—are an essential and defining feature of a eukaryotic cell. Now, researchers from Charles University in Prague and their colleagues are challenging this notion with their discovery of a eukaryotic organism,Monocercomonoides species PA203, which lacks mitochondria. The team’s phylogenetic analysis, published today (May 12) in Current Biology,suggests that Monocercomonoides—which belong to the Oxymonadida group of protozoa and live in low-oxygen environments—did have mitochondria at one point, but eventually lost the organelles.
“This is quite a groundbreaking discovery,” said Thijs Ettema, who studies microbial genome evolution at Uppsala University in Sweden and was not involved in the work.
“This study shows that mitochondria are not so central for all lineages of living eukaryotes,” Toni Gabaldonof the Center for Genomic Regulation in Barcelona, Spain, who also was not involved in the work, wrote in an email to The Scientist. “Yet, this mitochondrial-devoid, single-cell eukaryote is as complex as other eukaryotic cells in almost any other aspect of cellular complexity.”
Charles University’s Vladimir Hampl studies the evolution of protists. Along with Anna Karnkowska and colleagues, Hampl decided to sequence the genome of Monocercomonoides, a little-studied protist that lives in the digestive tracts of vertebrates. The 75-megabase genome—the first of an oxymonad—did not contain any conserved genes found on mitochondrial genomes of other eukaryotes, the researchers found. It also did not contain any nuclear genes associated with mitochondrial functions.
“It was surprising and for a long time, we didn’t believe that the [mitochondria-associated genes were really not there]. We thought we were missing something,” Hampl told The Scientist. “But when the data kept accumulating, we switched to the hypothesis that this organism really didn’t have mitochondria.”
Because researchers have previously not found examples of eukaryotes without some form of mitochondria, the current theory of the origin of eukaryotes poses that the appearance of mitochondria was crucial to the identity of these organisms.
“We now view these mitochondria-like organelles as a continuum from full mitochondria to very small . Some anaerobic protists, for example, have only pared down versions of mitochondria, such as hydrogenosomes and mitosomes, which lack a mitochondrial genome. But these mitochondrion-like organelles perform essential functions of the iron-sulfur cluster assembly pathway, which is known to be conserved in virtually all eukaryotic organisms studied to date.
Yet, in their analysis, the researchers found no evidence of the presence of any components of this mitochondrial pathway.
Like the scaling down of mitochondria into mitosomes in some organisms, the ancestors of modernMonocercomonoides once had mitochondria. “Because this organism is phylogenetically nested among relatives that had conventional mitochondria, this is most likely a secondary adaptation,” said Michael Gray, a biochemist who studies mitochondria at Dalhousie University in Nova Scotia and was not involved in the study. According to Gray, the finding of a mitochondria-deficient eukaryote does not mean that the organelles did not play a major role in the evolution of eukaryotic cells.
To be sure they were not missing mitochondrial proteins, Hampl’s team also searched for potential mitochondrial protein homologs of other anaerobic species, and for signature sequences of a range of known mitochondrial proteins. While similar searches with other species uncovered a few mitochondrial proteins, the team’s analysis of Monocercomonoides came up empty.
“The data is very complete,” said Ettema. “It is difficult to prove the absence of something but [these authors] do a convincing job.”
To form the essential iron-sulfur clusters, the team discovered that Monocercomonoides use a sulfur mobilization system found in the cytosol, and that an ancestor of the organism acquired this system by lateral gene transfer from bacteria. This cytosolic, compensating system allowed Monocercomonoides to lose the otherwise essential iron-sulfur cluster-forming pathway in the mitochondrion, the team proposed.
“This work shows the great evolutionary plasticity of the eukaryotic cell,” said Karnkowska, who participated in the study while she was a postdoc at Charles University. Karnkowska, who is now a visiting researcher at the University of British Columbia in Canada, added: “This is a striking example of how far the evolution of a eukaryotic cell can go that was beyond our expectations.”
“The results highlight how many surprises may await us in the poorly studied eukaryotic phyla that live in under-explored environments,” Gabaldon said.
Ettema agreed. “Now that we’ve found one, we need to look at the bigger picture and see if there are other examples of eukaryotes that have lost their mitochondria, to understand how adaptable eukaryotes are.”
Karnkowska et al., “A eukaryote without a mitochondrial organelle,” Current Biology,doi:10.1016/j.cub.2016.03.053, 2016.
•Monocercomonoides sp. is a eukaryotic microorganism with no mitochondria
•The complete absence of mitochondria is a secondary loss, not an ancestral feature
•The essential mitochondrial ISC pathway was replaced by a bacterial SUF system
The presence of mitochondria and related organelles in every studied eukaryote supports the view that mitochondria are essential cellular components. Here, we report the genome sequence of a microbial eukaryote, the oxymonad Monocercomonoides sp., which revealed that this organism lacks all hallmark mitochondrial proteins. Crucially, the mitochondrial iron-sulfur cluster assembly pathway, thought to be conserved in virtually all eukaryotic cells, has been replaced by a cytosolic sulfur mobilization system (SUF) acquired by lateral gene transfer from bacteria. In the context of eukaryotic phylogeny, our data suggest that Monocercomonoides is not primitively amitochondrial but has lost the mitochondrion secondarily. This is the first example of a eukaryote lacking any form of a mitochondrion, demonstrating that this organelle is not absolutely essential for the viability of a eukaryotic cell.
This method catches a bait protein together with its associated protein partners in virus-like particles that are budded from human cells. Like this, cell lysis is not needed and protein complexes are preserved during purification.
With his feet in both a proteomics lab and an interactomics lab, VIB/UGent professor Sven Eyckerman is well aware of the shortcomings of conventional approaches to analyze protein complexes. The lysis conditions required in mass spectrometry–based strategies to break open cell membranes often affect protein-protein interactions. “The first step in a classical study on protein complexes essentially turns the highly organized cellular structure into a big messy soup”, Eyckerman explains.
Inspired by virus biology, Eyckerman came up with a creative solution. “We used the natural process of HIV particle formation to our benefit by hacking a completely safe form of the virus to abduct intact protein machines from the cell.” It is well known that the HIV virus captures a number of host proteins during its particle formation. By fusing a bait protein to the HIV-1 GAG protein, interaction partners become trapped within virus-like particles that bud from mammalian cells. Standard proteomic approaches are used next to reveal the content of these particles. Fittingly, the team named the method ‘Virotrap’.
The Virotrap approach is exceptional as protein networks can be characterized under natural conditions. By trapping protein complexes in the protective environment of a virus-like shell, the intact complexes are preserved during the purification process. The researchers showed the method was suitable for detection of known binary interactions as well as mass spectrometry-based identification of novel protein partners.
Virotrap is a textbook example of bringing research teams with complementary expertise together. Cross-pollination with the labs of Jan Tavernier (VIB/UGent) and Kris Gevaert (VIB/UGent) enabled the development of this platform.
Jan Tavernier: “Virotrap represents a new concept in co-complex analysis wherein complex stability is physically guaranteed by a protective, physical structure. It is complementary to the arsenal of existing interactomics methods, but also holds potential for other fields, like drug target characterization. We also developed a small molecule-variant of Virotrap that could successfully trap protein partners for small molecule baits.”
Kris Gevaert: “Virotrap can also impact our understanding of disease pathways. We were actually surprised to see that this virus-based system could be used to study antiviral pathways, like Toll-like receptor signaling. Understanding these protein machines in their natural environment is essential if we want to modulate their activity in pathology.“
Trapping mammalian protein complexes in viral particles
Cell lysis is an inevitable step in classical mass spectrometry–based strategies to analyse protein complexes. Complementary lysis conditions, in situ cross-linking strategies and proximal labelling techniques are currently used to reduce lysis effects on the protein complex. We have developed Virotrap, a viral particle sorting approach that obviates the need for cell homogenization and preserves the protein complexes during purification. By fusing a bait protein to the HIV-1 GAG protein, we show that interaction partners become trapped within virus-like particles (VLPs) that bud from mammalian cells. Using an efficient VLP enrichment protocol, Virotrap allows the detection of known binary interactions and MS-based identification of novel protein partners as well. In addition, we show the identification of stimulus-dependent interactions and demonstrate trapping of protein partners for small molecules. Virotrap constitutes an elegant complementary approach to the arsenal of methods to study protein complexes.
Proteins mostly exert their function within supramolecular complexes. Strategies for detecting protein–protein interactions (PPIs) can be roughly divided into genetic systems1 and co-purification strategies combined with mass spectrometry (MS) analysis (for example, AP–MS)2. The latter approaches typically require cell or tissue homogenization using detergents, followed by capture of the protein complex using affinity tags3 or specific antibodies4. The protein complexes extracted from this ‘soup’ of constituents are then subjected to several washing steps before actual analysis by trypsin digestion and liquid chromatography–MS/MS analysis. Such lysis and purification protocols are typically empirical and have mostly been optimized using model interactions in single labs. In fact, lysis conditions can profoundly affect the number of both specific and nonspecific proteins that are identified in a typical AP–MS set-up. Indeed, recent studies using the nuclear pore complex as a model protein complex describe optimization of purifications for the different proteins in the complex by examining 96 different conditions5. Nevertheless, for new purifications, it remains hard to correctly estimate the loss of factors in a standard AP–MS experiment due to washing and dilution effects during treatments (that is, false negatives). These considerations have pushed the concept of stabilizing PPIs before the actual homogenization step. A classical approach involves cross-linking with simple reagents (for example, formaldehyde) or with more advanced isotope-labelled cross-linkers (reviewed in ref. 2). However, experimental challenges such as cell permeability and reactivity still preclude the widespread use of cross-linking agents. Moreover, MS-generated spectra of cross-linked peptides are notoriously difficult to identify correctly. A recent lysis-independent solution involves the expression of a bait protein fused to a promiscuous biotin ligase, which results in labelling of proteins proximal to the activity of the enzyme-tagged bait protein6. When compared with AP–MS, this BioID approach delivers a complementary set of candidate proteins, including novel interaction partners7, 8. Such particular studies clearly underscore the need for complementary approaches in the co-complex strategies.
The evolutionary stress on viruses promoted highly condensed coding of information and maximal functionality for small genomes. Accordingly, for HIV-1 it is sufficient to express a single protein, the p55 GAG protein, for efficient production of virus-like particles (VLPs) from cells9, 10. This protein is highly mobile before its accumulation in cholesterol-rich regions of the membrane, where multimerization initiates the budding process11. A total of 4,000–5,000 GAG molecules is required to form a single particle of about 145 nm (ref. 12). Both VLPs and mature viruses contain a number of host proteins that are recruited by binding to viral proteins. These proteins can either contribute to the infectivity (for example, Cyclophilin/FKBPA13) or act as antiviral proteins preventing the spreading of the virus (for example, APOBEC proteins14).
We here describe the development and application of Virotrap, an elegant co-purification strategy based on the trapping of a bait protein together with its associated protein partners in VLPs that are budded from the cell. After enrichment, these particles can be analysed by targeted (for example, western blotting) or unbiased approaches (MS-based proteomics). Virotrap allows detection of known binary PPIs, analysis of protein complexes and their dynamics, and readily detects protein binders for small molecules.
Concept of the Virotrap system
Classical AP–MS approaches rely on cell homogenization to access protein complexes, a step that can vary significantly with the lysis conditions (detergents, salt concentrations, pH conditions and so on)5. To eliminate the homogenization step in AP–MS, we reasoned that incorporation of a protein complex inside a secreted VLP traps the interaction partners under native conditions and protects them during further purification. We thus explored the possibility of protein complex packaging by the expression of GAG-bait protein chimeras (Fig. 1) as expression of GAG results in the release of VLPs from the cells9, 10. As a first PPI pair to evaluate this concept, we selected the HRAS protein as a bait combined with the RAF1 prey protein. We were able to specifically detect the HRAS–RAF1 interaction following enrichment of VLPs via ultracentrifugation (Supplementary Fig. 1a). To prevent tedious ultracentrifugation steps, we designed a novel single-step protocol wherein we co-express the vesicular stomatitis virus glycoprotein (VSV-G) together with a tagged version of this glycoprotein in addition to the GAG bait and prey. Both tagged and untagged VSV-G proteins are probably presented as trimers on the surface of the VLPs, allowing efficient antibody-based recovery from large volumes. The HRAS–RAF1 interaction was confirmed using this single-step protocol (Supplementary Fig. 1b). No associations with unrelated bait or prey proteins were observed for both protocols.
Figure 1: Schematic representation of the Virotrap strategy.
Expression of a GAG-bait fusion protein (1) results in submembrane multimerization (2) and subsequent budding of VLPs from cells (3). Interaction partners of the bait protein are also trapped within these VLPs and can be identified after purification by western blotting or MS analysis (4).
Virotrap for the detection of binary interactions
We next explored the reciprocal detection of a set of PPI pairs, which were selected based on published evidence and cytosolic localization15. After single-step purification and western blot analysis, we could readily detect reciprocal interactions between CDK2 and CKS1B, LCP2 and GRAP2, and S100A1 and S100B (Fig. 2a). Only for the LCP2 prey we observed nonspecific association with an irrelevant bait construct. However, the particle levels of the GRAP2 bait were substantially lower as compared with those of the GAG control construct (GAG protein levels in VLPs; Fig. 2a, second panel of the LCP2 prey). After quantification of the intensities of bait and prey proteins and normalization of prey levels using bait levels, we observed a strong enrichment for the GAG-GRAP2 bait (Supplementary Fig. 2).
…..
Virotrap for unbiased discovery of novel interactions
For the detection of novel interaction partners, we scaled up VLP production and purification protocols (Supplementary Fig. 5 and Supplementary Note 1 for an overview of the protocol) and investigated protein partners trapped using the following bait proteins: Fas-associated via death domain (FADD), A20 (TNFAIP3), nuclear factor-κB (NF-κB) essential modifier (IKBKG), TRAF family member-associated NF-κB activator (TANK), MYD88 and ring finger protein 41 (RNF41). To obtain specific interactors from the lists of identified proteins, we challenged the data with a combined protein list of 19 unrelated Virotrap experiments (Supplementary Table 1 for an overview). Figure 3 shows the design and the list of candidate interactors obtained after removal of all proteins that were found in the 19 control samples (including removal of proteins from the control list identified with a single peptide). The remaining list of confident protein identifications (identified with at least two peptides in at least two biological repeats) reveals both known and novel candidate interaction partners. All candidate interactors including single peptide protein identifications are given in Supplementary Data 2 and also include recurrent protein identifications of known interactors based on a single peptide; for example, CASP8 for FADD and TANK for NEMO. Using alternative methods, we confirmed the interaction between A20 and FADD, and the associations with transmembrane proteins (insulin receptor and insulin-like growth factor receptor 1) that were captured using RNF41 as a bait (Supplementary Fig. 6). To address the use of Virotrap for the detection of dynamic interactions, we activated the NF-κB pathway via the tumour necrosis factor (TNF) receptor (TNFRSF1A) using TNFα (TNF) and performed Virotrap analysis using A20 as bait (Fig. 3). This resulted in the additional enrichment of receptor-interacting kinase (RIPK1), TNFR1-associated via death domain (TRADD), TNFRSF1A and TNF itself, confirming the expected activated complex20.
Figure 3: Use of Virotrap for unbiased interactome analysis
Lysis conditions used in AP–MS strategies are critical for the preservation of protein complexes. A multitude of lysis conditions have been described, culminating in a recent report where protein complex stability was assessed under 96 lysis/purification protocols5. Moreover, the authors suggest to optimize the conditions for every complex, implying an important workload for researchers embarking on protein complex analysis using classical AP–MS. As lysis results in a profound change of the subcellular context and significantly alters the concentration of proteins, loss of complex integrity during a classical AP–MS protocol can be expected. A clear evolution towards ‘lysis-independent’ approaches in the co-complex analysis field is evident with the introduction of BioID6 and APEX25 where proximal proteins, including proteins residing in the complex, are labelled with biotin by an enzymatic activity fused to a bait protein. A side-by-side comparison between classical AP–MS and BioID showed overlapping and unique candidate binding proteins for both approaches7, 8, supporting the notion that complementary methods are needed to provide a comprehensive view on protein complexes. This has also been clearly demonstrated for binary approaches15 and is a logical consequence of the heterogenic nature underlying PPIs (binding mechanism, requirement for posttranslational modifications, location, affinity and so on).
In this report, we explore an alternative, yet complementary method to isolate protein complexes without interfering with cellular integrity. By trapping protein complexes in the protective environment of a virus-like shell, the intact complexes are preserved during the purification process. This constitutes a new concept in co-complex analysis wherein complex stability is physically guaranteed by a protective, physical structure. A comparison of our Virotrap approach with AP–MS shows complementary data, with specific false positives and false negatives for both methods (Supplementary Fig. 7).
The current implementation of the Virotrap platform implies the use of a GAG-bait construct resulting in considerable expression of the bait protein. Different strategies are currently pursued to reduce bait expression including co-expression of a native GAG protein together with the GAG-bait protein, not only reducing bait expression but also creating more ‘space’ in the particles potentially accommodating larger bait protein complexes. Nevertheless, the presence of the bait on the forming GAG scaffold creates an intracellular affinity matrix (comparable to the early in vitro affinity columns for purification of interaction partners from lysates26) that has the potential to compete with endogenous complexes by avidity effects. This avidity effect is a powerful mechanism that aids in the recruitment of cyclophilin to GAG27, a well-known weak interaction (Kd=16 μM (ref. 28)) detectable as a background association in the Virotrap system. Although background binding may be increased by elevated bait expression, weaker associations are readily detectable (for example, MAL—MYD88-binding study; Fig. 2c).
The size of Virotrap particles (around 145 nm) suggests limitations in the size of the protein complex that can be accommodated in the particles. Further experimentation is required to define the maximum size of proteins or the number of protein complexes that can be trapped inside the particles.
….
In conclusion, Virotrap captures significant parts of known interactomes and reveals new interactions. This cell lysis-free approach purifies protein complexes under native conditions and thus provides a powerful method to complement AP–MS or other PPI data. Future improvements of the system include strategies to reduce bait expression to more physiological levels and application of advanced data analysis options to filter out background. These developments can further aid in the deployment of Virotrap as a powerful extension of the current co-complex technology arsenal.
New Autism Blood Biomarker Identified
Researchers at UT Southwestern Medical Center have identified a blood biomarker that may aid in earlier diagnosis of children with autism spectrum disorder, or ASD
In a recent edition of Scientific Reports, UT Southwestern researchers reported on the identification of a blood biomarker that could distinguish the majority of ASD study participants versus a control group of similar age range. In addition, the biomarker was significantly correlated with the level of communication impairment, suggesting that the blood test may give insight into ASD severity.
“Numerous investigators have long sought a biomarker for ASD,” said Dr. Dwight German, study senior author and Professor of Psychiatry at UT Southwestern. “The blood biomarker reported here along with others we are testing can represent a useful test with over 80 percent accuracy in identifying ASD.”
ASD1 – was 66 percent accurate in diagnosing ASD. When combined with thyroid stimulating hormone level measurements, the ASD1-binding biomarker was 73 percent accurate at diagnosis
A Search for Blood Biomarkers for Autism: Peptoids
Autism spectrum disorder (ASD) is a neurodevelopmental disorder characterized by impairments in social interaction and communication, and restricted, repetitive patterns of behavior. In order to identify individuals with ASD and initiate interventions at the earliest possible age, biomarkers for the disorder are desirable. Research findings have identified widespread changes in the immune system in children with autism, at both systemic and cellular levels. In an attempt to find candidate antibody biomarkers for ASD, highly complex libraries of peptoids (oligo-N-substituted glycines) were screened for compounds that preferentially bind IgG from boys with ASD over typically developing (TD) boys. Unexpectedly, many peptoids were identified that preferentially bound IgG from TD boys. One of these peptoids was studied further and found to bind significantly higher levels (>2-fold) of the IgG1 subtype in serum from TD boys (n = 60) compared to ASD boys (n = 74), as well as compared to older adult males (n = 53). Together these data suggest that ASD boys have reduced levels (>50%) of an IgG1 antibody, which resembles the level found normally with advanced age. In this discovery study, the ASD1 peptoid was 66% accurate in predicting ASD.
….
Peptoid libraries have been used previously to search for autoantibodies for neurodegenerative diseases19 and for systemic lupus erythematosus (SLE)21. In the case of SLE, peptoids were identified that could identify subjects with the disease and related syndromes with moderate sensitivity (70%) and excellent specificity (97.5%). Peptoids were used to measure IgG levels from both healthy subjects and SLE patients. Binding to the SLE-peptoid was significantly higher in SLE patients vs. healthy controls. The IgG bound to the SLE-peptoid was found to react with several autoantigens, suggesting that the peptoids are capable of interacting with multiple, structurally similar molecules. These data indicate that IgG binding to peptoids can identify subjects with high levels of pathogenic autoantibodies vs. a single antibody.
In the present study, the ASD1 peptoid binds significantly lower levels of IgG1 in ASD males vs. TD males. This finding suggests that the ASD1 peptoid recognizes antibody(-ies) of an IgG1 subtype that is (are) significantly lower in abundance in the ASD males vs. TD males. Although a previous study14 has demonstrated lower levels of plasma IgG in ASD vs. TD children, here, we additionally quantified serum IgG levels in our individuals and found no difference in IgG between the two groups (data not shown). Furthermore, our IgG levels did not correlate with ASD1 binding levels, indicating that ASD1 does not bind IgG generically, and that the peptoid’s ability to differentiate between ASD and TD males is related to a specific antibody(-ies).
ASD subjects underwent a diagnostic evaluation using the ADOS and ADI-R, and application of the DSM-IV criteria prior to study inclusion. Only those subjects with a diagnosis of Autistic Disorder were included in the study. The ADOS is a semi-structured observation of a child’s behavior that allows examiners to observe the three core domains of ASD symptoms: reciprocal social interaction, communication, and restricted and repetitive behaviors1. When ADOS subdomain scores were compared with peptoid binding, the only significant relationship was with Social Interaction. However, the positive correlation would suggest that lower peptoid binding is associated with better social interaction, not poorer social interaction as anticipated.
The ADI-R is a structured parental interview that measures the core features of ASD symptoms in the areas of reciprocal social interaction, communication and language, and patterns of behavior. Of the three ADI-R subdomains, only the Communication domain was related to ASD1 peptoid binding, and this correlation was negative suggesting that low peptoid binding is associated with greater communication problems. These latter data are similar to the findings of Heuer et al.14 who found that children with autism with low levels of plasma IgG have high scores on the Aberrant Behavior Checklist (p < 0.0001). Thus, peptoid binding to IgG1 may be useful as a severity marker for ASD allowing for further characterization of individuals, but further research is needed.
It is interesting that in serum samples from older men, the ASD1 binding is similar to that in the ASD boys. This is consistent with the observation that with aging there is a reduction in the strength of the immune system, and the changes are gender-specific25. Recent studies using parabiosis26, in which blood from young mice reverse age-related impairments in cognitive function and synaptic plasticity in old mice, reveal that blood constituents from young subjects may contain important substances for maintaining neuronal functions. Work is in progress to identify the antibody/antibodies that are differentially binding to the ASD1 peptoid, which appear as a single band on the electrophoresis gel (Fig. 4).
……..
The ADI-R is a structured parental interview that measures the core features of ASD symptoms in the areas of reciprocal social interaction, communication and language, and patterns of behavior. Of the three ADI-R subdomains, only the Communication domain was related to ASD1 peptoid binding, and this correlation was negative suggesting that low peptoid binding is associated with greater communication problems. These latter data are similar to the findings of Heuer et al.14 who found that children with autism with low levels of plasma IgG have high scores on the Aberrant Behavior Checklist (p < 0.0001). Thus, peptoid binding to IgG1 may be useful as a severity marker for ASD allowing for further characterization of individuals, but further research is needed.
Titration of IgG binding to ASD1 using serum pooled from 10 TD males and 10 ASD males demonstrates ASD1’s ability to differentiate between the two groups. (B)Detecting IgG1 subclass instead of total IgG amplifies this differentiation. (C) IgG1 binding of individual ASD (n=74) and TD (n=60) male serum samples (1:100 dilution) to ASD1 significantly differs with TD>ASD. In addition, IgG1 binding of older adult male (AM) serum samples (n=53) to ASD1 is significantly lower than TD males, and not different from ASD males. The three groups were compared with a Kruskal-Wallis ANOVA, H = 10.1781, p<0.006. **p<0.005. Error bars show SEM. (D) Receiver-operating characteristic curve for ASD1’s ability to discriminate between ASD and TD males.
Association between peptoid binding and ADOS and ADI-R subdomains
Higher scores in any domain on the ADOS and ADI-R are indicative of more abnormal behaviors and/or symptoms. Among ADOS subdomains, there was no significant relationship between Communication and peptoid binding (z = 0.04, p = 0.966), Communication + Social interaction (z = 1.53, p = 0.127), or Stereotyped Behaviors and Restrictive Interests (SBRI) (z = 0.46, p = 0.647). Higher scores on the Social Interaction domain were significantly associated with higher peptoid binding (z = 2.04, p = 0.041).
Among ADI-R subdomains, higher scores on the Communication domain were associated with lower levels of peptoid binding (z = −2.28, p = 0.023). There was not a significant relationship between Social Interaction (z = 0.07, p = 0.941) or Restrictive/Repetitive Stereotyped Behaviors (z = −1.40, p = 0.162) and peptoid binding.
Computational Model Finds New Protein-Protein Interactions
Researchers at University of Pittsburgh have discovered 500 new protein-protein interactions (PPIs) associated with genes linked to schizophrenia.
Using a computational model they developed, researchers at the University of Pittsburgh School of Medicine have discovered more than 500 new protein-protein interactions (PPIs) associated with genes linked to schizophrenia. The findings, published online in npj Schizophrenia, a Nature Publishing Group journal, could lead to greater understanding of the biological underpinnings of this mental illness, as well as point the way to treatments.
There have been many genome-wide association studies (GWAS) that have identified gene variants associated with an increased risk for schizophrenia, but in most cases there is little known about the proteins that these genes make, what they do and how they interact, said senior investigator Madhavi Ganapathiraju, Ph.D., assistant professor of biomedical informatics, Pitt School of Medicine.
“GWAS studies and other research efforts have shown us what genes might be relevant in schizophrenia,” she said. “What we have done is the next step. We are trying to understand how these genes relate to each other, which could show us the biological pathways that are important in the disease.”
Each gene makes proteins and proteins typically interact with each other in a biological process. Information about interacting partners can shed light on the role of a gene that has not been studied, revealing pathways and biological processes associated with the disease and also its relation to other complex diseases.
Dr. Ganapathiraju’s team developed a computational model called High-Precision Protein Interaction Prediction (HiPPIP) and applied it to discover PPIs of schizophrenia-linked genes identified through GWAS, as well as historically known risk genes. They found 504 never-before known PPIs, and noted also that while schizophrenia-linked genes identified historically and through GWAS had little overlap, the model showed they shared more than 100 common interactors.
“We can infer what the protein might do by checking out the company it keeps,” Dr. Ganapathiraju explained. “For example, if I know you have many friends who play hockey, it could mean that you are involved in hockey, too. Similarly, if we see that an unknown protein interacts with multiple proteins involved in neural signaling, for example, there is a high likelihood that the unknown entity also is involved in the same.”
Dr. Ganapathiraju and colleagues have drawn such inferences on protein function based on the PPIs of proteins, and made their findings available on a website Schizo-Pi. This information can be used by biologists to explore the schizophrenia interactome with the aim of understanding more about the disease or developing new treatment drugs.
Schizophrenia interactome with 504 novel protein–protein interactions
(GWAS) have revealed the role of rare and common genetic variants, but the functional effects of the risk variants remain to be understood. Protein interactome-based studies can facilitate the study of molecular mechanisms by which the risk genes relate to schizophrenia (SZ) genesis, but protein–protein interactions (PPIs) are unknown for many of the liability genes. We developed a computational model to discover PPIs, which is found to be highly accurate according to computational evaluations and experimental validations of selected PPIs. We present here, 365 novel PPIs of liability genes identified by the SZ Working Group of the Psychiatric Genomics Consortium (PGC). Seventeen genes that had no previously known interactions have 57 novel interactions by our method. Among the new interactors are 19 drug targets that are targeted by 130 drugs. In addition, we computed 147 novel PPIs of 25 candidate genes investigated in the pre-GWAS era. While there is little overlap between the GWAS genes and the pre-GWAS genes, the interactomes reveal that they largely belong to the same pathways, thus reconciling the apparent disparities between the GWAS and prior gene association studies. The interactome including 504 novel PPIs overall, could motivate other systems biology studies and trials with repurposed drugs. The PPIs are made available on a webserver, called Schizo-Pi at http://severus.dbmi.pitt.edu/schizo-pi with advanced search capabilities.
Schizophrenia (SZ) is a common, potentially severe psychiatric disorder that afflicts all populations.1 Gene mapping studies suggest that SZ is a complex disorder, with a cumulative impact of variable genetic effects coupled with environmental factors.2 As many as 38 genome-wide association studies (GWAS) have been reported on SZ out of a total of 1,750 GWAS publications on 1,087 traits or diseases reported in the GWAS catalog maintained by the National Human Genome Research Institute of USA3 (as of April 2015), revealing the common variants associated with SZ.4 The SZ Working Group of the Psychiatric Genomics Consortium (PGC) identified 108 genetic loci that likely confer risk for SZ.5 While the role of genetics has been clearly validated by this study, the functional impact of the risk variants is not well-understood.6,7 Several of the genes implicated by the GWAS have unknown functions and could participate in possibly hitherto unknown pathways.8 Further, there is little or no overlap between the genes identified through GWAS and ‘candidate genes’ proposed in the pre-GWAS era.9
Interactome-based studies can be useful in discovering the functional associations of genes. For example,disrupted in schizophrenia 1 (DISC1), an SZ related candidate gene originally had no known homolog in humans. Although it had well-characterized protein domains such as coiled-coil domains and leucine-zipper domains, its function was unknown.10,11 Once its protein–protein interactions (PPIs) were determined using yeast 2-hybrid technology,12 investigators successfully linked DISC1 to cAMP signaling, axon elongation, and neuronal migration, and accelerated the research pertaining to SZ in general, and DISC1 in particular.13 Typically such studies are carried out on known protein–protein interaction (PPI) networks, or as in the case of DISC1, when there is a specific gene of interest, its PPIs are determined by methods such as yeast 2-hybrid technology.
Knowledge of human PPI networks is thus valuable for accelerating discovery of protein function, and indeed, biomedical research in general. However, of the hundreds of thousands of biophysical PPIs thought to exist in the human interactome,14,15 <100,000 are known today (Human Protein Reference Database, HPRD16 and BioGRID17 databases). Gold standard experimental methods for the determination of all the PPIs in human interactome are time-consuming, expensive and may not even be feasible, as about 250 million pairs of proteins would need to be tested overall; high-throughput methods such as yeast 2-hybrid have important limitations for whole interactome determination as they have a low recall of 23% (i.e., remaining 77% of true interactions need to be determined by other means), and a low precision (i.e., the screens have to be repeated multiple times to achieve high selectivity).18,19Computational methods are therefore necessary to complete the interactome expeditiously. Algorithms have begun emerging to predict PPIs using statistical machine learning on the characteristics of the proteins, but these algorithms are employed predominantly to study yeast. Two significant computational predictions have been reported for human interactome; although they have had high false positive rates, these methods have laid the foundation for computational prediction of human PPIs.20,21
We have created a new PPI prediction model called High-Confidence Protein–Protein Interaction Prediction (HiPPIP) model. Novel interactions predicted with this model are making translational impact. For example, we discovered a PPI between OASL and DDX58, which on validation showed that an increased expression of OASL could boost innate immunity to combat influenza by activating the RIG-I pathway.22 Also, the interactome of the genes associated with congenital heart disease showed that the disease morphogenesis has a close connection with the structure and function of cilia.23Here, we describe the HiPPIP model and its application to SZ genes to construct the SZ interactome. After computational evaluations and experimental validations of selected novel PPIs, we present here 504 highly confident novel PPIs in the SZ interactome, shedding new light onto several uncharacterized genes that are associated with SZ.
We developed a computational model called HiPPIP to predict PPIs (see Methods and Supplementary File 1). The model has been evaluated by computational methods and experimental validations and is found to be highly accurate. Evaluations on a held-out test data showed a precision of 97.5% and a recall of 5%. 5% recall out of 150,000 to 600,000 estimated number of interactions in the human interactome corresponds to 7,500–30,000 novel PPIs in the whole interactome. Note that, it is likely that the real precision would be higher than 97.5% because in this test data, randomly paired proteins are treated as non-interacting protein pairs, whereas some of them may actually be interacting pairs with a small probability; thus, some of the pairs that are treated as false positives in test set are likely to be true but hitherto unknown interactions. In Figure 1a, we show the precision versus recall of our method on ‘hub proteins’ where we considered all pairs that received a score >0.5 by HiPPIP to be novel interactions. In Figure 1b, we show the number of true positives versus false positives observed in hub proteins. Both these figures also show our method to be superior in comparison to the prediction of membrane-receptor interactome by Qi et al’s.24 True positives versus false positives are also shown for individual hub proteins by our method in Figure 1cand by Qi et al’s.23 in Figure 1d. These evaluations showed that our predictions contain mostly true positives. Unlike in other domains where ranked lists are commonly used such as information retrieval, in PPI prediction the ‘false positives’ may actually be unlabeled instances that are indeed true interactions that are not yet discovered. In fact, such unlabeled pairs predicted as interactors of the hub gene HMGB1 (namely, the pairs HMGB1-KL and HMGB1-FLT1) were validated by experimental methods and found to be true PPIs (See the Figures e–g inSupplementary File 3). Thus, we concluded that the protein pairs that received a score of ⩾0.5 are highly confident to be true interactions. The pairs that receive a score less than but close to 0.5 (i.e., in the range of 0.4–0.5) may also contain several true PPIs; however, we cannot confidently say that all in this range are true PPIs. Only the PPIs predicted with a score >0.5 are included in the interactome.
Computational evaluation of predicted protein–protein interactions on hub proteins: (a) precision recall curve. (b) True positive versus false positives in ranked lists of hub type membrane receptors for our method and that by Qi et al. True positives versus false positives are shown for individual membrane receptors by our method in (c) and by Qi et al. in (d). Thick line is the average, which is also the same as shown in (b). Note:x-axis is recall in (a), whereas it is number of false positives in (b–d). The range of y-axis is observed by varying the threshold from 1.0–0 in (a), and to 0.5 in (b–d).
SZ interactome
By applying HiPPIP to the GWAS genes and Historic (pre-GWAS) genes, we predicted over 500 high confidence new PPIs adding to about 1400 previously known PPIs.
Schizophrenia interactome: network view of the schizophrenia interactome is shown as a graph, where genes are shown as nodes and PPIs as edges connecting the nodes. Schizophrenia-associated genes are shown as dark blue nodes, novel interactors as red color nodes and known interactors as blue color nodes. The source of the schizophrenia genes is indicated by its label font, where Historic genes are shown italicized, GWAS genes are shown in bold, and the one gene that is common to both is shown in italicized and bold. For clarity, the source is also indicated by the shape of the node (triangular for GWAS and square for Historic and hexagonal for both). Symbols are shown only for the schizophrenia-associated genes; actual interactions may be accessed on the web. Red edges are the novel interactions, whereas blue edges are known interactions. GWAS, genome-wide association studies of schizophrenia; PPI, protein–protein interaction.
We have made the known and novel interactions of all SZ-associated genes available on a webserver called Schizo-Pi, at the addresshttp://severus.dbmi.pitt.edu/schizo-pi. This webserver is similar to Wiki-Pi33 which presents comprehensive annotations of both participating proteins of a PPI side-by-side. The difference between Wiki-Pi which we developed earlier, and Schizo-Pi, is the inclusion of novel predicted interactions of the SZ genes into the latter.
Despite the many advances in biomedical research, identifying the molecular mechanisms underlying the disease is still challenging. Studies based on protein interactions were proven to be valuable in identifying novel gene associations that could shed new light on disease pathology.35 The interactome including more than 500 novel PPIs will help to identify pathways and biological processes associated with the disease and also its relation to other complex diseases. It also helps identify potential drugs that could be repurposed to use for SZ treatment.
Functional and pathway enrichment in SZ interactome
When a gene of interest has little known information, functions of its interacting partners serve as a starting point to hypothesize its own function. We computed statistically significant enrichment of GO biological process terms among the interacting partners of each of the genes using BinGO36 (see online at http://severus.dbmi.pitt.edu/schizo-pi).
Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution
Massimo Stefani · Christopher M. Dobson
Abstract The deposition of proteins in the form of amyloid fibrils and plaques is the characteristic feature of more than 20 degenerative conditions affecting either the central nervous system or a variety of peripheral tissues. As these conditions include Alzheimer’s, Parkinson’s and the prion diseases, several forms of fatal systemic amyloidosis, and at least one condition associated with medical intervention (haemodialysis), they are of enormous importance in the context of present-day human health and welfare. Much remains to be learned about the mechanism by which the proteins associated with these diseases aggregate and form amyloid structures, and how the latter affect the functions of the organs with which they are associated. A great deal of information concerning these diseases has emerged, however, during the past 5 years, much of it causing a number of fundamental assumptions about the amyloid diseases to be reexamined. For example, it is now apparent that the ability to form amyloid structures is not an unusual feature of the small number of proteins associated with these diseases but is instead a general property of polypeptide chains. It has also been found recently that aggregates of proteins not associated with amyloid diseases can impair the ability of cells to function to a similar extent as aggregates of proteins linked with specific neurodegenerative conditions. Moreover, the mature amyloid fibrils or plaques appear to be substantially less toxic than the prefibrillar aggregates that are their precursors. The toxicity of these early aggregates appears to result from an intrinsic ability to impair fundamental cellular processes by interacting with cellular membranes, causing oxidative stress and increases in free Ca2+ that eventually lead to apoptotic or necrotic cell death. The ‘new view’ of these diseases also suggests that other degenerative conditions could have similar underlying origins to those of the amyloidoses. In addition, cellular protection mechanisms, such as molecular chaperones and the protein degradation machinery, appear to be crucial in the prevention of disease in normally functioning living organisms. It also suggests some intriguing new factors that could be of great significance in the evolution of biological molecules and the mechanisms that regulate their behaviour.
The genetic information within a cell encodes not only the specific structures and functions of proteins but also the way these structures are attained through the process known as protein folding. In recent years many of the underlying features of the fundamental mechanism of this complex process and the manner in which it is regulated in living systems have emerged from a combination of experimental and theoretical studies [1]. The knowledge gained from these studies has also raised a host of interesting issues. It has become apparent, for example, that the folding and unfolding of proteins is associated with a whole range of cellular processes from the trafficking of molecules to specific organelles to the regulation of the cell cycle and the immune response. Such observations led to the inevitable conclusion that the failure to fold correctly, or to remain correctly folded, gives rise to many different types of biological malfunctions and hence to many different forms of disease [2]. In addition, it has been recognised recently that a large number of eukaryotic genes code for proteins that appear to be ‘natively unfolded’, and that proteins can adopt, under certain circumstances, highly organised multi-molecular assemblies whose structures are not specifically encoded in the amino acid sequence. Both these observations have raised challenging questions about one of the most fundamental principles of biology: the close relationship between the sequence, structure and function of proteins, as we discuss below [3].
It is well established that proteins that are ‘misfolded’, i.e. that are not in their functionally relevant conformation, are devoid of normal biological activity. In addition, they often aggregate and/or interact inappropriately with other cellular components leading to impairment of cell viability and eventually to cell death. Many diseases, often known as misfolding or conformational diseases, ultimately result from the presence in a living system of protein molecules with structures that are ‘incorrect’, i.e. that differ from those in normally functioning organisms [4]. Such diseases include conditions in which a specific protein, or protein complex, fails to fold correctly (e.g. cystic fibrosis, Marfan syndrome, amyotonic lateral sclerosis) or is not sufficiently stable to perform its normal function (e.g. many forms of cancer). They also include conditions in which aberrant folding behaviour results in the failure of a protein to be correctly trafficked (e.g. familial hypercholesterolaemia, α1-antitrypsin deficiency, and some forms of retinitis pigmentosa) [4]. The tendency of proteins to aggregate, often to give species extremely intractable to dissolution and refolding, is of course also well known in other circumstances. Examples include the formation of inclusion bodies during overexpression of heterologous proteins in bacteria and the precipitation of proteins during laboratory purification procedures. Indeed, protein aggregation is well established as one of the major difficulties associated with the production and handling of proteins in the biotechnology and pharmaceutical industries [5].
Considerable attention is presently focused on a group of protein folding diseases known as amyloidoses. In these diseases specific peptides or proteins fail to fold or to remain correctly folded and then aggregate (often with other components) so as to give rise to ‘amyloid’ deposits in tissue. Amyloid structures can be recognised because they possess a series of specific tinctorial and biophysical characteristics that reflect a common core structure based on the presence of highly organised βsheets [6]. The deposits in strictly defined amyloidoses are extracellular and can often be observed as thread-like fibrillar structures, sometimes assembled further into larger aggregates or plaques. These diseases include a range of sporadic, familial or transmissible degenerative diseases, some of which affect the brain and the central nervous system (e.g. Alzheimer’s and Creutzfeldt-Jakob diseases), while others involve peripheral tissues and organs such as the liver, heart and spleen (e.g. systemic amyloidoses and type II diabetes) [7, 8]. In other forms of amyloidosis, such as primary or secondary systemic amyloidoses, proteinaceous deposits are found in skeletal tissue and joints (e.g. haemodialysis-related amyloidosis) as well as in several organs (e.g. heart and kidney). Yet other components such as collagen, glycosaminoglycans and proteins (e.g. serum amyloid protein) are often present in the deposits protecting them against degradation [9, 10, 11]. Similar deposits to those in the amyloidoses are, however, found intracellularly in other diseases; these can be localised either in the cytoplasm, in the form of specialised aggregates known as aggresomes or as Lewy or Russell bodies or in the nucleus (see below).
The presence in tissue of proteinaceous deposits is a hallmark of all these diseases, suggesting a causative link between aggregate formation and pathological symptoms (often known as the amyloid hypothesis) [7, 8, 12]. At the present time the link between amyloid formation and disease is widely accepted on the basis of a large number of biochemical and genetic studies. The specific nature of the pathogenic species, and the molecular basis of their ability to damage cells, are however, the subject of intense debate [13, 14, 15, 16, 17, 18, 19, 20]. In neurodegenerative disorders it is very likely that the impairment of cellular function follows directly from the interactions of the aggregated proteins with cellular components [21, 22]. In the systemic non-neurological diseases, however, it is widely believed that the accumulation in vital organs of large amounts of amyloid deposits can by itself cause at least some of the clinical symptoms [23]. It is quite possible, however, that there are other more specific effects of aggregates on biochemical processes even in these diseases. The presence of extracellular or intracellular aggregates of a specific polypeptide molecule is a characteristic of all the 20 or so recognised amyloid diseases. The polypeptides involved include full length proteins (e.g. lysozyme or immunoglobulin light chains), biological peptides (amylin, atrial natriuretic factor) and fragments of larger proteins produced as a result of specific processing (e.g. the Alzheimer βpeptide) or of more general degradation [e.g. poly(Q) stretches cleaved from proteins with poly(Q) extensions such as huntingtin, ataxins and the androgen receptor]. The peptides and proteins associated with known amyloid diseases are listed in Table 1. In some cases the proteins involved have wild type sequences, as in sporadic forms of the diseases, but in other cases these are variants resulting from genetic mutations associated with familial forms of the diseases. In some cases both sporadic and familial diseases are associated with a given protein; in this case the mutational variants are usually associated with early-onset forms of the disease. In the case of the neurodegenerative diseases associated with the prion protein some forms of the diseases are transmissible. The existence of familial forms of a number of amyloid diseases has provided significant clues to the origins of the pathologies. For example, there are increasingly strong links between the age at onset of familial forms of disease and the effects of the mutations involved on the propensity of the affected proteins to aggregate in vitro. Such findings also support the link between the process of aggregation and the clinical manifestations of disease [24, 25].
The presence in cells of misfolded or aggregated proteins triggers a complex biological response. In the cytosol, this is referred to as the ‘heat shock response’ and in the endoplasmic reticulum (ER) it is known as the ‘unfolded protein response’. These responses lead to the expression, among others, of the genes for heat shock proteins (Hsp, or molecular chaperone proteins) and proteins involved in the ubiquitin-proteasome pathway [26]. The evolution of such complex biochemical machinery testifies to the fact that it is necessary for cells to isolate and clear rapidly and efficiently any unfolded or incorrectly folded protein as soon as it appears. In itself this fact suggests that these species could have a generally adverse effect on cellular components and cell viability. Indeed, it was a major step forward in understanding many aspects of cell biology when it was recognised that proteins previously associated only with stress, such as heat shock, are in fact crucial in the normal functioning of living systems. This advance, for example, led to the discovery of the role of molecular chaperones in protein folding and in the normal ‘housekeeping’ processes that are inherent in healthy cells [27, 28]. More recently a number of degenerative diseases, both neurological and systemic, have been linked to, or shown to be affected by, impairment of the ubiquitin-proteasome pathway (Table 2). The diseases are primarily associated with a reduction in either the expression or the biological activity of Hsps, ubiquitin, ubiquitinating or deubiquitinating enzymes and the proteasome itself, as we show below [29, 30, 31, 32], or even to the failure of the quality control mechanisms that ensure proper maturation of proteins in the ER. The latter normally leads to degradation of a significant proportion of polypeptide chains before they have attained their native conformations through retrograde translocation to the cytosol [33, 34].
….
It is now well established that the molecular basis of protein aggregation into amyloid structures involves the existence of ‘misfolded’ forms of proteins, i.e. proteins that are not in the structures in which they normally function in vivo or of fragments of proteins resulting from degradation processes that are inherently unable to fold [4, 7, 8, 36]. Aggregation is one of the common consequences of a polypeptide chain failing to reach or maintain its functional three-dimensional structure. Such events can be associated with specific mutations, misprocessing phenomena, aberrant interactions with metal ions, changes in environmental conditions, such as pH or temperature, or chemical modification (oxidation, proteolysis). Perturbations in the conformational properties of the polypeptide chain resulting from such phenomena may affect equilibrium 1 in Fig. 1 increasing the population of partially unfolded, or misfolded, species that are much more aggregation-prone than the native state.
Fig. 1 Overview of the possible fates of a newly synthesised polypeptide chain. The equilibrium ① between the partially folded molecules and the natively folded ones is usually strongly in favour of the latter except as a result of specific mutations, chemical modifications or partially destabilising solution conditions. The increased equilibrium populations of molecules in the partially or completely unfolded ensemble of structures are usually degraded by the proteasome; when this clearance mechanism is impaired, such species often form disordered aggregates or shift equilibrium ② towards the nucleation of pre-fibrillar assemblies that eventually grow into mature fibrils (equilibrium ③). DANGER! indicates that pre-fibrillar aggregates in most cases display much higher toxicity than mature fibrils. Heat shock proteins (Hsp) can suppress the appearance of pre-fibrillar assemblies by minimising the population of the partially folded molecules by assisting in the correct folding of the nascent chain and the unfolded protein response target incorrectly folded proteins for degradation.
……
Little is known at present about the detailed arrangement of the polypeptide chains themselves within amyloid fibrils, either those parts involved in the core βstrands or in regions that connect the various β-strands. Recent data suggest that the sheets are relatively untwisted and may in some cases at least exist in quite specific supersecondary structure motifs such as β-helices [6, 40] or the recently proposed µ-helix [41]. It seems possible that there may be significant differences in the way the strands are assembled depending on characteristics of the polypeptide chain involved [6, 42]. Factors including length, sequence (and in some cases the presence of disulphide bonds or post-translational modifications such as glycosylation) may be important in determining details of the structures. Several recent papers report structural models for amyloid fibrils containing different polypeptide chains, including the Aβ40 peptide, insulin and fragments of the prion protein, based on data from such techniques as cryo-electron microscopy and solid-state magnetic resonance spectroscopy [43, 44]. These models have much in common and do indeed appear to reflect the fact that the structures of different fibrils are likely to be variations on a common theme [40]. It is also emerging that there may be some common and highly organised assemblies of amyloid protofilaments that are not simply extended threads or ribbons. It is clear, for example, that in some cases large closed loops can be formed [45, 46, 47], and there may be specific types of relatively small spherical or ‘doughnut’ shaped structures that can result in at least some circumstances (see below).
…..
The similarity of some early amyloid aggregates with the pores resulting from oligomerisation of bacterial toxins and pore-forming eukaryotic proteins (see below) also suggest that the basic mechanism of protein aggregation into amyloid structures may not only be associated with diseases but in some cases could result in species with functional significance. Recent evidence indicates that a variety of micro-organisms may exploit the controlled aggregation of specific proteins (or their precursors) to generate functional structures. Examples include bacterial curli [52] and proteins of the interior fibre cells of mammalian ocular lenses, whose β-sheet arrays seem to be organised in an amyloid-like supramolecular order [53]. In this case the inherent stability of amyloid-like protein structure may contribute to the long-term structural integrity and transparency of the lens. Recently it has been hypothesised that amyloid-like aggregates of serum amyloid A found in secondary amyloidoses following chronic inflammatory diseases protect the host against bacterial infections by inducing lysis of bacterial cells [54]. One particularly interesting example is a ‘misfolded’ form of the milk protein α-lactalbumin that is formed at low pH and trapped by the presence of specific lipid molecules [55]. This form of the protein has been reported to trigger apoptosis selectively in tumour cells providing evidence for its importance in protecting infants from certain types of cancer [55]. ….
Amyloid formation is a generic property of polypeptide chains ….
It is clear that the presence of different side chains can influence the details of amyloid structures, particularly the assembly of protofibrils, and that they give rise to the variations on the common structural theme discussed above. More fundamentally, the composition and sequence of a peptide or protein affects profoundly its propensity to form amyloid structures under given conditions (see below).
Because the formation of stable protein aggregates of amyloid type does not normally occur in vivo under physiological conditions, it is likely that the proteins encoded in the genomes of living organisms are endowed with structural adaptations that mitigate against aggregation under these conditions. A recent survey involving a large number of structures of β-proteins highlights several strategies through which natural proteins avoid intermolecular association of β-strands in their native states [65]. Other surveys of protein databases indicate that nature disfavours sequences of alternating polar and nonpolar residues, as well as clusters of several consecutive hydrophobic residues, both of which enhance the tendency of a protein to aggregate prior to becoming completely folded [66, 67].
……
Precursors of amyloid fibrils can be toxic to cells
It was generally assumed until recently that the proteinaceous aggregates most toxic to cells are likely to be mature amyloid fibrils, the form of aggregates that have been commonly detected in pathological deposits. It therefore appeared probable that the pathogenic features underlying amyloid diseases are a consequence of the interaction with cells of extracellular deposits of aggregated material. As well as forming the basis for understanding the fundamental causes of these diseases, this scenario stimulated the exploration of therapeutic approaches to amyloidoses that focused mainly on the search for molecules able to impair the growth and deposition of fibrillar forms of aggregated proteins. ….
Structural basis and molecular features of amyloid toxicity
The presence of toxic aggregates inside or outside cells can impair a number of cell functions that ultimately lead to cell death by an apoptotic mechanism [95, 96]. Recent research suggests, however, that in most cases initial perturbations to fundamental cellular processes underlie the impairment of cell function induced by aggregates of disease-associated polypeptides. Many pieces of data point to a central role of modifications to the intracellular redox status and free Ca2+ levels in cells exposed to toxic aggregates [45, 89, 97, 98, 99, 100, 101]. A modification of the intracellular redox status in such cells is associated with a sharp increase in the quantity of reactive oxygen species (ROS) that is reminiscent of the oxidative burst by which leukocytes destroy invading foreign cells after phagocytosis. In addition, changes have been observed in reactive nitrogen species, lipid peroxidation, deregulation of NO metabolism [97], protein nitrosylation [102] and upregulation of heme oxygenase-1, a specific marker of oxidative stress [103]. ….
Results have recently been reported concerning the toxicity towards cultured cells of aggregates of poly(Q) peptides which argues against a disease mechanism based on specific toxic features of the aggregates. These results indicate that there is a close relationship between the toxicity of proteins with poly(Q) extensions and their nuclear localisation. In addition they support the hypotheses that the toxicity of poly(Q) aggregates can be a consequence of altered interactions with nuclear coactivator or corepressor molecules including p53, CBP, Sp1 and TAF130 or of the interaction with transcription factors and nuclear coactivators, such as CBP, endowed with short poly(Q) stretches ([95] and references therein)…..
Concluding remarks
The data reported in the past few years strongly suggest that the conversion of normally soluble proteins into amyloid fibrils and the toxicity of small aggregates appearing during the early stages of the formation of the latter are common or generic features of polypeptide chains. Moreover, the molecular basis of this toxicity also appears to display common features between the different systems that have so far been studied. The ability of many, perhaps all, natural polypeptides to ‘misfold’ and convert into toxic aggregates under suitable conditions suggests that one of the most important driving forces in the evolution of proteins must have been the negative selection against sequence changes that increase the tendency of a polypeptide chain to aggregate. Nevertheless, as protein folding is a stochastic process, and no such process can be completely infallible, misfolded proteins or protein folding intermediates in equilibrium with the natively folded molecules must continuously form within cells. Thus mechanisms to deal with such species must have co-evolved with proteins. Indeed, it is clear that misfolding, and the associated tendency to aggregate, is kept under control by molecular chaperones, which render the resulting species harmless assisting in their refolding, or triggering their degradation by the cellular clearance machinery [166, 167, 168, 169, 170, 171, 172, 173, 175, 177, 178].
Misfolded and aggregated species are likely to owe their toxicity to the exposure on their surfaces of regions of proteins that are buried in the interior of the structures of the correctly folded native states. The exposure of large patches of hydrophobic groups is likely to be particularly significant as such patches favour the interaction of the misfolded species with cell membranes [44, 83, 89, 90, 91, 93]. Interactions of this type are likely to lead to the impairment of the function and integrity of the membranes involved, giving rise to a loss of regulation of the intracellular ion balance and redox status and eventually to cell death. In addition, misfolded proteins undoubtedly interact inappropriately with other cellular components, potentially giving rise to the impairment of a range of other biological processes. Under some conditions the intracellular content of aggregated species may increase directly, due to an enhanced propensity of incompletely folded or misfolded species to aggregate within the cell itself. This could occur as the result of the expression of mutational variants of proteins with decreased stability or cooperativity or with an intrinsically higher propensity to aggregate. It could also occur as a result of the overproduction of some types of protein, for example, because of other genetic factors or other disease conditions, or because of perturbations to the cellular environment that generate conditions favouring aggregation, such as heat shock or oxidative stress. Finally, the accumulation of misfolded or aggregated proteins could arise from the chaperone and clearance mechanisms becoming overwhelmed as a result of specific mutant phenotypes or of the general effects of ageing [173, 174].
The topics discussed in this review not only provide a great deal of evidence for the ‘new view’ that proteins have an intrinsic capability of misfolding and forming structures such as amyloid fibrils but also suggest that the role of molecular chaperones is even more important than was thought in the past. The role of these ubiquitous proteins in enhancing the efficiency of protein folding is well established [185]. It could well be that they are at least as important in controlling the harmful effects of misfolded or aggregated proteins as in enhancing the yield of functional molecules.
Nutritional Status is Associated with Faster Cognitive Decline and Worse Functional Impairment in the Progression of Dementia: The Cache County Dementia Progression Study1
Nutritional status may be a modifiable factor in the progression of dementia. We examined the association of nutritional status and rate of cognitive and functional decline in a U.S. population-based sample. Study design was an observational longitudinal study with annual follow-ups up to 6 years of 292 persons with dementia (72% Alzheimer’s disease, 56% female) in Cache County, UT using the Mini-Mental State Exam (MMSE), Clinical Dementia Rating Sum of Boxes (CDR-sb), and modified Mini Nutritional Assessment (mMNA). mMNA scores declined by approximately 0.50 points/year, suggesting increasing risk for malnutrition. Lower mMNA score predicted faster rate of decline on the MMSE at earlier follow-up times, but slower decline at later follow-up times, whereas higher mMNA scores had the opposite pattern (mMNA by time β= 0.22, p = 0.017; mMNA by time2 β= –0.04, p = 0.04). Lower mMNA score was associated with greater impairment on the CDR-sb over the course of dementia (β= 0.35, p < 0.001). Assessment of malnutrition may be useful in predicting rates of progression in dementia and may provide a target for clinical intervention.
Shared Genetic Risk Factors for Late-Life Depression and Alzheimer’s Disease
Background: Considerable evidence has been reported for the comorbidity between late-life depression (LLD) and Alzheimer’s disease (AD), both of which are very common in the general elderly population and represent a large burden on the health of the elderly. The pathophysiological mechanisms underlying the link between LLD and AD are poorly understood. Because both LLD and AD can be heritable and are influenced by multiple risk genes, shared genetic risk factors between LLD and AD may exist. Objective: The objective is to review the existing evidence for genetic risk factors that are common to LLD and AD and to outline the biological substrates proposed to mediate this association. Methods: A literature review was performed. Results: Genetic polymorphisms of brain-derived neurotrophic factor, apolipoprotein E, interleukin 1-beta, and methylenetetrahydrofolate reductase have been demonstrated to confer increased risk to both LLD and AD by studies examining either LLD or AD patients. These results contribute to the understanding of pathophysiological mechanisms that are common to both of these disorders, including deficits in nerve growth factors, inflammatory changes, and dysregulation mechanisms involving lipoprotein and folate. Other conflicting results have also been reviewed, and few studies have investigated the effects of the described polymorphisms on both LLD and AD. Conclusion: The findings suggest that common genetic pathways may underlie LLD and AD comorbidity. Studies to evaluate the genetic relationship between LLD and AD may provide insights into the molecular mechanisms that trigger disease progression as the population ages.
Association of Vitamin B12, Folate, and Sulfur Amino Acids With Brain Magnetic Resonance Imaging Measures in Older Adults: A Longitudinal Population-Based Study
Importance Vitamin B12, folate, and sulfur amino acids may be modifiable risk factors for structural brain changes that precede clinical dementia.
Objective To investigate the association of circulating levels of vitamin B12, red blood cell folate, and sulfur amino acids with the rate of total brain volume loss and the change in white matter hyperintensity volume as measured by fluid-attenuated inversion recovery in older adults.
Design, Setting, and Participants The magnetic resonance imaging subsample of the Swedish National Study on Aging and Care in Kungsholmen, a population-based longitudinal study in Stockholm, Sweden, was conducted in 501 participants aged 60 years or older who were free of dementia at baseline. A total of 299 participants underwent repeated structural brain magnetic resonance imaging scans from September 17, 2001, to December 17, 2009.
Main Outcomes and Measures The rate of brain tissue volume loss and the progression of total white matter hyperintensity volume.
Results In the multi-adjusted linear mixed models, among 501 participants (300 women [59.9%]; mean [SD] age, 70.9 [9.1] years), higher baseline vitamin B12 and holotranscobalamin levels were associated with a decreased rate of total brain volume loss during the study period: for each increase of 1 SD, β (SE) was 0.048 (0.013) for vitamin B12 (P < .001) and 0.040 (0.013) for holotranscobalamin (P = .002). Increased total homocysteine levels were associated with faster rates of total brain volume loss in the whole sample (β [SE] per 1-SD increase, –0.035 [0.015]; P = .02) and with the progression of white matter hyperintensity among participants with systolic blood pressure greater than 140 mm Hg (β [SE] per 1-SD increase, 0.000019 [0.00001]; P = .047). No longitudinal associations were found for red blood cell folate and other sulfur amino acids.
Conclusions and Relevance This study suggests that both vitamin B12 and total homocysteine concentrations may be related to accelerated aging of the brain. Randomized clinical trials are needed to determine the importance of vitamin B12supplementation on slowing brain aging in older adults.
Notes from Kurzweill
This vitamin stops the aging process in organs, say Swiss researchers
A potential breakthrough for regenerative medicine, pending further studies
Improved muscle stem cell numbers and muscle function in NR-treated aged mice: Newly regenerated muscle fibers 7 days after muscle damage in aged mice (left: control group; right: fed NR). (Scale bar = 50 μm). (credit: Hongbo Zhang et al./Science) http://www.kurzweilai.net/images/improved-muscle-fibers.png
EPFL researchers have restored the ability of mice organs to regenerate and extend life by simply administering nicotinamide riboside (NR) to them.
NR has been shown in previous studies to be effective in boosting metabolism and treating a number of degenerative diseases. Now, an article by PhD student Hongbo Zhang published in Science also describes the restorative effects of NR on the functioning of stem cells for regenerating organs.
As in all mammals, as mice age, the regenerative capacity of certain organs (such as the liver and kidneys) and muscles (including the heart) diminishes. Their ability to repair them following an injury is also affected. This leads to many of the disorders typical of aging.
Mitochondria —> stem cells —> organs
To understand how the regeneration process deteriorates with age, Zhang teamed up with colleagues from ETH Zurich, the University of Zurich, and universities in Canada and Brazil. By using several biomarkers, they were able to identify the molecular chain that regulates how mitochondria — the “powerhouse” of the cell — function and how they change with age. “We were able to show for the first time that their ability to function properly was important for stem cells,” said Auwerx.
Under normal conditions, these stem cells, reacting to signals sent by the body, regenerate damaged organs by producing new specific cells. At least in young bodies. “We demonstrated that fatigue in stem cells was one of the main causes of poor regeneration or even degeneration in certain tissues or organs,” said Zhang.
How to revitalize stem cells
Which is why the researchers wanted to “revitalize” stem cells in the muscles of elderly mice. And they did so by precisely targeting the molecules that help the mitochondria to function properly. “We gave nicotinamide riboside to 2-year-old mice, which is an advanced age for them,” said Zhang.
“This substance, which is close to vitamin B3, is a precursor of NAD+, a molecule that plays a key role in mitochondrial activity. And our results are extremely promising: muscular regeneration is much better in mice that received NR, and they lived longer than the mice that didn’t get it.”
Parallel studies have revealed a comparable effect on stem cells of the brain and skin. “This work could have very important implications in the field of regenerative medicine,” said Auwerx. This work on the aging process also has potential for treating diseases that can affect — and be fatal — in young people, like muscular dystrophy (myopathy).
So far, no negative side effects have been observed following the use of NR, even at high doses. But while it appears to boost the functioning of all cells, it could include pathological ones, so further in-depth studies are required.
Abstract of NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice
Adult stem cells (SCs) are essential for tissue maintenance and regeneration yet are susceptible to senescence during aging. We demonstrate the importance of the amount of the oxidized form of cellular nicotinamide adenine dinucleotide (NAD+) and its impact on mitochondrial activity as a pivotal switch to modulate muscle SC (MuSC) senescence. Treatment with the NAD+ precursor nicotinamide riboside (NR) induced the mitochondrial unfolded protein response (UPRmt) and synthesis of prohibitin proteins, and this rejuvenated MuSCs in aged mice. NR also prevented MuSC senescence in the Mdx mouse model of muscular dystrophy. We furthermore demonstrate that NR delays senescence of neural SCs (NSCs) and melanocyte SCs (McSCs), and increased mouse lifespan. Strategies that conserve cellular NAD+ may reprogram dysfunctional SCs and improve lifespan in mammals.
Discriminating the gene target of a distal regulatory element from other nearby transcribed genes is a challenging problem with the potential to illuminate the causal underpinnings of complex diseases. We present TargetFinder, a computational method that reconstructs regulatory landscapes from diverse features along the genome. The resulting models accurately predict individual enhancer–promoter interactions across multiple cell lines with a false discovery rate up to 15 times smaller than that obtained using the closest gene. By evaluating the genomic features driving this accuracy, we uncover interactions between structural proteins, transcription factors, epigenetic modifications, and transcription that together distinguish interacting from non-interacting enhancer–promoter pairs. Most of this signature is not proximal to the enhancers and promoters but instead decorates the looping DNA. We conclude that complex but consistent combinations of marks on the one-dimensional genome encode the three-dimensional structure of fine-scale regulatory interactions.
CRISPR-Cas9 Screening by Horizon Discovery, Cambridge, UK – HDx™ Reference Standards, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 2: CRISPR for Gene Editing and DNA Repair
CRISPR-Cas9 Screening by Horizon Discovery, Cambridge, UK – HDx™ Reference Standards
Reporters: David Orchard-Webb, PhD and Aviva Lev-Ari, PhD, RN
They have leveraged our cutting-edge technologies to build a ground-breaking platform for Immuno-Oncology drug discovery.
Their collection of high throughput assays allows you to rapidly identify single agents or combinations that affect T cell and natural killer (NK) cell function.
Gene-Editing in Immune Cells
Gene-editing in immune cells for your Immuno-Oncology project is now even faster than before. Leveraging the latest genome engineering tools, including CRISPR-Cas9, we’re able to precisely edit over 10 pre-characterized T-cell, B-cell, and monocytes lines.
Power Your Immune-Oncology Projects with Custom Engineered Immune Cells
Gene-editing in immune cell lines can be challenging due to low targeting efficiency and difficulties in single cell derivation of suspension cells. Horizon has validated 10+ immune cell lines including THP-1, Jurkat and NALM-6 cells for gene-editing projects. CRISPR, rAAV and ZFN gene-editing technologies are available depending on project requirements.
Take advantage of the largest panel of pre-characterized immune cell lines available and benefit from Horizon’s exceptional knowhow and experience in completing over 2,000 gene-editing projects.
Somatic Mutation Theory – Why it’s Wrong for Most Cancers, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 1: Next Generation Sequencing (NGS)
Somatic Mutation Theory – Why it’s Wrong for Most Cancers
Reporter: Aviva Lev-Ari, PhD, RN
Somatic Mutation Theory – Why it’s Wrong for Most Cancers.
Hysteron proteron reverses both temporal and logical order and this syllogism occurs in carcinogenesis and the somatic mutation theory (SMT): the first (somatic mutation) occurs only after the second (onset of cancer) and, therefore, observed somatic mutations in most cancers appear well after the early cues of carcinogenesis are in place. It is no accident that mutations are increasingly being questioned as the causal event in the origin of the vast majority of cancers as clinical data show little support for this theory when compared against the metrics of patient outcomes. Ever since the discovery of the double helical structure of DNA, virtually all chronic diseases came to be viewed as causally linked to one degree or another to mutations, even though we now know that genes are not simply blueprints, but rather an assemblage of alphabets that can, under non-genetic influences, be used to assemble a business letter or a work of Shakespearean literature. A minority of all cancers is indeed caused by mutations but the SMT has been applied to all cancers, and even to chemical carcinogenesis, in the absence of hard evidence of causality. Herein, we review the 100 year story of SMT and aspects that show why genes are not just blueprints, how radiation and mutation are associated in a more nuanced view, the proposed risk of cancer and bad luck, and the in vitro and in vivo evidence for a new cancer paradigm. This paradigm is scientifically applicable for the majority of non-heritable cancers and consists of a six-step sequence for the origin of cancer. This new cancer paradigm proclaims that somatic mutations are epiphenomena or later events occurring after carcinogenesis is already underway. This serves not just as a plausible alternative to SMT and explains the origin of the majority of cancers, but also provides opportunities for early interventions and prevention of the onset of cancer as a disease.
Conclusions
The incorrect interpretation of data can sometimes appear to be the more parsimonious explanation especially when it has acquired the mantle of a paradigm, as in the case of the SMT. Summa Cancerologica is not hypothetical or ontological. Its syllogism of carcinogenesis needs the consideration of all reasonable perspectives such as whether somatic mutations are later events or epiphenomena occurring at the end of the sequence of events in carcinogenesis. This mutatio praemissarum leads to a reflection of reasoned judgments of correct findings in cancer (mutations within tumors) together with clinical observations (relevance of such mutations to cancer therapy). An overemphasis of the SMT as the sole reason of the origin of carcinogenesis elevated it to the status of a dogma which downplays significant findings of mutations and genetics in different fields of nature, biology and science. However, there is hope that hereditary cancers can be treated in the near future as new technologies make it possible to manipulate proteins packaging DNA to turn on specific gene promoters and enhancers [164]. If this were applicable to the mass of non-hereditary cancers this approach would still be only symptomatic as the genesis of non-hereditary cancers is not caused by somatic mutations though somatic mutations occur within tumors. Focusing on the tumor cell without its origin including the microenvironment won’t be enough [165]. The reasoning on the origin of carcinogenesis, including different step-wise sequences, may help unmask mechanisms of the transition of a normal into a cancer cell (cancer genesis) as well as its different primary pathogenic stimulus, which can serve to prevent or retard cancers instead of concentrating on symptomatic strategies or for a cure for all cancers. It is scientifically valid based on in vitro and in vivo genetic findings that carcinogenesis consists of a six-step multi sequence process [17, 18]. This serves not just as a plausible alternative to the SMT to explain the origin of the majority of cancers, but could also suggest early interventions and thereby prevent the onset of cancer as a disease.
Genomics and epigenetics link to DNA structure, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 1: Next Generation Sequencing (NGS)
Genomics and epigenetics link to DNA structure
Larry H. Bernstein, MD, FCAP, Curator
LPBI
Sequence and Epigenetic Factors Determine Overall DNA Structure
Atomic-level simulations show electrostatic forces between each atom. [Alek Aksimentiev, University of Illinois at Urbana-Champaign]
The traditionally held hypothesis about the highly ordered organization of DNA describes the interaction of various proteins with DNA sequences to mediate the dynamic structure of the molecule. However, recent evidence has emerged that stretches of homologous DNA sequences can associate preferentially with one another, even in the absence of proteins.
Researchers at the University of Illinois Center for the Physics of Living Cells, Johns Hopkins University, and Ulsan National Institute of Science and Technology (UNIST) in South Korea found that DNA molecules interact directly with one another in ways that are dependent on the sequence of the DNA and epigenetic factors, such as methylation.
The researchers described evidence they found for sequence-dependent attractive interactions between double-stranded DNA molecules that neither involve intermolecular strand exchange nor are mediated by DNA-binding proteins.
“DNA molecules tend to repel each other in water, but in the presence of special types of cations, they can attract each other just like nuclei pulling each other by sharing electrons in between,” explained lead study author Hajin Kim, Ph.D., assistant professor of biophysics at UNIST. “Our study suggests that the attractive force strongly depends on the nucleic acid sequence and also the epigenetic modifications.”
The investigators used atomic-level supercomputer simulations to measure the forces between a pair of double-stranded DNA helices and proposed that the distribution of methyl groups on the DNA was the key to regulating this sequence-dependent attraction. To verify their findings experimentally, the scientists were able to observe a single pair of DNA molecules within nanoscale bubbles.
“Here we combine molecular dynamics simulations with single-molecule fluorescence resonance energy transfer experiments to examine the interactions between duplex DNA in the presence of spermine, a biological polycation,” the authors wrote. “We find that AT-rich DNA duplexes associate more strongly than GC-rich duplexes, regardless of the sequence homology. Methyl groups of thymine act as a steric block, relocating spermine from major grooves to interhelical regions, thereby increasing DNA–DNA attraction.”
The findings from this study were published recently in Nature Communications in an article entitled “Direct Evidence for Sequence-Dependent Attraction Between Double-Stranded DNA Controlled by Methylation.”
After conducting numerous further simulations, the research team concluded that direct DNA–DNA interactions could play a central role in how chromosomes are organized in the cell and which ones are expanded or folded up compactly, ultimately determining functions of different cell types or regulating the cell cycle.
“Biophysics is a fascinating subject that explores the fundamental principles behind a variety of biological processes and life phenomena,” Dr. Kim noted. “Our study requires cross-disciplinary efforts from physicists, biologists, chemists, and engineering scientists and we pursue the diversity of scientific disciplines within the group.”
Dr. Kim concluded by stating that “in our lab, we try to unravel the mysteries within human cells based on the principles of physics and the mechanisms of biology. In the long run, we are seeking for ways to prevent chronic illnesses and diseases associated with aging.”
Direct evidence for sequence-dependent attraction between double-stranded DNA controlled by methylation
Jejoong Yoo, Hajin Kim, Aleksei Aksimentiev, and Taekjip Ha Nature Communications 7 11045 (2016) DOI:10.1038/ncomms11045BibTex
Although proteins mediate highly ordered DNA organization in vivo, theoretical studies suggest that homologous DNA duplexes can preferentially associate with one another even in the absence of proteins. Here we combine molecular dynamics simulations with single-molecule fluorescence resonance energy transfer experiments to examine the interactions between duplex DNA in the presence of spermine, a biological polycation. We find that AT-rich DNA duplexes associate more strongly than GC-rich duplexes, regardless of the sequence homology. Methyl groups of thymine acts as a steric block, relocating spermine from major grooves to interhelical regions, thereby increasing DNA–DNA attraction. Indeed, methylation of cytosines makes attraction between GC-rich DNA as strong as that between AT-rich DNA. Recent genome-wide chromosome organization studies showed that remote contact frequencies are higher for AT-rich and methylated DNA, suggesting that direct DNA–DNA interactions that we report here may play a role in the chromosome organization and gene regulation.
Formation of a DNA double helix occurs through Watson–Crick pairing mediated by the complementary hydrogen bond patterns of the two DNA strands and base stacking. Interactions between double-stranded (ds)DNA molecules in typical experimental conditions containing mono- and divalent cations are repulsive1, but can turn attractive in the presence of high-valence cations2. Theoretical studies have identified the ion–ion correlation effect as a possible microscopic mechanism of the DNA condensation phenomena3, 4, 5. Theoretical investigations have also suggested that sequence-specific attractive forces might exist between two homologous fragments of dsDNA6, and this ‘homology recognition’ hypothesis was supported by in vitro atomic force microscopy7 and in vivo point mutation assays8. However, the systems used in these measurements were too complex to rule out other possible causes such as Watson–Crick strand exchange between partially melted DNA or protein-mediated association of DNA.
Here we present direct evidence for sequence-dependent attractive interactions between dsDNA molecules that neither involve intermolecular strand exchange nor are mediated by proteins. Further, we find that the sequence-dependent attraction is controlled not by homology—contradictory to the ‘homology recognition’ hypothesis6—but by a methylation pattern. Unlike the previous in vitro study that used monovalent (Na+) or divalent (Mg2+) cations7, we presumed that for the sequence-dependent attractive interactions to operate polyamines would have to be present. Polyamine is a biological polycation present at a millimolar concentration in most eukaryotic cells and essential for cell growth and proliferation9, 10. Polyamines are also known to condense DNA in a concentration-dependent manner2, 11. In this study, we use spermine4+(Sm4+) that contains four positively charged amine groups per molecule.
Sequence dependence of DNA–DNA forces
To characterize the molecular mechanisms of DNA–DNA attraction mediated by polyamines, we performed molecular dynamics (MD) simulations where two effectively infinite parallel dsDNA molecules, 20 base pairs (bp) each in a periodic unit cell, were restrained to maintain a prescribed inter-DNA distance; the DNA molecules were free to rotate about their axes. The two DNA molecules were submerged in 100mM aqueous solution of NaCl that also contained 20 Sm4+molecules; thus, the total charge of Sm4+, 80 e, was equal in magnitude to the total charge of DNA (2 × 2 × 20 e, two unit charges per base pair; Fig. 1a). Repeating such simulations at various inter-DNA distances and applying weighted histogram analysis12 yielded the change in the interaction free energy (ΔG) as a function of the DNA–DNA distance (Fig. 1b,c). In a broad agreement with previous experimental findings13, ΔG had a minimum, ΔGmin, at the inter-DNA distance of 25−30Å for all sequences examined, indeed showing that two duplex DNA molecules can attract each other. The free energy of inter-duplex attraction was at least an order of magnitude smaller than the Watson–Crick interaction free energy of the same length DNA duplex. A minimum of ΔG was not observed in the absence of polyamines, for example, when divalent or monovalent ions were used instead14, 15.
Figure 1: Polyamine-mediated DNA sequence recognition observed in MD simulations and smFRET experiments.
(a) Set-up of MD simulations. A pair of parallel 20-bp dsDNA duplexes is surrounded by aqueous solution (semi-transparent surface) containing 20 Sm4+ molecules (which compensates exactly the charge of DNA) and 100mM NaCl. Under periodic boundary conditions, the DNA molecules are effectively infinite. A harmonic potential (not shown) is applied to maintain the prescribed distance between the dsDNA molecules. (b,c) Interaction free energy of the two DNA helices as a function of the DNA–DNA distance for repeat-sequence DNA fragments (b) and DNA homopolymers (c). (d) Schematic of experimental design. A pair of 120-bp dsDNA labelled with a Cy3/Cy5 FRET pair was encapsulated in a ~200-nm diameter lipid vesicle; the vesicles were immobilized on a quartz slide through biotin–neutravidin binding. Sm4+ molecules added after immobilization penetrated into the porous vesicles. The fluorescence signals were measured using a total internal reflection microscope. (e) Typical fluorescence signals indicative of DNA–DNA binding. Brief jumps in the FRET signal indicate binding events. (f) The fraction of traces exhibiting binding events at different Sm4+ concentrations for AT-rich, GC-rich, AT nonhomologous and CpG-methylated DNA pairs. The sequence of the CpG-methylated DNA specifies the methylation sites (CG sequence, orange), restriction sites (BstUI, triangle) and primer region (underlined). The degree of attractive interaction for the AT nonhomologous and CpG-methylated DNA pairs was similar to that of the AT-rich pair. All measurements were done at [NaCl]=50mM and T=25°C. (g) Design of the hybrid DNA constructs: 40-bp AT-rich and 40-bp GC-rich regions were flanked by 20-bp common primers. The two labelling configurations permit distinguishing parallel from anti-parallel orientation of the DNA. (h) The fraction of traces exhibiting binding events as a function of NaCl concentration at fixed concentration of Sm4+ (1mM). The fraction is significantly higher for parallel orientation of the DNA fragments.
Unexpectedly, we found that DNA sequence has a profound impact on the strength of attractive interaction. The absolute value of ΔG at minimum relative to the value at maximum separation, |ΔGmin|, showed a clearly rank-ordered dependence on the DNA sequence: |ΔGmin| of (A)20>|ΔGmin| of (AT)10>|ΔGmin| of (GC)10>|ΔGmin| of (G)20. Two trends can be noted. First, AT-rich sequences attract each other more strongly than GC-rich sequences16. For example, |ΔGmin| of (AT)10 (1.5kcalmol−1 per turn) is about twice |ΔGmin| of (GC)10 (0.8kcalmol−1 per turn) (Fig. 1b). Second, duplexes having identical AT content but different partitioning of the nucleotides between the strands (that is, (A)20 versus (AT)10 or (G)20 versus (GC)10) exhibit statistically significant differences (~0.3kcalmol−1 per turn) in the value of |ΔGmin|.
To validate the findings of MD simulations, we performed single-molecule fluorescence resonance energy transfer (smFRET)17 experiments of vesicle-encapsulated DNA molecules. Equimolar mixture of donor- and acceptor-labelled 120-bp dsDNA molecules was encapsulated in sub-micron size, porous lipid vesicles18 so that we could observe and quantitate rare binding events between a pair of dsDNA molecules without triggering large-scale DNA condensation2. Our DNA constructs were long enough to ensure dsDNA–dsDNA binding that is stable on the timescale of an smFRET measurement, but shorter than the DNA’s persistence length (~150bp (ref. 19)) to avoid intramolecular condensation20. The vesicles were immobilized on a polymer-passivated surface, and fluorescence signals from individual vesicles containing one donor and one acceptor were selectively analysed (Fig. 1d). Binding of two dsDNA molecules brings their fluorescent labels in close proximity, increasing the FRET efficiency (Fig. 1e).
FRET signals from individual vesicles were diverse. Sporadic binding events were observed in some vesicles, while others exhibited stable binding; traces indicative of frequent conformational transitions were also observed (Supplementary Fig. 1A). Such diverse behaviours could be expected from non-specific interactions of two large biomolecules having structural degrees of freedom. No binding events were observed in the absence of Sm4+ (Supplementary Fig. 1B) or when no DNA molecules were present. To quantitatively assess the propensity of forming a bound state, we chose to use the fraction of single-molecule traces that showed any binding events within the observation time of 2min (Methods). This binding fraction for the pair of AT-rich dsDNAs (AT1, 100% AT in the middle 80-bp section of the 120-bp construct) reached a maximum at ~2mM Sm4+(Fig. 1f), which is consistent with the results of previous experimental studies2, 3. In accordance with the prediction of our MD simulations, GC-rich dsDNAs (GC1, 75% GC in the middle 80bp) showed much lower binding fraction at all Sm4+ concentrations (Fig. 1b,c). Regardless of the DNA sequence, the binding fraction reduced back to zero at high Sm4+ concentrations, likely due to the resolubilization of now positively charged DNA–Sm4+ complexes2, 3, 13.
Because the donor and acceptor fluorophores were attached to the same sequence of DNA, it remained possible that the sequence homology between the donor-labelled DNA and the acceptor-labelled DNA was necessary for their interaction6. To test this possibility, we designed another AT-rich DNA construct AT2 by scrambling the central 80-bp section of AT1 to remove the sequence homology (Supplementary Table 1). The fraction of binding traces for this nonhomologous pair of donor-labelled AT1 and acceptor-labelled AT2 was comparable to that for the homologous AT-rich pair (donor-labelled AT1 and acceptor-labelled AT1) at all Sm4+ concentrations tested (Fig. 1f). Furthermore, this data set rules out the possibility that the higher binding fraction observed experimentally for the AT-rich constructs was caused by inter-duplex Watson–Crick base pairing of the partially melted constructs.
Next, we designed a DNA construct named ATGC, containing, in its middle section, a 40-bp AT-rich segment followed by a 40-bp GC-rich segment (Fig. 1g). By attaching the acceptor to the end of either the AT-rich or GC-rich segments, we could compare the likelihood of observing the parallel binding mode that brings the two AT-rich segments together and the anti-parallel binding mode. Measurements at 1mM Sm4+ and 25 or 50mM NaCl indicated a preference for the parallel binding mode by ~30% (Fig. 1h). Therefore, AT content can modulate DNA–DNA interactions even in a complex sequence context. Note that increasing the concentration of NaCl while keeping the concentration of Sm4+ constant enhances competition between Na+ and Sm4+ counterions, which reduces the concentration of Sm4+ near DNA and hence the frequency of dsDNA–dsDNA binding events (Supplementary Fig. 2).
Methylation determines the strength of DNA–DNA attraction
Analysis of the MD simulations revealed the molecular mechanism of the polyamine-mediated sequence-dependent attraction (Fig. 2). In the case of the AT-rich fragments, the bulky methyl group of thymine base blocks Sm4+ binding to the N7 nitrogen atom of adenine, which is the cation-binding hotspot21, 22. As a result, Sm4+ is not found in the major grooves of the AT-rich duplexes and resides mostly near the DNA backbone (Fig. 2a,d). Such relocated Sm4+ molecules bridge the two DNA duplexes better, accounting for the stronger attraction16, 23, 24, 25. In contrast, significant amount of Sm4+ is adsorbed to the major groove of the GC-rich helices that lacks cation-blocking methyl group (Fig. 2b,e).
Figure 2: Molecular mechanism of polyamine-mediated DNA sequence recognition.
(a–c) Representative configurations of Sm4+ molecules at the DNA–DNA distance of 28Å for the (AT)10–(AT)10 (a), (GC)10–(GC)10 (b) and (GmC)10–(GmC)10 (c) DNA pairs. The backbone and bases of DNA are shown as ribbon and molecular bond, respectively; Sm4+ molecules are shown as molecular bonds. Spheres indicate the location of the N7 atoms and the methyl groups. (d–f) The average distributions of cations for the three sequence pairs featured in a–c. Top: density of Sm4+ nitrogen atoms (d=28Å) averaged over the corresponding MD trajectory and the z axis. White circles (20Å in diameter) indicate the location of the DNA helices. Bottom: the average density of Sm4+ nitrogen (blue), DNA phosphate (black) and sodium (red) atoms projected onto the DNA–DNA distance axis (x axis). The plot was obtained by averaging the corresponding heat map data over y=[−10, 10] Å. See Supplementary Figs 4 and 5 for the cation distributions at d=30, 32, 34 and 36Å.
If indeed the extra methyl group in thymine, which is not found in cytosine, is responsible for stronger DNA–DNA interactions, we can predict that cytosine methylation, which occurs naturally in many eukaryotic organisms and is an essential epigenetic regulation mechanism26, would also increase the strength of DNA–DNA attraction. MD simulations showed that the GC-rich helices containing methylated cytosines (mC) lose the adsorbed Sm4+ (Fig. 2c,f) and that |ΔGmin| of (GC)10 increases on methylation of cytosines to become similar to |ΔGmin| of (AT)10 (Fig. 1b).
To experimentally assess the effect of cytosine methylation, we designed another GC-rich construct GC2 that had the same GC content as GC1 but a higher density of CpG sites (Supplementary Table 1). The CpG sites were then fully methylated using M. SssI methyltransferase (Supplementary Fig. 3; Methods). As predicted from the MD simulations, methylation of the GC-rich constructs increased the binding fraction to the level of the AT-rich constructs (Fig. 1f).
The sequence dependence of |ΔGmin| and its relation to the Sm4+ adsorption patterns can be rationalized by examining the number of Sm4+ molecules shared by the dsDNA molecules (Fig. 3a). An Sm4+ cation adsorbed to the major groove of one dsDNA is separated from the other dsDNA by at least 10Å, contributing much less to the effective DNA–DNA attractive force than a cation positioned between the helices, that is, the ‘bridging’ Sm4+ (ref. 23). An adsorbed Sm4+ also repels other Sm4+ molecules due to like-charge repulsion, lowering the concentration of bridging Sm4+. To demonstrate that the concentration of bridging Sm4+ controls the strength of DNA–DNA attraction, we computed the number of bridging Sm4+ molecules, Nspm (Fig. 3b). Indeed, the number of bridging Sm4+ molecules ranks in the same order as |ΔGmin|: Nspm of (A)20>Nspm of (AT)10≈Nspm of (GmC)10>Nspm of (GC)10>Nspm of (G)20. Thus, the number density of nucleotides carrying a methyl group (T and mC) is the primary determinant of the strength of attractive interaction between two dsDNA molecules. At the same time, the spatial arrangement of the methyl group carrying nucleotides can affect the interaction strength as well (Fig. 3c). The number of methyl groups and their distribution in the (AT)10 and (GmC)10 duplex DNA are identical, and so are their interaction free energies, |ΔGmin| of (AT)10≈|ΔGmin| of (GmC)10. For AT-rich DNA sequences, clustering of the methyl groups repels Sm4+ from the major groove more efficiently than when the same number of methyl groups is distributed along the DNA (Fig. 3b). Hence, |ΔGmin| of (A)20>|ΔGmin| of (AT)10. For GC-rich DNA sequences, clustering of the cation-binding sites (N7 nitrogen) attracts more Sm4+ than when such sites are distributed along the DNA (Fig. 3b), hence |ΔGmin| is larger for (GC)10 than for (G)20.
Figure 3: Methylation modulates the interaction free energy of two dsDNA molecules by altering the number of bridging Sm4+.
(a) Typical spatial arrangement of Sm4+ molecules around a pair of DNA helices. The phosphates groups of DNA and the amine groups of Sm4+ are shown as red and blue spheres, respectively. ‘Bridging’ Sm4+molecules reside between the DNA helices. Orange rectangles illustrate the volume used for counting the number of bridging Sm4+ molecules. (b) The number of bridging amine groups as a function of the inter-DNA distance. The total number of Sm4+ nitrogen atoms was computed by averaging over the corresponding MD trajectory and the 10Å (x axis) by 20Å (y axis) rectangle prism volume (a) centred between the DNA molecules. (c) Schematic representation of the dependence of the interaction free energy of two DNA molecules on their nucleotide sequence. The number and spatial arrangement of nucleotides carrying a methyl group (T or mC) determine the interaction free energy of two dsDNA molecules.
Genome-wide investigations of chromosome conformations using the Hi–C technique revealed that AT-rich loci form tight clusters in human nucleus27, 28. Gene or chromosome inactivation is often accompanied by increased methylation of DNA29 and compaction of facultative heterochromatin regions30. The consistency between those phenomena and our findings suggest the possibility that the polyamine-mediated sequence-dependent DNA–DNA interaction might play a role in chromosome folding and epigenetic regulation of gene expression.
Rau, D. C., Lee, B. & Parsegian, V. A.Measurement of the repulsive force between polyelectrolyte molecules in ionic solution: hydration forces between parallel DNA double helices. Proc. Natl Acad. Sci. USA81, 2621–2625 (1984).
Raspaud, E., Olvera de la Cruz, M., Sikorav, J. L. & Livolant, F.Precipitation of DNA by polyamines: a polyelectrolyte behavior. Biophys. J.74, 381–393 (1998).
Grosberg, A. Y., Nguyen, T. T. & Shklovskii, B. I.The physics of charge inversion in chemical and biological systems. Rev. Mod. Phys.74, 329–345 (2002).
Thomas, T. & Thomas, T. J.Polyamines in cell growth and cell death: molecular mechanisms and therapeutic applications. Cell. Mol. Life Sci.58, 244–258 (2001).
Novel clinically-available comprehensive genomic profiling of both DNA and RNA in hematologic malignancies.
Profiling of 3696 clinical hematologic tumors identified somatic alterations that impact diagnosis, prognosis, and therapeutic selection.
Abstract
The spectrum of somatic alterations in hematologic malignancies includes substitutions, insertions/deletions (indels), copy number alterations (CNAs) and a wide range of gene fusions; no current clinically available single assay captures the different types of alterations. We developed a novel next-generation sequencing-based assay to identify all classes of genomic alterations using archived formalin-fixed paraffin-embedded (FFPE), blood and bone marrow samples with high accuracy in a clinically relevant timeframe, which is performed in our CLIA-certified CAP-accredited laboratory. Targeted capture of DNA/RNA and next-generation sequencing reliably identifies substitutions, indels, CNAs and gene fusions, with similar accuracy to lower-throughput assays which focus on specific genes and types of genomic alterations. Profiling of 3696 samples identified recurrent somatic alterations that impact diagnosis, prognosis and therapy selection. This comprehensive genomic profiling approach has proved effective in detecting all types of genomic alterations, including fusion transcripts, which increases the ability to identify clinically-relevant genomic alterations with therapeutic relevance.
In addition to the concordance analysis, genomic profiling of the 76 test samples using FoundationOne Heme also identified 126 additional somatic alterations including clinically relevant genomic alterations in KRAS, TET2, EZH2, and DNMT3A.
Importantly, the study also showed that the molecular information supplied by the test can help accurately match patients with a particular targeted therapy.
In the study Foundation Medicine shared clinical data from genomic profiling of 3,696 hematologic malignancies submitted to its CLIA-certified, NYS-approved lab.
More than 90 percent of the specimens — 3,433 out of 3696 — were successfully characterized. The test identified at least one driver alteration in 95 percent of the tumor specimens, and results showed that 77 percent of the cases harbored at least one alteration linked to a commercially available targeted therapy or one that is in clinical development, the MSKCC researchers reported.
In addition, 61 percent of the cases harbored at least one alteration with known prognostic relevance in that tumor type.
In discussion of the results, the study authors argued that clinical merit of the test was underscored by the demonstrated ability to identify genetic lesions with prognostic and therapeutic relevance in specific diseases.
For example, the authors wrote, “In the case of B-cell ALL … the challenge has been that the critical genes … can be altered by whole gene/intragenic deletions, DNA base-pair substitutions, and larger indels, as well as chromosomal, intergenic, and cryptic rearrangements, which lead to expression of fusion transcripts.”
“Currently, most centers use an amalgam of DNA, FISH, and gene-specific RNA approaches to identify a subset of the most critical genetic lesions in B-ALL. Our assay provides a single profiling platform that can reliably identify all known actionable disease alleles relevant to B-ALL to improve diagnosis and risk-adapted therapy for B-ALL patients,” they wrote.
Intratumor heterogeneity, it is known, can complicate cancer treatments. Now it appears the same may be true of tumor microenvironment heterogeneity. According to a new study from the Institute of Cancer Research (ICR), London, breast cancers that develop within an “ecologically diverse” breast cancer microenvironment are particularly likely to progress and lead to death.
The study took an unusual approach: It combined ecological scoring methods with genome-wide profiling data. This approach, besides showing clinical utility in the evaluation of breast cancer outcomes, demonstrated that even so contextual a discipline as genomics can benefit from being placed within a larger context. In this case, the context is essentially Darwinian, albeit at a small scale.
Natural selection is typically studied at the level of ecosystems consisting of animals and plants. In the current study, however, it was assessed at the level of the tumor microenvironment, which consists of cancer cells, immune system lymphocytes, and stromal cells.
The ICR scientists, led by Yinyin Yuan, Ph.D., presented their work February 16 in the journal PLoS Medicine, in an article entitled “Microenvironmental Heterogeneity Parallels Breast Cancer Progression: A Histology–Genomic Integration Analysis.” The article describes how the scientists developed a tumor ecosystem diversity index (EDI), a scoring system that indicates the degree of microenvironmental heterogeneity along three spatial dimensions in solid tumors. EDI scores take account of “fully automated histology image analysis coupled with statistical measures commonly used in ecology.”
“[EDI] was compared with disease-specific survival, key mutations, genome-wide copy number, and expression profiling data in a retrospective study of 510 breast cancer patients as a test set and 516 breast cancer patients as an independent validation set,” wrote the authors. “In high-grade (grade 3) breast cancers, we uncovered a striking link between high microenvironmental heterogeneity measured by EDI and a poor prognosis that cannot be explained by tumor size, genomics, or any other data types.”
By using the EDI, the ICR team was able to identify several particularly aggressive subgroups of breast cancer. In fact, the EDI was a stronger predictor of survival than many established markers for the disease.
The ICR researchers also looked at the EDI in addition to genetic factors. For example, the researchers found that the prognostic value of EDI was enhanced with the addition of TP53 mutation status. By integrating EDI data and genome-wide profiling data, the researchers identified losses of specific genes on 4p14 and 5q13 that were enriched in grade 3 tumors. These tumors, which showed high microenvironmental diversity, substratified patients into poor prognostic groups.
“Our findings show that mathematical models of ecological diversity can spot more aggressive cancers,” said Dr. Yuan. “By analyzing images of the environment around a tumor based on Darwinian natural selection principles, we can predict survival in some breast cancer types even more effectively than many of the measures used now in the clinic.
“In the future, we hope that by combining cell diversity scores with other factors that influence cancer survival, such as genetics and tumor size, we will be able to tell apart patients with more or less aggressive disease so we can identify those who might need different types of treatment.”
“This ingenious study…teaches us a valuable lesson,” added Paul Workman, Ph.D., chief executive of the ICR. “[We] should always remember that cancer cells are not developing and growing in isolation, but are part of a complex ecosystem that involves normal human cells, too. By better understanding these ecosystems, we aim to create new ways to diagnose, monitor and treat cancer.”
Microenvironmental Heterogeneity Parallels Breast Cancer Progression: A Histology–Genomic Integration Analysis
The intra-tumor diversity of cancer cells is under intense investigation; however, little is known about the heterogeneity of the tumor microenvironment that is key to cancer progression and evolution. We aimed to assess the degree of microenvironmental heterogeneity in breast cancer and correlate this with genomic and clinical parameters.
Methods and Findings
We developed a quantitative measure of microenvironmental heterogeneity along three spatial dimensions (3-D) in solid tumors, termed the tumor ecosystem diversity index (EDI), using fully automated histology image analysis coupled with statistical measures commonly used in ecology. This measure was compared with disease-specific survival, key mutations, genome-wide copy number, and expression profiling data in a retrospective study of 510 breast cancer patients as a test set and 516 breast cancer patients as an independent validation set. In high-grade (grade 3) breast cancers, we uncovered a striking link between high microenvironmental heterogeneity measured by EDI and a poor prognosis that cannot be explained by tumor size, genomics, or any other data types. However, this association was not observed in low-grade (grade 1 and 2) breast cancers. The prognostic value of EDI was superior to known prognostic factors and was enhanced with the addition of TP53 mutation status (multivariate analysis test set, p = 9 × 10−4, hazard ratio = 1.47, 95% CI 1.17–1.84; validation set, p = 0.0011, hazard ratio = 1.78, 95% CI 1.26–2.52). Integration with genome-wide profiling data identified losses of specific genes on 4p14 and 5q13 that were enriched in grade 3 tumors with high microenvironmental diversity that also substratified patients into poor prognostic groups. Limitations of this study include the number of cell types included in the model, that EDI has prognostic value only in grade 3 tumors, and that our spatial heterogeneity measure was dependent on spatial scale and tumor size.
Conclusions
To our knowledge, this is the first study to couple unbiased measures of microenvironmental heterogeneity with genomic alterations to predict breast cancer clinical outcome. We propose a clinically relevant role of microenvironmental heterogeneity for advanced breast tumors, and highlight that ecological statistics can be translated into medical advances for identifying a new type of biomarker and, furthermore, for understanding the synergistic interplay of microenvironmental heterogeneity with genomic alterations in cancer cells.
Background
The human body contains millions of cells, all of which grow, divide, and die in an orderly fashion to build tissues during early life and to replace worn-out or dying cells and repair injuries during adult life. Sometimes, however, normal cells acquire genetic changes (mutations) that allow them to divide uncontrollably and to move around the body (metastasize), resulting in cancer. Because any cell in the body can acquire the mutations needed for cancer development, there are many types of cancer. For example, breast cancer, the most common cancer in women, begins when the cells in the breast that normally make milk become altered. Moreover, different types of cancer progress and evolve differently—some cancers grow quickly and kill their “host” soon after diagnosis, whereas others can be successfully treated with drugs, surgery, or radiotherapy. The behavior of individual cancers depends both on the characteristics of the cancer cells within the tumor and on the interactions between the cancer cells and the normal stromal cells (the connective tissue cells of organs) and other cells (for example, immune cells) that surround and feed cancer cells (the tumor microenvironment).
Why Was This Study Done?
Although recent studies have highlighted the importance of the tumor microenvironment for disease-related outcomes, little is known about how the heterogeneity of the tumor microenvironment—the diversity of non-cancer cells within the tumor—affects outcomes. Mathematical modeling suggests that tumors with heterogeneous and homogeneous microenvironments have different growth patterns and that heterogeneous microenvironments are more likely to be associated with aggressive cancers than homogenous microenvironments. However, the lack of methods to quantify the spatial variability and cellular composition across solid tumors has prevented confirmation of these predictions. Here, the researchers develop a computational system for quantifying microenvironmental heterogeneity in breast cancer based on tumor morphology (shape and form) in histological sections (tissue samples taken from tumors that are examined microscopically). They then use this system to analyze the associations between clinical outcomes, molecular changes, and microenvironmental heterogeneity in breast cancer.
What Did the Researchers Do and Find?
The researchers used automated image analysis and statistical analysis to develop the ecosystem diversity index (EDI), a numerical measure of microenvironmental heterogeneity in solid tumors. They compared the EDI with prognosis (likely outcome), key mutations, genome-wide copy number (tumor cells often contain abnormal numbers of copies of specific genes), and expression profiling data (the expression of several key proteins is altered in tumors) in a test set of 510 samples from patients with breast cancer and in a validation set of 516 additional samples. Among high-grade breast cancers (grade 3 cancers; the grade of a cancer indicates what the cells look like; high-grade breast cancers have a poor prognosis), but not among low-grade breast cancers (grades 1 and 2), a high EDI (high microenvironmental heterogeneity) was associated with a poor prognosis. Specifically, patients with grade 3 tumors and a high EDI had a ten-year disease-specific survival rate of 51%, whereas the remaining patients with grade 3 tumors had a ten-year survival rate of 70%. Notably, the combination of a high EDI with specific DNA alterations—mutations in a gene called TP53 and loss of genes on Chromosomes 4p14 and 5q13—improved the accuracy of prognosis among patients with grade 3 breast cancer and stratified them into subgroups with disease-specific five-year survival rates of 35%, 9%, and 32%, respectively.
What Do These Findings Mean?
These findings establish a method for measuring the spatial heterogeneity of the microenvironment of solid tumors and suggest that the measurement of tumor microenvironmental heterogeneity can be coupled with information about genomic alterations to provide an accurate way to predict outcomes among patients with high-grade breast cancer. The association between EDI, specific genomic alterations, and outcomes needs to be confirmed in additional patients. However, these findings suggest that microenvironmental heterogeneity might provide an additional biomarker to help clinicians identify those patients with advanced breast cancer who have a particularly bad prognosis. The ability to identify these patients is important because it will help clinicians target aggressive treatments to individuals with a poor prognosis and avoid the overtreatment of patients whose prognosis is more favorable. Finally, and more generally, these findings describe a new way to investigate the interactions between the tumor microenvironment and genomic alterations in cancer cells.
Breast Cancer Now is a not-for-profit organization that provides up-to-date information about breast cancer (in English and Spanish)
The UK National Health Service Choices website has information and personal stories about breast cancer; the not-for-profit organization Healthtalkonline also provides personal stories about dealing with breast cancer
Wikipedia has a page about the tumor microenvironment (note that Wikipedia is a free online encyclopedia that anyone can edit; available in several languages)
Citation: Natrajan R, Sailem H, Mardakheh FK, Arias Garcia M, Tape CJ, Dowsett M, et al. (2016) Microenvironmental Heterogeneity Parallels Breast Cancer Progression: A Histology–Genomic Integration Analysis.
PLoS Med 13(2): e1001961. http://dx.doi.org:/10.1371/journal.pmed.1001961
Fig 1. In silico tumor dissection pipeline for quantifying spatial diversity in the tumor ecosystem. (A) Flow diagram depicting the overall study design. (B) Schematic of our pipeline for quantifying spatial diversity in pathological samples. H&E sections are morphologically classified and divided into regions to be spatially scored. The number of clusters k in the regional scores is indicative of the number of sub-populations of cell types in the tumor regions. (C) Examples of tumor regions with low and high diversity scores using the Shannon diversity index, accounting for cancer cells (outlined in green), lymphocytes (blue), and stromal cells (red). Cell classification is automated by image analysis. (D) The 3-D landscape of cell diversity scores on an example H&E section; the x- and y-axes are the geometric axes of the image, and the z-axis is cell diversity computed on a region-by-region basis. (E) The distribution of regional scores in a tumor from the METABRIC study with two regional clusters identified using Gaussian mixture clustering (grey shading: histogram; dashed black line: density; solid black lines: mixture components/clusters).
Fig 2. Application of EDI to 1,026 breast tumors from the METABRIC study. (A) The frequencies of EDI scores in breast tumors. (B) H&E staining, distribution of classified cells (green: cancer; blue: lymphocyte; red: stromal cells), and the heatmap of regional diversity scores for a tumor with the highest EDI score (EDI = 5). (C) Representative regions from each of the clusters k1–k5 are shown in a tumor with EDI = 5, with cluster k1 having the lowest diversity score and k5 the highest. By mapping regional clusters to the H&E image, we can begin to interpret these clusters with different cell diversity. We observed predominantly cancer cells in k1, increasingly more stromal cells and ductal in situ carcinoma cells (DCIS) in k2, and a vessel in k3. Cluster k4 features extensive stromal lymphocytes between ductal in situ carcinoma components, while k5 shows tumor-infiltrating lymphocytes (TIL) associated with invasive carcinoma cells.
Fig 3. Reproducibility, stability, and independence of the EDI-high group in 507 grade 3 breast tumors.
Fig 3. Reproducibility, stability, and independence of the EDI-high group in 507 grade 3 breast tumors. (A) Kaplan–Meier curves of disease-specific survival to illustrate the prognosis of EDI-high samples compared to other grade 3 samples in two independent patient cohorts. Shown below the graph are the number of patients (the number of disease-specific events) per group for EDI-low (grey) and EDI-high (red). (B) Agreement of the EDI subtyping between 100% data and resampling with progressively fewer tumor regions in 200 repeats. (C) Distribution of known subtypes in grade 3 tumors stratified by EDI; asterisks mark subtypes enriched in the EDI-high group. (D) Kaplan–Meier curves illustrating the duration of disease-specific survival according to tumor size (left) and improvement of stratification with the addition of EDI information (right).
Accumulating evidence suggests that the interactions of cancer cells and stromal cells within their microenvironment govern disease progression, metastasis, and, ultimately, the evolution of therapeutic resistance [1–3]. Recent reports have highlighted the significance of the contribution of stromal gene expression and morphological structure as powerful prognostic determinants for a number of tumor types, emphasizing the importance of the tumor microenvironment in disease-related outcomes [4–7]. In breast cancer, a number of studies have demonstrated the prognostic correlation of individual cell types, including the immune cell infiltrate that predicts response to therapy [8–10], and the high percentage of tumor stroma that predicts poor prognosis in triple-negative disease but good prognosis in estrogen receptor (ER)–positive disease [11,12]. Nevertheless, different types of cells coexist with varying degrees of heterogeneity within a tumor. This fundamental feature of human tumors and the combinatorial effects of cell types have been largely ignored, and the collective implications for clinical outcome remain elusive. Consistent observations from mathematical models have highlighted that tumors with diverse microenvironments show growth patterns dramatically different from those of tumors with homogeneous environments [13] and are more likely to be associated with aggressive cancer phenotypes [2] that select for cell migration and eventual metastasis by allowing cancer cells to evolve more rapidly [14]. These observations highlight the need to understand the collective physiological characteristics and heterogeneity of tumor microenvironments. However, there is a lack of methods to quantify the high spatial variability and diverse cellular composition across different solid tumors. Moreover, the interplay of genomic alterations in cancer cells and microenvironmental heterogeneity and its subsequent role in treatment response have not been explored. Our aims were (i) to develop a computational system for quantifying microenvironmental heterogeneity based on tumor morphology in routine histological sections, (ii) to define the clinical implications of microenvironmental heterogeneity, and (iii) to integrate this histologybased index with RNA gene expression and DNA copy number profiling data to identify molecular changes associated with microenvironmental heterogeneity.
The Ecosystem Diversity Index To characterize the tumor ecosystem based on cell compositions, we developed a new index to be used in conjunction with our image analysis tool [16]. First, we used our automated morphological classification method [16] to identify and classify cells into cancer, lymphocyte, or stromal cell classes in H&E sections (Fig 1B). We next divided sections into smaller spatial regions and quantified the diversity of the tumor ecosystem in a tumor region j using the Shannon diversity index: dj ¼ Sm i pi logpi ; ð1Þ where m is the number of cell types and pi is the proportion of the ith cell type (Fig 1B and 1C). A high value of the Shannon diversity index dj reports a heterogeneous environment populated by many cell types, whilst a low value indicates a homogeneous environment (Fig 1C). Compared to other methods such as the Simpson index, the Shannon diversity index accounts for rare species and, hence, is less dominated by main species [17]. Subsequently, we derived the ecosystem diversity index (EDI) by applying unsupervised clustering that identifies the optimum number of clusters in the dataset in an unbiased manner, in order to group tumor regions and quantify the degree of spatial heterogeneity. Let D = d1,d2,…,dn be the Shannon index for n regions in a tumor. We used Gaussian mixture models to fit data D: D SK k¼1okNðmk; s2 kÞ: ð2Þ where μk, ,s2 k, and ωk are the mean, variance, and weight of a Gaussian distribution k, and K is the number of clusters. The Bayesian information criterion was then used to select the best number of clusters K [18]. We used K = 1–5 as the range of K to avoid small EDI groups (S1 Text). The final value of K thus is a measurement of heterogeneity and the score of EDI for a tumor.
Fig 5. The relationship between ecological heterogeneity and cancer genomic aberrations in 507 grade 3 tumors. (A) Genome-wide copy number aberrations in grade 3 breast tumors and genomic coordinates of genes with copy number aberrations enriched in the EDI-high group. Lengths of black lines denote level of enrichment significance with copy number gains (above the horizontal line) or losses (below the horizontal line). (B) Kaplan–Meier curves illustrating the duration of disease-specific survival in grade 3 breast cancer patients according to copy number loss of the 4p14 region (left) and the EDI-high group with additional information of 4p14 copy number loss (right). (C) Kaplan–Meier curves illustrating the duration of disease-specific survival according to copy number loss of the 5q13 region (left) and the EDI-high group with additional information of 5q13 copy number loss (right).
This study has a number of limitations. The motivation for our computational development was to use a data-driven model and measure the degree of spatial heterogeneity in tumor pathological specimens. In this model, only three major cell types in breast tumors were considered. Further sub-classification of the different types of stromal and immune cells by immunohistochemistry may add additional discriminatory value to our model. For dissecting spatial heterogeneity, we chose to use square regions with equal sizes. We found that EDI was correlated with the size of the region chosen for calculation of the Shannon diversity index, and as such the spatial heterogeneity is scale dependent. This phenomenon has been well described in a number of studies in ecology that show that a scale needs to be chosen that is appropriate for the ecological process under study [38,39], further highlighting the analogy between tumor studies and ecology. Similar to the recent observation that breast cancer subclonal heterogeneity is correlated with tumor size [35], we also found an association between microenvironmental heterogeneity and tumor size; hence, EDI may have more limited value in smaller tumors. However, small tumors were present in the EDI-high group, and addition of EDI within tumors grouped by size further stratified their prognosis. We found that EDI was prognostic only in grade 3 tumors in our study, which could be a limitation, given the possible discordance in grading between pathologists.
The identification of additional biomarkers in subgroups of patients that identify them as high risk is important for patient management and to avoid overtreatment for low-risk patients. We envision that the use of our measure of microenvironment heterogeneity, together with key genomic alterations, will enable the diagnosis of patients at very high risk of relapse and facilitate the enrollment of these patients into additional clinical trials for novel therapies or treatment intensification. Our novel computational approach provides a fully automated tool that is relatively easy to implement. Integration of this measure with genomic profiling provides additional prognostic information independent of known clinical parameters. The results of this study highlight the possibility of a grade-3-specific prognostic tool that may aid in further classification of high-grade breast cancer patients beyond standard assays such as ER and HER2 status.
Article ID #201: Correspondence on Leadership in Genomics and other Gene Curations: Dr. Williams with Dr. Lev-Ari. Published on 2/18/2016
WordCloud Image Produced by Adam Tubman
Correspondence on Leadership in Genomics and other Gene Curations: Dr. Williams with Dr. Lev-Ari, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 1: Next Generation Sequencing (NGS)
Correspondence on Leadership in Genomics and other Gene Curations: Dr. Williams with Dr. Lev-Ari
Authors: Stephen J Williams, PhD and Aviva Lev-Ari, PhD, RN
Subject: Re: In light of — >>>>>> Leadership in Genomics: VarElect Variants in Disease and UCSC Genome Technology Center | Leaders in Pharmaceutical Business Intelligence
Leadership in Genomics: VarElect Variants in Disease and UCSC Genome Technology Center
Subject: re: In light of — >>>>>> Leadership in Genomics: VarElect Variants in Disease and UCSC Genome Technology Center | Leaders in Pharmaceutical Business Intelligence
Hi Aviva,
I am not sure what is being proposed here. In the cancer area, there are at least 1,200 genes implicated somehow in this disease and new ones are reported every day. This is a colossal task!
Subject: In light of — >>>>>> Leadership in Genomics: VarElect Variants in Disease and UCSC Genome Technology Center | Leaders in Pharmaceutical Business Intelligence
Dear Dr. Williams,
HERE I am thinking LOUD
Is it possible to go to the dashboard, all posts and click on your Name, you will get the Universe of ~200 articles that you published.
HOW one could search or one needs to visually glance at the title of each — so as to pull a subset of posts that are dedicated to a GENE.
Create an Excel File, place each gene inside and go to Weizmann Institute’s genecards.org and pullout from them respective data on that gene
By so doing we will have LPBI’s Gene Inventory which we could reference in the Drug Discovery process, we do more and more, as we are aggregating all Biologics under the Joint Venture with SBH Sciences, Inc.
In light of :
Leadership in Genomics: VarElect Variants in Disease and UCSC Genome Technology Center
1. HOW could we take this “to be create Excel File” to be published a PAGE, Password Protected as your Curation, it needs to have a Parent or a Hierarchy of Nesting in the Website architecture
And subject that to your our search into New Medicine, Inc. NM/OK DB for data complementarity compilation?
2. What Foundation Medicine, Now Roche, does have vs. Weizmann Institute’s genecards.org
3. Will Weizmann Institute’s genecards.org be interested in New Medicine, Inc., NM/OK DB?
4. I have explored with Foundation Medicine, Now Roche regarding New Medicine, Inc., NM/OK DB and their reply was that they focus ONLY on Genomics data in Cancer, thus,, no interest in New Medicine, Inc. NM/OK DB, there
5. What is in Weizmann Institute’s genecards.org that is NOT in UC Santa Cruz DBs ?
8. Doudna started her professorship at Yale University in 1994. While the group was able to grow high-quality crystals, they struggled with thephase problem due to unspecific binding of the metal ions. One of her early graduate students and later her husband, Jamie Cate decided to soak the crystals in osmiumhexamine to imitate magnesium. Using this strategy, they were able to solve the structure, the second solved folded RNA structure since tRNA.[9][10] The magnesium ions would cluster at the center of the ribozyme and would serve as a core for RNA folding similar to that of a hydrophobic core of a protein.[5]
9. In 2015, Doudna gave a TED Talk about the bioethics of using CRISPR. [13]
Precision medicines have the ability to transform healthcare and treat a myriad of unmet medical needs. The Caribou technology platform has the ability to generate transformative medicines in multiple different market segments.
Our current therapeutic areas of exploration include anti-microbials, animal health, and therapeutic bioproduction.
Human therapeutics
In 2014, Caribou co-founded Intellia Therapeutics to develop curative medicines utilizing the Caribou CRISPR-Cas9 platform. Rachel Haurwitz, President and Chief Executive Officer of Caribou, is a member of Intellia’s Board of Directors.
Intellia is developing human gene and cell therapies for both ex vivo and in vivo applications using CRISPR-Cas9 gene editing technology. Near-term ex vivo applications include the treatment of blood disorders and cancer. In January 2015, Intellia announced a five-year research and development collaboration with Novartis to accelerate the ex vivo development of new CRISPR-Cas9-based therapies using chimeric antigen receptor T cells (CARTs) and hematopoetic stem cells (HSCs).
Subject: Re: Leadership in Genomics: VarElect Variants in Disease and UCSC Genome Technology Center | Leaders in Pharmaceutical Business Intelligence
Every post I do that contains a gene in the post is curated with a link to genecards database so later it not only can be searched but is an integrated knowledge-analysis base integrated with a knowledge and fully integrated Omics database as gene cards . org also contains protein, structure and functional databases.
This is where I always felt the power of LPBI was in the genomic space, integration of a deep analysis curated database
Subject: Fwd: Leadership in Genomics: VarElect – Variants in Disease and UCSC Genome Technology Center | Leaders in Pharmaceutical Business Intelligence
Subject: Re: The Science Coming in 2016 – OpenMind
I want you to go to http://www.genecards.org/ then pick a gene and scroll down. You will see a database there for CRISPR products available from different distributors including Qiagen, Promega, Fisher Scientific, Santa Cruz as well as others. This seems to be already underway. It is possible to copy what these companies are already doing but I don’t see the business advantage in that. Please remember that 3D printing involves layering a of first and second dimension to a third dimension product. So for instance the cell would be the “first dimension” even though it is three dimensional but the effect of layering MULTIPLE layers of cells is what gives their 3D effect. The biomaterial you put in each tube is, in essence, your first dimension you are going to layer into a multilayered “3D” structure.
DNA can be made by synthesizers, there is no need to bioprint it, especially short fragments and in fact you wouldn’t. They can handle even longer material. Possibly if you want to replace a whole nucleosome but the chemistry is not there. That is fine working with Jennifer Duodna making a library of small guide RNA’s to be used in CRISPR however it seems to be in process as I said before. This would need to be done with her system and optimized for her system. You would also need a huge operation to do validation as well. In addition the number of mutations, SNPs, variants are extremely large and many are not disease specific.
Again each would have to be validated. In addition, unless you are doing embryo manipulation, you will need to partner with a company that has a good gene delivery system. This will cost $, probably around 500 million.
This gene fragment in red color — I am suggesting to build with 3D BioPrinting,
at the Oligonucleotide level.
Create a library of fragments for the most common mismatch in transcriptions, as well as on demand for rare deletions.
Per University of California, Santa Cruz, Database of Variations, prepare an INVENTORY of GENE REPAIR PARTS, manage the inventory by Analytics, where each part was implanted and monthly interval monitoring of segment incorporation and new function of protein folding achieved.
Trace the genetic therapy achieved by Gene editing.
Agilent was created as a spin off from Hewlett-Packard Company in 1999.
Agilent Technologies Inc. is engaged in the life sciences, diagnostics and applied chemical markets. The Company provides application focused solutions that include instruments, software, services and consumables for the entire laboratory workflow. The Company has three business segments:
the life sciences and applied markets business,
the diagnostics and genomics business, and
the Agilent Cross Lab business
The Company’s life sciences and applied markets business segment brings together the Company’s analytical laboratory instrumentation and informatics.
The Company’s diagnostics and genomics business segment consists of three businesses: the Dako business, the genomics business and the nucleic acid solutions business.
The Company’s Agilent Cross Lab business segment combines its analytical laboratory services and consumables business
CARPINTERIA, Calif.–(BUSINESS WIRE)–Dako, an Agilent Technologies company and a worldwide provider of cancer diagnostics, today announced the U.S. Food and Drug Administration has approved a new test that can identify PD-L1 expression levels on the surface of non-small cell lung cancer tumor cells and provide information on the survival benefit with OPDIVO® (nivolumab) for patients with non-squamous NSCLC.
Argentina | Australia | Austria | Brazil | Canada |Chile | China | Colombia | Czech Republic | Denmark | Ecuador | Finland | Germany |Hong Kong | Israel | Italy | Japan | Korea | Malaysia | Mexico | New Zealand | Norway | Paraguay | Peru| Philippines | Poland | Romania | Singapore | South Africa | Spain | Sweden |Switzerland | Taiwan ROC | Thailand | Turkey | United Kingdom | Uruguay | Vietnam
Gen9 is building on advances in synthetic biology to power a scalable fabrication capability that will significantly increase the world’s capacity to produce DNA content. The privately held company’s next-generation gene synthesis technology allows for the high-throughput, automated production of DNA constructs at lower cost and higher accuracy than previous methods on the market. Founded by world leaders in synthetic biology, Gen9 aims to ensure the constructive application of synthetic biology in industries ranging from enzyme and chemical production to pharmaceuticals and biofuels.
SERVICES
Synthetic Biology
Gene Synthesis Services
Variant Libraries
Gene Sequence Design Services
INVESTORS
Agilent Technologies : Private Equity
CAMBRIDGE, Mass. and SANTA CLARA, Calif. — April 24, 2013 —Gen9 Receives $21 Million Strategic Investment from Agilent Technologies
GenScript is the largest gene synthesis provider in the USA
GenScript Corporation, a biology contract research organization, provides biological research and drug discovery services to pharmaceutical companies, biotech firms, and research institutions in the United States, Europe, and Japan. It offers bio-reagent, custom molecular biology, custom peptide, protein production, custom antibody production, drug candidates testing, assay development and screening, lead optimization, antibody drug development, gene synthesis, and assay-ready cell line production services.
The company also offers molecular biology, peptide, protein, immunoassay, chemicals, and cell biology products. It offers its products through distributors in Tokyo, Japan; and Seoul, Korea. GenScript Corporation has a strategic partnership with Immunologix, Inc. The company was founded in 2002 and is based in Piscataway, New Jersey. It has subsidiaries in France, Japan, and China.
Note: As of October 24, 2011, Immunologix, Inc. was acquired by Intrexon Corporation. Immunologix, Inc. develops and produces antibody-based therapeutics for various biological targets. It produces human monoclonal antibodies against viral, bacterial, and tumor antigens, as well as human auto antigens.
Intrexon Corporation, founded in 1998, is a leader in synthetic biology focused on collaborating with companies in Health, Food, Energy, Environment and Consumer sectors to create biologically based products that improve quality of life and the health of the planet.
PRODUCTS AND SERVICES
Gene synthesis
Antibody services
Protein Services
Peptide services
INVESTORS
Note: The Balloch Group (‘TBG’) was established in 2001 by Howard Balloch (Canada‘s ambassador to China from 1996 to 2001). TBG has since grown from a market-entry consultancy working with North American clients in China to a leading advisory and merchant banking firm serving both domestic Chinese companies and multinational corporations. TBG was ranked as the number one boutique investment bank in China by ChinaVenture in 2008.
Monica Heger : SAN FRANCISCO (GenomeWeb) – Illumina today announced two new next-generation sequencing platforms, a targeted sequencing system called MiniSeq and a semiconductor sequencer that is still under development.
Illumina disclosed the initiatives during a presentation at the JP Morgan Healthcare conference held here today. During the presentation, Illumina CEO Jay Flatley also announced a new genotyping array called Infinium XT; a partnership with Bio-Rad to develop a single-cell sequencing workflow; preliminary estimates of its fourth-quarter 2015 revenues; and an update on existing products. The presentation followed the company’s announcement on Sunday that it has launched a new company called Grail to develop a next-generation sequencing test for early cancer detection from patient blood samples.
The MiniSeq system, which is based on Illumina’s current sequencing technology, will begin shipping early this quarter and has a list price of $49,500. It can perform a variety of targeted DNA and RNA applications, from single-gene to pathway sequencing, and promises “all-in” prices, including library prep and sequencing, of $200 to $300 per sample, Flatley said during the JP Morgan presentation.
Integrated DNA Technologies, Inc. (IDT), the global leader in nucleic acid synthesis, serving all areas of life sciences research and development, offers products for a broad range of genomics applications. IDT’s primary business is the production of custom, synthetic nucleic acids for molecular biology applications, including qPCR, sequencing, synthetic biology, and functional genomics. The company manufactures and ships an average of 44,000 custom nucleic acids per day to more than 82,000 customers worldwide. For more information, visit idtdna.com.
Dyes GMP for Molecular Diagnostics Large Scale Oligo Synthesis
Note : Skokie, IL – December 1, 2015. Integrated DNA Technologies Inc. (“IDT”), the global leader in custom nucleic acid synthesis, has entered into a definitive agreement to acquire the oligonucleotide synthesis business of AITbiotech Pte. Ltd. in Singapore (“AITbiotech”). With this acquisition, IDT expands its customer base across Southeast Asia making it possible for these additional customers to now have access to its broad range of products for genomic applications. AITbiotech will continue operations in its other core business areas.
With over 20 years of experience in oligonucleotide development and production, and over 1000 sequences manufactured, Avecia has played an integral role in the advancing oligo therapeutic market. Our mission is to continue to build value for our customers, as they progress through drug development into commercialization. And as a member of the Nitto Denko Corporation (nitto.com), Avecia is committed to the future of the oligonucleotide market. We are driven by innovative ideas and flexible solutions, designed to provide our customers with the best in service, quality, and technology.
OriGene Technologies, Inc. develops, manufactures, and sells genome wide research and diagnostic products for pharmaceutical, biotechnology, and academic research applications. The company offers cDNA clones, including TrueORF cDNA, viral ORF, destination vectors, TrueClones (human), TrueClones (mouse), organelle marker plasmids, MicroRNA tools, mutant and variant clones, plasmid purification kits, transfection reagents, and gene synthesis service; and HuSH shRNA, siRNA, miRNA, qPCR reagents, plasmid purification products, transfection reagents, PolyA+ and total RNA products, first-strand cDNA synthesis, and CRISPR/Cas9 genome products. It also provides proteins and lysates, such as purified human proteins, over-expression cell lysates, mass spectrometry standard proteins, and protein purification reagents; UltraMAB IHC antibodies, TrueMAB primary antibodies, anti-tag and fluorescent proteins, ELISA antibodies, luminex antibodies, secondary antibodies, and controls and others; and anatomic pathology products, including IHC antibodies, detection systems, and IHC accessories
The company offers luminex and ELISA antibody pairs, autoantibody profiling arrays, ELISA kits, cell assay kits, assay reagents, custom development, and fluorogenic cell assays; TissueFocus search tools; tissue sections; tissue microarrays, cancer protein lysate arrays, TissueScan cDNA arrays, tissue blocks, and quality control products, as well as tissue RNA, DNA, and protein lysates; and lab essentials. Its research areas include cancer biomarker research, RNAi, pathology IHC, stem cell research, ion channels, and protein kinase products. The company provides gene synthesis and molecular biology services, genome editing, custom cloning, custom shRNA, purified protein, monoclonal antibody development, and assay development. It sells its products through distributors worldwide, as well as online. OriGene Technologies, Inc. was incorporated in 1995 and is based in Rockville, Maryland.
Louis, MO – November 18, 2015 Merck KGaA, Darmstadt, Germany, Completes Sigma-Aldrich Acquisition
Merck KGaA today announced the completion of its $17 billion acquisition of Sigma-Aldrich, creating one of the leaders in the $130 billion global industry to help solve the toughest problems in life science.
Press Release: 18-Nov-2015
Letter to our Life Science Customers from Dr. Udit Batra
The life science business of Merck KGaA, Darmstadt, Germany brings together the world-class products and services, innovative capabilities and exceptional talent of EMD Millipore and Sigma-Aldrich to create a global leader in the life science industry.
“Everything we do starts with our shared purpose – to solve the toughest problems in life science by collaborating with the global scientific community.
This combination is built on complementary strengths, which will enable us to serve you even better as one organization than either company could alone.
This means providing a broader portfolio with a catalog of more than 300,000 products, including many of the most respected brands in the industry, greater geographic reach, and an unmatched combination of industry-leading capabilities.”
Thermo Fisher Scientific Inc. is a provider of analytical instruments, equipment, reagents and consumables, software and services for research, manufacturing, analysis, discovery and diagnostics. The company operates through four segments: Life Sciences Solutions, provides reagents, instruments and consumables used in biological and medical research, discovery and production of new drugs and vaccines as well as diagnosis of disease; Analytical Instruments, provides instruments, consumables, software and services that are used in the laboratory; Specialty Diagnostics, offers diagnostic test kits, reagents, culture media, instruments and associated products, and Laboratory Products and Services, offers self-manufactured and sourced products for the laboratory.
WALTHAM, Mass. & SANTA CLARA, Calif.–(BUSINESS WIRE)–Jan. 8, 2016– Thermo Fisher Scientific Inc. (NYSE:TMO), the world leader in serving science, and Affymetrix Inc. (NASDAQ:AFFX), a leading provider of cellular and genetic analysis products, today announced that their boards of directors have unanimously approved Thermo Fisher’s acquisition of Affymetrix for $14.00 per share in cash. The transaction represents a purchase price of approximately $1.3 billion.

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
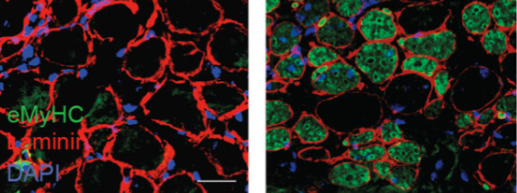{kind=link}
{kind=link}
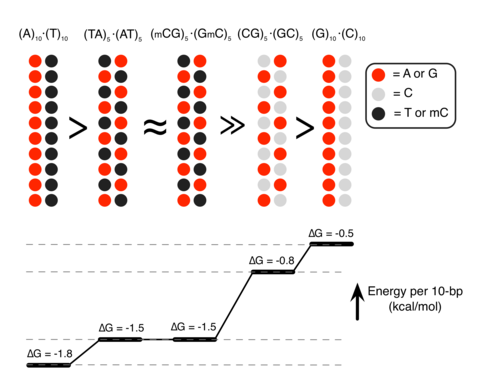{kind=link}
{kind=link}