
CRISPR/Cas9, Familial Amyloid Polyneuropathy ( FAP) and Neurodegenerative Disease
Curator: Larry H. Bernstein, MD, FCAP
CRISPR/Cas9 and Targeted Genome Editing: A New Era in Molecular Biology
The development of efficient and reliable ways to make precise, targeted changes to the genome of living cells is a long-standing goal for biomedical researchers. Recently, a new tool based on a bacterial CRISPR-associated protein-9 nuclease (Cas9) from Streptococcus pyogenes has generated considerable excitement (1). This follows several attempts over the years to manipulate gene function, including homologous recombination (2) and RNA interference (RNAi) (3). RNAi, in particular, became a laboratory staple enabling inexpensive and high-throughput interrogation of gene function (4, 5), but it is hampered by providing only temporary inhibition of gene function and unpredictable off-target effects (6). Other recent approaches to targeted genome modification – zinc-finger nucleases [ZFNs, (7)] and transcription-activator like effector nucleases [TALENs (8)]– enable researchers to generate permanent mutations by introducing doublestranded breaks to activate repair pathways. These approaches are costly and time-consuming to engineer, limiting their widespread use, particularly for large scale, high-throughput studies.
The Biology of Cas9
The functions of CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) and CRISPR-associated (Cas) genes are essential in adaptive immunity in select bacteria and archaea, enabling the organisms to respond to and eliminate invading genetic material. These repeats were initially discovered in the 1980s in E. coli (9), but their function wasn’t confirmed until 2007 by Barrangou and colleagues, who demonstrated that S. thermophilus can acquire resistance against a bacteriophage by integrating a genome fragment of an infectious virus into its CRISPR locus (10).
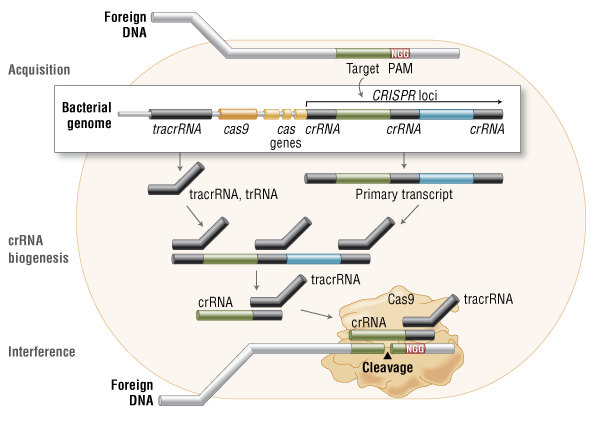
Three types of CRISPR mechanisms have been identified, of which type II is the most studied. In this case, invading DNA from viruses or plasmids is cut into small fragments and incorporated into a CRISPR locus amidst a series of short repeats (around 20 bps). The loci are transcribed, and transcripts are then processed to generate small RNAs (crRNA – CRISPR RNA), which are used to guide effector endonucleases that target invading DNA based on sequence complementarity (Figure 1) (11).
Figure 1. Cas9 in vivo: Bacterial Adaptive Immunity
https://www.neb.com/~/media/NebUs/Files/Feature%20Articles/Images/FA_Cas9_Fig1_Cas9InVivo.png
In the acquisition phase, foreign DNA is incorporated into the bacterial genome at the CRISPR loci. CRISPR loci is then transcribed and processed into crRNA during crRNA biogenesis. During interference, Cas9 endonuclease complexed with a crRNA and separate tracrRNA cleaves foreign DNA containing a 20-nucleotide crRNA complementary sequence adjacent to the PAM sequence. (Figure not drawn to scale.)
https://www.neb.com/~/media/NebUs/Files/Feature%20Articles/Images/FA_Cas9_GenomeEditingGlossary.png
One Cas protein, Cas9 (also known as Csn1), has been shown, through knockdown and rescue experiments to be a key player in certain CRISPR mechanisms (specifically type II CRISPR systems). The type II CRISPR mechanism is unique compared to other CRISPR systems, as only one Cas protein (Cas9) is required for gene silencing (12). In type II systems, Cas9 participates in the processing of crRNAs (12), and is responsible for the destruction of the target DNA (11). Cas9’s function in both of these steps relies on the presence of two nuclease domains, a RuvC-like nuclease domain located at the amino terminus and a HNH-like nuclease domain that resides in the mid-region of the protein (13).
To achieve site-specific DNA recognition and cleavage, Cas9 must be complexed with both a crRNA and a separate trans-activating crRNA (tracrRNA or trRNA), that is partially complementary to the crRNA (11). The tracrRNA is required for crRNA maturation from a primary transcript encoding multiple pre-crRNAs. This occurs in the presence of RNase III and Cas9 (12).
During the destruction of target DNA, the HNH and RuvC-like nuclease domains cut both DNA strands, generating double-stranded breaks (DSBs) at sites defined by a 20-nucleotide target sequence within an associated crRNA transcript (11, 14). The HNH domain cleaves the complementary strand, while the RuvC domain cleaves the noncomplementary strand.
The double-stranded endonuclease activity of Cas9 also requires that a short conserved sequence, (2–5 nts) known as protospacer-associated motif (PAM), follows immediately 3´- of the crRNA complementary sequence (15). In fact, even fully complementary sequences are ignored by Cas9-RNA in the absence of a PAM sequence (16).
Cas9 and CRISPR as a New Tool in Molecular Biology
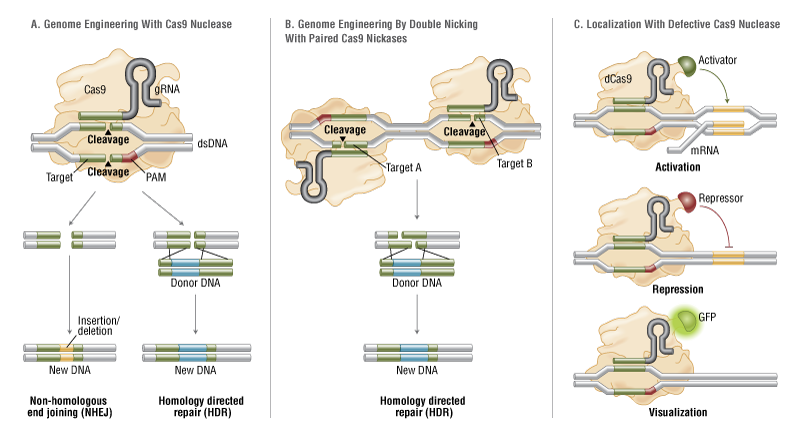
The simplicity of the type II CRISPR nuclease, with only three required components (Cas9 along with the crRNA and trRNA) makes this system amenable to adaptation for genome editing. This potential was realized in 2012 by the Doudna and Charpentier labs (11). Based on the type II CRISPR system described previously, the authors developed a simplified two-component system by combining trRNA and crRNA into a single synthetic single guide RNA (sgRNA). sgRNAprogrammed Cas9 was shown to be as effective as Cas9 programmed with separate trRNA and crRNA in guiding targeted gene alterations (Figure 2A).
To date, three different variants of the Cas9 nuclease have been adopted in genome-editing protocols. The first is wild-type Cas9, which can site-specifically cleave double-stranded DNA, resulting in the activation of the doublestrand break (DSB) repair machinery. DSBs can be repaired by the cellular Non-Homologous End Joining (NHEJ) pathway (17), resulting in insertions and/or deletions (indels) which disrupt the targeted locus. Alternatively, if a donor template with homology to the targeted locus is supplied, the DSB may be repaired by the homology-directed repair (HDR) pathway allowing for precise replacement mutations to be made (Figure 2A) (17, 18).
Cong and colleagues (1) took the Cas9 system a step further towards increased precision by developing a mutant form, known as Cas9D10A, with only nickase activity. This means it cleaves only one DNA strand, and does not activate NHEJ. Instead, when provided with a homologous repair template, DNA repairs are conducted via the high-fidelity HDR pathway only, resulting in reduced indel mutations (1, 11, 19). Cas9D10A is even more appealing in terms of target specificity when loci are targeted by paired Cas9 complexes designed to generate adjacent DNA nicks (20) (see further details about “paired nickases” in Figure 2B).
The third variant is a nuclease-deficient Cas9 (dCas9, Figure 2C) (21). Mutations H840A in the HNH domain and D10A in the RuvC domain inactivate cleavage activity, but do not prevent DNA binding (11, 22). Therefore, this variant can be used to sequence-specifically target any region of the genome without cleavage. Instead, by fusing with various effector domains, dCas9 can be used either as a gene silencing or activation tool (21, 23–26). Furthermore, it can be used as a visualization tool. For instance, Chen and colleagues used dCas9 fused to Enhanced Green Fluorescent Protein (EGFP) to visualize repetitive DNA sequences with a single sgRNA or nonrepetitive loci using multiple sgRNAs (27).
Figure 2. CRISPR/Cas9 System Applications
- Wild-type Cas9 nuclease site specifically cleaves double-stranded DNA activating double-strand break repair machinery. In the absence of a homologous repair template non-homologous end joining can result in indels disrupting the target sequence. Alternatively, precise mutations and knock-ins can be made by providing a homologous repair template and exploiting the homology directed repair pathway.
B. Mutated Cas9 makes a site specific single-strand nick. Two sgRNA can be used to introduce a staggered double-stranded break which can then undergo homology directed repair.
C. Nuclease-deficient Cas9 can be fused with various effector domains allowing specific localization. For example, transcriptional activators, repressors, and fluorescent proteins.
Targeting Efficiency and Off-target Mutations
Targeting efficiency, or the percentage of desired mutation achieved, is one of the most important parameters by which to assess a genome-editing tool. The targeting efficiency of Cas9 compares favorably with more established methods, such as TALENs or ZFNs (8). For example, in human cells, custom-designed ZFNs and TALENs could only achieve efficiencies ranging from 1% to 50% (29–31). In contrast, the Cas9 system has been reported to have efficiencies up to >70% in zebrafish (32) and plants (33), and ranging from 2–5% in induced pluripotent stem cells (34). In addition, Zhou and colleagues were able to improve genome targeting up to 78% in one-cell mouse embryos, and achieved effective germline transmission through the use of dual sgRNAs to simultaneously target an individual gene (35).
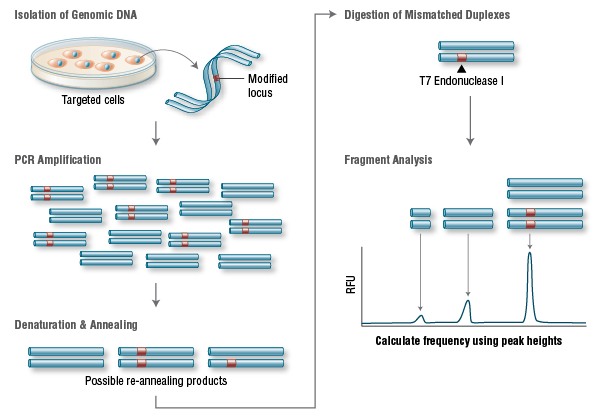
A widely used method to identify mutations is the T7 Endonuclease I mutation detection assay (36, 37) (Figure 3). This assay detects heteroduplex DNA that results from the annealing of a DNA strand, including desired mutations, with a wildtype DNA strand (37).
Figure 3. T7 Endonuclease I Targeting Efficiency Assay
Genomic DNA is amplified with primers bracketing the modified locus. PCR products are then denatured and re-annealed yielding 3 possible structures. Duplexes containing a mismatch are digested by T7 Endonuclease I. The DNA is then electrophoretically separated and fragment analysis is used to calculate targeting efficiency.
Another important parameter is the incidence of off-target mutations. Such mutations are likely to appear in sites that have differences of only a few nucleotides compared to the original sequence, as long as they are adjacent to a PAM sequence. This occurs as Cas9 can tolerate up to 5 base mismatches within the protospacer region (36) or a single base difference in the PAM sequence (38). Off-target mutations are generally more difficult to detect, requiring whole-genome sequencing to rule them out completely.
Recent improvements to the CRISPR system for reducing off-target mutations have been made through the use of truncated gRNA (truncated within the crRNA-derived sequence) or by adding two extra guanine (G) nucleotides to the 5´ end (28, 37). Another way researchers have attempted to minimize off-target effects is with the use of “paired nickases” (20). This strategy uses D10A Cas9 and two sgRNAs complementary to the adjacent area on opposite strands of the target site (Figure 2B). While this induces DSBs in the target DNA, it is expected to create only single nicks in off-target locations and, therefore, result in minimal off-target mutations.
By leveraging computation to reduce off-target mutations, several groups have developed webbased tools to facilitate the identification of potential CRISPR target sites and assess their potential for off-target cleavage. Examples include the CRISPR Design Tool (38) and the ZiFiT Targeter, Version 4.2 (39, 40).
Applications as a Genome-editing and Genome Targeting Tool
Following its initial demonstration in 2012 (9), the CRISPR/Cas9 system has been widely adopted. This has already been successfully used to target important genes in many cell lines and organisms, including human (34), bacteria (41), zebrafish (32), C. elegans (42), plants (34), Xenopus tropicalis (43), yeast (44), Drosophila (45), monkeys (46), rabbits (47), pigs (42), rats (48) and mice (49). Several groups have now taken advantage of this method to introduce single point mutations (deletions or insertions) in a particular target gene, via a single gRNA (14, 21, 29). Using a pair of gRNA-directed Cas9 nucleases instead, it is also possible to induce large deletions or genomic rearrangements, such as inversions or translocations (50). A recent exciting development is the use of the dCas9 version of the CRISPR/Cas9 system to target protein domains for transcriptional regulation (26, 51, 52), epigenetic modification (25), and microscopic visualization of specific genome loci (27).
The CRISPR/Cas9 system requires only the redesign of the crRNA to change target specificity. This contrasts with other genome editing tools, including zinc finger and TALENs, where redesign of the protein-DNA interface is required. Furthermore, CRISPR/Cas9 enables rapid genome-wide interrogation of gene function by generating large gRNA libraries (51, 53) for genomic screening.
The Future of CRISPR/Cas9
The rapid progress in developing Cas9 into a set of tools for cell and molecular biology research has been remarkable, likely due to the simplicity, high efficiency and versatility of the system. Of the designer nuclease systems currently available for precision genome engineering, the CRISPR/Cas system is by far the most user friendly. It is now also clear that Cas9’s potential reaches beyond DNA cleavage, and its usefulness for genome locus-specific recruitment of proteins will likely only be limited by our imagination.
Scientists urge caution in using new CRISPR technology to treat human genetic disease
By Robert Sanders, Media relations | MARCH 19, 2015
http://news.berkeley.edu/2015/03/19/scientists-urge-caution-in-using-new-crispr-technology-to-treat-human-genetic-disease/
http://news.berkeley.edu/wp-content/uploads/2015/03/crispr350.jpg
The bacterial enzyme Cas9 is the engine of RNA-programmed genome engineering in human cells. (Graphic by Jennifer Doudna/UC Berkeley)
A group of 18 scientists and ethicists today warned that a revolutionary new tool to cut and splice DNA should be used cautiously when attempting to fix human genetic disease, and strongly discouraged any attempts at making changes to the human genome that could be passed on to offspring.
Among the authors of this warning is Jennifer Doudna, the co-inventor of the technology, called CRISPR-Cas9, which is driving a new interest in gene therapy, or “genome engineering.” She and colleagues co-authored a perspective piece that appears in the March 20 issue of Science, based on discussions at a meeting that took place in Napa on Jan. 24. The same issue of Science features a collection of recent research papers, commentary and news articles on CRISPR and its implications. …..
A prudent path forward for genomic engineering and germline gene modification
David Baltimore1, Paul Berg2, …., Jennifer A. Doudna4,10,*, et al.
http://science.sciencemag.org/content/early/2015/03/18/science.aab1028.full
Science 19 Mar 2015. http://dx.doi.org:/10.1126/science.aab1028
Correcting genetic defects
Scientists today are changing DNA sequences to correct genetic defects in animals as well as cultured tissues generated from stem cells, strategies that could eventually be used to treat human disease. The technology can also be used to engineer animals with genetic diseases mimicking human disease, which could lead to new insights into previously enigmatic disorders.
The CRISPR-Cas9 tool is still being refined to ensure that genetic changes are precisely targeted, Doudna said. Nevertheless, the authors met “… to initiate an informed discussion of the uses of genome engineering technology, and to identify proactively those areas where current action is essential to prepare for future developments. We recommend taking immediate steps toward ensuring that the application of genome engineering technology is performed safely and ethically.”
Amyloid CRISPR Plasmids and si/shRNA Gene Silencers
http://www.scbt.com/crispr/table-amyloid.html
Santa Cruz Biotechnology, Inc. offers a broad range of gene silencers in the form of siRNAs, shRNA Plasmids and shRNA Lentiviral Particles as well as CRISPR/Cas9 Knockout and CRISPR Double Nickase plasmids. Amyloid gene silencers are available as Amyloid siRNA, Amyloid shRNA Plasmid, Amyloid shRNA Lentiviral Particles and Amyloid CRISPR/Cas9 Knockout plasmids. Amyloid CRISPR/dCas9 Activation Plasmids and CRISPR Lenti Activation Systems for gene activation are also available. Gene silencers and activators are useful for gene studies in combination with antibodies used for protein detection. Amyloid CRISPR Knockout, HDR and Nickase Knockout Plasmids
CRISPR-Cas9-Based Knockout of the Prion Protein and Its Effect on the Proteome
Mehrabian M, Brethour D, MacIsaac S, Kim JK, Gunawardana C.G, Wang H, et al.
PLoS ONE 2014; 9(12): e114594. http://dx.doi.org/10.1371/journal.pone.0114594
The molecular function of the cellular prion protein (PrPC) and the mechanism by which it may contribute to neurotoxicity in prion diseases and Alzheimer’s disease are only partially understood. Mouse neuroblastoma Neuro2a cells and, more recently, C2C12 myocytes and myotubes have emerged as popular models for investigating the cellular biology of PrP. Mouse epithelial NMuMG cells might become attractive models for studying the possible involvement of PrP in a morphogenetic program underlying epithelial-to-mesenchymal transitions. Here we describe the generation of PrP knockout clones from these cell lines using CRISPR-Cas9 knockout technology. More specifically, knockout clones were generated with two separate guide RNAs targeting recognition sites on opposite strands within the first hundred nucleotides of the Prnp coding sequence. Several PrP knockout clones were isolated and genomic insertions and deletions near the CRISPR-target sites were characterized. Subsequently, deep quantitative global proteome analyses that recorded the relative abundance of>3000 proteins (data deposited to ProteomeXchange Consortium) were undertaken to begin to characterize the molecular consequences of PrP deficiency. The levels of ∼120 proteins were shown to reproducibly correlate with the presence or absence of PrP, with most of these proteins belonging to extracellular components, cell junctions or the cytoskeleton.
Development and Applications of CRISPR-Cas9 for Genome Engineering
Patrick D. Hsu,1,2,3 Eric S. Lander,1 and Feng Zhang1,2,*
Cell. 2014 Jun 5; 157(6): 1262–1278. doi: 10.1016/j.cell.2014.05.010
Recent advances in genome engineering technologies based on the CRISPR-associated RNA-guided endonuclease Cas9 are enabling the systematic interrogation of mammalian genome function. Analogous to the search function in modern word processors, Cas9 can be guided to specific locations within complex genomes by a short RNA search string. Using this system, DNA sequences within the endogenous genome and their functional outputs are now easily edited or modulated in virtually any organism of choice. Cas9-mediated genetic perturbation is simple and scalable, empowering researchers to elucidate the functional organization of the genome at the systems level and establish causal linkages between genetic variations and biological phenotypes. In this Review, we describe the development and applications of Cas9 for a variety of research or translational applications while highlighting challenges as well as future directions. Derived from a remarkable microbial defense system, Cas9 is driving innovative applications from basic biology to biotechnology and medicine.
The development of recombinant DNA technology in the 1970s marked the beginning of a new era for biology. For the first time, molecular biologists gained the ability to manipulate DNA molecules, making it possible to study genes and harness them to develop novel medicine and biotechnology. Recent advances in genome engineering technologies are sparking a new revolution in biological research. Rather than studying DNA taken out of the context of the genome, researchers can now directly edit or modulate the function of DNA sequences in their endogenous context in virtually any organism of choice, enabling them to elucidate the functional organization of the genome at the systems level, as well as identify causal genetic variations.
Broadly speaking, genome engineering refers to the process of making targeted modifications to the genome, its contexts (e.g., epigenetic marks), or its outputs (e.g., transcripts). The ability to do so easily and efficiently in eukaryotic and especially mammalian cells holds immense promise to transform basic science, biotechnology, and medicine (Figure 1).
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4343198/bin/nihms659174f1.jpg
For life sciences research, technologies that can delete, insert, and modify the DNA sequences of cells or organisms enable dissecting the function of specific genes and regulatory elements. Multiplexed editing could further allow the interrogation of gene or protein networks at a larger scale. Similarly, manipulating transcriptional regulation or chromatin states at particular loci can reveal how genetic material is organized and utilized within a cell, illuminating relationships between the architecture of the genome and its functions. In biotechnology, precise manipulation of genetic building blocks and regulatory machinery also facilitates the reverse engineering or reconstruction of useful biological systems, for example, by enhancing biofuel production pathways in industrially relevant organisms or by creating infection-resistant crops. Additionally, genome engineering is stimulating a new generation of drug development processes and medical therapeutics. Perturbation of multiple genes simultaneously could model the additive effects that underlie complex polygenic disorders, leading to new drug targets, while genome editing could directly correct harmful mutations in the context of human gene therapy (Tebas et al., 2014).
Eukaryotic genomes contain billions of DNA bases and are difficult to manipulate. One of the breakthroughs in genome manipulation has been the development of gene targeting by homologous recombination (HR), which integrates exogenous repair templates that contain sequence homology to the donor site (Figure 2A) (Capecchi, 1989). HR-mediated targeting has facilitated the generation of knockin and knockout animal models via manipulation of germline competent stem cells, dramatically advancing many areas of biological research. However, although HR-mediated gene targeting produces highly precise alterations, the desired recombination events occur extremely infrequently (1 in 106–109 cells) (Capecchi, 1989), presenting enormous challenges for large-scale applications of gene-targeting experiments.
Genome Editing Technologies Exploit Endogenous DNA Repair Machinery
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4343198/bin/nihms659174f2.gif
To overcome these challenges, a series of programmable nuclease-based genome editing technologies have been developed in recent years, enabling targeted and efficient modification of a variety of eukaryotic and particularly mammalian species. Of the current generation of genome editing technologies, the most rapidly developing is the class of RNA-guided endonucleases known as Cas9 from the microbial adaptive immune system CRISPR (clustered regularly interspaced short palindromic repeats), which can be easily targeted to virtually any genomic location of choice by a short RNA guide. Here, we review the development and applications of the CRISPR-associated endonuclease Cas9 as a platform technology for achieving targeted perturbation of endogenous genomic elements and also discuss challenges and future avenues for innovation. ……
Figure 4 Natural Mechanisms of Microbial CRISPR Systems in Adaptive Immunity
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4343198/bin/nihms659174f4.gif
…… A key turning point came in 2005, when systematic analysis of the spacer sequences separating the individual direct repeats suggested their extrachromosomal and phage-associated origins (Mojica et al., 2005; Pourcel et al., 2005; Bolotin et al., 2005). This insight was tremendously exciting, especially given previous studies showing that CRISPR loci are transcribed (Tang et al., 2002) and that viruses are unable to infect archaeal cells carrying spacers corresponding to their own genomes (Mojica et al., 2005). Together, these findings led to the speculation that CRISPR arrays serve as an immune memory and defense mechanism, and individual spacers facilitate defense against bacteriophage infection by exploiting Watson-Crick base-pairing between nucleic acids (Mojica et al., 2005; Pourcel et al., 2005). Despite these compelling realizations that CRISPR loci might be involved in microbial immunity, the specific mechanism of how the spacers act to mediate viral defense remained a challenging puzzle. Several hypotheses were raised, including thoughts that CRISPR spacers act as small RNA guides to degrade viral transcripts in a RNAi-like mechanism (Makarova et al., 2006) or that CRISPR spacers direct Cas enzymes to cleave viral DNA at spacer-matching regions (Bolotin et al., 2005). …..
As the pace of CRISPR research accelerated, researchers quickly unraveled many details of each type of CRISPR system (Figure 4). Building on an earlier speculation that protospacer adjacent motifs (PAMs) may direct the type II Cas9 nuclease to cleave DNA (Bolotin et al., 2005), Moineau and colleagues highlighted the importance of PAM sequences by demonstrating that PAM mutations in phage genomes circumvented CRISPR interference (Deveau et al., 2008). Additionally, for types I and II, the lack of PAM within the direct repeat sequence within the CRISPR array prevents self-targeting by the CRISPR system. In type III systems, however, mismatches between the 5′ end of the crRNA and the DNA target are required for plasmid interference (Marraffini and Sontheimer, 2010). …..
In 2013, a pair of studies simultaneously showed how to successfully engineer type II CRISPR systems from Streptococcus thermophilus (Cong et al., 2013) andStreptococcus pyogenes (Cong et al., 2013; Mali et al., 2013a) to accomplish genome editing in mammalian cells. Heterologous expression of mature crRNA-tracrRNA hybrids (Cong et al., 2013) as well as sgRNAs (Cong et al., 2013; Mali et al., 2013a) directs Cas9 cleavage within the mammalian cellular genome to stimulate NHEJ or HDR-mediated genome editing. Multiple guide RNAs can also be used to target several genes at once. Since these initial studies, Cas9 has been used by thousands of laboratories for genome editing applications in a variety of experimental model systems (Sander and Joung, 2014). ……
The majority of CRISPR-based technology development has focused on the signature Cas9 nuclease from type II CRISPR systems. However, there remains a wide diversity of CRISPR types and functions. Cas RAMP module (Cmr) proteins identified in Pyrococcus furiosus and Sulfolobus solfataricus (Hale et al., 2012) constitute an RNA-targeting CRISPR immune system, forming a complex guided by small CRISPR RNAs that target and cleave complementary RNA instead of DNA. Cmr protein homologs can be found throughout bacteria and archaea, typically relying on a 5′ site tag sequence on the target-matching crRNA for Cmr-directed cleavage.
Unlike RNAi, which is targeted largely by a 6 nt seed region and to a lesser extent 13 other bases, Cmr crRNAs contain 30–40 nt of target complementarity. Cmr-CRISPR technologies for RNA targeting are thus a promising target for orthogonal engineering and minimal off-target modification. Although the modularity of Cmr systems for RNA-targeting in mammalian cells remains to be investigated, Cmr complexes native to P. furiosus have already been engineered to target novel RNA substrates (Hale et al., 2009, 2012). ……
Although Cas9 has already been widely used as a research tool, a particularly exciting future direction is the development of Cas9 as a therapeutic technology for treating genetic disorders. For a monogenic recessive disorder due to loss-of-function mutations (such as cystic fibrosis, sickle-cell anemia, or Duchenne muscular dystrophy), Cas9 may be used to correct the causative mutation. This has many advantages over traditional methods of gene augmentation that deliver functional genetic copies via viral vector-mediated overexpression—particularly that the newly functional gene is expressed in its natural context. For dominant-negative disorders in which the affected gene is haplosufficient (such as transthyretin-related hereditary amyloidosis or dominant forms of retinitis pigmentosum), it may also be possible to use NHEJ to inactivate the mutated allele to achieve therapeutic benefit. For allele-specific targeting, one could design guide RNAs capable of distinguishing between single-nucleotide polymorphism (SNP) variations in the target gene, such as when the SNP falls within the PAM sequence.
CRISPR/Cas9: a powerful genetic engineering tool for establishing large animal models of neurodegenerative diseases
Zhuchi Tu, Weili Yang, Sen Yan, Xiangyu Guo and Xiao-Jiang Li
Molecular Neurodegeneration 2015; 10:35 http://dx.doi.org:/10.1186/s13024-015-0031-x
Animal models are extremely valuable to help us understand the pathogenesis of neurodegenerative disorders and to find treatments for them. Since large animals are more like humans than rodents, they make good models to identify the important pathological events that may be seen in humans but not in small animals; large animals are also very important for validating effective treatments or confirming therapeutic targets. Due to the lack of embryonic stem cell lines from large animals, it has been difficult to use traditional gene targeting technology to establish large animal models of neurodegenerative diseases. Recently, CRISPR/Cas9 was used successfully to genetically modify genomes in various species. Here we discuss the use of CRISPR/Cas9 technology to establish large animal models that can more faithfully mimic human neurodegenerative diseases.
Neurodegenerative diseases — Alzheimer’s disease(AD),Parkinson’s disease(PD), amyotrophic lateral sclerosis (ALS), Huntington’s disease (HD), and frontotemporal dementia (FTD) — are characterized by age-dependent and selective neurodegeneration. As the life expectancy of humans lengthens, there is a greater prevalence of these neurodegenerative diseases; however, the pathogenesis of most of these neurodegenerative diseases remain unclear, and we lack effective treatments for these important brain disorders.
CRISPR/Cas9, Non-human primates, Neurodegenerative diseases, Animal model
There are a number of excellent reviews covering different types of neurodegenerative diseases and their genetic mouse models [8–12]. Investigations of different mouse models of neurodegenerative diseases have revealed a common pathology shared by these diseases. First, the development of neuropathology and neurological symptoms in genetic mouse models of neurodegenerative diseases is age dependent and progressive. Second, all the mouse models show an accumulation of misfolded or aggregated proteins resulting from the expression of mutant genes. Third, despite the widespread expression of mutant proteins throughout the body and brain, neuronal function appears to be selectively or preferentially affected. All these facts indicate that mouse models of neurodegenerative diseases recapitulate important pathologic features also seen in patients with neurodegenerative diseases.
However, it seems that mouse models can not recapitulate the full range of neuropathology seen in patients with neurodegenerative diseases. Overt neurodegeneration, which is the most important pathological feature in patient brains, is absent in genetic rodent models of AD, PD, and HD. Many rodent models that express transgenic mutant proteins under the control of different promoters do not replicate overt neurodegeneration, which is likely due to their short life spans and the different aging processes of small animals. Also important are the remarkable differences in brain development between rodents and primates. For example, the mouse brain takes 21 days to fully develop, whereas the formation of primate brains requires more than 150 days [13]. The rapid development of the brain in rodents may render neuronal cells resistant to misfolded protein-mediated neurodegeneration. Another difficulty in using rodent models is how to analyze cognitive and emotional abnormalities, which are the early symptoms of most neurodegenerative diseases in humans. Differences in neuronal circuitry, anatomy, and physiology between rodent and primate brains may also account for the behavioral differences between rodent and primate models.
Mitochondrial dynamics–fusion, fission, movement, and mitophagy–in neurodegenerative diseases
Hsiuchen Chen and David C. Chan
Human Molec Gen 2009; 18, Review Issue 2 R169–R176
http://dx.doi.org:/10.1093/hmg/ddp326
Neurons are metabolically active cells with high energy demands at locations distant from the cell body. As a result, these cells are particularly dependent on mitochondrial function, as reflected by the observation that diseases of mitochondrial dysfunction often have a neurodegenerative component. Recent discoveries have highlighted that neurons are reliant particularly on the dynamic properties of mitochondria. Mitochondria are dynamic organelles by several criteria. They engage in repeated cycles of fusion and fission, which serve to intermix the lipids and contents of a population of mitochondria. In addition, mitochondria are actively recruited to subcellular sites, such as the axonal and dendritic processes of neurons. Finally, the quality of a mitochondrial population is maintained through mitophagy, a form of autophagy in which defective mitochondria are selectively degraded. We review the general features of mitochondrial dynamics, incorporating recent findings on mitochondrial fusion, fission, transport and mitophagy. Defects in these key features are associated with neurodegenerative disease. Charcot-Marie-Tooth type 2A, a peripheral neuropathy, and dominant optic atrophy, an inherited optic neuropathy, result from a primary deficiency of mitochondrial fusion. Moreover, several major neurodegenerative diseases—including Parkinson’s, Alzheimer’s and Huntington’s disease—involve disruption of mitochondrial dynamics. Remarkably, in several disease models, the manipulation of mitochondrial fusion or fission can partially rescue disease phenotypes. We review how mitochondrial dynamics is altered in these neurodegenerative diseases and discuss the reciprocal interactions between mitochondrial fusion, fission, transport and mitophagy.
Applications of CRISPR–Cas systems in Neuroscience
Matthias Heidenreich & Feng Zhang
Nature Rev Neurosci 2016; 17:36–44 http://dx.doi.org:/10.1038/nrn.2015.2
Genome-editing tools, and in particular those based on CRISPR–Cas (clustered regularly interspaced short palindromic repeat (CRISPR)–CRISPR-associated protein) systems, are accelerating the pace of biological research and enabling targeted genetic interrogation in almost any organism and cell type. These tools have opened the door to the development of new model systems for studying the complexity of the nervous system, including animal models and stem cell-derived in vitro models. Precise and efficient gene editing using CRISPR–Cas systems has the potential to advance both basic and translational neuroscience research.
Cellular neuroscience, DNA recombination, Genetic engineering, Molecular neuroscience
Figure 3: In vitro applications of Cas9 in human iPSCs.close
http://www.nature.com/nrn/journal/v17/n1/carousel/nrn.2015.2-f3.jpg
a | Evaluation of disease candidate genes from large-population genome-wide association studies (GWASs). Human primary cells, such as neurons, are not easily available and are difficult to expand in culture. By contrast, induced pluripo…
Molecular Therapy 12 Jan 2016
Scientific Reports 31 Mar 2016
Scientific Reports 12 Nov 2015
Alzheimer’s Disease: Medicine’s Greatest Challenge in the 21st Century
https://www.physicsforums.com/insights/can-gene-editing-eliminate-alzheimers-disease/
The development of the CRISPR/Cas9 system has made gene editing a relatively simple task. While CRISPR and other gene editing technologies stand to revolutionize biomedical research and offers many promising therapeutic avenues (such as in the treatment of HIV), a great deal of debate exists over whether CRISPR should be used to modify human embryos. As I discussed in my previous Insight article, we lack enough fundamental biological knowledge to enhance many traits like height or intelligence, so we are not near a future with genetically-enhanced super babies. However, scientists have identified a few rare genetic variants that protect against disease. One such protective variant is a mutation in the APP gene that protects against Alzheimer’s disease and cognitive decline in old age. If we can perfect gene editing technologies, is this mutation one that we should be regularly introducing into embryos? In this article, I explore the potential for using gene editing as a way to prevent Alzheimer’s disease in future generations. Alzheimer’s Disease: Medicine’s Greatest Challenge in the 21st Century Can gene editing be the missing piece in the battle against Alzheimer’s? (Source: bostonbiotech.org) I chose to assess the benefit of germline gene editing in the context of Alzheimer’s disease because this disease is one of the biggest challenges medicine faces in the 21st century. Alzheimer’s disease is a chronic neurodegenerative disease responsible for the majority of the cases of dementia in the elderly. The disease symptoms begins with short term memory loss and causes more severe symptoms – problems with language, disorientation, mood swings, behavioral issues – as it progresses, eventually leading to the loss of bodily functions and death. Because of the dementia the disease causes, Alzheimer’s patients require a great deal of care, and the world spends ~1% of its total GDP on caring for those with Alzheimer’s and related disorders. Because the prevalence of the disease increases with age, the situation will worsen as life expectancies around the globe increase: worldwide cases of Alzheimer’s are expected to grow from 35 million today to over 115 million by 2050.
Despite much research, the exact causes of Alzheimer’s disease remains poorly understood. The disease seems to be related to the accumulation of plaques made of amyloid-β peptides that form on the outside of neurons, as well as the formation of tangles of the protein tau inside of neurons. Although many efforts have been made to target amyloid-β or the enzymes involved in its formation, we have so far been unsuccessful at finding any treatment that stops the disease or reverses its progress. Some researchers believe that most attempts at treating Alzheimer’s have failed because, by the time a patient shows symptoms, the disease has already progressed past the point of no return.
While research towards a cure continues, researchers have sought effective ways to prevent Alzheimer’s disease. Although some studies show that mental and physical exercise may lower ones risk of Alzheimer’s disease, approximately 60-80% of the risk for Alzheimer’s disease appears to be genetic. Thus, if we’re serious about prevention, we may have to act at the genetic level. And because the brain is difficult to access surgically for gene therapy in adults, this means using gene editing on embryos.
Reference https://www.physicsforums.com/insights/can-gene-editing-eliminate-alzheimers-disease/
Utilising CRISPR to Generate Predictive Disease Models: a Case Study in Neurodegenerative Disorders
Dr. Bhuvaneish.T. Selvaraj – Scottish Centre for Regenerative Medicine
- Introducing the latest developments in predictive model generation
- Discover how CRISPR is being used to develop disease models to study and treat neurodegenerative disorders
- In depth Q&A session to answer your most pressing questions
Turning On Genes, Systematically, with CRISPR/Cas9
Scientists based at MIT assert that they can reliably turn on any gene of their choosing in living cells. [Feng Zhang and Steve Dixon] http://www.genengnews.com/media/images/GENHighlight/Dec12_2014_CRISPRCas9GeneActivationSystem7838101231.jpg
With the latest CRISPR/Cas9 advance, the exhortation “turn on, tune in, drop out” comes to mind. The CRISPR/Cas9 gene-editing system was already a well-known means of “tuning in” (inserting new genes) and “dropping out” (knocking out genes). But when it came to “turning on” genes, CRISPR/Cas9 had little potency. That is, it had demonstrated only limited success as a way to activate specific genes.
A new CRISPR/Cas9 approach, however, appears capable of activating genes more effectively than older approaches. The new approach may allow scientists to more easily determine the function of individual genes, according to Feng Zhang, Ph.D., a researcher at MIT and the Broad Institute. Dr. Zhang and colleagues report that the new approach permits multiplexed gene activation and rapid, large-scale studies of gene function.
The new technique was introduced in the December 10 online edition of Nature, in an article entitled, “Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex.” The article describes how Dr. Zhang, along with the University of Tokyo’s Osamu Nureki, Ph.D., and Hiroshi Nishimasu, Ph.D., overhauled the CRISPR/Cas9 system. The research team based their work on their analysis (published earlier this year) of the structure formed when Cas9 binds to the guide RNA and its target DNA. Specifically, the team used the structure’s 3D shape to rationally improve the system.
In previous efforts to revamp CRISPR/Cas9 for gene activation purposes, scientists had tried to attach the activation domains to either end of the Cas9 protein, with limited success. From their structural studies, the MIT team realized that two small loops of the RNA guide poke out from the Cas9 complex and could be better points of attachment because they allow the activation domains to have more flexibility in recruiting transcription machinery.
Using their revamped system, the researchers activated about a dozen genes that had proven difficult or impossible to turn on using the previous generation of Cas9 activators. Each gene showed at least a twofold boost in transcription, and for many genes, the researchers found multiple orders of magnitude increase in activation.
After investigating single-guide RNA targeting rules for effective transcriptional activation, demonstrating multiplexed activation of 10 genes simultaneously, and upregulating long intergenic noncoding RNA transcripts, the research team decided to undertake a large-scale screen. This screen was designed to identify genes that confer resistance to a melanoma drug called PLX-4720.
“We … synthesized a library consisting of 70,290 guides targeting all human RefSeq coding isoforms to screen for genes that, upon activation, confer resistance to a BRAF inhibitor,” wrote the authors of the Nature paper. “The top hits included genes previously shown to be able to confer resistance, and novel candidates were validated using individual [single-guide RNA] and complementary DNA overexpression.”
A gene signature based on the top screening hits, the authors added, correlated with a gene expression signature of BRAF inhibitor resistance in cell lines and patient-derived samples. It was also suggested that large-scale screens such as the one demonstrated in the current study could help researchers discover new cancer drugs that prevent tumors from becoming resistant.
Susceptibility and modifier genes in Portuguese transthyretin V30M amyloid polyneuropathy: complexity in a single-gene disease
Miguel L. Soares1,2, Teresa Coelho3,6, Alda Sousa4,5, …, Maria Joa˜o Saraiva2,5 and Joel N. Buxbaum1
Human Molec Gen 2005; 14(4): 543–553 http://dx.doi.org:/10.1093/hmg/ddi051
https://www.researchgate.net/profile/Isabel_Conceicao/publication/8081351_Susceptibility_and_modifier_genes_in_Portuguese_transthyretin_V30M_amyloid_polyneuropathy_complexity_in_a_single-gene_disease/links/53e123d70cf2235f352733b3.pdf
Familial amyloid polyneuropathy type I is an autosomal dominant disorder caused by mutations in the transthyretin (TTR ) gene; however, carriers of the same mutation exhibit variability in penetrance and clinical expression. We analyzed alleles of candidate genes encoding non-fibrillar components of TTR amyloid deposits and a molecule metabolically interacting with TTR [retinol-binding protein (RBP)], for possible associations with age of disease onset and/or susceptibility in a Portuguese population sample with the TTR V30M mutation and unrelated controls. We show that the V30M carriers represent a distinct subset of the Portuguese population. Estimates of genetic distance indicated that the controls and the classical onset group were furthest apart, whereas the late-onset group appeared to differ from both. Importantly, the data also indicate that genetic interactions among the multiple loci evaluated, rather than single-locus effects, are more likely to determine differences in the age of disease onset. Multifactor dimensionality reduction indicated that the best genetic model for classical onset group versus controls involved the APCS gene, whereas for late-onset cases, one APCS variant (APCSv1) and two RBP variants (RBPv1 and RBPv2) are involved. Thus, although the TTR V30M mutation is required for the disease in Portuguese patients, different genetic factors may govern the age of onset, as well as the occurrence of anticipation.
Autosomal dominant disorders may vary in expression even within a given kindred. The basis of this variability is uncertain and can be attributed to epigenetic factors, environment or epistasis. We have studied familial amyloid polyneuropathy (FAP), an autosomal dominant disorder characterized by peripheral sensorimotor and autonomic neuropathy. It exhibits variation in cardiac, renal, gastrointestinal and ocular involvement, as well as age of onset. Over 80 missense mutations in the transthyretin gene (TTR ) result in autosomal dominant disease http://www.ibmc.up.pt/~mjsaraiv/ttrmut.html). The presence of deposits consisting entirely of wild-type TTR molecules in the hearts of 10– 25% of individuals over age 80 reveals its inherent in vivo amyloidogenic potential (1).
FAP was initially described in Portuguese (2) where, until recently, the TTR V30M has been the only pathogenic mutation associated with the disease (3,4). Later reports identified the same mutation in Swedish and Japanese families (5,6). The disorder has since been recognized in other European countries and in North American kindreds in association with V30M, as well as other mutations (7).
TTR V30M produces disease in only 5–10% of Swedish carriers of the allele (8), a much lower degree of penetrance than that seen in Portuguese (80%) (9) or in Japanese with the same mutation. The actual penetrance in Japanese carriers has not been formally established, but appears to resemble that seen in Portuguese. Portuguese and Japanese carriers show considerable variation in the age of clinical onset (10,11). In both populations, the first symptoms had originally been described as typically occurring before age 40 (so-called ‘classical’ or early-onset); however, in recent years, more individuals developing symptoms late in life have been identified (11,12). Hence, present data indicate that the distribution of the age of onset in Portuguese is continuous, but asymmetric with a mean around age 35 and a long tail into the older age group (Fig. 1) (9,13). Further, DNA testing in Portugal has identified asymptomatic carriers over age 70 belonging to a subset of very late-onset kindreds in whose descendants genetic anticipation is frequent. The molecular basis of anticipation in FAP, which is not mediated by trinucleotide repeat expansions in the TTR or any other gene (14), remains elusive.
Variation in penetrance, age of onset and clinical features are hallmarks of many autosomal dominant disorders including the human TTR amyloidoses (7). Some of these clearly reflect specific biological effects of a particular mutation or a class of mutants. However, when such phenotypic variability is seen with a single mutation in the gene encoding the same protein, it suggests an effect of modifying genetic loci and/or environmental factors contributing differentially to the course of disease. We have chosen to examine age of onset as an example of a discrete phenotypic variation in the presence of the particular autosomal dominant disease-associated mutation TTR V30M. Although the role of environmental factors cannot be excluded, the existence of modifier genes involved in TTR amyloidogenesis is an attractive hypothesis to explain the phenotypic variability in FAP. ….
ATTR (TTR amyloid), like all amyloid deposits, contains several molecular components, in addition to the quantitatively dominant fibril-forming amyloid protein, including heparan sulfate proteoglycan 2 (HSPG2 or perlecan), SAP, a plasma glycoprotein of the pentraxin family (encoded by the APCS gene) that undergoes specific calcium-dependent binding to all types of amyloid fibrils, and apolipoprotein E (ApoE), also found in all amyloid deposits (15). The ApoE4 isoform is associated with an increased frequency and earlier onset of Alzheimer’s disease (Ab), the most common form of brain amyloid, whereas the ApoE2 isoform appears to be protective (16). ApoE variants could exert a similar modulatory effect in the onset of FAP, although early studies on a limited number of patients suggested this was not the case (17).
In at least one instance of senile systemic amyloidosis, small amounts of AA-related material were found in TTR deposits (18). These could reflect either a passive co-aggregation or a contributory involvement of protein AA, encoded by the serum amyloid A (SAA ) genes and the main component of secondary (reactive) amyloid fibrils, in the formation of ATTR.
Retinol-binding protein (RBP), the serum carrier of vitamin A, circulates in plasma bound to TTR. Vitamin A-loaded RBP and L-thyroxine, the two natural ligands of TTR, can act alone or synergistically to inhibit the rate and extent of TTR fibrillogenesis in vitro, suggesting that RBP may influence the course of FAP pathology in vivo (19). We have analyzed coding and non-coding sequence polymorphisms in the RBP4 (serum RBP, 10q24), HSPG2 (1p36.1), APCS (1q22), APOE (19q13.2), SAA1 and SAA2 (11p15.1) genes with the goal of identifying chromosomes carrying common and functionally significant variants. At the time these studies were performed, the full human genome sequence was not completed and systematic singlenucleotide polymorphism (SNP) analyses were not available for any of the suspected candidate genes. We identified new SNPs in APCS and RBP4 and utilized polymorphisms in SAA, HSPG2 and APOE that had already been characterized and shown to have potential pathophysiologic significance in other disorders (16,20–22). The genotyping data were analyzed for association with the presence of the V30M amyloidogenic allele (FAP patients versus controls) and with the age of onset (classical- versus late-onset patients). Multilocus analyses were also performed to examine the effects of simultaneous contributions of the six loci for determining the onset of the first symptoms. …..
The potential for different underlying models for classical and late onset is supported by the MDR analysis, which produces two distinct models when comparing each class with the controls. One could view the two onset classes as unique diseases. If this is the case, then the failure to detect a single predictive genetic model is consistent with two related, but different, diseases. This is exactly what would be expected in such a case of genetic heterogeneity (28). Using this approach, a major gene effect can be viewed as a necessary, but not sufficient, condition to explain the course of the disease. Analyzing the cases but omitting from the analysis of phenotype the necessary allele, in this case TTR V30M, can then reveal a variety of important modifiers that are distinct between the phenotypes.
The significant comparisons obtained in our study cohort indicate that the combined effects mainly result from two and three-locus interactions involving all loci except SAA1 and SAA2 for susceptibility to disease. A considerable number of four-site combinations modulate the age of onset with SAA1 appearing in a majority of significant combinations in late-onset disease, perhaps indicating a greater role of the SAA variants in the age of onset of FAP.
The correlation between genotype and phenotype in socalled simple Mendelian disorders is often incomplete, as only a subset of all mutations can reliably predict specific phenotypes (34). This is because non-allelic genetic variations and/or environmental influences underlie these disorders whose phenotypes behave as complex traits. A few examples include the identification of the role of homozygozity for the SAA1.1 allele in conferring the genetic susceptibility to renal amyloidosis in FMF (20) and the association of an insertion/deletion polymorphism in the ACE gene with disease severity in familial hypertrophic cardiomyopathy (35). In these disorders, the phenotypes arise from mutations in MEFV and b-MHC, but are modulated by independently inherited genetic variation. In this report, we show that interactions among multiple genes, whose products are confirmed or putative constituents of ATTR deposits, or metabolically interact with TTR, modulate the onset of the first symptoms and predispose individuals to disease in the presence of the V30M mutation in TTR. The exact nature of the effects identified here requires further study with potential application in the development of genetic screening with prognostic value pertaining to the onset of disease in the TTR V30M carriers.
If the effects of additional single or interacting genes dictate the heterogeneity of phenotype, as reflected in variability of onset and clinical expression (with the same TTR mutation), the products encoded by alleles at such loci could contribute to the process of wild-type TTR deposition in elderly individuals without a mutation (senile systemic amyloidosis), a phenomenon not readily recognized as having a genetic basis because of the insensitivity of family history in the elderly.
Safety and Efficacy of RNAi Therapy for Transthyretin Amyloidosis
Coelho T, Adams D, Silva A, et al.
N Engl J Med 2013;369:819-29. http://dx.doi.org:/10.1056/NEJMoa1208760
Transthyretin amyloidosis is caused by the deposition of hepatocyte-derived transthyretin amyloid in peripheral nerves and the heart. A therapeutic approach mediated by RNA interference (RNAi) could reduce the production of transthyretin.
Methods We identified a potent antitransthyretin small interfering RNA, which was encapsulated in two distinct first- and second-generation formulations of lipid nanoparticles, generating ALN-TTR01 and ALN-TTR02, respectively. Each formulation was studied in a single-dose, placebo-controlled phase 1 trial to assess safety and effect on transthyretin levels. We first evaluated ALN-TTR01 (at doses of 0.01 to 1.0 mg per kilogram of body weight) in 32 patients with transthyretin amyloidosis and then evaluated ALN-TTR02 (at doses of 0.01 to 0.5 mg per kilogram) in 17 healthy volunteers.
Results Rapid, dose-dependent, and durable lowering of transthyretin levels was observed in the two trials. At a dose of 1.0 mg per kilogram, ALN-TTR01 suppressed transthyretin, with a mean reduction at day 7 of 38%, as compared with placebo (P=0.01); levels of mutant and nonmutant forms of transthyretin were lowered to a similar extent. For ALN-TTR02, the mean reductions in transthyretin levels at doses of 0.15 to 0.3 mg per kilogram ranged from 82.3 to 86.8%, with reductions of 56.6 to 67.1% at 28 days (P<0.001 for all comparisons). These reductions were shown to be RNAi mediated. Mild-to-moderate infusion-related reactions occurred in 20.8% and 7.7% of participants receiving ALN-TTR01 and ALN-TTR02, respectively.
ALN-TTR01 and ALN-TTR02 suppressed the production of both mutant and nonmutant forms of transthyretin, establishing proof of concept for RNAi therapy targeting messenger RNA transcribed from a disease-causing gene.
Alnylam May Seek Approval for TTR Amyloidosis Rx in 2017 as Other Programs Advance
Officials from Alnylam Pharmaceuticals last week provided updates on the two drug candidates from the company’s flagship transthyretin-mediated amyloidosis program, stating that the intravenously delivered agent patisiran is proceeding toward a possible market approval in three years, while a subcutaneously administered version called ALN-TTRsc is poised to enter Phase III testing before the end of the year.
Meanwhile, Alnylam is set to advance a handful of preclinical therapies into human studies in short order, including ones for complement-mediated diseases, hypercholesterolemia, and porphyria.
The officials made their comments during a conference call held to discuss Alnylam’s second-quarter financial results.
ATTR is caused by a mutation in the TTR gene, which normally produces a protein that acts as a carrier for retinol binding protein and is characterized by the accumulation of amyloid deposits in various tissues. Alnylam’s drugs are designed to silence both the mutant and wild-type forms of TTR.
Patisiran, which is delivered using lipid nanoparticles developed by Tekmira Pharmaceuticals, is currently in a Phase III study in patients with a form of ATTR called familial amyloid polyneuropathy (FAP) affecting the peripheral nervous system. Running at over 20 sites in nine countries, that study is set to enroll up to 200 patients and compare treatment to placebo based on improvements in neuropathy symptoms.
According to Alnylam Chief Medical Officer Akshay Vaishnaw, Alnylam expects to have final data from the study in two to three years, which would put patisiran on track for a new drug application filing in 2017.
Meanwhile, ALN-TTRsc, which is under development for a version of ATTR that affects cardiac tissue called familial amyloidotic cardiomyopathy (FAC) and uses Alnylam’s proprietary GalNAc conjugate delivery technology, is set to enter Phase III by year-end as Alnylam holds “active discussions” with US and European regulators on the design of that study, CEO John Maraganore noted during the call.
In the interim, Alnylam continues to enroll patients in a pilot Phase II study of ALN-TTRsc, which is designed to test the drug’s efficacy for FAC or senile systemic amyloidosis (SSA), a condition caused by the idiopathic accumulation of wild-type TTR protein in the heart.
Based on “encouraging” data thus far, Vaishnaw said that Alnylam has upped the expected enrollment in this study to 25 patients from 15. Available data from the trial is slated for release in November, he noted, stressing that “any clinical endpoint result needs to be considered exploratory given the small sample size and the very limited duration of treatment of only six weeks” in the trial.
Vaishnaw added that an open-label extension (OLE) study for patients in the ALN-TTRsc study will kick off in the coming weeks, allowing the company to gather long-term dosing tolerability and clinical activity data on the drug.
Enrollment in an OLE study of patisiran has been completed with 27 patients, he said, and, “as of today, with up to nine months of therapy … there have been no study drug discontinuations.” Clinical endpoint data from approximately 20 patients in this study will be presented at the American Neurological Association meeting in October.
As part of its ATTR efforts, Alnylam has also been conducting natural history of disease studies in both FAP and FAC patients. Data from the 283-patient FAP study was presented earlier this year and showed a rapid progression in neuropathy impairment scores and a high correlation of this measurement with disease severity.
During last week’s conference call, Vaishnaw said that clinical endpoint and biomarker data on about 400 patients with either FAC or SSA have already been collected in a nature history study on cardiac ATTR. Maraganore said that these findings would likely be released sometime next year.
Alnylam Presents New Phase II, Preclinical Data from TTR Amyloidosis Programs
https://www.genomeweb.com/rnai/alnylam-presents-new-phase-ii-preclinical-data-ttr-amyloidosis-programs
Amyloid disease drug approved
Nature Biotechnology 2012; (3) http://dx.doi.org:/10.1038/nbt0212-121b
The first medication for a rare and often fatal protein misfolding disorder has been approved in Europe. On November 16, the E gave a green light to Pfizer’s Vyndaqel (tafamidis) for treating transthyretin amyloidosis in adult patients with stage 1 polyneuropathy symptoms. [Jeffery Kelly, La Jolla]
Safety and Efficacy of RNAi Therapy for Transthyretin …
http://www.nejm.org/…/NEJMoa1208760?…
The New England Journal of Medicine
Aug 29, 2013 – Transthyretin amyloidosis is caused by the deposition of hepatocyte-derived transthyretin amyloid in peripheral nerves and the heart.
Alnylam’s RNAi therapy targets amyloid disease
Ken Garber
Nature Biotechnology 2015; 33(577) http://dx.doi.org:/10.1038/nbt0615-577a
RNA interference’s silencing of target genes could result in potent therapeutics.
http://www.nature.com/nbt/journal/v33/n6/images/nbt0615-577a-I1.jpg
The most clinically advanced RNA interference (RNAi) therapeutic achieved a milestone in April when Alnylam Pharmaceuticals in Cambridge, Massachusetts, reported positive results for patisiran, a small interfering RNA (siRNA) oligonucleotide targeting transthyretin for treating familial amyloidotic polyneuropathy (FAP). …
Nature Biotechnology 11 April 2016
Nature Biotechnology 02 March 2014
Nature Biotechnology 04 April 2016
Translational Neuroscience: Toward New Therapies
https://books.google.com/books?isbn=0262029863
Karoly Nikolich, Steven E. Hyman – 2015 – Medical
Tafamidis for Transthyretin Familial Amyloid Polyneuropathy: A Randomized, Controlled Trial. … Multiplex Genome Engineering Using CRISPR/Cas Systems.
Is CRISPR a Solution to Familial Amyloid Polyneuropathy?
Author and Curator: Larry H. Bernstein, MD, FCAP
Originally published as
http://scholar.aci.info/view/1492518a054469f0388/15411079e5a00014c3d
FAP is characterized by the systemic deposition of amyloidogenic variants of the transthyretin protein, especially in the peripheral nervous system, causing a progressive sensory and motor polyneuropathy.
FAP is caused by a mutation of the TTR gene, located on human chromosome 18q12.1-11.2.[5] A replacement of valine by methionine at position 30 (TTR V30M) is the mutation most commonly found in FAP.[1] The variant TTR is mostly produced by the liver.[citation needed] The transthyretin protein is a tetramer. ….












![Early A[bgr] accumulation in the parenchyma and blood vessels in a subset of eight patients with iCJD aged 36-51 years, but not in controls (stratum aged 36-51 years) of 19 prion diseases of other aetiologies, suggests human transmission.](https://i0.wp.com/www.nature.com/nature/journal/v525/n7568/carousel/nature15369-f3.jpg?resize=187%2C200)
![A[bgr] accumulation in central nervous system parenchyma and blood vessels (CAA) in iCJD.](https://i0.wp.com/www.nature.com/nature/journal/v525/n7568/images_article/nature15369-f1.jpg?w=500)






![EPPS ameliorates A[beta]-induced memory deficits in mice.](https://i0.wp.com/www.nature.com/ncomms/2015/151208/ncomms9997/images/ncomms9997-f1.jpg?w=500)
![EPPS disaggregates A[beta] plaques and oligomers in APP/PS1 mice.](https://i0.wp.com/www.nature.com/ncomms/2015/151208/ncomms9997/images/ncomms9997-f4.jpg?w=500)











![Anti-A[beta] antibody 3D6 reduces amyloid pathology but aggravates neuronal dysfunction.](https://i0.wp.com/www.nature.com/neuro/journal/vaop/ncurrent/carousel/nn.4163-F1.jpg?resize=405%2C200)
![Worsening of neuronal dysfunction by anti-A[beta] antibodies can occur independently of the effects on A[beta] pathology.](https://i0.wp.com/www.nature.com/neuro/journal/vaop/ncurrent/carousel/nn.4163-F2.jpg?resize=389%2C200)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}