Imaging Biomarker for Arterial Stiffness: Pathways in Pharmacotherapy for Hypertension and Hypercholesterolemia Management
Author, and Content Consultant to e-SERIES A: Cardiovascular Diseases: Justin Pearlman, MD, PhD, FACC
and
Article Curator: Aviva Lev-Ari, PhD, RN
This article has Four Parts:
Part 1:
Quantification of Arterial Stiffness selected for its Predictive Value for Cardiovascular (CV) Events.
Arterial stiffness can predict cardiovascular adverse events such as stroke and heart attack. While there are various ways to define and estimate arterial stiffness, relatively simple surrogates have clinical advantages and favorable reports regarding predictive accuracy. This article will review in particular carotid-femoral pulse wave velocity (cfPWV) as an imaging-based biomarker of arterial stiffness.
Part II:
Results for Advances and Recent Clinical Trials in Hypertension Management
Caution is required in the interpretation of trial results, due to the Hawthorne Effect: participation in a trial confers benefits to all groups. Usually the Hawthorne effect is attributed to the close attention and is considered transient, but it can have lasting impact. In a retrospective cohort study, the benefits of participation in clinical trials irrespective of the treatment allocation were illustrated by better persistence and adherence to prescribed medication long term.
- Participation in a clinical trial enhances adherence and persistence to treatment: a retrospective cohort study.
Hypertension . 2011 ; 58 : 573 – 578 .
- It is proving more and more difficult to show incremental benefit of new therapies over standard therapy in control groups that are on background therapy marked by high statin, antiplatelet, and other antihypertensive therapy rates, as well as more overweight and obesity and less tobacco use than in the past.
Participation in a Clinical Trial Enhances Adherence and Persistence to Treatment, A Retrospective Cohort Study Chronobiol Int . 2011 ; 28 : 601 – 610.
Cardiorenal end points in a trial of Aliskiren for type 2 diabetes. N Engl J Med . 2012 ; 367 : 2204 – 2213.
Part III:
Pharmacotherapy for Hypertension and Hypercholesterolemia Management: Mechanism of Action of Top 10 Cardio Drugs 2012, published on May 16, 2013. FiercePharma reports the top 10 drugs from expenditure standpoint:
Part IV: Management Aspects of the Global Pharmaceutical Industry
The 20 Highest-Paid Biopharma CEOs of 2012 are also reported by FiersePharma.
-
11
-
12
-
13
-
14
-
15
-
16
-
17
-
18
-
19
-
20
Part 1:
Quantification of Arterial Stiffness selected for its Predictive Value for Cardiovascular (CV) Events.
based on
Stéphane Laurent, Elie Mousseaux and Pierre Boutouyrie, Arterial Stiffness as an Imaging Biomarker : Are All Pathways Equal?
http://hyper.ahajournals.org/content/early/2013/05/20/HYPERTENSIONAHA.113.01372.citation
In a recent meta-analysis,2 Seventeen longitudinal studies totalizing 15,877 subjects with a mean follow-up of 7.7 years showed, for 1 SD increase in PWV, a risk ratio of 1.47 (1.31–1.64) for total mortality, 1.47 (1.29–1.66) for CV mortality, and 1.42 (1.29–1.58) for all-cause mortality.
Aortic stiffness, measured through cfPWV, can thus be considered as a novel imaging biomarker for predicting CV events, although its value as a true surrogate end point requires a large intervention trial to demonstrate that the reduction in arterial stiffness translates into a reduction in CV events.
Prediction of Occurrence of Cardiovascular Events Independently of Left Ventricular Mass in Hypertensive Patients: Monitoring of Timing of Korotkoff Sounds as Indicator of Arterial Stiffness
In this article by Gosse et al7 published in the present issue of Hypertension, the Authors provides an important contribution with regard to the predictive value of arterial stiffness for CV events for the following reasons:
- First, the authors reported that arterial stiffness, measured in a population of 793 patients with hypertension with a mean follow-up of 97 months, was independently related to all CV events, major CV events, and total mortality. Interestingly, the predictive value was significant in all subgroups of CV risk, defined by a low, medium, or high SCORE risk. These findings confirmed those of previous studies.
- Second, the authors took advantage of the simultaneous measurement of 24-hour blood pressure (BP) to include 24-hour mean BP in the multivariate Cox analysis, and this is a novelty. Thus, they were able to provide the demonstration that the predictive value of arterial stiffness is not only independent of office BP, as shown in most epidemiological studies, but also of 24-hour mean BP and pulse pressure (or alternatively 24-hour systolic and diastolic BPs) simultaneously measured.
- Third, among the 793 patients, 519 patients had baseline measurements of arterial stiffness before any antihypertensive treatment, and the remaining 274 patients had measurement during the follow- up period. The independent predictive value of arterial stiffness was significant whether measured before or after the administration of antihypertensive treatment.
- Finally, Gosse et al 7 showed, in a subgroup of 523 patients who had a measurement of left ventricular mass index, that the predictive value of arterial stiffness for major CV events was independent of left ventricular mass index. The authors thus confirmed the very few epidemiological studies which analyzed the predictive value of biomarkers of target organ damages (ie, left ventricular mass index, urinary albumin excretion rate, carotid intimamedia thickness, and arterial stiffness) and found that arterial stiffness retained a significant predictive value when adjusted either to left ventricular mass index6 or carotid intima-media thickness.5
- The method which has been used to determine arterial stiffness. Indeed, Gosse et al 8 proposed, 2 decades ago, to take advantage of an ambulatory measurement of BP and continuous monitoring of ECG >24 hours, to calculate the QKD interval. QKD is the time between the onset of the QRS on the ECG and the detection of the last Korotkoff sound by the microphone placed on the brachial artery. It has 2 components:
- the pre-ejection time, which is influenced by heart rate and
- the pulse transmission time, which is inversely related to PWV, and arterial stiffness.
- BP and QKD are measured repeatedly, and a stiffness parameter is derived from the linear regression of all the measurements of QKD, heart rate, and systolic BP >24 hours. The QKD interval is calculated for a 100-mm Hg BP, thus it gives an isobaric value of arterial stiffness, and for a 60-beats/min heart rate to reduce the influence of the pre-ejection time.
- Most importantly, the arterial pathway of pulse wave transmission includes the ascending aorta, the aortic arch, and muscular arteries (subclavian and brachial), and thus,
- differs from the carotid-femoral pathway of the cfPWV measurement, considered as gold standard for arterial stiffness.9
- cfPWV is calculated as the ratio of the transit time between the feet of the carotid and femoral pressure waveforms, and the carotid-femoral distance, a ratio which is undisputedly recognized as a stiffness parameter. Several studies and a consensus statement have determined the correction factor, which should be applied to the carotid-femoral distance, to take into account the fact that, when the pressure wave is recorded at the carotid level, it has already reached the descending thoracic aorta.
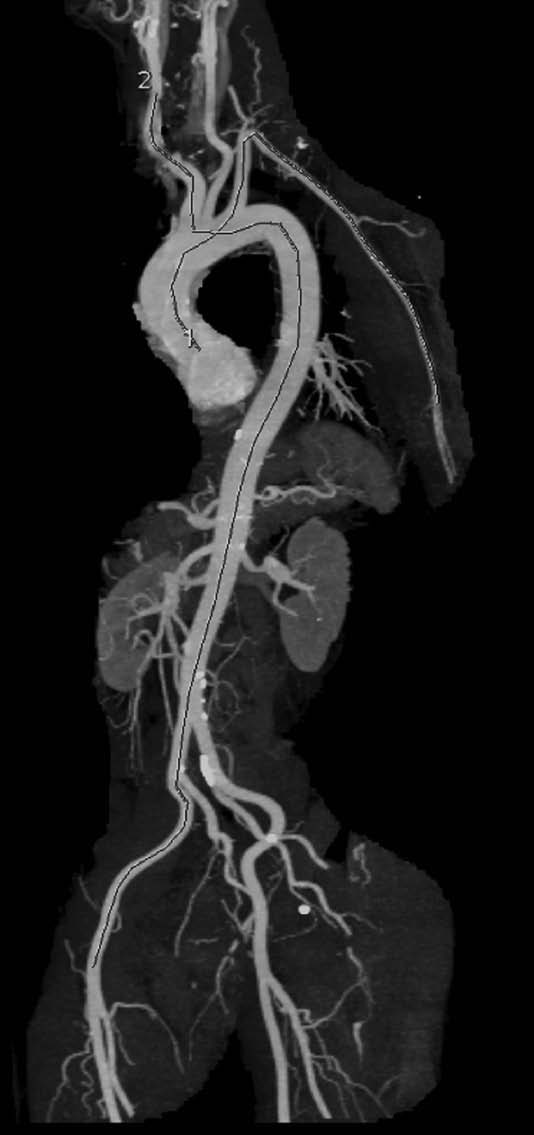
- The pressure wave travels mostly along an aortic segment, including the thoracic descending aorta and the abdominal aorta, and ultimately travels along the iliac and common femoral arteries. This is well exemplified by the Figure, which superimposes the trajectory of the pressure pulse wave on a normal angiogram obtained by magnetic resonance imaging.
VIEW FIGURE
The trajectories of the pressure pulse waves along the arterial segments are superimposed onto an angiogram obtained by computed tomography scan (left anterior oblique). The carotid-femoral pathway is described as dotted line, and the QKD pathway is described as dashed line.

FIGURE SOURCE:
http://hyper.ahajournals.org/content/early/2013/05/20/HYPERTENSIONAHA.113.01372.citation
The method developed by Gosse et al 7,8 measures the time delay between the onset of the QRS on the ECG and the detection of the last Korotkoff sound by the microphone placed on the brachial artery. Thus, the pressure pulse wave travels first along the ascending aorta and the aortic arch (ie, a short pathway of elastic arteries) and then along the subclavian and brachial arteries (ie, a much longer pathway of muscular arteries).
Because the stiffness of muscular arteries is little influenced by age and hypertension, Gosse et al8 attributed the difference in QKD duration to ascending aorta and aortic arch. However, a closer look at the Figure shows that the length of the ascending and aortic arch pathway represents a very small part of the total pathway and casts doubt about this statement.
Furthermore, in magnetic resonance imaging studies, the transit time of flow wave along the aortic arch (average 120 mm length) is often found ≈35 ms in young healthy subjects,10 a value which is far from the mean 206 ms QKD duration found in the present study. Thus, part of that QFD duration has to be further explained by both the preejection period and the transit time within muscular arteries.
Alternative Devices
- 2008 – The arteriograph system estimates PWV from a single-site determination of the suprasystolic waveform at the brachial artery site, and measures the time elapsed between the first wave ejected from the left ventricle to the aortic root, and its reflection from the bifurcation as the second systolic wave, with subtraction of the brachial artery transit time.
- 2010 – The Mobil-O-Graph system uses oscillometric recording of brachial artery pressure waveform and reconstructs the central pulse wave by applying a transfer function. Central pulse wave is then decomposed into forward and backward waves, and PWV isestimated from their time difference.
- Device |Method |Arterial Pathway |Predictive Value for CV Events | (Year of First Publication)
1984 Complior Mechanotransducer Carotid-femoral Yes (1999)
1990 Sphygmocor Tonometer Carotid-femoral Yes (2011)
1994 QKD ECG + Korotkoff sounds Aorta + brachial Yes (2005)
1997 Cardiov. Eng. Inc Tonometer Carotid-femoral Yes (2010)
2002 Doppler probes Doppler probe Aortic arch + descending aorta Yes (2002)
2002 VP-1000 Omron Brachial and ankle pressure cuffs Aorta + brachial + lower limbs Yes (2005)
2004 PulsePen Tonometer Carotid-femoral No
2006 CAVI-VaSera ECG + Brachial and ankle pressure cuffs Aorta + brachial + lower limbs No
2008 Arteriograph Arm pressure cuff Aorta + brachial No
2009 MRI-ArtFun MRI Aortic arch No
2009 Vicorder Cuffs Carotid-femoral No
2010 Mobil-O-Graph Arm pressure cuff Aorta No
Conclusions
The measurement of arterial stiffness is increasingly popular among physicians and researchers mainly because its predictive value for cardiovascular (CV) events has been well demonstrated. The largest amount of evidence has been given for aortic stiffness, measured through carotid-femoral pulse wave velocity (cfPWV). This has been initially reported in the late 1990s or early 2000s.1
Aortic stiffness is an independent predictor of all-cause and cardiovascular mortality in hypertensive patients. Hypertension. 2001;37:1236–1241.
European Network for Non-invasive Investigation of Large Arteries. Expert consensus document on arterial stiffness: methodological issues and clinical applications. Eur Heart J. 2006;27:2588–2605.
Arterial Stiffness as an Imaging Biomarker : Are All Pathways Equal? http://hyper.ahajournals.org/content/early/2013/05/20/HYPERTENSIONAHA.113.01372.citation
References for Imaging Biomarker for Arterial Stiffness, at the end of the paper
Part II:
Results for Advances and Recent Clinical Trials in Hypertension Management
Based on
Trends: tended to drive interest toward equivalence rather than efficacy studies (ie, trials designed to show an investigational agent is as good as, not better than, existing treatment), surrogate end points, including new blood pressure (BP) variables, and studies of combinations and algorithms rather than single interventions. Population studies around the world, however, continue to show that large numbers of people have hypertension that is not treated satisfactorily and are not achieving the goals set by the major national guidelines. These guidelines themselves are under continual scrutiny on the basis of recent data casting doubt on the validity of present BP goals. Guideline committees also face the issue that evidence based on expensive large-scale clinical trials is more often funded by the pharmaceutical or device industries than by government, leaving large evidence gaps in areas of public importance but no direct interest to industry funders. The purpose of the present article is to briefly review clinical trials of interventions in hypertension during the past 2 years.
Subject categories of Last Decade Clinical Trials on Hypertension
- Resistant Hypertension
- Resistant Hypertension and the Sympathetic
- Nervous System
- Trials of Pharmacotherapy
- Old Ground, New Findings
- Are Chlorthalidone and Nonthiazides the Best Diuretics for Treatment of Hypertension?
- BP Targets and Treatment
- Lifestyle and Nonpharmacological Approaches to Hypertension
- Sodium
- Trials of Nutrition and BP
- Resistance Exercise and BP
What Can Be Learned From Clinical Trials Reported in the Present Decade?
- Systems for blood pressure management in the community can be improved because a large treatment gap remains.
- Drug combinations from different classes with different modes of action are useful.
- Drug combinations that include drugs with similar mode of action do not generally enhance efficacy and come at a cost in adverse events.
- Small but important nutritional effects on blood pressure demand further examination.
- The sympathetic nervous system has returned as an important target for therapy of hypertension.
- Blood pressure targets and goals need refining, preferably on the basis of specifically designed clinical trials.
The scene for clinical trials of hypertension management is in transition. The era of mega trials may not be over but is certainly in decline, and in the past 2 years there have been no studies reporting primary outcome data the scale of the
- Antihypertensive and
- Lipid-Lowering Treatment
- Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT),
- The ONgoing Telmisartan Alone and in combination with Ramipril Global Endpoint Trial (ONTARGET),
- Anglo-Scandinavian Cardiac Outcomes Trial (ASCOT), and other
- major studies that marked clinical trial activity and informed guideline committees during the past 2 to 3 decades.
This reflects in part the view that the
- present benchmark pharmacological agents for treating hypertension are difficult to improve,
- some systemic issues affecting the pharmaceutical industry influencing the ability to make the large investment required to perform mega trials and
- the quality of the antihypertensive drug pipeline.
http://hyper.ahajournals.org/content/early/2013/05/20/HYPERTENSIONAHA.113.00863.citation
References for Clinical Trial on Hypertension, at the end of the paper
Part III:
Mechanism of Action of Top 10 Cardio Drugs 2012, published on May 16, 2013
The top 10 Cardio Drugs in 2012 belong to two drug classes
- Antihypertensive and
- Lipid-Lowering Treatment
Sales % Change 2012 vs 2011 by Drug Class
|
MOA
By
Drug Class
|
Drug Name
|
2011 Sales billion
|
2012 Sales billion
|
% change
|
|
Statins
|
Crestor
|
6.622
|
6.253
|
-6%
|
|
Lipitor
|
9.577
|
3.948
|
-59%
|
|
Zetia
|
2.428
|
2.567
|
+6%
|
|
Vytorin
|
1.882
|
1.747
|
-7%
|
|
Total Sales and % change Statins
|
|
20,509 |
14,515 |
-29.2% |
|
ARB
|
Diovan
|
5.665
|
4.417
|
-22%
|
|
ACEII
|
Benicar
|
2.602
|
2.446
|
-6%
|
|
ACEI
|
Micardis
|
2.217
|
2.098
|
-5%
|
|
ARB
|
Avapro
|
1.797
|
1.422
|
-30% (BMS)
|
|
ARB
|
Blopress
|
1.808
|
1.643
|
-9%
|
|
PAH
|
Tracleer
|
1.721
|
1.6
|
-7%
|
|
Total Sales and % change AntiHTN
|
|
15,810 |
13,626 |
-13.8% |
Data Source:
http://www.fiercepharma.com/special-reports/top-10-cardio-drugs-2012#ixzz2U9Axh8X4
1 Crestor
Crestor (AstraZeneca)
Patent expiry: July 2016
2012 sales: $6.253 billion
2011 sales: $6.622 billion
Change: (6%)
Crestor – FiercePharma http://www.fiercepharma.com/special-reports/crestor-0#ixzz2UACLZyaa
(rosuvastatin calcium) is indicated as an adjunct to diet to reduce elevated Total-C, LDL-C, ApoB, non-HDL-C, and triglycerides, and to increase HDL-C in adult patients with primary hyperlipidemia or mixed dyslipidemia and to slow the progression of atherosclerosis in adult patients as part of a treatment strategy to lower Total-C and LDL-C to target levels.1
2 Diovan
Diovan (Novartis)
Patent expiry: September 2012
2012 sales: $4.417 billion
2011 sales: $5.665 billion
Change: (22%)
Diovan – FiercePharma http://www.fiercepharma.com/special-reports/diovan#ixzz2UACdBCtZ
Valsartan (Angiotan or Diovan) is an angiotensin II receptor antagonist (more commonly called an “ARB”, or angiotensin receptor blocker), with particularly high affinity for the type I (AT1) angiotensin receptor. By blocking the action of angiotensin, valsartan dilates blood vessels and reduces blood pressure.[1] In the U.S., valsartan is indicated for treatment of high blood pressure, congestive heart failure (CHF), or post-myocardial infarction (MI).[2]
3 Lipitor
Lipitor (Pfizer)
Patent expiry: November 2011
2012 sales: $3.948 billion
2011 sales: $9.577 billion
Change: (59%)
Lipitor – FiercePharma http://www.fiercepharma.com/special-reports/lipitor-2#ixzz2UACsJ2Y2
(atorvastatin calcium) tablets are a prescription medicine that is used along with a low-fat diet. It lowers the LDL (“bad”) cholesterol and triglycerides in your blood. It can raise your HDL (“good”) cholesterol as well. LIPITOR can lower the risk for heart attack, stroke, certain types of heart surgery, and chest pain in patients who have heart disease or risk factors for heart disease such as age, smoking, high blood pressure, low HDL, or family history of early heart disease. LIPITOR can lower the risk for heart attack or stroke in patients with diabetes and risk factors such as diabetic eye or kidney problems, smoking, or high blood pressure.
LIPITOR is a member of the drug class known as statins, used for lowering blood cholesterol. It also stabilizes plaque and prevents strokes through anti-inflammatory and other mechanisms. Like all statins, atorvastatin works by inhibiting HMG-CoA reductase, an enzyme found in liver tissue that plays a key role in production of cholesterol in the body.
Atorvastatin was first synthesized in 1985 by Bruce Roth of Parke-Davis Warner-Lambert Company (now Pfizer). The best selling drug in pharmaceutical history, sales of Lipitor since it was approved in 1996 exceed US$125 billion, and the drug has topped the list of best-selling branded pharmaceuticals in the world for nearly a decade
4 Zetia
Zetia (Merck)
Patent expiry: December 2016
2012 sales: $2.567 billion
2011 sales: $2.428 billion
Change: 6%
Zetia – FiercePharma http://www.fiercepharma.com/special-reports/zetia#ixzz2UADFaGJ0
Ezetimibe (pron.: /ɛˈzɛtɨmɪb/) is a drug that lowers plasma cholesterol levels. It acts by decreasing cholesterol absorption in the intestine. It may be used alone (marketed as Zetia or Ezetrol), when other cholesterol-lowering medications are not tolerated, or together withstatins (e.g., ezetimibe/simvastatin, marketed as Vytorin and Inegy) when statins alone do not control cholesterol.
Ezetimibe decreases cholesterol levels, but has not been shown to improve outcomes in cardiovascular disease patients by decreasing atherosclerotic or vascular events compared to placebo. Ezetimibe is endorsed in the Canadian Lipid Guidelines and is considered a well-tolerated option for an add-on agent to statin, to help patients achieve their LDL (or bad cholesterol) targets. [1] Ezetimibe is the only add-on to statin therapy that has successfully shown cardiovascular benefit when combined with statin, but has not been proven to have an incremental benefit compared to statins alone. [2] Britain’s NICE statement, published in 2007, endorses its use for monotherapy if statins are not tolerated or as add-on therapy.[3]
5 Benicar
Benicar (Daiichi Sankyo)
Patent expiry: October 2016
2012 sales: $2.446 billion
2011 sales: $2.602 billion
Change: (6%)
Benicar – FiercePharma http://www.fiercepharma.com/special-reports/benicar#ixzz2UADYvld5
BENICAR and BENICAR HCT are prescription medicines used to lower high blood pressure (hypertension). They may be used alone or with other medicines used to treat high blood pressure. BENICAR HCT is not for use as the first medicine to treat high blood pressure.
Olmesartan is a prodrug that works by blocking the binding of angiotensin II to the AT1 receptors in vascular muscle; it is therefore independent of angiotensin II synthesis pathways, unlike ACE inhibitors. By blocking the binding rather than the synthesis of angiotensin II, olmesartan inhibits the negative regulatory feedback on renin secretion. As a result of this blockage, olmesartan reduces vasoconstriction and the secretion of aldosterone. This lowers blood pressure by producing vasodilation, and decreasing peripheral resistance.
6 Micardis
Micardis (Boehringer Ingelheim)
Patent Expiry: January 2014
2012 Sales: $2.098 billion
2011 Sales: $2.217 billion
Change: (5%)
Micardis – FiercePharma http://www.fiercepharma.com/special-reports/micardis#ixzz2UADpDZeO
Micardis® (telmisartan) tablets are a prescription medicine used to treat high blood pressure (hypertension). Additionally, MICARDIS 80 mg tablets are used in certain high-risk people aged 55 years and older who are unable to take a medicine called an angiotensin converting enzyme inhibitor (ACE-I) to help lower their risk of having certain cardiovascular problems such as stroke, heart attack, or death.
Micardis® (telmisartan) tablets are a prescription medicine used to treat high blood pressure (hypertension).
Telmisartan (INN) (pron.: /tɛlmɪˈsɑrtən/) is an angiotensin II receptor antagonist (angiotensin receptor blocker, ARB) used in the management of hypertension. It is marketed under thetrade name Micardis (by Boehringer Ingelheim), among others.
Telmisartan is an angiotensin II receptor blocker that shows high affinity for the angiotensin II receptor type 1 (AT1), with a binding affinity 3000 times greater for AT1 than AT2. It has the longest half-life of any ARB (24 hours)[1][4] and the largest volume of distribution.
In addition to blocking the RAs, telmisartan acts as a selective modulator of peroxisome proliferator-activated receptor gamma (PPAR-γ), a central regulator of insulin and glucose metabolism. It is believed that telmisartan’s dual mode of action may provide protective benefits against the vascular and renal damage caused by diabetes and cardiovascular disease (CVD).[4]
Telmisartan’s activity at the PPAR-γ receptor has prompted speculation around its potential as a sport doping agent as an alternative to GW 501516.[5] Telmisartan activates PPARδ receptors in several tissues. [6][7][8][9]
7 Avapro
Avapro (Sanofi)
Patent expiry: March 2012
Total 2012 sales: $1.925 billion
2012 sales Sanofi: $1.422 billion
2012 sales BMS: $503 million
Total 2011 sales: $2.749 billion
2011 sales Sanofi: $1.797 billion
2011 sales BMS: $952 million
Total Change: (30%)
Avapro – FiercePharma http://www.fiercepharma.com/special-reports/avapro#ixzz2UAE9iB2E
rbesartan (INN) (pron.: /ɜrbəˈsɑrtən/) is an angiotensin II receptor antagonist used mainly for the treatment of hypertension. Irbesartan was developed by Sanofi Research (now part ofsanofi-aventis). It is jointly marketed by sanofi-aventis and Bristol-Myers Squibb under the trade names Aprovel, Karvea, and Avapro.
As with all angiotensin II receptor antagonists, irbesartan is indicated for the treatment ofhypertension. Irbesartan may also delay progression of diabetic nephropathy and is also indicated for the reduction of renal disease progression in patients with type 2 diabetes,[1]hypertension and microalbuminuria (>30 mg/24 hours) or proteinuria (>900 mg/24 hours).[2]
A large randomized trial following 4100+ men and women with heart failure and normal ejection fraction (>=45%) over 4+ years found no improvement in study outcomes or survival with irbesartan as compared to
placebo.
[3]8 Vytorin
Vytorin (Merck)
Patent Expiry: April 2017
2012 sales: $1.747 billion
2011 sales: $1.882 billion
Change: (7%)
Vytorin – FiercePharma http://www.fiercepharma.com/special-reports/vytorin#ixzz2UAEQVcQr
Ezetimibe/simvastatin (pron.: /ɛˈzɛtɨmɪb ˌsɪmvəˈstætɨn/) is a drug combination used for the treatment of dyslipidemia. It is a combination of ezetimibe (best known as Zetia in the United States and Ezetrol elsewhere) and the statin drug simvastatin (best known as Zocor in the U.S.). The combination preparation is marketed by Merck & Co./Schering-PloughPharmaceuticals (joint venture) under the trade names Vytorin and Inegy.
Ezetimibe reduces blood cholesterol by inhibiting absorption of cholesterol by the small intestine by acting at the brush border of the small intestine and inhibits the absorption of cholesterol, leading to a decrease in the delivery of intestinal cholesterol to the liver.
Simvastatin is an HMG-CoA reductase inhibitor or statin. It works by blocking an enzymethat is necessary for the body to make cholesterol.
Even though ezetimibe decreases cholesterol levels, as of 2009 it has not been found to lead to improvement in real world outcomes.[1] The combination of simvastatin and ezetimibe has not been found to be any better than simvastatin alone. A panel of experts thus concluded in 2008 that it should “only be used as a last resort”.[2]
9 Blopress
Blopress (Takeda Pharmaceutical)
Patent expiry: June 2012
2012 sales: $1.643 billion
2011 sales: $1.808 billion
Change: (9%)
Blopress – FiercePharma http://www.fiercepharma.com/special-reports/blopress#ixzz2UAEnxyWy
Candesartan (rINN) (pron.: /ˌkændɨˈsɑrtən/) is an angiotensin II receptor antagonist used mainly for the treatment of hypertension. The prodrug candesartan cilexetil is marketed by AstraZeneca and Takeda Pharmaceuticals, commonly under the trade names Blopress,Atacand, Amias, and Ratacand
As all angiotensin II receptor antagonists, candesartan is indicated for the treatment of hypertension. Results from the CHARM study in the early 2000s demonstrated the morbidity and mortality reduction benefits of candesartan therapy in congestive heart failure.[1] Thus, while ACE inhibitors are still considered first-line therapy in heart failure, candesartan can be used in combination with an ACE to achieve improved mortality and morbidity vs. an ACE alone and additionally is an alternative in patients intolerant of ACE inhibitor therapy.
Prehypertension
In a four-year randomized controlled trial, candesartan was compared to placebo to see whether it could prevent or postpone the development of full-blown hypertension in people with so-called prehypertension. During the first two years of the trial, half of participants were given candesartan, and the others received placebo; candesartan reduced the risk of developing hypertension by nearly two-thirds during this period. In the last two years of the study, all participants were switched to placebo. By the end of the study, candesartan hadsignificantly reduced the risk of hypertension, by more than 15%. Serious side effects were actually more common among participants receiving placebo than in those given candesartan.[2]
Candesartan is also available in a combination formulation with a low dose thiazide diuretic, invariably hydrochlorothiazide, to achieve an additive antihypertensive effect. Candesartan/hydrochlorothiazide combination preparations are marketed under various trade names including Atacand HCT, Hytacand, Blopress Plus, Advantec and Ratacand Plus.
10 Tracleer
Tracleer (Actelion)
Patent expiry: November 2015
2012 sales: $1.600 billion
2011 sales: $1.721 billion
Change: (7%)
Tracleer – FiercePharma http://www.fiercepharma.com/special-reports/tracleer#ixzz2UAF2iIJB
Bosentan is a dual endothelin receptor antagonist used in the treatment of pulmonary artery hypertension (PAH). It is licensed in the United States, the European Union and other countries by Actelion Pharmaceuticals for the management of PAH under the trade name Tracleer.
Bosentan is a competitive antagonist of endothelin-1 at the endothelin-A (ET-A) and endothelin-B (ET-B) receptors. Under normal conditions, endothelin-1 binding of ET-A or ET-B receptors causes pulmonary vasoconstriction. By blocking this interaction, bosentan decreases pulmonary vascular resistance. Bosentan has a slightly higher affinity for ET-A than ET-B.
Clinical uses
Bosentan is indicated mainly for the treatment of pulmonary hypertension. In 2007, bosentan was approved in the European Union also for reducing the number of new digital ulcers in patients with systemic sclerosis and ongoing digital ulcer disease.
In the United States, bosentan is indicated for the treatment of pulmonary arterial hypertension (WHO Group I) in patients with WHO Class II-IV symptoms, to improve exercise capacity and decrease the rate of clinical worsening.[1]
http://www.fiercepharma.com/special-reports/top-10-cardio-drugs-2012
For years, cardio was king. The world’s all-time best-selling drug, Pfizer’s ($PFE) Lipitor, after all, is an antihyperlipidemic drug. Cardio drugs have traditionally made up one of the largest categories of therapeutic treatment in the drug universe.
According to EvaluatePharma‘s World Preview 2018 report, combined sales of antihypertensive drugs and antihyperlipidemics were more than $70 billion in 2011. That would put them at the top of the heap. Sales of antihypertensive drugs alone were more than $40 billion that year, making them the second-largest therapy area defined by the report, behind oncology drugs at $64.4 billion. The list, compiled by EvaluatePharma, includes the theraputic areas categorized as cardio, so it does not include some products sometimes used for heart disease but not in that therapeutic area, including blood thinners like Plavix.
But many of the top cardio drugs are long in the tooth, and generics are now eating their lunch. Did I mention Lipitor? Sales cratered last year, falling nearly 60%. Despite that, the drug placed third among the top 10 cardio drugs of 2012, a reminder of the stature it had achieved. Four of the top 10 have lost patent protection in the last two years, and most will be off patent by 2016, with only Merck’s ($MRK) Vytorin protected until 2017.
Last year, the top 10 cardio drugs racked up sales of $28.644 billion, down 23% from the $37.271 billion they sold in 2011. Still, the group has made a lot of money for its companies for years and, in some cases, completely changed the treatment of heart disease.
It is an interesting list. Only Merck has two drugs in the top 10. The other drugmakers make up a broad swath of the pharma industry. Read our report below, and if you have some insights you would like to share, please do.
Top 10 Cardio Drugs 2012 – FiercePharma http://www.fiercepharma.com/special-reports/top-10-cardio-drugs-2012#ixzz2UAByWR7s
Part IV:
20 Highest-Paid Biopharma CEOs of 2012
 Call it a rite of spring. Every year about this time, FiercePharma takes a look at executive compensation in the industry, and we rank the highest-paid CEOs. If you’re a regular reader, you’ll notice that this year’s list is longer than previous editions. And there’s a reason for that: curiosity.
Call it a rite of spring. Every year about this time, FiercePharma takes a look at executive compensation in the industry, and we rank the highest-paid CEOs. If you’re a regular reader, you’ll notice that this year’s list is longer than previous editions. And there’s a reason for that: curiosity.
As we were beginning to gather numbers from biopharma companies’ proxy statements and annual reports, news surfaced that Valeant Pharmaceuticals ($VRX) and Actavis ($ACT) had been in merger talks. The former CEO of Mylan ($MYL), one of Actavis’ rivals, regularly appeared on our highest-paid executives list, so we looked up the numbers on Actavis. No dice; CEO Paul Bisaro may have pulled off his biggest merger ever last year, but $8.66 million in compensation still didn’t qualify him for our ranking.
Then, we pulled out Valeant’s proxy statement. And while CEO Michael Pearson didn’t earn enough in 2012 to make the cutoff–his compensation just surpassed $6 million–he should have been at the top of the list last year. Pearson’s 2011 pay package broke $36 million. He collected more than $18 million in stock and option awards, plus a special $13.7 million dividend payment, stemming from agreements negotiated years before.
We hate to miss a scoop. Naturally. So, we vowed to avoid making the same mistake this time around. Rather than limit our executive-pay search to the biggest pharma companies and biotechs, plus the usual suspects who often make CEO-pay rankings, we used a bigger net. We collected compensation information from 50 companies, including numbers for CEOs, CFOs, R&D chiefs and other top executives.
Partly because of this search, but mostly because of big bonuses and awards at fast-growing Regeneron ($REGN), we have a brand-new No. 1 on our list. That’s Regeneron CEO Leonard Schleifer, whose 2012 compensation totaled $30.047 million. You’ll notice some other newbies, such as Leonard Bell from Alexion ($ALXN), whose pay bump put him in 12th place. And then there are familiar faces, such as Pfizer ($PFE) CEO Ian Read; Johnson & Johnson’s ($JNJ) former chairman and CEO, William Weldon; and Eli Lilly ($LLY) CEO John Lechleiter, who hung on in 10th place.
Many of the companies we researched pay their top people far less than the $10 million that served as our cutoff figure. Novo Nordisk ($NVO) CEO Lars Sorensen, who has presided over double-digit growth there for several years, collected a package of cash and stock awards worth about $5 million for 2012. GlaxoSmithKline ($GSK) CEO Andrew Witty made less than $6 million himself; he took a pay cut for the year because of Glaxo’s shortfall on certain performance targets.
And then there are others who would have made the list, had their titles been different. There’s Regeneron R&D chief George Yancopoulos, whose extraordinary $81 million in compensation shows how much the company appreciates its newly minted blockbuster, Eylea. There’s Mylan Chairman Robert Coury, who used to be a fixture on our list until Heather Bresch took over as CEO; he made more than $28 million last year. Novartis’ ($NVS) former chairman Daniel Vasella could have qualified for 12th place with his $13.98 million in compensation.
Vasella, then, gives us a quick segue to the ongoing debate over executive pay. In Switzerland, populist dismay at some high-profile compensation figures led to a public vote earlier this year. Citizens voted in new restrictions on common bonuses, such as golden parachutes, and gave shareholders a binding vote on executive pay. And local analysts figure that late-breaking news of Vasella’s behind-the-scenes noncompete agreement–worth some $78 million over 5 years–helped pay activists to get out the vote. (Vasella ended up refusing the deal, by the way.)
In the U.S., where executives are paid more than anywhere else in the world, shareholders at some companies have successfully lobbied for a greater emphasis on performance pay and against extraordinary bonuses, such as change-in-control payments that send top executives on their way with tens of millions after a merger. Other companies have instituted “say-on-pay” advisory votes for shareholders, but those often end up as rubber stamps for the status quo.
Now, we’re interested in what you have to say about executive compensation. Are the CEOs on this list worth their price? What’s a supersuccessful new drug worth? Should CEO pay be docked for R&D failures? What about failed launches? Should other, lower-paid executives earn more? Tweet your opinions to @FiercePharma using the hashtag #FPexecpay, leave your comments below or email us. We’ll collect your thoughts in a future article.
As always, feel free to send us your thoughts on our coverage. And if we missed a well-paid CEO, be sure to let us know.
— Tracy Staton (email | Twitter)
For more:
Top 10 Biotech CEO Pay Packages of 2012
Top 10 Pharma CEO salaries of 2010
Top 10 Pharma CEO salaries of 2009
2012’s 10 highest-paid Med Tech CEOs
Top 10 Medical Device Industry CEO Salaries for 2011
20 Highest-Paid Biopharma CEOs of 2012 – FiercePharma http://www.fiercepharma.com/special-reports/20-highest-paid-biopharma-ceos-2012#ixzz2UAGAlHay
REFERENCES FOR Part I: Arterial Stiffness
1. Laurent S, Boutouyrie P, Asmar R, Gautier I, Laloux B, Guize L,
Ducimetiere P, Benetos A. Aortic stiffness is an independent predictor
of all-cause and cardiovascular mortality in hypertensive patients.
Hypertension. 2001;37:1236–1241.
2. Vlachopoulos C, Aznaouridis K, Stefanadis C. Prediction of cardiovascular
events and all-cause mortality with arterial stiffness: a systematic
review and meta-analysis. J Am Coll Cardiol. 2010;55:1318–1327.
3. Boutouyrie P, Tropeano AI, Asmar R, Gautier I, Benetos A, Lacolley P,
Laurent S. Aortic stiffness is an independent predictor of primary coronary
events in hypertensive patients: a longitudinal study. Hypertension.
2002;39:10–15.
4. Mattace-Raso FU, van der Cammen TJ, Hofman A, van Popele NM, Bos
ML, Schalekamp MA, Asmar R, Reneman RS, Hoeks AP, Breteler MM,
Witteman JC. Arterial stiffness and risk of coronary heart disease and
stroke: the Rotterdam Study. Circulation. 2006;113:657–663.
5. Sehestedt T, Jeppesen J, Hansen TW, Rasmussen S, Wachtell K, Ibsen H,
Torp-Pedersen C, Olsen MH. Risk stratification with the risk chart from
the European Society of Hypertension compared with SCORE in the general
population. J Hypertens. 2009;27:2351–2357.
6. Mitchell GF, Hwang SJ, Vasan RS, Larson MG, Pencina MJ,
Hamburg NM, Vita JA, Levy D, Benjamin EJ. Arterial stiffness and
cardiovascular events: the Framingham Heart Study. Circulation. 2010;
121:505–511.
7. Gosse P, Cremer A, Papaioannou G, Yeim S. Arterial stiffness from monitoring
of timing of Korotkoff sounds predicts the occurrence of cardiovascular
events independently of left ventricular mass in hypertensive
patients. HYPERTENSIONAHA.113.01039 Published online before print May 20, 2013,doi: 10.1161/HYPERTENSIONAHA.113.01039
Hypertension. 2013;62:XX–XX.
http://hyper.ahajournals.org/content/early/2013/05/20/HYPERTENSIONAHA.113.01039.abstract.html?papetoc
8. Gosse P, Guillo P, Ascher G, Clementy J. Assessment of arterial distensibility
by monitoring the timing of Korotkoff sounds. Am J Hypertens.
1994;7:228–233.
9. Laurent S, Cockcroft J, Van Bortel L, Boutouyrie P, Giannattasio C,
Hayoz D, Pannier B, Vlachopoulos C, Wilkinson I, Struijker-Boudier
H; European Network for Non-invasive Investigation of Large Arteries.
Expert consensus document on arterial stiffness: methodological issues
and clinical applications. Eur Heart J. 2006;27:2588–2605.
10. Dogui A, Redheuil A, Lefort M, DeCesare A, Kachenoura N, Herment A,
Mousseaux E. Measurement of aortic arch pulse wave velocity in cardiovascular
MR: comparison of transit time estimators and description of a
new approach. J Magn Reson Imaging. 2011;33:1321–1329.
downloaded from
http://hyper.ahajournals.org/content/early/2013/05/20/HYPERTENSIONAHA.113.01372.citation
REFERENCES for Part II: Clinical Trials of Hypertension Management
1. Hanselin MR , Saseen JJ , Allen RR , Marrs JC , Nair KV . Description of
antihypertensive use in patients with resistant hypertension prescribed
four or more agents. Hypertension . 2011 ; 58 : 1008 – 1013 .
2. Bakris GL , Lindholm LH , Black HR , Krum H , Linas S , Linseman JV ,
Arterburn S , Sager P , Weber M . Divergent results using clinic and ambulatory
blood pressures: report of a darusentan-resistant hypertension trial.
Hypertension . 2010 ; 56 : 824 – 830 .
3. Smithwick RH , Thompson JE . Splanchnicectomy for essential hypertension;
results in 1,266 cases. J Am Med Assoc . 1953 ; 152 : 1501 – 1504 .
4. Esler M , Ferrier C , Lambert G , Eisenhofer G , Cox H , Jennings G .
Biochemical evidence of sympathetic hyperactivity in human hypertension.
Hypertension . 1991 ; 17 ( 4 Suppl ): III29 – III35 .
5. Esler MD , Krum H , Sobotka PA , Schlaich MP , Schmieder RE , Bohm
M . Symplicity HTN-2 investigators. Renal sympathetic denervation in
patients with treatment-resistant hypertension: a randomized controlled
trial. Lancet 2010 ; 376 : 1903 – 1909 .
6. Brinkmann J , Heusser K , Schmidt BM , Menne J , Klein G , Bauersachs J ,
Haller H , Sweep FC , Diedrich A , Jordan J , Tank J . Catheter-based renal
nerve ablation and centrally generated sympathetic activity in diffi cultto-
control hypertensive patients: prospective case series. Hypertension .
2012 ; 60 : 1485 – 1490 .
7. Mahfoud F , Cremers B , Janker J , et al . Renal hemodynamics and renal
function after catheter-based renal sympathetic denervation in patients
with resistant hypertension. Hypertension . 2012 ; 60 : 419 – 424 .
8. Lambert GW , Hering D , Esler MD , Marusic P , Lambert EA , Tanamas
SK , Shaw J , Krum H , Dixon JB , Barton DA , Schlaich MP . Health-related
quality of life after renal denervation in patients with treatment-resistant
hypertension. Hypertension . 2012 ; 60 : 1479 – 1484 .
9. Daugherty SL , Powers JD , Magid DJ , Masoudi FA , Margolis KL ,
O ’ Connor PJ , Schmittdiel JA , Ho PM . The association between medication
adherence and treatment intensifi cation with blood pressure control
in resistant hypertension. Hypertension . 2012 ; 60 : 303 – 309 .
10. Bobrie G , Frank M , Azizi M , Peyrard S , Boutouyrie P , Chatellier G ,
Laurent S , Menard J , Plouin PF . Sequential nephron blockade versus
sequential renin-angiotensin system blockade in resistant hypertension:
a prospective, randomized, open blinded endpoint study. J Hypertens .
2012 ; 30 : 1656 – 1664 .
11. Mancia G . Additional drug treatment in resistant hypertension: need for
randomized studies. J Hypertens . 2012 ; 30 : 1514 – 1515 .
12. Hermida RC , Ayala DE , Moj ó n A , Fontao MJ , Fern á ndez JR .
Chronotherapy with valsartan/hydrochlorothiazide combination in essential
hypertension: improved sleep-time blood pressure control with bedtime
dosing. Chronobiol Int . 2011 ; 28 : 601 – 610 .
13. Persu A , Renkin J , Thijs L , Staessen JA . Renal denervation: ultima
ratio or standard in treatment-resistant hypertension. Hypertension .
2012 ; 60 : 596 – 606 .
14. Lohmeier TE , Iliescu R . Chronic lowering of blood pressure by carotid
barorefl ex activation: mechanisms and potential for hypertension therapy.
Hypertension . 2011 ; 57 : 880 – 886 .
15. Bisognano JD , Bakris G , Nadim MK , Sanchez L , Kroon AA , Schafer J ,
de Leeuw PW , Sica DA . Barorefl ex activation therapy lowers blood pressure
in patients with resistant hypertension: results from the double-blind,
randomized, placebo-controlled rheos pivotal trial. J Am Coll Cardiol .
2011 ; 58 : 765 – 773 .
16. Parving HH , Brenner BM , McMurray JJ , de Zeeuw D , Haffner SM ,
Solomon SD , Chaturvedi N , Persson F , Desai AS , Nicolaides M , Richard
A , Xiang Z , Brunel P , Pfeffer MA ; ALTITUDE Investigators . Cardiorenal
end points in a trial of aliskiren for type 2 diabetes. N Engl J Med .
2012 ; 367 : 2204 – 2213 .
17. The ONTARGET Investigators . Telmisartan, ramipril, or both in patients
at high risk for vascular events. N Engl J Med . 2008 ; 358 : 1547 – 1559 .
18. Mancia G , Parati G , Bilo G , et al . Ambulatory blood pressure values in
the Ongoing Telmisartan Alone and in Combination with Ramipril Global
Endpoint Trial (ONTARGET). Hypertension . 2012 ; 60 : 1400 – 1406 .
19. Roush GC , Holford TR , Guddati AK . Chlorthalidone compared with
hydrochlorothiazide in reducing cardiovascular events: systematic review
and network meta-analyses. Hypertension . 2012 ; 59 : 1110 – 1117 .
20. Cushman WC , Bakris GL , White WB , Weber MA , Sica D , Roberts A ,
Lloyd E , Kupfer S . Azilsartan medoxomil plus chlorthalidone reduces
blood pressure more effectively than olmesartan plus hydrochlorothiazide
in stage 2 systolic hypertension. Hypertension . 2012 ; 60 : 310 – 318 .
21. Alderman MH , Piller LB , Ford CE , Probstfi eld JL , Oparil S , Cushman WC ,
Einhorn PT , Franklin SS , Papademetriou V , Ong ST , Eckfeldt JH , Furberg
CD , Calhoun DA , Davis BR ; Antihypertensive and Lipid-Lowering
Treatment to Prevent Heart Attack Trial Collaborative Research Group .
Clinical signifi cance of incident hypokalemia and hyperkalemia in treated
hypertensive patients in the antihypertensive and lipid-lowering treatment
to prevent heart attack trial. Hypertension . 2012 ; 59 : 926 – 933 .
22. Kostis WJ , Thijs L , Richart T , Kostis JB , Staessen JA . Persistence of mortality
reduction after the end of randomized therapy in clinical trials of
blood pressure-lowering medications. Hypertension . 2010 ; 56 : 1060 – 1068 .
23. van Onzenoort HA , Menger FE , Neef C , Verberk WJ , Kroon AA , de
Leeuw PW , van der Kuy PH . Participation in a clinical trial enhances
adherence and persistence to treatment: a retrospective cohort study.
Hypertension . 2011 ; 58 : 573 – 578 .
24. Sever PS . The Anglo-Scandinavian Cardiac Outcomes Trial: implications
and further outcomes. Hypertension . 2012 ; 60 : 248 – 259 .
25. Larstorp AC , Ariansen I , Gjesdal K , Olsen MH , Ibsen H , Devereux RB ,
Okin PM , Dahl ö f B , Kjeldsen SE , Wachtell K . Association of pulse pressure
with new-onset atrial fi brillation in patients with hypertension and
left ventricular hypertrophy: the Losartan Intervention For Endpoint
(LIFE) reduction in hypertension study. Hypertension . 2012 ; 60 : 347 – 353 .
26. Rossignol P , Cridlig J , Lehert P , Kessler M , Zannad F . Visit-to-visit blood
pressure variability is a strong predictor of cardiovascular events in hemodialysis:
insights from FOSIDIAL. Hypertension . 2012 ; 60 : 339 – 346 .
27. Matsui Y , O ’ Rourke MF , Hoshide S , Ishikawa J , Shimada K , Kario
K . Combined Effect of Angiotensin II Receptor Blocker and Either a
Calcium Channel Blocker or Diuretic on Day-by-Day Variability of Home
Blood Pressure: The Japan Combined Treatment With Olmesartan and a
Calcium-Channel Blocker Versus Olmesartan and Diuretics Randomized
Effi cacy Study. Hypertension . 2012 ; 59 : 1132 – 1138 .
28. Jennings GL , Sudhir K . Initial therapy of primary hypertension. Med J
Aust . 1990 ; 152 : 198 – 203 .
29. Egan BM , Bandyopadhyay D , Shaftman SR , Wagner CS , Zhao Y ,
Yu-Isenberg KS . Initial monotherapy and combination therapy and hypertension
control the fi rst year. Hypertension . 2012 ; 59 : 1124 – 1131 .
30. Bavry AA , Pepine CJ . Treatment of hypertension: lower is better, or is it?
Hypertension . 2012 ; 60 : 281 – 282 .
31. Mizuno R , Fujimoto S , Saito Y , Okamoto Y . Optimal antihypertensive
level for improvement of coronary microvascular dysfunction: the lower,
the better? Hypertension . 2012 ; 60 : 326 – 332 .
32. Roman MJ , Howard BV , Howard WJ , Mete M , Fleg JL , Lee ET ,
Devereux RB . Differential impacts of blood pressure and lipid lowering
on regression of ventricular and arterial mass: the Stop Atherosclerosis in
Native Diabetics Trial. Hypertension . 2011 ; 58 : 367 – 371 .
33. Bertoia ML , Waring ME , Gupta PS , Roberts MB , Eaton CB . Implications
of new hypertension guidelines in the United States. Hypertension .
2012 ; 60 : 639 – 644 .
34. Stewart S , Carrington MJ , Swemmer CH , et al .; VIPER-BP Study
Investigators . Effect of intensive structured care on individual blood pressure
targets in primary care: multicentre randomised controlled trial. BMJ .
2012 ; 345 : e7156 .
35. Dickinson BD , Havas S ; Council on Science and Public Health, American
Medical Association . Reducing the population burden of cardiovascular
disease by reducing sodium intake: a report of the Council on Science and
Public Health. Arch Intern Med . 2007 ; 167 : 1460 – 1468 .
36. O ’ Donnell MJ , Yusuf S , Mente A , Gao P , Mann JF , Teo K , McQueen M ,
Sleight P , Sharma AM , Dans A , Probstfi eld J , Schmieder RE . Urinary
sodium and potassium excretion and risk of cardiovascular events. JAMA .
2011 ; 306 : 2229 – 2238 .
37. Hummel SL , Seymour EM , Brook RD , Kolias TJ , Sheth SS , Rosenblum
HR , Wells JM , Weder AB . Low-sodium dietary approaches to stop
hypertension diet reduces blood pressure, arterial stiffness, and oxidative
stress in hypertensive heart failure with preserved ejection fraction.
Hypertension . 2012 ; 60 : 1200 – 1206 .
38. Soedamah-Muthu SS , Verberne LD , Ding EL , Engberink MF , Geleijnse JM .
Dairy consumption and incidence of hypertension: a dose-response metaanalysis
of prospective cohort studies. Hypertension . 2012 ; 60 : 1131 – 1137 .
39. West SG , Gebauer SK , Kay CD , Bagshaw DM , Savastano DM ,
Diefenbach C , Kris-Etherton PM . Diets containing pistachios reduce systolic
blood pressure and peripheral vascular responses to stress in adults
with dyslipidemia. Hypertension . 2012 ; 60 : 58 – 63 .
40. Zhang X , Qi Q , Liang J , Hu FB , Sacks FM , Qi L . Neuropeptide Y promoter
polymorphism modifi es effects of a weight-loss diet on 2-year
changes of blood pressure: the preventing overweight using novel dietary
strategies trial. Hypertension . 2012 ; 60 : 1169 – 1175 .
41. Meredith IT , Friberg P , Jennings GL , Dewar EM , Fazio VA , Lambert GW ,
Esler MD . Exercise training lowers resting renal but not cardiac sympathetic
activity in humans. Hypertension . 1991 ; 18 : 575 – 582 .
42. Cornelissen VA , Fagard RH , Coeckelberghs E , Vanhees L . Impact of resistance
training on blood pressure and other cardiovascular risk factors: a metaanalysis
of randomized, controlled trials. Hypertension . 2011 ; 58 : 950 – 958 .
Downloaded from
http://hyper.ahajournals.org/content/early/2013/05/20/HYPERTENSIONAHA.113.00863.citation
RELATED SOURCES:
Aortic pulse pressure is associated with the localization of coronary artery disease based on coronary flow lateralization. American journal of hypertension, 25(10), 1055-1063.
- Georges Khoueiry1,
- Basem Azab2,
- Estelle Torbey2,
- Nidal Abi Rafeh1,
- Jean-Paul Atallah2,
- Kathleen Ahern2,
- James Malpeso1,
- Donald McCord1 and
- Elie R. Chemaly3⇓
Author Affiliations
1Department of Cardiology, Staten Island University Hospital, Staten Island, New York, USA
2Department of Internal Medicine, Staten Island University Hospital, Staten Island, New York, USA
3Cardiovascular Institute, Mount Sinai School of Medicine, New York, New York, USA
Elie R. Chemaly (elie.chemaly@mssm.edu)
Abstract
Background Aortic pulse pressure (APP) is related to arterial stiffness and associated with the presence and extent of coronary artery disease (CAD). Besides, the left coronary artery (LCA) has a predominantly diastolic flow while the right coronary artery (RCA) receives systolic and diastolic flow. Thus, we hypothesized that increased systolic–diastolic pressure difference had a greater atherogenic effect on the RCA than on the LCA.
Methods A random sample of 433 CAD patients (145 females, 288 males, mean age 65.0 ± 11.1 years) undergoing coronary angiography at Staten Island University Hospital between January 2005 and May 2008 was studied. Coronary lesion was defined as a ≥50% luminal stenosis. Patients were divided into three groups, with isolated LCA lesions (n = 154), isolated RCA lesions (n = 36) or mixed LCA and RCA lesions (n = 243).
Results APP differed significantly between groups, being highest when the RCA alone was affected (67.6 ± 20.3 mm Hg for LCA vs. 78.8 ± 22.0 for RCA vs. 72.7 ± 22.6 for mixed, P = 0.008 for analysis of variance (ANOVA)). Age and gender were not associated with CAD location. Heart rate was associated with CAD location, lowest in RCA group, and negatively correlated with APP. However, left ventricular ejection fraction (LVEF) was lower in the mixed CAD group and positively correlated with APP. The association between APP and right-sided CAD persisted in multivariate logistic regression adjusting for confounders, including heart rate, LVEF and medication use. A similar but less significant pattern was seen with brachial arterial pressures.
Conclusions Aortic pulse pressure may affect CAD along with coronary flow phasic patterns.
The Relationship Between Diastolic Pressure and Coronary Collateral Circulation in Patients With Stable Angina Pectoris and Chronic Total Occlusion. Am J Hypertens (2013)doi: 10.1093/ajh/hps096
First published online: February 7, 2013
- Wang Shu1,
- Jing jing1,
- Liu Chang Fu1,
- Jiang Tie Min2,
- Yang Xiao Bo1,
- Zhou Ying1and
- Chen Yun Dai1,*
-
1 The Cardiovascular Medical Department of the General Hospital of the Chinese People’s Liberation Army, Beijing, China;
2 The Cardiovascular Medical Department of the Affiliated Hospital of the Chinese People’s Armed Police Logistics College, Tianjin, China.
- ↵Correspondence: Chen Yun Dai (chenyundai2002@163.com).
Abstract
BACKGROUND The most important biomechanical source of activation of the coronary collateral circulation (CCC) is increased tangential fluid shear stress at the arterial endothelial surface. The coronary circulation is unique in that most coronary blood flow occurs in diastole. Consequently, the diastolic blood pressure (DBP) may influence the tangential fluid shear stress on the arterial endothelial surface in diastole, therebyaffecting development of the CCC.
METHODS To investigate this, we conducted a study of 222 patients with stable angina pectoris and chronic total occlusion of coronary arteries. All of the patients had no history of coronary artery interventional therapy, coronary artery bypass surgery, cardiomyopathy, or congenital heart disease. The extent of the collateral vasculature of the area perfused by the artery affected by chronic total occlusion was graded as poor or well-developed according to Rentrop’s classification.
RESULTS Univariate analysis showed a significant difference between the study subgroup with poorly developed collaterals and that with well-developed collaterals in terms of high diastolic blood pressure (DBP) and mean DBP. Multivariate analysis revealed high DBP as the only independent positive predictor of a well-developed collateral circulation.
CONCLUSIONS High DBP is positively related to a well-developed CCC. Differences in development of the CCC may be one of the pathophysiologic mechanisms responsible for the J-curve phenomenon in the relationship between DBP and cardiovascular risk.
http://ajh.oxfordjournals.org/content/early/2013/02/06/ajh.hps096.abstract
Other related articles that were published on this Open Access Online Scientific Journal, include the following:
Artherogenesis: Predictor of CVD – the Smaller and Denser LDL Particles
Aviva Lev-Ari, PhD, RN 11/15/2012
http://pharmaceuticalintelligence.com/2012/11/15/artherogenesis-predictor-of-cvd-the-smaller-and-denser-ldl-particles/
Cardiovascular Diseases: Causes, Risks and Management, Volume Two, Risks of Cardiovascular Diseases
Justin D. Pearlman MD ME PhD MA FACC, Editor
http://pharmaceuticalintelligence.com/biomed-e-books/cardiovascular-diseases-risks-and-management/cvd-2-risk-assessment-of-cardiovascular-diseases/
Genetics of Conduction Disease: Atrioventricular (AV) Conduction Disease (block): Gene Mutations – Transcription, Excitability, and Energy Homeostasis
Aviva Lev-Ari, PhD, RN 4/28/2013
http://pharmaceuticalintelligence.com/2013/04/28/genetics-of-conduction-disease-atrioventricular-av-conduction-disease-block-gene-mutations-transcription-excitability-and-energy-homeostasis/
Genomics & Genetics of Cardiovascular Disease Diagnoses: A Literature Survey of AHA’s Circulation Cardiovascular Genetics, 3/2010 – 3/2013
Aviva Lev-Ari, PhD, RN and Larry H. Bernstein, MD, FCAP 3/7/2013
http://pharmaceuticalintelligence.com/2013/03/07/genomics-genetics-of-cardiovascular-disease-diagnoses-a-literature-survey-of-ahas-circulation-cardiovascular-genetics-32010-32013/
Hypertriglyceridemia concurrent Hyperlipidemia: Vertical Density Gradient Ultracentrifugation a Better Test to Prevent Undertreatment of High-Risk Cardiac Patients
Aviva Lev-Ari, PhD, RN 4/4/2013
http://pharmaceuticalintelligence.com/2013/04/04/hypertriglyceridemia-concurrent-hyperlipidemia-vertical-density-gradient-ultracentrifugation-a-better-test-to-prevent-undertreatment-of-high-risk-cardiac-patients/
Hypertension and Vascular Compliance: 2013 Thought Frontier – An Arterial Elasticity Focus
Justin D. Pearlman, MD, PhD and Aviva Lev-Ari, PhD, RN 5/11/2013
http://pharmaceuticalintelligence.com/2013/05/11/arterial-elasticity-in-quest-for-a-drug-stabilizer-isolated-systolic-hypertension-caused-by-arterial-stiffening-ineffectively-treated-by-vasodilatation-antihypertensives/
Read Full Post »







 DNA decoder: Knome’s software can tease out medically relevant changes in DNA that could disrupt individual gene function or even a whole molecular pathway, as is highlighted here—certain mutations in the BRCA2 gene, which affects the function of many other genes, can be associated with an increased risk of breast cancer.
DNA decoder: Knome’s software can tease out medically relevant changes in DNA that could disrupt individual gene function or even a whole molecular pathway, as is highlighted here—certain mutations in the BRCA2 gene, which affects the function of many other genes, can be associated with an increased risk of breast cancer.










 , where t is time, s is a complex parameter, and A is a complex scalar. Complex values simply mean two dimensional, e.g., magnitude (as in resistance) plus phase shift (to account for reactive components).
, where t is time, s is a complex parameter, and A is a complex scalar. Complex values simply mean two dimensional, e.g., magnitude (as in resistance) plus phase shift (to account for reactive components).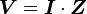 where V and I are the complex scalars in the voltage and current respectively and Z is the complex impedance.
where V and I are the complex scalars in the voltage and current respectively and Z is the complex impedance.

 British researchers are working to adapt technology from the aviation industry to help prevent complications among heart patients after surgery. Up to 1,000 sensors aboard aircraft help airlines determine when a plane requires maintenance,
British researchers are working to adapt technology from the aviation industry to help prevent complications among heart patients after surgery. Up to 1,000 sensors aboard aircraft help airlines determine when a plane requires maintenance, 





{kind=link}
{kind=link}