Healthcare analytics, AI solutions for biological big data, providing an AI platform for the biotech, life sciences, medical and pharmaceutical industries, as well as for related technological approaches, i.e., curation and text analysis with machine learning and other activities related to AI applications to these industries.
Biomolecular Condensates: A new approach to biology originated @MIT – Drug Discovery at DewPoint Therapeutics, Cambridge, MA gets new leaders, Ameet Nathwani, MD (ex-Sanofi, ex-Novartis) as Chief Executive Officer and Arie Belldegrun, PhD (ex-Kite Therapeutics) on R&D
“The real voyage of discovery consists, not in seeking new landscapes, but in having new eyes.” Marcel Proust
Starting with the study of P granules in C.elegans embryos in 2009, Tony Hyman, working with his collaborators like Frank Julicher, Cliff Brangwynne, Simon Alberti, Mike Rosen, and Rohit Pappu, began to unravel the mysteries of biomolecular condensates. These scientists realized that P granules behave like liquid droplets that form by phase separation (think of oil droplets in salad dressing) and called them condensates.
In subsequent studies, they found to their surprise that many compartments inside cells had the behavior of condensates: they are liquid-like and form by phase separation.
Inspired by the work of Tony and his colleagues, Richard Young, Phillip Sharp, and Arup Chakraborty at MIT applied these approaches to the study of gene expression, similarly shedding light on many important questions in gene control.
Other related article published in this Online Open Access Scientific Journal include:
Economic Potential of a Drug Invention (Prof. Zelig Eshhar, Weitzman Institute, registered the patent) versus a Cancer Drug in Clinical Trials: CAR-T as a Case in Point, developed by Kite Pharma, under Arie Belldegrun, CEO, acquired by Gilead for $11.9 billion, 8/2017.
UC Berkeley accelerates bio-preservation research as part NSF center
Reporter: Irina Robu, PhD
National Science Foundation funded a new research center which focuses on advancing methods for storing and preserving biological cells and tissues. The new center will be directed by John Bischof at the University of Minnesota and co-directed by Mehmet Toner at Massachusetts General Hospital in collaboration with researchers from UC Berkeley and UC Riverside. It is known that the inability to extend shelf life of biological tissues means that patients in Florida can’t receive a heart or lung from California. And when thawing any cell culture, the survival rate of the cells are about 60 percent, which is considered normal .
NSF Engineering Research Centers were founded to bring together academic and industry partners in interdisciplinary collaborations to tackle complex, long-range engineering challenges. The anticipation is that the centers will brood whole new industries with new systems-level technologies. In 2025, the award for ATP-Bio may be renewed for an additional five years.
This session will provide information regarding methodologic and computational aspects of proteogenomic analysis of tumor samples, particularly in the context of clinical trials. Availability of comprehensive proteomic and matching genomic data for tumor samples characterized by the National Cancer Institute’s Clinical Proteomic Tumor Analysis Consortium (CPTAC) and The Cancer Genome Atlas (TCGA) program will be described, including data access procedures and informatic tools under development. Recent advances on mass spectrometry-based targeted assays for inclusion in clinical trials will also be discussed.
Amanda G Paulovich, Shankha Satpathy, Meenakshi Anurag, Bing Zhang, Steven A Carr
Methods and tools for comprehensive proteogenomic characterization of bulk tumor to needle core biopsies
Shankha Satpathy
TCGA has 11,000 cancers with >20,000 somatic alterations but only 128 proteins as proteomics was still young field
CPTAC is NCI proteomic effort
Chemical labeling approach now method of choice for quantitative proteomics
Looked at ovarian and breast cancers: to measure PTM like phosphorylated the sample preparation is critical
Data access and informatics tools for proteogenomics analysis
Bing Zhang
Raw and processed data (raw MS data) with linked clinical data can be extracted in CPTAC
Python scripts are available for bioinformatic programming
Pathways to clinical translation of mass spectrometry-based assays
Meenakshi Anurag
· Using kinase inhibitor pulldown (KIP) assay to identify unique kinome profiles
· Found single strand break repair defects in endometrial luminal cases, especially with immune checkpoint prognostic tumors
· Paper: JNCI 2019 analyzed 20,000 genes correlated with ET resistant in luminal B cases (selected for a list of 30 genes)
· Validated in METABRIC dataset
· KIP assay uses magnetic beads to pull out kinases to determine druggable kinases
· Looked in xenografts and was able to pull out differential kinomes
· Matched with PDX data so good clinical correlation
· Were able to detect ESR1 fusion correlated with ER+ tumors
The adoption of omic technologies in the cancer clinic is giving rise to an increasing number of large-scale high-dimensional datasets recording multiple aspects of the disease. This creates the need for frameworks for translatable discovery and learning from such data. Like artificial intelligence (AI) and machine learning (ML) for the cancer lab, methods for the clinic need to (i) compare and integrate different data types; (ii) scale with data sizes; (iii) prove interpretable in terms of the known biology and batch effects underlying the data; and (iv) predict previously unknown experimentally verifiable mechanisms. Methods for the clinic, beyond the lab, also need to (v) produce accurate actionable recommendations; (vi) prove relevant to patient populations based upon small cohorts; and (vii) be validated in clinical trials. In this educational session we will present recent studies that demonstrate AI and ML translated to the cancer clinic, from prognosis and diagnosis to therapy.
NOTE: Dr. Fish’s talk is not eligible for CME credit to permit the free flow of information of the commercial interest employee participating.
Ron C. Anafi, Rick L. Stevens, Orly Alter, Guy Fish
Overview of AI approaches in cancer research and patient care
Rick L. Stevens
Deep learning is less likely to saturate as data increases
Deep learning attempts to learn multiple layers of information
The ultimate goal is prediction but this will be the greatest challenge for ML
ML models can integrate data validation and cross database validation
What limits the performance of cross validation is the internal noise of data (reproducibility)
Learning curves: not the more data but more reproducible data is important
Neural networks can outperform classical methods
Important to measure validation accuracy in training set. Class weighting can assist in development of data set for training set especially for unbalanced data sets
Discovering genome-scale predictors of survival and response to treatment with multi-tensor decompositions
Orly Alter
Finding patterns using SVD component analysis. Gene and SVD patterns match 1:1
Comparative spectral decompositions can be used for global datasets
Validation of CNV data using this strategy
Found Ras, Shh and Notch pathways with altered CNV in glioblastoma which correlated with prognosis
These predictors was significantly better than independent prognostic indicator like age of diagnosis
Identifying targets for cancer chronotherapy with unsupervised machine learning
Ron C. Anafi
Many clinicians have noticed that some patients do better when chemo is given at certain times of the day and felt there may be a circadian rhythm or chronotherapeutic effect with respect to side effects or with outcomes
ML used to determine if there is indeed this chronotherapy effect or can we use unstructured data to determine molecular rhythms?
Found a circadian transcription in human lung
Most dataset in cancer from one clinical trial so there might need to be more trials conducted to take into consideration circadian rhythms
Stratifying patients by live-cell biomarkers with random-forest decision trees
Stratifying patients by live-cell biomarkers with random-forest decision trees
Guy Fish CEO Cellanyx Diagnostics
Some clinicians feel we may be overdiagnosing and overtreating certain cancers, especially the indolent disease
This educational session focuses on the chronic wound healing, fibrosis, and cancer “triad.” It emphasizes the similarities and differences seen in these conditions and attempts to clarify why sustained fibrosis commonly supports tumorigenesis. Importance will be placed on cancer-associated fibroblasts (CAFs), vascularity, extracellular matrix (ECM), and chronic conditions like aging. Dr. Dvorak will provide an historical insight into the triad field focusing on the importance of vascular permeability. Dr. Stewart will explain how chronic inflammatory conditions, such as the aging tumor microenvironment (TME), drive cancer progression. The session will close with a review by Dr. Cukierman of the roles that CAFs and self-produced ECMs play in enabling the signaling reciprocity observed between fibrosis and cancer in solid epithelial cancers, such as pancreatic ductal adenocarcinoma.
Harold F Dvorak, Sheila A Stewart, Edna Cukierman
The importance of vascular permeability in tumor stroma generation and wound healing
Harold F Dvorak
Aging in the driver’s seat: Tumor progression and beyond
Sheila A Stewart
Why won’t CAFs stay normal?
Edna Cukierman
Tuesday, June 23
3:00 PM – 5:00 PM EDT
Other Articles on this Open Access Online Journal on Cancer Conferences and Conference Coverage in Real Time Include
The Relevance of Glycans in the Viral Pathology of COVID-19
Reporter: Ofer Markman, PhD
While we are constantly cautioning people from putting too high validity to the results seen in the bellow mentioned Study,
The ABO blood groups are a result of various and different glycans on the surfaces of red blood cells and their definition is related to Blood Immunology and Blood Typing classification used for Blood Donations. It was the hallmark of the 1930 Nobel prize of medicine.
Glycans are involved in the interaction of the flu virus to the host cell and the antiviral drug Tamiflu (Oseltamivir ) is based on the inhibition of that sort of interaction/modulation.
The Relevance of Glycans in the Viral Pathology of COVID-19
Even if true, the numbers in this paper show statistical significant difference but mildly significant difference in the risk profile suggesting we are to pay too much attention to the phenomena or worry, regardless of the fact these result have no significance on behavioral instructions nor would call for testing one’s blood type in regards to a viral infection. I would neither totally ignore the finding as it may shed light on viral pathology and infection mechanisms and understanding of the later may lead us to treatment or effective vaccines. But we are still early on this path.
yet a more relevant understanding of glycans in the biology of the virus:-
this week NEJM interview shed some more light on the issue in their 2nd part of the talk when they discussed again blood group risk factors in the covid-19 context.
Predicting the Protein Structure of Coronavirus: Inhibition of Nsp15 can slow viral replication and Cryo-EM – Spike protein structure (experimentally verified) vs AI-predicted protein structures (not experimentally verified) of DeepMind (Parent: Google) aka AlphaFold
Curators: Stephen J. Williams, PhD and Aviva Lev-Ari, PhD, RN
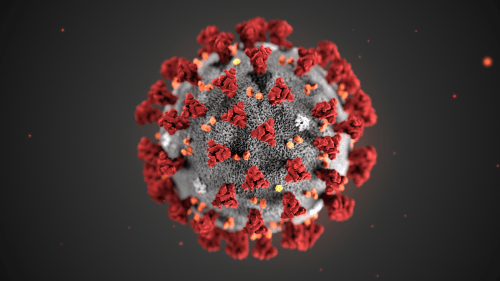
This illustration, created at the Centers for Disease Control and Prevention (CDC), reveals ultrastructural morphology exhibited by coronaviruses. Note the spikes that adorn the outer surface of the virus, which impart the look of a corona surrounding the virion, when viewed electron microscopically. A novel coronavirus virus was identified as the cause of an outbreak of respiratory illness first detected in Wuhan, China in 2019.
$The authors would like to note that the first eight authors are listed alphabetically.
Abstract
During its first month, the recently emerged 2019 Wuhan novel coronavirus (2019-nCoV) has already infected many thousands of people in mainland China and worldwide and took hundreds of lives. However, the swiftly spreading virus also caused an unprecedentedly rapid response from the research community facing the unknown health challenge of potentially enormous proportions. Unfortunately, the experimental research to understand the molecular mechanisms behind the viral infection and to design a vaccine or antivirals is costly and takes months to develop. To expedite the advancement of our knowledge we leverage the data about the related coronaviruses that is readily available in public databases, and integrate these data into a single computational pipeline. As a result, we provide a comprehensive structural genomics and interactomics road-maps of 2019-nCoV and use these information to infer the possible functional differences and similarities with the related SARS coronavirus. All data are made publicly available to the research community at http://korkinlab.org/wuhan
Figure 2. Structurally characterized non-structural proteins of 2019-nCoV. Highlighted in pink are mutations found when aligning the proteins against their homologs from the closest related coronaviruses: 2019-nCoV and human SARS, bat coronavirus, and another bat betacoronavirus BtRf-BetaCoV. The structurally resolved part of wNsp7 is sequentially identical to its homolog.
Figure 3. Structurally characterized structural proteins and an ORF of 2019-nCoV. Highlighted in pink are mutations found when aligning the proteins against their homologs from the closest related coronaviruses: 2019-nCoV and human SARS, bat coronavirus, and another bat betacoronavirus BtRf-BetaCoV. Highlighted in yellow are novel protein inserts found in wS.
Figure 4. Structurally characterized intra-viral and host-viral protein-protein interaction complexes of 2019-nCoV. Human proteins (colored in orange) are identified through their gene names. For each intra-viral structure, the number of subunits involved in the interaction is specified.
Figure 5. Evolutionary conservation of functional sites in 2019-nCoV proteins. A. Fully conserved protein binding sites (PBS, light orange) of wNsp12 in its interaction with wNsp7 and wNsp8 while other parts of the protein surface shows mutations (magenta); B. Both major monoclonal antibody binding site (light orange) and ACE2 receptor binding site (dark green) of wS are heavily mutated (binding site mutations are shown in red) compared to the same binding sites in other coronaviruses; mutations not located on the two binding sites are shown in magenta; C. Nearly intact protein binding site (light orange) of wNsp (papain-like protease PLpro domain) for its putative interaction with human ubiquitin-aldehyde (binding site mutations for the only two residues are shown in red, non-binding site mutations are shown in magenta); D. Fully conserved inhibitor ligand binding site (LBS, green) for wNsp5; non-binding site mutations are shown in magenta.
According to the World Health Organization, coronaviruses make up a large family of viruses named for the crown-like spikes found on their surface (Figure 1). They carry their genetic material in single strands of RNA and cause respiratory problems and fever. Like HIV, coronaviruses can be transmitted between animals and humans. Coronaviruses have been responsible for the Severe Acute Respiratory Syndrome (SARS) pandemic in the early 2000s and the Middle East Respiratory Syndrome (MERS) outbreak in South Korea in 2015. While the most recent coronavirus, COVID-19, has caused international concern, accessible and inexpensive sequencing is helping us understand COVID-19 and respond to the outbreak quickly.
Figure 1. Coronaviruses with the characteristic spikes as seen under a microscope.
First studies that explore genetic susceptibility to COVID-19 are now being published. The first results indicate that COVID-19 infects cells using the ACE2 cell-surface receptor. Genetic variants in the ACE2 receptor gene are thus likely to influence how effectively COVID-19 can enter the cells in our bodies. Researchers hope to discover genetic variants that confer resistance to a COVID-19 infection, similar to how some variants in the CCR5 receptor gene make people immune to HIV. At Nebula Genomics, we are monitoring the latest COVID-19 research and will add any relevant discoveries to the Nebula Research Library in a timely manner.
The Role of Genomics in Responding to COVID-19
Scientists in China sequenced COVID-19’s genome just a few weeks after the first case was reported in Wuhan. This stands in contrast to SARS, which was discovered in late 2002 but was not sequenced until April of 2003. It is through inexpensive genome-sequencing that many scientists across the globe are learning and sharing information about COVID-19, allowing us to track the evolution of COVID-19 in real-time. Ultimately, sequencing can help remove the fear of the unknown and allow scientists and health professionals to prepare to combat the spread of COVID-19.
Next-generation DNA sequencing technology has enabled us to understand COVID-19 is ~30,000 bases long. Moreover, researchers in China determined that COVID-19 is also almost identical to a coronavirus found in bats and is very similar to SARS. These insights have been critical in aiding in the development of diagnostics and vaccines. For example, the Centers for Disease Control and Prevention developed a diagnostic test to detect COVID-19 RNA from nose or mouth swabs.
Moreover, a number of different government agencies and pharmaceutical companies are in the process of developing COVID-19 vaccines to stop the COVID-19 from infecting more people. To protect humans from infection inactivated virus particles or parts of the virus (e.g. viral proteins) can be injected into humans. The immune system will recognize the inactivated virus as foreign, priming the body to build immunity against possible future infection. Of note, Moderna Inc., the National Institute of Allergy and Infectious Diseases, and Coalition for Epidemic Preparedness Innovations identified a COVID-19 vaccine candidate in a record 42 days. This vaccine will be tested in human clinical trials starting in April.
For more information about COVID-19, please refer to the World Health Organization website.
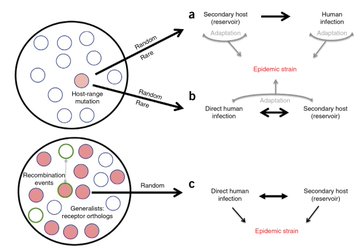
The problem w/ visionaries is that we don’t recognize them in a timely manner (too late) Ralph Baric @UNCpublichealth and Vineet Menachery deserve recognition for being 5 yrs ahead of #COVID19https://nature.com/articles/nm.3985…@NatureMedicinehttps://pnas.org/content/113/11/3048…@PNASNews via @hoondy
Senior, A.W., Evans, R., Jumper, J. et al.Improved protein structure prediction using potentials from deep learning. Nature577, 706–710 (2020). https://doi.org/10.1038/s41586-019-1923-7
Abstract
Protein structure prediction can be used to determine the three-dimensional shape of a protein from its amino acid sequence1. This problem is of fundamental importance as the structure of a protein largely determines its function2; however, protein structures can be difficult to determine experimentally. Considerable progress has recently been made by leveraging genetic information. It is possible to infer which amino acid residues are in contact by analysing covariation in homologous sequences, which aids in the prediction of protein structures3. Here we show that we can train a neural network to make accurate predictions of the distances between pairs of residues, which convey more information about the structure than contact predictions. Using this information, we construct a potential of mean force4 that can accurately describe the shape of a protein. We find that the resulting potential can be optimized by a simple gradient descent algorithm to generate structures without complex sampling procedures. The resulting system, named AlphaFold, achieves high accuracy, even for sequences with fewer homologous sequences. In the recent Critical Assessment of Protein Structure Prediction5 (CASP13)—a blind assessment of the state of the field—AlphaFold created high-accuracy structures (with template modelling (TM) scores6 of 0.7 or higher) for 24 out of 43 free modelling domains, whereas the next best method, which used sampling and contact information, achieved such accuracy for only 14 out of 43 domains. AlphaFold represents a considerable advance in protein-structure prediction. We expect this increased accuracy to enable insights into the function and malfunction of proteins, especially in cases for which no structures for homologous proteins have been experimentally determined7. https://doi.org/10.1038/s41586-019-1923-7
The scientific community has galvanised in response to the recent COVID-19 outbreak, building on decades of basic research characterising this virus family. Labs at the forefront of the outbreak response shared genomes of the virus in open access databases, which enabled researchers to rapidly develop tests for this novel pathogen. Other labs have shared experimentally-determined and computationally-predicted structures of some of the viral proteins, and still others have shared epidemiological data. We hope to contribute to the scientific effort using the latest version of our AlphaFold system by releasing structure predictions of several under-studied proteins associated with SARS-CoV-2, the virus that causes COVID-19. We emphasise that these structure predictions have not been experimentally verified, but hope they may contribute to the scientific community’s interrogation of how the virus functions, and serve as a hypothesis generation platform for future experimental work in developing therapeutics. We’re indebted to the work of many other labs: this work wouldn’t be possible without the efforts of researchers across the globe who have responded to the COVID-19 outbreak with incredible agility.
Knowing a protein’s structure provides an important resource for understanding how it functions, but experiments to determine the structure can take months or longer, and some prove to be intractable. For this reason, researchers have been developing computational methods to predict protein structure from the amino acid sequence. In cases where the structure of a similar protein has already been experimentally determined, algorithms based on “template modelling” are able to provide accurate predictions of the protein structure. AlphaFold, our recently published deep learning system, focuses on predicting protein structure accurately when no structures of similar proteins are available, called “free modelling”. We’ve continued to improve these methods since that publication and want to provide the most useful predictions, so we’re sharing predicted structures for some of the proteins in SARS-CoV-2 generated using our newly-developed methods.
It’s important to note that our structure prediction system is still in development and we can’t be certain of the accuracy of the structures we are providing, although we are confident that the system is more accurate than our earlier CASP13 system. We confirmed that our system provided an accurate prediction for the experimentally determined SARS-CoV-2 spike protein structure shared in the Protein Data Bank, and this gave us confidence that our model predictions on other proteins may be useful. We recently shared our results with several colleagues at the Francis Crick Institute in the UK, including structural biologists and virologists, who encouraged us to release our structures to the general scientific community now. Our models include per-residue confidence scores to help indicate which parts of the structure are more likely to be correct. We have only provided predictions for proteins which lack suitable templates or are otherwise difficult for template modeling. While these understudied proteins are not the main focus of current therapeutic efforts, they may add to researchers’ understanding of SARS-CoV-2.
Normally we’d wait to publish this work until it had been peer-reviewed for an academic journal. However, given the potential seriousness and time-sensitivity of the situation, we’re releasing the predicted structures as we have them now, under an open license so that anyone can make use of them.
Interested researchers can download the structures here, and can read more technical details about these predictions in a document included with the data. The protein structure predictions we’re releasing are for SARS-CoV-2 membrane protein, protein 3a, Nsp2, Nsp4, Nsp6, and Papain-like proteinase (C terminal domain). To emphasise, these are predicted structures which have not been experimentally verified. Work on the system continues for us, and we hope to share more about it in due course.
DeepMind has shared its results with researchers at the Francis Crick Institute, a biomedical research lab in the UK, as well as offering it for download from its website.
“Normally we’d wait to publish this work until it had been peer-reviewed for an academic journal. However, given the potential seriousness and time-sensitivity of the situation, we’re releasing the predicted structures as we have them now, under an open license so that anyone can make use of them,” it said. [ALA added bold face]
There are 93,090 cases of COVID-19, and 3,198 deaths, spread across 76 countries, according to the latest report from the World Health Organization at time of writing. ®
MHC content – The spike protein is thought to be the key to binding to cells via the angiotensin II receptor, the major mechanism the immune system uses to distinguish self from non-self
Preliminary Identification of Potential Vaccine Targets for the COVID-19 Coronavirus (SARS-CoV-2) Based on SARS-CoV Immunological Studies
Syed Faraz Ahmed 1,† , Ahmed A. Quadeer 1, *,† and Matthew R. McKay 1,2, *
1 Department of Electronic and Computer Engineering, The Hong Kong University of Science and
Technology, Hong Kong, China; sfahmed@connect.ust.hk
2 Department of Chemical and Biological Engineering, The Hong Kong University of Science and
Received: 9 February 2020; Accepted: 24 February 2020; Published: 25 February 2020
Abstract:
The beginning of 2020 has seen the emergence of COVID-19 outbreak caused by a novel coronavirus, Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). There is an imminent need to better understand this new virus and to develop ways to control its spread. In this study, we sought to gain insights for vaccine design against SARS-CoV-2 by considering the high genetic similarity between SARS-CoV-2 and SARS-CoV, which caused the outbreak in 2003, and leveraging existing immunological studies of SARS-CoV. By screening the experimentally determined SARS-CoV-derived B cell and T cell epitopes in the immunogenic structural proteins of SARS-CoV, we identified a set of B cell and T cell epitopes derived from the spike (S) and nucleocapsid (N) proteins that map identically to SARS-CoV-2 proteins. As no mutation has been observed in these identified epitopes among the 120 available SARS-CoV-2 sequences (as of 21 February 2020), immune targeting of these epitopes may potentially offer protection against this novel virus. For the T cell epitopes, we performed a population coverage analysis of the associated MHC alleles and proposed a set of epitopes that is estimated to provide broad coverage globally, as well as in China. Our findings provide a screened set of epitopes that can help guide experimental efforts towards the development of vaccines against SARS-CoV-2.
Re: Protein structure prediction has been done for ages…
Not quite, Natural Selection does not measure methods, it measures outputs, usually at the organism level.
Sure correct folding is necessary for much protein function and we have prions and chaperone proteins to get it wrong and right.
The only way NS measures methods and mechanisms is if they are very energetically wasteful. But there are some very wasteful ones out there. Beta-Catenin at the end of point of Wnt signalling comes particularly to mind.
“Determining the structure of the virus proteins might also help in developing a molecule that disrupts the operation of just those proteins, and not anything else in the human body.”
Well it might, but predicting whether a ‘drug’ will NOT interact with any other of the 20000+ protein in complex organisms is well beyond current science. If we could do that we could predict/avoid toxicity and other non-mechanism related side-effects & mostly we can’t.
There are 480 structures on PDBe resulting from a search on ‘coronavirus,’ the top hits from MERS and SARS. PR stunt or not, they did win the most recent CASP ‘competition’, so arguably it’s probably our best shot right now – and I am certainly not satisfied that they have been sufficiently open in explaining their algorithms though I have not checked in the last few months. No one is betting anyone’s health on this, and it is not like making one wrong turn in a series of car directions. Latest prediction algorithms incorporate contact map predictions, so it’s not like a wrong dihedral angle sends the chain off in the wrong direction. A decent model would give something to run docking algorithms against with a series of already approved drugs, then we take that shortlist into the lab. A confirmed hit could be an instantly available treatment, no two year wait as currently estimated. [ALA added bold face]
Re: these structure predictions have not been experimentally verified
Naaaah. Can’t possibly be a stupid marketing stunt.
Well yes, a good possibility. But it can also be trying to build on the open-source model of putting it out there for others to build and improve upon. Essentially opening that “peer review” to a larger audience quicker. [ALA added bold face]
What bothers me, besides the obvious PR stunt, is that they say this prediction is licensed. How can a prediction from software be protected by, I presume, patents? And if this can be protected without even verifying which predictions actually work, what’s to stop someone spitting out millions of random, untested predictions just in case they can claim ownership later when one of them is proven to work? [ALA added bold face]
AI-predicted protein structures could unlock vaccine for Wuhan coronavirus… if correct… after clinical trials It’s not quite DeepMind’s ‘Come with me if you want to live’ moment, but it’s close, maybe
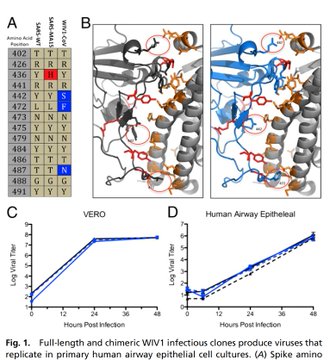
Experimentally derived by a group of scientists at the University of Texas at Austin and the National Institute of Allergy and Infectious Diseases, an agency under the US National Institute of Health. They both feature a “Spike protein structure.”
Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation
Other related articles published in this Open Access Online Scientific Journal include the following:
Group of Researchers @ University of California, Riverside, the University of Chicago, the U.S. Department of Energy’s Argonne National Laboratory, and Northwestern University solve COVID-19 Structure and Map Potential Therapeutics
Reporters: Stephen J Williams, PhD and Aviva Lev-Ari, PhD, RN
Group of Researchers @ University of California, Riverside, the University of Chicago, the U.S. Department of Energy’s Argonne National Laboratory, and Northwestern University solve COVID-19 Structure and Map Potential Therapeutics
Reporters: Stephen J Williams, PhD and Aviva Lev-Ari, PhD, RN
This illustration, created at the Centers for Disease Control and Prevention (CDC), reveals ultrastructural morphology exhibited by coronaviruses. Note the spikes that adorn the outer surface of the virus, which impart the look of a corona surrounding the virion, when viewed electron microscopically. A novel coronavirus virus was identified as the cause of an outbreak of respiratory illness first detected in Wuhan, China in 2019.
Image of newly mapped coronavirus protein, called Nsp15, which helps the virus replicate.
How UC is responding to the coronavirus (COVID-19)
The University of California is vigilantly monitoring and responding to new information about the coronavirus (COVID-19) outbreak, which has been declared a global health emergency.
The 3-D structure of a potential drug target in a newly mapped protein of COVID-19, or coronavirus, has been solved by a team of researchers from the University of California, Riverside, the University of Chicago, the U.S. Department of Energy’s Argonne National Laboratory, and Northwestern University.
The scientists said their findings suggest drugs previously developed to treat the earlier SARS outbreak could now be developed as effective drugs against COVID-19.
The initial genome analysis and design of constructs for protein synthesis were performed by the bioinformatic group of Adam Godzik, a professor of biomedical sciences at the UC Riverside School of Medicine.
The protein Nsp15 from Severe Acute Respiratory Syndrome Coronavirus 2, or SARS-CoV-2, is 89% identical to the protein from the earlier outbreak of SARS-CoV. SARS-CoV-2 is responsible for the current outbreak of COVID-19. Studies published in 2010 on SARS-CoV revealed inhibition of Nsp15 can slow viral replication.This suggests drugs designed to target Nsp15 could be developed as effective drugs against COVID-19.
Adam Godzik, UC Riverside professor of biomedical sciences Credit: Sanford Burnham Prebys Medical Discovery Institute
“While the SARS-CoV-19 virus is very similar to the SARS virus that caused epidemics in 2003, new structures shed light on the small, but potentially important differences between the two viruses that contribute to the different patterns in the spread and severity of the diseases they cause,” Godzik said.
The structure of Nsp15, which will be released to the scientific community on March 4, was solved by the group of Andrzej Joachimiak, a distinguished fellow at the Argonne National Laboratory, University of Chicago Professor, and Director of the Structural Biology Center at Argonne’s Advanced Photon Source, a Department of Energy Office of Science user facility.
“Nsp15 is conserved among coronaviruses and is essential in their lifecycle and virulence,” Joachimiak said. “Initially, Nsp15 was thought to directly participate in viral replication, but more recently, it was proposed to help the virus replicate possibly by interfering with the host’s immune response.”
Mapping a 3D protein structure of the virus, also called solving the structure, allows scientists to figure out how to interfere in the pathogen’s replication in human cells.
“The Nsp15 protein has been investigated in SARS as a novel target for new drug development, but that never went very far because the SARS epidemic went away, and all new drug development ended,” said Karla Satchell, a professor of microbiology-immunology at Northwestern, who leads the international team of scientists investigating the structure of the SARS CoV-2 virus to understand how to stop it from replicating. “Some inhibitors were identified but never developed into drugs. The inhibitors that were developed for SARS now could be tested against this protein.”
Rapid upsurge and proliferation of SARS-CoV-2 raised questions about how this virus could become so much more transmissible as compared to the SARS and MERS coronaviruses. The scientists are mapping the proteins to address this issue.
Over the past two months, COVID-19 infected more than 80,000 people and caused at least 2,700 deaths. Although currently mainly concentrated in China, the virus is spreading worldwide and has been found in 46 countries. Millions of people are being quarantined, and the epidemic has impacted the world economy. There is no existing drug for this disease, but various treatment options, such as utilizing medicines effective in other viral ailments, are being attempted.
Godzik, Satchell, and Joachimiak — along with the entire center team — will map the structure of some of the 28 proteins in the virus in order to see where drugs can throw a chemical monkey wrench into its machinery. The proteins are folded globular structures with precisely defined functions and their “active sites” can be targeted with chemical compounds.
The first step is to clone and express the genes of the virus proteins and grow them as protein crystals in miniature ice cube-like trays. The consortium includes nine labs across eight institutions that will participate in this effort.
Above is a modified version of the Northwestern University news release written by Marla Paul.
With an estimated global evaluation of around $14 billion US, synthetic biology is a rapidly accelerating market. Nonetheless while the growth of the market has been remarkable, the ttrue impact has not yet been seen. The era of AI will quickly increase the pace of discovery, and produce materials not seen in nature, through extrapolation and generative design. The extraordinary is now possible: producing spider silk without spiders, egg proteins without chickens and fragrances without flowers.
Synthetic biology companies are associating with fashion designers as well as forming ‘organism foundries. Rapidly, AI will utilize its learning of the natural world to make guided inferences which produce entirely new materials. From a technology perspective, we’re experiencing an explosion of capability that will be invasive in the next 3-5 years. Language models have come a long way, to the point where full models are being kept private so as not to endanger the public.
Already today, the average person has the ability to start their own commercial space venture for less than the cost of a juice franchise. PwC Australia’s Charmaine Green believes secret trends can hide among obvious ones. She outlines three trends leading to her hypothesis that Australia is well placed to become the global creative hub for video game development.
Economies like Australia are situated to capitalize on this trend, and video game development can become a permanent and substantial part of the economy. In Australia, Green argues, we have all the basic elements needed: high ingenuity, creative risk taking, and the freedom and flexibility that comes with the country’s small-to-mid studios.
Parkinson’s Disease (PD), characterized by both motor and non-motor system pathology, is a common neurodegenerative disorder affecting about 1% of the population over age 60. Its prevalence presents an increasing social burden as the population ages. Since its introduction in the 1960’s, dopamine (DA)-replacement therapy (e.g., L-DOPA) has remained the gold standard treatment. While improving PD patients’ quality of life, the effects of treatment fade with disease progression and prolonged usage of these medications often (>80%) results in side effects including dyskinesias and motor fluctuations. Since the selective degeneration of A9 mDA neurons (mDANs) in the substantia nigra (SN) is a key pathological feature of the disease and is directly associated with the cardinal motor symptoms, dopaminergic cell transplantation has been proposed as a therapeutic strategy.
Researchers showed that mammalian fibroblasts can be converted into embryonic stem cell (ESC)-like induced pluripotent stem cells (iPSCs) by introducing four transcription factors i.e., Oct4, Sox2, Klf4, and c-Myc. This was then accomplished with human somatic cells, reprogramming them into human iPSCs (hiPSCs), offering the possibility of generating patient-specific stem cells. There are several major barriers to implementation of hiPSC-based cell therapy for PD. First, probably due to the limited understanding of the reprogramming process, wide variability exists between the differentiation potential of individual hiPSC lines. Second, the safety of hiPSC-based cell therapy has yet to be fully established. In particular, since any hiPSCs that remain undifferentiated or bear sub-clonal tumorigenic mutations have neoplastic potential, it is critical to eliminate completely such cells from a therapeutic product.
In the present study the researchers established human induced pluripotent stem cell (hiPSC)-based autologous cell therapy. Researchers reported a platform of core techniques for the production of mDA progenitors as a safe and effective therapeutic product. First, by combining metabolism-regulating microRNAs with reprogramming factors, a method was developed to more efficiently generate clinical grade iPSCs, as evidenced by genomic integrity and unbiased pluripotent potential. Second, a “spotting”-based in vitro differentiation methodology was established to generate functional and healthy mDA cells in a scalable manner. Third, a chemical method was developed that safely eliminates undifferentiated cells from the final product. Dopaminergic cells thus produced can express high levels of characteristic mDA markers, produce and secrete dopamine, and exhibit electrophysiological features typical of mDA cells. Transplantation of these cells into rodent models of PD robustly restored motor dysfunction and reinnervated host brain, while showing no evidence of tumor formation or redistribution of the implanted cells.
Together these results supported the promise of these techniques to provide clinically applicable personalized autologous cell therapy for PD. It was recognized by researchers that this methodology is likely to be more costly in dollars and manpower than techniques using off-the-shelf methods and allogenic cell lines. Nevertheless, the cost for autologous cell therapy may be expected to decrease steadily with technological refinement and automation. Given the significant advantages inherent in a cell source free of ethical concerns and with the potential to obviate the need for immunosuppression, with its attendant costs and dangers, it was proposed that this platform is suitable for the successful implementation of human personalized autologous cell therapy for PD.
Engineered Bacteria used as Trojan Horse for Cancer Immunotherapy
Reporter: Irina Robu, PhD
Researchers are using synthetic biology— design and construction of new biological entities such as enzymes, genetic circuits, and cells or the redesign of existing biological systems—is changing medicine leading to innovative solution in molecular-based therapeutics. To address the issue of designing therapies that can induce a potent, anti-tumor immune response researchers at Columbia Engineering and Columbia Irving Medical Center engineered a strain of non-pathogenic bacteria that can colonize tumors in mice. The non-pathogenic bacteria act as Trojan Horse that can lead to complete tumor regression in a mouse model of lymphoma. Their results are currently published in Nature Medicine.
The scientists led by Nicholas Arpaia, used their expertise in synthetic biology and immunology to engineer a strain of bacteria able to grow and multiply in the necrotic core of tumors. The non-pathogenic E. coli are programmed to self-destruct when the bacteria numbers reach a critical threshold, allowing for actual release of therapeutics and averting them from causing havoc somewhere else in the body. Afterward, a small portion of bacteria survive lysis and repopulate the population which allows repeated rounds of drug delivery inside treated tumors.
In the present study, the scientists release a nanobody that targets CD47 protein, which defends cancer cells from being eaten by distinctive immune cells. The mutual effects of bacteria, induced local inflammation within the tumor and the blockage of the CD47 leads to better ingestion and activation of T-cells within the treated tumors. The team deduced that the treatment with their engineered bacteria not only cleared the treated tumors but also reduced the incidence of tumor metastasis.
Before moving to clinical trials, the team is performing proof-of-concept tests, safety and toxicology studies of their immunotherapeutic bacteria in a rand of advanced solid tumor settings in mouse models. They have currently collaborated with Gary Schwartz, deputy director of the Herbert Irving Comprehensive Cancer and have underway a company to translate their promising technology to patients.
“What it does is feeds misinformation to these regulatory elements, making them feel that there is too much carbon flow through both of these complexes, causing them to be inhibited,” Pardee said. “It simultaneously inhibits both complexes so tumor cells that are primarily driven by glucose cannot utilize glucose in the TCA cycle. Tumor cells that are primarily driven by glutamine usage cannot use glutamine-derived carbons in the TCA cycle. And, importantly, tumors cannot switch from one source to the other in the presence of CPI-613,” he explained.
He said that hitting two complexes simultaneously has many advantages. One is that the carbon source the tumor is primarily dependent on does not matter; another is that evolved resistance for both complexes simultaneously is very unlikely to happen.
Pardee said CPI-613’s key differentiators are that it is highly selective on the uptake and target level in cancer cells, which leads to less toxicity to healthy cells. This allows for patients to receive extended treatment courses and for the drug to be used in combination with other drugs.
CPI-613 is being administered in this clinical trial with a chemotherapy combination of fluorouracil, leucovorin, irinotecan, and oxaliplatin, called FOLFIRINOX.
New Targeted Cancer Therapy may be ‘Possible Hope’ for Some Pancreatic Cancer Patients
Pancreatic cancer is the 12th maximum common cancer and the fourth leading cause of cancer death. The cancer is often difficult to diagnose as there is no cost-effective ways to screen for the illness. For over 52% of people who are diagnosed after the cancer has spread and with a 5-year survival rate.
Scientists at Sheba Medical Center in Israel developed a targeted cancer therapy drug together with AstraZeneca and Merck which can offer a possible new solution for patients with a specific kind of pancreatic cancer by delaying the progression of the disease. To evaluate the safety and test the efficacy of a new drug treatment regimen based on Lynparza tablets. The tablets are a pharmacological inhibitor of the enzyme poly (ADP-ribose) polymerase which inhibit the enzyme. They were developed for a number of indications, but most prominently for the treatment of cancer, as numerous forms of cancer are more dependent for their development on the enzyme than regular cells are. This makes poly (ADP-ribose) polymerase an attractive target for cancer therapy.
Their study included 154 patients who were randomly assigned to get the tablets at a dose of 300 mg twice a day with metastatic pancreatic cancer who carried the genetic mutation called BRCA 1 and BRCA 2. BRCA1 and BRCA2 are human genes that produce proteins accountable for repairing damaged DNA and play a substantial role in preserving the genetic stability of cells. Once either of these genes is mutated, DNA damage can’t be repaired properly and cells become unstable. As a result, cells are more likely to develop additional genetic alterations that can lead to cancer.
Patients with these mutations make up six to seven percent of the metastatic pancreatic cancer patients. The trial using the using the medicine Lynparza offers possible hope for those who suffer from metastatic pancreatic cancer and have a BRCA mutation and slows down the disease progression. According to the researchers this is the first Phase 3 biomarker that is positive in pancreatic cancer and the drug gives incredible hope for patients with the advanced stage of the cancer.












{kind=link}
Paper in collection COVID-19 SARS-CoV-2 preprints from medRxiv and bioRxiv