Nanosensors for Protein Recognition and Gene Proteome Interaction
Larry H Bernstein, MD, FCAP, Curator
LPBI
Synthetic Antibody Detects Proteins
http://www.technologynetworks.com/Proteomics/news.aspx?ID=187242
Research could lead to nanosensors that recognize fibrinogen, insulin, or other biomarkers
Using carbon nanotubes, MIT chemical engineers have devised a new method for detecting proteins, including fibrinogen, one of the coagulation factors critical to the blood-clotting cascade.
This approach, if developed into an implantable sensor, could be useful for monitoring patients who are taking blood thinners, allowing doctors to make sure the drugs aren’t interfering too much with blood clotting.
The new method is the first to create synthetic recognition sites (similar to natural antibodies) for proteins and to couple them directly to a powerful nanosensor such as a carbon nanotube. The researchers have also made significant progress on a similar recognition site for insulin, which could enable better monitoring of patients with diabetes. It may also be possible to use this approach to detect proteins associated with cancer or heart disease, says Michael Strano, the Carbon P. Dubbs Professor in Chemical Engineering at MIT.
A targeted search
The new sensor is the latest example of a method developed in Strano’s lab, known asCorona Phase Molecular Recognition (CoPhMoRe).
This technique takes advantage of the interactions between a given polymer and a nanoparticle surface such as that of a fluorescent single-walled carbon nanotube, when the polymer is wrapped around the nanotube.
Certain regions of the polymers latch onto the nanoparticle surface like anchors, while other regions extend outwards into their environment. This outward-facing region, also known as the adsorbed phase or corona, has a 3-D structure that depends on the composition of the polymer.
CoPhMoRe works when a specific polymer adsorbs to the nanoparticle surface and creates a corona that recognizes the target molecule. These interactions are very specific, just like the binding between an antibody and its target. Binding of the target alters the carbon nanotubes’ natural fluorescence, allowing the researchers to measure how much of the target molecule is present.
Strano’s lab has previously used this approach to find recognition sites and develop nansensors for estradiol and riboflavin, among other molecules. The new paper represents their first attempt to identify corona phases that can detect proteins, which are larger, more complex, and more fragile than the molecules identified by their previous sensors.
For this study, Bisker began by screening carbon nanotubes wrapped in 20 different polymers including DNA, RNA, and polyethylene glycol (PEG), a polymer often added to drugs to increase their longevity in the bloodstream.
On their own, none of the polymers had any affinity for the 14 proteins tested, all taken from human blood. However, when the researchers tested polymer-wrapped nanotubes against the same proteins, they turned up a match between one of the modified nanotubes and fibrinogen.
“A chemist or a biologist would not be able to predict ahead of time that there should be any kind of affinity between fibrinogen and this corona phase,” Strano says. “It really is a new kind of molecular recognition.”
Fibrinogen, one of the most abundant proteins in human blood, is part of the blood-clotting cascade. When a blood vessel is damaged, an enzyme called thrombin converts fibrinogen into fibrin, a stringy protein that forms clots to seal the wound.
A sensor for fibrinogen could help doctors determine if patients who are taking blood thinners still have enough clotting capability to protect them from injury, and could allow doctors to calculate more finely tuned dosages. It could also be used to test patients’ blood clotting before they go into surgery, or to monitor wound healing, Bisker says.
Synthetic antibodies
The researchers believe their synthetic molecular recognition agents are an improvement over existing natural systems based on antibodies or DNA sequences known as aptamers, which are more fragile and tend to degrade over time.
“One of the advantages of this is that it’s a completely synthetic system that can have a much longer lifetime within the body,” Bisker says.
In 2013, researchers in Strano’s lab demonstrated that carbon nanotube sensors can remain active in mice for more than a year after being embedded in a polymer gel and surgically implanted under the skin.
In addition to insulin, the researchers are also interested in detecting troponin, a protein that is released by dying heart cells, or detecting proteins associated with cancer, which would be useful for monitoring the success of chemotherapy. These and other protein sensors could become critical components of devices that deliver drugs in response to a sign of illness.
“By measuring therapeutic markers in the human body in real time, we can enable drug delivery systems that are much smarter, and release drugs in precise quantities,” Strano says. “However, measurement of those biomarkers is the first step.”
New Device Uses Carbon Nanotubes to Snag Molecules
Nanotube “forest” in a microfluidic channel may help detect rare proteins and viruses.
Tuesday, December 22, 2015
| Nanotube “forest” in a microfluidic channel may help detect rare proteins and viruses. |
Engineers at MIT have devised a new technique for trapping hard-to-detect molecules, using forests of carbon nanotubes.
The team modified a simple microfluidic channel with an array of vertically aligned carbon nanotubes — rolled lattices of carbon atoms that resemble tiny tubes of chicken wire. The researchers had previously devised a method for standing carbon nanotubes on their ends, like trees in a forest. With this method, they created a three-dimensional array of permeable carbon nanotubes within a microfluidic device, through which fluid can flow.
Now the researchers have given the nanotube array the ability to trap certain particles. To do this, the team coated the array, layer by layer, with polymers of alternating electric charge.
“You can think of each nanotube in the forest as being concentrically coated with different layers of polymer,” says Brian Wardle, professor of aeronautics and astronautics at MIT. “If you drew it in cross-section, it would be like rings on a tree.”
Depending on the number of layers deposited, the researchers can create thicker or thinner nanotubes and thereby tailor the porosity of the forest to capture larger or smaller particles of interest.
The nanotubes’ polymer coating may also be chemically manipulated to bind specific bioparticles flowing through the forest. To test this idea, the researchers applied an established technique to treat the surface of the nanotubes with antibodies that bind to prostate specific antigen (PSA), a common experimental target. The polymer-coated arrays captured 40 percent more antigens, compared with arrays lacking the polymer coating.
Wardle says the combination of carbon nanotubes and multilayer coatings may help finely tune microfluidic devices to capture extremely small and rare particles, such as certain viruses and proteins.
“There are smaller bioparticles that contain very rich amounts of information that we don’t currently have the ability to access in point-of-care [medical testing] devices like microfluidic chips,” says Wardle, who is a co-author on the paper. “Carbon nanotube arrays could actually be a platform that could target that size of bioparticle.”
The paper’s lead author is Allison Yost, a former graduate student who is currently an engineer at Accion Systems. Others on the paper include graduate student Setareh Shahsavari; postdoc Roberta Polak; School of Engineering Professor of Teaching Innovation Gareth McKinley; professor of materials science and engineering Michael Rubner, and Raymond A. And Helen E. St. Laurent Professor of Chemical Engineering Robert Cohen.
A porous forest
Carbon nanotubes have been a subject of intense scientific study, as they possess exceptional electrical, mechanical, and optical properties. While their use in microfluidics has not been well explored, Wardle says carbon nanotubes are an ideal platform because their properties may be manipulated to attract certain nanometer-sized molecules. Additionally, carbon nanotubes are 99 percent porous, meaning a nanotube is about 1 percent carbon and 99 percent air.
“Which is what you need,” Wardle says. “You need to flow quantities of fluid through this material to shed all the millions of particles you don’t want to find and grab the one you do want to find.”
What’s more, Wardle says, a three-dimensional forest of carbon nanotubes would provide much more surface area on which target molecules may interact, compared with the two-dimensional surfaces in conventional microfluidics.
“The capture efficiency would scale with surface area,” Wardle notes.
A versatile array
The team integrated a three-dimensional array of carbon nanotubes into a microfluidic device by using chemical vapor deposition and photolithography to grow and pattern carbon nanotubes onto silicon wafers. They then grouped the nanotubes into a cylinder-shaped forest, measuring about 50 micrometers tall and 1 millimeter wide, and centered the array within a 3 millimeter-wide, 7-millimeter long microfluidic channel.
The researchers coated the nanotubes in successive layers of alternately charged polymer solutions in order to create distinct, binding layers around each nanotube. To do so, they flowed each solution through the channel and found they were able to create a more uniform coating with a gap between the top of the nanotube forest and the roof of the channel. Such a gap allowed solutions to flow over, then down into the forest, coating each individual nanotube. In the absence of a gap, solutions simply flowed around the forest, coating only the outer nanotubes.
After coating the nanotube array in layers of polymer solution, the researchers demonstrated that the array could be primed to detect a given molecule, by treating it with antibodies that typically bind to prostate specific antigen (PSA). They pumped in a solution containing small amounts of PSA and found that the array captured the antigen effectively, throughout the forest, rather than just on the outer surface of a typical microfluidic element.
Wardle says that the nanotube array is extremely versatile, as the carbon nanotubes may be manipulated mechanically, electrically, and optically, while the polymer coatings may be chemically altered to capture a wide range of particles. He says an immediate target may be biomarkers called exosomes, which are less than 100 nanometers wide and can be important signals of a disease’s progression.
“Science is really picking up on how much information these particles contain, and they’re sort of everywhere, but really hard to find, even with large-scale equipment,” Wardle says. “This type of device actually has all the characteristics and functionality that would allow you to go after bioparticles like exosomes and things that really truly are nanometer scale.”
This research was funded, in part, by the National Science Foundation.
|
A Natural Light Switch
| MIT scientists identify and map the protein behind a light-sensing mechanism. |
MIT scientists, working with colleagues in Spain, have discovered and mapped a light-sensing protein that uses vitamin B12 to perform key functions, including gene regulation.
The result, derived from studying proteins from the bacterium Thermus thermophilus, involves at least two findings of broad interest. First, it expands our knowledge of the biological role of vitamin B12, which was already understood to help convert fat into energy, and to be involved in brain formation, but has now been identified as a key part of photoreceptor proteins — the structures that allow organisms to sense and respond to light.
Second, the research describes a new mode of gene regulation, in which the light-sensing proteins play a key role. In so doing, the scientists observe, the bacteria have repurposed existing protein structures that use vitamin B12, and put them to work in new ways.

http://www.technologynetworks.com/images/videos/News%20Images/CR/MIT-Proteins-Light-1_0.jpg
“Nature borrowed not just the vitamin, but really the whole enzyme unit, and modified it … and made it a light sensor,” says Catherine Drennan, a professor of chemistry and biology at MIT
The paper describes the photoreceptors in three different states: in the dark, bound to DNA, and after being exposed to light.
“It’s wonderful that we’ve been able to get all the series of structures, to understand how it works at each stage,” Drennan says.
The paper has nine co-authors, including Drennan; graduate students Percival Yang-Ting Chen, Marco Jost, and Gyunghoon Kang of MIT; Jesus Fernandez-Zapata and S. Padmanabhan of the Institute of Physical Chemistry Rocasolano, in Madrid; and Monserrat Elias-Arnanz, Juan Manuel Ortiz-Guerreo, and Maria Carmen Polanco, of the University of Murcia, in Murcia, Spain.
The researchers used a combination of X-ray crystallography techniques and in-vitro analysis to study the bacteria. Drennan, who has studied enzymes that employ vitamin B12 since she was a graduate student, emphasizes that key elements of the research were performed by all the co-authors.
Jost performed crystallography to establish the shapes of the structures, while the Spanish researchers, Drennan notes, “did all of the control experiments to show that we were really thinking about this right,” among other things.

By studying the structures of the photoreceptor proteins in their three states, the scientists developed a more thorough understanding of the structures, and their functions, than they would have by viewing the proteins in just one state.
Microbes, like many other organisms, benefit from knowing whether they are in light or darkness. The photoreceptors bind to the DNA in the dark, and prevent activity pertaining to the genes of Thermus thermophilus. When light hits the microbes, the photoreceptor structures cleave and “fall apart,” as Drennan puts it, and the bacteria start producing carotenoids, which protect the organisms from negative effects of sunlight, such as DNA damage.
The research also shows that the exact manner in which the photoreceptors bind to the DNA is novel. The structures contain tetramers, four subunits of the protein, of which exactly three are bound to the genetic material — something Drennan says surprised her.
“That’s the best part about science,” Drennan says. “You see something novel, then you think it’s not really going to be that novel, but you do the experiments [and it is].”
Other scientists say the findings are significant. “It’s a very exciting development,” says Rowena Matthews, a professor emerita of biological chemistry at the University of Michigan, who has read the paper. Of the newly discovered use of vitamin B12 and a derivative of it, adenosylcobalamin, Matthews adds, “There was very limited knowledge of its versatility.”
Drennan adds that in the long run, the finding could have practical applications, such as the engineering of light-directed control of DNA transcription, or the development of controlled interactions between proteins.
“I would be very interested in … thinking about whether there could be practical applications of this,” Drennan says. |
HIV Protein Manipulates Hundreds of Human Genes
| Findings search for new or improved treatments for patients with AIDS. |
UT Southwestern Medical Center researchers have deciphered how a small protein made by the human immunodeficiency virus (HIV) that causes AIDS manipulates human genes to further its deadly agenda.
The findings, published in the online journal eLife, could aid in the search for new or improved treatments for patients with AIDS, or to the development of preventive strategies.
“We have identified the molecular mechanisms by which the Tat protein made by HIV interacts with the host cell to activate or repress several hundred human genes,” said Dr. Iván D’Orso, Assistant Professor of Microbiology at UT Southwestern and senior author of the study. “The findings clearly suggest that blocking Tat activity may be of therapeutic value to HIV patients.”
It has long been known that HIV causes AIDS by hijacking the body’s immune cells, transforming them into HIV factories and killing other immune cells that normally fight disease. HIV also hides in cells and continues to undermine the host’s immune system despite antiretroviral therapy that has improved the outlook of those with AIDS.
The latest data from the Centers for Disease Control and Prevention (CDC), in 2012, estimated 1.2 million Americans were living with HIV, including 156,300 whose infections had not been diagnosed. About 50,000 people in the U.S. are newly infected with HIV annually, the CDC projects. In 2013, the CDC estimated that over 26,000 Americans had the advanced form of HIV infection, AIDS.
Like all retroviruses, HIV has very few genes of its own and must take over the host’s cellular machinery in order to propagate and spread throughout the body. Although the broad aspects of that cellular hijacking were known, the nuances remain to be explored, Dr. D’Orso said.
“We observed that HIV methodically and precisely manipulates the host’s genes and cellular machinery. We also observed that HIV rewires cellular defensive pathways to benefit survival of the virus,” he added.
The study provides insights into HIV’s ability to survive despite antiretroviral therapy, findings that could lead to new therapeutic targets or ways to make current therapies more effective, he said.
“Our study indicates that this small viral protein, Tat, directly binds to about 400 human genes to generate an environment in which HIV can thrive. Then, this protein precisely turns off the body’s immune defense. It is striking that such a small viral protein has such a large impact,” Dr. D’Orso said. “The human genes and pathways that Tat manipulates correlate well with symptoms observed in these patients, such as immune system hyperactivation, then weakening, and accelerated aging,” Dr. D’Orso said, describing the situation in which HIV infection leads to AIDS.
Italy’s National Institute of Health in Rome recently completed a phase II clinical trial of an experimental vaccine that targets the Tat protein. That trial, which followed 87 HIV-positive patients for up to three years, reported that the vaccine was well-tolerated without significant side effects. However, it will take several years to determine if the vaccine works, Dr. D’Orso said.
Although someone can have HIV for years without showing symptoms, AIDS occurs when HIV blocks the body’s ability to fight off illness. The person then becomes overrun by the opportunistic infections and specific cancers that are hallmarks of AIDS.
|
New Light Shed on Genetic Regulation
A team of scientists has uncovered greater intricacy in protein signaling than was previously understood, shedding new light on the nature of genetic production.
Christine Vogel, an assistant professor in New York University’s Department of Biology and one of the study’s senior authors, explains that “to make a protein, we need to make a messenger RNA molecule from the gene encoded in the DNA, and then, in a second process, make proteins from these RNA molecules. Both processes are highly regulated and coupled.”
This coupling is similar to the coupling between a moving escalator and a person walking on it at the same time.
The research takes a closer look at how the two coupled processes change in the cell responding to an outside stimulus.
“Until recently, it has been very difficult to study these systems and researchers have thought that the movement of the escalator is most important during the cellular response,” Vogel explains. “We now show that is not necessarily the case, and under some circumstances, the person’s walking determines the overall outcome.”
In biology, this means that both of the processes—to make RNAs and proteins—play important roles, but with different patterns.
In their study, the scientists, who also included researchers from National University Singapore and Berlin’s Max Delbruck Center, took a closer look at how the two processes exactly respond over time.
Their results showed notable distinctions between DNA and mRNA in the nature of their signaling. Notably, the process of making RNA from DNA was pulse-like—a brief messaging over the studied period that returned to the normal levels by the end of the measurements. By contrast, the process of making a protein from RNA was akin to an on/off switch: once started, levels remained constant for consistent periods before reverting back to long stretches of dormancy.
While the reasons for these differences in cell behavior remain unknown, the researchers believe the answer may lie in the nature of the two tasks.
“It is very costly for the cell to make proteins, but making RNA messages from DNA is a relatively low-energy and simple process, so it makes sense that we see frequent, or pulsating, signaling at this stage,” observes Vogel. “By contrast, creating proteins is an intricate undertaking, requiring a great deal of time and energy. This may be why, once you decided to stop production of proteins, you do not turn it back on that easily—and the other way around.”
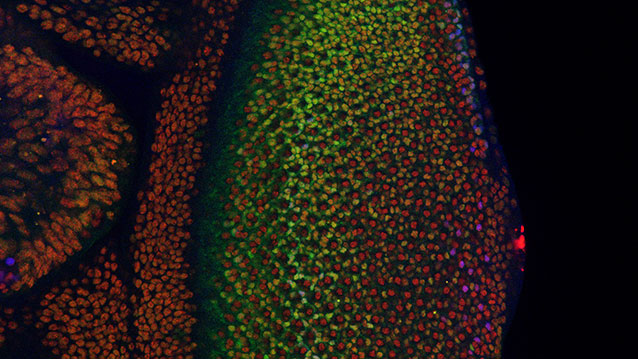
Where Cancer Cells May Begin
Scientists use fruit fly genetics to understand how things could go wrong in cancer.
Cancer cells are normal cells that go awry by making bad developmental decisions during their lives. In a study involving the fruit fly equivalent of an oncogene implicated in many human leukemias, Northwestern University researchers have gained insight into how developing cells normally switch to a restricted, or specialized, state and how that process might go wrong in cancer.
The fruit fly’s eye is an intricate pattern of many different specialized cells, such as light-sensing neurons and cone cells. Because flies share with humans many of the same cancer-causing genes, scientists use the precisely made compound eye of Drosophila melanogaster (the common fruit fly) as a workhorse to study what goes wrong in human cancer.

http://www.technologynetworks.com/images/videos/News%20Images/GT/retina638.jpg
A multidisciplinary team co-led by biologist Richard W. Carthew and engineer Luís A.N. Amaral studied normal cell behavior in the developing eye. The researchers were surprised to discover that the levels of an important protein called Yan start fluctuating wildly when the cell is switching from a more primitive, stem-like state to a more specialized state. If the levels don’t or can’t fluctuate, the cell doesn’t switch and move forward.
“This mad fluctuation, or noise, happens at the time of cell transition,” said Carthew, professor of molecular biosciences in Northwestern’s Weinberg College of Arts and Sciences. “For the first time, we see there is a brief time period as the developing cell goes from point A to point B. The noise is a state of ‘in between’ and is important for cells to switch to a more specialized state. This limbo might be where normal cells take a cancerous path.”
The researchers also found that a molecular signal received by a cell receptor called EGFR is important for turning the noise off. If that signal is not received, the cell remains in an uncontrolled state.
By pinpointing this noise and its “off” switch as important points in the normal process of cell differentiation, the Northwestern researchers provide targets for scientists studying how cells can go out of control and transform into cancer cells.
The “noisy” protein the Northwestern researchers studied is called Yan in the fly and Tel-1 in humans. (The protein is a transcription factor.) The Tel-1 protein instructs cells to turn into white blood cells; the gene that produces the protein, oncogene Tel-1, is frequently mutated in leukemia.
The EGFR protein that turns off the noise in flies is called Her-2 in humans. Her-2 is an oncogene that plays an important role in human breast cancer.
“On the surface, flies and humans are very different, but we share a remarkable amount of infrastructure,” said Carthew, a member of the Robert H. Lurie Comprehensive Cancer Center of Northwestern University. “We can use fruit fly genetics to understand how humans work and how things go wrong in cancer and other diseases.”
Fruit fly cells are small and closely packed together, making study of them challenging. Carthew and Amaral’s team of biologists, chemical and biological engineers, computer scientists and chemists together figured out how to identify and analyze thousands and thousands of individual cells in the flies’ eyes.
“In the past, people have built models of regulatory networks that control cell differentiation mostly by genetically perturbing one or two components of the network at a time and then compiling those results into models,” said Amaral, professor of chemical and biological engineering at the McCormick School of Engineering. “We instead measured the retina as it developed and found the unexpected behavior of the key regulatory factors Yan and EGFR.”
Nicolás Peláez, first author of the study and a Ph.D. candidate in interdisciplinary biological sciences working with Amaral and Carthew, built new tools to study this strange feature of noise in developing flies. His methods enabled the researchers to easily measure both the concentration of the Yan protein and its fluctuation (noise).
It takes 15 to 20 hours for a fruit fly cell to go from being an unrestricted cell to a restricted cell, Carthew said. Peláez determined the Yan protein is noisy, or fluctuating, for six to eight of those hours.
“Studying the dynamics of molecules regulating fly-eye patterning can inform us about human disease,” Peláez said. “Using model organisms such as fruit flies will help us understand quantitatively the basic biological principles governing differentiation in complex animals.”
Mechanism of Tumor Suppressing Gene Uncovered
The most commonly mutated gene in cancer,p53, works to prevent tumor formation by keeping mobile elements in check that otherwise lead to genomic instability, UT Southwestern Medical Center researchers have found.
The p53 gene long has been known to suppress tumor formation, but the mechanisms behind this function – and why disabling the gene allows tumors to form – were not fully understood.
Findings from the study answer some of these questions and could one day lead to new ways of diagnosing and treating cancer, said the study’s senior author, Dr. John Abrams, Professor of Cell Biology at UT Southwestern.
The investigators found that normal p53 gene action restrains transposons, mobile genetic elements called retroelements that can make copies of themselves and move to different positions on chromosomes. But, they discovered, when p53 is disabled by mutation, dramatic eruptions of these mobile elements occur. The study revealed that in mice with cancer and in human samples of two types of cancer (Wilms’ tumors and colon tumors) disabled for p53, transposons became very active.
In a healthy state, certain mechanisms work to keep these retroelements quiet and inactive, explained Dr. Abrams. One of those mechanisms is p53 action. Conversely, when p53 is mutated, retroelements can erupt.
“If you take the gene away, transposons can wreak havoc throughout the genome by causing it to become highly dysregulated, which can lead to disease,” Dr. Abrams said. “Our findings help explain why cancer genomes are so much more fluid and destabilized than normal genomes. They also provide a novel framework for understanding how normal cells become tumors.”
Although much more research is needed, Dr. Abrams said, the potential clinical implications of the team’s findings are significant.
“Understanding how p53 prevents tumors raises the prospect of therapeutic interventions to correct cases in which p53 is disabled,” Dr. Abrams said. “If retroelements are at the heart of certain p53-driven cancers, finding ways to suppress them could potentially allow us to prevent those cancers or intervene to keep them from progressing.”
This understanding also could lead to advances in diagnosing some cancers through biomarkers related to p53 and transposon activity.
“One possibility is that perhaps blood or urine tests could detect dysregulated retroelements that could be indicative of certain types of cancer,” Dr. Abrams said.
Like this:
Like Loading...
Read Full Post »











 . The power dissipated in this source is introduced back into the circuit in the power generated by the Nernst independent voltage sources,
. The power dissipated in this source is introduced back into the circuit in the power generated by the Nernst independent voltage sources,  and
and  . The power dissipated in the dependent voltage source Vloss models any additional power not used to perform chemical or electrical work. ……
. The power dissipated in the dependent voltage source Vloss models any additional power not used to perform chemical or electrical work. ……

 Thursday 15 October 2015, 12:00–13:00
Thursday 15 October 2015, 12:00–13:00










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}