Healthcare analytics, AI solutions for biological big data, providing an AI platform for the biotech, life sciences, medical and pharmaceutical industries, as well as for related technological approaches, i.e., curation and text analysis with machine learning and other activities related to AI applications to these industries.
Somatic Mutation Theory – Why it’s Wrong for Most Cancers, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 1: Next Generation Sequencing (NGS)
Somatic Mutation Theory – Why it’s Wrong for Most Cancers
Reporter: Aviva Lev-Ari, PhD, RN
Somatic Mutation Theory – Why it’s Wrong for Most Cancers.
Hysteron proteron reverses both temporal and logical order and this syllogism occurs in carcinogenesis and the somatic mutation theory (SMT): the first (somatic mutation) occurs only after the second (onset of cancer) and, therefore, observed somatic mutations in most cancers appear well after the early cues of carcinogenesis are in place. It is no accident that mutations are increasingly being questioned as the causal event in the origin of the vast majority of cancers as clinical data show little support for this theory when compared against the metrics of patient outcomes. Ever since the discovery of the double helical structure of DNA, virtually all chronic diseases came to be viewed as causally linked to one degree or another to mutations, even though we now know that genes are not simply blueprints, but rather an assemblage of alphabets that can, under non-genetic influences, be used to assemble a business letter or a work of Shakespearean literature. A minority of all cancers is indeed caused by mutations but the SMT has been applied to all cancers, and even to chemical carcinogenesis, in the absence of hard evidence of causality. Herein, we review the 100 year story of SMT and aspects that show why genes are not just blueprints, how radiation and mutation are associated in a more nuanced view, the proposed risk of cancer and bad luck, and the in vitro and in vivo evidence for a new cancer paradigm. This paradigm is scientifically applicable for the majority of non-heritable cancers and consists of a six-step sequence for the origin of cancer. This new cancer paradigm proclaims that somatic mutations are epiphenomena or later events occurring after carcinogenesis is already underway. This serves not just as a plausible alternative to SMT and explains the origin of the majority of cancers, but also provides opportunities for early interventions and prevention of the onset of cancer as a disease.
Conclusions
The incorrect interpretation of data can sometimes appear to be the more parsimonious explanation especially when it has acquired the mantle of a paradigm, as in the case of the SMT. Summa Cancerologica is not hypothetical or ontological. Its syllogism of carcinogenesis needs the consideration of all reasonable perspectives such as whether somatic mutations are later events or epiphenomena occurring at the end of the sequence of events in carcinogenesis. This mutatio praemissarum leads to a reflection of reasoned judgments of correct findings in cancer (mutations within tumors) together with clinical observations (relevance of such mutations to cancer therapy). An overemphasis of the SMT as the sole reason of the origin of carcinogenesis elevated it to the status of a dogma which downplays significant findings of mutations and genetics in different fields of nature, biology and science. However, there is hope that hereditary cancers can be treated in the near future as new technologies make it possible to manipulate proteins packaging DNA to turn on specific gene promoters and enhancers [164]. If this were applicable to the mass of non-hereditary cancers this approach would still be only symptomatic as the genesis of non-hereditary cancers is not caused by somatic mutations though somatic mutations occur within tumors. Focusing on the tumor cell without its origin including the microenvironment won’t be enough [165]. The reasoning on the origin of carcinogenesis, including different step-wise sequences, may help unmask mechanisms of the transition of a normal into a cancer cell (cancer genesis) as well as its different primary pathogenic stimulus, which can serve to prevent or retard cancers instead of concentrating on symptomatic strategies or for a cure for all cancers. It is scientifically valid based on in vitro and in vivo genetic findings that carcinogenesis consists of a six-step multi sequence process [17, 18]. This serves not just as a plausible alternative to the SMT to explain the origin of the majority of cancers, but could also suggest early interventions and thereby prevent the onset of cancer as a disease.
Fusion detection can be carried out with traditional opposing primer-based library preparation methods, which require target- and fusion-specific primers that define the region to be sequenced. With these methods, primers are needed that flank the target region and the fusion partner, so only known fusions can be detected. An alternative method, ArcherDX’ Anchored Multiplex PCR (AMP), can be used to detect the target of interest, plus any known and unknown fusion partners. This is because AMP uses target-specific unidirectional primers, along with reverse primers, that hybridize to the sequencing adapter that is ligated to each fragment prior to amplification.
In time, the narrow, tortuous paths followed by pioneers become wider and straighter, whether the pioneers are looking to settle new land or bring new biomarkers to the clinic.
In the case of biomarkers, we’re still at the stage where pioneers need to consult guides and outfitters or, in modern parlance, consultants and technology providers. These hardy souls tend to congregate at events like the Biomarker Conference, which was held recently in San Diego.
At this event, biomarker experts discussed ways to avoid unfortunate detours on the trail from discovery and development to clinical application and regulatory approval. Of particular interest were topics such as the identification of accurate biomarkers, the explication of disease mechanisms, the stratification of patient groups, and the development of standard protocols and assay platforms. In each of these areas, presenters reported progress.
Another crucial subject is the integration of techniques such as next-generation sequencing (NGS). This particular technique has been instrumental in advancing clinical cancer genomics and continues to be the most feasible way of simultaneously interrogating multiple genes for driver mutations.
Enriching nucleic acid libraries for target genes of interest prior to NGS greatly enhances the sensitivity of
detecting mutations, as the enriched regions are sequenced multiple times. This is particularly useful when analyzing clinical samples, which generate low amounts of poor-quality nucleic acids.
However, NGS has been limited in its ability to identify gene fusions and translocations, which underlie oncogenesis in a variety of cancers. “These challenges are largely related to the enrichment chemistry used to produce sequencing libraries,” commented Joshua Stahl, chief scientific officer and general manager, ArcherDX.
Most target-enrichment strategies require prior knowledge of both ends of the target region to be sequenced. Therefore, only gene fusions with known partners can be amplified for downstream NGS assays.
Archer’s Anchored Multiplex PCR (AMP™) technology overcomes this limitation, as it can enrich for novel fusions, while only requiring knowledge of one end of the fusion pair. At the heart of the AMP chemistry are unique Molecular Barcode (MBC) adapters, ligated to the 5′ ends of DNA fragments prior to amplification. The MBCs contain universal primer binding sites for PCR and a molecular barcode for identifying unique molecules. When combined with 3′ gene-specific primers, MBCs enable amplification of target regions with unknown 5′ ends.
“AMP is ideal for identifying gene fusions and other driver mutations from FFPE samples,” asserted Mr. Stahl. “Its robust utility was demonstrated for detection of gene fusions, point mutations, insertions, deletions, and copy number changes from low amounts of clinical formalin-fixed, paraffin-embedded (FFPE) RNA and DNA samples.
“Tagging each molecule of input nucleic acid with a unique molecular barcode allows for de-duplication, error correction, and quantitative analysis, resulting in high sequencing consensus. With its low error rate and low limits of detection, AMP is revolutionizing the field of cancer genomics.”
In a proof-of-concept study, a single-tube 23-plex panel was designed to amplify the kinase domains of ALK, RET, ROS1, and MUSK genes by AMP. This enrichment strategy enabled identification of gene fusions with multiple partners and alternative splicing events in lung cancer, thyroid cancer, and glioblastoma specimens by NGS.
Ignyta, a precision medicine company, adopted Archer’s AMP technology in Trailblaze Pharos™, a multiplex assay employed in their STARTRK-2 trial for identifying actionable NTRK, ROS1, and ALK gene rearrangements in solid tumors that can be treated with the novel kinase inhibitor, entrectinib. “Gene fusions are incredibly important in personalized medicine right now,” stated Mr. Stahl. “Archer’s FusionPlex assays are quickly becoming the new gold standard.”
Reading Cancer Signatures
This image, from the Massachusetts General Hospital Cancer Center, shows multicolor fluorescence in situ hybridization (FISH) analysis of cells from a patient with esophagogastric cancer. Remarkably, the FISH analysis revealed that co-amplification of the MET gene (red signal) and the EGFR gene (green signal) existed simultaneously in the same tumor cells. A chromosome 7 control probe is shown in blue.
“Each year 23,000 kidneys are transplanted, and over 175,000 kidney transplants are functional today,” noted Daniel R. Salomon, M.D., medical program director, Scripps Center for Organ Transplantation, Scripps Research Institute. “However, in just 5 years, 3 out of every 10 patients will be back on dialysis, and in 15 years, at least 75% of all patients will lose their kidney grafts.“Tumor biomarkers are critical for predicting and following patient responses to today’s cancer therapies,” said Darrell Borger, Ph.D., co-director of the Translational Research Laboratory and director of the Biomarker Laboratory, Massachussetts General Hospital (MGH) Cancer Center, Harvard Medical School. “If we understand what drives the malignancy in any given patient, we are able to match existing therapies to the patient’s genotype.”
Over the last decade, the Biomarker/Translational Research Laboratory has focused on developing clinical genotyping and fluorescent in situ hybridization (FISH) assays for rapid personalized genomic testing.
“Initially, we analyzed the most prevalent hotspot mutations, about 160 in 25 cancer genes,” continued Dr. Borger. “However, this approach revealed mutations in only half of our patients. With the advent of NGS, we are able to sequence 190 exons in 39 cancer genes and obtain significantly richer genetic fingerprints, finding genetic aberrations in 92% of our cancer patients.”
Using multiplexed approaches, Dr. Borger’s team within the larger Center for Integrated Diagnostics (CID) program at MGH has established high-throughput genotyping service as an important component of routine care. While only a few susceptible molecular alterations may currently have a corresponding drug, the NGS-driven analysis may supply new information for inclusion of patients into ongoing clinical trials, or bank the result for future research and development.
“A significant impediment to discovery of clinically relevant genomic signatures is our current inability to interconnect the data,” explained Dr. Borger. “On the local level, we are striving to compile the data from clinical observations, including responses to therapy and genotyping. Globally, it is imperative that comprehensive public databases become available to the research community.”
Tumor profiling at MGH have already yielded significant discoveries. Dr. Borger’s lab, in collaboration with oncologists at the MGH Cancer Center, found significant correlations between mutations in the genes encoding the metabolic enzymes isocitrate dehydrogenase (IDH1 and IDH2) and certain types of cancers, such as cholangiocarcinoma and acute myelogenous leukemia (AML).
Historically, cancer signatures largely focus on signaling proteins. Discovery of a correlative metabolic enzyme offered a promise of diagnostics based on metabolic byproducts that may be easily identified in blood. Indeed, the metabolite 2-hydroxyglutarate accumulates to high levels in the tissues of patients carrying IDH1 and IDH2 mutations. They have reported that circulating 2-hydroxyglutarate as measured in the blood correlates with tumor burden, and could serve as an important surrogate marker of treatment response.
“We believe that this is caused by chronic immune-mediated rejection. Failure of effective immunosuppression reduces functional life of these patients and adds in $9–13 billion in yearly healthcare costs.” Dr. Salomon emphasized that ineffective use of immunosuppressive drugs is partially due to the lack of an objective biomarker which could provide decision support for just-in-time adjustment in therapeutic regimens.
“Our research aims to provide that objective measure to clinicians,” explained Dr. Salomon.
To date, kidney transplant biopsies remain the gold standard, even though they are not suitable for continuous monitoring and have both costs and risks. Dr. Salomon’s team developed a minimally invasive diagnostic approach based on unbiased whole-genome expression profiling of blood samples. Using Affymetrix Human Genome U133 Plus 2.0 Gene Chips, the team analyzed 275 bloodsamples of kidney transplant patients with biopsy-proved acute rejection, acute dysfunction without rejection and transplant excellent phenotype.
The data was passed through several machine-learning algorithms to identify a group of about 250 classifiers that predict subacute or acute rejection with 80% accuracy. This signature is locked while the team continues to expand the core dataset aiming to reach a thousand samples by the end of this year.
“As opposed to classical approaches to biomarker discoveries limited to just a few classifiers, our methodology provides for the first use of unbiased whole-genome profiling in the identification of multiple molecular predictors,” declared Dr. Salomon. “We can use this molecular diagnostic strategy to reveal a subacute rejection prior to significant tissue injury leading to transplant dysfunction. Continuous monitoring would inform physicians on the balance between over-suppression and effective/optimal therapy.”
Dr. Salomon is a chief scientific advisor for Transplant Genomics (TGI), a start-up company created to translate the blood-based molecular diagnostics into clinical tests. In late 2016, TGI will begin providing its TruGraf blood tests for kidney transplant recipients for use by four to six U.S. transplant centers through an early-access program (EAP).
Additional tests designed to be used serially to diagnose and treat subclinical episodes of rejection including biopsy gene profiling are in the final stages of development. Validation and will be made available through the EAP in the upcoming months.
BioAgilytix’ MultiMuscle Analysis is a process that can split sample analysis into multiple parallel tracks to minimize antibody cross-reactivity and allow for use of the best-fit platform or kit for each biomarker analysis. The process may require only one tube of sample with only one F/T cycle.
Focusing on Large Molecules
BioAgilytix, a specialized bioanalytical laboratory, is a global leader in large molecule bioanalysis. The company’s business encompasses pharmacokinetic/pharmacodynamic (PK/PD) studies of large biomolecules, in addition to immunogenicity, biomarkers, and cell-based assays. In less than 10 years,BioAgilytix has grown from a start-up to an international powerhouse with over 100 employees—more than half possessing advanced scientific degrees—because of its team’s expertise in the complexities of large molecule drug development.
“In contrast to small molecule analysis, which has become more of a commodity due to its semiautomated and process-oriented nature, large molecule analysis is inherently challenging,” said Afshin Safavi, Ph.D., founder and chief science officer of BioAgilytix. “In large molecule bioanalysis, we rely heavily on analytical reagents, such as antibodies and recombinant proteins, which are known to show considerable variability from lot to lot.
BioAgilytix’ MultiMuscle Analysis is a process that can split sample analysis into multiple parallel tracks to minimize antibody cross-reactivity and allow for use of the best-fit platform or kit for each biomarker analysis. The process may require only one tube of sample with only one F/T cycle.
“Therefore, designing an effective analytical process for large biomolecules requires scientific personnel with years of experience. It also requires careful management of critical reagents, and a deep understanding of the capabilities and limitations of the platforms selected for use.”
Dr. Safavi explains that the biomarker field has been trending away from a gunshot approach traditionally favored by large pharma to more focused analyses of a few key biomarkers.
“Unlike several years ago, most biotech and pharma companies now perform careful due diligence and literature research before approaching us, to narrow down their investigation to just a handful of biomarkers,” he explained. Limited samples may drive the desire to multiplex as many biomarkers as possible, but a multiplex approach may often result in low quality data due to reagent cross-reactivity.
A recent process innovation developed by BioAgilytix, called MultiMuscle Analysis™, uses a customized parallel process to drastically reduce analytical process time and increase data quality. MultiMuscle Analysis splits the sample analysis into multiple parallel tracks, each performed on specialized equipment by scientists experienced in that particular platform.
“Say, for example, a customer requests measurements of 10 biomarkers,” ventured Dr. Safavi. “If we know some of the antibodies may cross-react, then we may, for example, end up with one heptaplex and three as uniplexes, all done in parallel.”
Using this approach, BioAgilytix is able to perform large biomarker analyses on a very large number of samples in near real-time. “We now receive samples from over 20 countries,” Dr. Safavi stated. “We have used the MultiMuscle approach successfully over and over.”
Feature ArticlesMore » May 1, 2016 (Vol. 36, No. 9)
Paving the Road for Clinical Biomarkers
Where Trackless Terrain Once Challenged Biomarker Development, Clearer Paths Are Emerging
Kate Marusina, Ph.D.
Focusing on Large Molecules
BioAgilytix’ MultiMuscle Analysis is a process that can split sample analysis into multiple parallel tracks to minimize antibody cross-reactivity and allow for use of the best-fit platform or kit for each biomarker analysis. The process may require only one tube of sample with only one F/T cycle.
BioAgilytix, a specialized bioanalytical laboratory, is a global leader in large molecule bioanalysis. The company’s business encompasses pharmacokinetic/pharmacodynamic (PK/PD) studies of large biomolecules, in addition to immunogenicity, biomarkers, and cell-based assays. In less than 10 years, BioAgilytix has grown from a start-up to an international powerhouse with over 100 employees—more than half possessing advanced scientific degrees—because of its team’s expertise in the complexities of large molecule drug development.
“In contrast to small molecule analysis, which has become more of a commodity due to its semiautomated and process-oriented nature, large molecule analysis is inherently challenging,” said Afshin Safavi, Ph.D., founder and chief science officer of BioAgilytix. “In large molecule bioanalysis, we rely heavily on analytical reagents, such as antibodies and recombinant proteins, which are known to show considerable variability from lot to lot.
“Therefore, designing an effective analytical process for large biomolecules requires scientific personnel with years of experience. It also requires careful management of critical reagents, and a deep understanding of the capabilities and limitations of the platforms selected for use.”
Dr. Safavi explains that the biomarker field has been trending away from a gunshot approach traditionally favored by large pharma to more focused analyses of a few key biomarkers.
“Unlike several years ago, most biotech and pharma companies now perform careful due diligence and literature research before approaching us, to narrow down their investigation to just a handful of biomarkers,” he explained. Limited samples may drive the desire to multiplex as many biomarkers as possible, but a multiplex approach may often result in low quality data due to reagent cross-reactivity.
A recent process innovation developed by BioAgilytix, called MultiMuscle Analysis™, uses a customized parallel process to drastically reduce analytical process time and increase data quality. MultiMuscle Analysis splits the sample analysis into multiple parallel tracks, each performed on specialized equipment by scientists experienced in that particular platform.
“Say, for example, a customer requests measurements of 10 biomarkers,” ventured Dr. Safavi. “If we know some of the antibodies may cross-react, then we may, for example, end up with one heptaplex and three as uniplexes, all done in parallel.”
Using this approach, BioAgilytix is able to perform large biomarker analyses on a very large number of samples in near real-time. “We now receive samples from over 20 countries,” Dr. Safavi stated. “We have used the MultiMuscle approach successfully over and over.”
Predicting Clotting or Hemorrhaging
Venous thromboembolism (VTE) is a disease that includes both deep vein thrombosis (DVT) and pulmonary embolism (PE). It is a common, lethal disorder, symptoms of which are often overlooked. VTE is the third most common cardiovascular illness after acute coronary syndrome and stroke.
Venous thrombi, composed predominately of red blood cells bound together by fibrin, form in sites of vessel damage and areas of stagnant blood flow. Once VTE is diagnosed, anticoagulation therapy is indicated.
A novel anticoagulant that reversibly and directly inhibits factor Xa, a key factor in the coagulation system, has been developed by Daiichi Sankyo. “Once on the path of development of an anticoagulant, we recognized the lack of a rapid and sensitive coagulation test that would not be affected by blood traces of anticoagulant therapies,” said Michele Mercuri, M.D., Ph.D., the company’s senior vice president. “An improved diagnostic test would speed up recognition and treatment of thrombosis, and would aid in development of reversing agents that reduce the effect of anticoagulant therapies when needed.”
When Daiichi Sankyo entered in collaboration with Perosphere to develop a novel broad-spectrum reversing agent, the company also supported development of a point-of-care coagulometer (still under development), a hand-held device designed for broad-spectrum monitoring of the activity of anticoagulants and their corresponding reversing agents, across drug classes. A single test requires only 10 µL of fresh or citrated whole blood from a venous draw or finger stick. It optically measures clotting starting with Factor XII activation to fibrin assembly.
Dr. Mercuri explains that none of the existing tests are able to predict whether a patient is at risk for either clotting or hemorrhaging. “Together with Prof. Zahi Fayad’s Team from the Icahn School of Medicine at Mt Sinai, we used magnetic resonance imaging with the gadolinium-based contrast reagent to detect the venous thrombi and follow their dissolution with edoxaban treatment,” reported Dr. Mercuri.
This study, the edoxaban Thrombus Reduction Imaging Study (eTRIS), was focused on developing and validating a magnetic resonance venography (MRV) image acquisition and analysis protocol for the quantification of thrombus volume in deep vein thrombosis. The multicenter study demonstrated excellent reproducibility of analysis of quantifying thrombus volume.
Sequence and Epigenetic Factors Determine Overall DNA Structure
Researchers at Ulsan National Institute of Science and Technology (UNIST) in South Korea found that DNA molecules directly interact with one another in ways that are dependent on the sequence of the DNA and epigenetic factors.
The researchers found evidence for sequence-dependent attractive interactions between double-stranded DNA molecules that neither involve intermolecular strand exchange nor are mediated by DNA-binding proteins.
“DNA molecules tend to repel each other in water, but in the presence of special types of cations, they can attract each other just like nuclei pulling each other by sharing electrons in between,” explained lead study author Hajin Kim, Ph.D., assistant professor of biophysics at UNIST. “Our study suggests that the attractive force strongly depends on the nucleic acid sequence and also the epigenetic modifications.”
The investigators used atomic-level simulations to measure forces between double-stranded DNA helices, proposing that the distribution of methyl groups on DNA were the key to regulating this sequence-dependent attraction.
The findings from this study were published recently in Nature Communications through an article entitled “Direct evidence for sequence-dependent attraction between double-stranded DNA controlled by methylation.”
The researchers surmised that direct DNA-DNA interactions could play a central role in how chromosomes are organized and packaged, determining the ultimate fate of many cell types.
Dr. Kim concluded by stating that “in our lab, we try to unravel the mysteries within human cells based on the principles of physics and the mechanisms of biology—seeking for ways to prevent chronic illnesses and diseases associated with aging.”
Searches Related to Direct evidence for sequence-dependent attraction between double-stranded DNA controlled by methylation
Although proteins mediate highly ordered DNA organization in vivo, theoretical studies suggest that homologous DNA duplexes can preferentially associate with one another even in the absence of proteins. Here we combine molecular dynamics simulations with single-molecule fluorescence resonance energy transfer experiments to examine the interactions between duplex DNA in the presence of spermine, a biological polycation. We find that AT-rich DNA duplexes associate more strongly than GC-rich duplexes, regardless of the sequence homology. Methyl groups of thymine acts as a steric block, relocating spermine from major grooves to interhelical regions, thereby increasing DNA–DNA attraction. Indeed, methylation of cytosines makes attraction between GC-rich DNA as strong as that between AT-rich DNA. Recent genome-wide chromosome organization studies showed that remote contact frequencies are higher for AT-rich and methylated DNA, suggesting that direct DNA–DNA interactions that we report here may play a role in the chromosome organization and gene regulation.
Formation of a DNA double helix occurs through Watson–Crick pairing mediated by the complementary hydrogen bond patterns of the two DNA strands and base stacking. Interactions between double-stranded (ds)DNA molecules in typical experimental conditions containing mono- and divalent cations are repulsive1, but can turn attractive in the presence of high-valence cations2. Theoretical studies have identified the ion–ion correlation effect as a possible microscopic mechanism of the DNA condensation phenomena3, 4, 5. Theoretical investigations have also suggested that sequence-specific attractive forces might exist between two homologous fragments of dsDNA6, and this ‘homology recognition’ hypothesis was supported by in vitro atomic force microscopy7 and in vivo point mutation assays8. However, the systems used in these measurements were too complex to rule out other possible causes such as Watson–Crick strand exchange between partially melted DNA or protein-mediated association of DNA.
Here we present direct evidence for sequence-dependent attractive interactions between dsDNA molecules that neither involve intermolecular strand exchange nor are mediated by proteins. Further, we find that the sequence-dependent attraction is controlled not by homology—contradictory to the ‘homology recognition’ hypothesis6—but by a methylation pattern. Unlike the previous in vitro study that used monovalent (Na+) or divalent (Mg2+) cations7, we presumed that for the sequence-dependent attractive interactions to operate polyamines would have to be present. Polyamine is a biological polycation present at a millimolar concentration in most eukaryotic cells and essential for cell growth and proliferation9, 10. Polyamines are also known to condense DNA in a concentration-dependent manner2, 11. In this study, we use spermine4+(Sm4+) that contains four positively charged amine groups per molecule.
Methylation determines the strength of DNA–DNA attraction
Analysis of the MD simulations revealed the molecular mechanism of the polyamine-mediated sequence-dependent attraction (Fig. 2). In the case of the AT-rich fragments, the bulky methyl group of thymine base blocks Sm4+ binding to the N7 nitrogen atom of adenine, which is the cation-binding hotspot21, 22. As a result, Sm4+ is not found in the major grooves of the AT-rich duplexes and resides mostly near the DNA backbone (Fig. 2a,d). Such relocated Sm4+ molecules bridge the two DNA duplexes better, accounting for the stronger attraction16, 23, 24, 25. In contrast, significant amount of Sm4+ is adsorbed to the major groove of the GC-rich helices that lacks cation-blocking methyl group (Fig. 2b,e).
Figure 2: Molecular mechanism of polyamine-mediated DNA sequence recognition.
(a–c) Representative configurations of Sm4+ molecules at the DNA–DNA distance of 28 Å for the (AT)10–(AT)10 (a), (GC)10–(GC)10 (b) and (GmC)10–(GmC)10 (c) DNA pairs. The backbone and bases of DNA are shown as ribbon and molecular bond, respectively; Sm4+ molecules are shown as molecular bonds. Spheres indicate the location of the N7 atoms and the methyl groups. (d–f) The average distributions of cations for the three sequence pairs featured in a–c. Top: density of Sm4+ nitrogen atoms (d=28 Å) averaged over the corresponding MD trajectory and the z axis. White circles (20 Å in diameter) indicate the location of the DNA helices. Bottom: the average density of Sm4+ nitrogen (blue), DNA phosphate (black) and sodium (red) atoms projected onto the DNA–DNA distance axis (x axis). The plot was obtained by averaging the corresponding heat map data over y=[−10, 10] Å. See Supplementary Figs 4 and 5 for the cation distributions at d=30, 32, 34 and 36 Å.
Genome-wide investigations of chromosome conformations using the Hi–C technique revealed that AT-rich loci form tight clusters in human nucleus27, 28. Gene or chromosome inactivation is often accompanied by increased methylation of DNA29 and compaction of facultative heterochromatin regions30. The consistency between those phenomena and our findings suggest the possibility that the polyamine-mediated sequence-dependent DNA–DNA interaction might play a role in chromosome folding and epigenetic regulation of gene expression.
Phenotypic and Biomarker-based Drug Discovery
Organizers: Michael Foley (Tri-Institutional Therapeutics Discovery Institute), Ralph Garippa (Memorial Sloan-Kettering Cancer Center), David Mark (F. Hoffmann-La Roche), Lorenz Mayr (Astra Zeneca), John Moffat (Genentech), Marco Prunotto (F. Hoffmann-La Roche), and Sonya Dougal (The New York Academy of Sciences)Presented by the Biochemical Pharmacology Discussion Group
Reported by Robert Frawley | Posted January 12, 2016
There are two major methods for designing pharmaceutical drugs. In traditional drug discovery (TDD), or empiric design, researchers target a particular domain or protein after working to understand its mechanisms and molecular biology. In phenotypic drug discovery (PDD), many different compounds are tested on a system until one results in an observable phenotype of success, and the compounds’ mechanisms of action are not considered. The Phenotypic and Biomarker-based Drug Discovery symposium, presented by the Academy’s Biochemical Pharmacology Discussion Group on October 27, 2015, featured current work in PDD and highlighted the need to bridge commercial and academic research to improve phenotypic drug design.
Phenotypic drug discovery—screening of thousands of substances for functional cellular outputs such as gene expression, growth arrest, and cancer cell death—has led to the development of more commercial drugs than TDD, the more common method of discovery. Indeed, as Jonathan A. Lee of Eli Lilly noted, spending on TDD is out of sync with the rate of new drugs reaching approval; the number of new drugs per billion dollars spent dropped sharply in the last few decades. He argued that the need for functionally validated drugs could be met through a renewed focus on PDD.
Bruce A. Posner started the morning session with a discussion of a phenotypic screen conducted at the University of Texas Southwestern Medical Center which identified two chemical scaffolds that are effective in killing non-small cell lung cancer (NSCLC) cells but are harmless to the non-cancer cells tested. In further studies, the group showed that an optimized analog of one scaffold arrested tumor growth in a mouse xenograft model of NSCLC. Both chemical scaffolds appear to work through a novel mechanism targeting stearoyl-CoA desaturase (SCD), which is known to be important in unsaturated fatty acid synthesis. These compounds were found to be specific, effective, and potent in NSCLC cell lines that express elevated levels of Cyp4F11 and/or related Cyp family members. The group also showed that these scaffolds function as prodrugs that are activated only in cancer cells expressing these Cyp isoforms and that the Cyps produce metabolites of the prodrug that bring about cancer-specific cell toxicity. The group is working to improve these scaffolds and to develop a putative biomarker based on Cyp expression.
The Broad Institute’s LINCS (Library of Network-based Cellular Signatures) database is designed to keep track of small-molecule therapeutics, collecting data on cellular responses to “perturbagens” (drugs, factors, and others stimuli). Data are generated using the L1000 assay, which assesses the expression of 1000 genes known to explain 80% of genetic variation in assayed cell lines. Aravind Subramanian explained that the technique can identify the majority of drug effects for a fraction of the cost of RNA sequencing. Although it examines only a subset of molecules and relies on measuring genetic responses, the technique can help predict the likelihood that new compounds will elicit desired effects.
Martin Main of AstraZeneca described phenotypic drug discovery at AstraZeneca. The company’s model for discovery is to check phenotypic markers at every step, as drugs are moved from cell lines to patients. Main’s team identified a molecule that enhances the regenerative function of cardiac myocytes after infarction. Using cells from several donors, the team validated a promising compound that increases proliferation of cardiac myocytes and drives epicardium-derived progenitor cells to assume a myocyte lineage. In another discovery, the team used islet β-cell regeneration as the phenotype, discovering a compound the researchers believe will reach clinical trials for type 2 diabetes.
Andras J. Bauer of Boehringer Ingelheim discussed a method to increase predictive strength in compound selection before phenotypic screening. By cataloging the structures of known target–reference compound binding pairs, the team can compare those structures to untested compounds, and then assess only the most promising compounds. The THICK (Target Hypothesis Information from Curated Knowledge bases) database gives interaction-probability scores to untested compounds on the basis of structure. Bauer also described a method to verify target–compound interaction without labeling the molecules, in which phenotypic results were verified with mass spectrometry.
In the afternoon session, Myles Fennell of Memorial Sloan-Kettering Cancer Center described his work testing small interfering RNA (siRNA) libraries to find siRNAs that alter macropinocytosis (MP), cell-surface ruffling that is seen in prostate cancer cells. The surface phenotype allows TMR-dextran uptake, which the researchers measured in the screen. MP is driven by RAS (a commonly affected gene family in cancers) and the pathways are already popular drug targets. The researchers tested two libraries of siRNAs, which block translation of specific proteins, using TMR as a marker to report MP severity, as well as sensitive single-cell assays to determine siRNA efficacy. The team identified promising target sequences and used a data-analysis pipeline called KNIME to define several hits, which the researchers are pursuing in therapeutic development.
TMR-dextran is able to work into cells undergoing macropinocytosis and thus these cells can be separated by phenotype as seen in the controls above. (Image courtesy of Myles Fennell)
Giulio Superti-Furga of the Austrian Academy of Sciences is a proponent of understanding the mechanisms of action (MOA) of candidate drugs. He began by explaining that the genome is an incomplete indicator of disease; epigenetics, altered protein function, metabolism, and other factors are also important. He then introduced pharmacoscopy and the “thermal shiftome” as methods to phenotypically screen compounds. Pharmacoscopy uses high-power automated microscopy to describe how compounds affect cell populations by using specific stains for different cell types; a computer then counts the cells expressing each stain, yielding results similar to those obtained via fluorescence-activated cell sorting but generated through an automated process. The thermal shiftome catalogs changes in thermal stability after protein binding in known reactions and is used to characterize the stability of new reactions. Superti-Furga offered a perspective that tempered the enthusiasm for pure PDD and advocated a mechanistic approach to drug discovery.
Michael R. Jackson, at one of the largest academic screening facilities, the Sanford Burnham Prebys Medical Discovery Institute, led a reexamination of drug screens performed by pharmaceutical companies. His team conducted millions of assays and accumulated a large data library with few new hits. However, the researchers were able to closely characterize the chemistry of one hit, an undisclosed interaction, and Jackson’s group is proceeding to develop a drug to modulate nuclear receptor signaling. The researchers also have a procedure that can screen for the differentiation of human induced pluripotent stem cells (iPSCs) into neurons for potential neuro-regenerative therapies. They developed high-throughput morphology, endpoint-measurement, and proliferation assays that generate tightly clustered, repeatable data. The team has produced consistent results screening 10 immune modulators and various cytokines to assess the reactivity and stability of the cells, providing reliable compound characterization. This success in human cells shows that a disease-relevant patient-derived screening platform to characterize differentiation and immune response is possible with robust assays.
In the next set of talks, Friedrich Metzger and Susanne Swalley described the parallel work of Hoffmann-La Roche and Novartis, respectively, toward treating spinal muscular atrophy (SMA). A devastating disease that leads to loss of motor function and affects motor nerve cells in the spinal cord, SMA presents a unique drug development opportunity. The condition is caused by the loss of function of a single gene product called survival of motor neuron (SMN1). Humans encode an unstable gene product, called SMN2, which is nearly homologous to SMN1.
Metzger explained that the inactive SMN2 variant is largely the same as active SMN1 but, missing exon 7, cannot compensate in its absence. The group from Hoffmann-La Roche aimed to stabilize SMN2 by promoting the inclusion of exon 7. The researchers conducted a phenotypic screen seeking a compound that could change the splicing in patient fibroblasts in vitro and produce a stable, functional SMN2 protein including exon 7. In studies with an SMN2Δ7 mouse model (lacking exon 7), mice drugged with the compound experienced full phenotypic rescue. The compound has been shown to induce alternative splicing of SMN2 to include exon 7 in healthy human volunteers; it was well tolerated and is moving to human patient trials.
Swalley discussed the target identification and MOA of the Novartis compound. After a screening process similar to Roche’s, Novartis moved its compound into animal models while also beginning parallel experimentation to find out why it worked. The group found that U1-snRNP, a spliceosome component required for the splicing process, is bound at two essential nucleotides by the compound. In the SMN2Δ7 mice, the compound improved survival and rescued full SMN2 protein expression. The Novartis compound stabilizes the appropriate spliceosome components to produce SMN2 with exon 7 intact. This novel mechanism demonstrates that a sequence-selective small molecule therapy can alter splicing activity to treat SMA. Together these talks demonstrated the power of PDD and the importance of validating drug mechanisms.
The final talk of the day was given by Hoffmann-La Roche’s Jitao David Zhang, who suggested that pathway reporter genes, which are only modulated when a specific signaling pathway is activated or inhibited, can be used as phenotypic readouts. It is known that gene expression data can predict cell phenotype. Using transcriptomics as a surrogate for downstream phenotypes, for example by using expression data from a gene subset to predict outcomes, would save time and effort. In an iPSC cardiomyocyte model of diabetic stress, machine learning (guided by pathway information) characterizes the response of the iPSCs to a library of compounds, highlighting compounds and pathways worthy of further investigation. This new platform for molecular phenotyping using pathway reporter genes, sequencing, and early analysis speeds compound characterization.
Use the tabs above to find multimedia from this event.
Presentations available from:
Andras J. Bauer, PhD, PharmD (Boehringer Ingelheim)
Myles Fennell, PhD (Memorial Sloan-Kettering Cancer Center)
Jonathan A. Lee, PhD (Eli Lilly)
Martin Main, PhD (AstraZeneca)
Yao Shen, PhD (Columbia University)
Susanne Swalley, PhD (Novartis Institutes for BioMedical Research)
Jitao David Zhang, PhD (F. Hoffmann-La Roche)
The medium ground finch (Geospiza fortis), shown here, diverged in beak size from the large ground finch (Geospiza magnirostris) on Daphne Major Island, Galápagos following a severe drought. Genomic screening of the genomes of medium ground finches revealed that a particular gene, HMGA2, played a large role in the rapid evolution of a smaller overall beak size in the medium ground finch. [Peter R. Grant]
An evolutionary phenomenon first described by Charles Darwin has the support of new and unusually strong supporting evidence. The phenomenon, called character displacement, may occur when species compete for the same food source. The species may evolve different body shapes, such as different beak sizes in the case of finches, diverging from each other until they relieve competitive stress.
Darwin developed the idea of character displacement after observing the finches of the Galápagos Islands. He proposed that changes in the size and form of the beak have enabled different species to utilize different food resources, such as insects, seeds, and nectar from cactus flowers, as well as blood from seabirds.
In a study of character displacement among Darwin’s finches, researchers from Uppsala University and Princeton University have now identified a gene that explains variation in beak size within and among species. The gene contributed to a rapid shift in beak size of the medium ground finch following a severe drought.
The details of the study appeared April 22 in the journal Science, in an article entitled, “A Beak Size Locus in Darwin’s Finches Facilitated Character Displacement during a Drought.” The article describes how the researchers alighted on a gene called HMGA2 after screening the genomes of medium ground finches that survived or died during a drought that occurred between 2004 and 2005. The researchers found that the HMGA2 gene comes in two forms: one is common in finches with small beaks, whereas the other is common in finches with large beaks. The proportion of the two forms in the birds’ genome changed as a result of the better survival of birds with small beaks.
“We used genomic analysis to investigate the genetic basis of a documented character displacement event in Darwin’s finches on Daphne Major in the Galápagos Islands,” wrote the authors. “We discovered a genomic region containing the HMGA2 gene that varies systematically among Darwin’s finch species with different beak sizes. Two haplotypes that diverged early in the radiation were involved in the character displacement event.”
In a previous study from the same team, the ALX1 gene was revealed to control beak shape (pointed or blunt). The HMGA gene that figures in the current study was previously associated with variation in body size in dogs and horses, and it is one of the genes that show the most consistent association with variation in stature in humans, a trait that is affected by hundreds of genes. HMGA2 has also a role in cancer biology as it affects the epithelial–mesenchymal transition (EMT) that is important for metastasis and cancer progression.
“Our data show that beak morphology is affected by many genes, as is the case for most biological traits,” said Sangeet Lamichhaney, the current study’s first author and a doctoral student in the laboratory of Leif Andersson, one of the study’s senior authors and a genomics professor at Uppsala. “However, we are convinced that we now have identified the two loci with the largest individual effects that have shaped the evolution of beak morphology among the Darwin’s finches.”
Andersson collaborated with Princeton researchers Peter Grant, the Class of 1877 Professor of Zoology, Emeritus, and B. Rosemary Grant, a senior research biologist, emeritus, in ecology and evolutionary biology.
“It was an exceptionally strong natural-selection event,” noted Peter Grant, who pointed out that that because Daphne Major is in an entirely natural state, the occurrence was completely unaffected by humans. “Now we have demonstrated that HMGA2 played a critical role in this evolutionary shift and that the natural selection acting on this gene during the drought is one of the highest yet recorded in nature.”
“This research tells us that a complex trait such as beak size can evolve significantly in a short time when the environment is stressful,” Rosemary Grant added. “We know that bacteria can evolve very quickly in the lab, but it is quite unusual to find a strong evolutionary change in a short time in a vertebrate animal.”
A beak size locus in Darwin’s finches facilitated character displacement during a drought
Sangeet Lamichhaney1, Fan Han1,Jonas Berglund1,…., B. Rosemary Grant2, Peter R. Grant2, Leif Andersson1,3,4,*
Observations of parallel evolution in the finches of the Galapagos, including body and beak size, contributed to Darwin’s theories. Lamichhaney et al. carried out whole-genome sequencing of 60 Darwin’s finches. These included small, medium, and large ground finches as well as small, medium, and large tree finches. A genomic region containing the HMGA2 gene correlated strongly with beak size across different species. This locus appears to have played a role in beak diversification throughout the radiation of Darwin’s finches.
Ecological character displacement is a process of morphological divergence that reduces competition for limited resources. We used genomic analysis to investigate the genetic basis of a documented character displacement event in Darwin’s finches on Daphne Major in the Galápagos Islands: The medium ground finch diverged from its competitor, the large ground finch, during a severe drought. We discovered a genomic region containing the HMGA2 gene that varies systematically among Darwin’s finch species with different beak sizes. Two haplotypes that diverged early in the radiation were involved in the character displacement event: Genotypes associated with large beak size were at a strong selective disadvantage in medium ground finches (selection coefficient s = 0.59). Thus, a major locus has apparently facilitated a rapid ecological diversification in the adaptive radiation of Darwin’s finches.
CRISPR/Cas9, Familial Amyloid Polyneuropathy (FAP) and Neurodegenerative Disease, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 2: CRISPR for Gene Editing and DNA Repair
CRISPR/Cas9, Familial Amyloid Polyneuropathy ( FAP) and Neurodegenerative Disease
Curator: Larry H. Bernstein, MD, FCAP
CRISPR/Cas9 and Targeted Genome Editing: A New Era in Molecular Biology
The development of efficient and reliable ways to make precise, targeted changes to the genome of living cells is a long-standing goal for biomedical researchers. Recently, a new tool based on a bacterial CRISPR-associated protein-9 nuclease (Cas9) from Streptococcus pyogenes has generated considerable excitement (1). This follows several attempts over the years to manipulate gene function, including homologous recombination (2) and RNA interference (RNAi) (3). RNAi, in particular, became a laboratory staple enabling inexpensive and high-throughput interrogation of gene function (4, 5), but it is hampered by providing only temporary inhibition of gene function and unpredictable off-target effects (6). Other recent approaches to targeted genome modification – zinc-finger nucleases [ZFNs, (7)] and transcription-activator like effector nucleases [TALENs (8)]– enable researchers to generate permanent mutations by introducing doublestranded breaks to activate repair pathways. These approaches are costly and time-consuming to engineer, limiting their widespread use, particularly for large scale, high-throughput studies.
The Biology of Cas9
The functions of CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) and CRISPR-associated (Cas) genes are essential in adaptive immunity in select bacteria and archaea, enabling the organisms to respond to and eliminate invading genetic material. These repeats were initially discovered in the 1980s in E. coli (9), but their function wasn’t confirmed until 2007 by Barrangou and colleagues, who demonstrated that S. thermophilus can acquire resistance against a bacteriophage by integrating a genome fragment of an infectious virus into its CRISPR locus (10).
Three types of CRISPR mechanisms have been identified, of which type II is the most studied. In this case, invading DNA from viruses or plasmids is cut into small fragments and incorporated into a CRISPR locus amidst a series of short repeats (around 20 bps). The loci are transcribed, and transcripts are then processed to generate small RNAs (crRNA – CRISPR RNA), which are used to guide effector endonucleases that target invading DNA based on sequence complementarity (Figure 1) (11).
Figure 1. Cas9 in vivo: Bacterial Adaptive Immunity
In the acquisition phase, foreign DNA is incorporated into the bacterial genome at the CRISPR loci. CRISPR loci is then transcribed and processed into crRNA during crRNA biogenesis. During interference, Cas9 endonuclease complexed with a crRNA and separate tracrRNA cleaves foreign DNA containing a 20-nucleotide crRNA complementary sequence adjacent to the PAM sequence. (Figure not drawn to scale.)
One Cas protein, Cas9 (also known as Csn1), has been shown, through knockdown and rescue experiments to be a key player in certain CRISPR mechanisms (specifically type II CRISPR systems). The type II CRISPR mechanism is unique compared to other CRISPR systems, as only one Cas protein (Cas9) is required for gene silencing (12). In type II systems, Cas9 participates in the processing of crRNAs (12), and is responsible for the destruction of the target DNA (11). Cas9’s function in both of these steps relies on the presence of two nuclease domains, a RuvC-like nuclease domain located at the amino terminus and a HNH-like nuclease domain that resides in the mid-region of the protein (13).
To achieve site-specific DNA recognition and cleavage, Cas9 must be complexed with both a crRNA and a separate trans-activating crRNA (tracrRNA or trRNA), that is partially complementary to the crRNA (11). The tracrRNA is required for crRNA maturation from a primary transcript encoding multiple pre-crRNAs. This occurs in the presence of RNase III and Cas9 (12).
During the destruction of target DNA, the HNH and RuvC-like nuclease domains cut both DNA strands, generating double-stranded breaks (DSBs) at sites defined by a 20-nucleotide target sequence within an associated crRNA transcript (11, 14). The HNH domain cleaves the complementary strand, while the RuvC domain cleaves the noncomplementary strand.
The double-stranded endonuclease activity of Cas9 also requires that a short conserved sequence, (2–5 nts) known as protospacer-associated motif (PAM), follows immediately 3´- of the crRNA complementary sequence (15). In fact, even fully complementary sequences are ignored by Cas9-RNA in the absence of a PAM sequence (16).
Cas9 and CRISPR as a New Tool in Molecular Biology
The simplicity of the type II CRISPR nuclease, with only three required components (Cas9 along with the crRNA and trRNA) makes this system amenable to adaptation for genome editing. This potential was realized in 2012 by the Doudna and Charpentier labs (11). Based on the type II CRISPR system described previously, the authors developed a simplified two-component system by combining trRNA and crRNA into a single synthetic single guide RNA (sgRNA). sgRNAprogrammed Cas9 was shown to be as effective as Cas9 programmed with separate trRNA and crRNA in guiding targeted gene alterations (Figure 2A).
To date, three different variants of the Cas9 nuclease have been adopted in genome-editing protocols. The first is wild-type Cas9, which can site-specifically cleave double-stranded DNA, resulting in the activation of the doublestrand break (DSB) repair machinery. DSBs can be repaired by the cellular Non-Homologous End Joining (NHEJ) pathway (17), resulting in insertions and/or deletions (indels) which disrupt the targeted locus. Alternatively, if a donor template with homology to the targeted locus is supplied, the DSB may be repaired by the homology-directed repair (HDR) pathway allowing for precise replacement mutations to be made (Figure 2A) (17, 18).
Cong and colleagues (1) took the Cas9 system a step further towards increased precision by developing a mutant form, known as Cas9D10A, with only nickase activity. This means it cleaves only one DNA strand, and does not activate NHEJ. Instead, when provided with a homologous repair template, DNA repairs are conducted via the high-fidelity HDR pathway only, resulting in reduced indel mutations (1, 11, 19). Cas9D10A is even more appealing in terms of target specificity when loci are targeted by paired Cas9 complexes designed to generate adjacent DNA nicks (20) (see further details about “paired nickases” in Figure 2B).
The third variant is a nuclease-deficient Cas9 (dCas9, Figure 2C) (21). Mutations H840A in the HNH domain and D10A in the RuvC domain inactivate cleavage activity, but do not prevent DNA binding (11, 22). Therefore, this variant can be used to sequence-specifically target any region of the genome without cleavage. Instead, by fusing with various effector domains, dCas9 can be used either as a gene silencing or activation tool (21, 23–26). Furthermore, it can be used as a visualization tool. For instance, Chen and colleagues used dCas9 fused to Enhanced Green Fluorescent Protein (EGFP) to visualize repetitive DNA sequences with a single sgRNA or nonrepetitive loci using multiple sgRNAs (27).
Wild-type Cas9 nuclease site specifically cleaves double-stranded DNA activating double-strand break repair machinery. In the absence of a homologous repair template non-homologous end joining can result in indels disrupting the target sequence. Alternatively, precise mutations and knock-ins can be made by providing a homologous repair template and exploiting the homology directed repair pathway.
B. Mutated Cas9 makes a site specific single-strand nick. Two sgRNA can be used to introduce a staggered double-stranded break which can then undergo homology directed repair.
C. Nuclease-deficient Cas9 can be fused with various effector domains allowing specific localization. For example, transcriptional activators, repressors, and fluorescent proteins.
Targeting Efficiency and Off-target Mutations
Targeting efficiency, or the percentage of desired mutation achieved, is one of the most important parameters by which to assess a genome-editing tool. The targeting efficiency of Cas9 compares favorably with more established methods, such as TALENs or ZFNs (8). For example, in human cells, custom-designed ZFNs and TALENs could only achieve efficiencies ranging from 1% to 50% (29–31). In contrast, the Cas9 system has been reported to have efficiencies up to >70% in zebrafish (32) and plants (33), and ranging from 2–5% in induced pluripotent stem cells (34). In addition, Zhou and colleagues were able to improve genome targeting up to 78% in one-cell mouse embryos, and achieved effective germline transmission through the use of dual sgRNAs to simultaneously target an individual gene (35).
A widely used method to identify mutations is the T7 Endonuclease I mutation detection assay (36, 37) (Figure 3). This assay detects heteroduplex DNA that results from the annealing of a DNA strand, including desired mutations, with a wildtype DNA strand (37).
Figure 3. T7 Endonuclease I Targeting Efficiency Assay
Genomic DNA is amplified with primers bracketing the modified locus. PCR products are then denatured and re-annealed yielding 3 possible structures. Duplexes containing a mismatch are digested by T7 Endonuclease I. The DNA is then electrophoretically separated and fragment analysis is used to calculate targeting efficiency.
Another important parameter is the incidence of off-target mutations. Such mutations are likely to appear in sites that have differences of only a few nucleotides compared to the original sequence, as long as they are adjacent to a PAM sequence. This occurs as Cas9 can tolerate up to 5 base mismatches within the protospacer region (36) or a single base difference in the PAM sequence (38). Off-target mutations are generally more difficult to detect, requiring whole-genome sequencing to rule them out completely.
Recent improvements to the CRISPR system for reducing off-target mutations have been made through the use of truncated gRNA (truncated within the crRNA-derived sequence) or by adding two extra guanine (G) nucleotides to the 5´ end (28, 37). Another way researchers have attempted to minimize off-target effects is with the use of “paired nickases” (20). This strategy uses D10A Cas9 and two sgRNAs complementary to the adjacent area on opposite strands of the target site (Figure 2B). While this induces DSBs in the target DNA, it is expected to create only single nicks in off-target locations and, therefore, result in minimal off-target mutations.
By leveraging computation to reduce off-target mutations, several groups have developed webbased tools to facilitate the identification of potential CRISPR target sites and assess their potential for off-target cleavage. Examples include the CRISPR Design Tool (38) and the ZiFiT Targeter, Version 4.2 (39, 40).
Applications as a Genome-editing and Genome Targeting Tool
Following its initial demonstration in 2012 (9), the CRISPR/Cas9 system has been widely adopted. This has already been successfully used to target important genes in many cell lines and organisms, including human (34), bacteria (41), zebrafish (32), C. elegans (42), plants (34), Xenopus tropicalis (43), yeast (44), Drosophila (45), monkeys (46), rabbits (47), pigs (42), rats (48) and mice (49). Several groups have now taken advantage of this method to introduce single point mutations (deletions or insertions) in a particular target gene, via a single gRNA (14, 21, 29). Using a pair of gRNA-directed Cas9 nucleases instead, it is also possible to induce large deletions or genomic rearrangements, such as inversions or translocations (50). A recent exciting development is the use of the dCas9 version of the CRISPR/Cas9 system to target protein domains for transcriptional regulation (26, 51, 52), epigenetic modification (25), and microscopic visualization of specific genome loci (27).
The CRISPR/Cas9 system requires only the redesign of the crRNA to change target specificity. This contrasts with other genome editing tools, including zinc finger and TALENs, where redesign of the protein-DNA interface is required. Furthermore, CRISPR/Cas9 enables rapid genome-wide interrogation of gene function by generating large gRNA libraries (51, 53) for genomic screening.
The Future of CRISPR/Cas9
The rapid progress in developing Cas9 into a set of tools for cell and molecular biology research has been remarkable, likely due to the simplicity, high efficiency and versatility of the system. Of the designer nuclease systems currently available for precision genome engineering, the CRISPR/Cas system is by far the most user friendly. It is now also clear that Cas9’s potential reaches beyond DNA cleavage, and its usefulness for genome locus-specific recruitment of proteins will likely only be limited by our imagination.
Scientists urge caution in using new CRISPR technology to treat human genetic disease
The bacterial enzyme Cas9 is the engine of RNA-programmed genome engineering in human cells. (Graphic by Jennifer Doudna/UC Berkeley)
A group of 18 scientists and ethicists today warned that a revolutionary new tool to cut and splice DNA should be used cautiously when attempting to fix human genetic disease, and strongly discouraged any attempts at making changes to the human genome that could be passed on to offspring.
Among the authors of this warning is Jennifer Doudna, the co-inventor of the technology, called CRISPR-Cas9, which is driving a new interest in gene therapy, or “genome engineering.” She and colleagues co-authored a perspective piece that appears in the March 20 issue of Science, based on discussions at a meeting that took place in Napa on Jan. 24. The same issue of Science features a collection of recent research papers, commentary and news articles on CRISPR and its implications. …..
A prudent path forward for genomic engineering and germline gene modification
Scientists today are changing DNA sequences to correct genetic defects in animals as well as cultured tissues generated from stem cells, strategies that could eventually be used to treat human disease. The technology can also be used to engineer animals with genetic diseases mimicking human disease, which could lead to new insights into previously enigmatic disorders.
The CRISPR-Cas9 tool is still being refined to ensure that genetic changes are precisely targeted, Doudna said. Nevertheless, the authors met “… to initiate an informed discussion of the uses of genome engineering technology, and to identify proactively those areas where current action is essential to prepare for future developments. We recommend taking immediate steps toward ensuring that the application of genome engineering technology is performed safely and ethically.”
Amyloid CRISPR Plasmids and si/shRNA Gene Silencers
Santa Cruz Biotechnology, Inc. offers a broad range of gene silencers in the form of siRNAs, shRNA Plasmids and shRNA Lentiviral Particles as well as CRISPR/Cas9 Knockout and CRISPR Double Nickase plasmids. Amyloid gene silencers are available as Amyloid siRNA, Amyloid shRNA Plasmid, Amyloid shRNA Lentiviral Particles and Amyloid CRISPR/Cas9 Knockout plasmids. Amyloid CRISPR/dCas9 Activation Plasmids and CRISPR Lenti Activation Systems for gene activation are also available. Gene silencers and activators are useful for gene studies in combination with antibodies used for protein detection. Amyloid CRISPR Knockout, HDR and Nickase Knockout Plasmids
CRISPR-Cas9-Based Knockout of the Prion Protein and Its Effect on the Proteome
The molecular function of the cellular prion protein (PrPC) and the mechanism by which it may contribute to neurotoxicity in prion diseases and Alzheimer’s disease are only partially understood. Mouse neuroblastoma Neuro2a cells and, more recently, C2C12 myocytes and myotubes have emerged as popular models for investigating the cellular biology of PrP. Mouse epithelial NMuMG cells might become attractive models for studying the possible involvement of PrP in a morphogenetic program underlying epithelial-to-mesenchymal transitions. Here we describe the generation of PrP knockout clones from these cell lines using CRISPR-Cas9 knockout technology. More specifically, knockout clones were generated with two separate guide RNAs targeting recognition sites on opposite strands within the first hundred nucleotides of the Prnp coding sequence. Several PrP knockout clones were isolated and genomic insertions and deletions near the CRISPR-target sites were characterized. Subsequently, deep quantitative global proteome analyses that recorded the relative abundance of>3000 proteins (data deposited to ProteomeXchange Consortium) were undertaken to begin to characterize the molecular consequences of PrP deficiency. The levels of ∼120 proteins were shown to reproducibly correlate with the presence or absence of PrP, with most of these proteins belonging to extracellular components, cell junctions or the cytoskeleton.
Recent advances in genome engineering technologies based on the CRISPR-associated RNA-guided endonuclease Cas9 are enabling the systematic interrogation of mammalian genome function. Analogous to the search function in modern word processors, Cas9 can be guided to specific locations within complex genomes by a short RNA search string. Using this system, DNA sequences within the endogenous genome and their functional outputs are now easily edited or modulated in virtually any organism of choice. Cas9-mediated genetic perturbation is simple and scalable, empowering researchers to elucidate the functional organization of the genome at the systems level and establish causal linkages between genetic variations and biological phenotypes. In this Review, we describe the development and applications of Cas9 for a variety of research or translational applications while highlighting challenges as well as future directions. Derived from a remarkable microbial defense system, Cas9 is driving innovative applications from basic biology to biotechnology and medicine.
The development of recombinant DNA technology in the 1970s marked the beginning of a new era for biology. For the first time, molecular biologists gained the ability to manipulate DNA molecules, making it possible to study genes and harness them to develop novel medicine and biotechnology. Recent advances in genome engineering technologies are sparking a new revolution in biological research. Rather than studying DNA taken out of the context of the genome, researchers can now directly edit or modulate the function of DNA sequences in their endogenous context in virtually any organism of choice, enabling them to elucidate the functional organization of the genome at the systems level, as well as identify causal genetic variations.
Broadly speaking, genome engineering refers to the process of making targeted modifications to the genome, its contexts (e.g., epigenetic marks), or its outputs (e.g., transcripts). The ability to do so easily and efficiently in eukaryotic and especially mammalian cells holds immense promise to transform basic science, biotechnology, and medicine (Figure 1).
For life sciences research, technologies that can delete, insert, and modify the DNA sequences of cells or organisms enable dissecting the function of specific genes and regulatory elements. Multiplexed editing could further allow the interrogation of gene or protein networks at a larger scale. Similarly, manipulating transcriptional regulation or chromatin states at particular loci can reveal how genetic material is organized and utilized within a cell, illuminating relationships between the architecture of the genome and its functions. In biotechnology, precise manipulation of genetic building blocks and regulatory machinery also facilitates the reverse engineering or reconstruction of useful biological systems, for example, by enhancing biofuel production pathways in industrially relevant organisms or by creating infection-resistant crops. Additionally, genome engineering is stimulating a new generation of drug development processes and medical therapeutics. Perturbation of multiple genes simultaneously could model the additive effects that underlie complex polygenic disorders, leading to new drug targets, while genome editing could directly correct harmful mutations in the context of human gene therapy (Tebas et al., 2014).
Eukaryotic genomes contain billions of DNA bases and are difficult to manipulate. One of the breakthroughs in genome manipulation has been the development of gene targeting by homologous recombination (HR), which integrates exogenous repair templates that contain sequence homology to the donor site (Figure 2A) (Capecchi, 1989). HR-mediated targeting has facilitated the generation of knockin and knockout animal models via manipulation of germline competent stem cells, dramatically advancing many areas of biological research. However, although HR-mediated gene targeting produces highly precise alterations, the desired recombination events occur extremely infrequently (1 in 106–109 cells) (Capecchi, 1989), presenting enormous challenges for large-scale applications of gene-targeting experiments.
Genome Editing Technologies Exploit Endogenous DNA Repair Machinery
To overcome these challenges, a series of programmable nuclease-based genome editing technologies have been developed in recent years, enabling targeted and efficient modification of a variety of eukaryotic and particularly mammalian species. Of the current generation of genome editing technologies, the most rapidly developing is the class of RNA-guided endonucleases known as Cas9 from the microbial adaptive immune system CRISPR (clustered regularly interspaced short palindromic repeats), which can be easily targeted to virtually any genomic location of choice by a short RNA guide. Here, we review the development and applications of the CRISPR-associated endonuclease Cas9 as a platform technology for achieving targeted perturbation of endogenous genomic elements and also discuss challenges and future avenues for innovation. ……
Figure 4Natural Mechanisms of Microbial CRISPR Systems in Adaptive Immunity
…… A key turning point came in 2005, when systematic analysis of the spacer sequences separating the individual direct repeats suggested their extrachromosomal and phage-associated origins (Mojica et al., 2005; Pourcel et al., 2005; Bolotin et al., 2005). This insight was tremendously exciting, especially given previous studies showing that CRISPR loci are transcribed (Tang et al., 2002) and that viruses are unable to infect archaeal cells carrying spacers corresponding to their own genomes (Mojica et al., 2005). Together, these findings led to the speculation that CRISPR arrays serve as an immune memory and defense mechanism, and individual spacers facilitate defense against bacteriophage infection by exploiting Watson-Crick base-pairing between nucleic acids (Mojica et al., 2005; Pourcel et al., 2005). Despite these compelling realizations that CRISPR loci might be involved in microbial immunity, the specific mechanism of how the spacers act to mediate viral defense remained a challenging puzzle. Several hypotheses were raised, including thoughts that CRISPR spacers act as small RNA guides to degrade viral transcripts in a RNAi-like mechanism (Makarova et al., 2006) or that CRISPR spacers direct Cas enzymes to cleave viral DNA at spacer-matching regions (Bolotin et al., 2005). …..
As the pace of CRISPR research accelerated, researchers quickly unraveled many details of each type of CRISPR system (Figure 4). Building on an earlier speculation that protospacer adjacent motifs (PAMs) may direct the type II Cas9 nuclease to cleave DNA (Bolotin et al., 2005), Moineau and colleagues highlighted the importance of PAM sequences by demonstrating that PAM mutations in phage genomes circumvented CRISPR interference (Deveau et al., 2008). Additionally, for types I and II, the lack of PAM within the direct repeat sequence within the CRISPR array prevents self-targeting by the CRISPR system. In type III systems, however, mismatches between the 5′ end of the crRNA and the DNA target are required for plasmid interference (Marraffini and Sontheimer, 2010). …..
In 2013, a pair of studies simultaneously showed how to successfully engineer type II CRISPR systems from Streptococcus thermophilus (Cong et al., 2013) andStreptococcus pyogenes (Cong et al., 2013; Mali et al., 2013a) to accomplish genome editing in mammalian cells. Heterologous expression of mature crRNA-tracrRNA hybrids (Cong et al., 2013) as well as sgRNAs (Cong et al., 2013; Mali et al., 2013a) directs Cas9 cleavage within the mammalian cellular genome to stimulate NHEJ or HDR-mediated genome editing. Multiple guide RNAs can also be used to target several genes at once. Since these initial studies, Cas9 has been used by thousands of laboratories for genome editing applications in a variety of experimental model systems (Sander and Joung, 2014). ……
The majority of CRISPR-based technology development has focused on the signature Cas9 nuclease from type II CRISPR systems. However, there remains a wide diversity of CRISPR types and functions. Cas RAMP module (Cmr) proteins identified in Pyrococcus furiosus and Sulfolobus solfataricus (Hale et al., 2012) constitute an RNA-targeting CRISPR immune system, forming a complex guided by small CRISPR RNAs that target and cleave complementary RNA instead of DNA. Cmr protein homologs can be found throughout bacteria and archaea, typically relying on a 5′ site tag sequence on the target-matching crRNA for Cmr-directed cleavage.
Unlike RNAi, which is targeted largely by a 6 nt seed region and to a lesser extent 13 other bases, Cmr crRNAs contain 30–40 nt of target complementarity. Cmr-CRISPR technologies for RNA targeting are thus a promising target for orthogonal engineering and minimal off-target modification. Although the modularity of Cmr systems for RNA-targeting in mammalian cells remains to be investigated, Cmr complexes native to P. furiosus have already been engineered to target novel RNA substrates (Hale et al., 2009, 2012). ……
Although Cas9 has already been widely used as a research tool, a particularly exciting future direction is the development of Cas9 as a therapeutic technology for treating genetic disorders. For a monogenic recessive disorder due to loss-of-function mutations (such as cystic fibrosis, sickle-cell anemia, or Duchenne muscular dystrophy), Cas9 may be used to correct the causative mutation. This has many advantages over traditional methods of gene augmentation that deliver functional genetic copies via viral vector-mediated overexpression—particularly that the newly functional gene is expressed in its natural context. For dominant-negative disorders in which the affected gene is haplosufficient (such as transthyretin-related hereditary amyloidosis or dominant forms of retinitis pigmentosum), it may also be possible to use NHEJ to inactivate the mutated allele to achieve therapeutic benefit. For allele-specific targeting, one could design guide RNAs capable of distinguishing between single-nucleotide polymorphism (SNP) variations in the target gene, such as when the SNP falls within the PAM sequence.
CRISPR/Cas9: a powerful genetic engineering tool for establishing large animal models of neurodegenerative diseases
Zhuchi Tu, Weili Yang, Sen Yan, Xiangyu Guo and Xiao-Jiang Li
Animal models are extremely valuable to help us understand the pathogenesis of neurodegenerative disorders and to find treatments for them. Since large animals are more like humans than rodents, they make good models to identify the important pathological events that may be seen in humans but not in small animals; large animals are also very important for validating effective treatments or confirming therapeutic targets. Due to the lack of embryonic stem cell lines from large animals, it has been difficult to use traditional gene targeting technology to establish large animal models of neurodegenerative diseases. Recently, CRISPR/Cas9 was used successfully to genetically modify genomes in various species. Here we discuss the use of CRISPR/Cas9 technology to establish large animal models that can more faithfully mimic human neurodegenerative diseases.
Neurodegenerative diseases — Alzheimer’s disease(AD),Parkinson’s disease(PD), amyotrophic lateral sclerosis (ALS), Huntington’s disease (HD), and frontotemporal dementia (FTD) — are characterized by age-dependent and selective neurodegeneration. As the life expectancy of humans lengthens, there is a greater prevalence of these neurodegenerative diseases; however, the pathogenesis of most of these neurodegenerative diseases remain unclear, and we lack effective treatments for these important brain disorders.
CRISPR/Cas9, Non-human primates, Neurodegenerative diseases, Animal model
There are a number of excellent reviews covering different types of neurodegenerative diseases and their genetic mouse models [8–12]. Investigations of different mouse models of neurodegenerative diseases have revealed a common pathology shared by these diseases. First, the development of neuropathology and neurological symptoms in genetic mouse models of neurodegenerative diseases is age dependent and progressive. Second, all the mouse models show an accumulation of misfolded or aggregated proteins resulting from the expression of mutant genes. Third, despite the widespread expression of mutant proteins throughout the body and brain, neuronal function appears to be selectively or preferentially affected. All these facts indicate that mouse models of neurodegenerative diseases recapitulate important pathologic features also seen in patients with neurodegenerative diseases.
However, it seems that mouse models can not recapitulate the full range of neuropathology seen in patients with neurodegenerative diseases. Overt neurodegeneration, which is the most important pathological feature in patient brains, is absent in genetic rodent models of AD, PD, and HD. Many rodent models that express transgenic mutant proteins under the control of different promoters do not replicate overt neurodegeneration, which is likely due to their short life spans and the different aging processes of small animals. Also important are the remarkable differences in brain development between rodents and primates. For example, the mouse brain takes 21 days to fully develop, whereas the formation of primate brains requires more than 150 days [13]. The rapid development of the brain in rodents may render neuronal cells resistant to misfolded protein-mediated neurodegeneration. Another difficulty in using rodent models is how to analyze cognitive and emotional abnormalities, which are the early symptoms of most neurodegenerative diseases in humans. Differences in neuronal circuitry, anatomy, and physiology between rodent and primate brains may also account for the behavioral differences between rodent and primate models.
Mitochondrial dynamics–fusion, fission, movement, and mitophagy–in neurodegenerative diseases
Neurons are metabolically active cells with high energy demands at locations distant from the cell body. As a result, these cells are particularly dependent on mitochondrial function, as reflected by the observation that diseases of mitochondrial dysfunction often have a neurodegenerative component. Recent discoveries have highlighted that neurons are reliant particularly on the dynamic properties of mitochondria. Mitochondria are dynamic organelles by several criteria. They engage in repeated cycles of fusion and fission, which serve to intermix the lipids and contents of a population of mitochondria. In addition, mitochondria are actively recruited to subcellular sites, such as the axonal and dendritic processes of neurons. Finally, the quality of a mitochondrial population is maintained through mitophagy, a form of autophagy in which defective mitochondria are selectively degraded. We review the general features of mitochondrial dynamics, incorporating recent findings on mitochondrial fusion, fission, transport and mitophagy. Defects in these key features are associated with neurodegenerative disease. Charcot-Marie-Tooth type 2A, a peripheral neuropathy, and dominant optic atrophy, an inherited optic neuropathy, result from a primary deficiency of mitochondrial fusion. Moreover, several major neurodegenerative diseases—including Parkinson’s, Alzheimer’s and Huntington’s disease—involve disruption of mitochondrial dynamics. Remarkably, in several disease models, the manipulation of mitochondrial fusion or fission can partially rescue disease phenotypes. We review how mitochondrial dynamics is altered in these neurodegenerative diseases and discuss the reciprocal interactions between mitochondrial fusion, fission, transport and mitophagy.
Applications of CRISPR–Cas systems in Neuroscience
Genome-editing tools, and in particular those based on CRISPR–Cas (clustered regularly interspaced short palindromic repeat (CRISPR)–CRISPR-associated protein) systems, are accelerating the pace of biological research and enabling targeted genetic interrogation in almost any organism and cell type. These tools have opened the door to the development of new model systems for studying the complexity of the nervous system, including animal models and stem cell-derived in vitro models. Precise and efficient gene editing using CRISPR–Cas systems has the potential to advance both basic and translational neuroscience research.
Cellular neuroscience, DNA recombination, Genetic engineering, Molecular neuroscience
Figure 3: In vitro applications of Cas9 in human iPSCs.close
a | Evaluation of disease candidate genes from large-population genome-wide association studies (GWASs). Human primary cells, such as neurons, are not easily available and are difficult to expand in culture. By contrast, induced pluripo…
The development of the CRISPR/Cas9 system has made gene editing a relatively simple task. While CRISPR and other gene editing technologies stand to revolutionize biomedical research and offers many promising therapeutic avenues (such as in the treatment of HIV), a great deal of debate exists over whether CRISPR should be used to modify human embryos. As I discussed in my previous Insight article, we lack enough fundamental biological knowledge to enhance many traits like height or intelligence, so we are not near a future with genetically-enhanced super babies. However, scientists have identified a few rare genetic variants that protect against disease. One such protective variant is a mutation in the APP gene that protects against Alzheimer’s disease and cognitive decline in old age. If we can perfect gene editing technologies, is this mutation one that we should be regularly introducing into embryos? In this article, I explore the potential for using gene editing as a way to prevent Alzheimer’s disease in future generations. Alzheimer’s Disease: Medicine’s Greatest Challenge in the 21st Century Can gene editing be the missing piece in the battle against Alzheimer’s? (Source: bostonbiotech.org) I chose to assess the benefit of germline gene editing in the context of Alzheimer’s disease because this disease is one of the biggest challenges medicine faces in the 21st century. Alzheimer’s disease is a chronic neurodegenerative disease responsible for the majority of the cases of dementia in the elderly. The disease symptoms begins with short term memory loss and causes more severe symptoms – problems with language, disorientation, mood swings, behavioral issues – as it progresses, eventually leading to the loss of bodily functions and death. Because of the dementia the disease causes, Alzheimer’s patients require a great deal of care, and the world spends ~1% of its total GDP on caring for those with Alzheimer’s and related disorders. Because the prevalence of the disease increases with age, the situation will worsen as life expectancies around the globe increase: worldwide cases of Alzheimer’s are expected to grow from 35 million today to over 115 million by 2050.
Despite much research, the exact causes of Alzheimer’s disease remains poorly understood. The disease seems to be related to the accumulation of plaques made of amyloid-β peptides that form on the outside of neurons, as well as the formation of tangles of the protein tau inside of neurons. Although many efforts have been made to target amyloid-β or the enzymes involved in its formation, we have so far been unsuccessful at finding any treatment that stops the disease or reverses its progress. Some researchers believe that most attempts at treating Alzheimer’s have failed because, by the time a patient shows symptoms, the disease has already progressed past the point of no return.
While research towards a cure continues, researchers have sought effective ways to prevent Alzheimer’s disease. Although some studies show that mental and physical exercise may lower ones risk of Alzheimer’s disease, approximately 60-80% of the risk for Alzheimer’s disease appears to be genetic. Thus, if we’re serious about prevention, we may have to act at the genetic level. And because the brain is difficult to access surgically for gene therapy in adults, this means using gene editing on embryos.
With the latest CRISPR/Cas9 advance, the exhortation “turn on, tune in, drop out” comes to mind. The CRISPR/Cas9 gene-editing system was already a well-known means of “tuning in” (inserting new genes) and “dropping out” (knocking out genes). But when it came to “turning on” genes, CRISPR/Cas9 had little potency. That is, it had demonstrated only limited success as a way to activate specific genes.
A new CRISPR/Cas9 approach, however, appears capable of activating genes more effectively than older approaches. The new approach may allow scientists to more easily determine the function of individual genes, according to Feng Zhang, Ph.D., a researcher at MIT and the Broad Institute. Dr. Zhang and colleagues report that the new approach permits multiplexed gene activation and rapid, large-scale studies of gene function.
The new technique was introduced in the December 10 online edition of Nature, in an article entitled, “Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex.” The article describes how Dr. Zhang, along with the University of Tokyo’s Osamu Nureki, Ph.D., and Hiroshi Nishimasu, Ph.D., overhauled the CRISPR/Cas9 system. The research team based their work on their analysis (published earlier this year) of the structure formed when Cas9 binds to the guide RNA and its target DNA. Specifically, the team used the structure’s 3D shape to rationally improve the system.
In previous efforts to revamp CRISPR/Cas9 for gene activation purposes, scientists had tried to attach the activation domains to either end of the Cas9 protein, with limited success. From their structural studies, the MIT team realized that two small loops of the RNA guide poke out from the Cas9 complex and could be better points of attachment because they allow the activation domains to have more flexibility in recruiting transcription machinery.
Using their revamped system, the researchers activated about a dozen genes that had proven difficult or impossible to turn on using the previous generation of Cas9 activators. Each gene showed at least a twofold boost in transcription, and for many genes, the researchers found multiple orders of magnitude increase in activation.
After investigating single-guide RNA targeting rules for effective transcriptional activation, demonstrating multiplexed activation of 10 genes simultaneously, and upregulating long intergenic noncoding RNA transcripts, the research team decided to undertake a large-scale screen. This screen was designed to identify genes that confer resistance to a melanoma drug called PLX-4720.
“We … synthesized a library consisting of 70,290 guides targeting all human RefSeq coding isoforms to screen for genes that, upon activation, confer resistance to a BRAF inhibitor,” wrote the authors of the Nature paper. “The top hits included genes previously shown to be able to confer resistance, and novel candidates were validated using individual [single-guide RNA] and complementary DNA overexpression.”
A gene signature based on the top screening hits, the authors added, correlated with a gene expression signature of BRAF inhibitor resistance in cell lines and patient-derived samples. It was also suggested that large-scale screens such as the one demonstrated in the current study could help researchers discover new cancer drugs that prevent tumors from becoming resistant.
Familial amyloid polyneuropathy type I is an autosomal dominant disorder caused by mutations in the transthyretin (TTR ) gene; however, carriers of the same mutation exhibit variability in penetrance and clinical expression. We analyzed alleles of candidate genes encoding non-fibrillar components of TTR amyloid deposits and a molecule metabolically interacting with TTR [retinol-binding protein (RBP)], for possible associations with age of disease onset and/or susceptibility in a Portuguese population sample with the TTR V30M mutation and unrelated controls. We show that the V30M carriers represent a distinct subset of the Portuguese population. Estimates of genetic distance indicated that the controls and the classical onset group were furthest apart, whereas the late-onset group appeared to differ from both. Importantly, the data also indicate that genetic interactions among the multiple loci evaluated, rather than single-locus effects, are more likely to determine differences in the age of disease onset. Multifactor dimensionality reduction indicated that the best genetic model for classical onset group versus controls involved the APCS gene, whereas for late-onset cases, one APCS variant (APCSv1) and two RBP variants (RBPv1 and RBPv2) are involved. Thus, although the TTR V30M mutation is required for the disease in Portuguese patients, different genetic factors may govern the age of onset, as well as the occurrence of anticipation.
Autosomal dominant disorders may vary in expression even within a given kindred. The basis of this variability is uncertain and can be attributed to epigenetic factors, environment or epistasis. We have studied familial amyloid polyneuropathy (FAP), an autosomal dominant disorder characterized by peripheral sensorimotor and autonomic neuropathy. It exhibits variation in cardiac, renal, gastrointestinal and ocular involvement, as well as age of onset. Over 80 missense mutations in the transthyretin gene (TTR ) result in autosomal dominant disease http://www.ibmc.up.pt/~mjsaraiv/ttrmut.html). The presence of deposits consisting entirely of wild-type TTR molecules in the hearts of 10– 25% of individuals over age 80 reveals its inherent in vivo amyloidogenic potential (1).
FAP was initially described in Portuguese (2) where, until recently, the TTR V30M has been the only pathogenic mutation associated with the disease (3,4). Later reports identified the same mutation in Swedish and Japanese families (5,6). The disorder has since been recognized in other European countries and in North American kindreds in association with V30M, as well as other mutations (7).
TTR V30M produces disease in only 5–10% of Swedish carriers of the allele (8), a much lower degree of penetrance than that seen in Portuguese (80%) (9) or in Japanese with the same mutation. The actual penetrance in Japanese carriers has not been formally established, but appears to resemble that seen in Portuguese. Portuguese and Japanese carriers show considerable variation in the age of clinical onset (10,11). In both populations, the first symptoms had originally been described as typically occurring before age 40 (so-called ‘classical’ or early-onset); however, in recent years, more individuals developing symptoms late in life have been identified (11,12). Hence, present data indicate that the distribution of the age of onset in Portuguese is continuous, but asymmetric with a mean around age 35 and a long tail into the older age group (Fig. 1) (9,13). Further, DNA testing in Portugal has identified asymptomatic carriers over age 70 belonging to a subset of very late-onset kindreds in whose descendants genetic anticipation is frequent. The molecular basis of anticipation in FAP, which is not mediated by trinucleotide repeat expansions in the TTR or any other gene (14), remains elusive.
Variation in penetrance, age of onset and clinical features are hallmarks of many autosomal dominant disorders including the human TTR amyloidoses (7). Some of these clearly reflect specific biological effects of a particular mutation or a class of mutants. However, when such phenotypic variability is seen with a single mutation in the gene encoding the same protein, it suggests an effect of modifying genetic loci and/or environmental factors contributing differentially to the course of disease. We have chosen to examine age of onset as an example of a discrete phenotypic variation in the presence of the particular autosomal dominant disease-associated mutation TTR V30M. Although the role of environmental factors cannot be excluded, the existence of modifier genes involved in TTR amyloidogenesis is an attractive hypothesis to explain the phenotypic variability in FAP. ….
ATTR (TTR amyloid), like all amyloid deposits, contains several molecular components, in addition to the quantitatively dominant fibril-forming amyloid protein, including heparan sulfate proteoglycan 2 (HSPG2 or perlecan), SAP, a plasma glycoprotein of the pentraxin family (encoded by the APCS gene) that undergoes specific calcium-dependent binding to all types of amyloid fibrils, and apolipoprotein E (ApoE), also found in all amyloid deposits (15). The ApoE4 isoform is associated with an increased frequency and earlier onset of Alzheimer’s disease (Ab), the most common form of brain amyloid, whereas the ApoE2 isoform appears to be protective (16). ApoE variants could exert a similar modulatory effect in the onset of FAP, although early studies on a limited number of patients suggested this was not the case (17).
In at least one instance of senile systemic amyloidosis, small amounts of AA-related material were found in TTR deposits (18). These could reflect either a passive co-aggregation or a contributory involvement of protein AA, encoded by the serum amyloid A (SAA ) genes and the main component of secondary (reactive) amyloid fibrils, in the formation of ATTR.
Retinol-binding protein (RBP), the serum carrier of vitamin A, circulates in plasma bound to TTR. Vitamin A-loaded RBP and L-thyroxine, the two natural ligands of TTR, can act alone or synergistically to inhibit the rate and extent of TTR fibrillogenesis in vitro, suggesting that RBP may influence the course of FAP pathology in vivo (19). We have analyzed coding and non-coding sequence polymorphisms in the RBP4 (serum RBP, 10q24), HSPG2 (1p36.1), APCS (1q22), APOE (19q13.2), SAA1 and SAA2 (11p15.1) genes with the goal of identifying chromosomes carrying common and functionally significant variants. At the time these studies were performed, the full human genome sequence was not completed and systematic singlenucleotide polymorphism (SNP) analyses were not available for any of the suspected candidate genes. We identified new SNPs in APCS and RBP4 and utilized polymorphisms in SAA, HSPG2 and APOE that had already been characterized and shown to have potential pathophysiologic significance in other disorders (16,20–22). The genotyping data were analyzed for association with the presence of the V30M amyloidogenic allele (FAP patients versus controls) and with the age of onset (classical- versus late-onset patients). Multilocus analyses were also performed to examine the effects of simultaneous contributions of the six loci for determining the onset of the first symptoms. …..
The potential for different underlying models for classical and late onset is supported by the MDR analysis, which produces two distinct models when comparing each class with the controls. One could view the two onset classes as unique diseases. If this is the case, then the failure to detect a single predictive genetic model is consistent with two related, but different, diseases. This is exactly what would be expected in such a case of genetic heterogeneity (28). Using this approach, a major gene effect can be viewed as a necessary, but not sufficient, condition to explain the course of the disease. Analyzing the cases but omitting from the analysis of phenotype the necessary allele, in this case TTR V30M, can then reveal a variety of important modifiers that are distinct between the phenotypes.
The significant comparisons obtained in our study cohort indicate that the combined effects mainly result from two and three-locus interactions involving all loci except SAA1 and SAA2 for susceptibility to disease. A considerable number of four-site combinations modulate the age of onset with SAA1 appearing in a majority of significant combinations in late-onset disease, perhaps indicating a greater role of the SAA variants in the age of onset of FAP.
The correlation between genotype and phenotype in socalled simple Mendelian disorders is often incomplete, as only a subset of all mutations can reliably predict specific phenotypes (34). This is because non-allelic genetic variations and/or environmental influences underlie these disorders whose phenotypes behave as complex traits. A few examples include the identification of the role of homozygozity for the SAA1.1 allele in conferring the genetic susceptibility to renal amyloidosis in FMF (20) and the association of an insertion/deletion polymorphism in the ACE gene with disease severity in familial hypertrophic cardiomyopathy (35). In these disorders, the phenotypes arise from mutations in MEFV and b-MHC, but are modulated by independently inherited genetic variation. In this report, we show that interactions among multiple genes, whose products are confirmed or putative constituents of ATTR deposits, or metabolically interact with TTR, modulate the onset of the first symptoms and predispose individuals to disease in the presence of the V30M mutation in TTR. The exact nature of the effects identified here requires further study with potential application in the development of genetic screening with prognostic value pertaining to the onset of disease in the TTR V30M carriers.
If the effects of additional single or interacting genes dictate the heterogeneity of phenotype, as reflected in variability of onset and clinical expression (with the same TTR mutation), the products encoded by alleles at such loci could contribute to the process of wild-type TTR deposition in elderly individuals without a mutation (senile systemic amyloidosis), a phenomenon not readily recognized as having a genetic basis because of the insensitivity of family history in the elderly.
Safety and Efficacy of RNAi Therapy for Transthyretin Amyloidosis
Transthyretin amyloidosis is caused by the deposition of hepatocyte-derived transthyretin amyloid in peripheral nerves and the heart. A therapeutic approach mediated by RNA interference (RNAi) could reduce the production of transthyretin.
Methods We identified a potent antitransthyretin small interfering RNA, which was encapsulated in two distinct first- and second-generation formulations of lipid nanoparticles, generating ALN-TTR01 and ALN-TTR02, respectively. Each formulation was studied in a single-dose, placebo-controlled phase 1 trial to assess safety and effect on transthyretin levels. We first evaluated ALN-TTR01 (at doses of 0.01 to 1.0 mg per kilogram of body weight) in 32 patients with transthyretin amyloidosis and then evaluated ALN-TTR02 (at doses of 0.01 to 0.5 mg per kilogram) in 17 healthy volunteers.
Results Rapid, dose-dependent, and durable lowering of transthyretin levels was observed in the two trials. At a dose of 1.0 mg per kilogram, ALN-TTR01 suppressed transthyretin, with a mean reduction at day 7 of 38%, as compared with placebo (P=0.01); levels of mutant and nonmutant forms of transthyretin were lowered to a similar extent. For ALN-TTR02, the mean reductions in transthyretin levels at doses of 0.15 to 0.3 mg per kilogram ranged from 82.3 to 86.8%, with reductions of 56.6 to 67.1% at 28 days (P<0.001 for all comparisons). These reductions were shown to be RNAi mediated. Mild-to-moderate infusion-related reactions occurred in 20.8% and 7.7% of participants receiving ALN-TTR01 and ALN-TTR02, respectively.
ALN-TTR01 and ALN-TTR02 suppressed the production of both mutant and nonmutant forms of transthyretin, establishing proof of concept for RNAi therapy targeting messenger RNA transcribed from a disease-causing gene.
Alnylam May Seek Approval for TTR Amyloidosis Rx in 2017 as Other Programs Advance
Officials from Alnylam Pharmaceuticals last week provided updates on the two drug candidates from the company’s flagship transthyretin-mediated amyloidosis program, stating that the intravenously delivered agent patisiran is proceeding toward a possible market approval in three years, while a subcutaneously administered version called ALN-TTRsc is poised to enter Phase III testing before the end of the year.
Meanwhile, Alnylam is set to advance a handful of preclinical therapies into human studies in short order, including ones for complement-mediated diseases, hypercholesterolemia, and porphyria.
The officials made their comments during a conference call held to discuss Alnylam’s second-quarter financial results.
ATTR is caused by a mutation in the TTR gene, which normally produces a protein that acts as a carrier for retinol binding protein and is characterized by the accumulation of amyloid deposits in various tissues. Alnylam’s drugs are designed to silence both the mutant and wild-type forms of TTR.
Patisiran, which is delivered using lipid nanoparticles developed by Tekmira Pharmaceuticals, is currently in a Phase III study in patients with a form of ATTR called familial amyloid polyneuropathy (FAP) affecting the peripheral nervous system. Running at over 20 sites in nine countries, that study is set to enroll up to 200 patients and compare treatment to placebo based on improvements in neuropathy symptoms.
According to Alnylam Chief Medical Officer Akshay Vaishnaw, Alnylam expects to have final data from the study in two to three years, which would put patisiran on track for a new drug application filing in 2017.
Meanwhile, ALN-TTRsc, which is under development for a version of ATTR that affects cardiac tissue called familial amyloidotic cardiomyopathy (FAC) and uses Alnylam’s proprietary GalNAc conjugate delivery technology, is set to enter Phase III by year-end as Alnylam holds “active discussions” with US and European regulators on the design of that study, CEO John Maraganore noted during the call.
In the interim, Alnylam continues to enroll patients in a pilot Phase II study of ALN-TTRsc, which is designed to test the drug’s efficacy for FAC or senile systemic amyloidosis (SSA), a condition caused by the idiopathic accumulation of wild-type TTR protein in the heart.
Based on “encouraging” data thus far, Vaishnaw said that Alnylam has upped the expected enrollment in this study to 25 patients from 15. Available data from the trial is slated for release in November, he noted, stressing that “any clinical endpoint result needs to be considered exploratory given the small sample size and the very limited duration of treatment of only six weeks” in the trial.
Vaishnaw added that an open-label extension (OLE) study for patients in the ALN-TTRsc study will kick off in the coming weeks, allowing the company to gather long-term dosing tolerability and clinical activity data on the drug.
Enrollment in an OLE study of patisiran has been completed with 27 patients, he said, and, “as of today, with up to nine months of therapy … there have been no study drug discontinuations.” Clinical endpoint data from approximately 20 patients in this study will be presented at the American Neurological Association meeting in October.
As part of its ATTR efforts, Alnylam has also been conducting natural history of disease studies in both FAP and FAC patients. Data from the 283-patient FAP study was presented earlier this year and showed a rapid progression in neuropathy impairment scores and a high correlation of this measurement with disease severity.
During last week’s conference call, Vaishnaw said that clinical endpoint and biomarker data on about 400 patients with either FAC or SSA have already been collected in a nature history study on cardiac ATTR. Maraganore said that these findings would likely be released sometime next year.
The first medication for a rare and often fatal protein misfolding disorder has been approved in Europe. On November 16, the E gave a green light to Pfizer’s Vyndaqel (tafamidis) for treating transthyretin amyloidosis in adult patients with stage 1 polyneuropathy symptoms. [Jeffery Kelly, La Jolla]
The most clinically advanced RNA interference (RNAi) therapeutic achieved a milestone in April when Alnylam Pharmaceuticals in Cambridge, Massachusetts, reported positive results for patisiran, a small interfering RNA (siRNA) oligonucleotide targeting transthyretin for treating familial amyloidotic polyneuropathy (FAP). …
FAP is characterized by the systemic deposition of amyloidogenic variants of the transthyretin protein, especially in the peripheral nervous system, causing a progressive sensory and motor polyneuropathy.
FAP is caused by a mutation of the TTR gene, located on human chromosome 18q12.1-11.2.[5] A replacement of valine by methionine at position 30 (TTR V30M) is the mutation most commonly found in FAP.[1] The variant TTR is mostly produced by the liver.[citation needed] The transthyretin protein is a tetramer. ….
Scientists at the Swiss Federal Institute of Technology (ETH) in Zurich have found an exciting new use for the cells that reside in the undesirable flabby tissue—creating pancreatic beta cells. The ETH researchers extracted stem cells from a 50-year-old test subject’s fatty tissue and reprogrammed them into mature, insulin-producing beta cells.
The findings from this study were published recently in Nature Communications in an article entitled “A Programmable Synthetic Lineage-Control Network That Differentiates Human IPSCs into Glucose-Sensitive Insulin-Secreting Beta-Like Cells.”
The investigators added a highly complex synthetic network of genes to the stem cells to recreate precisely the key growth factors involved in this maturation process. Central to the process were the growth factors Ngn3, Pdx1, and MafA; the researchers found that concentrations of these factors change during the differentiation process.
For instance, MafA is not present at the start of maturation. Only on day 4, in the final maturation step, does it appear, its concentration rising steeply and then remaining at a high level. The changes in the concentrations of Ngn3 and Pdx1, however, are very complex: while the concentration of Ngn3 rises and then falls again, the level of Pdx1 rises at the beginning and toward the end of maturation.
Senior study author Martin Fussenegger, Ph.D., professor of biotechnology and bioengineering at ETH Zurich’s department of biosystems science and engineering stressed that it was essential to reproduce these natural processes as closely as possible to produce functioning beta cells, stating that “the timing and the quantities of these growth factors are extremely important.”
The ETH researchers believe that their work is a real breakthrough, in that a synthetic gene network has been used successfully to achieve genetic reprogramming that delivers beta cells. Until now, scientists have controlled such stem cell differentiation processes by adding various chemicals and proteins exogenously.
“It’s not only really hard to add just the right quantities of these components at just the right time, but it’s also inefficient and impossible to scale up,” Dr. Fussenegger noted.
While the beta cells not only looked very similar to their natural counterparts—containing dark spots known as granules that store insulin—the artificial beta cells also functioned in a very similar manner. However, the researchers admit that more work needs to be done to increase the insulin output.
“At the present time, the quantities of insulin they secrete are not as great as with natural beta cells,” Dr. Fussenegger stated. Yet, the key point is that the researchers have for the first time succeeded in reproducing the entire natural process chain, from stem cell to differentiated beta cell.
In future, the ETH scientists’ novel technique might make it possible to implant new functional beta cells in diabetes sufferers that are made from their adipose tissue. While beta cells have been transplanted in the past, this has always required subsequent suppression of the recipient’s immune system—as with any transplant of donor organs or tissue.
“With our beta cells, there would likely be no need for this action since we can make them using endogenous cell material taken from the patient’s own body,” Dr. Fussenegger said. “This is why our work is of such interest in the treatment of diabetes.”
A programmable synthetic lineage-control network that differentiates human IPSCs into glucose-sensitive insulin-secreting beta-like cells
Synthetic biology has advanced the design of standardized transcription control devices that programme cellular behaviour. By coupling synthetic signalling cascade- and transcription factor-based gene switches with reverse and differential sensitivity to the licensed food additive vanillic acid, we designed a synthetic lineage-control network combining vanillic acid-triggered mutually exclusive expression switches for the transcription factors Ngn3 (neurogenin 3; OFF-ON-OFF) and Pdx1 (pancreatic and duodenal homeobox 1; ON-OFF-ON) with the concomitant induction of MafA (V-maf musculoaponeurotic fibrosarcoma oncogene homologue A; OFF-ON). This designer network consisting of different network topologies orchestrating the timely control of transgenic and genomic Ngn3, Pdx1 and MafA variants is able to programme human induced pluripotent stem cells (hIPSCs)-derived pancreatic progenitor cells into glucose-sensitive insulin-secreting beta-like cells, whose glucose-stimulated insulin-release dynamics are comparable to human pancreatic islets. Synthetic lineage-control networks may provide the missing link to genetically programme somatic cells into autologous cell phenotypes for regenerative medicine.
Cell-fate decisions during development are regulated by various mechanisms, including morphogen gradients, regulated activation and silencing of key transcription factors, microRNAs, epigenetic modification and lateral inhibition. The latter implies that the decision of one cell to adopt a specific phenotype is associated with the inhibition of neighbouring cells to enter the same developmental path. In mammals, insights into the role of key transcription factors that control development of highly specialized organs like the pancreas were derived from experiments in mice, especially various genetically modified animals1, 2, 3, 4. Normal development of the pancreas requires the activation of pancreatic duodenal homeobox protein (Pdx1) in pre-patterned cells of the endoderm. Inactivating mutations of Pdx1 are associated with pancreas agenesis in mouse and humans5, 6. A similar cell fate decision occurs later with the activation of Ngn3 that is required for the development of all endocrine cells in the pancreas7. Absence of Ngn3 is associated with the loss of pancreatic endocrine cells, whereas the activation of Ngn3 not only allows the differentiation of endocrine cells but also induces lateral inhibition of neighbouring cells—via Delta-Notch pathway—to enter the same pancreatic endocrine cell fate8. This Ngn3-mediated cell-switch occurs at a specific time point and for a short period of time in mice9. Thereafter, it is silenced and becomes almost undetectable in postnatal pancreatic islets. Conversely, Pdx1-positive Ngn3-positive cells reduce Pdx1 expression, as Ngn3-positive cells are Pdx1 negative10. They re-express Pdx1, however, as they go on their path towards glucose-sensitive insulin-secreting cells with parallel induction of MafA that is required for proper differentiation and maturation of pancreatic beta cells11. Data supporting these expression dynamics are derived from mice experiments1, 11, 12. A synthetic gene-switch governing cell fate decision in human induced pluripotent stem cells (hIPSCs) could facilitate the differentiation of glucose-sensitive insulin-secreting cells.
In recent years, synthetic biology has significantly advanced the rational design of synthetic gene networks that can interface with host metabolism, correct physiological disturbances13 and provide treatment strategies for a variety of metabolic disorders, including gouty arthritis14, obesity15 and type-2 diabetes16. Currently, synthetic biology principles may provide the componentry and gene network topologies for the assembly of synthetic lineage-control networks that can programme cell-fate decisions and provide targeted differentiation of stem cells into terminally differentiated somatic cells. Synthetic lineage-control networks may therefore provide the missing link between human pluripotent stem cells17 and their true impact on regenerative medicine18, 19, 20. The use of autologous stem cells in regenerative medicine holds great promise for curing many diseases, including type-1 diabetes mellitus (T1DM), which is characterized by the autoimmune destruction of insulin-producing pancreatic beta cells, thus making patients dependent on exogenous insulin to control their blood glucose21, 22. Although insulin therapy has changed the prospects and survival of T1DM patients, these patients still suffer from diabetic complications arising from the lack of physiological insulin secretion and excessive glucose levels23. The replacement of the pancreatic beta cells either by pancreas transplantation or by transplantation of pancreatic islets has been shown to normalize blood glucose and even improve existing complications of diabetes24. However, insulin independence 5 years after islet transplantation can only be achieved in up to 55% of the patients even when using the latest generation of immune suppression strategies25, 26. Transplantation of human islets or the entire pancreas has allowed T1DM patients to become somewhat insulin independent, which provides a proof-of-concept for beta-cell replacement therapies27, 28. However, because of the shortage of donor pancreases and islets, as well as the significant risk associated with transplantation and life-long immunosuppression, the rational differentiation of stem cells into functional beta-cells remains an attractive alternative29, 30. Nevertheless, a definitive cure for T1DM should address both the beta-cell deficit and the autoimmune response to cells that express insulin. Any beta-cell mimetic should be able to store large amounts of insulin and secrete it on demand, as in response to glucose stimulation29, 31. The most effective protocols for the in vitro generation of bonafide insulin-secreting beta-like cells that are suitable for transplantation have been the result of sophisticated trial-and-error studies elaborating timely addition of complex growth factor and small-molecule compound cocktails to human pancreatic progenitor cells32, 33, 34. The differentiation of pancreatic progenitor cells to beta-like cells is the most challenging part as current protocols provide inconsistent results and limited success in programming pancreatic progenitor cells into glucose-sensitive insulin-secreting beta-like cells35, 36, 37. One of the reasons for these observations could be the heterogeneity in endocrine differentiation and maturation towards a beta cell phenotype. Here we show that a synthetic lineage-control network programming the dynamic expression of the transcription factors Ngn3, Pdx1 and MafA enables the differentiation of hIPSC-derived pancreatic progenitor cells to glucose-sensitive insulin-secreting beta-like cells (Supplementary Fig. 1).
The differentiation pathway from pancreatic progenitor cells to glucose-sensitive insulin-secreting pancreatic beta-cells combines the transient mutually exclusive expression switches of Ngn3 (OFF-ON-OFF) and Pdx1 (ON-OFF-ON) with the concomitant induction of MafA (OFF-ON) expression10,11. Since independent control of the pancreatic transcription factors Ngn3, Pdx1 and MafA by different antibiotic transgene control systems responsive to tetracycline, erythromycin and pristinamycin did not result in the desired differential control dynamics (Supplementary Fig. 2), we have designed a vanillic acid-programmable synthetic lineage-control network that programmes hIPSC-derived pancreatic progenitor cells to specifically differentiate into glucose-sensitive insulin-secreting beta-like cells in a seamless and self-sufficient manner. The timely coordination of mutually exclusive Ngn3 and Pdx1 expression with MafA induction requires the trigger-controlled execution of a complex genetic programme that orchestrates two overlapping antagonistic band-pass filter expression profiles (OFF-ON-OFF and ON-OFF-ON), a positive band-pass filter for Ngn3 (OFF-ON-OFF) and a negative band-pass filter, also known as band-stop filter, for Pdx1 (ON-OFF-ON), the ramp-up expression phase of which is linked to a graded induction of MafA (OFF-ON).
The core of the synthetic lineage-control network consists of two transgene control devices that are sensitive to the food component and licensed food additive vanillic acid. These devices are a synthetic vanillic acid-inducible (ON-type) signalling cascade that is gradually induced by increasing the vanillic acid concentration and a vanillic acid-repressible (OFF-type) gene switch that is repressed in a vanillic acid dose-dependent manner (Fig. 1a,b). The designer cascade consists of the vanillic acid-sensitive mammalian olfactory receptor MOR9-1, which sequentially activates the G protein Sα (GSα) and adenylyl cyclase to produce a cyclic AMP (cAMP) second messenger surge38 that is rewired via the cAMP-responsive protein kinase A-mediated phospho-activation of the cAMP-response element-binding protein 1 (CREB1) to the induction of synthetic promoters (PCRE) containing CREB1-specific cAMP response elements (CRE; Fig. 1a). The co-transfection of pCI-MOR9-1 (PhCMV-MOR9-1-pASV40) and pCK53 (PCRE-SEAP-pASV40) into human mesenchymal stem cells (hMSC-TERT) confirmed the vanillic acid-adjustable secreted alkaline phosphatase (SEAP) induction of the designer cascade (>10nM vanillic acid; Fig. 1a). The vanillic acid-repressible gene switch consists of the vanillic acid-dependent transactivator (VanA1), which binds and activates vanillic acid-responsive promoters (for example, P1VanO2) at low and medium vanillic acid levels (<2μM). At high vanillic acid concentrations (>2μM), VanA1 dissociates from P1VanO2, which results in the dose-dependent repression of transgene expression39 (Fig. 1b). The co-transfection of pMG250 (PSV40-VanA1-pASV40) and pMG252 (P1VanO2-SEAP-pASV40) into hMSC-TERT corroborated the fine-tuning of the vanillic acid-repressible SEAP expression (Fig. 1b).
Figure 1: Design of a vanillic acid-responsive positive band-pass filter providing an OFF-ON-OFF expression profile.
a) Vanillic acid-inducible transgene expression. The constitutively expressed vanillic acid-sensitive olfactory G protein-coupled receptor MOR9-1 (pCI-MOR9-1; PhCMV-MOR9-1-pA) senses extracellular vanillic acid levels and triggers G protein (Gs)-mediated activation of the membrane-bound adenylyl cyclase (AC) that converts ATP into cyclic AMP (cAMP). The resulting intracellular cAMP surge activates PKA (protein kinase A), whose catalytic subunits translocate into the nucleus to phosphorylate cAMP response element-binding protein 1 (CREB1). Activated CREB1 binds to synthetic promoters (PCRE) containing cAMP-response elements (CRE) and induces PCRE-driven expression of human placental secreted alkaline phosphatase (SEAP; pCK53, PCRE-SEAP-pA). Co-transfection of pCI-MOR9-1 and pCK53 into human mesenchymal stem cells (hMSC-TERT) grown for 48h in the presence of increasing vanillic acid concentrations results in a dose-inducible SEAP expression profile. (b) Vanillic acid-repressible transgene expression. The constitutively expressed, vanillic acid-dependent transactivator VanA1(pMG250, PSV40-VanA1-pA, VanA1, VanR-VP16) binds and activates the chimeric promoter P1VanO2 (pMG252, P1VanO2-SEAP-pA) in the absence of vanillic acid. In the presence of increasing vanillic acid concentrations, VanA1 is released from P1VanO2, and transgene expression is shut down. Co-transfection of pMG250 and pMG252 into hMSC-TERT grown for 48h in the presence of increasing vanillic acid concentrations results in a dose-repressible SEAP expression profile. (c) Positive band-pass expression filter. Serial interconnection of the synthetic vanillic acid-inducible signalling cascade (a) with the vanillic acid-repressible transcription factor-based gene switch (b) by PCRE-mediated expression of VanA1 (pSP1, PCRE-VanA1-pA) results in a two-level feed-forward cascade. Owing to the opposing responsiveness and differential sensitivity to vanillic acid, this synthetic gene network programmes SEAP expression with a positive band-pass filter profile (OFF-ON-OFF) as vanillic acid levels are increased. Medium vanillic acid levels activate MOR9-1, which induces PCRE-driven VanA1 expression. VanA1remains active and triggers P1VanO2-mediated SEAP expression in feed-forward manner, which increases to maximum levels. At high vanillic acid concentrations, MOR9-1 maintains PCRE-driven VanA1 expression, but the transactivator dissociates from P1VanO2, which shuts SEAP expression down. Co-transfection of pCI-MOR9-1, pSP1 and pMG252 into hMSC-TERT grown for 48h in the presence of increasing vanillic acid concentrations programmes SEAP expression with a positive band-pass profile (OFF-ON-OFF). Data are the means±s.d. of triplicate experiments (n=9).
The opposing responsiveness and differential sensitivity of the control devices to vanillic acid are essential to programme band-pass filter expression profiles. Upon daisy-chaining the designer cascade (pCI-MOR9-1; PhCMV-MOR9-1-pASV40; pSP1, PCRE-VanA1-pASV40) and the gene switch (pSP1, PCRE-VanA1-pASV40; pMG252, P1VanO2-SEAP-pASV40) in the same cell, the network executes a band-pass filter SEAP expression profile when exposed to increasing concentrations of vanillic acid (Fig. 1c). Medium vanillic acid levels (10nM to 2μM) activate MOR9-1, which induces PCRE-driven VanA1 expression. VanA1 remains active within this concentration range and, in a feed-forward amplifier manner, triggers P1VanO2-mediated SEAP expression, which gradually increases to maximum levels (Fig. 1c). At high vanillic acid concentrations (2μM to 400μM), MOR9-1 maintains PCRE-driven VanA1 expression, but the transactivator is inactivated and dissociates from P1VanO2, which results in the gradual shutdown of SEAP expression (Fig. 1c).
For the design of the vanillic acid-programmable synthetic lineage-control network, constitutive MOR9-1 expression and PCRE-driven VanA1 expression were combined with pSP12 (pASV40-Ngn3cm←P3VanO2mFT-miR30Pdx1g-shRNA-pASV40) for endocrine specification and pSP17(PCREm-Pdx1cm-2A-MafAcm-pASV40) for maturation of developing beta-cells (Fig. 2a,b). ThepSP12-encoded expression unit enables the VanA1-controlled induction of the optimized bidirectional vanillic acid-responsive promoter (P3VanO2) that drives expression of a codon-modified Ngn3cm, the nucleic acid sequence of which is distinct from its genomic counterpart (Ngn3g) to allow for quantitative reverse transcription–PCR (qRT–PCR)-based discrimination. In the opposite direction, P3VanO2 transcribes miR30Pdx1g-shRNA, which exclusively targets genomicPdx1 (Pdx1g) transcripts for RNA interference-based destruction and is linked to the production of a blue-to-red medium fluorescent timer40 (mFT) for precise visualization of the unit’s expression dynamics in situ.pSP17 contains a dicistronic expression unit in which the modified high-tightness and lower-sensitivity PCREm promoter (see below) drives co-cistronic expression of Pdx1cm andMafAcm, which are codon-modified versions producing native transcription factors that specifically differ from their genomic counterparts (Pdx1g, MafAg) in their nucleic acid sequence. After individual validation of the vanillic acid-controlled expression and functionality of all network components (Supplementary Figs 2–9), the lineage-control network was ready to be transfected into hIPSC-derived pancreatic progenitor cells. These cells are characterized by high expression of Pdx1g and Nkx6.1 levels and the absence of Ngn3g and MafAg production32, 33, 34 (day 0:Supplementary Figs 10–16).
(a) Schematic of the synthetic lineage-control network. The constitutively expressed, vanillic acid-sensitive olfactory G protein-coupled receptor MOR9-1 (pCI-MOR9-1; PhCMV-MOR9-1-pA) senses extracellular vanillic acid levels and triggers a synthetic signalling cascade, inducing PCRE-driven expression of the transcription factor VanA1 (pSP1, PCRE-VanA1-pA). At medium vanillic acid concentrations (purple arrows), VanA1 binds and activates the bidirectional vanillic acid-responsive promoter P3VanO2 (pSP12, pA-Ngn3cm←P3VanO2mFT-miR30Pdx1g-shRNA-pA), which drives the induction of codon-modified Neurogenin 3 (Ngn3cm) as well as the coexpression of both the blue-to-red medium fluorescent timer (mFT) for precise visualization of the unit’s expression dynamics and miR30pdx1g-shRNA (a small hairpin RNA programming the exclusive destruction of genomic pancreatic and duodenal homeobox 1 (Pdx1g) transcripts). Consequently, Ngn3cm levels switch from low to high (OFF-to-ON), and Pdx1g levels toggle from high to low (ON-to-OFF). In addition, Ngn3cm triggers the transcription of Ngn3g from its genomic promoter, which initiates a positive-feedback loop. At high vanillic acid levels (orange arrows), VanA1 is inactivated, and both Ngn3cm and miR30pdx1g-shRNA are shut down. At the same time, the MOR9-1-driven signalling cascade induces the modified high-tightness and lower-sensitivity PCREm promoter that drives the co-cistronic expression of the codon-modified variants of Pdx1 (Pdx1cm) and V-maf musculoaponeurotic fibrosarcoma oncogene homologue A (MafAcm; pSP17, PCREm-Pdx1cm-2A-MafAcm-pA). Consequently, Pdx1cm and MafAcm become fully induced. As Pdx1cm expression ramps up, it initiates a positive-feedback loop by inducing the genomic counterparts Pdx1g and MafAg. Importantly, Pdx1cm levels are not affected by miR30Pdx1g-shRNA because the latter is specific for genomic Pdx1g transcripts and because the positive feedback loop-mediated amplification of Pdx1gexpression becomes active only after the shutdown of miR30Pdx1g-shRNA. Overall, the synthetic lineage-control network provides vanillic acid-programmable, transient, mutually exclusive expression switches for Ngn3 (OFF-ON-OFF) and Pdx1 (ON-OFF-ON) as well as the concomitant induction of MafA (OFF-ON) expression, which can be followed in real time (Supplementary Movies 1 and 2). (b) Schematic illustrating the individual differentiation steps from human IPSCs towards beta-like cells. The colours match the cell phenotypes reached during the individual differentiation stages programmed by the lineage-control network shown in a.
Following the co-transfection of pCI-MOR9-1 (PhCMV-MOR9-1-pASV40), pSP1 (PCRE-VanA1-pASV40), pSP12 (pASV40-Ngn3cm←P3VanO2mFT-miR30Pdx1g-shRNA-pASV40) and pSP17(PCREm-Pdx1cm-2A-MafAcm-pASV40) into hIPSC-derived pancreatic progenitor cells, the synthetic lineage-control network should override random endogenous differentiation activities and execute the pancreatic beta-cell-specific differentiation programme in a vanillic acid remote-controlled manner. To confirm that the lineage-control network operates as programmed, we cultivated network-containing and pEGFP-N1-transfected (negative-control) cells for 4 days at medium (2μM) and then 7 days at high (400μM) vanillic acid concentrations and profiled the differential expression dynamics of all of the network components and their genomic counterparts as well as the interrelated transcription factors and hormones in both whole populations and individual cells at days 0, 4, 11 and 14 (Figs 2 and 3 and Supplementary Figs 11–17).
Figure 3: Dynamics of the lineage-control network.
(a,b) Quantitative RT–PCR-based expression profiling of the pancreatic transcription factors Ngn3cm/g, Pdx1cm/g and MafAcm/g in hIPSC-derived pancreatic progenitor cells containing the synthetic lineage-control network at days 4 and 11. Data are the means±s.d. of triplicate experiments (n=9). (c–g) Immunocytochemistry of pancreatic transcription factors Ngn3cm/g, Pdx1cm/g and MafAcm/g in hIPSC-derived pancreatic progenitor cells containing the synthetic lineage-control network at days 4 and 11. hIPSC-derived pancreatic progenitor cells were co-transfected with the lineage-control vectors pCI-MOR9-1 (PhCMV-MOR9-1-pA), pSP1 (PCRE-VanA1-pA), pSP12 (pA-Ngn3cm←P3VanO2mFT-miR30Pdx1g-shRNA-pA) and pSP17 (PCREm-Pdx1cm-2A-MafAcm) and immunocytochemically stained for (c) VanA1 and Pdx1 (day 4), (d) VanA1 and Ngn3 (day 4), (e) VanA1 and Pdx1 (day 11), (f) MafA and Pdx1 (day 11) as well as (g) VanA1 and insulin (C-peptide) (day 11). The cells staining positive for VanA1 are containing the lineage-control network. DAPI, 4′,6-diamidino-2-phenylindole. Scale bar, 100μm.
…….
Multicellular organisms, including humans, consist of a highly structured assembly of a multitude of specialized cell phenotypes that originate from the same zygote and have traversed a preprogrammed multifactorial developmental plan that orchestrates sequential differentiation steps with high precision in space and time19, 51. Because of the complexity of terminally differentiated cells, the function of damaged tissues can for most medical indications only be restored via the transplantation of donor material, which is in chronically short supply52.
Despite significant progress in regenerative medicine and the availability of stem cells, the design of protocols that replicate natural differentiation programmes and provide fully functional cell mimetics remains challenging29, 53. For example, efforts to generate beta-cells from human embryonic stem cells (hESCs) have led to reliable protocols involving the sequential administration of growth factors (activin A, bone morphogenetic protein 4 (BMP-4), basic fibroblast growth factor (bFGF), FGF-10, Noggin, vascular endothelial growth factor (VEGF) and Wnt3A) and small-molecule compounds (cyclopamine, forskolin, indolactam V, IDE1, IDE2, nicotinamide, retinoic acid, SB−431542 and γ-secretase inhibitor) that modulate differentiation-specific signalling pathways31, 54, 55. In vitro differentiation of hESC-derived pancreatic progenitor cells into beta-like cells is more challenging and has been achieved recently by a complex media formulation with chemicals and growth factors32, 33, 34.
hIPSCs have become a promising alternative to hESCs; however, their use remains restricted in many countries56. Most hIPSCs used for directed differentiation studies were derived from a juvenescent cell source that is expected to show a higher degree of differentiation potential compared with older donors that typically have a higher need for medical interventions37, 57, 58. We previously succeeded in producing mRNA-reprogrammed hIPSCs from adipose tissue-derived mesenchymal stem cells of a 50-year-old donor, demonstrating that the reprogramming of cells from a donor of advanced age is possible in principle59.
Recent studies applying similar hESC-based differentiation protocols to hIPSCs have produced cells that release insulin in response to high glucose32, 33, 34. This observation suggests that functional beta-like cells can eventually be derived from hIPSCs32, 33. In our hands, the growth-factor/chemical-based technique for differentiating human IPSCs resulted in beta-like cells with poor glucose responsiveness. Recent studies have revealed significant variability in the lineage specification propensity of different hIPSC lines35, 60 and substantial differences in the expression profiles of key transcription factors in hIPSC-derived beta-like cells33. Therefore, the growth-factor/chemical-based protocols may require further optimization and need to be customized for specific hIPSC lines35. Synthetic lineage-control networks providing precise dynamic control of transcription factor expression may overcome the challenges associated with the programming of beta-like cells from different hIPSC lines.
Rather than exposing hIPSCs to a refined compound cocktail that triggers the desired differentiation in a fraction of the stem cell population, we chose to design a synthetic lineage-control network to enable single input-programmable differentiation of hIPSC-derived pancreatic progenitor cells into glucose-sensitive insulin-secreting beta-like cells. In contrast with the use of growth-factor/chemical-based cocktails, synthetic lineage-control networks are expected to (i) be more economical because of in situ production of the required transcription factors, (ii) enable simultaneous control of ectopic and chromosomally encoded transcription factor variants, (iii) tap into endogenous pathways and not be limited to cell-surface input, (iv) display improved reversibility that is not dependent on the removal of exogenous growth factors via culture media replacement, (v) provide lateral inhibition, thereby reducing the random differentiation of neighbouring cells and (vi) enable trigger-programmable and (vii) precise differential transcription factor expression switches.
The synthetic lineage-control network that precisely replicates the endogenous relative expression dynamics of the transcription factors Pdx-1, Ngn3 and MafA required the design of a new network topology that interconnects a synthetic signalling cascade and a gene switch with differential and opposing sensitivity to the food additive vanillic acid. This differentiation device provides different band-pass filter, time-delay and feed-forward amplifier topologies that interface with endogenous positive-feedback loops to orchestrate the timely expression and repression of heterologous and chromosomally encoded Ngn3, Pdx1 and MafA variants. The temporary nature of the engineering intervention, which consists of transient transfection of the genetic lineage-control components in the absence of any selection, is expected to avoid stable modification of host chromosomes and alleviate potential safety concerns. In addition, the resulting beta-cell mass could be encapsulated inside vascularized microcontainers28, a proven containment strategy in prototypic cell-based therapies currently being tested in animal models of prominent human diseases14, 15, 16, 61, 62 as well as in human clinical trials28.
The hIPSC-derived beta-like cells resulting from this trigger-induced synthetic lineage-control network exhibited glucose-stimulated insulin-release dynamics and capacity matching the human physiological range and transcriptional profiling, flow cytometric analysis and electron microscopy corroborated the lineage-controlled stem cells reached a mature beta-cell phenotype. In principle, the combination of hIPSCs derived from the adipose tissue of a 50-year-old donor59 with a synthetic lineage-control network programming glucose-sensitive insulin-secreting beta-like cells closes the design cycle of regenerative medicine63. However, hIPSCs that are derived from T1DM patients, differentiated into beta-like cells and transplanted back into the donor would still be targeted by the immune system, as demonstrated in the transplantation of segmental pancreatic grafts from identical twins64. Therefore, any beta-cell-replacement therapy will require complementary modulation of the immune system either via drugs30, 65, engineering or cell-based approaches66, 67 or packaging inside vascularizing, semi-permeable immunoprotective microcontainers28.
Capitalizing on the design principles of synthetic biology, we have successfully constructed and validated a synthetic lineage-control network that replicates the differential expression dynamics of critical transcription factors and mimicks the native differentiation pathway to programme hIPSC-derived pancreatic progenitor cells into glucose-sensitive insulin-secreting beta-like cells that compare with human pancreatic islets at a high level. The design of input-triggered synthetic lineage-control networks that execute a preprogrammed sequential differentiation agenda coordinating the timely induction and repression of multiple genes could provide a new impetus for the advancement of developmental biology and regenerative medicine.
Other related articles published in this Open Access Online Scientific Journal include the following:
Adipocyte Derived Stroma Cells: Their Usage in Regenerative Medicine and Reprogramming into Pancreatic Beta-Like Cells
Genomics and epigenetics link to DNA structure, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 1: Next Generation Sequencing (NGS)
Genomics and epigenetics link to DNA structure
Larry H. Bernstein, MD, FCAP, Curator
LPBI
Sequence and Epigenetic Factors Determine Overall DNA Structure
Atomic-level simulations show electrostatic forces between each atom. [Alek Aksimentiev, University of Illinois at Urbana-Champaign]
The traditionally held hypothesis about the highly ordered organization of DNA describes the interaction of various proteins with DNA sequences to mediate the dynamic structure of the molecule. However, recent evidence has emerged that stretches of homologous DNA sequences can associate preferentially with one another, even in the absence of proteins.
Researchers at the University of Illinois Center for the Physics of Living Cells, Johns Hopkins University, and Ulsan National Institute of Science and Technology (UNIST) in South Korea found that DNA molecules interact directly with one another in ways that are dependent on the sequence of the DNA and epigenetic factors, such as methylation.
The researchers described evidence they found for sequence-dependent attractive interactions between double-stranded DNA molecules that neither involve intermolecular strand exchange nor are mediated by DNA-binding proteins.
“DNA molecules tend to repel each other in water, but in the presence of special types of cations, they can attract each other just like nuclei pulling each other by sharing electrons in between,” explained lead study author Hajin Kim, Ph.D., assistant professor of biophysics at UNIST. “Our study suggests that the attractive force strongly depends on the nucleic acid sequence and also the epigenetic modifications.”
The investigators used atomic-level supercomputer simulations to measure the forces between a pair of double-stranded DNA helices and proposed that the distribution of methyl groups on the DNA was the key to regulating this sequence-dependent attraction. To verify their findings experimentally, the scientists were able to observe a single pair of DNA molecules within nanoscale bubbles.
“Here we combine molecular dynamics simulations with single-molecule fluorescence resonance energy transfer experiments to examine the interactions between duplex DNA in the presence of spermine, a biological polycation,” the authors wrote. “We find that AT-rich DNA duplexes associate more strongly than GC-rich duplexes, regardless of the sequence homology. Methyl groups of thymine act as a steric block, relocating spermine from major grooves to interhelical regions, thereby increasing DNA–DNA attraction.”
The findings from this study were published recently in Nature Communications in an article entitled “Direct Evidence for Sequence-Dependent Attraction Between Double-Stranded DNA Controlled by Methylation.”
After conducting numerous further simulations, the research team concluded that direct DNA–DNA interactions could play a central role in how chromosomes are organized in the cell and which ones are expanded or folded up compactly, ultimately determining functions of different cell types or regulating the cell cycle.
“Biophysics is a fascinating subject that explores the fundamental principles behind a variety of biological processes and life phenomena,” Dr. Kim noted. “Our study requires cross-disciplinary efforts from physicists, biologists, chemists, and engineering scientists and we pursue the diversity of scientific disciplines within the group.”
Dr. Kim concluded by stating that “in our lab, we try to unravel the mysteries within human cells based on the principles of physics and the mechanisms of biology. In the long run, we are seeking for ways to prevent chronic illnesses and diseases associated with aging.”
Direct evidence for sequence-dependent attraction between double-stranded DNA controlled by methylation
Jejoong Yoo, Hajin Kim, Aleksei Aksimentiev, and Taekjip Ha Nature Communications 7 11045 (2016) DOI:10.1038/ncomms11045BibTex
Although proteins mediate highly ordered DNA organization in vivo, theoretical studies suggest that homologous DNA duplexes can preferentially associate with one another even in the absence of proteins. Here we combine molecular dynamics simulations with single-molecule fluorescence resonance energy transfer experiments to examine the interactions between duplex DNA in the presence of spermine, a biological polycation. We find that AT-rich DNA duplexes associate more strongly than GC-rich duplexes, regardless of the sequence homology. Methyl groups of thymine acts as a steric block, relocating spermine from major grooves to interhelical regions, thereby increasing DNA–DNA attraction. Indeed, methylation of cytosines makes attraction between GC-rich DNA as strong as that between AT-rich DNA. Recent genome-wide chromosome organization studies showed that remote contact frequencies are higher for AT-rich and methylated DNA, suggesting that direct DNA–DNA interactions that we report here may play a role in the chromosome organization and gene regulation.
Formation of a DNA double helix occurs through Watson–Crick pairing mediated by the complementary hydrogen bond patterns of the two DNA strands and base stacking. Interactions between double-stranded (ds)DNA molecules in typical experimental conditions containing mono- and divalent cations are repulsive1, but can turn attractive in the presence of high-valence cations2. Theoretical studies have identified the ion–ion correlation effect as a possible microscopic mechanism of the DNA condensation phenomena3, 4, 5. Theoretical investigations have also suggested that sequence-specific attractive forces might exist between two homologous fragments of dsDNA6, and this ‘homology recognition’ hypothesis was supported by in vitro atomic force microscopy7 and in vivo point mutation assays8. However, the systems used in these measurements were too complex to rule out other possible causes such as Watson–Crick strand exchange between partially melted DNA or protein-mediated association of DNA.
Here we present direct evidence for sequence-dependent attractive interactions between dsDNA molecules that neither involve intermolecular strand exchange nor are mediated by proteins. Further, we find that the sequence-dependent attraction is controlled not by homology—contradictory to the ‘homology recognition’ hypothesis6—but by a methylation pattern. Unlike the previous in vitro study that used monovalent (Na+) or divalent (Mg2+) cations7, we presumed that for the sequence-dependent attractive interactions to operate polyamines would have to be present. Polyamine is a biological polycation present at a millimolar concentration in most eukaryotic cells and essential for cell growth and proliferation9, 10. Polyamines are also known to condense DNA in a concentration-dependent manner2, 11. In this study, we use spermine4+(Sm4+) that contains four positively charged amine groups per molecule.
Sequence dependence of DNA–DNA forces
To characterize the molecular mechanisms of DNA–DNA attraction mediated by polyamines, we performed molecular dynamics (MD) simulations where two effectively infinite parallel dsDNA molecules, 20 base pairs (bp) each in a periodic unit cell, were restrained to maintain a prescribed inter-DNA distance; the DNA molecules were free to rotate about their axes. The two DNA molecules were submerged in 100mM aqueous solution of NaCl that also contained 20 Sm4+molecules; thus, the total charge of Sm4+, 80 e, was equal in magnitude to the total charge of DNA (2 × 2 × 20 e, two unit charges per base pair; Fig. 1a). Repeating such simulations at various inter-DNA distances and applying weighted histogram analysis12 yielded the change in the interaction free energy (ΔG) as a function of the DNA–DNA distance (Fig. 1b,c). In a broad agreement with previous experimental findings13, ΔG had a minimum, ΔGmin, at the inter-DNA distance of 25−30Å for all sequences examined, indeed showing that two duplex DNA molecules can attract each other. The free energy of inter-duplex attraction was at least an order of magnitude smaller than the Watson–Crick interaction free energy of the same length DNA duplex. A minimum of ΔG was not observed in the absence of polyamines, for example, when divalent or monovalent ions were used instead14, 15.
Figure 1: Polyamine-mediated DNA sequence recognition observed in MD simulations and smFRET experiments.
(a) Set-up of MD simulations. A pair of parallel 20-bp dsDNA duplexes is surrounded by aqueous solution (semi-transparent surface) containing 20 Sm4+ molecules (which compensates exactly the charge of DNA) and 100mM NaCl. Under periodic boundary conditions, the DNA molecules are effectively infinite. A harmonic potential (not shown) is applied to maintain the prescribed distance between the dsDNA molecules. (b,c) Interaction free energy of the two DNA helices as a function of the DNA–DNA distance for repeat-sequence DNA fragments (b) and DNA homopolymers (c). (d) Schematic of experimental design. A pair of 120-bp dsDNA labelled with a Cy3/Cy5 FRET pair was encapsulated in a ~200-nm diameter lipid vesicle; the vesicles were immobilized on a quartz slide through biotin–neutravidin binding. Sm4+ molecules added after immobilization penetrated into the porous vesicles. The fluorescence signals were measured using a total internal reflection microscope. (e) Typical fluorescence signals indicative of DNA–DNA binding. Brief jumps in the FRET signal indicate binding events. (f) The fraction of traces exhibiting binding events at different Sm4+ concentrations for AT-rich, GC-rich, AT nonhomologous and CpG-methylated DNA pairs. The sequence of the CpG-methylated DNA specifies the methylation sites (CG sequence, orange), restriction sites (BstUI, triangle) and primer region (underlined). The degree of attractive interaction for the AT nonhomologous and CpG-methylated DNA pairs was similar to that of the AT-rich pair. All measurements were done at [NaCl]=50mM and T=25°C. (g) Design of the hybrid DNA constructs: 40-bp AT-rich and 40-bp GC-rich regions were flanked by 20-bp common primers. The two labelling configurations permit distinguishing parallel from anti-parallel orientation of the DNA. (h) The fraction of traces exhibiting binding events as a function of NaCl concentration at fixed concentration of Sm4+ (1mM). The fraction is significantly higher for parallel orientation of the DNA fragments.
Unexpectedly, we found that DNA sequence has a profound impact on the strength of attractive interaction. The absolute value of ΔG at minimum relative to the value at maximum separation, |ΔGmin|, showed a clearly rank-ordered dependence on the DNA sequence: |ΔGmin| of (A)20>|ΔGmin| of (AT)10>|ΔGmin| of (GC)10>|ΔGmin| of (G)20. Two trends can be noted. First, AT-rich sequences attract each other more strongly than GC-rich sequences16. For example, |ΔGmin| of (AT)10 (1.5kcalmol−1 per turn) is about twice |ΔGmin| of (GC)10 (0.8kcalmol−1 per turn) (Fig. 1b). Second, duplexes having identical AT content but different partitioning of the nucleotides between the strands (that is, (A)20 versus (AT)10 or (G)20 versus (GC)10) exhibit statistically significant differences (~0.3kcalmol−1 per turn) in the value of |ΔGmin|.
To validate the findings of MD simulations, we performed single-molecule fluorescence resonance energy transfer (smFRET)17 experiments of vesicle-encapsulated DNA molecules. Equimolar mixture of donor- and acceptor-labelled 120-bp dsDNA molecules was encapsulated in sub-micron size, porous lipid vesicles18 so that we could observe and quantitate rare binding events between a pair of dsDNA molecules without triggering large-scale DNA condensation2. Our DNA constructs were long enough to ensure dsDNA–dsDNA binding that is stable on the timescale of an smFRET measurement, but shorter than the DNA’s persistence length (~150bp (ref. 19)) to avoid intramolecular condensation20. The vesicles were immobilized on a polymer-passivated surface, and fluorescence signals from individual vesicles containing one donor and one acceptor were selectively analysed (Fig. 1d). Binding of two dsDNA molecules brings their fluorescent labels in close proximity, increasing the FRET efficiency (Fig. 1e).
FRET signals from individual vesicles were diverse. Sporadic binding events were observed in some vesicles, while others exhibited stable binding; traces indicative of frequent conformational transitions were also observed (Supplementary Fig. 1A). Such diverse behaviours could be expected from non-specific interactions of two large biomolecules having structural degrees of freedom. No binding events were observed in the absence of Sm4+ (Supplementary Fig. 1B) or when no DNA molecules were present. To quantitatively assess the propensity of forming a bound state, we chose to use the fraction of single-molecule traces that showed any binding events within the observation time of 2min (Methods). This binding fraction for the pair of AT-rich dsDNAs (AT1, 100% AT in the middle 80-bp section of the 120-bp construct) reached a maximum at ~2mM Sm4+(Fig. 1f), which is consistent with the results of previous experimental studies2, 3. In accordance with the prediction of our MD simulations, GC-rich dsDNAs (GC1, 75% GC in the middle 80bp) showed much lower binding fraction at all Sm4+ concentrations (Fig. 1b,c). Regardless of the DNA sequence, the binding fraction reduced back to zero at high Sm4+ concentrations, likely due to the resolubilization of now positively charged DNA–Sm4+ complexes2, 3, 13.
Because the donor and acceptor fluorophores were attached to the same sequence of DNA, it remained possible that the sequence homology between the donor-labelled DNA and the acceptor-labelled DNA was necessary for their interaction6. To test this possibility, we designed another AT-rich DNA construct AT2 by scrambling the central 80-bp section of AT1 to remove the sequence homology (Supplementary Table 1). The fraction of binding traces for this nonhomologous pair of donor-labelled AT1 and acceptor-labelled AT2 was comparable to that for the homologous AT-rich pair (donor-labelled AT1 and acceptor-labelled AT1) at all Sm4+ concentrations tested (Fig. 1f). Furthermore, this data set rules out the possibility that the higher binding fraction observed experimentally for the AT-rich constructs was caused by inter-duplex Watson–Crick base pairing of the partially melted constructs.
Next, we designed a DNA construct named ATGC, containing, in its middle section, a 40-bp AT-rich segment followed by a 40-bp GC-rich segment (Fig. 1g). By attaching the acceptor to the end of either the AT-rich or GC-rich segments, we could compare the likelihood of observing the parallel binding mode that brings the two AT-rich segments together and the anti-parallel binding mode. Measurements at 1mM Sm4+ and 25 or 50mM NaCl indicated a preference for the parallel binding mode by ~30% (Fig. 1h). Therefore, AT content can modulate DNA–DNA interactions even in a complex sequence context. Note that increasing the concentration of NaCl while keeping the concentration of Sm4+ constant enhances competition between Na+ and Sm4+ counterions, which reduces the concentration of Sm4+ near DNA and hence the frequency of dsDNA–dsDNA binding events (Supplementary Fig. 2).
Methylation determines the strength of DNA–DNA attraction
Analysis of the MD simulations revealed the molecular mechanism of the polyamine-mediated sequence-dependent attraction (Fig. 2). In the case of the AT-rich fragments, the bulky methyl group of thymine base blocks Sm4+ binding to the N7 nitrogen atom of adenine, which is the cation-binding hotspot21, 22. As a result, Sm4+ is not found in the major grooves of the AT-rich duplexes and resides mostly near the DNA backbone (Fig. 2a,d). Such relocated Sm4+ molecules bridge the two DNA duplexes better, accounting for the stronger attraction16, 23, 24, 25. In contrast, significant amount of Sm4+ is adsorbed to the major groove of the GC-rich helices that lacks cation-blocking methyl group (Fig. 2b,e).
Figure 2: Molecular mechanism of polyamine-mediated DNA sequence recognition.
(a–c) Representative configurations of Sm4+ molecules at the DNA–DNA distance of 28Å for the (AT)10–(AT)10 (a), (GC)10–(GC)10 (b) and (GmC)10–(GmC)10 (c) DNA pairs. The backbone and bases of DNA are shown as ribbon and molecular bond, respectively; Sm4+ molecules are shown as molecular bonds. Spheres indicate the location of the N7 atoms and the methyl groups. (d–f) The average distributions of cations for the three sequence pairs featured in a–c. Top: density of Sm4+ nitrogen atoms (d=28Å) averaged over the corresponding MD trajectory and the z axis. White circles (20Å in diameter) indicate the location of the DNA helices. Bottom: the average density of Sm4+ nitrogen (blue), DNA phosphate (black) and sodium (red) atoms projected onto the DNA–DNA distance axis (x axis). The plot was obtained by averaging the corresponding heat map data over y=[−10, 10] Å. See Supplementary Figs 4 and 5 for the cation distributions at d=30, 32, 34 and 36Å.
If indeed the extra methyl group in thymine, which is not found in cytosine, is responsible for stronger DNA–DNA interactions, we can predict that cytosine methylation, which occurs naturally in many eukaryotic organisms and is an essential epigenetic regulation mechanism26, would also increase the strength of DNA–DNA attraction. MD simulations showed that the GC-rich helices containing methylated cytosines (mC) lose the adsorbed Sm4+ (Fig. 2c,f) and that |ΔGmin| of (GC)10 increases on methylation of cytosines to become similar to |ΔGmin| of (AT)10 (Fig. 1b).
To experimentally assess the effect of cytosine methylation, we designed another GC-rich construct GC2 that had the same GC content as GC1 but a higher density of CpG sites (Supplementary Table 1). The CpG sites were then fully methylated using M. SssI methyltransferase (Supplementary Fig. 3; Methods). As predicted from the MD simulations, methylation of the GC-rich constructs increased the binding fraction to the level of the AT-rich constructs (Fig. 1f).
The sequence dependence of |ΔGmin| and its relation to the Sm4+ adsorption patterns can be rationalized by examining the number of Sm4+ molecules shared by the dsDNA molecules (Fig. 3a). An Sm4+ cation adsorbed to the major groove of one dsDNA is separated from the other dsDNA by at least 10Å, contributing much less to the effective DNA–DNA attractive force than a cation positioned between the helices, that is, the ‘bridging’ Sm4+ (ref. 23). An adsorbed Sm4+ also repels other Sm4+ molecules due to like-charge repulsion, lowering the concentration of bridging Sm4+. To demonstrate that the concentration of bridging Sm4+ controls the strength of DNA–DNA attraction, we computed the number of bridging Sm4+ molecules, Nspm (Fig. 3b). Indeed, the number of bridging Sm4+ molecules ranks in the same order as |ΔGmin|: Nspm of (A)20>Nspm of (AT)10≈Nspm of (GmC)10>Nspm of (GC)10>Nspm of (G)20. Thus, the number density of nucleotides carrying a methyl group (T and mC) is the primary determinant of the strength of attractive interaction between two dsDNA molecules. At the same time, the spatial arrangement of the methyl group carrying nucleotides can affect the interaction strength as well (Fig. 3c). The number of methyl groups and their distribution in the (AT)10 and (GmC)10 duplex DNA are identical, and so are their interaction free energies, |ΔGmin| of (AT)10≈|ΔGmin| of (GmC)10. For AT-rich DNA sequences, clustering of the methyl groups repels Sm4+ from the major groove more efficiently than when the same number of methyl groups is distributed along the DNA (Fig. 3b). Hence, |ΔGmin| of (A)20>|ΔGmin| of (AT)10. For GC-rich DNA sequences, clustering of the cation-binding sites (N7 nitrogen) attracts more Sm4+ than when such sites are distributed along the DNA (Fig. 3b), hence |ΔGmin| is larger for (GC)10 than for (G)20.
Figure 3: Methylation modulates the interaction free energy of two dsDNA molecules by altering the number of bridging Sm4+.
(a) Typical spatial arrangement of Sm4+ molecules around a pair of DNA helices. The phosphates groups of DNA and the amine groups of Sm4+ are shown as red and blue spheres, respectively. ‘Bridging’ Sm4+molecules reside between the DNA helices. Orange rectangles illustrate the volume used for counting the number of bridging Sm4+ molecules. (b) The number of bridging amine groups as a function of the inter-DNA distance. The total number of Sm4+ nitrogen atoms was computed by averaging over the corresponding MD trajectory and the 10Å (x axis) by 20Å (y axis) rectangle prism volume (a) centred between the DNA molecules. (c) Schematic representation of the dependence of the interaction free energy of two DNA molecules on their nucleotide sequence. The number and spatial arrangement of nucleotides carrying a methyl group (T or mC) determine the interaction free energy of two dsDNA molecules.
Genome-wide investigations of chromosome conformations using the Hi–C technique revealed that AT-rich loci form tight clusters in human nucleus27, 28. Gene or chromosome inactivation is often accompanied by increased methylation of DNA29 and compaction of facultative heterochromatin regions30. The consistency between those phenomena and our findings suggest the possibility that the polyamine-mediated sequence-dependent DNA–DNA interaction might play a role in chromosome folding and epigenetic regulation of gene expression.
Rau, D. C., Lee, B. & Parsegian, V. A.Measurement of the repulsive force between polyelectrolyte molecules in ionic solution: hydration forces between parallel DNA double helices. Proc. Natl Acad. Sci. USA81, 2621–2625 (1984).
Raspaud, E., Olvera de la Cruz, M., Sikorav, J. L. & Livolant, F.Precipitation of DNA by polyamines: a polyelectrolyte behavior. Biophys. J.74, 381–393 (1998).
Grosberg, A. Y., Nguyen, T. T. & Shklovskii, B. I.The physics of charge inversion in chemical and biological systems. Rev. Mod. Phys.74, 329–345 (2002).
Thomas, T. & Thomas, T. J.Polyamines in cell growth and cell death: molecular mechanisms and therapeutic applications. Cell. Mol. Life Sci.58, 244–258 (2001).
Pull at Cancer’s Levers, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 1: Next Generation Sequencing (NGS)
Pull at Cancer’s Levers
Curator: Larry H. Bernstein, MD, FCAP
Driving Cancer Immunotherapy
The Stakes in Immuno-Oncology Are Too High for Researchers to Pull at Cancer’s Levers Blindly. Researchers Need a System.
Within the past decade or so, a revolutionary idea has emerged in the minds of scientists, physicians, and medical experts. Instead of using man-made chemicals to treat cancer, let us instead unleash the power of our own bodies upon the malignancy.
This idea is the inspiration behind cancer immunotherapy, which is, according to most experts, a therapeutic approach that involves training the immune system to fight off cancer. In the words of one expert, cancer immunotherapy means “taking the immune system’s inherent properties and turbo charging those to fight cancer.”
Cancer immunotherapy technologies are being developed to accomplish
several tasks:
Enhance the molecular targeting of cancer cells
Report the rate of killing by specific immune agents
Direct immune cells toward tumor destruction.
Since its inception, the field has evolved, and it continues to do so. It began with in vivo investigations of tumor growth and development, and it progressed through laboratory investigations of cellular morphology and survival curves. And now it is adopting pathway analysis to guide therapeutic development and improve patient care.
To begin to understand cancer immunotherapy, one must understand how the immune system targets tumor cells. One of the prominent adaptive components of the immune system is the T cell, which responds to perceived threats through the massive increase in clonal T cells targeted in some way toward the diseased cell or pathogen.
The T-Cell Repertoire
Adaptive Biotechnologies’ immunoSEQ Assay, a high-throughput research platform for immune system profiling, is designed to generate sequencer-ready libraries using highly optimized primer sets in a multiplex PCR format that targets T- and B-cell receptor genes. This image depicts how the assay’s two-step PCR process can be used to quantify the clonal diversity of immune cells.
Immunologists call this process VJD rearrangement. It happens during T-lymphocyte development and affects three gene regions, the variable (V), the diversity (D), and the joining (J) regions. This rearrangement of the genetic code allow for the structural diversity in T-cell receptors responsible for antigenic specificity including antigenic targets on tumor cells. In the case of cancer, specificity is complicated because the tumor is actually part of the body itself, one of the reasons cancers naturally evade detection.
The specificity problem would always hinder attempts to goad the immune system into attacking cancer, scientists realized, unless technologies emerged that could efficiently track the clonal diversity of T cells inside patients. Existing technologies, such as spectratyping, were inadequate. In 2007, when Dr. Robins and his collaborators began developing the technology, only 10,000 T-cell receptor sequences had been reported in all the literature using older methodologies.
“The immunology field of the time had no connection with high-throughput sequencing,” notes Dr. Robins, recalling his days as a computational biologist for the Fred Hutchinson Cancer Research Center. “It became clear that instead of using this old technology to look at T-cell receptors, we could just directly sequence them—if we could amplify them correctly.”
With its first experiment, Dr. Robins’ team ended up with six million T-cell receptor sequences. “Our approach,” Dr. Robins modestly suggests, “kind of changed the scale of what we were able to do.” The team went on to develop advanced multiplex sequencing technology, doing work that essentially started the field of immune sequencing. “Previously,” maintains Dr. Robins, “no one had ever been able to quantitatively do a multiplex PCR.”
Adaptive Biotechnologies’ product, the ImmunoSEQ® assay, uses several hundred primer pairs to quantify the clonal diversity of T cells. Using this technology, researchers and clinicians can focus on T-cell clones that are expanded specifically in or near a tumor or that are circulating in the blood stream.
“You obviously can’t get a serial sample of the tumor,” explains Dr. Robins, “but you can get serial samples of blood,” allowing for immune cell repertoire tracking during the progression of a disease. The technology is already being used to assess leukemias in the clinic, directly tracking the leukemia itself based on the massive clonal expansion of a single cancerous B or T cell.
Eventually, Dr. Robins’ team hopes to monitor serial changes in T cell clones before, during, and after therapeutic intervention. The team has even developed a tumor infiltrating lymphocyte (TIL) assay to examine clones that are attracted to tumors.
Circulating Tumor Cells
“Years ago, they were just interested in what was happening in the tumor,” says Daniel Adams, senior research scientist at Creatv MicroTech. “Now people have realized that the immune system is reacting to the tumor.”
Scientists such as Adams have been tracking tumor cells and tumor-modified stromal cells, as well as components of the non-adaptive immune system, directly within the bloodstream to examine changes that occur over time.
“We can’t go back in to re-biopsy the patient every year, or every time there is a recurrence,” says Adams, “It’s just not feasible.”
That is why Creatv MicroTech, with locations in Maryland and New Jersey, has developed the CellSieve, a mechanical cell filter. The CellSieve, which improves on older technology through better polymers and engineering, isolates circulating tumor cells (CTCs) and stromal cells in order to capture them for further clinical analysis.
Isolation, culture and expansion of cells isolated on CellSieve™. (A) MCF-7 cells spiked into vacutainers, isolated by filtration and cultured on CellSieve for 2-3 weeks. A 3 dimensional cluster attributed to this cell line is seen on the filter. (green=anti-cytokeratin, blue=DAPI) (B) PANC-1 cells spiked into vacutainers, isolated by filtration and grown on CellSieve for 2-3 weeks. PANC-1 is seen growing as a monolayer on the filter. (C) SKBR3 cells are spiked into blood, filtered by CellSieve. The CTCs are identified by presence of anti-cytokeratin and anti-EpCAM, and absence of anti-CD45. After CTCs are counted, cells are subtyped by HER2 FISH. (D) SKBR3 cells are spiked into vacutainers, isolated by filtration and grown on CellSieve for 2-3 weeks. Expanded colonies were directly analyzed as a whole colony and as individual cells, molecularly by HER2/CR17 FISH probes. (E) Circulating stromal cell, e.g. a 70 µm giant cancer associated macrophage can be identified for clinical use, myeloid marker in red. (F) A cell of interest can be identified and restained with immunotherapeutic biomarkers, e.g. PD-L1 (green) and PD-1 (purple). (G) After filtration, cells were identified with histopathological stains (e.g. H&E) for cytological analysis. (H) After H&E, external cell structures were analyzed by SEM. [Creatv MicroTech].
“As a patient goes through therapy, the patient’s resistance builds, and the cancer recurs in different subpopulations,” states Adams. “And after a few years, the original tumor mass is no longer applicable to what is growing in the patient farther down the road.”
Although CTCs are exceedingly rare in the bloodstream, with just one or two in every 5 to 10 mL of blood, and although these cells have a very low viability, the surviving CTCs have a high prognostic value.
“We looked at 30 to 40 breast cancer patients over two years,” reports Adams. “And we showed that if you have a dividing CTC, you have a 90% chance of dying in two years and a 100% chance of dying within two and half years.”
Furthermore, the immune system response can be tracked, says Adams, by examining stromal cells, which can also be collected with the CellSieve filtration device. That is, these cells can be collected serially. Much recent evidence supports the conclusion that stromal cells in the tumor environment co-evolve with the tumor, suggesting that stromal marker changes reflect tumor changes.
“There is this plethora of stromal cells and tumor cells out there in the circulation for you to look at,” declares Adams. “Once the cells are isolated, you can subject them to pathological approaches, biomarker approaches, or molecular approaches—or all of the above.”
A MicroTech Creatv study published in the Royal Society of Chemistry showed the efficacy of following up CTC isolation with techniques such as fluorescence in situ hybridization (FISH), histopathological analysis, and cell culture.
Cancer-Killing Assays
Diverse mechanisms are at play in cancer biology. Our understanding of these mechanisms contributes to a couple of virtuous cycles. It strengthens and is strengthened by diagnostic approaches, such as immune- and tumor-cell monitoring. The same could be said of therapeutic approaches. Cancer biology will inform and be informed by cancer immunotherapies such as adoptive cell transfer. To maintain the virtuous cycle, however, it will be necessary to conduct in vitro testing.
“There is no doubt that immunotherapy is going to play a major role in the treatment of cancer,” says Brandon Lamarche, Ph.D., technical communicator and scientist at ACEA Biosciences. “Regardless of what the route is, what is going to have to happen in terms of the research area is that you need an effective cell-killing assay.”
ACEA Biosciences, a San Diego-based company, has developed a microtiter plate that is coated with gold electrodes across 75% of the well bottoms. When the microtiter plates are placed in the company’s xCELLigence plate reader, the electrodes enable the detection of changes in cell morphology and viability through electrical impedance.
“The instrument provides a weak electric potential to the electrodes on the plate, so you get electrons flowing between these electrodes,” explains Dr. Lamarche. Researchers can then apply reagents or non-adherent immune cell suspensions to adherent cancer cells and examine the effect.
Dr. Lamarche asserts that the xCELLigence system overcomes problems that bedevil competing cell-killing assays. These problems include leaky and radioactive labels, such as chromium 51, and assays that can only provide users with an endpoint for cell killing. “With xCELLigence,” he insists, “you’re getting the full spectrum of what’s happening, and there’s all kinds of subtleties in the cell-killing curves that are very informative in terms of the biology.”
ACEA would like to see the xCELLigence system become the new standard in cell-killing assays from standard research to clinical testing on patient tumors. Dr. Lamarche envisions a day when patient tumor cells are quickly screened with therapeutic scenarios to determine the most efficacious killing option. “xCELLigence technology,” he suggests, “enables you to quickly sample a broad spectrum of conditions with a very simple workflow.”
Bioinformatics of Immuno-Oncology
From monitoring to treatment modalities, the field of cancer immunotherapy is aided by bioinformatics-minded data-mining experts, such as the analysts at Thompson-Reuters who are compiling data archives and applying advanced analytics to find new targets. “Essentially,” says Richard Harrison Ph.D., the company’s chief scientific officer for the life science division, “for every stage within pharmaceutical drug development, we have a database associated with that.”
The analysts at Thompson-Reuters curate and compile databases such as MedaCore and Cortellis, which they provide to their clients to help them with their research and clinical studies. “We can take customer data, and using our tools and our pathway maps, we can help them understand what their data is telling them,” explains Dr. Harrison.
Matt Wampole, Ph.D., a solutions scientist at Thompson-Reuters, spends his days reaching out and working with customers to help them understand and better use the company’s products. “Bench researchers,” he points out, “don’t necessarily know what is upstream of whatever expression change might be leading to a particular change in regulation.” Dr. Wampole indicates that he is part of a “solution team” that aids clients in determining important signaling cascades, regulators, and so on.
“We have a group of individuals who are very ‘skilling’ experts in the field,” Dr. Wampole continues, “including experts in the areas such as biostatistics, data curation, and data analytics. These experts help clients identify models, stratify patients, understand mechanisms, and look into disease mechanisms.”
Dr. Harrison sums up the Thompson-Reuters approach as follows: “We look for master regulators that can serve as both targets and biomarkers.” By examining the gene signatures from both the patient and from curated datasets, in the case of cancer immunotherapy, they hope to segregate patients according to what drugs will work best for them.
“We are working with a number of pharmaceutical companies to put our approach into practice for clinical trials,” informs Dr. Harrison. The approach has already been applied in several studies, including one that used data analysis of cell lines to help predict drug response in patients. Another study helped stratify glioblastoma patients.
Tumor-Targeted Delivery Platform
PsiOxus Therapeutics, which is focused on immune therapeutics in oncology, has developed a patented platform for tumor-targeted delivery based on its oncolytic vaccine, Enadenotucirev (EnAd), which can be delivered systemically via intravenous administration.
According to company officials, EnAd’s anti-cancer scope can be expanded by adding new genes, thereby enabling the creation of a broad range of unique immuno-oncology therapeutics. In a recent study conducted at the University of Oxford, researchers led by Philip G. Jakeman, Ph.D., sought to improve the models for evaluating cancer therapeutics by introducing ex vivo methodologies for research into colorectal cancer.
The ex vivo approach utilized was able to exploit a major advantage by preserving the three-dimensional architecture of the tumor and its associated compartments, including immune cells. The study, which was presented at the International Summit on Oncolytic Viral Therapeutics in Quebec, showed the tissue slice model can provide a novel means to assessing an oncolytic vaccine in a system that more accurately recapitulates human tumors, provide a more stringent test for oncolytic viruses, such as EnAd, and allow study of the human immune cells within the tumor 3D context.
By maintaining the components of the tumor immune microenvironment, this new methodology could become useful in analyzing anti-viral responses within tumors, or even in evaluating therapeutics that target immunosuppressive tumor micro-environments, noted the Oxford team.
Deciphering the Cancer Transcriptome
A Rogue’s Gallery of Malignant Outliers May Hide in Transcriptome Profiles That Emphasize Averages
The key link between genomic instability and cancer progression is transcriptome dynamics. The shifts in transcriptome dynamics that contribute to cancer evolution may come down to statistical outliers. [iStock/zmeel]
In recent years, scientists have adopted a gene-centric view of cancer, a tendency to see each malignant transformation as the consequence of alterations in a discrete number of genes or pathways. These alterations are, fortunately, absent from healthy cells, but they pervert malignant cells.
The gene-centric view takes in molecular landscapes illuminated by genomic and transcriptomic technologies. For example, genomes can be cost-effectively sequenced within hours. Such capabilities have made it possible to interrogate associations between genotypes and phenotypes for increasing numbers of conditions, and to collect data from progressively larger patient groups.
As genomic and transcriptomic technologies rise, they reveal much—but much remains hidden, too. Perhaps these technologies are less like the sun and more like the proverbial streetlight, the one that narrows our searches because we’re inclined to stay in the light, even though what we hope to find may lie in the shadows.
“Each individual study that looks at the cancer transcriptome is impressive and tells a convincing story, but if we put several high-quality papers together, there are very few genes that overlap,” says Henry H. Heng, Ph.D., professor of molecular medicine, genetics, and pathology at Wayne State University. “This shows that something is wrong.”
Distinct Karyotypes
One of the major observations in Dr. Heng’s lab is that the intra- and intertumor cellular heterogeneity results in nearly every cancer cell having a unique, distinct karyotype, that is, an important but often ignored genotype. “Biological systems need a lot of heterogeneity,” notes Dr. Heng. “People like to think that this is noise, but heterogeneity is a fundamental buffer system for biological function to be achievable. Moreover, it is the key agent for cellular adaptation.”
To capture the degree of genomic heterogeneity at the genome level and its impact on cancer cell growth, Dr. Heng and colleagues performed serial dilutions to isolate single mouse ovarian surface epithelial cells that had undergone spontaneous transformation. Spectral karyotyping revealed that within a short timeframe each of these unstable cells exhibited a very distinct karyotype. In these unstable cells, cloning at the level of the karyotype was not possible.
Stable cells exhibited a normal growth distribution, i.e., no subset of stable cells contributed disproportionately to the overall growth of the cell population. In contrast, unstable cell populations showed a non-normal growth distribution, with few cells contributing most to the cell population’s growth. For example, a single unstable colony contributed more than 70% to the cell population’s growth. This finding suggests that although average profiles can be used to describe non-transformed cells, they cannot be taken to represent the biology of malignant cells.
“Most people who study the transcriptome want to get rid of the noise, but the noise is in fact the strategy that cancer uses to be successful,” explains Dr. Heng. “Each individual cancer cell is very weak but together the entity becomes very robust.”
In a recent model that Dr. Heng and colleagues proposed, system inheritance visualizes chromosomes not merely as the vehicle for transmitting genetic information, but as the genetic network organizer that shapes the physical interactions between genes in the three-dimensional space. Based on this model, individual genes represent parts of the system. The same genes can be reorganized to form different systems, and chromosomal instability becomes more important than the contribution of individual genes and pathways to cancer biology.
The vital link between genomic instability and cancer progression is transcriptome dynamics, and the shifts in those dynamics that contribute to cancer evolution may come down to statistical outliers.
“Transcriptome studies rarely focus on single-cell analyses, which means important outliers are frequently ignored,” declares Dr. Heng. “This preoccupation with uninformative averages explains why we have learned so little despite having examined so many transcriptomes.”
Chimeras and Fusion Genes
“Our focus is on chimeric RNA molecules,” says Laising Yen, Ph.D, assistant professor of pathology at Baylor College of Medicine. “This category of RNAs is very special because their sequences come from different genes.”
In a study that was designed to capture chimeric RNAs in prostate cancer, Dr. Yen and his colleagues performed high-throughput sequencing of the transcriptomes from human prostate cancer samples. “We found far more chimeric RNAs, in terms of abundance, and a number of species that are not seen in normal tissue,” reports Dr. Yen. This approach identified over 2,300 different chimeric RNA species. Some of these chimeras were present in prostate cancer cell lines, but not in primary human prostate epithelium cells, which points toward their relevance in cancer.
“Most of these chimeric RNAs do not have a genomic counterpart, which means that they could be produced by trans-splicing,” explains Dr. Yen. During trans-splicing, individual RNAs are generated and trans-spliced together as a single RNA, which provides a mechanism for generating a chimera.
“The other possibility is that in cancer cells, where gene–gene boundaries are known to become broken, chimeras can be formed by cis-splicing from a very long transcript that encodes several neighboring genes located on the same chromosome,” informs Dr. Yen. Chimeric RNAs formed by either of these two mechanisms can potentially translate into fusion proteins, and these aberrant proteins may have oncogenic consequences.
Another effort in Dr. Yen’s laboratory focuses on chromosomal aberrations in ovarian cancer. One of the hallmarks of ovarian cancer is the high degree of genomic rearrangement and the increased genomic instability.
“When we looked at ovarian cancers, we did not find as many chimeric RNAs,” notes Dr. Yen. “But we found many fusion genes.” Gene fusions, similarly to chimeric RNAs, increase the diversity of the cellular proteome, which could be used selectively by cancer cells to increase their rates of proliferation, survival, and migration.
A recent study in Dr. Yen’s lab identified BCAM-AKT2, a recurrent fusion gene that is specific and unique to high-grade serous ovarian cancer. BCAM-AKT2 is the only fusion gene in this malignancy that was proved to be translated into a fusion kinase in patients, which points toward its functional significance and potential therapeutic value.
“Recurrent fusion genes, which are repeatedly found in many patients in precisely made forms, indicate that there is a reason that they are present,” concludes Dr. Yen. “This might have important therapeutic implications.”
Context-Specific Patterns
“We contributed to a study of tumor gene expresssion that we are currently revisiting because so much more data has become available,” says Barbara Stranger, Ph.D., assistant professor, Institute for Genomics and Systems Biology, University of Chicago. “The data is being processed in homogenized analytic pipelines, and we can look at many more tumor types across the Cancer Genome Atlas than a few years ago.”
Previously, Dr. Stranger and colleagues performed expression quantitative trait loci (eQTL) analyses to examine mRNA and miRNA expression in breast, colon, kidney, lung, and prostate cancer samples. This approach identified 149 known cancer risk loci, 42 of which were significantly associated with expression of at least one transcript.
Causal alleles are being prioritized using a fine-mapping strategy that integrated the eQTL analysis with genome-wide DNAseI hypersensitivity profiles obtained from ENCODE data. These analyses are focusing on capturing differences across tumors and on performing comparisons with normal tissue, and one of the challenges is the lack of normal tissue from the same patients.
“But still there is a lot of power in these analyses because they are based on large-scale genomic datasets. Also, these tumor datasets can be compared with large-scale normal tissue genomics datasets, such as the NIH’s Genotype-Tissue Expression (GTEx) project,” clarifies Dr. Stranger. “This helps us characterize differences between those tumors and normal tissue in terms of the genetics of gene regulation.”
An ongoing effort in Dr. Stranger’s laboratory involves elucidating how the effect of genetic polymorphisms is shaped by context. Stimulated cellular states, cell-type differences, cellular senescence, and disease are some of the contexts that are known to impact genetic polymorphisms.
“We have seen a lot of context specificity,” states Dr. Stranger. “Our observations suggest that a genetic polymorphism can have a specific effect in regulating a particular gene or transcript in one context, and another effect in another context.”
Another example of cellular context is sex, and an active area of investigation in Dr. Stranger’s lab proposes to dissect the manner in which sex differences shape the regulatory effects of genetic polymorphisms.
“Thinking about sex-specific differences is not very different from thinking about a different cellular environment,” notes Dr. Stranger.
The expression of specific transcription factors can be determined by sex; consequently, a polymorphism that interacts with a transcription factor may have functional outcomes that can be seen in only one of the sexes.
“There are gene-level and gene-splicing differences that we see in normal tissues between males and females, and we want to take the same approach and look at the cancer context to see whether the genetic regulation of gene expression and transcript splicing is different between individuals and whether it has a sex bias,” concludes Dr. Stranger. “Finally, we want to see how that differs in cancer relative to normal tissues.”
Early Clinical Impact
An increasing number of clinicians are adding the cancer transcriptome to their precision medicine program. They have found that the transcriptome is important in identifying clinically impactful results. [iStock/DeoSum]
“Over the last two years,” says Andrew Kung, M.D., Ph.D., chief of the Division of Pediatric Hematology, Oncology, and Stem Cell Transplantation at Columbia University Medical Center, “we have included the cancer transcriptome as part of our precision medicine program.” Dr. Kung and colleagues developed a clinical genomics test that includes whole-exome sequencing of tumors and normal tissue and RNA-seq of the tumor.
“Our results show that the transcriptome is very important in identifying clinically impactful results,” asserts Dr. Kung. “The technology has really moved from a research tool to real clinical application.” In fact, the test has been approved by New York State for use in cancer patients.
The data from transcriptome profiling has enabled identification of translocations, verification of somatic alterations, and assessment of expression levels of cancer genes. Dr. Kung and his colleagues are using genomic information for initial diagnosis and prognostic decisions, as well as the investigation of potentially actionable alterations and the monitoring of disease response.
To gain insight into gene-expression changes, transcriptome analysis usually compares two different types of tissues or cells. For example, analyses may attempt to identify differentially expressed genes in cancer cells and normal cells.
“In patients with cancer, we usually do not have access to the normal cell of origin, making it harder to identify the genes that are over- or under-expressed,” explains Dr. Kung. “Fortunately, the vast amounts of existing gene-expression data allow us to identify genes whose expression are most changed relative to models built on the expression data aggregated across large existing datasets.”
These genomic technologies were first used to augment the care of pediatric patients at Columbia. The technologies were so successful that they attracted philanthropic funding, which is being used to expand access to genomic testing to all children with high-risk cancer across New York City.
Other related articles published in this Open Access Online Scientific Journal include the following:
Immunotherapy in Combination, 2016 MassBio Annual Meeting 03/31/2016 8:00 AM – 04/01/2016 3:00 PM Royal Sonesta Hotel, Cambridge, MA
Epigenetics, Environment and Cancer: Articles of Note @PharmaceuticalIntelligence.com
Author and Curator: Larry H. Bernstein, MD, FCAP and Curator: Aviva Lev-Ari, PhD, RN
Introduction
Author: Larry H. Bernstein, MD, FCAP
The following discussions are presented in two series. The first set of discussions is mainly concerned with the role of genomics in the rapidly emerging research domain of genomics and medicine. The recent advances in genomic research at the end of the 20th century brought into the new millennium a seminal accomplishment because of the mapping of the human genome. This development required advances in technology that touches on biochemistry, organic chemistry, physical chemistry, mathematics and computational sciences that have been followed by a surge of innovation for the last 15 years. This was an accomplishment of basic science research that can be ascribed to substantial leadership from the National Institutes of Health, and to a diversity of research centers within the United States, England, France, and Germany, and Israel among others.
In looking back at this development, it might appear to be weighted heavily in a concentrated work on the genetic code. This was predated by the discovery of genetic inborn errors of metabolism that was at least a half century precedent. Thus a model was constructed for the accounting for many human conditions that are expressed in-utero, perinatal, postnatal, and at critical life stages. However, even allowing for over-simplification of a model of life reduced to the expression of a genetic code, this has led to the genesis of a concept of genetic clarification of life “maladies”, diagnostic, therapeutic, and prognostic implications. The concept of a “personalized medicine” emerges from such a construct.
I have already ceded considerable ground in an argument of what occurs in life, illness, and death at the cellular, organ, and organ system level. There are indeed gene amplifications and downregulation of genes that are expressed or have an “on-off” nature in transcription, which becomes a major driver of metabolic control. In this respect, the classic model of gene-RNA-protein has been superseded by a much more complicated model, but still in the realm of personalized medicine. The classic model of metabolism is tied to anabolic and catabolic pathways, glycolytic and mitochondrial substrates, amino acids, proteins and 3D-protein aggregates that have functional roles, and that is controlled by allosteric interactions, ion transport, membrane affinity, signaling pathways, and hydrophilic and hydrophobic effects. This leads to the second part of the discussion about epigenetics and environmental impacts on cellular function. It is by no means irrelevant because the evolution of organisms from sea to land, and the existence of living forms in mountainous and desert regions imposed restrictions that required adaptation. A full understanding of these factors is required in the immersion in personalized medicine.
The preceding chapters have provided a substantial insight into the growth and acceleration of work related to translational medicine and personalized medicine. I make note of the fact that a substantial knowledge has been from basic research using animal models, including C. Eligans. The amount of knowledge is quite impressive. Let me review some major points gained from these presentations.
Non-coding areas of our DNA are far from being without function. But the ensuing work with RNAs is captivating. Whether regulating gene expression and transcription, or providing protein attachment sites, this once-dismissed part of the genome is vital for all life.
There are two basic categories of nitrogenous bases: the purines (adenine [A] and guanine [G]), each with two fused rings, and the pyrimidines (cytosine [C], thymine [T], and uracil [U]), each with a single ring. Furthermore, it is now widely accepted that RNA contains only A, G, C, and U (no T), whereas DNA contains only A, G, C, and T (no U).
There is no uncertainty about the importance of “Junk DNA”. It is both an evolutionary remnant, and it has a role in cell regulation. Further, the role of histones in their relationship the oligonucleotide sequences is not understood. We now have a large output of research on noncoding RNA, including siRNA, miRNA, and others with roles other than transcription. This requires major revision of our model of cell regulatory processes. The classic model is solely transcriptional.
DNA-> RNA-> Amino Acid in a protein.
Redrawn we have
DNA-> RNA-> DNA and
DNA->RNA-> protein-> DNA.
DNA is involved mainly with genetic information storage, while RNA molecules—mRNA, rRNA, tRNA, miRNA, and others—are engaged in diverse structural, catalytic, and regulatory activities, in addition to translating genes into proteins. RNA’s multitasking prowess, at the heart of the RNA World hypothesis implicating RNA as the first molecule of life, likely spurred the evolution of numerous modified nucleotides. This enabled the diversified complementarity and secondary structures that allow RNA species to specifically interact with other components of the cellular machinery such as DNA and proteins. The alphabet of RNA consists of at least 140 alternative nucleotide forms.
Among the 140 modified RNA nucleotide variants identified, methylation of adenosine at the N6 position (m6A) is the most prevalent epigenetic mark in eukaryotic mRNA. Identified in bacterial rRNAs and tRNAs as early as the 1950s, this type of methylation was subsequently found in other RNA molecules, including mRNA, in animal and plant cells as well. In 1984, researchers identified a site that was specifically methylated—the 3′ untranslated region (UTR) of bovine prolactin mRNA.1 As more sites of m6A modification were identified, a consistent pattern emerged: the methylated A is preceded by A or G and followed by C (A/G—methylated A—C).
Although the identification of m6A in RNA is 40 years old, until recently researchers lacked efficient molecular mapping and quantification methods to fully understand the functional implications of the modification. In 2012, we (D.D. and G.R.) combined the power of next-generation sequencing (NGS) with traditional antibody-mediated capture techniques to perform high-resolution transcriptome-wide mapping of m6A, an approach we termed m6A-seq.2 Briefly, the transcriptome is randomly fragmented and an anti-m6A antibody is used to fish out the methylated RNA fragments; the m6A-containing fragments are then sequenced and aligned to the genome, thus allowing us to locate the positions of methylation marks.
The work of Warburg and Meyerhoff, followed by that of Krebs, Kaplan, Chance, and others built a solid foundation in the knowledge of enzymes, coenzymes, adenine and pyridine nucleotides, and metabolic pathways, not to mention the importance of Fe3+, Cu2+, Zn2+, and other metal cofactors.
Of huge importance was the work of Jacob, Monod and Changeux, and the effects of cooperativity in allosteric systems and of repulsion in tertiary structure of proteins related to hydrophobic and hydrophilic interactions, which involves the effect of one ligand on the binding or catalysis of another, demonstrated by the end-product inhibition of the enzyme, L-threonine deaminase (Changeux 1961), L-isoleucine, which differs sterically from the reactant, L-threonine whereby the former could inhibit the enzyme without competing with the latter. The current view based on a variety of measurements (e.g., NMR, FRET, and single molecule studies) is a ‘‘dynamic’’ proposal by Cooper and Dryden (1984) that the distribution around the average structure changes in allostery affects the subsequent (binding) affinity at a distant site.
Present day applications of computational methods to biomolecular systems, combined with structural, thermodynamic, and kinetic studies, make possible an approach to that question, so as to provide a deeper understanding of the requirements for allostery. The current view is that a variety of measurements (e.g., NMR, FRET, and single molecule studies) are providing additional data beyond that available previously from structural, thermodynamic, and kinetic results. These should serve to continue to improve our understanding of the molecular mechanism of allostery
Metal-mediated formation of free radicals causes various modifications to DNA bases, enhanced lipid peroxidation, and altered calcium and sulfhydryl homeostasis. The measurement of free radicals has increased awareness of radical-induced impairment of the oxidative/antioxidative balance, essential for an understanding of disease progression. Metal-mediated formation of free radicals causes various modifications to DNA bases, enhanced lipid peroxidation, and altered calcium and sulfhydryl homeostasis. Lipid peroxides, formed by the attack of radicals on polyunsaturated fatty acid residues of phospholipids, can further react with redox metals finally producing mutagenic and carcinogenic malondialdehyde, 4-hydroxynonenal and other exocyclic DNA adducts (etheno and/or propano adducts). The unifying factor in determining toxicity and carcinogenicity for all these metals is the generation of reactive oxygen and nitrogen species. Various studies have confirmed that metals activate signaling pathways and the carcinogenic effect of metals has been related to activation of mainly redox sensitive transcription factors, involving NF-kappaB, AP-1 and p53.
There is heterogeneity in the immediate interstices between cancer cells, which may seem surprising, but it should not be. This refers to the complexity of the cells arranged as tissues and to their immediate environment, which I shall elaborate on. Integration with genome-wide profiling data identified losses of specific genes on 4p14 and 5q13 that were enriched in grade 3 tumors with high microenvironmental diversity that also substratified patients into poor prognostic groups.
IDH1 mutations have been identified at the Arg132 codon. Mutations in IDH2 have been identified at the Arg140 codon, as well as at Arg172, which is aligned with IDH1 Arg132. IDH1 and IDH2 mutations are heterozygous in cancer, and they catalyze the production of α-2-hydroxyglutarate. The study found human IDH1 transitions between an inactive open, an inactive semi-open, and a catalytically active closed conformation. In the inactive open conformation, Asp279 occupies the position where the isocitrate substrate normally forms hydrogen bonds with Ser94. This steric hindrance by Asp279 to isocitrate binding is relieved in the active closed conformation.
There are allelic variations that underlie common diseases and complete genome sequencing for many individuals with and without disease is required. However, there are advantages and disadvantages as we can carry out partial surveys of the genome by genotyping large numbers of common SNPs in genome-wide association studies but there are problems such as computing the data efficiently and sharing the information without tempering privacy.
Since the first report of p53 as a non-histone target of a histone acetyltransferase (HAT), there has been a rapid proliferation in the description of new non-histone targets of HATs. Of these,
transcription factors comprise the largest class of new targets.
The substrates for HATs extend to
cytoskeletal proteins,
molecular chaperones and
nuclear import factors.
Deacetylation of these non-histone proteins by histone deacetylases (HDACs) opens yet another exciting new field of discovery in
the role of the dynamic acetylation and deacetylation on cellular function.
We capture the dynamic interactions between the systems under stress that are elicited by cytokine-driven hormonal responses, long thought to be circulatory and multisystem, that affect the major compartments of fat and lean body mass, and are as much the drivers of metabolic pathway changes that emerge as epigenetics, without disregarding primary genetic diseases.
The greatest difficulty in organizing such a work is in whether it is to be merely a compilation of cancer expression organized by organ systems, or whether it is to capture developing concepts of underlying stem cell expressed changes that were once referred to as “dedifferentiation”. In proceeding through the stages of neoplastic transformation, there occur adaptive local changes in cellular utilization of anabolic and catabolic pathways, and a retention or partial retention of functional specificities.
This effectively results in the same cancer types not all fitting into the same “shoe”. There is a sequential loss of identity associated with cell migration, cell-cell interactions with underlying stroma, and metastasis., but cells may still retain identifying “signatures” in microRNA combinatorial patterns. The story is still incomplete, with gaps in our knowledge that challenge the imagination.
What we have laid out is a map with substructural ordered concepts forming subsets within the structural maps. There are the traditional energy pathways with terms aerobic and anaerobic glycolysis, gluconeogenesis, triose phosphate branch chains, pentose shunt, and TCA cycle vs the Lynen cycle, the Cori cycle, glycogenolysis, lipid peroxidation, oxidative stress, autosomy and mitosomy, and genetic transcription, cell degradation and repair, muscle contraction, nerve transmission, and their involved anatomic structures (cytoskeleton, cytoplasm, mitochondria, liposomes and phagosomes, contractile apparatus, synapse.
We are a magnificent “magical” experience in evolutionary time, functioning in a bioenvironment, put rogether like a truly complex machine, and with interacting parts. What are those parts – organelles, a genetic message that may be constrained and it may be modified based on chemical structure, feedback, crosstalk, and signaling pathways. This brings in diet as a source of essential nutrients, exercise as a method for delay of structural loss (not in excess), stress oxidation, repair mechanisms, and an entirely unexpected impact of this knowledge on pharmacotherapy.
Despite what we have learned, the strength of inter-molecular interactions, strong and weak chemical bonds, essential for 3-D folding, we know little about the importance of trace metals that have key roles in catalysis and because of their orbital structures, are essential for organic-inorganic interplay. This will not be coming soon because we know almost nothing about the intracellular, interstitial, and intravesicular distributions and how they affect the metabolic – truly metabolic events.
We must translate the sequence information from genomics locus of the genes to function with related polymorphism of these genes so that possible patterns of the gene expression and disease traits can be matched. Then, we may develop precision technologies for:
Diagnostics
Targeted Drugs and Treatments
Biomarkers to modulate cells for correct functions
With the knowledge of:
gene expression variations
insight in the genetic contribution to clinical endpoints ofcomplex disease and
their biological risk factors,
share etiologic pathways
which requires an understanding of both:
the structure and
the biology of the genome.
A new paradigm is summarized in a sequence of six steps:
“(1) A pathogenic stimulus (biological or chemical) leads at first to a normal reaction seen in wound healing, namely, inflammation. When the inflammatory stimulus is too great or too prolonged, the healing process is unsuccessful, and that results in
(2) chronic inflammation.
“That’s just the beginning. When chronic inflammation persists,
(3) fibrosis [thickening and scarring of the connective tissue,] develops. The fibrosis, with its ongoing alteration of the cellular microenvironment is different and creates
(4) a precancerous niche, resulting in a chronically stressed cellular matrix. In such a situation, the organism deploys
(5) a chronic stress escape strategy. But if this attempt fails to resolve the precancerous state, then
(6) a normal cell is transformed into a cancerous cell.”
Keep in mind:
Nutritional resources that have been available and made plentiful over generations are not abundant in some climates.
Despite the huge impact that genomics has had on biological progress over the last century, there is a huge contribution not to be overlooked in epigenetics, metabolomics, and pathways analysis.
I have provided mechanisms explanatory for regulation of the cell that go beyond the classic model of metabolic pathways associated with the cytoplasm, mitochondria, endoplasmic reticulum, and lysosome, such as, the cell death pathways, expressed in apoptosis and repair. Nevertheless, there is still a missing part of this discussion that considers the time and space interactions of the cell, cellular cytoskeleton and extracellular and intracellular substrate interactions in the immediate environment.
Signal transduction occurs when an extracellular signaling[1] molecule activates a specific receptor located on the cell surface or inside the cell. In turn, this receptor triggers a biochemical chain of events inside the cell, creating a response.[2] Depending on the cell, the response alters the cell’s metabolism, shape, gene expression, or ability to divide.[3] The signal can be amplified at any step. Thus, one signaling molecule can cause many responses.[4]
In 1970, Martin Rodbell examined the effects of glucagon on a rat’s liver cell membrane receptor. He noted that guanosine triphosphate disassociated glucagon from this receptor and stimulated the G-protein, which strongly influenced the cell’s metabolism. Thus, he deduced that the G-protein is a transducer that accepts glucagon molecules and affects the cell.[5] For this, he shared the 1994 Nobel Prize in Physiology or Medicine with Alfred G. Gilman.
Signal transduction involves the binding of extracellular signaling molecules and ligands to cell-surface receptors that trigger events inside the cell. The combination of messenger with receptor causes a change in the conformation of the receptor, known as receptor activation. This activation is always the initial step (the cause) leading to the cell’s ultimate responses (effect) to the messenger. Despite the myriad of these ultimate responses, they are all directly due to changes in particular cell proteins. Intracellular signaling cascades can be started through cell-substratum interactions; examples are the integrin that binds ligands in the extracellular matrix and steroids.[13] Most steroid hormones have receptors within the cytoplasm and act by stimulating the binding of their receptors to the promoter region of steroid-responsive genes.[14] Examples of signaling molecules include the hormone melatonin,[15] the neurotransmitter acetylcholine[16] and the cytokineinterferon γ.[17]
Various environmental stimuli exist that initiate signal transmission processes in multicellular organisms; examples include photons hitting cells in the retina of the eye,[20] and odorants binding to odorant receptors in the nasal epithelium.[21] Certain microbial molecules, such as viral nucleotides and protein antigens, can elicit an immune system response against invading pathogens mediated by signal transduction processes. This may occur independent of signal transduction stimulation by other molecules, as is the case for the toll-like receptor. It may occur with help from stimulatory molecules located at the cell surface of other cells, as with T-cell receptor signaling.
Unraveling the multitude of
nutrigenomic,
proteomic, and
metabolomic patterns
that arise from the ingestion of foods or their
bioactive food components
will not be simple but is likely to provide insights into a tailored approach to diet and health. The use of new and innovative technologies, such as
microarrays,
RNA interference, and
nanotechnologies,
will provide needed insights into molecular targets for specific bioactive food components and
how they harmonize to influence individual phenotypes(1).
Oct4 has a critical role in committing pluripotent cells into the somatic cellular pathway. When embryonic stem cells overexpress Oct4, they undergo rapid differentiation and then lose their ability for pluripotency. Other studies have shown that Oct4 expression in somatic cells reprograms them for transformation into a particular germ cell layer and also gives rise to induced pluripotent stem cells (iPSCs) under specific culture conditions.
Oct4 is the gatekeeper into and out of the reprogramming expressway. By modifying experimental conditions, Oct4 plus additional factors can induce formation of iPSCs, epiblast stem cells, neural cells, or cardiac cells. Dr. Schöler suggests that Oct4 a potentially key factor not only for inducing iPSCs but also for transdifferention. “Therapeutic applications might eventually focus less on pluripotency and more on multipotency,
Epigenetics is getting a big attention recently to understand genomics and provide better results. However, this field is studied for many years under functional genomics and developmental biology for cellular and molecular biology. Stem cells have a free drive that we have not figured out yet. So genomics must be studied essentially with people training in developmental biology and comparative molecular genetics knowledge to make heads and tail for translational medicine.
There are three main routes of epigenetic modifications one
histone modifications via acetylation and methylation and the other is
DNA methylation, which are two classical mechanisms in epigenetics.
The third factor is
non-coding RNAs that are usually underestimated even not included.
In 1993, Kavai group showed brain development assays of mice showed that only 0.7% genome has tissue and cellular specificity, and 1.7% of genome was able to turn on and off. This conclusion is relevant to genome sequencing data. Also, previous studies in genome and RNA biology presented that RNA directed DNA modifications lead into splicing and transcriptional silencing for gene regulation in Arapsidosis, mice, and Drosophila. (Borge, F. and. Martiensse, R.A. 2013; Di Croce L, Raker VA, Corsaro M, et al. 2002; Piferrer, F, 2013; Jun Kawai1 et al. 1993)
The environment creates the epigenerators including temperature, differentiation signals and metabolites that trigger the cell membrane proteins for development of signal transduction within the cell to activate gene(s) and to create cellular response. These changes can be modulated but they are not necessary for modulation. The second step involves epigenetic initiators that require precise coordination to recognize specific sequences on a chromatin in response to epigenerator signals. These molecules are
DNA binding proteins and
non coding RNAs.
After they are involved they are on for life and controlled by autoregulatory mechanisms, like Sxl (sex lethal) RNA binding protein in somatic sex determination and ovo DNA binding protein in germline sex determination of fruit fly. Both have autoregulation mechanisms, cross talks, differential signals and cross reacting genes since after the final update made the soma has to maintain the decision to stay healthy and develop correctly. Then, this brings the third level mechanism called epigenetic maintainers that are DNA methylating enzymes, histone modifying enzymes and histone variants. The good news is they can be reversed. As a result the phonotype establishes either a
short term phenotype, transient for transcription,
DNA replication and repair or
long term phenotype outcomes that are chromatin conformation and heritable markers.
Early in development things are short term and stop after the development seized but be able to maintain the short term phenotype during wound healing, coagulation, trauma, disease and immune responses.
The metabolome for each organism is unique, but from an evolutionary perspective has metabolic pathways in common, and expressed in concert with the environment that these living creatures exist. The metabolome of each has adaptive accommodation with suppression and activation of pathways that are functional and necessary in balance, for its existence.
Most interesting is a recent report from Johns Hopkins in Mar 28, PNAS on breast cancer and stem cell physiology. “Aggressive cancers contain regions where the cancer cells are starved for oxygen and die off, yet patients with these tumors generally have the worst outcome,” Semenza said in a release. “Our new findings tell us that low oxygen conditions actually encourage certain cancer stem cells to multiply through the same mechanism used by embryonic stem cells.”
One of the genes responsible for initiating a stem cell fate under low oxygen conditions is called NANOG. This gene is one of many turned on in oxygen-poor conditions by proteins called hypoxia-inducible factors, or HIFs. NANOG in turn instructs cells to become stem cells to resist the poor conditions and help survival.
NANOG levels can be artificially lowered in embryonic stem cells by experimentally methylating the respective mRNA transcript at the sixth position of its adenine nucleotide. Since this methylation is otherwise thought to stabilize the transcript from degradation, this may help NANOG abandon its proposed stem cell fate for the cell.
In addition to the basic essential nutrients and their metabolic utilization, they are under cellular metabolic regulation that is tied to signaling pathways. In addition, the genetic expression of the organism is under regulatory control by the interaction of RNAs that interact with the chromatin genetic framework, with exosomes, and with protein modulators. This is referred to as epigenetics, but there are also drivers of metabolism that are shaped by the interactions between enzymes and substrates, and are related to the tertiary structure of a protein. The framework for diseases in and Pharmaceutical interventions that are designed to modulate specific metabolic targets are addressed as the pathways are unfolded.
Personalized Medicine is here now
Two years ago AJP was found to have a positive test for BRCA1, carrying an 87 percent risk for breast cancer and a 50 percent risk for ovarian cancer. At that time she had a preventive mastectomy. The decision was not easy, but it also brought into consideration that her mother and grandmother both died of breast cancer. She did not have an oophorectomy at that time because on considering the advice of medical experts, she would have been left with no estrogen support. She wanted to delay her early vegetative senescence. She has reached the age of 39 years and on the advice of medical expert opinion, she proceeded with salpingo-oophorectomy, at age 39 years, a decade before her mother had developed cancer. But her delay was to allow her to recover and adjust emotionally to her ongoing situation, with a remaining risk for ovarian cancer.
in a report in Carcinogenesis back in 2005[3] Lorena Losi, Benedicte Baisse, Hanifa Bouzourene and Jean Benhatter had shown some similar results in colorectal cancer as their abstract described:
“In primary colorectal cancers (CRCs), intratumoral genetic heterogeneity was more often observed in early than in advanced stages, at 90 and 67%, respectively. All but one of the advanced CRCs were composed of one predominant clone and other minor clones, whereas no predominant clone has been identified in half of the early cancers. A reduction of the intratumoral genetic heterogeneity for point mutations and a relative stability of the heterogeneity for allelic losses indicate that, during the progression of CRC, clonal selection and chromosome instability continue, while an increase cannot be proven.”
An article written by Drs. Andrei Krivtsov and Scott Armstrong entitled “Can One Cell Influence Cancer Heterogeneity”[4] commented on a study by Friedman-Morvinski[5] in Inder Verma’s laboratory discussed how genetic lesions can revert differentiated neurons and glial cells to an undifferentiated state [an important phenotype in development of glioblastoma multiforme].
In particular it is discussed that epigenetic state of the transformed cell may contribute to the heterogeneity of the resultant tumor. Indeed many investigators (initially discovered and proposed by Dr. Beatrice Mintz of the Institute for Cancer Research, later to be named the Fox Chase Cancer Center) show the cellular microenvironment influences transformation and tumor development [6-8].
The mechanism by which tissue microecology influences invasion and metastasis is largely unknown. Recent studies have indicated differences in the molecular architecture of the metastatic lesion compared to the primary tumor, however, systemic analysis of the alterations within the activated protein signaling network has not been described. Using laser capture microdissection, protein microarray technology, and a unique specimen collection of 34 matched primary colorectal cancers (CRC) and synchronous hepatic metastasis, the quantitative measurement of the total and activated/phosphorylated levels of 86 key signaling proteins was performed. Activation of the EGFR-PDGFR-cKIT network, in addition to PI3K/AKT pathway, was found uniquely activated in the hepatic metastatic lesions compared to the matched primary tumors. If validated in larger study sets, these findings may have potential clinical relevance since many of these activated signaling proteins are current targets for molecularly targeted therapeutics. Thus, these findings could lead to liver metastasis specific molecular therapies for CRC.
Not only was 2015 the warmest worldwide since records began, it shattered the previous record
Reporter: Aviva Lev-Ari, PhD
Global temperatures in 2015 were by far the hottest in modern times, according to new data from American science agencies.
Not only was 2015 the warmest worldwide since records began in 1880, it shattered the previous record held in 2014 by the widest margin ever observed, a report by the National Oceanic and Atmospheric Administration (NOAA) said. “During 2015, the average temperature across global land and ocean surfaces was 0.90 Celsius above the 20th century average,” the NOAA report said.
“This was the highest among all years in the 1880 to 2015 record [and also] the largest margin by which the annual global temperature record has been broken.”
The US space agency NASA, which monitors global climate using a fleet of satellites and weather stations, confirmed that last year broke records for heat in contemporary times. NASA said that the temperature changes are largely driven by increased carbon dioxide and other human-made emissions into the atmosphere.
“Climate change is the challenge of our generation,” NASA administrator Charles Bolden said. “Today’s announcement not only underscores how critical NASA’s Earth observation program is, it is a key data point that should make policy makers stand up and take notice — now is the time to act on climate.”




















{kind=link}
{kind=link}
{kind=link}
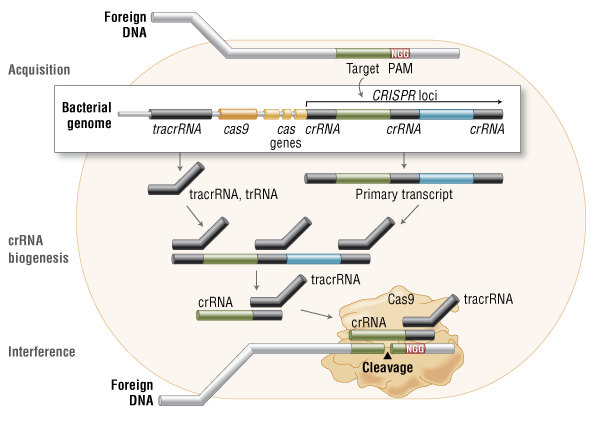{kind=link}
{kind=link}
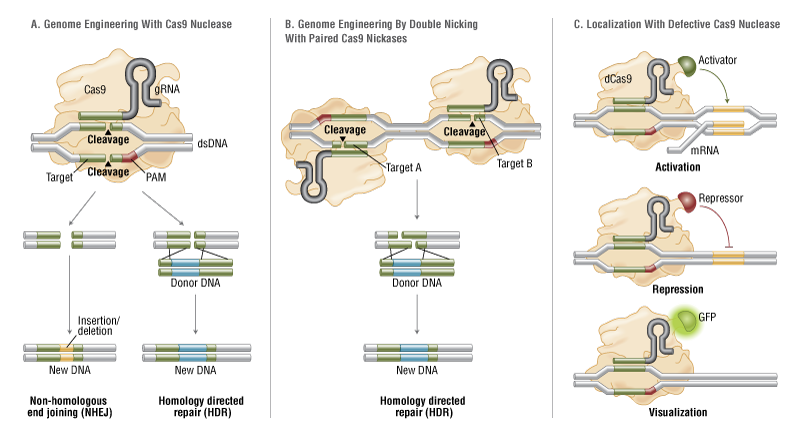{kind=link}
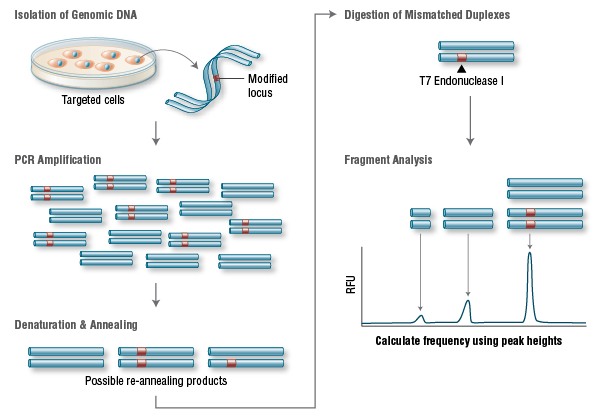{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
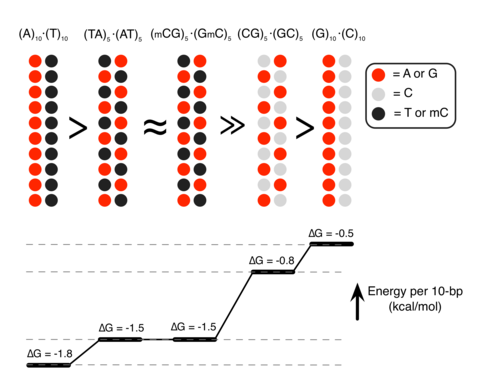{kind=link}