Healthcare analytics, AI solutions for biological big data, providing an AI platform for the biotech, life sciences, medical and pharmaceutical industries, as well as for related technological approaches, i.e., curation and text analysis with machine learning and other activities related to AI applications to these industries.
RNA from the SARS-CoV-2 virus taking over the cells it infects: Virulence – Pathogen’s ability to infect a Resistant Host: The Imbalance between Controlling Virus Replication versus Activation of the Adaptive Immune Response
Curator: Aviva Lev-Ari, PhD, RN – I added colors and bold face
UPDATED on 9/8/2020
What bats can teach us about developing immunity to Covid-19 | Free to read
Another duality and paradox in the Treatment of COVID-19 Patients in ICUs was expressed by Mike Yoffe, MD, PhD, David H. Koch Professor of Biology and Biological Engineering, Massachusetts Institute of Technology. Dr. Yaffe has a joint appointment in Acute Care Surgery, Trauma, and Surgical Critical Care, and in Surgical Oncology @BIDMC
on 6/29 at SOLUTIONS with/in/sight at Koch Institute @MIT
How Are Cancer Researchers Fighting COVID-19? (Part II)”Jun 29, 2020 11:30 AM EST
In COVID-19 patients: two life threatening conditions are seen in ICUs:
Blood Clotting – Hypercoagulability or Thrombophilia
Cytokine Storm – immuno-inflammatory response
The coexistence of 1 and 2 – HINDERS the ability to use effectively tPA as an anti-clotting agent while the cytokine storm is present.
Mike Yoffe’s related domain of expertise:
Signaling pathways and networks that control cytokine responses and inflammation
Misregulation of cytokine feedback loops, along with inappropriate activation of the blood clotting cascade causes dysregulation of cell signaling pathways in innate immune cells (neutrophils and macrophages), resulting in tissue damage and multiple organ failure following trauma or sepsis. Our research is focused on understanding the role of the p38-MK2 pathway in cytokine control and innate immune function, and on cross-talk between cytokines, clotting factors, and neutrophil NADPH oxidase-derived ROS in tissue damage, coagulopathy, and inflammation, using biochemistry, cell biology, and mouse knock-out/knock-in models. We recently discovered a particularly important link between abnormal blood clotting and the complement pathway cytokine C5a which causes excessive production of extracellular ROS and organ damage by neutrophils after traumatic injury.
SARS-CoV-2 infection induces low IFN-I and -III levels with a moderate ISG response
Strong chemokine expression is consistent across in vitro, ex vivo, and in vivo models
Low innate antiviral defenses and high pro-inflammatory cues contribute to COVID-19
Summary
Viral pandemics, such as the one caused by SARS-CoV-2, pose an imminent threat to humanity. Because of its recent emergence, there is a paucity of information regarding viral behavior and host response following SARS-CoV-2 infection. Here we offer an in-depth analysis of the transcriptional response to SARS-CoV-2 compared with other respiratory viruses. Cell and animal models of SARS-CoV-2 infection, in addition to transcriptional and serum profiling of COVID-19 patients, consistently revealed a unique and inappropriate inflammatory response. This response is defined by low levels of type I and III interferons juxtaposed to elevated chemokines and high expression of IL-6. We propose that reduced innate antiviral defenses coupled with exuberant inflammatory cytokine production are the defining and driving features of COVID-19.
Defining the Transcriptional Response to SARS-CoV-2 Relative to Other Respiratory Viruses
To compare the transcriptional response of SARS-CoV-2 with other respiratory viruses, including MERS-CoV, SARS-CoV-1, human parainfluenza virus 3 (HPIV3), respiratory syncytial virus (RSV), and IAV, we first chose to focus on infection in a variety of respiratory cell lines (Figure 1). To this end, we collected poly(A) RNA from infected cells and performed RNA sequencing (RNA-seq) to estimate viral load. These data show that virus infection levels ranged from 0.1% to more than 50% of total RNA reads (Figure 1A).
Discussion
In the present study, we focus on defining the host response to SARS-CoV-2 and other human respiratory viruses in cell lines, primary cell cultures, ferrets, and COVID-19 patients. In general, our data show that the overall transcriptional footprint of SARS-CoV-2 infection was distinct in comparison with other highly pathogenic coronaviruses and common respiratory viruses such as IAV, HPIV3, and RSV. It is noteworthy that, despite a reduced IFN-I and -III response to SARS-CoV-2, we observed a consistent chemokine signature. One exception to this observation is the response to high-MOI infection in A549-ACE2 and Calu-3 cells, where replication was robust and an IFN-I and -III signature could be observed. In both of these examples, cells were infected at a rate to theoretically deliver two functional virions per cell in addition to any defective interfering particles within the virus stock that were not accounted for by plaque assays. Under these conditions, the threshold for PAMP may be achieved prior to the ability of the virus to evade detection through production of a viral antagonist. Alternatively, addition of multiple genomes to a single cell may disrupt the stoichiometry of viral components, which, in turn, may itself generate PAMPs that would not form otherwise. These ideas are supported by the fact that, at a low-MOI infection in A549-ACE2 cells, high levels of replication could also be achieved, but in the absence of IFN-I and -III induction. Taken together, these data suggest that, at low MOIs, the virus is not a strong inducer of the IFN-I and -III system, as opposed to conditions where the MOI is high.
Taken together, the data presented here suggest that the response to SARS-CoV-2 is imbalanced with regard to controlling virus replication versus activation of the adaptive immune response. Given this dynamic, treatments for COVID-19 have less to do with the IFN response and more to do with controlling inflammation. Because our data suggest that numerous chemokines and ILs are elevated in COVID-19 patients, future efforts should focus on U.S. Food and Drug Administration (FDA)-approved drugs that can be rapidly deployed and have immunomodulating properties.
One of the features distinguishing SARS-CoV-2 from its more pathogenic counterpart SARS-CoV is the presence of premature stop codons in its ORF3b gene. Here, we show that SARS-CoV-2 ORF3b is a potent interferon antagonist, suppressing the induction of type I interferon more efficiently than its SARS-CoV ortholog. Phylogenetic analyses and functional assays revealed that SARS-CoV-2-related viruses from bats and pangolins also encode truncated ORF3b gene products with strong anti-interferon activity. Furthermore, analyses of more than 15,000 SARS-CoV-2 sequences identified a natural variant, in which a longer ORF3b reading frame was reconstituted. This variant was isolated from two patients with severe disease and further increased theability of ORF3b to suppress interferon induction. Thus, our findings not only help to explain the poor interferon response in COVID-19 patients, but also describe a possibility of the emergence of natural SARS-CoV-2 quasi-species with extended ORF3b that may exacerbate COVID-19 symptoms.
Highlights
ORF3b of SARS-CoV-2 and related bat and pangolin viruses is a potent IFN antagonist
SARS-CoV-2 ORF3b suppresses IFN induction more efficiently than SARS-CoV ortholog
The anti-IFN activity of ORF3b depends on the length of its C-terminus
An ORF3b with increased IFN antagonism was isolated from two severe COVID-19 cases
RNA (in green) from the SARS-CoV-2 virus is shown taking over the cells it infects.ICAHN SCHOOL OF MEDICINE AT MOUNT SINAI
A deep dive into how the new coronavirus infects cells has found that it orchestrates a hostile takeover of their genes unlike any other known viruses do, producing what one leading scientist calls “unique” and “aberrant” changes.Recent studies show that in seizing control of genes in the human cells it invades, the virus changes how segments of DNA are read, doing so in a way that might explain why the elderly are more likely to die of Covid-19 and why antiviral drugs might not only save sick patients’ lives but also prevent severe disease if taken before infection.“It’s something I have never seen in my 20 years of” studying viruses, said virologist Benjamin tenOever of the Icahn School of Medicine at Mount Sinai, referring to how SARS-CoV-2, the virus that causes Covid-19, hijacks cells’ genomes.The “something” he and his colleagues saw is how SARS-CoV-2 blocks one virus-fighting set of genes but allows another set to launch, a pattern never seen with other viruses. Influenza and the original SARS virus (in the early 2000s), for instance, interfere with both arms of the body’s immune response — what tenOever dubs “call to arms” genes and “call for reinforcement” genes.The first group of genes produces interferons. These proteins, which infected cells release, are biological semaphores, signaling to neighboring cells to activate some 500 of their own genes that will slow down the virus’ ability to make millions of copies of itself if it invades them. This lasts seven to 10 days, tenOever said, controlling virus replication and thereby buying time for the second group of genes to act.This second set of genes produce their own secreted proteins, called chemokines, that emit a biochemical “come here!” alarm. When far-flung antibody-making B cells and virus-killing T cells sense the alarm, they race to its source. If all goes well, the first set of genes holds the virus at bay long enough for the lethal professional killers to arrive and start eradicating viruses.
“Most other viruses interfere with some aspect of both the call to arms and the call for reinforcements,” tenOever said. “If they didn’t, no one would ever get a viral illness”: The one-two punch would pummel any incipient infection into submission.
SARS-CoV-2, however, uniquely blocks one cellular defense but activates the other, he and his colleagues reported in a study published last week in Cell. They studied healthy human lung cells growing in lab dishes, ferrets (which the virus infects easily), and lung cells from Covid-19 patients. In all three, they found that within three days of infection, the virus induces cells’ call-for-reinforcement genes to produce cytokines. But it blocks their call-to-arms genes — the interferons that dampen the virus’ replication.
The result is essentially no brakes on the virus’s replication, but a storm of inflammatory molecules in the lungs, which is what tenOever calls an “unique” and “aberrant” consequence of how SARS-CoV-2 manipulates the genome of its target.
In another new study, scientists in Japan last week identified how SARS-CoV-2 accomplishes that genetic manipulation. Its ORF3b gene produces a protein called a transcription factor that has “strong anti-interferon activity,” Kei Sato of the University of Tokyo and colleagues found — stronger than the original SARS virus or influenza viruses. The protein basically blocks the cell from recognizing that a virus is present, in a way that prevents interferon genes from being expressed.
In fact, the Icahn School team found no interferons in the lung cells of Covid-19 patients. Without interferons, tenOever said, “there is nothing to stop the virus from replicating and festering in the lungs forever.”
That causes lung cells to emit even more “call-for-reinforcement” genes, summoning more and more immune cells. Now the lungs have macrophages and neutrophils and other immune cells “everywhere,” tenOever said, causing such runaway inflammation “that you start having inflammation that induces more inflammation.”
At the same time, unchecked viral replication kills lung cells involved in oxygen exchange. “And suddenly you’re in the hospital in severe respiratory distress,” he said.
In elderly people, as well as those with diabetes, heart disease, and other underlying conditions, the call-to-arms part of the immune system is weaker than in younger, healthier people, even before the coronavirus arrives. That reduces even further the cells’ ability to knock down virus replication with interferons, and imbalances the immune system toward the dangerous inflammatory response.
The discovery that SARS-CoV-2 strongly suppresses infected cells’ production of interferons has raised an intriguing possibility: that taking interferons might prevent severe Covid-19 or even prevent it in the first place, said Vineet Menachery of the University of Texas Medical Branch.
In a study of human cells growing in lab dishes, described in a preprint (not peer-reviewed or published in a journal yet), he and his colleagues also found that SARS-CoV-2 “prevents the vast amount” of interferon genes from turning on. But when cells growing in lab dishes received the interferon IFN-1 before exposure to the coronavirus, “the virus has a difficult time replicating.”
After a few days, the amount of virus in infected but interferon-treated cells was 1,000- to 10,000-fold lower than in infected cells not pre-treated with interferon. (The original SARS virus, in contrast, is insensitive to interferon.)
Ending the pandemic and preventing its return is assumed to require an effective vaccine to prevent infectionand antiviral drugs such as remdesivir to treat the very sick, but the genetic studies suggest a third strategy: preventive drugs.
It’s possible that treatment with so-called type-1 interferon “could stop the virus before it could get established,” Menachery said.
Giving drugs to healthy people is always a dicey proposition, since all drugs have side effects — something considered less acceptable than when a drug is used to treat an illness. “Interferon treatment is rife with complications,” Menachery warned. The various interferons, which are prescribed for hepatitis, cancers, and many other diseases, can cause flu-like symptoms.
But the risk-benefit equation might shift, both for individuals and for society, if interferons or antivirals or other medications are shown to reduce the risk of developing serious Covid-19 or even make any infection nearly asymptomatic.
Interferon “would be warning the cells the virus is coming,” Menachery said, so such pretreatment might “allow treated cells to fend off the virus better and limit its spread.” Determining that will of course require clinical trials, which are underway.
Other related articles in this Open Access Online Scientific Journal include the following:
Structure-guided Drug Discovery: (1) The Coronavirus 3CL hydrolase (Mpro) enzyme (main protease) essential for proteolytic maturation of the virus and (2) viral protease, the RNA polymerase, the viral spike protein, a viral RNA as promising two targets for discovery of cleavage inhibitors of the viral spike polyprotein preventing the Coronavirus Virion the spread of infection
Predicting the Protein Structure of Coronavirus: Inhibition of Nsp15 can slow viral replication and Cryo-EM – Spike protein structure (experimentally verified) vs AI-predicted protein structures (not experimentally verified) of DeepMind (Parent: Google) aka AlphaFold
Curators: Stephen J. Williams, PhD and Aviva Lev-Ari, PhD, RN
Glycobiology vs Proteomics: Glycobiologists Prespective in the effort to explain the origin, etiology and potential therapeutics for the Coronavirus Pandemic (COVID-19).
Actemra, immunosuppressive which was designed to treat rheumatoid arthritis but also approved in 2017 to treat cytokine storms in cancer patients SAVED the sickest of all COVID-19 patients
The Accelerating COVID-19 Therapeutic Interventions and Vaccines (ACTIV) Partnership on May 18, 2020: Leadership of AbbVie, Amgen, AstraZeneca, Bristol Myers Squibb, Eisai, Eli Lilly, Evotec, Gilead, GlaxoSmithKline, Johnson & Johnson, KSQ Therapeutics, Merck, Novartis, Pfizer, Roche, Sanofi, Takeda, and Vir. We also thank multiple NIH institutes (especially NIAID), the FDA, BARDA, CDC, the European Medicines Agency, the Department of Defense, the VA, and the Foundation for NIH
Tweets & Retweets 2020 World Medical Innovation Forum – COVID-19, AI and the Future of Medicine, Featuring Harvard and Industry Leader Insights – MGH & BWH, Virtual Event: Monday, May 11, 8:15 a.m. – 5:15 p.m. ET
A Series of Recently Published Papers Report the Development of SARS-CoV2 Neutralizing Antibodies and Passive Immunity toward COVID19
Curator: Stephen J. Williams, Ph.D.
Passive Immunity and Treatment of Infectious Diseases
The ability of one person to pass on immunity to another person (passive immunity) is one of the chief methods we develop immunity to many antigens. For instance, maternal antibodies are passed to the offspring in the neonatal setting as well as in a mother’s milk during breast feeding. In the clinical setting this is achieved by transferring antibodies from one patient who has been exposed to an antigen (like a virus) to the another individual. However, the process of purifying the most efficacious antibody as well as its mass production is limiting due to its complexity and cost and can be prohibitively long delay during a pandemic outbreak, when therapies are few and needed immediately. Regardless, the benefits of developing neutralizing antibodies to confer passive immunity versus development of a vaccine are evident, as the former takes considerable less time than development of a safe and effective vaccine. For a good review on the development and use of neutralizing antibodies and the use of passive immunity to treat infectious diseases please read the following review:
Antibodies have been used for over a century in the prevention and treatment of infectious disease. They are used most commonly for the prevention of measles, hepatitis A, hepatitis B, tetanus, varicella, rabies, and vaccinia. Although their use in the treatment of bacterial infection has largely been supplanted by antibiotics, antibodies remain a critical component of the treatment of diptheria, tetanus, and botulism. High-dose intravenous immunoglobulin can be used to treat certain viral infections in immunocompromised patients (e.g., cytomegalovirus, parvovirus B19, and enterovirus infections). Antibodies may also be of value in toxic shock syndrome, Ebola virus, and refractory staphylococcal infections. Palivizumab, the first monoclonal antibody licensed (in 1998) for an infectious disease, can prevent respiratory syncytial virus infection in high-risk infants. The development and use of additional monoclonal antibodies to key epitopes of microbial pathogens may further define protective humoral responses and lead to new approaches for the prevention and treatment of infectious diseases.
TABLE 1
Summary of the efficacy of antibody in the prevention and treatment of infectious diseases
A Great Explanation of Active versus Passive Immunity by Dr. John Campbell, one of the pioneers in the field of immunology:Antibodies have been used for over a century in the prevention and treatment of infectious disease. They are used most commonly for the prevention of measles, hepatitis A, hepatitis B, tetanus, varicella, rabies, and vaccinia. Although their use in the treatment of bacterial infection has largely been supplanted by antibiotics, antibodies remain a critical component of the treatment of diptheria, tetanus, and botulism. High-dose intravenous immunoglobulin can be used to treat certain viral infections in immunocompromised patients (e.g., cytomegalovirus, parvovirus B19, and enterovirus infections). Antibodies may also be of value in toxic shock syndrome, Ebola virus, and refractory staphylococcal infections. Palivizumab, the first monoclonal antibody licensed (in 1998) for an infectious disease, can prevent respiratory syncytial virus infection in high-risk infants. The development and use of additional monoclonal antibodies to key epitopes of microbial pathogens may further define protective humoral responses and lead to new approaches for the prevention and treatment of infectious diseases.
However, developing successful neutralizing antibodies can still be difficult but with the latest monoclonal antibody technology, as highlighted by the following papers, this process has made much more efficient. In addition, it is not feasable to isolate antibodies from the plasma of covalescent patients in a scale that is needed for a worldwide outbreak.
When fighting off foreign invaders, our bodies make antibodies precisely produced for the task. The reason vaccines offer such long-lasting protection is they train the immune system to identify a pathogen, so immune cells remember and are ready to attack the virus when it appears. Monoclonal antibodies for coronavirus would take the place of the ones our bodies might produce to fight the disease. The manufactured antibodies would be infused into the body to either tamp down an existing infection, or to protect someone who has been exposed to the virus. However, these drugs are synthetic versions of the convalescent plasma treatments that rely on antibodies from people who have recovered from infection. But the engineered versions are easier to scale because they’re manufactured in rats, rather than from plasma donors.
The following papers represent the latest published work on development of therapeutic and prophylactic neutralizing antibodies to the coronavirus SARS-CoV2
1. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody.
SARS-CoV-2 is a newly emerged coronavirus responsible for the current COVID-19 pandemic that has resulted in more than 3.7 million infections and 260,000 deaths as of 6 May 20201,2. Vaccine and therapeutic discovery efforts are paramount to curb the pandemic spread of this zoonotic virus. The SARS-CoV-2 spike (S) glycoprotein promotes entry into host cells and is the main target of neutralizing antibodies. Here we describe multiple monoclonal antibodies targeting SARS-CoV-2 S identified from memory B cells of an individual who was infected with SARS-CoV in 2003. One antibody, named S309, potently neutralizes SARS-CoV-2 and SARS-CoV pseudoviruses as well as authentic SARS-CoV-2 by engaging the S receptor-binding domain. Using cryo-electron microscopy and binding assays, we show that S309 recognizes a glycan-containing epitope that is conserved within the sarbecovirus subgenus, without competing with receptor attachment. Antibody cocktails including S309 along with other antibodies identified here further enhanced SARS-CoV-2 neutralization and may limit the emergence of neutralization-escape mutants. These results pave the way for using S309- and S309-containing antibody cocktails for prophylaxis in individuals at high risk of exposure or as a post-exposure therapy to limit or treat severe disease.
2. Potent neutralizing antibodies against SARS-CoV-2 identified by high-throughput single-cell sequencing of convalescent patients’ B cells
The COVID-19 pandemic urgently needs therapeutic and prophylactic interventions. Here we report the rapid identification of SARS-CoV-2 neutralizing antibodies by high-throughput single-cell RNA and VDJ sequencing of antigen-enriched B cells from 60 convalescent patients. From 8,558 antigen-binding IgG1+ clonotypes, 14 potent neutralizing antibodies were identified with the most potent one, BD-368-2, exhibiting an IC50 of 1.2 ng/mL and 15 ng/mL against pseudotyped and authentic SARS-CoV-2, respectively. BD-368-2 also displayed strong therapeutic and prophylactic efficacy in SARS-CoV-2-infected hACE2-transgenic mice. Additionally, the 3.8Å Cryo-EM structure of a neutralizing antibody in complex with the spike-ectodomain trimer revealed the antibody’s epitope overlaps with the ACE2 binding site. Moreover, we demonstrated that SARS-CoV-2 neutralizing antibodies could be directly selected based on similarities of their predicted CDR3H structures to those of SARS-CoV neutralizing antibodies. Altogether, we showed that human neutralizing antibodies could be efficiently discovered by high-throughput single B-cell sequencing in response to pandemic infectious diseases.
3. A human monoclonal antibody blocking SARS-CoV-2 infection
The emergence of the novel human coronavirus SARS-CoV-2 in Wuhan, China has caused a worldwide epidemic of respiratory disease (COVID-19). Vaccines and targeted therapeutics for treatment of this disease are currently lacking. Here we report a human monoclonal antibody that neutralizes SARS-CoV-2 (and SARS-CoV) in cell culture. This cross-neutralizing antibody targets a communal epitope on these viruses and may offer potential for prevention and treatment of COVID-19.
Extra References on Development of Neutralizing antibodies for COVID19 {Sars-CoV2} published this year (2020) [1-4]
Fan P, Chi X, Liu G, Zhang G, Chen Z, Liu Y, Fang T, Li J, Banadyga L, He S et al: Potent neutralizing monoclonal antibodies against Ebola virus isolated from vaccinated donors. mAbs 2020, 12(1):1742457.
Dussupt V, Sankhala RS, Gromowski GD, Donofrio G, De La Barrera RA, Larocca RA, Zaky W, Mendez-Rivera L, Choe M, Davidson E et al: Potent Zika and dengue cross-neutralizing antibodies induced by Zika vaccination in a dengue-experienced donor. Nature medicine 2020, 26(2):228-235.
Young CL, Lyons AC, Hsu WW, Vanlandingham DL, Park SL, Bilyeu AN, Ayers VB, Hettenbach SM, Zelenka AM, Cool KR et al: Protection of swine by potent neutralizing anti-Japanese encephalitis virus monoclonal antibodies derived from vaccination. Antiviral research 2020, 174:104675.
IBM Releases Novel AI-Powered Technologies to Help Health and Research Community Accelerate the Discovery of Medical Insights and Treatments for COVID-19
IBM Research has been actively developing new cloud and AI-powered technologies that can help researchers across a variety of scientific disciplines accelerate the process of discovery. As the COVID-19 pandemic unfolds, we continue to ask how these technologies and our scientific knowledge can help in the global battle against coronavirus.
Today, we are making available multiple novel, free resources from across IBM to help healthcare researchers, doctors and scientists around the world accelerate COVID-19 drug discovery: from gathering insights, to applying the latest virus genomic information and identifying potential targets for treatments, to creating new drug molecule candidates.
Though some of the resources are still in exploratory stages, IBM is making them available to qualifying researchers at no charge to aid the international scientific investigation of COVID-19.
Healthcare agencies and governments around the world have quickly amassed medical and other relevant data about the pandemic. And, there are already vast troves of medical research that could prove relevant to COVID-19. Yet, as with any large volume of disparate data sources, it is difficult to efficiently aggregate and analyze that data in ways that can yield scientific insights.
To help researchers access structured and unstructured data quickly, we are offering a cloud-based AI research resource that has been trained on a corpus of thousands of scientific papers contained in the COVID-19 Open Research Dataset (CORD-19), prepared by the White House and a coalition of research groups, and licensed databases from the DrugBank, Clinicaltrials.gov and GenBank. This tool uses our advanced AI and allows researchers to pose specific queries to the collections of papers and to extract critical COVID-19 knowledge quickly. Please note, access to this resource will be granted only to qualified researchers. To learn more and request access, please click here.
Aiding the Hunt for Treatments
The traditional drug discovery pipeline relies on a library of compounds that are screened, improved, and tested to determine safety and efficacy. In dealing with new pathogens such as SARS-CoV-2, there is the potential to enhance the compound libraries with additional novel compounds. To help address this need, IBM Research has recently created a new, AI-generative framework which can rapidly identify novel peptides, proteins, drug candidates and materials.
We have applied this AI technology against three COVID-19 targets to identify 3,000 new small molecules as potential COVID-19 therapeutic candidates. IBM is releasing these molecules under an open license, and researchers can study them via a new interactive molecular explorer tool to understand their characteristics and relationship to COVID-19 and identify candidates that might have desirable properties to be further pursued in drug development.
To streamline efforts to identify new treatments for COVID-19, we are also making the IBM Functional Genomics Platform available for free for the duration of the pandemic. Built to discover the molecular features in viral and bacterial genomes, this cloud-based repository and research tool includes genes, proteins and other molecular targets from sequenced viral and bacterial organisms in one place with connections pre-computed to help accelerate discovery of molecular targets required for drug design, test development and treatment.
Select IBM collaborators from government agencies, academic institutions and other organizations already use this platform for bacterial genomic study. And now, those working on COVID-19 can request the IBM Functional Genomics Platform interface to explore the genomic features of the virus. Access to the IBM Functional Genomics Platform will be prioritized for those conducting COVID-19 research. To learn more and request access, please click here.
Drug and Disease Information
Clinicians and healthcare professionals on the frontlines of care will also have free access to hundreds of pieces of evidence-based, curated COVID-19 and infectious disease content from IBM Micromedex and EBSCO DynaMed. Using these two rich decision support solutions, users will have access to drug and disease information in a single and comprehensive search. Clinicians can also provide patients with consumer-friendly patient education handouts with relevant, actionable medical information. IBM Micromedex is one of the largest online reference databases for medication information and is used by more than 4,500 hospitals and health systems worldwide. EBSCO DynaMed provides peer-reviewed clinical content, including systematic literature reviews in 28 specialties for comprehensive disease topics, health conditions and abnormal findings, to highly focused topics on evaluation, differential diagnosis and management.
The scientific community is working hard to make important new discoveries relevant to the treatment of COVID-19, and we’re hopeful that releasing these novel tools will help accelerate this global effort. This work also outlines our long-term vision for the future of accelerated discovery, where multi-disciplinary scientists and clinicians work together to rapidly and effectively create next generation therapeutics, aided by novel AI-powered technologies.
Grant will allow company to accelerate access to its AI solutions and use of ultrasound in COVID-19 emergency settings
TEL AVIV, Israel, May 12, 2020 /PRNewswire-PRWeb/ — DiA Imaging Analysis, a leading provider of AI based ultrasound analysis solutions, today announced that it has received a government grant from the Israel Innovation Authority (IIA) to develop solutions for ultrasound imaging analysis of COVID-19 patients using Artificial Intelligence (AI).Using ultrasound in point of care emergency settings has gained momentum since the outbreak of COVID-19 pandemic. In these settings, which include makeshift hospital COVID-19 departments and triage “tents,” portable ultrasound offers clinicians diagnostic decision support, with the added advantage of being easier to disinfect and eliminating the need to transport patients from one room to another.However, analyzing ultrasound images is a process that it is still mostly done visually, leading to a growing market need for automated solutions and decision support.As the leading provider of AI solutions for ultrasound analysis and backed by Connecticut Innovations, DiA makes ultrasound analysis smarter and accessible to both new and expert ultrasound users with various levels of experience. The company’s flagship LVivo Cardio Toolbox for AI-based cardiac ultrasound analysis enables clinicians to automatically generate objective clinical analysis, with increased accuracy and efficiency to support decisions about patient treatment and care.
The IIA grant provides a budget of millions NIS to increase access to DiA’s solutions for users in Israel and globally, and accelerate R&D with a focus on new AI solutions for COVID-19 patient management. DiA solutions are vendor-neutral and platform agnostic, as well as powered to run in low processing, mobile environments like handheld ultrasound.Recent data highlights the importance of looking at the heart during the progression of COVID-19, with one study citing 20% of patients hospitalized with COVID-19 showing signs of heart damage and increased mortality rates in those patients. DiA’s LVivo cardiac analysis solutions automatically generate objective, quantified cardiac ultrasound results to enable point-of-care clinicians to assess cardiac function on the spot, near patients’ bedside.
According to Dr. Ami Applebaum, the Chairman of the Board of the IIA, “The purpose of IIA’s call was to bring solutions to global markets for fighting COVID-19, with an emphasis on relevancy, fast time to market and collaborations promising continuity of the Israeli economy. DiA meets these requirements with AI innovation for ultrasound.”DiA has received several FDA/CE clearances and established distribution partnerships with industry leading companies including GE Healthcare, IBM Watson and Konica Minolta, currently serving thousands of end users worldwide.”We see growing use of ultrasound in point of care settings, and an urgent need for automated, objective solutions that provide decision support in real time,” said Hila Goldman-Aslan, CEO and Co-founder of DiA Imaging Analysis, “Our AI solutions meet this need by immediately helping clinicians on the frontlines to quickly and easily assess COVID-19 patients’ hearts to help guide care delivery.”
About DiA Imaging Analysis:
DiA Imaging Analysis provides advanced AI-based ultrasound analysis technology that makes ultrasound accessible to all. DiA’s automated tools deliver fast and accurate clinical indications to support the decision-making process and offer better patient care. DiA’s AI-based technology uses advanced pattern recognition and machine-learning algorithms to automatically imitate the way the human eye detects image borders and identifies motion. Using DiA’s tools provides automated and objective AI tools, helps reduce variability among users, and increases efficiency. It allows clinicians with various levels of experience to quickly and easily analyze ultrasound images.
Almost 60,000 viewed the AACR 2020 Virtual meeting for the April 27 session
The following speakers were the first cancer researchers treating patients at the epicenters of the pandemic even though nothing was known about the virus
The experience of treating patients with cancer during the COVID-19 pandemic in China Li Zhang, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology
reporting a retrospective study from three hospitals from Wuhan
2.2% of Wuhan cancer patients were COVID positive; most were lung cancers and most male; 35% were stage four
most have hospital transmission of secondary infection; had severe events when admitted
74% were prescribed antivirals like ganciclovir and others; iv IgG was given to some
mortailtiy rate of 26%; by April 4 54% were cured and discharged; median time of infection to severe event was 7 days; clinical presentation SARS sepsis, and shock
by day 10 in lung cancer patients, see lung path but after supportive therapy improved
cancer patients at stage four who did not receive therapy were at higher risk
cancer patients who had received chemo in last 14 days had higher risk of infection
they followed up with cancer patients on I/O inhibitors; it seemed there was only one patient that contracted COVID19 so there may not be as much risk with immune checkpoint inhibitors
TERAVOLT (Thoracic cancERs international coVid 19 cOLlaboraTion): First results of a global collaboration to address the impact of COVID-19 in patients with thoracic malignancies
Dr Marina Chiara Garassino is the Chief of the Thoracic Oncology Unit at Istituto Nazionale dei Tumori, Milan, Italy. She leads the strategy for clinical and translational research in advanced and locally advanced NSCLC, SCLC, mesothelioma and thymic malignancies. Istituto Nazionale dei Tumori in Milan is the most important comprehensive cancer in Italy and one of the most important in Europe. As a medical oncologist, she has done research in precision medicine and in immuno-oncology. Her main research interests have been mainly development of new drugs and therapeutical strategies and biomarkers. She has contributed to over 150 peer-reviewed publications, including publications as first or last author in the New England Journal of Medicine, Lancet Oncology, Journal of Clinical Oncology, Annals of Oncology. She has delivered many presentations at international congresses, including AACR, ASCO, ECCO, ESMO, WCLC. Her education includes a degree and further specialization in Medical Oncology at Università degli Studi in Milan. She achieved a Master Degree in Oncology management at University of Economics “Luigi Bocconi”. She completed her training with an ESMO Clinical fellowship in 2009 at Christie’s Hospital in Manchester (UK). She was a member of the EMA SAG (Scientific Advisory Group). She is serving as ESMO Council member as the Chair of the National Societies Committee. She was the ESMO National Representative for Italy for 5 years (2011-2017). She is serving on several ESMO Committees (Public Policy extended Committee, Press Committee, Women for Oncology Committee, Lung Cancer faculty, Membership Committee).She used to be an active member of the Young Oncologist Committee. She’s serving on both ESMO, WCLC and ASCO annual congress Lung Cancer Track (2019, and 2020), Chair of ESMO National Societies, from 2019. She is the founder and president of Women for Oncology Italy.
2 million confirmed cases but half of patients are asymptomatic and not tested; pooled prevalance of COVID in cancer patients in Italy was 2%; must take them as high risk patients
they were not prepared for pandemic lasting for months instead of days; March 15 in middle of outbreak they started TERAVOLT registry; by March 26 had IRB approval; they are accruing 17 new patients per week; Ontario also joined in and has become worldwide (21 countries involved); in registry they also included radiologic exams and COVID testing result
most patients were males and many smokers; 75% had SCLC; 83% of cases had one comorbility like hypertension and one third had at least one comorbility; 73.9% of patients were on treatment (they see this in their clinic: 30% on chemo or TKI alone; other patients were just on folowup
most of symptoms overlap with symptoms of lung cancer like pneumonia and pneumonitits and multi organ failure; most were hospitalized
unexpected high mortality among lung cancer patients with COVID19; this mortality seems due to COVID and not to cancer;
study had some limitations like short followup and some surgical cases so some bias may be present
she stresses don’t go it alone and make your own registry JOIN A REGISTRY
Outcome of cancer patients infected with COVID-19, including toxicity of cancer treatments
Fabrice Barlesi @barlesi
Gustave Roussy Cancer Campus
Professor Fabrice Barlesi
As a specialist in lung cancer, precision medicine and cancer immunology, Prof. Fabrice Barlesi is a major contributor to research in the field of novel oncological therapies. He was apppointed General Director of Gustave Roussy in January 2020.
Fabrice Barlesi is Professor of Medicine at the University of Aix-Marseille. He has been head of the Multidisciplinary Oncology and Innovative Therapies Department of the Nord Hospital in Marseille (Marseille Public Hospitals) and the Marseille Centre for Early Trials in Oncology (CLIP2) which were established by him. He holds a doctorate in Sciences and Management with methods of analysis of health care systems, together with an ESSEC (international business school) master’s degree in general hospital management.
Professor Barlesi was also a co-founder of the Marseille Immunopôle French Immunology network, which aims to coordinate immunological expertise in the Aix-Marseille metropolitan area. In this context, he has organised PIONeeR (Investment in the future RHU 2017), the major international Hospital-University research project whose objective is to improve understanding of resistance to immunotherapy – anti-PD1(L1) – in lung cancer and help to prevent and overcome it. He was also vice-chair of the PACA (Provence, Alps and Côte d’Azur) Region Cancer Research Directorate.
Professor Barlesi is the author and co-author of some 300 articles in international journals and specialist publications. In 2018, the European Society of Medical Oncology (ESMO) and the International Association for the Study of Lung Cancer (IASLC) awarded him the prestigious Heine H. Hansen prize. He appears in the 2019 world list of most influential researchers (Highly cited researchers, Web of Science Group).
March 14 started protective measures and at peak had increased commited beds at highest rate
12% of cancer patients tested positive for COVID; (by RTPCR); they curated data across different chemo regimens used
they retrospectively collected data; primary endpoint was clinical worsening; median of disease 13 days;
they actually had more breast cancer patients and other solid malignancies; 23% of covid cases no symptoms; 83% finally did have the symptoms after followup; diarhea actually in 10% of cases so clinics are seeing this as a symptom
CT scan showed 66% cases had pneumonitits like display; 25% patients were managed as outpatient
24% patients worsened during treatment but 75% were able to go home (treated at home or well)
I/O did not have negative outcome and you can use these drugs without increasing risk to COVID
although many clinical trials have been hindered they are actively recruiting for COVID-cancer studies
outcomes with respect to death and symptoms are comparable to worldwide stats
Adapting oncologic practice to COVID19 outbreak: From outpatient triage to risk assessment for specific treatment in Madrid, Spain Carlos Gomez-Martin
Octubre University Hospital
MOST slides were DO NOT POST so as requested data will not be shown; this study will be published soon
Summary is that Spain is seeing statistics like other European countries and similar results
Tocilizumab, the IL6 antagonists had been suggested as a treatment for cytokine storm and they are involved in a trial with this agent; results will be published
Experience in using oncology drugs in patients with COVID-19
Paolo A. Ascierto
Istituto Nazionale Tumori IRCCS Fondazione Pascale
giving surgery only for patients at highest risk of cancer mortality so using neoadjuvant therapy more often
telemedicine is a viable strategy for patient consult
for metastatic melanoma they are given highest priority for treatment
they are conducting a tocilizumab clinical trial and have accrued over 300 patients
results are in press so please look for publication soon
also can use TNF inhibitor, JAK inhibitor, IL1 inhibitor to treat cytokine storm
COVID-19 and cancer: Flattening the curve but widening disparities Louis P. Voigt
Memorial Sloan Kettering Cancer Center
Sloan has performed about 5000 COVID tests; 78 patients needed hospitilization; 15 died; 40% still in ICU
they do see many African American patients
mortality rates in US (published) have been around 50-60 % for cancer patients with COVID; Sloan prelim results are lower but still accruing data
Patients with cancer appear more vulnerable to SARS-COV-2: A multi-center study during the COVID-19 outbreak Hongbing Cai
Zhongnan Hospital of Wuhan University
metastatic cancer showed much higher risk than non cancer but non metastatic showed increased risk too
main criteria of outcome was ICU admission
patients need to be isolated and personalized treatment plans need to be made
many comparisons were between non cancer and cancer which was clearest significance; had not looked at cancer types or stage grade or treatment
it appears that there are more questions right now than answers so data collection is a priority
Each sheet in the workbook is separated by current COVID-19 vaccine trials, currents COVID-19 trials with the IL6R (interleukin 6 receptor) antagonist tocilizumab, and all COVID related trials. The Excel spreadsheet also contains links to more information about the trials.
As of April 15, 2020 the number of listed trials are as follows:
clinicaltrials.gov search terms
Number of results
Number of completed trials
Number of trials currently recruiting
COVID-19 or SARS-CoV-2
410
5 completed
5 withdrawn
192
1st row terms + vaccine
28
0
15
1st row terms + tocilizumab
16
0
10
1st row terms + hydroxychloroquine
61
1
22
A few highlights of the COVID related trials on clinicaltrials.gov
This is an open label, randomized, controlled, pilot clinical study in patients with COVID-19, to obtain preliminary biologic, physiologic, and clinical data in patients with COVID-19 treated with rhACE2 or control patients, to help determine whether a subsequent Phase 2B trial is warranted.
Condition or disease
Intervention/treatment
Phase
COVID-19
Drug: Recombinant human angiotensin-converting enzyme 2 (rhACE2)
Not Applicable
Detailed Description:
This is a small pilot study investigating whether there is any efficacy signal that warrants a larger Phase 2B trial, or any harm that suggests that such a trial should not be done. It is not expected to produce statistically significant results in the major endpoints. The investigators will examine all of the biologic, physiological, and clinical data to determine whether a Phase 2B trial is warranted.
Primary efficacy analysis will be carried only on patients receiving at least 4 doses of active drug. Safety analysis will be carried out on all patients receiving at least one dose of active drug.
It is planned to enroll more than or equal to 24 subjects with COVID-19. It is expected to have at least 12 evaluable patients in each group.
Experimental group: 0.4 mg/kg rhACE2 IV BID and standard of care Control group: standard of care
Intervention duration: up to 7 days of therapy
No planned interim analysis.
Study was withdrawn before participants were enrolled.
Gut dysbiosis co-exists in patients with coronavirus pneumonia. Some of these patients would develop secondary bacterial infections and antibiotic-associated diarrhea (AAD). The recent study on using washed microbiota transplantation (WMT) as rescue therapy in critically ill patients with AAD demonstrated the important clinical benefits and safety of WMT. This clinical trial aims to evaluate the outcome of WMT combining with standard therapy for patients with 2019-novel coronavirus pneumonia, especially for those patients with dysbiosis-related conditions.
Detailed Description:
An ongoing outbreak of 2019 novel coronavirus was reported in Wuhan, China. 2019-nCoV has caused a cluster of pneumonia cases, and posed continuing epidemic threat to China and even global health. Unfortunately, there is currently no specific effective treatment for the viral infection and the related serious complications. It is in urgent need to find a new specific effective treatment for the 2019-nCoV infection. According to Declaration of Helsinki and International Ethical Guidelines for Health-related Research Involving Humans, the desperately ill patients with 2019-nCov infection during disease outbreaks have a moral right to try unvalidated medical interventions (UMIs) and that it is therefore unethical to restrict access to UMIs to the clinical trial context.
There is a vital link between the intestinal tract and respiratory tract, which was exemplified by intestinal complications during respiratory disease and vice versa. Some of these patients can develop secondary bacterial infections and antibiotic-associated diarrhea (AAD). The recent study on using washed microbiota transplantation (WMT) as rescue therapy in critically ill patients with AAD demonstrated the important clinical benefits and safety of WMT. Additionally, the recent animal study provided direct evidence supporting that antibiotics could decrease gut microbiota and the lung stromal interferon signature and facilitate early influenza virus replication in lung epithelia. Importantly, the above antibiotics caused negative effects can be reversed by fecal microbiota transplantation (FMT) which suggested that FMT might be able to induce a significant improvement in the respiratory virus infection. Another evidence is that the microbiota could confer protection against certain virus infection such as influenza virus and respiratory syncytial virus by priming the immune response to viral evasion. The above results suggested that FMT might be a new therapeutic option for the treatment of virus-related pneumonia. The methodology of FMT recently was coined as WMT, which is dependent on the automatic facilities and washing process in a laboratory room. Patients underwent WMT with the decreased rate of adverse events and unchanged clinical efficacy in ulcerative colitis and Crohn’s disease. This clinical trial aims to evaluate the outcome of WMT combining with standard therapy for patients with novel coronavirus pneumonia, especially for those patients with dysbiosis-related conditions.
Responsible Party:
Faming Zhang, Director of Medical Center for Digestive Diseases, The Second Hospital of Nanjing Medical University
The 2019 novel coronavirus pneumonia outbroken in Wuhan, China, which spread quickly to 26 countries worldwide and presented a serious threat to public health. It is mainly characterized by fever, dry cough, shortness of breath and breathing difficulties. Some patients may develop into rapid and deadly respiratory system injury with overwhelming inflammation in the lung. Currently, there is no effective treatment in clinical practice. The present clinical trial is to explore the safety and efficacy of Human Umbilical Cord Mesenchymal Stem Cells (UC-MSCs) therapy for novel coronavirus pneumonia patients.
Detailed Description:
Since late December 2019, human pneumonia cases infected by a novel coronavirus (2019-nCoV) were firstly identified in Wuhan, China. As the virus is contagious and of great epidemic, more and more cases have found in other areas of China and abroad. Up to February 24, a total of 77, 779 confirmed cases were reported in China. At present, there is no effective treatment for patients identified with novel coronavirus pneumonia. Therefore, it’s urgent to explore more active therapeutic methods to cure the patients.
Recently, some clinical researches about the 2019 novel coronavirus pneumonia published in The Lancet and The New England Journal of Medicine suggested that massive inflammatory cell infiltration and inflammatory cytokines secretion were found in patients’ lungs, alveolar epithelial cells and capillary endothelial cells were damaged, causing acute lung injury. It seems that the key to cure the pneumonia is to inhibit the inflammatory response, resulting to reduce the damage of alveolar epithelial cells and endothelial cells and repair the function of the lung.
Mesenchymal stem cells (MSCs) are widely used in basic research and clinical application. They are proved to migrate to damaged tissues, exert anti-inflammatory and immunoregulatory functions, promote the regeneration of damaged tissues and inhibit tissue fibrosis. Studies have shown that MSCs can significantly reduce acute lung injury in mice caused by H9N2 and H5N1 viruses by reducing the levels of proinflammatory cytokines and the recruitment of inflammatory cells into the lungs. Compared with MSCs from other sources, human umbilical cord-derived MSCs (UC-MSCs) have been widely applied to various diseases due to their convenient collection, no ethical controversy, low immunogenicity, and rapid proliferation rate. In our recent research, we confirmed that UC-MSCs can significantly reduce inflammatory cell infiltration and inflammatory factors expression in lung tissue, and significantly protect lung tissue from endotoxin (LPS) -induced acute lung injury in mice.
The purpose of this clinical study is to investigate safety and efficiency of UC-MSCs in treating pneumonia patients infected by 2019-nCoV. The investigators planned to recruit 48 patients aged from 18 to 75 years old and had no severe underlying diseases. In the cell treatment group, 24 patients received 0.5*10E6 UC-MSCs /kg body weight intravenously treatment 4 times every other day besides conventional treatment. In the control group, other 24 patients received conventional treatment plus 4 times of placebo intravenously. The lung CT, blood biochemical examination, lymphocyte subsets, inflammatory factors, 28-days mortality, etc will be evaluated within 24h and 1, 2, 4, 8 weeks after UC-MSCs treatment.
Sponsor:
Puren Hospital Affiliated to Wuhan University of Science and Technology
Collaborator:
Wuhan Hamilton Bio-technology Co., Ltd
Study was withdrawn before participants were enrolled.
There are currently no clinical studies reporting clinical characteristics difference between the hypertension patients with and without ACEI treatment when suffered with novel coronavirus infection in China
Detailed Description:
At present, the outbreak of the new coronavirus (2019-nCoV) infection in Wuhan and Hubei provinces has attracted great attention from the medical community across the country. Both 2019-nCoV and SARS viruses are coronaviruses, and they have a large homology.
Published laboratory studies have suggested that SARS virus infection and its lung injury are related to angiotensin-converting enzyme 2 (ACE2) in lung tissue. And ACE and ACE2 in the renin-angiotensin system (RAS) are vital central links to maintain hemodynamic stability and normal heart and kidney function in vivo.
A large amount of evidence-based medical evidence shows that ACE inhibitors are the basic therapeutic drugs for maintaining hypertension, reducing the risk of cardiovascular, cerebrovascular, and renal adverse events, improving quality of life, and prolonging life in patients with hypertension. Recent experimental studies suggest that treatment with ACE inhibitors can significantly reduce pulmonary inflammation and cytokine release caused by coronavirus infection.
ACEI treatment
hypertension patients with ACEI treatment when suffered with novel coronavirus infection in China
Control
hypertension patients without ACEI treatment when suffered with novel coronavirus infection in China
Locations
China
The First Affiliated Hospital of Chongqing Medical University Chongqing, China
Sponsors and Collaborators Chongqing Medical University
Responsible PI:
Dongying Zhang, Associate Professor, Chongqing Medical University
Withdrawn (Similar projects have been registered, and it needs to be withdrawn.)
Structure-guided Drug Discovery: (1) The Coronavirus 3CL hydrolase (Mpro) enzyme (main protease) essential for proteolytic maturation of the virus and (2) viral protease, the RNA polymerase, the viral spike protein, a viral RNA as promising two targets for discovery of cleavage inhibitors of the viral spike polyprotein preventing the Coronavirus Virion the spread of infection
Curators and Reporters: Stephen J. Williams, PhD and Aviva Lev-Ari, PhD, RN
Therapeutical options to coronavirus (2019-nCoV) include consideration of the following:
(a) Monoclonal and polyclonal antibodies
(b) Vaccines
(c) Small molecule treatments (e.g., chloroquinolone and derivatives), including compounds already approved for other indications
(d) Immuno-therapies derived from human or other sources
Structure of the nCoV trimeric spike
The World Health Organization has declared the outbreak of a novel coronavirus (2019-nCoV) to be a public health emergency of international concern. The virus binds to host cells through its trimeric spike glycoprotein, making this protein a key target for potential therapies and diagnostics. Wrapp et al. determined a 3.5-angstrom-resolution structure of the 2019-nCoV trimeric spike protein by cryo–electron microscopy. Using biophysical assays, the authors show that this protein binds at least 10 times more tightly than the corresponding spike protein of severe acute respiratory syndrome (SARS)–CoV to their common host cell receptor. They also tested three antibodies known to bind to the SARS-CoV spike protein but did not detect binding to the 2019-nCoV spike protein. These studies provide valuable information to guide the development of medical counter-measures for 2019-nCoV. [Bold Face Added by ALA]
The outbreak of a novel coronavirus (2019-nCoV) represents a pandemic threat that has been declared a public health emergency of international concern. The CoV spike (S) glycoprotein is a key target for vaccines, therapeutic antibodies, and diagnostics. To facilitate medical countermeasure development, we determined a 3.5-angstrom-resolution cryo–electron microscopy structure of the 2019-nCoV S trimer in the prefusion conformation. The predominant state of the trimer has one of the three receptor-binding domains (RBDs) rotated up in a receptor-accessible conformation. We also provide biophysical and structural evidence that the 2019-nCoV S protein binds angiotensin-converting enzyme 2 (ACE2) with higher affinity than does severe acute respiratory syndrome (SARS)-CoV S. Additionally, we tested several published SARS-CoV RBD-specific monoclonal antibodies and found that they do not have appreciable binding to 2019-nCoV S, suggesting that antibody cross-reactivity may be limited between the two RBDs. The structure of 2019-nCoV S should enable the rapid development and evaluation of medical countermeasures to address the ongoing public health crisis.
SOURCE
Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation
Recent emergence of the COVID-19 coronavirus has resulted in a WHO-declared public health emergency of international concern. Research efforts around the world are working towards establishing a greater understanding of this particular virus and developing treatments and vaccines to prevent further spread.
While PDB entry 6lu7 is currently the only public-domain 3D structure from this specific coronavirus, the PDB contains structures of the corresponding enzyme from other coronaviruses. The 2003 outbreak of the closely-related Severe Acute Respiratory Syndrome-related coronavirus (SARS) led to the first 3D structures, and today there are more than 200 PDB structures of SARS proteins. Structural information from these related proteins could be vital in furthering our understanding of coronaviruses and in discovery and development of new treatments and vaccines to contain the current outbreak.
The coronavirus 3CL hydrolase (Mpro) enzyme, also known as the main protease, is essential for proteolytic maturation of the virus. It is thought to be a promising target for discovery of small-molecule drugs that would inhibit cleavage of the viral polyprotein and prevent spread of the infection.
Comparison of the protein sequence of the COVID-19 coronavirus 3CL hydrolase (Mpro) against the PDB archive identified 95 PDB proteins with at least 90% sequence identity. Furthermore, these related protein structures contain approximately 30 distinct small molecule inhibitors, which could guide discovery of new drugs. Of particular significance for drug discovery is the very high amino acid sequence identity (96%) between the COVID-19 coronavirus 3CL hydrolase (Mpro) and the SARS virus main protease (PDB 1q2w). Summary data about these closely-related PDB structures are available (CSV) to help researchers more easily find this information. In addition, the PDB houses 3D structure data for more than 20 unique SARS proteins represented in more than 200 PDB structures, including a second viral protease, the RNA polymerase, the viral spike protein, a viral RNA, and other proteins (CSV).
Public release of the COVID-19 coronavirus 3CL hydrolase (Mpro), at a time when this information can prove most vital and valuable, highlights the importance of open and timely availability of scientific data. The wwPDB strives to ensure that 3D biological structure data remain freely accessible for all, while maintaining as comprehensive and accurate an archive as possible. We hope that this new structure, and those from related viruses, will help researchers and clinicians address the COVID-19 coronavirus global public health emergency.
Update: Released COVID-19-related PDB structures include
PDB structure 6lu7 (X. Liu, B. Zhang, Z. Jin, H. Yang, Z. Rao Crystal structure of COVID-19 main protease in complex with an inhibitor N3 doi: 10.2210/pdb6lu7/pdb) Released 2020-02-05
PDB structure 6vsb (D. Wrapp, N. Wang, K.S. Corbett, J.A. Goldsmith, C.-L. Hsieh, O. Abiona, B.S. Graham, J.S. McLellan (2020) Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation Science doi: 10.1126/science.abb2507) Released 2020-02-26
PDB structure 6lxt (Y. Zhu, F. Sun Structure of post fusion core of 2019-nCoV S2 subunit doi: 10.2210/pdb6lxt/pdb) Released 2020-02-26
PDB structure 6lvn (Y. Zhu, F. Sun Structure of the 2019-nCoV HR2 Domain doi: 10.2210/pdb6lvn/pdb) Released 2020-02-26
PDB structure 6vw1
J. Shang, G. Ye, K. Shi, Y.S. Wan, H. Aihara, F. Li Structural basis for receptor recognition by the novel coronavirus from Wuhan doi: 10.2210/pdb6vw1/pdb
Released 2020-03-04
PDB structure 6vww
Y. Kim, R. Jedrzejczak, N. Maltseva, M. Endres, A. Godzik, K. Michalska, A. Joachimiak, Center for Structural Genomics of Infectious Diseases Crystal Structure of NSP15 Endoribonuclease from SARS CoV-2 doi: 10.2210/pdb6vww/pdb
Released 2020-03-04
PDB structure 6y2e
L. Zhang, X. Sun, R. Hilgenfeld Crystal structure of the free enzyme of the SARS-CoV-2 (2019-nCoV) main protease doi: 10.2210/pdb6y2e/pdb
Released 2020-03-04
PDB structure 6y2f
L. Zhang, X. Sun, R. Hilgenfeld Crystal structure (monoclinic form) of the complex resulting from the reaction between SARS-CoV-2 (2019-nCoV) main protease and tert-butyl (1-((S)-1-(((S)-4-(benzylamino)-3,4-dioxo-1-((S)-2-oxopyrrolidin-3-yl)butan-2-yl)amino)-3-cyclopropyl-1-oxopropan-2-yl)-2-oxo-1,2-dihydropyridin-3-yl)carbamate (alpha-ketoamide 13b) doi: 10.2210/pdb6y2f/pdb
Released 2020-03-04
PDB structure 6y2g
L. Zhang, X. Sun, R. Hilgenfeld Crystal structure (orthorhombic form) of the complex resulting from the reaction between SARS-CoV-2 (2019-nCoV) main protease and tert-butyl (1-((S)-1-(((S)-4-(benzylamino)-3,4-dioxo-1-((S)-2-oxopyrrolidin-3-yl)butan-2-yl)amino)-3-cyclopropyl-1-oxopropan-2-yl)-2-oxo-1,2-dihydropyridin-3-yl)carbamate (alpha-ketoamide 13b) doi: 10.2210/pdb6y2g/pdb
Released 2020-03-04
Coronavirus disease 2019 (COVID-19) is a global pandemic impacting nearly 170 countries/regions and more than 285,000 patients worldwide. COVID-19 is caused by the Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2), which invades cells through the angiotensin converting enzyme 2 (ACE2) receptor. Among those with COVID-19, there is a higher prevalence of cardiovascular disease and more than 7% of patients suffer myocardial injury from the infection (22% of the critically ill). Despite ACE2 serving as the portal for infection, the role of ACE inhibitors or angiotensin receptor blockers requires further investigation. COVID-19 poses a challenge for heart transplantation, impacting donor selection, immunosuppression, and post-transplant management. Thankfully there are a number of promising therapies under active investigation to both treat and prevent COVID-19. Key Words: COVID-19; myocardial injury; pandemic; heart transplant
Towler P, Staker B, Prasad SG, Menon S, Tang J, Parsons T, Ryan D, Fisher M, Williams D, Dales NA, Patane MA, Pantoliano MW (Apr 2004). “ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis”. The Journal of Biological Chemistry. 279 (17): 17996–8007. doi:10.1074/jbc.M311191200. PMID14754895.
Turner AJ, Tipnis SR, Guy JL, Rice G, Hooper NM (Apr 2002). “ACEH/ACE2 is a novel mammalian metallocarboxypeptidase and a homologue of angiotensin-converting enzyme insensitive to ACE inhibitors”. Canadian Journal of Physiology and Pharmacology. 80 (4): 346–53. doi:10.1139/y02-021. PMID12025971.
Zhang, Haibo; Penninger, Josef M.; Li, Yimin; Zhong, Nanshan; Slutsky, Arthur S. (3 March 2020). “Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target”. Intensive Care Medicine. Springer Science and Business Media LLC. doi:10.1007/s00134-020-05985-9. ISSN0342-4642. PMID32125455.
^Gurwitz, David (2020). “Angiotensin receptor blockers as tentative SARS‐CoV‐2 therapeutics”. Drug Development Research. doi:10.1002/ddr.21656. PMID32129518.
ACE2 receptors have been shown to be the entry point into human cells for some coronaviruses, including the SARSvirus.[10] A number of studies have identified that the entry point is the same for SARS-CoV-2,[11] the virus that causes COVID-19.[12][13][14][15]
Some have suggested that a decrease in ACE2 could be protective against Covid-19 disease[16], but others have suggested the opposite, that Angiotensin II receptor blocker drugs could be protective against Covid-19 disease via increasing ACE2, and that these hypotheses need to be tested by datamining of clinical patient records.[17]
We need your help! Folding@home is joining researchers around the world working to better understand the 2019 Coronavirus (2019-nCoV) to accelerate the open science effort to develop new life-saving therapies. By downloading Folding@Home, you can donate your unused computational resources to the Folding@home Consortium, where researchers working to advance our understanding of the structures of potential drug targets for 2019-nCoV that could aid in the design of new therapies. The data you help us generate will be quickly and openly disseminated as part of an open science collaboration of multiple laboratories around the world, giving researchers new tools that may unlock new opportunities for developing lifesaving drugs.
2019-nCoV is a close cousin to SARS coronavirus (SARS-CoV), and acts in a similar way. For both coronaviruses, the first step of infection occurs in the lungs, when a protein on the surface of the virus binds to a receptor protein on a lung cell. This viral protein is called the spike protein, depicted in red in the image below, and the receptor is known as ACE2. A therapeutic antibody is a type of protein that can block the viral protein from binding to its receptor, therefore preventing the virus from infecting the lung cell. A therapeutic antibody has already been developed for SARS-CoV, but to develop therapeutic antibodies or small molecules for 2019-nCoV, scientists need to better understand the structure of the viral spike protein and how it binds to the human ACE2 receptor required for viral entry into human cells.
Proteins are not stagnant—they wiggle and fold and unfold to take on numerous shapes. We need to study not only one shape of the viral spike protein, but all the ways the protein wiggles and folds into alternative shapes in order to best understand how it interacts with the ACE2 receptor, so that an antibody can be designed. Low-resolution structures of the SARS-CoV spike protein exist and we know the mutations that differ between SARS-CoV and 2019-nCoV. Given this information, we are uniquely positioned to help model the structure of the 2019-nCoV spike protein and identify sites that can be targeted by a therapeutic antibody. We can build computational models that accomplish this goal, but it takes a lot of computing power.
This is where you come in! With many computers working towards the same goal, we aim to help develop a therapeutic remedy as quickly as possible. By downloading Folding@home here [LINK] and selecting to contribute to “Any Disease”, you can help provide us with the computational power required to tackle this problem. One protein from 2019-nCoV, a protease encoded by the viral RNA, has already been crystallized. Although the 2019-nCoV spike protein of interest has not yet been resolved bound to ACE2, our objective is to use the homologous structure of the SARS-CoV spike protein to identify therapeutic antibody targets.
This illustration, created at the Centers for Disease Control and Prevention (CDC), reveals ultrastructural morphology exhibited by coronaviruses. Note the spikes that adorn the outer surface of the virus, which impart the look of a corona surrounding the virion, when viewed electron microscopically. A novel coronavirus virus was identified as the cause of an outbreak of respiratory illness first detected in Wuhan, China in 2019.
Structures of the closely related SARS-CoV spike protein bound by therapeutic antibodies may help rapidly design better therapies. The three monomers of the SARS-CoV spike protein are shown in different shades of red; the antibody is depicted in green. [PDB: 6NB7 https://www.rcsb.org/structure/6nb7]
I am reposting the following Science blog post from Derrick Lowe as is and ask people go browse through the comments on his Science blog In the Pipeline because, as Dr. Lowe states that in this current crisis it is important to disseminate good information as quickly as possible so wanted the readers here to have the ability to read his great posting on this matter of Covid-19. Also i would like to direct readers to the journal Science opinion letter concerning how important it is to rebuild the trust in good science and the scientific process. The full link for the following In the Pipeline post is: https://blogs.sciencemag.org/pipeline/archives/2020/03/06/covid-19-small-molecule-therapies-reviewed
A Summary of current potential repurposed therapeutics for COVID-19 Infection from In The Pipeline: A Science blog from Derick Lowe
Let’s take inventory on the therapies that are being developed for the coronavirus epidemic. Here is a very thorough list of at Biocentury, and I should note that (like Stat and several other organizations) they’re making all their Covid-19 content free to all readers during this crisis. I’d like to zoom in today on the potential small-molecule therapies, since some of these have the most immediate prospects for use in the real world.
The ones at the front of the line are repurposed drugs that are already approved for human use, for a lot of obvious reasons. The Biocentury list doesn’t cover these, but here’s an article at Nature Biotechnology that goes into detail. Clinical trials are a huge time sink – they sort of have to be, in most cases, if they’re going to be any good – and if you’ve already done all that stuff it’s a huge leg up, even if the drug itself is not exactly a perfect fit for the disease. So what do we have? The compound that is most advanced is probably remdesivir from Gilead, at right. This has been in development for a few years as an RNA virus therapy – it was originally developed for Ebola, and has been tried out against a whole list of single-strand RNA viruses. That includes the related coronaviruses SARS and MERS, so Covid-19 was an obvious fit.
The compound is a prodrug – that phosphoramide gets cleaved off completely, leaving the active 5-OH compound GS-44-1524. It mechanism of action is to get incorporated into viral RNA, since it’s taken up by RNA polymerase and it largely seems to evade proofreading. This causes RNA termination trouble later on, since that alpha-nitrile C-nucleoside is not exactly what the virus is expecting in its genome at that point, and thus viral replication is inhibited.
There are five clinical trials underway (here’s an overview at Biocentury). The NIH has an adaptive-design Phase II trial that has already started in Nebraska, with doses to be changed according to Bayesian readouts along the way. There are two Phase III trials underway at China-Japan Friendship Hospital in Hubei, double-blinded and placebo-controlled (since placebo is, as far as drug therapy goes, the current standard of care). And Gilead themselves are starting two open-label trials, one with no control arm and one with an (unblinded) standard-of-care comparison arm. Those might read out first, depending on when they get off the ground, but will be only rough readouts due to the fast-and-loose trial design. The two Hubei trials and the NIH one will add some rigor to the process, but I’m not sure when they’re going to report. My personal opinion is that I like the chances of this drug more than anything else on this list, but it’s still unlikely to be a game-changer.
There’s an RNA polymerase inhibitor (favipiravir) from Toyama, at right, that’s in a trial in China. It’s a thought – a broad-spectrum agent of this sort would be the sort of thing to try. But unfortunately, from what I can see, it has already turned up as ineffective in in vitro tests. The human trial that’s underway is honestly the sort of thing that would only happen under circumstances like the present: a developing epidemic with a new pathogen and no real standard of care. I hold out little hope for this one, but given that there’s nothing else at present, it probably should be tried. As you’ll see, this is far from the only situation like this.
One of the screens of known drugs in China that also flagged remdesivir noted that the old antimalarial drug chloroquine seemed to be effective in vitro. It had been reported some years back as a possible antiviral, working through more than one mechanism, probably both at viral entry and intracellularly thereafter. That part shouldn’t be surprising – chloroquine’s actual mode(s) of action against malaria parasites are still not completely worked out, either, and some of what people thought they knew about it has turned out to be wrong. There are several trials underway with it at Chinese facilities, some in combination with other agents like remdesivir. Chloroquine has of course been taken for many decades as an antimalarial, but it has a number of liabilities, including seizures, hearing damage, retinopathy and sudden effects on blood glucose. So it’s going to be important to establish just how effective it is and what doses will be needed. Just as with vaccine candidates, it’s possible to do more harm with a rushed treatment than the disease is doing itself
There are several other known antiviral drugs are being tried in China, but I don’t have too much hope for those, either. The neuraminidase inhibitors such as oseltamivir (better known as Tamiflu) were tried against SARS and were ineffective; there is no reason to expect anything versus Covid-19 although these drugs are a component of some drug cocktail trials. The HIV protease therapies such as darunavir and the combination therapy Kaletra are in trials, but that’s also a rather desperate long shot, since there’s no particular reason to think that they will have any such protease inhibition against what this new virus has to offer (and indeed, such agents weren’t much help against SARS in the end, either). The classic interferon/ribavirin combination seems to have had some activity against SARS and MERS, and is in two trials from what I can see. That’s not an awful idea by any means, but it’s not a great one, either: if your viral disease has interferon/ribavirin as a front line therapy, it generally means that there’s nothing really good available. No, unless we get really lucky none of these ideas are going to slow the disease down much.
There are a few other repurposed-protease-inhibitors ideas out there, such as this one. (Edit: I had seen this paper but couldn’t track it down, so thanks to those who sent it along). This paper suggests that the TMPRSS2 protease is important for viral entry on the human-cell-side of the process, a pathway that has been noted for other coronaviruses. And it points out that there is a an approved inhibitor (in Japan) for this enzyme (camostat), so that would definitely seem to be worth a trial, probably in combination with remdesivir.
That’s about it for the existing small molecules, from what I can see. What about new ones? Don’t hold your breath, is all I can say. A drug discovery program from scratch against a new pathogen is, as many readers here well know, not a trivial exercise. As this Bloomberg article details, many such efforts in the past (small molecules and vaccines alike) have come to grief because by the time they had anything to deliver the epidemic itself had passed. Indeed, Gilead’s remdesivir had already been dropped as a potential Ebola therapy.
You will either need to have a target in mind up front or go phenotypic. For the former, what you’d see are better characterizations of the viral protease and more extensive screens against it. Two other big target areas are viral entry (which involves the “spike” proteins on the virus surface and the ACE2 protein on human cells) and viral replication. To the former, it’s worth quickly noting that ACE2 is so much unlike the more familiar ACE protein that none of the cardiovascular ACE inhibitors do anything to it at all. And targeting the latter mechanisms is how remdesivir was developed as a possible Ebola agent, but as you can see, that took time, too. Phenotypic screens are perfectly reasonable against viral pathogens as well, but you’ll need to put time and effort into that assay up front, just as with any phenotypic effort, because as anyone who does that sort of work will tell you, a bad phenotypic screen is a complete waste of everyone’s time.
One of the key steps for either route is identifying an animal model. While animal models of infectious disease can be extremely well translated to human therapy, that doesn’t happen by accident: you need to choose the right animal. Viruses in general (and coronaviruses are no exception) vary widely in their effects in different species, and not just across the gaps of bird/reptile/human and the like. No, you’ll run into things where even the usual set of small mammals are acting differently from each other, with some of them not even getting sick at all. This current virus may well have gone through a couple of other mammalian species before landing on us, but you’ll note that dogs (to pick one) don’t seem to have any problem with it.
All this means that any new-target new-chemical-matter effort against Covid-19 (or any new pathogen) is going to take years, and there is just no way around that. Update: see here for just such an effort to start finding fragment hits for the viral protease. This puts small molecules in a very bimodal distribution: you have the existing drugs that might be repurposed, and are presumably available right now. Nothing else is! At the other end, for completely new therapies you have the usual prospects of drug discovery: years from now, lots of money, low success rate, good luck to all of us. The gap between these two could in theory be filled by vaccines and antibody therapies (if everything goes really, really well) but those are very much their own area and will be dealt with in a separate post.
Either way, the odds are that we (and I mean “we as a species” here) are going to be fighting this epidemic without any particularly amazing pharmacological weapons. Eventually we’ll have some, but I would advise people, pundits, and politicians not to get all excited about the prospects for some new therapies to come riding up over the hill to help us out. The odds of that happening in time to do anything about the current outbreak are very small. We will be going for months, years, with the therapeutic options we have right now. Look around you: what we have today is what we have to work with.
Other related articles published in this Open Access Online Scientific Journal include the following:
Group of Researchers @ University of California, Riverside, the University of Chicago, the U.S. Department of Energy’s Argonne National Laboratory, and Northwestern University solve COVID-19 Structure and Map Potential Therapeutics
Reporters: Stephen J Williams, PhD and Aviva Lev-Ari, PhD, RN
Predicting the Protein Structure of Coronavirus: Inhibition of Nsp15 can slow viral replication and Cryo-EM – Spike protein structure (experimentally verified) vs AI-predicted protein structures (not experimentally verified) of DeepMind (Parent: Google) aka AlphaFold
Curators: Stephen J. Williams, PhD and Aviva Lev-Ari, PhD, RN
Effective humoral immune responses to infection and immunization are defined by high-affinity antibodies generated as a result of B cell differentiation and selection that occurs within germinal centers (GC). Within the GC, B cells undergo affinity maturation, an iterative and competitive process wherein B cells mutate their immunoglobulin genes (somatic hypermutation) and undergo clonal selection by competing for T cell help. Balancing the decision to remain within the GC and continue participating in affinity maturation or to exit the GC as a plasma cell (PC) or memory B cell (MBC) is critical for achieving optimal antibody avidity, antibody quantity, and establishing immunological memory in response to immunization or infection. Humoral immune responses during chronic infections are often dysregulated and characterized by hypergammaglobulinemia, decreased affinity maturation, and delayed development of neutralizing antibodies. Previous studies have suggested that poor antibody quality is in part due to deletion of B cells prior to establishment of the GC response.
In fact the impact of chronic infections on B cell fate decisions in the GC remains poorly understood. To address this question, researchers used single-cell transcriptional profiling of virus-specific GC B cells to test the hypothesis that chronic viral infection disrupted GC B cell fate decisions leading to suboptimal humoral immunity. These studies revealed a critical GC differentiation checkpoint that is disrupted by chronic infection, specifically at the point of dark zone re-entry. During chronic viral infection, virus-specific GC B cells were shunted towards terminal plasma cell (PC) or memory B cell (MBC) fates at the expense of continued participation in the GC. Early GC exit was associated with decreased B cell mutational burden and antibody quality. Persisting antigen and inflammation independently drove facets of dysregulation, with a key role for inflammation in directing premature terminal GC B cell differentiation and GC exit. Thus, the present research defines GC defects during chronic viral infection and identify a critical GC checkpoint that is short-circuited, preventing optimal maturation of humoral immunity.
Together, these studies identify a key GC B cell differentiation checkpoint that is dysregulated during chronic infection. Further, it was found that the chronic inflammatory environment, rather than persistent antigen, is sufficient to drive altered GC B cell differentiation during chronic infection even against unrelated antigens. However, the data also indicate that inflammatory circuits are likely linked to perception of antigen stimulation. Nevertheless, this study reveals a B cell-intrinsic program of transcriptional skewing in chronic viral infection that results in shunting out of the cyclic GC B cell process and early GC exit with consequences for antibody quality and hypergammaglobulinemia. These findings have implications for vaccination in individuals with pre-existing chronic infections where antibody responses are often ineffective and suggest that modulation of inflammatory pathways may be therapeutically useful to overcome impaired humoral immunity and foster affinity maturation during chronic viral infections.
Once herpes simplex infects a person, the virus goes into hiding inside nerve cells, hibernating there for life, periodically waking up from its sleep to reignite infection, causing cold sores or genital lesions to recur. Research from Harvard Medical School showed that the virus uses a host protein called CTCF, or cellular CCCTC-binding factor, to display this type of behavior. Researchers revealed with experiments on mice that CTCF helps herpes simplex regulate its own sleep-wake cycle, enabling the virus to establish latent infections in the body’s sensory neurons where it remains dormant until reactivated. Preventing that latency-regulating protein from binding to the virus’s DNA, weakened the virus’s ability to come out of hiding.
Herpes simplex virus’s ability to go in and out of hiding is a key survival strategy that ensures its propagation from one host to the next. Such symptom-free latency allows the virus to remain out of the reach of the immune system most of the time, while its periodic reactivation ensures that it can continue to spread from one person to the next. On one hand, so-called latency-associated transcript genes, or LAT genes, turn off the transcription of viral RNA, inducing the virus to go into hibernation, or latency. On the other hand, a protein made by a gene called ICP0 promotes the activity of genes that stimulate viral replication and causes active infection.
Based on these earlier findings, the new study revealed that this balancing act is enabled by the CTCF protein when it binds to the viral DNA. Present during latent or dormant infections, CTCF is lost during active, symptomatic infections. The researchers created an altered version of the virus that lacked two of the CTCF binding sites. The absence of the binding sites made no difference in early-stage or acute infections. Similar results were found in infected cultured human nerve cells (trigeminal ganglia) and infected mice model. The researchers concluded that the mutant virus was found to have significantly weakened reactivation capacity.
Taken together, the experiments showed that deleting the CTCF binding sites weakened the virus’s ability to wake up from its dormant state thereby establishing the evidence that the CTCF protein is a key regulator of sleep-wake cycle in herpes simplex infections.
Includes FDA Approved Drugs for Infections and Infectious Diseases: Bacterial Infection, Viral Infection, Fungal Infection, Allergy-related Infections and Other, 1995 – 2016
VOLUME 2: covers the frontier of research on Infectious Diseases and the Human Immune System. The Immune Response, Disease Specific Immune Response, Immunodiagnostics and Immunotherapy, Immunotherapy and Autoimmunity,
Bacterial Infections, Bacteria Types, Antibactirial Therapeutics, FDA Approved Drugs for Infections and Infectious Diseases: Bacterial Infection, 1995 – 2016. Viral Infection: Virus Types, Antiviral Therapeutics, and FDA Approved Drugs for Infections and Infectious Diseases: Viral Infection, Fungal Infections, Allergy-related Infections, Other Infections,1995 – 2016,
VOLUME 3: covers the state of Science on the Historical Perspective of Immunology, Development of the Immune System, Signaling and Immunology, Cellular Immunity, Immunology and Inflammatory Response. Antibody-based Immunity, Vaccines and Microbiome, Immuno-Pharmaceutics, Cancer Immunotherapy, Immunomodulation and Neuro-Immunology.
Volume 2: Summary
The material that has been covered is a considerable material on the basic types of infections – bacterial, viral, and fungal, and diseases related to immune mechanisms. There has been a substantial coverage of the drugs and the manufacturers. This material brings to the discussion an international problem of drug resistance that applies much to bacteria, and a considerable amount of material on advances in drug development that takes into consideration protein structure and protein-protein interactions. The coverage of virus diseases brings to the forefront vaccines. However, in such cases as the influenza virus, a rapid genetic change of the virus makes the use of vaccines an issue for continuing revision.
Volume 3: Summary
The second volume is only concerned with the pathobiology of the inflammatory response, including sepsis, and it does not leave out hematopoiesis, and it lays out the difference between the B-clles and the T-cells that are related to the Toll receptor. Here we have looked closely at two immune disorders, Inflammatory Bowel Disease (Crohn’s Disease) and Rheumatoid Arthritis. Here we have discussed immunomodulation and signaling of the pathways involved, and the programmed cell death response. We have also covered the relationship of the immune response to autoimmune disorders and to cancer. The treatment of cancer now heavily leans toward the blocking of destructive processes in the immunomodulatory pathways.
Epilogue – Volume 2
Volume 2 has covered the most common bacterial and viral diseases that we find widely, or sporadically. It detailed the development of sepsis, and the immune response factor. The immune response involves local cellular invasion of lymphocytes related to initiation of T-cells and macrophages, and also the proteomic generated B-cell antibodies. These reactions are both local and systemic, as bacterial invasion is local and usually related to the tissue of residence (large intestine, oral, lung, genital). In the case of virus, the site of entry is often respiratory or by food intake, but these agents may rapidly become systemic. The other matter of the immune response is autoimmune, a reaction against the self. It is not entirely clear how this is initiated, but it has been related to failure to develop immunity in the prenatal or postnatal period. The only other possibility that might be considered would be by the mechanism of cell remodeling by an apoptotic related mechanism. The other chapters deal with therapeutics.
Epilogue – Volume 3
These two volumes have traversed a large knowledge-base. The first was directed largely at the well known bacterial, virus, fungal diseases, as well as autoimmunity. It specified recent FDA approved recommendations of pharmaceutics for these conditions. It also gives some attention to the immune response in inflammatory and autoimmune diseases, but not cancer. The second volume gives a concise history of development of Leukemias, Lymphomas pathology.
The Immune System, Stress Signaling, Infectious Diseases and Therapeutic Implications: VOLUME 2: Infectious Diseases and Therapeutics and VOLUME 3: The Immune System and Therapeutics (Series D: BioMedicine & Immunology) Kindle Edition – on Amazon.com since 9/4/2017





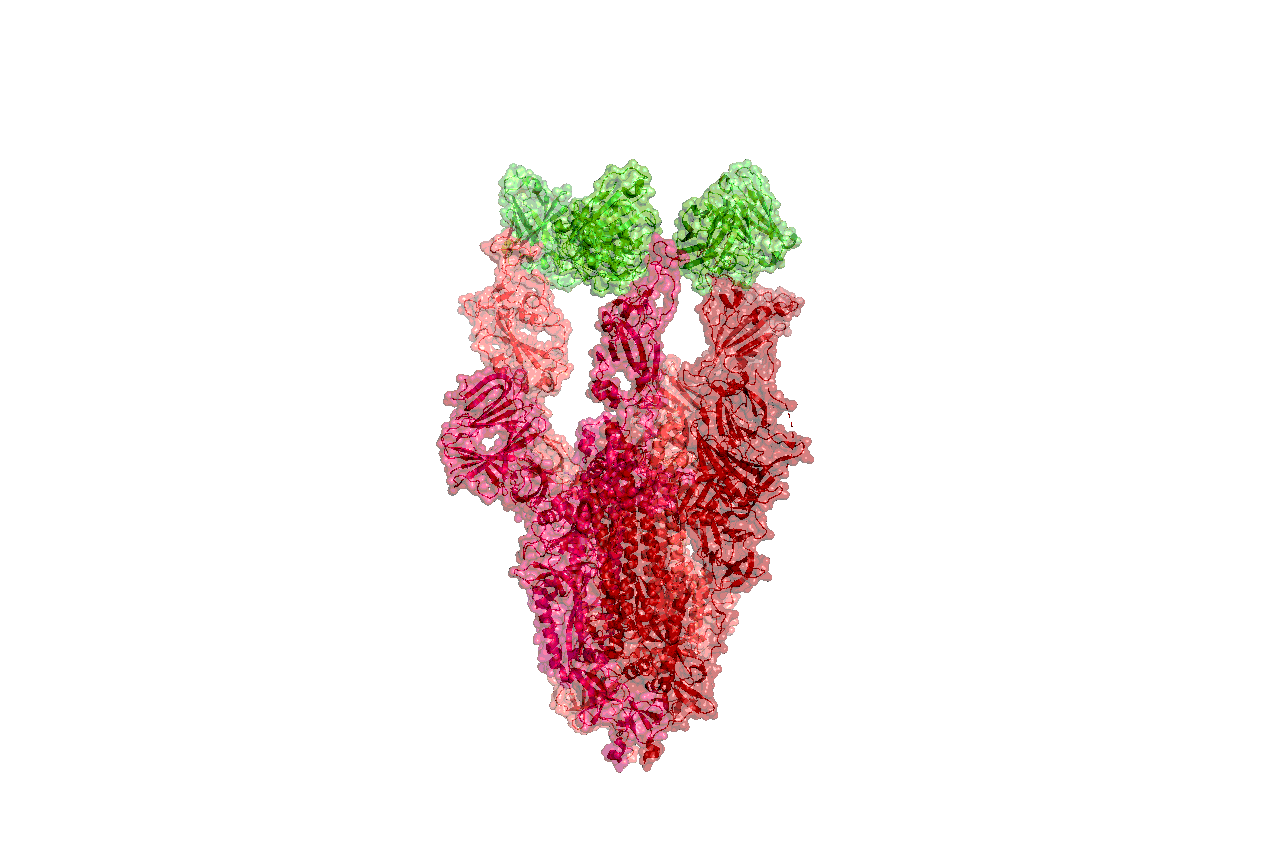








30489-X&id=fx1.jpg){kind=link}