Assessing effects of antimetastatic treatment
Larry H. Bernstein, MD, FCAP, Curator
LPBI
Combining Kinetic Ligand Binding and 3D Tumor Invasion Technologies to Assess Drug Residence Time and Anti-metastatic Effects of CXCR4 Inhibitors
Application Note 3D Cell Culture, ADME/Tox, Cell Imaging, Cell-Based Assays
BioTek Instruments, Inc. P.O. Box 998, Highland Park, Winooski, Vermont 05404-0998
Brad Larson and Leonie Rieger, BioTek Instruments, Inc., Winooski, VT
Nicolas Pierre, Cisbio US, Inc., Bedford, MA
Hilary Sherman, Corning Incorporated, Life Sciences, Kennebunk, ME
http://vertassets.blob.core.windows.net/download/ba9da411/ba9da411-a56c-42d3-a1a0-8c128224947f/cisbio_residence_time_app_note_final.pdf
Metastasis, the spread of cancer cells from the original tumor to secondary locations within the body, is linked to approximately 90% of cancer deaths1 . The expression of chemokine receptors, such as CXCR4 and CCR7, is tightly correlated with the metastatic properties of breast cancer cells. In vivo, neutralizing the interaction of CXCR4 and its known ligand, SDF1-α (CXCL12), significantly impaired the metastasis of breast cancer cells and cell migration2 . Traditionally, the discovery of novel agents has been guided by the affinity of the ligand for the receptor under equilibrium conditions, largely ignoring the kinetic aspects of the ligandreceptor interaction. However, awareness of the importance of binding kinetics has started to increase due to accumulating evidence3, 4, 5, 6 suggesting that the in vivo effectiveness of ligands may be attributed to the time a particular ligand binds to its receptor (drug-target residence time).
Similarly, appropriate in vitro cell models have also been lacking to accurately assess the ability of novel therapies to inhibit tumor invasion. Tumors in vivo exist as a three-dimensional (3D) mass of multiple cell types, including cancer and stromal cells7 . Therefore, incorporating a 3D spheroid-type cellular structure that includes co-cultured cell types forming a tumoroid, provides a more predictive model than the use of individual cancer cells cultured on the bottom of a well in traditional two-dimensional (2D) format.
Here we examine the drug-target residence time of various CXCR4 inhibitors using a direct, homogeneous ligand binding assay and CXCR4 expressing cell line in a kinetic format. This inhibitor panel was further tested in a 3D tumor invasion assay to determine whether there is a correlation between the molecule’s CXCR4 residence time and inhibition of the phenotypic effect of tumor invasion. MDA-MB-231 breast adenocarcinoma cells, known to be invasive, and metastasize to lung from primary mammary fat pad tumors8 , were included, in addition to primary human dermal fibroblasts. Cellular analysis algorithms provided accurate quantification of changes to the original tumoroid structure, as well as invadopodia development. The combination presents an accurate, yet easy-to-use method to assess target-based and phenotypic effects of new, potential anti-metastatic drugs.
……
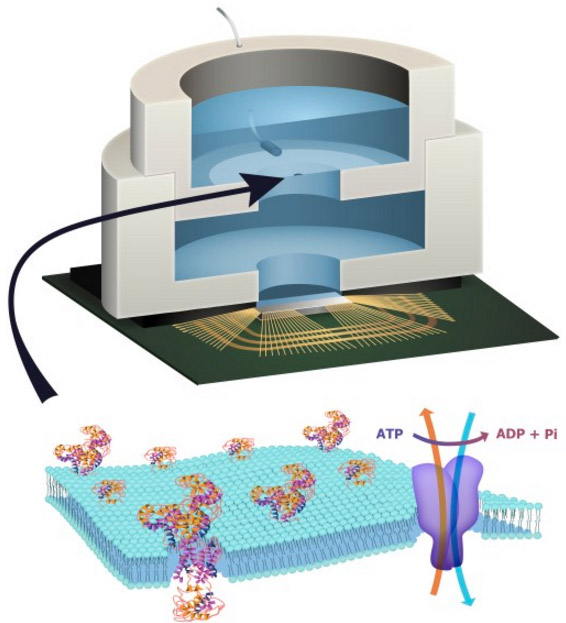
Cytation™ 5 Cell Imaging Multi-Mode Reader Cytation 5 is a modular multi-mode microplate reader that combines automated digital microscopy and microplate detection. Cytation 5 includes filter- and monochromator-based microplate reading; the microscopy module provides high resolution microscopy in fluorescence, brightfield, color brightfield and phase contrast. With special emphasis on live-cell assays, Cytation 5 features temperature control to 65 °C, CO2 / O2 gas control and dual injectors for kinetic assays. Shaking and Gen5 software are also standard. The instrument was used to image spheroids, as well as individual cell invasion through the Matrigel matrix.
Tag-lite® Receptor Ligand Binding Assay
Figure 1. Tag-lite® Receptor Ligand Binding Assay Procedure. The Tag-lite CXCR4 assay relies on a fully functional SNAP-tag fused CXCR4 receptor and fluorescently labeled ligand SDF1-α. Being homogeneous, the binding assay allows for binding events to be precisely recorded in time. The assay can be used to derive the kinetic binding parameters of unlabeled compounds by application of the Motulsky and Mahan equations.
……
Results and Discussion
Drug-Target Residence Time
Determination Association Kinetics of SDF1-α-d2 Labeled Ligand
The final Drug-Target Residence Time value takes into account the observed on and off rates of the unlabeled inhibitors as well as the labeled SDF1-α-d2 ligand, and is computed by incorporation of the Motulsky and Mahan equation9 . The first step to calculate the final value was to perform an associative binding experiment using a concentration range of 0-100 nM of the d2 acceptor fluor labeled ligand. Binding was monitored kinetically over a period of 40 minutes.
Figure 2. Association binding graph of SDF1-α-d2. Observed associative binding curves calculated from HTRF ratios of wells containing SDF1-α-d2 ligand concentrations ranging from 0-100 nM. Non-specific binding values subtracted from total ratios to determine observed specific binding.
Binding increases over time until it plateaus after several minutes (Figure 2). The plateau in an association experiment depends on the concentration of labeled SDF1-α used. Higher plateaus will be obtained with higher concentrations. Fitting of the curves with Graph Pad Prism yields the observed association rate values for all concentrations tested or kobs.
The Kd value of the labeled ligand was also determined by plotting the HTRF ratios generated after a binding equilibrium was reached with the different concentrations of ligand tested.
Figure 3. SDF1-α-d2 saturation binding curve. HTRF ratios generated upon the achievement of binding equilibrium of tested [SDF1-α-d2].
In a saturation binding experiment, increasing concentrations of labeled SDF1-α result in increased binding. Saturation is obtained when no further binding can be recorded. The ligand concentration that binds to half the receptor sites at equilibrium or Kd was 29 nM.
An assessment of whether the labeled SDF1-α ligand follows the Law of Mass action can also be carried out. If the system does follow the Law of Mass action then kobs increases linearly with increasing concentrations of SDF1-α.
Due to the linear shape of the curve, and an R2 value >0.9, Law of Mass Action was proven for the labeled SDF1-α ligand. This allowed for the use of Graph Pad Prism software to derive association and dissociation rate constants from the linear regression line. The rate constant values experimentally found or mathematically derived are summarized in Table 1. kon,SDF1-α-d2 and koff ,SDF1-α-d2 were 0.001 nM-1.s-1 and 0.04 s-1, respectively
Table SDF1-α-d2 Kinetic Binding Characterization
Association Kinetics of SDF1-α-d2 Labeled Ligand In the theory developed by Motulsky and Mahan, an unlabeled competitor is co-incubated with a labeled ligand during a kinetic association experiment. Here, a single concentration of the SDF1-α-d2 ligand, 25 nM, was co-incubated with multiple concentrations of the unlabeled SDF1-α competitors in the presence of the CXCR4 expressing cells. Kinetic binding of the labeled ligand was then monitored over time.
Figure 5. Kinetics of Competitive Binding. Plot of specific binding HTRF ratios over time for the SDF1-α-d2 ligand when in the presence of 100, 10, or 1 nM concentrations of (A.) AMD 3100, (B.) AMD 3465, or (C.) IT1t.
From the curve fitting of the observed SDF1-α-d2 kinetic binding, and incorporation of the Law of Mass Action linear regression line, k(off) (Min-1) values were then calculated. Final residence time (R) values could then be determined using the following formula:
R = 1/k(off)
Therefore, molecules having a lower k(off) rate reside at the target receptor for longer periods of time.
Table 2. SDF1-α Competitor Dissociation Rate and Residence Time Values.
From the shape of the curves in Figure 5, and a comparison of the residence time values generated for the labeled ligand and unlabeled competitors (Table 2), qualitative and quantitative assumptions regarding the various competitors can then be made. First, if the competitor dissociates faster from its target than the ligand (smaller R value), such as is seen with AMD 3100 (Figure 5A), the specific binding of the ligand will slowly and monotonically approach its equilibrium in time. However, when the competitor dissociates slower (larger R value), the association curve of the ligand consists of two phases, starting with a typical “overshoot” and then a decline until a new equilibrium is reached. Competitors whose residence times are greater than that of the SDF1-α-d2 ligand, such as AMD 3465 and IT1t (Figure 5B and C), may then exhibit a stronger inhibitory response when used in the confirmatory phenotypic 3D tumor invasion assay.
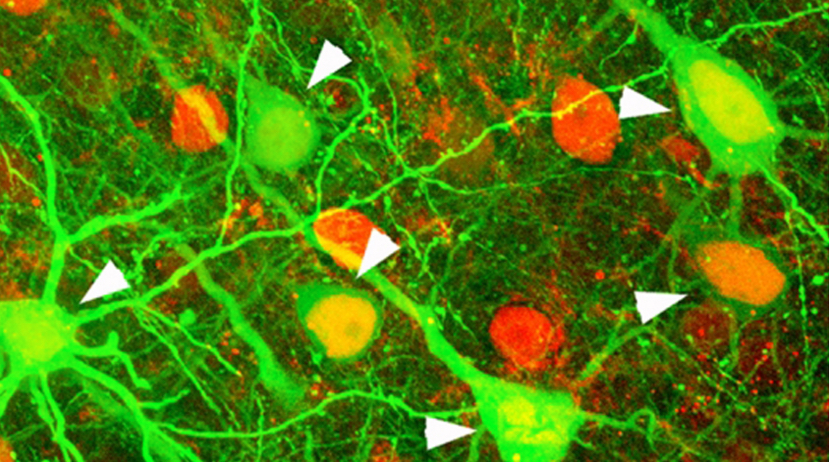
Interruption of Invasion via SDF1-α Ligand Binding Inhibition As stated previously, interruption of the interaction between CXCR4 and its known ligand, SDF1-α, impairs metastasis of breast cancer and cell migration2 . Therefore, a phenotypic assessment of the CXCR4 inhibitor panel was then performed to determine whether changes in the level of tumor migration could be detected, and more importantly, if compounds exhibiting longer residence times compared to SDF1-α-d2 exhibited a higher inhibitory effect on migration through the 3D matrix. MDA-MB-231 breast adenocarcinoma cells, co-cultured with human dermal fibroblasts, were used as the in vitro tumor model. This breast cancer cell line has been previously shown to express the CXCR4 receptor10.
Figure 6. Image-based Monitoring of MDA-MB-231/Fibroblast Tumor Invasion. Overlaid brightfield and fluorescent images captured using a 4x objective, after a 0 and 5 day incubation period with AMD 3465, IT1t, and CTCE 9908. Imaging channel representation: Brightfield – Total cells and invadopodia; GFP – MDA-MB-231 cells; RFP – Fibroblasts.
Figure 7. Quantification of Invasive Tumor Area. 4x overlaid images captured following 5 day (A.) 100 and (B.) 0 μM IT1t incubation with tumoroids. Object masks automatically drawn by Gen5 using the following criteria: Threshold: 5000 RFU; Min. Object Size: 400 μm; Max. Object Size: 1500 μm; Image Smoothing Strength: 0; Background Flattening Size: Auto.
Cellular analysis is performed with the Cytation 5 using the brightfield signal to quantify the extent of invasion. Minimum and maximum object sizes, as well as brightfield threshold values are set such that a precise object mask is automatically drawn around each tumoroid in its entirety (Figure 7A and B). The same criteria are used for all images evaluated during the experiment. This allows for a quantitative comparison of the area covered within each object mask to be completed.
Figure 8. Tumor Invasion Inhibition Determination. Graphs of individual tumoroid areas on day 0, and subsequent to five day invasion period in the presence of inhibitor concentrations.
The 4x images displayed (Figure 6), as well as the graphs in Figure 8, demonstrating total tumoroid area coverage before and after the incubation period illustrate the ability of CXCR4 inhibitors to interrupt tumor invasion consistent with the previously determined residence time. AMD 3465 and IT1t, which exhibit a residence time longer than SDF1-α-d2, effectively minimize tumor invasion in a dose dependent manner. The decrease in MDAMB-231 GFP and fibroblast RFP expression exhibited after a 5 day 100 μM IT1t incubation, also seen after a 7 day AMD 3465 incubation of the same concentration (data not shown), may also indicate the chronic cytotoxic effects that elevated dosing of these compounds can have on both cancer and stromal cells. All other compounds show little to no effect on the ability of the tumoroid to migrate through the 3D matrix. While AMD 3465 and ITt1 display the same sub-nanomolar potency, AMD3465 prevails as a CXCR4 inhibitor due to its greater residence time.
Conclusions The Tag-lite CXCR4 ligand binding assay provides a simple, yet robust cell-based approach to determine kinetic binding of known receptor ligands, as well as competitive binding of test molecules. The simultaneous dual emission capture and injection capabilities of the Synergy Neo allow accurate calculations of kinetic association and dissociation rates to be made when used in conjunction with the Tag-lite® assay. Corning Spheroid Microplates then provide an easy-to-use, consistent method to perform spheroid aggregation and confirmatory 3D tumor invasion assays. Imaging of spheroid formation, as well as invading structures can be performed by the Cytation™ 5 using brightfield or fluorescent channels to easily track tumoroid invasion. The flexible cellular analysis capacity of the Gen5™ Data Analysis Software also allows for accurate assessment of 3D tumor invasion during the entire incubation period. The combination of assay chemistry, cell model, kinetic microplate and image-based monitoring, in addition to cellular analysis provide an ideal method to better understand the target-based and phenotypic effects of potential inhibitors of tumor invasion and metastasis.
References
1. Saxe, Charles. ‘Unlocking The Mysteries Of Metastasis’. ExpertVoices 2013. http://www.cancer.org/ cancer/news/expertvoices/post/2013/01/23/unlockingthe-mysteries-of-metastasis.aspx. Accessed 16 Mar. 2015.
2. Müller, A., Homey, B., Soto, H., Ge, N., Catron, D., Buchanan, M., McClanahan, T., Mruphy, E., Yuan, W., Wagner, S., Barrera, J., Mohar, A., Verástegui, E., Zlotnik, A. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001, 410, 50-56.
3. Swinney, D. Biochemical mechanisms of drug action: what does it take for success? Nat Rev Drug Discov. 2004, 3, 801-808.
4. Copeland, R., Pompliano, D., Meek, T. Drugtarget residence time and its implications for lead optimization. Nat Rev Drug Discov. 2006,5, 730-739.
5. Tummino, P., Copeland, R. Residence time of receptor-ligand complexes and its effect on biological function. Biochemistry. 2008, 47, 5481-5492.
6. Zhang, R., Monsma, F. The importance of drug-target residence time. Curr Opin Drug Discov Devel. 2009, 12, 488-496.
7. Mao, Y., Keller, E., Garfield, D., Shen, K., Wang, J. Stromal cells in tumor microenvironment and breast cancer. Cancer Metast Rev. 2013, 32, 303-315.
8. Kamath, L., Meydani, A., Foss, F., Kuliopulos, A. Signaling from protease-activated receptor-1 inhibits migration and invasion of breast cancer cells. Cancer Res. 2001, 61, 5933-5940.
9. Motulsky, H., Mahan, L. The kinetics of competitive radioligand binding predicted by the law of mass action. Mol Pharmacol. 1984, 25, 1-9.
10. Sun, Y., Mao, X, Fan, C, Liu, C., Guo, A., Guan, S., Jin, Q., Li, B., Yao, F., Jin, F. CXCL12-CXCR4 axis promotes the natural selection of breast cancer cell metastasis. Tumor Biol. 2014, 35, 7765-7773.
Like this:
Like Loading...
Read Full Post »










 . The power dissipated in this source is introduced back into the circuit in the power generated by the Nernst independent voltage sources,
. The power dissipated in this source is introduced back into the circuit in the power generated by the Nernst independent voltage sources,  and
and  . The power dissipated in the dependent voltage source Vloss models any additional power not used to perform chemical or electrical work. ……
. The power dissipated in the dependent voltage source Vloss models any additional power not used to perform chemical or electrical work. ……

 Thursday 15 October 2015, 12:00–13:00
Thursday 15 October 2015, 12:00–13:00













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}