Healthcare analytics, AI solutions for biological big data, providing an AI platform for the biotech, life sciences, medical and pharmaceutical industries, as well as for related technological approaches, i.e., curation and text analysis with machine learning and other activities related to AI applications to these industries.
Jing Chen of Emory University School of Medicine, Hualiang Jiang of the Shanghai Institute of Materia Medica of the Chinese Academy of Sciences, Chuan He of the University of Chicago, and coworkers have now developed a selective way of blocking copper transport in cancer cells (Nat. Chem. 2015, DOI: 10.1038/nchem.2381). By screening a database of 200,000 druglike small molecules, the researchers discovered a promising compound, DC_AC50, for cancer treatment. They zeroed in on the compound by testing how well database hits inhibited a protein-protein interaction leading to copper transport and reduced proliferation of cancer cells.
licensed DC_AC50 to Suring Therapeutics, in Suzhou, China
INNOVATORS Jing Chen of Emory University School of Medicine, Hualiang Jiang of the Shanghai Institute of Materia Medica of the Chinese Academy of Sciences, Chuan He of the University of Chicago, and coworkers
Chaperone proteins (green) transfer copper ions to copper-dependent proteins (lilac) via ligand exchange between two cysteines (-SH groups) on each protein. DC_AC50 binds the chaperone and inhibits this interaction.
Jing Chen of Emory University School of Medicine, Hualiang Jiang of the Shanghai Institute of Materia Medica of the Chinese Academy of Sciences,Chuan He of the University of Chicago, and coworkers have now developed a selective way of blocking copper transport in cancer cells (Nat. Chem. 2015, DOI: 10.1038/nchem.2381). By screening a database of 200,000 druglike small molecules, the researchers discovered a promising compound, DC_AC50, for cancer treatment. They zeroed in on the compound by testing how well database hits inhibited a protein-protein interaction leading to copper transport and reduced proliferation of cancer cells.
Scientists had already found a molecule, tetrathiomolybdate, that interferes with copper trafficking and have tested it in clinical trials against cancer. But tetrathiomolybdate is a copper chelator: It inhibits copper transport in cells by nonselectively sequestering copper ions. Sometimes, the chelator snags too much copper, inhibiting essential copper-based processes in normal cells and causing side effects.
In contrast, DC_AC50 works by inhibiting interactions between proteins in the copper-trafficking pathway: It prevents chaperone proteins, called Atox1 and CCS, from passing copper ions to enzymes that use them to run vital cellular processes. Cancer cells are heavy users of Atox1 and CCS, so DC_AC50 affects cancer cells selectively.
The team has licensed DC_AC50 to Suring Therapeutics, in Suzhou, China, for developing anticancer therapies. The group also plans to further tweak DC_AC50 to develop more-potent versions.
Thomas O’Halloran of Northwestern University, who has studied tetrathiomolybdate, comments that “the challenge in drug design is hitting one of these copper-dependent processes without messing with housekeeping functions that normal cells depend upon. DC_AC50 appears to block the function of copper metallochaperone proteins without interacting directly with their cargo, copper ions. As the first member of a new class of inhibitors, it provides a new way to interrogate the physiology of copper trafficking disorders and possibly intervene.”
ZYDPLA1 is a novel compound in the Gliptin class of antidiabetic agents. It works by blocking the enzyme Dipeptidyl Peptidase-4 (DPP-4), which inactivates the Incretin hormone GLP-1. By increasing the GLP-1 levels, ZYDPLA1 glucose-dependently increases insulin secretion and lowers glucagon secretion.
Zydus announces data presentations on ZYDPLA1 “A once-weekly small molecule DPP-IV inhibitor for treating diabetes”, at the ENDO conference in Chicago, Illinois, USA. Ahmedabad, India June 9, 2014 The Zydus group will be presenting data on its molecule ZYDPLA1 a novel compound in the Gliptin class of anti-diabetic agents during the joint meeting of the International Society of Endocrinology and the Endocrine Society: ICE/ENDO 2014 to be held from June 21-24, 2014 in Chicago, Illinois.
ZYDPLA1, currently in Phase I clinical evaluation in USA, is an orally active, small molecule NCE, discovered and developed by the Zydus Research Centre. ZYDPLA1 works by blocking the enzyme Dipeptidyl Peptidase-4 (DPP-4), which inactivates the Incretin hormone GLP-1. By increasing the GLP- 1 levels, ZYDPLA1 glucose-dependently increases insulin secretion. This results in an overall improvement in the glucose homoeostasis, including reduction in HbA1c and blood sugar levels.
The Chairman & Managing Director of Zydus, Mr. Pankaj R. Patel said, “Currently, all available DPP-4 inhibitors are dosed once-daily. ZYDPLA1 with a once-a-week dosing regimen would provide diabetic patients with a more convenient treatment alternative. ZYDPLA1 will offer sustained action, which will result in an improved efficacy profile.”
The number of diabetics in the world is estimated to be over 360 million. In 2025 nearly half of the world’s diabetic population will be from India, China, Brazil, Russia and Turkey. The sales of the DPP IV inhibitors is expected to peak at almost $14 billion by 2022. Research in the field of anti-diabetic therapy seeks to address the problems of hypoglycemia, GI side effects, lactic acidosis, weight gain, CV risks, edema, potential immunogenicity etc., which pose a major challenge in the treatment of diabetes.
About Zydus
Headquartered in Ahmedabad, India, Zydus Cadila is an innovative, global pharmaceutical company that discovers, manufactures and markets a broad range of healthcare therapies. The group employs over 16,000 people worldwide including over 1100 scientists engaged in R & D and is dedicated to creating healthier communities globally. As a leading healthcare provider, it aims to become a global researchbased pharmaceutical company by 2020. The group has a strong research pipeline of NCEs, biologics and vaccines which are in various stages of clinical trials including late stage.
About Zydus Research Centre
The Zydus Research Centre has over 20 discovery programmes in the areas of cardio-metabolic disorders, pain, inflammation and oncology. Zydus has in-house capabilities to conduct discovery research from concept to IND-enabling pre-clinical development and human proof-of-concept clinical trials. The Zydus Research group had identified and developed Lipaglyn™ (Saroglitazar) which has now become India’s first NCE to reach the market. Lipaglyn™ is a breakthrough therapy in the treatment of diabetic dyslipidemia and Hypertriglyceridemia. The company recently announced the commencement of Phase III trials of LipaglynTM (Saroglitazar) in patients suffering from Lipodystrophy.
Zydus announces US FDA approval for initiating Phase I clinical trials of ‘ZYDPLA1’ – a novel next generation orally active, small molecule DPP-4 inhibitor to treat Type 2 Diabetes Ahmedabad, October 23, 2013
• Zydus strengthens its cardiometabolic pipeline with the addition of ZYDPLA1
• Novel next generation New Chemical Entity (NCE) would offer once-a-week oral treatment option, a significant benefit to Type-2 diabetic patients Close on the heels of launching Lipaglyn, the breakthrough therapy to treat diabetic dyslipidemia and India’s first NCE to reach the market, the Zydus group announced the Phase I clinical trial approval from the USFDA for ZYDPLA1 – a Next Generation, long-acting DPP-4 Inhibitor.
ZYDPLA1 is an orally active, small molecule NCE, discovered and developed by the Zydus Research Centre, the NCE research wing of Zydus. ZYDPLA1 is a novel compound in the Gliptin class of antidiabetic agents. It works by blocking the enzyme Dipeptidyl Peptidase-4 (DPP-4), which inactivates the Incretin hormone GLP-1. By increasing the GLP-1 levels, ZYDPLA1 glucose-dependently increases insulin secretion and lowers glucagon secretion. This results in an overall improvement in the glucose homoeostasis, including reduction in HbA1c and blood sugar levels.
Currently, all available DPP-4 inhibitors are dosed once-daily. ZYDPLA1 with a once-a-week dosing regimen, would provide diabetic patients with a more convenient treatment alternative. ZYDPLA1 will offer sustained action, which will result in an improved efficacy profile.
Speaking on the new development, Mr. Pankaj R. Patel, Chairman and Managing Director, Zydus Group, said, “After a promising start with Lipaglyn, we take another big leap forward in the area of diabetic research and long term management of Type 2 diabetes. The IND approval by USFDA is another major regulatory milestone for us. We believe that ZYDPLA1 holds promise and would take us closer to our mission of reducing the burden of chronic diseases and addressing unmet medical needs in the treatment of diabetes.”
The number of diabetics in the world is estimated to be over 360 million. In 2025 nearly half of the world’s diabetic population will be from India, China, Brazil, Russia and Turkey. The sales of the DPPIV inhibitors is expected to peak at almost $14 billion by 2022. Research in the field of anti-diabetic therapy seeks to address the problems of hypoglycemia, GI side effects, lactic acidosis, weight gain, CV risks, edema, potential immunogenicity etc., which pose a major challenge in the treatment of diabetes.
Zydus is the only Indian pharma company to launch its own patented NCE – Lipaglyn™, the world’s first drug to be approved for the treatment of diabetic dyslipidemia. It aims to be a leading global healthcare provider with a robust product pipeline, achieve sales of over $3 billion by 2015 and be a research-based pharmaceutical company by 2020.
The Zydus Research Centre has over 20 discovery programmes ongoing with several candidates in the pre-clinical development stage focused on metabolic, cardiovascular, pain, inflammation and oncology therapeutic areas. With over 400 research professionals spearheading its research programme, Zydus has inhouse capabilities to conduct discovery research from concept to IND-enabling pre-clinical development and human proof-of-concept clinical trials. ZYDPLA1 is the latest addition to the group’s strong research pipeline of 6 NCEs which are in various stages of clinical trials. For more information, please visit: http://www.zyduscadila.com
Advinus Therapeutics announced that it has successfully completed a 14-day POC study in 60 Type II diabetic patients on its lead molecule, GKM-001, a glucokinase activator. The results of the trial show effective glucose lowering across all doses tested without any incidence of hypoglycemia or any other clinically relevant adverse events.
GKM-001 is differentiated from most other GK molecules that are in development, or have been discontinued, due to its novel liver selective mechanism of action.
GKM-001 belongs to a novel class of molecules for treatment of type II diabetes. It is an activator of Glucokinase (GK), a glucose-sensing enzyme found mainly in the liver and pancreas. Being liver selective, GKM-001 mostly activates GK in the liver and not in pancreas, which is its key differentiation from most competitor molecules that activate GK in pancreas as well.
A glucokinase activator for treatment of type II diabetes, currently in PI. Advinus is actively exploring partnership options to expedite further development and WW marketing of GKM-001.
Examples C2 and C3 were prepared in analogues manner of example (Cl) from the appropriate chiral intermediate:
Example Dl : (+)-{5-Chloro-2-[2-(4-cyclopropanesulfonylphenyl)-2-(2,4- difluorophenoxy)acetylamino]thiazol-4-yl}acetic acid, ethyl ester
Advinus’ GK-activator Achieves Early POC for Diabetes
November 29 2011
Partnership Dialog Actively Underway
Advinus Therapeutics, a research-based pharmaceutical company founded by globally experienced industry executives and promoted by the TATA Group, announced that it has successfully completed a 14-day POC study in 60 Type II diabetic patients on its lead molecule, GKM-001, a glucokinase activator. The results of the trial show effective glucose lowering across all doses tested without any incidence of hypoglycemia or any other clinically relevant adverse events.
The clinical trials on GKM-001 validate the company’s pre-clinical hypothesis that a liver selective Glucokinase activator would not cause hypoglycemia (very low blood sugar), while showing robust efficacy.
“GKM-001 is differentiated from most other GK molecules that are in development, or have been discontinued, due to its novel liver selective mechanism of action. GKM-001 has a prolonged pharmacological effect and a half-life that should support a once a day dosing as both mono and combination therapy.” said Dr. Rashmi Barbhaiya, MD & CEO, Advinus Therapeutics. He added that Advinus is actively exploring partnership options to expedite further development and global marketing of GKM-001.
GKM-001 belongs to a novel class of molecules for treatment of type II diabetes. It is an activator of Glucokinase (GK), a glucose-sensing enzyme found mainly in the liver and pancreas. Being liver selective, GKM-001 mostly activates GK in the liver and not in pancreas, which is its key differentiation from most competitor molecules that activate GK in pancreas as well. The resulting increase in insulin secretion creates a potential for hypoglycemia-a risk GKM-001 is designed to avoid. Advinus has the composition of matter patent on GKM-001 for all major markets globally. Both the Single Ascending Dose data, in healthy and type II diabetics, and the Multiple Ascending Dose Study in Type II diabetics has shown that the molecule shows effective glucose lowering in a dose dependent manner and has excellent safety and tolerability profile over a 40-fold dose range. The pharmacokinetic properties of the molecule support once a day dosing. GKM-001 has the potential to be “First-in-Class” drug to address this large, growing and yet poorly addressed market.
Advinus also has identified a clinical candidate as a back-up to GKM-001, which is structurally different. In its portfolio, the company has a growing pipeline for COPD, sickle cell disease, inflammatory bowel disease, type 2 diabetes, acute and chronic pain and rheumatoid arthritis in various stages of late discovery and pre-clinical development.
The first glucokinase modulator discovered and developed in India
A new concept for the management of diabetes for patients, globally
100 per cent ‘made in India’ molecule for the treatment of diabetes
IND approved by DGCI, Phase I clinical trial shows excellent safety and tolerance profiles with efficacy
Bangalore: Advinus Therapeutics (Advinus), the research-based pharmaceutical company founded by leading global pharmaceutical executives and promoted by the Tata group, today, announced the discovery of a novel molecule for the treatment of type II diabetes — GKM-001.The molecule is an activator of glucokinase; an enzyme that regulates glucose balance and insulin secretion in the body.
GKM-001 is a completely indigenously developed molecule and the initial clinical trials have shown excellent results for both safety and efficacy.
“Considering past failures of other companies on this target, our discovery programme primarily focused on identifying a molecule that would be efficacious without causing hypoglycaemia; a side effect associated with most compounds developed for this target.
“Recently completed Phase I data indicate that Advinus’ GKM–001 is a liver selective molecule that has overcome the biggest clinical challenge of hypoglycaemia. GKM-001 is differentiated from most other GK molecules in development due to this novel mechanism of action,” said Dr Rashmi Barbhaiya, MD and CEO, Advinus Therapeutics.
He further added, “We are very proud that GKM-001 is 100 per cent Indian. Advinus’s discovery team in Pune discovered the molecule and entire preclinical development was carried out at our centre in Bangalore. The Investigational New Drug (IND) application was filed with the DGCI for approval to initiate clinical trials in India within 34 months of initiation of the discovery programme. Subsequent to the approval of the IND, we have completed the Phase I Single Ascending Dose study in India within two months.”
GKM-001 is a novel molecule for the treatment of type II diabetes. It is the first glucokinase modulator discovered and developed in India and has potential to be both first or best in class. The success in discovering GKM-001 is attributed to the science-driven efforts in Advinus laboratories and ‘breaking the conventional mold’ for selection of a drug candidate. Advinus has ‘composition of matter’ patent on the molecule for all major markets globally. Glucokinase as a class of target is considered to be novel as currently there is no product in the market or in late clinical trials. The strategy for early clinical development revolved around assessing safety (particularly hypoglycaemia) and early assessment of therapeutic activity (glucose lowering and other biomarkers) in type II diabetics. The Phase I data, in both healthy and type II diabetics, shows excellent safety and tolerability over a 40-fold dose range and desirable pharmacokinetic properties consistent with ‘once a day’ dosing. The next wave of clinical studies planned continues on this strategy of early testing in type II diabetics.
Right behind the lead candidate GKM-001, Advinus has a rich pipeline of back up compounds on the same target. These include several structurally different compounds with diverse potency, unique pharmacology and tissue selectivity. Having discovered the molecule with early indication of wide safety margins, desired efficacy and pharmacokinetic profiles, the company now seeks to out-licence GKM-001 and its discovery portfolio.
FDA Cellular & Gene Therapy Guidances: Implications for CRSPR/Cas9 Trials, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 2: CRISPR for Gene Editing and DNA Repair
FDA Cellular & Gene Therapy Guidances: Implications for CRSPR/Cas9 Trials
Reporter: Stephen J. Williams, PhD
The recent announcement by Editas CEO Katrine Bosley to pursue a CRSPR/Cas9 gene therapy trial to correct defects in an yet to be disclosed gene to treat one form of a rare eye disease called Leber congenital amaurosis (multiple mutant genes have been linked to the disease) have put an interesting emphasis on the need for a regulatory framework to initiate these trials. Indeed at the 2015 EmTechMIT Conference Editas CEO Katrine Bosley had mentioned this particular issue: the need for discourse with FDA and regulatory bodies to establish guidelines for design of clinical trials using the CRSPR gene editing tool.
To this effect, I have listed below, the multiple FDA Guidance Documents surrounding gene therapy to show that, in the past year, the FDA has shown great commitment to devise a regulatory framework for this therapeutic area.
FDA Guidance On Source Animal, Product, Preclinical and Clinical Issues Concerning the Use of Xenotranspantation Products in Humans – Implications for 3D BioPrinting of Regenerative Tissue
Reporter: Stephen J. Williams, Ph.D.
The FDA has submitted Final Guidance on use xeno-transplanted animal tissue, products, and cells into human and their use in medical procedures. Although the draft guidance was to expand on previous guidelines to prevent the introduction, transmission, and spread of communicable diseases, this updated draft may have implications for use of such tissue in the emerging medical 3D printing field.
This document is to provide guidance on the production, testing and evaluation of products intended for use in xenotransplantation. The guidance includes scientific questions that should be addressed by sponsors during protocol development and during the preparation of submissions to the Food and Drug Administration (FDA), e.g., Investigational New Drug Application (IND) and Biologics License Application (BLA). This guidance document finalizes the draft guidance of the same title dated February 2001.
For the purpose of this document, xenotransplantation refers to any procedure that involves the transplantation, implantation, or infusion into a human recipient of either (a) live cells, tissues, or organs from a nonhuman animal source, or (b) human body fluids, cells, tissues or organs that have had ex vivo contact with live nonhuman animal cells, tissues or organs. For the purpose of this document, xenotransplantation products include live cells, tissues or organs used in xenotransplantation. (See Definitions in section I.C.)
This document presents issues that should be considered in addressing the safety of viable materials obtained from animal sources and intended for clinical use in humans. The potential threat to both human and animal welfare from zoonotic or other infectious agents warrants careful characterization of animal sources of cells, tissues, and organs. This document addresses issues such as the characterization of source animals, source animal husbandry practices, characterization of xenotransplantation products, considerations for the xenotransplantation product manufacturing facility, appropriate preclinical models for xenotransplantation protocols, and monitoring of recipients of xenotransplantation products. This document recommends specific practices intended to prevent the introduction and spread of infectious agents of animal origin into the human population. FDA expects that new methods proposed by sponsors to address specific issues will be scientifically rigorous and that sufficient data will be presented to justify their use.
Examples of procedures involving xenotransplantation products include:
transplantation of xenogeneic hearts, kidneys, or pancreatic tissue to treat organ failure,
implantation of neural cells to ameliorate neurological degenerative diseases,
administration of human cells previously cultured ex vivo with live nonhuman animal antigen-presenting or feeder cells, and
extracorporeal perfusion of a patient’s blood or blood component perfused through an intact animal organ or isolated cells contained in a device to treat liver failure.
Imagine stripping out the failing components of an old car — the engine, transmission, exhaust system and all of those parts — leaving just the old body and other structural elements. Replace those old mechanical parts with a brand new electric, hydrogen powered, biofuel, nuclear or whatever kind of engine you want and now you have a brand new car. It has an old frame, but that’s okay. The frame wasn’t causing the problem, and it can live on for years, undamaged.
When challenged to design internal organs, tissue engineers are taking a similar approach, particularly with the most complex organs, like the heart, liver and kidneys. These organs have three dimensional structures that are elaborate, not just at the gross anatomic level, but in microscopic anatomy too. Some day, their complex connective tissue scaffolding, the stroma, might be synthesized from the needed collagen proteins with advanced 3-D printing. But biomedical engineering is not there yet, so right now the best candidate for organ scaffolding comes from one of humanity’s favorite farm animals: the pig.
Chimera alarmists connecting with anti-biotechnology movements might cringe at the thought of building new human organs starting with pig tissue, but if you’re using only the organ scaffolding and building a working organ from there, pig organs may actually be more desirable than those donated by humans.
How big is the anti-chimerite movement?
Unlike anti-GMO and anti-vaccination activists, there really aren’t too many anti-chemerites around. Nevertheless, there is a presence on the web of people who express concern about mixing of humans and non-human animals. Presently, much of their concern is focussed on the growing of human organs inside non-human animals, pigs included. One anti-chemerite has written that it could be a problem for the following reason:
Once a human organ is grown inside a pig, that pig is no longer fully a pig. And without a doubt, that organ will no longer be a fully human organ after it is grown inside the pig. Those receiving those organs will be allowing human-animal hybrid organs to be implanted into them. Most people would be absolutely shocked to learn some of the things that are currently being done in the name of science.
The blog goes on to express alarm about the use of human genes in rice and from there morphs into an off the shelf garden variety anti-GMO tirade, though with an an anti-chemeric current running through it. The concern about making pigs a little bit human and humans a little bit pig becomes a concern about making rice a little bit human. But the concern about fusing tissues and genes of humans and other species does not fit with the trend in modern medicine.
Utilization of pig tissue enters a new age
A porcine human ear for xenotransplantation. source: The Scientist
For decades, pig, bovine and other non-human tissues have been used in medicine. People are walking around with pig and cow heart valves. Diabetics used to get a lot of insulin from pigs and cows, although today, thanks to genetic engineering, they’re getting human insulin produced by microorganisms modified genetically to make human insulin, which is safer and more effective.
When it comes to building new organs from old ones, however, pig organs could actually be superior for a couple of reasons. For one thing, there’s no availability problem with pigs. Their hearts and other organs also have all of the crucial components of the extracellular matrix that makes up an organ’s scaffolding. But unlike human organs, the pig organs don’t tend to carry or transfer human diseases. That is a major advantage that makes them ideal starting material. Plus there is another advantage: typically, the hearts of human cadavers are damaged, either because heart disease is what killed the human owner or because resuscitation efforts aimed at restarting the heart of a dying person using electrical jolts and powerful drugs.
Rebuilding an old organ into a new one
How then does the process work? Whether starting with a donated human or pig organ, there are several possible methods. But what they all have in common is that only the scaffolding of the original organ is retained. Just like the engine and transmission of the old car, the working tissue is removed, usually using detergents. One promising technique that has been applied to engineer new hearts is being tested by researchers at the University of Pittsburgh. Detergents pumped into the aorta attached to a donated heart (donated by a human cadaver, or pig or cow). The pressure keeps the aortic valve closed, so the detergents to into the coronary arteries and through the myocardial (heart muscle) and endocardial (lining over the muscle inside the heart chambers) tissue, which thus gets dissolved over the course of days. What’s left is just the stroma tissue, forming a scaffold. But that scaffold has signaling factors that enable embryonic stem cells, or specially programed adult pleuripotent cells to become all of the needed cells for a new heart.
Eventually, 3-D printing technology may reach the point when no donated scaffolding is needed, but that’s not the case quite yet, plus with a pig scaffolding all of the needed signaling factors are there and they work just as well as those in a human heart scaffold. All of this can lead to a scenario, possibly very soon, in which organs are made using off-the-self scaffolding from pig organs, ready to produce a custom-made heart using stem or other cells donated by new organ’s recipient.
David Warmflash is an astrobiologist, physician, and science writer. Follow @CosmicEvolution to read what he is saying on Twitter.
And a Great Article in The Scientist by Dr. Ed Yong Entitled
To cope with a growing shortage of hearts, livers, and lungs suitable for transplant, some scientists are genetically engineering pigs, while others are growing organs in the lab.
.. where Joseph Vacanti and David Cooper figured that using
“engineered pigs without the a-1,3-galactosyltransferase gene that produces the a-gal residues. In addition, the pigs carry human cell-membrane proteins such as CD55 and CD46 that prevent the host’s complement system from assembling and attacking the foreign cells”
… thereby limiting rejection of the xenotransplated tissue.
In addition to issues related to animal virus transmission the issue of optimal scaffolds for organs as well as the advantages which 3D Printing would have in mass production of organs is discussed:
To Vacanti, artificial scaffolds are the future of organ engineering, and the only way in which organs for transplantation could be mass-produced. “You should be able to make them on demand, with low-cost materials and manufacturing technologies,” he says. That is relatively simple for organs like tracheas or bladders, which are just hollow tubes or sacs. Even though it is far more difficult for the lung or liver, which have complicated structures, Vacanti thinks it will be possible to simulate their architecture with computer models, and fabricate them with modern printing technology. (See “3-D Printing,” The Scientist, July 2012.) “They obey very ordered rules, so you can reduce it down to a series of algorithms, which can help you design them,” he says. But Taylor says that even if the architecture is correct, the scaffold would still need to contain the right surface molecules to guide the growth of any added cells. “It seems a bit of an overkill when nature has already done the work for us,” she says.
Other articles of FDA Guidance and 3D Bio Printing on this Open Access Journal Include:
New FDA Draft Guidance On Homologous Use of Human Cells, Tissues, and Cellular and Tissue-Based Products – Implications for 3D BioPrinting of Regenerative Tissue
Reporter: Stephen J. Williams, Ph.D.
The FDA recently came out with a Draft Guidance on use of human cells, tissues and cellular and tissue-based products (HCT/P) {defined in 21 CFR 1271.3(d)} and their use in medical procedures. Although the draft guidance was to expand on previous guidelines to prevent the introduction, transmission, and spread of communicable diseases, this updated draft may have implications for use of such tissue in the emerging medical 3D printing field.
In 21 CFR 1271.10, the regulations identify the criteria for regulation solely under section 361 of the PHS Act and 21 CFR Part 1271. An HCT/P is regulated solely under section 361 of the PHS Act and 21 CFR Part 1271 if it meets all of the following criteria (21 CFR 1271.10(a)):
The HCT/P is minimally manipulated;
The HCT/P is intended for homologous use only, as reflected by the labeling, advertising, or other indications of the manufacturer’s objective intent;
The manufacture of the HCT/P does not involve the combination of the cells or tissues with another article, except for water, crystalloids, or a sterilizing, preserving, or storage agent, provided that the addition of water, crystalloids, or the sterilizing, preserving, or storage agent does not raise new clinical safety concerns with respect to the HCT/P; and
Either:
The HCT/P does not have a systemic effect and is not dependent upon the metabolic activity of living cells for its primary function; or
The HCT/P has a systemic effect or is dependent upon the metabolic activity of living cells for its primary function, and:
Is for autologous use;
Is for allogeneic use in a first-degree or second-degree blood relative; or
Is for reproductive use.
If an HCT/P does not meet all of the criteria in 21 CFR 1271.10(a), and the establishment that manufactures the HCT/P does not qualify for any of the exceptions in 21 CFR 1271.15, the HCT/P will be regulated as a drug, device, and/or biological product under the Federal Food, Drug and Cosmetic Act (FD&C Act), and/or section 351 of the PHS Act, and applicable regulations, including 21 CFR Part 1271, and pre-market review will be required.
1 Examples of HCT/Ps include, but are not limited to, bone, ligament, skin, dura mater, heart valve, cornea, hematopoietic stem/progenitor cells derived from peripheral and cord blood, manipulated autologous chondrocytes, epithelial cells on a synthetic matrix, and semen or other reproductive tissue. The following articles are not considered HCT/Ps: (1) Vascularized human organs for transplantation; (2) Whole blood or blood components or blood derivative products subject to listing under 21 CFR Parts 607 and 207, respectively; (3) Secreted or extracted human products, such as milk, collagen, and cell factors, except that semen is considered an HCT/P; (4) Minimally manipulated bone marrow for homologous use and not combined with another article (except for water, crystalloids, or a sterilizing, preserving, or storage agent, if the addition of the agent does not raise new clinical safety concerns with respect to the bone marrow); (5) Ancillary products used in the manufacture of HCT/P; (6) Cells, tissues, and organs derived from animals other than humans; (7) In vitro diagnostic products as defined in 21 CFR 809.3(a); and (8) Blood vessels recovered with an organ, as defined in 42 CFR 121.2 that are intended for use in organ transplantation and labeled “For use in organ transplantation only.” (21 CFR 1271.3(d))
Contains Nonbinding Recommendations Draft – Not for Implementation
Section 1271.10(a)(2) (21 CFR 1271.10(a)(2)) provides that one of the criteria for an HCT/P to be regulated solely under section 361 of the PHS Act is that the “HCT/P is intended for homologous use only, as reflected by the labeling, advertising, or other indications of the manufacturer’s objective intent.” As defined in 21 CFR 1271.3(c), homologous use means the repair, reconstruction, replacement, or supplementation of a recipient’s cells or tissues with an HCT/P that performs the same basic function or functions in the recipient as in the donor. This criterion reflects the Agency’s conclusion that there would be increased safety and effectiveness concerns for HCT/Ps that are intended for a non-homologous use, because there is less basis on which to predict the product’s behavior, whereas HCT/Ps for homologous use can reasonably be expected to function appropriately (assuming all of the other criteria are also met).2 In applying the homologous use criterion, FDA will determine what the intended use of the HCT/P is, as reflected by the the labeling, advertising, and other indications of a manufacturer’s objective intent, and will then apply the homologous use definition.
FDA has received many inquiries from manufacturers about whether their HCT/Ps meet the homologous use criterion in 21 CFR 1271.10(a)(2). Additionally, transplant and healthcare providers often need to know this information about the HCT/Ps that they are considering for use in their patients. This guidance provides examples of different types of HCT/Ps and how the regulation in 21 CFR 1271.10(a)(2) applies to them, and provides general principles that can be applied to HCT/Ps that may be developed in the future. In some of the examples, the HCT/Ps may fail to meet more than one of the four criteria in 21 CFR 1271.10(a).
III. QUESTIONS AND ANSWERS
What is the definition of homologous use?
Homologous use means the repair, reconstruction, replacement, or supplementation of a recipient’s cells or tissues with an HCT/P that performs the same basic function or functions in the recipient as in the donor (21 CFR 1271.3(c)), including when such cells or tissues are for autologous use. We generally consider an HCT/P to be for homologous use when it is used to repair, reconstruct, replace, or supplement:
Recipient cells or tissues that are identical (e.g., skin for skin) to the donor cells or tissues, and perform one or more of the same basic functions in the recipient as the cells or tissues performed in the donor; or,
Recipient cells that may not be identical to the donor’s cells, or recipient tissues that may not be identical to the donor’s tissues, but that perform one or more of the same basic functions in the recipient as the cells or tissues performed in the donor.3
3“Establishment Registration and Listing for Manufacturers of Human Cellular and Tissue-Based Products” 63 FR 26744 at 26749 (May 14, 1998).
Contains Nonbinding Recommendations Draft – Not for Implementation
1-1. A heart valve is transplanted to replace a dysfunctional heart valve. This is homologous use because the donor heart valve performs the same basic function in the donor as in the recipient of ensuring unidirectional blood flow within the heart.
1-2. Pericardium is intended to be used as a wound covering for dura mater defects. This is homologous use because the pericardium is intended to repair or reconstruct the dura mater and serve as a covering in the recipient, which is one of the basic functions it performs in the donor.
Generally, if an HCT/P is intended for use as an unproven treatment for a myriad of
diseases or conditions, the HCT/P is likely not intended for homologous use only.4
What does FDA mean by repair, reconstruction, replacement, or supplementation of a recipient’s cells or tissues?
Repair generally means the physical or mechanical restoration of tissues, including by covering or protecting. For example, FDA generally would consider skin removed from a donor and then transplanted to a recipient in order to cover a burn wound to be a homologous use. Reconstruction generally means surgical reassembling or re-forming. For example, reconstruction generally would include the reestablishment of the physical integrity of a damaged aorta.5 Replacement generally means substitution of a missing tissue or cell, for example, the replacement of a damaged or diseased cornea with a healthy cornea or the replacement of donor hematopoietic stem/progenitor cells in a recipient with a disorder affecting the hematopoietic system that is inherited, acquired, or the result of myeloablative treatment. Supplementation generally means to add to, or complete. For example, FDA generally would consider homologous uses to be the implantation of dermal matrix into the facial wrinkles to supplement a recipient’s tissues and the use of bone chips to supplement bony defects. Repair, reconstruction, replacement, and supplementation are not mutually exclusive functions and an HCT/P could perform more than one of these functions for a given intended use.
What does FDA mean by “the same basic function or functions” in the definition of homologous use?
For the purpose of applying the regulatory framework, the same basic function or functions of HCT/Ps are considered to be those basic functions the HCT/P performs in the body of the donor, which, when transplanted, implanted, infused, or transferred, the HCT/P would be expected to perform in the recipient. It is not necessary for the HCT/P in the recipient to perform all of the basic functions it performed in the donor, in order to
4 “Human Cells, Tissues, and Cellular and Tissue-Based Products; Establishment Registration and Listing” 66 FR 5447 at 5458 (January 19, 2001).
5 “Current Good Tissue Practice for Human Cell, Tissue, and Cellular and Tissue-Based Product Establishments; Inspection and Enforcement” 69 FR 68612 at 68643 (November 24, 2004) states, “HCT/Ps with claims for “reconstruction or repair” can be regulated solely under section 361 of the PHS Act, provided the HCT/P meets all the criteria in § 1271.10, including minimal manipulation and homologous use.”
Contains Nonbinding Recommendations Draft – Not for Implementation
meet the definition of homologous use. However, to meet the definition of homologous use, any of the basic functions that the HCT/P is expected to perform in the recipient must be a basic function that the HCT/P performed in the donor.
A homologous use for a structural tissue would generally be to perform a structural function in the recipient, for example, to physically support or serve as a barrier or conduit, or connect, cover, or cushion.
A homologous use for a cellular or nonstructural tissue would generally be a metabolic or biochemical function in the recipient, such as, hematopoietic, immune, and endocrine functions.
3-1. The basic functions of hematopoietic stem/progenitor cells (HPCs) include to form and to replenish the hematopoietic system. Sources of HPCs include cord blood, peripheral blood, and bone marrow.6
HPCs derived from peripheral blood are intended for transplantation into an individual with a disorder affecting the hematopoietic system that is inherited, acquired, or the result of myeloablative treatment. This is homologous use because the peripheral blood product performs the same basic function of reconstituting the hematopoietic system in the recipient.
HPCs derived from bone marrow are infused into an artery with a balloon catheter for the purpose of limiting ventricular remodeling following acute myocardial infarction. This is not homologous use because limiting ventricular remodeling is not a basic function of bone marrow.
A manufacturer provides HPCs derived from cord blood with a package insert stating that cord blood may be infused intravenously to differentiate into neuronal cells for treatment of cerebral palsy. This is not homologous use because there is insufficient evidence to support that such differentiation is a basic function of these cells in the donor.
3-2. The basic functions of the cornea include protecting the eye by forming its outermost layer and serving as the refracting medium of the eye. A corneal graft is transplanted to restore sight in a patient with corneal blindness. This is homologous use because a corneal graft performs the same basic functions in the donor as in the recipient.
3-3. The basic functions of a vein or artery include serving as a conduit for blood flow throughout the body. A cryopreserved vein or artery is used for arteriovenous access during hemodialysis. This is homologous use because the vein or artery is supplementing the vessel as a conduit for blood flow.
3-4. The basic functions of amniotic membrane include covering, protecting, serving as a selective barrier for the movement of nutrients between the external and in utero
6 Bone marrow meets the definition of an HCT/P only if is it more than minimally manipulated; intended by the manufacturer for a non-homologous use, or combined with certain drugs or devices.
Contains Nonbinding Recommendations Draft – Not for Implementation
environment, and to retain fluid in utero. Amniotic membrane is used for bone tissue replacement to support bone regeneration following surgery to repair or replace bone defects. This is not a homologous use because bone regeneration is not a basic function of amniotic membrane.
3-5. The basic functions of pericardium include covering, protecting against infection, fixing the heart to the mediastinum, and providing lubrication to allow normal heart movement within chest. Autologous pericardium is used to replace a dysfunctional heart valve in the same patient. This is not homologous use because facilitating unidirectional blood flow is not a basic function of pericardium.
Does my HCT/P have to be used in the same anatomic location to perform the same basic function or functions?
An HCT/P may perform the same basic function or functions even when it is not used in the same anatomic location where it existed in the donor.7 A transplanted HCT/P could replace missing tissue, or repair, reconstruct, or supplement tissue that is missing or damaged, either when placed in the same or different anatomic location, as long as it performs the same basic function(s) in the recipient as in the donor.
4-1. The basic functions of skin include covering, protecting the body from external force, and serving as a water-resistant barrier to pathogens or other damaging agents in the external environment. The dermis is the elastic connective tissue layer of the skin that provides a supportive layer of the integument and protects the body from mechanical stress.
An acellular dermal product is used for supplemental support, protection, reinforcement, or covering for a tendon. This is homologous use because in both anatomic locations, the dermis provides support and protects the soft tissue structure from mechanical stress.
An acellular dermal product is used for tendon replacement or repair. This is not homologous use because serving as a connection between muscle and bone is not a basic function of dermis.
4-2. The basic functions of amniotic membrane include serving as a selective barrier for the movement of nutrients between the external and in utero environment and to retain fluid in utero. An amniotic membrane product is used for wound healing of dermal ulcers and defects. This is not homologous use because wound healing of dermal lesions is not a basic function of amniotic membrane.
4-3. The basic functions of pancreatic islets include regulating glucose homeostasis within the body. Pancreatic islets are transplanted into the liver through the portal vein,
7 “Human Cells, Tissues, and Cellular and Tissue-Based Products; Establishment Registration and Listing” 66 FR 5447 at 5458 (January 19, 2001).
6
Contains Nonbinding Recommendations Draft – Not for Implementation
for preservation of endocrine function after pancreatectomy. This is homologous use because the regulation of glucose homeostasis is a basic function of pancreatic islets.
What does FDA mean by “intended for homologous use” in 21 CFR 1271.10(a)(2)?
The regulatory criterion in 21 CFR 1271.10(a)(2) states that the HCT/P is intended for homologous use only, as reflected by the labeling, advertising, or other indications of the manufacturer’s objective intent.
Labeling includes the HCT/P label and any written, printed, or graphic materials that supplement, explain, or are textually related to the product, and which are disseminated by or on behalf of its manufacturer.8 Advertising includes information, other than labeling, that originates from the same source as the product and that is intended to supplement, explain, or be textually related to the product (e.g., print advertising, broadcast advertising, electronic advertising (including the Internet), statements of company representatives).9
An HCT/P is intended for homologous use when its labeling, advertising, or other indications of the manufacturer’s objective intent refer to only homologous uses for the HCT/P. When an HCT/P’s labeling, advertising, or other indications of the manufacturer’s objective intent refer to non-homologous uses, the HCT/P would not meet the homologous use criterion in 21 CFR 1271.10(a)(2).
What does FDA mean by “manufacturer’s objective intent” in 21 CFR 1271.10(a)(2)?
A manufacturer’s objective intent is determined by the expressions of the manufacturer or its representatives, or may be shown by the circumstances surrounding the distribution of the article. A manufacturer’s objective intent may, for example, be shown by labeling claims, advertising matter, or oral or written statements by the manufacturer or its representatives. It may be shown by the circumstances that the HCT/P is, with the knowledge of the manufacturer or its representatives, offered for a purpose for which it is neither labeled nor advertised.
Cyclin-dependent kinases (CDKs), in complex with their cyclin partners, modulate the transition through phases of the cell division cycle. Cyclin D–CDK complexes are important in cancer progression, especially for certain types of breast cancer. Fassl et al. discuss advances in understanding the biology of cyclin D–CDK complexes that have led to new concepts about how drugs that target these complexes induce cancer cell cytostasis and suggest possible combinations to widen the types of cancer that can be treated. They also discuss progress in overcoming resistance to cyclin D–CDK inhibitors and their possible application to diseases beyond cancer. —GKA
Structured Abstract
BACKGROUND
Cyclins and cyclin-dependent kinases (CDKs) drive cell division. Of particular importance to the cancer field are D-cyclins, which activate CDK4 and CDK6. In normal cells, the activity of cyclin D–CDK4/6 is controlled by the extracellular pro-proliferative or inhibitory signals. By contrast, in many cancers, cyclin D–CDK4/6 kinases are hyperactivated and become independent of mitogenic stimulation, thereby driving uncontrolled tumor cell proliferation. Mouse genetic experiments established that cyclin D–CDK4/6 kinases are essential for growth of many tumor types, and they represent potential therapeutic targets. Genetic and cell culture studies documented the dependence of breast cancer cells on CDK4/6. Chemical CDK4/6 inhibitors were synthesized and tested in preclinical studies. Introduction of these compounds to the clinic represented a breakthrough in breast cancer treatment and will likely have a major impact on the treatment of many other tumor types.
ADVANCES
Small-molecule CDK4/6 inhibitors (palbociclib, ribociclib, abemaciclib) showed impressive results in clinical trials for patients with hormone receptor–positive breast cancers. Addition of CDK4/6 inhibitors to standard endocrine therapy substantially extended median progression-free survival and prolonged median overall survival. Consequently, all three CDK4/6 inhibitors have been approved for treatment of women with advanced or metastatic hormone receptor–positive breast cancers. In the past few years, the renewed interest in CDK4/6 biology has yielded several surprising discoveries. The emerging concept is that CDK4/6 kinases regulate a much wider set of cellular functions than anticipated. Consequently, CDK4/6 inhibitors, beyond inhibiting tumor cell proliferation, affect tumor cells and the tumor environment through mechanisms that are only beginning to be elucidated. For example, inhibition of CDK4/6 affects antitumor immunity acting both on tumor cells and on the host immune system. CDK4/6 inhibitors were shown to enhance the efficacy of immune checkpoint blockade in preclinical mouse cancer models. These new concepts are now being tested in clinical trials.
OUTLOOK
Palbociclib, ribociclib, and abemaciclib are being tested in more than 300 clinical trials for more than 50 tumor types. These trials evaluate CDK4/6 inhibitors in combination with a wide range of therapeutic compounds that target other cancer-relevant pathways. Several other combination treatments were shown to be efficacious in preclinical studies and will enter clinical trials soon. Another CDK4/6 inhibitor, trilaciclib, is being tested for its ability to shield normal cells of the host from cytotoxic effects of chemotherapy. New CDK4/6 inhibitors have been developed and are being assessed in preclinical and clinical trials. The major impediment in the therapeutic use of CDK4/6 inhibitors is that patients who initially respond to treatment often develop resistance and eventually succumb to the disease. Moreover, a substantial fraction of tumors show preexisting, intrinsic resistance to CDK4/6 inhibitors. One of the main challenges will be to elucidate the full range of resistance mechanisms. Even with the current, limited knowledge, one can envisage the principles of new, improved approaches to overcome known resistance mechanisms. Another largely unexplored area for future study is the possible involvement of CDK4/6 in other pathologic states beyond cancer. This will be the subject of intense studies, and it may extend the utility of CDK4/6 inhibitors to the treatment of other diseases.
Targeting cyclin D–CDK4/6 for cancer treatment.
D-cyclins (CycD) activate CDK4 and CDK6 in G1 phase of the cell cycle and promote cell cycle progression by phosphorylating the retinoblastoma protein RB1. RB1 inhibits E2F transcription factors; phosphorylation of RB1 activates E2F-driven transcription. In many cancers, CycD-CDK4/6 is constitutively activated and drives uncontrolled cell proliferation. The development of small-molecule CDK4/6 inhibitors provided a therapeutic tool to repress constitutive CycD-CDK4/6 activity and to inhibit cancer cell proliferation. As with several targeted therapies, tumors eventually develop resistance and resume cell proliferation despite CDK4/6 inhibition. New combination treatments, involving CDK4/6 inhibitors plus inhibition of other pathways, are being tested in the clinic to delay or overcome the resistance.
Cyclin-dependent kinases 4 and 6 (CDK4 and CDK6) and their activating partners, D-type cyclins, link the extracellular environment with the core cell cycle machinery. Constitutive activation of cyclin D–CDK4/6 represents the driving force of tumorigenesis in several cancer types. Small-molecule inhibitors of CDK4/6 have been used with great success in the treatment of hormone receptor–positive breast cancers and are in clinical trials for many other tumor types. Unexpectedly, recent work indicates that inhibition of CDK4/6 affects a wide range of cellular functions such as tumor cell metabolism and antitumor immunity. We discuss how recent advances in understanding CDK4/6 biology are opening new avenues for the future use of cyclin D–CDK4/6 inhibitors in cancer treatment.
Cyclin D1, the activator of CDK4 and CDK6, was discovered in the early 1990s (1, 2). The role of cyclin D1 in oncogenesis was already evident at the time of its cloning, as it was also identified as the protein product of the PRAD1 oncogene, which is rearranged and overexpressed in parathyroid adenomas (3), and of the BCL1 oncogene, which is rearranged in B-lymphocytic malignancies (4). Subsequently, the remaining two D-type cyclins, D2 and D3, were discovered on the basis of their homology to cyclin D1 (1).
Cyclins serve as regulatory subunits of cyclin-dependent kinases (CDKs) (5). Shortly after the discovery of D-cyclins, CDK4 and CDK6 were identified as their kinase partners (6). Mouse gene knockout studies revealed that CDK4 and CDK6 play redundant roles in development, and combined ablation of CDK4 and CDK6 was found to result in embryonic lethality (7). The essentially identical phenotype was seen in cyclin D–knockout mice, thereby confirming the role of D-cyclins as CDK4/6 activators in vivo (8). Surprisingly, these analyses revealed that many normal nontransformed mammalian cell types can proliferate without any cyclin D–CDK4/6 activity (7, 8).
CDK4 and CDK6 are expressed at constant levels throughout the cell cycle. By contrast, D-cyclins are labile proteins that are transcriptionally induced upon stimulation of cells with growth factors. For this reason, D-cyclins are regarded as links between the cellular environment and the cell cycle machinery (6).
Cell cycle inhibitors play an important role in regulating the activity of cyclin D–CDK4/6 (Fig. 1). The INK inhibitors (p16INK4A, p15INK4B, p18INK4C, p19INK4D) bind to CDK4 or CDK6 and prevent their interaction with D-type cyclins, thereby inhibiting cyclin D–CDK4/6 kinase activity. By contrast, KIP/CIP inhibitors (p27KIP1, p57KIP2, p21CIP1), which inhibit the activity of CDK2-containing complexes, serve as assembly factors for cyclin D–CDK4/6 (6, 9). This was demonstrated by the observation that mouse fibroblasts devoid of p27KIP1 and p21CIP1 fail to assemble cyclin D–CDK4/6 complexes (10).
Fig. 1. Molecular events governing progression through the G1 phase of the cell cycle.
The mammalian cell cycle can be divided into G1, S (DNA synthesis), G2, and M (mitosis) phases. During G1 phase, cyclin D (CycD)–CDK4/6 kinases together with cyclin E (CycE)–CDK2 phosphorylate the retinoblastoma protein RB1. This activates the E2F transcriptional program and allows entry of cells into S phase. Members of the INK family of inhibitors (p16INK4A, p15INK4B, p18INK4C, and p19INK4D) inhibit cyclin D–CDK4/6; KIP/CIP proteins (p21CIP1, p27KIP1, and p57KIP2) inhibit cyclin E–CDK2. Cyclin D–CDK4/6 complexes use p27KIP1 and p21CIP1 as “assembly factors” and sequester them away from cyclin E–CDK2, thereby activating CDK2. Proteins that are frequently lost or down-regulated in cancers are marked with green arrows, overexpressed proteins with red arrows.
p27KIP1 can bind cyclin D–CDK4/6 in an inhibitory or noninhibitory mode, depending on p27KIP1 phosphorylation status. Cyclin D–p27KIP1-CDK4/6 complexes are catalytically inactive unless p27KIP1 is phosphorylated on Tyr88 and Tyr89 (11). Two molecular mechanisms may explain this switch. First, Tyr88/Tyr89 phosphorylation may dislodge the helix of p27KIP1 from the CDK active site and allow adenosine triphosphate (ATP) binding (12). Second, the presence of tyrosine-unphosphorylated p27KIP1 within the cyclin D–CDK4 complex prevents the activating phosphorylation of CDK4’s T-loop by the CDK-activating kinase (CAK) (12). Brk has been identified as a physiological kinase of p27KIP1 (13); Abl and Lyn can phosphorylate p27KIP1 in vitro, but their in vivo importance remains unclear (11, 14).
The activity of cyclin D–CDK4/6 is also regulated by proteolysis. Cyclin D1 is an unstable protein with a half-life of less than 30 min. At the end of G1 phase, cyclin D1 is phosphorylated at Thr286 by GSK3β (15). This facilitates association of cyclin D1 with the nuclear exportin CRM1 and promotes export of cyclin D1 from the nucleus to the cytoplasm (16). Subsequently, phosphorylated cyclin D1 becomes polyubiquitinated by E3 ubiquitin ligases, thereby targeting it for proteasomal degradation. Several substrate receptors of E3 ubiquitin ligases have been implicated in recognizing phosphorylated cyclin D1, including F-box proteins FBXO4 (along with αB crystallin), FBXO31, FBXW8, β-TrCP1/2, and SKP2 (17). The anaphase-promoting complex/cyclosome (APC/C) was also proposed to target cyclin D1 while F-box proteins FBXL2 and FBXL8 target cyclins D2 and D3 (17, 18). Surprisingly, the level and stability of cyclin D1 was unaffected by depletion of several of these proteins, indicating that some other E3 plays a rate-limiting role in cyclin D1 degradation (19). Indeed, recent studies reported that D-cyclins are ubiquitinated and targeted for proteasomal degradation by the E3 ubiquitin ligase CRL4, which uses AMBRA1 protein as its substrate receptor (20–22).
Cyclin D–CDK4/6 in cancer
Genomic aberrations of the cyclin D1 gene (CCND1) represent frequent events in different tumor types. The t(11;14)(q13;q32) translocation juxtaposing CCND1 with the immunoglobulin heavy-chain (IGH) locus represents the characteristic feature of mantle-cell lymphoma and is frequently observed in multiple myeloma or plasma cell leukemia (23, 24). Amplification of CCND1 is seen in many other malignancies—for example, in 13 to 20% of breast cancers (23, 24), more than 40% of head and neck squamous cell carcinomas, and more than 30% of esophageal squamous cell carcinomas (23). A higher proportion of cancers (e.g., up to 50% of mammary carcinomas) overexpress cyclin D1 protein (24). Also, cyclins D2 and D3, CDK4, and CDK6 are overexpressed in various tumor types (5, 9). Cyclin D–CDK4/6 can also be hyperactivated through other mechanisms such as deletion or inactivation of INK inhibitors, most frequently p16INK4A (5, 9, 23). Altogether, a very large number of human tumors contain lesions that hyperactivate cyclin D–CDK4/6 (5).
An oncogenic role for cyclin D–CDK4/6 has been supported by mouse cancer models. For example, targeted overexpression of cyclin D1 in mammary glands of transgenic mice led to the development of mammary carcinomas (25). Also, overexpression of cyclin D2, D3, or CDK4, or loss of p16INK4a resulted in tumor formation (9).
Conversely, genetic ablation of D-cyclins, CDK4, or CDK6 decreased tumor sensitivity (9). For instance, Ccnd1– or Cdk4-null mice, or knock-in mice expressing kinase-inactive cyclin D1–CDK4/6, were resistant to develop human epidermal growth factor receptor 2 (HER2)–driven mammary carcinomas (26–29). An acute, global shutdown of cyclin D1 in mice bearing HER2-driven tumors arrested tumor growth and triggered tumor-specific senescence while having no obvious impact on normal tissues (30). Likewise, an acute ablation of CDK4 arrested tumor cell proliferation and triggered tumor cell senescence in a KRAS-driven non–small-cell lung cancer (NSCLC) mouse model (31). These observations indicated that CDK4 and CDK6 might represent excellent therapeutic targets in cancer treatment.
CDK4/6 functions in cell proliferation and oncogenesis
The best-documented function of cyclin D–CDK4/6 in driving cell proliferation is phosphorylation of the retinoblastoma protein, RB1, and RB-like proteins, RBL1 and RBL2 (5, 6) (Fig. 1). Unphosphorylated RB1 binds and inactivates or represses E2F transcription factors. According to the prevailing model, phosphorylation of RB1 by cyclin D–CDK4/6 partially inactivates RB1, leading to release of E2Fs and up-regulation of E2F-transcriptional targets, including cyclin E. Cyclin E forms a complex with its kinase partner, CDK2, and completes full RB1 phosphorylation, leading to activation of the E2F transcriptional program and facilitating S-phase entry (5, 6). In normal, nontransformed cells, the activity of cyclin D–CDK4/6 is tightly regulated by the extracellular mitogenic milieu. This links inactivation of RB1 with mitogenic signals. In cancer cells carrying activating lesions in cyclin D–CDK4/6, the kinase is constitutively active, thereby decoupling cell division from proliferative and inhibitory signals (5).
This model has been questioned by the demonstration that RB1 exists in a monophosphorylated state throughout G1 phase and becomes inactivated in late G1 by cyclin E–CDK2, which “hyperphosphorylates” RB1 on multiple residues (32). However, recent single-cell analyses revealed that cyclin D–CDK4/6 activity is required for the hyperphosphorylation of RB1 throughout G1, whereas cyclin E/A–CDK maintains RB1 hyperphosphorylation in S phase (33). Moreover, phosphorylation of RB1 by cyclin D–CDK4/6 was shown to be required for normal cell cycle progression (34).
In addition to this kinase-dependent mechanism, up-regulation of D-cyclin expression and formation of cyclin D–CDK4/6 complexes lead to redistribution of KIP/CIP inhibitors from cyclin E–CDK2 complexes (which are inhibited by these proteins) to cyclin D–CDK4/6 (which use them as assembly factors), thereby activating the kinase activity of cyclin E–CDK2 (6). Cyclin E–CDK2 in turn phosphorylates RB1 and other cellular proteins and promotes cell cycle progression.
Cyclin D1–CDK4/6 directly phosphorylates, stabilizes, and activates the transcription factor FOXM1. This promotes cell cycle progression and protects cancer cells from entering senescence (35). Cyclin D–CDK4 also phosphorylates and inactivates SMAD3, which mediates transforming growth factor–β (TGF-β) antiproliferative response. CDK4/6-dependent phosphorylation of SMAD3 inhibits its transcriptional activity and disables the ability of TGF-β to induce cell cycle arrest (36). FZR1/CDH1, an adaptor protein of the APC complex, is another phosphorylation substrate of CDK4. Depletion of CDH1 in human cancer cells partially rescued the proliferative block upon CDK4/6 inhibition, and it cooperated with RB1 depletion in restoring full proliferation (37).
Cyclin D–CDK4/6 also phosphorylates and inactivates TSC2, a negative regulator of mTORC1, thereby resulting in mTORC1 activation. Conversely, inhibition of CDK4/6 led to decreased mTORC1 activity and reduced protein synthesis in cells representing different human tumor types. It was proposed that through TSC2 phosphorylation, activation of cyclin D–CDK4/6 couples cell growth with cell division (38). Consistent with this, the antiproliferative effect of CDK4/6 inhibition was reduced in cells lacking TSC2 (38).
MEP50, a co-regulatory factor of protein arginine-methyltransferase 5 (PRMT5), is phosphorylated by cyclin D1–CDK4. Through this mechanism, cyclin D1–CDK4/6 increases the catalytic activity of PRMT5/MEP50 (39). It was proposed that deregulation of cyclin D1–CDK4 kinase in tumor cells, by increasing PRMT5/MEP50 activity, reduces the expression of CUL4, a component of the E3 ubiquitin-ligase complex, and stabilizes CUL4 targets such as CDT1 (39). In addition, by stimulating PRMT5/MEP50-dependent arginine methylation of p53, cyclin D–CDK4/6 suppresses the expression of key antiproliferative and pro-apoptotic p53 target genes (40). Another study proposed that PRMT5 regulates splicing of the transcript encoding MDM4, a negative regulator of p53. CDK4/6 inhibition reduced PRMT5 activity and altered the pre-mRNA splicing of MDM4, leading to decreased levels of MDM4 protein and resulting in p53 activation. This, in turn, up-regulated the expression of a p53 target, p21CIP1, that blocks cell cycle progression (41).
During oncogenic transformation of hematopoietic cells, chromatin-bound CDK6 phosphorylates the transcription factors NFY and SP1 and induces the expression of p53 antagonists such as PRMT5, PPM1D, and MDM4 (42). Also, in acute myeloid leukemia cells expressing constitutively activated FLT3, CDK6 binds the promoter region of the FLT3 gene as well as the promoter of PIM1 pro-oncogenic kinase and stimulates their expression. Treatment of FLT3-mutant leukemic cells with a CDK4/6 inhibitor decreased FLT3 and PIM1 expression and triggered cell cycle arrest and apoptosis (43). The relevance of these various mechanisms in the context of human tumors is unclear and requires further study.
Mechanism of action of CDK4/6 inhibitors
Three small-molecule CDK4/6 inhibitors have been extensively characterized in preclinical studies: palbociclib and ribociclib, which are highly specific CDK4/6 inhibitors, and abemaciclib, which inhibits CDK4/6 and other kinases (Table 1). It has been assumed that these compounds act in vivo by directly inhibiting cyclin D–CDK4/6 (9). This simple model has been recently questioned by observations that palbociclib inhibits only cyclin D–CDK4/6 dimers, but not trimeric cyclin D–CDK4/6-p27KIP1 (44). However, it is unlikely that substantial amounts of cyclin D–CDK4 dimers ever exist in cells, because nearly all cyclin D–CDK4 in vivo is thought to be complexed with KIP/CIP proteins (11, 14, 44). Palbociclib also binds monomeric CDK4 (44). Surprisingly, treatment of cancer cells with palbociclib for 48 hours failed to inhibit CDK4 kinase, despite cell cycle arrest, but it inhibited CDK2 (44). Hence, palbociclib might prevent the formation of active CDK4-containing complexes (through binding to CDK4) and indirectly inhibit CDK2 by liberating KIP/CIP inhibitors. This model needs to be reconciled with several observations. First, treatment of cells with CDK4/6 inhibitors results in a rapid decrease of RB1 phosphorylation on cyclin D–CDK4/6-dependent sites, indicating an acute inhibition of CDK4/6 (45–47). Moreover, CDK4/6 immunoprecipitated from cells can be inhibited by palbociclib (48) and p21CIP-associated cyclin CDK4/6 kinase is also inhibited by treatment of cells with palbociclib (49). Lastly, CDK2 is dispensable for proliferation of several cancer cell lines (50, 51), hence the indirect inhibition of CDK2 alone is unlikely to be responsible for cell cycle arrest.
Name of compound
IC50
Other known targets
Stage of clinical development
Palbociclib (PD-0332991)
D1-CDK4, 11 nM;
D2-CDK6, 15 nM;
D3-CDK4, 9 nM
FDA-approved for HR+/HER2– advanced
breast cancer in combination with
endocrine therapy; phase 2/3 trials
for several other tumor types
Ribociclib (LEE011)
D1-CDK4, 10 nM;
D3-CDK6, 39 nM
FDA-approved for HR+/HER2– advanced
breast cancer in combination with
endocrine therapy; phase 2/3 trials
for several other tumor types
Abemaciclib (LY2835219)
D1-CDK4, 0.6 to 2 nM;
D3-CDK6, 8 nM
Cyclin T1–CDK9, PIM1, HIPK2, CDKL5,
CAMK2A, CAMK2D, CAMK2G,
GSK3α/β, and (at higher doses)
cyclin E/A–CDK2 and cyclin B–CDK1
FDA-approved for early (adjuvant) and
advanced HR+/HER2– breast cancer in
combination with endocrine therapy;
FDA-approved as monotherapy in advanced
HR+/HER2– breast cancer; phase 2/3 trials
for several other tumor types
Trilaciclib (G1T28)
D1-CDK4, 1 nM;
D3-CDK6, 4 nM
FDA-approved for small-cell lung cancer
to reduce chemotherapy-induced bone
marrow suppression; phase 2/3 trials
for other solid tumors
Lerociclib (G1T38)
D1-CDK4, 1 nM;
D3-CDK6, 2 nM
Phase 1/2 trials for HR+/HER2– advanced
breast cancer and EGFR-mutant
non–small-cell lung cancer
SHR6390
CDK4, 12 nM;
CDK6, 10 nM
Phase 1/2/3 trials for HR+/HER2– advanced
breast cancer and other solid tumors
PF-06873600
CDK4, 0.13 nM (Ki),
CDK6, 0.16 nM (Ki)
CDK2, 0.09 nM (Ki)
Phase 2 trials for HR+/HER2– advanced
breast cancer and other solid tumors
FCN-437
D1-CDK4, 3.3 nM;
D3-CDK6, 13.7 nM
Phase 1/2 trials for HR+/HER2– advanced
breast cancer and other solid tumors
Birociclib (XZP-3287)
Not reported
Phase 1/2 trials for HR+/HER2– advanced
breast cancer and other solid tumors
HS-10342
Not reported
Phase 1/2 trials for HR+/HER2– advanced
breast cancer and other solid tumors
This table lists major inhibitors of CDK4 and CDK6, half-maximal inhibitory concentration (IC50) for different cyclin D–CDK4/6 complexes (if known), other known targets, and the stage of clinical development. Ki, inhibitory constant.
Palbociclib, ribociclib, and abemaciclib were shown to block binding of CDK4 and CDK6 to CDC37, the kinase-targeting subunit of HSP90, thereby preventing access of CDK4/6 to the HSP90-chaperone system (52). Because the HSP90-CDC37 complex stabilizes several kinases (53), these observations suggest that CDK4/6 inhibitors, by disrupting the interaction between CDC37 and CDK4 or CDK6, might promote degradation of CDK4 and CDK6. However, depletion of CDK4/6 is typically not observed upon treatment with CDK4/6 inhibitors (54). More studies are needed to resolve these conflicting reports and to establish how CDK4/6 inhibitors affect the cell cycle machinery in cancer cells.
Validation of CDK4/6 inhibitors as anticancer agents
Consistent with the notion that RB1 represents the major rate-limiting substrate of cyclin D–CDK4/6 in cell cycle progression (55–57), palbociclib, ribociclib, and abemaciclib were shown to block proliferation of several RB1-positive cancer cell lines, but not cell lines that have lost RB1 expression (46, 58, 59). Breast cancer cell lines representing the luminal, estrogen receptor–positive (ER+) subtype were shown to be most susceptible to cell proliferation arrest upon palbociclib treatment (45). Palbociclib, ribociclib, abemaciclib, and another CDK4/6 inhibitor, lerociclib, were demonstrated to display potent antitumor activity in xenografts of several tumor types, including breast cancers (46, 60–62). Palbociclib and abemaciclib cross the blood-brain barrier and inhibit growth of intracranial glioblastoma (GBM) xenografts, with abemaciclib being more efficient in reaching the brain (63, 64). Recently, additional CDK4/6 inhibitors were shown to exert therapeutic effects in mouse xenograft models of various cancer types, including SHR6390 (65), FCN-437 (66), and compound 11 (67); the latter two were reported to cross the blood-brain barrier. In most in vivo studies, the therapeutic effect was dependent on expression of intact RB1 protein in tumor cells (46, 63). However, antitumor effects of palbociclib were also reported in bladder cancer xenografts independently of RB1 status; this was attributed to decreased phosphorylation of FOXM1 (68).
Tumor cell senescence upon CDK4/6 inhibition
In addition to blocking cell proliferation, inhibition of CDK4/6 can also trigger tumor cell senescence (63), which depends on RB1 and FOXM1 (35, 54). The role of RB1 in enforcing cellular senescence is well established (69). In addition, cyclin D–CDK4/6 phosphorylates and activates FOXM1, which has anti-senescence activity (35, 70). Senescence represents a preferred therapeutic outcome to cell cycle arrest, as it may lead to a durable inhibition of tumor growth.
It is not clear what determines the extent of senescence upon treatment of cancer cells with CDK4/6 inhibitors. A recent study showed that inhibition of CDK4/6 leads to an RB1-dependent increase in reactive oxygen species (ROS) levels, resulting in activation of autophagy, which mitigates the senescence of breast cancer cells in vitro and in vivo (71). Co-treatment with palbociclib plus autophagy inhibitors strongly augmented the ability of CDK4/6 inhibitors to induce tumor cell senescence and led to sustained inhibition of cancer cell proliferation in vitro and of xenograft growth in vivo (71). Decreased mTOR signaling after long-term CDK4/6 inhibition was shown to be essential for the induction of senescence in melanoma cells, and activation of mTORC1 overrode palbociclib-induced senescence (72). Others postulated that expression of the chromatin-remodeling enzyme ATRX and degradation of MDM2 determines the choice between quiescence and senescence upon CDK4/6 inhibition (73). Inhibition of CDK4 causes dissociation of the deubiquitinase HAUSP/USP7 from MDM2, thereby driving autoubiquitination and proteolytic degradation of MDM2, which in turn promotes senescence. This mechanism requires ATRX, which suggests that expression of ATRX can be used to predict the senescence response (73). Two additional proteins that play a role in this process are PDLIM7 and type II cadherin CDH18. Expression of CDH18 correlated with a sustained response to palbociclib in a phase 2 trial for patients with liposarcoma (74).
Markers predicting response to CDK4/6 inhibition
Only tumors with intact RB1 respond to CDK4/6 inhibitor treatment by undergoing cell cycle arrest or senescence (9, 58). In addition, “D-cyclin activating features” (CCND1 translocation, CCND2 or CCND3 amplification, loss of the CCND1-3 3′-untranslated region, and deletion of FBXO31 encoding an F-box protein implicated in cyclin D1 degradation) were shown to confer a strong response to abemaciclib in cancer cell lines (58). Moreover, co-deletion of CDKN2A and CDKN2C (encoding p16INK4A/p19ARF and p18INK4C, respectively) confers palbociclib sensitivity in glioblastoma (75). Thr172 phosphorylation of CDK4 and Tyr88 phosphorylation of p27KIP1 (both associated with active cyclin D–CDK4) correlate with sensitivity of breast cancer cell lines or tumor explants to palbociclib (76, 77). Surprisingly, in PALOMA-1, PALOMA-2, and PALOMA-3 trials (78–80), and in another independent large-scale study (81), CCND1 gene amplification or elevated levels of cyclin D1 mRNA or protein were not predictive of palbociclib efficacy. Conversely, overexpression of CDK4, CDK6, or cyclin E1 is associated with resistance of tumors to CDK4/6 inhibitors (see below).
Synergy of CDK4/6 inhibitors with other compounds
Several preclinical studies have documented the additive or synergistic effects of combining CDK4/6 inhibitors with inhibitors of the receptor tyrosine kinases as well as phosphoinositide 3-kinase (PI3K), RAF, or MEK (Table 2). This synergism might be because these pathways impinge on the cell cycle machinery through cyclin D–CDK4/6 (82–86). In some cases, the effect was seen in the presence of specific genetic lesions, such as EGFR, BRAFV600E, KRAS, and PIK3CA mutations (59, 87–89) (Table 2). When comparing different dosing regimens, continuous treatment with a MEK inhibitor with intermittent palbociclib resulted in more complete tumor responses than other combination schedules (90). Treatment with CDK4/6 inhibitors sensitized cancer cells to ionizing radiation (63) or cisplatin (68). The synergism with platinum-based chemotherapy was attributed to the observation that upon this treatment, CDK6 phosphorylates and stabilizes the FOXO3 transcription factor, thereby promoting tumor cell survival. Consequently, inhibition of CDK6 increases platinum sensitivity by enhancing tumor cell death (91).
In several instances, co-treatment with CDK4/6 inhibitors prevented the development of resistance to other compounds or inhibited the proliferation of resistant tumor cells. Co-treatment of melanoma patient-derived xenografts (PDXs) with ribociclib plus the RAF inhibitor encorafenib delayed or prevented development of encorafenib resistance (92). PDXs that acquired encorafenib resistance remained sensitive to the combination of encorafenib plus ribociclib (59). Treatment of BRAFV600E-mutant melanoma xenografts with palbociclib plus the BRAFV600E inhibitor PLX4720 prevented development of resistance (89). BRAFV600E-mutant melanoma cell lines that acquired resistance to the BRAFV600E inhibitor vemurafenib remained sensitive to palbociclib or abemaciclib, and xenografts underwent senescence and tumor regression upon CDK4/6 inhibition (72, 93). Treatment of ALK-mutant, ALK kinase inhibitor–resistant neuroblastoma xenografts with palbociclib restored the sensitivity to these compounds (94). A combination of PI3K and CDK4/6 inhibitors overcame the intrinsic and acquired resistance of breast cancers to PI3K inhibitors and resulted in regression of PIK3CA-mutant xenografts (88).
Up-regulation of cyclin D1 expression was shown to mediate acquired resistance of HER2+ tumors to anti-HER2 therapies in a mouse breast cancer model (95). Treatment of mice bearing trastuzumab-resistant tumors or PDXs of resistant HER2+ mammary carcinomas with abemaciclib restored the sensitivity of tumors to HER2 inhibitors and inhibited tumor cell proliferation. Moreover, in the case of treatment-naïve tumors, co-administration of abemaciclib significantly delayed the development of resistance to anti-HER2 therapies (95).
Several anticancer treatments, such as chemotherapy, target dividing cells. Because CDK4/6 inhibitors block tumor cell proliferation, they might impede the effects of chemotherapy. Indeed, several reports have documented that co-administration of CDK4/6 inhibitors antagonized the antitumor effects of compounds that act during S phase (doxorubicin, gemcitabine, methotrexate, mercaptopurine) or mitosis (taxanes) (96, 97). However, some authors reported synergistic effects (98, 99), although the molecular underpinnings are unclear.
A recent report documented that administration of CDK4/6 inhibitors prior to taxanes inhibited tumor cell proliferation and impeded the effect of taxanes (100). By contrast, administration of taxanes first (or other chemotherapeutic compounds that act on mitotic cells or cells undergoing DNA synthesis), followed by CDK4/6 inhibitors, had a strong synergistic effect. The authors showed that by repressing the E2F-dependent transcriptional program, CDK4/6 inhibitors impaired the expression of genes required for DNA-damage repair via homologous recombination. Because treatment of cancer cells with chemotherapy triggers DNA damage, the impairment of DNA-damage repair induced cytotoxicity, thereby explaining the synergistic effect (100).
Cells with impaired homologous recombination rely on poly-(ADP-ribose) polymerase (PARP) for double-stranded DNA-damage repair, which renders them sensitive to PARP inhibition. Indeed, a strong synergistic effect has been demonstrated between CDK4/6 inhibitors and PARP inhibitors in PDX-derived cell lines (100). Such synergy was also reported for ovarian cancer cells (101). Another study found that inhibition of CDK4/6 resulted in down-regulation of PARP levels (102).
Protection against chemotherapy-induced toxicity
Administration of palbociclib to mice induced reversible quiescence in hematopoietic stem/progenitor cells (HSPCs). This effect protected mice from myelosuppression after total-body irradiation. Moreover, treatment of tumor-bearing mice with CDK4/6 inhibitors together with irradiation mitigated radiation-induced toxicity without compromising the therapeutic effect (103). Co-administration of a CDK4/6 inhibitor, trilaciclib, with cytotoxic chemotherapy (5-FU, etoposide) protected animals from chemotherapy-induced exhaustion of HSPCs, myelosuppression, and apoptosis of bone marrow (60, 104). These observations led to phase 2 clinical trial, which evaluated the effects of trilaciclib administered prior to etoposide and carboplatin for treatment of small-cell lung cancer. Trilaciclib improved myelopreservation while having no adverse effect on antitumor efficacy (105). A similar phase 2 clinical trial investigating trilaciclib in combination with gemcitabine and carboplatin chemotherapy in patients with metastatic triple-negative breast cancer (TNBC) did not observe a significant difference in myelosuppression. However, this study demonstrated an overall survival benefit of the combination therapy (106, 107).
Metabolic function of CDK4/6 in cancer cells
The role of CDK4/6 in tumor metabolism is only starting to be appreciated (Fig. 2A). Treatment of pancreatic cancer cells with CDK4/6 inhibitors was shown to induce tumor cell metabolic reprogramming (108). CDK4/6 inhibition increased the numbers of mitochondria and lysosomes, activated mTOR, and increased the rate of oxidative phosphorylation, likely through an RB1-dependent mechanism (108). Combined inhibition of CDK4/6 and mTOR strongly suppressed tumor cell proliferation (108). Moreover, CDK4/6 can phosphorylate and inactivate TFEB, the master regulator of lysosomogenesis, and through this mechanism reduce lysosomal numbers. Conversely, CDK4/6 inhibition activated TFEB and increased the number of lysosomes (109). Another mechanism linking CDK4/6 and lysosomes was provided by the observation that treatment of TNBC cells with CDK4/6 inhibitors decreased mTORC1 activity and impaired the recruitment of mTORC1 to lysosomes (110). Consistent with the idea that mTORC1 inhibits lysosomal biogenesis, CDK4/6 inhibition increased the number of lysosomes in tumor cells. Because an increased lysosomal biomass underlies some cases of CDK4/6 inhibitor resistance (see below) (111), stimulation of lysosomogenesis by CDK4/6 inhibitors might limit their clinical efficacy by inducing resistance.
Fig. 2. CDK4 and CDK6: More than cell cycle kinases.
Although the role of CDK4 and CDK6 in cell cycle progression has been well documented, both kinases regulate several other functions that are only now starting to be unraveled. (A) Inhibition of CDK4/6 (CDK4/6i) affects lysosome and mitochondrial numbers as well as oxidative phosphorylation. Cyclin D3–CDK6 phosphorylates glycolytic enzymes 6-phosphofructokinase (PFKP) and pyruvate kinase M2 (PKM2), thereby controlling ROS levels via the pentose phosphate (PPP) and serine synthesis pathways. (B) Inhibition of CDK4/6 affects antitumor immunity, acting both within cancer cells and on the immune system of the host. In tumor cells, inhibition of CDK4/6 impedes expression of an E2F target, DNA methyltransferase (DNMT). DNMT inhibition reduces methylation of endogenous retroviral genes (ERV) and increases intracellular levels of double-stranded RNA (dsRNA) (114). In effector T cells, inhibition of CDK4/6 stimulates NFAT transcriptional activity and enhances secretion of IFN-γ and interleukin 2 (IL-2) (115).
Lastly, CDK4/6 inhibition impaired lysosomal function and the autophagic flux in cancer cells. It was argued that this lysosomal dysfunction was responsible for the senescent phenotype in CDK4/6 inhibitor–treated cells (110). Because lysosomes are essential for autophagy, the authors co-treated TNBC xenografts with abemaciclib plus an AMPK activator, A769662 (which induces autophagy), and found that this led to cancer cell death and subsequent regression of tumors (110).
Cyclin D3–CDK6 phosphorylates and inhibits two rate-limiting glycolytic enzymes, 6-phosphofructokinase and pyruvate kinase M2. This redirects glycolytic intermediates into the pentose phosphate pathway (PPP) and serine synthesis pathway. Through this mechanism, cyclin D3–CDK6 promotes the production of reduced nicotinamide adenine dinucleotide phosphate (NADPH) and reduced glutathione (GSH) and helps to neutralize ROS (112). Treatment of tumors expressing high levels of cyclin D3–CDK6 (such as leukemias) with CDK4/6 inhibitors reduced the PPP- and serine-synthesis pathway flow, thereby depleting the antioxidants NADPH and GSH. This increased ROS levels and triggered tumor cell apoptosis (112).
Another link between cyclin D–CDK4/6 in metabolism and cancer was provided by the observation that livers of obese/diabetic mice up-regulate cyclin D1 expression (113). Treatment of these mice with an antidiabetic compound, metformin, reduced liver cyclin D1 levels and largely protected mice against development of hepatocellular carcinoma. Also, genetic ablation of cyclin D1 protected obese/diabetic mice from liver cancer, and administration of palbociclib inhibited liver cancer progression. These treatments had no effect on tumors in nonobese animals (113). These observations raise the possibility of using antidiabetic compounds with CDK4/6 inhibitors for treatment of liver cancers in obese patients.
CDK4/6 inhibitors and antitumor immune responses
Several recent reports have started to unravel how inhibition of CDK4/6 influences antitumor immune responses, acting both on tumor cells as well as on the tumor immune environment (Fig. 2B). Treatment of breast cancer–bearing mice or breast cancer cells with abemaciclib activated expression of endogenous retroviral elements in tumor cells, thereby increasing the levels of double-stranded RNA. This, in turn, stimulated production of type III interferons and increased presentation of tumor antigens. Hence, CDK4/6 inhibitors, by inducing viral gene expression, trigger antiviral immune responses that help to eliminate the tumor (114).
Inhibition of CDK4/6 also affects the immune system by impeding the proliferation of CD4+FOXP3+ regulatory T cells (Tregs), which normally inhibit the antitumor response. Because cytotoxic CD8+ T cells are less affected by CDK4/6 inhibition, abemaciclib treatment decreases the Treg/CD8+ ratio of intratumoral T cells and facilitates tumor cell killing by cytotoxic CD8+ T cells (114).
Inhibition of CDK4/6 also resulted in activation of T cells through derepression of NFAT signaling. NFAT4 (and possibly other NFATs) are phosphorylated by cyclin D3–CDK6 (115). Inhibition of CDK4/6 decreased phosphorylation of NFATs, resulting in their nuclear translocation and enhanced transcriptional activity. This caused up-regulation of NFAT targets, resulting in T cell activation, which enhanced the antitumor immune response. In addition, CDK4/6 inhibitors increased the infiltration of effector T cells into tumors, likely because of elevated levels of chemokines CXCL9 and CXCL10 after CDK4/6 inhibitor treatment (115). Abemaciclib treatment also induced inflammatory and activated T cell phenotypes in tumors and up-regulated the expression of immune checkpoint proteins CD137, PD-L1, and TIM-3 on CD4+ and CD8+ cells (116).
CDK4/6 inhibition also caused up-regulation of PD-L1 protein expression in tumor cells (117, 118). This effect was shown to be independent of RB1 status in the tumor. Mechanistically, CDK4/6 phosphorylates and stabilizes SPOP, which promotes PD-L1 polyubiquitination and degradation (118). Cyclin D–CDK4 also represses expression of PD-L1 through RB1. Specifically, cyclin D–CDK4/6-mediated phosphorylation of RB1 on S249/T252 promotes binding of RB1 to NF-κB protein p65, and this represses the expression of a subset NF-κB–regulated genes, including PD-L1 (119).
These observations prompted tests of the efficacy of combining CDK4/6 inhibitors with antibodies that elicit immune checkpoint blockade. Indeed, treatment of mice bearing autochthonous breast cancers, or cancer allografts, with CDK4/6 inhibitors together with anti-PD-1/PD-L1 antibodies enhanced the efficacy of immune checkpoint blockade and led to complete tumor regression in a high proportion of animals (114, 115, 118). Conversely, activation of the cyclin D–CDK4 pathway by genomic lesions in human melanomas correlated with resistance to anti–PD-1 therapy (117).
Some authors did not observe synergy when abemaciclib was administered concurrently with immune checkpoint inhibitors in allograft tumor models (116, 120). However, a strong synergistic antitumor effect was detected when abemaciclib was administered first (and continued) and anti–PD-L1 antibody was administered later. The combined treatment induced immunological memory, as mice that underwent tumor regression were resistant to rechallenge with the same tumor (116). Abemaciclib plus anti–PD-L1 treatment increased infiltration of CD4+ and CD8+ T cells into tumors, and increased the expression of major histocompatibility complex class I (MHC-I) and MHC-II on tumor cells and on macrophages and MHC-I on dendritic cells (116). In the case of anti–CTLA-4 plus anti–PD-1 treatment in melanoma allograft model, the synergistic effect was observed when immune checkpoint inhibitor treatment was started first, followed by abemaciclib (120).
The synergistic antitumor effect of PI3K and CDK4/6 inhibitors in TNBC is mediated, in part, by enhancement of tumor immunogenicity (121). Combined treatment of TNBC cells with ribociclib plus the PI3K inhibitor apelisib synergistically up-regulated the expression of immune-related pathways in tumor cells, including proteins involved in antigen presentation. Co-treatment of tumor-bearing mice also decreased proliferation of CD4+FOXP3+ Treg cells, increased activation of intratumoral CD4+ and CD8+ T cells, increased the frequency of tumor-infiltrating NKT cells, and decreased the numbers of intratumoral immunosuppressive myeloid-derived suppressor cells. Moreover, combined treatment strongly augmented the response to immune checkpoint therapy with PD-1 and CTLA-4 antibodies (121).
Single-cell RNA sequencing of human melanomas identified an immune resistance program expressed by tumor cells that correlates with T cell exclusion from the tumor mass and immune evasion by tumor cells. The program can predict the response of tumors to immune checkpoint inhibitors. Treatment of human melanoma cells with abemaciclib repressed this program in an RB1-dependent fashion (120).
Together, these findings indicate that CDK4/6 inhibitors may convert immunologically “cold” tumors into “hot” ones. The most pressing issue is to validate these findings in a clinical setting. The utility of combining CDK4/6 inhibitors with PD-1 or PD-L1 antibodies is currently being evaluated in several clinical trials. Note that the effects of CDK4/6 inhibition on the immune system of the host are independent of tumor cell RB1 status, raising the possibility of using CDK4/6 inhibitors to also boost the immune response against RB1-negative tumors.
CDK4/6 inhibitors in clinical trials
Table 3 summarizes major clinical trials with CDK4/6 inhibitors. Given early preclinical data indicating that breast cancers—in particular, the hormone receptor–positive ones—are very sensitive to CDK4/6 inhibition (as discussed above), many clinical trials have focused on this cancer type. Most studies have evaluated CDK4/6 inhibitors administered together with anti-estrogens (the aromatase inhibitors letrozole or anastrozole, or the estrogen receptor antagonist fulvestrant) for treatment of advanced/metastatic HR+/HER2– breast cancers in postmenopausal women. Addition of CDK4/6 inhibitors significantly extended median progression-free survival (78, 122–130) and prolonged median overall survival (131–134). Moreover, abemaciclib has shown clinical activity when administered as a single agent (135). Consequently, palbociclib, ribociclib, and abemaciclib have been approved by the US Food and Drug Administration (FDA) for treatment of patients with advanced/metastatic HR+/HER2– breast cancer (Box 1). A recent phase 3 clinical trial, MonarchE, evaluated abemaciclib plus standard endocrine therapy in treatment of patients with early-stage, high-risk, lymph node–positive HR+/HER2– breast cancer. Addition of abemaciclib reduced the risk of breast cancer recurrence (136). This is in contrast to the similar PALLAS study reported this year, which found no benefit of adding palbociclib to endocrine therapy for women with early-stage breast cancer (137). Analysis of patient populations in these two trials may help to explain the different outcomes. It is also possible that the favorable outcome of the MonarchE study reflects a broader spectrum of kinases inhibited by abemaciclib. The utility of CDK4/6 inhibitors in early-stage breast cancer remains unclear and is being addressed in ongoing clinical trials (PALLAS, PENELOPE-B, EarLEE-1, MonarchE) (138).
CDK4/6 inhibitor
Trial name
Trial details
Treatment
Patients
Outcome
Ref.
Other outcomes
Palbociclib
PALOMA-1
Randomized
phase 2
Aromatase inhibitor
letrozole alone
(standard of care)
versus letrozole
plus palbociclib
Postmenopausal women
with advanced ER+/HER2–
breast cancer who had
not received any systemic
treatment for their
advanced disease
Addition of palbociclib markedly
increased median PFS from
10.2 months in the
letrozole group to
20.2 months in the
palbociclib plus
letrozole group
On the basis of this result, palbociclib
received a “Breakthrough Therapy”
designation status from FDA and was
granted accelerated approval, in
combination with letrozole, for the
treatment of ER+/HER2– metastatic
breast cancer
Palbociclib
PALOMA-2
Double-blind
phase 3
Palbociclib plus
letrozole as first-
line therapy
Postmenopausal women
with ER+/HER2–
breast cancer
Addition of palbociclib strongly
increased median PFS:
14.5 months in the placebo-
letrozole group versus
24.8 months in the
palbociclib-letrozole group
Palbociclib was equally efficacious in
patients with luminal A and B breast
cancers, and there was no single
biomarker associated with the lack of
clinical benefit, except for RB1 loss;
CDK4 amplification was associated
with endocrine resistance, but this
was mitigated by addition of
palbociclib; tumors with high levels
of FGFR2 and ERBB3 mRNA
displayed greater PFS gain
after addition of palbociclib (79)
Palbociclib
PALOMA-3
Randomized
phase 3
Estrogen receptor
antagonist
fulvestrant plus
placebo versus
fulvestrant plus
palbociclib
Women with HR+/HER2–
metastatic breast cancer
that had progressed on
previous endocrine therapy
The study demonstrated a
substantial prolongation
of median PFS in the palbociclib-
treated group: 4.6 months in the
placebo plus fulvestrant group
versus 9.5 months in the
palbociclib plus fulvestrant
group; addition of palbociclib
also extended median overall
survival from 28.0 months
(placebo-fulvestrant) to
34.9 months (palbociclib-
fulvestrant); estimated rate
of survival at 3 years was
41% versus 50%, respectively
Palbociclib in
patients with
early breast
cancer at high
risk of recurrence
Ongoing
Ribociclib
MONA
LEESA-2
Randomized
phase 3
Ribociclib plus
letrozole versus
placebo plus
letrozole
First-line treatment for
postmenopausal women
with HR+/HER2– recurrent
or metastatic breast
cancer who had not
received previous
systemic therapy for
advanced disease
At 18 months, PFS
was 42.2% in the
placebo-letrozole
group and 63.0%
in the ribociclib-
letrozole group
Patients with advanced
(metastatic or recurrent)
HR+/HER2– breast cancer
who have either received no
treatment for the advanced
disease or previously
received a single line of
endocrine therapy for the
advanced disease
Addition of ribociclib significantly
extended median PFS, from
12.8 months (placebo-fulvestrant)
to 20.5 months (ribociclib-
fulvestrant); overall survival at
42 months was also extended
from 45.9% (placebo-fulvestrant)
to 57.8% (ribociclib-fulvestrant)
Ribociclib versus
placebo together
with an anti-
estrogen tamoxifen
or an aromatase
inhibitor (letrozole
or anastrozole)
Premenopausal and
perimenopausal women
with HR+/HER2– advanced
breast cancer who had not
received previous treatment
with CDK4/6 inhibitors
Ribociclib significantly increased
median PFS from 13.0 months in
the placebo-endocrine therapy
group to 23.8 months in the
ribociclib-endocrine therapy
group; overall survival was also
strongly prolonged in the ribociclib
group (estimated overall survival
at 42 months was 46.0% for the
placebo group and 70.2% in the
ribociclib group)
Ribociclib in the
treatment of early-
stage, high-risk
HR+/HER2–
breast cancers
Ongoing
Abemaciclib
MONARCH 1
Phase 2 trial
Abemaciclib as a
single agent
Women with HR+/HER2–
metastatic breast cancer
who had progressed on or
after prior endocrine therapy
and had 1 or 2 chemotherapy
regimens in the metastatic
setting
Abemaciclib exhibited promising activity
in these heavily pretreated patients
with poor prognosis; median
PFS was 6.0 months and overall
survival 17.7 months
The most common adverse events
were diarrhea, fatigue, and
nausea (136)
Abemaciclib
MONARCH 2
Double-blind
phase 3
Abemaciclib in
combination
with fulvestrant
Women with HR+/HER2– breast
cancer who had progressed
while receiving endocrine
therapy, or while receiving
first-line endocrine therapy for
metastatic disease
Addition of abemaciclib significantly
increased PFS from 9.3 months in
the placebo-fulvestrant to 16.4 in
the abemaciclib-fulvestrant group;
median overall survival was also
extended from 37.3 months
to 46.7 months
Abemaciclib plus
an aromatase
inhibitor
(anastrozole
or letrozole)
Postmenopausal women
with advanced HR+/HER2–
breast cancer who had
no prior systemic therapy
in the advanced setting
Addition of abemaciclib prolonged
PFS from 14.8 months (in
the placebo-aromatase
inhibitor group) to 28.2 months
(abemaciclib-aromatase
inhibitor group)
Patients with HR+/HER2–
lymph node–positive,
high-risk early
breast cancer
Preliminary analysis indicates that
addition of abemaciclib resulted
in a significant improvement of
invasive disease-free survival
and of distant relapse-
free survival
Chemotherapy alone
(gemcitabine and
carboplatin),
versus concurrent
administration of
trilaciclib plus
chemotherapy,
versus
administration of
trilaciclib prior to
chemotherapy
(to mitigate the
cytotoxic effect of
chemotherapy on
bone marrow)
Patients with recurrent or
metastatic triple-negative
breast cancer who had no
more than two previous
lines of chemotherapy
Addition of trilaciclib did not offer
detectable myeloprotection, but
resulted in increased overall
survival (from 12.8 months in the
chemotherapy-only group to
20.1 months in the concurrent
trilaciclib and chemotherapy
group and 17.8 months in trilaciclib
before chemotherapy group)
Approved by FDA in 2016, in combination with fulvestrant for the treatment of hormone receptor–positive, HER2-negative (HR+/HER2–) advanced or metastatic breast cancer in women with disease progression following endocrine therapy. Approved in 2017 for the treatment of HR+/HER2– advanced or metastatic breast cancer in combination with an aromatase inhibitor as initial endocrine-based therapy in postmenopausal women.
Palbociclib is administered at a dose of 125 mg (given orally) daily for 3 weeks followed by 1 week off, or 200 mg daily for 2 weeks followed by 1 week off. The rate-limiting toxicities are neutropenia, thrombocytopenia, and anemia.
Ribociclib
Approved by FDA in 2017, in combination with an aromatase inhibitor as initial endocrine-based therapy for the treatment of postmenopausal women with HR+/HER2– advanced or metastatic breast cancer. In 2018, the FDA expanded the indication for ribociclib in combination with an aromatase inhibitor for pre/perimenopausal women with HR+/HER2– advanced or metastatic breast cancer, as initial endocrine-based therapy. FDA also approved ribociclib in combination with fulvestrant for postmenopausal women with HR+/HER2– advanced or metastatic breast cancer, as initial endocrine-based therapy or following disease progression on endocrine therapy.
Ribociclib is administered at a dose of 600 mg (given orally) daily for 3 weeks followed by 1 week off. The main toxicities are neutropenia and thrombocytopenia.
Abemaciclib
Approved by FDA in 2017, in combination with fulvestrant for women with HR+/HER2– advanced or metastatic breast cancer with disease progression following endocrine therapy. In addition, abemaciclib was approved as monotherapy for women and men with HR+/HER2– advanced or metastatic breast cancer with disease progression following endocrine therapy and prior chemotherapy in the metastatic setting. Approved by FDA in 2018 in combination with an aromatase inhibitor as initial endocrine-based therapy for postmenopausal women with HR+/HER2– advanced or metastatic breast cancer. Approved by FDA in 2021 for adjuvant treatment of early-stage HR+/HER2– breast cancer in combination with endocrine therapy.
Abemaciclib is administered at a dose of 200 mg (given orally) every 12 hours. The dose-limiting toxicity is fatigue. Neutropenia is also observed but is not rate-limiting. Other severe side effects include diarrhea and nausea.
Currently, palbociclib is being used in 164 active or recruiting clinical trials, ribociclib in 69 trials, and abemaciclib in 98 trials for more than 50 tumor types (139). These trials evaluate combinations of CDK4/6 inhibitors with a wide range of compounds (Table 4). Trials with trilaciclib test the benefit of this compound in preserving bone marrow and the immune system.
Additional target
Inhibitor
Immune checkpoint inhibitor
Tumor type
Trial identifier
Palbociclib
Aromatase
Letrozole, anastrozole,
exemestane
HR+ breast cancer, HR+ ovarian
cancer, metastatic breast cancer,
metastatic endometrial cancer
tumors with ERK1/2
mutations, glioblastoma,
metastatic cancer
NCT04534283,
NCT04391595,
NCT02857270
Trilaciclib
Proliferating cells
Chemotherapy
SCLC: This trial evaluates the
potential clinical benefit of
trilaciclib in preventing
chemotherapy-induced
myelosuppression in patients
receiving chemotherapy
NCT04504513
Proliferating cells +
PD-L1
Carboplatin + etoposide
Atezolizumab
SCLC: This trial investigates the
potential clinical benefit of trilaciclib
in preserving the bone marrow and
the immune system, and enhancing
antitumor efficacy when
administered with chemotherapy
NCT03041311
Proliferating cells
Topotecan
SCLC: This trial investigates the
potential clinical benefit of
trilaciclib in preserving the
bone marrow and the immune
system, and enhancing the
antitumor efficacy of chemotherapy
when administered prior
to chemotherapy
NCT02514447
Proliferating cells
Carboplatin + gemcitabine
Metastatic TNBC: This study
investigates the potential
clinical benefit of trilaciclib in
preserving the bone marrow
and the immune system, and
enhancing the antitumor efficacy
of chemotherapy when administered
prior to chemotherapy
NCT02978716
Lerociclib
ER
ER antagonist: fulvestrant
HR+/HER2– metastatic
breast cancer
NCT02983071
EGFR
Osimertinib
EGFR mutant NSCLC
NCT03455829
SHR6390
ER
ER antagonist: fulvestrant
HR+/HER2– recurrent/
metastatic breast cancer
NCT03481998
Aromatase
Letrozole, anastrozole
HR+/HER2– recurrent/
metastatic breast cancer
NCT03966898,
NCT03772353
EGFR/HER2
Pyrotinib
HER2+ gastric cancer, HER2+
metastatic breast cancer
NCT04095390,
NCT03993964
AR
AR antagonists: SHR3680
metastatic TNBC
NCT03805399
PF-06873600
Endocrine therapy
Single agent and then
in combination with
endocrine therapy
HR+/HER2– metastatic breast
cancer, ovarian and fallopian tube
cancer, TNBC and other tumors
Although CDK4/6 inhibitors represent very effective agents in cancer treatment, nearly all patients eventually develop resistance and succumb to the disease. Moreover, a substantial fraction of tumors show intrinsic resistance to treatment with CDK4/6 inhibitors (Fig. 3).
Fig. 3. Mechanisms of cancer cell resistance to CDK4/6 inhibition.
Known mechanisms include loss of RB1, activation of pathways impinging on CycD-CDK4/6, amplification of the CDK4/6 genes and overexpression of CDK6 protein, activation of CycE-CDK2, and lysosomal sequestration of CDK4/6 inhibitors. Blank pieces of the puzzle denote additional mechanisms that remain to be discovered.
The best-documented mechanism of preexisting and acquired resistance is the loss of RB1 (71, 81, 140). Acquired RB1 loss has been detected in PDXs (141), in circulating tumor DNA (ctDNA) (142, 143), and in tumors from patients treated with CDK4/6 inhibitors (144, 145). However, RB1 mutations are likely subclonal and are seen in only 5 to 10% of patients (143, 145).
Increased expression of CDK6 was shown to underlie acquired resistance to CDK4/6 inhibitors. Amplification of the CDK6 gene and the resulting overexpression of CDK6 protein were found in abemaciclib-resistant ER+ breast cancer cells (146) and in ctDNA of patients with ER+ breast cancers that progressed during treatment with palbociclib plus endocrine therapy (147). Also, CDK4 gene amplification conferred insensitivity to CDK4/6 inhibition in GBM and sarcomas (148–150), whereas overexpression of CDK4 protein was associated with resistance to endocrine therapy in HR+ breast cancers (79).
Resistant breast cancer cells can also up-regulate the expression of CDK6 through suppression of the TGF-β/SMAD4 pathway by the microRNA miR-432-5p. In this mechanism, exosomal expression of miR-432-5p mediates the transfer of the resistance phenotype between neighboring cell populations (151). Another mechanism of CDK6 up-regulation in ER+ breast cancers is the loss of FAT1, which represses CDK6 expression via the Hippo pathway. Loss of FAT1 triggers up-regulation of CDK6 expression by the Hippo pathway effectors TAZ and YAP. Moreover, genomic alterations in other components of the Hippo pathway, although rare, are also associated with reduced sensitivity to CDK4/6 inhibitors (81).
Genetic lesions that activate pathways converging on D-type cyclins can cause resistance to CDK4/6 inhibitors. These include (i) FGFR1/2 gene amplification or mutational activation, detected in ctDNA from patients with ER+ breast cancers that progressed upon treatment with palbociclib plus endocrine therapy (147); (ii) hyperactivation of the MAPK pathway in resistant prostate adenocarcinoma cells, possibly due to increased production of EGF by cancer cells (152); and (iii) increased secretion of FGF in palbociclib-resistant KRAS-mutant NSCLC cells, which stimulates FGFR1 signaling in an autocrine or paracrine fashion, resulting in activation of ERK1/2 and mTOR as well as up-regulation of D-cyclin, CDK6, and cyclin E expression (153). Analyses of longitudinal tumor biopsies from a melanoma patient revealed an activating mutation in the PIK3CA gene that conferred resistance to ribociclib plus MEK inhibitor treatment (154). It is possible that these lesions elevate the cellular levels of active cyclin D–CDK4/6 complexes, thereby increasing the threshold for CDK4/6 inhibition.
Formation of a noncanonical cyclin D1–CDK2 complex was shown to represent another mechanism of acquired CDK4/6 inhibitor resistance. Such a complex was observed in palbociclib-treated ER+ breast cancer cells and was implicated in overcoming palbociclib-induced cell cycle arrest (141). Also, depletion of AMBRA1 promoted the interaction of D-cyclins with CDK2, resulting in resistance to CDK4/6 inhibitors (20, 22); it remains to be seen whether this represents an intrinsic or acquired resistance mechanism in human tumors.
Genetic analyses revealed that activation of cyclin E can bypass the requirement for cyclin D–CDK4/6 in development and tumorigenesis (155, 156). Hence, it comes as no surprise that increased activity of cyclin E–CDK2 is responsible for a large proportion of intrinsic and acquired resistance to CDK4/6 inhibitors. Several different mechanisms can activate cyclin E–CDK2 kinase in resistant tumor cells: (i) Down-regulation of KIP/CIP inhibitors results in increased activity of cyclin E–CDK (54, 157). (ii) Loss of PTEN expression, which activates AKT signaling, leads to nuclear exclusion of p27KIP1. This in turn prevents access of p27KIP1 to CDK2, resulting in increased CDK2 kinase activity (144). (iii) Activation of the PI3K/AKT pathway causes decreased levels of p21CIP1. Co-treatment of melanoma PDXs with MDM2 inhibitors (which up-regulate p21CIP1 via p53) sensitized intrinsically resistant tumor cells to CDK4/6 inhibitors (158). (iv) Up-regulation of cyclin D1 levels triggers sequestration of KIP/CIP inhibitors from cyclin E–CDK2 to cyclin D–CDK4/6, thereby activating the former (158). (v) Amplification of the CCNE1 gene and increased levels of cyclin E1 protein result in elevated activity of E-CDK2 kinase (141). (vi) mTOR signaling has been shown to up-regulate cyclin E1 (and D1) in KRAS-mutated pancreatic cancer cells; CDK2 activity was essential for CDK4/6 inhibitor resistance in this setting (159). (vii) Up-regulation of PDK1 results in activation of the AKT pathway, which increases the expression of cyclins E and A and activates CDK2 (160). (viii) In CDK4/6 inhibitor–resistant melanoma cells, high levels of RNA-binding protein FXR1 increase translation of the amino acid transporter SLC36A1. Up-regulation of SLC36A1 expression activates mTORC1, which in turn increases CDK2 expression (161). All these lesions are expected to allow cell proliferation, despite CDK4/6 inhibition, as a consequence of the activation of the downstream cell cycle kinase CDK2.
The role for cyclin E–CDK2 in CDK4/6 inhibitor resistance has been confirmed in clinical trials. In patients with advanced ER+ breast cancer treated with palbociclib and letrozole or fulvestrant, the presence of proteolytically cleaved cytoplasmic cyclin E in tumor tissue conferred strongly shortened progression-free survival (71). Moreover, analyses of PALOMA-3 trial for patients with ER+ breast cancers revealed lower efficacy of palbociclib plus fulvestrant in patients displaying high cyclin E mRNA levels in metastatic biopsies (80). Amplification of the CCNE1 gene was detected in ctDNA of patients with ER+ breast cancers that progressed on palbociclib plus endocrine therapy (147). Also, amplification of the CCNE2 gene (encoding cyclin E2) was seen in a fraction of CDK4/6 inhibitor–resistant HR+ mammary carcinomas (145, 162).
Collectively, these analyses indicate that resistant cells may become dependent on CDK2 for cell cycle progression. Indeed, depletion of CDK2 or inhibition of CDK2 kinase activity in combination with CDK4/6 inhibitors blocked proliferation of CDK4/6 inhibitor–resistant cancer cells (111, 141, 158–161). Recently, two CDK2-specific inhibitors, PF-07104091 (163) and BLU0298 (164), have been reported. PF-07104091 is now being tested in a phase 2 clinical trial in combination with palbociclib plus antiestrogens. Another recent study identified a novel compound, PF-3600, that inhibits CDK4/6 and CDK2 (165). PF3600 had potent antitumor effects against xenograft models of intrinsic and acquired resistance to CDK4/6 inhibition (165). A phase 2 clinical trial is currently evaluating this compound as a single agent and in combination with endocrine therapy in patients with HR+/HER2– breast cancer and other cancer types.
Whole-exome sequencing of 59 HR+/HER2– metastatic breast tumors from patients treated with CDK4/6 inhibitors and anti-estrogens revealed eight alterations that likely conferred resistance: RB1 loss; amplification of CCNE2 or AURKA; activating mutations or amplification of AKT1, FGFR2, or ERBB2; activating mutations in RAS genes; and loss of ER expression. The frequent activation of AURKA (in 27% of resistant tumors) raises the possibility of combining CDK4/6 inhibitors with inhibitors of Aurora A kinase to overcome resistance (145).
In contrast to ER+ mammary carcinomas, TNBCs are overall resistant to CDK4/6 inhibition (45). A subset of TNBCs display high numbers of lysosomes, which causes sequestration of CDK4/6 inhibitors into the expanded lysosomal compartment, thereby preventing their action on nuclear CDK4/6. Preclinical studies revealed that lysosomotropic agents that reverse the lysosomal sequestration (such as chloroquine, azithromycin, or siramesine) render TNBC cells fully sensitive to CDK4/6 inhibition (71, 111). These observations now need to be tested in clinical trials for TNBC patients.
Outlook
Although D-cyclins and CDK4/6 were discovered 30 years ago, several aspects of cyclin D–CDK4/6 biology, such as their role in antitumor immunity, are only now starting to be appreciated. The full range of cyclin D–CDK4/6 functions in tumor cells remains unknown. It is likely that these kinases play a much broader role in cancer cells than is currently appreciated. Hence, the impact of CDK4/6 inhibition on various aspects of tumorigenesis requires further study. Also, treatment of patients with CDK4/6 inhibitors likely affects several aspects of host physiology, which may be relevant to cancer progression.
In the next years, we will undoubtedly witness the development and testing of new CDK4/6 inhibitors. Because activation of CDK2 represents a frequent CDK4/6 inhibitor resistance mechanism, compounds that inhibit CDK4/6 and CDK2 may prevent or delay the development of resistance. Conversely, selective compounds that inhibit CDK4 but not CDK6 may allow more aggressive dosing, as they are expected not to result in bone marrow toxicity caused by CDK6 inhibition. New, less basic CDK4/6 inhibitor compounds (111) may escape lysosomal sequestration and may be efficacious against resistant cancer types such as TNBC. Degrader compounds, which induce proteolysis of cyclin D rather than inhibit cyclin D–CDK4/6 kinase, may have superior properties, as they would extinguish both CDK4/6-dependent and -independent functions of D-cyclins in tumorigenesis. Moreover, dissolution of cyclin D–CDK4/6 complexes is expected to liberate KIP/CIP inhibitors, which would then inhibit CDK2. D-cyclins likely play CDK-independent functions in tumorigenesis—for example, by regulating gene expression (166). However, their role in tumor biology and the utility of targeting these functions for cancer treatment remain largely unexplored.
An important challenge will be to test and identify combinatorial treatments involving CDK4/6 inhibitors for the treatment of different tumor types. CDK4/6 inhibitors trigger cell cycle arrest of tumor cells and, in some cases, senescence. It will be essential to identify combination treatments that convert CDK4/6 inhibitors from cytostatic compounds to cytotoxic ones, which would unleash the killing of tumor cells. Genome-wide high-throughput screens along with analyses of mouse cancer models and PDXs will help to address this point. Another largely unexplored area of cyclin D–CDK4/6 biology is the possible involvement of these proteins in other pathologies, such as metabolic disorders. Research in this area may extend the use of CDK4/6 inhibitors to treatment of other diseases. All these unresolved questions ensure that CDK4/6 biology will remain an active area of basic, translational, and clinical research for several years to come.
CDK inhibitors and Breast Cancer
The U.S. Food and Drug Administration today granted accelerated approval to Ibrance (palbociclib) to treat advanced (metastatic) breast cancer inr postmenopausal women with estrogen receptor (ER)-positive, human epidermal growth factor receptor 2 (HER2)-negative metastatic breast cancer who have not yet received an endocrine-based therapy. It is to be used in combination with letrozole, another FDA-approved product used to treat certain kinds of breast cancer in postmenopausal women.
See Dr. Melvin Crasto’s blog posts on the announcement of approval of Ibrance (palbociclib) at
Palbociclib and LY2835219 are both cyclin-dependent kinase (CDK) 4/6 inhibitors. CDK4 and CDK6 are kinases that, together with cyclin D1, facilitate the transition of dividing cells from the G1 to the S (synthesis) phase of the cell cycle. Preclinical studies have shown that breast cancer cells rely on CDK4 and CDK6 for division and growth, and that selective CDK4/6 inhibitors can arrest the cells at this G1/S phase checkpoint.
The results of the phase II trial of palbociclib and phase I trial of LY2835219 both indicated that hormone receptor (HR)-positive disease appears to be the best marker to predict patient response.
LY2835219 Phase I Trial Demonstrates Early Activity
The CDK4/6 inhibitor LY2835219 has demonstrated early activity in heavily pretreated women with metastatic breast cancer. Nineteen percent of these women (9 out of 47) had a partial response and 51% (24 out of 47) had stable disease following monotherapy with the oral CDK4/6 inhibitor. Patients had received a median of seven prior therapies, and 75% had metastatic disease in the lung, liver, or brain. The median age of patients was 55 years.
All of the partial responses were in patients with HR-positive disease. The overall response rate for this patient subset was 25% (9 of 36 patients). Twenty of the patients with stable disease had HR-positive disease, with 13 patients having stable disease lasting 24 weeks or more.
Despite treatment, disease progression occurred in 23% of the patients.
These results were presented at a press briefing by Amita Patnaik, MD, associate director of clinical research at South Texas Accelerated Research Therapeutics in San Antonio, Texas, at the 2014 American Association for Cancer Research (AACR) Annual Meeting, held April 5–9, in San Diego.
The phase I trial of LY2835219 enrolled 132 patients with five different tumor types, including metastatic breast cancer. Patients received 150-mg to 200-mg doses of the oral drug every 12 hours.
The overall disease control rate was 70% for all patients and 81% among the 36 HR-positive patients.
The median progression-free survival (PFS) was 5.8 months for all patients and 9.1 months for HR-positive patients. Patnaik noted that the median PFS is still a moving target, as 18 patients, all with HR-positive disease, remain on therapy.
“The data are rather encouraging for a very heavily pretreated patient population,” said Patnaik during the press briefing.
Even though the trial was not designed to compare efficacy based on breast cancer subpopulations, the results in HR-positive tumors are particularly encouraging, according to Patnaik.
Common adverse events thought to be treatment-related were diarrhea, nausea, fatigue, vomiting, and neutropenia. These adverse events occurred in 5% or less of patients at grade 3 or 4 toxicity, except neutropenia, which occurred as a grade 3 or 4 toxicity in 11% of patients. Patnaik noted during the press briefing that the neutropenia was uncomplicated and did not result in discontinuation of therapy by any of the patients.
Palbociclib Phase II Data “Impressive”
The addition of the oral CDK4/6 inhibitor palbociclib resulted in an almost doubling of PFS in first-line treatment of postmenopausal metastatic breast cancer patients with HR-positive disease compared with a control population. The patients in this trial were not previously treated for their metastatic breast cancer, unlike the patient population in the phase I LY2835219 trial.
Patients receiving the combination of palbociclib at 125 mg once daily plus letrozole at 2.5 mg once daily had a median PFS of 20.2 months compared with a median of 10.2 months for patients treated with letrozole alone (hazard ratio = 0.488; P = .0004).
Richard S. Finn, MD, assistant professor of medicine at the University of California, Los Angeles, presented the data from the phase II PALOMA-1 trial at a press briefing at the AACR Annual Meeting.
A total of 165 patients were randomized 1:1 to either the experimental arm or control arm.
Forty-three percent of patients in the combination arm had an objective response compared with 33% of patients in the control arm.
Overall survival (OS), a secondary endpoint in this trial, was encouraging but the results are still preliminary, said Finn during the press briefing. The median OS was 37.5 months in the palbociclib arm compared with 33.3 months in the letrozole alone arm (P = .21). Finn noted that long-term follow-up is necessary to establish the median OS. “This first look of the survival data is encouraging. This is a front-line study, and it is encouraging that there is early [separation] of the curves,” he said.
No new toxicities were reported since the interim trial results. Common adverse events included leukopenia, neutropenia, and fatigue. The neutropenia could be quickly resolved and was uncomplicated and not accompanied by fever, said Finn.
Palbociclib is currently being tested in two phase III clinical trials: The PALOMA-3 trial is testing the combination of palbociclib with letrozole and fulvestrant in late-stage metastatic breast cancer patients who have failed endocrine therapy. The PENELOPE-B trial is testing palbociclib in combination with standard endocrine therapy in HR-positive breast cancer patients with residual disease after neoadjuvant chemotherapy and surgery.
References
Patnaik A, Rosen LS, Tolaney SM, et al. Clinical activity of LY2835219, a novel cell cycle inhibitor selective for CDK4 and CDK6, in patients with metastatic breast cancer. American Association for Cancer Research Annual Meeting 2014; April 5–9, 2014; San Diego. Abstr CT232.
Finn RS, Crown JP, Lang I, et al. Final results of a randomized phase II study of PD 0332991, a cyclin-dependent kinase (CDK)-4/6 inhibitor, in combination with letrozole vs letrozole alone for first-line treatment of ER+/HER2-advanced breast cancer (PALOMA-1; TRIO-18). American Association for Cancer Research Annual Meeting 2014; April 5–9, 2014; San Diego. Abstr CT101.
This article has been cited by other articles in PMC.
Cyclin-dependent kinases (CDKs) drive cell cycle progression and control transcriptional processes. The dysregulation of multiple CDK family members occurs commonly in human cancer; in particular, the cyclin D-CDK4/6-retinoblastoma protein (RB)-INK4 axis is universally disrupted, facilitating cancer cell proliferation and prompting long-standing interest in targeting CDK4/6 as an anticancer strategy. Most agents that have been tested inhibit multiple cell cycle and transcriptional CDKs and have carried toxicity. However, several selective and potent inhibitors of CDK4/6 have recently entered clinical trial. PD0332991, the first to be developed, resulted from the introduction of a 2-aminopyridyl substituent at the C2-position of a pyrido(2,3-d)pyrimidin-7-one backbone, affording exquisite selectivity toward CDK4/6.1 PD0332991 arrests cells in G1 phase by blocking RB phosphorylation at CDK4/6-specfic sites and does not inhibit the growth of RB-deficient cells.2 Phase I studies conducted in patients with advanced RB-expressing cancers demonstrated mild side effects and dose-limiting toxicities of neutropenia and thrombocytopenia, with prolonged stable disease in 25% of patients.3,4 In cyclin D1-translocated mantle cell lymphoma, PD0332991 extinguished CDK4/6 activity in patients’ tumors, resulting in markedly reduced proliferation, and translating to more than 1 year of stability or response in 5 of 17 cases.5
Two recent papers from the Knudsen laboratory make several important observations that will help guide the continued clinical development of CDK4/6 inhibitors. In the study by Dean et al., surgically resected patient breast tumors were grown on a tissue culture matrix in the presence or absence of PD0332991. Crucially, these cultures retained associated stromal components known to play important roles in cancer pathogenesis and therapeutic sensitivities, as well as key histological and molecular features of the primary tumor, including expression of ER, HER2 and Ki-67. Similar to results in breast cancer cell lines,6 the authors demonstrate that only RB-positive tumors have growth inhibition in response to PD0332991, irrespective of ER or HER2 status, while tumors lacking RB were completely resistant. This result underscores RB as the predominant target of CDK4/6 in breast cancer cells and the primary marker of drug response in primary patient-derived tumors. As expected, RB-negative tumors routinely demonstrated robust expression of p16INK4A; however, p16INK4A expression was not always a surrogate marker for RB loss, supporting the importance of direct screening of tumors for RB expression to select patients appropriate for CDK4/6 inhibitor clinical trials.
In the second study, McClendon et al. investigated the efficacy of PD0332991 in combination with doxorubicin in triple-negative breast cancer cell lines. Again, RB functionality was paramount in determining response to either PD0332991 monotherapy or combination treatment. In RB-deficient cancer cells, CDK4/6 inhibition had no effect in either instance. However, in RB-expressing cancer cells, CDK4/6 inhibition and doxorubicin provided a cooperative cytostatic effect, although doxorubicin-induced cytotoxicity was substantially reduced, assessed by markers for mitotic catastrophe and apoptosis. Additionally, despite cytostatic cooperativity, CDK4/6 inhibition maintained the viability of RB-proficient cells in the presence of doxorubicin, which repopulated the culture after removal of drug. These results reflect previous data demonstrating that ectopic expression of p16INK4A can protect cells from the lethal effects of DNA damaging and anti-mitotic chemotherapies.7 Similar results have been reported in MMTV-c-neu mice bearing RB-proficient HER2-driven tumors, where PD0332991 compromised carboplatin-induced regressions,8 suggesting that DNA-damaging treatments should not be combined concomitantly with CDK4/6 inhibition in RB-proficient tumors.
To combine CDK4/6 inhibition with cytotoxics, sequential treatment may be considered, in which CDK4/6 inhibition is followed by DNA damaging chemotherapy; cells relieved of G1 arrest may synchronously enter S phase, where they may be most susceptible to agents disrupting DNA synthesis. Release of myeloma cells from a prolonged PD0332991-mediated G1 block leads to S phase synchronization; interestingly, all scheduled gene expression is not completely restored (including factors critical to myeloma survival such as IRF4), further favoring apoptotic responses to cytotoxic agents.9 Furthermore, in RB-deficient tumors, CDK4/6 inhibitors may be used to maximize the therapeutic window between transformed and non-transformed cells treated with chemotherapy. In contrast to RB-deficient cancer cells, RB-proficient non-transformed cells arrested in G1 in response to PD0332991 are afforded protection from DNA damaging agents, thereby reducing associated toxicities, including bone marrow suppression.8
In summary, the current work provides evidence for RB expression as a determinant of response to CDK4/6 inhibition in primary tumors and highlights the complexity of combining agents targeting the cell cycle machinery with DNA damaging treatments.
To model the heterogeneity of breast cancer as observed in the clinic, we employed an ex vivo model of breast tumor tissue. This methodology maintained the histological integrity of the tumor tissue in unselected breast cancers, and importantly, the explants retained key molecular markers that are currently used to guide breast cancer treatment (e.g., ER and Her2 status). The primary tumors displayed the expected wide range of positivity for the proliferation marker Ki67, and a strong positive correlation between the Ki67 indices of the primary and corresponding explanted tumor tissues was observed. Collectively, these findings indicate that multiple facets of tumor pathophysiology are recapitulated in this ex vivo model. To interrogate the potential of this preclinical model to inform determinants of therapeutic response, we investigated the cytostatic response to the CDK4/6 inhibitor, PD-0332991. This inhibitor was highly effective at suppressing proliferation in approximately 85% of cases, irrespective of ER or HER2 status. However, 15% of cases were completely resistant to PD-0332991. Marker analyses in both the primary tumor tissue and the corresponding explant revealed that cases resistant to CDK4/6 inhibition lacked the RB-tumor suppressor. These studies provide important insights into the spectrum of breast tumors that could be treated with CDK4/6 inhibitors, and defines functional determinants of response analogous to those identified through neoadjuvant studies.
Keywords: ER, PD0332991, breast cancer, cell cycle, ex vivo
Breast cancer is a highly heterogeneous disease.1–4 Such heterogeneity is known to influence patient response to both standard of care and experimental therapeutics. In regards to biomarker-driven treatment of breast cancers, it was initially recognized that the presence of the estrogen receptor α (ER) in a fraction of breast cancer cells was associated with the response to tamoxifen and similar anti-estrogenic therapies.5,6 Since this discovery, subsequent marker analyses and gene expression profiling studies have further divided breast cancer into a series of distinct subtypes that harbor differing and often divergent therapeutic sensitivities.1–3 While clearly important in considering the use of several current standard of care therapies, these markers, or molecular sub-types, do not necessarily predict the response to new therapeutic approaches that are currently undergoing clinical development. Thus, there is the continued need for functional analyses of drug response and the definition of new markers that can be used to direct treatment strategies.
Currently, all preclinical cancer models are associated with specific limitations. It is well known that cell culture models lack the tumor microenvironment known to have a significant impact on tumor biology and therapeutic response.7–9 Xenograft models are dependent on the host response for the engraftment of tumor cells in non-native tissues, which do not necessarily recapitulate the nuances of complex tumor milieu.10 In addition, genetically engineered mouse models, while enabling the tumor to develop in the context of the host, can develop tumors that do not mirror aspects of human disease.10 Furthermore, it remains unclear whether any preclinical model truly represents the panoply of breast cancer subtypes that are observed in the clinic. Herein, we utilized a primary human tumor explant culture approach to interrogate drug response, as well as specific determinants of therapeutic response, in an unselected series of breast cancer cases.
Triple-negative breast cancer (TNBC) is an aggressive disease that lacks established markers to direct therapeutic intervention. Thus, these tumors are routinely treated with cytotoxic chemotherapies (e.g., anthracyclines), which can cause severe side effects that impact quality of life. Recent studies indicate that the retinoblastoma tumor suppressor (RB) pathway is an important determinant in TNBC disease progression and therapeutic outcome. Furthermore, new therapeutic agents have been developed that specifically target the RB pathway, potentially positioning RB as a novel molecular marker for directing treatment. The current study evaluates the efficacy of pharmacological CDK4/6 inhibition in combination with the widely used genotoxic agent doxorubicin in the treatment of TNBC. Results demonstrate that in RB-proficient TNBC models, pharmacological CDK4/6 inhibition yields a cooperative cytostatic effect with doxorubicin but ultimately protects RB-proficient cells from doxorubicin-mediated cytotoxicity. In contrast, CDK4/6 inhibition does not alter the therapeutic response of RB-deficient TNBC cells to doxorubicin-mediated cytotoxicity, indicating that the effects of doxorubicin are indeed dependent on RB-mediated cell cycle control. Finally, the ability of CDK4/6 inhibition to protect TNBC cells from doxorubicin-mediated cytotoxicity resulted in recurrent populations of cells specifically in RB-proficient cell models, indicating that CDK4/6 inhibition can preserve cell viability in the presence of genotoxic agents. Combined, these studies suggest that while targeting the RB pathway represents a novel means of treatment in aggressive diseases such as TNBC, there should be a certain degree of caution when considering combination regimens of CDK4/6 inhibitors with genotoxic compounds that rely heavily on cell proliferation for their cytotoxic effects.
Click on Video Link for Dr. Tolaney slidepresentation of recent data with CDK4/6 inhibitor trial results https://youtu.be/NzJ_fvSxwGk
Sara Tolaney, MD, MPH, a breast oncologist with the Susan F. Smith Center for Women’s Cancers at Dana-Farber Cancer Institute, gives an overview of phase I clinical trials and some of the new drugs being tested to treat breast cancer. This talk was originally given at the Metastatic Breast Cancer Forum at Dana-Farber on Oct. 5, 2013.
A great article on current clinical trials and explanation of cdk inhibitors by Sneha Phadke, DO; Alexandra Thomas, MD at the site OncoLive
cdk4/6 inhibitor Ibrance Has Favorable Toxicity and Adverse Event Profile
As discussed in earlier posts and the Introduction to this chapter on Cytotoxic Chemotherapeutics, most anti-cancer drugs developed either to target DNA, DNA replication, or the cell cycle usually have similar toxicity profile which can limit their therapeutic use. These toxicities and adverse events usually involve cell types which normally exhibit turnover in the body, such as myeloid and lymphoid and granulocytic series of blood cells, epithelial cells lining the mucosa of the GI tract, as well as follicular cells found at hair follicles. This understandably manifests itself as common toxicities seen with these types of agents such as the various cytopenias in the blood, nausea vomiting diarrhea (although there are effects on the chemoreceptor trigger zone), and alopecia.
It was felt that the cdk4/6 inhibitors would show serious side effects similar to other cytotoxic agents and this definitely may be the case as outlined below:
For full details, please see Pharmacology/Toxicology review by Dr. Wei Chen The nonclinical studies adequately support the safety of oral administration of palbociclib for the proposed indication and the recommendation from the team is for approval. Non-clinical studies of palbociclib included safety pharmacology studies, genotoxicity
studies, reproductive toxicity studies, pharmacokinetic studies, toxicokinetic studies and repeat-dose general toxicity studies which were conducted in rats and dogs. The pivotal toxicology studies were conducted in compliance with Good Laboratory Practice regulation.
Pharmacology:
As described above, palbociclib is an inhibitor of CDK4 and CDK6. Palbociclib modulates downstream targets of CDK4 and CDK6 in vitro and induces G1 phase cell cycle arrest and therefore acts to inhibit DNA synthesis and cell proliferation. Combination of palbociclib with anti-estrogen agents demonstrated synergistic inhibition
of cell proliferation in ER+ breast cancer cells. Palbociclib showed anti-tumor efficacy in animal tumor model studies. Safety pharmacology studies with palbociclib demonstrated adverse effects on both the respiratory and cardiovascular function of dogs at a dose of 125mg/day (four times and 50-times the human clinical exposure
respectively) based on mean unbound Cmax.
General toxicology:
Palbociclib was studied in single dose toxicity studies and repeated dose studies in rats and dogs. Adverse effects in the bone marrow, lymphoid tissues, and male reproductive organs were observed at clinically relevant exposures. Partial to complete reversibility of toxicities to the hematolymphopoietic and male reproductive systems was demonstrated following a recovery period (4-12 weeks), with the exception of the male reproductive organ findings in dogs. Gastrointestinal, liver, kidney, endocrine/metabolic (altered glucose metabolism), respiratory, ocular, and adrenal effects were also seen.
Genetic toxicology:
Palbociclib was evaluated for potential genetic toxicity in in vitro and in vivo studies. The Ames bacterial mutagenicity assay in the presence or absence of metabolic activation demonstrated non-mutagenicity. In addition, palbociclib did not induce chromosomal aberrations in cultured human peripheral blood lymphocytes in the presence or absence of metabolic activation. Palbociclib was identified as aneugenic based on kinetochore analysis of micronuclei formation in an In vitro assay in CHO-WBL cells. In addition, palbociclib was shown to induce micronucleus formation in male rats at doses 100
mg/kg/day (10x human exposure at the therapeutic dose) in an in vivo rat micronucleus assay.
Reproductive toxicology: No effects on estrous cycle and no reproductive toxicities were noticed in standard assays.
Pharmacovigilance (note please see PDF for more information)
Deaths Associated With Trials: Although a few deaths occurred during some trials no deaths were attributed to the drug.
Non-Serious Adverse Events:
(note a reviewers comment below concerning incidence of pulmonary embolism is a combination trial with letrazole)
Other article in this Open Access Journal on Cell Cycle and Cancer Include:
Curation of Recently Halted Oncology Trials Due to Serious Adverse Events – 2015
Curator: Stephen J. Williams, Ph.D.
The following is reports of oncology clinical trials in 2015 which have been halted for Serious Adverse Events (SAE), in most instances of an idiopathic nature. For comparison I have listed (as of this writing) the oncology drug approvals (8) for 2015. (from CenterWatch.com)
Oncology Drugs Approved in 2015
Farydak (panobinostat); Novartis; For the treatment of multiple myeloma, Approved February 2015
Ibrance (palbociclib); Pfizer; For the treatment of ER-positive, HER2-negative breast cancer, Approved February 2015
Lenvima (lenvatinib); Eisai; For the treatment of thyroid cancer, Approved February 2015
October 7, 2015
By Alex Keown, BioSpace.com Breaking News Staff
PRINCETON, N.J. – Following the death of a patient, the U.S. Food and Drug Administration (FDA) placed a hold on Advaxis (ADXS)’s experimental cancer treatment axalimogene filolisbac, which is currently in mid-stage trials.
In a statement issued this morning, Advaxis maintains the patient’s death was a result of the severity of her cancer and not due to the company’s experimental cancer treatment. It is seeking proof from the FDA that the drug was not a factor in the death. Still, the hold on the experimental cancer drug will cause the company to halt four clinical trials, Advaxis said. Other clinical trials, including those with the experimental ADXS-PSA and ADXS-HER2, are not affected by this hold. The company said it will continue to actively enroll and dose patients.
Halozyme Therapeutics acknowledged today that the FDA placed a formal clinical hold on its troubled Study 202 assessing its experimental drug PEGPH20 in patients with pancreatic cancer—less than a week after the company temporarily halted enrolling and dosing patients in the ongoing Phase II trial.
The agency told Halozyme it placed the clinical hold following the company’s pause in study activity. The trial’s independent data monitoring committee is evaluating data from the trial to learn why patients treated with PEGPH20 as well as nab-paclitaxel and gemcitabine saw a higher rate of blood clots and other thromboembolic events compared with patients treated with nab-paclitaxel and gemcitabine alone.
“We will be providing this information to the data monitoring committee and the FDA in parallel so they can complete their respective assessments,” Helen Torley, M.B. Ch.B., M.R.C.P., Halozyme’s president and CEO, said in a statement.
“Pancreatic cancer has one of the lowest survival rates of any cancer. We remain committed to evaluating PEGPH20 as a possible therapy to address this devastating disease,” Dr. Torley added.
As with Halozyme’s statement last week, the company’s latest remarks did not indicate when Halozyme expects to resume enrolling and dosing patients in Study 202, or how many patients had been enrolled and dosed when the temporary halt occurred.
The trial was envisioned as having 124 subjects, divided evenly between a treatment arm of PEGPH20 and nab-paclitaxel, and a gemcitabine arm, preceded by eight subject “run-in” phase assessing safety and tolerability, according to Study 202’s page on ClinicalTrials.gov (NCT01839487), last updated on January 27.
The study is one of two Phase II trials for PEGPH20; the other, SWOG, also aims to assess the drug for pancreatic cancer.
PEGPH20 is an investigational PEGylated form of Halozyme’s FDA-approved recombinant human hyaluronidase rHuPH20 (marketed as Hylenex®), designed to dramatically increases the half-life of the compound in the blood and allow for intravenous administration.
The temporary halt for Study 202 came two months after Halozyme publicly cited “potential acceleration of the PEGPH20 program” among several R&D programs for which it raised funds through a public offering of common stock that closed in February and generated approximately $107.8 million in net proceeds.
CytRx ($CYTR) has run into an unexpected roadblock with its cancer drug conjugate aldoxorubicin, slamming the brakes on new patient recruitment in all their clinical trials after the FDA dropped a partial clinical hold on the program. According to the biotech the hold was forced by the death of a patient who was given the drug through a compassionate use program.
LA-based CytRx execs say that patients already enrolled in the studies will continue to receive the therapy as investigators added new safety measures, retooling trial protocols to include an “appropriate inclusion/exclusion criteria, an additional patient screening assessment and an evaluation of serum electrolytes prior to aldoxorubicin administration.” The patient who died, they added, had not qualified for any of its studies.
As it stands now, the biotech doesn’t know exactly how long the partial hold will last, but their announcement sought to calm jumpy investors, saying they expected to resolve the FDA’s demands “expeditiously” and can stick to their current timelines. CytRx says it expects to report preliminary results from their mid-stage study of Kaposi’s sarcoma in the second quarter of 2015 and preliminary results from the ongoing Phase II clinical trial of aldoxorubicin in glioblastoma multiforme in the first half of 2015. The company added that it is committed to completing enrollment in their Phase III trial by the end of next year.
hat reassurance appears to have helped with investors, who seemed to count this as more of a temporary setback than a catastrophe. Shares for CytRx were down about 9% in mid-morning trading.
Aldoxorubicin uses a linker molecule to attach to albumin in the blood and concentrate in tumors, where the acidic environment releases the chemotherapy doxorubicin in doses up to four times higher than what’s used now. Late last year their stock soared after their drug scored promising results for progression-free survival in a Phase IIb trial.
This case illustrates one reason why biotechs often quietly squirm under the pressure of compassionate use programs. They can be expensive to operate, time-consuming and raise fresh concerns when a patient dies or experiences a setback. On the other hand, if regulators take action like this following the death of an advanced stage cancer patient, there may have been something about the case that triggered broader concerns for the entire patient population
Eli Lilly and Co.’s experimental lung cancer drug has raised concerns with U.S. regulators that it may increase patients’ risk of suffering potentially deadly blood clots.
The drug, known as necitumumab, improved patients’ overall chances of survival, yet people taking the medicine also experienced more risk, Food and Drug Administration staff said in a report Tuesday. Indianapolis-based Lilly is seeking to sell the medicine to treat a subset of the most common type of lung cancer.
FDA advisers will meet Thursday to discuss the risks and benefits of necitumumab for patients with advanced squamous non-small cell lung cancer, in combination with chemotherapy. The FDA is expected to decide if Lilly can sell the drug by the end of the year.
While the safety of necitumumab reflects that of similar drugs, the increased danger of clotting “in this already high risk population is of concern,” FDA staff wrote.
One study showed that out of 538 patients taking necitumumab and chemotherapy, 9 percent experienced a serious clot, compared with 5 percent of 541 patients given only chemotherapy, according to the staff report.
Squamous lung cancer accounts for 25 percent to 30 percent of all lung cancer, according to the American Cancer Society.
Patients in a clinical trial who took necitumumab lived a median of 11.5 months, 1.6 months longer than those who got only chemotherapy, the FDA staff report said.
Opdivo (nivolumab) is a human monoclonal antibody used to treat patients with unresectable or metastatic melanoma and disease progression following ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor; and to treat metastatic squamous non-small cell lung cancer with progression on or after platinum-based chemotherapy. Common side effects of Opdivo include fatigue, rash, itching, cough, upper respiratory tract infection, swelling of the extremities, shortness of breath, muscle pain, decreased appetite, nausea, vomiting, constipation, diarrhea, weakness, swelling, fever, abdominal pain, chest pain, joint pain, and weight loss.
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The data described in the WARNINGS AND PRECAUTIONS section and below reflect exposure to OPDIVO in Trial 1, a randomized trial in patients with unresectable or metastatic melanoma and in Trial 3, a single-arm trial in patients with metastatic squamous non-small cell lung cancer (NSCLC).
Clinically significant adverse reactions were evaluated in a total of 691 patients enrolled in Trials 1, 3, or an additional dose finding study (n=306) administering OPDIVO at doses of 0.1 to 10 mg/kg every 2 weeks [see WARNINGS AND PRECAUTIONS].
Unresectable or Metastatic Melanoma
The safety of OPDIVO was evaluated in Trial 1, a randomized, open-label trial in which 370 patients with unresectable or metastatic melanoma received OPDIVO 3 mg/kg every 2 weeks (n=268) or investigator’s choice of chemotherapy (n=102), either dacarbazine 1000 mg/m² every 3 weeks or the combination of carboplatin AUC 6 every 3 weeks plus paclitaxel 175 mg/m² every 3 weeks [see Clinical Studies]. The median duration of exposure was 5.3 months (range: 1 day to 13.8+ months) with a median of eight doses (range: 1 to 31) in OPDIVO-treated patients and was 2 months (range: 1 day to 9.6+ months) in chemotherapy treated patients. In this ongoing trial, 24% of patients received OPDIVO for greater than 6 months and 3% of patients received OPDIVO for greater than 1 year.
In Trial 1, patients had documented disease progression following treatment with ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor. The trial excluded patients with autoimmune disease, prior ipilimumab-related Grade 4 adverse reactions (except for endocrinopathies) or Grade 3 ipilimumab-related adverse reactions that had not resolved or were inadequately controlled within 12 weeks of the initiating event, patients with a condition requiring chronic systemic treatment with corticosteroids ( > 10 mg daily prednisone equivalent) or other immunosuppressive medications, a positive test for hepatitis B or C, and a history of HIV.
The study population characteristics in the OPDIVO group and the chemotherapy group were similar: 66% male, median age 59.5 years, 98% white, baseline ECOG performance status 0 (59%) or 1 (41%), 74% with M1c stage disease, 73% with cutaneous melanoma, 11% with mucosal melanoma, 73% received two or more prior therapies for advanced or metastatic disease, and 18% had brain metastasis. There were more patients in the OPDIVO group with elevated LDH at baseline (51% vs. 38%).
OPDIVO was discontinued for adverse reactions in 9% of patients. Twenty-six percent of patients receiving OPDIVO had a drug delay for an adverse reaction. Serious adverse reactions occurred in 41% of patients receiving OPDIVO. Grade 3 and 4 adverse reactions occurred in 42% of patients receiving OPDIVO. The most frequent Grade 3 and 4 adverse reactions reported in 2% to less than 5% of patients receiving OPDIVO were abdominal pain, hyponatremia, increased aspartate aminotransferase, and increased lipase.
– Press release from Eisai (NOTE: TRIAL NOT HALTED)
Feb 13, 2015
WOODCLIFF LAKE, N.J., Feb. 13, 2015 /PRNewswire/ — Eisai Inc. announced today that the U.S. Food and Drug Administration (FDA) approved the company’s receptor tyrosine kinase inhibitor LENVIMA™ (lenvatinib) for the treatment of locally recurrent or metastatic, progressive, radioactive iodine-refractory differentiated thyroid cancer (RAI-R DTC). LENVIMA was approved following a priority review by the FDA, which is designated for drugs the FDA believes have the potential to provide a significant improvement in the treatment of a serious condition. LENVIMA demonstrated a statistically significant progression-free survival (PFS) prolongation and response rate in patients with progressive, differentiated thyroid cancer who had become refractory to radioactive iodine (RAI) therapy.
In the clinical trial, adverse events led to dose reductions in 68% of patients who received LENVIMA and 5% of patients who received placebo. Some patients will need to discontinue treatment for serious adverse reactions. In the trial, 18% of patients treated with LENVIMA and 5% who received placebo discontinued treatment. The most common adverse reactions (at least 10%) that resulted in dose reductions of LENVIMA were hypertension (13%), proteinuria (11%), decreased appetite (10%), and diarrhea (10%).
“AstraZeneca ($AZN) is pressing pause on trials combining two of its most important pipeline cancer treatments after tracking reports of lung disease, halting enrollment as it gathers more information.
The company is testing a combination of AZD9291 and durvalumab, formerly MEDI4736, in two studies involving patients with non-small cell lung cancer. Late last month, AstraZeneca hit the brakes on enrollment in both trials due to an increase in reports of interstitial lung disease, which can lead to dangerous scarring and impaired pulmonary function. The pauses are temporary, the company stressed in an emailed statement, and patients already enrolled in the study will be given new consent forms to ensure they understand the risks before choosing whether keep getting treatment.”
Other posts on this site on Cytotoxicity and Cancer include
Location: MassBio, 300 Technology Sq., 8th Floor, Cambridge, MA
This interactive Forum will help attendees understand the FDA and how to achieve maximum success and avoid common pitfalls when working with the agency in early drug development. Our panel of experts, including a former FDA investigator, will cover the following topics:
Overview of the FDA’s structure and function
How did we get here? The Evolution of FDA
Getting into the Clinic: The IND and Case Studies
Chemistry, Manufacturing and Controls – Why is CMC is so Important to FDA?
Come join the discussion and take away a deeper understanding of FDA expectations and valuable tips for improved communication.
Drew Barlow has over 15 years of experience in CMC regulatory affairs & quality compliance. After a stint in clinical research at Wake Forest University, Drew began his tenure with the Food and Drug Administration in the Office of Regulatory Affairs. With the Agency Drew carried out investigations, inspections and other assignments over FDA regulated industries, specializing in pharmaceutical manufacturers and GMP compliance. Joining Vertex Pharmaceuticals in 2006, Drew established the post approval CMC regulatory group and oversaw numerous small molecule development projects in a variety of disease areas. He then brought his regulatory CMC strategy expertise to Alkermes plc where he led multiple global late phase programs. Drew joined the Syner-G team in January of 2015. His areas of expertise include devising & implementing CMC strategies for worldwide marketing applications, agency meeting planning & facilitating, post approval regulatory reporting, IND and IMPD preparation, GMP inspection readiness, and Quality by Design implementation. Drew received a B.S in Biology from Mount Saint Mary’s College in Maryland and a Master’s in Public Health from The University of North Carolina at Greensboro.
David S. Mantus, Ph.D., Vice President Regulatory Affairs & Quality Assurance, BIND Therapeutics
Dave Mantus has been working in Regulatory Affairs for 23 years both in industry, as a consultant and as a professor. Dr. Mantus served as Vice President, Regulatory Affairs at Cubist Pharmaceuticals and held various regulatory roles at Sention Inc., Shire Biologics, PAREXEL, the Massachusetts Public Health Laboratory, and Procter and Gamble Pharmaceuticals. He is a co-author of a book on regulatory affairs, “FDA Regulatory Affairs: A Guide for Prescription Drugs, Medical Devices, and Biologics,” that is in its third edition, and has published numerous scientific papers and done presentations around the world. In 2009, Dr. Mantus was named Citizen Schools (www.citizenschools.org) “Citizen Teacher of the Year” for his work teaching at the Edwards Middle School in Charlestown, MA. Dr. Mantus received his B.S. in Chemistry at the College of William and Mary, his M.S. and Ph.D. in Chemistry from Cornell University and was a post-doctoral research fellow in Biomedical Engineering at the University of Washington.
Julia Morteo, Director, Regulatory Affairs, Stealth BioTherapeutics
Julia has nearly 10 years of extensive Regulatory Affairs experience, serving functions in Regulatory Operations, Regulatory-CMC, and Regulatory Strategy. Julia spent 8 years at Cubist Pharmaceuticals, where she helped bring eCTD submission capabilities in house, was responsible for the CMC content of marketing applications for CUBICIN® in over 50 countries worldwide, and provided Regulatory support on pre- and post-approval programs. For the past several years, Julia has functioned as the Global Regulatory Strategy lead on more than 10 development programs ranging from pre-clinical to Phase 3 across multiple therapeutic areas. Julia has led numerous successful IND submissions, IND amendments, and FDA meetings. Julia has been with Stealth BioTherapeutics since 2014, where she is the Regulatory lead on Stealth’s Phase 2 neurologic rare disease and CV/Renal programs. Julia graduated from Mount Holyoke College, where she was a double major in Religion and a self-designed major in Science and Medicine in Cultural Perspective.





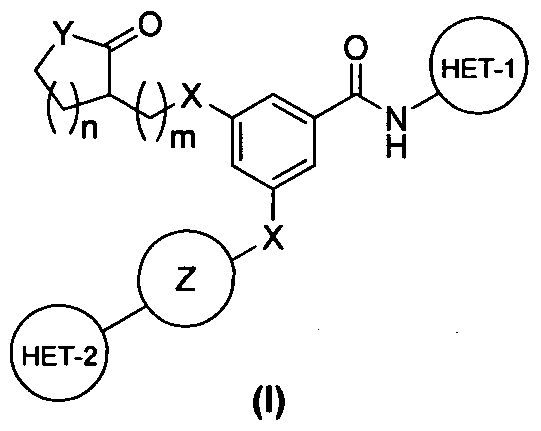












{kind=link}
{kind=link}
{kind=link}
{kind=link}