Healthcare analytics, AI solutions for biological big data, providing an AI platform for the biotech, life sciences, medical and pharmaceutical industries, as well as for related technological approaches, i.e., curation and text analysis with machine learning and other activities related to AI applications to these industries.
One of the unexpected findings of the Human Genome Project was that over 98% of the human genome does not encode for proteins. Once dismissed as “junk” genomic material, non-protein-coding DNA is now appraised more highly.
Or to be more precise, at least some portions of non-protein-coding DNA are thought to serve important biological functions.
For example, some stretches of DNA give rise to a noncoding but still functional kind of RNA called microRNA. MicroRNAs have increasingly emerged in recent years as key regulators of biological processes and pathways.
During the years since their discovery, a key question in the biology of microRNAs has focused on the number of microRNAs encoded in the genome. Between 1993 and 2015, approximately 1,900 human genome loci were discovered to produce microRNAs and were added to miRBbase, the public database that catalogues and annotates microRNA molecules.
The cataloguing of microRNAs work has been pursued with extra urgency since 2004, the year the connection between microRNAs and human disease was first demonstrated. “When this connection was made, it launched a whole new field,” says Isidore Rigoutsos, Ph.D., professor of pathology, anatomy, and cell biology and director of the Computational Medicine Center at Thomas Jefferson University.
Another Set of MicroRNAs Emerge
“We wanted to know how many microRNA-producing loci really exist in humans,” recalls Dr. Rigoutsos. In a study published in 2015, Dr. Rigoutsos and colleagues analyzed datasets from 1,323 individuals that represented 13 different tissues and identified an additional 3,356 such genomic loci that produce (at least) 3,707 novel microRNs
“We basically tripled the number of locations in the human genome that are now known to encode microRNAs,” asserts Dr. Rigoutsos. Considering that each microRNA regulates up to hundreds of different mRNAs, and that each mRNA is regulated by tens of microRNAs, this finding adds a new layer of complexity to the regulatory dynamics of the human transcriptome.
The newly unveiled microRNAs and previously characterized microRNAs have distinct expression patterns. While 50–60% of the microRNAs previously deposited into the miRBase are expressed in multiple tissues, only about 10% of the newly discovered microRNAs are shared across multiple tissue types. Also, most of the newly found microRNAs show tissue-specific expression.
Using Argonaute CLIP-seq data, Dr. Rigoutsos and colleagues showed that similar percentages of the two sets of microRNAs were in complex with Argonaute proteins. “This shows that these novel microRNAs participate in RNA interference just as frequently as the miRBase microRNAs,” contends Dr. Rigoutsos.
In a comparative analysis between the human microRNA datasets and the chimpanzee, gorilla, orangutan, macaque, mouse, fruit fly, and mouse genomes, Dr. Rigoutsos and colleagues discovered that almost 95% of the newly unveiled microRNAs were primate-specific, and over 56% of them were found only in humans.
“We are seeing many human microRNAs that do not exist in the mouse,” states Dr. Rigoutsos. “This means that the mouse models engineered to capture human disease cannot recapitulate the interactions mediated by these microRNAs.
Interest in IsomiRs Grows
In the years since the biology of microRNAs started receiving increasing attention, the conventional view has been that one microRNA locus generates one microRNA. However, once deep sequencing became widely available, microRNA variants that showed differences at their 5′- or 3′-termini have been described.
“It was initially presumed that these variants were likely the result of the enzyme Dicer not being sufficiently accurate when processing microRNA precursors,” notes Dr. Rigoutsos. Subsequent research revealed that microRNAs are more dynamic than previously thought, with each precursor being able to generate multiple mature microRNA species known as isomiRs.
To gain insight into the biology of isomiRs, Dr. Rigoutsos and colleagues analyzed genomic datasets from 452 individuals participating in the 1000 Genomes Project. The datasets comprised five different populations and two races. In addition, each population was represented by an even number of men and women.
This collection allowed the abundance of microRNA isoforms to be examined with respect to population, gender, and race. “We found that isomiRs have expression profiles that are population-, race-, and gender-dependent,” informs Dr. Rigoutsos.
All the transcriptome data that this analysis was based on came from immortalized B cells. “These are cells that normally are not associated with gender differences, but molecularly we found, in these cells, differences between men and women of the same population and race,” explains Dr. Rigoutsos.
Expanding these observations to disease states, Dr. Rigoutsos and colleagues collected isomiR profiles from tissue affected by breast cancer, and compared them with isomiR profiles from control breast tissue. The investigators found that the isomiR profiles also depend on tissue state (healthy vs. diseased), on disease subtype, and on the patient’s race.
For example, their analysis identified several miR-183-5p isoforms that were upregulated in white triple-negative breast cancer patients compared to control breast samples, but not in black/African-American triple-negative breast cancer patients. In an in vitro phase of this study, three isoforms of this microRNA species were overexpressed in human breast cancer cell lines.
“We found very little overlap in the gene sets that were affected by each of these isoforms,” emphasizes Dr. Rigoutsos. Despite being generated simultaneously by the same locus, each of the three isoforms affected distinct groups of genes, thus exerting different effects on the transcriptome.
“As the relative abundance of these isoforms changes ever so slightly from patient to patient, it will affect the corresponding gene groups slightly differently,” concludes Dr. Rigoutsos. “In the process, it creates a new molecular background in each patient.”
MicroRNAs Point to Therapeutic Strategies against Colorectal Cancer
“We are using microRNAs as modulators to overcome chemotherapy resistance in colorectal cancer,” says Jingfang Ju, Ph.D., associate professor of pathology and co-director of translational research at Stony Brook University School of Medicine. Resistance to chemotherapy is one of the major challenges in the clinical management of malignancies, including colorectal cancer. Chemotherapy is usually unable to eliminate cancer stem cells, which may become even more resistant over time, and several microRNAs have been implicated in this process. “We reasoned that we could provide new modulatory approaches to target this small cell population and allow chemotherapy, radiotherapy, or immunotherapy to eliminate resistant populations or at least prolong long-term survival,” Dr. Ju said.
This image shows how miR-129 may function as a tumor suppressor in colorectal cancer. In this model, which has been proposed by researchers at Stony Brook University’s Translational Research Laboratory, miR-129 suppresses the protein expression of three critical targets—BCL2, TS, and E2F3. Downregulation of BCL2 activates the intrinsic apoptosis pathway by cleaving caspase-9 and caspase-3. Downregulation of TS and E2F3 inhibits cell proliferation by impacting the cell cycle. Consequently, miR-129 exerts a strong antitumor phenotype by induction of apoptosis and impairment of proliferation in tumor cells. [Mihriban Karaayvaz, Haiyan Zhai, Jingfang Ju]
In a retrospective study in which colorectal patient samples were used, Dr. Ju and colleagues revealed that hsa-miR-140-5p expression progressively decreases from normal tissues to primary colorectal cancer tissue, and that it shows a further decrease in liver and lymph node metastases. The experimental overexpression of hsa-miR-140-5p inhibited colorectal cancer stem cell growth by disrupting autophagy, and in a mouse model of disease it abolished tumor formation and metastasis.
In addition to hsa-miR-140-5p, Dr. Ju and colleagues recently identified hsa-miR-129 and found that it, too, has therapeutic potential. Specifically, they showed that hsa-miR-129 enhanced the sensitivity of colorectal cancer cells to 5-fluorouracil, pointing toward its ability to function as a tumor suppressor.
One of the mechanisms implicated in this process was the ability of miR-192 to inhibit protein translation of several important targets. These include Bcl-2 (B-cell lymphoma 2), a key anti-apoptotic protein; E2F3, a major cell cycle regulator; and thymidylate synthase, an enzyme that is inhibited by 5-fluorouracil.
The NIH recently awarded a $3 million grant to establish the Long Island Bioscience Hub (LIBH), which is part of the NIH’s Research Evaluation and Commercialization Hub (REACH) program and represents a partnership between the Center for Biotechnology, Stony Brook University, Cold Spring Harbor Laboratory, and Brookhaven National Laboratory. One of the technology development grants, as part of the first funding cycle of this initiative, will support a feasibility investigation of hsa-miR-129-based therapeutics in colon cancer, an effort led by Dr. Ju. “We are further exploring this novel mechanism,” states Dr. Ju. “We anticipate conducting pharmacokinetic studies and moving to a clinical trial in the future.”
MicroRNA Insights Gleaned from Host-Virus Interactions
At Mount Sinai Hospital’s Icahn School of Medicine, researchers used a codon-optimized version of VP55 produced from an adenovirus-based vector to study the impact of microRNA deletion on the response to virus infection. This image shows RNA in situ hybridization of fibroblasts expressing VP55 (top left), and that of mock-treated fibroblasts (bottom right). Ribosomal RNA, DNA, and microRNAs (miR-26) are depicted by red, blue (DAPI), and green fluorophores, respectively.
“We observed that when a poxvirus is artificially engineered to encode a microRNA, the microRNA is destroyed along with all the microRNAs from the host cell,” says Benjamin R. tenOever, Ph.D., professor of microbiology at the Icahn School of Medicine, Mount Sinai Hospital. Previously, Dr. tenOever’s group reported that a single vaccinia virus-encoded gene product, VP55, is sufficient to achieve this effect. The group also found that the protein adds nontemplate adenosines to the 3′-end of microRNAs associated with the RNA-induced silencing complex.
biology,” asserts Dr. tenOever.
In a recent study, Dr. tenOever and colleagues used a codon-optimized version of VP55 produced from an adenovirus-based vector to study the impact microRNA deletion would have on our normal response to virus infection. “We found that after administration of the vector and rapid ablation of microRNA expression, there is very little that happens over the first one to two days,” informs Dr. tenOever. During the first 24–48 hours after VP55 delivery, the elimination of cellular microRNAs impacted less than 0.35% of the over 11,000 genes expressed in the cell. After 9 days, however, almost 20% of the genes showed significant changes in expression.
“MicroRNAs are very powerful and influential in controlling the biology of the cell but they do so over the long term,” declares Dr. tenOever. These findings are in agreement with knowledge that has accumulated over the years about microRNA biology, which established that microRNAs play a central role in determining how cells differentiate during development.
“While microRNAs can act on hundreds of mRNAs, their action requires several days of fine-tuning to have long-term consequences,” adds Dr. tenOever. This finding suggests miRNAs are unable to significantly contribute to the acute response to virus infection.
The one exception to this observation was that, even though very few genes were affected in the first 48 hours after VP55 delivery, several genes encoding chemokines were impacted. These included chemokines responsible for recruiting antigen-presenting cells, neutrophils, and other immune cells.
An in vivo analysis of mouse lung tissue 48 hours after vector administration confirmed that several cytokines were specifically upregulated, resulting in immune cell infiltration following the degradation of all microRNAs. These results indicate that the acute viral infection is largely independent of microRNAs, and that microRNAs are primarily involved in the adaptive response to infection and other longer term processes.
MicroRNA Biomarkers Reveal Molecular Pathways of Kidney Damage
“Our approach involves looking at microRNAs from the perspective of biomarkers as a readout for kidney damage,” says Vishal S. Vaidya, Ph.D., associate professor of medicine and environmental health at Brigham and Women’s Hospital, Harvard Medical School, and Harvard T.H. Chan School of Public Health. “At the same time, we are exploring their utility as therapeutics.”
A large number of medications and occupational toxins cause kidney damage, but many tests to assess kidney function and damage are not sufficiently sensitive or specific, opening the need for novel diagnostic strategies. MicroRNAs, which are differentially expressed between healthy and diseased states, are promising as early biomarkers for impaired renal function.
“MicroRNAs can also provide information about which pathways are active and which targets can be druggable,” points out Dr. Vaidya.
In a study that used microRNAs and proteins to provide a combined biomarker signature, Dr. Vaidya and colleagues examined two patient cohorts, one presenting with acetaminophen-induced kidney injury and the other one with cisplatin-induced kidney damage. “Protein biomarkers provide sensitivity, and microRNAs offer mechanistic insight,” explains Dr. Vaidya.
This approach helped visualize metabolic pathways that are altered in the kidney during toxic injury. “The biggest challenge, from a therapeutic perspective, is that microRNAs regulate many mRNAs and, therefore, impact many proteins,” concludes Dr. Vaidya.
Curators: Larry H Bernstein, MD, FCAP and Aviva Lev-Ari, PhD, RN
Revised 2/14/2016
The following presentation explores the application of antisense oligonucleotide agents that modulate the activity of Il17 and Il23 signaling activity in the cell.
IL 17 & 23
United States Patent
9,238,042
Schnell , et al.
January 19, 2016
Antisense modulation of interleukins 17 and 23 signaling
Provided are antisense oligonucleotides and other agents that target and modulate IL-17 and/or IL-23 signaling activity in a cell, compositions that comprise the same, and methods of use thereof. Also provided are animal models for identifying agents that modulate 17 and/or IL-23 signaling activity.
Abes et al., “Arginine-rich cell penetrating peptides: Design, structure-activity, and applications to alter pre-mRNA splicing by steric-block oligonucleotides,” J Pept Sci 14: 455-460, 2008. cited by applicant .
Abes et al., “Delivery of steric block morpholino oligomers by (R-X-R).sub.4 peptides: structure-activity studies,” Nucleic Acids Research 36(20): 6343-6354, Sep. 16, 2008. cited by applicant .
Abes et al., “Vectorization of morpholino oligomers by the (R-Ahx-R).sub.4 peptide allows efficient splicing correction in the absence of endosomolytic agents,” Journal of Controlled Release 116: 304-313, 2006. cited by applicant .
Lebleu et al., “Cell penetrating peptide conjugates of steric block oligonucleotides,” Advanced Drug Delivery Reviews 60: 517-529, 2008. cited by applicant .
Marshall et al., “Arginine-rich cell-penetrating peptides facilitate delivery of antisense oligomers into murine leukocytes and alter pre-mRNA splicing,” Journal of Immunological Methods 325: 114-126, 2007. cited by applicant .
Moulton et al., “Cellular Uptake of Antisense Morpholino Oligomers Conjugated to Arginine-Rich Peptides,” Bioconjugate Chem 15: 290-299, 2004. cited by applicant .
Summerton et al., “Morpholino Antisense Oligomers: Design, Preparation, and Properties,” Antisense & Nucleic Acid Drug Development 7: 187-195, 1997. cited by applicant .
Wright et al., “The Human IL-17F/IL-17A Heterodimeric Cytokine Signals through the IL-17RA/IL-17RC Receptor Complex,” The Journal of Immunology 181: 2799-2805, 2008. cited by applicant .
Immunity. 2015 Oct 20;43(4):739-50. doi: 10.1016/j.immuni.2015.08.019. Epub 2015 Sep 29.
Differential Roles for Interleukin-23 and Interleukin-17 in Intestinal Immunoregulation.
Interleukin-23 (IL-23) and IL-17 are cytokines currently being targeted in clinical trials. Although inhibition of both of these cytokines is effective for treating psoriasis, IL-12 and IL-23 p40 inhibition attenuates Crohn’s disease, whereas IL-17A or IL-17 receptor A (IL-17RA) inhibition exacerbates Crohn’s disease. This dichotomy between IL-23 and IL-17 was effectively modeled in the multidrug resistance-1a-ablated (Abcb1a(-/-)) mouse model of colitis. IL-23 inhibition attenuated disease by decreasing colonic inflammation while enhancing regulatory T (Treg) cell accumulation. Exacerbation of colitis by IL-17A or IL-17RA inhibition was associated with severe weakening of the intestinal epithelial barrier, culminating in increased colonic inflammation and accelerated mortality. These data show that IL-17A acts on intestinal epithelium to promote barrier function and provide insight into mechanisms underlying exacerbation of Crohn’s disease when IL-17A or IL-17RA is inhibited.
Immunity. 2015 Oct 20;43(4):727-38. doi: 10.1016/j.immuni.2015.09.003. Epub 2015 Sep 29.
Interleukin-23-Independent IL-17 Production Regulates Intestinal Epithelial Permeability.
Whether interleukin-17A (IL-17A) has pathogenic and/or protective roles in the gut mucosa is controversial and few studies have analyzed specific cell populations for protective functions within the inflamed colonic tissue. Here we have provided evidence for IL-17A-dependent regulation of the tight junction protein occludin during epithelial injury that limits excessive permeability and maintains barrier integrity. Analysis of epithelial cells showed that in the absence of signaling via the IL-17 receptor adaptor protein Act-1, the protective effect of IL-17A was abrogated and inflammation was enhanced. We have demonstrated that after acute intestinal injury, IL-23R(+) γδ T cells in the colonic lamina propria were the primary producers of early, gut-protective IL-17A, and this production of IL-17A was IL-23 independent, leaving protective IL-17 intact in the absence of IL-23. These results suggest that IL-17-producing γδ T cells are important for the maintenance and protection of epithelial barriers in the intestinal mucosa.
Gastroenterology. 2008 Apr;134(4):1038-48. doi: 10.1053/j.gastro.2008.01.041. Epub 2008 Jan 17.
Regulation of gut inflammation and th17 cell response by interleukin-21.
Interleukin (IL)-21, a T-cell-derived cytokine, is overproduced in inflammatory bowel diseases (IBD), but its role in the pathogenesis of gut inflammation remains unknown. We here examined whether IL-21 is necessary for the initiation and progress of experimental colitis and whether it regulates specific pathways of inflammation.
Both dextran sulfate sodium colitis and trinitrobenzene sulfonic acid-relapsing colitis were induced in wild-type and IL-21-deficient mice. CD4(+)CD25(-) T cells from wild-type and IL-21-deficient mice were differentiated in T helper cell (Th)17-polarizing conditions, with or without IL-21 or an antagonistic IL-21R/Fc. We also examined whether blockade of IL-21 by anti-IL-21 antibody reduced IL-17 in cultures of IBD lamina propria CD3(+) T lymphocytes. Cytokines were evaluated by real-time polymerase chain reaction and/or enzyme-linked immunosorbent assay.
High IL-21 was seen in wild-type mice with dextran sulfate sodium- and trinitrobenzene sulfonic acid-relapsing colitis. IL-21-deficient mice were largely protected against both colitides and were unable to up-regulate Th17-associated molecules during gut inflammation, thus suggesting a role for IL-21 in controlling Th17 cell responses. Indeed, naïve T cells from IL-21-deficient mice failed to differentiate into Th17 cells. Treatment of developing Th17 cells from wild-type mice with IL-21R/Fc reduced IL-17 production. Moreover, in the presence of transforming growth factor-beta1, exogenous IL-21 substituted for IL-6 in driving IL-17 induction. Neutralization of IL-21 reduced IL-17 secretion by IBD lamina propria lymphocytes.
These results indicate that IL-21 is a critical regulator of inflammation and Th17 cell responses in the gut.
Neurochem Res. 2010 Jun;35(6):940-6. doi: 10.1007/s11064-009-0091-9. Epub 2009 Nov 14.
Synergy of IL-23 and Th17 cytokines: new light on inflammatory bowel disease.
Inflammatory bowel diseases (IBDs), including Crohn’s disease and ulcerative colitis, involve an interplay between host genetics and environmental factors including intestinal microbiota. Animal models of IBD have indicated that chronic inflammation can result from over-production of inflammatory responses or deficiencies in key negative regulatory pathways. Recent research advances in both T-helper 1 (Th1) and T-helper 17 (Th17) effect responses have offered new insights on the induction and regulation of mucosal immunity which is linked to the development of IBD. Th17 cytokines, such as IL-17 and IL-22, in combination with IL-23, play crucial roles in intestinal protection and homeostasis. IL-23 is expressed in gut mucosa and tends to orchestrate T-cell-independent pathways of intestinal inflammation as well as T cell dependent pathways mediated by cytokines produced by Th1 and Th17 cells. Th17 cells, generally found to be proinflammatory, have specific functions in host defense against infection by recruiting neutrophils and macrophages to infected tissues. Here we will review emerging data on those cytokines and their related regulatory networks that appear to govern the complex development of chronic intestinal inflammation; we will focus on how IL-23 and Th17 cytokines act coordinately to influence the balance between tolerance and immunity in the intestine.
The role of IL-23 and IL-17 in the response to fungal infection has been the focus of recent reports. In this issue of the European Journal of Immunology there is an article that reports an important role for IL-23 and IL-17 in limiting fungal control, promoting neutrophillic inflammation and regulating the killing activity of neutrophils. In the fungal model it appears that IL-23 and IL-17 are counter-productive for protection.
IL-12 and IL-23 cytokines: from discovery to targeted therapies for immune-mediated inflammatory diseases
The cytokine interleukin-12 (IL-12) was thought to have a central role in T cell–mediated responses in inflammation for more than a decade after it was first identified. Discovery of the cytokine IL-23, which shares a common p40 subunit with IL-12, prompted efforts to clarify the relative contribution of these two cytokines in immune regulation. Ustekinumab, a therapeutic agent targeting both cytokines, was recently approved to treat psoriasis and psoriatic arthritis, and related agents are in clinical testing for a variety of inflammatory disorders. Here we discuss the therapeutic rationale for targeting these cytokines, the unintended consequences for host defense and tumor surveillance and potential ways in which these therapies can be applied to treat additional immune disorders.
IL-12 and IL-23 are produced by inflammatory myeloid cells and influence the development of TH1 cell and IL-17–producing T helper (TH17) cell responses, respectively. The rationale for developing IL-12 antagonists was prompted by observations that mice deficient in IL-12p40 are resistant to experimentally induced autoimmune conditions, including paralysis induction after immunization with brain-derived antigens, arthritis inflammation after immunization with a joint antigen, ocular disease after immunization with a retinal antigen and multiple gut disease models. This suggested that IL-12 could be an effective therapeutic target1, 2, 3, 4, 5. Studies of neutralizing antibodies to IL-12p40 in multiple mouse strains seemed to confirm the importance of therapeutically targeting IL-12 to decrease immune pathology6, 7. However, mice deficient in the other IL-12 subunit, IL-12p35, showed no protection or showed exacerbated disease in some models1, 2. Following the recognition, in 2000, that IL-12 and IL-23 share the IL-12p40 subunit but only IL-23 uses the p19 subunit8, it was determined that mice deficient in IL-23 but not IL-12 are resistant to experimental immune-mediated disease1, 2, 3, 4, 5. By 2000, the first anti–IL-12p40 therapy targeting IL-12—subsequently recognized to target IL-23 as well—was under evaluation in patients with Crohn’s disease9. Currently, at least 10 therapeutic agents targeting IL-12, IL-23 or IL-17A are being tested in the clinic for more than 17 immune-mediated diseases (Table 1). Here we discuss the preclinical and clinical data validating these therapeutic strategies and the potential consequences of targeting these immune pathways.
Figure 1: Schematic representation of IL-12 and IL-23, and their receptors and downstream signaling pathways
IL-12 is made up of the IL-12/23p40 and IL-12p35 subunits, and IL-23 comprises IL-23p19 and IL-12/23p40. IL-12 signals through the IL-12Rβ1 and IL-12Rβ2 subunits, and IL-23 signals through IL-12Rβ1 and IL-23R. IL-12 stimulation of JAK2…
Figure 4: Schematic representation of the mechanisms by which IL-23 indirectly or directly promotes tumorigenesis, growth and metastasis.
IL-23 is produced by myeloid cells in response to exogenous or endogenous signals such as damage-associated molecular patterns (DAMPs), pathogen-associated molecular patterns (PAMPs) or tumor-secreted factors such as prostaglandin E2 (PGE2). IL-23 can act directly on tumor cells to promote their transformation, proliferation and/or metastasis. In mice, IL-23R is expressed on several innate and adaptive immune cell types, which are found in various proportions in tumors. Stimulation of IL-23R on these immune cells leads to production of cytokines such as IL-17 and/or IL-22, which can have direct proliferative effects on stromal or tumor cells. IL-17 and/or IL-22 also elicit a range of factors from various hematopoietic and nonhematopoietic cells, which can have direct effects on tumor proliferation and metastasis or induce the production of additional inflammatory cytokines, chemokines and mediators such as IL-6, IL-8, matrix metallopeptidases (MMPs) and vascular endothelial growth factor (VEGF), all of which can contribute to the generation of a tumor microenvironment in which CD8 and NK cell effector functions are suppressed. DC, dendritic cell; Mφ, macrophage.
Nature Medicine 21, 719–729 (2015) doi:10.1038/nm.3895
Familial genetic studies, large-scale genome-wide association studies (GWAS) and next-generation sequencing approaches have highlighted therapeutic indications where IL-23 may contribute to inflammatory disease risk. For example, a psoriasis GWAS reported a protective association for the single-nucleotide polymorphism (SNP) rs11209026 (c.1142G>A; p.Arg381Gln) residing in the IL-23R protein-coding sequence with a modest odds ratio (OR) of 0.67 (P = 7 × 10−7)25. A GWAS in ileal Crohn’s disease also showed an association with rs11209026 (ref. 26), with the minor glutamine variant protective for Crohn’s disease risk with an OR of 0.26–0.45. The protective association of this variant (and other SNPs in linkage disequilibrium with it) in Crohn’s disease was also shown in ulcerative colitis27, 28, 29, 30, 31,32, 33, 34, 35, 36, 37, 38, 39, 40, 41. The largest meta-analysis of all inflammatory bowel disease GWAS to date (~40,000 cases and ~40,000 controls) indicates that carriage of the glutamine variant gives a modest reduction for disease risk (OR = 0.43, P = 8 × 10−161) (ref. 36). The rs11209026 allele is also associated with protection from ankylosing spondylitis42, 43, psoriatic arthritis44, 45, 46, 47 and graft-versus-host disease48, 49, 50, 51. Notably, this IL-23R variant has not been reliably associated with other common inflammatory diseases such as rheumatoid arthritis, type 1 diabetes or multiple sclerosis in GWAS powered to detect protective effects similar to those seen in Crohn’s disease and psoriasis52, 53, 54. Although these GWAS findings are compelling, it is important to keep in mind the limitations of such studies; these common loci tend to additively explain only a small proportion of the narrow-sense heritability of disease risk55.
Treatment of inflammatory disease with any immunosuppressive agent carries the theoretical risk of impaired host defense responses to pathogens and/or decreased tumor surveillance. Emerging data from human loss-of-function variants and mouse preclinical studies have informed the relative risks of targeting IL-12 and/or IL-23.
The theoretical risk of compromised immunity are of particular concern owing to immune defects discovered in patients with autosomal recessive deficiencies in IL-12/23p40 and IL-12Rβ1 (refs.105,106,107) (Fig. 3). Both deficiencies are genetic etiologies of Mendelian susceptibility to mycobacterial disease (MSMD) (genes involved in MSMD are listed at http://www.biobase-international.com), a rare condition in otherwise healthy patients who have a selective infection predisposition to weakly virulent mycobacteria such as Bacillus Calmette-Guerin (BCG) vaccines, nontuberculous environmental mycobacteria and virulent Mycobacterium tuberculosis (OMIM209950)108, 109, 110, 111, 112, 113. Half of patients with MSMD also have nontyphoidal and, to a lesser extent, typhoidal Salmonella infection.
Owing to the roles of IL-12 and/or IL-23 in host defense and tumor surveillance, particular attention has been focused on infectious disease–related adverse events after anti–IL-12/23p40 treatment in humans. Meta-analysis of briakinumab’s phase 2, phase 3 and open-label extension (OLE) psoriasis databases in 2010 identified 14 cases of candidiasis (including mucocutaneous esophageal and oral candidiasis); no reports of mycobacteria or Salmonella were noted. With regard to the roles of IL-12 and/or IL-23 in tumorigenesis, malignancies were observed at a rate of 1.7 events per 100 patient years (PY), and were cancers commonly seen in the general population.
Concluding remarks
Clinical testing of IL-23 and IL-17A inhibitors have confirmed the initial hypotheses that IL-23–TH17 pathways are indispensable in promoting immune-mediated diseases, and agents targeting these pathways work particularly well in specific disease settings. However, it is not clear why IL-17A and IL-17RA antagonists work well for psoriasis but exacerbate Crohn’s disease95, 96. It appears that different classes of inhibitor targeting IL-23 and IL-17 pathways may have unique nonoverlapping attributes in different clinical settings. Investigators are still learning where the overlap occurs and what the differences are between targeting IL-23 and targeting other related pathway cytokines. For example, mouse innate lymphoid cells constitutively produce gut protective IL-17A and IL-22 in an IL-23–independent manner. The constitutive IL-17A and IL-22 expression levels generated in response to commensal gut organisms seem to be crucial for maintenance of epithelial barrier function185 and tight junction formation (D.J.C., unpublished observation). However, high levels of IL-17A and IL-22 induced by IL-23 can be pathogenic during tissue injury responses in the presence of additional inflammatory cytokines such as IL-1, IL-6, GM-CSF and TNF. Therefore, targeting IL-23 via anti–IL-23p19 will partially suppress IL-17A and reduce inflammation, whereas anti–IL-17A therapy will neutralize all protective IL-17A.
The immune system’s function is to maintain balance in the face of insult from external pathogens and accumulation of genetic errors leading to cancer. Disruption of this balance toward immune-exuberance can lead to autoimmunity and immunopathology after infection, whereas inadequate immunity can allow pathogen evasion and breakdown in tumor surveillance. The common thread that connects autoimmunity, infection and cancer is inflammation, and the drivers of inflammation are intercellular messengers that enable cross-talk between immune cells and surrounding stromal tissues. We have underscored the importance of innate cell-produced IL-12 and IL-23 as intermediaries that act on T cells and NK cells to promote inflammation and highlighted that IL-12 and IL-23 have overlapping cellular immune functions. Whereas IL-12 is important in driving STAT1- and STAT4-mediated immune surveillance against specific intracellular pathogens and immunity against neoplasm, IL-23 promotes STAT3-dependent antifungal immunity and drives ‘sterile’ wound-healing responses in psoriatic lesions, which have a gene signature similar to that of many autoinflammatory conditions186, 187. Strikingly, this signature of uncontrolled wound-healing response is also observed in many cancers188. Although there is insufficient clinical data to determine the long-term safety of IL-23 inhibitors, preclinical models suggest that IL-23 paradoxically promotes tumorigenesis by enhancing skin and mucosal tissue inflammation associated with immune evasion mechanisms.
As the roles of IL-12 and IL-23 were elucidated in preclinical models, there was concern that inhibiting these factors could lead to profound immune suppression. Is it better to target factors capable of regulating a broad range of immune function and may leave patients unprotected against pathogens and cancers or to aim for a restricted pathway that may have limited efficacy for treatment of immune disorders? Although the efficacy and safety profiles of IL-12/23p40, IL-23p19 and IL-17A and IL-17RA therapies become clearer with each clinical trial, the decisions to progress these targets were made many years in advance, on the basis of limited data. Animal studies are important for elucidating the cellular and molecular mechanisms, but clinical testing is required to determine whether a specific disease mechanism also operates in humans. Immunological research is at an inflection point, where the basic concepts of molecular and cellular immunology are being translated into effective therapies for diseases that were considered intractable only a few years ago. Despite the challenges, efforts to translate basic disease mechanisms to the clinic are finally paying off. Although much work remains to be done, the fundamental question of which immune target will benefit which patient population is now being clarified. We optimistically await the answers that will change the lives of patients with serious immune-mediate conditions.
Increasing evidence points to a pathologic role for cytokines in Crohn’s colitis. Levels of cytokines are increased in diseased segments of colon in Crohn’s colitis, but no one has studied the concentration of cytokines in clinically and histologically nondiseased segments.
Mucosal biopsies were obtained from 7 patients with active segmental Crohn’s colitis and from 7 controls without inflammatory bowel disease. The concentration of Interleukin (IL)-1 beta, IL-2, IL-6, and IL-8 in patients and controls were determined using enzyme linked immunosorbent assay and compared. Histologic sections were also performed to confirm diseased and nondiseased segments of colon.
The concentrations of IL-1 beta, IL-6, and IL-8 were significantly higher in the involved segments of colon (10.3 +/- 4.1, 3.7 +/- 1.0, 34.4 +/- 6.9 picograms [pg] per mg) when compared to controls (1.8 +/- 0.5, 1.1 +/- 0.5, 5.3 +/- 1.0 pg/mg). The concentrations of IL-1 beta, IL-2, and IL-8 (8.5 +/- 2.9, 5.3 +/- 1.2, 26.3 +/- 8.8 pg/mg) in normal appearing segments of colon of patients with Crohn’s colitis were also significantly higher than in controls, whose IL-2 level was 2.0 +/- 0.5 pg/mg. IL-1 beta and IL-8 were significantly more concentrated in both the involved and uninvolved colonic segments of patients with Crohn’s colitis compared to controls. IL-2 and IL-6 were also more concentrated in Crohn’s patients than in controls, but not significantly. The differences in interleukin concentrations between involved and uninvolved segments of colon in patients with segmental Crohn’s colitis were not significant.
Although Crohn’s colitis is often a segmental disease, concentrations of IL-1 beta and IL-8 are increased throughout the entire colon. These observations reinforce the hypothesis that Crohn’s colitis involves the whole colon even when this is not apparent clinically or histologically.
Inflammatory bowel diseases (IBD) are characterized by a sustained inflammatory cascade that gives rise to the release of mediators capable of degrading and modifying bowel wall structure. Our aims were (i) to measure the production of matrix metalloproteinase-3 (MMP-3), and its tissue inhibitor, tissue inhibitor of metalloproteinase-1 (TIMP-1), by inflamed and uninflamed colonic mucosa in IBD, and (ii) to correlate their production with that of proinflammatory cytokines and the anti-inflammatory cytokine, IL-10. Thirty-eight patients with IBD, including 25 with Crohn’s disease and 13 with ulcerative colitis, were included. Ten controls were also studied. Biopsies were taken from inflamed and uninflamed regions and inflammation was graded both macroscopically and histologically. Organ cultures were performed for 18 h. Tumour necrosis factor-alpha (TNF-alpha), IL-6, IL-1beta, IL-10, MMP-3 and TIMP-1 concentrations were measured using specific immunoassays. The production of both MMP-3 and the TIMP-1 were either undetectable or below the sensitivity of our immunoassay in the vast majority of uninflamed samples either from controls or from those with Crohn’s disease or ulcerative colitis. In inflamed mucosa, the production of these mediators increased significantly both in Crohn’s disease (P < 0.01 and 0.001, respectively) and ulcerative colitis (P < 0.001 and 0.001, respectively). Mediator production in both cases was significantly correlated with the production of proinflammatory cytokines and IL-10, as well as with the degree of macroscopic and microscopic inflammation. Inflamed mucosa of both Crohn’s disease and ulcerative colitis show increased production of both MMP-3 and its tissue inhibitor, which correlates very well with production of IL-1beta, IL-6, TNF-alpha and IL-10.
Inflammatory cytokines, including tumour necrosis factor-alpha (TNF-alpha) and interleukin (IL)-1 beta, have been implicated as primary mediators of intestinal inflammation in inflammatory bowel disease.
To investigate the in vitro effects of oxpentifylline (pentoxifylline; PTX; a phosphodiesterase inhibitor) on inflammatory cytokine production (1) by peripheral mononuclear cells (PBMCs) and (2) by inflamed intestinal mucosa cultures from patients with Crohn’s disease and patients with ulcerative colitis.
PBMCs and mucosal biopsy specimens were cultured for 24 hours in the absence or presence of PTX (up to 100 micrograms/ml), and the secretion of TNF-alpha, IL-1 beta, IL-6, and IL-8 determined by enzyme linked immunosorbent assays (ELISAs).
PTX inhibited the release of TNF-alpha by PBMCs from patients with inflammatory bowel disease and the secretion of TNF-alpha and IL-1 beta by organ cultures of inflamed mucosa from the same patients. Secretion of TNF-alpha by PBMCs was inhibited by about 50% at a PTX concentration of 25 micrograms/ml (IC50). PTX was equally potent in cultures from controls, patients with Crohn’s disease, and those with ulcerative colitis. The concentrations of IL-6 and IL-8 were not significantly modified in PBMCs, but IL-6 increased slightly in organ culture supernatants.
PTX or more potent related compounds may represent a new family of cytokine inhibitors, potentially interesting for treatment of inflammatory bowel disease.
IL-23/T(H)17 inflammatory responses are regarded as central to the pathogenesis of inflammatory bowel disease, but clinically IL-17A antibodies have shown low efficacy and increased infections in Crohn’s disease. Hence, we decided to closely examine the role of the IL-23/T(H)17 axis in 3 models of colitis.
IL-17A(-/-) and IL-17Ra(-/-) T cells were transferred into Rag1 and RaW mice to assess the role of IL-17A-IL-17Ra signaling in T cells during colitis. In Winnie mice with spontaneous colitis due to an epithelial defect, we studied the progression of colitis in the absence of IL-17A and the efficacy of neutralizing antibodies against the IL-17A or IL-23p19 cytokines.
In transfer colitis models, IL-17A-deficient T cells failed to ameliorate disease, and IL-17Ra-deficient T cells were more colitogenic than wild-type T cells. In Winnie mice with an epithelial defect and spontaneous T(H)17-dominated inflammation, genetic deficiency of IL-17A did not suppress initiation of colitis but limited colitis progression. Furthermore, inhibition of IL-17A by monoclonal antibodies did not reduce colitis severity. In contrast, neutralizing IL-23 using an anti-p19 antibody significantly alleviated both emerging and established colitis, downregulating T(H)17 proinflammatory cytokine expression and diminishing neutrophil infiltration.
Our results support clinical studies showing that IL-17 neutralization is not therapeutic but that targeting IL-23 suppresses intestinal inflammation. Effects of IL-23 distinct from its effects on maturation of IL-17A-producing lymphocytes may underlie the protection from inflammatory bowel disease conveyed by hypomorphic IL-23 receptor polymorphisms and contribute to the efficacy of IL-23 neutralizing antibodies in inflammatory bowel disease.
Luger, D.et al. Either a Th17 or a Th1 effector response can drive autoimmunity: conditions of disease induction affect dominant effector category. J. Exp. Med.205, 799–810 (2008).
Our previous data suggested that IL-17A contributes to the inhibition of Th1 cell function in the gut. However, the underlying mechanisms remain unclear. Here we demonstrate that IL-17A signaling in colonic epithelial cells (CECs) increases TNF-α-induced PI3K-AKT and ERK phosphorylation and inhibits TNF-α induced expression of IL-12P35 and of a Th1 cell chemokine, CXCL11 at mRNA level. In a co-culture system using HT-29 cells and PBMCs, IL-17A inhibited TNF-α-induced IL-12P35 expression by HT-29 cells and led to decreased expression of IFN-γ and T-bet by PBMCs. Finally, adoptive transfer of CECs from mice with Crohn’s Disease (CD) led to an enhanced Th1 cell response and exacerbated colitis in CD mouse recipients. The pathogenic effect of CECs derived from CD mice was reversed by co-administration of recombinant IL-17A. Our data demonstrate a new IL-17A-mediated regulatory mechanism in CD. A better understanding of this pathway might shed new light on the pathogenesis of CD.
IL-17A and IL-17F, produced by the Th17 CD4(+) T cell lineage, have been linked to a variety of inflammatory and autoimmune conditions. We recently reported that activated human CD4(+) T cells produce not only IL-17A and IL-17F homodimers but also an IL-17F/IL-17A heterodimeric cytokine. All three cytokines can induce chemokine secretion from bronchial epithelial cells, albeit with different potencies. In this study, we used small interfering RNA and Abs to IL-17RA and IL-17RC to demonstrate that heterodimeric IL-17F/IL-17A cytokine activity is dependent on the IL-17RA/IL-17RC receptor complex. Interestingly, surface plasmon resonance studies indicate that the three cytokines bind to IL-17RC with comparable affinities, whereas they bind to IL-17RA with different affinities. Thus, we evaluated the effect of the soluble receptors on cytokine activity and we find that soluble receptors exhibit preferential cytokine blockade. IL-17A activity is inhibited by IL-17RA, IL-17F is inhibited by IL-17RC, and a combination of soluble IL-17RA/IL-17RC receptors is required for inhibition of the IL-17F/IL-17A activity. Altogether, these results indicate that human IL-17F/IL-17A cytokine can bind and signal through the same receptor complex as human IL-17F and IL-17A. However, the distinct affinities of the receptor components for IL-17A, IL-17F, and IL-17F/IL-17A heterodimer can be exploited to differentially affect the activity of these cytokines.
Increasing evidence points to a pathologic role for cytokines in Crohn’s colitis. Levels of cytokines are increased in diseased segments of colon in Crohn’s colitis, but no one has studied the concentration of cytokines in clinically and histologically nondiseased segments.
Mucosal biopsies were obtained from 7 patients with active segmental Crohn’s colitis and from 7 controls without inflammatory bowel disease. The concentration of Interleukin (IL)-1 beta, IL-2, IL-6, and IL-8 in patients and controls were determined using enzyme linked immunosorbent assay and compared. Histologic sections were also performed to confirm diseased and nondiseased segments of colon.
The concentrations of IL-1 beta, IL-6, and IL-8 were significantly higher in the involved segments of colon (10.3 +/- 4.1, 3.7 +/- 1.0, 34.4 +/- 6.9 picograms [pg] per mg) when compared to controls (1.8 +/- 0.5, 1.1 +/- 0.5, 5.3 +/- 1.0 pg/mg). The concentrations of IL-1 beta, IL-2, and IL-8 (8.5 +/- 2.9, 5.3 +/- 1.2, 26.3 +/- 8.8 pg/mg) in normal appearing segments of colon of patients with Crohn’s colitis were also significantly higher than in controls, whose IL-2 level was 2.0 +/- 0.5 pg/mg. IL-1 beta and IL-8 were significantly more concentrated in both the involved and uninvolved colonic segments of patients with Crohn’s colitis compared to controls. IL-2 and IL-6 were also more concentrated in Crohn’s patients than in controls, but not significantly. The differences in interleukin concentrations between involved and uninvolved segments of colon in patients with segmental Crohn’s colitis were not significant.
Although Crohn’s colitis is often a segmental disease, concentrations of IL-1 beta and IL-8 are increased throughout the entire colon. These observations reinforce the hypothesis that Crohn’s colitis involves the whole colon even when this is not apparent clinically or histologically.
Interleukin-17A (IL-17A) and its receptor (IL-17RA) are prototype members of IL-17 ligand/receptor family firstly identified in CD4+ T cells, which comprises six ligands (IL-17A to IL- 17F) and five receptors (IL-17RA to IL-17RE). IL-17A is predominantly secreted by T helper 17 (Th17) cells, and plays important roles in the development of autoimmune and inflammatory diseases. IL-17RA is widely expressed, and forms a complex with IL-17RC. Binding of IL-17A to this receptor complex triggers the activation of several intracellular signaling pathways. In this review, we aimed to summarize literature data about molecular features of IL-17A and IL-17RA from gene to mature protein. We are also providing insight into regulatory mechanisms, protein structural conformation, including ligand-receptor interaction, and an overview of signaling pathways. Our aim was to compile the data on molecular characteristics of IL-17A and IL-17RA which may help in the understanding of their functions in health and disease.
Gut. 2014 Dec;63(12):1902-12. doi: 10.1136/gutjnl-2013-305632. Epub 2014 Feb 17.
Involvement of interleukin-17A-induced expression of heat shock protein 47 in intestinal fibrosis in Crohn’s disease.
Intestinal fibrosis is a clinically important issue in Crohn’s disease (CD). Heat shock protein (HSP) 47 is a collagen-specific molecular chaperone involved in fibrotic diseases. The molecular mechanisms of HSP47 induction in intestinal fibrosis related to CD, however, remain unclear. Here we investigated the role of interleukin (IL)-17A-induced HSP47 expression in intestinal fibrosis in CD.
Expressions of HSP47 and IL-17A in the intestinal tissues of patients with IBD were determined. HSP47 and collagen I expressions were assessed in intestinal subepithelial myofibroblasts (ISEMFs) isolated from patients with IBD and CCD-18Co cells treated with IL-17A. We examined the role of HSP47 in IL-17A-induced collagen I expression by administration of short hairpin RNA (shRNA) to HSP47 and investigated signalling pathways of IL-17A-induced HSP47 expression using specific inhibitors in CCD-18Co cells.
Gene expressions of HSP47 and IL-17A were significantly elevated in the intestinal tissues of patients with active CD. Immunohistochemistry revealed HSP47 was expressed in α-smooth muscle actin (α-SMA)-positive cells and the number of HSP47-positive cells was significantly increased in the intestinal tissues of patients with active CD. IL-17A enhanced HSP47 and collagen I expressions in ISEMFs and CCD-18Co cells. Knockdown of HSP47 in these cells resulted in the inhibition of IL-17A-induced collagen I expression, and analysis of IL-17A signalling pathways revealed the involvement of c-Jun N-terminal kinase in IL-17A-induced HSP47 expression.
IL-17A-induced HSP47 expression is involved in collagen I expression in ISEMFs, which might contribute to intestinal fibrosis in CD.
Intestinal fibrosis is a clinically important issue of inflammatory bowel disease (IBD). It is unclear whether or not heat shock protein 47 (HSP47), a collagen-specific molecular chaperone, plays a critical role in intestinal fibrosis. The aim of this study is to investigate the role of HSP47 in intestinal fibrosis of murine colitis.
HSP47 expression and localization were evaluated in interleukin-10 knockout (IL-10KO) and wild-type (WT, C57BL/6) mice by immunohistochemistry. Expression of HSP47 and transforming growth factor-β1 (TGF-β1) in colonic tissue was measured. In vitro studies were conducted in NIH/3T3 cells and primary culture of myofibroblasts separated from colonic tissue of IL-10KO (PMF KO) and WT mice (PMF WT) with stimulation of several cytokines. We evaluated the inhibitory effect of administration of small interfering RNA (siRNA) targeting HSP47 on intestinal fibrosis in IL-10KO mice in vivo.
Immunohistochemistry revealed HSP47 positive cells were observed in the mesenchymal and submucosal area of both WT and IL-10 KO mice. Gene expressions of HSP47 and TGF-β1 were significantly higher in IL-10KO mice than in WT mice and correlated with the severity of inflammation. In vitro experiments with NIH3T3 cells, TGF-β1 only induced HSP47 gene expression. There was a significant difference of HSP47 gene expression between PMF KO and PMF WT. Administration of siRNA targeting HSP47 remarkably reduced collagen deposition in colonic tissue of IL-10KO mice.
Our results indicate that HSP47 plays an essential role in intestinal fibrosis of IL-10KO mice, and may be a potential target for intestinal fibrosis associated with IBD.
Peritoneal fibrosis is a serious complication in patients on continuous ambulatory peritoneal dialysis (CAPD), but the molecular mechanism of this process remains unclear. Heat shock protein 47 (HSP47), a collagen-specific molecular chaperone, is essential for biosynthesis and secretion of collagen molecules, and is expressed in the tissue of human peritoneal fibrosis. In the present study, we examined the effect of HSP47 antisense oligonucleotides (ODNs) on the development of experimental peritoneal fibrosis induced by daily intraperitoneal injections of chlorhexidine gluconate (CG).
HSP47 antisense or sense ODNs were injected simultaneously with CG from day 14, after injections of CG alone. Peritoneal tissue was dissected out 28 days after CG injection. The expression patterns of HSP47, type I and type III collagen, alpha-smooth muscle actin (alpha-SMA), as a marker of myofibroblasts, ED-1 (as a marker of macrophages), and factor VIII were examined by immunohistochemistry.
In rats treated with CG alone, the submesothelial collagenous compact zone was thickened, where the expression levels of HSP47, type I and type III collagen and alpha-SMA were increased. Marked macrophage infiltration was also noted and the number of vessels positively stained for factor VIII increased in the CG-treated group. Treatment with antisense ODNs, but not sense ODNs, abrogated CG-induced changes in the expression of HSP47, type I and III collagen, alpha-SMA, and the number of infiltrating macrophages and vessels.
Our results indicate the involvement of HSP47 in the progression of peritoneal fibrosis and that inhibition of HSP47 expression might merit further clinical investigation for the treatment of peritoneal fibrosis in CAPD patients.
Heat shock protein 47 (HSP47) is a collagen-specific molecular chaperone that is required for molecular maturation of various types of collagens. Recent studies have shown a close association between increased expression of HSP47 and excessive accumulation of collagens in scar tissues of various human and experimental fibrotic diseases. It is presumed that the increased levels of HSP47 in fibrotic diseases assist in excessive assembly and intracellular processing of procollagen molecules and, thereby, contribute to the formation of fibrotic lesions. Studies have also shown that suppression of HSP47 expression can reduce accumulation of collagens to delay the progression of fibrotic diseases in experimental animal models. Because HSP47 is a specific chaperone for collagen synthesis, it provides a selective target to manipulate collagen production, a phenomenon that might have enormous clinical impact in controlling a wide range of fibrotic diseases. Here, we outline the fibrogenic role of HSP47 and discuss the potential usefulness of HSP47 as an anti-fibrotic therapeutic target.
Levels of interleukin-17A (IL-17A) have been found to be increased in synovial fluid from individuals with systemic sclerosis (SSc). This study was undertaken to investigate whether IL-17A-producing cells are present in affected SSc skin, and whether IL-17A exerts a role in the transdifferentiation of myofibroblasts.
Skin biopsy samples were obtained from the involved skin of 8 SSc patients and from 8 healthy control donors undergoing plastic surgery. Immunohistochemistry and multicolor immunofluorescence techniques were used to identify and quantify the cell subsets in vivo, including IL-17A+, IL-4+, CD3+, tryptase-positive, α-smooth muscle actin (α-SMA)-positive, myeloperoxidase-positive, and CD1a+ cells. Dermal fibroblast cell lines were generated from all skin biopsy samples, and quantitative polymerase chain reaction, Western blotting, and solid-phase assays were used to quantify α-SMA, type I collagen, and matrix metalloproteinase 1 (MMP-1) production by the cultured fibroblasts.
IL-17A+ cells were significantly more numerous in SSc skin than in healthy control skin (P = 0.0019) and were observed to be present in both the superficial and deep dermis. Involvement of both T cells and tryptase-positive mast cells in the production of IL-17A was observed. Fibroblasts positive for α-SMA were found adjacent to IL-17A+ cells, but not IL-4+ cells. However, IL-17A did not induce α-SMA expression in cultured fibroblasts. In the presence of IL-17A, the α-SMA expression induced in response to transforming growth factor β was decreased, while MMP-1 production was directly enhanced. Furthermore, the frequency of IL-17A+ cells was higher in the skin of SSc patients with greater severity of skin fibrosis (lower global skin thickness score).
IL-17A+ cells belonging to the innate and adaptive immune system are numerous in SSc skin. IL-17A participates in inflammation while exerting an inhibitory activity on myofibroblast transdifferentiation. These findings are consistent with the notion that IL-17A has a direct negative-regulatory role in the development of dermal fibrosis in humans.
Gut. 2014 Dec;63(12):1902-12. doi: 10.1136/gutjnl-2013-305632. Epub 2014 Feb 17.
Involvement of interleukin-17A-induced expression of heat shock protein 47 in intestinal fibrosis in Crohn’s disease.
Intestinal fibrosis is a clinically important issue in Crohn’s disease (CD). Heat shock protein (HSP) 47 is a collagen-specific molecular chaperone involved in fibrotic diseases. The molecular mechanisms of HSP47 induction in intestinal fibrosis related to CD, however, remain unclear. Here we investigated the role of interleukin (IL)-17A-induced HSP47 expression in intestinal fibrosis in CD.
Expressions of HSP47 and IL-17A in the intestinal tissues of patients with IBD were determined. HSP47 and collagen I expressions were assessed in intestinal subepithelial myofibroblasts (ISEMFs) isolated from patients with IBD and CCD-18Co cells treated with IL-17A. We examined the role of HSP47 in IL-17A-induced collagen I expression by administration of short hairpin RNA (shRNA) to HSP47 and investigated signalling pathways of IL-17A-induced HSP47 expression using specific inhibitors in CCD-18Co cells.
Gene expressions of HSP47 and IL-17A were significantly elevated in the intestinal tissues of patients with active CD. Immunohistochemistry revealed HSP47 was expressed in α-smooth muscle actin (α-SMA)-positive cells and the number of HSP47-positive cells was significantly increased in the intestinal tissues of patients with active CD. IL-17A enhanced HSP47 and collagen I expressions in ISEMFs and CCD-18Co cells. Knockdown of HSP47 in these cells resulted in the inhibition of IL-17A-induced collagen I expression, and analysis of IL-17A signalling pathways revealed the involvement of c-Jun N-terminal kinase in IL-17A-induced HSP47 expression.
IL-17A-induced HSP47 expression is involved in collagen I expression in ISEMFs, which might contribute to intestinal fibrosis in CD.
Peritoneal fibrosis is a serious complication in patients on continuous ambulatory peritoneal dialysis (CAPD), but the molecular mechanism of this process remains unclear. Heat shock protein 47 (HSP47), a collagen-specific molecular chaperone, is essential for biosynthesis and secretion of collagen molecules, and is expressed in the tissue of human peritoneal fibrosis. In the present study, we examined the effect of HSP47 antisense oligonucleotides (ODNs) on the development of experimental peritoneal fibrosis induced by daily intraperitoneal injections of chlorhexidine gluconate (CG).
HSP47 antisense or sense ODNs were injected simultaneously with CG from day 14, after injections of CG alone. Peritoneal tissue was dissected out 28 days after CG injection. The expression patterns of HSP47, type I and type III collagen, alpha-smooth muscle actin (alpha-SMA), as a marker of myofibroblasts, ED-1 (as a marker of macrophages), and factor VIII were examined by immunohistochemistry.
In rats treated with CG alone, the submesothelial collagenous compact zone was thickened, where the expression levels of HSP47, type I and type III collagen and alpha-SMA were increased. Marked macrophage infiltration was also noted and the number of vessels positively stained for factor VIII increased in the CG-treated group. Treatment with antisense ODNs, but not sense ODNs, abrogated CG-induced changes in the expression of HSP47, type I and III collagen, alpha-SMA, and the number of infiltrating macrophages and vessels.
Our results indicate the involvement of HSP47 in the progression of peritoneal fibrosis and that inhibition of HSP47 expression might merit further clinical investigation for the treatment of peritoneal fibrosis in CAPD patients.
To determine the effect of heat shock protein 47 (HSP47) on the expression of collagen I induced by transforming growth factor beta(1) (TGF-beta(1)) in hepatic stellate cell-T6 (HSC-T6) cells.
We used 1 ng/mL and 10 ng/mL recombinant human TGF-beta(1) to stimulate the cultured HSC-T6 cells. Heat shock response (HSR) and antisense oligonucleotides of HSP47 were used to induce and block the expression of HSP47, respectively. The expressions of HSP47 and collagen I were detected by Western blot and the cell viability was observed by MTT assay.
Both HSP47 and collagen I were expressed in normal HSC-T6 cells. Collagen I and HSP47 expression could be induced by both 1 ng/mL and 10 ng/mL TGF-beta(1) and collagen I was expressed the most after the treatment with 10 ng/mL TGF-beta(1). Although HSR could not affect the synthesis of collagen I as it induced the HSP47 expression, HSR could promote the expression of collagen I induced by TGF-beta(1). With no effect on the cell viability, antisense oligonucleotides could significantly inhibit HSR-mediated HSP47 expression and TGF-beta(1)-induced collagen I synthesis.
Over-expression of HSP47 enhances TGF-beta(1)-induced expression of collagen I in HSC-T6 cells, and HSP47 may play important roles in the process of hepatic fibrosis
Interleukin (IL)-17A and IL-17E (also known as IL-25) have been implicated in fibrosis in various tissues. However, the role of these cytokines in the development of intestinal strictures in Crohn’s disease (CD) has not been explored. We investigated the levels of IL-17A and IL-17E and their receptors in CD strictured and non-strictured gut, and the effects of IL-17A and IL-17E on CD myofibroblasts.
IL-17A was significantly overexpressed in strictured compared with non-strictured CD tissues, whereas no significant difference was found in the expression of IL-17E or IL-17A and IL-17E receptors (IL-17RC and IL-17RB, respectively) in strictured and non-strictured CD areas. Strictured CD explants released significantly higher amounts of IL-17A than non-strictured explants, whereas no difference was found as for IL-17E, IL-6, or tumor necrosis factor-α production. IL-17A, but not IL-17E, significantly inhibited myofibroblast migration, and also significantly upregulated matrix metalloproteinase (MMP)-3, MMP-12, tissue inhibitor of metalloproteinase-1 and collagen production by myofibroblasts from strictured CD tissues.
Our results suggest that IL-17A, but not IL-17E, is pro-fibrotic in CD. Further studies are needed to clarify whether the therapeutic blockade of IL-17A through the anti-IL-17A monoclonal antibody secukinumab is able to counteract the fibrogenic process in CD.
Int J Colorectal Dis. 2013 Jul;28(7):915-24. doi: 10.1007/s00384-012-1632-2. Epub 2012 Dec 28.
Role of N-acetylcysteine and GSH redox system on total and active MMP-2 in intestinal myofibroblasts of Crohn’s disease patients.
Intestinal subepithelial myofibroblasts (ISEMFs)(1) are the predominant source of matrix metalloproteinase-2 (MMP-2) in gut, and a decrease in glutathione/oxidized glutathione (GSH/GSSG) ratio, intracellular redox state index, occurs in the ISEMFs of patients with Crohn’s disease (CD). The aim of this study is to demonstrate a relationship between MMP-2 secretion and activation and changes of GSH/GSSG ratio in ISEMFs stimulated or not with tumor necrosis factor alpha (TNFα).
ISEMFs were isolated from ill and healthy colon mucosa of patients with active CD. Buthionine sulfoximine, GSH synthesis inhibitor, and N-acetylcysteine (NAC), precursor of GSH synthesis, were used to modulate GSH/GSSG ratio. GSH and GSSG were measured by HPLC and MMP-2 by ELISA Kit.
In cells, stimulated or not with TNFα, a significant increase in MMP-2 secretion and activation, related to increased oxidative stress, due to low GSH/GSSG ratio, was detected. NAC treatment, increasing this ratio, reduced MMP-2 secretion and exhibited a direct effect on the secreted MMP-2 activity. In NAC-treated and TNFα-stimulated ISEMFs of CD patients’ MMP-2 activity were restored to physiological value. The involvement of c-Jun N-terminal kinase pathway on redox regulation of MMP-2 secretion has been demonstrated.
For the first time, in CD patient ISEMFs, a redox regulation of MMP-2 secretion and activation related to GSH/GSSG ratio and inflammatory state have been demonstrated. This study suggests that compounds able to maintain GSH/GSSG ratio to physiological values can be useful to restore normal MMP-2 levels reducing in CD patient intestine the dysfunction of epithelial barrier.
BMC Pulm Med. 2012 Jun 13;12:24. doi: 10.1186/1471-2466-12-24.
Pirfenidone inhibits TGF-β1-induced over-expression of collagen type I and heat shock protein 47 in A549 cells.
Pirfenidone is a novel anti-fibrotic and anti-inflammatory agent that inhibits the progression of fibrosis in animal models and in patients with idiopathic pulmonary fibrosis (IPF). We previously showed that pirfenidone inhibits the over-expression of collagen type I and of heat shock protein (HSP) 47, a collagen-specific molecular chaperone, in human lung fibroblasts stimulated with transforming growth factor (TGF)-β1 in vitro. The increased numbers of HSP47-positive type II pneumocytes as well as fibroblasts were also diminished by pirfenidone in an animal model of pulmonary fibrosis induced by bleomycin. The present study evaluates the effects of pirfenidone on collagen type I and HSP47 expression in the human alveolar epithelial cell line, A549 cells in vitro.
The expression of collagen type I, HSP47 and E-cadherin mRNAs in A549 cells stimulated with TGF-β1 was evaluated by Northern blotting or real-time PCR. The expression of collagen type I, HSP47 and fibronectin proteins was assessed by immunocytochemical staining.
TGF-β1 stimulated collagen type I and HSP47 mRNA and protein expression in A549 cells, and pirfenidone significantly inhibited this process. Pirfenidone also inhibited over-expression of the fibroblast phenotypic marker fibronectin in A549 cells induced by TGF-β1.
We concluded that the anti-fibrotic effects of pirfenidone might be mediated not only through the direct inhibition of collagen type I expression but also through the inhibition of HSP47 expression in alveolar epithelial cells, which results in reduced collagen synthesis in lung fibrosis. Furthermore, pirfenidone might partially inhibit the epithelial-mesenchymal transition.
Intestinal Inflammatory Pharmaceutics, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 2: CRISPR for Gene Editing and DNA Repair
February 10, 2016 | This morning, AbbVie announced a partnership with Synlogic of Cambridge, Mass., to create microbiome-based therapies for the treatment of inflammatory bowel disease (IBD). The two companies have sketched out a suggested three-year timeline for preclinical research and development, after which AbbVie will take over advancing any drug candidates into clinical trials.
Drugs inspired by the microbes that live in the human gut are a hot topic in biotech. Companies like Seres Health and Vedanta Biosciences are pursuing the idea from a variety of angles, from making traditional small molecule drugs that interact with the microbiome, to creating probiotics or microbial cocktails that restore a healthy balance to the gut ecosystem. IBD, including Crohn’s disease and ulcerative colitis, is an especially popular target for these companies, thanks to strong suggestions that bacterial populations can affect the course of the disease. Already, Second Genome and Coronado Biosciences have taken prospective treatments into the clinic (though the latter has been dealt serious setbacks in Phase II trials).
But even among this peculiar batch of startups, Synlogic’s approach to drug design is exquisitely odd. The company calls its products “synthetic biotics”―in fact, they’re genetically engineered bacteria whose DNA contains intricately designed “gene circuits,” built to start producing therapeutic molecules when and only when the patient needs them.
“We are not looking at correcting the dysregulation of microbes in the gut, like other microbiome companies,” CEO José-Carlos Gutiérrez-Ramos tells Bio-IT World. “We have one bacterium, and it’s engineered to do different functions.”
Synlogic was founded in 2013 by two synthetic biologists at MIT, Timothy Lu and Jim Collins. (Bio-IT World has previously spoken with Lu about his academic work on bacterial gene circuits.) Gutiérrez-Ramos joined almost two years later, leaving a position as the head of Pfizer’s BioTherapeutics R&D group, where he had plenty of opportunity to turn emerging biotechnology ideas into drug candidates ready for submission to the FDA.
Still, synthetic biotics are a good deal more unusual than the biologic drugs he worked on at Pfizer.
His new company doesn’t quite spin functions for its microbes out of whole cloth. All the genes the company uses are copied either from the human genome, or from the bacteria living inside us. But by recombining those genes into circuits, Gutiérrez-Ramos believes Synlogic can finely control whether and when genes are expressed, giving its synthetic biotics the same dosage control as a traditional drug. Meanwhile, choosing the right bacterium to engineer―the current favorite is a strain called E. coli Nissle―ensures the biotics do not form stable colonies in the gut, but can be cleared out as soon as a patient stops treatment.
“We’re pharma guys,” he says. “What we want is to have pharmacologically well-defined products.”
The Molecular Circuit Board
Even before the partnership with AbbVie, Synlogic had a pipeline of drug candidates in development, all meant to treat rare genetic disorders caused by single mutations that shut down the activity of a crucial gene. In principle, there seems to be no reason that bacteria carrying the right genes couldn’t pick up the slack. “We know the patient is missing a function that is typically performed by the liver, or the kidney, or the pancreas,” says Gutiérrez-Ramos. “What we do is shift that function from an organ to a stable fraction of the microbiome.”
The approach is in some ways analogous to gene therapy, where a corrected version of a broken gene is inserted into a patient’s own DNA. “We don’t use that word, but the fact is it’s a non-somatic gene therapy,” Gutiérrez-Ramos says. “And if something goes wrong, you can control it just by stopping treatment.” The most advanced synthetic biotic in Synlogic’s pipeline targets urea cycle disorder, exactly the sort of disease that might otherwise be addressed by gene therapy: patients are missing a single enzyme that helps remove nitrogen from the body and prevent it from forming ammonia in the bloodstream. Synlogic will meet with the FDA this March to discuss whether and how this first product can be tested in humans.
The new IBD program with AbbVie, however, adds a whole new level of complexity. Executives from the two companies have been in discussions for around six months, and both agree that no single mechanism will be enough to provide significant relief for patients. Crohn’s and ulcerative colitis are painful autoimmune diseases that involve both a weakening of the epithelial lining in the stomach, and a buildup of inflammatory molecules. The development plan that AbbVie and Synlogic have agreed on includes three separate methods of attack to relieve these symptoms.
“One approach AbbVie is very interested in is for our synthetic biotics to produce substances that could tighten the epithelial barrier,” says Gutiérrez-Ramos. “Another approach is to degrade pro-inflammatory molecules”―the same tack taken by AbbVie’s current leading IBD drug, Humira, which targets the inflammatory protein TNFα. “Finally, we can produce anti-inflammatory molecules.”
Uniquely, synthetic biotics can perform all three functions at once; it’s just a matter of inserting the right genes. But that alone might not be a decisive advantage over some sort of combination therapy. The biggest selling point of Synlogic’s microbes is not the genes they can be engineered to express―what you might call the “output” of their gene circuits―but the input, the DNA elements called “inducible promoters” that decide when those genes should be activated.
The core idea is that patients will have a constant population of synthetic biotics in their bodies, taken daily―but those microbes will only generate their therapeutic payloads when needed. In IBD, Gutiérrez-Ramos explains, “it’s not that the patient is always inflamed, but they have flares. Our vision, and AbbVie’s vision, is that the bacteria that you take every day sense when the flare is coming, and then trigger the genetic output.”
This would be a major improvement over a drug like Humira, which after all is constantly inhibiting a part of the immune system. Patients taking Humira, or one of the many other immunosuppressant drugs for IBD, are at a constantly heightened risk of infection; tuberculosis is a particular specter for these patients. If Synlogic can find a genetic “on-switch” that responds to a reliable indicator of IBD flares, it could potentially create a much more precisely administered treatment, while still giving patients the simple dosing schedule of one pill every day.
The company has leads on two inducible promoters that might do the trick: one that reacts to nitric oxide, and another tied to reactive oxygen species. Of course, there’s no guarantee that either will respond sensitively to IBD flares in a real clinical setting. “This is an early time for the technology,” says Gutiérrez-Ramos. “We have demonstrated this in animals, but we have to demonstrate it in humans.”
Although it’s far too early to say if synthetic biotics will become an ordinary part of the pharma toolkit, AbbVie’s decision to invest in the technology offers the means to test this approach on a large scale. Synlogic expects to raise its own funding for trials of its rare disease products, which the FDA does not expect to enroll huge numbers of patients, but IBD is a problem of a very different order.
“We are very honored to work with truly the leader in treatment of inflammatory bowel disease,” says Gutiérrez-Ramos. With the backing of big pharma, it will be possible to trial microbiome-based therapies for the kinds of common, chronic diseases that are the biggest drain on our healthcare system. What’s more, the AbbVie partnership is an important signal of the industry’s faith in synthetic biology as an approach to treating disease.
Signaling of Immune Response in Colon Cancer, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 1: Next Generation Sequencing (NGS)
Signaling of Immune Response in Colon Cancer
Larry H. Bernstein, MD, FCAP, Curator
LPBI
Revised 1/13/2016
STING Protein May Serve as Biomarker for Colorectal and Other Cancers
Scientists at University of Miami Miller School of Medicine’s Sylvester Comprehensive Cancer Center say they have discovered how the stimulator of interferon genes (STING) signaling pathway may play an important role in alerting the immune system to cellular transformation. They believe their finding will shed further light on the immune system’s response to cancer development.
In 2008, Glen N. Barber, Ph.D., leader of the viral oncology program at Sylvester, and professor and chairman of cell biology at the Miller School of Medicine, and colleagues published in Nature (“STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling”) the discovery of STING as a new cellular molecule that recognizes virus and bacteria infection to initiate host defense and immune responses. In the new study, published in Cell Reports (“Deregulation of STING Signaling in Colorectal Carcinoma Constrains DNA Damage Responses and Correlates With Tumorigenesis”), they describe STING’s role in the potential suppression of colorectal cancer.
“Since 2008 we’ve known that STING is crucial for antiviral and antibacterial responses,” said Dr. Barber. “But until now, little had been known about its function in human tumors. In this study we show, for the first time, that STING signaling is repressed in colorectal carcinoma and other cancers, an event which may enable transformed cells to evade the immune system.”
Colorectal cancer currently affects around 1.2 million people in the U.S. and 150,000 new cases are diagnosed every year, making it the third most common cancer in both men and women. Since most colon cancers develop from benign polyps, they can be treated successfully when detected early. However, if the tumor has already spread, survival rates are generally low.
Using disease models of colorectal cancer, the team of Sylvester scientists showed that loss of STING signaling negatively affected the body’s ability to recognize DNA-damaged cells. In particular, certain cytokines that facilitate tissue repair and antitumor priming of the immune system were not sufficiently produced to initiate a significant immune response to eradicate the colorectal cancer.
“We were able to show that impaired STING responses may enable damaged cells to elude the immune system,” continued Dr. Barber. “And if the body doesn’t recognize and attack cancer cells, they will multiply and, ultimately, spread to other parts of the body.”
He and his colleagues suggest evaluating STING signaling as a prognostic marker for the treatment of colorectal as well as other cancers. For example, Dr. Barber’s study showed that cancer cells with defective STING signaling were particularly prone to attack by oncolytic viruses presently being used as cancer therapies.
“Impaired STING responses may enable damaged cells to evade host immunosurveillance processes, although they provide a critical prognostic measurement that could help predict the outcome of effective oncoviral therapy,” wrote the investigators.
This is the first detailed examination of how the stimulator of interferon genes (STING) signaling pathway, discovered by Glen N. Barber, Ph.D., Leader of the Viral Oncology Program at Sylvester Comprehensive Cancer Center, may play an important role in alerting the immune system to cellular transformation.
In 2008, Barber, who is also Professor and Chairman of Cell Biology at the University of Miami Miller School of Medicine, and colleagues published in Nature the discovery of STINGas a new cellular molecule that recognizes virus and bacteria infection to initiate host defense and immune responses. In the new study they describe STING’s role in the potential suppression of colorectal cancer.
“Since 2008 we’ve known that STING is crucial for antiviral and antibacterial responses,” said Barber. “But until now, little had been known about its function in human tumors. In this study we show, for the first time, that STING signaling is repressed in colorectal carcinoma and other cancers, an event which may enable transformed cells to evade the immune system.”
Colorectal cancer currently affects around 1.2 million people in the United States and 150.000 new cases are diagnosed every year, making it the third most common cancer in both men and women. Since most colon cancers develop from benign polyps, they can be treated successfully when detected early. However, if the tumor has already spread, survival rates are generally low.
Using disease models of colorectal cancer, the team of Sylvester scientists showed that loss of STING signaling negatively affected the body’s ability to recognize DNA-damaged cells. In particular, certain cytokines – small proteins important for cell signaling – that facilitate tissue repair and anti-tumor priming of the immune system were not sufficiently produced to initiate a significant immune response to eradicate the colorectal cancer.
“We were able to show that impaired STING responses may enable damaged cells to elude the immune system,” added Barber. “And if the body doesn’t recognize and attack cancer cells, they will multiply and, ultimately, spread to other parts of the body.”
Barber and his colleagues suggest evaluating STING signaling as a prognostic marker for the treatment of colorectal as well as other cancers. For example, Barber’s study showed that cancer cells with defective STING signaling were particularly prone to attack by oncolytic viruses presently being used as cancer therapies. Alternate studies with colleagues have also shown that activators of STING signaling are potent stimulators of anti-tumor immune responses. Collectively, the control of STING signaling may have important implications for cancer development as well as cancer treatment.
Every step you take: STING pathway key to tumor immunity
A recently discovered protein complex known as STING plays a crucial role in detecting the presence of tumor cells and promoting an aggressive anti-tumor response by the body’s innate immune system, according to two separate studies published in the Nov. 20 issue of the journal Immunity.
The studies, both from University of Chicago-based research teams, have major implications for the growing field of cancer immunotherapy. The findings show that when activated, the STING pathway triggers a natural immune response against the tumor. This includes production of chemical signals that help the immune system identify tumor cells and generate specific killer T cells. The research also found that targeted high-dose radiation therapy dials up the activation of this pathway, which promotes immune-mediated tumor control.
These findings could “enlarge the fraction of patients who respond to immunotherapy with prolonged control of the tumor,” according to a commentary on the papers by the University of Verona’s Vincenzo Bronte, MD. “Enhancing the immunogenicity of their cancers might expand the lymphocyte repertoire that is then unleashed by interference with checkpoint blockade pathways,” such as anti-PD-1.
STING, short for STimulator of INterferon Genes complex, is a crucial part of the process the immune system relies on to detect threats — such as infections or cancer cells — that are marked by the presence of DNA that is damaged or in the wrong place, inside the cell but outside the nucleus.
Detection of such “cytosolic” DNA initiates a series of interactions that lead to the STING pathway. Activating the pathway triggers the production of interferon-beta, which in turn alerts the immune system to the threat, helps the system detect cancerous or infected cells, and ultimately sends activated T cells into the battle.
“We have learned
“Innate immune sensing via the host STING pathway is critical for tumor control by checkpoint blockade,” Gajewski’s team noted in their paper. They found promising drugs known as checkpoint inhibitors — such as anti PD-1 or anti PD-L1, which can take the brakes off of an immune response — were not effective in STING-deficient mice. New agents that stimulate the STING pathway are being developed as potential cancer therapeutics.
A second University of Chicago team, led by cancer biologistYang-Xin Fu, MD, PhD, professor of pathology, and Ralph Weichselbaum, MD, chairman of radiation and cellular oncology and co-director of the Ludwig Center for Metastasis Research, found that high-dose radiation therapy not only kills targeted cancer cells but the resulting DNA damage drives a systemic immune response.
a great deal recently about what we call checkpoints, the stumbling blocks that prevent the immune system from ultimately destroying cancers,” said Thomas Gajewski, MD, PhD, professor of medicine and pathology at the University of Chicago and senior author of one of the studies. “Blockade of immune checkpoints, such as with anti-PD-1, is therapeutic in a subset of patients, but many individuals still don’t respond. Understanding the role of the STING pathway provides insights into how we can ‘wake up’ the immune response against tumors. This can be further boosted by checkpoint therapies.”
The two published studies, he said, help move this approach forward.
In a series of experiments in mice, both research teams found tumor cell-derived DNA could initiate an immune response against cancers. But when tested in mice that lacked a functional gene for STING, the immune system did not effectively respond.
“This result unifies traditional studies of DNA damage with newly identified DNA sensing of immune responses,” Fu said.
“This is a previously unknown mechanism,” Weichselbaum added.
In mice that lacked STING, however, there was no therapeutic immune response. The induction of interferons by radiation and consequent cancer cell killing, they conclude, depends on STING-pathway signaling.
They did find that combining cyclic guanosine monophosphate-adenosine monophosphate (cGAMP), an earlier step in the STING pathway, with radiation, could greatly enhance the antitumor efficacy of radiation.
“This opens a new avenue to develop STING-related agonists for patients with radiation-resistant cancers,” Fu said.
STING-Dependent Cytosolic DNA Sensing Mediates Innate Immune Recognition of Immunogenic Tumors
• Spontaneous T cell responses against tumors require the host STING pathway in vivo
• Tumor-derived DNA can induce type I interferon production via STING
• Tumor DNA can be identified in host APCs in the tumor microenvironment in vivo
Summary
Spontaneous T cell responses against tumors occur frequently and have prognostic value in patients. The mechanism of innate immune sensing of immunogenic tumors leading to adaptive T cell responses remains undefined, although type I interferons (IFNs) are implicated in this process. We found that spontaneous CD8+ T cell priming against tumors was defective in mice lacking stimulator of interferon genes complex (STING), but not other innate signaling pathways, suggesting involvement of a cytosolic DNA sensing pathway. In vitro, IFN-β production and dendritic cell activation were triggered by tumor-cell-derived DNA, via cyclic-GMP-AMP synthase (cGAS), STING, and interferon regulatory factor 3 (IRF3). In the tumor microenvironment in vivo, tumor cell DNA was detected within host antigen-presenting cells, which correlated with STING pathway activation and IFN-β production. Our results demonstrate that a major mechanism for innate immune sensing of cancer occurs via the host STING pathway, with major implications for cancer immunotherapy.
Immunity Erratum STING-Dependent Cytosolic DNA Sensing Mediates Innate Immune Recognition of Immunogenic Tumors
Seng-Ryong Woo, Mercedes B. Fuertes, Leticia Corrales, Stefani Spranger, Michael J. Furdyna, Michael Y.K. Leung, Ryan Duggan, Ying Wang, Glen N. Barber, Katherine A. Fitzgerald, Maria-Luisa Alegre, and Thomas F. Gajewski* *Correspondence: tgajewsk@medicine.bsd.uchicago.eduhttp://dx.doi.org/10.1016/j.immuni.2014.12.015 (Immunity 41, 830–842; November 20, 2014)
The original Figure 3C accidentally contained a duplicated panel in the bright-field column, third row down, and this has now been replaced with the correct data. The change does not alter the conclusions of the paper. This mistake has now been corrected online, and the authors regret the error.
Cytosolic DNA Sensors (CDSs): a STING in the tail – Review
The innate immune system provides the first line of defense against infectious pathogens and serves to limit their early proliferation. It is also vital in priming and activating the adaptive immune system.
Innate immune detection of intracellular DNA derived from viruses and invasive bacteria is important to initiate an effective protective response. This crucial step depends on cytosolic DNA sensors (CDSs), which upon activation trigger the production of type I interferons (IFNs) and the induction of IFN-responsive genes and proinflammatory chemokines.
Although the identity of these CDSs is not fully uncovered, much progress has been made in understanding the signaling pathways triggered by these sensors.
Cytosolic DNA-mediated production of type I IFNs (mainly IFN-β) requires the transcription factor IFN regulatory factor 3 (IRF3), which is activated upon phosphorylation by TANK-binding-kinase-1 (TBK1) [1].
STING in DNA sensing
Recently, a new molecule, STING (stimulator of IFN genes), has been shown to be essential for the TBK1-IRF3- dependent induction of IFN-β by transfected DNA ligands and intracellular DNA produced by pathogens after infection [2, 3].
STING (also known as MITA, MPYS and ERIS) is a transmembrane protein that resides in the endoplasmic reticulum (ER) [2-6]. In response to cytosolic DNA, STING forms dimers and translocates from the ER to the Golgi then to punctate cytosolic structures where it colocalizes with TBK-1, leading to the phosphorylation of IRF3.
How STING stimulates TBK1-dependent IRF3 activation was recently elucidated by Tanaka and Chen. They found that, upon cytosolic DNA sensing, the C-terminal tail of STING acts as a scaffold protein to promote the phosphorylation of IRF3 by TBK1 [7].
STING in the host response to intracellular pathogens. Linking type I IFN response and autophagy for better defense
Until very recently, STING in addition to its role as an adaptor protein was also thought to function as a sensor of cyclic dinucleotides.
Burdette et al. first demonstrated that STING binds directly to the bacterial molecule cyclic diguanylate monophosphate (c-di-GMP) [8]. This finding was confirmed by several teams who examined the structure of STING bound to c-di-GMP [9-11], including Cheng and colleagues, however their data suggest that STING is not the primary sensor of c-di-GMP [12]. Rather, they indicate that DDX41, an identified CDS, functions as a direct receptor for cyclic dinucleotides upstream of STING. The authors hypothesized that DDX41 binds to c-di-GMP then forms a complex with STING to activate the IFN response.
STING induces autophagy
Exciting new developments reveal that STING participates in another aspect of innate immunity, autophagy.
Autophagy plays a critical role in host defense responses to pathogens by promoting the elimination of microbes that enter into the cytosol by their sequestration into autophagosomes and their delivery to the lysosome.
Recent studies have reported that DNA viruses and intracellular bacteria induce autophagy and that this process is dependent on cytosolic genomic DNA and STING [13-15]. Robust induction of autophagy was also observed after transfection of various double stranded (ds) DNA species, such as poly(dA:dT), poly(dG:dC) or plasmid DNA, but not single stranded (ss) DNA, dsRNA or ssRNA [16].
Interestingly, activated STING was shown to relocate to unidentified membrame-bound compartments where it colocalizes with LC3, a hallmark of autophagy, and ATg9a. The latter protein was reported to regulate the interaction between STING and TBK1 after dsDNA stimulation [16]. The E3 ubiquitin ligases TRIM56 and TRIM32
were also found to regulate STING by mediating its dimerization through K63-linked ubiquitination [17, 18].
Several cytosolic DNA sensors upstream of STING have been proposed. DNA-dependent activator of IRFs (DAI) was the first CDS discovered based on the ability of transfected poly(dA:dT) to induce IFN-β [19]. However, the role of DAI has been shown to be very cell-type specific and cells derived from DAI-deficient mice responded normally to dsDNA ligands [20].
While analyzing immune responses to dsDNA regions derived from vaccinia virus (VACV-70) or Herpes simplex virus 1 (HSV-60) genomes, Unterholzner et al. identified IFI16 as a DNA binding protein mediating IFN-β induction [21]. Interestingly, IFI16 belongs to a new family of pattern recognition receptors that contain the pyrin and HIN domain (PYHIN), termed AIM2-like receptors (ALRs).
AIM2 is a STING-independent cytosolic DNA sensor that forms an inflammasome with ASC to trigger caspase-1 activation and the secretion of the proinflammatory cytokines IL-1β and IL-18 [20].
Members of the DExD/H-box helicase superfamily have also been reported to function as cytosolic DNA sensors. While DHX36 and DHX9 were identified as STING-independent but MyD88-dependent sensors of CpG-containing DNA in plasmacytoid dendritic cells, DDX41 was found to bind various dsDNA ligands and localize with STING to promote IFN-β expression [22]. Other CDSs have been reported to function independently of STING: RNA Pol III, LRRFIP1 and Ku70 [20].
Unlike cytosolic RNA sensors (RIG-I, MDA-5), which detect structural moieties specific to pathogen RNA, such as 5’-triphosphates, it is not clear whether cytosolic DNA sensors can recognize any particular structural motif of DNA that would discriminate between self and non-self. This suggests that CDSs may have a role not only in anti-microbial innate immune responses but also in autoimmunity. A multitude of CDSs have been described but whether they are all true receptors remains an open question.
1. Stetson DB & Medzhitov R. 2006. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity. 24(1):93-103.
2. Ishikawa H. & Barber GN., 2008. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 455(7213):674-8.
3. Ishikawa H. et al., 2009. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 461(7265):788-92.
4. Zhong B. et al., 2008. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity. 29(4):538-50.
5. Jin L. et al., 2008. MPYS, a novel membrane tetraspanner, is associated with major histocompatibility complex class II and mediates transduction of apoptotic signals. Mol Cell Biol. 28(16):5014-26.
UV Light Potentiates STING (Stimulator of Interferon Genes)-dependent Innate Immune Signaling through Deregulation of ULK1 (Unc51-like Kinase 1).
The mechanism by which ultraviolet (UV) wavelengths of sunlight trigger or exacerbate the symptoms of the autoimmune disorder lupus erythematosus is not known but may involve a role for the innate immune system. Here we show that UV radiation potentiates STING (stimulator of interferon genes)-dependent activation of the immune signaling transcription factor interferon regulatory factor 3 (IRF3) in response to cytosolic DNA and cyclic dinucleotides in keratinocytes and other human cells. Furthermore, we find that modulation of this innate immune response also occurs with UV-mimetic chemical carcinogens and in a manner that is independent of DNA repair and several DNA damage and cell stress response signaling pathways. Rather, we find that the stimulation of STING-dependent IRF3 activation by UV is due to apoptotic signaling-dependent disruption of ULK1 (Unc51-like kinase 1), a pro-autophagic protein that negatively regulates STING. Thus, deregulation of ULK1 signaling by UV-induced DNA damage may contribute to the negative effects of sunlight UV exposure in patients with autoimmune disorders.
STING and the innate immune response to nucleic acids in the cytosol
Cytosolic detection of pathogen-derived nucleic acids is critical for the initiation of innate immune defense against diverse bacterial, viral and eukaryotic pathogens. Conversely, inappropriate responses to cytosolic nucleic acids can produce severe autoimmune pathology. The host protein STING has been identified as a central signaling molecule in the innate immune response to cytosolic nucleic acids. STING seems to be especially critical for responses to cytosolic DNA and the unique bacterial nucleic acids called ‘cyclic dinucleotides’. Here we discuss advances in the understanding of STING and highlight the many unresolved issues in the field.
The detection of pathogen-derived nucleic acids is a central strategy by which the innate immune system senses microbes to then initiate protective responses1. Conversely, inappropriate recognition of self nucleic acids can result in debilitating autoimmune diseases such as systemic lupus erythematosus2. It is therefore important to understand the molecular basis of the detection of nucleic acids by the innate immune system. Studies have established that nucleic acids derived from extracellular sources are sensed mainly by endosomal Toll-like receptors (TLRs), such as TLR3, TLR7 and TLR9, whereas cytosolic nucleic acids are detected independently of TLRs by a variety of less-well-characterized mechanisms1.
Studies have identified STING (‘stimulator of interferon genes’; also known as TMEM173, MPYS, MITA and ERIS) as a critical signaling molecule in the innate response to cytosolic nucleic-acid ligands. STING was first described as a protein that interacts with major histocompatibility complex class II molecules3, but the relevance of this interaction remains unclear. Subsequent studies have instead focused on the role of STING in the transcriptional induction of type I interferons and coregulated genes in response to nucleic acids in the cytosol. Several groups have independently isolated STING by screening for proteins able to induce interferon-B (IFN-B) when overexpressed4–6. Studies of STING-deficient mice have subsequently confirmed the essential role of STING in innate responses to cytosolic nucleic-acid ligands, particularly double-stranded DNA (dsDNA) and unique bacterial nucleic acids called ‘cyclic dinucleotides’7–9. Several studies have also linked STING to the interferon response to cytosolic RNA5–7, but this has not been found consistently7,8,10,11; thus, we focus here on the role of STING in response to DNA and cyclic dinucleotides.
Protein Stimulator of interferon genes protein
Gene TMEM173
Organism Homo sapiens (Human)
Facilitator of innate immune signaling that acts as a sensor of cytosolic DNA from bacteria and viruses and promotes the production of type I interferon (IFN-alpha and IFN-beta). Innate immune response is triggered in response to non-CpG double-stranded DNA from viruses and bacteria delivered to the cytoplasm. Acts by recognizing and binding cyclic di-GMP (c-di-GMP), a second messenger produced by bacteria, and cyclic GMP-AMP (cGAMP), a messenger produced in response to DNA virus in the cytosol: upon binding of c-di-GMP or cGAMP, autoinhibition is alleviated and TMEM173/STING is able to activate both NF-kappa-B and IRF3 transcription pathways to induce expression of type I interferon and exert a potent anti-viral state. May be involved in translocon function, the translocon possibly being able to influence the induction of type I interferons. May be involved in transduction of apoptotic signals via its association with the major histocompatibility complex class II (MHC-II). Mediates death signaling via activation of the extracellular signal-regulated kinase (ERK) pathway. Essential for the induction of IFN-beta in response to human herpes simplex virus 1 (HHV-1) infection. Exhibits 2′,3′ phosphodiester linkage-specific ligand recognition. Can bind both 2′-3′ linked cGAMP and 3′-3′ linked cGAMP but is preferentially activated by 2′-3′ linked cGAMP (PubMed:26300263)
Stimulator of interferon genes protein (IPR029158)
Transmembrane protein 173, also known as stimulator of interferon genes protein (STING) or endoplasmic reticulum interferon stimulator (ERIS), is a transmembrane adaptor protein which is involved in innate immune signalling processes. It induces expression of type I interferons (IFN-alpha and IFN-beta) via the NF-kappa-B and IRF3, pathways in response to non-self cytosolic RNA and dsDNA [PMID: 18724357, PMID: 19776740,PMID: 18818105, PMID: 19433799].
Neutrophil Serine Proteases in Disease and Therapeutic Considerations
Larry H. Bernstein, MD, FCAP, Curator
LPBI
SERPINB1 Regulates the activity of the neutrophil proteases elastase, cathepsin G, proteinase-3, chymase,
chymotrypsin, and kallikrein-3. Belongs to the serpin family. Ov-serpin subfamily. Note: This description may
include information from UniProtKB.
Monocyte/neutrophil elastase inhibitor (EI) is a protein of approximately 42,000 Mr with serpin-like functional properties. Remold-O’Donnell et al. (1992) cloned EI cDNA and identified 3 EI mRNA species of 1.5, 1.9, and 2.6 kb in monocyte-like cells
and no hybridizing mRNA in lymphoblastoid cells lacking detectable EI enzymatic activity. The cDNA open reading frame encoded
a 379-amino acid protein. Its sequence established EI as a member of the serpin superfamily. Sequence alignment indicated that
the reactive center P1 residue is cys-344, consistent with abrogation of elastase inhibitory activity by iodoacetamide and making
EI a naturally occurring cys-serpin.
Mapping
In the course of studying 4 closely linked genes encoding members of the ovalbumin family of serine proteinase inhibitors
(Ov-serpins) located on 18q21.3, Schneider et al. (1995) investigated the mapping of elastase inhibitor. They prepared PCR
primer sets of the gene, and by using the NIGMS monochromosomal somatic cell hybrid panel, showed that the EI gene maps
to chromosome 6.
By amplifying DNA of a somatic cell hybrid panel, Evans et al. (1995) unambiguously localized ELANH2 to chromosome 6.
With the use of a panel of radiation and somatic cell hybrids specific for chromosome 6, they refined the localization to
the short arm telomeric of D6S89, F13A (134570), and D6S202 at 6pter-p24.
Evans, E., Cooley, J., Remold-O’Donnell, E. Characterization and chromosomal localization of ELANH2, the gene encoding human
monocyte/neutrophil elastase inhibitor. Genomics 28: 235-240, 1995. [PubMed: 8530031, related citations] [Full Text]
Remold-O’Donnell, E., Chin, J., Alberts, M. Sequence and molecular characterization of human monocyte/neutrophil elastase inhibitor.
Proc. Nat. Acad. Sci. 89: 5635-5639, 1992. [PubMed: 1376927, related citations][Full Text]
Schneider, S. S., Schick, C., Fish, K. E., Miller, E., Pena, J. C., Treter, S. D., Hui, S. M., Silverman, G. A. A serine proteinase inhibitor locus at
18q21.3 contains a tandem duplication of the human squamous cell carcinoma antigen gene. Proc. Nat. Acad. Sci. 92: 3147-3151, 1995.
[PubMed: 7724531,related citations] [Full Text]
Leukocyte elastase inhibitor is also known as serpin B1. Serpins (SERine Proteinase INhibitors) belong to MEROPS inhibitor family I4 (clan ID)
[PMID: 14705960].
Serpin B1 regulates the activity of neutrophil serine proteases such as elastase, cathepsin G and proteinase-3 and may play a regulatory role to
limit inflammatory damage due to proteases of cellular origin [PMID: 11747453]. It also functions as a potent intracellular inhibitor of granzyme
H [PMID: 23269243]. In mouse, four different homologues of human serpin B1 have been described [PMID: 12189154].
The neutrophil serine protease inhibitor SerpinB1 protects against inflammatory lung injury and morbidity in influenza virus infection
SerpinB1 is an efficient inhibitor of neutrophil serine proteases. SerpinB1-/- mice fail to clear bacterial lung infection with increased inflammation and neutrophil death. Here, we investigated the role of serpinB1 in influenza virus infection, where infiltrating neutrophils and monocytes facilitate virus clearance but can also cause tissue injury. Influenza virus (H3N2 A/Phil/82) infection caused greater and more protracted body weight loss in serpinB1-/- vs. WT mice (20% vs. 15%; nadir on day 4 vs. day 3). Increased morbidity was not associated with defective virus clearance. Cytokines (IFN, TNF, IL-17, IFN, G-CSF) and chemokines (MIP-1, KC, MIP-2) were increased in serpinB1-/- mice vs. WT on days 2-7 post-infection but not on day 1. In WT mice, histology indicated large infiltration of neutrophils peaking on day 1 and maximal airway injury on day 2 that resolved on day 3 coincident with the influx of monocytes/macrophages. In serpinB1-/- mice, neutrophils also peaked on day 1; epithelial injury was severe and sustained with accumulation of dead cells on day 2 and 3. Immunophenotyping of lung digests on day 2 and 3 showed delayed recruitment of monocytes, macrophages and DC in serpinB1-/- mice, but increase of activated CD4 (day 2-3) and CD8 (day 3) T cells. Our findings demonstrate that serpinB1 protects against morbidity and inflammatory lung injury associated with influenza infection.
The neutrophil serine protease inhibitor serpinb1 preserves lung defense functions in Pseudomonas aeruginosainfection
Neutrophil serine proteases (NSPs; elastase, cathepsin G, and proteinase-3) directly kill invading microbes. However, excess NSPs in the lungs play a central role in the pathology of inflammatory pulmonary disease. We show that serpinb1, an efficient inhibitor of the three NSPs, preserves cell and molecular components responsible for host defense against Pseudomonas aeruginosa. On infection, wild-type (WT) and serpinb1-deficient mice mount similar early responses, including robust production of cytokines and chemokines, recruitment of neutrophils, and initial containment of bacteria. However, serpinb1−/− mice have considerably increased mortality relative to WT mice in association with late-onset failed bacterial clearance. We found that serpinb1-deficient neutrophils recruited to the lungs have an intrinsic defect in survival accompanied by release of neutrophil protease activity, sustained inflammatory cytokine production, and proteolysis of the collectin surfactant protein–D (SP-D). Coadministration of recombinant SERPINB1 with the P. aeruginosa inoculum normalized bacterial clearance inserpinb1−/− mice. Thus, regulation of pulmonary innate immunity by serpinb1 is nonredundant and is required to protect two key components, the neutrophil and SP-D, from NSP damage during the host response to infection.
Neutrophils are the first and most abundant phagocytes mobilized to clear pathogenic bacteria during acute lung infection. Prominent among their antimicrobial weapons, neutrophils carry high concentrations of a unique set of serine proteases in their granules, including neu trophil elastase (NE), cathepsin G (CG), and proteinase-3. These neutrophil serine proteases (NSPs) are required to kill phagocytosed bacteria and fungi (1, 2). Indeed, neutrophils lacking NE fail to kill phagocytosed pathogens, and mice deficient for NE and/or CG have increased mortality after infection with pulmonary pathogens (3, 4). However, NSPs in the lung airspace can have a detrimental effect in severe inflammatory lung disease through degradation of host defense and matrix proteins (5–7). Thus, understanding of the mechanisms that regulate NSP actions during lung infections associated with neutrophilia will help identify strategies to balance host defense and prevent infection-induced tissue injury.
SERPINB1, also known as monocyte NE inhibitor (8), is an ancestral serpin super-family protein and one of the most efficient inhibitors of NE, CG, and proteinase-3 (9, 10). SERPINB1 is broadly expressed and is at particularly high levels in the cytoplasm of neutrophils (11, 12). SERPINB1 has been found complexed to neutro phil proteases in lung fluids of cystic fibrosis patients and in a baboon model of bronchopulmonary dysplasia (13, 14). Although these studies suggest a role for SERPINB1 in regulating NSP activity, it is unclear whether these complexes reflect an important physiological role for SERPINB1 in the lung air space.
RESULTS
To define the physiological importance of SERPINB1 in shaping the outcome of bacterial lung infection, we generated mice deficient for serpinb1 (serpinb1−/−) by targeted mutagenesis in embryonic stem (ES) cells (Fig. 1, A–C). Crossings of heterozygous mice produced WT (+/+), heterozygous (+/−), and KO (−/−) mice for serpinb1 at expected Mendelian ratios (25% +/+, 51% +/−, and 24% −/−; n = 225; Fig. 1 D), indicating no embryonic lethality. Bone marrow neutrophils of serpinb1−/− mice lacked expression of the protein, whereas heterozygous serpinb1+/− mice had reduced levels compared with WT mice (Fig. 1 E). Importantly, levels of the cognate neutrophil proteases NE and CG, measured as antigenic units, were not altered by deletion of serpinb1 (Fig. 1 F). When maintained in a specific pathogen-free environment, serpinb1−/− mice did not differ from WT littermates in growth, litter size, or life span (followed up to 12 mo), and no gross or histopathological defects were observed at necropsy in 8-wk-old mice.
6–8-wk-old animals were intranasally inoculated with the nonmucoid Pseudomonas aeruginosa strain PAO1. Using two infection doses (3 × 106 and 7 × 106 CFU/mouse),serpinb1−/− mice had a significantly lower survival probability and a shorter median survival time compared with WT mice (Fig. 2 A). Further groups of infected mice were used to evaluate bacterial clearance. At 6 h after infection, the bacteria were similarly restricted in mice of the two genotypes, suggesting that the serpinb1−/− mice have a normal initial response to infection. At 24 h, the median bacterial count in the lungs of serpinb1−/− mice was five logs higher than that of the WT mice (P < 0.001), and the infection had spread systemically in serpinb1−/− mice but not in WT mice, as shown by high median CFU counts in the spleen (Fig. 2 B). Histological examination at 24 h after infection revealed abundant neutrophil infiltration in the lungs of both WT and serpinb1−/− mice, and consistent with the bacteriological findings, numerous foci of bacterial colonies and large areas of alveolar exudates were found in serpinb1−/− mice only (Fig. 2 C). When challenged with the mucoid P. aeruginosa clinical strain PA M57-15 isolated from a cystic fibrosis patient, WT mice cleared >99.9% of the inoculum within 24 h, whereas serpinb1-deficient mice failed to clear the infection (Fig. 2 D). Thus, the NSP inhibitor serpinb1 is essential for maximal protection against pneumonia induced by mucoid and nonmucoid strains of P. aeruginosa.
Figure 2.
Serpinb1−/− mice fail to clear P. aeruginosalung infection. (A) Kaplan-Meier survival curves of WT (+/+) and serpinb1-deficient (−/−) mice intranasally inoculated with nonmucoid P. aeruginosa strain PAO1. Increased mortality of serpinb1−/− mice was statistically significant (P = 0.03 at 3 × 106CFU/mouse; P < 0.0001 at 7 × 106CFU/mouse). (B) CFUs per milligram of lung (left) and splenic (right) tissue determined 6 and 24 h after inoculation with 3 × 106 CFUP. aeruginosa PAO1 in WT (+/+, filled circles) and serpinb1−/− (−/−, open circles) mice. Each symbol represents a value for an individual mouse. Differences between median values (horizontal lines) were analyzed by the Mann-Whitney U test. Data below the limit of detection (dotted line) are plotted as 0.5 CFU × dilution factor. (C) Lung sections stained with hematoxylin and eosin show bacterial colonies (arrowheads) and alveolar exudate in lungs of serpinb1−/− mice 24 h after infection with P. aeruginosa PAO1. Bars, 50 μm. (D) Total CFUs in the lung and spleen 24 h after inoculation with 2 × 108 CFU of the mucoid P. aeruginosa strain PA M57-15 in WT (+/+, filled circles) and serpinb1−/− (−/−, open circles) mice. Differences between median values (horizontal lines) were analyzed by the Mann-Whitney U test.
To verify specificity of the gene deletion, we tested whether delivering rSERPINB1 would correct the defective phenotype. Indeed, intranasal instillation of rSERPINB1 to serpinb1−/− mice at the time of inoculation significantly improved clearance of P. aeruginosa PAO1 from the lungs assessed at 24 h and reduced bacteremia compared with infectedserpinb1−/− mice that received PBS instead of the recombinant protein (Fig. S1 A, available at http://www.jem.org/cgi/content/full/jem.20070494/DC1). We have previously demonstrated that rSERPINB1 has no effect on the growth of P. aeruginosa in vitro (15) and does not induce bacterial aggrega tion (16). Also, rSERPINB1 mixed with PAO1 had no effect on adherence of the bacteria to human bronchial epithelial and corneal epithelial cell lines (unpublished data). Therefore, the improved bacterial clearance in treated serpinb1−/− mice is not related to a direct antibacterial role for rSERPINB1 but rather to reducing injury induced by excess neutrophil proteases. In addition, previous in vivo studies in WT rats showed that rSERPINB1 can protect against elastase-induced lung injury (17) and accelerate bacterial clearance two- to threefold in the Pseudomonas agar bead model (15).
Evidence of excess NSP action was examined in the lungs of infected serpinb1−/− mice by measuring surfactant protein–D (SP-D). SP-D, a multimeric collagenous C-type lectin produced by alveolar epithelial cells, is highly relevant as a host defense molecule, because it functions as an opsonin in microbial clearance (18) and acts on alveolar macrophages to regulate pro- and antiinflammatory cytokine production (19). SP-D is also relevant as an NSP target because it is degraded in vitro by trace levels of each of the NSPs (16, 20). SP-D levels in lung homogenates of WT and serpinb1−/− mice were similar 6 h after P. aeruginosa infection. At 24 h, SP-D levels were reduced in the lungs ofserpinb1−/− mice compared with WT mice, as indicated by immunoblots. A lower molecular mass band indicative of proteolytic degradation is also apparent (Fig. 3 A). Densitometry analysis of the 43-kD SP-D band relative to β-actin indicated that the reduction of SP-D level was statistically significant (+/+, 45 ± 6 [n = 8]; −/−, 10 ± 2 [n = 8]; P < 0.0001 according to the Student’s t test). Furthermore, rSERPINB1 treatment ofP. aeruginosa–infected serpinb1−/− mice partly prevented the degradation of SP-D in lung homogenates compared with nontreated mice (Fig. S1 B). As a further test of the impact of serpinb1 deletion on NSP activity, isolated neutrophils of serpinb1−/− mice were treated with LPS and FMLP and tested for their ability to cleave recombinant rat SP-D (rrSP-D) in vitro. The extent of rrSP-D cleavage by serpinb1−/− neutrophils was fourfold greater than by WT neutrophils, as determined by densitometry. The cleavage was specific for NSPs because it was abrogated by rSERPINB1 and diisopropyl fluorophosphate (Fig. 3 B). Collectively, these findings indicate a direct role for serpinb1 in regulating NSP activity released by neutrophils and in preserving SP-D, an important-host defense molecule.
Efficient clearance of P. aeruginosa infection requires an early cytokine and chemokine response coordinated by both resident alveolar macrophages and lung parenchymal cells (21, 22). The IL-8 homologue keratinocyte-derived chemokine (KC) and the cytokines TNF-α, IL-1β, and G-CSF were measured in cell-free bronchoalveolar (BAL) samples. Although the tested cytokines were undetectable in sham-infected mice of both genotypes (unpublished data), comparable induc tion of these cytokines was observed in BAL of WT and serpinb1−/− mice at 6 h after infection, demonstrating that there is no early defect in cytokine production in serpinb1−/− mice. At 24 h, levels of TNF-α, KC, and IL-1β were sustained or increased in serpinb1−/− mice and significantly higher than cytokine levels in WT mice. G-CSF levels at 24 h were elevated to a similar extent in BAL of WT and KO mice (Fig. 3 C). However, G-CSF levels were significantly higher in the serum of serpinb1−/− mice (WT, 336 ± 80 ng/ml; KO, 601 ± 13 ng/ml; n = 6 of each genotype; P < 0.01). In addition, serpinb1−/− mice that were treated at the time of infection with rSERPINB1 had cytokine levels in 24-h lung homogenates that were indistinguishable from those of infected WT mice (Fig. S1 C). The increased cytokine production in the lungs of infected serpinb1−/− mice may be caused by failed bacterial clearance but also by excess NSPs, which directly induce cytokine and neutrophil chemokine production in pulmonary parenchymal cells and alveolar macrophages (23, 24).
Neutrophil recruitment to the lungs was next examined as a pivotal event of the response to P. aeruginosa infection (25). Lung homogenates were assayed for the neutrophil-specific enzyme myeloperoxidase (MPO) to quantify marginating, interstitial, and alveolar neutrophils. Neutrophils in BAL fluid were directly counted as a measure of neutrophil accumulation in the alveolar and airway lumen. MPO in lung homo genates was undetectable in uninfected mice and was comparably increased in mice of both genotypes at 6 h, suggesting normal early serpinb1−/− neutrophil margination and migration into the interstitium. However, by 24 h after infection, MPO levels in lung homogenates remained high in WT mice but were significantly decreased in serpinb1−/− mice (Fig. 4 A). Importantly, the content of MPO per cell was the same for isolated neutrophils of WT andserpinb1−/− mice (+/+, 369 ± 33 mU/106 cells; −/−, 396 ± 27 mU/106 cells). The numbers of neutrophils in BAL were negligible in uninfected mice and were similarly increased in WT and serpinb1−/− mice at 6 h after infection. Neutrophil counts in BAL further increased at 24 h, but the mean BAL neutrophil numbers were significantly lower in serpinb1−/− mice compared with WT mice (Fig. 4 B). The evidence from the 6-h quantitation of MPO in homogenates and neutrophils in BAL strongly suggests that neutrophil recruitment is not defective in infected serpinb1−/− mice. Moreover, the high levels of cytokines and neutrophil chemoattractant KC in serpinb1−/− mice at 24 h (Fig. 3 C) also suggest that, potentially, more neutrophils should be recruited. Therefore, to examine neutrophil recruitment in serpinb1−/− mice, we used a noninfectious model in which neutrophils are mobilized to migrate to the lung after intranasal delivery of P. aeruginosa LPS. MPO levels in lung homogenate and neutrophil numbers in BAL were not statistically different in WT and serpinb1−/− mice 24 h after LPS instillation (Fig. 4, C and D). Furthermore, the number of circulating blood neutrophils and recruited peritoneal neutrophils after injection of sterile irritants glycogen and thioglycollate did not differ in WT and serpinb1−/− mice (unpublished data). Alveolar macrophage numbers were similar in uninfected mice of both genotypes (∼5 × 105 cells/mouse) and did not substantially change upon infection. Collectively, these findings show that neutrophil recruitment to the lungs in response to P. aeruginosa infection is not defective in serpinb1−/− mice, and therefore, the recovery of lower numbers of serpinb1−/− neutrophils at 24 h after infection suggests their decreased survival.
To examine the putative increased death of serpinb1−/− neutrophils in the lungs after P. aeruginosa infection, lung sections were analyzed by immunohistochemistry. Caspase-3–positive leukocytes were more relevant in the alveolar space of serpinb1−/− mice compared with WT mice at 24 h after infection, suggesting increased neutrophil apoptosis (Fig. 5 A). The positive cells were counted in 50 high power fields (hpf’s), and mean numbers of caspase-3–stained cells were increased in the lungs of serpinb1−/− mice (1.8 ± 0.2 cells/hpf) compared with WT mice (0.4 ± 0.1 cells/hpf; P < 0.0001). To characterize neutrophils in the alveoli and airways, neutrophils in BAL were identified in flow cytometry by forward scatter (FSC) and side scatter and were stained with annexin V (AnV) and propidium iodide (PI). At 24 h after infection, the proportion of late apoptotic/necrotic neutrophils (AnV+PI+) was increased at the expense of viable neutrophils (AnV−PI−) in the BAL of serpinb1−/− mice compared with WT mice (Fig. 5 B). Neutrophil fragments in BAL were also identified in flow cytometry by low FSC (FSClow) within the neutrophil population defined by the neutrophil marker Gr-1. The number of neutrophil fragments (FSClow, Gr-1+) relative to intact neutrophils was increased two- to threefold at 24 h after infection for serpinb1−/− compared with WT mice (Fig. 5 C). Moreover, free MPO in BAL supernatants was increased in serpinb1−/− mice compared with WT mice at 24 h after infection, indicating increased PMN lysis or degranulation (Fig. 5 D).
Finally, we questioned whether the enhanced death of serpinb1−/− pulmonary neutrophils was a primary effect of gene deletion or a secondary effect caused by, for example, bacteria or components of inflammation. To address this, neutrophils were collected using the noninfectious LPS recruitment model and were cultured in vitro to allow for spontaneous cell death. After 24 h, the percentages of apoptotic and necrotic neutrophils evaluated by microscopy were increased in serpinb1−/− neutrophils compared with WT neutrophils (Fig. 6, A–C). A similar increase in apoptotic cells was observed using AnV/PI staining and measurements of hypodiploid DNA (unpublished data). Moreover, live cell numbers from serpinb1−/− mice remaining in culture after 24 h were significantly decreased compared with WT mice (Fig. 6 D). The in vitro findings indicate that enhanced death of pulmonary neutrophils of infected serpinb1−/− mice is at least in part a cell-autonomous defect likely mediated by unchecked NSP actions.
In this paper, we have demonstrated that serpinb1, an intracellular serpin family member, regulates the innate immune response and protects the host during lung bacterial infection. Serpinb1 is among the most potent inhibitors of NSPs and is carried at high levels within neutrophils. Serpinb1-deficient mice fail to clear P. aeruginosa PAO1 lung infection and succumb from systemic bacterial spreading. The defective immune function in serpinb1−/− mice stems at least in part from an increased rate of neutrophil necrosis, reducing the number of phagocytes and leading to increased NSP activity in the lungs with proteolysis of SP-D. In addition, serpinb1-deficient mice also have impaired clearance of the mucoid clinical strain PA M57-15. Interestingly, mucoid strains of P. aeruginosa are cleared with a very high efficiency from the lungs of WT and cystic fibrosis transmembrane conductance regulator–deficient mice (26). The phenotype of serpinb1−/− mice reproduces major pathologic features of human pulmonary diseases characterized by excessive inflammation, massive neutrophil recruitment to the air space, and destruction of cellular and molecular protective mechanisms. Importantly, serpinb1 deficiency may be helpful as an alternative or additional model of the inflammatory lung pathology of cystic fibrosis.
The present study documents a key protective role for serpinb1 in regulating NSP actions in the lung. This role has previously been attributed to the NSP inhibitors α1-antitrypsin and secretory leukocyte protease inhibitor, which are found in the airway and alveolar lining fluid (27, 28). However, patients with α1-antitrypsin deficiency do not present with pulmonary infection secondary to innate immune defects despite increased NSP activity that leads to reduced lung elasticity and emphysema. Moreover, there is so far no evidence that deficiency in secretory leukocyte protease inhibitor results in failure to clear pulmonary infection. Because synthesis and storage of NSPs in granules is an event that exclusively takes place in bone marrow promyelocytes (29), the regulation of NSPs in the lung relies entirely on NSP inhibitors. Thus, the extent of the innate immune defect inserpinb1−/− mice and the normalization of bacterial clearance with topical rSERPINB1 treatment indicate that serpinb1 is required to regulate NSP activity in the airway fluids and that, during acute lung infection associated with high neutrophilic recruitment, there is insufficient compensation by other NSP inhibitors. The devastating effects of NSPs when released in the lungs by degranulating and necrotic neutrophils are well documented in human pulmonary diseases (5, 6, 30). Therefore, our findings clearly establish a physiological and nonredundant role for serpinb1 in regulating NSPs during pulmonary infection.
NSPs also cleave molecules involved in apoptotic cell clearance, including the surfactant protein SP-D and the phosphatidylserine receptor on macrophages (31, 32), thereby tipping the balance further toward a detrimental outcome. The increased numbers of leukocytes with active caspase-3 in the alveolar space of P. aeruginosa–infectedserpinb1−/− mice suggest that the removal of apoptotic cells may be inadequate during infection. SP-D has been shown to stimulate phagocytosis of P. aeruginosa by alveolar macrophages in vitro (33), and SP-D–deficient mice were found to have defective early (6-h) clearance of P. aeruginosa from the lung (34). Although the destruction of SP-D alone may not entirely account for the defective phenotype of serpinb1−/− mice, loss of SP-D likely diminishes bacterial clearance and removal of apop totic neutrophils.
Given that NSPs also mediate bacterial killing, why would NSP excess lead to a failed bacterial clearance? In the NE KO mice, the decreased killing activity of neutrophils is a direct consequence of the loss of the bactericidal activity of NE. The absence of an early bacterial clearance defect at 6 h after infection in serpinb1−/− mice suggests that there is initially normal bacterial killing. The current understanding is that the compartmentalization of the NSPs is crucial to the outcome of their actions: on the one hand, NSPs are protective when killing microbes within phagosomes, and on the other hand, extracellular NSPs destroy innate immune defense molecules such as lung collectins, immunoglobulins, and complement receptors. We have shown that the regulation of NSP activity is essential and that cytoplasmic serpinb1 provides this crucial shield. Neutrophils undergoing cell death gradually transition from apoptosis, characterized by a nonpermeable plasma membrane, to necrosis and lysis, where cellular and granule contents, including NSPs, are released. The increased pace of serpinb1−/− neutrophil cell death strongly suggests that unopposed NSPs may precipitate neutrophil demise and, therefore, reduce the neutrophil numbers leading to a late-onset innate immune defect. High levels of G-CSF, a prosurvival cytokine for neutrophils, also indicate that increased cell death is likely independent or downstream of G-CSF.
In conclusion, serpinb1 deficiency unleashes unbridled proteolytic activity during inflammation and thereby disables two critical components of the host response to bacterial infection, the neutrophil and the collectin SP-D. The phenotype of the infectedserpinb1-deficient mouse, characterized by a normal early antibacterial response that degenerates over time, highlights the delicate balance of protease–antiprotease systems that protect the host against its own defenses as well as invading microbes during infection-induced inflammation.
Proteinase 3 and neutrophil elastase enhance inflammation in mice by inactivating antiinflammatory progranulin
Neutrophil granulocytes form the body’s first line of antibacterial defense, but they also contribute to tissue injury and noninfectious, chronic inflammation. Proteinase 3 (PR3) and neutrophil elastase (NE) are 2 abundant neutrophil serine proteases implicated in antimicrobial defense with overlapping and potentially redundant substrate specificity. Here, we unraveled a cooperative role for PR3 and NE in neutrophil activation and noninfectious inflammation in vivo, which we believe to be novel. Mice lacking both PR3 and NE demonstrated strongly diminished immune complex–mediated (IC-mediated) neutrophil infiltration in vivo as well as reduced activation of isolated neutrophils by ICs in vitro. In contrast, in mice lacking just NE, neutrophil recruitment to ICs was only marginally impaired. The defects in mice lacking both PR3 and NE were directly linked to the accumulation of antiinflammatory progranulin (PGRN). Both PR3 and NE cleaved PGRN in vitro and during neutrophil activation and inflammation in vivo. Local administration of recombinant PGRN potently inhibited neutrophilic inflammation in vivo, demonstrating that PGRN represents a crucial inflammation-suppressing mediator. We conclude that PR3 and NE enhance neutrophil-dependent inflammation by eliminating the local antiinflammatory activity of PGRN. Our results support the use of serine protease inhibitors as antiinflammatory agents.
Neutrophils belong to the body’s first line of cellular defense and respond quickly to tissue injury and invading microorganisms (1). In a variety of human diseases, like autoimmune disorders, infections, or hypersensitivity reactions, the underlying pathogenic mechanism is the formation of antigen-antibody complexes, so-called immune complexes (ICs), which trigger an inflammatory response by inducing the infiltration of neutrophils (2). The subsequent stimulation of neutrophils by C3b-opsonized ICs results in the generation of ROS and the release of intracellularly stored proteases leading to tissue damage and inflammation (3). It is therefore important to identify the mechanisms that control the activation of infiltrating neutrophils.
Neutrophils abundantly express a unique set of neutrophil serine proteases (NSPs), namely cathepsin G (CG), proteinase 3 (PR3; encoded by Prtn3), and neutrophil elastase (NE; encoded by Ela2), which are stored in the cytoplasmic, azurophilic granules. PR3 and NE are closely related enzymes, with overlapping and potentially redundant substrate specificities different from those of CG. All 3 NSPs are implicated in antimicrobial defense by degrading engulfed microorganisms inside the phagolysosomes of neutrophils (4–8). Among many other functions ascribed to these enzymes, PR3 and NE were also suggested to play a fundamental role in granulocyte development in the bone marrow (9–11).
While the vast majority of the enzymes is stored intracellularly, minor quantities of PR3 and NE are externalized early during neutrophil activation and remain bound to the cell surface, where they are protected against protease inhibitors (12, 13). These membrane presented proteases were suggested to act as path clearers for neutrophil migration by degrading components of the extracellular matrix (14). This notion has been addressed in a number of studies, which yielded conflicting results (15–17). Thus, the role of PR3 and NE in leukocyte extravasation and interstitial migration still remains controversial.
Emerging data suggest that externalized NSPs can contribute to inflammatory processes in a more complex way than by simple proteolytic tissue degradation (18). For instance, recent observations using mice double-deficient for CG and NE indicate that pericellular CG enhances IC-mediated neutrophil activation and inflammation by modulating integrin clustering on the neutrophil cell surface (19, 20). Because to our knowledge no Prtn3–/– mice have previously been generated, the role of this NSP in inflammatory processes has not been deciphered. Moreover, NE-dependent functions that can be compensated by PR3 in Ela2–/–animals are still elusive.
One mechanism by which NSPs could upregulate the inflammatory response has recently been proposed. The ubiquitously expressed progranulin (PGRN) is a growth factor implicated in tissue regeneration, tumorigenesis, and inflammation (21–23). PGRN was previously shown to directly inhibit adhesion-dependent neutrophil activation by suppressing the production of ROS and the release of neutrophil proteases in vitro (23). This antiinflammatory activity was degraded by NE-mediated proteolysis of PGRN to granulin (GRN) peptides (23). In contrast, GRN peptides may enhance inflammation (23) and have been detected in neutrophil-rich peritoneal exudates (24). In short, recent studies proposed PGRN as a regulator of the innate immune response, but the factors that control PGRN function are still poorly defined and its relevance to inflammation needs to be elucidated in vivo.
In the present study, we generated double-deficient Prtn3–/–Ela2–/– mice to investigate the role of these highly similar serine proteases in noninfectious neutrophilic inflammation. We established that PR3 and NE are required for acute inflammation in response to subcutaneous IC formation. The proteases were found to be directly involved in early neutrophil activation events, because isolated Prtn3–/–Ela2–/– neutrophils were poorly activated by ICs in vitro. These defects in Prtn3–/–Ela2–/– mice were accompanied by accumulation of PGRN. We demonstrated that PGRN represents a potent inflammation-suppressing factor that is cleaved by both PR3 and NE. Our data delineate what we believe to be a previously unknown proinflammatory role for PR3 and NE, which is accomplished via the local inactivation of antiinflammatory PGRN.
Generation of Prtn3–/–Ela2–/– mice.
To analyze the role of PR3 and NE in neutrophilic inflammation, we generated a Prtn3–/–Ela2–/– mouse line by targeted gene disruption in embryonic stem cells (see Supplemental Figure 1; supplemental material available online with this article; doi: 10.1172/JCI34694DS1). Positive recombination of the Prtn3/Ela2locus was proven by Southern blotting of embryonic stem cell clones (Figure (Figure1A).1A). Prtn3–/–Ela2–/– mice showed no expression of mRNA for PR3 and NE in bone marrow cells, as assessed by RT-PCR (Figure (Figure1B).1B). The successful elimination of PR3 and NE was confirmed at the level of proteolytic activity in neutrophil lysates using a PR3/NE-specific chromogenic substrate (Supplemental Figure 3) as well as by casein zymography (Figure (Figure1C).1C). The substantially reduced casein degradation by heterozygous neutrophils indicates gene-dosage dependence of PR3/NE activities. Furthermore, PR3 and NE deficiency was proven by Western blotting using cell lysates from bone marrow–derived neutrophils, while other enzymes stored in azurophilic granula, such as CG and myeloperoxidase (MPO), were normally detected (Figure (Figure1D).1D). Crossing of heterozygous Prtn3+/–Ela2+/– mice resulted in regular offspring of WT, heterozygous, and homozygous genotype according to the Mendelian ratio. Despite the absence of 2 abundant serine proteases, and in contrast to expectations based on previous reports (9–11), we found unchanged neutrophil morphology (Figure (Figure1E)1E) and regular neutrophil populations in the peripheral blood of the mutant mice, the latter as assessed via flow cytometry to determine the differentiation markers CD11b and Gr-1 (Figure (Figure1F)1F) (25, 26). Moreover, Prtn3–/–Ela2–/– mice demonstrated normal percentages of the leukocyte subpopulations in the peripheral blood, as determined by the Diff-Quick staining protocol and by hemocytometric counting (Supplemental Figure 2, A and B). Hence, the proteases are not crucially involved in granulopoiesis, and ablating PR3 and NE in the germ line represents a valid approach to assess their biological significance in vivo.
Generation and characterization of Prtn3–/–Ela2–/– mice.
PR3 and NE are dispensable for neutrophil extravasation and interstitial migration.
To examine neutrophil infiltration into the perivascular tissue, we applied phorbol esters (croton oil) to the mouse ears. At 4 h after stimulation, we assessed the neutrophil distribution in relation to the extravascular basement membrane (EBM) by immunofluorescence microscopy of fixed whole-mount specimens (Figure (Figure2A).2A). We found that Prtn3–/–Ela2–/– neutrophils transmigrated into the interstitium without retention at the EBM (Figure (Figure2B),2B), resulting in quantitatively normal and widespread neutrophil influx compared with WT mice (Figure (Figure2C).2C). Moreover, we analyzed chemotactic migration of isolated neutrophils through a 3-dimensional collagen meshwork in vitro (Supplemental Video 1) and found unhampered chemotaxis toward a C5a gradient, based on the directionality (Figure (Figure2D)2D) and velocity (Figure (Figure2E)2E) of Prtn3–/–Ela2–/–neutrophils. These findings led us to conclude that PR3 and NE are not principally required for neutrophil extravasation or interstitial migration.
PR3 and NE are not principally required for neutrophil extravasation and interstitial migration.
Reduced inflammatory response to ICs in Prtn3–/–Ela2–/– mice.
The formation of ICs represents an important trigger of neutrophil-dependent inflammation in many human diseases (2). To determine the role of PR3 and NE in this context, we induced a classic model of subcutaneous IC-mediated inflammation, namely the reverse passive Arthus reaction (RPA) (27). At 4 h after RPA induction, we assessed the cellular inflammatory infiltrates by histology using H&E-stained skin sections (Figure (Figure3A).3A). Neutrophils, which were additionally identified by Gr-1 immunohistochemistry, made up the vast majority of all cellular infiltrates (Figure (Figure3A).3A). We found that neutrophil infiltration to the sites of IC formation was severely diminished in Prtn3–/–Ela2–/– mice. Indeed, histological quantification revealed significantly reduced neutrophil influx in Prtn3–/–Ela2–/– mice compared with WT mice, while Ela2–/– mice showed marginally reduced neutrophil counts (Figure (Figure3B).3B). These results indicate that PR3 and NE fulfill an important proinflammatory function during IC-mediated inflammation.
Impaired inflammatory response to locally formed ICs inPrtn3–/–Ela2–/– mice.
(A) Representative photomicrographs of inflamed skin sections 4 h after IC formation. Neutrophils were identified morphologically (polymorphic nucleus) in H&E stainings and by Gr-1 staining (red). The cellular infiltrates were located to the adipose tissue next to the panniculus carnosus muscle (asterisks) and were primarily composed of neutrophil granulocytes. Scale bars: 200 μm. (B) Neutrophil infiltrates in lesions from Prtn3–/–Ela2–/– mice were significantly diminished compared with Ela2–/– mice and WT mice. Neutrophil influx in Ela2–/–mice was slightly, but not significantly, diminished compared with WT mice. Results are mean ± SEM infiltrated neutrophils per HPF. *P < 0.05.
PR3 and NE enhance neutrophil activation by ICs in vitro.
PR3 and NE enhance neutrophil activation by ICs in vitro.
Because PR3 and NE were required for the inflammatory response to IC (Figure (Figure3),3), but not to phorbol esters (Figure (Figure2),2), we considered the enzymes as enhancers of the neutrophil response to IC. We therefore assessed the oxidative burst using dihydrorhodamine as a readout for cellular activation of isolated, TNF-α–primed neutrophils in the presence of ICs in vitro. While both WT and Prtn3–/–Ela2–/– neutrophils showed a similar, approximately 20-min lag phase before the oxidative burst commenced, the ROS production over time was markedly reduced, by 30%–40%, in the absence of PR3 and NE (Figure (Figure4A).4A). In contrast, oxidative burst triggered by 25 nM PMA was not hindered in Prtn3–/–Ela2–/– neutrophils (Figure (Figure4B),4B), which indicated no general defect in producing ROS. We also performed a titration series ranging from 0.1 to 50 nM PMA and found no reduction in oxidative burst activity in Prtn3–/–Ela2–/– neutrophils at any PMA concentration used (Supplemental Figure 4). These data are consistent with our in vivo experiments showing that neutrophil influx to ICs was impaired (Figure (Figure3),3), whereas the inflammatory response to phorbol esters was normal (Figure (Figure2,2, A–C), in Prtn3–/–Ela2–/– mice. To compare neutrophil priming in WT and Prtn3–/–Ela2–/–neutrophils, we analyzed cell surface expression of CD11b after 30 min of incubation at various concentrations of TNF-α and found no difference (Supplemental Figure 5). Moreover, we observed normal neutrophil adhesion to IC-coated surfaces (Supplemental Figure 6A) and unaltered phagocytosis of opsonized, fluorescently labeled E. coli bacteria (Supplemental Figure 6, B and C) in the absence of both proteases. We therefore hypothesized that PR3 and NE enhance early events of adhesion-dependent neutrophil activation after TNF-α priming and binding of ICs. It is important to note that Ela2–/– neutrophils were previously shown to react normally in the same setup (20). Regarding the highly similar cleavage specificities of both proteases, we suggested that PR3 and NE complemented each other during the process of neutrophil activation and inflammation.
Impaired oxidative burst and PGRN degradation by IC-activatedPrtn3–/–Ela2–/– neutrophils.
Oxidative burst as the readout for neutrophil activation by ICs was measured over time. (A) While no difference was observed during the initial 20-min lag phase of the oxidative burst, Prtn3–/–Ela2–/– neutrophils exhibited diminished ROS production over time compared with WT neutrophils. (B) Bypassing receptor-mediated activation using 25 nM PMA restored the diminished oxidative burst of Prtn3–/–Ela2–/–neutrophils. Results are presented as normalized fluorescence in AU (relative to maximum fluorescence produced by WT cells). Data (mean ± SD) are representative of 3 independent experiments each conducted in triplicate. (C) Isolated mouse neutrophils were activated by ICs in vitro and tested for PGRN degradation by IB. In the cellular fraction, the PGRN (~80 kDa) signal was markedly increased in Prtn3–/–Ela2–/–cells compared with WT and Ela2–/– neutrophils. Intact PGRN was present in the supernatant (SN) of IC-activated Prtn3–/–Ela2–/–neutrophils only, not of WT or Ela2–/– cells. (D and E) Exogenous administration of 100 nM PGRN significantly reduced ROS production of neutrophils activated by ICs (D), but not when activated by PMA (E). Data (mean ± SD) are representative of 3 independent experiments each conducted in triplicate.
Antiinflammatory PGRN is degraded by PR3 and NE during IC-mediated neutrophil activation.
PGRN inhibits neutrophil activation by ICs in vitro.
PGRN is a potent inhibitor of IC-stimulated inflammation in vivo.
PR3 and NE cleave PGRN during inflammation in vivo.
Finally, we aimed to demonstrate defective PGRN degradation in Prtn3–/–Ela2–/– mice during neutrophilic inflammation in vivo. For practical reasons, we harvested infiltrated neutrophils from the inflamed peritoneum 4 h after casein injection and subjected the lysates of these cells to anti-PGRN Western blot. Intact, inhibitory PGRN was detected in Prtn3–/–Ela2–/– neutrophils, but not in WT cells (Figure (Figure6D).6D). These data prove that neutrophilic inflammation is accompanied by proteolytic removal of antiinflammatory PGRN and that the process of PGRN degradation is essentially impaired in vivo in the absence of PR3 and NE.
Chronic inflammatory and autoimmune diseases are often perpetuated by continuous neutrophil infiltration and activation. According to the current view, the role of NSPs in these diseases is mainly associated with proteolytic tissue degradation after their release from activated or dying neutrophils. However, recent observations suggest that NSPs such as CG may contribute to noninfectious diseases in a more complex manner, namely as specific regulators of inflammation (18). Here, we demonstrate that PR3 and NE cooperatively fulfilled an important proinflammatory role during neutrophilic inflammation. PR3 and NE directly enhanced neutrophil activation by degrading oxidative burst–suppressing PGRN. These findings support the use of specific serine protease inhibitors as antiinflammatory agents.
Much attention has been paid to the degradation of extracellular matrix components by NSPs. We therefore expected that ablation of both PR3 and NE would cause impaired neutrophil extravasation and interstitial migration. Surprisingly, we found that the proteases were principally dispensable for these processes:Prtn3–/–Ela2–/– neutrophils migrated normally through a dense, 3-dimensional collagen matrix in vitro and demonstrated regular extravasation in vivo when phorbol esters were applied (Figure (Figure2).2). This finding is in agreement with recent reports that neutrophils preferentially and readily cross the EBM through regions of low matrix density in the absence of NE (28).
Conversely, we observed that PR3 and NE were required for the inflammatory response to locally formed ICs (Figure (Figure3).3). Even isolated Prtn3–/–Ela2–/– neutrophils were challenged in performing oxidative burst after IC stimulation in vitro (Figure (Figure4A),4A), showing that the proteases directly enhanced the activation of neutrophils also in the absence of extracellular matrix. However, when receptor-mediated signal transduction was bypassed by means of PMA, neutrophils from Prtn3–/–Ela2–/– mice performed normal oxidative burst (Figure (Figure4B),4B), indicating that the function of the phagocyte oxidase (phox) complex was not altered in the absence of PR3 and NE. These findings substantiate what we believe to be a novel paradigm: that all 3 serine proteases of azurophilic granules (CG, PR3, and NE), after their release in response to IC encounter, potentiate a positive autocrine feedback on neutrophil activation.
In contrast to CG, the highly related proteases PR3 and NE cooperate in the effacement of antiinflammatory PGRN, leading to enhanced neutrophil activation. Previous studies already demonstrated that PGRN is a potent inhibitor of the adhesion-dependent oxidative burst of neutrophils in vitro, which can be degraded by NE (23). Here, we showed that PR3 and NE play an equally important role in the regulation of PGRN function. Ela2–/– neutrophils were sufficiently able to degrade PGRN. Only in the absence of both PR3 and NE was PGRN degradation substantially impaired, resulting in the accumulation of antiinflammatory PGRN during neutrophil activation in vitro (Figure (Figure4C)4C) and neutrophilic inflammation in vivo (Figure (Figure6D).6D). Moreover, we provided in vivo evidence for the crucial role of PGRN as an inflammation-suppressing mediator, because administration of recombinant PGRN potently inhibited the neutrophil influx to sites of IC formation (Figure (Figure6,6, A–C). Hence, the cooperative degradation of PGRN by PR3 and NE is a decisive step for the establishment of neutrophilic inflammation.
The molecular mechanism of PGRN function is not yet completely understood, but it seems to interfere with integrin (CD11b/CD18) outside-in signaling by blocking the function of pyk2 and thus dampens adhesion-related oxidative burst even when added after the initial lag phase of oxidase activation (23). PGRN is produced by neutrophils and stored in highly mobile secretory granules (29). It was recently shown that PGRN can bind to heparan-sulfated proteoglycans (30), which are abundant components of the EBM and various cell surfaces, including those of neutrophils. Also, PR3 and NE are known to interact with heparan sulfates on the outer membrane of neutrophils, where the enzymes appear to be protected against protease inhibitors (12, 13, 31). These circumstantial observations support the notion that PGRN cleavage by PR3 and NE takes place at the pericellular microenvironment of the neutrophil cell surface.
Impaired outside-in signaling most likely reduced the oxidative burst in Prtn3–/–Ela2–/– neutrophils adhering to ICs. In support of this hypothesis, we excluded an altered response to TNF-α priming (Supplemental Figure 5) as well as reduced adhesion to immobilized ICs and defective endocytosis of serum-opsonized E. coli in Prtn3–/–Ela2–/– neutrophils (Supplemental Figure 6). MPO content and processing was also unchanged in Prtn3–/–Ela2–/– neutrophils (Figure (Figure1D);1D); hence, the previously discussed inhibitory effect of MPO on phox activity (32, 33) does not appear to be stronger in neutrophils lacking PR3 and NE. Because there was no difference in the lag phase of the oxidative burst, initial IC-triggered receptor activation was probably not affected by either PRGN or PR3/NE. Our concept is consistent with all these observations and takes into account that PGRN unfolds its suppressing effects in the second phase, when additional membrane receptors, endogenous PGRN, and some PR3/NE from highly mobile intracellular pools are translocated to the cell surface. The decline and cessation of ROS production suggested to us that outside-in signaling was not sustained and that active oxidase complexes were no longer replenished in the absence of PR3 and NE. Our present findings, however, do not allow us to exclude other potential mechanisms, such as accelerated disassembly of the active oxidase complex.
Proposed function of PR3 and NE in IC-mediated inflammation.
TNF-α–primed neutrophils extravasate from blood vessels, translocate PR3/NE to the cellular surface, and discharge PGRN to the pericellular environment (i). During transmigration of interstitial tissues (ii), neutrophil activation is initially suppressed by relatively high pericellular levels of antiinflammatory PGRN (green shading), which is also produced locally by keratinocytes and epithelial cells of the skin. Until IC depots are reached, neutrophil activation is inhibited by PGRN. Surface receptors (e.g., Mac-1) recognize ICs, which results in signal transduction (black dotted arrow) and activation of the phox. The molecular pathway of PGRN-mediated inhibition is not completely understood, but it may interfere with integrin signaling after IC encounter (green dotted line inside the cell). Adherence of neutrophils to ICs (iii) further increases pericellular PR3 and NE activity. PR3 and NE cooperatively degrade PGRN in the early stage of neutrophilic activation to facilitate optimal neutrophil activation (red shading), resulting in sustained integrin signaling (red arrow) and robust production of ROS by the phox system. Subsequently, neutrophils release ROS together with other proinflammatory mediators and chemotactic agents, thereby enhancing the recruitment of further neutrophils and establishing inflammation (iv). In the absence of PR3/NE, the switch from inflammation-suppressing (ii) to inflammation-enhancing (iii) conditions is substantially delayed, resulting in diminished inflammation in response to ICs (iv).
NSPs are strongly implicated as effector molecules in a large number of destructive diseases, such as emphysema or the autoimmune blistering skin disease bullous pemphigoid (14, 35–37). Normally, PR3/NE activity is tightly controlled by high plasma levels of α1-antitrypsin. This balance between proteases and protease inhibitors is disrupted in patients with genetic α1-antitrypsin deficiency, which represents a high risk factor for the development of emphysema and certain autoimmune disorders (38). The pathogenic effects of NSPs in these diseases have so far been associated with tissue destruction by the proteases after their release from dying neutrophils. Our findings showed that PR3 and NE were already involved in much earlier events of the inflammatory process, because the enzymes directly regulated cellular activation of infiltrating neutrophils by degrading inflammation-suppressing PGRN. This concept is further supported by previous studies showing increased inflammation in mice lacking serine protease inhibitors such as SERPINB1 or SLPI (39, 40). Blocking PR3/NE activity using specific inhibitors therefore represents a promising therapeutic strategy to treat chronic, noninfectious inflammation. Serine protease inhibitors as antiinflammatory agents can interfere with the disease process at 2 different stages, because they attenuate both early events of neutrophil activation and proteolytic tissue injury caused by released NSPs.
Editorial: Serine proteases, serpins, and neutropenia
Cyclic neutropenia and severe congenital neutropenia are autosomal-dominant diseases usually attributable to mutations in the gene for neutrophil elastase orELANE. Patients with these diseases are predisposed to recurrent and life-threatening infections [1]. Neutrophil elastase, the product of the ELANE gene, is a serine protease that is synthesized and packaged in the primary granules of neutrophils. These granules are formed at the promyelocytes stage of neutrophil development. Synthesis of mutant neutrophil elastase in promyelocytes triggers the unfolded protein response and a cascade of intracellular events, which culminates in death of neutrophil precursors through apoptosis [2]. This loss of cells causes the marrow abnormality often referred to as “maturation arrest” [3, 4].
Neutrophil elastase is one of the serine proteases normally inhibited by serpinB1. In this issue of JLB, Benarafa and coauthors [5] present their intriguing studies of serpinB1 expression in human myeloid cells and their extensive investigations ofSERPINB1−/− mice. They observed that serpinB1 expression parallels protease expression. The peak of serpinB1 expression occurs in promyelocytes. Benarafa et al. [5] found that SERPINB1−/− mice have a deficiency of postmitotic neutrophils in the bone marrow. This change was accompanied by an increase in the plasma levels of G-CSF. The decreased supply of marrow neutrophils reduced the number of neutrophils that could be mobilized to an inflammatory site. Using colony-forming cell assays, they determined that the early myeloid progenitor pool was intact. Separate assays showed that maturing myeloid cells were being lost through accelerated apoptosis of maturing neutrophils in the marrow. The authors concluded that serpinB1 is required for maintenance of a healthy reserve of marrow neutrophils and a normal acute immune response [5].
This paper provides new and fascinating insights for understanding the mechanism for neutropenia. It also suggests opportunities to investigate potential therapies for patients with neutropenia and prompts several questions. As inhibition of the activity of intracellular serine proteases is the only known function of serpinB1, the findings reported by Benarafa et al. [5] suggest that uninhibited serine proteases perturbed neutrophil production severely. The SERPINB1−/− mice used in their work have accelerated apoptosis of myeloid cells, a finding suggesting that uninhibited serine proteases or mutant neutrophil elastase perturb myelopoiesis by similar mechanisms. It is now important to determine whether the defect in the SERPINB1−/− mice is, indeed, attributable to uninhibited activity of normal neutrophil elastase, other neutrophil proteases, or another mechanism. ″Double-knockout″ studies in mice deficient in neutrophil elastase and serpinB1 might provide an answer.
This report provides evidence regarding the intracellular mechanisms for the apoptosis of myeloid cells and indicates that other studies are ongoing. The key antiapoptotic proteins, Mcl-1, Bcl-XL, and A1/Bfl-I, are apparently not involved. A more precise understanding of the mechanisms of cell death is important for development of targeted therapies for neutropenia. It is also important to discover whether only cells of the neutrophil lineage are involved or whether monocytes are also affected. In cyclic and congenital neutropenia, patients failed to produce neutrophils, but they can produce monocytes; in fact, they overproduce monocytes and have significantly elevated blood monocyte counts. Neutropenia with monocytosis is probably attributable to differences in the expression of ELANE in the two lineages. Benarafa et al. [5] reported that human bone marrow monocytes contain substantially less serpinB1 than marrow neutrophils, suggesting that the expression of serpinB1 and the serine proteases are closely coordinated.
This report shows the importance of the marrow neutrophil reserves in the normal response to infections. Compared with humans, healthy mice are always neutropenic, but they have a bigger marrow neutrophil reserve, and their mature neutrophils in the marrow and blood look like human band neutrophils. These differences are well known, but they are critical for considering the clinical inferences that can be made from this report. For example, although theSERPINB1−/− mice were not neutropenic, human SERPINB1−/− might cause neutropenia because of physiological differences between the species. If some but not all mutations in SERPINB1 cause neutropenia, we might gain a better understanding about how serpinB1 normally inhibits the neutrophil’s serine proteases.
We do not know if some or all of the mutant neutrophil elastases can be inhibited by serpinB1. We do not know whether cyclic or congenital neutropenia are attributable to defects in this interaction. However, we do know that there are chemical inhibitors of neutrophil elastase that can abrogate apoptosis of myeloid cells in a cellular model for congenital neutropenia [6]. It would be interesting to see if these chemical inhibitors can replace the natural inhibitor and normalize neutrophil production in the SERPINB1−/− mice. This would provide evidence to support use of chemical protease inhibitors as a treatment for cyclic and congenital neutropenia.
Concerns with the use of G-CSF for the treatment of cyclic and congenital neutropenia are how and why some of these patients are at risk of developing leukemia. Are the SERPINB1−/− mice with a hyperproliferative marrow and high G-CSF levels also at risk of developing myeloid leukemia?
This is a very provocative paper, and much will be learned from further studies of the SERPINB1−/− mice.
SerpinB1 is critical for neutrophil survival through cell-autonomous inhibition of cathepsin G
Mathias Baumann1,2, Christine T. N. Pham3, and Charaf Benarafa1
Serine protease inhibitor serpinB1 protects neutrophils by inhibition of their own azurophil granule protease cathepsin G.
Granule permeabilization in neutrophils leads to cathepsin G–mediated death upstream and independent of apoptotic caspases.
Abstract
Bone marrow (BM) holds a large reserve of polymorphonuclear neutrophils (PMNs) that are rapidly mobilized to the circulation and tissues in response to danger signals. SerpinB1 is a potent inhibitor of neutrophil serine proteases neutrophil elastase (NE) and cathepsin G (CG). SerpinB1 deficiency (sB1−/−) results in a severe reduction of the BM PMN reserve and failure to clear bacterial infection. Using BM chimera, we found that serpinB1 deficiency in BM cells was necessary and sufficient to reproduce the BM neutropenia ofsB1−/− mice. Moreover, we showed that genetic deletion of CG, but not NE, fully rescued the BM neutropenia in sB1−/− mice. In mixed BM chimera and in vitro survival studies, we showed that CG modulates sB1−/− PMN survival through a cell-intrinsic pathway. In addition, membrane permeabilization by lysosomotropic agent L-leucyl-L-leucine methyl ester that allows cytosolic release of granule contents was sufficient to induce rapid PMN death through a CG-dependent pathway. CG-mediated PMN cytotoxicity was only partly blocked by caspase inhibition, suggesting that CG cleaves a distinct set of targets during apoptosis. In conclusion, we have unveiled a new cytotoxic function for the serine protease CG and showed that serpinB1 is critical for maintaining PMN survival by antagonizing intracellular CG activity.
Introduction
Polymorphonuclear neutrophil (PMN) granulocytes are essential components of the innate immune response to infection. PMNs are relatively short-lived leukocytes that originate from hematopoietic stem cells in the bone marrow (BM) in a process called granulopoiesis. Granulopoiesis proceeds through a proliferative phase followed by a maturation phase. After maturation, the BM retains a large reserve of mature PMNs, which includes over 90% of the mature PMNs in the body while only a small proportion (1%-5%) is in the blood.1,2 Even in noninflammatory conditions, granulopoiesis is remarkable as >1011 PMNs are produced daily in an adult human, only to be disposed of, largely unused, a few hours later.3 There is evidence that the majority of PMNs produced never reach circulation and die within the BM.4 Congenital or acquired forms of neutropenia are associated with the highest risks of bacterial and fungal infection,5 indicating a strong evolutionary pressure to maintain granulopoiesis at high levels and sustain a large mobilizable pool of PMNs in the BM.
In steady state, PMNs die by apoptosis, a form of programmed cell death that allows for the safe disposal of aging PMNs and their potentially toxic cargo. Like in other cells, caspases participate in the initiation, amplification, and execution steps of apoptosis in PMNs.6,7 Interestingly, noncaspase cysteine proteases calpain and cathepsin D were reported to induce PMN apoptosis through activation of caspases.8⇓⇓–11 In addition, PMNs carry a unique set of serine proteases (neutrophil serine proteases [NSPs]) including elastase (NE), cathepsin G (CG), and proteinase-3 (PR3) stored active in primary granules. There is strong evidence for a role of NSPs in killing pathogens and inducing tissue injury when released extracellularly.12⇓–14 In contrast, the function of NSPs in PMN homeostasis and cell death remains elusive. In particular, no defects in granulopoiesis or PMN homeostasis have been reported in mice deficient in cathepsin G (CG−/−),15 neutrophil elastase (NE−/−),16,17 or dipeptidylpeptidase I (DPPI−/−), which lack active NSPs.18 We have recently shown that mice lacking the serine protease inhibitor serpinB1 (sB1−/−) have reduced PMN survival in the lungs following Pseudomonas infection and that these mice have a profound reduction in mature PMN numbers in the BM.19,20SerpinB1, also known as monocyte NE inhibitor, is expressed at high levels in the cytoplasm of PMNs and is one of the most potent inhibitors of NE, CG, and PR3.21,22 In this study, we tested the hypothesis that serpinB1 promotes PMN survival by inhibiting 1 or several NSPs, and we discovered a novel regulatory pathway in PMN homeostasis in vivo.
Defective PMN reserve in BM chimera depends on serpinB1 deficiency in the hematopoietic compartment. Flow cytometry analysis of major BM leukocyte subsets of lethally irradiated mice was performed 8 to 10 weeks after BM transfer. (A) Irradiated WT (CD45.1) mice were transferred with WT (●) or sB1−/− (○) BM cells. (B) Irradiated WT (●) andsB1−/− (○) mice both CD45.2 were transferred with WT (CD45.1) BM cells. Each circle represents leukocyte numbers for 1 mouse and horizontal line indicates the median. Median subsets numbers were compared by the Mann-Whitney test (*P < .05; ***P < .001).
CG regulates neutrophil numbers in the BM
Because serpinB1 is an efficient inhibitor of NE, CG, and PR3, we then examined PMN numbers in mice deficient in 1 or several NSPs in combination with serpinB1 deletion. As expected, sB1−/− mice had significantly reduced numbers and percentage of mature PMNs in the BM compared with WT and heterozygous sB1+/− mice. In addition, PMN numbers were normal in mice deficient in either DPPI, NE, or CG (Figure 2A). DPPI is not inhibited by serpinB1 but is required for the activation of all NSPs, and no NSP activity is detectable in DPPI−/− mice.18,23 PMN counts in DPPI−/−.sB1−/− BM were significantly higher than in sB1−/− BM, suggesting that 1 or several NSPs contribute to the PMN survival defect. To examine the role of NSPs in this process, we crossed several NSP-deficient strains with sB1−/− mice. We found that NE.CG.sB1−/− mice had normal PMN numbers indicating that these NSPs play a key role in the defective phenotype of sB1−/− PMNs (Figure 2A). Furthermore, CG.sB1−/− mice showed normal PMN numbers whereasNE.sB1−/− mice retained the BM neutropenia phenotype indicating that CG, but not NE, plays a significant role in the death of sB1−/− PMNs (Figure 2A). In addition, the double-deficient NE.sB1−/− mice had significantly lower BM myelocyte numbers than sB1−/− mice while the myelocyte numbers in singly deficient NE−/− and sB1−/− BM were normal (Figure 2B). These results suggest that NE may promote myeloid cell proliferation, an activity that is revealed only when serpinB1 is absent. This complex interaction between sB1 and NE requires further investigation. On the other hand, B-cell and monocyte numbers and relative percentage in the BM were largely similar in all genotypes (supplemental Figure 2). Total numbers of blood leukocytes, erythrocytes, and platelets were normal in mice deficient in NSPs and/or serpinB1 (supplemental Figure 3). PMN numbers in blood were normal insB1−/− mice in steady state and combined deficiency of NSPs did not significantly alter these numbers (Figure 2C). Taken together, our results indicate that serpinB1 likely sustains the survival of postmitotic PMNs through its interaction with CG.
Figure 2
PMN and myelocyte numbers in BM and blood of mice deficient in NSPs and serpinB1.
Granule membrane permeabilization induces CG-mediated death in PMNs
To test whether granule disruption contributes to the serpinB1-regulated CG-dependent cell death, BM cells were treated with the lysosomotropic agent LLME. LLME accumulates in lysosomes where the acyl transferase activity of DPPI generates hydrophobic (Leu-Leu)n-OMe polymers that induce lysosomal membrane permeabilization (LMP) and cytotoxicity in granule-bearing cells such as cytotoxic T lymphocytes, NK cells, and myeloid cells.29,30
G-CSF therapy increases sB1−/− PMN numbers via enhanced granulopoiesis
G-CSF therapy is an effective long-term treatment in many cases of severe congenital neutropenia and it is also used to prevent chemotherapy-induced febrile neutropenia by enhancing PMN production. In addition, G-CSF delays neutrophil apoptosis by differentially regulating proapoptotic and antiapoptotic factors.10 To test whether G-CSF could rescue sB1−/− PMN survival defect, WT and sB1−/− mice were treated with therapeutic doses of G-CSF or saline for 5 days and BM and blood PMNs were analyzed 24 hours after the last injection. Total counts of myelocytes and PMNs were significantly increased in the BM of treated mice compared with their respective untreated genotype controls (Figure 6A-B). The increase in myelocyte numbers was identical in G-CSF–treated WT and sB1−/− mice, indicating that G-CSF–induced granulopoiesis proceeds normally in sB1−/−myeloid progenitors (Figure 6B).
Figure 6
In vivo G-CSF therapy increases PMN numbers in BM of sB1−/− mice.
SerpinB1 is a member of the clade B serpins, a subfamily composed of leaderless proteins with nucleocytoplasmic localization. Clade B serpins are often expressed in cells that also carry target proteases, which led to the hypothesis that intracellular serpins protect against misdirected granule proteases and/or protect bystander cells from released proteases.31 We previously reported that deficiency in serpinB1 is associated with reduced PMN survival in the BM and at inflammatory sites.19,20 The evidence presented here demonstrates that the cytoprotective function of serpinB1 in PMNs is based on the inhibition of granule protease CG. Deficiency in CG was sufficient to rescue the defect of sB1−/− mice as illustrated by normal PMN counts in the BM of double knockout CG.sB1−/− mice. We also showed that the protease-serpin interaction occurred within PMNs. Indeed, WT PMNs had a greater survival over sB1−/− PMNs in mixed BM chimera, whereas the survival of CG.sB1−/− PMNs was similar to WT PMNs after BM transfer. SerpinB1 is an ancestral clade B serpin with a conserved specificity determining reactive center loop in all vertebrates.32 Furthermore, human and mouse serpinB1 have the same specificity for chymotrypsin-like and elastase-like serine proteases.21,22 Likewise, human and mouse CG have identical substrate specificities and the phenotype of CG−/− murine PMN can be rescued by human CG.33 Therefore, it is highly likely that the antagonistic functions of CG and serpinB1 in cellular homeostasis observed in mice can be extended to other species.
Extracellular CG was previously reported to promote detachment-induced apoptosis (anoikis) in human and mouse cardiomyocytes.34 This activity is mediated through the shedding and transactivation of epidermal growth factor receptor and downregulation of focal adhesion signaling.35,36 In our study, exogenous human CG also induced PMN death in vitro but these effects were not enhanced in sB1−/− PMNs and the neutropenia associated with serpinB1 deficiency was principally cell intrinsic. How intracellular CG induces PMN death remains to be fully investigated. However, our studies provide some indications on the potential pathways. Like other NSPs, the expression of CG is transcriptionally restricted to the promyelocyte stage during PMN development and NSPs are then stored in active form in primary azurophil granules.37 Because serpinB1 is equally efficient at inhibiting NE, CG, and PR3, it was surprising that deletion of CG alone was sufficient to achieve a complete reversal of the PMN survival defect in CG.sB1−/− mice. A possible explanation would be that CG gains access to targets more readily than other granule proteases. There is evidence that binding to serglycin proteoglycans differs between NE and CG resulting in altered sorting of NE but not CG into granules of serglycin-deficient PMNs.38 Different interactions with granule matrix may thus contribute to differential release of CG from the granules compared with other NSPs. However, because sB1−/− PMNs have similar levels of CG and NE as WT PMNs20 and because LLME-induced granule permeabilization likely releases all granule contents equally, we favor an alternative interpretation where CG specifically targets essential cellular components that are not cleaved by the other serpinB1-inhibitable granule proteases. Upon granule permeabilization, we found that CG can induce cell death upstream of caspases as well as independent of caspases. CG was previously shown to activate caspase-7 in vitro and it functions at neutral pH, which is consistent with a physiological role in the nucleocytoplasmic environment.39 Cell death induced by lysosomal/granule membrane permeabilization has previously been linked to cysteine cathepsins in other cell types. However, these proteases appear to depend on caspase activation to trigger apoptosis and they function poorly at neutral pH, questioning their potential role as regulators of cell death.40 In contrast, CG-mediated cell death is not completely blocked by caspase inhibition, which is a property reminiscent of granzymes in cytotoxic T cells.41 In fact, CG is phylogenetically most closely related to serine proteases granzyme B and H.42 Granzymes have numerous nuclear, mitochondrial, and cytoplasmic target proteins leading to cell death41 and we anticipate that this may also be the case for CG.
……
G-CSF therapy is successfully used to treat most congenital and acquired neutropenia through increased granulopoiesis, mobilization from the BM, and increased survival of PMNs. Prosurvival effects of G-CSF include the upregulation of antiapoptotic Bcl-2 family members, which act upstream of the mitochondria and the activation of effector caspases. In sB1−/− mice, G-CSF levels in serum are fourfold higher than in WT mice in steady state and this is accompanied by an upregulation of the antiapoptotic Bcl-2 family member Mcl-1 in sB1−/− PMNs.19 Here, G-CSF therapy significantly increased granulopoiesis in both WT and sB1−/− mice. However, the PMN numbers in treated sB1−/− BM and blood were significantly lower than those of treated WT mice, indicating only a partial rescue of the survival defect. This is consistent with our findings that CG-mediated death can proceed independent of caspases and can thus bypass antiapoptotic effects mediated by G-CSF.
CG has largely been studied in association with antimicrobial and inflammatory functions due to its presence in PMNs.12⇓–14,49 In this context, we have previously shown that serpinB1 contributes to prevent increased mortality and morbidity associated with production of inflammatory cytokines upon infection with Pseudomonas aeruginosa and influenza A virus.20,50 In this study, we demonstrate that serpinB1 inhibition of the primary granule protease CG in PMNs is essential for PMN survival and this ultimately regulates PMN numbers in vivo. Our findings also extend the roles of CG from antimicrobial and immunoregulatory functions to a novel role in inducing cell death.
Neutrophil Elastase, Proteinase 3, and Cathepsin G as Therapeutic Targets in Human Diseases
Polymorphonuclear neutrophils are the first cells recruited to inflammatory sites and form the earliest line of defense against invading microorganisms. Neutrophil elastase, proteinase 3, and cathepsin G are three hematopoietic serine proteases stored in large quantities in neutrophil cytoplasmic azurophilic granules. They act in combination with reactive oxygen species to help degrade engulfed microorganisms inside phagolysosomes. These proteases are also externalized in an active form during neutrophil activation at inflammatory sites, thus contributing to the regulation of inflammatory and immune responses. As multifunctional proteases, they also play a regulatory role in noninfectious inflammatory diseases. Mutations in the ELA2/ELANE gene, encoding neutrophil elastase, are the cause of human congenital neutropenia. Neutrophil membrane-bound proteinase 3 serves as an autoantigen in Wegener granulomatosis, a systemic autoimmune vasculitis. All three proteases are affected by mutations of the gene (CTSC) encoding dipeptidyl peptidase I, a protease required for activation of their proform before storage in cytoplasmic granules. Mutations of CTSC cause Papillon-Lefèvre syndrome. Because of their roles in host defense and disease, elastase, proteinase 3, and cathepsin G are of interest as potential therapeutic targets. In this review, we describe the physicochemical functions of these proteases, toward a goal of better delineating their role in human diseases and identifying new therapeutic strategies based on the modulation of their bioavailability and activity. We also describe how nonhuman primate experimental models could assist with testing the efficacy of proposed therapeutic strategies.
Human polymorphonuclear neutrophils represent 35 to 75% of the population of circulating leukocytes and are the most abundant type of white blood cell in mammals (Borregaard et al., 2005). They are classified as granulocytes because of their intracytoplasmic granule content and are characterized by a multilobular nucleus. Neutrophils develop from pluripotent stem cells in the bone marrow and are released into the bloodstream where they reach a concentration of 1.5 to 5 × 109 cells/liter. Their half-life in the circulation is only on the order of a few hours. They play an essential role in innate immune defense against invading pathogens and are among the primary mediators of inflammatory response. During the acute phase of inflammation, neutrophils are the first inflammatory cells to leave the vasculature, where they migrate toward sites of inflammation, following a gradient of inflammatory stimuli. They are responsible for short-term phagocytosis during the initial stages of infection (Borregaard and Cowland, 1997; Hampton et al., 1998; Segal, 2005). Neutrophils use complementary oxidative and nonoxidative pathways to defend the host against invading pathogens (Kobayashi et al., 2005).
The three serine proteases neutrophil elastase (NE1), proteinase 3 (PR3), and cathepsin G (CG) are major components of neutrophil azurophilic granules and participate in the nonoxidative pathway of intracellular and extracellular pathogen destruction. These neutrophil serine proteases (NSPs) act intracellularly within phagolysosomes to digest phagocytized microorganisms in combination with microbicidal peptides and the membrane-associated NADPH oxidase system, which produces reactive oxygen metabolites (Segal, 2005). An additional extracellular antimicrobial mechanism, neutrophil extracellular traps (NET), has been described that is made of a web-like structure of DNA secreted by activated neutrophils (Papayannopoulos and Zychlinsky, 2009) (Fig. 1). NETs are composed of chromatin bound to positively charged molecules, such as histones and NSPs, and serve as physical barriers that kill pathogens extracellularly, thus preventing further spreading. NET-associated NSPs participate in pathogen killing by degrading bacterial virulence factors extracellularly (Brinkmann et al., 2004;Papayannopoulos and Zychlinsky, 2009).
Polymorphonuclear neutrophil. Quiescent (A) and chemically activated (B) neutrophils purified from peripheral blood. C, PMA-activated neutrophils embedded within NET and neutrophil spreading on insoluble elastin.
In addition to their involvement in pathogen destruction and the regulation of proinflammatory processes, NSPs are also involved in a variety of inflammatory human conditions, including chronic lung diseases (chronic obstructive pulmonary disease, cystic fibrosis, acute lung injury, and acute respiratory distress syndrome) (Lee and Downey, 2001; Shapiro, 2002; Moraes et al., 2003; Owen, 2008b). In these disorders, accumulation and activation of neutrophils in the airways result in excessive secretion of active NSPs, thus causing lung matrix destruction and inflammation. NSPs are also involved in other human disorders as a consequence of gene mutations, altered cellular trafficking, or, for PR3, autoimmune disease. Mutations in the ELA2/ELANE gene encoding HNE are the cause of human cyclic neutropenia and severe congenital neutropenia (Horwitz et al., 1999, 2007). Neutrophil membrane-bound proteinase 3 (mPR3) is the major target antigen of anti-neutrophil cytoplasmic autoantibodies (ANCA), which are associated with Wegener granulomatosis (Jenne et al., 1990). All three proteases are affected by mutation of the gene (CTSC) encoding dipeptidyl peptidase I (DPPI), which activates several granular hematopoietic serine proteases (Pham and Ley, 1999; Adkison et al., 2002). Mutations of CTSC cause Papillon-Lefèvre syndrome and palmoplantar keratosis (Hart et al., 1999; Toomes et al., 1999).
…….
Fully processed mature HNE, PR3, and CG isolated from azurophilic granules contain, respectively, 218 (Bode et al., 1986; Sinha et al., 1987), 222 (Campanelli et al., 1990b), and 235 (Salvesen et al., 1987; Hof et al., 1996) residues. They are present in several isoforms depending on their carbohydrate content, with apparent mass of 29 to 33 kDa upon SDS-polyacrylamide gel electrophoresis (Twumasi and Liener, 1977; Watorek et al., 1993). HNE and PR3 display two sites of N-glycosylation, whereas CG possesses only one. NSPs are stored mainly in neutrophil azurophilic granules, but HNE is also localized in the nuclear envelope, as revealed by immunostaining and electron microscopy (Clark et al., 1980;Benson et al., 2003), whereas PR3 is also found in secretory vesicles (Witko-Sarsat et al., 1999a). Upon neutrophil activation, granular HNE, PR3, and CG are secreted extracellularly, although some molecules nevertheless remain at the cell surface (Owen and Campbell, 1999; Owen, 2008a). The mechanism through which NSPs are sorted from the trans-Golgi network to the granules has not been completely defined, even though an intracellular proteoglycan, serglycin, has been identified as playing a role in elastase sorting and packaging into azurophilic granules (Niemann et al., 2007). Unlike HNE and CG, PR3 is constitutively expressed on the membranes of freshly isolated neutrophils (Csernok et al., 1990; Halbwachs-Mecarelli et al., 1995). Stimulation of neutrophils at inflammatory sites triggers intracytoplasmic granules to translocate to the phagosomes and plasma membrane, thereby liberating their contents. The first step of the translocation to the target membrane depends on cytoskeleton remodeling and microtubule assembly (Burgoyne and Morgan, 2003). This is followed by a second step of granule tethering and docking, which are dependent on the sequential intervention of SNARE proteins (Jog et al., 2007).
…….
Exposure of neutrophils to cytokines (TNF-α), chemoattractants (platelet-activating factor, formyl-Met-Leu-Phe, or IL-8), or bacterial lipopolysaccharide leads to rapid granule translocation to the cell surface with secretion of HNE, PR3, and CG into the extracellular medium (Owen and Campbell, 1999). A fraction of secreted HNE, PR3, and CG is detected at the surface of activated neutrophils (Owen et al., 1995a, 1997; Campbell et al., 2000). Resting purified neutrophils from peripheral blood express variable amounts of PR3 on their surface. A bimodal, apparently genetically determined, distribution has been observed with two populations of quiescent neutrophils that express or do not express the protease at their surface (Halbwachs-Mecarelli et al., 1995; Schreiber et al., 2003). The percentage of mPR3-positive neutrophils ranges from 0 to 100% of the total neutrophil population within individuals. Furthermore, the percentage of mPR3-positive neutrophils remains stable over time and is not affected by neutrophil activation (Halbwachs-Mecarelli et al., 1995).
The mechanism through which HNE and CG are associated with the outer surface of the plasma membrane of neutrophils mainly involves electrostatic interactions with the sulfate groups of chondroitin sulfate- and heparan sulfate-containing proteoglycans (Campbell and Owen, 2007). These two proteases are released from neutrophil cell surfaces by high concentrations of salt (Owen et al., 1995b, 1997;Korkmaz et al., 2005a) and after treatment with chondroitinase ABC and heparinase (Campbell and Owen, 2007). Membrane PR3 is not solubilized by high salt concentrations, which means that its membrane association is not charge dependant (Witko-Sarsat et al., 1999a; Korkmaz et al., 2009). Unlike HNE and CG, PR3 bears at its surface a hydrophobic patch formed by residues Phe166, Ile217, Trp218, Leu223, and Phe224 that is involved in membrane binding (Goldmann et al., 1999; Hajjar et al., 2008) (Fig. 3B). Several membrane partners of PR3 have been identified, including CD16/FcγRIIIb (David et al., 2005; Fridlich et al., 2006), phospholipid scramblase-1, a myristoylated membrane protein with translocase activity present in lipid rafts (Kantari et al., 2007), CD11b/CD18 (David et al., 2003), and human neutrophil antigen NB1/CD177 (von Vietinghoff et al., 2007; Hu et al., 2009), a 58- to 64-kDa glycosyl-phosphatidylinositol anchored surface receptor belonging to the urokinase plasminogen activator receptor superfamily (Stroncek, 2007). NB1 shows a bimodal distribution that superimposes with that of PR3 on purified blood neutrophils (Bauer et al., 2007). Active, mature forms of PR3 but not pro-PR3 can bind to the surface of NB1-transfected human embryonic kidney 293 cells (von Vietinghoff et al., 2008) and Chinese hamster ovary cells (Korkmaz et al., 2008b). Interaction involves the hydrophobic patch of PR3 because specific amino acid substitutions disrupting this patch in the closely related gibbon PR3 prevent binding to NB1-transfected cells (Korkmaz et al., 2008b). Decreased interaction of pro-PR3 with NB1-transfected cells is explained by the topological changes affecting the activation domain containing the hydrophobic patch residues. Together, these results support the hydrophobic nature of PR3-membrane interaction.
……..
Roles in Inflammatory Process Regulation
NSPs are abundantly secreted into the extracellular environment upon neutrophil activation at inflammatory sites. A fraction of the released proteases remain bound in an active form on the external surface of the plasma membrane so that both soluble and membrane-bound NSPs are able to proteolytically regulate the activities of a variety of chemokines, cytokines, growth factors, and cell surface receptors. Secreted proteases also activate lymphocytes and cleave apoptotic and adhesion molecules (Bank and Ansorge, 2001; Pham, 2006; Meyer-Hoffert, 2009). Thus, they retain pro- and anti-inflammatory activities, resulting in a modulation of the immune response at sites of inflammation.
…….
Processing of Cytokines, Chemokines, and Growth Factors.
Processing and Activation of Cellular Receptors.
Induction of Apoptosis by Proteinase 3.
Physiological Inhibitors of Elastase, Proteinase 3, and Cathepsin G
During phagocytosis and neutrophil turnover, HNE, PR3, and CG are released into the extracellular space as active proteases. The proteolytic activity of HNE, PR3, and CG seems to be tightly regulated in the extracellular and pericellular space to avoid degradation of connective tissue proteins including elastin, collagen, and proteoglycans (Janoff, 1985). Protein inhibitors that belong to three main families, the serpins, the chelonianins, and the macroglobulins, ultimately control proteolytic activity of HNE, PR3, and CG activities. The individual contributions of these families depend on their tissue localization and that of their target proteases. The main characteristics of HNE, PR3, and CG physiological inhibitors are presented in Table 2.
Serine Protease Inhibitors
Serpins are the largest and most diverse family of protease inhibitors; more than 1000 members have been identified in human, plant, fungi, bacteria, archaea, and certain viruses (Silverman et al., 2001; Mangan et al., 2008). They share a similar highly conserved tertiary structure and similar molecular weight of approximately 50 kDa. Human serpins belong to the first nine clades (A–I) of the 16 that have been described based on phylogenic relationships (Irving et al., 2000; Silverman et al., 2001; Mangan et al., 2008). For historical reasons, α1-protease inhibitor (α1-PI) was assigned to the first clade. Clade B, also known as the ov-serpin clan because of the similarity of its members to ovalbumin (a protein that belongs to the serpin family but lacks inhibitory activity), is the second largest clan in humans, with 15 members identified so far. Ov-serpin clan members are generally located in the cytoplasm and, to a lesser extent, on the cell surface and nucleus (Remold-O’Donnell, 1993).
Serpins play important regulatory functions in intracellular and extracellular proteolytic events, including blood coagulation, complement activation, fibrinolysis, cell migration, angiogenesis, and apoptosis (Potempa et al., 1994). Serpin dysfunction is known to contribute to diseases such as emphysema, thrombosis, angioedema, and cancer (Carrell and Lomas, 1997; Lomas and Carrell, 2002). Most inhibitory serpins target trypsin-/chymotrypsin-like serine proteases, but some, termed “cross-class inhibitors,” have been shown to target cysteine proteases (Annand et al., 1999). The crystal structure of the prototype plasma inhibitor α1-PI revealed the archetype native serpin fold (Loebermann et al., 1984). All serpins typically have three β-sheets (termed A, B, and C) and eight or nine α-helices (hA–hI) arranged in a stressed configuration. The so-called reactive center loop (RCL) of inhibitory molecules determines specificity and forms the initial encounter complex with the target protease (Potempa et al., 1994; Silverman et al., 2001). Serpins inhibit proteases by a suicide substrate inhibition mechanism. The protease initially recognizes the serpin as a potential substrate using residues of the reactive center loop and cleaves it between P1 and P1′ This cleavage allows insertion of the cleaved RCL into the β-sheet A of the serpin, dragging the protease with it and moving it over 71 Å to the distal end of the serpin to form a 1:1 stoichiometric covalent inhibitory complex (Huntington et al., 2000). Such cleavage generates a ∼4-kDa C-terminal fragment that remains noncovalently bound to the cleaved serpin. Displacement of the covalently attached active site serine residue from its catalytic partner histidine explains the loss of catalytic function in the covalent complex. The distortion of the catalytic site structure prevents the release of the protease from the complex, and the structural disorder induces its proteolytic inactivation (Huntington et al., 2000). Covalent complex formation between serpin and serine proteases triggers a number of conformational changes, particularly in the activation domain loops of the bound protease (Dementiev et al., 2006).
………
Pathophysiology of Elastase, Proteinase 3 and Cathepsin G in Human Diseases
In many instances, the initiation and propagation of lung damage is a consequence of an exaggerated inappropriate inflammatory response, which includes the release of proteases and leukocyte-derived cytotoxic products (Owen, 2008b;Roghanian and Sallenave, 2008). Inflammation is a physiological protective response to injury or infection consisting of endothelial activation, leukocyte recruitment and activation, vasodilation, and increased vascular permeability. Although designed to curtail tissue injury and facilitate repair, the inflammatory response sometimes results in further injury and organ dysfunction. Inflammatory chronic lung diseases, chronic obstructive pulmonary disease, acute lung injury, acute respiratory distress syndrome, and cystic fibrosis are syndromes of severe pulmonary dysfunction resulting from a massive inflammatory response and affecting millions of people worldwide. The histological hallmark of these chronic inflammatory lung diseases is the accumulation of neutrophils in the microvasculature of the lung. Neutrophils are crucial to the innate immune response, and their activation leads to the release of multiple cytotoxic products, including reactive oxygen species and proteases (serine, cysteine, and metalloproteases). The physiological balance between proteases and antiproteases is required for the maintenance of the lung’s connective tissue, and an imbalance in favor of proteases results in lung injury (Umeki et al., 1988; Tetley, 1993). A number of studies in animal and cell culture models have demonstrated a contribution of HNE and related NSPs to the development of chronic inflammatory lung diseases. Available preclinical and clinical data suggest that inhibition of NSP in lung diseases suppresses or attenuates the contribution of NSP to pathogenesis (Chughtai and O’Riordan, 2004; Voynow et al., 2008; Quinn et al., 2010). HNE could also participate in fibrotic lung remodeling by playing a focused role in the conversion of latent transforming growth factor-β into its biologically active form (Chua and Laurent, 2006; Lungarella et al., 2008).
ANCA-associated vasculitides encompasses a variety of diseases characterized by inflammation of blood vessels and by the presence of autoantibodies directed against neutrophil constituents. These autoantibodies are known as ANCAs (Kallenberg et al., 2006). In Wegener granulomatosis (WG), antibodies are mostly directed against PR3. WG is a relatively uncommon chronic inflammatory disorder first described in 1931 by Heinz Karl Ernst Klinger as a variant of polyarteritis nodosa (Klinger, 1931). In 1936, the German pathologist Friedrich Wegener described the disease as a distinct pathological entity (Wegener, 1936, 1939). WG is characterized by necrotizing granulomatous inflammation and vasculitis of small vessels and can affect any organ (Fauci and Wolff, 1973; Sarraf and Sneller, 2005). The most common sites of involvement are the upper and lower respiratory tract and the kidneys. WG affects approximately 1 in 20,000 people; it can occur in persons of any age but most often affects those aged 40 to 60 years (Walton, 1958; Cotch et al., 1996). Approximately 90% of patients have cold or sinusitis symptoms that fail to respond to the usual therapeutic measures and that last considerably longer than the usual upper respiratory tract infection. Lung involvement occurs in approximately 85% of the patients. Other symptoms include nasal membrane ulcerations and crusting, saddle-nose deformity, inflammation of the ear with hearing problems, inflammation of the eye with sight problems, and cough (with or without hemoptysis).
Hereditary Neutropenias
Neutropenia is a hematological disorder characterized by an abnormally low number of neutrophils (Horwitz et al., 2007). The normal neutrophil count fluctuates across human populations and within individual patients in response to infection but typically lies in the range of 1.5 to 5 × 109 cells/liter. Neutropenia is categorized as severe when the cell count falls below 0.5 × 109 cells/liter. Hence, patients with neutropenia are more susceptible to bacterial infections and, without prompt medical attention, the condition may become life-threatening. Common causes of neutropenia include cancer chemotherapy, drug reactions, autoimmune diseases, and hereditary disorders (Berliner et al., 2004; Schwartzberg, 2006).
Papillon-Lefèvre Syndrome
……….
New Strategies for Fighting Neutrophil Serine Protease-Related Human Diseases
Administration of therapeutic inhibitors to control unwanted proteolysis at inflammation sites has been tested as a therapy for a variety of inflammatory and infectious lung diseases (Chughtai and O’Riordan, 2004). Depending on the size and chemical nature of the inhibitors, they may be administered orally, intravenously, or by an aerosol route. Whatever the mode of administration, the access of therapeutic inhibitors to active proteases is often hampered by physicochemical constraints in the extravascular space and/or by the partitioning of proteases between soluble and solid phases.
……….
Concluding Remarks
NSPs were first recognized as protein-degrading enzymes but have now proven to be multifunctional components participating in a variety of pathophysiological processes. Thus, they appear as potential therapeutic targets for drugs that inhibit their active site or impair activation from their precursor. Overall, the available preclinical and clinical data suggest that inhibition of NSPs using therapeutic inhibitors would suppress or attenuate deleterious effects of inflammatory diseases, including lung diseases. Depending on the size and chemical nature of inhibitors, those may be administered orally, intravenously, or by aerosolization. But the results obtained until now have not been fully convincing because of the poor knowledge of the biological function of each protease, their spatiotemporal regulation during the course of the disease, the physicochemical constraints associated with inhibitor administration, or the use of animal models in which NSP regulation and specificity differ from those in human. Two different and complementary approaches may help bypass these putative problems. One is to target active proteases by inhibitors at the inflammatory site in animal models in which lung anatomy and physiology are close to those in human to allow in vitro and in vivo assays of human-directed drugs/inhibitors. The other is to prevent neutrophil accumulation at inflammatory sites by impairing production of proteolytically active NSPs using an inhibitor of their maturation protease, DPPI. Preventing neutrophil accumulation at the inflammatory sites by therapeutic inhibition of DPPI represents an original and novel approach, the exploration of which has just started (Méthot et al., 2008). Thus pharmacological inactivation of DPPI in human neutrophils could well reduce membrane binding of PR3 and, as a consequence, neutrophil priming by pathogenic auto-antibodies in WG. In addition, it has been recognized that the intracellular level of NSPs depends on their correct intracellular trafficking. In the future, pharmacological targeting of molecules specifically involved in the correct intracellular trafficking of each NSP could possibly regulate their production and activity, a feature that could be exploited as a therapeutic strategy for inflammatory diseases.
Current knowledge on the effects of nut consumption on human health has rapidly increased in recent years and it now appears that nuts may play a role in the prevention of chronic age-related diseases. Frequent nut consumption has been associated with better metabolic status, decreased body weight as well as lower body weight gain over time and thus reduce the risk of obesity. The effect of nuts on glucose metabolism, blood lipids, and blood pressure are still controversial. However, significant decreased cardiovascular risk has been reported in a number of observational and clinical intervention studies. Thus, findings from cohort studies show that increased nut consumption is associated with a reduced risk of cardiovascular disease and mortality (especially that due to cardiovascular-related causes). Similarly, nut consumption has been also associated with reduced risk of certain cancers, such as colorectal, endometrial, and pancreatic neoplasms. Evidence regarding nut consumption and neurological or psychiatric disorders is scarce, but a number of studies suggest significant protective effects against depression, mild cognitive disorders and Alzheimer’s disease. The underlying mechanisms appear to include antioxidant and anti-inflammatory actions, particularly related to their mono- and polyunsaturated fatty acids (MUFA and PUFA, as well as vitamin and polyphenol content. MUFA have been demonstrated to improve pancreatic beta-cell function and regulation of postprandial glycemia and insulin sensitivity. PUFA may act on the central nervous system protecting neuronal and cell-signaling function and maintenance. The fiber and mineral content of nuts may also confer health benefits. Nuts therefore show promise as useful adjuvants to prevent, delay or ameliorate a number of chronic conditions in older people. Their association with decreased mortality suggests a potential in reducing disease burden, including cardiovascular disease, cancer, and cognitive impairments.
Global life expectancy has increased from 65 years in 1990 to about 71 years in 2013 [1]. As life expectancy has increased, the number of healthy years lost due to disability has also risen in most countries, consistent with greater morbidity [2]. Reduction of mortality rates in developed countries has been associated with a shift towards more chronic non-communicable diseases [1]. Cardiovascular diseases (CVDs) and related risk factors, such as hypertension, diabetes mellitus, hypercholesterolemia, and obesity are the top causes of death globally, accounting for nearly one-third of all deaths worldwide [3]. Equally, the estimated incidence, mortality, and disability- adjusted life-years (DALYs) for cancer rose to 14.9 million incident cancer cases, 8.2 million deaths, and 196.3 million DALYs, with the highest impact of prostate and breast cancer in men and women, respectively [4]. Depression is a leading cause of disability worldwide (in terms of total years lost due to disability), especially in high-income countries, increasing from 15th to 11th rank (37% increase) and accounting for 18% of total DALYs (almost 100 million DALYs) [5]. Overall, the global rise in chronic non-communicable diseases is congruent with a similar rise in the elderly population. The proportion of people over the age of 60 is growing faster than any other age group and is estimated to double from about 11% to 22% within the next 50 years [6]. Public health efforts are needed to face this epidemiological and demographic transition, both improving the healthcare systems, as well as assuring a better health in older people. Accordingly, a preventive approach is crucial to dealing with an ageing population to reduce the burden of chronic disease.
In this context, lifestyle behaviors have demonstrated the highest impact for older adults in preventing and controlling the morbidity and mortality due to non- communicable diseases [7]. Unhealthy behaviors, such as unbalanced dietary patterns, lack of physical activity and smoking, play a central role in increasing both cardiovascular and cancer risk [7]. Equally, social isolation and depression in later life may boost health decline and significantly contribute to mortality risk [8]. The role of diet in prevention of disability and death is a well-established factor, which has an even more important role in geriatric populations. Research has focused on the effect of both single foods and whole dietary patterns on a number of health outcomes, including mortality, cardiovascular disease (CVD), cancer and mental health disorders (such as cognitive decline and depression) [9-13]. Plantbased dietary patterns demonstrate the most convincing evidence in preventing chronic non-communicable diseases [14-17]. Among the main components (including fruit and vegetables, legumes and cereals), only lately has attention focused on foods such as nuts. Knowledge on the effect of nut consumption on human health has increased rapidly in recent years. The aim of this narrative review is to examine recent evidence regarding the role of nut consumption in preventing chronic disease in older people.
Tree nuts are dry fruits with an edible seed and a hard shell. The most popular tree nuts are almonds (Prunus amigdalis), hazelnuts (Corylus avellana), walnuts (Juglans regia), pistachios (Pistachia vera), cashews (Anacardium occidentale), pecans (Carya illinoiensis), pine nuts (Pinus pinea), macadamias (Macadamia integrifolia), Brazil nuts (Bertholletia excelsa), and chestnuts (Castanea sativa). When considering the “nut” group, researchers also include peanuts (Arachis hypogea), which technically are groundnuts. Nuts are nutrient dense foods, rich in proteins, fats (mainly unsaturated fatty acids), fiber, vitamins, minerals, as well as a number of phytochemicals, such as phytosterols and polyphenols [18]. Proteins account for about 10-25% of energy, including individual aminoacids, such as L-arginine, which is involved in the production of nitric oxide (NO), an endogenous vasodilatator [19].
The fatty acids composition of nuts involves saturated fats for 415% and unsaturated fatty acids for 30-60% of the content. Unsaturated fatty acids are different depending on the nut type, including monounsaturated fatty acids (MUFA, such as oleic acid in most of nuts, whereas polyunsaturated fatty acids (PUFA, such as alpha-linolenic acid) in pine nuts and walnuts [20]. Also fiber content is similar among most nut types (about 10%), although pine nuts and cashews hold the least content. Vitamins contained in nuts are group B vitamins, such as B6 (involved in many aspects of macronutrient metabolism) and folate (necessary for normal cellular function, DNA synthesis and metabolism, and homocysteine detoxification), as well as tocopherols, involved in anti-oxidant mechanisms [21]. Among minerals contained in vegetables, nuts have an optimal content in calcium, magnesium, and potassium, with an extremely low amount of sodium, which is implicated on a number of pathological conditions, such as bone demineralization, hypertension and insulin resistance[22]. Nuts are also rich in phytosterols, non-nutritive components of certain plant-foods that exert both structural (at cellular membrane phospholipids level) and hormonal (estrogen-like) activities [23]. Finally, nuts have been demonstrated to be a rich source of polyphenols, which account for a key role in their antioxidant and anti-inflammatory effects.
Metabolic disorders are mainly characterized by obesity, hypertension, dyslipidemia, and hyperglycemia/ hyperinsulinemia/type-2 diabetes, all of which act synergistically to increase morbidity and mortality of aging population.
Obesity Increasing high carbohydrate and fat food intake in the last decades has contributed significantly to the rise in metabolic disorders. Nuts are energy-dense foods that have been thought to be positively associated with increased body mass index (BMI). As calorie-dense foods, nuts may contain 160–200 calories per ounce. The recommendation from the American Heart Association to consume 5 servings per week (with an average recommended serving size of 28 g) corresponds to a net increase of 800–1000 calories per week, which may cause weight gain. However, an inverse relation between the frequency of nut consumption and BMI has been observed in large cohort studies [24]. Pooling the baseline observations of BMI by category of nut consumption in 5 cohort studies found a significant decreasing trend in BMI values with increasing nut intake [24]. While the evidence regarding nut consumption and obesity is limited, findings so far are encouraging [25, 26]. When the association between nut consumption and body weight has been evaluated longitudinally over time, nut intake was associated with a slightly lower risk of weight gain and obesity [25]. In the Nurses’ Health Study II (NHS II), women who eat nuts ≥2 times per week had slightly less weight gain (5.04 kg) than did women who rarely ate nuts (5.55 kg) and marginally significant 23% lower risk of obesity after 9-year follow-up [25]. Further evaluation of the NHS II data and the Physicians’ Health Study (PHS) comprising a total of 120,877 US women and men and followed up to 20 years revealed that 4-y weight change was inversely associated with a 1-serving increment in the intake of nuts (20.26 kg) [27]. In the “Seguimiento Universidad de Navarra” (SUN) cohort study, a significant decreased weight change has been observed over a period of 6 years [26]. After adjustment for potential confounding factors the analysis was no longer significant, but overall no weight gain associated with >2 servings per week of nuts has been observed. Finally, when considering the role of the whole diet on body weight, a meta-analysis of 31 clinical trials led to the conclusion of a null effect of nut intake on body weight, BMI, and waist circumference [28].
Glucose metabolism and type-2 diabetes The association between nut consumption and risk of type-2 diabetes in prospective cohort studies is controversial [29-32]. A pooled analysis relied on the examination of five large cohorts, including the NHS, the Shanghai Women’s Health Study, the Iowa Women’s Health Study, and the PHS, and two European studies conducted in Spain (the PREDIMED trial) and Finland including a total of more than 230,000 participants and 13,000 cases, respectively. Consumption of 4 servings per week was associated with 13% reduced risk of type-2 diabetes without effect modification by age [29]. In contrast, other pooled analyses showed non-significant reduction of risk for increased intakes of nuts, underlying that the inverse association between the consumption of nuts and diabetes was attenuated after adjustment for confounding factors, including BMI [30]. However, results from experimental studies showed promising results. Thus, nut consumption has been demonstrated to exert beneficial metabolic effects due to their action on post-prandial glycemia an insulin sensitivity. A number of RCTs have demonstrated positive effects of nut consumption on post-prandial glycemia in healthy individuals [33-38]. Moreover, a meta-analysis of RCTs on the effects of nut intake on glycemic control in diabetic individuals including 12 trials and a total of 450 participants showed that diets with an emphasis on nuts (median dose = 56 g/d) significantly lowered HbA1c (Mean Difference [MD] : -0.07%; 95% confidence interval [CI]: -0.10, -0.03%; P = 0.0003) and fasting glucose (MD : -0.15 mmol/L; 95% CI: -0.27, -0.02 mmol/L; P = 0.03) compared with control diets [39]. No significant treatment effects were observed for fasting insulin and homeostatic model assessment (HOMA-IR), despite the direction of effect favoring diet regimens including nuts.
Blood lipids and hypertension Hypertension and dyslipidemia are major risk factors for CVD. Diet alone has a predominant role in blood pressure and plasma lipid homeostasis. One systematic review [40] and 3 pooled quantitative analyses of RCTs [41-43] evaluated the effects of nut consumption on lipid profiles. A general agreement was relevant on certain markers, as daily consumption of nuts (mean = 67 g/d) induced a pooled reduction of total cholesterol concentration (10.9 mg/dL [5.1% change]), low-density lipoprotein cholesterol concentration (LDL-C) (10.2 mg/dL [7.4% change]), ratio of LDL-C to high-density lipoprotein cholesterol concentration (HDL-C) (0.22 [8.3% change]), and ratio of total cholesterol concentration to HDL-C (0.24 [5.6% change]) (P <0.001 for all) [42]. All meta-analyses showed no significant effects of nut (including walnut) consumption on HDL cholesterol or triglyceride concentrations in healthy individuals [41], although reduced plasma triglyceride levels were found in individuals with hypertriglyceridemia [42]. Interestingly, the effects of nut consumption were dose related, and different types of nuts had similar effects on blood lipid concentrations.
There is only limited evidence from observational studies to suggest that nuts have a protective role on blood pressure. A pooled analysis of prospective cohort studies on nut consumption and hypertension reported a decreased risk associated with increased intake of nuts [32]. Specifically, only a limited number of cohort studies have been conducted exploring the association between nut consumption and hypertension (n = 3), but overall reporting an 8% reduced risk of hypertension for individuals consuming >2 servings per week (Risk Ratio [RR] = 0.92, 95% CI: 0.87-0.97) compared with never/rare consumers, whereas consumption of nuts at one serving per week had similar risk estimates (RR = 0.97, 95% CI: 0.83, 1.13) [32]. These findings are consistent with results obtained in a pooled analysis of 21 experimental studies reporting the effect of consuming single or mixed nuts (in doses ranging from 30 to 100 g/d) on systolic (SBP) and diastolic blood pressure (DBP) [44]. A pooled analysis found a significant reduction in SBP in participants without type2 diabetes [MD: -1.29 mmHg; 95% CI: -2.35, -0.22; P = 0.02] and DBP (MD: -1.19; 95% CI: -2.35, -0.03; P = 0.04), whereas subgroup analyses of different nut types showed that pistachios, but not other nuts, significantly reduced SBP (MD: -1.82; 95% CI: -2.97, -0.67; P = 0.002) and SBP (MD: -0.80; 95% CI: -1.43, -0.17; P = 0.01) [44].
Nut consumption and CVD risk Clustering of metabolic risk factors occurs in most obese individuals, greatly increasing risk of CVD. The association between nut consumption and CVD incidence [29-31] and mortality [24] has been explored in several pooled analyses of prospective studies. The overall risk calculated for CVD on a total of 8,862 cases was reduced by 29% for individuals consuming 7 servings per week (RR = 0.71, 95% CI: 0.59, 0.85) [30]. A meta-analysis including 9 studies on coronary artery disease (CAD) including 179,885 individuals and 7,236 cases, reporting that 1-serving/day increment would reduce risk of CAD of about 20% (RR = 0.81, 95% CI: 0.72, 0.91) [31]. Similar risk estimates were calculated for ischemic heart disease (IHD), with a comprehensive reduced risk of about 25-30% associated with a daily intake of nuts [29, 30]. Findings from 4 prospective studies have been pooled to estimate the association between nut consumption and risk of stroke, and a non-significant/borderline reduced risk was found [29-31, 45]. CVD mortality was explored in a recent meta-analysis including a total of 354,933 participants, 44,636 cumulative incident deaths, and 3,746,534 cumulative person-years [24]. One serving of nuts per week and per day resulted in decreased risk of CVD mortality (RR = 0.93, 95% CI: 0.88, 0.99 and RR =0.61, 95% CI: 0.42, 0.91, respectively], primarily driven by decreased coronary artery disease (CAD) deaths rather than stroke deaths [24]. Overall, all pooled analyses demonstrated a significant association between nut consumption and cardiovascular health. However, it has been argued that nut consumption was consistently associated with healthier background characteristics reflecting overall healthier lifestyle choices that eventually lead to decreased CVD mortality risk.
Nut consumption and cancer risk Cancer is one of the leading causes of death in the elderly population. After the evaluation of the impact on cancer burden of food and nutrients, it has been concluded that up to one third of malignancies may be prevented by healthy lifestyle choices. Fruit and vegetable intake has been the focus of major attention, but studies on nut consumption and cancer are scarce. A recent metaanalysis pooled together findings of observational studies on cancer incidence, including a total of 16 cohort and 20 casecontrol studies comprising 30,708 cases, compared the highest category of nut consumption with the lowest category and found a lower risk of any cancer of 25% (RR = 0.85, 95% CI: 0.86, 0.95) [46]. When the analysis was conducted by cancer site, highest consumption of nuts was associated with decreased risk of colorectal (RR = 0.76, 95% CI: 0.61, 0.96), endometrial (RR = 0.58, 95% CI: 0.43, 0.79), and pancreatic cancer (RR = 0.71, 95% CI: 0.51, 0.99), with only one cohort study was conducted on the last [46]. The potential protective effects of nut consumption on cancer outcomes was supported also by pooled analysis of 3 cohort studies [comprising the PREDIMED, the NHS, the HPS, and the Health Professionals Follow-Up Study (HPFS) cohorts] showing a decreased risk of cancer death for individuals consuming 3-5 servings of nuts per week compared with never eaters (RR = 0.86, 95% CI: 0.75, 0.98) [24]. The analysis was recently updated by including results from the Netherlands Cohort Study reaching a total of 14,340 deaths out of 247,030 men and women observed, confirming previous results with no evidence of between-study heterogeneity (RR = 0.85, 95% CI: 0.77, 0.93) [47]. However, a dose- response relation showed the non-linearity of the association, suggesting that only moderate daily consumption up to 5 g reduced risk of cancer mortality, and extra increased intakes were associated with no further decreased risk.
Nut consumption and affective/cognitive disorders Age-related cognitive decline is one of the most detrimental health problems in older people. Cognitive decline is a paraphysiological process of aging, but timing and severity of onset has been demonstrated to be affected by modifiable lifestyle factors, including diet. In fact, the nature of the age- related conditions leading to a mild cognitive impairment (MCI) differs by inflammation-related chronic neurodegenerative diseases, such as dementia, Alzheimer’s disease, Parkinson’s disease and depression. Evidence restricted to nut consumption alone is scarce, but a number of studies have been conducted on dietary patterns including nuts as a major component. A pooled analysis synthesizing findings of studies examining the association between adherence to a traditional Mediterranean diet and risk of depression (n = 9), cognitive decline (n = 8), and Parkinson’s disease (n = 1) showed a reduction of risk of depression (RR = 0.68, 95% CI: 0.54, 0.86) and cognitive impairment (RR = 0.60, 95% CI: 0.43, 0.83) in individuals with increased dietary adherence [10].
The study that first found a decreased risk of Alzheimer’s disease in individuals highly adherent to the Mediterranean diet was conducted in over 2,000 individuals in the Washington/Hamilton Heights-Inwood Columbia Aging Project (WHICAP), a cohort of non-demented elders aged 65 and older living in a multi-ethnic community of Northern Manhattan in the US (Hazard Ratio [HR] = 0.91, 95% CI: 0.83, 0.98) [48]. These results have been replicated in further studies on the Mediterranean diet, however nut consumption was not documented [49, 50]. A number of observational studies also demonstrated a significant association between this dietary pattern and a range of other cognitive outcomes, including slower global cognitive decline [51]. However, evidence from experimental studies is limited to the PREDIMED trial, providing interesting insights on the association between the Mediterranean diet supplemented with mixed nuts and both depression and cognitive outcomes. Regarding depression, the nutritional intervention with a Mediterranean diet supplemented with nuts showed a lower risk of about 40% in participants with type-2 diabetes (RR = 0.59, 95% CI: 0.36, 0.98) compared with the control diet [52]. However the effect was not significant in the whole cohort overall [52]. Regarding cognitive outcomes after a mean follow-up of 4.1 years, findings from the same trial showed significant improvements in memory and global cognition tests for individuals allocated to the Mediterranean diet supplemented with nuts [adjusted differences: -0.09 (95% CI: -0.05, 0.23), P = 0.04 and -0.05 (95% CI: -0.27, 0.18), P = 0.04, respectively], compared to control group, showing that Mediterranean diet plus mixed nuts is associated with improved cognitive function [53].
Potential mechanisms of protection of nut consumption Despite the exact mechanisms by which nuts may ameliorate human health being largely unknown, new evidence has allowed us to start to better understand the protection of some high-fat, vegetable, energy-dense foods such as nuts. Non- communicable disease burden related with nutritional habits is mainly secondary to exaggerated intakes of refined sugars and saturated fats, such as processed and fast- foods. Nuts provide a number of nutrient and non-nutrient compounds and it is only recently that scientists have tried to examine their effects on metabolic pathways.
Metabolic and cardiovascular protection With special regard to body weight and their potential effects in decreasing the risk of obesity (or weight gain, in general), nuts may induce satiation (reduction in the total amount of food eaten in a single meal) and satiety (reduction in the frequency of meals) due to their content in fibers and proteins, which are associated with increased release of glucagon-like protein 1 (GLP-1) and cholecystokinin (CCK), gastrointestinal hormones with satiety effects [54, 55]. The content in fiber of nuts may also increase thermogenesis and resting energy expenditure, and reduce post- prandial changes of glucose, thus ameliorating inflammation and insulin resistance. Moreover, the specific content profile of MUFA and PUFA provides readily oxidized fats than saturated or trans fatty acids, leading to reduced fat accumulation [56, 57]. The beneficial effects of nuts toward glucose metabolism may be provided by their MUFA content that improves the efficiency of pancreatic beta-cell function by enhancing the secretion of GLP1, which in turn helps the regulation of postprandial glycemia and insulin sensitivity [58]. MUFA and PUFA are also able to reduce serum concentrations of the vasoconstrictor thromboxane 2, which might influence blood pressure regulation. Together with polyphenols and anti-oxidant vitamins, nuts may also ameliorate inflammatory status at the vascular level, reducing circulating levels of soluble cellular adhesion molecules, such as intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), and E-selectin, which are released from the activated endothelium and circulating monocytes [59]. Moreover, nuts may improve vascular reactivity due to their content in L-arginine, which is a potent precursor of the endogenous vasodilator nitric oxide. Nuts content in microelements is characterized by a mixture that may exert a direct effect in modulating blood pressure, including low content of sodium and richness in magnesium, potassium and calcium, which may interact to beneficially influence blood pressure
Despite the exact mechanisms by which nuts may ameliorate human health being largely unknown, new evidence has allowed us to start to better understand the protection of some high-fat, vegetable, energy-dense foods such as nuts. Non- communicable disease burden related with nutritional habits is mainly secondary to exaggerated intakes of refined sugars and saturated fats, such as processed and fast- foods. Nuts provide a number of nutrient and non-nutrient compounds and it is only recently that scientists have tried to examine their effects on metabolic pathways.
Cancer protection The potential mechanisms of action of nuts that may intervene in the prevention of cancer have not been totally elucidated. Numerous hypotheses have been proposed on the basis of basic research exploring the antioxidant and anti-inflammatory compounds characterizing nuts [61]. Vitamin E can regulate cell differentiation and proliferation, whereas polyphenols (particularly flavonoids such as quercetin and stilbenes such as resveratrol) have been shown to inhibit chemically-induced carcinogenesis [62]. Polyphenols may regulate the inflammatory response and immunological activity by acting on the formation of the prostaglandins and pro-inflammatory cytokines, which may be an important mechanism involved in a number of cancers, including colorectal, gastric, cervical and pancreatic neoplasms [62]. Among other compounds contained in nuts, dietary fiber may exert protective effects toward certain cancers (including, but not limited to colorectal cancer) by the aforementioned metabolic effects as well as increasing the volume of feces and anaerobic fermentation, and reducing the length of intestinal transit. As a result, the intestinal mucosa is exposed to carcinogens for a reduced time and the carcinogens in the colon are diluted [62]. Finally, there is no specific pathway demonstrating the protective effect of PUFA intake against cancer, but their interference with cytokines and prostaglandin metabolism may inhibit a state of chronic inflammation that may increase cancer risk [63].
Cognitive aging and neuro-protection There is no universal mechanism of action for nuts with regard to age-related conditions. A number of systemic biological conditions, such as oxidative stress, inflammation, and reduced cerebral blood flow have been considered as key factors in the pathogenesis of both normal cognitive ageing and chronic neurodegenerative disease [64]. Nuts, alone or as part of healthy dietary patterns, may exert beneficial effects due to their richness in antioxidants, including vitamins, polyphenols and unsaturated fatty acids, that may be protective against the development of cognitive decline and depression [65, 66]. Both animal studies and experimental clinical trials demonstrated vascular benefits of nuts, including the aforementioned lowering of inflammatory markers and improved endothelial function, which all appear to contribute to improved cognitive function [67]. The antioxidant action may affect the physiology of the ageing brain directly, by protecting neuronal and cell-signaling function and maintenance. Moreover, certain compounds contained in nuts may directly interact with the physiology and functioning of the brain. For instance, walnuts are largely composed of PUFA, especially ALA, which have been suggested to induce structural change in brain areas associated with affective experience [66]. Moreover, PUFA have been associated with improved symptoms in depressed patients, suggesting an active role in the underlying pathophysiological mechanisms [68]. Thus, the mechanisms of action of nut consumption on age-related cognitive and depressive disorders are complex, involving direct effects on brain physiology at the neuronal and cellular level and indirect effects by influencing inflammation.
Summary From an epidemiological point of view, nut eaters have been associated with overall healthier lifestyle habits, such as increased physical activity, lower prevalence of smoking, and increased consumption of fruits and vegetables [24]. These variables represent strong confounding factors in determining the effects of nuts alone on human health and final conclusions cannot be drawn. Nevertheless, results from clinical trials are encouraging. Nuts show promise as useful adjuvants to prevent, delay or ameliorate a number of chronic conditions in older people.
The National Institutes of Health recently designated inflammation a priority. CREDIT ILLUSTRATION BY CHAD HAGEN
Several years ago, I fell at the gym and ripped two tendons in my wrist. The pain was excruciating, and within minutes my hand had swollen grotesquely and become hot to the touch. I was reminded of a patient I’d seen early in medical school, whose bacterial infection extended from his knee to his toes. Latin was long absent from the teaching curriculum, but, as my instructor examined the leg, he cited the four classic symptoms of inflammation articulated by the Roman medical writer Celsus in the first century: rubor, redness; tumor, swelling; calor, heat; and dolor, pain. In Latin, inflammatio means “setting on fire,” and as I considered the searing pain in my injured hand I understood how the condition earned its name.
Inflammation occurs when the body rallies to defend itself against invading microbes or to heal damaged tissue. The walls of the capillaries dilate and grow more porous, enabling white blood cells to flood the damaged site. As blood flows in and fluid leaks out, the region swells, which can put pressure on surrounding nerves, causing pain; inflammatory molecules may also activate pain fibres. The heat most likely results from the increase in blood flow.
The key white blood cell in inflammation is called a macrophage, and for decades it has been a subject of study in my hematology laboratory and in many others. Macrophages were once cast as humble handmaidens of the immune system, responsible for recognizing microbes or debris and gobbling them up. But in recent years researchers have come to understand that macrophages are able to assemble, within themselves, specialized platforms that pump out the dozens of molecules that promote inflammation. These platforms, called inflammasomes, are like pop-up factories—quickly assembled when needed and quickly dismantled when the crisis has passed.
For centuries, scientists have debated whether inflammation is good or bad for us. Now we believe that it’s both: too little, and microbes fester and spread in the body, or wounds fail to heal; too much, and nearby healthy tissue can be degraded or destroyed. The fire of inflammation must be tightly controlled—turned on at the right moment and, just as critically, turned off. Lately, however, several lines of research have revealed that low-level inflammation can simmer quietly in the body, in the absence of overt trauma or infection, with profound implications for our health. Using advanced technologies, scientists have discovered that heart attacks, diabetes, and Alzheimer’s disease may be linked to smoldering inflammation, and some researchers have even speculated about its role in psychiatric conditions.
As a result, understanding and controlling inflammation has become a central goal of modern medical investigation. The internal research arm of the National Institutes of Health recently designated inflammation a priority, mobilizing several hundred scientists and hundreds of millions of dollars to better define its role in health and disease; in 2013, the journal Science devoted an entire issue to the subject. This explosion in activity has captured the public imagination. In best-selling books and on television and radio talk shows, threads of research are woven into cure-all tales in which inflammation is responsible for nearly every malady, and its defeat is the secret to health and longevity. New diets will counter the inflammation simmering in your gut and restore your mental equilibrium. Anti-inflammatory supplements will lift your depression and ameliorate autism. Certain drugs will tamp down the silent inflammation that degrades your tissues, improving your health and extending your life. Everything, and everyone, is inflamed.
Such claims aside, there is genuine evidence that inflammation plays a role in certain health conditions. In atherosclerosis, blood flow to the heart or the brain is blocked, resulting in a heart attack or a stroke. For a long time, atherosclerosis was thought to result mainly from eating fatty foods, which clogged the arteries. “Atherosclerosis was all about fats and grease,” Peter Libby, a professor at Harvard Medical School and a cardiologist at Brigham and Women’s Hospital, in Boston, told me recently. “Most physicians saw atherosclerosis as a straight plumbing problem.”
During his cardiology training, Libby studied immunology, and he became fascinated with the work of Rudolf Virchow, a nineteenth-century German pathologist. Virchow speculated that atherosclerosis might be an active process, caused by inflamed blood vessels, not one caused simply by the accumulation of fat. In the mid-nineteen-nineties, in studies with mice, Libby, working in parallel with other groups of scientists, found that low-density lipoproteins—LDLs, those particles of “bad” cholesterol—can work their way into the lining of arteries. There, they sometimes trigger an inflammatory response, which can cause blood clots that block the artery. Libby and others began to understand that atherosclerosis wasn’t a mere plumbing problem but also an immune disease—“our body’s defenses turned against ourselves,” he told me.
Paul Ridker, a cardiovascular expert and a colleague of Libby’s at Harvard and Brigham and Women’s, moved the research beyond the laboratory. He found that many patients who’d had heart attacks, despite lacking factors such as high blood pressure, high cholesterol, and a history of smoking, had an elevated level of C-reactive protein, a molecule produced in response to inflammation, in their blood. After demonstrating, in a separate study, that cholesterol-reducing statins could also reduce C-reactive-protein levels, Ridker launched the Jupiter trial, in which people with elevated levels of C-reactive protein but normal cholesterol levels were given a placebo or a statin medication. In 2008, the published results showed that the subjects who received the statin saw their levels of C-reactive protein drop and were less likely three and a half years later to suffer a heart attack. This suggested that elevated cholesterol isn’t the only factor at work in cardiovascular disease, and that in some cases statins, acting as anti-inflammatory agents, could be used to treat the condition.
The benefit was modest: the statin treatment reduced the risk of heart attack in only about one per cent of the patients. Still, that figure is statistically significant, and for one in every hundred patients—a hundred in every ten thousand—it’s meaningful. An independent safety-monitoring board ended the study early, saying that it was unethical to continue once it was clear that statins provided a benefit not available to the subjects on the placebo. (Critics argue that shortening the trial, which was funded by a drug company, exaggerated the potential benefits and underestimated long-term harm, but the researchers strongly disagree.) The N.I.H. and other scientific groups are funding new studies to further explore whether anti-inflammatory drugs—for example, low doses of immunomodulatory agents that are used for treating severe arthritis—can help prevent cardiovascular disease.
Another chronic condition that has been linked to inflammation is Type II diabetes. People with this condition can’t adequately use insulin, a molecule that enables the body’s cells to take glucose out of the bloodstream and derive energy from it. Their organs fail and glucose builds to dangerous levels in the blood. Recently, researchers have found macrophages in the pancreases of people with Type II diabetes. The macrophages release inflammatory molecules that are thought to impair insulin activity. One of these inflammatory molecules is called interleukin-1, and in 2007 the New England Journal of Medicine reported on a clinical trial in which an interleukin-1 blocker proved to be modestly effective at lowering blood-sugar levels in Type II diabetics. This suggests that, by blocking inflammation, it might be possible to restore insulin activity and alleviate some of the symptoms of diabetes.
Alzheimer’s disease, too, seems to show a link to inflammation. Alzheimer’s results from the buildup of amyloid and tau proteins in the brain; specialized cells called glial cells, which are related to macrophages, recognize these proteins as debris and release inflammatory molecules to get rid of them. This inflammation is thought to further impair the working of neurons, worsening Alzheimer’s. The connection is tantalizing, but it’s important to note that it doesn’t mean that inflammation causes Alzheimer’s. Nor is there strong evidence that inflammation contributes to other forms of dementia where the brain isn’t filled with protein debris. And in clinical trials anti-inflammatory drugs like naproxen and ibuprofen have failed to ameliorate or prevent Alzheimer’s.
On September 18, 2015, scientists at the N.I.H. convened a meeting to publicly present their research priorities, one of which is to decipher the consequences of inflammation. It’s increasingly apparent that inflammation plays some role in many health conditions, but scientists are far from grasping the nature of that relationship, the mechanisms involved, or the extent to which treating inflammation is helpful.
“We really don’t know how much inflammation contributes to diabetes, Alzheimer’s, depression, and other disorders,” Michael Gottesman, a director of research at the N.I.H., told me. “We know a lot about the mouse and its immune response. Much, much less is understood in humans. As we learn more, we see how much more we need to learn.” Gottesman pointed out that, of the thousand or so proteins circulating in our bloodstream, about a third are involved in inflammation and in our immune response, so simply detecting their presence doesn’t reveal much about their potential involvement in any particular disease. “Correlation is not causation,” he emphasized. “Because you find an inflammatory protein in a certain disorder, it doesn’t mean that it is causing that disorder.”
This lack of certainty hasn’t dampened the enthusiasm of a growing number of doctors who believe that inflammation is the source of a wide range of conditions, including dementia, depression, autism, A.D.H.D., and even aging. One of the most prominent such voices is that of Mark Hyman, whose books—including “The Blood Sugar Solution 10-Day Detox Diet”—are best-sellers. Hyman serves as a personal health adviser to Bill and Hillary Clinton and to the King and Queen of Jordan. Recently, he was recruited by the Cleveland Clinic with millions of dollars in funding to establish a center based on his ideas. Trained in family medicine, Hyman told me that he considers himself a new type of doctor. “I am a doctor who treats root causes and addresses the body as a dynamic system,” he wrote in an e-mail. “Being an inflammalogist is part of that.”
Studies with human subjects clearly indicate that, in cases where inflammation underlies a chronic condition, the inflammation is local: in the arteries (heart disease); or in the brain (Alzheimer’s); or in the pancreas (diabetes). And though there are associations between various forms of inflammatory disease—for example, people with psoriasis or periodontal disease have a somewhat higher risk of heart disease—it has not been proved that there is a causal connection. Hyman and other doctors, such as the neurologist David Perlmutter, promote a more radical idea: that certain foods and environmental toxins cause smoldering inflammation, which somehow spreads to other areas of the body, including the brain, degrading one’s health, mental acuity, and life span.
The notion of a gut-brain connection seems to derive from studies with mice, including one that showed that introducing a bacterium into a mouse’s gastrointestinal tract led to behavioral changes, such as a reluctance to navigate mazes. But there’s scant evidence that anything similar happens in people, or any rigorous study to show that “anti-inflammatory diets” reduce depression. Earlier this year, the journal Brain, Behavior, and Immunity published a meta-analysis of more than fifty clinical studies that found inflammatory molecules in patients with depression. The paper revealed that there was little consistency from study to study about which molecules correlated to the condition. Steven Hyman, a former director of the National Institute of Mental Health and now the head of the Stanley Center at the Broad Institute (and no relation to Mark Hyman), in Cambridge, Massachusetts, noted that depression is “one of those topics where exuberant theorization vastly outstrips the data.”
Nonetheless, Mark Hyman holds fast to his view. “Inflammation is the final common pathway for pretty much all chronic diseases,” he told me. His recommended solution is an “anti-inflammatory diet”—omitting sugar, caffeine, beans, dairy, gluten, and processed foods, as well as taking a variety of supplements, including probiotics, fish oil, Vitamins C and D, and curcumin, a key molecule in turmeric. Hyman introduced me to one of the patients he had treated with his anti-inflammatory diet and supplements, a forty-seven-year-old hedge-fund manager in Cambridge named Jim Silverman. Two decades ago, Silverman began noticing blood in his stool. A colonoscopy resulted in a diagnosis of ulcerative colitis. In the ensuing years, Silverman was treated by gastroenterologists with aspirin-based medication, anti-inflammatory suppositories, and even corticosteroids, but the problem persisted. Then, five years ago, on a flight home from a business conference, he happened to sit next to Hyman, who told him that he could cure colitis.
“I thought, What a bullshitter,” Silverman said. He travelled anyway to Hyman’s UltraWellness Center, in Lenox, Massachusetts, to consult with him. Hyman told him that dairy was inflaming his bowel. Silverman was skeptical, but he kept track of his diet and bleeding episodes, and ultimately concluded that restricting dairy products resulted in long periods without bleeding. He now thinks that he could be suffering from a dairy allergy. In addition to avoiding dairy products, he continues to follow the anti-inflammatory regimen of supplements prescribed by Hyman. “I’m just taking it because I’m doing well,” he said. “I have no idea if it’s doing anything, but I don’t want to rock the boat.”
I asked Gary Wu, a professor of gastroenterology at the Perelman School of Medicine, at the University of Pennsylvania, and one of the world’s experts on the gut microbiome, about the alleged value of treating inflammatory bowel disease by restricting specific foods. Recently, in the journal Gastroenterology, Wu and his colleagues published a comprehensive review of scientific studies on diet and inflammatory bowel disease. They found only two dietary interventions that had been proved to reduce inflammation: an “elemental diet,” which is a liquid mixture of amino acids, simple sugars, and triglycerides, and a slightly more complex liquid diet. The liquid mixtures are typically administered with a tube placed through the nose. “The diet is not palatable,” Wu said. “And you don’t eat during the day. There is no intake of whole foods at all.”
David Agus, a cancer specialist and a professor of medicine and engineering at the University of Southern California, is equally skeptical of Hyman’s claims for the anti-inflammation diet. Agus, who is perhaps best known for being the doctor on “CBS This Morning,” recently received a multimillion-dollar grant from the National Cancer Institute to study how inflammation may spur the growth of tumors. “This notion that foods cause inflammation and foods can block inflammation, there’s zero data that it changes clinical outcomes,” he told me. “If the idea gets people to eat fruits and vegetables, I love it, but it’s not real.” Agus noted that vitamins don’t counter inflammation, and that it’s been shown, in rigorous clinical trials, that they may increase one’s risk of developing cancer.
Still, Agus views inflammation as a component not only of cancer but also of chronic diseases like diabetes and dementia. Rather than special diets, he supports preventively taking approved anti-inflammatory medications, such as aspirin and statins, and scrupulously scheduling the standard vaccinations in order to prevent infections. In “The End of Illness,” Agus encourages the reader to “reduce your daily dose of inflammation” by, among other things, not wearing high heels, since these can inflame your feet and the inflammation could possibly affect your vital organs. When I pressed him on that suggestion, he told me, “What I meant is that if your feet hurt all day it’s probably not a good thing. The downside is you just wear a different pair of shoes. The upside is it gave you an understanding of inflammation and its role in disease.”
Mark Hyman, at times, acknowledges the possible limits of his paradigm. When I asked him about the alleged link among gut inflammation, diet, and psychological disorders, he conceded that some of his evidence was anecdotal, derived from his own clinical practice. He mentioned the case of a child with asthma, eczema, and A.D.H.D., whom he treated with “an elimination diet, taking him off processed foods, and giving him supplements.” The child’s allergic problems improved and his behavior was markedly better, Hyman said: “It was a light-bulb moment. I saw secondary effects on the brain that came out of treating physical problems.”
He also cited studies of patients with rheumatoid arthritis, a painful and debilitating auto-immune condition that inflames and erodes the joints, who became less depressed after being treated with inflammatory blockers. But had the anti-inflammatory treatment directly lifted their depression, or had their mood improved simply because they were more mobile and in less pain? I told Hyman that it was hard to connect the dots. “For sure,” he said.
Connecting the dots is a challenge even for scientists who are actively involved in inflammation research. One afternoon, I visited Ramnik Xavier, the chair of gastroenterology at Massachusetts General Hospital and an expert in Crohn’s disease and ulcerative colitis. The bowel is inflamed in both conditions: ulcerative colitis affects the colon, whereas Crohn’s disease can affect any part of the digestive system. But the nature of inflammation varies almost from person to person and involves interactions among DNA, many kinds of gastrointestinal cells, and the peculiarities of the gut microbiome. “Lots of cells, lots of genes, lots of bugs,” Xavier said.
Xavier, a compact man with a laconic manner and thick black hair marked by streaks of gray, initially studied the role of specialized white blood cells, known as T-cells and B-cells, in defending the body against the development of colitis. Eventually, with Mark Daly, a geneticist at the Broad Institute, Xavier began to search for genes that predispose people to inflammatory bowel disease and for genes that might protect them against it. The two scientists, as part of an international consortium, have identified at least a hundred and sixty areas of DNA that are associated with an increased risk of inflammatory bowel disease; Xavier’s lab has zeroed in on about two dozen genes within these regions of DNA.
One of the frustrations of treating inflammation is that our weapons against it are so imprecise. Drugs like naproxen and ibuprofen are the equivalent of peashooters. At the other extreme, cannon-like steroids shut down the immune system, raising the risk of infection, eroding the bones, predisposing the patient to diabetes, and causing mood swings. Even the peashooters can cause collateral damage: aspirin may help to protect against colon cancer, heart attack, and stroke, but it also raises the risk of gastrointestinal bleeding. Ibuprofen, naproxen, and similar drugs were labelled by the F.D.A. as increasing the risk of heart attack and stroke in people who’ve never suffered either condition, and clinical trials failed to show that they prevent or ameliorate dementia. (Although these drugs reduce inflammation, they may also alter the lining of blood vessels and increase the risk of clots.) Statins lower the chance of a heart attack, but there is growing concern not only about the side effect of muscle pain but also about increasing the likelihood of diabetes. And the absolute benefits of these preventive medications is slight, measured in single digits.
In the lab at the Broad Institute, Xavier and his team were trying to discover new treatments that can block inflammation in a targeted manner. The day I visited, they were assessing molecules associated with colitis, especially one called interleukin-10, or IL-10, which is known to decrease inflammation. In a cavernous room, I watched as a robotic arm moved among racks of plastic plates, each containing hundreds of small wells in which chemical compounds were being tested. Some people with Crohn’s disease have genetic mutations that disable the salubrious effects of IL-10. Xavier is trying to identify molecules that can compensate for this deficiency, in the hope that such molecules might eventually be turned into drugs to treat this subset of patients.
But other patients suffer from a different manifestation of Crohn’s—they can’t fully clear debris from cells in their gut, so it builds up, triggering inflammation. In a neighboring lab, members of Xavier’s research team were trying to develop drugs for that condition, too. A robotic arm was handling plates that contained genetically engineered cells and moving them under a fluorescent microscope. The images appeared on a computer screen—fields of cells studded with yellow and green dots, like the sky in van Gogh’s “Starry Night.”
On another visit, Xavier took me to his clinic at Mass General. Patients, ranging from the very young to the elderly, were reclining in Barcaloungers as nurses and physicians intravenously administered potent anti-inflammatory drugs. Later, I spoke by phone to one of Xavier’s patients, a forty-nine-year-old woman named Maria Ray, who received a diagnosis of colitis in 1998. She was treated with sulfa drugs and corticosteroids, which controlled the problem for several years, but in 2004, after a series of flare-ups, she underwent surgery to remove her colon. Soon after, she developed ulcers on her skin, arthritis of her knees and elbows, and inflammation in both eyes. Xavier prescribed other drugs, and for two years her condition improved, but lately her skin lesions and eye inflammation have returned. “We hoped surgery would cure her ulcerative colitis,” Xavier said. “But we don’t really understand why there is such an overactive immune system now inflaming these other parts of her body.”
At the very least, the fact that Ray has symptoms in many organs, despite the removal of her colon, complicates the simplistic view that treating the gut will suppress inflammation elsewhere. Moreover, there’s no evidence that patients with Crohn’s or colitis are more likely than average to develop dementia and other cognitive disorders. “What we see in mice is not always reproduced in people,” Xavier said.
Perhaps no aspect of inflammation is more compelling, or illusory, than the idea that it may be responsible for aging. An internist friend in Manhattan told me that healthy patients occasionally come in to her office carrying Mark Hyman’s books, eager to live longer by following his anti-inflammation life style. When I asked Hyman if he could introduce me to someone who follows his longevity regimen, he readily offered himself. “I’m aiming to live to a hundred and twenty,” he said.
The notion stems from grains of evidence, such as studies that have shown an increase in inflammation with age. The genesis of aging is still a mystery. It may occur for a host of reasons—a waning of the energy generated by the mitochondria within cells, the tendency of DNA to grow fragile and more mutation-prone over time—and it’s much too simplistic to attribute the process to inflammation alone. Luigi Ferrucci, the scientific director of the National Institute on Aging, conducted some of the early research on inflammation and aging, and for a while, he told me, he believed the avenue held promise. On the morning we spoke, he had just finished his daily six-mile run. Sixty-one years old, born in Livorno, on the coast of Tuscany, Ferrucci is an animated man with a stubbly beard who favors crew-neck sweaters. In the past four decades, he has studied thousands of people in order to identify the biological processes that result in aging. He measured scores of molecules in the blood, hoping to find clues that would lead him to the cause of aging’s hallmarks, particularly sarcopenia, or loss of muscle mass, and cognitive decline.
His most illuminating studies involved people in late middle age who showed no sign of heart disease, diabetes, dementia, or other conditions that might be associated with inflammation. He found that a single inflammatory molecule, called interleukin-6, was the most powerful predictor of who would eventually become disabled. Healthy patients with high levels of the IL-6 molecule aged more quickly and grew sicker than those without the inflammatory molecule. “I thought I had discovered the cause of aging and was going to win the Nobel Prize,” Ferrucci said, laughing.
But then he found other subjects with no evidence of inflammation, and without elevated levels of IL-6 or other inflammatory molecules, whose bodies nevertheless began to decline. “We are looking at the layer, not at the core of the problem,” he said. “Inflammation may accelerate aging in some people—but it is a manifestation of something that is occurring underneath.” He reiterated the point that correlation is not causation. “If you have the curiosity of the scientist, you can’t stop there, because you want to know why,” he said. “You want to break the toy so you can see how it’s working inside.”
Toward that end, Ferrucci recently organized a large team of collaborators and launched a new clinical study, GESTALT, which stands for Genetic and Epigenetic Signatures of Translational Aging Laboratory Testing. Groups of healthy people will be studied intensively as they age, with detailed analyses of their DNA, RNA, proteins, metabolic capacity, and other sophisticated parameters, every two years for at least a decade. “Then we can say what mechanisms account for increased inflammation with aging, and the loss of muscle mass, or the loss of memory, or the loss of energy capacity or fitness,” Ferrucci said. “These have never really been addressed on a deep level in humans.”
In the meantime, he sticks to a Mediterranean diet, mainly out of fealty to his heritage. (Ferrucci is known among his N.I.H. colleagues as a gourmet Italian cook.) The media recently gave much attention to a study, published in 2013 in the New England Journal of Medicine, on the benefits of a Mediterranean diet in preventing heart attack or stroke. But, as Ferrucci noted, the benefits weren’t clearly related to inflammation and they accrued to a very small percentage of the subjects on the diet. “Believe me, if there were a diet that prevented aging, I would be on it,” he said.
We’d all like a simple solution for complex medical problems. We’re desperate to feel in command of our lives, particularly as we age and see friends and family afflicted by Alzheimer’s, stroke, and heart failure. “My patients, understandably, are very focussed on the foods they eat, wanting control, hoping they won’t have to take immune-suppressive treatments,” Gary Wu, the University of Pennsylvania gastroenterologist, told me.
Some years ago, I became obsessed with a restrictive diet—no bread, cheese, ice cream, cookies—in an attempt to lower my cholesterol levels. (My father died of a heart attack in his fifties, and I was haunted by his fate.) After nearly six months, I’d lost some fifteen pounds, but my cholesterol level had hardly budged, and I’d become so vigilant about everything I ate that I stopped enjoying meals. Gradually, I resumed a balanced and more reasonable diet and regained an appreciation for one of life’s fundamental pleasures.
Scientists may yet discover that inflammation contributes to disease in unexpected ways. But it’s important to remember, too, that inflammation serves a vital role in the body. “We are playing with one of the primary mechanisms selected by nature to maintain the integrity of our body against the thousand environmental attacks that we receive every day,” Ferrucci said. “Inflammation is part of our maintenance and repair system. Without it, we can’t heal.”
This year’s American College of Rheumatology (ACR) meeting, which took place in San Francisco from November 6 to 11, reflected an unprecedented level of innovation, including numerous advances in the area of rheumatoid arthritis (RA).
BioPharma Dive spoke with executives from Pfizer and Eli Lilly to discuss the new treatment landscape and ongoing innovation in this therapeutic space.
Old innovation makes way for new innovation
Twenty years ago, the standard of care for RA was some combination of basic NSAIDS, along with methotrexate. Caregivers focused on symptom relief, and it was widely understood that many patients would fail to achieve remission. As the disease developed, patients would eventually develop severely life-limiting disabilities as their disease progressed.
During this period, researchers presenting at conferences grew excited about data on a new class of drugs known as anti-tumor necrosis factor (TNF) antibodies. In an article published in Acta Orthopaedica Scandinavica in 1995, two physician-researchers wrote the following:
“Primary results have recently been published on the use of anti-TNF monoclonal antibodies. In a controlled trial these antibodies were able to significantly influence a number of disease-activity variables in RA. An important observation was that the clinical effect lasted from weeks to, in some cases, months. Although the potential of these agents for clinical use is still uncertain, these observations suggest that interfering with certain targets of the immune-inflammatory process is possible, effective and so far without side effects.”
About four years after Drs. Van de Putte and Van Riel extolled the virtues of disease-modifying biologics in clinical trials, the first anti-TNF antibody, Remicade (infliximab) was approved in 1999. At that point, the standard of care for RA improved significantly, forever changing the treatment paradigm for patients with RA.
The expanding class of JAK inhibitors
At this year’s ACR meeting, researchers focused on anti-inflammatory antibodies and a relatively new class of oral drugs known as janus kinase (JAK) inhibitors. Interest in JAK inhibitors has spiked since the approval of Pfizer’s oral medication Xeljanz (tofacitinib) —the first, and currently the only, JAK inhibitor approved for the treatment of moderate-to-severe RA.JAK inhibitors have garnered interest because of the role they can play in expanding a treatment area dominated by synthetic and biologic disease-modifying anti-rheumatic drugs (DMARDs). Could JAK inhibitors provide the breakthrough in RA that the anti-TNF antibodies provided almost 20 years ago?
Currently, Eli Lilly and Incyte are in late-stage development of baricitinib, a JAK1/JAK2 inhibitor for treatment of RA. Until last December, Johnson & Johnson (J&J) and Astellas were working jointly on another JAK inhibitor, known as ASPO15K, but J&J exercised its opt-out option and left the partnership. Astellas vowed to go it alone or look for a new partner, but there have not been many updates on ASPO15K within the last year.
Innovation means understanding and responding to unmet needs
Like many other therapeutic areas, RA treatments are often used in combination. For some patients, the combination of methotrexate and a powerful biologic, such as Remicade (infliximab), will help a patient achieve remission Yet others will either not respond to methotrexate and Remicade, or will have a negative reaction. Understanding how to help nonresponders achieve relief has become a key area of research in RA.
According to Terence Rooney, MD, Medical Director at Lilly Bio-Medicines, “A substantial proportion of patients treated with methotrexate – commonly used across the disease continuum for 25 years – do not achieve satisfactory disease control, signaling a need for more effective RA treatment options. In addition, studies have shown that some patients who initially respond to biologics lose response over time, and approximately 40 percent of patients with high disease activity never respond adequately to TNF antagonist biologics.”
Innovative clinical trial design
As Lilly and Incyte approach the end of the development process for baricitinib, they have been collecting results from clinical trials designed to both establish basic efficacy and safety in placebo-controlled and comparator trials, and to obtain data on targeted patient populations.
According to Rooney, “The baricitinib phase three program investigated the benefit of baricitinib across the spectrum of patients with rheumatoid arthritis, including newly diagnosed patients, patients who had failed to respond to conventional DMARDs, and patients who had failed multiple injectable biologic DMARD therapies.”
“In addition, the phase 3 program included two 52-week studies that incorporated either methotrexate or adalimumab as active comparators to provide useful information for therapeutic positioning of baricitinib. In these studies, baricitinib was statistically superior to methotrexate and to adalimumab in improving signs and symptoms, physical function, and important patient-reported outcomes including pain, fatigue and stiffness.”
Rooney also pointed out that there is additional data establishing baricitinib as a DMARD that significantly inhibits progressive radiographic joint damage.
Experience plus evidence equals more innovation
As has become the norm, companies at ACR often highlight new data confirming the efficacy and safety of already approved drugs in larger patient populations and in real-world settings..
Lilly currently has data on more than 40,000 patients worldwide, reflecting its global ambitions. Assuming that baricitinib is approved next year (the goal is to file at the end of the year), Lilly will continue to present data at ACR in the coming years highlighting the results of its long-term extension study, RA-BEYOND.
Pfizer’s up-to-date Xeljanz data presentation at ACR
Although Xeljanz has been on the market for three years in more than 40 countries, Pfizer continues to focus on collecting new data and using it to expand use of Xeljanz. In fact, Pfizer had 20 abstracts focused solely on Xeljanz at ACR 2015.
According to Rory O’Connor, MD, Senior Vice President and Head of Global Medical Affairs, Global Innovative Pharmaceuticals Business, Pfizer, “Ongoing clinical trials and long-term extension studies provide important information about the safety and efficacy of Xeljanz in RA. We are focused on continuing to build on our knowledge of the clinical application of Xeljanz in real-world settings.”
Pfizer was also able to highlight new data that supports their recent NDA for Xeljanz XR, a once-daily formulation of Xeljanz, which is currently approved as a twice-daily dosing formulation.
JAK inhibition beyond RA
One of the most exciting things about the progress with JAK inhibitors is the possibility to innovate treatments beyond RA. Lilly has been exploring the role of JAK-dependent cytokines in the pathogenesis of numerous inflammatory and autoimmune diseases. The company also plans to meet with regulatory authorities to develop a pediatric program for juvenile RA and idiopathic arthritis.
Meanwhile, Pfizer has developed a broad portfolio of various JAK inhibitors and therapies with new modes of action. Already, Pfizer researchers have completed two phase three studies in ulcerative colitis and the top-line results have been positive.
Medical meetings are exciting, because they provide a forum for discussing breakthroughs and portending a future in which the standard of care improves. For companies like Lilly, Incyte, and Pfizer, continual development of more novel approaches to serious diseasesis like a call-response echo chamber in which innovation drives more innovation, resulting in better long-term outcomes for patients.
The JAK/STAT signaling pathway
Jason S. Rawlings, Kristin M. Rosler, Douglas A. Harrison
The Janus kinase/signal transducers and activators of transcription (JAK/STAT) pathway is one of a handful of pleiotropic cascades used to transduce a multitude of signals for development and homeostasis in animals, from humans to flies. In mammals, the JAK/STAT pathway is the principal signaling mechanism for a wide array of cytokines and growth factors. JAK activation stimulates cell proliferation, differentiation, cell migration and apoptosis. These cellular events are critical to hematopoiesis, immune development, mammary gland development and lactation, adipogenesis, sexually dimorphic growth and other processes. Predictably, mutations that reduce JAK/STAT pathway activity affect these processes (reviewed by Igaz et al., 2001; O’Shea et al., 2002). Conversely, mutations that constitutively activate or fail to regulate JAK signaling properly cause inflammatory disease, erythrocytosis, gigantism and an array of leukemias. Here we present a general overview of the JAK/STAT pathway and illustrate the primary mechanisms of activation and regulation of this essential signaling cascade.
Mechanistically, JAK/STAT signaling is relatively simple, with only a few principal components (for reviews, see Aaronson and Horvath, 2002; Heinrich et al., 2003; Kisseleva et al., 2002; O’Shea et al., 2002). As described above, a variety of ligands and their receptors stimulate the JAK/STAT pathway. Intracellular activation occurs when ligand binding induces the multimerization of receptor subunits. For some ligands, such as erythropoietin and growth hormone, the receptor subunits are bound as homodimers while, for others, such as interferons and interleukins, the receptor subunits are heteromultimers. For signal propagation through either homodimers or heteromultimers, the cytoplasmic domains of two receptor subunits must be associated with JAK tyrosine kinases. JAKs are distinctive in that they have tandem kinase-homologous domains at the C-terminus. The first is a non-catalytic regulatory domain, whereas the second has tyrosine kinase activity. In mammals, the JAK family comprises four members: JAK1, JAK2, JAK 3 and Tyk2. JAK activation occurs upon ligand-mediated receptor multimerization because two JAKs are brought into close proximity, allowing trans-phosphorylation. The activated JAKs subsequently phosphorylate additional targets, including both the receptors and the major substrates, STATs. STATs are latent transcription factors that reside in the cytoplasm until activated. The seven mammalian STATs bear a conserved tyrosine residue near the C-terminus that is phosphorylated by JAKs. This phosphotyrosine permits the dimerization of STATs through interaction with a conserved SH2 domain. Phosphorylated STATs enter the nucleus by a mechanism that is dependent on importin α-5 (also called nucleoprotein interactor 1) and the Ran nuclear import pathway. Once in the nucleus, dimerized STATs bind specific regulatory sequences to activate or repress transcription of target genes. Thus the JAK/STAT cascade provides a direct mechanism to translate an extracellular signal into a transcriptional response.
In addition to the principal components of the pathway, other effector proteins have been identified that contribute to at least a subset of JAK/STAT signaling events. STAMs (signal-transducing adapter molecules) are adapter molecules with conserved VHS and SH3 domains (Lohi and Lehto, 2001). STAM1 and STAM2A can be phosphorylated by JAK1-JAK3 in a manner that is dependent on a third domain present in some STAMs, the ITAM (inducible tyrosine-based activation motif). Through a poorly understood mechanism, the STAMs facilitate the transcriptional activation of specific target genes, including MYC. A second adapter that facilitates JAK/STAT pathway activation is StIP (stat-interacting protein), a WD40 protein. StIPs can associate with both JAKs and unphosphorylated STATs, perhaps serving as a scaffold to facilitate the phosphorylation of STATs by JAKs. A third class of adapter with function in JAK/STAT signaling is the SH2B/Lnk/APS family. These proteins contain both pleckstrin homology and SH2 domains and are also substrates for JAK phosphorylation. Both SH2-Bβ and APS associate with JAKs, but the former facilitates JAK/STAT signaling while the latter inhibits it. The degree to which each of these adapter families contributes to JAK/STAT signaling is not yet well understood, but it is clear that various proteins outside the basic pathway machinery influence JAK/STAT signaling.
In addition to JAK/STAT pathway effectors, there are three major classes of negative regulator: SOCS (suppressors of cytokine signaling), PIAS (protein inhibitors of activated stats) and PTPs (protein tyrosine phosphatases) (reviewed by Greenhalgh and Hilton, 2001). Perhaps the simplest are the tyrosine phosphatases, which reverse the activity of the JAKs. The best characterized of these is SHP-1, the product of the mouse motheaten gene. SHP-1 contains two SH2 domains and can bind to either phosphorylated JAKs or phosphorylated receptors to facilitate dephosphorylation of these activated signaling molecules. Other tyrosine phosphatases, such as CD45, appear to have a role in regulating JAK/STAT signaling through a subset of receptors.
SOCS proteins are a family of at least eight members containing an SH2 domain and a SOCS box at the C-terminus (reviewed by Alexander, 2002). In addition, a small kinase inhibitory region located N-terminal to the SH2 domain has been identified for SOCS1 and SOCS3. The SOCS complete a simple negative feedback loop in the JAK/STAT circuitry: activated STATs stimulate transcription of the SOCS genes and the resulting SOCS proteins bind phosphorylated JAKs and their receptors to turn off the pathway. The SOCS can affect their negative regulation by three means. First, by binding phosphotyrosines on the receptors, SOCS physically block the recruitment of signal transducers, such as STATs, to the receptor. Second, SOCS proteins can bind directly to JAKs or to the receptors to specifically inhibit JAK kinase activity. Third, SOCS interact with the elongin BC complex and cullin 2, facilitating the ubiquitination of JAKs and, presumably, the receptors. Ubiquitination of these targets decreases their stability by targeting them for proteasomal degradation.
The third class of negative regulator is the PIAS proteins: PIAS1, PIAS3, PIASx and PIASy. These proteins have a Zn-binding RING-finger domain in the central portion, a well-conserved SAP (SAF-A/Acinus/PIAS) domain at the N-terminus, and a less-well-conserved carboxyl domain. The latter domains are involved in target protein binding. The PIAS proteins bind to activated STAT dimers and prevent them from binding DNA. The mechanism by which PIAS proteins act remains unclear. However, PIAS proteins have recently been demonstrated to associate with the E2 conjugase Ubc9 and to have E3 conjugase activity for sumoylation that is mediated by the RING finger domain (reviewed by Jackson, 2001). Although there is evidence that STATs can be modified by sumoylation (Rogers et al., 2003), the function of that modification in negative regulation is not yet known.
Although the mechanism of JAK/STAT signaling is relatively simple in theory, the biological consequences of pathway activation are complicated by interactions with other signaling pathways (reviewed by Heinrich et al., 2003; Rane and Reddy, 2000; Shuai, 2000). An understanding of this cross-talk is only beginning to emerge, but the best characterized interactions of the JAK/STAT pathway are with the receptor tyrosine kinase (RTK)/Ras/MAPK (mitogen-activated protein kinase) pathway. The relationship between these cascades is complex and their paths cross at multiple levels, each enhancing activation of the other. First, activated JAKs can phosphorylate tyrosines on their associated receptors that can serve as docking sites for SH2-containing adapter proteins from other signaling pathways. These include SHP-2 and Shc, which recruit the GRB2 adapter and stimulate the Ras cascade. The same mechanism stimulates other cascades, such as the recruitment and JAK phosphorylation of insulin receptor substrate (IRS) and p85, which results in the activation of the phosphoinositide 3-kinase (PI3K) pathway [for more on PI3K signaling, see Foster et al. (Foster et al., 2003)]. JAK/STAT signaling also indirectly promotes Ras signaling through the transcriptional activation of SOCS3. SOCS3 binds RasGAP, a negative regulator of Ras signaling, and reduces its activity, thereby promoting activation of the Ras pathway. Reciprocally, RTK pathway activity promotes JAK/STAT signaling by at least two mechanisms. First, the activation of some RTKs, including EGFR and PDGFR, results in the JAK-independent tyrosine phosphorylation of STATs, probably by the Src kinase. Second, RTK/Ras pathway stimulation causes the downstream activation of MAPK. MAPK specifically phosphorylates a serine near the C-terminus of most STATs. While not absolutely necessary for STAT activity, this serine phosphorylation dramatically enhances transcriptional activation by STAT. In addition to RTK and PI3K interactions with JAK/STAT signaling, multiple levels of cross-talk with the TGF-β signaling pathway have been recently reported [for a review of TGF-β, see (Moustakas, 2002)]. Furthermore, the functions of activated STATs can be altered through association with other transcription factors and cofactors that are regulated by other signaling pathways. Thus the integration of input from many signaling pathways must be considered if we are to understand the biological consequences of cytokine stimulation.
Agaisse, H., Petersen, U. M., Boutros, M., Mathey-Prevot, B. and Perrimon, N. (2003). Signaling role of hemocytes in Drosophila JAK/STAT-dependent response to septic injury. Dev. Cell5, 441-450.
The JAK/STAT secondary messenger signaliing pathway..
Presented by: Joseph Farahany, M.D
Jak/Stat Signaling Pathway
Jaks and Stats are critical components of many cytokine receptor systems; regulating growth, survival, differentiation, and pathogen resistance. An example of these pathways is shown for the IL-6 (or gp130) family of receptors, which coregulate B cell differentiation, plasmacytogenesis, and the acute phase reaction. Cytokine binding induces receptor dimerization, activating the associated Jaks, which phosphorylate themselves and the receptor. The phosphorylated sites on the receptor and Jaks serve as docking sites for the SH2-containing Stats, such as Stat3, and for SH2-containing proteins and adaptors that link the receptor to MAP kinase, PI3K/Akt, and other cellular pathways.
Phosphorylated Stats dimerize and translocate into the nucleus to regulate target gene transcription. Members of the suppressor of cytokine signaling (SOCS) family dampen receptor signaling via homologous or heterologous feedback regulation. Jaks or Stats can also participate in signaling through other receptor classes, as outlined in the Jak/Stat Utilization Table. Researchers have found Stat3 and Stat5 to be constitutively activated by tyrosine kinases other than Jaks in several solid tumors
The Jak/Stat pathway mediates the effects of cytokines, like erythropoietin, thrombopoietin, and G-CSF, which are protein drugs for the treatment of anemia, thrombocytopenia, and neutropenia, respectively. The pathway also mediates signaling by interferons, which are used as antiviral and antiproliferative agents. Researchers have found that dysregulated cytokine signaling contributes to cancer. Aberrant IL-6 signaling contributes to the pathogenesis of autoimmune diseases, inflammation, and cancers such as prostate cancer and multiple myeloma. Jak inhibitors currently are being tested in models of multiple myeloma. Stat3 can act as an oncogene and is constitutively active in many tumors. Crosstalk between cytokine signaling and EGFR family members is seen in some cancer cells. Research has shown that in glioblastoma cells overexpressing EGFR, resistance to EGFR kinase inhibitors is induced by Jak2 binding to EGFR via the FERM domain of the former [Sci. Signal. (2013) 6, ra55].
Activating Jak mutations are major molecular events in human hematological malignancies. Researchers have found a unique somatic mutation in the Jak2 pseudokinase domain (V617F) that commonly occurs in polycythemia vera, essential thrombocythemia, and idiopathic myelofibrosis. This mutation results in the pathologic activation Jak2, associated with receptors for erythropoietin, thrombopoietin, and G-CSF, which control erythroid, megakaryocytic, and granulocytic proliferation and differentiation. Researchers have also shown that somatic acquired gain-of-function mutations of Jak1 are found in adult T cell acute lymphoblastic leukemia. Somatic activating mutations in Jak1, Jak2, and Jak3 have also been identified in pediatric acute lymphoblastic leukemia (ALL). Furthermore, Jak2 mutations have been detected around pseudokinase domain R683 (R683G or DIREED) in Down syndrome childhood B-ALL and pediatric B-ALL.
Universal and essential to cytokine receptor signaling, the JAK-STAT pathway is one of the best understood signal transduction cascades. Almost 40 cytokine receptors signal through combinations of four JAK and seven STAT family members, suggesting commonality across the JAK-STAT signaling system. Despite intense study, there remain substantial gaps in understanding how the cascades are activated and regulated. Using the examples of the IL-6 and IL-10 receptors, I will discuss how diverse outcomes in gene expression result from regulatory events that effect the JAK1-STAT3 pathway, common to both receptors. I also consider receptor preferences by different STATs and interpretive problems in the use of STAT-deficient cells and mice. Finally, I consider how the suppressor of cytokine signaling (SOCS) proteins regulate the quality and quantity of STAT signals from cytokine receptors. New data suggests that SOCS proteins introduce additional diversity into the JAK-STAT pathway by adjusting the output of activated STATs that alters downstream gene activation.
The mammalian JAK and STAT family members have been extensively, and seemingly exhaustively, analyzed in the mouse and human systems. All four JAK and seven STAT family members have been deleted in the mouse, in addition to the creation of conditional alleles for genes whose loss of function leads to embryonic or perinatal lethality (Stat3, combined deficiency of Stat5a and Stat5b, and Jak2). In humans, detailed genetic studies have been performed in people bearing mutant Jak or Stat genes. Specific Abs to phospho-forms of each protein are used to study how the JAK-STAT cascade is activated by cytokine receptors. Crystallographic studies have illuminated structural information for multiple STAT family members in different forms. Pharmacological inhibitors have been developed for clinical use where JAK-STAT signaling is implicated in disease pathology and progression. Finally, in most cases, a specific JAK-STAT combination has been paired with each cytokine receptor, and this information translated into cell-type specific patterns of cytokine responsiveness and gene expression.
Major questions remain concerning how the JAK-STAT cascade functions to control specific gene expression patterns, and how the cascades are regulated. I will describe three elements of JAK-STAT signaling that require experimental investigation. First, I will address an unexpected experimental complication that arises from the analysis of mice and cells that lack one or more STAT family member. Second, I will use JAK1-STAT3 signaling from the IL-10R and IL-6R systems to illustrate that we lack detailed understanding of how specificity in gene expression is generated by receptors that use identical JAK-STAT members. Third, we have yet to explain how STAT activation is negatively regulated. Although the suppressor of cytokine signaling (SOCS)3 proteins are the best understood negative regulators of the JAK-STAT pathway, the biochemical mechanism of SOCS-mediated inhibition is unexplained. Moreover, additional inhibitory pathways have also been proposed to block the production of activated STATs. Collectively, I will argue that our understanding of the pathway from cytokine receptor to gene expression profile is in its infancy, but remains one of the best opportunities to dissect signal transduction.
Overview of the proximal JAK-STAT activation mechanism
The current model of JAK-STAT signaling holds that cytokine receptor engagement activates the associated JAK combination, which in turn phosphorylates the receptor cytoplasmic domain to allow recruitment of a STAT, which in turn is phosphorylated, dimerizes and moves to the nucleus to bind specific sequences in the genome and activate gene expression. Cytoplasmic domains of cytokine receptors associate with JAKs via JAK binding sites located close to the membrane (1). The postulated role of JAKs in trafficking or chaperoning the receptors to the cell surface is debated (2, 3, 4, 5, 6). Regardless of the when and where cytokine receptors and JAKs associate, their close apposition at the membrane is required to stimulate the kinase activity of the JAK following cytokine binding. At this stage in the activation of the pathway, we understand next to nothing about the structural basis of the JAK-receptor interaction, how receptor intracellular domains reorient upon cytokine binding and physically contact the JAK to receive the phosphorylation modification.
JAK-mediated phosphorylation of the receptor creates binding sites for the Src homology 2 (SH2) domains of the STATs. STAT recruitment is followed by tyrosine, and in some cases, serine phosphorylation on key residues (by the JAKs and other closely associated kinases) that leads to transit into the nucleus. This brief summary of the activation of the JAK-STAT pathway omits numerous unresolved details: the STAT monomer to dimer transition has been questioned, as has the role of phosphorylation in dimerization and nuclear transit (7). Furthermore, it is unclear how many configurations of STAT homo- and heterocomplexes are present in cells before, during, and after cytokine stimulation (8, 9,10). We do not understand the detailed structural basis for the preference of one SH2 domain for a given receptor, and we have little knowledge of how other non-JAK kinases are recruited to the receptors and phosphorylate the STATs.
Many receptors signal through a small number of JAKs
Cytokine receptors signal through two types of pathways: the JAK-STAT pathway and other pathways that usually involve the activation of the MAP kinase cascade. Although the latter will not be discussed here, it is worth noting that elegant genetic studies have demonstrated the importance of these pathways in various pathological systems (11, 12,13, 14). There are now ∼36 cytokine receptor combinations that respond to ∼38 cytokines (counting the type I IFNs as one because they all signal through the IFN-αβR). Different cells and tissues express distinct receptor combinations that respond to cytokine combinations unique to the microenvironment or systemic response of the organism. Hence, at any given time, a single cell may integrate signals from multiple cytokine receptors. Genetic studies have established that the cytokine receptor system is restrictive in that different classes of receptors preferentially use one JAK or JAK combination (7): receptors required for hemopoietic cell development and proliferation use JAK2, common γ-chain receptors use JAK1 and JAK3 whereas other receptors use only JAK1 (Fig. 1⇓). Unexplained is the selective use of these combinations: why the IFN-γR rigidly uses the JAK1, JAK2 combination is unknown as is the restricted use of TYK2. Compared with JAK1–3, TYK2 is unusual in that loss of function mutations in the mouse have shown obligate, but not absolute, requirements in IFN-αβR and IL-12R signaling (15, 16). In contrast, human TYK2 seems to be essential for signaling through a broader range of cytokine receptors (17).
FIGURE 1.
The majority of cytokine receptors use three JAK combinations. Shown are well-studied cases where JAK usage by each cytokine receptor has been established by genetic and biochemical studies. Exceptions shown are the G-CSFR (∗) where it is currently unclear whether both JAK1 and JAK2 are required together. Additionally, the IL-12R (†) and IL-23R (†) require TYK2 but the requirement for JAK2 has not been definitively determined. Receptors that use JAK2 and JAK3, JAK3 alone, TYK2 alone, or JAK3 and TYK2 have not been described.
The preferential association of JAKs to certain receptor classes raises several issues. First, how did the JAK-receptor combinations evolve? Because the number of receptors is relatively large, why has the number of JAKs remained small? Why have the combinations of JAK pairs also remained small given that there are 10 possible combinations that can be used (Fig. 1⇑)? Second, how flexible is the cytokine receptor-JAK pair? That is, can receptors be engineered for interchangeable JAK use, or is a given JAK combination fixed for a specific receptor class? For example, can JAK1, JAK3, or TYK2 activate erythropoietin receptor (EpoR) signaling (if so engineered) or is JAK2 obligatory for signaling? These questions allude to a fundamental issue that concerns the function of the JAK in cytokine receptor activation: if the only function of the JAKs is to phosphorylate tyrosine resides on the cytoplasmic domain of the receptors, then it should be possible to trade JAK-receptor pairs. If these receptors retain identical downstream gene expression profiles, then the signal generated by the JAK is generic and functions primarily to activate the receptor (6). Conversely, it is also possible that each receptor-JAK combination retains crucial specificity functions and swapping, for example, JAK1 for JAK2 on the EpoR will modify or destroy a specific function in erythrogenesis. These questions can be addressed experimentally by replacing one preferred JAK binding site for another in genes encoding different receptors. The EpoR is a good test example because the activity of the receptor and its signaling pathway is essential for life and erythropoiesis is readily assayed.
Core versus cell-type specific STAT signaling
Microarray experiments designed to monitor changes in gene expression induced by JAK-STAT signaling have revealed that both cell-type specific transcription and core, or stereotypic, mRNA profiles are induced by activated cytokine receptors in different cell types (Fig. 2⇓). For example, IFN-γ, via STAT1, induces the expression of a similar cohort of genes regardless of the cell type tested (18). These genes are often termed the “IFN signature” and overlap with the gene expression pattern induced by IFN-αβ signaling that also involves STAT1, in cooperation with STAT2 and IRF9. The IFN signature is readily observed in microarray experiments and is indicative of STAT1 activity. The STAT6 pathway activated by IL-4 or IL-13 provides an example of a cell-type specific response. IL-4-regulated genes in T cells have a distinct signature compared with IL-4/IL-13 signaling in macrophages or other non-lymphocytes (19, 20, 21, 22). In the latter, genes such as Arg1(encoding arginase 1) are often induced >100-fold but are silent in T cells (23, 24, 25, 26,27). Collectively these data argue that STATs activate defined gene sets, depending on their genomic accessibility, and possibly on cofactors that further refine gene expression profiles. STAT3 signaling illustrates a more complex system and will be discussed below to illustrate the distinctions between IL-6 and IL-10 signaling.
FIGURE 2.
Core signaling by STATs. Representative examples of gene expression induced by STAT signaling in different tissues. The examples were extracted and edited from numerous microarray and empirical studies.
Interpreting experiments using STAT loss-of-function systems
Experiments with the different STAT knockout mice, and cells derived from these animals, have been critical for understanding specific requirements of individual STATs in gene expression following cytokine receptor signaling. The interpretation of these experiments is generally straightforward. For example, STAT5a and STAT5b are essential for the expression of genes that promote hemopoietic survival (28, 29, 30) whereas STAT1 is required for the expression of IFN-regulated genes that are involved in the protection against pathogens (18). However, by EMSA and immunoblotting experiments, most cytokines have been shown to activate multiple STATs, prompting experiments to determine transcriptional responses that can be activated in the absence of a given STAT. An initial example of this type of approach was performed by Schreiber and colleagues who interrogated gene expression profiles induced by IFN-γ signaling in the absence of STAT1 (31, 32). In these experiments, IFN-γ was used to stimulate STAT1-deficient bone marrow-derived macrophages and fibroblasts. Numerous genes were induced by IFN-γ in the absence of STAT1, leading to the conclusion that the IFN-γR activates a STAT1-independent gene expression program. However, inspection of the genes induced by IFN-γ signaling in STAT1-deficient cells shows many to be STAT3-regulated genes such asSocs3, Gadd45, and Cebpb. STAT3 phosphorylation is normally induced by IFN-γ in wild-type cells but in the absence of STAT1, STAT3 signaling is dominant. What is the mechanism of this effect? We now know from experiments using STAT-deficient cells that receptor occupancy, or lack of occupancy by the dominant STAT that binds the receptor, causes a switch from one activated STAT to another (33). A converse example is the conversion of IL-6R signaling to a dominant STAT1 activation in STAT3-deficient cells (34). This switch causes the downstream induction of the IFN gene expression pathway just as IFN-γ would cause in wild-type cells.
A related example is observed when IL-6 signaling is tested in the absence of SOCS3. SOCS3 is induced by STAT signaling from different cytokine receptors and functions as a feedback inhibitor of the IL-6R (and the G-CSFR, LIFR, and leptinR) by binding to phosphorylated Y757 on the gp130 cytoplasmic domain (see below). However in the absence of SOCS3, STAT3 phosphorylation is greatly increased (35, 36, 37). At the same time however, STAT1 phosphorylation is also induced, leading to a dominant IFN-like gene expression signature (35, 36). Thus SOCS3 regulates both the quantity and type of STAT signal generated from the IL-6R. Although the mechanism of the SOCS3 effect is unclear, the promiscuity of different receptors for different STATs argues that loss-of-function experiments must be carefully examined for the activation of other STAT molecules that fill the “hole” created by the loss of one STAT. These data also suggest that different cytokine receptors have evolved selectivity for different classes of STATs. Although STAT1 and STAT3 can apparently interchangeably bind the IL-6R or IFN-γR when either molecule is missing, signaling in wild-type cells shows a strong preference for one STAT over the other. Likewise, other receptors may have evolved to bind only one STAT, and in the absence of the key STAT, the other STATs cannot bind and/or be activated by the receptor.
The above examples primarily describe experiments using STAT1–STAT3-activating receptors but these are not isolated cases. In T cells stimulated by IL-12, STAT4 is activated and drives IFN-γ production. This pathway is a central regulatory event in the development of the Th1 type T cell responses. IFN-αβ, via the IFN-αβR, also activates STAT4 (in addition to STAT1 and STAT2 that forms a complex with IRF-9 to mediate anti-viral gene expression) but cannot activate strong IFN-γ production and therefore cannot drive Th1 development (38). However, in the absence of STAT1, IFN-αβ causes a large increase in IFN-γ production, especially in vivo during viral infection (39, 40). These data were originally interpreted to mean that STAT1 normally suppressed IFN-γ production. However, the data can just as easily be resolved when we consider that STAT4 activation from the IFN-αβR is increased in the absence of STAT1. Recent data confirm this interpretation but also show that STAT4 activation by the IFN-αβR, although increased, cannot sustain IFN-γ production from T cells when compared with IL-12 (38). This is probably because of the stronger differential activity of SOCS1 on the IFN-αβR versus the IL-12R (discussed below). I would predict that an IFN-αβR that is refractory to SOCS1 (or active in a Socs1−/− background) would behave identically to the IL-12R in the absence of STAT1.
Although loss of gene expression may be observed in a given STAT knockout, a corresponding increase in the ectopic activation of another STAT pathway may confound the interpretation of results in both in vitro and in vivo systems. Because specific Abs are available for each tyrosine-phosphorylated STAT molecule, a simple solution is to first measure which other STATs are activated by a given receptor in the absence of the STAT of interest. Experiments using STAT knockout systems should also be supported by additional data that uses complimentary mutations in the receptor that ablate STAT recruitment, or complete loss of the receptor. Finally, it is worth noting that the loss of a STAT pathway from a receptor signaling system can cause additional loss of key negative regulatory systems including feedback loops such as SOCS induction as presently debated for G-CSFR signaling and receptor systems discussed below (41, 42, 43, 44, 45).
Proposed differential STAT activation by IL-10 or IL-6. Shown are three classes of genes activated by STAT3 where Socs3 is a representative “common” gene induced by both receptors. In the absence of SOCS3, the IL-6R can activate the anti-inflammatory genes in the same way as the IL-10R. The mechanism of this effect remains to be established.
JAK/STAT Activation Inhibitors
The JAK/STAT pathway plays an important role in cytokine receptor-mediated signal transduction via activation of downstream signal transducers and activators of transcription (STAT), phosphatidylinositol 3-kinase (PI3K), and mitogen-activated protein kinase (MAPK) pathways.
These inhibitors are useful tools for exploring the contribution of JAK/STAT-mediated signaling.
The JAK/STAT pathway transduces signals from multiple cytokines and controls haematopoiesis, immunity and inflammation. In addition, pathological activation is seen in multiple malignancies including the myeloproliferative neoplasms (MPNs). Given this, drug development efforts have targeted the pathway with JAK inhibitors such as ruxolitinib. Although effective, high costs and side effects have limited its adoption. Thus, a need for effective low cost treatments remains.
Methods & Findings
We used the low-complexity Drosophila melanogaster pathway to screen for small molecules that modulate JAK/STAT signalling. This screen identified methotrexate and the closely related aminopterin as potent suppressors of STAT activation. We show that methotrexate suppresses human JAK/STAT signalling without affecting other phosphorylation-dependent pathways. Furthermore, methotrexate significantly reduces STAT5 phosphorylation in cells expressing JAK2 V617F, a mutation associated with most human MPNs. Methotrexate acts independently of dihydrofolate reductase (DHFR) and is comparable to the JAK1/2 inhibitor ruxolitinib. However, cells treated with methotrexate still retain their ability to respond to physiological levels of the ligand erythropoietin.
Conclusions
Aminopterin and methotrexate represent the first chemotherapy agents developed and act as competitive inhibitors of DHFR. Methotrexate is also widely used at low doses to treat inflammatory and immune-mediated conditions including rheumatoid arthritis. In this low-dose regime, folate supplements are given to mitigate side effects by bypassing the biochemical requirement for DHFR. Although independent of DHFR, the mechanism-of-action underlying the low-dose effects of methotrexate is unknown. Given that multiple pro-inflammatory cytokines signal through the pathway, we suggest that suppression of the JAK/STAT pathway is likely to be the principal anti-inflammatory and immunosuppressive mechanism-of-action of low-dose methotrexate. In addition, we suggest that patients with JAK/STAT-associated haematological malignancies may benefit from low-dose methotrexate treatments. While the JAK1/2 inhibitor ruxolitinib is effective, a £43,200 annual cost precludes widespread adoption. With an annual methotrexate cost of around £32, our findings represent an important development with significant future potential.
Citation: Thomas S, Fisher KH, Snowden JA, Danson SJ, Brown S, Zeidler MP (2015) Methotrexate Is a JAK/STAT Pathway Inhibitor. PLoS ONE 10(7): e0130078. http://dx.doi.org:/10.1371/journal.pone.0130078
Cell Death Pathway Insights, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 2: CRISPR for Gene Editing and DNA Repair
Cell Death Pathway Insights
Larry H. Bernstein, MD, FCAP, Curator
LPBI
Phosphorylation and activation of ubiquitin-specific protease-14 by Akt regulates the 1 ubiquitin-proteasome system
Daichao Xu1,2, Bing Shan1,4, Byung-Hoon Lee3,4, Kezhou Zhu1,4, Tao Zhang1,4, Huawang Sun1, 4 Min Liu1, Linyu Shi1, Wei Liang1, et al.
eLife 2015;10.7554/eLife.10510 DOI: http://dx.doi.org/10.7554/eLife.10510
In this study, we report that USP14 is an Akt substrate and that this phosphorylation activates the DUB activity of USP14 both in vitro and in cells. We also demonstrate that phosphorylation of USP14 is critical for Akt to control UPS and consequentially global protein degradation via the UPS.
Regulation of ubiquitin-proteasome system (UPS), which controls the turnover of short-lived proteins in eukaryotic cells, is critical in maintaining cellular proteostasis. Here we show that 40 USP14, a major deubiquitinating enzyme that regulates the UPS, is a substrate of Akt, a serine/threonine-specific protein kinase critical in mediating intracellular signaling transducer for growth factors. We report that Akt-mediated phosphorylation of USP14 at Ser432, which normally blocks its catalytic site in the inactive conformation, activates its deubiquitinating activity in vitro and in cells. We also demonstrate that phosphorylation of USP14 is critical for Akt to regulate proteasome activity and consequently global protein degradation. Since Akt can be activated by a wide range of growth factors and is under negative control by phosphoinosotide phosphatase PTEN, we suggest that regulation of UPS by Akt-mediated phosphorylation of USP14 may provide a common mechanism for growth factors to control global proteostasis and for promoting tumorigenesis in PTEN-negative cancer cells.
The ubiquitin-proteasome system (UPS), a major degradative mechanism in eukaryotic cells, is involved in the degradation of short-lived proteins as well as misfolded and damaged proteins 69 (Komander and Rape, 2012). The 26S proteasome specifically targets and degrades proteins conjugated to ubiquitin. Regulation of protein deubiquitination by deubiquitinating enzymes (DUBs) is recognized as an important regulatory step in the ubiquitin-proteasome system. USP14, a deubiquitinating enzyme reversibly associated with the proteasome, negatively regulates the activity of proteasomes by trimming ubiquitin chains on proteasome-bound substrates (Borodovsky et al., 2001; Koulich et al., 2008; Lee et al., 2010). Purified recombinant USP14 is largely inactive and can be highly activated when in association with proteasome (Hu 76 et al., 2005; Koulich et al., 2008; Lee et al., 2010). However, a significant fraction of USP14 is present intracellularly in a proteasome-free state (Koulich et al., 2008) and it is not clear if and how proteasome-free USP14 might serve a significant physiological function. Akt, a serine/threonine-specific protein kinase and an important intracellular signaling transducer for growth factors such as insulin, is involved in regulating cell proliferation, metabolism, transcription, migration and apoptosis (Manning and Cantley, 2007). The activity of Akt is regulated by PI(3,4,5)P3, a lipid product of the phosphoinositide 3-kinases (PI3Ks). The intracellular levels of PI(3,4,5)P3 are negatively regulated by phosphatases such as SHIP1/2 and PTEN. The latter, a phosphoinoside phosphatase, is encoded by a tumor suppressor gene that is mutated in human cancers at high frequency (Cantley and Neel, 1999). Akt has been reported to mediate the phosphorylation of many substrates that in turn regulate cell proliferation, metabolism, transcription, migration and apoptosis. However, very little is known about its role in the UPS, and furthermore no mechanistic link between Akt and UPS has been elucidated.
Two forms of USP14 have been determined crystallographically: the inactive free form and an adduct between Ub-aldehyde (Ubal) and USP14, which provides insight into the catalytically active state (Hu et al., 2005). The key difference between these two structures is in the position of the blocking loops, BL1 and BL2, which project over the catalytic cleft of USP14 and block the access of the C-terminal residues of ubiquitin in the inactive form (Figure 1A). In Ubal-modified USP14, BL1 and BL2 are rearranged, thus exposing the cleft. In particular, Ser432, located within BL2, shifts its position over a distance of 3-5Å between the two states (Hu et al., 2005) (Figure 1B). Since Ser432 residue is located very close to a highly negatively charged patch (Figure 107 1C), we reasoned that when Ser432 residue was phosphorylated, the negatively charged phosphate group might induce a repulsive force, thereby inducing rearrangement of the BL2 loop and removing the inhibitory effect of this loop on the activity of USP14. The amino acid 110 sequences around Ser432 are highly evolutionarily conserved among USP14 orthologues 111 (Figure 1D) and Ser432 is predicted to be an Akt substrate by Scansite (http://scansite3.mit.edu/#home). We therefore tested the possibility that USP14 might be a substrate of activated Akt. We first examined the interaction between USP14 and Akt using a co-immunoprecipitation assay. As shown in Figure1-figure supplement 1A, when USP14 and Akt were overexpressed in HEK293T cells, their interaction was readily detectable. To test whether Akt could phosphorylate USP14, we overexpressed USP14 and an activated Akt (Myr-Akt) in HEK293T cells, and performed a quantitative phosphoproteomic analysis (Figure 118 1-figure supplement 1B). We identified four phosphorylation sites on USP14 when it was 119 expressed alone: Ser143, Ser230, Thr235, and Ser432 (Figure 1-figure supplement 1C-D). Notably, the phosphorylation levels of two of the four sites, Ser143 and Ser432, were increased considerably in cells expressing activated Akt (Figure 1E).
Figure 1. Structural basis of USP14 activation by phosphorylation of Ser432. (A) Detailed view of blocking loop 2 (BL2), which occludes the active site of USP14 (PDB access code 2AYN). The BL2 loop, which contains Ser432, is shown in stick model, in the apo form. (B) Combined ribbon representation and stick model showing a comparison of the conformations of the BL2 loop containing in the apo form (blue, PDB access code 2AYN) and in the USP14-Ubal adduct (orange, PDB access code 2AYO). In this drawing, the Ser432 and Cys114 residues are 504 shown in stick model, and the bound Ubal (a ubiquitin derivative in which the C-terminal 505 carboxylate is replaced by an aldehyde) in the complex is drawn in green. (C) A surface charge potential representation (contoured at ±7 kT/eV; blue/red) of USP14 (PDB accession 2AYN) showing that the S432 residue is very close to a highly negatively charged patch mainly formed by the acidic E188, D199 and E202 residues. When S432 is phosphorylated, the negatively charged phosphate group may induce a repulsive force, thereby relieving inhibition of the catalytic activity of USP14. (D) USP14 domain organization and sequence alignment of the Akt 511 phosphorylation site within USP14 orthologues from different species. Two blocking loops (BL1 512 and BL2) covering the USP14 active site are shown. The Akt phosphorylation site in USP14 from different species as predicted by Scansite. (E) S432 is the major phosphorylation site in USP14. HEK293T cells were treated as in Figure 1-figure supplement 1B, followed by ESI-MS analysis. Spectral counts were determined by ESI-MS. (F) Akt phosphorylates USP14 in vitro. Bacterially expressed and purified USP14 was incubated with active Akt in the presence of ATP. Reaction products were resolved by SDS-PAGE, and phosphorylated species were detected by a phospho-Ser antibody.
Since Ser432 residue is located very close to a highly negatively charged patch (Figure 107 1C), we reasoned that when Ser432 residue was phosphorylated, the negatively charged phosphate group might induce a repulsive force, thereby inducing rearrangement of the BL2 loop and removing the inhibitory effect of this loop on the activity of USP14. The amino acid sequences around Ser432 are highly evolutionarily conserved among USP14 orthologues (Figure 1D) and Ser432 is predicted to be an Akt substrate by Scansite (http://scansite3.mit.edu/#home). We therefore tested the possibility that USP14 might be a substrate of activated Akt. We first examined the interaction between USP14 and Akt using a co-immunoprecipitation assay. As shown in Figure1-figure supplement 1A, when USP14 and Akt were overexpressed in HEK293T cells, their interaction was readily detectable. To test whether Akt could phosphorylate USP14, we overexpressed USP14 and an activated Akt (Myr-Akt) in HEK293T cells, and performed a quantitative phosphoproteomic analysis (Figure 1-figure supplement 1B). We identified four phosphorylation sites on USP14 when it was expressed alone: Ser143, Ser230, Thr235, and Ser432 (Figure 1-figure supplement 1C-D). Notably, the phosphorylation levels of two of the four sites, Ser143 and Ser432, were increased considerably in cells expressing activated Akt (Figure 1E).
To examine whether USP14 is a direct substrate for Akt, we conducted an in vitro kinase 123 assay using activated recombinant Akt and purified recombinant USP14 expressed in E. coli. We 124 found that co-incubation of USP14 and Akt led to modification of USP14 as detected by a pan phospho-Ser antibody (Figure 1F), suggesting that USP14 is a substrate for Akt.
Figure 1-figure supplement 1. Akt phosphorylates USP14. (A) Akt interacts with USP14. HEK293T cells were transfected with indicated plasmids for 24 h. The cell lysates were collected for co-immunoprecipitation and western blotting analysis. (B) Schematic representation of mass spectrometry assay to determine USP14 phosphorylation sites by Akt. (C) Four phosphorylation sites of USP14 were determined by mass spectrometry. (D) The representative MS/MS spectrum of phosphorylated tryptic peptide ‘SSSphosSGHYVSWVK’ of human USP14 protein. The peptide sequence ‘SSSphosSGHYVSWVK’ containing phosphorylated S432 was identified by shotgun analysis using mass spectrometry when USP14 was co-expressed with Myr-Akt in HEK293T cells. Fragmentation ion of the amide bond of the peptide result in formation of ‘b’ ion and ‘y’ ion series corresponding to the N-terminal and C-terminal fragments respectively. Representative ions with phosphorylation and H2O loss were manually labeled in red on the spectrum.
To determine if Ser143 and Ser432 were indeed phosphorylated by Akt, we used this pan phospho-Ser antibody as above and found phosphorylation of WT USP14, but not of S143A/S432A mutant USP14, after incubating with activated Akt in a kinase assay (Figure 2A). To differentiate the relative importance of Ser143 and Ser432 as phosphorylation sites by Akt, we overexpressed activated Akt (Myr-Akt) in HEK293T cells with WT, S143A, S432A or double S143A/S432A (AA) mutants. We found that S143A mutant showed partially reduced phosphorylation as compared to that of WT, whereas phosphorylation of the USP14 S432A mutant was significantly decreased and that of AA double mutant was completely eliminated (Figure 2B). These results suggested S432 as a major and S143 as a minor phosphorylation site of Akt.
The phosphorylation of USP14 by Akt was further confirmed using an Akt phosphorylation-consensus motif (R××S/T) antibody (Figure 2-figure supplement 1A). The reactivity of USP14 with pan phospho-Ser antibody was eliminated after incubation with lambda phosphatase (Figure 2C). Notably, the phosphorylation levels of USP14 were decreased in cells when treated with MK2206, an inhibitor of Akt (Figure 2D), or when serum deprived, a condition known to inactivate endogenous Akt (Zhang et al., 2015) (Figure 2D).
To further verify the phosphorylation of USP14 S432 by Akt, we developed a phospho-Ser432 specific antibody. Phosphorylation of S432 can be detected after incubation of WT, but not S432A mutant USP14, with recombinant activated Akt in a kinase reaction (Figure 145 2E). This was further confirmed by using phos-tag electrophoresis which can specifically retard the migration of phosphorylated protein species (Kinoshita et al., 2009) (Figure 2E). Expression of Myr-Akt also led to S342 phosphorylation of endogenous USP14 (Figure 2F). Treatment with either MK2206 or AZD5363, two structurally unrelated Akt inhibitors, led to decrease of USP14 S432 phosphorylation levels (Figure 2-figure supplement 1B-C). Moreover, treatment with PI3K inhibitors, either Wortmannin or GDC0941, but not ERK1/2 inhibitor U0126, also significantly decreased the phosphorylation levels of USP14 S432 (Figure 2-figure supplement 152 1D-E). In addition, we tested growth factors such as IGF-1 or EGF, both of which are known to promote activation of Akt. We found that the treatment of IGF-1 or EGF resulted in phosphorylation of USP14 S432, which was blocked in cells pre-treated with MK2206 (Figure 155 2G-H). Finally, USP14 S432 is dramatically more phosphorylated in PTEN knockout mouse embryonic fibroblasts (MEFs), which carry high levels of Akt activity, than that of WT MEFs as determined by western blotting using the phospho-USP14(S432) antibody and phos-tag electrophoresis (Figure 2I), and the phosphorylation of USP14 S432 was blocked by Akt inhibitors (Figure 2-figure supplement 1F). From these results, we conclude that Ser432 of USP14 is a major phosphorylation site by Akt.
Figure 2. USP14 is phosphorylated at Ser432 by activated Akt. (A) In vitro phosphorylation 521 of USP14 at S432 by Akt. Bacterially expressed and purified wide type USP14 or AA mutant incubated with active Akt in the presence of ATP. Reaction products were resolved by SDS-PAGE, and phosphorylation was detected by the phospho-Ser antibody. (B) Akt phosphorylates USP14 at S432 in vivo. Western blot analysis of whole cell lysate and immunoprecipitates derived from HEK293T cells transfected with wild type USP14, USP14 S143A, USP14 S432A and USP14 S143A/S432A (AA) constructs using the phospho-Ser antibody. L.E., long exposure. (C) Immunoprecipitation (IP) and IB analysis of HEK293T cells transfected with HA-USP14 and Myr-Akt and preincubated with or without λ-phosphatase as indicated. (D) Inhibition of Akt decreased exogenous USP14 phosphorylation. HEK293T cells were transfected with Myc-USP14 for 20 h then treated with 1 μM MK2206 or deprived of serum for another 4 h before harvest. (E) In vitro kinase assay to detect Akt phosphorylation of USP14 by phospho-Ser432 specific antibody and phos-tag-containing gels. Bacterially expressed and purified wide type USP14 or S432A mutant was incubated with active Akt in the presence of ATP. The reaction products were resolved by SDS-PAGE, and USP14 phosphorylation was detected using an antibody that specifically recognizes Ser432 phosphorylation of USP14 or determined by differential migration on phos-tag gels. (F) In vivo detection of endogenous USP14 Ser432 phosphorylation by anti-p-Ser432 specific antibody. Western blot analysis of immunoprecipitates derived from H4 cells transfected with or without Myr-Akt plasmids using the anti-p-Ser432 specific antibody. (G, H) Phosphorylation of endogenous USP14 S432 upon 540 stimulation with IGF-1 or EGF. HEK293T cells were serum-starved and pre-treated with Akt inhibitor MK2206 (1 μM) for 30 min before stimulation with IGF-1 (100 ng/mL) for 30 min (G) or EGF (100 ng/mL) for 1 h (H). The cell lysates were immunoprecipitated with USP14 antibody and western-blotted with anti-p-S432 antibody. (I) Phosphorylation of endogenous USP14 S432 in Pten knockout cells with high activity of Akt. Lysates from MEFs with indicated genotypes 545 were immunoprecipitated with USP14 antibody and then western-blotted with p-S432 antibody. The differential migration of phospho-USP14 on phos-tag-containing gels was determined as shown in the bottom panel.
Activation of USP14 by Akt mediated phosphorylation Because bacterially expressed and purified USP14 protein exhibits very low catalytic activity (Lee et al., 2010), we tested whether Akt-mediated phosphorylation might activate the DUB activity of USP14. We compared the activity of recombinant USP14 in a Ub-AMC (ubiquitin-7-amido-4-methylcoumarin, a fluorogenic substrate) hydrolysis assay in the presence or absence of Akt. Bacterially expressed and purified USP14 (Figure 3-figure supplement 1) showed trace hydrolyzing activity towards Ub-AMC as reported (Lee et al., 2010), while USP14 incubated with Akt showed high activity (Figure 3A). To validate Akt-mediated activation of USP14 in cells, we co-expressed USP14 and Myr-Akt in HEK293T cells. USP14 immunoprecipitated from cells co-expressing activated Akt showed higher activity in Ub-AMC assay than that expressed alone (Figure 3B). On the other hand, USP14 isolated from HEK293T cells incubated with Akt inhibitor MK2206 showed reduced activity in Ub-AMC assay (Figure 3C). Moreover, USP14 isolated from HEK293T cells stimulated with IGF-1 showed higher
activity, which was suppressed when cells were pre-treated with MK2206 (Figure 3D). To determine the specific contribution of Ser432, we compared the activity of USP14 S432A mutant protein in Ub-AMC assay with that of WT in the presence of Akt, and found that the stimulating effect of Akt on the hydrolyzing activity of USP14 was largely blocked by S432A mutation (Figure 3E), but not by S143A mutation (Figure 3-figure supplement 2B).
To further characterize the effect of Ser432 phosphorylation, we expressed and purified recombinant S432E USP14 protein, which mimics the phosphorylation state of USP14, from E. coli (Figure 3-figure supplement 1) and analyzed its activity by Ub-AMC assay. Interestingly, we found that USP14 S432E mutant protein alone showed high levels of Ub-AMC hydrolyzing activity (Figure 3F). Consistent with S432 as the major phosphorylation site by Akt, double E mutant (S143E/S432E) showed almost the same levels of hydrolyzing activity as that of S432E single mutant and S143E mutation had no significant impact on the activity of USP14 (Figure 3-figure supplement 2C-D). To determine its enzyme kinetics, we incubated USP14 S432E mutant protein with increasing amounts of Ub-AMC (Figure 3-figure supplement 2E) and determined the Km value (Km = 26 μM) from the slope of a Lineweaver-Burk plot (Figure 3G).
We characterized the distributions of p-S432 USP14 and total USP14 with that of proteasome in Pten-/- MEFs using glycerol gradient centrifugation (Koulich et al., 2008). We found that majority of p-S432 USP14 was distributed in the fractions with lower molecular weight proteins and distinguishable from the fractions where larger protein complexes, such as proteasomes, were localized. On the other hand, unphosphorylated USP14 was found in the fractions where larger molecular weight complexes, such as proteasome, are known to be localized (Figure 3-figure supplement 2F). Thus, S432 phosphorylated and unphosphorylated USP14 might be distributed differently in the cells. We next determined whether phospho-mimetic mutant of USP14 could be further activated by interacting with proteasome. Interestingly, we found that the Ub-AMC hydrolytic activity of S432E mutant could be further 200 activated when incubated with proteasome in vitro (Figure 3H). Taken together, these results suggest that S432 phosphorylation and intraction with proteasome may be two different
regulatory mechanisms for USP14.
Figure 3. Phosphorylation of USP14 by Akt activates USP14 DUB activity. (A) Akt activates USP14 DUB activity in vitro. USP14 protein (1μg) was incubated with or without active Akt (1 μg) in kinase assay buffer in a total volume of 50 μL for 1 h at 30oC, then the reaction mixtures were subjected to Ub-AMC assay. RFU, relative fluorescence units. (B, C) Akt activates USP14 in cells. USP14 was immunoprecipitated from HEK293T cells co-expressed with activated Akt (B) or treated with 10 μM MK2206 for 4h (C) and then eluted with HA-peptide following Ub-AMC hydrolysis assay. (D) Activation of USP14 by stimulating cells with IGF-1. HEK293T cells were serum-starved and pre-treated with or without Akt inhibitor MK2206 (1 μM) for 30 min before stimulation with IGF-1 (100ng/mL) for 30 min. USP14 was then immunoprecipitated and eluted with HA-peptide. The activity of USP14 was determined using Ub-AMC hydrolysis assay. (E) USP14 activation by Akt is blocked by S432A mutation. Ub-AMC hydrolysis assay of wide type USP14 or S432A mutant in the presence or absence of active Akt. (F) Ub-AMC hydrolysis assay of bacterially expressed and purified wide type USP14 or S432E mutant. (G) Lineweaver-Burk analysis of USP14 S432E, obtained by measuring the initial rates at varying Ub-AMC concentrations (see Figure 3-figure supplement 2E for reference). (H) The activity of phospho-mimetic USP14 mutant can be further stimulated by the presence of proteasome. Ub-AMC hydrolysis assay of wild type USP14 or S432E mutant in the presence or absence of Ub-VS-treated human proteasome [VS-proteasome (see Lee et al., 2010); 1 nM]. Ptsm, 26S proteasome.
Phosphorylation of USP14 promotes both K48 and K63 deubiquitination activity To assess the impact of USP14 phosphorylation on its selectivity towards different types of 206 ubiquitin linkages, we incubated USP14 WT and S432E mutant protein with diubiquitin species of K48, K63 and linear linkages. Conversion to monomeric Ub was monitored via SDS-PAGE followed by western blotting. We observed significantly increased hydrolytic activity of S432E mutant, as compared to that of WT, towards both Lys48 and Lys63 diubiquitin, while linear diubiquitin was not readily cleaved by WT or mutant USP14 (Figure 4A-B and Figure 4-figure supplement 1A). Similarly, immunoprecipitated USP14 from cells showed significant activity toward both Lys48 and Lys63 diubiquitin, but not linear diubiquitin (Figure 4-figure supplement 1B-C). In contrast, S432A mutant immunoprecipitated from cells showed lower activity towards both Lys48 and Lys63 diubiquitin than that of WT (Figure 4C). Regulation of ubiquitin-proteasome system by Akt depends on phosphorylation of USP14. Since USP14 is a negative regulator of the UPS (Koulich et al., 2008; Lee et al., 2010; Lee et al., 2011) and we found USP14 can be phosphorylated and activated by Akt, we reasoned that 219 Akt-mediated activation of USP14 might lead to inhibition of the ubiquitin-proteasome system (UPS) and generally enhance the stability of many proteins. To this end, we generated a stable 221 cell line expressing GFP-CL1 (also known as GFPu), an engineered ubiquitin-dependent proteasome substrate widely used as a reporter for UPS activity (Bence et al., 2001; Kelly et al., 2007; Li et al., 2013; Liu et al., 2014) (Figure 5-figure supplement 1A-C). Treatment of cells with Akt inhibitors or serum deprivation or PI3K inhibitor, all of which can block Akt activity (Zhang et al., 2015), led to reduced level of GFP-CL1 as detected by both western blotting and fluorescence microscopy (Figure 5A-C and Figure 5-figure supplement 1D). Conversely, the expression of activated Akt (Myr-Akt) led to increased levels of GFP-CL1 protein. Treatment of WT H4 cells with IGF-1 or EGF also led to increased levels of GFP-CL1 protein (Figure 5D-G and Figure 5-figure supplement 1E). In contrast, in USP14 knockout H4 cells (generated using CRISPR/Cas9 technology, Figure 5-figure supplement 2A-D), the expression of Myr-Akt did not affect the levels of GFP-CL1 (Figure 5H). From these results, we conclude that Akt 232 negatively regulates the UPS in an USP14-dependent manner.
We next tested the importance of USP14 phosphorylation for Akt to regulate UPS. We found that in contrast to USP14 WT reconstituted H4 cells, USP14 AA mutant reconstituted H4 cells showed no increase in the accumulation of GFP-CL1 in response to the expression of activated Akt (Figure 5-figure supplement 2E and Figure 5I). As a control, we found that the expression of Akt had no effect on a ubiquitin-independent substrate of the proteasome, C-terminal ornithine decarboxylase-GFP (GFP-cODC) (Hoyt et al., 2005; Kelly et al., 2007; Lee et al., 2010) (Figure 5-figure supplement 2F-G), suggesting that Akt does not inhibit the UPS through a general inhibition of the proteasome itself. Taken together, these data show that 241 phosphorylation of USP14 by Akt is important for this kinase to negatively regulate the UPS in a ubiquitin-dependent manner.
Phosphorylation of USP14 regulates global protein degradation To further understand the physiological roles of Akt-mediated USP14 phosphorylation and subsequently activation, we sought to study the impact of USP14 phosphorylation on global protein degradation. Since the loss of USP14 accelerates cellular proteolysis (Koulich et al., 2008; Lee et al., 2010), we performed a quantitative proteomic analysis to determine the levels of proteins in WT H4 cells, H4 USP14-KO cells, and H4 USP14-KO cells complemented with WT USP14, S143A/S432A (AA) or S143D/S432D (DD) mutants. Using an isobaric TMT labeling approach, our mass spectrometry analysis identified 18,400 peptides with high confidence (q<0.01), corresponding to 3,648 proteins with a minimum of two peptides from each protein. 2,763 proteins, which were quantified in at least 2 replicates, were subjected to further analysis. We found the global protein patterns of H4 USP14-KO cells were similar to those of H4 USP14 KO-AA cells, but distinct from those of WT H4 cells. We identified a common set of 87 proteins that were reduced in H4 KO cells as compared to H4 WT cells or to H4 KO cells complemented with WT USP14 (KO-WT) (Figure 6, Lane1-2). The levels of these proteins were also significantly reduced in H4 KO-AA cells (Figure 6, Lane 3). Importantly, the levels of this set of 87 proteins in H4 KO-DD cells were significantly higher than that of H4 KO-AA cells (Figure 6, Lane 4).
Figure 6. Phosphorylation of USP14 regulates global protein degradation. The quantitative 605 analysis of proteome change in USP14 knockout or USP14 mutant cells were performed by 606 TMT-isobaric labeling followed by shotgun analysis. The heat map was plotted based on the set of 87 proteins that are down-regulated greater than or equal to 1.2 fold in H4 KO cells compared to H4 WT cells or to H4 KO cells complemented with WT USP14 (KO-WT). The log base 2 of average ratios was plotted as indicated.
To verify that the identified changes in protein abundance were due to proteasomal degradation, we treated H4 KO-AA cells with proteasome inhibitor MG132 and analyzed the protein level change of these 87 proteins. We found that the levels of these proteins increased significantly in MG132-treated KO-AA cells compared to that of control KO-AA cells (Figure 6, Lane 5), suggesting that these proteins were indeed subject to an increased rate of proteasome degradation with expression of non-phosphorylatable USP14. Interestingly, the top hit on this list of 87 proteins that were differentially regulated upon the loss of USP14 is mTOR, a central established regulator of cellular metabolism and tumorigenesis. We confirmed the role of USP14 on the levels of mTOR by western blotting. We found that the levels of mTOR were reduced inH4 KO and H4 KO cells complemented with USP14 AA mutant, but restored upon the expression of USP14 DD mutant (Figure 6-figure supplement 1). Taken together, our results suggest that phosphorylation of USP14 may provide a mechanism for Akt to regulate global protein degradation through the proteasome, which in turn may control key cellular pathways involved in regulating metabolism and tumorigenesis.
NF-kB-Independent Role of IKKa/IKKb in Preventing RIPK1 Kinase-Dependent Apoptotic and Necroptotic Cell Death during TNF Signaling
IKKa/IKKb prevent RIPK1 kinase-dependent death independently of NF-kB activation
IKKa/IKKb directly phosphorylate RIPK1 in TNFR1 complex I
Impaired phosphorylation of RIPK1 correlates with enhanced binding to FADD/caspase-8
IKK kinase inhibition induces TNF-mediated RIPK1 kinasedependent cell death in vivo
In Brief Dondelinger et al. describe an unexpected NF-kB-independent function of the IKK complex in protecting against TNF-induced RIPK1 kinase-dependent cell death. In TNFR1 complex I, IKKa/ IKKb directly phosphorylates RIPK1, leading to a reduction in RIPK1’s ability to bind FADD/caspase-8 and to induce apoptosis.
TNF is a master pro-inflammatory cytokine. Activation of TNFR1 by TNF can result in both RIPK1-independent apoptosis and RIPK1 kinase-dependent apoptosisornecroptosis.Thesecelldeathoutcomes are regulated by two distinct checkpoints during TNFR1 signaling. TNF-mediated NF-kB-dependent induction of pro-survival or anti-apoptotic molecules is a well-known late checkpoint in the pathway, protecting cells from RIPK1-independent death. On the other hand, the molecular mechanism regulating the contribution of RIPK1 to cell death is far less understood. We demonstrate here that the IKK complex phosphorylates RIPK1 at TNFR1 complex I and protects cells from RIPK1 kinase-dependent death, independent of its function in NF-kB activation. We provide in vitro and in vivo evidence that inhibition of IKKa/IKKb or its upstream activators sensitizes cells to death by inducing RIPK1 kinase-dependent apoptosis or necroptosis. We therefore report on an unexpected, NF-kB-independent role for the IKK complex in protecting cells from RIPK1-dependent death downstream of TNFR1.
The IkB kinase (IKK) complex, composed of the regulatory subunit NEMO (also known as IKKg) and the two catalytic subunits IKKa and IKKb, plays a central role in the induction of immune and inflammatory responses as well as in promoting cell survival and tumorigenesis (Baldwin, 2012; Baud and Karin, 2009; Hayden and Ghosh, 2012; Liu et al., 2012). Its activation constitutes the ignition phase of the canonical NF-kB pathway, which
ultimately results in the translocation of NF-kB dimers to the nucleus, where they promote transcription of a myriad of genes involved in inflammation, survival, and tumorigenesis.
TNF is a master pro-inflammatory cytokine, and inappropriate TNF signaling has been demonstrated to drive many inflammatory diseases. Activation of TNFR1 by TNF promotes inflammation either directly by activating the canonical NF-kB pathway or indirectly by promoting cell death, which exacerbates inflammation by releasing damage-associated molecular patterns (DAMPs) as well as by affecting the permeability of the bodily barriers to microbes (Pasparakis and Vandenabeele, 2015). In most cell types, activation of TNFR1 does not induce death but triggers canonical NF-kB-dependent transcriptional upregulation of genes encoding pro-survival and pro-inflammatory molecules. Ligation of TNF to trimeric TNFR1 induces the rapid assembly of a plasma membrane-bound signaling complex, known as complex I, that contains TRADD, RIPK1, and the E3 ubiquitin ligases TRAF2, cIAP1, cIAP2, and LUBAC (Walczak, 2011). The conjugation of ubiquitin chains to RIPK1 by cIAP1/ cIAP2 generates binding sites for TAB2/TAB3 and NEMO and allows further recruitment and activation of TAK1 and IKKa/ IKKb (Bertrand et al., 2008; Ea et al., 2006; Gerlach et al., 2011; Kanayama et al., 2004; Mahoney et al., 2008; Wu et al., 2006). TAK1 activates the IKK complex by phosphorylation, resulting in the rapid and selective IKK-mediated phosphorylation of IkBa and in its subsequent ubiquitylation-dependent proteasomal degradation. IkBa degradation then permits translocation of the NF-kB heterodimer p50/p65 to the nucleus, where it induces transcription of multiple responsive genes, including pro-survival genes such as cFLIP (Hayden and Ghosh, 2014). The anti-apoptotic potential of cFLIP resides in its ability to counteract activation of caspase-8 from a cytosolic TRADD-FADD-caspase-8 cytosolic complex, named complex IIa, which is believed to originate from complex I internalization (Irmler et al., 1997; Micheau and Tschopp, 2003; Wang et al., 2008; Wilson et al., 2009). Accordingly, TNFR1-mediated RIPK1-independent apoptosis requires inhibition of the NF-kB response (Van Antwerp et al., 1996), commonly obtained in vitro by the use of pharmacological inhibitors of transcription or translation, respectively, Actinomycin D (ActD) and cycloheximide (CHX).
The NF-kB-mediated induction of pro-survival/anti-apoptotic molecules is, however, not the only cell death checkpoint in the TNFR1 pathway (O’Donnell and Ting, 2011). Indeed, altering activation of the canonical NF-kB pathway by inhibiting components located upstream of IkBa, namely, cIAP1/cIAP2, TAK1, and NEMO, was reported to further sensitize cells to death by additionally inducing RIPK1-dependent death (Dondelinger et al., 2013; Legarda-Addison et al., 2009; O’Donnell et al., 2012). Depending on the cellular context, activated RIPK1 accelerates cell death either by promoting assembly of a RIPK1FADD-caspase-8 cytosolic apoptotic complex, referred to as complex IIb (Wilson et al., 2009), or by promoting necroptosis via activation of the RIPK3-MLKL pathway (Pasparakis and Vandenabeele, 2015). Although initiated by cIAP1/cIAP2-mediated ubiquitylation of RIPK1 in complex I, the last molecular step in the regulation of this early RIPK1 kinase-dependent cell death checkpoint is currently unknown. In this study, we demonstrate that RIPK1 is a bona fide substrate of IKKa and IKKb and that IKKa/IKKb-mediated phosphorylation of RIPK1 in complex I protects cells from RIPK1 kinase-dependent death.
NEMO Deficiency and IKKa/IKKb Double Deficiencies Induce TNFR1-Mediated RIPK1 Kinase-Dependent Apoptosis We previously reported that the ubiquitin chains conjugated to RIPK1 by cIAP1/cIAP2 do not constitute the ultimate step regulating the contribution of RIPK1 to TNF-induced cell death. Indeed, genetic or pharmacological inhibition of TAK1 also drivesRIPK1-dependentdeathwithoutaffectingRIPK1ubiquitylation in complex I (Dondelinger et al., 2013). In this study, we investigated the role of the IKK complex in the regulation of this cell death checkpoint. Indeed, the IKK complex lies between TAK1andIkBainthepathway,andalthoughexpressionofaproteasome-resistant form of IkBa (IkBaSR) induces RIPK1-independent apoptosis (Dondelinger et al., 2013), NEMO deficiency was reported to sensitize cells to TNF-induced death by additionally promoting RIPK1-dependent apoptosis (Legarda-Addison et al., 2009). In absence of cIAP1/cIAP2 or TAK1, TNF-mediated RIPK1-dependent apoptosis was shown to rely on RIPK1 kinase activity (Dondelinger et al., 2013; Wang et al., 2008). To test whether this is also true in absence of NEMO, we first stimulated NEMO-deficient mouse embryonic fibroblasts (MEFs) with TNF in the absence or presence of Nec-1, a RIPK1 kinase inhibitor. Interestingly, we found that Nec-1 greatly, but not entirely,protectedNemo/MEFsfromTNF-induced apoptosis, as monitored by cell permeability, caspase-3 activity, and caspase-3 and caspase-8 processing (Figures 1A–1D, 1K, and 1L). These results indicated that, similarly as cIAP1/cIAP2 and TAK1, NEMO also regulates both RIPK1 kinase-dependent and RIPK1-independent cell death checkpoints downstream of TNFR1. To test whether this protective function of NEMO
reflects its role as adaptor protein recruiting IKKa and IKKb to TNFR1 complex I, we next stimulated Ikka/, Ikkb/, and Ikka//Ikkb/ MEFs with TNF. Interestingly, while IKKa or IKKb single deficiency had little effect on apoptosis induction (Figures 1E–1H), their combined depletion mimicked the phenotype observed in the Nemo/ MEFs (Figures 1I–1L), suggesting redundant roles of IKKa and IKKb downstream of NEMO in preventing RIPK1-dependent apoptosis. To exclude the possibility that the phenotypes observed in the various MEF genotypes were originating from intrinsic defects due to clonal expansion, we confirmed our findings in Ripk1+/+ and Ripk1/ MEFs depleted of IKK proteins by siRNA (Figure S1). Of note, NEMO siRNA had little effect on cell death induction in these experiments, probably due to the poor efficiency in repressing NEMO.
Figure 1. NEMO Deficiency and IKKa/IKKb Double Deficiencies Induce TNFR1-Mediated RIPK1 Kinase-Dependent Apoptosis (A–L)MEFsoftheindicatedgenotypesweretreatedwith20ng/mlhTNFinthepresenceorabsenceofNec-1,andcelldeath(A,C,E,G,andI)andcaspaseactivity (B, D, F, H, and J) were measured in function of time, respectively, by SytoxGreen positivity and DEVD-AMC fluorescence. Protein levels were determined by immunoblotting in unstimulated cells (K) or 15 hr poststimulation with the indicated compounds (L). Forthe celldeath results, error bars represent theSEM ofthreeindependent experiments. Forthe caspase-3activity results, error bars represent SDof triplicates of one representative experiment. See also Figure S1.
IKKa and IKKb Mediate Their Protective Effect on RIPK1 via Their Enzymatic Activities Because IKKa and IKKb are serine/threonine kinases, we next evaluated the requirement of their enzymatic activities for their ability to repress RIPK1-dependent apoptosis. To do so, we tested the effect of five different IKK inhibitors on TNF-induced death and found that all of them led to a combination of RIPK1 kinase-dependent and RIPK1-independent death, as observed in the Ikka//Ikkb/ MEFs (Figures S2A and S2B). We further confirmed RIPK1 kinase-dependent apoptosis induction using TPCA-1 (Figures 2A–2C), as this inhibitor had no effect on TNF-induced death in Ikka//Ikkb/ MEFs (Figure S2B). TPCA-1 was used at 5 mM, a concentration reported to inhibit both IKKa and IKKb kinase activities (IC50 = 400 nM and 17.9 nM for IKKa and IKKb, respectively) (Podolin et al., 2005). We demonstrated that the apoptotic cell death was mostly depending on RIPK1 kinase activity by either co-incubating cells with Nec-1 (Figures 2A–2C) or by stimulating RIPK1 kinase-dead-expressing MEFs (Ripk1 K45A)(Figures 2D and 2E) (Berger et al., 2014). Importantly, Nec-1 had no effect in Ripk1 K45A MEFs, excluding any off-target effect (Figures S2E andS2F). Of note, similar results were obtained upon pharmacological inhibition of cIAP1/cIAP2 or TAK1 (Figures 2F, 2G, S2C, and S2D). In line with a role of IKKa and IKKb downstream of cIAP1/cIAP2, TAK1, and NEMO in the pathway, we tested the effect of TPCA-1 on TNF-induced death in ciap1/2/, Tak1/, and Nemo/ MEFs and found no additional effect (Figures 2H–2K). Together, these results indicate that the kinase activities of IKKa/IKKb regulate, downstream of cIAP1/cIAP2, TAK1, and NEMO, both RIPK1 kinasedependent and RIPK1-independent cell death checkpoints.
Figure 2. IKKa and IKKb Mediate Their Protective Effect on RIPK1 via Their Enzymatic Activities (A, B,and D–K)Ripk1+/+ or MEFsof theindicated genotypes weretreated with20ng/ml hTNF inthepresenceof theindicated compounds, and celldeath (A,D,F, H, I, J, K) and caspase-3 activity (B, E, G) were measured in function of time, respectively, by SytoxGreen positivity and DEVD-AMC fluorescence. (C) Protein levels in wild-type MEFs determined by immunoblotting 4 hr poststimulation. Forthecell death results,error bars represent the SEMof three independent experiments. Forthe caspase-3 activity results, error bars represent SDoftriplicates of one representative experiment. See also Figure S2.
IKKa/IKKb Protect Cells from RIPK1-Dependent Apoptosis Independently of NF-kB We previously demonstrated that, in absence of cIAP1/cIAP2 or TAK1, RIPK1 contribution to TNF-induced death is regulated independently of a defect in the canonical NF-kB-dependent upregulation of pro-survival genes (Dondelinger et al., 2013). Moreover, NEMO was also reported to inhibit RIPK1 activation in an NF-kB-independent manner (Legarda-Addison et al., 2009; O’Donnell et al., 2012). IKKa and IKKb are best known for their roles in NF-kB activation, but NF-kB-independent functions have also been reported (Hinz and Scheidereit, 2014). To confirm that IKKa and IKKb regulate RIPK1 activation independently of the canonical NF-kB response, we took two different approaches. In the first one, we tested the effect of inhibiting IKKa/IKKb in conditions where the NF-kB response is prevented by incubating the cells with the translational inhibitor CHX. In the second, we used p65/ MEFs, which are defective for canonical NF-kB activation (Beg et al., 1995). As previously reported (Wang et al., 2008), apoptosis induced by TNF+CHX occurred with a slow kinetic and independently of RIPK1 kinase activity (Figures 3A–3C). Remarkably, a pretreatment with TPCA-1 greatly sensitized cells to apoptosis, and this sensitization was prevented by Nec-1 (Figures 3A– 3C, S3A, and S3B). Similar results were obtained when stimu
lating NF-kB-deficient p65/ MEFs with TNF and TPCA-1 (Figures 3D–3F) or in combination with TAK1 and cIAP1/ cIAP2 inhibitors (Figures S3C and S3D). Together, these results demonstrate that RIPK1-independent and -dependent apoptotic pathways are regulated by two different cell death checkpoints downstream of TNFR1 and that IKKa/IKKb regulate both of them in NF-kB-dependent and -independent manners, respectively.
Figure 3. IKKa/IKKb Protect Cells from RIPK1-Dependent Apoptosis Independently of NF-kB (A,B,D,andE)Ripk1+/+ (AandB)orp65/(DandE)MEFswerestimulatedwith20ng/mlhTNFinthepresenceoftheindicatedcompounds,andcelldeath(Aand D) and caspase activity (B and E) were measured in function of time, respectively, by SytoxGreen positivity and DEVD-AMC fluorescence. (C and F) Ripk1+/+ (C) or p65/ (F) MEFs were stimulated for, respectively, 15 hr and 8 hr with the indicated compounds, and protein levels were determined by immunoblotting. Forthe celldeath results, error bars represent theSEM ofthreeindependent experiments. Forthe caspase-3activity results, error bars represent SDof triplicates of one representative experiment. See also Figure S3.
Defective RIPK1 Phosphorylation in Complex I Correlates with RIPK1 Kinase-Dependent Contribution to TNF-Induced Apoptosis Knowing that the IKK complex physically interacts with RIPK1 in complex I, we hypothesized that the kinase-dependent role of IKKa/IKKb in preventing RIPK1 kinase-dependent apoptosis results from its ability to phosphorylate RIPK1. To test this hypothesis, we analyzed whether RIPK1 is phosphorylated in complex I and whether its phosphorylation state is altered in conditions affecting activation of IKKa/IKKb, but not when the pathway is inhibited downstream of IKKa/IKKb. Because RIPK1 is highly ubiquitylated in complex I (which prevents the detection by immunoblot of potential mobility shifts resulting from its phosphorylation), we removed the ubiquitin chains conjugated to RIPK1 by incubating complex I, pulled-down using FLAG-TNF, with the deubiquitylase USP2 (Figure 4A). By doing so, we observed that the pool of deubiquitylated RIPK1 was running at a higher molecular weight than normal and confirmed, by l-phosphatase treatment, that this mobility shift was resulting from phosphorylation, but not auto-phosphorylation since it was not inhibited by Nec-1 or in Ripk1 K45A MEFs (Figures 4A–4C). Remarkably, and in line with the model of cIAP1/ cIAP2-mediated ubiquitylation-dependent recruitment of TAK1 and NEMO/IKKa/IKKb to complex I and with our cell death results, we found that RIPK1 phosphorylation in complex I is affected in ciap1/2/, Tak1/, Nemo/, and Ikka//Ikkb/, but not in Ikka/, Ikkb/, and p65/ MEFs or in MEFs preincubated with CHX (Figures 4D, 4E, 4G, and 4H). Importantly, IKK activity is greatly affected (as observed by IkBa phosphorylation) in all conditions in which we observed impaired RIPK1 phosphorylation, thereby further demonstrating the link between RIPK1 phosphorylation and IKK enzymatic activities (Figure 4E). Defective RIPK1 phosphorylation in complex I was also observed following pharmacological inhibition of cIAP1/ cIAP2, TAK1, or IKKa/IKKb (Figure 4F).
Figure 4. Defective RIPK1 Phosphorylation in Complex I Correlates with RIPK1 Kinase-Dependent Contribution to TNF-Induced Apoptosis (A–H) Ripk1+/+ MEFs (A, B, F, and H) or MEFs with the indicated genotype (C, D, E, and G) were stimulated for 5 min with 2 mg/ml FLAG-hTNF in the presence or absence of the indicated compounds. TNFR1 complex I was then FLAG immunoprecipitated, incubated with the deubiquitylating enzyme USP2 or lambda phosphatase (l PPase) when indicated, and RIPK1 ubiquitylation and phosphorylation finally analyzed by immunoblotting. * indicates an aspecific band. See also Figure S7
Direct Phosphorylation of RIPK1 by IKKa/IKKb Prevents RIPK1 from Integrating Complex IIb To test the direct contribution of IKKa/IKKb to RIPK1 phosphorylation, we next performed in vitro kinase assays using recombinant proteins and included Nec-1 in the reactions to prevent RIPK1 autophosphorylation. We found that both IKKa and IKKb directly phosphorylated full-length RIPK1 or a mutated version lacking the death and RHIM domain (RIPK11–479)(Figures 5A, 5B, and S4A). Of note, RIPK1 phosphorylation by IKKb induced a mobility shift of RIPK1 not detected when using IKKa (Figures 5A and 5B), suggesting some specificity in the residues phosphorylated by both kinases. In line with our cellular data, TPCA-1 repressed, although with different efficiencies, the direct phosphorylation of RIPK1 by IKKa and IKKb. In contrast, recombinant TAB1/TAK1 did notlead to detectable RIPK1 phosphorylation by autoradiography (Figure 5C).
We next tested the consequence of genetic or pharmacological inhibition of IKKa/IKKb, and of the resulting defective phosphorylation of RIPK1 in complex I, on the ability of RIPK1 to integrate the cytosolic caspase-8-activating complex IIb. We found, by performing FADD and caspase-8 immunoprecipitations, that inhibition of IKKa/IKKb enzymatic activities resulted in the binding of RIPK1 to FADD and caspase-8, a process relying on RIPK1 kinase activity (Figures 5D–5G). In contrast, CHX pre-treatment, which does not affect phosphorylation of RIPK1 in complex I (Figure 4H), led to much less recruitment of RIPK1 to FADD/caspase-8, and this recruitment was not inhibited by Nec-1 (Figures 5F, 5G, and S4B). Of note, association of TRADD with FADD/caspase-8 was not observed under these conditions. Together, these results suggest that IKKa/IKKb-mediated phosphorylation of RIPK1 either represses RIPK1 kinase activity or interferes with RIPK1’s ability to bind complex IIb components.
Figure 5. Direct Phosphorylation of RIPK1 by IKKa/IKKb Prevents RIPK1 from Integrating Complex IIb (A–C) Recombinant GST-IKKa, GST-IKKb, or GST-TAB1-TAK1 fusion protein was incubated with a recombinant truncated (GST-RIPK11–479) or full-length (GST-RIPK1FL) form of RIPK1 in a radioactive in vitro kinase assay in the presence of the indicated inhibitors. Phosphorylation was revealed by SDS-PAGE followed by autoradiography. (D–G) MEFs with the indicated genotype (D and E) or Ripk1+/+ MEFs (F and G) were pre-incubated with zVAD-fmk and with the indicated compounds for 30 min and then stimulated with 20 ng/ml hTNF. After 4 hr, complex II was isolated by FADD or caspase-8 immunoprecipitation and RIPK1 binding revealed by immunoblotting. See also Figure S4.
IKKa/IKKb Mediate In Vivo Protection to RIPK1 KinaseDependent Death To test the in vivo relevance of our in vitro findings, we evaluated the contribution of RIPK1 kinase activity to two different mouse models of TNF-induced death. In the first one, we injected Ripk1K45A/K45A and Ripk1+/+ littermates with TNF in association with D-galactosamine. In this well-known model of acute hepatitis, TNF-mediated hepatocyte apoptosis is reported to result from transcriptional inhibition (Decker and Keppler, 1974), thereby affecting the NF-kB pathway downstream of IKKa/ IKKb. In accordance with our in vitro results, we found that Ripk1K45A/K45A mice were not protected from TNF-induced lethality and apoptotic liver damage, as monitored by survival curves, blood levels of aspartate transaminase/alanine transaminase (AST/ALT), caspase-3 activation in the liver by DEVDase assays, and active caspase-3 staining (Figures 6A–6E). Blood levels of lactate dehydrogenase (LDH), a marker of necrosis, were not upregulated by TNF+GalN injection (Figure 6F).
In the next model, we injected Ripk1K45A/K45A and Ripk1+/+ littermates with a sub-lethal dose of TNF (5 mg) in presence or absence of TPCA-1 (10 mg/kg) to inhibit the canonical NF-kB pathway at the level of IKKa/IKKb. Remarkably, while TPCA-1 hadnotoxicity onitsown(FiguresS5AandS5B),itscombination with TNF resulted in the rapid death of all Ripk1+/+, but no Ripk1K45A/K45A, mice (Figure 6G). Accordingly, Ripk1K45A/K45A mice were protected from TNF-induced hypothermia and had no increase in serum levels of ASL/ALT or caspase-3 activation in the liver (Figures 6H–6L). In contrast to TNF+GalN injection, TNF+TPCA-1 led to a substantial increase of LDH levels in the serum that was also absent in Ripk1K45A/K45A mice, suggesting additional necroptosis induction (Figure 6M). Importantly, RIPK1 kinase inhibition by co-administration of Nec-1s (Degterev et al., 2013), a modified and more stable version of Nec-1, in C57BL/6J mice also significantly delayed the death and injury induced by TNF+TPCA-1 injection (Figures S5C–S5I).
Together, these in vivo results demonstrate TNF-mediated RIPK1-independent and RIPK1 kinase-dependent hepatocyte apoptosis in condition of NF-kB inhibition downstream or at the level of IKKa/IKKb, respectively.
Figure 6. IKKa/IKKb Mediate In Vivo Protection to RIPK1 Kinase-Dependent Death (A) Cumulative survival rates of littermate Ripk1+/+ and Ripk1K45A/K45A C57BL/6J females injected with GalN 15 min prior to injection with mTNF (n = 5). (B, C, and F) Blood AST (B), ALT (C), and LDH (F) levels determined 3 hr post-TNF injection (Ripk1+/+ n = 3, and Ripk1K45A/K45A n = 4). (D and E) Caspase-3 activity in liver samples (Ripk1+/+ n = 3, and Ripk1K45A/K45A n = 4) isolated 3 hr post-TNF injection and determined by Ac-DEVD-AMC fluorescence assay (D) or anti-cleaved caspase-3 staining (E). (G) Cumulative survival rates of littermate Ripk1+/+ and Ripk1K45A/K45A C57BL/6J females injected with TPCA-1 20 min prior to injection with mTNF (n = 5). (H) Body temperature as a function of time. (I, J, and M) Blood AST (I), ALT (J), and LDH (M) levels determined 3 hr post-TNF injection (n = 4). (K and L) Caspase-3 activity in liver samples (n = 4) isolated 3 hr post-TNF injection and determined by Ac-DEVD-AMC fluorescence assay (K) or anti-cleaved caspase-3 staining (L). Scale bar, 25 mm. Error bars represent the SEM of the indicated n values. See also Figure S5.
IKKa/IKKb Protect Cells from RIPK1 Kinase-Dependent Necroptosis Independently of NF-kB Our in vivo results suggested that TNF+TPCA-1 additionally induced necroptosis in the injected mice. To test the possibility that IKKa/IKKb also regulates RIPK1 kinase-dependent necroptosis independently of NF-kB, we in vitro stimulated MEFs with TNF+CHX in the presence of the pan caspase inhibitor zVAD-fmk and of TPCA-1. As shown in Figure 7A,TNF-mediated
necroptosis induced by TNF+CHX+zVAD is fully repressed by Nec-1 but still greatly enhanced by additionally inhibiting IKKa/ IKKb with TPCA-1 (Figures 7A and S6A). The mouse fibrosarcoma cell line L929sAhFAS is a prototypic model for necroptosis since these cells succumb by necroptosis upon single TNF stimulation. While inhibiting NF-kB by CHX sensitized these cells to necroptosis, the sensitization was again enhanced when IKKa/ IKKb was additionally inhibited by TPCA-1 (Figures 7B and S6B). Our results therefore demonstrate that IKKa/IKKb prevent RIPK1 kinase-dependent apoptosis and necroptosis downstream of TNFR1 independently of their known function in protecting the cells from death by mediating NF-kB-dependent upregulation of pro-survival/anti-death genes.
RIPK1 kinase-dependent necroptosis relies on the downstream activation of the RIPK3-MLKL pathway (Cho et al., 2009; He et al., 2009; Pasparakis and Vandenabeele, 2015;Sun et al., 2012; Zhang et al., 2009; Zhao et al., 2012). To further characterize the contribution of RIPK3 to the lethality resulting from the in vivo injection of TNF+TPCA-1, we challenged Ripk3+/+ and Ripk3/ littermates with this trigger. Contrary to Ripk1K45A/K45A mice, Ripk3/ mice were greatly, but not entirely, protected from death and hypothermia induced by TNF+TPCA-1 (Figures 7C and 7D). Interestingly, the protection was not originating from the liver, as RIPK3 deficiency did not prevent liver damage (Figures 7E–7H). Instead, RIPK3 deficiency prevented the increased of LDH levels in the blood, resulting from necrosis of undefined organ(s) (Figure 7I). These in vivo results therefore suggest that the lethality induced by TNF+TPCA-1 results from both RIPK1 kinase-dependent apoptosis and necroptosis.
Figure 7. IKKa/IKKb Protect Cells from RIPK1 Kinase-Dependent Necroptosis Independently of NF-kB (A and B) Ripk1+/+ MEFs (A) and L929sAhFas cells (B) were stimulated with hTNF (20 ng/ml in A and 33 pg/ml in B) in the presence of the indicated compounds, and cell death was measured as a function of time by SytoxGreen positivity. (C) Cumulative survival rates of littermate Ripk3+/+ and Ripk3/ C57BL/6J females injected with TPCA-1 20 min prior to injection with mTNF (Ripk3+/+ n = 4, and Ripk3/ n = 7). (D) Body temperature as a function of time. (E, F, and I) Blood AST (E), ALT (F), and LDH (I) levels determined 3 hr post-TNF injection (Ripk3+/+ n = 4, and Ripk3/ n = 3). (G and H) Caspase-3 activity in liver samples (Ripk3+/+ n = 4, and Ripk3/n = 3) isolated 3 hr post-TNF injection and determined by Ac-DEVD-AMC fluorescence assay (G) or anti-cleaved caspase-3 staining (H). Scale bar,25mm.Fortheinvitrocell death results,error bars represent the SEM of three independent experiments. For the in vivo results,error bars represent the SEM of the indicated n values
Sensing of TNF by TNFR1 at the cell surface can paradoxically result in the activation of signaling pathways with opposite consequences: cell survival or cell death. The fact that survival is the dominant outcome in most cell types indicates the existence of molecular mechanisms actively repressing TNFR1-mediated cell death.Two major mechanisms have been reported to control cell death downstream of TNFR1 (O’Donnell and Ting, 2011).The first identified one is well characterized and consists in a relatively slow process involving the NF-kB-dependent induction of pro-survival/anti-death molecules, such as cFLIP (Karin and Lin, 2002; Kreuz et al., 2001; Liu et al., 1996; Micheau et al., 2001; Panayotova-Dimitrova et al., 2013; Van Antwerp et al., 1996; Wang et al., 1998). The second one, which is less understood and more recently reported, is believed to take place at an earlier stage following TNFR1 activation and is shown to be independent of the NF-kB response (Dondelinger et al., 2013; Legarda-Addison et al., 2009; O’Donnell et al., 2007, 2012; Wang et al., 2008). Interestingly, while the first checkpoint regulates slow apoptosis by inhibiting activation of complex IIa (TRADD-FADD-caspase-8), the second one regulates the contribution of RIPK1 to cell death by either preventing RIPK1 from integrating the apoptotic complex IIb (RIPK1-FADD-casapase-8) or by limiting its contribution to the necrosome (RIPK1RIPK3-MLKL) (Cho et al., 2009; He et al., 2009; Sun et al., 2012; Vanlangenakker et al., 2011; Wang et al., 2008; Wilson et al., 2009; Zhang et al., 2009; Zhao et al., 2012). It has long been thought that IKKa/IKKb inhibits TNF-induced cell death through activation of the NF-kB pathway. In this study, we provide evidences that IKKa and IKKb also regulate cell death by direct phosphorylation of RIPK1 at the level of TNFR1 complex I.
TNF-induced RIPK1-dependent apoptosis was first described in conditions affecting cIAP1/cIAP2-mediated RIPK1 ubiquitylation (Bertrand et al., 2008; O’Donnell et al., 2007; Petersen et al., 2007; Wang et al., 2008), which led to the hypothesis that the ubiquitin chains on RIPK1 were directly preventing its binding to FADD, keeping RIPK1 in a survival modus. This‘‘direct’’effect, however, has later been challenged. Binding of the adaptor proteins TABs and NEMO to RIPK1 ubiquitin chains allows recruitment of TAK1 and of IKKa/IKKb to TNFR1 complex I (Ea et al., 2006; Li et al., 2006; Wu et al., 2006), and TAK1 inhibition was shown to result in TNF-mediated RIPK1-dependent apoptosis without affecting RIPK1 ubiquitylation status (Dondelinger et al., 2013). We show here that RIPK1 is phosphorylated in complex I and that affecting RIPK1 ubiquitylation by cIAP1/ cIAP2depletion directlyimpactsitsphosphorylation.Incontrast, TAK1, NEMO, or IKKa/IKKb depletion affects RIPK1 phosphorylation and induces RIPK1-dependent cell death but does not alter its ubiquitylation state in complex I. Together, these results indicate that ubiquitylation and phosphorylation of RIPK1 occur sequentially and that RIPK1 phosphorylation regulates its killing potential. Because activation of the IKK complex lies downstream of TAK1 but upstream of IkBa and p65, our results suggest a model in which IKKa/IKKb constitute the last step in the regulation of the RIPK1 cell death checkpoint (graphical abstract). Indeed, p65 deletion, expression of IkBaSR, or CHX pre-treatment induces TNF-mediated RIPK1-independent apoptosis and does not alter RIPK1 ubiquitylation or phosphorylation in complex I.
RIPK1 enzymatic activity is needed for the integration of RIPK1 to complex IIb and to the necrosome, which respectively drives apoptosis or necroptosis under TNF-stimulated conditions (Cho et al., 2009; Dondelinger et al., 2013; He et al., 2009; Wang et al., 2008). The precise role of RIPK1 kinase activity in these processes remains unclear but may involve autophosphorylation-driven conformational changes allowing increased binding of RIPK1 to the death complex components. The kinase activity of RIPK1 therefore requires active repression to avoid unnecessary cell death. A recent report suggests that RIPK1 phosphorylation on Ser89 suppresses its kinase activity (McQuade et al., 2013). It is therefore tempting to speculate that IKK-mediated phosphorylation of RIPK1 in complex I affects RIPK1 kinase activity. Alternatively, the phosphorylation of RIPK1 by IKKs may directly affect binding of RIPK1 to the death complex components or facilitate its dissociation from complex I. We performed mass spectrometry analysis to identify the residues of RIPK1 phosphorylated by IKKa and IKKb and found several sites, but not Ser89 (Figures S4C and S4D). Unfortunately, we were unable to demonstrate the direct physiological relevance of the identified phosphorylation sites due to the fact that all Ripk1/ reconstituted MEFs, even those with WT RIPK1 (irrespective of RIPK1 expression levels), started to succumb upon single TNF stimulation (data not shown), a problem previously reported (Gentleetal., 2011). The fact that the combined repression of IKKa and IKKb is needed to induce RIPK1 kinase-dependent death, and that the phosphorylation by each kinase results in different RIPK1 mobility shifts when run on gels, may indicate that phosphorylation on several residues is required to negatively regulate RIPK1.
IKKa and IKKb are best known for their roles in NF-kB activation, but NF-kB-independent functions have also been reported, some of which are even implicated in cell fate decisions (Hinz and Scheidereit, 2014). Using pharmacological inhibition of IKKa/IKKb in a p65-deficient background, or together with CHX, we demonstrated an NF-kB-independent function of IKKa/IKKb in protecting cells from TNF-induced RIPK1 kinasedependent apoptosis and necroptosis. In vivo, we demonstrate that TNF induces apoptosis of hepatocytes independently of RIPK1 when the NF-kB pathway is affected downstream of IKKa/IKKb (TNF+GalN). In contrast, pharmacological inhibition of the NF-kB pathway at the level of IKKa/IKKb (TNF+TPCA-1) sensitizesmicetoTNF-inducedshock,whichisaccompaniedby RIPK1 kinase-dependent, but RIPK3-independent, apoptosis of hepatocytes and RIPK1/RIPK3-dependent cellular death, presumably necroptosis, in undefined organs. We can indeed not formally rule out the possibility that the increase in serum LDH levels originates from secondary necrosis of apoptotic cells. Importantly, genetic and chemical inhibition of RIPK1 enzymatic activity protected the mice from TNF-induced cellular damage and death. These results therefore demonstrate the in vivo roles of IKKa/IKKb in protecting cells from RIPK1 kinase-dependent death.
The LUBAC complex, which includes its component Sharpin, is recruited to complex I during TNF signaling, and the inactivating mouse Sharpin cpdm mutation was reported to cause multi-organinflammationresultingfromTNF-mediated RIPK1kinase-dependent death (Berger et al., 2014; Kumari et al., 2014; Rickard et al., 2014). In line with our results, we found that TNF-mediated RIPK1 kinase-dependent death of mouse dermal fibroblasts (MDFs) isolated from Sharpincpdm mice is associated with defective RIPK1 phosphorylation in complex I (Figures S7A and S7B), probably resulting from the altered recruitment of IKK proteins to complex I, as previously reported for other LUBAC components (Haas et al., 2009). The genetic disruption of Nemo, Ikka, Ikkb, orIkka/Ikkb in mice results in early lethality with massive cellular death in several organs, such as the liver, the skin, and, in the case of Ikka//b/ mice, the nervous system (Hu et al., 1999; Li et al., 1999, 2000; Rudolph et al., 2000; Takeda et al., 1999). So far, these phenotypes have exclusively been explained by defects in NF-kB activation, but our study indicates that RIPK1 activation probably contributes to these pathological conditions. In the same line, RIPK1 kinase-dependent apoptosis may drive the spontaneous development of hepatocellular carcinoma observed in mice ablated of Nemo in the liver parenchymal cells (Luedde et al., 2007). Testing the contribution of RIPK1 to those phenotypes is an exciting future challenge, which may open doors for the use of chemical inhibitors of RIPK1 in the treatment of human diseases associated with IKK malfunctions, such as incontinentia pigmenti (Conte et al., 2014).
Translocation of interleukin-1β into a vesicle intermediate in autophagy-mediated secretion
Min Zhang1, Sam Kenny2, Liang Ge1, Ke Xu2 and Randy Schekman1*
eLife 2015;10.7554/eLife.11205 http://dx.doi.org/10.7554/eLife.11205
In this study, we probed the organelle association and molecular requirements for the secretion of one such unconventional cargo protein, IL-1β. Using surrogate cell lines rather than macrophages to reconstitute autophagy-mediated secretion of IL-1β (Figure 1), we find mature IL-1β localized to the lumen of the membrane in early intermediates and mature autophagosomes (Figures 2-4, 6). This surprising location may help to explain how mature IL-1β is secreted in a soluble form to the cell surface (Figure 9C). However, localization to the lumen between the two membranes of the autophagosome would require that IL-1β is translocated from the cytoplasm across the membrane precursor of a phagophore, rather than being engulfed as the phagophore membrane matures by closure into an autophagosome.
The exact route by which the autophagosome delivers mature IL-1β to the cell surface as well as how it avoids fusion with degradative lysosome remains obscure, possibly involving interaction with the multi-vesicular body or some form of lysosome as a prelude to fusion at the cell surface (Figure 9C), and this process may require selective recruitment of membrane sorting and targeting factors such as Rabs and SNAREs.
Recent evidence suggests that autophagy facilitates the unconventional secretion of the pro-inflammatory cytokine interleukin 1β (IL-1β). Here, we reconstituted an autophagy-regulated secretion of mature IL-1β (m-IL-1β) in non-macrophage cells. We found that cytoplasmic IL-1β associates with the autophagosome and m-IL-1β enters into the lumen of a vesicle intermediate but not into the cytoplasmic interior formed by engulfment of the autophagic membrane. In advance of secretion, m-IL-1β appears to be translocated across a membrane in an event that may require m-IL-1β to be unfolded or remain conformationally flexible and is dependent on two KFERQ-like motifs essential for the association of IL-1β with HSP90. A vesicle, possibly a precursor of the phagophore, contains translocated m-IL-1β and later turns into an autophagosome in which m-IL-1β resides within the intermembrane space of the double-membrane structure. Completion of IL-1β secretion requires Golgi reassembly and stacking proteins (GRASPs) and multi-vesicular body (MVB) formation.
Most eukaryotic secretory proteins with an N-terminal signal peptide are delivered through the classical secretion pathway involving an endoplasmic reticulum (ER)-to-Golgi apparatus itinerary (Lee et al., 2004; Schatz and Dobberstein, 1996). However, a substantial number of secretory proteins lack a classical signal peptide, called leaderless cargoes, and are released by unconventional means of secretion (Nickel and Rabouille, 2009; Nickel and Seedorf, 2008). The range of unconventional secretory cargoes encompasses angiogenic growth factors, inflammatory cytokines and extracellular matrix components etc. most of which play essential roles for development, immune surveillance and tissue organization (Nickel, 2003; Rabouille et al., 2012). Unlike a unified route for classical protein secretion, leaderless cargoes undergoing unconventional secretion employ multiple means of protein delivery, the details of which are largely unknown (Ding et al., 2012; Nickel, 2010; Rabouille et al., 2012; Zhang and Schekman, 2013).
IL-1β is one of the most intensely investigated cargoes of unconventional secretion. A biologically inactive 31 kDa precursor, pro-IL-1β, is made following initiation of the NF-κB signaling cascade. Pro-IL-1β is subsequently converted into the active form, the 17 kDa mature IL-1β, by the pro-inflammatory protease caspase-1 which is activated, in response to extracellular stimuli, after its recruitment to a multi-protein complex called the inflammasome (Burns et al., 2003; Cerretti et al., 1992; Rathinam et al., 2012; Thornberry et al., 1992). Interpretation of the mechanism of unconventional secretion of IL-1β is complicated by the fact that one of the physiologic reservoirs of this cytokine, macrophages, undergoes pyroptotic death and cell lysis under conditions of inflammasome activation of caspase-1. Indeed, many reports including two recent publications make the case for cell lysis as a means of release of mature IL-1β (Liu et al., 2014; Shirasaki et al., 2014). In contrast, other reports demonstrate proper secretion of mature IL-1β without cell lysis in, for example, neutrophils, which are nonetheless dependent on the inflammasome response to activate caspase-1 and secrete mature IL-1β (Chen et al., 2014).
Macroautophagy (hereafter autophagy) is a fundamental mechanism for bulk turnover of intracellular components in response to stresses such as starvation, oxidative stress and pathogen invasion (Mizushima and Levine, 2010; Yang and Klionsky, 2010). The process is characterized by the formation of a double-membrane vesicle, called the autophagosome, through the elongation and closure of a cup-shaped membrane precursor, termed the phagophore, to engulf cytoplasmic cargoes (Hamasaki et al., 2013; Lamb et al., 2013). Completion of autophagosome formation requires a sophisticated protein-vesicle network organized by autophagic factors, such as autophagy-related (ATG) proteins, and target membranes (Feng et al., 2014; Mizushima et al., 2011). Besides the degradative function, autophagy or ATG proteins have recently been implicated in multiple secretory pathways including the delivery of leaderless cargoes undergoing unconventional secretion, such as the mammalian pro-inflammatory cytokines IL-1β and IL-18, the nuclear factor HMGB1, and the yeast acyl coenzyme A-binding protein Acb1, to the extracellular space (Bruns et al., 2011; Dupont et al., 2011; Duran et al., 2010; Manjithaya and Subramani, 2011; Pfeffer, 2010; Subramani and Malhotra, 2013). The Golgi reassembly and stacking protein(s) GRASP(s) (GRASP55 and GRASP65 in mammals, dGRASP in Drosophila, GrpA in Dictyostelium and Grh1 in yeast) are required for autophagy-regulated unconventional secretion (Giuliani et al., 2011; Kinseth et al., 2007; Levi and Glick, 2007; Manjithaya et al., 2010).
Dupont et al. (2011) documented a role for autophagy in the secretion of mature IL-1β (Dupont et al., 2011), but how a protein sequestered within an autophagosome could be exported as a soluble protein was unexplained. Here, we sought to understand how conditions of starvation-induced autophagy could localize IL-1β into an autophagosomal membrane. We reconstituted the autophagy-regulated secretion of IL-1β in cultured cell lines and detected a vesicle intermediate, possibly an autophagosome precursor, containing mature IL-1β. Three-dimensional (3D) Stochastic Optical Reconstruction Microscopy (STORM) demonstrated that, after entering into the autophagosome, IL-1β colocalizes with LC3 on the autophagosomal membrane, which, together with an antibody accessibility assay and observations from biochemical assays, implies a topological distribution in the intermembrane space of the autophagosome. This distribution of IL-1β explains the mechanism accounting for its secretion as a soluble protein through either a direct fusion of autophagosome with the plasma membrane or via the MVB pathway. Quite aside from the possible complication of cell lysis, another body of work has suggested an unconventional pathway for the proper secretion of IL-1β. Pro-IL-1β lacks a typical signal peptide and the propeptide is processed in the cytosol rather than the ER (Rubartelli et al., 1990; Singer et al., 1988). Although mature IL-1β appears to be incorporated into a vesicular transport system, secretion is not blocked by Brefeldin A, a drug that blocks the traffic of standard secretory proteins form the Golgi apparatus (Rubartelli et al., 1990). Multiple mechanisms have been implicated in the unconventional secretion of IL-1β, including autophagy, secretory lysosomes, multi-vesicular body (MVB) formation and micro-vesicle shedding (Andrei et al., 1999; Andrei et al., 2004; Brough et al., 2003; Lopez-Castejon and Brough, 2011; MacKenzie et al., 2001; Qu et al., 2007; Verhoef et al., 2003). However, a clear demonstration of the mechanism for the entry of IL-1β into a vesicular carrier, e.g. the autophagosome, is lacking.
Reconstitution of autophagy-regulated IL-1β secretion
A dual effect of autophagy has been proposed on the secretion of IL-1β in macrophages (Deretic et al., 2012; Jiang et al., 2013). On one hand, induction of autophagy directly promotes IL-1β secretion after inflammasome activation by incorporating it into the autophagosomal carrier (Dupont et al., 2011). On the other hand, autophagy indirectly dampens IL-1β secretion by degrading components of the inflammasome as well as reducing endogenous triggers for inflammasome assembly, including reactive oxygen species (ROS) and damaged components, which are required for the activation of caspase-1 and the production of active IL-1β (Harris et al., 2011; Nakahira et al., 2011; Shi et al., 2012; Zhou et al., 2011).
To focus our study specifically on the role of autophagy in IL-1β secretion, we reconstituted a stage of IL-1β secretion downstream of inflammasome activation by co-expressing pro-IL-1β (p-IL-1β) and pro-caspase-1 (p-caspase-1) in non-macrophage cells. As shown in Figure 1A, the generation and secretion (~5%) of mature IL-1β (m-IL-1β) was achieved by co-expression of p-IL-1β and p-caspase-1 in HEK293T cells. Mature IL-1β was not produced or secreted without p-caspase-1, whereas a low level of secreted p-IL-1β (~0.2%) was detected with or without the expression of p-caspase-1. Furthermore, little cell lysis occurred during the treatment we used to induce IL-1β secretion: Much less precursor than mature IL-1β and little cytoplasmic tubulin was detected released into the cell supernatant during the 2 h incubation in starvation medium (Figure 1A). Starvation, a condition that stimulates autophagy, enhanced IL-1β secretion (~3 fold) and reduced the level of IL-1β in the cell lysates (Figure 1A, B). Inhibition of autophagy by the phosphatidylinositol 3-kinase (PI3K) inhibitors 3-methyladenine (3-MA) or wortmannin (Wtm) blocked IL-1β secretion activated by starvation and caused the accumulation of mature IL-1β in the cell (Figure 1B). Likewise, in an autophagy-deficient cell line, Atg5 knockout (KO) mouse embryo fibroblasts (MEFs) (Mizushima et al., 2001), IL-1β secretion was reduced and failed to respond to starvation (Figure 1C). Moreover, IL-1β secretion was also inhibited in a dose-dependent manner in the presence of an ATG4B mutant (C74A) (Fujita et al., 2008), or after the depletion of ATG2A and B (Velikkakath et al., 2012), or FIP200 (Hara et al., 2008), which block autophagosome biogenesis at different stages (Figure 1D-F). Therefore, the reconstituted system recapitulates the autophagy-regulated secretion of IL-1β.
In macrophages, MVB formation and GRASP proteins are required for IL-1β secretion (Dupont et al., 2011; Qu et al., 2007). Inhibiting MVB formation by depletion of the ESCRT components, hepatocyte growth factor receptor substrate (Hrs) or TSG101, compromised secretion of IL-1β and CD63, an exosome marker (Figure 1-figure supplement 1A). Knockdown of the GRASP55 or GRASP65 also led to the reduction of IL-1β secretion (Figure 1- figure supplement 1B). Therefore, in addition to functions required for autophagy, the secretion of IL-1β in HEK293T cells depends on GRASP proteins and at least two proteins implicated in MVB formation, as reported previously (Dupont et al., 2011; Qu et al., 2007).
Figure 1 Reconstitution of autophagy-regulated IL-1β secretion in cultured cells (A) Reconstitution of starvation-induced IL-1β secretion in HEK293T cells. HEK293T cells were transfected with a single plasmid encoding p-IL-1β or together with the p-caspase-1 plasmid. After transfection (24 h), the cells were either treated in regular (DMEM) or starvation (EBSS) medium for 2 h. The medium and cells were collected separately and immunoblot was performed to determine the level of indicated proteins. (B) PI3K inhibitors 3-methyladenine (3-MA) or wortmannin (Wtm) inhibit IL-1β secretion. HEK293T cells transfected with p-IL-1β and p-caspase-1 plasmids were cultured in DMEM, EBSS, or EBSS containing 10 mM 3-MA or 20 nM wortmannin for 2 h. The medium and cells were collected separately and immunoblot was performed as shown in (A). (C) IL-1β secretion is blocked in Atg5 KO MEFs. Control WT or Atg5 KO MEFs were transfected with p-IL-1β and p-caspase-1 plasmids. After transfection (24 h), the cells were either cultured in DMEM or EBSS for 2 h followed by immunoblot as shown in (A). (D) IL-1β secretion is inhibited by the ATG4B mutant (C74A). HEK293T cells were transfected with plasmids encoding p-IL-1β, p-caspase-1 and different amounts of ATG4B (C74A) plasmid DNA as indicated. After transfection (24 h), cells were starved in EBSS for 2 h followed by immunoblot as shown in (A). (E) Knockdown of Atg2 reduces IL-1β secretion. HEK293T cells were transfected with control siRNA or siRNAs against Atg2A, Atg2B alone or both. After transfection (48 h), the cells were transfected with p-IL-1β and p-caspase-1 plasmids. After another 24 h, the cells were starved in EBSS for 2 h followed by immunoblot as shown in (A). (F) Knockdown of FIP200 reduces IL-1β secretion. HEK293T cells were transfected with control siRNA or FIP200 siRNA. IL-1β secretion under starvation conditions was determined as shown in (E). Quantification of IL-1β secretion was calculated as the ratio between the amount of IL-1β in the medium and the total amount (the sum of IL-1β in both medium and lysate).
Figure 1- figure supplement 1 Depletion of ESCRT or GRASPs affects IL-1β secretion HEK293T cells were transfected with indicated siRNAs (Hrs (ESCRT-0) (A), Tsg101 (ESCRT-I) (A), GRASP55 (B) or GRASP65 (B)). After transfection (48 h), the cells were transfected with p-IL-1β and p-caspase-1 plasmids. After another 24 h, the cells were starved in EBSS for 2 h followed by immunoblot as shown in Figure 1A. Quantification of IL-1β secretion was calculated as the ratio between the amount of IL-1β in the medium and the total amount (the sum of IL-1β in both medium and lysate).
IL-1β transits through an autophagosomal carrier during secretion.
To study if autophagy directly regulates IL-1β secretion, we employed a three-step membrane fractionation procedure as described previously (Figure 2A)(Ge et al., 2013). We first performed a differential centrifugation to obtain 3k, 25k and 100k membrane pellet fractions. Both IL-1β and the lipidated form of LC3 (LC3-II), a protein marker of autophagosome, were mainly enriched in the 25k membrane fraction (Figure 2B). We then separated the 25k membrane through a sucrose step gradient ultracentrifugation where both IL-1β and LC3-II co-distributed in the L fraction at the boundary between 0.25 M and 1.1 M layer of sucrose (Figure 2B). Further fractionation of the L fraction using an OptiPrep gradient showed co-fractionation of IL-1β with LC3-II (Figure 2C). To confirm the presence of IL-1β in the autophagosome, we performed immunoisolation of LC3-positive autophagosomes from the 25k fraction and found that IL-1β, especially the mature form, co-sedimented with autophagosomes (Figure 2D). Consistent with our observations, a recent study also showed a colocalization of IL-1β and LC3 in the form of puncta in macrophages (Dupont et al., 2011). These data demonstrate that at least a fraction of intracellular mature IL-1β associates with the autophagosome, possibly related to its role in IL-1β secretion.
Figure 2 IL-1β vesicles co-fractionate with LC3 vesicles (A) Membrane fractionation scheme. Briefly, HEK293T cells transfected with p-IL-1β and p-caspase-1 plasmids were starved in EBSS for 2 h, collected and homogenized. Cell lysates were subjected to differential centrifugations at 3,000×g (3k), 25,000×g (25k) and 100,000×g (100k). The level of IL-1β in each membrane fraction was determined by immunoblot. The 25k pellet, in which IL-1β was mainly enriched, was selected and a sucrose gradient ultracentrifugation was performed to separate membranes in the 25k pellet to the L (light) and P (pellet) fractions. The L fraction, which contained the majority of IL-1β, was further resolved on an OptiPrep gradient after which ten fractions from the top were collected. (B, C) Immunoblot was performed to examine the distribution of IL-1β, LC3 as well as the indicated membrane markers in the indicated membrane fractions. T, top; B, bottom (D) HEK293T cells transfected with p-IL-1β, p-caspase-1 and FLAG-tagged LC3-I plasmids were starved in EBSS for 2 h. LC3 positive membranes were immunoisolated with anti-FLAG agarose from the 25k pellet and the presence of IL-1β was determined by immunoblot analysis. FT, flowthrough.
To determine if IL-1β is localized to the phagophore in the absence of autophagosome completion, we fractionated membranes from ATG2-depleted cells, which are deficient in phagophore elongation and therefore fail to form mature autophagosomes (Velikkakath et al., 2012), and examined the distribution of LC3-II, which remains attached to immature phagophore membranes, and mature and precursor IL-1β. We performed the three-step fractionation described above. In control cells, IL-1β co-distributed with LC3-II in all three steps (Figure 3). Depletion of ATG2 did not affect the co-fractionation of IL-1β and LC3-II (Figure 3), indicating that IL-1β enters into the phagophore membrane before the completion of the autophagosome.
Figure 3 IL-1β co-distributes with LC3 in Atg2-depleted cells (A) HEK293T cells were transfected with siRNAs against Atg2A and Atg2B followed with p-IL-1β and 739 p-caspase-1 plasmids as shown in Figure 1E. The cells were starved in EBSS for 2 h. Membrane fractions (3k, 25k, 100k (×g), L and P) were separated from the post-nuclear supernatant as depicted in Figure 2B. (B) Ten membrane fractions were collected from the OptiPrep gradient ultracentrifugation as depicted in Figure 2C. Immunoblot was performed to examine the distribution of IL-1β, LC3 as well as the indicated membrane markers. T, top; B, bottom.
Autophagosome formation is not required for entry of IL-1β into vesicles
We asked how IL-1β enters into the autophagosome. One possibility is engulfment through the closure of the phagophore membrane during autophagosome maturation as in the capture of autophagic cargo. In this scenario, closure of the phagophore to complete autophagosome formation would be required to sequester IL-1β away from the cytoplasm. Alternatively, we considered the possibility that IL-1β may be translocated through a membrane into the lumen of the phagophore envelope and be sequestered from the cytoplasm even before the mature autophagosome is sealed. To test this possibility, we performed proteinase K protection experiments with the membranes from ATG2-depleted cells (Figure 4A). In control cells, p62 (an autophagic cargo) and a fraction of LC3-II (which was encapsulated after autophagosome completion), as well as mature IL-1β, were largely resistant to proteinase K digestion similar to the ER luminal protein, protein disulfide isomerase (PDI). In contrast, SEC22B, a membrane anchored SNARE protein exposed to the cytoplasm, was sensitive to proteinase K digestion (Figure 4A). Triton X-100 treatment permeabilized the membrane and rendered all proteins tested sensitive to proteinase K digestion (Figure 4A). This demonstrated that the majority of membrane localized IL-1β was sequestered within an organelle, likely the autophagosome, as demonstrated by the fractionation results of Figures 2 and 3. However, the result did not pinpoint where within the autophagosome IL-1β was housed. In ATG2-depleted cells, p62 and LC3-II remained sensitive to proteinase K digestion, consistent with the hypothesis that ATG2 is essential for maturation and closure of the autophagosome (Figure 4A). However, in the same samples the majority of IL-1β resisted degradation by proteinase K treatment (Figure 4A), except on addition of Triton X-100 to permeabilize membranes. Although the precursor form of IL-1β remained associated with isolated autophagosome and phagophore membranes (Figure 3), the protein was degraded when membranes from normal and ATG2-depleted cells were treated with protease in the presence or absence of Triton X-100 (data not shown). Thus, the mature but not the precursor IL-1β appears to be transported into the phagophore.
A most recent study showed that small, closed double-membrane structures could be observed in ATG2-depleted cells (Kishi-Itakura et al., 2014). To rule out the possibility that IL-1β was engulfed by the small closed autophagosomes, we employed Atg5 KO MEFs in which the phagophore could not be closed (Kishi-Itakura et al., 2014; Mizushima et al., 2001). Similar to what we observed in ATG2-depleted cells, IL-1β was protected from proteinase K digestion in membranes from Atg5 KO MEFs (Figure 4B). In addition, IL-1β was sequestered within vesicles in FIP200 (another early factor in phagophore development (Hara et al., 2008)) knockdown cells (Figure 4C). These data indicate that the entry of IL-1β into the vesicle carrier is not dependent on the formation of the autophagosome. These results are inconsistent with a role for engulfment of IL-1β by the maturing phagophore and suggest instead that IL-1β may be translocated across a membrane into a vesicle precursor of the phagophore, possibly at a very early stage in the development of the organelle.
Figure 4 Closure of the autophagosome is not required for the entry of IL-1β into vesicles (A) HEK293T cells were transfected with siRNAs against Atg2A and Atg2B followed by transfection with p-IL-1β and p-caspase-1 plasmids as shown in Figure 1E. The cells were starved in EBSS for 2 h and proteinase K digestion was performed with the 25k membrane fractions. (B) Atg5 WT, KO MEFs were transfected with p-IL-1β and p-caspase-1 plasmids as shown in Figure 1B. The cells were starved in EBSS for 2 h followed by proteinase K digestion as shown in (A). 752 (C) HEK293T cells were transfected with siRNA against FIP200 followed by analysis of membrane entry of 753 IL-1β as shown in (A). The level of proteinase K protection was calculated as the percentage of the total protein. Error bars represent standard deviations of at least three experiments.
Entry of IL-1β into the vesicle carrier requires protein conformational flexibility
We then sought to test if IL-1β could directly translocate across the membrane of a vesicle carrier. As protein unfolding is usually required for protein translocation, we adopted an approach used in many other circumstances wherein a targeted protein is fused to dihydrofolate reductase (DHFR), an enzyme whose three-dimensional structure is stabilized by the folate derivative aminopterin, hence providing a chemical ligand to impede the unfolding process (Backhaus et al., 2004; Eilers and Schatz, 1986; Wienhues et al., 1991). We first determined the secretion of the DHFR-fused IL-1β. As shown in Figure 5A, secretion of a mature IL-1β-DHFR fusion protein was enhanced by starvation similar to the untagged counterpart. Importantly, IL-1β-DHFR secretion was reduced in a dose-dependent manner in the presence of aminopterin (Figure 5B). Of notice, treatment of aminopterin did not completely abolish IL-1β secretion perhaps due to a cell death-induced release of IL-1β at high concentrations of aminopterin, as indicated by the release of a low level of tubulin into the medium fraction (Figure 5B). As a control, aminopterin did not reduce the secretion of untagged IL-1β, confirming its specific effect on DHFR (Figure 5- figure supplement 1). Fractionation of cells 185 incubated with aminopterin showed a reduced level of IL-1β in the membrane fraction with a corresponding 186 increase in the cytosol fraction (Figure 5C). The residual DHFR-tagged IL-1β associated with membranes from aminopterin-treated cells was sensitive to proteinase K digestion (Figure 5D), indicating that this pool of membrane-associated IL-1β did not translocate into the lumen of the vesicle. The data suggest that entry of IL-1β into a vesicle carrier involves a process of protein unfolding and translocation.
Figure 5 Protein unfolding is required for the entry of IL-1β into vesicles (A) Secretion of DHFR-tagged IL-1β. HEK293T cells were transfected with p-IL-1β-DHFR and p-caspase-1 plasmids. After transfection (24 h), the cells were treated with DMEM or EBSS for 2 h. Release of IL-1β was determined as shown in Figure 1. (B) Secretion of IL-1β-DHFR was inhibited by aminopterin. HEK293T cells were transfected with p-IL-1β-DHFR and p-caspase-1 plasmids. After transfection (24 h), the cells were treated with EBSS, or EBSS containing different concentrations of aminopterin as indicated for 15 min followed by determination of IL-1β secretion as shown in (A). Quantification of IL-1β secretion was calculated as the ratio between the amount of IL-1β in the medium and the total amount (the sum of IL-1β in both medium and lysate). (C) Less IL-1β enters into membrane in the presence of aminopterin. HEK293T cells were transfected with p-IL-1β-DHFR and p-caspase-1 plasmids. After transfection (24 h), the cells were either untreated or treated with 5 μM aminopterin in EBSS for 2 h. The membrane fraction was collected from the top fractions of a Nycodenz density gradient resolved from membranes in a 25k pellet as described in Material and Methods. The cytosolic fraction was collected as the supernatant after 100k×g centrifugation. All fractions were analyzed by immunoblotting using indicated antibodies. (D) IL-1β-DHFR is not protected from proteinase K in the presence of aminopterin. Nycodenz -floated membrane fraction collected as shown in (C) was subjected to proteinase K digestion and then analyzed by immunoblotting using indicated antibodies.
Figure 5- figure supplement 1 Secretion of IL-1β is not affected by aminopterin HEK293T cells were transfected with p-IL-1β and p-caspase-1 plasmids. After transfection (24 h), the cells were treated with EBSS, or EBSS containing different concentrations of aminopterin as indicated for 15 min followed by determination of IL-1β secretion as shown in Figure 1 (A).
IL-1β colocalizes with LC3 on the autophagosome envelope
If IL-1β is directly translocated across the membrane of a vesicle intermediate, fusion of these vesicles to form a double-membrane autophagosome would deposit IL-1β in the lumen between the two membranes of the autophagosome. To visualize the subcellular localization of IL-1β, we employed U2OS cells, which formed 194 large and distinct autophagosomes after starvation. U2OS cells co-expressing p-IL-1β and p-caspase-1 secreted IL-1β in a starvation-enhanced and PI3K-dependent manner similar to HEK293T cells (Figure 6- figure supplement 1). To prepare for the subsequent fluorescence imaging, we also employed a FLAG-tagged m-IL-1β, which allowed us to directly determine the topological localization of the m-IL-1β. Secretion of m-IL-1β-FLAG from U2OS cells was stimulated by starvation and dependent on PI3K (Figure 6- figure supplement 1).
To determine the topological distribution of IL-1β, we first performed confocal immunofluorescence labeling experiments. After starvation, cells were exposed to 40 μg/ml of digitonin to permeabilize the plasma membrane, harvested and washed with cold PBS to remove the excess cytosolic m-IL-1β-FLAG. In cells expressing either p-IL-1β and p-caspase-1, or m-IL-1β alone, LC3 and IL-1β were observed by confocal microscopy to localize together or adjacent to one another on the edge of ring-shaped autophagosomes (Figure 6- figure supplement 2). To further resolve these ring structures, we employed 3D STORM (Huang et al., 2008; Rust et al., 2006) super-resolution microscopy (Hell, 2007; Huang et al., 2010) (Figure 6 and Figure 6- figure supplements 3, 4 and Videos 1 and 2). Ring-shaped autophagosomes positive for LC3 (cyan) formed after starvation. Some IL-1β (magenta) also organized in ring-shaped structures that co-localized with LC3 (Figure 6 and Figure 6- figure supplement 3). Around 18 ring structures of IL-1β accounting for ~5% of the total IL-1β signal were observed in each cell. A 3D virtual Z-stack analysis confirmed the spatial co-distribution of LC3 and IL-1β on a ball-shaped vesicle (Video 1 and 2). The diameter of the structures double-labeled with LC3 and IL-1β are ~700 nm (larger structures up to 2 μm in diameter were also found) which is comparable to the size of the autophagosome. Occasionally, we also found IL-1β localized in the center of the ring structure, where cytoplasmic autophagic cargoes fill, surrounded by LC3 (Figure 6-figure supplement 4). This portion of IL-1β was possibly being engulfed by the autophagosome.
The visual detection of IL-1β localized to ring-shaped autophagosomes is consistent with our biochemical assays that place IL-1β in the intermembrane space between the outer and inner membrane of the autophagosome. We devised a further visual test of this conclusion using selective permeabilization of cell surface and intracellular membranes with digitonin and saponin, respectively (Figure 6-figure supplement 5). We compared antibody accessibility to IL-1β and DFCP1, a marker located on the cytosolic surface of the omegasome (a harbor for the phagophore) in both WT and Atg5 KO cells. Consistent with a cytosolic surface localization, DFCP1 was readily labeled in cells treated with digitonin alone (selectively permeabilizes the plasma membrane) in both WT and Atg5 KO cells (Figure 6-figure supplement 5A-E). In contrast, IL-1β was accessible to the antibody only after treatment with digitonin and saponin (gently permeabilizes the endomembrane) (Figure 6-figure supplement 5A, B and F) in WT cells. This by itself would not distinguish localization of IL-1β to the intermembrane space vs the cytoplasmic enclosed space of a mature autophagosome. However, in Atg5 KO cells where the phagophore precursor envelope remains open and exposed to the cytosol, saponin treatment was necessary to expose IL-1β to antibody and roughly half of the labeled structures coincided with the phagophore marker DFCP1 (Figure 6-figure supplement 5C, D and F). This visual assay further confirms the intermembrane localization of IL-1β in the phagophore and 231 autophagosome.
Figure 6 Topological localization of IL-1β in the autophagosomal carrier determined by STORM U2OS cells were transfected with a plasmid containing the expression cassette of FLAG-tagged mature IL-1β (m-IL-1β-FLAG). After transfection (24 h), the cells were starved in EBSS for 1 h followed by immunofluorescence labeling with mouse monoclonal anti-LC3 and rabbit polyclonal anti-FLAG antibodies. STORM analysis imaging and data analysis were performed as described in Materials and Methods. Cyan, LC3; Magenta, IL-1β; Bars: 2 μm (original image) and 500 nm (magnified inset)
Figure 6- figure supplement 1 Secretion of IL-1β in U2OS cells 795 U2OS cells were transfected with plasmids encoding the p-IL-1β and p-caspase-1 (first 4 lanes) or m-IL-1β-FLAG (last 4 lanes). After transfection (24 h), the cells were untreated or starved in the absence or presence of indicated PI3K inhibitors (3-MA or wortmannin (Wtm)) followed by measurement of secretion as indicated in Figure 1 (A) and (B). α-m-IL-1β, IL-1b antibody; α-FLAG, FLAG antibody
Figure 6- figure supplement 2 Localization of IL-1β determined by confocal microscopy U2OS cells were transfected with plasmids encoding the p-IL-1β and p-caspase-1 (A) or m-IL-1β-FLAG (B). After transfection (24 h), the cells were starved for 1 h followed by immunofluorescence labeling and confocal 804 microscopy analysis. Bar: 10 μm
Figure 6- figure supplement 3 Extra images for Figure 6 Bars: 2 μm (original image) and 500 nm (magnified inset)
Figure 6- figure supplement 4 A minority of IL-1β engulfed by autophagosome U2OS cells were transfected and treated followed by STORM analysis as shown in Figure 6. Arrow head points to the autophagosome with engulfed IL-1β. Bar: 2 μm
Figure 6- figure supplement 5 Determination of the topological localization of IL-1β in the autophagosome and phagophore (A, C) Diagrams of autophagosome (A)/phagophore (B) and omegasome, antibody accessibility for each possible situation of IL-1β localization, and summaries of the antibody accessibility of m-IL-1β-FLAG (red) and EGFP-DFCP1 (green) are illustrated. (B, D) U2OS cells (B) and Atg5 KO MEFs (D) were transfected with plasmids encoding the m-IL-1β-FLAG and EGFP-DFCP1. After transfection (24 h), the cells were starved in EBSS for 1 h followed by digitonin treatment and fixation (see Materials and Methods). The cells were either labeled with anti-FLAG (to label IL-1β) and anti-EGFP (to label EGFP-DFCP1) antibodies (Digitonin) or further treated with Saponin followed by antibody labeling (Digitonin+Saponin). Images were acquired by confocal microscopy. Bar: 10 μm 825 (E) Quantification of the percentage of EGFP-DFCP1 labeled by EGFP antibody. Percentage was counted by 826 the ratio of puncta numbers of antibody labeled EGFP-DFCP1 and EGFP-DFCP1 according to the EGFP signal. Error bars are standard deviations of more than 50 cells in two independent experiments. (F) Quantification of the puncta number for m-IL-1β-FLAG puncta (red) and those colocalized with DFCP1 (yellow). Error bars are standard deviations of more than 50 cells in two independent experiments.
Video 1 832 3D section of the magnified structure in Figure 6 (upper one) 833 The virtual Z-section thickness is 150 nm, and the step size is 50 nm. Cyan, LC3; Magenta, IL-1β; Bar 500 nm 834 835 Video 2 836 3D section of the magnified structure in Figure 6 (lower one) 837 The virtual Z-section thickness is 150 nm, and the step size is 50 nm. Cyan, LC3; Magenta, IL-1β; Bar 500 nm
Two KFERQ-like motifs are required for the entry of IL-1β into the vesicle carrier
In chaperone-mediated autophagy (CMA), cargoes are recognized by a KFERQ sequence motif for transport into the lysosome (Dice et al., 1986; Kaushik and Cuervo, 2012). We analyzed the primary sequence of IL-1β and found three KFERQ-like motifs on IL-1β including 127LRDEQ131, 132QKSLV136 and 198QLESV202 (Figure 7A). We mutated the glutamine, which has been shown to be essential for the function of the motif, as well as an adjacent amino acid in each motif (E130Q131, Q132K133 and Q198L199) to alanines and examined the secretion efficiency of these mutants. The 130-131AA mutant did not affect secretion of IL-1β (Figure 7B). However, the Q132K133 and Q198L199 mutations were both defective in secretion of mature IL-1β which instead accumulated in the cytoplasmic fraction (Figure 7B). A low level of release of the pro-forms persisted as seen with WT and mutant protein (Figure 7B). The cytoplasmic mature forms of the mutant proteins were less abundant in the membrane fraction compared with the WT mature IL-1β (Figure 7C, compare the lanes without proteinase K treatment). In addition, the membrane associated mutant IL-1β remained proteinase K accessible (less than 10% of protection compared with ~45% of WT IL-1β), demonstrating that these two KFERQ-like motifs are required for the membrane translocation of IL-1β (Figure 7C). Equal amounts of WT and mutant p-IL-1β associated with the membrane but both remained largely proteinase K accessible (Figure 7C).
Figure 7 Mutation of the KFERQ-like motif affects IL-1β secretion and entry into vesicles (A) Protein sequence of IL-1β. The yellow region indicates mature IL-1β. Three KFERQ-like motifs (aa127-131, aa132-136 and aa198-202) are highlighted in red underlined bold. (B) Secretion of IL-1β mutants. HEK293T cells were transfected with p-IL-1β-DHFR and p-caspase-1 plasmids. After transfection (24 h), the cells were either treated with DMEM or EBSS for 2 h. Secretion of 845 IL-1β mutant proteins was detected by immunoblot. (C) IL-1β mutant 132-133AA or 198-199AA is accessible to proteinase K digestion. HEK293T cells were transfected with plasmids encoding p-caspase-1 and IL-1β mutant 132-133AA or 198-199AA. After transfection (24 h), the cells were treated with EBSS for 2 h. The 25k membrane fraction was collected and subjected to proteinase K digestion assay and then analyzed by immunoblot using indicated antibodies. The level of proteinase K protection was calculated as the percentage of the total protein. Error bars represent standard deviations of at least three experiments.
HSP90 is required for the entry of IL-1β into the vesicle intermediate
The chaperone protein HSC70 and HSP90 have been reported to function in chaperone-mediated autophagy (CMA) (Kaushik and Cuervo, 2012; Majeski and Dice, 2004). HSP70 has also been implicated in autophagy and stress responses (Murphy, 2013). We performed shRNA-mediated knockdown of the three chaperone proteins to assess their potential role in the membrane translocation of IL-1β. Knockdown of Hsp90, but not of Hsp70 or Hsc70 substantially reduced IL-1β secretion (Figure 8A). As a control, knockdown of Hsc70 compromised CMA as indicated by the stabilization of a CMA cargo, GAPDH (Figure 8-figure supplement 1A). Moreover, secretion of mature IL-1β was inhibited in a dose-dependent manner by an HSP90 inhibitor geldanamycin (Figure 8B). In both experiments, mature IL-1β accumulated in the cytosol fraction at the expense of secretion. Knockdown of Hsp90 also rendered IL-1β accessible to proteinase K digestion (Figure 8C), consistent with a role for HSP90 in the translocation of IL-1β as opposed to some later secretion event. Furthermore, in a co-immunoprecipitation assay, HSP90 associated with m-IL-1β but not the translocation-deficient mutants Q132K133 and Q198L199 (Figure 8D). Although p-IL-1β also formed a complex with HSP90, the efficiency appeared lower than for m-IL-1β. These results suggest that HSP90 binds to a region of the mature IL-1β, including the essential residues Q132K133 and Q198L199, to promote the translocation event. Cleavage of p-IL-1β by caspase-1 may potentiate the recruitment of HSP90 to the mature form of IL-1β however chaperone binding is not required for this proteolytic event (Figures 8D and 7B).
In the CMA pathway, HSC70 and HSP90 play different roles. HSC70 binds to cargoes and delivers them into 266 the lysosome as well as disassembling LAMP2A oligomers, whereas HSP90 is required for the oligomerization and stability of LAMP2A (Bandyopadhyay et al., 2008; Chiang et al., 1989). Co-immunoprecipitation indicated that IL-1β associates with HSP90 but not HSC70 (Figure 8-figure supplement 1B). In addition, knockdown of Lamp2A compromised CMA but did not affect the secretion of IL-1β, and disruption of the lysosome did not result in the release of IL-1β from the membrane carrier (Figure 8-figure supplement 1C-E). These data suggest that the translocation of IL-1β into the vesicle carrier is mechanistically distinct from CMA.
Figure 8 HSP90 is involved in the entry of IL-1β into vesicles (A) Knockdown of Hsp90 inhibits IL-1β secretion. HEK293T cells were transduced with lentivirus carrying control (Ctrl) shRNA or shRNA against Hsc70, Hsp90 or Hsp70. Then the cells were transfected with p-IL-1β and p-caspase-1 plasmids. After transfection (24 h), the cells were cultured in EBSS for 2 h followed by determination of IL-1β secretion by immunoblot. (B) IL-1β secretion is reduced in the presence of HSP90 inhibitor geldanamycin. HEK293T cells were transfected with p-IL-1β and p-caspase-1 plasmids. After transfection (24 h), the cells were treated with EBSS containing different concentrations of geldanamycin as indicated. Immunoblot was performed as shown in Figure 1. Quantification of IL-1β secretion was calculated as the ratio between the amount of IL-1β in the medium and the total amount (the sum of IL-1β in both medium and lysate). (C) IL-1β remains accessible to proteinase K in Hsp90 knockdown cells. HEK293T cells were transduced with lentivirus carrying control (Ctrl) shRNA or shRNA against Hsp90. Then the cells were transfected with p-IL-1β and p-caspase-1 plasmids. After transfection (24 h), the cells were cultured in EBSS for 2 h. The 25k membrane fraction was collected and digested with proteinase K and then analyzed by immunoblotting using indicated antibodies. (D) Association of HSP90 with IL-1β WT and mutants. HEK293T cells transfected with p-caspase-1 and IL-1β mutant 132-133AA or 198-199AA were starved in EBSS for 2 h. Immunoprecipitation (IP) with anti-HSP90 antibody coupled to protein G-agarose was performed, followed by an immunoblot with anti-IL-1β and anti-HSP90 antibodies.
Figure 8- figure supplement 1 Translocation of IL-1β is mechanistically different from CMA (A) Knockdown of Hsc70 reduces CMA. HEK293T cells transduced with lentivirus carrying control (Ctrl) shRNA or shRNA against Hsc70 were incubated with regular medium (-CMA) or DMEM (+CMA) in the presence of 20 μg/ml cycloheximide for 24 h. The cells were lysed and analyzed by immunoblotting using indicated antibodies. For quantification, the ratio of GAPDH and tubulin was calculated and normalized by that in control (-CMA) treatment which was set as one. (B) Co-immunoprecipitation of HSC70 or HSP90 with IL-1β. HEK293T cells transfected with m-IL-1β-FLAG were starved in EBSS for 2 h. Immunoprecipitation (IP) with anti-HSC70 or anti-HSP90 antibody coupled to protein A/G-agarose was performed, followed by an immunoblot with indicated antibodies. (C) Knockdown of Lamp2 blocks CMA. HEK293T cells were transfected with control or LAMP2 siRNA. After transfection (48 h), the cells were trypsinized and plated. After 24 h, siRNA transfection was repeated. After another 48 h, the cells were trypsinized and plated. After 24 h, the cells were incubated with regular medium (-CMA) or DMEM (+CMA) in the presence of 20 μg/ml cycloheximide for 24 h. The cells were lysed and analyzed by immunoblotting using indicated antibodies. For quantification, the ratio of GAPDH and Tubulin was calculated and normalized by that in control (-CMA) treatment which was set as one. (D) Knockdown of LAMP2 does not affect IL-1β secretion. HEK293T cells were transfected with control or LAMP2 siRNA as show in (C). After the second siRNA transfection (24h), the cells were transfected with m-IL-1β-FLAG plasmid. After transfection (24 h), the cells were either cultured in DMEM or EBSS for 2 h followed by determination of IL-1β secretion by immunoblot as shown in Figure 1A. Quantification of IL-1β secretion was calculated as the ratio between the amount of IL-1β in the medium and the total amount (the sum of IL-1β in both medium and lysate). (E) Level of IL-1β in the membrane fraction was not affected by lysosome disruption. HEK293T cells 897 transfected with m-IL-1β were cultured in EBSS for 2 h and then treated with DMSO or 0.5 mM glycyl-L-phenylalanine-2-naphthylamide (GPN) for 10 min. The membrane fraction was collected from the top fractions of a Nycodenz density gradient resolved from membranes in a 25k pellet as described in Material and Methods. Both membrane fraction and cell lysis were analyzed by immunoblotting using indicated antibodies.
We next asked if starvation regulated the association between HSP90 and IL-1β. We performed an HSP90 co-immunoprecipitation experiment with cytosol prepared from cells grown in nutrient-rich or starvation conditions (Figure 9A). Starvation led to a ~2.5 fold increase of the association of HSP90 and IL-1β (Figure 9A). This increase was likely not due to starvation-stimulated processing of p-IL-1β because starvation had no effect on the cleavage of mutant forms of IL-1β unable to bind HSP90 (Figure 7B). Starvation led to a ~ 2 fold increase in the membrane localization and cytosolic depletion of mature IL-1β (Figure 9B). Starvation may stimulate the recruitment of a complex of m-IL-1β/HSP90 to the membrane responsible for IL-1β translocation (Figure 9B).
Figure 9 Induction of autophagy enhances the membrane incorporation of IL-1β (A) Starvation enhances the association of IL-1β with HSP90. HEK293T cells transfected with p-IL-1β and p-caspase-1 were cultured in DMEM or EBSS for 2 h. Immunoprecipitation with anti-HSP90 antibody was performed followed by an immunoblot with anti-IL-1β and anti-HSP90 antibodies. (B) Starvation promotes the entry of IL-1β into the membrane fraction. HEK293T cells transfected with p-IL-1β and p-caspase-1 were cultured in DMEM or EBSS for 2 h. The membrane fraction was collected from the top fractions of a Nycodenz density gradient resolved from membranes in a 25k pellet as described in Material and Methods. The cytosolic fraction was collected as the supernatant after 100k×g centrifugation. Immunoblot was performed to determine the levels of IL-1β in both fractions. (C) A proposed model for autophagy-mediated IL-1β secretion. Cytosolic IL-1β associates with HSP90 which facilitates the translocation of IL-1β into the lumen of a vesicle carrier which later either turns into a phagophore and an autophagosome or fuses with them. IL-1β localizes between the outer and inner membrane after the double membrane autophagosome forms. The topological distribution ensures the secretion of IL-1β in a soluble form. The IL-1β-containing autophagosome may directly fuse with the plasma membrane or further fuse with a MVB followed by fusion with the plasma membrane.
Genetic and cell biological studies have implicated autophagy in the transport of several leaderless cargoes to the extracellular space (Bruns et al., 2011; Dupont et al., 2011; Duran et al., 2010; Manjithaya et al., 2010). Unconventional secretory cargoes, such as IL-1β and Acb1, have been shown to have overlapping requirements with formation of the autophagosome or its precursor suggesting that the autophagosome may physically convey these cargo proteins to the cell surface. A key question is if and how these cargoes engage the autophagosome and how this structure exports soluble cargo molecules. In this study, we probed the organelle association and molecular requirements for the secretion of one such unconventional cargo protein, IL-1β. Using surrogate cell lines rather than macrophages to reconstitute autophagy-mediated secretion of IL-1β (Figure 1), we find mature IL-1β localized to the lumen of the membrane in early intermediates and mature autophagosomes (Figures 2-4, 6). This surprising location may help to explain how mature IL-1β is secreted in a soluble form to the cell surface (Figure 9C). However, localization to the lumen between the two membranes of the autophagosome would require that IL-1β is translocated from the cytoplasm across the membrane precursor of a phagophore, rather than being engulfed as the phagophore membrane matures by closure into an autophagosome. Our evidence suggests that IL-1β must unfold or be held in an unfolded state to promote membrane translocation (Figure 5) and that a complex sorting signal in the mature portion of IL-1β interacts with HSP90 to deliver the chaperone and its cargo to a site on a phagophore precursor membrane where the mature species is translocated (Figures 7-9).
The unconventional secretory cargo fibroblast growth factor 2 (FGF2) has been shown to directly translocate across the plasma membrane as a folded protein without the apparent aid of chaperones (Backhaus et al., 2004; Steringer et al., 2015). Unlike FGF2, the entry of IL-1β into the autophagosomal carrier appears to be dependent on protein unfolding in a conformational state that may be fostered by the association of HSP90 with two KFERQ-like sequences within the mature portion of IL-1β (Figure 5 and 8). This translocation mechanism appears superficially similar to another delivery process termed HSC70-dependent CMA in which autophagic cargoes bearing KFERQ targeting motifs are directed into the lysosome for degradation. Indeed, using a cell-free approach to study the import of CMA cargo into isolated lysosomes, Salvador et al. (2000) reported that DHFR fused to a CMA cargo is blocked in translocation by addition of methotrexate, a drug that stabilizes DHFR to unfolding, just as we have shown that IL-1β fused to DHFR is blocked in cells treated with a cell permeable folate analog, aminopterin (Wei et al., 2013). In our fractionation study, IL-1β distributed in LC3-positive autophagosomal carriers that were separated from the lysosome marker LAMP2, the proposed receptor or channel for uptake of CMA cargo (Kaushik and Cuervo, 2012)(Figure 2B). This observation, together with the involvement of a different chaperone i.e. HSP90, suggests distinct routes for IL-1β and cargoes of the CMA pathway.
The target membrane for IL-1β translocation may be a vesicle that could fuse with or form the autophagosome. We find that mature IL-1β can be detected within protease inaccessible membranes in cells blocked early in the autophagic pathway (e.g. ATG5 null cells and cells depleted of FIP200, both of which block at a stage prior to the lipidation of LC3). The identity of the vesicle carrier is unknown and could be any one of those reported to supply membrane to the formation of the autophagosome (Ge et al., 2014a; Lamb et al., 2013). Although we have ruled out the involvement of LAMP2A IL-1β translocation, it is likely that a membrane receptor locating on the membrane of the vesicle carrier, perhaps a functional equivalent of LAMP2A, recruits the protein complex of HSP90 and IL-1β, therefore designating the correct membrane targeting of IL-1β. In addition, a protein conducting channel may be involved in the translocation of IL-1β into the membrane. It seems unlikely that a standard translocation channel, such as SEC61, is involved in this import process, but no current evidence bears on this point.
The exact route by which the autophagosome delivers mature IL-1β to the cell surface as well as how it avoids fusion with degradative lysosome remains obscure, possibly involving interaction with the multi-vesicular body or some form of lysosome as a prelude to fusion at the cell surface (Figure 9C), and this process may require selective recruitment of membrane sorting and targeting factors such as Rabs and SNAREs. Fusion of the autophagosome directly with the plasma membrane would lead to the release of soluble IL-1β available to trigger an inflammatory response in the surrounding tissue. If mature IL-1β were engulfed within the cytoplasmic interior of the autophagosome, fusion of this organelle at the cell surface might release an intact vesicle corresponding to the inner membrane-enclosed cytoplasmic compartment of the autophagosome. We found mature IL-1β secreted by macrophages or in our surrogate cell system to be completely soluble, thus inconsistent with the engulfment model (data not shown). An alternative possibility may be that the autophagosome fuses with another intracellular organelle such as the MVB or the lysosome under conditions where the inner membrane of the autophagosome is degraded. If so, mature IL-1β would be available for secretion if the combined organelle (amphisome, Figure 9C) fused with the plasma membrane. However, for this model to be viable, the mature IL-1β released on dissolution of the autophagosome inner membrane would have to withstand proteolytic attack such as may be encountered in an amphisome or lysosome. Because mature IL-1β is clearly sensitive to proteolysis (Figure 4), thus any compartment engaged in presenting autophagosomal content to the cell surface must be depleted of proteases. The nature of the organelle that delivers autophagosome content to the plasma membrane may be probed by selective ablation of different Rab proteins, e.g. Rab11, Rab27 and Rab35, which appear to be required for fusion of the MVB with the cell surface (Hsu et al., 2010; Ostrowski et al., 2010; Savina et al., 2002), or Rab27a and Rab38, implicated in the fusion of lysosomes at the cell surface (Blott and Griffiths, 2002; Hume et al., 2001; Jager et al., 2000.



















{kind=link}
{kind=link}
{kind=link}
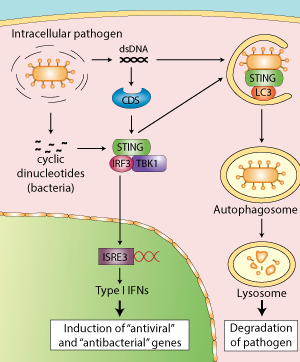{kind=link}
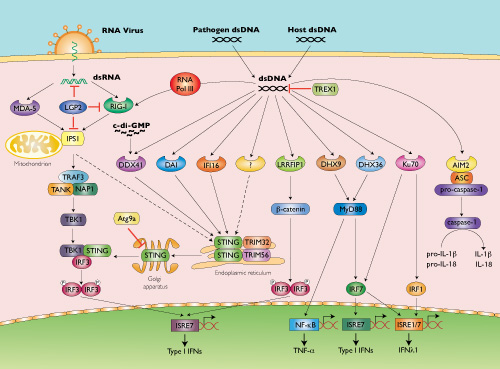{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}