Cholesterol metabolism in pancreatic cancer
Larry H. Bernstein, MD, FCAP, Curator
LPBI
New Pancreatic Treatment Shows Promise
http://www.genengnews.com/gen-news-highlights/new-pancreatic-treatment-shows-promise/81252686/

Study demonstrates how controlling cholesterol metabolism in pancreatic cancer cells reduces metastasis. [NIH]. http://www.genengnews.com/Media/images/GENHighlight/thumb_May4_2016_NIH_PancreaticCancerCells8616346835.jpg
Scientists say they have shown how controlling cholesterol metabolism in pancreatic cancer cells reduces metastasis, pointing to a potential new treatment using drugs previously developed for atherosclerosis.
“We show for the first time that if you control the cholesterol metabolism you could reduce pancreatic cancer spread to other organs,” said Ji-Xin Cheng, Ph.D., a professor in Purdue University’s Weldon School of Biomedical Engineering and Department of Chemistry. “We chose pancreatic cancer to test this approach because it is the most aggressive disease of all the cancers.”
Dr. Cheng had previously led a team of researchers discovering a link between prostate cancer’s aggressiveness and the accumulation of a compound produced when cholesterol is metabolized in cells, findings that could bring new diagnostic and treatment methods. The new study involved researchers at the Purdue Center for Cancer Research and School of Biomedical Engineering, the Indiana University Simon Cancer Center and School of Medicine, and Purdue’s Department of Biological Sciences, Department of Comparative Pathobiology, and Department of Biochemistry.
The findings, detailed in a paper (“Abrogating Cholesterol Esterification Suppresses Growth and Metastasis of Pancreatic Cancer”) just published in Oncogene, suggest that a class of drugs previously developed to treat atherosclerosis could be repurposed for treatment of pancreatic cancer and other forms of cancer. Atherosclerosis is the buildup of fats, cholesterol, and other substances in arteries, restricting blood flow.
The researchers found accumulations of the compound cholesteryl ester in human pancreatic cancer specimens and cell lines, demonstrating a link between cholesterol esterification and metastasis. Excess quantities of cholesterol result in cholesteryl ester being stored in lipid droplets within cancer cells.
“The results of this study demonstrate a new strategy for treating metastatic pancreatic cancer by inhibiting cholesterol esterification,” said Jingwu Xie, Ph.D., the Jonathan and Jennifer Simmons Professor at the Indiana University School of Medicine and a researcher at the Indiana University Melvin and Bren Simon Cancer Center.
The paper’s lead author is Purdue postdoctoral fellow Junjie Li, Ph.D. Purdue researchers have developed an analytical tool, Raman spectromicroscopy, that allows compositional analysis of single lipid droplets in living cells.
“We identified an aberrant accumulation of cholesteryl ester in human pancreatic cancer specimens and cell lines,” Dr. Li said. “Depletion of cholesterol esterification significantly reduced pancreatic tumor growth and metastasis in mice.”
Findings show that drugs like avasimibe, previously developed for treatment of atherosclerosis, reduced the accumulation of cholesteryl ester. Pancreatic cancer usually kills within a few months of diagnosis. It is hoped the potential new treatment might extend life of these patients for a year, Cheng said.
The accumulation of cholesteryl ester is controlled by an enzyme called acyl-coenzyme A acyltransferase-1 (ACAT-1), and findings have correlated a higher expression of the enzyme with a poor survival rate for patients. The researchers analyzed tissue samples from pancreatic cancer patients and then tested the drug treatment in a type of laboratory mice referred to as an orthotopic mouse model, developed at the IU School of Medicine. Specimens of human pancreatic tissues were obtained from the Simon Cancer Center Solid Tissue Bank.
Imaging showed a decrease of the number of lipid droplets, and Raman spectral analysis verified a significant reduction of cholesteryl ester in the lipid droplets, suggesting that avasimibe acted by blocking cholesterol esterification. The drug did not induce weight loss, and there was no apparent organ toxicity in the liver, kidney, lung and spleen, Dr. Cheng said.
Findings also showed that blocking storage of cholesteryl ester causes cancer cells to die, specifically due to damage to the endoplasmic reticulum, a workhorse of protein and lipid synthesis.
“By using avasimibe, a potent inhibitor of ACAT-1, we found that pancreatic cancer cells were much more sensitive to ACAT-1 inhibition than normal cells,” added Dr. Cheng.
Additional research confirmed that the anticancer effect of avasimibe is specific to ACAT-1 inhibition. The researchers performed various biochemical assays and “genetic ablation” to confirm the drug’s anticancer effect.
“The results showed that avasimibe treatment for four weeks remarkably suppressed tumor size and largely reduced tumor growth rate,” said paper co-author Timothy Ratliff, the Robert Wallace Miller Director of Purdue’s Center for Cancer Research. “Metastatic lesions in lymph nodes and distant organs also were assessed at the end of the study. A much higher number of metastatic lesions in lymph nodes were detected in the control group than the avasimibe-treated group.”
Each mouse in the control group showed at least one metastatic lesion in the liver. In contrast, only three mice in the avasimibe-treated group showed single lesion in liver.
Abrogating cholesterol esterification suppresses growth and metastasis of pancreatic cancer
J Li1, D Gu2, S S-Y Lee1, B Song1, S Bandyopadhyay3, S Chen4, S F Konieczny3,5, T L Ratliff5,6, X Liu5,7, J Xie2 and J-X Cheng1,5
Oncogene 2 May 2016; http://dx.doi.org:/10.1038/onc.2016.168
Cancer cells are known to execute reprogramed metabolism of glucose, amino acids and lipids. Here, we report a significant role of cholesterol metabolism in cancer metastasis. By using label-free Raman spectromicroscopy, we found an aberrant accumulation of cholesteryl ester in human pancreatic cancer specimens and cell lines, mediated by acyl-CoA cholesterol acyltransferase-1 (ACAT-1) enzyme. Expression of ACAT-1 showed a correlation with poor patient survival. Abrogation of cholesterol esterification, either by an ACAT-1 inhibitor or by shRNA knockdown, significantly suppressed tumor growth and metastasis in an orthotopic mouse model of pancreatic cancer. Mechanically, ACAT-1 inhibition increased intracellular free cholesterol level, which was associated with elevated endoplasmic reticulum stress and caused apoptosis. Collectively, our results demonstrate a new strategy for treating metastatic pancreatic cancer by inhibiting cholesterol esterification.
Metastasis is the major cause of cancer-related mortality. Though localized tumors can often be treated by surgery or other therapies, treatment options for metastatic diseases are limited. Cancer metastasis has been revealed to be a multiple step process, including cancer cell migration, local invasion, intravasation, circulation through blood and lymph vessels, extravasation, survival and colonization in distant organs.1, 2, 3Mediators identified in these processes have provided the basis for the development of therapies to target metastasis. Current therapeutic strategies for treating metastatic tumors mainly focus on targeting the adhesive molecules and extracellular proteases.4However, these therapeutics have not been proven to be effective in clinical trials, partially owing to the various escape mechanisms used by the metastatic cancer cells.2, 5, 6 Thus, an unmet need exists to develop new therapeutic strategies for treating metastatic cancers.
Recent advances in cancer metabolism have unveiled many potential therapeutic targets for cancer treatment. Metabolic reprogramming, a strategy used by cancer cells to adapt to the rapid proliferation, is being recognized as a new hallmark of cancer.7 Substantial studies have found increased glycolysis, glutaminolysis, nucleotide and lipid synthesis in cancer cells.7, 8, 9,10 Considering that altered metabolic pathways only happen in cancer cells but not in normal cells, targeting these pathways may provide cancer-specific treatments. A number of inhibitors of metabolic enzymes, such as glycolysis inhibitors, are under clinical trials as targeted cancer therapeutics.11
Of various metabolic pathways, lipid metabolism has been suggested to have an important role in cancer cell migration, invasion and metastasis.12 A recent study reported that surrounding adipocytes provide energy source for ovarian cancer cells to promote its rapid growth and metastasis.13 Blocking lipidde novo synthesis pathway has been shown to suppress tumor regrowth and metastasis after anti-angiogenesis treatment withdrawal.14 In parallel, lipolysis by the enzyme monoacylglycerol lipase was shown to regulate the fatty acid network, which promotes cancer cell migration, invasion and growth.15
Cholesterol, a critical component of the plasma membrane, is also implied to be correlated to cancer metastasis.16 It has been shown that prostate cancer bone metastases contain a high level of cholesterol.17 Modulation of cholesterol level in plasma membrane was shown to regulate the capability of cell migration.18, 19Moreover, cholesterol-enriched lipid rafts were shown to have an essential role in cancer cell adhesion and migration.20 Mammalian cells obtain cholesterol either from de novo synthesis or from the uptake of low-density lipoprotein (LDL).21 Inside cells, excess free cholesterol is esterified and stored as cholesteryl ester (CE) in lipid droplets (LDs), which is mediated by acyl-CoA cholesterol acyltransferase (ACAT).22 Increased CE level has been reported in breast cancer,23 leukemia,24 glioma25 and prostate cancer.26Despite these advances, the role of cholesterol esterification in cancer progression, especially in cancer metastasis, is not well understood.
In this article, we report a link between cholesterol esterification and metastasis in pancreatic cancer. Using stimulated Raman scattering (SRS) microscopy and Raman spectroscopy to map LDs stored inside single cells and analyze the composition of individual LDs, we identified an aberrant accumulation of CE in human pancreatic cancer specimens and cell lines. Abrogation of cholesterol esterification, either by inhibiting ACAT-1 enzyme activity or by shRNA knockdown of ACAT-1 expression, significantly reduced pancreatic tumor growth and metastasis in an orthotopic mouse model. Mechanistically, inhibition of cholesterol esterification disturbed cholesterol homeostasis by increasing intracellular free cholesterol level, which was associated with elevated endoplasmic reticulum (ER) stress and eventually led to apoptosis.
In this study, we revealed a link between CE accumulation and pancreatic cancer metastasis. Accumulation of CE via ACAT-1 provides a mechanism to keep high metabolic activity and avoid toxicity from excess free cholesterol. Previously, CE has been reported in breast cancer,23 leukemia,24 glioma25 and prostate cancer.26 Inhibition of cholesterol esterification was shown to suppress tumor growth or cancer cell proliferation.24, 25, 26 Here, we demonstrate that inhibition of cholesterol esterification can be used to treat metastatic pancreatic cancer.
Cholesterol is an essential lipid having important roles in membrane construction, hormone production and signaling.21Aberrant cholesterol metabolism is known to be associated with cardiovascular diseases and cancers.35, 36 Statins, inhibitors of HMG-CoA reductase, have been explored as potential therapies for pancreatic cancer.37 However, statins were not associated with a reduced risk of pancreatic cancer in clinical trials.38 One possible reason is that HMG-CoA reductase is also required for downstream protein prenylation, a critical process for protein activation.39Thus, the effect of statin is not just inhibiting cholesterol synthesis, but also other pathways which may render toxicity to normal cells. This non-specific toxicity is a possible reason for the limited anti-cancer outcome of statin in clinical trials.
Our study identified cholesterol esterification as a novel target for suppression of pancreatic cancer proliferation and metastasis. Inhibitors of ACAT-1 are expected to have great value as cancer-targeting therapeutics, as CE accumulation only occurs in cancer tissues or cell lines. Our animal studies with avasimibe treatment showed no adverse effect to the animals at a dosage of 15 mg/kg. More importantly, modulation of cholesterol esterification suppressed not only tumor growth but also tumor metastasis. These results are expected to stimulate further biological studies to fully appreciate the role of cholesterol metabolism in cancer initiation and progression. As CE accumulation happens in several types of aggressive cancer, blocking cholesterol esterification could be pursued as a therapeutic strategy for other types of cancers. By combining with existing chemotherapies, such as gemcitabine, we believe this metabolic treatment possesses high possibilities to extend patients’ survival time by retarding cancer progression and metastasis.
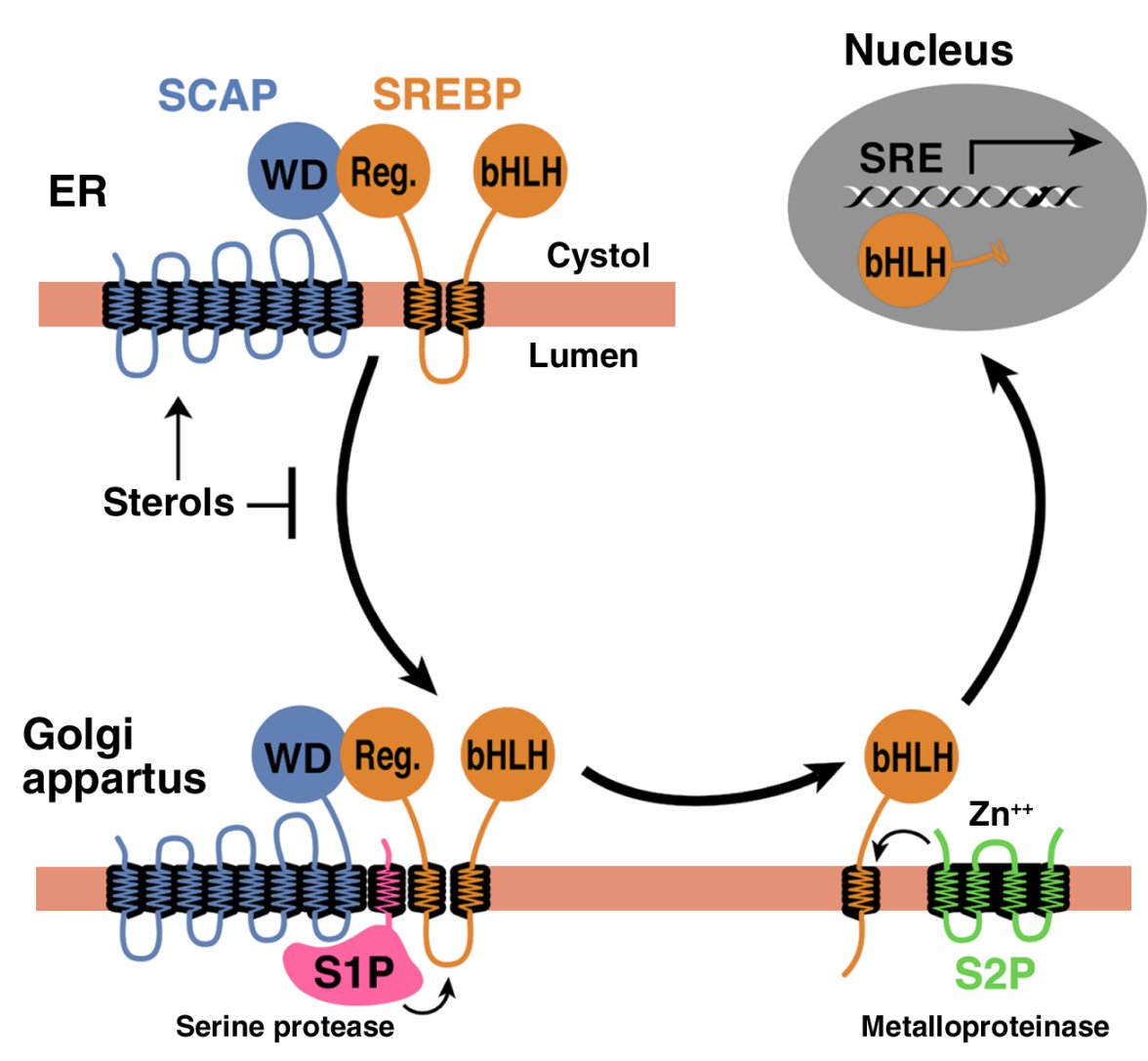
The molecular mechanism that links CE accumulation to cancer aggressiveness needs further studies. One possible mechanism is that cholesterol esterification keeps signaling pathways active by maintaining a low free cholesterol environment. One of the possible targets is the caveolin-1 signaling pathway. Caveolin-1, a regulator of cellular cholesterol homeostasis, is considered as a marker for pancreatic cancer progression.11 Particularly, a promoting role of caveolin-1 in pancreatic cancer metastasis has been reported.40 Our preliminary studies showed ACAT-1 inhibition reduced the expression level of SREBP1, caveolin-1 and phosphorylated ERK1/2 (unpublished data). The effect on caveolin-1 is probably mediated by SREBP1, which senses the intracellular cholesterol homeostasis.41 Meanwhile, caveolin-1 may have an important role in mediating the action of SREBP1 on MAPK pathways,42, 43 which are known to have essential roles in cancer cell metastasis.44 Therefore, it is possible that increased free cholesterol level induced by ACAT-1 inhibition inactivates SREBP1, leading to downregulation of caveolin-1/MAPK pathway, which contributes to the reduced cancer aggressiveness.
Besides the caveolin-1/MAPK signaling, other possibilities include the potential alteration of the membrane composition, such as lipid rafts, by ACAT-1 inhibition. Lipid rafts are known to provide platforms for multiple cellular signaling pathways.20 Thus, modulation of cholesterol metabolism is likely to have more profound effects via other signaling pathways. Future studies are needed to fully elucidate the molecular mechanism.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}