Praluent FDA Approved
Larry H. Bernstein, MD, FCAP, Curator
LPBI
|
Posted in Cardiovascular Pharmacogenomics, Cardiovascular Research, Clinical & Translational, Curation, Molecular Genetics & Pharmaceutical, Pharmacotherapy of Cardiovascular Disease, tagged Cardiovascular disease, LDL cholesterol lowering, statins on October 31, 2015| Leave a Comment »
Praluent FDA Approved
Larry H. Bernstein, MD, FCAP, Curator
LPBI
|
Posted in Curation, Genetics & Innovations in Treatment, Genetics & Pharmaceutical, Genome Biology, Innovations, Methods, Molecular Genetics & Pharmaceutical, Personalized and Precision Medicine & Genomic Research, tagged Bioanalytical Chemistry, DNA repair, genomics on October 31, 2015| Leave a Comment »
DNA Repair
Larry H. Bernstein, MD, FCAP, Curator
LPBI
In celebration of the 2015 Nobel Prize in Chemistry
http://blogs.rsc.org/cc/2015/10/27/in-celebration-of-the-2015-nobel-prize-in-chemistry/

Reviews
Finding needles in a basestack: recognition of mismatched base pairs in DNA by small molecules
Anton Granzhan, Naoko Kotera and Marie-Paule Teulade-Fichou
Chem. Soc. Rev., 2014, 43, 3630-3665
DOI: http://dx.doi.org:/10.1039/C3CS60455
The chemical biology of sirtuins
Bing Chen, Wenwen Zang, Juan Wang, Yajun Huang, Yanhua He, Lingling Yan, Jiajia Liu and Weiping Zheng
Chem. Soc. Rev., 2015, 44, 5246-5264
DOI: http://dx.doi.org:/10.1039/C4CS00373J
Luminescent oligonucleotide-based detection of enzymes involved with DNA repair
Chung-Hang Leung, Hai-Jing Zhong, Hong-Zhang He, Lihua Lu, Daniel Shiu-Hin Chan and Dik-Lung Ma
Chem. Sci., 2013, 4, 3781-3795
DOI: http://dx.doi.org:/10.1039/C3SC51228B
Research articles
A label-free and sensitive fluorescent method for the detection of uracil-DNA glycosylase activity
Jing Tao, Panshu Song, Yusuke Sato, Seiichi Nishizawa, Norio Teramae, Aijun Tong and Yu Xiang
Chem. Commun., 2015, 51, 929-932
DOI: http://dx.doi.org:/10.1039/C4CC06170E
DNA-mediated supercharged fluorescent protein/graphene oxide interaction for label-free fluorescence assay of base excision repair enzyme activity
Zhen Wang, Yong Li, Lijun Li, Daiqi Li, Yan Huang, Zhou Nie and Shouzhuo Yao
Chem. Commun., 2015, 51, 13373-13376
DOI: http://dx.doi.org:/10.1039/C5CC04759E
A fluorescent G-quadruplex probe for the assay of base excision repair enzyme activity
Chang Yeol Lee, Ki Soo Park and Hyun Gyu Park
Chem. Commun., 2015, 51, 13744-13747
DOI: http://dx.doi.org:/10.1039/C5CC05010C
A chemical probe targets DNA 5-formylcytosine sites and inhibits TDG excision, polymerases bypass, and gene expression
Liang Xu, Ying-Chu Chen, Satoshi Nakajima, Jenny Chong, Lanfeng Wang, Li Lan, Chao Zhang and Dong Wang
Chem. Sci., 2014, 5, 567-574
DOI: http://dx.doi.org:/10.1039/C3SC51849C
Sensitive detection of polynucleotide kinase using rolling circle amplification-induced chemiluminescence
Wei Tang, Guichi Zhu and Chun-yang Zhang
Chem. Commun., 2014, 50, 4733-4735
DOI: 10.1039/C4CC00256C
Rescuing DNA repair activity by rewiring the H-atom transfer pathway in the radical SAM enzyme, spore photoproduct lyase
Alhosna Benjdia, Korbinian Heil, Andreas Winkler, Thomas Carell and Ilme Schlichting
Chem. Commun., 2014, 50, 14201-14204
DOI: http://dx.doi.org:/10.1039/C4CC05158K
Expanding DNAzyme functionality through enzyme cascades with applications in single nucleotide repair and tunable DNA-directedassembly of nanomaterials
Yu Xiang, Zidong Wang, Hang Xing and Yi Lu
Chem. Sci., 2013, 4, 398-404
DOI: http://dx.doi.org:/10.1039/C2SC20763J
Detection of base excision repair enzyme activity using a luminescent G-quadruplex selective switch-on probe
Ka-Ho Leung, Hong-Zhang He, Victor Pui-Yan Ma, Hai-Jing Zhong, Daniel Shiu-Hin Chan, Jun Zhou, Jean-Louis Mergny, Chung-Hang Leung and Dik-Lung Ma
Chem. Commun., 2013, 49, 5630-5632
DOI: http://dx.doi.org:/10.1039/C3CC41129J
Endonuclease IV discriminates mismatches next to the apurinic/apyrimidinic site in DNA strands: constructing DNA sensing platforms with extremely high selectivity
Xianjin Xiao, Yang Liu and Meiping Zhao
Chem. Commun., 2013, 49, 2819-2821
DOI: http://dx.doi.org:/10.1039/C3CC40902C
Top 15 most downloaded Chem Soc Rev articles in Q3, 2015
Ultra-stable organic fluorophores for single-molecule research
Qinsi Zheng, Manuel F. Juette, Steffen Jockusch, Michael R. Wasserman, Zhou Zhou, Roger B. Altman and Scott C. Blanchard
DOI: 10.1039/C3CS60237K, Review Article
The chemistry of graphene oxide
Daniel R. Dreyer, Sungjin Park, Christopher W. Bielawski and Rodney S. Ruoff
DOI: 10.1039/B917103G, Critical Review
Selection of boron reagents for Suzuki–Miyaura coupling
Alastair J. J. Lennox and Guy C. Lloyd-Jones
DOI: 10.1039/C3CS60197H, Review Article
Physical and chemical tuning of two-dimensional transition metal dichalcogenides
Haotian Wang, Hongtao Yuan, Seung Sae Hong, Yanbin Li and Yi Cui
DOI: 10.1039/C4CS00287C, Review Article
Advances on structuring, integration and magnetic characterization of molecular nanomagnets on surfaces and devices
N. Domingo, E. Bellido and D. Ruiz-Molina
DOI: 10.1039/C1CS15096K, Critical Review
Heterogeneous photocatalyst materials for water splitting
Akihiko Kudo and Yugo Miseki
DOI: 10.1039/B800489G, Critical Review
Shape control in gold nanoparticle synthesis
Marek Grzelczak, Jorge Pérez-Juste, Paul Mulvaney and Luis M. Liz-Marzán
DOI: 10.1039/B711490G, Tutorial Review
An overview of nanoparticles commonly used in fluorescent bioimaging
Otto S. Wolfbeis
DOI: 10.1039/C4CS00392F, Review Article
Heterogeneous catalysis for sustainable biodiesel production via esterification and transesterification
Adam F. Lee, James A. Bennett, Jinesh C. Manayil and Karen Wilson
DOI: 10.1039/C4CS00189C, Review Article
A review of electrode materials for electrochemical supercapacitors
Guoping Wang, Lei Zhang and Jiujun Zhang
DOI: 10.1039/C1CS15060J, Critical Review
MOF positioning technology and device fabrication
Paolo Falcaro, Raffaele Ricco, Cara M. Doherty, Kang Liang, Anita J. Hill and Mark J. Styles
DOI: 10.1039/C4CS00089G, Review Article
Microfluidic lab-on-a-chip platforms: requirements, characteristics and applications
Daniel Mark, Stefan Haeberle, Günter Roth, Felix von Stetten and Roland Zengerle
DOI: 10.1039/B820557B, Critical Review
Noble metal-free hydrogen evolution catalysts for water splitting
Xiaoxin Zou and Yu Zhang
DOI: 10.1039/C4CS00448E, Review Article
“Green” electronics: biodegradable and biocompatible materials and devices for sustainable future
Mihai Irimia-Vladu
DOI: 10.1039/C3CS60235D, Review Article
Synthetic biology for the directed evolution of protein biocatalysts: navigating sequence space intelligently
Andrew Currin, Neil Swainston, Philip J. Day and Douglas B. Kell
DOI: 10.1039/C4CS00351A, Review Article
Posted in Clinical Diagnostics, Clinical Genomics, Commercialization, Computational Biology/Systems and Bioinformatics, Curation, Genetics & Pharmaceutical, Genome Biology, Health Economics and Outcomes Research, Health Law & Patient Safety, Innovations, Intellectual Property, Innovations, Commercialization, Investment in technological breakthrough, Law and Medicine Conflicts, Metabolomics, Molecular Genetics & Pharmaceutical, Patents, Personalized and Precision Medicine & Genomic Research, Pharmaceutical Analytics, Population Health Management, Genetics & Pharmaceutical, Regulated Clinical Trials: Design, Methods, Components and IRB related issues, RNA Biology, Cancer and Therapeutics, tagged discrepancy between the FDA and the manufacturer, Identify causal variants from human sequencing data, pathway and network analysis, Pathway Genomics on October 23, 2015| Leave a Comment »
pathway and network analysis of complex ‘omics data
Larry H. Bernstein, MD, FCAP, Curator
LPBI
While blood tests can be used to detect some cancers, the FDA said a San Diego company has no proof its blood test works in patients who have not already been diagnosed with some form of the disease.
WASHINGTON, Sept. 25 (UPI) — A San Diego company selling an early cancer detection test was notified by the U.S. Food and Drug Administration it can find no evidence the test actually works, and is concerned it could prove to be harmful for some people.
Pathway Genomics debuted its CancerIntercept test in early September with claims it can detect cancer cell DNA in the blood, picking up mutations linked to as many as 10 different cancers. The goal is to catch cancer early in people who are “otherwise healthy” and not showing symptoms of the disease.
“Based on our review of your promotional materials and the research publication cited above, we believe you are offering a high risk test that has not received adequate clinical validation and may harm the public health,” said FDA Deputy Director James L. Woods in a letter to the company.
CancerIntercept is billed by the company as a blood test looking for DNA fragments in the bloodstream and testing them for 96 genomic markers it says are found in several specific tumor types.
The direct-to-consumer test can be purchased through the Pathway Genomics website, with programs ranging from a one-time test to a quarterly “subscription” for people who want regular testing.
The company states, in several sections of its website, “the presence of one or more of these genomic markers in a patient’s bloodstream may indicate that the patient has a previously undetected cancer. However, the test is not diagnostic, and thus, follow-up screening and clinical testing would be required to confirm the presence or absence of a specific cancer in the patient.”
The FDA is concerned that people may seek treatment for tumors that do not require medical attention, or spend money and possibly seek out treatment they do not need at all — in either case, unnecessary treatment for cancer is potentially harmful to people, the agency said.
CancerIntercept has not been approved by the FDA for use as a medical device, nor has it been subjected to peer review as most tests of its type would be. The company published a white paper on its website which outlines how the test works, supporting its efficacy with references to several clinical trials on detection of mutated DNA in the bloodstream.
Glenn Braunstein, Chief Medical Officer at Pathway Genomics, told The VergePathway had validated its tests with “hundreds” of patients, though those patients had well-defined, often advanced cancers.
In the letter from the FDA, Woods requests the company provide a timeline for meeting with the agency to review plans for future longitudinal studies on the product and specific details on studies that have been conducted before it was made available to consumers.
The clinical laboratory is an essential player in the treatment of cancer providing a diagnostic, potentially a prognostic, and follow-up treatment armamentarium. The laboratory diagnostics industry has grown over the last half century into a highly accurate, well regulated industry with highly automated and point of care technologies. Prior to introduction, the tests that are put on the market have to be validated prior to introduction.
How are they validated?
The most common approach is for the test to be used concomitantly with treatment in a clinical trial. Measurements may be made prior to surgical biopsy and treatment, and at a month or 6 months to a year later. The pharmaceutical and diagnostics industries are independent, even though a large company may have both pharmaceutical and diagnostic divisions. Consequently, the integration of diagnostics and therapeutics occurs on the front lines of patient care.
How this discrepancy between the FDA and the manufacturer could occur is not clear because prior to introduction, the test would have to be rigorously reviewed by the American Association for Clinical Chemistry, the largest and most competent organization to cover the scientific work, having industry-based committees. The only problem is that the companies may have products that are patented and have competing claims or interests. This is perhaps most likely to be problematic in the competitive environment of genomics testing.
The company here reported on is Pathway Genomics, that offers Ingenuity for pathway and variant analysis. There is no concern about the analysis methods, that are well studied. The concern is the validation of such method for screening of patients without prior diagnosis.
Model, analyze, and understand the complex biological and chemical systems at the core of life science research with IPA
QIAGEN’S Ingenuity Pathway Analysis (IPA) has been broadly adopted by the life science research community and is cited in thousands of peer-reviewed journal articles.
https://youtu.be/_HDkjuxYRcY?t=25

http://www.ingenuity.com/wp-content/uploads/2014/01/variant-analyisis-interpretation.png
Rapidly Identify and Prioritize Variants
Ingenuity Variant Analysis combines analytical tools and integrated content to help you rapidly identify and prioritize variants by drilling down to a small, targeted subset of compelling variants based both upon published biological evidence and your own knowledge of disease biology. With Variant Analysis, you can interrogate your variants from multiple biological perspectives, explore different biological hypotheses, and identify the most promising variants for follow-up.
Variant Analysis used in NCI-60 Interpretation of Genomic Variants
The NCI-60 Data Set offers tremendous promise in the development and prescription of cancer drugs
97% of surveyed researchers are satisfied with the ease of use of Ingenuity Variant Analysis and we are honored that they chose to share the data through our Publish tool.
See the research verified by TechValidate
“Being a bioinformatician, I appreciated the speed and the complexity of analysis. Without Variant Analysis, I couldn’t have completed the analysis of 700 exomes in such a short time …. I found Variant Analysis very intuitive and easy to use.”
Posted in Cancer and Current Therapeutics, CANCER BIOLOGY & Innovations in Cancer Therapy, Clinical Genomics, Curation, Innovations, Molecular Genetics & Pharmaceutical, Personalized and Precision Medicine & Genomic Research, Pharmaceutical Analytics, Pharmaceutical Drug Discovery, Pharmacogenomics, tagged Cancer - General, genomics, Personalized medicine, pharmacogenomics on October 9, 2015| Leave a Comment »
Curator: Larry H Bernstein, MD, FCAP
Rodríguez-Antona C1, Taron M.
J Intern Med. 2015 Feb; 277(2):201-17
http://dx.doi.org:/10.1111/joim.12321
Personalized medicine involves the selection of the safest and most effective pharmacological treatment based on the molecular characteristics of the patient. In the case of anticancer drugs, tumor cell alterations can have a great impact on drug activity and, in fact, most biomarkers predicting response originate from these cells. On the other hand, the risk of developing severe toxicity may be related to the genetic background of the patient. Thus, understanding the molecular characteristics of both the tumor and the patient, and establishing their relation with drug outcomes will be critical for the identification of predictive biomarkers and to provide the basis for individualized treatments. This is a complex scenario where multiple genes as well as pathophysiological and environmental factors are important; in addition, tumors exhibit large inter- and intraindividual variability in space and time. Against this background, the huge amounts of biological and genetic data generated by the high-throughput technologies will facilitate pharmacogenomic progress, suggest novel druggable molecules and support the design of future strategies aimed at disease control. Here, we will review the current challenges and opportunities for pharmacogenomic studies in oncology, as well as the clinically established biomarkers. Lung and renal cancer, two areas in which huge progress has been made in the last decade, will be used to illustrate advances in personalized cancer treatment; we will review EGFR mutation as the paradigm of targeted therapies in lung cancer, and discuss the dissection of lung cancer into clinically relevant molecular subsets and novel advances that suggest an important role of single nucleotide polymorphisms in the response to antiangiogenic agents, as well as the challenges that remain in these fields. Finally, we will present new approaches and future prospects for personalizing medicine in oncology.
Posted in Academic Publishing, Biological Networks, Cell Biology, Chemical Biology and its relations to Metabolic Disease, Chemical Genetics, CRISPR/Cas9 & Gene Editing, Curation, Developmental biology, Disease Biology, Experimental validation, Genetics & Pharmaceutical, Genome Biology, Innovations, Methods, Molecular Genetics & Pharmaceutical, Pharmaceutical Analytics, Pharmaceutical Drug Discovery, Signaling & Cell Circuits, Technology Advance Assessment of, tagged CRISPR-Cas9, gene targeting, genetic engineering on September 8, 2015| Leave a Comment »
Curator: Larry H. Bernstein, MD, FCAP
UPDATED on 10/9/2015

The microbial adaptive immune system CRISPR mediates defense against foreign genetic elements through two classes of RNA-guided nuclease effectors. Class 1 effectors utilize multi-protein complexes, whereas class 2 effectors rely on single-component effector proteins such as the well-characterized Cas9. Here, we report characterization of Cpf1, a putative class 2 CRISPR effector. We demonstrate that Cpf1 mediates robust DNA interference with features distinct from Cas9. Cpf1 is a single RNA-guided endonuclease lacking tracrRNA, and it utilizes a T-rich protospacer-adjacent motif. Moreover, Cpf1 cleaves DNA via a staggered DNA double-stranded break. Out of 16 Cpf1-family proteins, we identified two candidate enzymes from Acidaminococcus and Lachnospiraceae, with efficient genome-editing activity in human cells. Identifying this mechanism of interference broadens our understanding of CRISPR-Cas systems and advances their genome editing applications.
VIEW IMAGE and SOURCE
http://www.cell.com/cell/abstract/S0092-8674%2815%2901200-3
SOURCE
Series E. 2: 7.5
Rudolf Jaenisch
One-Step Generation of Mice Carrying Mutations in Multiple Genes by CRISPR/Cas-Mediated Genome Engineering
Haoyi Wang6, Hui Yang6, Chikdu S. Shivalila6, Meelad M. Dawlaty, Albert W. Cheng, Feng Zhang, Rudolf Jaenisch
Cell May 2013; 153: 4(9), p910–918 http://dx.doi.org/10.1016/j.cell.2013.04.025
Highlights
Summary
Mice carrying mutations in multiple genes are traditionally generated by sequential recombination in embryonic stem cells and/or time-consuming intercrossing of mice with a single mutation. The CRISPR/Cas system has been adapted as an efficient gene-targeting technology with the potential for multiplexed genome editing. We demonstrate that CRISPR/Cas-mediated gene editing allows the simultaneous disruption of five genes (Tet1, 2, 3, Sry, Uty – 8 alleles) in mouse embryonic stem (ES) cells with high efficiency. Coinjection of Cas9 mRNA and single-guide RNAs (sgRNAs) targeting Tet1 and Tet2 into zygotes generated mice with biallelic mutations in both genes with an efficiency of 80%. Finally, we show that coinjection of Cas9 mRNA/sgRNAs with mutant oligos generated precise point mutations simultaneously in two target genes. Thus, the CRISPR/Cas system allows the one-step generation of animals carrying mutations in multiple genes, an approach that will greatly accelerate the in vivo study of functionally redundant genes and of epistatic gene interactions.
Generating genetically modified mice using CRISPR/Cas-mediated genome engineering
Hui Yang, Haoyi Wang & Rudolf Jaenisch
Nature Protocols 2014; 9, 1956–1968. http://dx.doi.org:/10.1038/nprot.2014.134
Mice with specific gene modifications are valuable tools for studying development and disease. Traditional gene targeting in mice using embryonic stem (ES) cells, although suitable for generating sophisticated genetic modifications in endogenous genes, is complex and time-consuming. We have recently described CRISPR/Cas-mediated genome engineering for the generation of mice carrying mutations in multiple genes, endogenous reporters, conditional alleles or defined deletions. Here we provide a detailed protocol for embryo manipulation by piezo-driven injection of nucleic acids into the cytoplasm to create gene-modified mice. Beginning with target design, the generation of gene-modified mice can be achieved in as little as 4 weeks. We also describe the application of the CRISPR/Cas technology for the simultaneous editing of multiple genes (five genes or more) after a single transfection of ES cells. The principles described in this protocol have already been applied in rats and primates, and they are applicable to sophisticated genome engineering in species in which ES cells are not available.
Our long-range goals are to understand epigenetic regulation of gene expression in mammalian development and disease. An important question is to understand the different epigenetic conformations that distinguish differentiated cell states and to define strategies to transdifferentiate one differentiated cell type into another. Embryonic stem cells are of major significance because they have the potential to generate any cell type in the body and, therefore, are of great interest for regenerative medicine. A major focus of our work is to understand the molecular mechanisms that allow the reprogramming of somatic cells to an embryonic pluripotent state and to use the potential of patient specific pluripotent cells to study complex human diseases.
One-Step Generation of Mice Carrying Mutations in Multiple Genes by CRISPR/Cas-Mediated Genome Engineering
Haoyi Wang1, 6, Hui Yang1, 6, Chikdu S. Shivalila1, 2, 6, Meelad M. Dawlaty1, Albert W. Cheng1, 3, Feng Zhang4, 5, Rudolf Jaenisch1, 3,
Cell May 2013; 153(4): 910–918 http://dx.doi.org:/10.1016/j.cell.2013.04.025
Highlights
Mice carrying mutations in multiple genes are traditionally generated by sequential recombination in embryonic stem cells and/or time-consuming intercrossing of mice with a single mutation. The CRISPR/Cas system has been adapted as an efficient gene-targeting technology with the potential for multiplexed genome editing. We demonstrate that CRISPR/Cas-mediated gene editing allows the simultaneous disruption of five genes (Tet1, 2, 3, Sry, Uty – 8 alleles) in mouse embryonic stem (ES) cells with high efficiency. Coinjection of Cas9 mRNA and single-guide RNAs (sgRNAs) targeting Tet1 and Tet2 into zygotes generated mice with biallelic mutations in both genes with an efficiency of 80%. Finally, we show that coinjection of Cas9 mRNA/sgRNAs with mutant oligos generated precise point mutations simultaneously in two target genes. Thus, the CRISPR/Cas system allows the one-step generation of animals carrying mutations in multiple genes, an approach that will greatly accelerate the in vivo study of functionally redundant genes and of epistatic gene interactions.
Genetically modified mice represent a crucial tool for understanding gene function in development and disease. Mutant mice are conventionally generated by insertional mutagenesis (Copeland and Jenkins, 2010; Kool and Berns, 2009) or by gene-targeting methods (Capecchi, 2005). In conventional gene-targeting methods, mutations are introduced through homologous recombination in mouse embryonic stem (ES) cells. Targeted ES cells injected into wild-type (WT) blastocysts can contribute to the germline of chimeric animals, generating mice containing the targeted gene modification (Capecchi, 2005). It is costly and time consuming to produce single-gene knockout mice and even more so to make double-mutant mice. Moreover, in most other mammalian species, no established ES cell lines are available that contribute efficiently to chimeric animals, which greatly limits the genetic studies in many species.
Alternative methods have been developed to accelerate the process of genome modification by directly injecting DNA or mRNA of site-specific nucleases into the one-cell embryo to generate DNA double-strand break (DSB) at a specified locus in various species (Bogdanove and Voytas, 2011; Carroll et al., 2008; Urnov et al., 2010). DSBs induced by these site-specific nucleases can then be repaired by error-prone nonhomologous end joining (NHEJ) resulting in mutant mice and rats carrying deletions or insertions at the cut site (Carbery et al., 2010; Geurts et al., 2009; Sung et al., 2013;Tesson et al., 2011). If a donor plasmid with homology to the ends flanking the DSB is coinjected, high-fidelity homologous recombination can produce animals with targeted integrations (Cui et al., 2011; Meyer et al., 2010). Because these methods require the complex designs of zinc finger nucleases (ZNFs) or Transcription activator-like effector nucleases (TALENs) for each target gene and because the efficiency of targeting may vary substantially, no multiplexed gene targeting in animals has been reported to date. To dissect the functions of gene family members with redundant functions or to analyze epistatic relationships in genetic pathways, mice with two or more mutated genes are required, prompting the development of efficient technology for the generation of animals carrying multiple mutated genes.
Recently, the type II bacterial CRISPR/Cas system has been demonstrated as an efficient gene-targeting technology with the potential for multiplexed genome editing. Bacteria and archaea have evolved an RNA-based adaptive immune system that uses CRISPR (clustered regularly interspaced short palindromic repeat) and Cas (CRISPR-associated) proteins to detect and destroy invading viruses and plasmids (Horvath and Barrangou, 2010; Wiedenheft et al., 2012). Cas proteins, CRISPR RNAs (crRNAs), andtrans-activating crRNA (tracrRNA) form ribonucleoprotein complexes, which target and degrade foreign nucleic acids, guided by crRNAs ( Gasiunas et al., 2012; Jinek et al., 2012). It was shown that the Cas9 endonuclease from Streptococcus pyogenes type II CRISPR/Cas system can be programmed to produce sequence-specific DSB in vitro by providing a synthetic single-guide RNA (sgRNA) consisting of a fusion of crRNA and tracrRNA ( Jinek et al., 2012). More intriguingly, Cas9 and sgRNA are the only components necessary and sufficient for induction of targeted DNA cleavage in cultured human cells ( Cho et al., 2013; Cong et al., 2013; Mali et al., 2013) as well as in zebrafish (Chang et al., 2013; Hwang et al., 2013). A recent report also demonstrated disruption of a GFP transgene in mice using the CRISPR/Cas system ( Shen et al., 2013). The ease of design, construction, and delivery of multiple sgRNAs suggest the possibility of multiplexed genome editing in mammals. Indeed, one study demonstrated that two loci separated by 119 bp could be cleaved simultaneously in cultured human cells at a low efficiency ( Cong et al., 2013). The extent of achievable multiplexed genome editing has yet to be demonstrated in stem cells as well as in animals. Here, we use the CRISPR/Cas system to drive both NHEJ-based gene disruption and homology directed repair (HDR)-based precise gene editing to achieve highly efficient and simultaneous targeting of multiple genes in stem cells and mice.
Simultaneous Targeting up to Five Genes in ES Cells
To test whether the CRISPR/Cas system could produce targeted cleavage in the mouse genome, we transfected plasmids expressing both the mammalian-codon-optimized Cas9 and a sgRNA targeting each gene ( Cong et al., 2013; Mali et al., 2013) into mouse ES cells and determined the targeted cleavage efficiency by the Surveyor assay ( Guschin et al., 2010). All three Cas9-sgRNA transfections produced cleavage at target loci with high efficiency of 36% at Tet1, 48% atTet2, and 36% at Tet3 ( Figure 1B). Because each target locus contains a restriction enzyme recognition site ( Figure 1A), we PCR amplified an ∼500 bp fragment around each target site and digested the PCR products with the respective enzyme. A correctly targeted allele will lose the restriction site, which can be detected by failure to cleave upon enzyme treatment. Using this restriction fragment length polymorphism (RFLP) assay, we screened 48 ES cell clones from each single-targeting experiment. Consistent with the Surveyor analysis, a high percentage of ES cell clones were targeted, with a high probability of having both alleles mutated ( Figure S1A available online). The results summarized in Table 1 demonstrate that between 65% and 81% of the tested ES cell clones carried mutations in the Tet genes with up to 77% having mutations in both alleles.
Figure 1.
Multiplexed Gene Targeting in mouse ES cells
(A) Schematic of the Cas9/sgRNA-targeting sites in Tet1, 2, and 3. The sgRNA-targeting sequence is underlined, and the protospacer-adjacent motif (PAM) sequence is labeled in green. The restriction sites at the target regions are bold and capitalized. Restriction enzymes used for RFLP and Southern blot analysis are shown, and the Southern blot probes are shown as orange boxes.
(B) Surveyor assay for Cas9-mediated cleavage at Tet1, 2, and 3 loci in ES cells.
(C) Genotyping of triple-targeted ES cells, clones 51, 52, and 53 are shown. Upper: RFLP analysis. Tet1PCR products were digested with SacI, Tet2 PCR products were digested with EcoRV, and Tet3 PCR products were digested with XhoI. Lower: Southern blot analysis. For the Tet1 locus, SacI digested genomic DNA was hybridized with a 5′ probe. Expected fragment size: WT = 5.8 kb, TM (targeted mutation) = 6.4 kb. For the Tet2 locus, SacI, and EcoRV double-digested genomic DNA was hybridized with a 3′ probe. Expected fragment size: WT = 4.3 kb, TM = 5.6 kb. For the Tet3 locus, BamHI and XhoI double-digested genomic DNA was hybridized with a 5′ probe. Expected fragment size: WT = 3.2 kb, TM = 8.1 kb.
(D) The sequence of six mutant alleles in triple-targeted ES cell clone 14 and 41. PAM sequence is labeled in red.
(E) Analysis of 5hmC levels in DNA isolated from triple-targeted ES cell clones by dot blot assay using anti-5hmC antibody. A previously characterized DKO clone derived using traditional method is used as a control. See also Figure S1.
Figure S1.
Single-, Triple-, and Quintuple-Gene Targeting in mES Cells, Related to Figure 1
(A) RFLP analysis of clones from each single-targeting experiment (1 to 17 are shown).
(B) RFLP analysis of triple-gene-targeted clones (37 to 53 are shown). Tet1 PCR products were digested with SacI, Tet2 PCR products were digested with EcoRV, and Tet3 PCR products were digested with XhoI. WT control is shown in the last lane. Genotyping of clone 51, 52, and 53 are also shown in Figure 1C.
(C) Schematic of the Cas9/sgRNA-targeting sites in Sry and Uty. The sgRNA-targeting sequence is underlined, and the protospacer-adjacent motif (PAM) sequence is labeled in green. The restriction sites at the target regions are bold and capitalized. Restriction enzymes used for RFLP analysis are shown.
(D) RFLP analysis of quintuple-gene-targeted clones (1 to 10 are shown). Sry PCR products were digested with BsaJI, Uty PCR products were digested with AvrII. WT control is shown in the last lane. RFLP analysis of Tet1, 2, 3 loci are not shown.
Table 1.
CRISPR/Cas-Mediated Gene Targeting in V6.5 ES Cells
| Mutant Alleles per Clone / Total Clones Tested | |||||||
| Gene | 6 | 5 | 4 | 3 | 2 | 1 | 0 |
| Tet1 | N/A | 27/48 | 4/48 | 17/48 | |||
| Tet2 | 37/48 | 2/48 | 9/48 | ||||
| Tet3 | 32/48 | 3/48 | 13/48 | ||||
| Tet1+ Tet2 + Tet3 | 20/96 | 16/96 | 2/96 | 2/96 | 1/96 | 0/96 | 55/96 |
Plasmids encoding Cas9 and sgRNAs targeting Tet1, Tet2, and Tet3 were transfected separately (single targeting) or in a pool (triple targeting) into ES cells. The number of total alleles mutated in each ES cell clone is listed from 0 to 2 for single-targeting experiment, and 0 to 6 for triple-targeting experiment. The number of clones containing each specific number of mutated alleles is shown in relation to the total number of clones screened in each experiment. See also Table S1.
The high efficiency of single-gene modification prompted us to test the possibility of targeting all three genes simultaneously. For this we cotransfected ES cells with the constructs expressing Cas9 and three sgRNAs targeting Tet1, 2, and 3. Of 96 clones screened using the RFLP assay, 20 clones were identified as having mutations in all six alleles of the three genes ( Figures 1C and S1B and Table 1). To exclude that a PCR bias could give false positive results, we performed Southern blot analysis and confirmed complete agreement with the RFLP results ( Figure 1C). We subcloned and sequenced the PCR products of Tet1-, Tet2-, and Tet3-targeted regions to verify that all of eight tested clones carried biallelic mutations in all three genes with most clones displaying two mutant alleles for each gene with small insertions or deletions (indels) at the target site ( Figure 1D). To test whether these mutant alleles would abolish the function of Tet proteins, we compared the 5hmC level of targeted clones to WT ES cells. Previously, we reported a depletion of 5hmC in Tet1/Tet2 double-knockout ES cells derived using traditional gene-targeting methods ( Dawlaty et al., 2013). As expected from loss of function alleles, we found a significant reduction of 5hmC levels in all clones carrying biallelic mutations in the three genes ( Figure 1E).
To further test the potential of multiplexed gene targeting by CRISPR/Cas system, we designed sgRNAs targeting two Y-linked genes, Sry and Uty ( Figure S1C). Short PCR products encoding sgRNAs targeting all five genes (Tet1, Tet2, Tet3, Sry, and Uty) were pooled and cotransfected with a Cas9 expressing plasmid and the PGK puroR cassette into ES cells. Of 96 clones that were screened using the RFLP assay, 10% carried mutations in all eight alleles of the five genes ( Figure S1D and Table S1), demonstrating the capacity of the CRISP/Cas9 system for highly efficient multiplexed gene targeting.
One-Step Generation of Single-Gene Mutant Mice by Zygote Injection
We tested whether mutant mice could be generated in vivo by direct embryo manipulation. Capped polyadenylated Cas9 mRNA was produced by in vitro transcription and coinjected with sgRNAs. Initially, to determine the optimal concentration of Cas9 mRNA for targeting in vivo, we microinjected varying amounts of Cas9-encoding mRNA with Tet1 targeting sgRNA at constant concentration (20 ng/μl) into pronuclear (PN) stage one-cell mouse embryos and assessed the frequency of altered alleles at the blastocyst stage using the RFLP assay. As expected, higher concentration of Cas9 mRNA led to more efficient gene disruption ( Figure S2A). Nevertheless, even embryos injected with the highest amount of Cas9 mRNA (200 ng/μl) showed normal blastocyst development, suggesting low toxicity.
Figure S2.
One-Step Generation of Single-Gene Mutant Mice by Zygote Injection, Related to Figure 2
(A) RFLP analysis of blastocysts injected with different concentration of Cas9 mRNA and Tet1 sgRNA at 20 ng/μl. Tet1 PCR products were digested with SacI.
(B) Commonly recovered Tet1 and Tet2 alleles resulted from MMEJ. PAM sequence of each targeting sequence is labeled in green. Microhomology flanking the DSB is bold and underlined in WT sequence.
(C) RFLP analysis of eight Tet3-targeted blastocysts demonstrated high targeting efficiency (embryo 3 and 5 failed to amplify). Tet3 PCR products were digested with XhoI.
(D) Some Tet3-targeted mice show smaller size and all homozygous mutants died within 1 day after birth.
(E) RFLP analysis of Tet3 single-targeted newborn mice. Mouse 8 and 14 survived after birth. Sample 2 and 6 failed to amplify.
(F) Sequences of both Tet3 alleles of surviving Tet3-targeted mouse 14. PAM sequences are labeled in red.
To investigate whether postnatal mice carrying targeted mutations could be generated, we coinjected sgRNAs targeting Tet1or Tet2 with different concentrations of Cas9 mRNA. Blastocysts derived from the injected embryos were transplanted into foster mothers and newborn pups were obtained. As summarized in Table 2, about 10% of the transferred blastocysts developed to birth independent of the RNA concentrations used for injection suggesting low fetal toxicity of the Cas9 mRNA and sgRNA. RFLP, Southern blot, and sequencing analysis demonstrated that between 50 and 90% of the postnatal mice carried biallelic mutations in either target gene ( Figures 2A, 2B, and 2C and Table 2).
Table 2.
CRISPR/Cas-Mediated Single-Gene Targeting in BDF2 Mice
| Gene | Cas9/sg RNA (ng/μl) | Blastocysts/Injected Zygotes | Transferred Embryos (Recipients) | Newborns (Dead) | Mutant Alleles per Mouse/Total Mice Testeda | ||
| 2 | 1 | 0 | |||||
| Tet1 | 200/20 | 38/50 | 19 (1) | 2 (0) | 2/2 | 0/2 | 0/2 |
| 100/20 | 50/60 | 25 (1) | 3 (0) | 2/3 | 0/3 | 1/3 | |
| 50/20 | 40/50 | 40 (2) | 8 (3) | 4/7 | 2/7 | 1/7 | |
| 100/50 | 167/198 | 60 (3) | 12 (2) | 9/11 | 1/11 | 1/11 | |
| Tet2 | 100/50 | 176/203 | 108 (5) | 22 (3) | 19/20 | 0/20 | 1/20 |
| Tet3 | 100/50 | 85/112 | 64 (4) | 15 (13) | 9/13 | 2/13 | 2/13 |
Cas9 mRNA and sgRNAs targeting Tet1, Tet2, or Tet3 were injected into fertilized eggs. The blastocysts derived from injected embryos were transplanted into foster mothers and newborn pups were obtained and genotyped. The number of total alleles mutated in each mouse is listed from 0 to 2. The number of mice containing each specific number of mutated alleles is shown in relation to the total number of mice screened in each experiment. See also Table S2. A Some of the pups were cannibalized.
http://ars.els-cdn.com/content/image/1-s2.0-S0092867413004674-gr2.jpg
Surprisingly, specific Δ9 Tet1 and specific Δ8 and Δ15 Tet2 mutant alleles were repeatedly recovered in independently derived mice. Preferential generation of these alleles is likely caused by a short sequence repeat flanking the DSB (see Figure S2B) consistent with previous reports demonstrating that perfect microhomology sequences flanking the cleavage sites can generate microhomology-mediated precise deletions by end repair mechanism (MMEJ) ( McVey and Lee, 2008; Symington and Gautier, 2011) (Figure S2B). A similar observation was also made when TALEN mRNA was injected into one-cell rat embryos ( Tesson et al., 2011).
We also derived blastocysts from zygotes injected with Cas9 mRNA and Tet3 sgRNA. Genotyping of the blastocysts demonstrated that of eight embryos three were homozygous and three were heterozygous Tet3 mutants (two failed to amplify) (Figure S2C). Some blastocysts were implanted into foster mothers and, upon C section, we readily identified multiple mice of smaller size ( Figure S2D), many of which died soon after delivery. Genotyping shown in Figure S2E indicated that all pups with mutations in both Tet3 alleles died neonatally. Only 2 out of 15 mice survived that were either Tet3heterozygous mutants or WT ( Figure S2F). These results are consistent with the lethal neonatal phenotype of Tet3 knockout mice generated using traditional methods ( Gu et al., 2011), although we have not yet established which of the Tet3 mutations produced loss of function rather than hypomorphic alleles.
One-Step Generation of Double-Gene Mutant Mice by Zygote Injection
To test whether Tet1/Tet2 double-mutant mice could be produced from single embryos, we coinjected Tet1 and Tet2 sgRNAs with 20 or 100 ng/μl Cas9 mRNA into zygotes. A total of 28 pups were born from 144 embryos transferred into foster mothers (21% live-birth rate) that had been injected at the zygote stage with high concentrations of RNA (Cas9 mRNA at 100 ng/μl, sgRNAs at 50 ng/μl), consistent with low or no toxicity of the Cas9 mRNA and sgRNAs ( Table 3). RFLP, Southern blot analysis, and sequencing identified 22 mice carrying targeted mutations at all four alleles of the Tet1 and Tet2genes ( Figures 2D and 2E) with the remaining mice carrying mutations in a subset of alleles ( Table 3). Injection of zygotes with low concentration of RNA (Cas9 mRNA at 20 ng/μl, sgRNAs at 20 ng/μl) yielded 19 pups from 75 transferred embryos (25% live-birth rate), which is a higher survival rate than from embryos injected with 100 ng/μl of Cas9 RNA. Nevertheless, more than 50% of the pups were biallelic Tet1/Tet2 double mutants ( Table 3). These results demonstrate that postnatal mice carrying biallelic mutations in two different genes can be generated within one month with high efficiency (Figure 2F).
Table 3.
CRISPR/Cas-Mediated Double-Gene Targeting in BDF2 Mice
| Gene | Cas9/sgRNA (ng/μl) | Blastocyst/Injected Zygotes | Transferred Embryos (Recipients) | Newborns (Dead) | Mutant Alleles per Mouse/Total Mice Testeda | ||||
| 4 | 3 | 2 | 1 | 0 | |||||
| Tet1 +Tet2 | 100 / 50 | 194/229 | 144(7) | 31(8) | 22/28 | 4/28 | 1/28 | 1/28 | 0/28 |
| 20 / 20 | 92/109 | 75(5) | 19(3) | 11/19 | 1/19 | 2/19 | 3/19 | 2/19 | |
Cas9 mRNA and sgRNAs targeting Tet1and Tet2 were coinjected into fertilized eggs. The blastocysts derived from the injected embryos were transplanted into foster mothers and newborn pups were obtained and genotyped. The number of total alleles mutated in each mouse is listed from 0 to 4 for Tet1 and Tet2. The number of mice containing each specific number of mutated alleles is shown in relation to the number of total mice screened in each experiment. A Some of the pups were cannibalized.
Although the high live-birth rate and normal development of mutant mice suggest low toxicity of CRISPR/Cas9 system, we sought to determine the off-target effects in vivo. Previous work in vitro, in bacteria, and in cultured human cells suggested that the protospacer-adjacent motif (PAM) sequence NGG and the 8 to 12 base “seed sequence” at the 3′ end of the sgRNA are most important for determining the DNA cleavage specificity (Cong et al., 2013; Jiang et al., 2013; Jinek et al., 2012). Based on this rule, only three and four potential off targets exist in mouse genome for Tet1 and Tet2 sgRNA, respectively ( Table S2 and Experimental Procedures), with each of them perfectly matching the 12 bp seed sequence at the 3′ end and the NGG PAM sequence of the sgRNA (there is no potential off-target site for Tet3 sgRNA using this prediction rule). From seven double-mutant mice produced from injection with high RNA concentration we PCR amplified 400 to 500 bp fragments from all seven potential off-target loci and found no cleavage in the Surveyor assay ( Figure S3), suggesting a high specificity of CRISPR/Cas system.
Figure S3.
Off-Target Analysis of Double-Mutant Mice, Related to Figure 2
(A) Three potential off targets of Tet1 sgRNA and four potential off targets of Tet2 sgRNA are shown. The 12 bp perfect matching seed sequence is labeled in blue, and NGG PAM sequence is labeled in red.
(B) Surveyor assay of all seven potential off-target loci in seven double-mutant mice derived with high concentration of Cas9 mRNA (100 ng/μl) injection. WT control is included as the eighth sample. The weak cleavage activity at Ubr1 locus is not due to off-target effect because sequences of these PCR products show no mutations.
http://ars.els-cdn.com/content/image/1-s2.0-S0092867413004674-figs3.jpg
Multiplexed Precise HDR-Mediated Genome Editing In Vivo
The NHEJ-mediated gene mutations described above produced mutant alleles with different and unpredictable insertions and deletions of variable size. We explored the possibility of precise homology directed repair (HDR)-mediated genome editing by coinjecting Cas9 mRNA, sgRNAs, and single-stranded DNA oligos into one-cell embryos. For this we designed an oligo targeting Tet1 so as to change two base pairs of a SacI restriction site and creating instead an EcoRI site and a second oligo targetingTet2 with two base pair changes that would convert an EcoRV site into an EcoRI site (Figure 3A). Blastocysts were derived from zygotes injected with Cas9 mRNA and sgRNAs and oligos targeting Tet1 or Tet2, respectively. DNA was isolated, amplified, and digested with EcoRI to detect oligo-mediated HDR events. Six out of nine Tet1-targeted embryos and 9 out of 15 Tet2-targeted embryos incorporated an EcoRI site at the respective target locus, with several embryos having both alleles modified (Figure S4A). When Cas9 mRNA, sgRNAs, and single-stranded DNA oligos targeting both Tet1 and Tet2 were coinjected into zygotes, out of 14 embryos, four were identified that were targeted with the oligo at the Tet1 locus, seven that were targeted with the oligo at the Tet2 locus and one embryo (2) that had one allele of each gene correctly modified (Figure S4B). All four alleles of embryo 2 were sequenced, confirming that one allele of each gene contained the 2 bp changes directed by the oligo, whereas the other alleles were disrupted by NHEJ-mediated deletion and insertion ( Figure S4C).
Figure 3.
Multiplexed HDR-Mediated Genome Editing In Vivo
(A) Schematic of the oligo-targeting sites at Tet1 and Tet2 loci. The sgRNA-targeting sequence is underlined, and the PAM sequence is labeled in green. Oligo targeting each gene is shown under the target site, with 2 bp changes labeled in red. Restriction enzyme sites used for RFLP analysis are bold and capitalized.
(B) RFLP analysis of double oligo injection mice with HDR-mediated targeting at the Tet1 and Tet2 loci.
(C) The sequences of both alleles of Tet1 and Tet2 in mouse 5 and 7 show simultaneously HDR-mediated targeting at one allele or two alleles of each gene, and NHEJ-mediated disruption at the other alleles. See also Figure S4.
http://ars.els-cdn.com/content/image/1-s2.0-S0092867413004674-gr3.jpg
Figure S4.
Multiplexed Precise HDR-Mediated Genome Editing In Vivo, Related to Figure 3
(A) RFLP analysis of single oligo injection embryos with HDR-mediated targeting at Tet1 and Tet2 locus.
(B) RFLP analysis of double oligo injection embryos with multiplexed HDR-mediated targeting at both Tet1and Tet2 loci.
(C) Sequences of both alleles of Tet1 and Tet2 in embryo 2 show simultaneously HDR-mediated targeting at one allele of both genes, and NHEJ-mediated gene disruption at the other allele of each gene.
http://ars.els-cdn.com/content/image/1-s2.0-S0092867413004674-figs4.jpg
Blastocysts with double oligo injections were implanted into foster mothers and a total of 10 pups were born from 48 embryos transferred (21% live-birth rate). Upon RFLP analysis using EcoRI, we identified seven mice containing EcoRI sites at the Tet1 locus and eight mice containing EcoRI sites at the Tet2 locus, with six mice containing EcoRI sites at both Tet1 and Tet2 loci ( Figure 3B). We also applied RFLP analysis using SacI and EcoRV to Tet1 and Tet2 loci, respectively, showing that all alleles not targeted by oligos contained disruptions, which is in consistent with the high biallelic mutation rate by Cas9 mRNA and sgRNAs injection. These results were confirmed by sequencing demonstrating mutations in all four alleles of mouse 5 and 7 ( Figure 3C). Our results demonstrate that mice with HDR-mediated precise mutations in multiple genes can be generated in one step by CRISPR/Cas-mediated genome editing.
Discussion
The genetic manipulation of mice is a crucial approach for the study of development and disease. However, the generation of mice with specific mutations is labor intensive and involves gene targeting by homologous recombination in ES cells, the production of chimeric mice, and, after germline transmission of the targeted ES cells, the interbreeding of heterozygous mice to produce the homozygous experimental animals, a process that may take 6 to 12 months or longer (Capecchi, 2005). To produce mice carrying mutations in several genes requires time-consuming intercrossing of single-mutant mice. Similarly, the generation of ES cells carrying homozygous mutations in several genes is usually achieved by sequential targeting, a process that is labor intensive, necessitating multiple consecutive cloning steps to target the genes and to delete the selectable markers.
As summarized in Figure 4, we have established three different approaches for the generation of mice carrying multiple genetic alterations. We demonstrate that CRISPR/Cas-mediated genome editing in ES cells can generate the simultaneous mutations of several genes with high efficiency, a single-step approach allowing the production of cells with mutations in five different genes (Figure 4A). We chose the threeTet genes as targets because the respective mutant phenotypes have been well defined previously ( Dawlaty et al., 2011, 2013; Gu et al., 2011). Cells mutant for Tet1, 2 and 3were depleted of 5hmC as would be expected for loss of function mutations of the genes (Dawlaty et al., 2013). However, we have not as yet established, which of the Cas9-mediated gene mutations produced loss of function rather than hypomorphic alleles.
Figure 4.
Mutiplexed Genome Editing in ES Cells and Mouse
(A) Multiple gene targeting in ES cells.
(B) One-step generation of mice with multiple mutations. Upper: multiple targeted mutations with random indels introduced through NHEJ. Lower: multiple predefined mutations introduced through HDR-mediated repair.
We also show that mouse embryos can be directly modified by injection of Cas9 mRNA and sgRNA into the fertilized egg resulting in the efficient production of mice carrying biallelic mutations in a given gene. More significantly, coinjection of Cas9 with Tet1 andTet2 sgRNAs into zygotes produced mice that carried mutations in both genes (Figure 4B, upper). We found that up to 95% of newborn mice were biallelic mutant in the targeted gene when single sgRNA was injected and when coinjected with two different sgRNAs, up to 80% carried biallelic mutations in both targeted genes. Thus, mice carrying multiple mutations can be generated within 4 weeks, which is a much shorter time frame than can be achieved by conventional consecutive targeting of genes in ES cells and avoids time-consuming intercrossing of single-mutant mice.
The introduction of DSBs by CRISPR/Cas generates mutant alleles with varying deletions or insertions in contrast to designed precise mutations created by homologous recombination. The introduction of point mutations into human ES cells, cancer cell lines, and mouse by ZNF or TALEN along with DNA oligo has been demonstrated previously (Chen et al., 2011; Soldner et al., 2011; Wefers et al., 2013). We demonstrate that CRISPR/Cas-mediated targeting is useful to generate mutant alleles with predetermined alterations, and coinjection of single-stranded oligos can introduce designed point mutations into two target genes in one step, allowing for multiplexed gene editing in a strictly controlled manner (Figure 4B, lower). It will be of great interest to assess whether this targeting system allows for the production of conditional alleles, or precise insertion of larger DNA fragments such as GFP markers so as to generate conditional knockout and reporter mice for specific genes.
There are several potential limitations of the CRISPR/Cas technology. First, the requirement for a NGG PAM sequence of S. pyogenes Cas9 limits the target space in the mouse genome. It has been shown that the Streptococcus thermophilus LMD-9 Cas9 using different PAM sequence can also induce targeted DNA cleavage in mammalian cells ( Cong et al., 2013). Therefore, exploiting different Cas9 proteins may enable to target most of the mouse genome. Second, although the sgRNAs used here showed high targeting efficiency, much work is needed to elucidate the rules for designing sgRNAs with consistent high targeting efficiency, which is essential for multiplexed genome engineering. Third, although our off-target analysis for the seven most likely off targets of Tet1 and Tet2 sgRNAs failed to detect mutations in these loci, it is possible that other mutations were induced following as yet unidentified rules. A more thorough sequencing analysis for a large number of sgRNAs will provide more information about the potential off-target cleavage of the CRISPR/Cas system and lead to a better prediction of potential off-target sites. Last, oligo-mediated repair allows for precise genome editing, but the other allele is often mutated through NHEJ ( Figures 3B, 3C, andS4C). We have shown that using lower Cas9 mRNA concentration generates more mice with heterozygous mutations. Therefore, it may be possible to optimize the system for more efficient generation of mice with only one oligo -modified allele. In addition, employment of Cas9 nickase will likely avoid this complication because it mainly induces DNA single-strand break, which is typically repaired through HDR ( Cong et al., 2013;Mali et al., 2013).
It is likely that a much larger number of genomic loci than targeted in the present work can be modified simultaneously when pooled sgRNAs are introduced. The methods presented here open up the possibility of systematic genome engineering in mice, facilitating the investigation of entire signaling pathways, of synthetic lethal phenotypes or of genes that have redundant functions. A particularly interesting application is the possibility to produce mice carrying multiple alterations in candidate loci that have been identified in GWAS studies to play a role in the genesis of multigenic diseases. In summary, CRISPR/Cas-mediated genome editing makes possible the generation of ES cells and mice carrying multiple genetic alterations and will facilitate the genetic dissection of development and complex diseases.
One-Step Generation of Mice Carrying Reporter and Conditional Alleles by CRISPR/Cas-Mediated Genome Engineering
Hui Yang1, 4, Haoyi Wang1, 4, Chikdu S. Shivalila1, 2, 4, Albert W. Cheng1, 3, Linyu Shi1, Rudolf Jaenisch1, 3
Cell Sep 2013; 154(6):1370-1379 http://dx.doi.org:/10.1016/j.cell.2013.08.022
Highlights
The type II bacterial CRISPR/Cas system is a novel genome-engineering technology with the ease of multiplexed gene targeting. Here, we created reporter and conditional mutant mice by coinjection of zygotes with Cas9 mRNA and different guide RNAs (sgRNAs) as well as DNA vectors of different sizes. Using this one-step procedure we generated mice carrying a tag or a fluorescent reporter construct in the Nanog, the Sox2, and the Oct4 gene as well as Mecp2 conditional mutant mice. In addition, using sgRNAs targeting two separate sites in the Mecp2 gene, we produced mice harboring the predicted deletions of about 700 bps. Finally, we analyzed potential off-targets of five sgRNAs in gene-modified mice and ESC lines and identified off-target mutations in only rare instances.
Mice with specific gene modification are valuable tools for studying development and disease. Traditional gene targeting in embryonic stem (ES) cells, although suitable for generating sophisticated genetic modifications in endogenous genes, is complex and time-consuming (Capecchi, 2005). The production of genetically modified mice and rats has been greatly accelerated by novel approaches using direct injection of DNA or mRNA of site-specific nucleases into the one-cell-stage embryo, generating DNA double-strand breaks (DSB) at specified sequences leading to targeted mutations (Carbery et al., 2010, Geurts et al., 2009, Shen et al., 2013, Sung et al., 2013, Tesson et al., 2011 and Wang et al., 2013). Coinjection of a single-stranded or double-stranded DNA template containing homology to the sequences flanking the DSB can produce mutant alleles with precise point mutations or DNA inserts (Brown et al., 2013, Cui et al., 2011, Meyer et al., 2010, Wang et al., 2013 and Wefers et al., 2013). Recently, pronuclear injection of two pairs of ZFNs and two double-stranded donor vectors into rat fertilized eggs produced rat containing loxP-flanked (floxed) alleles (Brown et al., 2013). However, the complex and time-consuming design and generation of ZFNs and double-stranded donor vectors limit the application of this method.
CRISPR (clustered regularly interspaced short palindromic repeat) and Cas (CRISPR-associated) proteins function as the RNA-based adaptive immune system in bacteria and archaea (Horvath and Barrangou, 2010 and Wiedenheft et al., 2012). The type II bacterial CRISPR/Cas system has been demonstrated as an efficient gene-targeting technology that facilitates multiplexed gene targeting (Cong et al., 2013 and Wang et al., 2013). Because the binding of Cas9 is guided by the simple base-pair complementarities between the engineered single-guide RNA (sgRNA) and a target genomic DNA sequence, it is possible to direct Cas9 to any genomic locus by providing the engineered sgRNA (Cho et al., 2013, Cong et al., 2013, Gilbert et al., 2013, Hwang et al., 2013, Jinek et al., 2012, Jinek et al., 2013, Mali et al., 2013b, Qi et al., 2013 and Wang et al., 2013).
Previously, we used the type II bacterial CRISPR/Cas system as an efficient tool to generate mice carrying mutations in multiple genes in one step (Wang et al., 2013). However, this study left a number of issues unresolved. For example, neither the efficiency of using the CRISPR/Cas gene-editing approach for the insertion of DNA constructs into endogenous genes nor its utility to create conditional mutant mice was clarified. Here, we report the one-step generation of mice carrying reporter constructs in three different genes as well as the derivation of conditional mutant mice. In addition, we performed an extensive off-target cleavage analysis and show that off-target mutations are rare in targeted mice and ES cells derived from CRISPR/Cas zygote injection.
Results
Targeted Insertion of Short DNA Fragments
http://ars.els-cdn.com/content/image/1-s2.0-S0092867413010167-figs1.jpg
Mice with Reporters in the Endogenous Nanog, Sox2, and Oct4 Genes
http://ars.els-cdn.com/content/image/1-s2.0-S0092867413010167-gr1.jpg
http://ars.els-cdn.com/content/image/1-s2.0-S0092867413010167-gr2.jpg
http://ars.els-cdn.com/content/image/1-s2.0-S0092867413010167-figs2.jpg
Finally, we designed sgRNA targeting the Oct4 3′ UTR, which was coinjected with a published donor vector designed to integrate the 3 kb transgene cassette (IRES-eGFP-loxP-Neo-loxP; Figure 2D) at the 3′ end of the Oct4 gene ( Lengner et al., 2007). Blastocysts were derived from injected zygotes, inspected for GFP expression, and explanted to derive ES cells. About 20% (47/254) of the blastocysts displayed uniform GFP expression in the ICM region. Three of nine derived ES cell lines expressed GFP (Figure 2E), including one showed mosaic expression ( Table S2). Three out of ten live-born mice contained the targeted allele ( Table 1). Correct targeting in mice and ES cell lines was confirmed by Southern blot analysis ( Figure 2F).
Conventionally, transgenic mice are generated by pronuclear instead of cytoplasmic injection of DNA. To optimize the generation of CRISPR/Cas9-targeted embryos, we compared different concentrations of RNA and the Nanog-mCherry or the Oct4-GFP DNA vectors as well as three different delivery modes: (1) simultaneous injection of all constructs into the cytoplasm, (2) simultaneous injection of the RNA and the DNA into the pronucleus, and (3) injection of Cas9/sgRNA into the cytoplasm followed 2 hr later by pronuclear injection of the DNA vector. Table S1 shows that simultaneous injection of all constructs into the cytoplasm at a concentration of 100 ng/μl Cas9 RNA, 50 ng/μl of sgRNA and 200 ng/μl of vector DNA was optimal, resulting in 9% (86/936) to 19% (47/254) of targeted blastocysts. Similarly, the simultaneous injection of 5 ng/μl Cas9 RNA, 2.5 ng/μl of sgRNA, and 10 ng/μl of DNA vector into the pronucleus yielded between 9% (7/75) and 18% (13/72) targeted blastocysts. In contrast, the two-step procedure with Cas9 and sgRNA simultaneous injected into the cytoplasm followed 2 hr later by pronuclear injection of different concentrations of DNA vector yielded no or at most 3% (1/34) positive blastocysts. Thus, our results suggest that simultaneous injection of RNA and DNA into the cytoplasm or nucleus is the most efficient procedure to achieve targeted insertion.
Conditional Mecp2 Mutant Mice
http://ars.els-cdn.com/content/image/1-s2.0-S0092867413010167-figs3.jpg
http://ars.els-cdn.com/content/image/1-s2.0-S0092867413010167-gr3.jpg
A total of 98 E13.5 embryos and mice were generated from zygotes injected with Cas9 mRNA, sgRNAs, and DNA oligos targeting the L2 and R1 sites. Genomic DNA was digested with both NheI and EcoRI, and analyzed by Southern blot using exon 3 and exon 4 probes (Figures 3A and 3B). The L2 and R1 oligos contained, in addition to the loxP site, different restriction sites (NheI or EcoRI). Thus, single loxP site integration at L2 or R1 will produce either a 3.9 kb or a 2 kb band, respectively, when hybridized with the exon3 probe (Figures 3A and B). We found that about 50% (45/98) of the embryos and mice carried a loxP site at the L2 site and about 25% (25/98) at the R1 site. Importantly, integration of both loxP sites on the same DNA molecule, generating a floxed allele, produces a 700 bp band as detected by exon 3 probe hybridization (Figures 3A and 3B). RFLP analysis, sequencing (Figures S4A and S4B) and Southern blot analysis (Figure 3B) showed that 16 out of the 98 mice tested contained two loxP sites flanking exon 3 on the same allele. Table 2 summarizes the frequency of all alleles and shows that the overall insertion frequency of an L2 or R1 insertion was slightly higher in females (21/38) than in males (28/60) consistent with the higher copy number of the X-linkedMecp2 gene in females. To confirm that the floxed allele was functional, we used genomic DNA for in vitro Cre-mediated recombination. Upon Cre treatment, both the deletion and circular products were detected by PCR in targeted mice, but not in DNA from wild-type mice ( Figure 3C). The PCR products were sequenced and confirmed the precise Cre-loxP-mediated recombination ( Figure S4C).
http://ars.els-cdn.com/content/image/1-s2.0-S0092867413010167-figs4.jpg
Mosaicism
Off-Target Analysis
In this study, we demonstrate that CRISPR/Cas technology can be used for efficient one-step insertions of a short epitope or longer fluorescent tags into precise genomic locations, which will facilitate the generation of mice carrying reporters in endogenous genes. Mice and embryos carrying reporter constructs in the Sox2, the Nanog and the Oct4 gene were derived from zygotes injected with Cas9 mRNA, sgRNAs, and DNA oligos or vectors encoding a tag or a fluorescent marker. Moreover, microinjection of two Mecp2-specific sgRNAs, Cas9 mRNA, and two different oligos encoding loxP sites into fertilized eggs allowed for the one-step generation of conditional mutant mice. In addition, we show that the introduction of two spaced sgRNAs targeting the Mecp2 gene can produce mice carrying defined deletions of about 700 bp. Though all RNA and DNA constructs were injected into the cytoplasm or nucleus of zygotes, the gene modification events could happen at the one-cell stage or later. Indeed, Southern analyses revealed mosaicism in 17% (1/6) to 40% (20/49) of the targeted mice and ES cell lines indicating that the insertion of the transgenes had occurred after the zygote stage ( Table S2).
More…
In summary, CRISPR/Cas-mediated genome editing represents an efficient and simple method of generating sophisticated genetic modifications in mice such as conditional alleles and endogenous reporters in one step. The principles described in this study could be directly adapted to other mammalian species, opening the possibility of sophisticated genome engineering in many species where ES cells are not available.
Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system
Albert W Cheng1,2,*, Haoyi Wang1,*, Hui Yang1, Linyu Shi1, Yarden Katz1,3, Thorold W Theunissen1, Sudharshan Rangarajan1, Chikdu S Shivalila1,4, Daniel B Dadon1,4 and Rudolf Jaenisch1,4
Cell Research (2013) 23:1163–1171. http://dx.doi.org:/10.1038/cr.2013.122
Technologies allowing for specific regulation of endogenous genes are valuable for the study of gene functions and have great potential in therapeutics. We created the CRISPR-on system, a two-component transcriptional activator consisting of a nuclease-dead Cas9 (dCas9) protein fused with a transcriptional activation domain and single guide RNAs (sgRNAs) with complementary sequence to gene promoters. We demonstrate that CRISPR-on can efficiently activate exogenous reporter genes in both human and mouse cells in a tunable manner. In addition, we show that robust reporter gene activation in vivo can be achieved by injecting the system components into mouse zygotes. Furthermore, we show that CRISPR-on can activate the endogenous IL1RN, SOX2, and OCT4genes. The most efficient gene activation was achieved by clusters of 3-4 sgRNAs binding to the proximal promoters, suggesting their synergistic action in gene induction. Significantly, when sgRNAs targeting multiple genes were simultaneously introduced into cells, robust multiplexed endogenous gene activation was achieved. Genome-wide expression profiling demonstrated high specificity of the system.
Gene expression is strictly controlled in many biol-ogical processes, such as development and diseases. Transcription factors regulate gene expression by binding to specific DNA sequences at the enhancer and promoter regions of target genes, and modulate transcription through their effector domains1. Based on the same principle, artificial transcription factors (ATFs) have been generated by fusing various functional domains to a DNA binding domain engineered to bind to the genes of interest, thereby modulating their expression2,3. The capability of regulating endogenous gene expression using ATFs may facilitate the study of the transcriptional network underlying complex biological processes and provide new therapeutic options for diseases. Significant efforts and progress have been made to engineer DNA binding domains with defined specificities. The decipherment of the “code” of DNA binding specificity of zinc finger proteins and transcription activator-like effectors (TALE) has led to the rational design of DNA binding domains to recognize specific nucleotides with certain probability4,5,6,7,8,9,10. However, binding specificity of these ATFs is usually degenerate, can be difficult to predict and the complex and time-consuming design and generation limits their applications. To study the transcriptional network in a systematic manner, regulating multiple endogenous genes is required, prompting the development of efficient technology for simultaneous regulation of multiple endogenous genes.
CRISPR (clustered regularly interspaced short palin-dromic repeat) and Cas (CRISPR-associated) proteins are utilized by bacteria and archea to defend against viral pathogens11,12. Because the binding of Cas protein is guided by the simple base-pair complementarities between the engineered single guide RNA (sgRNA) and a target genomic DNA sequence, Cas9 could be directed to specific genomic locus or multiple loci simultaneously, by providing the engineered sgRNAs13,14,15,16,17,18,19,20. A recent study described the CRISPRi (CRISPR interference) system, in which the nuclease-deficient dCas9 (D10A; H840A) proteins blocked the transcription apparatus when directed to promoters or gene bodies in bacteria21. A subsequent study demonstrated a more efficient gene repression in eukaryotes by dCas9 fused with a transcription repression domain or exogenous transgene activation when fused with an activation domain22. Two most recent studies showed single endogenous gene activation using dCas9-based activators9,10. To what extent multiple endogenous genes could be regulated simultaneously has not been explored. In this study we report the generation of an RNA-programmable CRISPR-on system, which enables the simultaneous activation of multiple endogenous genes with a defined stoichiometry.
…
We show here that the CRISPR-on system can be used for the simultaneous induction of at least three different endogenous genes. More significantly, we demonstrated that the stoichiometry of gene induction of multiple genes can be tuned by adjusting the relative amount of their cognate sgRNAs. Simultaneous activation of multiple endogenous genes with defined stoichiometry opens up novel opportunities for systems biology as it allows for the predictable manipulation of transcriptional networks.
Finally, with the ease of design and synthesis, a library of sgRNAs could be generated. When introduced into a cell line constitutively expressing dCas9 activator, gene activation screens mediated by RNA (RNAa) could be achieved. As the specificity components (sgRNA) can be separately designed and constructed from the effector component (Cas fusion proteins), the same library of sgRNAs could be used with different dCas9 fusions (e.g., VP160 domain for transactivation, KRAB domain for transcriptional repression, chromatin modifier domains for specific histone modification) to exert different functions at particular genomic loci.
….
Our long-range goals are to understand epigenetic regulation of gene expression in mammalian development and disease. An important question is to understand the different epigenetic conformations that distinguish differentiated cell states and to define strategies to transdifferentiate one differentiated cell type into another. Embryonic stem cells are of major significance because they have the potential to generate any cell type in the body and, therefore, are of great interest for regenerative medicine. A major focus of our work is to understand the molecular mechanisms that allow the reprogramming of somatic cells to an embryonic pluripotent state and to use the potential of patient specific pluripotent cells to study complex human diseases.
Three years ago, no one knew or cared about much about a protein called Cpf1 produced by a bacterial gene. Now, it shows potential for making a fast-developing genome editing technique called CRISPR easier and more accurate. Bioinformaticians identified this protein and its potential connection to CRISPR by scanning the public database of genome sequences. Their colleagues now show that two of 16 versions of this protein tested can delete a gene in a human cell. Cpf1 has other advantages as well-being smaller than one of the popular Cas9 proteins used and depending on a smaller piece of RNA to find its target DNA. But its utility for editing genomes of human and other cells needs further testing.
Posted in Curation, Disease Biology, Genetics & Innovations in Treatment, Genetics & Pharmaceutical, Genome Biology, Innovations, Interviews with Scientific Leaders, Methods, Molecular Genetics & Pharmaceutical, Mutant Gene Expression, Personalized and Precision Medicine & Genomic Research, Pharmaceutical Analytics, Small Molecules in Development of Therapeutic Drugs, tagged Genome engineering, targeted gene modification on September 8, 2015| Leave a Comment »
Larry H Bernstein, MD, FCAP, Curator
Leaders in Pharmaceutical Intelligence

Article ID #183: Targeted gene modification. Published on 9/8/2015
WordCloud Image Produced by Adam Tubman
Series E. 2: 7.8
Mario R. Capecchi won a 2007 Nobel Prize for his work on targeted gene modification.
Born in Italy in 1937, scientist Mario R. Capecchi emigrated to the United States after World War II and later became a geneticist and professor. His groundbreaking work on targeted gene modification won him a Nobel Prize in 2007.
The Making of a Scientist II
In 1996, as a Kyoto Prize laureate, I was asked to write an autobiographical sketch of my early upbringing. Through this exercise, shared by all of the laureates, the hope was to uncover potential influences or experiences that may have been key to fostering the creative spirit within us. In my own case, what I saw was that, despite the complete absence of an early nurturing environment, the intrinsic drive to make a difference in our world is not easily quenched and that given an opportunity, early handicaps can be overcome and dreams achieved. This was intended as a message of hope for those who have struggled early in their lives. As I have previously noted, our ability to identify the genetic and environmental factors that contribute to talents such as creativity are too complex for us to currently predict. In the absence of such wisdom our only recourse is to provide all children with the opportunities to pursue their passions and dreams. Our understanding of human development is too meager to allow us to predict the next Beethoven, Modigliani, or Martin Luther King.
The content of the autobiographical sketch was based on my own memories, on conversations with my aunt and uncle, who raised me once I arrived in the United States, and on conversations with my mother. Because of the added exposure resulting from the winning of the Nobel Prize, I have received letters from people who knew me in Italy during those formative early years. In addition members of the press have taken an interest in my story and have sought independent corroboration. An amazing and wonderful surprise is that they have discovered a half-sister of whom I was completely unaware. She is two years younger than I, and was given up for adoption before she was one year old. Most recently I had the opportunity to meet my half-sister. She was a very nice person, as a sister should be. I am grateful for all of these new sources of information and revelation. Where appropriate, I will weave the new information into this retelling of my story.
Autobiographical Sketch
I was born in Verona, Italy on October 6, 1937. Fascism, Nazism, and Communism were raging through the country. My mother, Lucy Ramberg, was a poet; my father, Luciano Capecchi, an officer in the Italian Air Force. This was a time of extremes, turmoil and juxtapositions of opposites. They had a passionate love affair, and my mother wisely chose not to marry him. This took a great deal of courage on her part. It embittered my father.
 |
| Figure 1. A photograph of my mother, Lucy Ramberg, at age 19. |
I have only a few pictures of my mother. She was a beautiful woman with a passion for languages and a flair for the dramatic (see Figure 1). This picture was taken when she was 19. She grew up, with her two brothers, in a villa in Florence, Italy. There were magnificent gardens, a nanny, gardeners, cooks, house cleaners, and private tutors for languages, literature, history, and the sciences. She was fluent in half a dozen languages. Her father, Walter Ramberg, was an archeologist specializing in Greek antiquities, born and trained in Germany. Her mother was a painter born and raised in Oregon, USA. In her late teens, my grandmother, Lucy Dodd, packed up her steamer trunks and sailed with her mother from Oregon to Florence, Italy, where they settled. My grandmother was determined to become a painter. This occurred near the end of the 19th century, a time when young women were not expected to set off on their own with strong ambitions of developing their own careers.
 |
| Figure 2. A painting done by my grandmother, Lucy Dodd Ramberg, of her three children, left to right, Edward, Lucy, and Walter. It was painted at their villa in Florence, Italy in 1913. |
 |
| Figure 3. A painting by Lucy Dodd Ramberg of my mother, Lucy, and uncle Edward having tea at the villa in Florence, Italy (1913). |
My grandmother became a very gifted painter. Let me share with you a couple of her paintings, which also illustrate the young lives of her children. These paintings are very large, approximately seven feet by five feet. The first painting (Figure 2) is the center panel of a triptych depicting my mother and her two brothers Walter and Edward (both of whom became physicists) surrounded by olive trees at the villa in Florence. The influence of the French impressionist painters is evident. The second painting (Figure 3) is of my mother, age 8, and her younger brother Edward, age 6, having a tea party, again at the villa in Florence. Their father, the German archeologist, was killed as a young man in World War I. My grandmother finished raising her three children on her own by painting, mostly portraits, and by converting the family villa into a finishing school for young women, primarily from the United States.
 |
| Figure 4. A photograph of the chalet where my mother and I lived in Wolfgrübben just north of Bolzano, Italy. In the foreground is my mother, Lucy. |
My mother’s love and passion was poetry. She published in German. She received her university training at the Sorbonne in Paris and was a lecturer at that university in literature and languages. At that time, she joined with a group of poets, known as the Bohemians, who were prominent for their open opposition to Fascism and Nazism. In 1937, my mother moved to the Tyrol, the Italian Alps. Figure 4 shows the chalet north of Bolzano, in Wolfgrübben, with my mother in the foreground. We lived in this chalet until I was 3½ years old. In the spring of 1941, German officers came to our chalet and arrested my mother. This is one of my earliest memories. My mother had taught me to speak both Italian and German, and I was quite aware of what was happening. I sensed that I would not see my mother again for many years, if ever. She was incarcerated as a political prisoner in Germany.
I have believed that her place of incarceration was Dachau. This was based on conversations with my uncle Edward, my mother’s younger brother. During World War II, my uncle lived in the United States. Throughout these war years, he made many attempts to locate where my mother was being held. The most reliable information indicated that the location was near Munich. Dachau is located near Munich and was built to hold political prisoners. My mother survived her captivity, but after the war, despite my prodding, she refused to talk about her war experiences.
Reporters from the Associated Press (AP) have found records that my mother was indeed a prisoner during the war in Germany. In fact, they have found records of German interest in my mother’s political activities preceding 1939. In that year, they had her arrested by the Italian authorities and jailed in Perugia and subsequently released. However, the AP reporters did not find records indicating that my mother was incarcerated in Dachau. Though Germans were noted for their meticulous record keeping, it would be difficult now to evaluate the accuracy of the existing war records, particularly for cases where data is missing. It is clear, however, that exactly where in Germany my mother was held has not yet been determined. Regardless of which prison camp was involved, her experiences were undoubtedly more horrific than mine. She had aged beyond recognition during those five years of internment. Following her release, though she lived until she was 82 years old, she never psychologically recovered from her wartime experiences.
My mother had anticipated her arrest by German authorities. Prior to their arrival, she had sold most of her possessions and gave the proceeds to an Italian peasant family in the Tyrol so that they could take care of me. I lived on their farm for one year. It was a very simple life. They grew their own wheat, harvested it, and took it to the miller to be ground. From the flour they made bread which they took to the baker to be baked. During this time, I spent most of my time with the women of the farm. In the late fall, the grapes were harvested by hand and put into enormous wooden vats. The children, including me, stripped, jumped into the vats and mashed the grapes with our feet. We became squealing masses of purple energy. I still remember the pungent odor and taste of the fresh grapes. Most recently, members of the Dolomiten Press have located this farm and I had the opportunity to visit it. It is still owned by the same family that occupied it when I was there. The old farm house has been taken down and a new one erected. However, the pictures of the old farm house, as well as the surrounding land are remarkably consistent with my memories.
World War II was now fully under way. The American and British forces had landed in Southern Italy and were proceeding northward. Bombings of northern Italian cities were a daily occurrence. As constant reminders of the war, curfews and blackouts were in effect every night; no lights were permitted. In the night we could hear the drone of presumed American and British reconnaissance planes which we nicknamed “Pepe.” One hot afternoon, American planes swooped down from the sky and began machine gunning the peasants in the fields. A senseless exercise. A bullet grazed my leg, fortunately not breaking any bones. I still have the scar, which, many years later my daughter proudly had me display to her third-grade class in Utah.
For reasons that have never been clear to me, my mother’s money ran out after one year and, at age 4½, I set off on my own. I headed south, sometimes living in the streets, sometimes joining gangs of other homeless children, sometimes living in orphanages, and most of the time being hungry. My recollections of those four years are vivid but not continuous, rather like a series of snapshots. Some of them are brutal beyond description, others more palatable.
There are records in the archives of Ritten, a region of the Southern Alps of Italy, that I left Bozen to go to Reggio Emilia on July 18, 1942. AP reporters exploring this history have suggested that my father came to the farm, picked me up, and that we went together to Reggio Emilia where he was living. I have no memory of his coming to the farm, nor of having travelled with him to Reggio Emilia. I have recently received a letter from a man who remembers me as the youngest member of his street gang operating in Bolzano, which is on the way to Reggio Emilia.
I did end up in Reggio Emilia, which is approximately 160 miles south of Bolzano. I knew that my father lived in Reggio Emilia and I have previously noted that I had lived with him a couple of times from 1942-1946, for a total period of approximately three weeks. The question has been raised why I didn’t live with him for a much longer period. The reason was that he was extremely abusive. Amidst all of the horrors of war, perhaps the most difficult for me to accept as a child was having a father who was brutal to me.
Recently, I have also received a very nice letter from the priest in Reggio Emilia who ran the orphanage in which I was eventually placed. I remember him because he was one of the very few men I encountered in Reggio Emilia who showed compassion for the children and took an interest in me. I am surprised, but pleased, that after all these years he still remembers me among the thousands of children he was responsible for over the years. Further, I believe I was at that orphanage for only several months, the first time in the fall of 1945, after which I ran away, followed by a second period, in the same orphanage, in the spring of 1946. But his memory is genuine, for he recounts incidents consistent with my memories that could only have been known through our common experience.
In the spring of 1945, Munich was liberated by the American troops. My mother had survived her captivity and set out to find me. In October 1946, she succeeded. As an example of her flair for the dramatic, she found me on my ninth birthday, and I am sure that this was by design. I did not recognize her. In five years she had aged a lifetime. I was in a hospital when she found me. All of the children in this hospital were there for the same reasons: malnutrition, typhoid, or both. The prospects for most of those children ever leaving that hospital were slim because they had no nourishing food. Our daily diet consisted of a bowl of chicory coffee and a small crust of old bread. I had been in that hospital in Reggio Emilia for what seemed like a year. Scores of beds lined the rooms and corridors of the hospital, one bed touching the next. There were no sheets or blankets. It was easier to clean without them. Our symptoms were monotonously the same. In the morning we awoke fairly lucid. The nurse, Sister Maria, would take our temperature. She promised me that if I could go through one day without a high fever, I could leave the hospital. She knew that without any clothes I was not likely to run away. By late morning, the high, burning fever would return and we would pass into oblivion. Consistent with the diagnosis of typhoid, many years later I received a typhoid/paratyphoid shot, went into shock, and passed out.
 |
| Figure 5. A photograph of my uncle Edward Ramberg working in his laboratory at RCA Princeton, New Jersey. |
The same day that my mother arrived at the hospital, she bought me a full set of new clothes, a Tyrolean outfit complete with a small cap with a feather in it. I still have the hat. We went to Rome to process papers, where I had my first bath in six years, and then on to Naples. My mother’s younger brother, Edward, had sent her money to buy two boat tickets to America. I was expecting to see roads paved with gold in America. As it turned out, I found much more: opportunities.
On arriving in America, my mother and I lived with my uncle and aunt, Edward and Sarah Ramberg. Edward, my mother’s younger brother was a brilliant physicist. He was a Ph.D. student in quantum mechanics with Arnold Sommerfeld and translated one of Sommerfeld’s major texts into English. Among Edward’s many contributions was his discovery of how to focus electrons, knowledge which he used in helping to build the first electron microscope at RCA. Edward’s books on electron optics have been published in many languages. During my visit to Japan to celebrate the Kyoto Prize, several Japanese physicists approached me to express how grateful they were for my uncle’s texts from which they learned electron optics. Another achievement, of which he was less proud was being a principal contributor to the development of both black and white and color television. While I grew up in his home, television was not allowed. Figure 5 shows a photograph of my uncle working in his laboratory.
My aunt and uncle were Quakers and they did not support violence as solutions to political problems anywhere in the world. During World War II, my uncle did alternative service rather than bear arms. He worked in a mental institution in New Hampshire, cleared swamps in the south, and was a guinea pig for the development of vaccines against tropical diseases. After the war he settled in a commune in Pennsylvania, called Bryn Gweled, which he helped found. People of all races and religious affiliations were welcomed in this community. It was a marvelous place for children: it contained thick woods for exploration and had communal activities of all kinds – painting, dance, theater, sports, electronics, and many sessions devoted to the discussion of the major religious philosophies of the world. Every week there were communal work parties, putting in roads, phone lines, and electrical lines, building a community center and so on.
The contrast between living primarily alone in the streets of Italy and living in an intensely cooperative and supportive community in Pennsylvania was enormous. Time was needed for healing and for erasing the images of war from my mind. I remember that for many years after coming to the United States I would go to sleep tossing and turning with such force that by morning the sheets were torn and the bed frame broken. This activity disturbed my aunt and uncle to the extent that Sarah would take me from one child psychologist or psychiatrist, to another. These professionals were not very helpful, but the support of the community was. The nightly activity eventually subsided. There may be lessons to be learned from such experiences for the treatment of the children from Darfur, the Congo, and now Kenya.
Sarah and Edward took on the challenge of converting me into a productive human being. This, I am sure, was a very formidable task. I had received little or no formal education or training for living in a social environment. Quakers do not believe in frills, but rather in a life of service. My aunt and uncle taught me by example. I was given few material goods, but every opportunity to develop my mind and soul. What I made of myself would be entirely up to me. The day after I arrived in America, I went to school. I started in the third grade in the Southampton public school system. Sarah also took on the task of teaching me to read, starting from the very beginning.
The first task was to learn English. I had a marvelous third grade teacher. She was patient and encouraging. The class was studying Holland, so I started participation in class functions by painting a huge mural on butcher block paper with tulips, windmills, children ice skating, children in Dutch costumes, and ships. It was a collage of activities and colors. This did not require verbal communication.
I was a good, but not serious, student in grade school and high school. Academics came easily to me. I attended an outstanding high school, George School, a Quaker school north of Philadelphia. The teachers were superb, challenging, enthusiastic, competent, and caring. They enjoyed teaching. The campus was also magnificent, particularly in the spring when the cherry and dogwood trees were bursting with blossoms. An emphasis on Quaker beliefs permeated all of the academic and sports programs. A favorite period for many, including me, was Quaker meeting, a time set aside for silent meditation, and taking stock of where we were going. My wife and I sent our daughter to George School for her own last two years in high school so that she might also benefit from the personal virtues it promotes, and we think she has.
Sports were very important to me at George School, and physical activity has remained an important activity for me to this day. I played varsity football, soccer, and baseball, and wrestled. I was particularly proficient at wrestling. I enjoyed the drama of a single opponent, as well as the physical and psychological challenges of the sport. After George School, I went to Antioch, a small liberal arts college in Ohio.
At Antioch College I became a serious student, converting to academics all of the energy I had previously devoted to sports. Coming from George School, I carried the charge of making this a better, more equitable world for all people. Most of the problems appeared to be political, so I started out at Antioch majoring in political science. However, I soon became disillusioned with political science since there appeared to be little science to this discipline, so I switched to the physical sciences – physics and chemistry. I found great pleasure in the simplicity and elegance of mathematics and classical physics. I took almost every mathematics, physics, and chemistry course offered at Antioch, including Boolean algebra and topology, electrodynamics, and physical chemistry.
Although I found physics and mathematics intellectually satisfying, it was becoming apparent that what I was learning came from the past. The newest physics that was taught at Antioch was quantum mechanics, a revolution that had occurred in the 1920’s and earlier. Also, many frontiers of experimental physics, particularly experimental particle physics, were requiring the use of larger and larger accelerators, which involved bigger and bigger teams of scientists and support groups to execute the experiments. I was looking for a science in which the individual investigator had a more intimate, hands-on involvement with the experiments. Fortunately, Antioch had an outstanding work-study program; one quarter we studied on campus, the next was spent working on jobs related to our fields of interest. The jobs, in my case laboratory jobs, were maintained all over the country, and every three months we packed up our bags and set off for a new city and a new work experience. So one quarter off I went to Boston and the Massachusetts Institute of Technology (MIT).
There I encountered molecular biology as the field was being born (late 1950’s). This was a new breed of science and scientist. Everything was new. There were no limitations. Enthusiasm permeated this field. Devotees from physics, chemistry, genetics, and biology joined its ranks. The common premises were that the most complex biological phenomena could, with persistence, be understood in molecular terms and that biological phenomena observed in simple organisms, such as viruses and bacteria, were mirrored in more complex ones. Implicit corollaries to this premise were that whatever was learned in one organism was likely to be directly relevant to others and that similar approaches could be used to study biological phenomena in many organisms. Genetics, along with molecular biology, became the principal means for dissecting complex biological phenomena into workable subunits. Soon all organisms came under the scrutiny of these approaches.
I became a product of the molecular biology revolution. The next generation. As an Antioch college undergraduate, I worked several quarters in Alex Rich’s laboratory at MIT. He was an x-ray crystallographer, with very broad interests in molecular biology. While at MIT I was also fortunate to be influenced bySalvador Luria, Cyrus Leventhal and Boris Magasanik, through courses, seminars, and personal discussions. At that time Sheldon Penman and Jim Darnell were also working in Alex Rich’s laboratory. When placed in the same room, these two were particularly boisterous, providing comic relief to the fast moving era.
After Antioch, I set off for what I perceived as the “Mecca” of molecular biology, Harvard University. I had interviewed with Professor James D. Watson, of “Watson and Crick” fame, and asked him where should I do my graduate studies. His reply was curt and to the point: “Here. You would be fucking crazy to go anywhere else.” The simplicity of the message was very persuasive.
 |
| Figure 6. A photograph of James D. Watson. |
James D. Watson had a profound influence on my career (see Figure 6). He was my mentor. He did not teach me how to do molecular biology; because of my Antioch job experiences, I had already become a proficient experimenter. Jim instead taught me the process of science – how to extract the questions in a field that are critical to it and at the same time approachable through current technology. As an individual, he personified molecular biology, and, as his students, we were its eager practitioners. His bravado encouraged self-confidence in those around him. His stark honesty made our quest for truth uncompromising. His sense of justice encouraged compassion. He taught us not to bother with small questions, for such pursuits were likely to produce small answers. At a critical time, when I was contemplating leaving Harvard as a faculty member and going to Utah, he, being familiar with my self-sufficiency, counseled me that I could do good science anywhere. The move turned out to be a good decision. In Utah I had the luxury to pursue long-term projects that were not readily possible at Harvard, which, in too many cases had become a bastion of short-term gratification.
Doing science in Jim’s laboratory was exhilarating. As a graduate student, I was provided with what appeared to be limitless resources. I could not be kept out of the laboratory. Ninety-hour weeks were common. The lab was filled with talented students, each working on his or her own set of projects. Represented was a mixture of genetics, molecular biology, and biochemistry. We were cracking the genetic code, determining how proteins were synthesized, and isolating and characterizing the enzymatic machinery required for transcription. At this time, Walter Gilbert was also working in Jim’s laboratory. He was then a member of the physics department, but had also been bitten by the molecular biology bug. Jim and Wally complemented each other brilliantly, because they approached science from very different perspectives. Jim was intuitive, biological; Wally quantitative, with a physicist’s perspective. They were both very competitive. As students, we received the benefit of both, but also their scrutiny. They were merciless, but fair. You had to have a tough hide, but you learned rigor, both with respect to your science and your presentations. Once you made it through Jim’s laboratory, the rest of the world seemed a piece of cake. It was excellent training. Despite the toughness, which at times was hard, Jim was extremely supportive. He also made sure that you, the student, received full credit for your work. Despite the fact that Jim was responsible for all of the resources needed to run his laboratory, if you did all of the work for a given paper, then you were the sole author on that paper. Among all of the laboratory heads in the world, I believe that Jim Watson was among very few in implementing this policy.
The summer before I started graduate school, Marshall Nirenberg had announced that polyU directs the synthesis of polyphenylalanine in a cell free protein synthesizing extract. That paper was a bombshell! I decided I would generate a cell-free extract capable of synthesizing real, functional proteins. Jim’s laboratory had started working on the RNA bacteriophage, R17. Its genome also served as messenger RNA to direct the synthesis of its viral proteins. That would be my message. The cell-free protein synthesizing extract worked beautifully. Authentic viral coat protein and replicase were shown to be synthesized in the extract1. Further, the coat protein was functional, it bound to a specific sequence of the R17 genome, thereby modulating the synthesis of the replicase. To this day, the high affinity of the viral coat protein for this RNA sequence is exploited as a general reporter system to track RNA trafficking within living cells and neuronal axons. In collaboration with Gary Gussin, also a graduate student in Jim’s laboratory, this system was used to determine the molecular mechanism of genetic suppression of nonsense mutations2. In collaboration with Jerry Adams, another graduate student in Jim’s laboratory, the system was also used to determine that initiation of the synthesis of all proteins in bacteria proceeded through the use of formyl-methionine-tRNA3,4. A similar mechanism is involved in the initiation of protein synthesis in all eukaryotic organisms. Finally, I used the same in vitro system to show that termination of protein synthesis unexpectedly utilized protein factors, rather than tRNA, to accomplish this end5,6. Jim Watson would later offer the very complimentary comment “that Capecchi accomplished more as a graduate student than most scientists accomplish in a lifetime.” It was, indeed, a productive time, but it wasn’t work; it was sheer joy.
While a graduate student in Jim’s laboratory, I was invited to become a junior fellow of the Society of Fellows at Harvard. Being a junior fellow was very special. The society’s membership, junior and senior fellows, represented a broad spectrum of disciplines; all the members were talented, and most of them were much more verbal than I. Social discourse centered around meals, prepared by an exquisite French chef and ending with fine brandy and Cuban cigars. Frequent guests at these dinners were the likes of Leonard Bernstein. Surreal maybe, but also very special.
 |
| Figure 7. A photograph of Karl G. Lark. |
From Jim’s laboratory, I joined the faculty in the Department of Biochemistry at Harvard Medical School, across the river in Boston. During my four years at Harvard Medical School I quickly rose through the ranks, but then, I unexpectedly decided to go to Utah. I was looking for something different. There were excellent scientists in the department I was in at Harvard Medical School, but the department was not built with synergy in mind. Each research group was an island onto itself. At that time, they were also unwilling to hire additional young faculty and thereby provide the department with a more youthful, energetic character. At the University of Utah, I would be joining a newly formed department that was being assembled by a very talented scientist and administrator, Karl G. Lark (Figure 7). He had excellent taste in scientists and a vision of assembling a faculty that would enjoy working together and striving together for excellence. I could be a participant in the growth of that department and help shape its character. Furthermore, the University’s administration, led then by President David P. Gardner, was in synchrony with this vision and a strong supporter. Gordon had already attracted Baldomero (Toto) Olivera, Martin Rechsteiner, Sandy Parkinson, and Larry Okun to Utah. After I arrived at Utah, we were able to bring to Utah such outstanding scientists as Ray Gesteland, John Roth, and Mary Beckerle. Utah also provided wide open space, an entirely new canvas upon which to create a new career (see Figures 8). These are views from one of the homes in Utah which I have shared with my wife, Laurie Fraser, and daughter, Misha. The air is clean, and I can look for long distances. The elements of nature are all around us. What a place to begin a new life!
 |
| Figure 8. Views from one of our homes in Utah and a photograph of my wife, Laurie Fraser, and daughter, Misha, just after she was born. Misha is now graduating from the University of California, Santa Cruz as an arts major. |
From Les Prix Nobel. The Nobel Prizes 2007, Editor Karl Grandin, [Nobel Foundation], Stockholm, 2008
This autobiography/biography was written at the time of the award and later published in the book series Les Prix Nobel/ Nobel Lectures/The Nobel Prizes. The information is sometimes updated with an addendum submitted by the Laureate.
Copyright © The Nobel Foundation 2007
Dr. Capecchi is a member of the National Academy of Sciences (1991) and the European Academy of Sciences (2002). He has won numerous awards, including the Bristol-Myers Squibb Award for Distinguished Achievement in Neuroscience Research (1992), the Gairdner Foundation International Award for Achievements in Medical Sciences (1993), the General Motors Corporation’s Alfred P. Sloan Jr. Prize for Outstanding Basic Science Contributions to Cancer Research (1994), the German Molecular Bioanalytics Prize, (1996), the Kyoto Prize in Basic Sciences (1996), the Franklin Medal for Advancing Our Knowledge of the Physical Sciences (1997), the Feodor Lynen Lectureship (1998), the Rosenblatt Prize for Excellence (1998), the Baxter Award for Distinguished Research in the Biomedical Sciences (1998), the Helen Lowe Bamberger Colby and John E. Bamberger Presidential Endowed Chair in the University of Utah Health Sciences Center (1999), lectureship in the Life Sciences for the Collège de France (2000), the Horace Mann Distinguished Alumni Award, Antioch College (2000), the Italian Premio Phoenix-Anni Verdi for Genetics Research Award (2000), the Spanish Jiménez-Diáz Prize (2001), the Pioneers of Progress Award (2001), the Albert Lasker Award for Basic Medical Research (2001), the National Medal of Science (2001), the John Scott Medal Award (2002), the Massry Prize (2002), the Pezcoller Foundation-AACR International Award for Cancer Research (2003), the Wolf Prize in Medicine (2002/03), the March of Dimes Prize in Developmental Biology (2005),and the Nobel Prize in Physiology and Medicine (2007) with Oliver Smithies and Martin Evans.
Research interests include: the molecular genetic analysis of early mouse development, neural development in mammals, production of murine models of human genetic diseases, gene therapy, homologous recombination and programmed genomic rearrangements in the mouse.
http://www.hhmi.org/news/making-scientist
Mario Capecchi received a Kyoto Prize from the Inamori Foundation in 1996. The lecture he delivered when he accepted the prize in Japan in November 1996 tells the story of his remarkable life. The text of the lecture has been edited for length.
Radoslav Bozov commented on Targeted gene modification
Targeted gene modification Larry H Bernstein, MD, FCAP, Curator Leaders in Pharmaceutical Intelligence Series E. 2: …
Larry, same thing, data redundancy of data mining issues, of what data is in reality of physics beyond nano space in time! Working on something that does not exits in space and time, but computable mass of ‘designed’ energy formulated systems: Hox gene does not exist: It is a piece of time we percive through some kind of imagination +1,
The data generated through m/z methods is space-time unaccurate! Guess what is double R doing here instead of double Y, wonder why miRNA are obejctive to polymer degradation process ??!! what are we really seeing is not what is really in there!
Score Expect Method Identities Positives Gaps
19.2 bits(38) 0.076 Composition-based stats. 8/17(47%) 10/17(58%) 0/17(0%)
Query 511 LTEDRRAFAARMAEIGE 527
LT DRR AR+ + E
Sbjct 38 LTRDRRYEVARLLNLTE 54
Breaking news about genomic engineering, T2DM and cancer treatments
Larry H. Bernstein, MD, FCAP, Curator
Posted in Cell Biology, Signaling & Cell Circuits, Chemical Biology and its relations to Metabolic Disease, Chemical Genetics, Curation, Disease Biology, Small Molecules in Development of Therapeutic Drugs, Gene Regulation, Genetics & Pharmaceutical, Human Immune System in Health and in Disease, Immuno-Oncology & Genomics, Infectious Disease Immunodiagnostics, Innovation in Immunology Diagnostics, Metabolism, Molecular Genetics & Pharmaceutical, Pharmaceutical Analytics, Pharmaceutical Drug Discovery, Proteins, Proteomics, Signaling & Cell Circuits, Transcriptomics, Translational Science, tagged B cell-derived coactivator, B-cell signaling, immunoglobulins, octamer-binding transcription factors, POU dimer on September 7, 2015| Leave a Comment »
Roeder – the coactivator OCA-B, the first cell-specific coactivator, discovered by Roeder in 1992, is unique to immune system B cells
Larry H Bernstein, MD, FCAP, Curator
Leaders in Pharmaceutical Intelligence
The B-cell-specific transcription coactivator OCA-B/OBF-1/Bob-1 is essential for normal production of immunoglobulin isotypes
Unkyu Kim*, Xiao-Feng Qin†, Shiaoching Gong†, Sean Stevens*, Yan Luo*, Michel Nussenzweig† & Robert G. Roeder*
Nature 383, 542 – 547 (10 October 1996); http://dx.doi.org:/10.1038/383542a0
* Laboratory of Biochemistry and Molecular Biology, and † Laboratory of Molecular Immunology, Howard Hughes Medical Institute, The Rockefeller University, New York, New York 10021, USA
OCA-B was initially identified as a B-cell-restricted coactivator that functions with octamer binding transcription factors (Oct-1 and Oct-2) to mediate efficient cell type-specific transcription of immunoglobulin promoters in vitro 1–3. Subsequent cloning studies led to identification of the coactivator as a single poly-peptide, designated either as OCA-B (ref. 3), OBF-1 (ref. 4) or Bob-1 (ref. 5). OCA-B itself does not bind to DNA directly, but interacts with either Oct-1 or Oct-2 to potentiate transcriptional activation1–5. To determine the biological role of OCA-B, we generated OCA-B-deficient mice by gene targeting. Mice lacking OCA-B undergo normal antigen-independent, B-cell differentiation, including appropriate expression of both immunoglobulin genes and other early B-cell-restricted genes. However, antigen-dependent maturation of B cells is greatly affected. The pro- liferative response to surface IgM crosslinking is impaired, and there is a severe deficiency in the production of secondary immunoglobulin isotypes including IgGl, IgG2a, IgG2b, IgG3, IgA and IgE in OCA-B-deficient B cells. This defect is not due to a failure of the isotype switching process, but rather to reduced levels of transcription from normally switched immunoglobulin heavy-chain loci. In accord with the defective isotype production, germinal centre formation is absent in these mutant mice.
References
| 1. | Pierani, A., Heguy, A., Fujii, H. & Roeder, R. G. Mol. Cell. Biol. 10, 6204−6215 (1990). | PubMed | ChemPort | |
| 2. | Luo, Y., Fujii, H., Gerster, T. & Roeder, R. G. Cell 71, 231−241 (1992). | Article | PubMed | ISI | ChemPort | |
| 3. | Luo, Y. & Roeder, R. G. Mol. Cell. Biol. 15, 4115−4124 (1995). | PubMed | ISI | ChemPort | |
| 4. | Strubin, M., Newell, J. W. & Matthias, P. Cell 80, 497−506 (1995). | Article | PubMed | ISI | ChemPort | |
| 5. | Gstaiger, M., Knoepfel, L., Georgiev, O., Schaffner, W. & Hovens, C. M. Nature 373, 360−362 (1995). | Article | PubMed | ISI | ChemPort | |
| 6. | Yancopoulos, G. D. & Alt, F. W. Cell 40, 271−281 (1985). | Article | PubMed | ISI | ChemPort | |
| 7. | Yancopoulos, G. D. & Alt, F. W. Annu. Rev. Immunol. 4, 339−368 (1986). | Article | PubMed | ISI | ChemPort | |
| 8. | Rajewsky, K. Curr. Opin. Immunol. 4, 171−176 (1992). | Article | PubMed | ChemPort | |
| 9. | Rolink, A. & Melchers, F. Adv. Immunol. 53, 123−156 (1993). | PubMed | ISI | ChemPort | |
| 10. | Gold, M. R. & DeFranco, A. L. Adv. Immunol. 55, 221−295 (1994). | PubMed | ISI | ChemPort | |
| 11. | Coffman, R. L., Lebman, D. A. & Rothman, P. Adv. Immunol. 54, 229−270 (1993). | PubMed | ISI | ChemPort | |
| 12. | Parker, D. C. Annu. Rev. Immunol. 11, 331−360 (1993). | Article | PubMed | ISI | ChemPort | |
| 13. | Wang, H. Y., Paul, W. E. & Keegan, A. Immunity 4, 113−122 (1996). | Article | PubMed | ISI | ChemPort | |
| 14. | McLennan, I. C. Annu. Rev. Immunol. 12, 117−139 (1994). | Article | PubMed | ISI | ChemPort | |
| 15. | Lutzker, S. & Alt, F. W. Mol. Cell. Biol. 8, 1849−1852 (1988). | PubMed | ISI | ChemPort | |
| 16. | Stavnezer, J. et al. Proc. Natl Acad. Sci. USA 85, 7704−7708 (1988). | PubMed | ChemPort | |
| 17. | Staudt, L. M. & Lenardo, M. J. Annu. Rev. Immunol. 9, 373−398 (1991). | Article | PubMed | ISI | ChemPort | |
| 18. | Dariavach, P., Williams, G. T., Campbell, K., Pettersson, S. & Neuberger, M. S. Eur. J. Immunol. 21, 1499−1504 (1991). | PubMed | ISI | ChemPort | |
| 19. | Grant, P. A., Thompson, C. B. & Pettersson, S. EMBO J. 14, 4501−4513 (1995). | PubMed | ISI | ChemPort | |
| 20. | Bain, G., Gruenwald, S. & Murre, C. Mol. Cell. Biol. 13, 3522−3529 (1993). | PubMed | ISI | ChemPort | |
| 21. | Tybulewicz, V. L., Crawford, C. E., Jackson, P. K., Bronson, R. T. & Mulligan, R. C. Cell 65, 1153−1163 (1991). | Article | PubMed | ISI | ChemPort | |
| 22. | DeFranco, A. L. J. Exp. Med. 155, 1523−1536 (1982). | Article | PubMed | ISI | ChemPort | |
| 23. | Bottaro, A. et al. EMBO J. 13, 665−674 (1994). | PubMed | ISI | ChemPort | |
| 24. | Cogne, M. et al. Cell 77, 737−747 (1994). | Article | PubMed | ISI | |
| 25. | Gong, S. & Nussenzweig, M. C. Science 272, 411−414 (1996). | PubMed | ISI | ChemPort | |
| 26. | Qin, X. F. et al. EMBO J. 13, 5967−5976 (1994). | PubMed | ISI | ChemPort | |
| 27. | Li, S. C. et al. Int. Immunol. 6, 491−497 (1994). | PubMed | ChemPort | |
Cloning, Functional Characterization, and Mechanism of Action of the B-Cell-Specific Transcriptional Coactivator
Oca-B Yan Luo & Robert G. Roeder*
Laboratory of Biochemistry and Molecular Biology, The Rockefeller University, New York, New York 10021
Molecular And Cellular Biology, Aug. 1995; 15(8):4115–4124 0270-7306/95/
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC230650/pdf/154115.pdf
Biochemical purification and cognate cDNA cloning studies have revealed that the previously described transcriptional coactivator OCA-B consists of a 34- or 35-kDa polypeptide with sequence relationships to known coactivators that function by protein-protein interactions. Studies with a recombinant protein have proved that a single OCA-B polypeptide is the main determinant for B-cell-specific activation of immunoglobulin (Ig) promoters and provided additional insights into its mechanism of action. Recombinant OCA-B can function equally well with Oct-1 or Oct-2 on an Ig promoter, but while corresponding POU domains are sufficient for OCA-B interaction, and for octamer-mediated transcription of a histone H2B promoter, an additional Oct-1 or Oct-2 activation domain(s) is necessary for functional synergy with OCA-B. Further studies show that Ig promoter activation by Oct-1 and OCA-B requires still other general (USA-derived) cofactors and also provide indirect evidence that distinct Oct-interacting cofactors regulate H2B transcription.
Identification of transcription coactivator OCA-B dependent genes involved in antigen-dependent B cell differentiation by cDNA array analyses
Unkyu Kim*, Rachael Siegel*, Xiaodi Ren*, Cary S. Gunther*, Terry Gaasterland†, and Robert G. Roeder*‡
*Laboratory of Biochemistry and Molecular Biology and †Laboratory of Computational Genomics, The Rockefeller University, 1230 York Avenue, New York, NY 10021
PNAS http://www.pnas.org/content/100/15/8868.full.pdf
The tissue-specific transcriptional coactivator OCA-B is required for antigen-dependent B cell differentiation events, including germinal center formation. However, the identity of OCA-B target genes involved in this process is unknown. This study has used large-scale cDNA arrays to monitor changes in gene expression patterns that accompany mature B cell differentiation. B cell receptor ligation alone induces many genes involved in B cell expansion, whereas B cell receptor and helper T cell costimulation induce genes associated with B cell effector function. OCA-B expression is induced by both B cell receptor ligation alone and helper T cell costimulation, suggesting that OCA-B is involved in B cell expansion as well as B cell function. Accordingly, several genes involved in cell proliferation and signaling, such as Lck, Kcnn4, Cdc37, cyclin D3, B4galt1, and Ms4a11, have been identified as OCA-B-dependent genes. Further studies on the roles played by these genes in B cells will contribute to an understanding of B cell differentiation.
Identification and Characterization of a Novel OCA-B Isoform: Implications for a Role in B Cell Signaling Pathways
Xin Yu, Lu Wang†, Yan Luo, Robert G. Roeder
Immunity Feb 2001; 14(2): 157–167 http://dx.doi.org:/10.1016/S1074-7613(01)00099-1
OCA-B is a B lymphocyte–specific transcription coactivator that mediates tissue- and stage-restricted transcription of immunoglobulin genes. Earlier genetic studies revealed that OCA-B is essential for germinal center formation and production of secondary immunoglobulin isotypes. Biochemically purified OCA-B contains p35 and p34 isoforms, and a further analysis has now revealed that p35 is derived from a newly found isoform, p40. More importantly, it has been found that p35 is myristoylated in vivo and that this leads to dramatic changes (including localization to membrane compartments) in its properties. These results suggest that the p35 isoform of OCA-B has functions distinct from those of the nuclear p34 and that it might be a component of a signaling pathway that is required for late-stage B cell development.
The B cell–restricted function of immunoglobulin (Ig) promoters is mediated mainly by an octamer element (5′-ATGCAAAT-3′) that is conserved in virtually all Ig heavy (H) and light (L) chain gene promoters, as well as in some Ig enhancers (reviewed by Staudt and Lenardo, 1990). However, this same element is also a key central element for transcription of differentially regulated genes that include ubiquitously expressed small nuclear RNA genes (snRNA) and cell cycle-regulated histone H2B genes (reviewed inLuo et al., 1992). The regulatory functions of octamer elements, therefore, are likely dependent on transcription factors that bind this DNA sequence. The well-characterized octamer binding transcription factors include the ubiquitous Oct-1 and the B cell–enriched Oct-2, both of which belong to the POU family and share a conserved DNA binding structure called the POU domain (reviewed by Herr et al. 1988 and Wegner et al. 1993). It was originally thought that Oct-2 would account for the tissue-specific activity of Ig promoters, whereas Oct-1 would facilitate transcription of the ubiquitously expressed genes regulated through octamer elements (e.g., snRNA and histone H2B genes) Staudt et al. 1986, Cockerill and Klinken 1990 and Murphy et al. 1992. However, subsequent biochemical Pierani et al. 1990 and Luo et al. 1992 and genetic (Corcoran et al., 1993)analyses clearly demonstrated that this was not the case. Instead, the promoter specificity was shown to be due to an Oct-1 interacting factor called OCA-B (Luo et al., 1992), and the purification of related p35 and p34 isoforms with apparently equivalent activity in vitro (Luo and Roeder, 1995) set the stage for further studies of the structure and function of OCA-B.
Subsequent to the biochemical identification of OCA-B and its mechanism of action, cognate cDNAs were cloned using both biochemical (Luo and Roeder, 1995) and genetic screening Gstaiger et al. 1995 and Strubin et al. 1995 methods. Analyses of recombinant OCA-B (p34) function in cell-free systems and in transfection assays confirmed both physical and functional interactions with Oct-1 and Oct-2 (via their POU domains) on Ig promoters Gstaiger et al. 1995, Luo and Roeder 1995 and Strubin et al. 1995 and led to the definition of an N-terminal OCA-B domain that interacts with the Oct POU domainCepek et al. 1996, Gstaiger et al. 1996, Babb et al. 1997 and Chaseman et al. 1999 and a C-terminal activation domain that acts synergistically with Oct activation domains to recruit additional coactivators (Luo et al., 1998).
The physiological roles of OCA-B were further investigated by genetic disruption of OCA-B expression in mice Kim et al. 1996, Nielson et al. 1996 and Schubart et al. 1996. These studies showed that, although not required for early B cell development, OCA-B functions are essential both for germinal center formation and for efficient secondary Ig isotype production (including IgGs, IgA, and IgE). In accordance with the biochemical function of OCA-B in activating Ig promoter transcription, it has been found that the decrease of secondary antibody production in OCA-B-deficient mice is largely due to reduced levels of transcription from normally switched IgH chain loci, rather than a reduced capacity for class switching events per se Kim et al. 1996 and Schubart et al. 1996. Recent results further demonstrated that OCA-B plays an essential role in efficient transcription from switched IgH loci by directly regulating 3′ IgH enhancer function in conjunction with Oct-1 or Oct-2 Tang and Sharp 1999 and Stevens et al. 2000b. On the other hand, the lack of germinal center formation in OCA-B-deficient mice cannot be explained by reduced Ig isotype production, since these are two independent events in B cell development (Vajdy et al., 1995). Therefore, OCA-B may regulate germinal center formation by activating the expression of other target genes or by mediating signal pathways that in turn trigger a specific genetic program. At least two lines of evidence support this idea: (1) B cells lacking OCA-B are defective in the proliferative response to surface IgM cross-linking (Kim et al., 1996); (2) OCA-B expression, which is very low in early B cells but high in activated B cells in vivo, can be dramatically and synergistically induced in naive B cells by B cell stimuli (CD40L, Ig cross-linking, and IL4) that are required for germinal center formation (Qin et al., 1998).
Our findings in this report raise the possibility that OCA-B may be directly involved in B cell signaling pathways through novel mechanisms. We report the presence of a novel isoform of OCA-B (p40) that results from utilization of an upstream alternative translation initiation codon and that serves as a precursor to the p35 isoform of OCA-B. Relative to the conventional p34 OCA-B isoform, p35 shows distinct protein modification, subcellular localization, and transcriptional coactivator properties. The unique features of p35 suggest a novel function for this molecule in signal transduction.
Synergism with the Coactivator OBF-1 (OCA-B, BOB-1) Is Mediated by a Specific POU Dimer Configuration
Alexey Tomilin1, 2, #, Attila Reményi2, 4, #, Katharina Lins1, Hanne Bak2, Sebastian Leidel1, Gerrit Vriend3, Matthias Wilmanns4, Hans R Schöler1, 2
Cell Dec 2000; 103(6):853–864 doi:10.1016/S0092-8674(00)00189-6
POU domain proteins contain a bipartite DNA binding domain divided by a flexible linker that enables them to adopt various monomer configurations on DNA. The versatility of POU protein operation is additionally conferred at the dimerization level. The POU dimer formed on the PORE (ATTTGAAATGCAAAT) can recruit the transcriptional coactivator OBF-1, whereas POU dimers formed on the consensus MORE (ATGCATATGCAT) or on MOREs from immunoglobulin heavy chain promoters (AT[G/A][C/A]ATATGCAA) fail to interact. An interaction with OBF-1 is precluded since the same Oct-1 residues that form the MORE dimerization interface are also used for OBF-1/Oct-1 interactions on the PORE. Our findings provide a paradigm of how specific POU dimer assemblies can differentially recruit a coregulatory activity with distinct transcriptional readouts.
Development of multicellular organisms is characterized by an intricate series of genetic and epigenetic events that generate the complex adult body from the unicellular zygote. A refined and sophisticated regulatory network that is established during embryogenesis reflects the complexity of organisms. Although embryonic development is a multistep process characterized by the sequential activation and repression of many genes, only a relatively small number of transcription factors are responsible for regulating the expression of developmental genes. This diversity in transcriptional control by a limited array of transcription factors is achieved through a complex network of interactions between these proteins and specific DNA sequences found in promoters and enhancers of developmental genes. The primary structure of these DNA elements defines the composition and architecture of the transcriptional activation complexes that ultimately control gene expression in the appropriate temporo-spatial context of the developing organism. For example, nonsteroid members of the nuclear receptor superfamily that possess a zinc-finger DNA binding domain operate by binding to the hormone response elements (HREs). HREs consist of two minimal core hexad sequences, AGGTCA, which can be configured into various functional motifs. The orientation and spacing between these two hexamers as well as subtle differences in their sequence dictate the identity and the mode (monomer, hetero-, or homodimer) of nuclear receptor binding that results in diverse effects on transcription (Mangelsdorf and Evans 1995).
The operation of members of the POU domain family of transcription factors is also highly dependent on the nature of cognate DNA elements. The 160 amino-acid-long DNA binding domain of these proteins is composed of two structurally independent subdomains: the POU-type homeodomain (POU-homeo or POUH), and the POU-specific domain (POUS) that are connected by a flexible linker region (27 and 36). POU domain proteins demonstrate impressive versatility in how they regulate transcription. This is due to several, often interdependent, factors: (1) flexible amino acid–base interaction, (2) variable orientation, spacing, and positioning of DNA-tethered POU subdomains relative to each other, (3) posttranslational modification, and (4) interaction with heterologous proteins (Herr and Cleary 1995).
POU domain proteins are able to bind to DNA cooperatively, thus conferring additional functional variability. The homo- and heterodimerization of Oct-1 and Oct-2 on immunoglobulin (Ig) heavy chain promoters (VH) provided evidence of cooperativity, with a yet unknown dimer arrangement (13, 16 and 23). The cis-elements are considered to consist of low-affinity heptamer and high-affinity octamer sites separated by two nucleotides (![]() AT
AT![]() ).
).
The pituitary-specific POU domain protein Pit-1 binds to DNA either as a homodimer or as a heterodimer with Oct-1 (Voss et al. 1991). Crystallographic studies determined the structure of a Pit-1 homodimer assembled on the synthetic motif ATGTATATACAT (referred to here as PitD) that had been derived from the natural Pit-1 cognate element within the prolactin gene promoter (ATATATATTCAT) (Jacobson et al. 1997). The structure of the Pit-1 POUS and POUH domains, and their docking onto DNA, are very similar to that observed in the cocrystal of the Oct-1 POU domain monomer with the octamer site (ATGCAAAT, Klemm et al. 1994). The Oct-1 POUS domain recognizes the ATGC subsite whereas the Pit-1 POUS domain binds to the sequence ATAC. However, the latter subsite lies on the opposite strand and, as a consequence, the orientation of POUS relative to the POUH domain is inverted (Jacobson et al. 1997).
Another mechanism outlining cooperative DNA binding by POU proteins was recently determined during the course of an Oct-4 target gene characterization (Botquin et al. 1998). The P alindromic O ct factor R ecognition E lement (PORE), ATTTGAAATGCAAAT (15 bp), of the Osteopontin (OPN) enhancer interacts with an Oct-4 dimer, thereby mediating strong transcriptional activation in preimplantation mouse embryos. Homo- and heterodimerization of other Oct factors like Oct-1 and Oct-6 on the PORE has also been demonstrated.
The aforementioned examples provide evidence of the various ways in which POU domain proteins are able to cooperatively bind to substrate DNA. The particular mode of binding employed is primarily defined by the DNA sequence. To address the question of whether diversity in cooperative binding is reflected in transcriptional regulation, we have assessed and compared the ability of two different types of POU dimers to interact with the coactivator OBF-1 (OCA-B, Bob-1). This coactivator synergistically interacts with Oct-1 and Oct-2 monomers bound to the octamer motif (18, 9, 17 and 33). We have investigated one type of POU dimer that is formed on the PORE and another that is formed on another palindromic DNA motif called MORE (M ore P ORE), ATGCATATGCAT. The data presented in this study provide an example of how POU domain molecules that bind to DNA in the same stoichiometry but in different configurations can differentially recruit a transcriptional coactivator to the promoter resulting in differential transcriptional activation.
B-cell-specific Coactivator OCA-B: Biochemical Aspects, Role in B-Cell Development and Beyond
Cold Spring Harb Symp Quant Biol 1999 64: 119-132;
http://dx.doi.org:/10.1101/sqb.1999.64.119
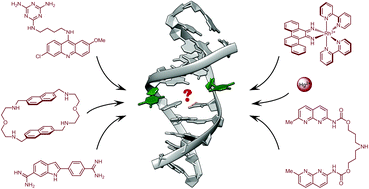
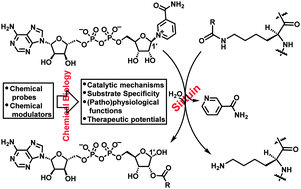
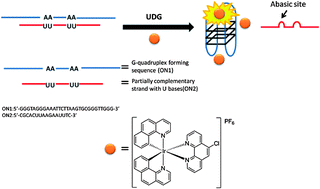
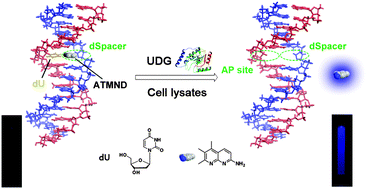
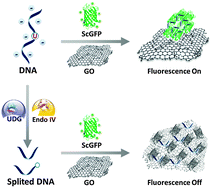
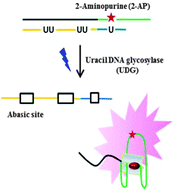
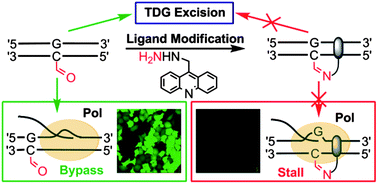
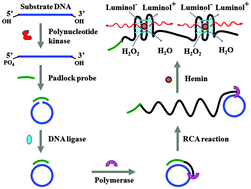
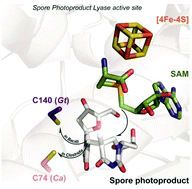
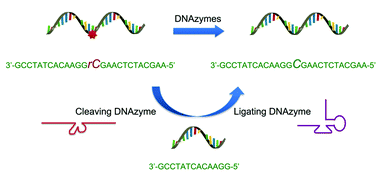
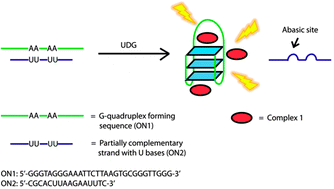
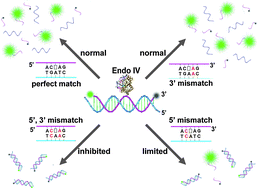

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}