Pathology Emergence in the 21st Century
Author and Curator: Larry Bernstein, MD, FCAP
This discussion follows the series on DNA and its replication, the code of life, and immediately follows a an up to date survey of RNA, it’s many discovered forms, their function, and the transcription of RNA, intermediate to protein synthesis. This will comprise a series of articles, including the chemistry, structure, and function of proteins, before turning to the “metabolome”. This discussion is about the development of the scientific profession of pathology in three notable phases. The first phase might be considered the gross anatomical discussion developed in Vienna, under Rokitansky, whose greatest student was the Hungarian pathologist, Semelweis, who began the insistence on handwashing prior to visiting the delivery room based on the observation that deliver was safer done by midwifes than by physicians. This was prior to the great discoveries in microbiology. The Rokitansky procedure is distinctive in removing organ systems in order to examine postmortem anatomical changes. His document on the pathology of the organs was monumental. It was refined by Rudolf Virchow, who removed one organ at a time, but he also had the now developing field of microscopic anatomy (also describing leukemia),


Rudolph Virchow 1821-1902
that would also be embellished by a generation of histologists who introduced staining techniques. The most remarkable anatomist to emerge in that period was John Hunter, the Scottish born anatomist and surgeon in London, who taught medical students from England and United States, and who was the physician for Sir David Hume. When he served in the 30 years war, he pulled wounded soldiers out of the mud to a clearing to care for their wounds.
The 20th century saw the introduction of a new medical school education system under advisement by the Carnegie Commission. [Abraham Flexner Report] This led to the teaching of basic sciences of anatomy, physiology, pharmacology, pathology, and later genetics and microbiology as prerequisite to clinical teaching. Moreover, teaching was done by formal systems of disease, which later developed into rotations in Obstetrics and Gynecology, Medicine, and Surgery, Pediatrics, to which endocrinology and neurology and psychiatry were added. The first great Medical School was actually Johns Hopkins, that paved the groundwork. Harvard, Cornell, Columbia, and NYU followed, as did the University of California, San Francisco, and the University of Chicago. This was a period dominated by many important discoveries, and a domination of the Nobel Prizes in Medicine and Physiology, Chemistry and Physics (rivalled only by Germany and United Kingdom). The Uniqueness of Pathology lies in its placement in the first year of medical school, in preparation for the formal study of medicine, with a foot in both basic sciences and the other in clinical practice. The pathologist received all tissues removed from surgical procedures, and performed autopsies to determine the cause of death and comorbid conditions. The development of a substantial knowledge of the kidney gave rise to the specialty of nephrology, and at the same time drew pathologists into a “phase” of molecular biology with the introduction of the electron microscope. However, the field of immunology developed, and the need for transfusion hastened the emergence of clinical antigen-antibody testing, the crossmatch, blood banking, and later, the emergence of organ transplantation. In addition, clinical microbiology became a part of clinical pathology, and included fungi and tick-borne diseases, and nemitodes.
We have entered the third phase of pathology with the completion of the human genetic code, and with the development of target pharmaceutical development in the 21st century.
Modern Techniques of Molecular Pathology
A look at clinical laboratory science and its expected progress over the next decade
Molecular Diagnostics 2013; 22 (2), p 35Promising forecasts project great expectations for medical sciences in the year 2013 and beyond. These predictions follow a decade after completion of the Human Genome Project, and are accompanied by immense breakthroughs in computational and applied mathematics. In my view, they are:
- Genomic and allied -omics technology
- Innovation in mathematical classification (complexity)
- Nanotechnology
- Synthetic chemistry from physics, organic and inorganic chemistry
It is not my intention to go deeply into the exponential group of these advanced and integrative sciences; rather, I want to raise awareness of an emerging new world that will open to the clinical laboratory scientist, and signal the need in the next generation of laboratory personnel to embrace knowledge domains that will be critical for their careers.
All of these breakthroughs are tied together by a search for personalized and integrative medicine. These breakthroughs will reinvent nutritional and pharmaceutical medicine as well as medical devices and restructure clinical laboratory and imaging applications to cardiology, oncology, radiology and anatomical pathology.
Metabolomics
What does metabolomics and metabolic profiling have to do with this? Metabolomics is the measurement of small molecules that interact with membrane receptors1 that are involved with regulation of genomic transcription and cellular regulation and upregulation or downregulation of metabolic processes essential to health. As well, these small molecules may provide targets for disease treatments, and as they are investigated, also provide further “analytes for diagnosis and, moreover, prediction of short-term or long-term outcomes.”2
As a result, the laboratory will become a more significant factor in measuring health and disease and in guiding health or disease maintenance. As our population has reached increased age limits, the laboratory has been a contributor in the public health sphere, and will have a greater role as a result of:
- Improved tie in with provision of information to not only the healthcare workers, but also the patient
- Achieve turnaround times for critical results through better workflow
- Greater control and access to a next generation of point-of-care technology integrated with the laboratory database, and a restructured electronic health record
Despite the hype about the “big data” revolution, this is achievable in the system proposed because there is a published model to achieve this.2
Familiar Methods
Either individually or grouped as a profile, metabolites are detected by nuclear magnetic resonance spectroscopy or mass spectrometry, providing a basis for uses of metabolome findings extended to the early detection and diagnosis of cancer and as both a predictive and pharmacodynamics marker of drug effect. We can expect it to become the link between the laboratory and the clinic. The methods used in genomics are microarrays, and for proteomics they are the already familiar chromatographic principles that species migrate at different rates through a column matrix based on their volatility, or carries out a separation as the molecules differ by their adsorption to and elution from a solid matrix, dependent on the binding to the matrix and solubility in the solvent eluate, modified by pH, ionic concentration, and specific conditions needed for recovery. Powerful mathematical tools are used to analyze the data.3
Cardiovascular Disease
Although coronary thrombosis is the final event in acute coronary syndromes, increasing evidence suggests that inflammation also plays a key role in development of atherosclerosis and its clinical manifestations, such as myocardial infarction, stroke and peripheral vascular disease. The inflammatory component was indicated by epidemiological studies of elevated serum levels of high sensitivity C-reactive protein. That eventually led to the demonstration of a benefit from reduction of CRP in individuals without characteristic lipidemia in a major clinical trial, which drew a relationship between diabetes, obesity and disordered inflammatory response in the causation of coronary artery disease, aortic valve and artery disease, carotid artery and peripheral vascular disease.
Cancer
Because cancer cells are known to possess a highly unique metabolic phenotype, development of specific biomarkers in oncology is possible and might be used in identifying fingerprints, profiles or signatures to detect the presence of cancer, determine prognosis and/or assess the pharmacodynamic effects of therapy.4
HDM2, a negative regulator of the tumor suppressor p53, is over-expressed in many cancers that retain wild-type p53. Consequently, the effectiveness of chemotherapies that induce p53 might be limited, and inhibitors of the HDM2-p53 interaction are being sought as tumor-selective drugs.5
Coagulation
Blood coagulation plays a key role among numerous mediating systems activated in inflammation. Receptors of the PAR family serve as sensors of serine proteinases of the blood clotting system in the target cells involved in inflammation. Activation of PAR_1 by thrombin and of PAR_2 by factor Xa leads to a rapid expression and exposure on the membrane of endothelial cells of both adhesive proteins that mediate an acute inflammatory reaction and of the tissue factor that initiates the blood coagulation cascade.
The details of evolving methods are avoided in order to build the argument that a very rapid expansion of discovery has been evolving depicting disease, disease mechanisms, disease associations, metabolic biomarkers, study of effects of diet and diet modification, and opportunities for targeted drug development.
- Bernstein is CEO of Triplex Medical Science and CSO of Leaders in Pharmaceutical Intelligence. He has been involved in a collaborative project on reducing the noise that exists in complex data and developing a primary evidence-based classification since retiring from a career in pathology supning four decades.
References
1. Bernstein LH. Metabolomics, metabonomics and functional nutrition: The next step in nutritional metabolism and biotherapeutics. Journal of Pharmacy and Nutrition Sciences, 2012;2.
2. David G, Bernstein LH, Coifman RR. Generating evidence-based interpretation of hematology screens via anomaly characterization. The Open Clinical Chemistry Journal 2011;4: 10-16.
3. Grainger DJ. Megavariate statistics meets high data-density analytical methods: The future of medical diagnostics? IRTL Reviews 2003;1:1-6.
4. Spratlin JL, Serkova NJ, Eckhardt SG. Clinical applications of metabolomics in oncology: A review. Clin Cancer Res 2009;15; 15(2): 431-440.
5. Fischer PM, Lane DP. Small molecule inhibitors of thep53 suppressor HDM2: Have protein-protein interactions come of age as drug targets? Trends in Pharm Sci 2004;25(7):343-346. |
Directions for Genomics in Personalized Medicine
Author: Larry H. Bernstein, MD, FCAP
Purpose
This discussion will identify the huge expansion of genomic technology in the search for biopharmacotherapeutic targets that continue to be explored involving different levels and interacting signaling pathways. There are several methods of analyzing gene expression that will be discussed. Great primary emphasis required investigation of combinations of mutations expressed in different cancer types.
James Watson has proposed a major hypothesis that expresses the need to focus on “central” “driver mutations” that correspond with the regulation of gene expression, cell proliferation, and cell metabolism with a critical rejection of antioxidant benefits. What hasn’t been know is why drug resistance develops and whether the cellular migration and aerobic glycolysis can be redirected after cell metastasis occurs. I attempt to bring out the complexities of current efforts.
Introduction
- This discussion is a continuation of a previous discussion on the role of genomics if discovery of therapeutic targets for cancer, each somewhat different, but all related to:
- The reversal of carcinoma by targeting a key driver of multiple signaling pathways that activate cell proliferation
- Pinpointing a stage in a multistage process at which tumor progression links to changes in morphology from basal cells to invasive carcinoma with changes in polarity and loss of glandular architecture
- Reversal of the carcinoma through using a small molecule that either is covalently bound to a nanoparticle delivery system that blocks or reverses tumor development
- Synthesis of a small molecule that interacts with the translation of the genome either by substitution of a key driver molecule or by blocking at the mRNA stage of translation
- Blocking more than one signaling pathway that are links to carcinogenesis and cellular proliferation and invasion
Difficulty of the problem
A problem expressed by James Watson is that the investigations that are ongoing
- are following a pathway that is not driven by attacking the “primary” driver of carcinogenesis.
He uses the Myc gene as an example, as noted in the previous discussion. The problem may be more complicated than he envisions.
- The most consistent problem in chemotherapy, irrespective of the design and the target has been cancer remission for a short time followed by recurrence, and then
- switching to another drug, or combination chemotherapy.
It is common to “clean” the field at the time of resection using radiotherapy before chemotherapy.
- But the goal is understood to be “palliation”, not cure.
This raises a serious issue in the hypothesis posed by Watson. The issue is
- whether there is a core locus of genetic regulation that is common to carcinogenesis irrespective of tissue metabolic expression.
- This is supported by the observation that tissue specific expression is lost in cancer cells by de-differentiation.
Historical Perspective
AEROBIC GLYCOLYSIS
In 1967 Otto Warburg published his view in a paper “The prime cause and prevention of cancer”.
There are primary and secondary causes of all diseases
- plague – primary: plague bacillus
- plague – secondary: filth, rats, and fleas

Otto Heinrich Warburg (Photo credit: Wikipedia)
cancer, above all diseases,
- has countless seconday causes
- primary – replacement of respiration of oxygen in normal body tissue by fermentation of glucose with conversion from obligate aerobic to anaerobic, as in bacterial cells
The cornerstone to understanding cancer is in study of the energetics of life
This thinking came out of decades of work in the Dahlem Institute Kaiser Wilhelm pre WWII and Max Planck Institute after WWII, supported by the Rockefeller Foundation.
- The oxygen- and hydrogen-transferring enzymes were discovered and isolated.
- The methods were elegant for that time, using a manometer that improved on the method used by Haldane, that did not allow the leakage of O2 or CO2.
- The interest was initiated by the increased growth of Sea Urchin eggs after fertization, which turned out to be not comparable to the rapid growth of cancer cells.
- Warburg used both normal and cancer tissue and measured the utilization of O2. He found
- that the normal tissue did not accumulate lactic acid.
- Cancer tissue generated lactic acid
- the rate of O2 consumption the same as normal tissue, but
- the rate of lactate formation far exceeded any tissue, except the retina.
- This was a discovery studied by “Pasteur” 60 years earlier (facultative aerobes), which he called thePasteur effect.
- Hematopoietic cells of bone marrow develop aerobic glycolysis when exposed to a low oxygen condition.
He then followed on an observation by Otto Meyerhoff (Embden-Myerhoff cycle) that in muscle
- the consumption of one molecule of oxygen generates two molecules of lactate, but in aerobic glycolysis, the relationship disappears.
- He expressed the effectiveness of respiration by the ‘Meyerhoff quotient’.
- He found that cancer cells didn’t have a quotient of ’2′
The role of the allosteric enzyme phosphofructokinase (PFK) not then known, would tie together the glycolytic and gluconeogenic pathways.
He used a heavy metal ion chelator ethylcarbylamine to
- sever the link and turn on aerobic glycolysis.
The explanation for this was provided years later by the work fleshed out by Lynen, Bucher, Lowry, Racker, and Sols.
- The rate-limiting enzyme, PFK is regulated by the concentrations of ATP, ADP, and inorganic phosphate. The ethylcarbylamide was an ‘uncoupler’ of oxidative phosphorylation.
Warburg understood that when normal cells switched to aerobic glycolysis
- it is a re-orientation of normal cell expression.
- this provides the basis for the inference that neoplastic cells become more like each other than their cell of origin.
- embryonic cells can be transformed into cancer cells under hypoxic conditions
- re-exposure to higher oxygen did not cause reversion back to normal cells.
Warburg publically expressed the rejected view in 1954 (at age 83) that restriction of chemical wastes, food additives, and air pollution would substantially reduce cancer rates.
His emphasis on the impairment of respiration was inadequate.
- the prevailing view today is loss of controlled growth of normal cells in cancer cells.
Otto Warburg: Cell Physiologist, Biochemist, and Eccentric. Hans Krebs, in collaboration with Roswitha Schmid. Clarendon Press, Oxford. 1991.ISBN 0-19-858171-8.
A multiphoton fluorescence microscope (MFM) is a specialized optical microscope.
The MFM uses pulsed long-wavelength light to excite fluorophores within the specimen being observed. The fluorophore absorbs the energy from two long-wavelength photons which must arrive simultaneously in order to excite an electron into a higher energy state, from which it can decay, emitting a fluorescence signal. It differs from traditional fluorescence microscopy in which the excitation wavelength is shorter than the emission wavelength, as the summed energies of two long-wavelength exciting photons will produce an emission wavelength shorter than the excitation wavelength.[1]
Multiphoton fluorescence microscopy has similarities to confocal laser scanning microscopy. Both use focused laser beams scanned in a raster pattern to generate images, and both have an optical sectioning effect. Unlike confocal microscopes, multiphoton microscopes do not contain pinhole apertures, which give confocal microscopes their optical sectioning quality. The optical sectioning produced by multiphoton microscopes is a result of the point spread function formed where the pulsed laser beams coincide. The multiphoton point spread function is typically dumbbell-shaped (longer in the x-y plane), compared to the upright rugby-ball shaped point spread function of confocal microscopes. However, in many interesting cases the shape of the spot and its size can be designed to realize specific desired goals.[2]
The longer wavelength, low energy (typically infra-red) excitation lasers of multiphoton microscopes are well-suited to use in imaging live cells as they cause less damage than short-wavelength lasers, so cells may be observed for longer periods with fewer toxic effects. Many researchers are currently working toward better and higher resolution multiphoton imaging developments.
Two-photon excitation microscopy is a fluorescence imaging technique that allows imaging of living tissue up to a very high depth, up to about one millimeter. Being a special variant of the multiphoton fluorescence microscope, it uses red-shifted excitation light which can also excite fluorescent dyes. However, for each excitation, two photons of infrared light are absorbed. Using infrared light minimizes scattering in the tissue. Due to the multiphoton absorption, the background signal is strongly suppressed. Both effects lead to an increased penetration depth for these microscopes. Two-photon excitation can be a superior alternative to confocal microscopy due to its deeper tissue penetration, efficient light detection, and reduced phototoxicity.[1]
Two-photon excitation employs two-photon absorption, a concept first described by Maria Goeppert-Mayer (1906–1972) in her doctoral dissertation in 1931,[2] and first observed in 1961 in a CaF2:Eu2+ crystal using laser excitation by Wolfgang Kaiser.[3] Isaac Abella showed in 1962 in cesium vapor that two-photon excitation of single atoms is possible.[4]
The concept of two-photon excitation is based on the idea that two photons of comparably lower energy than needed for one photon excitation can also excite a fluorophore in one quantum event. Each photon carries approximately half the energy necessary to excite the molecule. An excitation results in the subsequent emission of a fluorescence photon, typically at a higher energy than either of the two excitatory photons. The probability of the near-simultaneous absorption of two photons is extremely low. Therefore a high flux of excitation photons is typically required, usually from a femtosecond laser. The purpose of employing the two-photon effect is that the axial spread of the point-spread-function is substantially lower than for single-photon excitation. As a result, the resolution along the z dimension is improved, allowing for thin optical sections to be cut. In addition, in many interesting cases the shape of the spot and its size can be designed to realize specific desired goals.[5] Two-photon microscopes are less damaging to the sample than a single-photon confocal microscope.
The most commonly used fluorophores have excitation spectra in the 400–500 nm range, whereas the laser used to excite the two-photon fluorescence lies in the ~700–1000 nm (infrared) range. If the fluorophore absorbs two infrared photons simultaneously, it will absorb enough energy to be raised into the excited state. The fluorophore will then emit a single photon with a wavelength that depends on the type of fluorophore used (typically in the visible spectrum). Because two photons are absorbed during the excitation of the fluorophore, the probability for fluorescent emission from the fluorophores increases quadratically with the excitation intensity. Therefore, much more two-photon fluorescence is generated where the laser beam is tightly focused than where it is more diffuse. Effectively, excitation is restricted to the tiny focal volume (~1 femtoliter), resulting in a high degree of rejection of out-of-focus objects. This localization of excitation is the key advantage compared to single-photon excitation microscopes, which need to employ additional elements such as pinholes to reject out-of-focus fluorescence. The fluorescence from the sample is then collected by a high-sensitivity detector, such as a photomultiplier tube. This observed light intensity becomes one pixel in the eventual image; the focal point is scanned throughout a desired region of the sample to form all the pixels of the image. The scan head is typically composed of two mirrors, the angles of which can be rapidly altered with a galvanometer.
.Multiphoton microscopy: a potential “optical biopsy” tool for real-time evaluation of lung tumors without the need for exogenous contrast agents.
Jain M1, Narula N, Aggarwal A, Stiles B, Shevchuk MM, Sterling J, Salamoon B, Chandel V, Webb WW, Altorki NK, Mukherjee S.
From the Departments of Urology (Dr Jain), Pathology and Laboratory Medicine (Drs Narula and Shevchuk), Biochemistry (Drs Aggarwal and Mukherjee, Mr Sterling, and Mr Salamoon), Thoracic Surgery (Drs Stiles and Altorki), and Surgery (Mr Chandel), Weill Cornell Medical College, New York, New York; and the School of Applied and Engineering Physics, Cornell University, Ithaca, New York (Dr Webb). Dr Aggarwal is now with the Department of Science, Borough of Manhattan Community College, New York.
Arch Pathol Lab Med. 2014 Aug;138(8):1037-47. http://dx.doi.org:/10.5858/arpa.2013-0122-OA. Epub 2013 Nov 7
Context.-Multiphoton microscopy (MPM) is an emerging, nonlinear, optical-biopsy technique, which can generate subcellular-resolution images from unprocessed and unstained tissue in real time.
Objective.-To assess the potential of MPM for lung tumor diagnosis. Design.-Fresh sections from tumor and adjacent nonneoplastic lung were imaged with MPM and then compared with corresponding hematoxylin-eosin slides.
Results.-Alveoli, bronchi, blood vessels, pleura, smokers’ macrophages, and lymphocytes were readily identified with MPM in nonneoplastic tissue. Atypical adenomatous hyperplasia (a preinvasive lesion) was identified in tissue adjacent to the tumor in one case. Of the 25 tumor specimens used for blinded pathologic diagnosis, 23 were diagnosable with MPM. Of these 23 cases, all but one adenocarcinoma (15 of 16; 94%) was correctly diagnosed on MPM, along with their histologic patterns. For squamous cell carcinoma, 4 of 7 specimens (57%) were correctly diagnosed. For the remaining 3 squamous cell carcinoma specimens, the solid pattern was correctly diagnosed in 2 additional cases (29%), but it was not possible to distinguish the squamous cell carcinoma from adenocarcinoma. The other squamous cell carcinoma specimen (1 of 7; 14%) was misdiagnosed as adenocarcinoma because of pseudogland formation. Invasive adenocarcinomas with acinar and solid pattern showed statistically significant increases in collagen. Interobserver agreement for collagen quantification (among 3 observers) was 80%.
Conclusions.-Our pilot study provides a proof of principle that MPM can differentiate neoplastic from nonneoplastic lung tissue and identify tumor subtypes. If confirmed in a future, larger study, we foresee real-time intraoperative applications of MPM, using miniaturized instruments for directing lung biopsies, assessing their adequacy for subsequent histopathologic analysis or banking, and evaluating surgical margins in limited lung resections. PMID: 24199831
Lung cancer is the most common cause of cancer-related mortality worldwide in both men and women, with 226 160 new cases and 160 340 deaths estimated in the United States alone in 2012.1 Lung tumors are currently detected on chest radiography and computed tomography imaging, but definitive diagnosis, especially distinguishing the various subtypes of lung cancer, requires cytologic or histopathologic examination. Although considered the gold standard in establishing diagnosis, histopathology requires time-consuming tissue processing and can sometimes require repeat biopsies if the initial specimen was nondiagnostic. To overcome some of the obstacles associated with histopathologic processing, efforts have been made to develop high-resolution “optical biopsy” imaging techniques.
In this proof-of-principle pilot study, we explored the use of multiphoton microscopy (MPM) as a promising, new optical biopsy tool for the detection and diagnosis of lung tumors in real time.
Multiphoton microscopy relies on the simultaneous absorption of 2 or 3 low-energy (near-infrared) photons to cause a nonlinear excitation, equivalent to that created by a single photon of shorter wavelength light. By using 2-photon excitation in the 700- to 800-nm range, MPM enables both in vivo and ex vivo imaging of fresh, unprocessed, and unstained tissue at histologic resolution via generating intrinsic tissue emissions. Intrinsic tissue emission signals used in this study included autofluorescence and second harmonic generation (SHG).2–4
Twenty-five adult subjects diagnosed with lung cancer and undergoing lobectomies at our institution participated in this Institutional Review Board–approved study.
An Olympus FluoView FV1000MPE imaging system (Olympus America, Center Valley, Pennsylvania) was used for all MPM imaging. For detailed description of MPM imaging conditions, please see Supplementary Methods (supplemental digital content for this article is available at www.archivesofpathology.org in the August 2014 table of contents). Briefly, fresh (unprocessed and unstained) specimens were excited using 780 nm light from a tunable titanium-sapphire laser (Mai Tai DeepSee, Spectra-Physics, Irvine, California). Three distinct intrinsic tissue-emission signals were collected using photomultiplier tubes and were then color coded by using MetaMorph (version 7.0, revision 4, Molecular Devices, Sunnyvale, California) as follows: (1) SHG (360–400 nm, color-coded red), a nonlinear scattering signal originating from tissue collagen; (2) short wavelength autofluorescence (420–490 nm, color-coded green), originating in part from reduced nicotinamide adenine dinucleotide and flavin adenine dinucleotide in normal epithelial, neoplastic, and inflammatory cells, and from elastin in the alveolar septa; and (3) long wavelength autofluorescence (550–650 nm, color-coded blue), originating in part from carbon-laden macrophages.

Arch Path MPM
http://www.archivesofpathology.org/na101/home/literatum/publisher/pinnacle/journals/content/arpa/2014/15432165-138.8/arpa.2013-0122-oa/20140721/images/medium/i1543-2165-138-8-1037-f01.gif
http://www.archivesofpathology.org/na101/home/literatum/publisher/pinnacle/journals/content/arpa/2014/15432165-138.8/arpa.2013-0122-oa/20140721/images/large/i1543-2165-138-8-1037-f01.jpeg
Figure 1. Comparative multiphoton microscopy (MPM) and hematoxylin-eosin images of nonneoplastic and smoker lung. A and B, Low-magnification images show lung parenchyma composed of alveoli (arrows) surrounded by pleura (arrowheads). Inset in MPM shows pleura with collagen (red) and elastin (green) components. C and D, High-magnification images show primarily elastin fibers, with some collagen in the septal wall (arrowheads) of the alveoli (arrows). E and F, Low-magnification images show bronchus (*) with cartilage (arrowheads) and a medium-sized blood vessel (arrows). G and H, High-magnification images show columnar lining of the bronchus (arrows) and underlying connective tissue (arrowheads). I and J, Alveoli filled with carbon-laden macrophages (arrows; blue in MPM) and noncarbon-laden macrophages (arrowheads; green in MPM). K and L, A collection of small lymphocytes (arrowheads and inset), along with smoker’s macrophages (arrows). Some loss and thickening of the alveolar septa is shown (I through L) (MPM, original magnifications ×48 [A and E], ×96 [A inset], ×300 [C, G, I, and K], ×600 [K inset]; hematoxylin-eosin, original magnifications ×40 [B, F] and ×200 [D, H, J, and L]).
To assess the diagnostic potential of MPM, a blinded analysis was conducted. The attending pulmonary pathologist and an attending general surgical pathologist first familiarized themselves to the histologic features seen on MPM images in both nonneoplastic and neoplastic (adenocarcinoma and squamous cell carcinoma [SCC]) lung tissue. Because, in our study, multiple images were acquired from different areas of a given tumor, we used some of those images for the training set and did not include them in the blinded test set. Subsequently, test MPM images from all lung tumor specimens were assessed in a blinded fashion and were categorized according to subtype and pattern using the routine histopathologic criteria.9,10 These diagnoses were then correlated with diagnoses made by the same pathologist, based on the corresponding H&E sections prepared from the same specimens.
Visualizing Lung From Smoker With MPM
Using MPM to Identify Invasive and Preinvasive Adenocarcinoma
Figure 2, A through B, shows an example of a lesion with atypical adenomatous hyperplasia, which was an incidental finding on MPM in “tumor-free” lung tissue. It shows the proliferation of atypical pneumocytes, along the preexisting alveolar wall. Gaps between the cells (discontinuous layer of pneumocytes) support the diagnosis of atypical adenomatous hyperplasia. Adenocarcinoma of lung with lepidic-predominant pattern, in contrast, shows continuous proliferation of tumor cells along the alveolar wall.

Arch Path progression from atypical lesion to invasive adenocarcinoma
Figure 2. Comparative multiphoton microscopy and hematoxylin-eosin images showing progression from atypical lesion to various patterns of invasive adenocarcinoma of lung. A and B, Images of atypical adenomatous hyperplasia shows a focus of pneumocyte proliferation (cuboidal cells with gaps between them) along the alveolar wall (arrows and insets). C and D, Images of adenocarcinoma of lung with lepidic-predominant pattern (arrows) and a few clusters of free-floating tumor cells (arrowheads). E and F, Images of adenocarcinoma of lung with acinar-predominant pattern (arrows). G and H, Images showing solid pattern (arrows) with suggestion of gland formation (arrowheads). I and J, Images showing papillary pattern (papillae with fibrovascular core; arrows). K and L, Images showing micropapillary pattern, with complete destruction of normal lung parenchyma. The airspace shows small papillary clusters of tumor cells (arrows) lacking true fibrovascular cores (multiphoton microscopy, original magnifications ×300 [A, C, E, G, I, and K] and ×600 [inset]; hematoxylin-eosin, originalmagnifications ×200 [B, D, F, H, J, and L] ×400 [inset]).

Arch Path SCC
http://www.archivesofpathology.org/na101/home/literatum/publisher/pinnacle/journals/content/arpa/2014/15432165-138.8/arpa.2013-0122-oa/20140721/images/medium/i1543-2165-138-8-1037-f02.gif
http://www.archivesofpathology.org/na101/home/literatum/publisher/pinnacle/journals/content/arpa/2014/15432165-138.8/arpa.2013-0122-oa/20140721/images/large/i1543-2165-138-8-1037-f02.jpeg
Adenocarcinoma with a papillary-predominant pattern shows clear papillary projections composed of cuboidal to columnar cells, which line collagen-rich fibrovascular cores (Figure 2, I and J). Micropapillary adenocarcinoma, on the other hand, shows small papillary clusters of malignant cells within the airspace, with no true fibrovascular cores (Figure 2, K and L).
Using MPM to Identify SCC of Lung
Figure 3 shows high-magnification images acquired from the tumor mass in subjects with a diagnosis of SCC. Figure 3, A and B, shows sheets of malignant cells with a complete loss of the normal architecture of the lung parenchyma. These cells are seen as arranged in a pavementlike fashion with a high nuclear to cytoplasmic ratio (indicating a high-grade tumor). Pavementlike arrangement of cells is a characteristic of SCC that helps to differentiate it from the solid-predominant pattern of adenocarcinoma. We also observed an increased amount of stroma and mononuclear inflammatory cells surrounding the tumor (Figure 3, A, inset). These mononuclear inflammatory cells were confirmed as lymphocytes on H&E. This case was correctly diagnosed as SCC on MPM. Figure 3, C and D, shows images from the tumor mass of another subject, also diagnosed with SCC on H&E. However, it was misdiagnosed as adenocarcinoma on MPM, primarily because the central necrosis in the tumor nest was interpreted as gland formation. When the images were reanalyzed with knowledge of the SCC diagnosis, the pavementlike arrangement of cells was identified. We thus expect that, with a larger sample of SCC specimens and more experience, our ability to correctly diagnose SCC will improve significantly. Also, in the future, any specimen with a likely SCC diagnosis will be imaged over a larger area by using image tiling and by taking multiple stacks from various areas in the lesion, so as not to be misled by occasional, spurious, histologic features.
Figure 3. Comparative multiphoton microscopy and hematoxylin-eosin images of squamous cell carcinoma of the lung. A and B, Images of squamous cell carcinoma (SCC) of the lung showing sheets of malignant cells with high nuclear to cytoplasmic ratio (arrows), surrounded by lymphocytes (arrowheads) interspersed in collagen bundles (inset). C and D, Images of SCC of the lung showing pavementlike arrangement of the cells (arrows). Also shown is a nest of squamous cells with focal necrosis (arrowheads) forming pseudoglands, leading to a misdiagnosis of adenocarcinoma (multiphoton microscopy, original magnifications ×300 [A and C] and ×600 [inset]; hematoxylin-eosin, original magnifications ×200 [B and D]).
http://www.archivesofpathology.org/na101/home/literatum/publisher/pinnacle/journals/content/arpa/2014/15432165-138.8/arpa.2013-0122-oa/20140721/images/medium/i1543-2165-138-8-1037-f03.gif
http://www.archivesofpathology.org/na101/home/literatum/publisher/pinnacle/journals/content/arpa/2014/15432165-138.8/arpa.2013-0122-oa/20140721/images/large/i1543-2165-138-8-1037-f03.jpeg
Using MPM to Assess the Degree of Collagen in Lung Carcinoma
Previous studies have reported the amount of collagen as a prognostic factor in small, peripheral lung adenocarcinomas5,6,8 and SCCs.7 In our study, we categorized adenocarcinomas into 3 groups:
(1) well-differentiated, adenocarcinomas with lepidic-predominant patterns;
(2) moderately differentiated, invasive adenocarcinomas with acinar-predominant patterns; and
(3) poorly differentiated, adenocarcinomas with solid-predominant patterns.
A well-preserved lung architecture (Figure 4, A through C), with slight alveolar septal thickening from collagen deposition, was seen in low-magnification images of a well-differentiated adenocarcinoma (with a lepidic-predominant pattern). In contrast, invasive adenocarcinomas with both acinar-predominant (Figure 4, D through F) and solid-predominant (Figure 4, G through I) patterns showed increases in collagen content.
Our proof-of-principle pilot study indicates the potential utility of MPM for differentiating nonneoplastic from neoplastic lung tissue in fresh, ex vivo specimens without the use of exogenous contrast agents. Furthermore, study pathologists successfully identified the histologic subtypes of tumor and recognized inflammatory cells, such as lymphocytes and smoker macrophages. We also performed collagen quantification in adenocarcinomas and demonstrated its correlation with the degree of differentiation.
Indeed, the diagnostic potential of MPM for differentiating malignant from benign/inflammatory lesions has been previously investigated in multiple organ systems in both animal models and human tissues.3,12–19 Specifically, normal and diseased lung have been investigated in both small animals and in ex vivo human tissue using MPM.20–26 However, most studies focused on extracellular matrix remodeling associated with lung pathologies.22,23,25,26 To date, few studies have explored the potential of MPM for differentiating benign lesions from neoplastic ones.
. Our study is the first, to our knowledge, to present not only a detailed histology of normal human lung tissue obtained with MPM but also to show the histologic features that can be used to identify a variety of inflammatory, preneoplastic, and neoplastic lesions, in accordance with World Health Organization10 and International Association for the Study of Lung Cancer9 criteria. Furthermore, we demonstrated the ability of a pulmonary pathologist and a general surgical pathologist to differentiate between lesion subtypes in a blinded fashion with high reliability
The Human Genome Project

J. Craig Venter (Photo credit: Wikipedia)
The Human Genome Project, driven by Francis Collins at NIH, and by Craig Venterat the Institute for Genome Research (TIGR) had parallel projects to map the human chromosome, completed in 2003. It originally aimed to map the nucleotides contained in a human haploid reference genome (more than three billion). TIGR was the first complete genomic sequencing of a free living organism, Haemophilus influenzae, in 1995. This used a shotgun sequencing technique pioneered earlier, but which had never been used for a whole bacterium.
Venter broke away from the HGP and started Celera in 1998 because of resistance to the shotgun sequency method, and his team completed the genome sequence in three years – seven years’ less time than the HGP timetable (using the gene of Dr. Venter). TIGR eventually sequenced and analyzed more than 50 microbial genomes. Its bioinformatics group developed
- pioneering software algorithms that were used to analyze these genomes,
- including the automatic gene finder GLIMMER and
- the sequence alignment program MUMmer.
In 2002, Venter created and personally funded the J. Craig Venter Institute (JCVI) Joint Technology Center (JTC), which specialized in high throughput sequencing. The JTC, in the top ranks of scientific institutions worldwide, sequenced nearly 100 million base pairs of DNA per day for its affiliated institutions (JCVI) .
He received his his Ph.D. degree in physiology and pharmacology from the University of California, San Diego in 1975 under biochemist Nathan O. Kaplan. A full professor at the State University of New York at Buffalo, he joined the National Institutes of Health in 1984. There he learned of a technique for rapidly identifying all of the mRNAs present in a cell and began to use it to identify human brain genes. The short cDNA sequence fragments discovered by this method are calledexpressed sequence tags (ESTs), a name coined by Anthony Kerlavage at TIGR.
Venter believed that shotgun sequencing was the fastest and most effective way to get useful human genome data. There was a belief that shotgun sequencing was less accurate than the clone-by-clone method chosen by the HGP, but the technique became widely accepted by the scientific community and is still the de facto standard used today.
References
Shreeve, James (2004). The Genome War: How Craig Venter Tried to Capture the Code of Life and Save the World. Knopf. ISBN 0375406298.
Sulston, John (2002). The Common Thread: A Story of Science, Politics, Ethics and the Human Genome. Joseph Henry Press. ISBN 0309084091.
“The Human Genome Project Race”. Center for Biomolecular Science & Engineering,UC Santa Cruz. Retrieved 20 March 2012.
Venter, J. Craig (2007). A Life Decoded: My Genome: My Life. Viking Adult. ISBN 0670063584.
Use of a Fluorophor Probe
An article has been discussed by Dr. Tilda Barilya on use of a sensitive fluorescent probe in the near IR spectrum at > 700 nm to identify malignant ovarian cells in-vivo in abdominal exploration
- by tagging an overexpressed FR-α (folate-FITA)
The author makes the point that:
- In ovarian cancer, the FR-α appears to constitute a good target because it is overexpressed in 90–95% of malignant tumors, especially serous carcinomas.
- Targeting ligand, folate, is attractive as it is nontoxic, inexpensive and relatively easily conjugated to a fluorescent dye to create a tumor-specific fluorescent contrast agent.
- The report is identified as “ the first in-human proof-of-principle of the use of intraoperative tumor-specific fluorescence imaging in staging and debulking surgery for ovarian cancer using the systemically administered targeted fluorescent agent folate-FITC.”
While this does invoke possibilities for prognosis, the decision to perform the surgery,
- whether laparoscopic or open, is late in the discovery process. However,
it does suggest the possibility that the discovery and the treatment might be combined
- if the biomarker itself had the fluorescence to identify the overexpression, but
- it also is combined with a tag to block the overexpession. This hypothetical possibility is now expressed below.
http://pharmaceuticalintelligence.com/2013/01/19/ovarian-cancer-and-fluorescence-guided-surgery-a-report/
Gene Editing

Aviva Lev-Ari, PhD, RD
Dr. Aviva Lev-Ari reports that a new technique developed at MIT Broad Institute and the Rockefeller University can edit DNA in precise locations
taken from Science News titled “Editing Genome With High Precision: New Method to Insert Multiple Genes in Specific Locations, Delete Defective Genes”.
Using this system, scientists can alter
- several genome sites simultaneously and
- can achieve much greater control over where new genes are inserted
According to Feng Zhang, this is an improvement beyond splicing the gene in specific locations and
insertion of complexes difficult to assemble known as
transcription activator-like effector nucleases (TALENs).
- The researchers create DNA-editing complexes
- using naturally occurring bacterial protein-RNA systems
- that recognize and snip viral DNA, including
- a nuclease called Cas9 bound to short RNA sequences.
- they target specific locations in the genome, and
- when they encounter a match, Cas9 cuts the DNA.
This approach can be used either to
- disrupt the function of a gene or
- to replace it with a new one.
- To replace the gene, a DNA template for the new gene has to be copied into the genome after the DNA is cut. The method is also very precise –
- if there is a single base-pair difference between the RNA targeting sequence and the genome sequence, Cas9 is not activated.
In its first iteration, it appears comparable in efficiency to what
zinc finger nucleases and TALENs have to offer.
The research team has deposited the necessary genetic components with a nonprofit called Addgene, and they have also created a website with tips and tools for using this new technique.
The above story is reprinted from materials provided by Massachusetts Institute of Technology.
The original article was written by Anne Trafton. Le Cong, F. Ann Ran, David Cox, Shuailiang Lin, Robert Barretto, Naomi Habib, Patrick D. Hsu, Xuebing Wu, Wenyan Jiang, Luciano Marraffini, and Feng Zhang.
Multiplex Genome Engineering Using CRISPR/Cas Systems.
Science, 3 January 2013 DOI: 10.1126/science.1231143.
http://Science.com. Editing genome with high precision: New method to insert multiple genes in specific locations, delete defective genes. ScienceDaily. Retrieved January 20, 2013, from http://www.sciencedaily.com /releases/2013/01/130103143205.htm? goback=%2Egde_4346921_member_205356312.
Dr. Lev-Ari also reports on a study of early detection of breast cancer in “Mechanism involved in Breast Cancer Cell Growth: Function in Early Detection & Treatment“, by Dr. Rotem Karni and PhD student Vered Ben Hur at the Institute for Medical Research Israel-Canada of the Hebrew University.
http://pharmaceuticalintelligence.com/2013/01/17/mechanism-involved-in-breast-cancer-cell-growth-function-in-early-detection-treatment/
These researchers have discovered a new mechanism by which breast cancer cells switch on their aggressive cancerous behavior. The discovery provides a valuable marker for the early diagnosis and follow-up treatment of malignant growths.
The method they use is
- RNA splicing and insertion.
- The information needed for the production of a mature protein is encoded in segments called exons .
- In the splicing process, the non-coding segments of the RNA (introns) are spliced from the pre-mRNA and
- the exons are joined together.
Alternative splicing is when a specific ”scene” (or exon) is either inserted or deleted from the movie (mRNA), thus changing its meaning.
- Over 90 percent of the genes in our genome undergo alternative splicing of one or more of their exons, and
- the resulting changes in the proteins encoded by these different mRNAs are required for normal function.
- the normal process of alternative splicing is altered in cancer, and
- ”bad” protein forms are generated that aid cancer cell proliferation and survival.
The researchers reported in online Cell Reports that breast cancer cells
- change the alternative splicing of an important enzyme, calledS6K1, which is
- a protein involved in the transmission of information into the cell.
- when this happens, breast cancer cells start to produce shorter versions of this enzyme and
- these shorter versions transmit signals ordering the cells to grow, proliferate, survive and invade other tissues (otherwise proliferation is suppressed)
The application to biotherapeutics would be to ”reverse” the alternative splicing of S6K1 in cancer cells back to the normal situation as a novel anti-cancer therapy.
Imaging Mass Cytometry
This literature review shows how researchers used CyTOF mass cytometry to obtain spatial resolution of cell samples.
The authors used mass cytometry to measure heterogeneity in breast cancer tumors using FFPE breast cancer samples. [© Kheng Guan Toh – Fotolia.com]
The integration of mass spectrometry (specifically laser ablation of the sample in combination with inductively coupled plasma mass spectrometry) with flow cytometry instrumentation along with sensitive and rare earth metal labels
- has enabled multiplexing of up to 32 cell markers (see Assay Drug Dev Technol 2011;9:567 commentary “Flow cytometry goes atomic;” CyTOF system sold by Fluidigm, formerly DVS Sciences).
This process employs typical immunocytochemistry techniques, but the antibodies are tagged with rare earth metal isotopes (predominately lanthanides)
- that act as specific reporters of cellular proteins.
Comparison of these rare earth–labeled antibodies to typical fluorescent antibody labels supports that
- these labels do not affect the specificity or sensitivity of the antibodies. In this article the authors* extend this method to obtain spatial resolution of cell samples.

mass cytometry to measure heterogeneity in breast cancer tumors using FFPE

is correlated with the position of the laser spot as it is scanned across the sample with 1 μm resolution.In the method, the signals from the rare earth reporters following laser ablation of the sample
The limit of detection is determined to be ∼500 molecules. The data can then be plotted based on the position of each ion spot for each rare earth reporter, and these images
- are then overlaid to create a high-dimensional image that can be analyzed (Figure).
Measurement of a 0.5 mm × 0.5 mm area at 1 μm resolution takes ∼5 h. The system is capable of measuring 100 analytes simultaneously, but only 32 rare earth metal chelates are currently available. The authors applied this method
- to measure heterogeneity in breast cancer tumors using formalin-fixed, paraffin-embedded (FFPE) breast cancer samples.
A total of 21 FFPE samples were analyzed using 32-plex imaging mass cytometry covering cell markers and phosphoproteins. Differences in expression even within the same tumor sample were noted, and
- the subpopulations branch points often contained markers used for patient classification. Some exceptions occurred; for example,
- Her2 was detected and confirmed in one triple-negative case.
This high-dimensional imaging should increase our understanding of tumor biology and pathologies.
*Abstract from Nature Methods 2014, Vol. 11: 403–406
Mass cytometry enables high-dimensional, single-cell analysis of cell type and state. In mass cytometry, rare earth metals are used as reporters on antibodies. Analysis of metal abundances using the mass cytometer
- allows determination of marker expression in individual cells. Mass cytometry has previously been applied only to cell suspensions.
To gain spatial information, we have coupled
- immunohistochemical and immunocytochemical methods
- with high-resolution laser ablation to CyTOF mass cytometry.
This approach enables the simultaneous imaging of 32 proteins and protein modifications at subcellular resolution;
- with the availability of additional isotopes, measurement of over 100 markers will be possible.
We applied imaging mass cytometry to human breast cancer samples, allowing delineation of cell subpopulations and cell–cell interactions and highlighting tumor heterogeneity. Imaging mass cytometry
- complements existing imaging approaches.
It will enable basic studies of tissue heterogeneity and function and
- support the transition of medicine toward individualized molecularly targeted diagnosis and therapies.
Preventing a cellular identity crisis
http://news.sciencemag.org/sites/default/files/styles/thumb_article_l/public/sn-criticalgenesH.jpg?itok=0wmD-o4a
Staying true.

neural progenitor cells keep their identities
Researchers have discovered a molecular signature in the genome that might help cells like these neural progenitor cells keep their identities throughout their lives.
By Mitch Leslie 31 July 2014
Cells rely on different ways to establish who they are and what they do. A novel mechanism
- marks the identities of different kinds of cells in the human body—
- and prevents them from transforming into another type altogether.
Scientists learned decades ago to read the basic genetic code by which cells convert a string of DNA bases into a protein’s amino acids. But for more than 10 years,
- they’ve been trying to crack what’s known as the histone code, a more complex cipher embedded within organisms’ genomes.
Histones are the proteins that DNA coils around in chromosomes. Chemically tweaking histones in a variety of ways can
- adjust the activity of genes, turning them up or down. For example,
- cells shut off genes by attaching three methyl groups to a specific spot on a histone type known as H3.
- affixing three methyl groups to another H3 location, a modification known as H3K4me3, has a different effect.
Cells typically add the H3K4me3 tags to histones in small sections of the genome, but researchers noticed that
- sometimes the tag can sprawl across much larger areas,
- modifying broad swaths of histones.
To find out whether these large blocks of histones carrying H3K4me3 tags convey a message in the histone code, molecular geneticist Anne Brunet of Stanford University in Palo Alto, California, and colleagues
- traced their occurrence in more than 20 different cell types.
They found that the longest stretches pinpoint different sets of genes in different types of cells. As a result, the researchers realized
- they could discriminate liver cells from, say, muscle cells or kidney cells
- based only on the chromosomal locations of the largest H3K4me3 blocks. In addition,
- they noticed that these stretches tended to mark genes that are crucial for a cell type’s function or
- that help make it distinct. In embryonic stem cells, for instance,
- they occur on genes that control the cells’ capacity to specialize.
The researchers further demonstrated that the labels mark cell identity genes by
- using a technique called RNA interference (RNAi) in adult neural progenitor cells,
- which can morph into any cell type in the brain.
As the researchers revealed online today in Cell,
- they applied RNAi to dial down the genes that carried large blocks of H3K4me3 tags and
- found that it impaired the cells’ ability to reproduce and to spawn neurons. However,
- the progenitor cells could still divide normally if the researchers quieted genes that had only short sections of H3K4me3 tags or none at all.
The presence of long stretches of H3K4me3 markers
- might help cells keep their identities for life.
- “we’ve discovered a new signature,” Brunet says.
Although many other scientists have studied H3K4me3 tags, “the concept that this one mark
- can distinguish all these cell types
- the discovery could allow quick identification of cell types,
- which would be useful in situations such as cancer diagnosis.
Posted in Biology
Gene Deletion Slows Aging and Reduces Cancer Risk

gene deletion
Source: © Gernot Krautberger – Fotolia.com
Scientists at the Wistar Institute say they have discovered that
- mice lacking a specific protein live longer lives with fewer age-related illnesses.
The mice that lack the TRAP-1 protein, demonstrated less age-related tissue degeneration, obesity, and spontaneous tumor formation when compared to normal mice. Their findings could change how scientists view the metabolic networks within cells. In healthy cells,
TRAP-1 is an important regulator of metabolism and
- regulates energy production in mitochondria, organelles that generate chemically useful energy for the cell.
In the mitochondria of cancer cells, TRAP-1 is universally overproduced.
The Wistar team’s report (“Deletion of the Mitochondrial Chaperone TRAP-1 Uncovers Global Reprogramming of Metabolic Networks”), which appears in Cell Reports, shows how ”
- knockout” mice bred to lack the TRAP-1 protein compensate for this loss by switching to alternative cellular mechanisms for making energy.
“We see this astounding change in TRAP-1 knockout mice, where they show fewer signs of aging and are less likely to develop cancers,” said Dario C. Altieri. M.D., director of the Wistar Institute’s National Cancer Institute-designated Cancer Center. “Our findings provide
- an unexpected explanation for how TRAP-1 and related proteins regulate metabolism within our cells.
- we didn’t expect to see healthier mice with fewer tumors.
Dr. Altieri and his colleagues created the TRAP-1 knockout mice as part of their
- ongoing investigation into the drug Gamitrinib, which targets the protein in the mitochondria of tumor cells.
TRAP-1 is a member of the heat shock 90 (HSP90) protein family, which are
- chaperone proteins that guide the physical formation of other proteins and
- serve a regulatory function within mitochondria.
- Tumors use HSP90 proteins like TRAP-1 to help survive therapeutic attack.
In tumors,
- the loss of TRAP-1 is devastating, triggering a host of catastrophic defects, including
- metabolic problems that ultimately result in in death of the tumor cells,, BUT
- Mice that lack TRAP-1 from the start have three weeks in the womb to compensate for the loss of the protein.”
In the knockout mice, the loss of TRAP-1
- causes mitochondrial proteins to misfold, which then
- triggers a compensatory response that causes cells to consume more oxygen and metabolize more sugar. which
- causes mitochondria in knockout mice to produce deregulated levels of ATP,
- the chemical used as an energy source to power all the everyday molecular reactions that allow a cell to function.
This increased mitochondrial activity actually creates a moderate boost in oxidative stress (free radical damage) and the associated DNA damage. While DNA damage may seem counterproductive to longevity and good health,
- the low level of DNA damage actually reduces cell proliferation—slowing growth down to allow the cell’s natural repair mechanisms to take effect.
“TRAP-1−/− mice are viable and showed reduced incidence of age-associated pathologies including – obesity, inflammatory tissue degeneration, dysplasia, and spontaneous tumor formation,- accompanied by
- global upregulation of oxidative phosphorylation and glycolysis transcriptomes, causing
- deregulated mitochondrial respiration,
- oxidative stress,
- impaired cell proliferation, and
- a switch to glycolytic metabolism in vivo.
These data identify TRAP-1 as
- a central regulator of mitochondrial bioenergetics, and
- this pathway could contribute to metabolic rewiring in tumors.”
“Our findings strengthen the case
- for targeting HSP90 in tumor cells, but it
- may have implications for metabolism and longevity,” explained Dr. Altieri.
GEN News 8-1-2014
The role of the Wnt signaling pathway in cancer stem cells: prospects for drug development
Yong-Mi Kim, Michael Kahn
1Children’s Hospital Los Angeles, Division of Hematology and Oncology, Department of Pediatrics and Pathology, 2Department of Biochemistry and Molecular Biology, Keck School of Medicine of University of Southern California, 3Norris Comprehensive Cancer Research Center, University of Southern California, Los Angeles, CA, USA
Research and Reports in Biochemistry July 2014; 4:1—12
http://dx.doi.org/10.2147/RRBC.S53823
Abstract: Cancer stem cells (CSCs), also known as tumor initiating cells are now considered to be
- the root cause of most if not all cancers, evading treatment and giving rise to disease relapse.
They have become a central focus in new drug development.
- Prospective identification,
- understanding the key pathways that maintain CSCs, and
- being able to target CSCs, particularly
- if the normal stem cell population could be spared, could offer an incredible therapeutic advantage.
The Wnt signaling cascade is critically important in stem cell biology, both
- in homeostatic maintenance of tissues and organs through their respective somatic stem cells and
- in the CSC/tumor initiating cell population.
Aberrant Wnt signaling is associated with a wide array of tumor types. Therefore, the ability to
- safely target the Wnt signaling pathway offers enormous promise to target CSCs. However,
- just like the sword of Damocles, significant risks and concerns regarding targeting such a critical pathway in normal stem cell maintenance and tissue homeostasis remain ever present.
With this in mind, we review recent efforts in modulating the Wnt signaling cascade and critically analyze therapeutic approaches at various stages of development.
Keywords: beta-catenin, CBP, p300, wnt inhibition
A*STAR Scientists Pinpoint Genetic Changes that Spell Cancer: Fruit flies light the way for scientists to uncover genetic changes.
With a new approach, researchers may rapidly distinguish the range of
- genetic changes that are causally linked to cancer (i.e.“driver” mutations)
- versus those with limited impact on cancer progression.
This study published in the prestigious journal Genes & Development could pave the way
- to design more targeted treatment against different cancer types, based on
- the specific cancer-linked mutations present in the patient,
- an advance in the development of personalized medicine.
Signaling pathways involved in tumour formation are conserved from fruit flies to humans. In fact, about 75 percent of known human disease genes have a recognizable match in the genome of fruit flies.
Leveraging on their genetic similarities, Dr Hector Herranz, a post-doctorate from the Dr. Stephen Cohen’s team developed an innovative strategy to genetically screen the whole fly genome for “cooperating” cancer genes.
- These genes appear to have little or no impact on cancer.
- However, they cooperate with other cancer genes, so that
- the combination causes aggressive cancer, which
- neither would cause alone.
In this study, the team was specifically looking for genes that
- could cooperate withEGFR “driver” mutation,
- a genetic change commonly associated with breast and lung cancers in humans.
- SOCS5 (reported in this paper) is one of the several new “cooperating” cancer genes to be identified.
Already, there are indications that levels of SOCS5 expression are
- reduced in breast cancer, and
- patients with low levels of SOCS5 have poor prognosis.”
The IMCB team is preparing to explore the use of SOCS5 as a biomarker in diagnosis for cancer.
Probing What Fuels Cancer
http://genes&development.com
‘Altered cellular metabolism is a hallmark of cancer,’ says Dr Patrick Pollard, in the Nuffield Department of Clinical Medicine at Oxford.
Most cancer cells get the energy they need predominantly through
- a high rate of glycolysis – allowing cancer cells deal with the low oxygen levels that tend to be present in a tumour. But
- whether dysfunctional metabolism causes cancer, as Warburg believed, or is something that happens afterwards is a different question.
- In the meantime, gene studies rapidly progressed and indicated that genetic changes occur in cancer.
DNA mutations spring up all the time in the body’s cells, but
- most are quickly repaired.
- Alternatively the cell might shut down or be killed off (apoptosis) before any damage is caused. However, the repair machinery is not perfect.
- If changes occur that bypass parts of the repair machinery or sabotage it,
- the cell can escape the body’s normal controls on growth and
- DNA changes can begin to accumulate as the cell becomes cancerous.
Patrick believes certain changes in cells can’t always be accounted for by ‘genetics.’
He is now collaborating with Professor Tomoyoshi Soga’s large lab at Keio University in Japan, which has been at the forefront of developing the technology for metabolomics research over the past couple of decades.
The Japanese lab’s ability to
- screen samples for thousands of compounds and metabolites at once, and
- the access to tumour material and cell and animal models of disease
- enables them to probe the metabolic changes that occur in cancer.
There is reason to believe that
- dysfunctional cell metabolism is important in cancer.
- genes with metabolic functions are associated with some cancers
- changes in the function of a metabolic enzyme have been implicated in the development of gliomas.
These results have led to the idea that
- some metabolic compounds, or metabolites, when they accumulate in cells, can cause changes to metabolic processes and set cells off on a path towards cancer.
Patrick Pollard and colleagues have now published a perspective article in the journal Frontiers in Molecular and Cellular Oncology that proposes
- fumarate as such an ‘oncometabolite’. Fumarate is a standard compound involved in cellular metabolism.
The researchers summarize evidence that shows how
- accumulation of fumarate when an enzyme goes wrong affects various biological pathways in the cell.
- It shifts the balance of metabolic processes and disrupts the cell in ways that could favour development of cancer.
Patrick and colleagues write in their latest article that the shift in focus of cancer research to include cancer cell metabolism ‘has highlighted how woefully ignorant we are about the complexities and interrelationships of cellular metabolic pathways’.
NATURE GENETICS | BRIEF COMMUNICATION
Recurrent SMARCA4 mutations in small cell carcinoma of the ovary
Nature Genetics (2014); 46: 424–426 http://dx.doi.org:/10.1038/ng.2922
Small cell carcinoma of the ovary, hypercalcemic type (SCCOHT) is a rare, highly aggressive form of ovarian cancer primarily diagnosed in young women. We identified
- inactivating biallelic SMARCA4 mutations in 100% of the 12 SCCOHT tumors examined.
Protein studies confirmed loss of SMARCA4 expression, suggesting a key role for the SWI/SNF chromatin-remodeling complex in SCCOHT.
At a glance
Figures
Figure 1: SMARCA4 mutations in SCCOHT and TCGA samples.close

SMARCA4 mutations in SCCOHT and TCGA samples.close
(a) Domain structure of the SMARCA4 protein (UniProt, SMCA4_HUMAN) overlaid with the alterations identified in 11 of the 12 SCCOHT cases in this study (case numbers in parentheses; case 103 with exon deletion is not shown). SNF2_N, SNF…
http://www.nature.com/ng/journal/v46/n5/carousel/ng.2922-F1.jpg
Figure 2: Analyses of the splice-site mutation in case 102.close

Analyses of the splice-site mutation in case 102.close
(a) Immunoblotting with antibody to the N terminus of SMARCA4. A high-grade serous ovarian cancer cell line (PEO4) and frozen tumor samples from two individuals with high-grade serous ovarian cancer (HGOC) were used as positive control…
http://www.nature.com/ng/journal/v46/n5/carousel/ng.2922-F2.jpg
References
- Estel, R., Hackethal, A., Kalder, M. & Munstedt, K. Gynecol. Obstet.284, 1277–1282(2011).
- Young, R.H., Oliva, E. & Scully, R.E. J. Surg. Pathol.18, 1102–1116 (1994).
- Harrison, M.L. et al. Oncol.100, 233–238 (2006).
- Seidman, J.D. Oncol.59, 283–287 (1995).
- Pautier, P. et al. Oncol.18, 1985–1989 (2007).
- McCluggage, W.G. Anat. Pathol.11, 288–296 (2004).
- Hendricks, K.B., Shanahan, F. & Lees, E. Cell. Biol.24, 362–376 (2004).
- Napolitano, M.A. et al. Cell Sci.120, 2904–2911 (2007).
- Kupryjańczyk, J. et al. J. Pathol.64, 238–246 (2013).
- Longy, M., Toulouse, C., Mage, P., Chauvergne, J. & Trojani, M. Med. Genet.33, 333–335(1996).
- McDonald, J.M. et al. Pediatr. Surg.47, 588–592 (2012).
- Reisman, D., Glaros, S. & Thompson, E.A. Oncogene28, 1653–1668 (2009).
- Wilson, B.G. & Roberts, C.W. Rev. Cancer11, 481–492 (2011).
- Oike, T. et al. Cancer Res.73, 5508–5518 (2013).
- Guan, B., Wang, T.L. & Shih, I.-M. Cancer Res.71, 6718–6727 (2011).
- Won, H.H., Scott, S.N., Brannon, A.R., Shah, R.H. & Berger, M.F. Vis. Exp.80, e50710(2013).
- Wagle, N. et al. Cancer Discov.2, 82–93 (2012).
- Li, H. & Durbin, R. Bioinformatics25, 1754–1760 (2009).
- DePristo, M.A. et al. Genet.43, 491–498 (2011).
- Cibulskis, K. et al. Biotechnol.31, 213–219 (2013).
- Robinson, J.T. et al. Biotechnol.29, 24–26 (2011).
- Cerami, E. et al. Cancer Discov.2, 401–404 (2012).
- Gao, J. et al. Signal.6, pl1 (2013).
Download references
Primary authors
These authors contributed equally to this work.
Petar Jelinic & Jennifer J Mueller, Department of Surgery
Petar Jelinic, Jennifer J Mueller, Narciso Olvera, Fanny Dao & Douglas A Levine
Department of Surgery
Sasinya N Scott, Ronak Shah, Robert A Soslow & Michael F Berger
Department of Pathology,
JianJiong Gao & Nikolaus Schultz
Computational Biology Program,
Mithat Gonen Department of Epidemiology and Biostatistics, Memorial Sloan-Kettering Cancer Center, New York, New York, USA.
Corresponding author
Douglas A Levine
Supplementary information
Supplementary Figures
- Supplementary Figure 1: Sequence analyses forSMARCA4 in SCCOHT cases. (715 KB)
Next-generation sequence coverage demonstrating identified variants (top panels) and validation through Sanger sequencing (bottom panels).
- Supplementary Figure 2:SMARCA4 gene expression across TCGA tumors for cases with available mutation and RNA-seq data (RSEM). (366 KB)
A correlation is seen between inactivating SMARCA4 mutations and decreased gene expression across various solid tumors. A two-sided Student’s t test was used to compare samples with non-missense mutations and other samples without mutations or with only missense mutations. For all TCGA samples, the mean RNA-seq RSEM (2,050, s.d. of 1,760) was less in samples with non-missense mutations than in other samples without mutations or with only missense mutations (3,724, s.d. of 1,692; P = 8.7 × 10−4). For TCGA lung adenocarcinoma samples, the mean RNA-seq RSEM (601, s.d. of 370) was less in samples with non-missense mutations than in other samples without mutations or with only missense mutations (3,330, s.d. of 1,524; P = 2 × 10−8).
- Supplementary Figure 3: Immunohistochemistry for SMARCA4 in SCCOHT cases. (566 KB)
High-grade serous ovarian carcinoma is used as a positive control. Case numbers are indicated in each panel. Immunohistochemistry results are provided in Supplementary Table 1. Note the intense staining of blood vessels and stromal cell nuclei as internal controls.
- Supplementary Figure 4: Analysis of homozygous deletion in case 103. (109 KB)
Next-generation sequence coverage demonstrating that exons 25 and 26 are deleted. An electropherogram from Sanger sequencing of cDNA validating that the deletion retains an ORF from exon 24 to exon 27 (Panel A). One-step RT-PCR confirms that tumor tissue yields a single band with primers that span exons 24 and 27 (Panel B; *, nonspecific band). One-step RT-PCR with primers targeting regions upstream and downstream from the deletion site show equal expression, demonstrating continuation of transcription downstream from the deletion (Panel C).
- Supplementary Figure 5: Analysis of splice-site variant in case 102. (90 KB)
One-step RT-PCR confirms that the exon-intron band is preferentially expressed over the exon-exon band in tumor tissue (Panel A). One-step RT-PCR with primers targeting regions upstream and downstream from the mutation site show equal expression, demonstrating continuation of transcription downstream from the mutation (Panel B). Immunoblots are shown in Figure 2b. The exon-exon primers detected weaker bands, reflecting loss of expression in tumor tissues compared with normal tissues in cases with splice-site mutations. The exon-intron primers demonstrated equivalent to greater expression of the retained intron in the tumor tissues. As SMARCA4 introns may be retained in non-cancer tissues, some intronic expression is expected in normal tissues. These data taken together indicate preferential intronic expression, as expected, in cDNA sequenced from tumor samples with splice-site mutations.
- Supplementary Figure 6: Sequence analyses forSMARCA4 in the H1299 cell line. (134 KB)
An electropherogram from Sanger sequencing of genomic DNA validating a 69-nt deletion in the ORF of this control cell line that results in loss of protein expression, as shown inFigure 2b.
- Supplementary Figure 7: SMARCA4 effect on cell proliferation. (131 KB)
SMARCA4 overexpression in H1299 cells. Representative immunoblot from three biologic replicates demonstrates a correlation between increased SMARCA4 and p21 expression (Panel A). Cell growth assessment in H1299 cells overexpressing SMARCA4. Mean cell counts from three biologic replicates (Panel B). Representative immunoblot confirmed SMARCA4 knockdown in 293T cells using shRNA. As a control, shNTC (Non-Targeting Control) was used (Panel C). XTT proliferation assay in 293T cells depleted of SMARCA4. Means represent three independent experiments (Panel D).
- Supplementary Figure 8: Overall survival among lung adenocarcinoma TCGA cases based on inactivatingSMARCA4 (174 KB)
Median overall survival was 11.6 months among 6 patients with inactivating SMARCA4mutations compared with 44.6 months for 197 patients without inactivating mutations.
- Supplementary Figure 9: Histopathological features of an SCCOHT. (979 KB)
The typical histopathological features of SCCOHT, including a combination of small neoplastic cells forming a pseudofollicular space and larger rhabdoid cells, are visible in a sample obtained from 1 of 12 tumors that were subjected to target capture and massively parallel DNA sequencing (hematoxylin and eosin).
PDF files
- Supplementary Text and Figures (5,357 KB)
Supplementary Figures 1–9 and Supplementary Tables 1–5
CRISPR-Cas9 Foundational Technology originated at UC, Berkeley & UCSF, Broad Institute is developing Biotech Applications — Intellectual Property emerging as Legal Potential Dispute
Curator and Reporter: Aviva Lev-Ari, PhD, RN
http://pharmaceuticalintelligence.com/2014/06/18/crispr-cas9-foundational-technology-originated-at-uc-berkeley-ucsf-broad-institute-is-developing-biotech-applications-intellectual-property-emerging-as-legal-potential-dispute/
CRISPR-Cas9 Foundational Technology – The definition of “Prior Art” is at a very high stack, June 2014.
On 6/16/2014 Dr. Aviva Lev-Ari published the following two articles:
Lecture Contents delivered at Koch Institute for Integrative Cancer Research, Summer Symposium 2014: RNA Biology, Cancer and Therapeutic Implications, June 13, 2014 @MIT
http://pharmaceuticalintelligence.com/2014/06/16/lecture-contents-delivered-at-koch-institute-for-integrative-cancer-research-summer-symposium-2014-rna-biology-cancer-and-therapeutic-implications-june-13-2014-mit/
Prediction of the Winner RNA Technology, the FRONTIER of SCIENCE on RNA Biology, Cancer and Therapeutics & The Start Up Landscape in Boston
http://pharmaceuticalintelligence.com/2014/06/16/prediction-of-the-winner-rna-technology-the-frontier-of-science-on-rna-biology-cancer-and-therapeutics-the-start-up-landscape-in-boston/
Other related articles on CRISPR-Cas9 Technology published on this Open Access Online Scientific Journal include the following:
2:15 – 2:45, 6/13/2014, Jennifer Doudna “The biology of CRISPRs: from genome defense to genetic engineering”
http://pharmaceuticalintelligence.com/2014/06/13/215-245-6132014-jennifer-doudna-the-biology-of-crisprs-from-genome-defense-to-genetic-engineering/
Ribozymes and RNA Machines – Work of Jennifer A. Doudna
http://pharmaceuticalintelligence.com/2013/04/15/ribozymes-and-rna-machines-work-of-jennifer-a-doudna/
CRISPR @MIT – Genome Surgery
http://pharmaceuticalintelligence.com/2014/04/21/crispr-mit-genome-surgery/
Gene Therapy and the Genetic Study of Disease: @Berkeley and @UCSF – New DNA-editing technology spawns bold UC initiative as Crispr Goes Global
http://pharmaceuticalintelligence.com/2014/03/27/gene-therapy-and-the-genetic-study-of-disease-berkeley-and-ucsf-new-dna-editing-technology-spawns-bold-uc-initiative-as-crispr-goes-global/
Diagnosing Diseases & Gene Therapy: Precision Genome Editing and Cost-effective microRNA Profiling
http://pharmaceuticalintelligence.com/2013/03/28/diagnosing-diseases-gene-therapy-precision-genome-editing-and-cost-effective-microrna-profiling/
An expanded-DNA Biology from Scripps Research Institute: Beyond A-T and C-G: Applications for new Medicines and Nanotechnology
http://pharmaceuticalintelligence.com/2014/05/11/an-expanded-dna-biology-from-scripps-research-institute-beyond-a-t-and-c-g-applications-for-new-medicines-and-nanotechnology/
Evaluate your Cas9 Gene Editing Vectors: CRISPR/Cas Mediated Genome Engineering – Is your CRISPR gRNA optimized for your cell lines?
http://pharmaceuticalintelligence.com/2014/03/25/evaluate-your-cas9-gene-editing-vectors-crisprcas-mediated-genome-engineering-is-your-crispr-grna-optimized-for-your-cell-lines/
2:15 – 2:45, 6/13/2014, Jennifer Doudna “The biology of CRISPRs: from genome defense to genetic engineering”
http://pharmaceuticalintelligence.com/2014/06/13/215-245-6132014-jennifer-doudna-the-biology-of-crisprs-from-genome-defense-to-genetic-engineering/
About CRISPR “this technology will revolutionize biology in the same way PCR did,” Rudolf Jaenisch introducing Jennifer Doudna
Top CRISPR Related Publications
http://blog.appliedstemcell.com/top-crispr-related-publications/
Capturing key concepts of Prof. Jennifer Doudna’s Lecture @ KI Symposium:
- acquired immunity in bacteria
- three steps:
- adaptation
- biogenesis
- interference
Big Pharma is using its venture cash to outsource early R&D to biotech
July 31, 2014 | By John Carroll
Analysts at Silicon Valley Bank have been crunching the numbers on biotech investing, and they have found that
- a group of busy corporate venture arms has fundamentally changed the landscape for startups and
- the entire field of early-stage drug development–
- with some big implications for the current crop of industry upstarts.
Over the past two years corporate venture funding for biotech companies has surged back to 2008 levels, the bank’s analysts conclude, and
- it now adds up to a much larger portion of the total amount of investment cash that’s available to biotechs.
Last year these corporate financing arms accounted for slightly
- more than a third of all the cash that flowed into biotech, according to SVB. And
- the corporate VCs have a big appetite for investing in early-stage rounds.

Courtesy of Silicon Valley Bank
| Courtesy of Silicon Valley Bank |
“I think we’ve reached a healthy level of funding in the sector right now,” says Jon Norris, the author of the report and managing director at Silicon Valley Bank. He adds that
- with the IPO window still open to biotechs, a lot of early- or mid-stage companies are now choosing to jump through
- to the public market rather than make a deal with pharma.
The IPO alternative has also made it possible to drive up the value of biotech assets, which now command record payments.
But that’s a trend that can’t run forever.
“This can’t run out too much longer into 2015,” says Norris. “I can see it starting to close.”
As the window shuts, he adds, you can expect to see the number of M&A deals rise. And the biggest biotech deals will likely be worth more, as big exits–defined as deals with an upfront of $75 million or more–jumped to an average record high of $549 million last year, a 10% spike over 2012.

Courtesy of Silicon Valley Bank 2
| Courtesy of Silicon Valley Bank |
In its analysis, Silicon Valley Bank concludes that the early-stage investment gamble now
- amounts to a strategic move by the top Big Pharma companies to outsource a considerable portion of their early-stage R&D work,
- priming the cash pump directly through their own venture arms as well as by investing in many of the new venture funds filling up with risk capital. And
- the change-up is likely to continue to drive partnering as well as Big Pharma
- forges a new round of development pacts and M&A deals with their venture colleagues involved in biotech.
“We’ve all seen over the last few years the pullback in overall R&D spending by pharma and biotech,” says Norris. “There’s a tendency for these (pharma) folks to outsource their innovation.”
Not surprisingly, experimental
- cancer drugs are attracting the bulk of Big Pharma’s attention and corporate cash, followed by
- platform technologies that generate new leads, metabolics, ophthalmology, cardiovascular, CNS, dermatology, GI and inflammation, says SVB.
The leading corporate venture investors in the industry include Novartis ($NVS),Astellas, Pfizer ($PFE), S.R. One ($GSK), Amgen ($AMGN) and J&J Development Corp. ($JNJ). And nearly 90% of top corporate investment deals are directed at Series A or B rounds. More than half of these new investments, says SVB, were in preclinical or Phase I companies.
Extensive Promoter-Centered Chromatin Interactions Provide a Topological Basis for Transcription Regulation
(Li G, Ruan X, Auerbach RK, Sandhu KS, et al.) Cell 2012; 148(1-2): 84-98. http://cell.com
http://FrontiersMolecularCellularOncology.com
Using genome-wide Chromatin Interaction Analysis with Paired-End-Tag sequencing (ChIA-PET),
mapped long-range chromatin interactions associated with RNA polymerase II in human cells
uncovered widespread promoter-centered intragenic, extragenic, and intergenic interactions.
- These interactions further aggregated into higher-order clusters
- proximal and distal genes were engaged through promoter-promoter interactions.
- most genes with promoter-promoter interactions were active and transcribed cooperatively
- some interacting promoters could influence each other implying combinatorial complexity of transcriptional controls.
Comparative analyses of different cell lines showed that
- cell-specific chromatin interactions could provide structural frameworks for cell-specific transcription,
- and suggested significant enrichment of enhancer-promoter interactions for cell-specific functions.
- genetically-identified disease-associated noncoding elements were spatially engaged with corresponding genes through long-range interactions.
Overall, our study provides insights into transcription regulation by
- three-dimensional chromatin interactions for both housekeeping and
- cell-specific genes in human cells.
New Nucleoporin: Regulator of Transcriptional Repression and Beyond.
NJ Sarma and K Willis
Nucleus 2012; 3(6): 1–8; http://Nucleus.com © 2012 Landes Bioscience
Transcriptional regulation is a complex process that requires the integrated action of many multi-protein complexes.
The way in which a living cell coordinates the action of these complexes in time and space is still poorly understood.
- nuclear pores, well known for their role in 3′ processing and export of transcripts, also participate in the control of transcriptional initiation.
- nuclear pores interface with the well-described machinery that regulates initiation.
This work led to the discovery that
- specific nucleoporins are required for binding of the repressor protein Mig1 to its site in target promoters.
- Nuclear pores are involved in repressing, as well as activating, transcription.
Here we discuss in detail the main models explaining our result and consider what each implies about the roles that nuclear pores play in the regulation of gene expression.
Computational Design of Targeted Inhibitors of Polo-Like Kinase 1 ( lk1).
(KS Jani and DS Dalafave) Bioinformatics and Biology Insights 2012:6 23–31.
http://dx.doi.org:/10.4137/BBI.S8971
Computational design of small molecule putative inhibitors of Polo-like kinase 1 (Plk1) is presented. Plk1, which regulates the cell cycle, is often over expressed in cancers.
- Down regulation of Plk1 has been shown to inhibit tumor progression.
- Most kinase inhibitors interact with the ATP binding site on Plk1, which is highly conserved.
- This makes the development of Plk1-specific inhibitors challenging, since different kinases have similar ATP sites.
However, Plk1 also contains a unique region called the polo-box domain (PBD), which is absent from other kinases.
- the PBD site was used as a target for designed Plk1 putative inhibitors.
- Common structural features of several experimentally known Plk1 ligands were first identified.
- The findings were used to design small molecules that specifically bonded Plk1.
- Drug likeness and possible toxicities of the molecules were investigated.
- Molecules with no implied toxicities and optimal drug likeness values were used for docking studies.
- Several molecules were identified that made stable complexes only with Plk1 and LYN kinases, but not with other kinases.
- One molecule was found to bind exclusively the PBD site of Plk1.
Possible utilization of the designed molecules in drugs against cancers with over expressed Plk1 is discussed.
Conclusions
The previous discussions reviewed the status of an evolving personalized medicine multicentered and worldwide enterprise. It is also clear from these reports that the search for targeted drugs matched to a cancer profile or signature has identified several approaches that show great promise.
- We know considerably more about metabolic pathways and linked changes in transcription that occur in neoplastic development.
- There are several methods used to do highly accurate insertions in gene sequences that are linked to specific metabolic changes, and
- some may have significant implications for therapeutics, if
- the link is a change that is associated with a driver mutation
- the link can be identified by a fluorescent or other probe
- the link is tied to a mRNA or peptide product that is a biomarker measured in the circulation
- We have probes to genetic links to the control of many and interacting signaling pathways.
- We know more about transcription through mRNA.
- We are closer to the possibility that metabolic substrates, like ‘fumarate’ (a key intermediate in the TCA cycle), may provide a means to reverse regulate the neoplastic cells.
- We may also find metabolic channels that drive the cells from proliferation to apoptosis or normal activity.
Summary
This discussion identified the huge expansion of genomic technology in the investigation of biopharmacotherapeutic targets that have been identified involving different levels and interacting signaling pathways. There are several methods of analyzing gene expression, and a primary emphasis is given to combinations of mutations expressed in different cancer types. There is a major hypothesis that expresses the need to focus on “central” “driver mutations” that correspond with the regulation of gene expression, cell proliferation, and cell metabolism. What hasn’t been know is why drug resistance develops and whether the cellular migration and aerobic glycolysis can be redirected after cell metastasis occurs.

mutation in the matched nucleotides
.
A slight mutation in the matched nucleotides can lead to chromosomal aberrations and unintentional genetic rearrangement. (Photo credit: Wikipedia)

Phosphofructokinase mechanism
Deutsch: Regulation der Phosphofructokinase (Photo credit: Wikipedia)
Additional Related articles
Universal Language: The Pistoia Alliance Takes on Indescribable Biology
By Aaron Krol
July 18, 2014 | The Pistoia Alliance, founded after a meeting between members of Pfizer, AstraZeneca, Novartis and GlaxoSmithKline, has come to resemble a United Nations of the life sciences industry. Now in its fifth year, the Alliance’s membership has grown to include nearly all the largest pharma companies (Eli Lilly is the only holdout in the top ten) plus a huge assortment of publishers, IT vendors, small biotechs and academic groups. It makes for a complicated network of business partners and competitors, but they do have some basic needs in common. In particular, the Pistoia Alliance exists to build IT architectures that serve the precompetitive stages of research and development.
“The key to the Pistoia Alliance is that, as time has gone by, most companies have figured out that you can’t go it alone,” says Sergio Rotstein, the Director of Research Business Technologies at Pfizer and a member of the Alliance’s board of directors. “Even the tightest of companies has opened up its walls quite a bit to collaboration… The idea of me asking my buddy from Merck, how did you solve that problem, and by the way would you mind giving me the solution — ten years ago, that would have gotten me laughed out of the room.”
The Pistoia Alliance has previously sponsored new methods for querying databases and the scientific literature, and a more effective algorithm for compressing and sharing genetic sequencing data. Over the past year, another Pistoia project, HELM, has entered the public domain after gradual development by an assortment of Alliance members. An open source language and set of editing tools for working with large biomolecules, HELM has already become a foundational part of research in at least three large pharmaceutical companies.
At the Bio-IT World Best Practices Awards this April, the HELM project won the Pistoia Alliance a top prize in the category of Informatics. These awards recognize advances in information technology and good management strategies at all levels of the biomedical industry. While the Best Practices Awards always seek to highlight programs that could be widely replicated, Bio-IT World rarely has the opportunity to single out a project that has been adopted so quickly across so many organizations as the Pistoia Alliance’s efforts around HELM.
A Loss for Words
HELM addresses a problem at the root level of drug discovery. Pharmaceutical and biotech companies are looking at increasingly complex molecules in the search for new therapeutics, testing out RNA- and peptide-based compounds that tap directly into cellular pathways. The trouble is that these large molecules, which are often hybrids of RNA, amino acids and other chemical structures, are difficult to concisely describe, even when their structures are perfectly known. They are too large and ungainly to represent atom-by-atom, but not uniform enough to be reduced to nucleotides and peptide chains.
“There have been a number of ways to represent small molecules,” says Rotstein. “That’s been the bread and butter of a number of companies for a long time, and that’s the realm of cheminformatics. And there’s been a lot of methodology for dealing with sequence-based entities, like genes and proteins, which is the realm of bioinformatics. The issue is that the types of molecules that we are targeting fall in between these two.”
This isn’t just a semantic issue; not having a standard language for biomolecules has practical consequences. It’s hard to register these molecules in databases, and even harder to conduct searches for them or share their structures with collaborators. The problem has recently come to a head, as growing knowledge of interlocking cellular systems has led researchers to therapies that increasingly resemble the body’s own tangled biology. “It follows the natural progression of science itself,” says Rotstein. “The application of peptides with unnatural amino acids, and the area of antibody -drug conjugates, has been growing a great deal over the past few years. A lot of the companies that traditionally worked in the small molecule space, nowadays are looking for a diverse portfolio.”
In 2008, Rotstein was part of an oligonucleotide unit at Pfizer that set out to build a new language to describe the compounds it was working with. The language would be similar to the small molecule notation SMILES (the Simplified Molecular-Input Line-Entry System), which renders a chemical structure as a continuous string of characters, while using symbols from the ASCII alphabet to resolve properties like where bonds occur and how molecules branch. Instead of using atoms as the smallest units in the chain, however, much larger groups — monomers like nucleotides and amino acids — would receive short, unique IDs that could be strung together into polymers. The amino acid cysteine, for instance, could be represented simply as “C.” New monomers would be registered with new IDs in a central database, and every ID would be linked to a complete description in small molecule notation.

oligonucleotide conplex
A complex oligonucleotide peptide conjugate, featuring amino acids, RNA, and other chemical structures. The molecule is rendered as both a monomer graph, and in HELM notation. Reproduced from the Journal of Chemical Information and Modeling with permission of the author
The language was called HELM, the Hierarchical Editing Language for Macromolecules: “hierarchical” because strings of monomers are built into simple polymers, which in turn are joined into complex polymers. HELM was easy to use and unambiguous, and was soon adopted in many more departments at Pfizer. For the first time, it was possible to quickly enter a new macromolecule in Pfizer’s registry, check for uniqueness, and receive a corporate ID to take the project forward.
A Living Language
At the same time that Rotstein’s team was developing HELM at Pfizer, other pharma companies and informatics vendors were struggling with the same problem. The software provider Accelrys (now BIOVIA), for instance, had modified the Molfile chemical table format to deal with hybrid macromolecules, in a system the company called the Self-Contained Sequence Representation (SCSR). There was a danger of proliferating standards, which would not only create redundant work at each company writing its own language, but also threaten the ability of these organizations to share information with each other.
Meanwhile, a member survey at the Pistoia Alliance flagged the representation of complex biomolecules as one of the industry’s top three non-competitive problems. Since Pfizer had already published a paper on HELM and built a software toolkit around the system, the company volunteered to make the entire program open source and continue its development with other members of the Alliance.
“We saw an opportunity for Pfizer,” says Rotstein. “If this did indeed become a standard, and the open source tools continued to evolve through contributions of the whole community, that would help us too.” All told, 24 companies sent volunteers to work on HELM, untangling the code from Pfizer’s internal systems, making it public, and extending the tools that serve the language.
The entire HELM project is now available on GitHub, and uses the permissive MIT open source license, which gives anyone the right to download and modify the code without requiring any contribution back to the project. That should encourage vendors to build commercial software on top of HELM, helping to foster compatibility across the industry.
The basic HELM toolkit includes search functions and uniqueness checks, as well as the HELM Editor, a platform for drawing chemical structures. The HELM Editor lets users plug in or draw monomers, then move up the scale to polymers made from those building blocks. It can be used simply as a translation tool, taking existing structures and giving them names in HELM notation, but Rotstein says it would also be a preferred platform for making new molecules from scratch.

HELM photo of siRNA
A screenshot from the HELM Editor, showing a siRNA molecule under construction. Image credit: Pistoia Alliance
Since HELM was released to the public last year, development has continued at various partner organizations. Roche was one of the first adopters, and has been relentlessly adding functionality to the toolkit. “Roche created a custom antibody-drawing capability on top of the HELM Editor, and it’s truly phenomenal,” says Rotstein. “They are now putting the finishing touches on that, and as soon as they’re done, they are pushing it right back out into the open source.”
He adds that Pfizer plans to start using Roche’s antibody drawing tool itself. “That tool alone will probably return our entire investment on externalizing HELM.”
Most recently, this month the Pistoia Alliance released Exchangeable HEL M, another big push for interoperability. While some basic monomers, like the natural amino acids and nucleotides, have universal IDs in HELM, most monomer IDs are unique to each user, stored in an internal database. That’s a necessary feature to make HELM flexible to the needs of every user, but it means that most molecules only make sense in the context of the databases against which they were designed.
Exchangeable HELM provides a file format that includes both the larger HELM sequence of a macromolecule, and separately, the chemical structure of each monomer inside it. That makes it easy for collaborators — say, a large pharma company and a CRO hired for a specific project — to send molecular structures back and forth. Exchangeable HELM also offers a tool to “translate” between databases, if two organizations have different internal IDs for the same monomer.
The Lingua Franca
So far, Pfizer, Roche, and Lundbeck are the largest drug companies to switch their systems over to HELM, and Rotstein says a “robust pipeline of other companies” is preparing to adopt the language. Meanwhile, vendors that serve the drug industry are preparing for a widespread change. NextMove Software and ChemAxon are both working in HELM, and even BIOVIA, which plans to continue using SCSR internally, has made its systems compatible with HELM to more easily share large molecules with clients and partners.
The adoption of HELM will be buoyed by public resources in the life sciences that are turning to the language as the obvious choice for representing complex molecules. One big supporter is the European Bioinformatics Institute, whose ubiquitous ChEMBL database of chemical compounds will include HELM notations in its next release.
Increasingly, says Rotstein, the Pistoia Alliance is speaking of a HELM ecosystem. “We want to have content providers that have structures in HELM format. We want vendors whose software can read and write HELM. We want companies that use HELM as their standard, we want CROs that can use HELM to exchange information with those companies, and next on our list are downstream things like scientific journals and regulatory agencies.” Large publishers and regulators would be especially important adopters, because they are such frequent and public ports of call for companies sending macromolecular structures outside their walls. If the FDA or Nature Publishing Group began accepting HELM structures, it could be a major convenience when applying for clinical trials and publications. “It would be much easier to just send a file that says, ‘here’s exactly what my structure is,’” says Rotstein, “rather than having to verbally explain the structure.”
Having HELM in place as a widely-shared language could also benefit other Pistoia Alliance projects. For example, the Controlled Substance Compliant Services Project is currently building a database of compounds that are regulated or restricted in various countries around the world, so companies can quickly refer to the local legislation affecting compounds they want to work with. If large biomolecules are subject to regulations, HELM would be a convenient way to make those policies searchable.
Like other Pistoia Alliance initiatives, HELM is designed to run smoothly in the background. Defining the structure of macromolecules in a standard format, is not a process that should offer any company an edge in drug discovery, but a basic feature at the foundation of the life sciences. In an ideal world, says Rotstein, “this should be a non-issue. The ability to represent these molecules, and get them in and out of our system so we can store them, search them, and run calculations on them, should be trivial.”
Pathology Practiced Todat
How doctors group non Hodgkin lymphomas
There are many different types of non Hodgkin lymphoma. Doctors estimate that there are more than 60 subtypes. Understanding how the different types of NHL are grouped, or classified, can be difficult. A variety of systems for classifying lymphomas have been used over the years. The latest is the World Health Organisation classification of 2008. We give a simple description of the groups on this page.
The pathologist will examine the cells to see
Grade of NHL
Doctors put non Hodgkin lymphomas into 2 groups depending on how quickly they are likely to grow and spread
- Low grade (indolent) – these tend to grow very slowly
- High grade (aggressive) – these tend to grow more quickly
The different grades of non Hodgkin lymphoma are treated in slightly different ways.
Type of white blood cell
One way of classifying NHL is by the type of white blood cells (lymphocytes) affected – B cells or T cells. Most people with NHL have B cell lymphomas.
What the lymphoma cells look like
Your doctor will be able to give your type of non Hodgkin lymphoma a name depending on the appearance of the lymphoma cells. These names are quite complicated. But they are useful to doctors because the different types can behave differently. Different treatments are used for the different types. So knowing the type helps the doctor know how to treat them. In the laboratory a pathologist looks at the cells to see if they are
- Large or small
- Grouped together in structures called follicles (follicular type) or spread out (diffuse type)
Low grade non Hodgkin lymphomas tend to have small cells that are grouped together.
Low grade (slowly growing) NHL
Low grade lymphomas tend to grow very slowly. Doctors call them indolent lymphomas. They include
Small lymphocytic lymphoma
Small lymphocytic lymphoma is also called chronic lymphocytic leukaemia (CLL). It makes up about 6 out of 100 lymphomas in the UK (6%). In theory, lymphoma is an illness that starts in the lymph nodes and leukaemia is an illness of the blood. But leukaemia and lymphoma have many similarities and often affect the body in similar ways. Chronic lymphocytic leukaemia is the term used for this condition if many of the abnormal cells are in the blood. Doctors call it small lymphocytic lymphoma when the disease involves the lymph nodes in particular.
The B-cell lymphomas are types of lymphoma affecting B cells. Lymphomas are “blood cancers” in the lymph glands. They develop more frequently in older adults and in immunocompromised individuals.
B-cell lymphomas include both Hodgkin’s lymphomas and most non-Hodgkins lymphomas. They are often divided into indolent (slow-growing) lymphomas and aggressive lymphomas. Indolent lymphomas respond rapidly to treatment and are kept under control (in remission) with long-term survival of many years, but are not cured. Aggressive lymphomas usually require intensive treatments, but have good prospects for a permanent cure.[1]
Prognosis and treatment depends on the specific type of lymphoma as well as the stage and grade. Treatment includes radiation and chemotherapy. Early-stage indolent B-cell lymphomas can often be treated with radiation alone, with long-term non-reoccurrence. Early-stage aggressive disease is treated with chemotherapy and often radiation, with a 70-90% cure rate.[1] Late-stage indolent lymphomas are sometimes left untreated and monitored until they progress. Late-stage aggressive disease is treated with chemotherapy, with cure rates of over 70%.[1]
Small cell Lymphocytic lymphoma (overlaps with Chronic lymphocytic leukemia)
Indolent NHL. These types of lymphoma grow very slowly. As a result, people with indolent NHL may not need to start treatment when it is first diagnosed. They are followed closely, and treatment is only started when they develop symptoms or the disease begins to change; this is called watchful waiting. When indolent lymphoma is located only in one area (called localized disease, stages I and II; see the Stages section), radiation therapy may eliminate the NHL.
Subtyping
In addition to determining if the NHL is indolent or aggressive and whether it is B-cell, T-cell, of NK-cell lymphoma, it is very important to determine the subtype of NHL because each subtype can behave differently and may require different treatments. There are about 35 subtypes of NHL.
Small lymphocytic lymphoma. This type of lymphoma is very closely related to a disease called B-cell chronic lymphocytic leukemia (CLL), and about 5% of people with NHL have this subtype. It is considered an indolent lymphoma. Patients with small lymphocytic lymphoma may receive a combination of chemotherapy, monoclonal antibodies, and/or radiation therapy, or they may be followed closely with watchful waiting.
Lymphoma – B cell neoplasms
Non-Hodgkin Lymphoma
Cytogenetics
Reviewer: Nikhil Sangle, M.D., University of Utah and ARUP Laboratories (see Reviewers page)
Revised: 17 February 2011, last major update February 2011
Copyright: (c) 2001-2011, PathologyOutlines.com, Inc.
List of translocations
==============================================
Relatively common translocations are listed below
See each topic for more complete lists:
● t(1;14)(p32;q11): SCL (tal-1) and T cell receptor delta/alpha; preT ALL (15-30%)
● deletion of 11q23: CLL (10-20%)
● t(11;14)(q13;q32): bcl1/PRAD1 and IgH; mantle cell lymphoma (90%), B cell prolymphocytic leukemia (20%), myeloma (3%)
● Trisomy 12: B-CLL (30%)
● deletion 13q14: B -CLL (25-50%)
● t(14;19)(q32;q13): IgH and bcl3; B-CLL
● t(16;22);(q23;q11): cmaf and Ig lambda; multiple myeloma
● Trisomy 18: common in marginal zone lymphoma, MALT type
Lymphoma – B cell neoplasms
B cell lymphoma subtypes
Chronic lymphocytic leukemia – features that differ from SLL
Reviewer: Nikhil Sangle, M.D., University of Utah and ARUP Laboratories (see Reviewers page)
Revised: 20 September 2012, last major update February 2011
Copyright: (c) 2001-2012, PathologyOutlines.com, Inc
Terminology
==============================================
- Leukemic disorder of CD5+ CD23+ tumor cells, usually B cell, that are small, round, low grade, with soccer ball appearance
Terminology
==============================================
- Called CLL/chronic lymphocytic leukemia if leukemic involvement at diagnosis (5K or more of monoclonal B cell lymphocytosis per microliter)
● Less than 5K per microliter is termed monoclonal B lymphocytosis or possibly low stage CLL
● CLL with increased prolymphocytes (CLL/prolymphocytic leukemia): 10-55% prolymphocytes
● Prolymphocytic leukemia: >55% prolymphocytes
Lymphoma – B cell neoplasms
B cell lymphoma subtypes
Small lymphocytic lymphoma
Reviewer: Nikhil Sangle, M.D., University of Utah and ARUP Laboratories (see Reviewers page)
Revised: 6 February 2012, last major update February 2011
Copyright: (c) 2001-2012, PathologyOutlines.com, Inc
Definition
==============================================
- Common low grade B cell lymphoma with pseudofollicles composed of mature lymphocytes resembling soccer balls in peripheral blood; cells are CD5+, CD23+
Clinical features
==============================================
- Usually older patients (median age 60 years), 2/3 male, with disease in bone marrow, lymph nodes, spleen, liver
● Often presents with leukemia, although patients may be asymptomatic
● SLL may progress to blood (leukemic) involvement, but if so, there is usually less leukocytosis than cases with initial diagnosis of CLL
● Almost all cases are B cell origin
● Associated with hypogammaglobulinemia, monoclonal immunoglobulin spikes in some, infections; also autoantibodies to red blood cells and platelets causing hemolytic anemia and thrombocytopenia
● Median survival 4-6 years; indolent unless it transforms
Poor prognostic factors
==============================================
- 17p deletions, 11q22-23 deletion, non-mutated immunoglobulin genes, aberrant expression of CD2, CD7, CD10, CD13, CD33 or CD34 (Am J Clin Pathol 2003;119:824)
In 2013, the North American market was valued at $128.9 million and accounted for the largest share of the global digital pathology market, followed by Europe and Asia. The North American market is expected to grow at a healthy growth rate over the next five years. This high growth can be attributed to the favorable reimbursement scenario in the U.S. and the use of digital pathology to improve the quality of cancer diagnosis in Canada. However, lack of FDA approvals for digital pathology to be used for primary diagnosis acts as a major barrier for the North American market.
Other posts related to this discussion were published on this Open Source Online Scientific Journal from Leaders in Pharmaceutical Business Intelligence:
Big Data in Genomic Medicine, LHB
http://pharmaceuticalintelligence.com/2012/12/17/big-data-in-genomic-medicine/
A New Therapy for Melanoma, LHB
http://pharmaceuticalintelligence.com/2012/09/15/a-new-therapy-for-melanoma/
BRCA1 a tumour suppressor in breast and ovarian cancer – functions in transcription, ubiquitination and DNA repair, S Saha
http://pharmaceuticalintelligence.com/2012/12/04/brca1-a-tumour-suppressor-in-breast-and-ovarian-cancer-functions-in-transcription-ubiquitination-and-dna-repair/
Judging ‘Tumor response’-there is more food for thought, R Saxena
http://pharmaceuticalintelligence.com/2012/12/04/judging-the-tumor-response-there-is-more-food-for-thought/
Computational Genomics Center: New Unification of Computational Technologies at Stanford, A. Lev-Ari
http://pharmaceuticalintelligence.com/2012/12/03/computational-genomics-center-new-unification-of-computational-technologies-at-stanford/
Ovarian Cancer and fluorescence-guided surgery: A report, T. Barliya
http://pharmaceuticalintelligence.com/2013/01/19/ovarian-cancer-and-fluorescence-guided-surgery-a-report/
Personalized medicine gearing up to tackle cancer , R. Saxena
http://pharmaceuticalintelligence.com/2013/01/07/personalized-medicine-gearing-up-to-tackle-cancer/
Exploring the role of vitamin C in Cancer therapy, R. Saxena
http://pharmaceuticalintelligence.com/2013/01/15/exploring-the-role-of-vitamin-c-in-cancer-therapy/
Differentiation Therapy – Epigenetics Tackles Solid Tumors, SJ Williams
http://pharmaceuticalintelligence.com/2013/01/03/differentiation-therapy-epigenetics-tackles-solid-tumors/
Mechanism involved in Breast Cancer Cell Growth: Function in Early Detection & Treatment, A. Lev-Ari
http://pharmaceuticalintelligence.com/2013/01/17/mechanism-involved-in-breast-cancer-cell-growth-function-in-early-detection-treatment/
Personalized Medicine: Cancer Cell Biology and Minimally Invasive Surgery (MIS), A. Lev-Ari
http://pharmaceuticalintelligence.com/2012/12/01/personalized-medicine-cancer-cell-biology-and-minimally-invasive-surgery-mis/
Role of Primary Cilia in Ovarian Cancer, A. Awan
http://pharmaceuticalintelligence.com/2013/01/15/role-of-primary-cilia-in-ovarian-cancer-2/
The Molecular Pathology of Breast Cancer Progression, T. Bailiya`
http://pharmaceuticalintelligence.com/2013/01/10/the-molecular-pathology-of-breast-cancer-progression/
Stanniocalcin: A Cancer Biomarker, A. Awan
http://pharmaceuticalintelligence.com/2012/12/25/stanniocalcin-a-cancer-biomarker/
Nanotechnology, personalized medicine and DNA sequencing, T. Barliya
http://pharmaceuticalintelligence.com/2013/01/09/nanotechnology-personalized-medicine-and-dna-sequencing/
Gastric Cancer: Whole-genome reconstruction and mutational signatures, A. Lev-Ari
http://pharmaceuticalintelligence.com/2012/12/24/gastric-cancer-whole-genome-reconstruction-and-mutational-signatures-2/
Paradigm Shift in Human Genomics – Predictive Biomarkers and Personalized Medicine – Part 1, A. Lev-Ari
http://pharmaceuticalintelligence.com/2013/01/13/paradigm-shift-in-human-genomics-predictive-biomarkers-and-personalized-medicine-part-1/
LEADERS in Genome Sequencing of Genetic Mutations for Therapeutic Drug Selection in Cancer Personalized Treatment: Part 2, A. Lev-Ari
http://pharmaceuticalintelligence.com/2013/01/13/leaders-in-genome-sequencing-of-genetic-mutations-for-therapeutic-drug-selection-in-cancer-personalized-treatment-part-2/
Personalized Medicine: An Institute Profile – Coriell Institute for Medical Research: Part 3, A. Lev-Ari
http://pharmaceuticalintelligence.com/2013/01/13/personalized-medicine-an-institute-profile-coriell-institute-for-medical-research-part-3/
The Consumer Market for Personal DNA Sequencing: Part 4, A. Lev-Ari
http://pharmaceuticalintelligence.com/2013/01/13/consumer-market-for-personal-dna-sequencing-part-4/
Harnessing Personalized Medicine for Cancer Management, Prospects of Prevention and Cure: Opinions of Cancer Scientific Leaders @http://pharmaceuticalintelligence.com A. Lev-Ari
http://pharmaceuticalintelligence.com/2013/01/13/7000/
GSK for Personalized Medicine using Cancer Drugs needs Alacris systems biology model to determine the in silico effect of the inhibitor in its “virtual clinical trial” A Lev-Ari
http://pharmaceuticalintelligence.com/2012/11/14/gsk-for-personalized-medicine-using-cancer-drugs-needs-alacris-systems-biology-model-to-determine-the-in-silico-effect-of-the-inhibitor-in-its-virtual-clinical-trial/
Recurrent somatic mutations in chromatin-remodeling and ubiquitin ligase complex genes in serous endometrial tumors, S. Saha
http://pharmaceuticalintelligence.com/2012/11/19/recurrent-somatic-mutations-in-chromatin-remodeling-and-ubiquitin-ligase-complex-genes-in-serous-endometrial-tumors/
Metabolic drivers in aggressive brain tumors, pkandala
http://pharmaceuticalintelligence.com/2012/11/11/metabolic-drivers-in-aggressive-brain-tumors/
Personalized medicine-based cure for cancer might not be far away, R. Saxena
http://pharmaceuticalintelligence.com/2012/11/20/personalized-medicine-based-cure-for-cancer-might-not-be-far-away/
Response to Multiple Cancer Drugs through Regulation of TGF-β Receptor Signaling: a MED12 Control, A. Lev-Ari
http://pharmaceuticalintelligence.com/2012/11/21/response-to-multiple-cancer-drugs-through-regulation-of-tgf-%CE%B2-receptor-signaling-a-med12-control/
Human Variome Project: encyclopedic catalog of sequence variants indexed to the human genome sequence, A. Lev-Ari
http://pharmaceuticalintelligence.com/2012/11/24/human-variome-project-encyclopedic-catalog-of-sequence-variants-indexed-to-the-human-genome-sequence/
Prostate Cancer Cells: Histone Deacetylase Inhibitors Induce Epithelial-to-Mesenchymal Transition, SJ Williams
http://pharmaceuticalintelligence.com/2012/11/30/histone-deacetylase-inhibitors-induce-epithelial-to-mesenchymal-transition-in-prostate-cancer-cells/
Tumor Imaging and Targeting: Predicting Tumor Response to Treatment: Where we stand?, R. Saxena
http://pharmaceuticalintelligence.com/2012/12/13/imaging-and-targeting-the-tumor-predicting-tumor-response-where-we-stand/
Nanotechnology: Detecting and Treating metastatic cancer in the lymph node, T. Barliya
http://pharmaceuticalintelligence.com/2012/12/19/nanotechnology-detecting-and-treating-metastatic-cancer-in-the-lymph-node/
Heroes in Medical Research: Barnett Rosenberg and the Discovery of Cisplatin, SJ Williams
http://pharmaceuticalintelligence.com/2013/01/12/heroes-in-medical-research-barnett-rosenberg-and-the-discovery-of-cisplatin/
Inspiration From Dr. Maureen Cronin’s Achievements in Applying Genomic Sequencing to Cancer Diagnostics, A. Lev-Ari
http://pharmaceuticalintelligence.com/2013/01/10/inspiration-from-dr-maureen-cronins-achievements-in-applying-genomic-sequencing-to-cancer-diagnostics/
The “Cancer establishments” examined by James Watson, co-discoverer of DNA w/Crick, 4/1953, A. Lev-Ari
http://pharmaceuticalintelligence.com/2013/01/09/the-cancer-establishments-examined-by-james-watson-co-discover-of-dna-wcrick-41953/
Nanotech Therapy for Breast Cancer. T. Barlyia
http://pharmaceuticalintelligence.com/2012/12/09/naotech-therapy-for-breast-cancer/
Dasatinib in Combination With Other Drugs for Advanced, Recurrent Ovarian Cancer, pkandala
http://pharmaceuticalintelligence.com/2012/12/08/dasatinib-in-combination-with-other-drugs-for-advanced-recurrent-ovarian-cancer/
Squeezing Ovarian Cancer Cells to Predict Metastatic Potential: Cell Stiffness as Possible Biomarker, pkandala
http://pharmaceuticalintelligence.com/2012/12/08/squeezing-ovarian-cancer-cells-to-predict-metastatic-potential-cell-stiffness-as-possible-biomarker/
Hypothesis – following on James Watson, LHB
http://pharmaceuticalintelligence.com/2013/01/27/novel-cancer-h…ts-are-harmful/
Otto Warburg, A Giant of Modern Cellular Biology, LHB
http://pharmaceuticalintelligence.com/2012/11/02/otto-warburg-a-giant-of-modern-cellular-biology/
Is the Warburg Effect the cause or the effect of cancer: A 21st Century View? LHB
http://pharmaceuticalintelligence.com/2012/10/17/is-the-warburg-effect-the-cause-or-the-effect-of-cancer-a-21st-century-view/
Remembering a Great Scientist among Mentors, LHB
http://pharmaceuticalintelligence.com/2013/01/26/remembering-a-great-scientist-among-mentors/
Portrait of a great scientist and mentor: Nathan Oram Kaplan, LHB
http://pharmaceuticalintelligence.com/2013/01/26/portrait-of-a-great-scientist-and-mentor-nathan-oram-kaplan/
Predicting Tumor Response, Progression, and Time to Recurrence, LHB
http://pharmaceuticalintelligence.com/2012/12/20/predicting-tumor-response-progression-and-time-to-recurrence/
Directions for genomics in personalized medicine, LHB
http://pharmaceuticalintelligence.com/2013/01/27/directions-for-genomics-in-personalized-medicine/
How mobile elements in “Junk” DNA promote cancer. Part 1: Transposon-mediated tumorigenesis, Sjwilliams
http://pharmaceuticalintelligence.com/2012/10/31/how-mobile-elements-in-junk-dna-prote-cacner-part1-transposon-mediated-tumorigenesis/
Novel Cancer Hypothesis Suggests Antioxidants Are Harmful, LHB
http://pharmaceuticalintelligence.com/2013/01/27/novel-cancer-hypothesis-suggests-antioxidants-are-harmful/
Mitochondria: Origin from oxygen free environment, role in aerobic glycolysis, metabolic adaptation, LHB
http://pharmaceuticalintelligence.com/2012/09/26/mitochondria-origin-from-oxygen-free-environment-role-in-aerobic-glycolysis-metabolic-adaptation/
Advances in Separations Technology for the “OMICs” and Clarification of Therapeutic Targets, LHB
http://pharmaceuticalintelligence.com/2012/10/22/advances-in-separations-technology-for-the-omics-and-clarification-of-therapeutic-targets/
Cancer Innovations from across the Web, LHB
http://pharmaceuticalintelligence.com/2012/11/02/cancer-innovations-from-across-the-web/
Mitochondrial Damage and Repair under Oxidative Stress, LHB
http://pharmaceuticalintelligence.com/2012/10/28/mitochondrial-damage-and-repair-under-oxidative-stress/
Mitochondria: More than just the “powerhouse of the cell” R. Saxena
http://pharmaceuticalintelligence.com/2012/07/09/mitochondria-more-than-just-the-powerhouse-of-the-cell/
Mitochondria and Cancer: An overview of mechanisms, R. Saxena
http://pharmaceuticalintelligence.com/2012/09/01/mitochondria-and-cancer-an-overview/
Mitochondrial fission and fusion: potential therapeutic targets? R. Saxena
http://pharmaceuticalintelligence.com/2012/10/31/mitochondrial-fission-and-fusion-potential-therapeutic-target/
Mitochondrial mutation analysis might be “1-step” away, R. Saxena
http://pharmaceuticalintelligence.com/2012/08/14/mitochondrial-mutation-analysis-might-be-1-step-away/
β Integrin emerges as an important player in mitochondrial dysfunction associated Gastric Cancer, R. Saxena
http://pharmaceuticalintelligence.com/2012/09/10/%CE%B2-integrin-emerges-as-an-important-player-in-mitochondrial-dysfunction-associated-gastric-cancer/
mRNA interference with cancer expression, LHB
http://pharmaceuticalintelligence.com/2012/10/26/mrna-interference-with-cancer-expression/
What can we expect of tumor therapeutic response? LHB
http://pharmaceuticalintelligence.com/2012/12/05/what-can-we-expect-of-tumor-therapeutic-response/
Expanding the Genetic Alphabet and linking the genome to the metabolome, LHB
http://pharmaceuticalintelligence.com/2012/09/24/expanding-the-genetic-alphabet-and-linking-the-genome-to-the-metabolome/
Breast Cancer, drug resistance, and biopharmaceutical targets, LHB
http://pharmaceuticalintelligence.com/2012/09/18/breast-cancer-drug-resistance-and-biopharmaceutical-targets/
Breast Cancer: Genomic Profiling to Predict Survival: Combination of Histopathology and Gene Expression Analysis, A. Lev-Ari
http://pharmaceuticalintelligence.com/2012/12/24/breast-cancer-genomic-profiling-to-predict-survival-combination-of-histopathology-and-gene-expression-analysis/
Ubiquinin-Proteosome pathway, autophagy, the mitochondrion, proteolysis and cell apoptosis, LHB
http://pharmaceuticalintelligence.com/2012/10/30/ubiquinin-proteosome-pathway-autophagy-the-mitochondrion-proteolysis-and-cell-apoptosis/
Identification of Biomarkers that are Related to the Actin Cytoskeleton, LHB
http://pharmaceuticalintelligence.com/2012/12/10/identification-of-biomarkers-that-are-related-to-the-actin-cytoskeleton/
Nitric Oxide has a ubiquitous role in the regulation of glycolysis -with a concomitant influence on mitochondrial function, LHB
http://pharmaceuticalintelligence.com/2012/09/16/nitric-oxide-has-a-ubiquitous-role-in-the-regulation-of-glycolysis-with-a-concomitant-influence-on-mitochondrial-function/
Genomic Analysis: FLUIDIGM Technology in the Life Science and Agricultural Biotechnology, A. Lev-Ari http://pharmaceuticalintelligence.com/2012/08/22/genomic-analysis-fluidigm-technology-in-the-life-science-and-agricultural-biotechnology/
Nanotechnology: Detecting and Treating metastatic cancer in the lymph node, T. Barliya
http://pharmaceuticalintelligence.com/2012/12/19/nanotechnology-detecting-and-treating-metastatic-cancer-in-the-lymph-node/
Reporter: Aviva Lev-Ari, PhD, RN
Crohn’s disease driven by inflammation – not genetics, reports study Aug. 15, 2012
Inflammation — not genetic susceptibility —
- drives the growth of intestinal bacteria and invasive E. coli linked to Crohn’s disease (CD), reports a new Cornell study.
Scientists have long wondered about the role of bacteria in CD. Recent studies have shown marked changes in the composition of the intestinal bacteria in people
- with CD, leading researchers to ask: Are microbial abnormalities a direct consequence of genetic abnormalities linked to Crohn’s and precede and initiate inflammation, or does intestinal inflammation bring on the bugs?
This study also reports that a common therapy directed against intestinal inflammation decreases dysbiosis. In addition, the study found that
- the lack of a receptor that helps recruit T cells, which are needed for cell-mediated immunity, to the gut also decreases inflammation and dysbiosis, offering a new option for therapeutic intervention.Inflammation, in fact,
- drives microbial imbalances (dysbiosis) and
the proliferation of a specific type of E. coli that is adherent, invasive and found in the ileum, reported Cornell researchers July 31 in PLoS (7[7]).
CD is a chronic debilitating inflammatory bowel disease that involves a complex interaction of
- host genes,
- the immune system,
- the intestinal microbiome and
- the environment.
To mirror the complex nature of the disease, Simpson’s team designed a study that
- incorporated inflammatory triggers related to relapse of CD and ileal inflammation.
The team focused on ileal dysbiosis, which is prevalent in 70 percent of CD cases and
- used a variety of contemporary techniques to generate a comprehensive picture of the composition and spatial distribution of the ileal microbiome.
Particular attention was paid to pinpointing
- the number,
- pathotype and
- location
of E. coli associated with intestinal inflammation in people, dogs and mice.
The findings demonstrate that
- inflammation drives ileal dysbiosis and proliferation of CD-associated adherent invasive E. coli.
- the host genotype and therapeutically blocking inflammation both impact the onset and extent of ileal dysbiosis.
The investigation leveraged the knowledge and resources of researchers in the labs of Erik Denker, Dwight Bowman and Sean McDonough labs. Building on findings in patients with Crohn’s disease evaluated by Dr. Ellen Scherl’s group at Weill Cornell Medical College, this collaboration shed new light on this debilitating disease.
This work was supported by NewYork-Presbyterian Hospital/Weill Cornell Medical Center, the Jill Roberts Center for Inflammatory Bowel Disease and the National Institutes of Health.
http://www.news.cornell.edu/stories/Aug12/Inflammation.html
Functional Proteomics Related to Energy Metabolism of Synaptosomes
from iTRAQ-Based Quantitative Proteomics Analysis Revealed Alterations of Carbohydrate Metabolism Pathways and Mitochondrial Proteins in a Male Sterile Cybrid Pummelo
Bei-Bei Zheng †, Yan-Ni Fang †, Zhi-Yong Pan †, Li Sun †, Xiu-Xin Deng †, Jude W. Grosser ‡, andWen-Wu Guo *†
† Key Laboratory of Horticultural Plant Biology (Ministry of Education), Huazhong Agricultural University, Wuhan 430070, China
‡ Citrus Research and Education Center, University of Florida, 700 Experiment Station Road, Lake Alfred, Florida 33850, United States
- Proteome Res., May 13, 2014, 13(6), pp 2998–3015 http://dx.doi.org:/10.1021/pr500126g
Plant Biochemistry
Comprehensive and quantitative proteomic information on citrus floral bud is significant for understanding
- male sterility of the cybrid pummelo (G1+HBP) with nuclear genome of HBP and foreign mitochondrial genome of G1.
Scanning electron microscopy and transmission electron microscopy analyses of the anthers showed that
- the development of pollen wall in G1+HBP was severely defective with a lack of exine and sporopollenin formation.
Proteomic analysis was used to identify the differentially expressed proteins between male sterile G1+HBP and fertile type (HBP)
- with the aim to clarify their potential roles in another development and male sterility.
On the basis of iTRAQ quantitative proteomics, we identified 2235 high-confidence protein groups, 666 of which showed
- differentially expressed profiles in one or more stages.
Proteins up- or down-regulated in G1+HBP were mainly involved in
- carbohydrate and energy metabolism (e.g., pyruvate dehydrogenase, isocitrate dehydrogenase, ATP synthase, and malate dehydrogenase),
- nucleotide binding (RNA-binding proteins),
- protein synthesis and degradation (e.g., ribosome proteins and proteasome subunits).
Additionally, the proteins located in mitochondria also showed changed expression patterns. These findings provide a valuable inventory of proteins involved in floral bud development and contribute to elucidate the mechanism of cytoplasmic male sterility in the cybrid pummelo.
Keywords: cybrid; male sterility; mitochondria; proteome; transcriptome; primary metabolites
BIMSB Proteomics / Metabolomics
Overview
Within the past decades biochemical data of single processes, metabolic and signaling pathways were collected and advances in technology
- led to improvements of sensitivity and resolution of bioanalytical techniques.
These achievements build the bases for the so called ‘genome wide biochemistry’. High throughput techniques are the tool for large scale ‘-omics’ studies
- allowing the obtainment of a nearly complete picture of a determinate cell state, concerning its metabolites, proteins and transcripts.
However, a single level study of a living organism cannot give a complete understanding of the mechanisms regulating biological functions.
The integration of transcriptomics, proteomics and metabolomics data with existing knowledge allows connecting biological processes which were treated as independent so far. In this context the aim of our group is
- to apply metabolomics and proteomics techniques for absolute quantification and
- to analyze turnover rates of proteins and metabolites using stable isotopes. In addition,
- the development of data analysis workflows and integrative strategies are in the focus of our interest.
The central metabolism is the principal source of energy and building blocks for cell growth and survival. It is highly flexible and adjusted to the physiological program of the cell, organ and organism. In a healthy state
- cellular metabolism is tightly regulated to guarantee physiological function but also efficient usage of available recourses.
Metabolic dys-regulations are cause or response to many diseases. An impaired metabolic activity can lead to
- the loss of the physiological activity, cell damage or inefficient substrate usage. However,
- the underlying mechanisms leading to metabolic dys-functions are not well understood.
The regulation of metabolism is complex, because
- it acts at all biological layers – transcriptional, translational and post-translational.
Thus the metabolic activity of a cell, organ or organism inherits the information of regulatory layers in a multidimensional manner. I guess only the use of integrative mathematical approaches will enable us to decode such complex information.
In this regard, decoding the metabolic composition of biofluids e.g. blood serum
- may allow to determine a systems status, to identify diseases, predict drug responsiveness and to follow the success of medical treatments. This is a step towards personalized medicine.
http://www.mdc-berlin.de/20902775/en/research/core_facilities/cf_massspectromety_bimsb
Coordination of bacterial proteome with metabolism by cyclic AMP signaling
Conghui You, Hiroyuki Okano, Sheng Hui, Zhongge Zhang, Minsu Kim, et al.
Nature (15 August 2013); 500, 301–306 http://dx.doi.org:/10.1038/nature12446
The cyclic AMP (cAMP)-dependent catabolite repression effect in Escherichia coli is among the most intensely studied regulatory processes in biology. However,
- the physiological function(s) of cAMP signalling and its molecular triggers remain elusive.
Here we use a quantitative physiological approach to show that
- cAMP signalling tightly coordinates the expression of catabolic proteins with biosynthetic and ribosomal proteins,
- in accordance with the cellular metabolic needs during exponential growth.
The expression of carbon catabolic genes increased linearly
- with decreasing growth rates upon limitation of carbon influx,
- but decreased linearly with decreasing growth rate upon limitation of nitrogen or sulphur influx.
In contrast, the expression of biosynthetic genes showed the opposite linear growth-rate dependence as the catabolic genes. A coarse-grained mathematical model provides a quantitative framework for understanding and predicting
- gene expression responses to catabolic and anabolic limitations.
A scheme of integral feedback control featuring the inhibition of cAMP signalling by metabolic precursors is proposed and validated. These results reveal a key physiological role of
- cAMP-dependent catabolite repression: to ensure that proteomic resources are spent on distinct metabolic sectors as needed
- in different nutrient environments.
Our findings underscore the power of quantitative physiology in unravelling the underlying functions of complex molecular signalling networks.
Exosomes from bone marrow mesenchymal stem cells contain a microRNA that promotes dormancy in metastatic breast cancer cells
Makiko Ono1, Nobuyoshi Kosaka1, Naoomi Tominaga1, Yusuke Yoshioka1, Fumitaka Takeshita1, et al.
Sci. Signal., July 2014; 7(332), p. ra63 http://dx.doi.org:/10.1126/scisignal.2005231
Breast cancer patients often develop metastatic disease years after resection of the primary tumor. The patients are asymptomatic because the disseminated cells appear to become dormant and are undetectable. Because the proliferation of these cells is slowed, dormant cells are often unresponsive to traditional chemotherapies that exploit the rapid cell cycling of most cancer cells. We generated a bone marrow–metastatic human breast cancer cell line (BM2) by tracking and isolating fluorescent-labeled MDA-MB-231 cells that disseminated to the bone marrow in mice. Coculturing BM2 cells with bone marrow mesenchymal stem cells (BM-MSCs) isolated from human donors revealed that BM-MSCs suppressed the proliferation of BM2 cells, decreased the abundance of stem cell–like surface markers, inhibited their invasion through Matrigel Transwells, and decreased their sensitivity to docetaxel, a common chemotherapy agent. Acquisition of these dormant phenotypes in BM2 cells was also observed by culturing the cells in BM-MSC–conditioned medium or with exosomes isolated from BM-MSC cultures, which were taken up by BM2 cells. Among various microRNAs (miRNAs) increased in BM-MSC–derived exosomes compared with those from adult fibroblasts, overexpression of miR-23b in BM2 cells induced dormant phenotypes through the suppression of a target gene, MARCKS, which encodes a protein that promotes cell cycling and motility. Metastatic breast cancer cells in patient bone marrow had increased miR-23b and decreasedMARCKS expression. Together, these findings suggest that exosomal transfer of miRNAs from the bone marrow may promote breast cancer cell dormancy in a metastatic niche.
Citation:
- Ono, N. Kosaka, N. Tominaga, Y. Yoshioka, F. Takeshita, R. Takahashi, M. Yoshida, H. Tsuda, K. Tamura, and T. Ochiya, Exosomes from bone marrow mesenchymal stem cells contain a microRNA that promotes dormancy in metastatic breast cancer cells. Sci. Signal.7, ra63 (2014).
Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis.
Andrabi SA1, Umanah GK2, Chang C3, Stevens DA4, Karuppagounder SS2, Gagné JP5, Poirier GG5, Dawson VL6, Dawson TM7.
Proc Natl Acad Sci U S A. 2014 Jul 15; 111(28):10209-14. http://dx.doi.org:/10.1073/pnas.1405158111
Excessive poly(ADP-ribose) (PAR) polymerase-1 (PARP-1) activation kills cells via a cell-death process designated “parthanatos” in which PAR induces the mitochondrial release and nuclear translocation of apoptosis-inducing factor to initiate chromatinolysis and cell death. Accompanying the formation of PAR are the reduction of cellular NAD(+) and energetic collapse, which have been thought to be caused by the consumption of cellular NAD(+) by PARP-1. Here we show that the bioenergetic collapse following PARP-1 activation is not dependent on NAD(+) depletion. Instead PARP-1 activation initiates glycolytic defects via PAR-dependent inhibition of hexokinase, which precedes the NAD(+) depletion in N-methyl-N-nitroso-N-nitroguanidine (MNNG)-treated cortical neurons. Mitochondrial defects are observed shortly after PARP-1 activation and are mediated largely through defective glycolysis, because supplementation of the mitochondrial substrates pyruvate and glutamine reverse the PARP-1-mediated mitochondrial dysfunction. Depleting neurons of NAD(+) with FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, does not alter glycolysis or mitochondrial function. Hexokinase, the first regulatory enzyme to initiate glycolysis by converting glucose to glucose-6-phosphate, contains a strong PAR-binding motif. PAR binds to hexokinase and inhibits hexokinase activity in MNNG-treated cortical neurons. Preventing PAR formation with PAR glycohydrolase prevents the PAR-dependent inhibition of hexokinase. These results indicate that bioenergetic collapse induced by overactivation of PARP-1 is caused by PAR-dependent inhibition of glycolysis through inhibition of hexokinase.
PMID:24987120 PMCID: PMC4104885 [Available on 2015/1/15]
Aim24 stabilizes respiratory chain supercomplexes and is required for efficient respiration
Deckers M1, Balleininger M1, Vukotic M1, Römpler K1, Bareth B1, Juris L1, Dudek J2.
FEBS Lett. 2014 Jun 10. pii: S0014-5793(14)00458-X. http://dx.doi.org:/10.1016/j.febslet.2014.06.006
The mitochondrial respiratory chain is essential for the conversion of energy derived from the oxidation of metabolites into the membrane potential, which drives the synthesis of ATP. The electron transporting complexes bc1 complex and the cytochrome c oxidase assemble into large supercomplexes, allowing efficient energy transduction. Currently, we have only limited information about what determines the structure of the supercomplex. Here, we characterize Aim24 in baker’s yeast as a protein, which is integrated in the mitochondrial inner membrane and is required for the structural integrity of the supercomplex. Deletion of AIM24 strongly affects activity of the respiratory chain and induces a growth defect on non-fermentable medium. Our data indicate that Aim24 has a function in stabilizing the respiratory chain supercomplexes. PMID: 24928273
KEYWORDS: Aim24; Membrane protein; Metabolism; Mitochondria; Respiration; Supercomplex
Read Full Post »















































































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
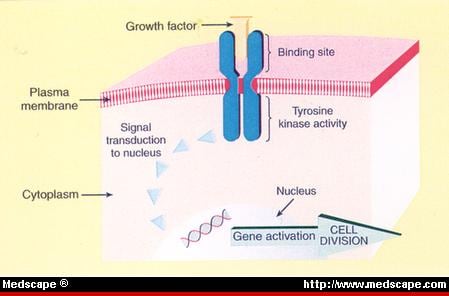{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
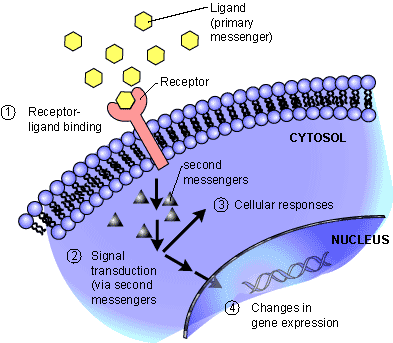{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}