Nutrition and Aging
Curator: Larry H Bernstein, MD, FCAP
UPDATED 10/26/2015
Hospital Malnutrition: Prevalence, Identification and Impact on Patients and the Healthcare System
Lisa A. Barker,1,* Belinda S. Gout,1 and Timothy C. Crowe2
Author information ► Article notes ► Copyright and License information ►
Int J Environ Res Public Health. 2011 Feb; 8(2): 514–527.
Published online 2011 Feb 16. doi: 10.3390/ijerph8020514
This article has been cited by other articles in PMC.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3084475/
Malnutrition is a debilitating and highly prevalent condition in the acute hospital setting, with Australian and international studies reporting rates of approximately 40%. Malnutrition is associated with many adverse outcomes including depression of the immune system, impaired wound healing, muscle wasting, longer lengths of hospital stay, higher treatment costs and increased mortality. Referral rates for dietetic assessment and treatment of malnourished patients have proven to be suboptimal, thereby increasing the likelihood of developing such aforementioned complications. Nutrition risk screening using a validated tool is a simple technique to rapidly identify patients at risk of malnutrition, and provides a basis for prompt dietetic referrals. In Australia, nutrition screening upon hospital admission is not mandatory, which is of concern knowing that malnutrition remains under-reported and often poorly documented. Unidentified malnutrition not only heightens the risk of adverse complications for patients, but can potentially result in foregone reimbursements to the hospital through casemix-based funding schemes. It is strongly recommended that mandatory nutrition screening be widely adopted in line with published best-practice guidelines to effectively target and reduce the incidence of hospital malnutrition.
Keywords: diagnosis-related groups, economics, hospital, malnutrition, nutrition assessment, screening
What Is Malnutrition?
Malnutrition is a broad term that can be used to describe any imbalance in nutrition; from over-nutrition often seen in the developed world, to under-nutrition seen in many developing countries, but also in hospitals and residential care facilities in developed nations. Malnutrition can develop as a consequence of deficiency in dietary intake, increased requirements associated with a disease state, from complications of an underlying illness such as poor absorption and excessive nutrient losses, or from a combination of these aforementioned factors [1,2]. Malnutrition is associated with negative outcomes for patients, including higher infection and complication rates [3–6], increased muscle loss [6–8], impaired wound healing [4,9], longer length of hospital stay [10–12] and increased morbidity and mortality [13–17].
Recently, the definition of malnutrition has been clarified by the European Society of Parenteral and Enteral Nutrition (ESPEN) to highlight the differences between cachexia, sarcopenia (loss of muscle mass and function) and malnutrition [18]. Cachexia can be defined as a “multifactorial syndrome characterized by severe body weight, fat and muscle loss and increased protein catabolism due to underlying disease(s)” [18]. Therefore, malnutrition seen in hospitalised patients is often a combination of cachexia (disease-related) and malnutrition (inadequate consumption of nutrients) as opposed to malnutrition alone. Within the context of this review, the definition of malnutrition adopted refers to the complex interplay between underlying disease, disease-related metabolic alterations and the reduced availability of nutrients (because of reduced intake, impaired absorption and/or increased losses or a combination of these) which is a combination of cachexia and malnutrition [18].
In 1859, Florence Nightingale wrote about hospitalised soldiers during the Crimea war, starving amongst plenty of food [19]. Over 100 years later, beginning from the 1970s, numerous authors have reported malnutrition rates in hospital patients to be approximately 35%, with 30 to 55% of patients entering acute hospitals being at risk of malnutrition [20–24]. Studies have also reported on factors which contribute to malnutrition (see Table 1), consequences of malnutrition and the benefit nutrition support can offer malnourished patients [15,25–27].
Protein-Energy Malnutrition in Elderly Medical Patients
Constans MD†,*, Y. Bacq MD‡, J.-F. Bréchot MD§, J.-L. Guilmot MD‡, P. Choutet MD‡and F. Lamisse MD‡
Journal of the American Geriatrics Society Mar 1992; Volume 40, Issue 3: 263–268
online: 27 APR 2015 http://dx.doi.org:/10.1111/j.1532-5415.1992.tb02080.x
Consecutive sample of 324 hospitalized patients ≥70 years (86.4% of eligible patients). Norms of measurements were obtained from a referred sample of healthy control subjects (26 males and 36 females).
Main Outcome Measures Mid-arm circumference, triceps skinfold thickness, serum albumin, prealbumin, and retinol-binding protein levels were measured in patients at admission and on the 15th day.
Results (1) Prevalence of PEM was 30% in male and 41% in female patients. (2) Both mid-arm circumference and serum albumin level decreased over the first 15 days of hospital stay (53 patients, paired t test, P < 0.05). Triceps skinfold thickness did not change. (3) A step-wise discriminant-function analysis determined the utility of the parameters at admission as predictors of in-hospital mortality before the 15th day. Mid-arm circumference, triceps skinfold thickness, albumin, and prealbumin levels, as well as age, are predictors of in-hospital mortality, with 73% sensitivity, 69% specificity, and 70% of correctly classified patients of both sexes.
Conclusions Parameters used are predictors for short-term in-hospital mortality of elderly patients hospitalized in an acute medical unit. The lean body mass is preferentially mobilized for energy during hospitalization.
Downsizing of Lean Body Mass is a Key Determinant of Alzheimer’s Disease
Yves Ingenbleek,∗ and Larry H. Bernstein
Laboratory of Nutrition, Faculty of Pharmacy, University Louis Pasteur, Strasbourg, France; Laboratory of Clinical Pathology, New York Methodist Hospital, Weill-Cornell University, New York, NY, US
Journal of Alzheimer’s Disease 44 (2015) 745–754 http://dx.doi.org:/10.3233/JAD-141950
Lean body mass (LBM) encompasses all metabolically active organs distributed into visceral and structural tissue compartments and collecting the bulk of N and K stores of the human body. Transthyretin (TTR) is a plasma protein mainly secreted by the liver within a trimolecular TTR-RBP-retinol complex revealing from birth to old age strikingly similar evolutionary patterns with LBM in health and disease. TTR is also synthesized by the choroid plexus along distinct regulatory pathways. Chronic dietary methionine (Met) deprivation or cytokine-induced inflammatory disorders generates LBM downsizing following differentiated physiopathological processes. Met-restricted regimens downregulate the transsulfuration cascade causing upstream elevation of homocysteine (Hcy) safeguarding Met homeostasis and downstream drop of hydrogen sulfide (H2S) impairing anti-oxidative capacities. Elderly persons constitute a vulnerable population group exposed to increasing Hcy burden and declining H2S protection, notably in plant-eating communities or in the course of inflammatory illnesses. Appropriate correction of defective protein status and eradication of inflammatory processes may restore an appropriate LBM size allowing the hepatic production of the retinol circulating complex to resume, in contrast with the refractory choroidal TTR secretory process. As a result of improved health status, augmented concentrations of plasma-derived TTR and retinol may reach the cerebrospinal fluid and dismantle senile amyloid plaques, contributing to the prevention or the delay of the onset of neurodegenerative events in elderly subjects at risk of Alzheimer’s disease.
Plasma transthyretin (TTR) was initially proposed as an index of protein-depleted states following field surveys undertaken in Senegal (West Africa) on children suffering from varying stages of malnutrition ranging from cachectic marasmus to edematous kwashiorkor [1]. The serum analyte is now widely measured in developing areas for the nutritional follow-up of underprivileged populations [2, 3] and in developed countries to screen hospitalized patients who require dietary management [4, 5]. Several neurological investigations have recently reported the innovative observation that the same TTR biomarker impacts on the outcome of Alzheimer’s disease (AD) [6,7], raising the basic premise that alterations of protein status might be implicated in neurodegenerative disorders. Preliminary studies have indeed suggested that the reliability of the TTR indicator is based on its accurately identifying loss of lean body mass (LBM) [8] effecting metabolically active tissues in health and disease. The below review describes the unrecognized correlations linking LBM entity to TTR fluctuations and the mechanisms whereby LBM downsizing, as determined by declining TTR plasma concentrations, generates significant public health consequences in neurodeteriorating morbidities, taking AD as exemplary.
Plasma transthyretin as biomarker of lean body mass and catabolic states
Yves Ingenbleek, 1 MD PhD & Larry H. Bernstein, 2 MD
3Laboratory of Nutrition, Faculty of Pharmacy, University Louis Pasteur, Strasbourg, France; and 4Laboratory of Clinical Pathology, New York Methodist Hospital, Weill-Cornell University, New York, NY
Adv Nutr 2015; 6:1–9.
Plasma transthyretin (TTR) is a plasma protein secreted by the liver which circulates bound to retinol-binding protein (RBP4) and its retinol ligand. TTR is the sole plasma protein revealing from birth to old age evolutionary patterns closely superimposable to those of lean body mass (LBM) and working as its best surrogate analyte. Any alteration in energy- to-protein balance impairs the accretion of LBM reserves and causes early depression of TTR production. In acute inflammatory states, cytokines induce urinary leakage of nitrogenous catabolites, deplete LBM stores and cause abrupt drop of TTR-RBP4 values. As a result, thyroxine and retinol ligands are released in free form, creating a second frontline strengthening that primarily initiated by cytokines. Malnutrition and inflammation thus keep 10. in check TTR and RBP4 secretion using distinct and unrelated physiological pathways but operate in concert to downregulate LBM stores. The TTR biomarker integrates these opposite mechanisms at any time, constituting an ideally suited tool to grade residual LBM resources still available for metabolic responses, hence predicting outcome of the most interwoven disease conditions.
Cognitive Impairments in Elderly Diabetic Patients: Understanding the Risks for Better Management
Medscape Medical News from the
Visit Medscape in Hall B Booth #B13:31
Medscape Diabetes & Endocrinology
COMMENTARY
Lyse Bordier, MD
http://www.medscape.com/viewarticle/852112
Editor’s Note: The following is an edited, translated transcript of a presentation by Professor Lyse Bordier, a diabetologist at Military Hospital Bégin, Saint-Mandé, France, summarizing her lecture at the European Association for the Study of Diabetes (EASD) 2015 AnnualMeeting in Stockholm, Sweden.
Hello. I am Professor Lyse Bordier. I work at the Bégin Military Hospital, in Saint-Mandé, France, and I had the pleasure of participating in a symposium organized by the EASD 2015 conference in Stockholm on elderly patients, specifically on cognitive impairments.
A Public Health Problem
Dementia and cognitive impairments are a major problem; Alzheimer disease accounts for 70% of all cases of dementia. The other main causes are vascular dementias and mixed dementias. They are a real public health problem; it is estimated that, in the United States, 5.2 million people have this condition, and worldwide, every 7 seconds, a new case of dementia is diagnosed.[1,2] In France, for example, it was estimated in 2010 that 750,000-850,000 people had dementia and that this figure will increase by a factor of 2.4 by the year 2050.
Diabetes is an important contributor to the development of cognitive impairments, all the way up to dementia. In Europe, it is estimated that nearly 25% of people over age 85 years have dementia. Its prevalence and incidence are higher in women than in men.[2] We know that the complications of diabetes have changed over the years and that acute metabolic complications are, in the end, much less important. With the improvement in life expectancy in our diabetic patients, who are now better treated thanks to better therapeutic management, new complications have arisen, such as renal failure, heart failure, and, of course, geriatric complications, which are, in large part, cognitive disorders.[3]
Prevalence Underestimated by Physicians
These cognitive impairments are common and largely underestimated. This was clearly shown in the GERODIAB study,[4] which included a cohort of 987 patients over the age of 70 years. At inclusion, the physicians reported that 11% of their patients had cognitive impairments and that 3% had dementia. In actual fact, 25% of the patients had impaired cognitive functions, with a Mini-Mental State Examination (MMSE) score under 25. The prevalence is therefore significantly underestimated by physicians.
Cognitive impairments are more prevalent and more severe in diabetics than in nondiabetics. It is estimated that the risk for cognitive impairments and that for dementia are 20% to 70% and 60% higher, respectively, in the presence of diabetes.[5] Furthermore, the risk for Alzheimer dementia is considerable, it being 40% higher in diabetics. As expected (given the combination of the other cardiovascular risk factors), the increase in the risk is even greater for vascular dementia, with an odds ratio of 2.38.[6]
Mechanisms
What are the mechanisms in the development of cognitive impairments and dementia? There are many mechanisms, and they are often poorly understood. Hyperglycemia plays a very important role as a direct result of oxidative stress, of advanced glycation end-products, but also as a result of micro- and macroangiopathy, hypertension, and dyslipidemia.[7,8] Other major factors, such as hypoglycemia,[9-12]play an extremely important role in the development of cognitive impairments. As well, a great deal of literature has been published lately on the role of inflammation[13] and genetic factors. Another widely known aspect is insulin resistance, which increases the risk for dementia at a fairly early stage by 40%[14,15]; this already during the metabolic syndrome, even before the onset of type 2 diabetes.
http://img.medscape.com/article/852/112/852112-Figure1.jpg
Figure. Multiple and poorly understood mechanisms of cognitive impairments and dementia. HTA = arterial hypertension. Adapted from Buysschaert M, et al.[16]
What Are the Consequences of Cognitive Impairments?
Cognitive impairments lead to a number of complications, including a reduction in life expectancy. In the GERODIAB cohort, we found, after 2 years of follow-up, that the mortality rate was twice as high in the patients with an MMSE score <24 compared with those with an MMSE score >24. In this study, the patients with a lower MMSE score had less well-controlled diabetes, were usually treated with insulin, and had heart failure and cerebrovascular complications more often. Very surprisingly, hypoglycemia was not more prevalent in these patients, perhaps because, being less independent, they were better managed by care teams.[17]
Cognitive impairments lead to geriatric complications, such as malnutrition, falls, and a loss of autonomy. They also promote social and family isolation and iatrogenic accidents, as well as depression, which can both mask cognitive impairments and exacerbate an underlying dementia. Another important aspect is that cognitive impairments increase the risk for hypoglycemia. This has been shown very clearly in all of the studies. There is, in fact, a bidirectional link between dementia and hypoglycemia: Hypoglycemia doubles the risk for dementia, and dementia triples the risk for hypoglycemia.[18]
Screening and Management
What do we do when a patient presents with cognitive impairments? First, they should be identified so that they can be managed. We need to be vigilant for certain little signs: changes in the patient’s behavior (eg, a patient who forgets his appointments, whose personal hygiene has declined, who is less diligent in keeping his blood glucose diary, and, lastly, who has an unexplained diabetic imbalance). We should also know how to use simple tests, such as the MMSE, which provides an overall assessment of space-time orientation, cognitive functions, language functions, and calculation, and how to assess the patient’s autonomy and loss of autonomy.[19] Next, we should, as per the recommendations of the American Diabetes Association[20] and the EASD, individualize the glycemic goals, taking into account, in the most fragile, elderly patients, cognitive status, the level of autonomy, depression, nutritional status—in particular, sarcopenia, which can coexist with obesity, and the risk for hypoglycemia.[21]
We should therefore avoid overtreating the most fragile patients (those at greatest risk for hypoglycemia), but neither should we undertreat patients who have a long life expectancy and who could develop micro- and macroangiopathic complications.
One last aspect, which is very important, is the family. Help needs to be provided to prevent the patient’s loss of autonomy.[21] Lastly, I think that cognitive decline should be added to the already long list of degenerative complications of diabetes.
Transthyretin Blocks Retinol Uptake and Cell Signaling by the Holo-Retinol-Binding Protein Receptor STRA6
+Author Affiliations
- aDepartments of Pharmacology
- bNutrition, Case Western Reserve University School of Medicine, Cleveland, Ohio, USA
- cInstitut de Génétique et de Biologie Moléculaire et Cellulaire, Centre National de la Recherche Scientifique (UMR7104), Institut National de la Santé et de la Recherche Médicale U964, Université de Strasbourg, Illkirch, France
Vitamin A is secreted from cellular stores and circulates in blood bound to retinol-binding protein (RBP). In turn, holo-RBP associates in plasma with transthyretin (TTR) to form a ternary RBP-retinol-TTR complex. It is believed that binding to TTR prevents the loss of RBP by filtration in the kidney. At target cells, holo-RBP is recognized by STRA6, a plasma membrane protein that serves a dual role: it mediates uptake of retinol from extracellular RBP into cells, and it functions as a cytokine receptor that, upon binding holo-RBP, triggers a JAK/STAT signaling cascade. We previously showed that STRA6-mediated signaling underlies the ability of RBP to induce insulin resistance. However, the role that TTR, the binding partner of holo-RBP in blood, plays in STRA6-mediated activities remained unknown. Here we show that TTR blocks the ability of holo-RBP to associate with STRA6 and thereby effectively suppresses both STRA6-mediated retinol uptake and STRA6-initiated cell signaling. Consequently, TTR protects mice from RBP-induced insulin resistance, reflected by reduced phosphorylation of insulin receptor and glucose tolerance tests. The data indicate that STRA6 functions only under circumstances where the plasma RBP level exceeds that of TTR and demonstrate that, in addition to preventing the loss of RBP, TTR plays a central role in regulating holo-RBP/STRA6 signaling.
INTRODUCTION
Vitamin A (retinol [ROH]) plays critical roles both in the embryo and in the adult, where it regulates multiple cellular processes and is essential for embryonic development, reproduction, immune function, and vision (29, 32, 33). The vitamin exerts many of its biological activities by giving rise to active metabolites: the visual chromophore 11-cis-retinaldehyde and retinoic acid (RA), which regulates gene transcription by activating specific nuclear receptors (11, 27). ROH is stored in various tissues, including white adipose tissue (WAT), lung, and retinal pigment epithelium in the eye, but its main storage site is the liver. ROH is secreted from storage into the circulation bound to retinol-binding protein (RBP), a 21-kDa polypeptide that contains one binding site for ROH. In most mammals, ROH-bound RBP (holo-RBP) does not circulate alone but is associated with another protein called transthyretin (TTR), a 56-kDa homotetramer that, in addition to associating with RBP, functions as a carrier for thyroid hormones (23, 24). ROH thus reaches target tissues bound in a holo-RBP-TTR complex that, under normal circumstances, displays a 1:1 molar stoichiometry. It is believed that binding of RBP to TTR serves to prevent the loss of the smaller protein from blood by filtration in the glomeruli. The concentration of the holo-RBP-TTR complex in plasma is kept constant at 1 to 2 μM except in extreme cases of vitamin A deficiency or in disease states. Notably, RBP levels are markedly elevated in blood of obese mice and humans, and it was reported that, under these circumstances, the protein induces insulin resistance (35).
Association with the TTR-RBP complex allows the poorly soluble ROH to circulate in blood, but the vitamin dissociates from RBP prior to entering cells. It was proposed that, due to its hydrophobic nature, ROH can readily move from extracellular RBP into cells by diffusion across the plasma membranes at fluxes that are dictated by its extracellular-to-intracellular concentration gradient (10,14, 20, 21). However, it has also been suggested that uptake of ROH from circulating holo-RBP is mediated by a cell surface receptor (13, 28). Indeed, a plasma membrane protein termed STRA6 (stimulated by retinoic acid 6) was found to bind holo-RBP and transport ROH into cells (15). Our recent studies revealed that, in addition to its function as an ROH transporter, STRA6 is a cytokine receptor. We thus found that binding of holo-RBP triggers phosphorylation of a tyrosine residue in the cytosolic domain of STRA6, resulting in recruitment and activation of the Janus kinase JAK2 and, in a cell-dependent manner, the transcription factors STAT3 or STAT5. Holo-RBP thus activates STRA6-mediated signaling that culminates in upregulation of STAT target genes (2, 4). As STAT target genes in white adipose tissue and muscle include Suppressor of cytokine signaling 3 (Socs3), a potent inhibitor of insulin signaling (8), these findings suggested a rationale for understanding how elevated serum levels of RBP in obese animals induce insulin resistance (35). Additional studies showed that activation of STRA6 is triggered not simply by binding of holo-RBP but by a STRA6-mediated translocation of ROH from extracellular holo-RBP to an intracellular acceptor, the retinol-binding protein CRBP-I. Importantly, this movement was found to be critically linked to the intracellular metabolism of ROH (5). The data further established that ROH uptake and signaling by STRA6 are interdependent, i.e., that activation of a JAK2/STAT cascade by the receptor requires ROH uptake and, conversely, that phosphorylation of STRA6 is essential for enabling ROH transport to proceed (5).
While these recent studies provided surprising new insights into the involvement of STRA6 in vitamin A biology, the role that TTR, the binding partner of holo-RBP in blood, may play in STRA6-mediated functions remained unknown. Here, we show that TTR blocks the ability of holo-RBP to associate with STRA6 and thereby effectively suppresses both STRA6-mediated ROH uptake and STRA6-initiated cell signaling. We show further that, consequently, TTR protects mice from RBP-induced insulin resistance. The data indicate that, in addition to preventing the loss of RBP by filtration in the kidney, TTR plays a central role in regulating holo-RBP/STRA6 signaling.
….
TTR inhibits STRA6-mediated uptake of ROH from holo-RBP. In most mammals, holo-RBP circulates in blood in complex with transthyretin (TTR). To begin to examine the effect of TTR on STRA6 function, hepatocarcinoma HepG2 cells, which endogenously express STRA6, were used to compare the cellular uptake of ROH from holo-RBP and from TTR-bound holo-RBP. Recombinant RBP and TTR were expressed in E. coli and purified (see Materials and Methods). HepG2 cells were treated with RBP complexed with [3H]retinol at a 1 μM concentration, similar to the serum RBP level, or with 1 μM [3H]retinol-labeled RBP complexed with TTR at a 1:1 molar stoichiometry, similar to that found in blood (24). Media were removed, cells washed, and organic compounds extracted from the cells into ethanol, and the amount of [3H]retinol taken up within the incubation period was measured by scintillation counting. The rates of uptake of retinol under the assay conditions were constant during the initial 5 min (Fig. 1a), and subsequent experiments were carried out with a single 3-min time point, well within the initial linear rate. The rate of ROH uptake from the holo-RBP-TTR complex was lower than that of the uptake from holo-RBP alone (Fig. 1a). Moreover, increasing the TTR/RBP ratio by increasing the concentration of TTR inhibited ROH uptake in a dose-dependent manner (Fig. 1b). The dose response of the initial rate of ROH transport from holo-RBP showed a two-phase behavior comprised of an initial saturable component, likely attributable to STRA6-mediated uptake, followed by a nonsaturable phase, reflecting passive diffusion of ROH across the plasma membranes (Fig. 1c). In contrast, uptake of ROH from the holo-RBP-TTR complex displayed a single, nonsaturable phase (Fig. 1c). These observations suggest that TTR does not impede the ability of ROH to enter cells by passive diffusion but effectively blocks ROH transport mediated by STRA6. In agreement with this notion, increasing the expression level of STRA6 in HepG2 cells (Fig. 1d) facilitated ROH uptake from holo-RBP in a dose-responsive manner but had no effect on transport of ROH from TTR-bound holo-RBP (Fig. 1e). Also in agreement, decreasing the expression of STRA6 in HepG2 cells (Fig. 1f) or in NIH 3T3-L1 adipocytes (Fig. 1g) reduced the rate of ROH uptake from holo-RBP but did not affect uptake from TTR-bound holo-RBP. The observation that, in both cell lines, rates of uptake from the holo-RBP-TTR complex were similar to those observed in the absence of STRA6 supports the conclusion that TTR specifically inhibits STRA6-mediated transport.
View larger version:
FIG 1
TTR inhibits STRA6-mediated uptake of ROH from holo-RBP. (a) Uptake of [3H]ROH by HepG2 cells treated with RBP-[3H]ROH or RBP-[3H]ROH-TTR (1 μM) for denoted times. (b) Uptake of [3H]ROH by HepG2 cells treated with the denoted concentrations of RBP-[3H]ROH or RBP-[3H]ROH-TTR for 3 min. (c) Uptake of [3H]ROH by HepG2 cells following a 3-min incubation with 1 μM RBP-[3H]ROH in the presence of denoted concentrations of TTR. (d) Levels of STRA6 mRNA in HepG2 cells transfected with various amounts of STRA6 cDNA. (e) Effect of increasing the expression level of STRA6 in HepG2 cells on uptake of [3H]ROH from RBP-[3H]ROH or RBP-[3H]ROH-TTR (1 μM, 3 min). (f) Top, expression level of STRA6 in HepG2 cells transfected with an empty vector (e.v.) or vector harboring STRA6shRNA. Bottom, effect of decreasing the expression level of STRA6 in HepG2 cells on uptake of [3H]ROH from RBP-[3H]ROH or from RBP-[3H]ROH-TTR (1 μM, 3 min). (g) Top, expression level of STRA6 in NIH 3T3-L1 cells transfected with an empty vector (e.v.) or a vector harboring STRA6shRNA. Bottom, effect of decreasing the expression level of STRA6 in NIH 3T3-L1 adipocytes on uptake of [3H]ROH from RBP-[3H]ROH or from RBP-[3H]ROH-TTR (1 μM, 3 min). (h) Twelve-week-old WT and STRA6-null male mice were injected intraperitoneally with RBP-[3H]ROH (100 μl, 0.1 mCi, 1 μM). Two hours later, tissues were isolated, weighed, and homogenized, and [3H]ROH was quantified. Data are means ± standard errors of the means; *, P < 0.01 for RBP-ROH-treated versus RBP-ROH-TTR-treated groups. All P values were calculated using a two-tailed Student t test.
The effect of TTR on ROH uptake from holo-RBP was then examined in vivo using our newly generated STRA6-null mice (26). Twelve-week-old wild-type (WT) and STRA6-null male mice were injected intraperitoneally with [3H]ROH-labeled holo-RBP or with holo-RBP complexed with TTR, and ROH uptake into tissues was assessed 2 h later. Uptake of ROH into the STRA6-expressing tissues WAT, skeletal muscle, and the eye was modestly but significantly lower in STRA6-null than in WT mice (Fig. 1h), reflecting that the contribution of STRA6 to overall vitamin A uptake by tissues in vivo is small. ROH uptake from TTR-bound holo-RBP was all but identical to that observed in STRA6−/− animals (Fig. 1h). Neither ablation of STRA6 nor the presence of TTR affected ROH uptake by the liver, an organ that does not express STRA6 (Fig. 1h). Hence, TTR specifically inhibits STRA6-mediated uptake of ROH in vivo.
TTR inhibits the association of holo-RBP with STRA6.STRA6 may bind the ternary RBP-ROH-TTR complex or, alternatively, it may recognize only free holo-RBP. To dissect these possibilities, we considered that, unlike in most mammals, holo-RBP in zebrafish (Danio rerio) does not associate with TTR. Thus, presumably, zebrafish STRA6 does not contain a TTR-binding region, and while ROH uptake by the mammalian STRA6 may involve recognition of TTR, ROH uptake by zebrafish STRA6 (dSTRA6) will not. In these experiments, NIH 3T3 fibroblasts, which do not endogenously express STRA6, were used. We previously showed that ROH metabolism is essential both for STRA6-mediated ROH transport and for holo-RBP-induced STRA6 signaling (5). Hence, to enable STRA6 action in these cells, an NIH 3T3 line in which ROH metabolism is enhanced by stably overexpressing lecithin:ROH-acyltransferase (LRAT), which catalyzes ROH esterification, was generated. Ectopic overexpression of either hSTRA6 or dSTRA6 in LRAT-expressing NIH 3T3 fibroblasts enhanced ROH uptake from holo-RBP to a similar extent, and introduction of TTR similarly decreased the rate of uptake (Fig. 2a andb). The similarity of the response of dSTRA6, which is unlikely to contain a TTR-binding capability, to that of hSTRA6 suggests that STRA6 in both species recognizes only free and not TTR-bound holo-RBP.
View larger version:
FIG 2
STRA6 does not bind the holo-RBP-TTR complex. (a) NIH 3T3 cells stably overexpressing LRAT were transfected with an empty vector (e.v.) or with expression vectors encoding human (hSTRA6) or zebrafish (dSTRA6) STRA6, resulting in similar levels of mRNAs. (b) Uptake of [3H]ROH from RBP-[3H]ROH or RBP-[3H]ROH-TTR (1 μM, 3 min) by cells expressing hSTRA6 or dSTRA6. (c) RBP-ROH (R-R) or RBP-ROH-TTR (R-R + TTR) (1 μM) was incubated with the chemical cross-linker bis(sulfosuccinimidyl) suberate (0.5 mM) for 14 h. Proteins were resolved by SDS-PAGE and visualized by Coomassie blue staining. (d) Cross-linked complexes and additional cross-linker (0.5 mM) were added to HepG2 cells transfected with an e.v. or with a vector encoding histidine-tagged STRA6. Following a 15-min incubation, his-STRA6 was immunoprecipitated using antibodies against the tag, and precipitated RBP and STRA6 were visualized by immunoblotting. (e) Fluorescence titrations of RBP and its F96A/L97A mutant (1 μM) with ROH. Progress of titrations was monitored by following the increase in ROH fluorescence upon binding to the protein (λex = 330 nm; λem = 460 nm). (f) Fluorescence anisotropy titrations of holo-RBP and holo-RBP-F96A/L97A (3 μM) with TTR. Progress of titrations was monitored by measuring the fluorescence anisotropy of bound ROH (λex = 330 nm; λem = 460 nm). (g) Uptake of [3H]ROH from holo-RBP-F96A/L97A (1 μM, 3 min) in the presence or absence of TTR. Data are means ± standard errors of the means (SEM). *, P < 0.01 versus cells transfected with an empty vector; **, P = 0.01 versus cells transfected with an empty vector and treated with RBP-ROH. All Pvalues were calculated using a two-tailed Student t test.
The question of whether STRA6 binds free or TTR-bound holo-RBP was then directly addressed. Recombinant holo-RBP was incubated alone or in the presence of TTR with the chemical cross-linker bis(sulfosuccinimidyl) suberate (0.5 mM, 14 h), resulting in efficient cross-linking of the holo-RBP-TTR complex (Fig. 2c). The mixtures and additional cross-linker were added to NIH 3T3 cells ectopically overexpressing histidine-tagged STRA6. STRA6 was immunoprecipitated, and precipitated proteins were resolved by SDS-PAGE and immunoblotted for RBP-containing complexes (Fig. 2d). Cross-linking of cells with holo-RBP resulted in the appearance of a band with a molecular mass of ∼100 kDa, corresponding to that of an RBP-bound STRA6. No such band was observed in cells cross-linked with the RBP-ROH-TTR complex, and no bands that might correspond to a STRA6-RBP-TTR (∼150 kDa) appeared. The data thus indicate that STRA6 associates only with free holo-RBP and that the presence of TTR prevents the association.
To further examine whether TTR inhibits STRA6-mediated ROH uptake by preventing holo-RBP from binding to the receptor, an RBP mutant defective in its ability to bind TTR was generated. The reported three-dimensional crystal structure of the holo-RBP-TTR complex suggests that the interactions between the two proteins are mediated by several residues, including Phe96 and Leu97 (18). An RBP mutant in which these residues were replaced with alanines (RBP-F96A/L97A) was thus generated. The mutations did not alter the affinity of RBP for retinol (Fig. 2e), indicating that the overall fold of the mutant is intact. As expected, the F96A/L97A mutations disrupted the association of RBP with TTR (Fig. 2f). Measurements of ROH uptake showed that, in contrast with its inhibitory activity on ROH uptake from WT-RBP, TTR had no effect on ROH uptake from RBP-F96A/L97A (Fig. 2g). These observations further establish that TTR inhibits STRA6-mediated ROH uptake by sequestering holo-RBP and not by direct association with the receptor.
TTR inhibits holo-RBP-induced STRA6 signaling.The effect of TTR on RBP-induced STRA6 signaling was then examined using NIH 3T3-L1 adipocytes. We previously showed that in these cells, activation of STRA6 by holo-RBP triggers a JAK2/STAT5 cascade to induce the STAT target genes SOCS3 and PPARγ and inhibit insulin responses (2). Preadipocytes NIH 3T3-L1 cells were grown 2 days past confluence and induced to differentiate using a standard hormone mix (10 μg/ml insulin, 0.5 mM 3-isobutyl-1-methylxanthine [IBMX], 0.25 mM dexamethasone). Three days later, media were replaced and cells grown for 4 days. Differentiation was verified by monitoring lipid accumulation and by examining the expression of the adipocyte marker FABP4 (3). As expected, treatment of differentiated adipocytes with holo-RBP (R-R) increased the phosphorylation levels of JAK2 and STAT5 (Fig. 3a). In contrast, the holo-RBP-TTR complex did not alter the phosphorylation status of these proteins (Fig. 3a). Accordingly, TTR-bound holo-RBP failed to induce the expression of SOCS3 and PPARγ (Fig. 3b). To examine the effect of TTR on the ability of holo-RBP to suppress insulin responses, cells were pretreated with holo-RBP or holo-RBP-TTR for 8 h and treated with insulin for 15 min, and the levels of phosphorylation of the insulin receptor (IR) and its downstream effector AKT were monitored. The data show that inhibition of insulin-induced phosphorylation of IR and AKT by holo-RBP was blunted in the presence of TTR (Fig. 3c). TTR also inhibited the ability of holo-RBP, but not of holo-RBP-F96A/L97A, defective in TTR binding, to trigger STAT5 phosphorylation (Fig. 3d) or to induce the expression of SOCS3 in NIH 3T3-L1 adipocytes (Fig. 3e) or in HepG2 cells (Fig. 3f).
View larger version:
FIG 3
TTR blocks activation of STRA6 signaling by holo-RBP. (a) NIH 3T3-L1 adipocytes were treated with 1 μM RBP-ROH (R-R), TTR, or RBP-ROH-TTR (R-R-TTR) for 15 min. Cells were lysed, and phosphorylated JAK2 (pJAK2) and STAT5 (pSTAT5) were visualized by immunoblotting. (b) NIH 3T3-L1 adipocytes cells were treated with 1 μM RBP-ROH, TTR, or RBP-ROH-TTR for 4 h, and levels of SOCS3 and PPARγ mRNA were assessed by Q-PCR. Data are means ± SEM. *, P < 0.001 versus nontreated cells. (c) NIH 3T3-L1 adipocytes were pretreated with 1 μM RBP-ROH, TTR, or RBP-ROH-TTR for 8 h and then treated with insulin (25 nM, 15 min.). Phosphorylated IR (pIR) and AKT (pAKT) were visualized by immunoblotting. Bottom, quantitation of band intensities. Means of two independent experiments. (d) NIH 3T3-L1 adipocytes were treated with RBP-ROH or RBP-F96A/L97A-ROH (RBP96/97-R) in the presence or absence of TTR (1 μM each, 15 min). Lysates were immunoblotted for pSTAT5. (e) NIH 3T3-L1 adipocytes were treated with RBP-ROH or RBP-F96A/L97A-ROH in the presence or absence of TTR (1 μM each, 4 h). Levels of SOCS3 mRNA were assessed by Q-PCR. Data are means ± SEM. *, P < 0.001 versus nontreated cells; **, P < 0.001 versus R-R-TTR-treated cells. (f) HepG2 cells were treated with RBP-ROH in the presence or absence of TTR (1 μM each, 4 h). Levels of SOCS3 mRNA were assessed by Q-PCR. Data are means ± SEM. *, P < 0.001 versus nontreated cells. (g) The phosphotyrosine motifs in mouse, human, and zebrafish STRA6 (mSTRA6, hSTRA6, and dSTRA6). (h) NIH 3T3 fibroblasts stably expressing LRAT were transfected with zebrafish and human STRA6 and treated with 1 μM RBP-ROH or RBP-ROH-TTR for 15 min, and lysates were immunoblotted for pSTAT3. (i) NIH 3T3 fibroblasts stably overexpressing LRAT were transfected with dSTRA6 or hSTRA6 and treated with 1 μM RBP-ROH or RBP-ROH-TTR for 4 h. Levels of SOCS3 mRNA were assessed by Q-PCR. Data are means ± SEM. *, P < 0.001 versus nontreated cells. All P values were calculated using a two-tailed student t test.
The effect of TTR on signaling by the zebrafish STRA6 was then examined. Notably, the phosphotyrosine in the cytosolic domain of STRA6, the STAT recruitment site of the receptor, is present in the dSTRA6, suggesting evolutionary conservation of STRA6 signaling (Fig. 3g). In these experiments, NIH 3T3 fibroblasts that ectopically overexpress LRAT were transfected with expression vectors for either hSTRA6 or dSTRA6. Treatment of cells expressing either hSTRA6 or dSTRA6 with holo-RBP induced phosphorylation of STAT3, the preferred STRA6-activated STAT in these cells (Fig. 3h), and upregulation of SOCS3 (Fig. 3i). TTR suppressed the ability of holo-RBP to induce STAT3 phosphorylation and to upregulate SOCS3 expression in cells expressing either hSTRA6 or dSTRA6 (Fig. 3hand i).
TTR inhibits the ability of holo-RBP to suppress insulin responses in vivo.The effect of TTR on the ability of holo-RBP to promote insulin resistance in vivo was then investigated. Eight-week-old mice were injected with recombinant holo-RBP, TTR, or holo-RBP-TTR. Mice were injected three times at 2-h intervals and sacrificed an hour after the last injection. The treatments resulted in respective elevation of serum levels of RBP, TTR, or both (Fig. 4a and b). As expected, treatment of mice with holo-RBP reduced the phosphorylation levels of the insulin receptor and AKT and induced the expression of SOCS3 and PPARγ in WAT (Fig. 4cand f) and skeletal muscle (Fig. 4d and g) but not in liver (Fig. 4e and h). In contrast, treatment with RBP-ROH-TTR did not affect the phosphorylation of IR and AKT or the expression levels of the STAT target genes (Fig. 4c to h).
View larger version:
FIG 4
TTR suppresses activation of STRA6 by holo-RBP in vivo. Mice were injected three times with 0.1 μmol RBP-ROH or 0.1 μmol RBP-ROH complexed with TTR and sacrificed 1 h after the last injection. (a, b) Immunoblots of RBP (a) and TTR (b) in serum following the respective injections. Blots from 2 mice of each group are shown. (c to e) Immunoblots of phosphorylated insulin receptor (pIR), AKT (pAKT), and STAT5 (pSTAT5) in WAT (c), skeletal muscle (d), and liver (e) of mice treated as denoted. Total IR served as a loading control. (f to h) Levels of mRNA of SOCS3 and PPARγ in WAT (f), skeletal muscle (g), and liver (h) of treated mice. Data are means ± SEM. *, P < 0.001 for buffer-treated versus RBP-ROH-treated mice.
The observations that only free and not TTR-bound holo-RBP activates STRA6 suggest that the serum RBP/TTR ratio is crucial for regulating STRA6 signaling. In agreement with the report that expression of RBP in adipose tissue increases in obese rodents and humans, resulting in elevation of serum RBP levels (35), feeding mice a high-fat, high-sucrose (HFHS) diet for 10 weeks resulted in upregulation of the expression of RBP in WAT but not in liver (Fig. 5a). In contrast, TTR expression in these organs was not affected by the diet (Fig. 5b). Accordingly, the serum level of RBP was markedly elevated, while the serum level of TTR remained unchanged in obese mice (Fig. 5c). Hence, the RBP/TTR ratio is significantly higher in blood of obese than of lean mice.
View larger version:
FIG 5
TTR is protective against holo-RBP-induced insulin resistance. (a and b) Levels of mRNA of RBP (a) and TTR (b) in WAT and liver of lean mice and of mice fed an HFHS diet for 10 weeks (obese). (c) Immunoblots of RBP and TTR in serum of mice fed an HFHS diet for 0, 3, 6, and 10 weeks. (d to j) Mice were implanted with an Alzet pump that contained buffer, 0.1 μM holo-RBP, or 0.1 μM holo-RBP complexed with TTR. Implants were replaced once a week for 3 weeks. (d) Immunoblots of RBP (top) and TTR (bottom) in serum following 3 weeks of denoted treatments. (e, f) Immunoblots of pIR and pSTAT5 in WAT (e) and skeletal muscle (f) of mice treated as denoted. (g to i) Levels of SOCS3 mRNA in WAT (g), skeletal muscle (h), and liver (i) of mice treated as denoted. (j) Glucose tolerance tests carried out following 3 weeks of denoted treatments. Data are means ± SEM. *, P < 0.001 for lean versus obese mice; **, P < 0.001 for buffer-treated versus RBP-ROH-treated mice. All P values were calculated using a two-tailed Student ttest.
To directly determine if TTR prevents holo-RBP-induced insulin resistance, mice were treated with holo-RBP or holo-RBP-TTR for 3 weeks prior to the glucose tolerance tests (GTT). Mice were treated by implanting Alzet osmotic pumps containing the appropriate proteins (1 μM), thereby delivering constant amounts of proteins over the 3-week period. Similar to what was seen in the short-term treatments (Fig. 4), 3-week treatment of mice with holo-RBP induced phosphorylation of STAT5, reduced the activation level of IR, and upregulated SOCS3 and PPARγ in WAT (Fig. 5e and g) and muscle (Fig. 5f and h) but not in the liver (Fig. 5i). In contrast, treatment with TTR-bound holo-RBP had no effect on the phosphorylation of STAT5 or IR and did not alter the expression levels of the STAT target genes (Fig. 5e to i). Accordingly, while holo-RBP treatment resulted in a sluggish response in GTT, reflecting the development of insulin resistance, treatment with the holo-RBP-TTR complex did not alter the insulin responses of the mice (Fig. 5j). Hence, association with TTR suppresses the ability of holo-RBP to interfere with insulin signaling.
DISCUSSION
Upon binding of extracellular holo-RBP, STRA6 transports ROH into cells, and it activates a signaling cascade culminating in induction of STAT target genes (4, 5). The observations described here reveal that the binding partner of RBP in blood, TTR, effectively blocks association of holo-RBP with STRA6. Consequently, STRA6 mediates cellular ROH uptake only from free and not from TTR-bound holo-RBP. The data further show that, even in the presence of free holo-RBP, STRA6-mediated ROH uptake by tissues comprises only a small fraction of total uptake by target tissues in vivo (Fig. 1h). The observations thus support the previously proposed model whereby supply of ROH from circulating holo-RBP or holo-RBP-TTR to cells occurs primarily by diffusion through the plasma membranes (10, 14,20, 21). Taken together with the observations that ROH transport by STRA6 is critical for enabling activation of STRA6 signaling (5), the data indicate that, with the exception of the eye (26), the main role of ROH transport by STRA6 is not to provide the vitamin to cells but to couple sensing of circulating free holo-ROH levels to cell signaling. It is worth noting that even in the eye, morphological changes and reduction in visual function in Stra6-null mice are mild, indicating that STRA6 is not the only pathway by which ROH enters the retinal pigment epithelium (26).
The data reveal that, in addition to its function in preventing filtration of the 21-kDa RBP in the kidney, TTR plays an important role in protecting cells from holo-RBP-induced signaling mediated by STRA6. The observations that STRA6 “senses” only free and not TTR-bound RBP establish that the receptor functions only under circumstances in which the serum RBP level exceeds that of TTR. Such circumstances are encountered, for example, in obese animals in which the serum level of RBP is elevated while the TTR level is not (Fig. 5c). The circumstances under which the plasma RBP concentration exceeds that of TTR in healthy lean animals remain to be clarified. In this regard, it is interesting that it has long been known that insulin responsiveness varies in a circadian fashion (17, 31). The molecular basis for these diurnal variations is incompletely understood, but the data presented here raise the intriguing possibility that they may arise from diurnal variations in the plasma RBP/TTR ratio.
The RBP/TTR ratio in blood may be altered by changes in the expression level of RBP, or TTR, or both. TTR is expressed in the central nervous system and in the liver, with the latter serving as the main source for the protein in serum (9). Expression of hepatic TTR is downregulated, and consequently, the serum TTR level dramatically decreases during the acute-phase response (APR), a process characterized by rapid reprogramming of gene expression and metabolism in response to inflammatory cytokine signaling (1, 22). The low serum level of TTR associated with APR may release holo-RBP, thereby activating STRA6. Hence, STRA6 signaling may play a role in APR. It has also been reported that hepatic TTR expression is regulated by sex hormones (12) and is directly controlled by hepatocyte nuclear factor 4α (HNF-4α) (30). The expression of RBP in brown adipose tissue and liver was reported to be regulated by cyclic AMP-mediated pathways and by the nuclear receptors PPARα and PPARγ (6, 25). Whether, by controlling TTR or RBP expression, these factors regulate the RBP-TTR ratio in blood and thus STRA6 signaling remains to be clarified.
Notably, as free holo-RBP is rapidly excreted by glomerular filtration, its lifetime in serum is short. Holo-RBP thus functions like a classical cytokine: its availability to its membrane receptor is tightly regulated, and its signaling activities are constrained by a short half-life in the circulation. These characteristics of the signaling activities of holo-RBP strikingly differ from the characteristics of its role as a shuttling protein that mobilizes ROH from liver stores. Unlike in the former capacity, where holo-RBP functions on its own, delivery of ROH to target tissues is mediated by the holo-RBP-TTR complex. The plasma level of this complex is under tight homeostatic control, and it provides ROH to target cells to support tissue requirement for vitamin A without the need for a specialized receptor.
Biochim Biophys Acta. 1996 May 2; 1294(1):48-54.
Retinoid binding to retinol-binding protein and the interference with the interaction with transthyretin.
The retinol carrier retinol-binding protein (RBP) forms a complex with the thyroid hormone binding protein transthyretin in the plasma of a number of vertebrate species. The interactions of retinoid-RBP complexes, as well as of unliganded RBP, with transthyretin have been investigated by means of fluorescence anisotropy studies. The presence of two independent and equivalent RBP binding sites per transthyretin molecule has been established for proteins purified from species distant in evolution. Although the natural ligand retinol participates in the interaction between retinol-RBP and transthyretin, its binding to RBP is not a prerequisite for protein-protein interaction. The dissociation constants of human transthyretin binding liganded and unliganded forms of human RBP were determined to be: all-trans retinol-RBP, Kd approximately 0.2 microM; apoRBP, Kd approximately 1.2 microM; all-trans retinoic acid-RBP, Kd approximately 0.8 microM; all-trans retinyl methyl ether-RBP, Kd approximately 6 microM. The complex of RBP with the synthetic retinoid fenretinide, which bears the bulky hydroxyphenyl end group, exhibits negligible affinity for transthyretin. The replacement of RBP-bound retinol with synthetic retinoids affects RBP-transthyretin recognition to an extent that appears to be well correlated with the nature and steric hindrance of the groups substituting the retinol hydroxyl group, consistent with their location at the interface between the contact areas of RBP and transthyretin.
Methods Mol Biol. 2010; 652:189-207. doi: 10.1007/978-1-60327-325-1_11.
The interaction between retinol-binding protein and transthyretin analyzed by fluorescence anisotropy.
The retinol carrier retinol-binding protein (RBP) forms in blood a complex with the thyroid hormone carrier transthyretin (TTR). The interactions of retinoid-RBP complexes, as well as of unliganded RBP, with TTR can be investigated by means of fluorescence anisotropy. RBP represents the prototypic lipocalin, in the internal cavity of which the retinol molecule is accommodated. Due to the tight binding of retinol within a substantially apolar binding site, an intense fluorescence emission characterizes the RBP-bound vitamin. The addition of TTR to the retinol-RBP complex (holoRBP) causes a marked increase in the fluorescence anisotropy of the RBP-bound retinol within the system, due to the formation of the holoRBP-TTR complex, which allows the interaction between the two proteins to be monitored. The fluorescence anisotropy technique is also suitable to study the interaction of TTR with apoRBP and RBP in complex with non-fluorescent retinoids. In the latter cases, the fluorescence signal is provided by a fluorescent probe covalently linked to TTR rather than by RBP-bound retinol. We report here on the preparation of recombinant human RBP and TTR, the covalent labeling of TTR with the fluorescent dansyl probe, and fluorescence anisotropy titrations for RBP and TTR.
Vitam Horm. 2004; 69:271-95.
Plasma retinol-binding protein: structure and interactions with retinol, retinoids, and transthyretin.
Retinol-binding protein (RBP) is the retinol-specific transport protein present in plasma. The available crystal structures of different forms of RBP have provided details of the interactions of this binding protein with retinol, retinoids, and transthyretin (TTR, one of the plasma carriers of thyroid hormones). The core of RBP is a beta-barrel, the cavity of which accommodates retinol, establishing with its buried portions apolar contacts. Instead, the retinol hydroxyl is near the protein surface, in the region of the entrance loops surrounding the opening of the binding cavity, and participates in polar interactions. The stability of the retinol-RBP complex appears to be further enhanced when holo-RBP is bound to TTR. Accordingly, the region of the entrance loops represents the contact area of RBP interacting with the TTR counterpart, such that the hydroxyl of the RBP-bound vitamin becomes fully buried in the holo-RBP-TTR complex. Limited protein conformational changes affecting the entrance loops, which lead to a decrease or loss of the binding affinity of RBP for TTR, have been demonstrated for apo-RBP and RBP in complex with retinoids modified in the area of the retinol hydroxyl. A relatively small number of amino acid residues of RBP, essentially confined to the region of the entrance loops, and of TTR appear to play a critical role in the formation of the RBP-TTR complex, as established by crystallographic studies, mutational analysis, and amino acid sequence analysis of phylogenetically distant RBPs and TTRs. Overall, the available evidence indicates the existence of a high degree of complementarity between RBP and TTR, the contact areas of which are highly sensitive to conformational changes and amino acid replacements.
Biochim Biophys Acta. 2004 Dec 1; 1703(1):1-9.
Interactions amongst plasma retinol-binding protein, transthyretin and their ligands: implications in vitamin A homeostasis and transthyretin amyloidosis.
Retinol transport complex consisting of retinol-binding protein (RBP) and transthyretin (TTR) is involved in the transport of retinol (vitamin A) and thyroxine (T(4)) in the human plasma. RBP is a 21-kDa single polypeptide chain protein, synthesized in the liver, which binds and transports retinol to the target organs. The circulating RBP binds to another protein called TTR, a 55-kDa homotetrameric T(4) transport protein. Such protein-protein complex formation is thought to prevent glomerular filtration of low molecular mass RBP. Misfolding and aggregation of TTR is implicated in amyloid disorders such as familial amyloid polyneuropathy (FAP) and senile systemic amyloidosis (SSA). Recent observations suggest that both RBP and T(4), the physiological ligands of TTR, prevent its misfolding and amyloid fibril formation, suggesting yet another structure-function relationship to this protein-protein complex. TTR2, a poorly characterized protein, was also found bound to RBP in human and pig plasma but its significance remains to be understood. Furthermore, knockout models of both RBP and TTR unequivocally demonstrated the importance of this protein-protein complex in retinoid transport. Thus, interactions amongst multiple components of retinol transport play critical roles in vitamin A homeostasis and TTR amyloidosis
Retinol Binding Protein and Its Interaction with Transthyretin
Marcia E Newcomer* and David E. Ong
…………..———————–
Protein Sci. 2001 Nov; 10(11): 2301–2316.
doi: 10.1110/ps.22901
PMCID: PMC2374051
Role of conserved residues in structure and stability: Tryptophans of human serum retinol-binding protein, a model for the lipocalin superfamily
Lesley H. Greene,1,3 Evangelia D. Chrysina,2 Laurence I. Irons,2 Anastassios C. Papageorgiou,2,4 K. Ravi Acharya,2and Keith Brew1,
The flexible loop and, in particular, Trp 67 is known to be involved in molecular packing interactions at the interface of the human RBP-TTR (transthyretin) complex (Monaco et al. 1995; Naylor and Newcomer 1999). In the two structures reported for this complex, Trp67 and Trp91 have critical roles in heterodimer stabilization, but a more detailed examination of these particular residues is not possible because of the low resolution (3.1 Å). Thus, when the rRBP67L/91H structure is compared with the structure of RBP in the complex, only gross structural differences can be ideied. The most profound differences between the recombinant apo-RBP and RBP-TTR complex are those around residue 62 and at the C terminus (root mean square, 1.05 Å; Fig. 2 ▶); both regions are implicated in interactions with TTR in the complex. In the rRBP structure, Trp67 is disordered, as are the rest of the residues that form the flexible loop, whereas in the rRBP67L/91H structure, the effect of the sequence substitution at position 67 was not investigated in detail because of the poor electron density in this region.
Trp24 is a component of the first (A) of the strands that form the β-barrel, whereas another highly conserved residue, Arg139, is located at the end of the final H strand. The interactions between these two residues contribute significantly to the formation of the barrel cylinder and closing of its base (Tables 1, 22).).
Retinol-binding studies with selected tryptophan mutants indicated that the mutations did not eliminate the ability of the protein to bind all-trans retinol (data not shown). After folding and purification, the mutants were isolated in yields of ∼20 mg/L, except for those with substitutions for Trp24 or Arg139, in which the yields were 4- to 20-fold lower.
The folding behavior of RBP has a specific biological interest because only the holo form of the protein is secreted after biosynthesis in mammalian and other cells; RBP molecules that do not acquire a retinol ligand within the cell are retained in the endoplasmic reticulum (Melhus et al. 1992) and appear not to be fully folded (Kaji and Lodish 1993). Previously determined structures for human and bovine apo-RBP show close similarity to the holo-protein. These apo-proteins were prepared from natural holo-protein after extraction with ethyl ether to remove the bound ligand (Zanotti et al. 1993a). Here we find that recombinant human apo-RBP produced by in vitro folding of material extracted from inclusion bodies has a structure and spectroscopic properties that are closely similar to those of apo and holo forms of natural human holo-RBP (Cowan et al. 1990), holo and apo bovine RBP (Zanotti et al. 1993a,c), and RBP in complexes with different retinoids (Zanotti et al. 1993b). Because our preparations of apo-rRBP have never bound a retinol ligand, we can conclude that although retinol may enhance folding yields, the ligand is not necessary for an irreversible maturation step in folding. Thus, the degradation of apo-RBP in vivo must be linked to some specific structural feature or property of the apo- versus holo-protein.
The results described here are necessary for the design and interpretation of unfolding and folding kinetics of rRBP because they establish conditions in which the folded and unfolded conformers are most populated. They also allow comparisons of the effects of mutations on the stability of the transition states for folding and unfolding with those of the native and unfolded states. The spectroscopic properties of the mutants indicate signals that provide information about the structure formation in different parts of the RBP molecule during folding processes. A major focus of our work is to determine if there is a relationship between folding and sequence conservation in functionally divergent paralogous proteins. This is a concept that is recently receiving attention from experimentalists (Martinez and Serrano 1999; Hamill et al. 2000; Nishimura et al. 2000; Plaxco et al. 2000). rRBP, as an experimental model for the large lipocalin superfamily, represents an ideal system to further our understanding between evolution and folding. Toward this end, kinetic and additional X-ray crystallographic studies are in progress with recombinant RBP and mutants.
Protein Synthesis at the Blood-Brain Barrier THE MAJOR PROTEIN SECRETED BY AMPHIBIAN CHOROID PLEXUS IS A LIPOCALIN*
Marc G. Achen, Paul J. Harms, Tim Thomas, Samantha J. Richardson, Richard E. H. Wettenhall, and Gerhard SchreiberS
From the Russell Grimwade School of Biochemistry, University of Melbourne, Parkuille, Victoria 3052, Australia
THE JOURNAL OF BIOLOGICAL CHEMISTRY Nov 15, 1992; 267(32): 23170-23174
Epithelial cells, located at the barriers between extracellular compartments, synthesize and secrete proteins required in these compartments. Examples of such cells are the hepato- cytes providing plasma proteins (for review see Ref. l), the Sertoli cells in the testes synthesizing and secreting cerulo- plasmin (2) and transferrin (3), and the choroid plexus pro- ducing proteins, many of which have transport functions, for the extracellular environment in the brain (Ref. 4, for review see Ref. 1). The epithelial cells of the mammalian (1, 5, 6) and avian (7, 8) choroid plexus, forming the blood-cerebro- spinal fluid barrier, are highly specialized in the synthesis of one particular protein, namely transthyretin. This transthy- retin is secreted exclusively toward the brain (9) and has been proposed to mediate the transport of thyroxine from the bloodstream to the brain (9, 10). Expression of the transthy- retin gene is initiated in the choroid plexus Anlage very early in life (11, 12). Analysis of the proteins synthesized by in vitro incubated choroid plexus from various species showed abundant trans- thyretin synthesis and secretion by choroid plexus from mam- mals, birds, and reptiles (6). The choroid plexus from an amphibian, the cane toad, also synthesized and secreted one predominant polypeptide product (6). However, the size of this product was larger than that of the transthyretin subunit (6). In the following, we describe isolation, properties, cloning, structural analysis of the protein and cDNA, and tissue specificity of expression for the polypeptide most abundantly synthesized and secreted by amphibian choroid plexus. The obtained data also allow a more precise determination of the stage at which the high transthyretin gene expression first occurred in the evolution of the vertebrate brain.
Among the proteins secreted by choroid plexus of vertebrates, one protein is much more abundant than all others. In mammals, birds, and reptiles this protein is transthyretin, a tetramer of identical 15-kDa sub- units. In this study choroid plexus from frogs, tadpoles, and toads incubated in vitro were found to synthesize and secrete one predominant protein. However, this consisted of one single 20-kDa polypeptide chain. It was expressed throughout amphibian metamorphosis. Part of its amino acid sequence was determined and used for construction of oligonucleotides for polymer- ase chain reaction. The amplified DNA was used to screen a toad choroid plexus cDNA library. Full-length cDNA clones were isolated and sequenced. The derived amino acid sequence for the encoded protein was 183 amino acids long, including a 20-amino acid preseg- ment. The calculated molecular weight of the mature protein was 18,500. Sequence comparison with other proteins showed that the protein belonged to the lipo- calin superfamily. Its expression was highest in cho- roid plexus, much lower in other brain areas, and absent from liver. Since no transthyretin was detected in proteins secreted from amphibian choroid plexus, abundant synthesis and secretion of transthyretin in choroid plexus must have evolved only after the stage of the amphibians.
Comparison of the Structure of the Major Protein Synthesized and Secreted by Cane Toad Choroid Plexus with Those of Other Proteins-A protein database search showed that the major amphibian choroid plexus secreted protein belongs to the superfamily of the lipocalins. Lipocalins are small proteins (160-190 residues), most of which are secreted. They possess a common three-dimensional structure (calyx) with a hydro- phobic pocket (25, 26). The binding of small hydrophobic molecules is also a common feature of lipocalins (27). There was variability in the extent of the similarity in amino acid sequence of the cane toad 20-kDa protein with other lipocalins. The most similar was glutathione-independ ent brain prostaglandin D synthetase from humans (28) and rats (29), with this protein from both species exhibiting 41% amino acid identity and 84% similarity for conservative amino acid substitutions to the cane toad 20-kDa protein
FIG. 4. Tissue specificity of the expression of the gene for the major cane toad choroid plexus protein. Northern analysis of mRNAs of different organs. Total cellular RNA from the cane toad, 20 pg from liver, 5 pg from brain without choroid plexus, and 0.1 pg from choroid plexus, was subjected to Northern analysis as describedu nder “Experimental Procedures” using the cDNA de- scribed in Fig. 3 as probe. Autoradiographic exposure was for 48 h at -70 “C. The positions of 28 and 18 S ribosomalR NAb ands are indicated on the right.
However, in contradistinction to the 20-kDa protein described here and to most other lipocalins which are secreted, prostaglandin D synthetase has only been localized intracel- lularly (30). The amino acid sequence identities with some other lipocalins were: rat a2,,-globulin, 32% (31); y component of human complement C8,28% (32); human a,,-globulin, 27% (33); chicken CH21 protein, 25% (34); mouse major urinary protein, 25% (35); and a protein from frog olfactory neuro- epithelium, 25% (36). The sequences for these other lipocalins are not shown in a figure. No other group or single protein, including transthyretins, showed any significant sequence similarity.
Functional and Phylogenetic Implications-The brain possesses its own extracellular environment of specific composition. The blood-brain barrier and the blood-cerebrospinal fluid barrier separate the extracellular spaces of the brain from those in the rest of the body. The cerebrospinal fluid, filling the ventricles and paracerebral spaces in the brain and communicating by bulk-exchange with the fluid in the inter- stitial space of the brain (37), is produced by the choroid plexus, the site of the blood-cerebrospinal fluid barrier. The choroid plexus has been reported to synthesize a number of transport proteins, such as transthyretin (38, 40, 41), trans- ferrin (4, 39, 42), ceruloplasmin (2), and retinol-binding pro- tein (43). Of the proteins synthesized and secreted by the choroid plexus in mammals, birds, and reptiles, transthyretin is by far the most abundant. It is secreted exclusively toward the brain (9) and has been proposed to mediate the transfer of thyroxine to the brain (9, 10).
The data presented in this paper demonstrate that, also in amphibians, the choroid plexus is highly specialized for synthesis and secretion of a specific protein. However, this protein is not transthyretin but is a member of the lipocalin superfamily. Such a lipocalin could possibly have at ransport function across the blood-brain barrier. The absence of transthyretin secretion by the choroid plexus in amphibians, as shown in this paper, suggests that the high expression and secretion of transthyretin, as seen in mammals, birds, and reptiles must have evolved only after the stage of the amphibians. The function in the amphibian brain of the 20-kDa lipocalin abundantly synthesized and secreted by choroid plexus is yet to be elucidated.
Transthyretin and Lean Body Mass in Stable and Stressed State
A Second Look at the Transthyretin Nutrition Inflammatory Conundrum
TTR – amyloidosis
J Neurol Neurosurg Psychiatry 2015;86:159-167 http://dx.doi.org:/10.1136/jnnp-2014-308107
CNS involvement in V30M transthyretin amyloidosis: clinical, neuropathological and biochemical findings
Luís F Maia1,2,3, Rui Magalhães4, Joel Freitas2, Ricardo Taipa5, Manuel Melo Pires5, Hugo Osório6, Daniel Dias7, Helena Pessegueiro8, Manuel Correia2, Teresa Coelho1,9
Correspondence to – Dr Luís F Maia, Serviço de Neurologia, Hospital de Santo António—CHP, Largo Prof. Abel Salazar, Porto 4099-001, Portugal; luis.lf.maia@gmail.com
Online First 4 August 2014
Objectives Since liver transplant (LT) was introduced to treat patients with familial amyloid polyneuropathy carrying the V30M mutation (ATTR-V30M), ocular and cardiac complications have developed. Long-term central nervous system (CNS) involvement was not investigated. Our goals were to: (1) identify and characterise focal neurological episodes (FNEs) due to CNS dysfunction in ATTR-V30M patients; (2) characterise neuropathological features and temporal profile of CNS transthyretin amyloidosis.
Methods We monitored the presence and type of FNEs in 87 consecutive ATTR-V30M and 35 non-ATTR LT patients. FNEs were investigated with CT scan, EEG and extensive neurovascular workup. MRI studies were not performed because all patients had cardiac pacemakers as part of the LT protocol. We characterised transthyretin amyloid deposition in the brains of seven ATTR-V30M patients, dead 3–13 years after polyneuropathy onset.
Results FNEs occurred in 31% (27/87) of ATTR-V30M and in 5.7% (2/35) of the non-ATTR transplanted patients (OR=7.0, 95% CI 1.5 to 33.5). FNEs occurred on average 14.6 years after disease onset (95% CI 13.3 to 16.0) in ATTR-V30M patients, which is beyond the life expectancy of non-transplanted ATTR-V30M patients (10.9, 95% CI 10.5 to 11.3). ATTR-V30M patients with FNEs had longer disease duration (OR=1.24; 95% CI 1.07 to 1.43), renal dysfunction (OR=4.65; 95% CI 1.20 to 18.05) and were men (OR=3.57; 95% CI 1.02 to 12.30). CNS transthyretin amyloidosis was already present 3 years after polyneuropathy onset and progressed from the meninges and its vessels towards meningocortical vessels and the superficial brain parenchyma, as disease duration increased.
Conclusions Our findings indicate that CNS clinical involvement occurs in ATTR-V30M patients regardless of LT. Longer disease duration after LT can provide the necessary time for transthyretin amyloidosis to progress until it becomes clinically relevant. Highly sensitive imaging methods are needed to identify and monitor brain ATTR. Disease modifying therapies should consider brain TTR as a target.
Transthyretin-type cerebral amyloid angiopathy: a serious complication in post-transplant patients with familial amyloid polyneuropathy
Yoshiki Sekijima1,2
1Department of Medicine (Neurology and Rheumatology), Shinshu University School of Medicine, Matsumoto, Japan
2Institute for Biomedical Sciences, Shinshu University, Matsumoto, Japan
J Neurol Neurosurg Psychiatry 2015;86:124 http://dx.doi.org:/10.1136/jnnp-2014-308576
Correspondence to
Dr Yoshiki Sekijima, Department of Medicine (Neurology and Rheumatology), Shinshu University School of Medicine, 3-1-1 Asahi, Matsumoto 390-8621, Japan; sekijima@shinshu-u.ac.jp
Online First 11 August 2014
Liver transplantation is a well-established treatment for transthyretin (TTR)-type familial amyloid polyneuropathy (TTR-FAP).1 According to data in the Familial Amyloidotic Polyneuropathy World Transplant Registry (http://www.fapwtr.org/ram_fap.htm), more than 2000 liver transplantations have been performed to date in 19 countries. Transplantation replaces the variant TTR gene with the wild-type gene in the liver, the main source of serum circulating TTR. The serum concentration of variant TTR decreases rapidly, reaching almost zero after the operation. The effects of liver transplantation on neuropathy are evident as its progression is …
Linköping Studies in Science and technology Dissertation No. 1179
Molecular Aspects of Transthyretin Amyloid Disease
Karin Sörgjerd
Biochemistry Department of Physics, Chemistry and Biology
Linköping University, SE- 58183 Linköping, Sweden Linköping 2008
ISBN 978-91-7393-906-5 http://liu.diva-portal.org/smash/get/diva2:1717/FULLTEXT01.pdf
This thesis was made to get a deeper understanding of how chaperones interact with unstable, aggregation prone, misfolded proteins involved in human disease. Over the last two decades, there has been much focus on misfolding diseases within the fields of biochemistry and molecular biotechnology research. It has become obvious that proteins that misfold (as a consequence of a mutation or outer factors), are the cause of many diseases. Molecular chaperones are proteins that have been defined as agents that help other proteins to fold correctly and to prevent aggregation. Their role in the misfolding disease process has been the subject for this thesis.
Transthyretin (TTR) is a protein found in human plasma and in cerebrospinal fluid. It works as a transport protein, transporting thyroxin and holo-retinol binding protein. The structure of TTR consists of four identical subunits connected through hydrogen bonds and hydrophobic interactions. Over 100 point mutations in the TTR gene are associated with amyloidosis often involving peripheral neurodegeneration (familial amyloidotic polyneuropathy (FAP)). Amyloidosis represents a group of diseases leading to extra cellular deposition of fibrillar protein known as amyloid. We used human SH-SY5Y neuroblastoma cells as a model for neurodegeneration. Various conformers of TTR were incubated with the cells for different amounts of time. The experiments showed that early prefibrillar oligomers of TTR induced apoptosis when neuroblastoma cells were exposed to these species whereas mature fibrils were not cytotoxic. We also found increased expression of the molecular chaperone BiP in cells challenged with TTR oligomers.
Point mutations destabilize TTR and result in monomers that are unstable and prone to aggregate. TTR D18G is naturally occurring and the most destabilized TTR mutant found to date. It leads to central nervous system (CNS) amyloidosis. The CNS phenotype is rare for TTR amyloid disease. Most proteins associated with amyloid disease are secreted proteins and secreted proteins must pass the quality control check within the endoplasmic reticulum (ER). BiP is a Hsp70 molecular chaperone situated in the ER. BiP is one of the most important components of the quality control system in the cell. We have used TTR D18G as a model for understanding how an extremely aggregation prone protein is handled by BiP. We have shown that BiP can selectively capture TTR D18G during co-expression in both E. coli and during over expression in human 293T cells and collects the mutant in oligomeric states. We have also shown that degradation of TTR D18G in human 293T cells occurs slower in presence of BiP, that BiP is present in amyloid deposition in human brain and mitigates cytotoxicity of TTR D18G oligomers.
Included papers
Paper I: Detection and characterization of aggregates, prefibrillar amyloidogenic oligomers, and protofibrils using fluorescence spectroscopy., Lindgren M, Sörgjerd K, Hammarström P., Biophys J. 2005 Jun;88(6):4200-12.
Paper II: Prefibrillar Amyloid Aggregates and Cold Shocked Tetrameric Wild Type Transthyretin are Cytotoxic. Sörgjerd K, Klingstedt T, Lindgren M, Kågedal K, Hammarström P. In manuscript.
Paper III: Retention of misfolded mutant transthyretin by the chaperone BiP/GRP78 mitigates amyloidogenesis., Sörgjerd K, Ghafouri B, Jonsson BH, Kelly JW, Blond SY, Hammarström P., J Mol Biol. 2006 Feb 17;356(2):469-82.
Paper IV: BiP can function as a molecular shepherd that alleviates oligomer toxicity and amass amyloid. Sörgjerd K.,Wiseman R.L, Kågedal K., Berg I., Klingstedt T., Budka H, Nilsson K.P.R., Ron D., Hammarström P. In manuscript.
Abbreviations
ANS 8-anilino-1-naphthalene sulfonic acid
Bis-ANS 4-4-bis-1-phenylamino-8- naphthalene sulfonate
CNS central nervous system
CtD C-terminal domain
DCVJ 4-(dicyanovinyl)-julolidine
ER endoplasmic reticulum
FAP familial amyloidotic polyneuropathy
LCP luminescent conjugated polymer
MTT 3[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide
NBC neuroblastoma cells NtD N-terminal domanin
RBP retinol binding protein
SDS-PAGE sodium dodecyl sulphate polyacrylamide gel electrophoresis
SSA senile systemic amyloidosis
ThT thioflavin T
T4 thyroxin
TEM transmission electron microscopy
TTR transthyretin
TTR D18G transthyretin with amino acid substitution from aspartic acid to glycine at position 18
UPR unfolded protein response
Table of contents
1 Introduction
3 Proteins
5 Protein production- the background story
6 Protein folding
8 Genetic mutations
9 Protein misfolding
10 Transthyretin (TTR)
13 The TTR D18G mutation
18 Molecular chaperones
21 BiP
22 Structure and mechanism of BiP
23 A role for BiP during translocation
25 The role of molecular chaperones in misfolding diseases
25 The ER, cellular stress and cell death
27 The unfolded protein response (UPR)
28 Apoptosis
31 Caspases
31 Methods
33 Cloning, mutagenesis
33 SDS-PAGE and Western blotting
33 Circular Dichroism
33 Fluorescence spectroscopy
34 Chemical cross-linking
35 Transmission Electron Microscopy (TEM) 3
35 Affinity chromatography and immunoprecipitation
36 Analytical ultracentrifugation
36 Results
37 Paper I_
38 Cross-linking to probe formation of aggregates
38 Size and morphology of aggregates and protofibrils
39 Characterization of TTR conformers using molecular probes
40 Different kinetics for different probes
41 Paper II
42 Early oligomeric species of TTR kill human cells
42 Early oligomeric species of TTR induce ER stress
44 Paper III_
45 BiP selectively binds to destabilized variants of TTR
45 Composition of the BiP- TTR D18G complex
46 The BiP- TTR D18G interaction
46 BiP plays a protective role against the toxic effects of TTR D18G
Paper IV
49 BiP interacts with TTR D18G in the mammalian ER
49 The degradation rate of TTR D18G is slowed down in the presence of BiP
49 The BiP- TTR D18G complex was present in a wide distribution of molecular weights
51 BiP can protect cells from TTR D18G cytotoxicity
52 BiP is found in TTR D18G aggregates in patient tissue
53 Conclusions ________________________________________________________55
55 References ________________________________________________________ 57
This thesis summarizes what I have been working on for the past five years and what conclusions I have made from my findings. My main characters are two proteins called BiP; which is a molecular chaperone believed to play a protective role in cells, and transthyretin (TTR); which is associated with human misfolding disease. It has been known for a long time that TTR misfolding disease starts with TTR denaturation and leads to aggregation and fibrillation of TTR, which accumulates in tissues and organs in patients suffering from the disease. Still, there are no cures for most of these kinds of diseases and the pathogenesis and mechanisms are not fully understood.
The aims with my studies have been to elucidate what role BiP plays in TTR misfolding diseases. I have specifically studied a mutant of TTR called TTR D18G, since that mutant is the most destabilized and most unusual form of TTR. I have also aimed to follow the mechanisms for TTR misfolding and to study the consequences in human cells when exposed to misfolded variants of TTR.
I have included four papers in this thesis. In the first, the aggregation process of transthyretin is described, and the different states in the process are characterized. In the second paper, the effect of different conformational states of aggregated TTR variants on human cells has been studied. In the third paper, the interactions of TTR D18G with BiP are characterized and hypothesizes about what role BiP plays in TTR misfolding diseases have been made. In the fourth paper, the BiP TTR D18G interaction is studied from a mammalian point of view and the effects of BiP and TTR D18G on human cells are elucidated.
Proteins are responsible for most of the reactions occurring in the human body such as transport of nutrients and oxygen, defense against microorganisms, control of gene expression, transmission of signals etc. In all organisms and in each cell, they exist and they work. In the human body, over 30 000 different proteins (and around 30 000 protein coding genes) [1] with almost as many different functions are present, however, most of them still have unknown functions. The building blocks for proteins are called amino acids. There are 20 amino acids used for protein synthesis and 12 of them can be produced by human cells whereas eight need to be supplied with the diet. Thus, a versatile diet is important for the body to work properly with all its > 30 000 different proteins to be properly synthesized. An average protein consists of hundreds of amino acids, linked together in different sequences by the peptide bond, and it is the order of the amino acids that dictate the final shape of the protein. The amino acids, the building blocks in a polypeptide, have different properties; they can be polar, non-polar or charged and the hydrophobic ones are usually buried in the interior of the folded protein. The structures of the proteins, i.e., their conformations, differ due to different types of secondary structures, called α-helices or β-sheet and how these structural elements are arranged. It is the conformation that dictates the protein function. Every single amino acid in the folded protein can contribute to and play a role for protein function. A substitution of one amino acid to another might in some cases lead to re-arrangements of the whole protein structure, and thereby induce a new behavior of the protein (often leading to destabilization and degradation). In other cases, an amino acid substitution does not influence the conformation at all. 6
Figure 1) The primary structure of a protein showing amino acids as a string of pearls. The side chains of the amino acids, denoted with R can be polar, non-polar or charged. When the amino acids are connected to form a poly peptide chain, the COO- group of one amino acid reacts with the NH3 + of another, and a peptide bond is formed with release of a water molecule.
Protein molecules can interact with each other, and protein-protein interactions are necessary for many biological functions. Interactions can be prolonged, when a complex is formed, or transient, i.e. when signals are transferred within or between cells. Interactions can also be non-preferred, such as protein aggregation.
Protein production- the background story All proteins begin as linear sequences of amino acids linked together as a string of pearls (Figure 1). The information about the amino acid sequence of the protein, leading to their different conformations is encoded in the deoxyribonucleic acid (DNA), in the specific genes for the proteins of interest, called the genetic code. When synthesis of a polypeptide begins, the DNA information is transferred to a piece of messenger RNA (mRNA). Formation of RNA from DNA is a process called transcription and occurs with help from an enzyme called RNA polymerase. DNA was identified in 1944 and its double helical conformation was revealed in 1953 [2]. Since DNA is situated in the cell nucleus whereas the protein synthesis occurs in the cytoplasm, an intermediate needs to be involved in the transcription. In the fifties, Primary protein structure Amino acid C COONH3 + R H Primary protein structure Amino acid C COONH3 + R H C COONH3 + R H C COONH3 + R H 7 there were discussions about that intermediate and it was proposed that another nucleic acid, single stranded ribonucleic acid (RNA), would be the intermediate responsible for transferring information from the cell nucleus to the cytoplasm. Later, it was formulated that the genetic information in the DNA is transcribed to RNA and then translated from RNA into a protein. The idea was developed over time and in the sixties, it was proposed that a gene is transcribed into a specific RNA species, called mRNA and that a short-lived, non-ribosomal RNA directs the synthesis of proteins. In 1965, Francois Jacob, Jacques Monod and André Lwoff received the Nobel Prize for their research about mRNA. In the seventies, it became known that mRNA could be spliced after transcription, resulting in that the primary transcript can generate different mRNAs and different proteins. In the 1980s, it was found that small RNA molecules could bind to a complementary sequence in mRNA and inhibit translation [3]. mRNA is single stranded and its sequence is called sence because it results in a protein. The complementary sequence is called antisense. When sence mRNA base pair with anti sence mRNA, translation is blocked. The mechanism has not been fully understood until Craig C. Mello coined the term RNA interference in his work from 1997, published in Cell [4]. In 2006, Andrew Z. Fire and Craig C. Mello received the Nobel Prize for uncovering the mechanism of RNA interference. They discovered that genes could be silenced, i.e. gene activity could be turned off, by double-stranded RNA [5].
Protein production is intitiated by transcription from DNA to mRNA. The transcription starts with binding of RNA polymerase to the DNA which, together with different cofactors, unwinds the DNA. The unwinding helps the RNA polymerase to bind the single stranded DNA template. But there is also need for different transcription factors to make the interaction possible. Once RNA polymerase has bound, the elongation starts, which means that an RNA copy of the DNA template is made as RNA polymerase is traversing along the template strand. The copy (mRNA) is transported to the cytoplasm once it is finished. In the cytoplasm, the sequence is translated into amino acids with help from the ribosome. Ribosomes consist of different subunits that surrounds mRNA and use its sequence as a template for amino acid synthesis where the ribosome is constantly fed with amino acids from transport RNA (tRNA) molecules, each specific for one amino acid (Figure 2). When the amino acid sequence is finished, it is released from the ribosome and folds into a three dimensional structure and is transported to its predestined destination.
Figure 2) From DNA to protein. In the nucleus, mRNA is made, which is a copy of the DNA template containing all the genetic information. Protein synthesis is performed in the cytoplasm by the ribosomes.
Translation
Cytoplasm
Mature protein
Folding
Nucleus
DNA
Transcription to mRNA
Mature mRNA
Transport to cytoplasm
Ribosome
Amino acids
Protein folding The normal protein function does not appear until the polypeptide has developed its final three dimensional conformation, i.e. the protein has been folded. Protein folding involves interactions of the amino acids within the polypeptide to form different kinds of secondary structures; α-helices and β-sheet (Figure 3). The secondary structure elements can arrange into a tertiary structure mediated by side chain interactions. The folding process occurs right after the synthesis of the polypeptide and is normally a relatively fast process. It can even occur on a milli or micro second time scale.
American biochemist Christian Anfinsen was the first to show that the order of amino acids in the primary structure is what dictates the final protein conformation [6]. He also found that if a folded protein was denatured, i.e. the non-covalent native Hbonds, the charge-charge interactions and the hydrophobic interactions were broken and the protein became unfolded, it could again find its folded, native conformation, i.e. refold, under permissive conditions. Anfinsen was awarded the Nobel Prize in 1972. However, for many proteins, the process is not as simple as it was initially described by Anfinsen. Some proteins can fold, unfold and refold spontaneously in vitro (usually the smallest ones) in a one-step process [7], some fold through one or many intermediates and most need assistance to adopt their ultimate conformation. The cells are provided with molecules whose major function is to help proteins to fold correctly, these molecules are also proteins, and are called molecular chaperones.
Genetic mutations …
Figure 3. Illustration of secondary and tertiary structures of a protein. a) α-helix b) β-sheet, within a β-hairpin c) tertiary structure of a folded protein consisting of both α-helices and β-sheets and the spacial orientation of the secondary structure elements are dictated by side chain interactions (www.pdb.org).
Protein misfolding When errors occur in the protein folding machinery, it can result in protein misfolding and misfolding disease. Misfolding diseases are often associated with amyloidosis [8]. The reasons for protein misfolding could be mutations in the genes that code for the proteins that are to be misfolded, or outer factors like stress. For some cases, it is unknown why proteins start to misfold. The consequences could be harmful to the surrounding cells and to the organism in general. Protein misfolding diseases strike many people during their lifetime, and it seems like the phenomenon has become more prevalent during the past years. One early disease of this kind was described in 1906, when the German neurologist Alois Alzheimer found a form of amyloidosis that affects the brain. The disease was later named Alzheimer´s disease (AD). Most AD patients get the disease sporadically, i.e., it is usually not inherited. The symptoms for disease include memory disturbance and loss of other intellectual abilities, the symptoms are also called dementia. To date, more than 20 million people are believed to suffer from dementia [9]. In the fifties, the first form of transmissible human amyloid disesase, named kuru, was found among people practicing cannibalism in Papua New Guinea. Shortly afterwards, a disease with similar pathology was discovered in Europe and United States; transmissible spongiform encephalopathy (TSE), among individuals that had been treated with growth hormones extracted from human cadavers. The TSEs include Bovine Spongiform Encephalopathy or Mad Cow Disease (BSE), Creutzfeldt Jakob Disease (vCJD), Gerstmann-Straussler-Scheinker (GSS) disease and fatal familial insomnia (FFI). Certain misfolded proteins, called prions, are implicated in these diseases. The word “prion” stands for “proteinaceous infectious particle”, referring to its pathogenic variants. Stanley B. Pruisiner was the first to identify the molecular mechanisms of prions [10-13] and he was awarded the Nobel Prize in Medicine in 1997 for discovering a new infectious agent- a protein.
All misfolding diseases, whether they are sporadic, inherited or transmissible, are associated with deposition of proteins in different organs, depending on the disease (Table 1). The proteins involved are normally soluble but have become insoluble and aggregated and have developed fibril-like structures. Remarkably, the final fibrils are strikingly similar regardless of the precursor protein, consisting of cross-β-sheet structure with twisted morphology. The mechanisms for developing fibrils are also similar; starting with a conformational change in the protein which becomes a building block for aggregates or clusters that develop long fibril like structures over time and accumulate in tissues or organs in the body with consequences like impaired organ function and cell death [14].
Alzheimer´s, Parkinson´s and Creutzfeldt Jakob disease are examples of notorious misfolding diseases, but there are also less known diseases like Familiar amyloidotic polyneuropathy (FAP) with related pathology (Table 1). The precursor protein for FAP is transthyretin (TTR).
Table 1. A selection of diseases coupled to protein misfolding and amyloidosis and their precursor proteins.
Clinical syndrome Precursor protein
Alzheimer´s disease Aβ-protein
Primary Systemic Amyloidosis Ig Light Chain
Secondary Systemic Amyloidosis Serum Amyloid A
Senile Systemic Amyloidosis (SSA) Transthyretin
Familial Amyloid Polyneuropathy (FAP) Transthyretin
Finnish Hereditary Systemic Amyloidosis Gelsolin
Type II Diabetes Islet Amyloid Peptide
Non-Neuropathic Systemic Amyloidosis Lysozyme
Cerebral Amyloid Angiopathy Cystatin C
Atrial Amyloidosis Atrial Natriuretic Factor
Familial Amyloidosis Type III Apolipoprotein A-1
Hereditary Renal Amyloidosis Fibrinogen
Transthyretin (TTR) Transthyretin (TTR) was discovered in 1942, it became known as prealbumin and was by then detected in the cerebrospinal fluid. In the fifties, prealbumin was identified as a thyroxine (T4) binding protein by Sidney Harold Ingbar, which was published in 1958 in Endocrinology [15]. Kanai et al [16] characterized prealbumin as a retinol binding protein and published their finding in a paper in Journal of Molecular Biology, and prealbumin became Retinol Binding Prealbumin (RBPA). The structure of RBPA was described by Blake and colleagues in 1978 [17] and in 1981, the name transthyretin was accepted [18].
TTR has a molecular weight of 55 kDa and its structure (studied by X-ray crystallography) is a homotetramer with four identical, monomeric subunits, composed of 127 amino acids [19]. Each subunit has a molecular weight of 14 kDa and contains eight β-strands denoted A-H and a helix between strands E and F. The β- strands in each monomer form a β-barrel consisting of two antiparallel four stranded β-sheets containing the DAGH and CBEF strands. Association of two monomers to a dimer forms a β-sandwich stabilized by hydrogen bonds between the H-H (Figure 4) and F-F strands. Association of another dimeric β-sandwich produces the tetrameric conformation. The dimers are connected through hydrophobic interactions between the A-B loop of one monomer and the H-strand of the opposite dimer.
Figure 4. The dimeric form of TTR. The dimer is held together and stabilized by hydrogen bonds between strands H and H and F and F (not shown) in the TTR structure (www.pdb.org, pdb code 1DVQ).
Tetrameric TTR contains two identical T4- binding sites (Figure 5) located in a channel at the center of the molecule [20]. If one of the binding sites is occupied with T4, it becomes harder for the second T4-molecule to bind because of an allosteric effect (negative cooperativity) that takes place in the molecule upon binding of T4. T4 is a thyroid hormone and it plays a role in the metabolism, but is also important for neuronal development [21]. Free T4 is metabolically active. Plasma TTR functions as a transporter of T4 in the blood and transports 15-20 % of serum-T4 and around 80% of CNS-T4 [22]. Other T4 binding proteins and transporters include albumin and thyroxine-binding globulin. TTR is also involved in the transportation of retinol (vitamin A) in complex with retinol binding protein (RBP). The TTR–RBP–retinol complex is formed in the endoplasmic reticulum (ER) of hepatocytes. In TTR, there are four binding sites for RBP, however, only two molecules can bind at the same time because of steric hindrance. In the plasma, most of the TTR does not bind RBP [23, 24]. The source for plasma TTR is the liver. In human plasma, TTR is present at a concentration of 0.25 g/l [23]. The major sites for TTR production are in the liver, the choroid plexus of the brain and the retinal pigment epithelium in the eye.
There are over 100 known mutations in the TTR gene that are associated with amyloid deposition, with varying phenotype depending on the mutation [25] (some are shown in Figure 8). The most common neurodegenerative disease associated with TTR mutations is familial amyloidotic polyneuropathy (FAP) [23, 26-28], which first was described in 1952 by Corino Andrade [29] in Portugal. For more than 20 years, Andrade and colleagues had observed 74 cases from different families suffering from a progressive and mortal, by then unknown disease. ….
Figure 5. Tetrameric TTR (from rat). The binding sites for T4 are pointed out with the arrows (www.pdb.org, pdb code 1IE4).
Although TTR amyloid deposit disease is associated with TTR variants, senile systemic amyloidosis (SSA) is a disease associated with TTR wild type and affects up to 25% of people over the age of 80 and is characterized by amyloid deposits in the heart [36, 37]. The primary structure of TTR is therefore not the only explanation for development of TTR amyloidosis [38]. SSA is usually benign and without symptoms, and mostly men are severely affected [39]. Analysis of the amyloid fibril deposits in SSA patients have revealed that the amyloids contain fragments of TTR and those fragments dominate over full length TTR [40, 41]. The fragmentation has occurred at certain positions (predominantly at positions 46, 49 and 52) in the molecule which makes it tempting to believe that the cleavage of TTR is the cause of disease since the cleavage might expose sequences that are prone to aggregate. However, the cleavage mechanism is not fully understood.
Formation of TTR amyloids starts with dissociation of the tetramer into monomers, that in turn partly unfold and develop aggregates and amyloid fibrils over time [42- 44] (Figure 6). While the structure and properties of amyloid fibrils have been in the focus for diagnosing and understanding the pathogenesis for amyloid disease, there is now increasing evidence that the intermediate states in the amyloid formation pathway, are the most toxic species [45-48].
Another role for TTR, which recently has been published, is the ability to bind the Aβ-protein, which plays the major role in the pathology of Alzheimer´s disease. TTR can act in a chaperone like manner and thereby prevent formation of Aβ amyloid aggregates and thereby possibly halt progression of Alzheimer disease [49].
Figure 6. The amyloid formation process of TTR. The native tetrameric structure of TTR is destabilized and form a rearranged structure that dissociates into monomers. The monomeric species are unstable and aggregation prone and can mature into long, inert fibril structures or unfold. Unfolded TTR can also rearrange to a molten globule like structure (A-state) that has very similar properties as the monomeric amyloidogenic intermediates.
The TTR D18G mutation TTR D18G is a naturally occurring mutation in the TTR gene. The mutation was originally discovered in a Hungarian family, where four definite and three probable affected members were identified. It leads to amyloidosis in the central nervous system (CNS) with disease onset at an average age of 44. The affected family members had extensive amyloid deposition in the leptomeningeal vessels and in the subarachnoid membrane. ….
TTR D18G was identified as the most destabilized TTR mutant found to date. Recently, it has been demonstrated that a combination of thermodynamic and kinetic stability of TTR mutants is strongly correlated to disease progression. …
Analysis of serum and cerebrospinal fluid (CSF) of a heterozygote D18G patient revealed that only TTR wt could be detected [53], which is an indication of degradation or accumulation of D18G within the cell or rapid degradation post secretion. This could explain why patients do not develop disease until 44 years of age.
Figure 7. Position of D18 in the TTR monomer and residues believed to influence the tetramer stability. A) Structure of a TTR monomer. Residues within a radius of B) 5Å C) 7Å
TTR D18G is monomeric (Figure 7) and unable to form tetramers under physiological conditions. The mutant is aggregation prone and aggregates 1000-fold faster that TTR wt under physiological conditions [53]. The location of the D18G mutation is at the end of the A-strand of TTR. The neighboring residues (Figure 7) are known to stabilize the tetrameric structure. For example, the A25T mutation results in destabilization of the TTR tetramer and cause CNS amyloidosis [54], the V20I mutation leads to destabilization of the TTR tetramer and cause cardiac amyloidosis [55], the F87M/L110M mutations engineer the TTR molecule to be monomeric [56] and the L111M mutation leads to cardiac amyloidosis [57].
The region with the D18 mutation obviously has high impact on TTR tetramerization and stability. TTR D18G cannot efficiently form hybrid tetramers with TTR wt. T4 binding was found to facilitate tetramerization of D18G in the choroid plexus, where concentrations of T4 is high, but not in the CSF where the concentrations are lower. That could explain the TTR D18G prevalence to accumulate in the CNS. Expression of TTR D18G in E.coli leads to the formation of inclusion bodies [53].
Figure 8. Primary sequence of TTR with naturally found mutations marked below the wild type sequence (Hou et al 2007).
Molecular chaperones The term “molecular chaperone” was coined by Ron Laskey in 1978. Laskey observed that a nuclear protein, called nucleoplasmin, could solve a misassembly problem during the assembly of histone proteins, termed nucleosomes, in amphibian eggs. Nucleosomes bind DNA by electrostatic interactions. If the interactions are broken, e.g. by changes in the physiological conditions, the nucleoplasmin is not able to rebind the DNA, even if the physiological conditions are readapted, which leads to aggregation of the protein. This can be prevented by the presence of nucleoplasmin, is able to bind the nucleosomes and protect them [58].
The molecular chaperones are today known as folding helpers and it is believed that they are essential for cell survival and for life processes in general. They are present in the mitochondria, the Golgi, the ER and in the cytoplasm of all cells. The chaperones can correct mistakes in the folding machinery, unfold and send misfolded species to be degraded, or hold on to proteins that cannot be folded in a productive way, thereby preventing escapes of misfolded proteins that could cause damage. The chaperones direct their substrates into productive folding, transport or degradation pathways, but they do not become parts of the final structures of the proteins they interact with [59]. The majority of newly synthesized proteins need assistance to adopt their final conformation. Molecular chaperones stabilize non-native proteins, unfold incorrectly folded proteins and send abnormal proteins for degradation. They do not interact with native proteins, only the unfolded or partially unfolded ones. They are interacting with the proteins for a finite time and thereafter release their substrates, often mediated through ATP hydrolysis. Some chaperones interact with a wide variety of polypeptide chains whereas others are very restrictive and only bind to specific targets.
The heat shock response was first discovered in 1962 in Drosophila flies and the heat shock proteins (HSP) were identified as a set of proteins whose expression was induced when the cells were exposed to elevated temperature [60]. Shortly after they had been discovered, it became evident that their synthesis was not only due to elevated temperatures in cells but also to other forms of outer stresses, like radiation (UV or gamma-irradiation), oxidative stress, exposure to heavy metals, amino acid analogues etc. Protein misfolding and aggregation can lead to acute or chronic stress and activation of inappropriate signaling pathways. HSPs have strong cytoprotective effects [61] and are thought to restore the cellular homeostasis when it is disturbed.
Mammalian HSPs are classified according to their molecular weights (in kilodaltons) and are divided into two main groups, the high molecular weight HSPs and the small molecular weight HSPs. The first group includes three major families: Hsp60, Hsp70 and Hsp90. The first group (the heavy HSPs) consists of ATP dependent chaperones whereas the second group (the light HSPs) consists of ATP independent chaperones.
The Hsp70 family is the most studied HSP family, containing proteins from 66 to 78 kDa. Some of the Hsp70 proteins are localized in the cytosol (Hsp70 and Hsp72), one is found in the mitochondrion (mtHsp70) and one in the ER (BiP).
BiP BiP was first defined as glucose regulated protein with a molecular weight of 78 kDa (GRP78) or immunoglobulin heavy chain binding protein. Its function as a molecular chaperone was established by Munro et al [62] in 1986, who demonstrated that BiP is an ATP dependent member of the Hsp70 family, located in the ER lumen. BiP interacts with newly synthesized proteins and chaperones them during transport through the cell and is believed to be one of the most important components in facilitating folding in the ER. BiP has a molecular mass of 74 kDa and its 3D structure is not known but it has been defined by X-ray crystallography for DnaK (an E.coli Hsp70 and BiP homologue).
Structure and mechanism of BiP All the Hsp70 family members have the same structural organization with a 44 kDa N-terminal ATPase domain (NtD), a 18 kDa C-terminal substate binding domain (CtD) (Figure 9) and a third domain, belonging to the C-terminal domain whose function is unknown. The NtD and the CtD communicate allosterically with each other. If the NtD is occupied by an ATP molecule, the affinity for the substrate in the CtD is low but if the NtD is occupied by an ADP molecule, the affinity for substrate is high. If the CtD binding site is occupied by a substrate, the rate of ATP hydrolysis in the NtD increases [63, 64]. Thus, an unfolded polypeptide captured by BiP, can undergo cycles of binding and release, cycles that will proceed until BiP binding motifs no longer are present in the released and folded polypeptide. BiP recognizes a wide variety of nascent polypeptides with no obvious sequence similarity. However, experiments that have been done in order to find sequences that BiP preferentially binds to, have shown that the binding motifs consist of a high proportion of hydrophobic residues, normally located in the interior of a folded protein. It has also been shown that those motifs preferably consist of seven amino acids [63].
Figure 9. The NtD and the CtD of the bacterial BiP homologue DnaK. The NtD binds ATP/ADP (ADP is marked with black, left figure) whereas the CtD is substrate binding (substrate (NRLLLTG) is marked with black, right figure). The domains communicate allosterically with each other. When ATP is bound to the NtD, the CtD releases its substrate (www.pdb.org, pdb code 1S3X for left figure and 1Q5L for right figure).
…..
BiP can self associate into different oligomeric species and it is the CtD that is responsible for oligomerization. The more oligomeric BiP is, the less active is it [65]. BiP can also be post-translationally modified by phosphorylation and by ADP ribosylation. These modifications are believed to play a role in the synthesis and the polypeptide binding of BiP. Accumulation of unfolded proteins in the ER leads to an decreased amount of modified BiP whereas unmodified, monomeric BiP increases [64].
Many chaperones need co-chaperones to be effective. Hsp70 chaperones often need Jdomain containing Hsp40 proteins. The function for the Hsp40 proteins is to stimulate the ATPase activity which is crucial for Hsp70 chaperone activity. BiP can interact with different J-domain proteins [72] which are necessary for the chaperone function. ….
A role for BiP during translocation ….
The role of molecular chaperones in misfolding diseases A current opinion is that the chaperones play important roles in the protein misfolding diseases since they are parts of the control system in the cell [76, 77]. All proteins associated with the classical amyloid diseases are secreted proteins and will therefore pass the quality control checks within the ER, where they interact with a number of proteins facilitating protein folding. In some cases, misfolded proteins are accumulated in the ER [5]. This accumulation causes “ER-stress”, a condition that normal cells respond to by increasing the transcription of genes encoding ERchaperones, such as BiP, to facilitate protein folding or by suppressing the mRNA translation to synthesize proteins. These mechanisms are called “the unfolded protein response” (UPR). Once proteins are aggregated into extracellular amyloid deposits they are quite resistant to degradation.
The ER, cellular stress and cell death The ER is a membrane bound cellular organelle, consisting of tubules, vesicles and cisternae. The environment is oxidizing, which facilitates formation of disulphide bonds in maturing proteins and thereby stabilizing their structures. ER is involved in protein translation, folding, post translational modifications and quality control of proteins that are to be secreted from the cell. The majority of secreted or plasma membrane proteins enter the ER and fold within it. The vesicles of the ER are responsible for transport of proteins to be used in the cell membrane or to be secreted from the cell. Molecular chaperones and folding enzymes assist nascent proteins to fold inside the ER and correctly folded proteins are transported to the Golgi apparatus. Proteins that are not able to fold or that are misfolded, are accumulated in the ER since they cannot be exported. There are different mechanisms responding to accumulation of unfolded or misfolded proteins inside the ER. One of the mechanisms is termed ER-associated degradation (ERAD), which recognizes the misfolded proteins and retrotranslocates them to the cytoplasm and send them for degradation by the ubiquitin-proteasome degradation machinery [78]. Another mechanism that responds to accumulation of unfolded proteins in the ER is the unfolded protein response (UPR). Accumulation of unfolded proteins in the ER may also lead to cell death (apoptosis), if the condition is prolonged and cannot be solved (Figure 10). ER chaperones and ER components play a crucial role for recognition of unfolded proteins and are continuously expressed in the ER. [79].
Figure 10. The ER functions. Proteins entering the ER are facing different destinies. The correctly folded proteins are sent for export, whereas proteins that are not able to fold are sent for degradation. Accumulation of incorrectly folded proteins leads to ER stress, which in turn can result in apoptosis if the condition is prolonged. Most ER processes involve several chaperone systems as indicated in the figure.
The unfolded protein response (UPR) ER has a certain loading capacity, which varies between different cell types and during a cell’s life. When unfolded proteins are accumulated in the ER, the cell becomes stressed and the folding machinery gets perturbed. Unfolded proteins have hydrophobic residues exposed, which normally are buried in the interior of the folded protein. These hydrophobic parts tend to form (protein) aggregates that are toxic to cells. ER stress can also occur as a result of starvation, virus infections or heat, and other outer factors that influence cells negatively, and the condition is either transient or permanent. The cells respond to the stress by activating a pathway of signals leading to transcription of more chaperones, e.g BiP. Simultaneously, the translation of new proteins and the loading of proteins into ER are reduced, and further accumulation of unfolded proteins is decreased. …
ER stress leads mainly to three sets of responses: first, the amount of unfolded proteins that enters the ER is reduced (lowered protein synthesis and translocation into the ER); second, the ER folding capacity is increased (transcriptional activation of UPR target genes) and third, if the homeostasis has not been re-established, cell death (the cells commit suicide (apoptosis) to protect the organism). ER stress leads to activation of different signaling pathways, mediated by trans-membrane proteins, so called stress transducers, which sense the ER overload and transmit a signal to the cytosol where the transcription and translation of proteins take place. Three pathways have been identified (Figure 11), mediated by inositol-requiring protein-1 (IRE1), activating transcription factor-6 (ATF6) or protein kinase RNA (PKR)-like ER kinase (PERK) [82]. ….
Figure 11. The unfolded protein response with a central role for BiP.
Apoptosis Sometimes, cells have to die. They can do it in different ways and for different reasons. One reason for cell death is tissue damage, which results in a process called necrosis. During necrosis, damaged cells swell and burst and release their contents to the surrounding area, which in turn can damage the neighbouring cells and give rise to an inflammation. ….
Caspases Caspases is a family of calcium dependent cysteine proteases and they are able to cleave their substrates after aspartate residues. Robert Horwitz and colleagues identified a gene (in C.elegans) called Ced-3, which coded for a protein with similar properties to the, by then, only known caspase (caspase 1) and what they found was required for cell death [88]. After that discovery, other caspases in different organisms were soon identified and their roles were surveyed [89]. Caspases contain three domains; an N-terminal domain (which vary in size between different caspases), a large domain containing the active site and a small C-terminal domain. ….
Figure 12. The inactive procaspase and the active caspase.
Methods …
Results The findings will be presented in four papers, which are summarized below. The first paper is a study about TTR and its misfolding and fibrillation pathway. The oligomeric intermediates in the process were studied and characterized. In paper II, the different states in the TTR oligomerization pathway were captured, and added to neuroblastoma cells to elucidate which species were toxic to cells when applied from the outside. We could see that early oligomers were toxic to neuroblastoma cells. This resulted in apoptosis and release of BiP into the cytoplasm. In the third paper, the misfolding of TTR is studied from a disease point of view and the role of BiP in misfolding diseases is discussed. The most unstable TTR variant found to date, TTR D18G, was used as a model for the study and we found that BiP strongly interacted with this mutant, which was not the case for TTR wt or other mutants. Paper IV is a study of the role of BiP for TTR D18G misfolding within a eukaryotic cell. Most of the work done previously was in vitro studies and measurements were performed on purified proteins, made in E.coli cells. However, the fourth study was done in an in vivo context, to get a cell biologigal aspect of the work. Human kidney cells were used to express proteins and the interactions inside the cells were studied. We could see that the in vivo results in human cells correlated well to what we had seen earlier from E.coli expressed complexes.
Paper I The purpose with the study in the first paper was to characterize and understand the aggregation process of TTR. We used different techniques to detect structural changes in the aggregation process. An in vitro protocol for creating TTR oligomers was used. Oligomers were studied by using fluorescence spectroscopy, circular dichroism, chemical cross-linking and transmission electron microscopy. ….
Figure 16. Different probes were used to follow the TTR misfolding reaction. Aliquots of the aggregation reaction of TTR were withdrawn and assayed at 2μM probe + 2 μM TTR. Symbols: ANS (circles), Bis-ANS (inverted triangles), DCVJ (triangles) and ThT (squares). The fluorescence intensity of the different probes in the presence of the unfolded monomer state and the burst amplitude from the fit is indicated with horizontal lines labeled with the letter U and “burst” in colors corresponding to the probe.
Paper II In paper I, we characterized different states in the TTR aggregation pathway. In paper II, we harvested the different states in the aggregation pathway (during fibrillation) and challenged with neuroblastoma cells with these species. We exposed the cells to both early, oligomeric TTR species, long and mature fibrils of TTR and native TTR wt. We also wanted to investigate if BiP was upregulated in cells as a marker for UPR activation when exposed to TTR oligomers and used immunostaining for BiP to detect BiP localization in cells.
Early oligomeric species of TTR kill human cells Phase contrast images of human SH-SY5Y neuroblastoma cells exposed to early oligomers or mature fibrils of TTR in 48 hours, revealed that cells that had been exposed to oligomers were dying, whereas cells exposed to mature fibrils were still alive and healthy (Figure 17). …
Figure 17. Phase contrast pictures of cell viability after exposure to TTR. Cells exposed to early TTR oligomers, which had aggregated for 5 min, demonstrated apoptotic morphology, such as decreased cell size and more sparse population compared to non exposed cells (C) or cells exposed to native TTR wt, 22˚C (wt). Cells exposed to TTR that had aggregated for longer than 1 h were more dense. Cells exposed to TTR that had aggregated for more than one day (24 h and 1 week (1 w)) showed similar morphology as control cells (C).
Figure 18. BiP is upregulated in cells stressed with TTR oligomers and cold, native TTR wt. A) Confocal microscopy images of cells immunostained for BiP (green). Top micrograph: cells exposed to vehicle (C). Middle micrograph: cells exposed to early TTR oligomers (5 min). Bottom micrograph: cells exposed to cold native TTR wt (wt 4 °C). B) Western blot analysis of BiP expression in cells following exposure to early oligomers of TTR or cold native TTR wt (4 °C). GAPDH was used as a protein loading control to quantify the level of BiP expression.
Paper III To understand how the chaperone BiP could interact with an unstable, aggregation prone, protein mutant like TTRD18G, plasmids containing genes for His6-BiP or FT2- TTRD18G were introduced into E.coli cells grown in LB media. The proteins were thereafter expressed with IPTG, the cells were harvested and the protein containing lysate was purified on a Ni-NTA-affinity chromatography column for capturing His6- BiP. As controls, BiP was also expressed with FT2-TTRA25T, FT2-TTRL55P or FT2- TTRwt and were treated in a similar way. The protein containing cell lysate was separated on the column and fractions containing column flow through, wash buffer, and protein eluate were analyzed by SDS-PAGE.
BiP selectively binds to destabilized variants of TTR….
Figure 19. Comparison of different TTR mutants in their ability to bind to BiP. TTR wt was compared with the mutants TTR D18G, TTR A25T and TTR L55P. FT= Flow Through, w1= wash 1, w2= wash 2, el= eluate, MW= Molecular Weight marker. ….
Figure 20. The binding site for BiP in the TTR molecule. The F-strand in the TTR molecule was found to be its binding site for BiP. ….
BiP plays a protective role against the toxic effects of TTR D18G ….
Figure 21. BiP protects from TTR D18G cytotoxicity by keeping it in a soluble form. A) Micrographs of TTR D18G complex before (upper left) and after (upper right) addition of ATP. ATP releases substrates from BiP, and release of TTR D18G results in aggregation. B) ThT fluoresecence of BiP/TTR D18G (filled bars) and BiP (open bars) before and after addition of ATP or addition of the competitive peptide 88-103 TTR. Without incubation (w/o inc), without ATP, 37˚C incubation for 18 h (37˚C no add), with ATP, 37˚C, 18h (37˚C ATP), with 88-103 TTR peptide, 37˚C, 18h (37˚C 88-103).
Paper IV In this paper, the idea was to obtain a cell biological aspect of BiP/TTR D18G binding and to investigate if the E.coli derived complexes could be confirmed in a human cellular system. We wanted to study complex formation between BiP and TTR D18G in vivo and used human 293T kidney cells to overexpress the proteins. We also wanted to investigate if BiP would influence the degradation rate of TTR D18G. We could clearly see that the complex was formed in vivo in human cells, which confirmed our previous results. It is also known from before that BiP has the ability to oligomerize. We found that BiP did not prevent aggregation of TTR D18G, but rather oligomerized with it, both in soluble and insoluble aggregates. Surprisingly, aggregates seemed to accumulate inside the ER. We also found that the degradation of TTR D18G was altered in presence of a high amount of BiP.
BiP interacts with TTR D18G in the mammalian ER ….
The degradation rate of TTR D18G is slowed down in the presence of BiP ….
Figure 22. Selective binding of mutant TTR. Cells expressing Flag-BiP and/or TTRwt or TTR D18G were lysed with triton. The triton supernatants (containing the soluble protein fractions) and the triton pellets (containing the insoluble protein fractions) were collected. Immunoprecipitation with anti-TTR antibody shows that Flag-BiP is pulled down with TTR D18G and to a small extent with TTR wt. The amount of soluble TTR D18G seemed to increase when BiP was overexpressed.
Figure 23. The TTR D18G degradation rate is slowed down in presence of BiP. Pulse chase analysis results showed that the degradation of TTR D18G alone (circles) occurred faster than when BiP was overexpressed in the cells (squares).
The BiP- TTR D18G complex was present in a wide distribution of molecular weights ….
Figure 24. Distribution of oligomers in cells expressing TTR D18G alone or TTR D18G and BiP.
BiP can protect cells from TTR D18G cytotoxicity ….
Figure 25. BiP can rescue cells from dying in apoptosis. Phase contrast pictures of human NBC exposed to cold TTR wt (wt), TTR D18G (D18G) or TTR D18G in complex with BiP (D18G + BiP) showed that cells treated with TTR D18G were dead after 48 hours (decreased cell size, modified morphology and sparse population) whereas TTR D18G in complex with BiP were more viable (more dense and normal morphology). ….
BiP is found in TTR D18G aggregates in patient tissue ….
Figure 26. BiP co-localizes with TTR containing amyloid in the brain. Amyloid staining (LCP), TTR, and BiP are all co-localized, however there are patters where the individual signals dominates, indicating that the amyloid composition is layered within the deposits (indicated with arrows, right panel) ….
Conclusions From the studies in this thesis, it could be concluded that:
Fluorescence spectroscopy used intelligently (various molecular probes and time resolves techniques) is a very powerful tool to assay formation of amyloid and prefibrillar oligomers.
Formation of TTR oligomers during acidic conditions from the A-state occurs very fast (within minutes) after the process has been initiated. The formation of amyloid like fibrils occurs via oligomeric intermediates.
Oligomeric, intermediate species in the TTR aggregation pathway are toxic to neuroblastoma cells and cause apoptosis. Mature fibrils are less toxic. Cold stored native, tetrameric TTR is also cytotoxic suggesting an additional pathway for a labile tetramer or monomer to be toxic.
The ER chaperone BiP selectively captures the pathogenic, misfolding prone mutant of TTR; TTR D18G.
The binding site for BiP in TTR is the part in TTR that is involved in formation of the tetrameric structure and is possibly an elongation site in fibrils, comprising residues 88-103. Hence, BiP maintains TTR D18G in a soluble oligomeric form which should be a protection mechanism against oligomer toxicity.
BiP co-aggregates with TTR D18G. The larger the TTR containing aggregates are, the fewer BiP are in the complex. We ascribe this collection role for BiP as a molecular shepherd.
The degradation process of TTR D18G is slowed down in the presence of BiP.
BiP can escape the ER in complexes with TTR D18G and accumulate as extracellular amyloid in human brain.
New Studies Bring Scientists Closer to Combating Dangerous Unstable Proteins
Madeline McCurry-Schmidt
http://www.scripps.edu/newsandviews/e_20141027/wiseman.html
Scientists at The Scripps Research Institute (TSRI) have discovered a way to decrease deadly protein deposits in the heart, kidney and other organs associated with a group of human diseases called the systemic amyloid diseases.
“If we can develop a strategy to reduce the load that’s coming from these proteins, then we can open up treatment options that could be broadly applied to treat multiple systemic amyloid diseases,” said Luke Wiseman, assistant professor at TSRI and a senior author of the new research.
In related studies published recently in the journals Proceedings of the National Academy of Sciences (PNAS) and Chemistry & Biology, Wiseman and his colleagues described a process that can catch unstable proteins before they are released from the cell and form deposits. The process involves a “transcription factor” (which controls genetic expression) called ATF6 that may provide a drug target for future therapies.
Systemic amyloid diseases are caused by the buildup of unstable protein in extracellular environments such as the blood. The accumulation of these proteins damages organs such as the heart, kidney and gut, leading to organ malfunction and, eventually, death. Currently, treatment options for these diseases are limited.
“There has been a lot of work on ATF6, but people haven’t yet asked the functional question—can ATF6 be therapeutically accessed?” said Wiseman.
The Root of the Disease
In the recent PNAS study, the Wiseman lab, in collaboration with Jeffery Kelly’s lab at TSRI, focused on a systemic amyloid disease called light chain amyloidosis, where the unstable proteins are called light chain immunoglobulins.
Current treatments for light chain amyloidosis involve chemotherapy to kill the dysfunctional cells that secrete the disease-associated proteins, but about 30 percent of patients have significant buildup of light chain in the heart, making them too weak for this treatment. The researchers sought to develop a strategy to reduce the buildup of these proteins and increase treatment options for these patients.
Wiseman, Kelly and their teams went to the source of the unstable proteins: a part of the cell called the endoplasmic reticulum (ER). In the ER, proteins, such as immunoglobulin light chains, fold into structures that are then secreted into the blood where they perform important functions in the body. In light chain amyloidosis, mutations in immunoglobulin light chains make the proteins unstable, allowing them to unfold in the blood and form toxic clusters (aggregates) that damage the heart.
Using human cells, the researchers used a library of compounds that target specific biologic pathways to identify mechanisms that would reduce the secretion of unstable light chains from the ER. This approach identified an ER mechanism called the Unfolded Protein Response, or UPR, as a pathway whose activation preferentially reduces secretion of disease-associated light chains.
The UPR regulates ER function through the increased expression of proteins, such as “chaperones,” that directly influence the folding and secretion of destabilized proteins. Although sustained activation of the UPR is toxic, the team wondered if specific aspects of this pathway could be targeted to help cells “catch” these unstable light chains before they are secreted to the blood, where they can cause damage.
Using a chemical biologic approach, the researchers showed that activation of the UPR-associated protein ATF6 increases expression of many ER proteins involved in regulating protein folding and trafficking and reduces secretion of disease-associated light chains without causing toxic consequences. Furthermore, they showed that activating ATF6 decreases the extracellular aggregation of light chains by about 75 percent, suggesting the potential to reduce disease.
“This is an approach to treat light chain amyloidosis that ‘cuts it off at the source’ by not allowing the disease-associated immunoglobulin light chain to get out of cells to aggregate,” said TSRI Research Associate Christina Cooley, co-first author of the new study with Lisa M. Ryno, now an assistant professor at Oberlin College.
One Strategy, Multiple Diseases
In a second study, published October 23 online ahead of print by the journalChemistry and Biology, Wiseman and his team asked if ATF6 activation could be similarly used to reduce secretion and aggregation of transthyretin—a protein that aggregates in association with other systemic amyloid diseases referred to as the transthyretin amyloidoses.
Using a similar approach, the Wiseman lab showed that ATF6 activation reduced the secretion and extracellular aggregation of disease-associated transthyretin variants. Interestingly, the team reported that ATF6 activation increases the ability of the cell to “read” the stability of proteins.
Detecting small variations in stability is crucial because some misfolded proteins can evade the cell’s protective responses. These proteins are only slightly misfolded, and they can slip past the degradation proteins and form dangerous aggregates in the blood.
Wiseman hopes researchers will be able to design therapeutics to take advantage of the body’s natural ability to use AFT6 to decrease secretion and aggregation of multiple amyloid disease-associated proteins.
“It’s very exciting to see if we can treat multiple diseases with one drug, which would really offset the cost of developing a specific drug for each amyloid disease,” said TSRI graduate student John Chen, co-first author of the Chemistry and Biology study with Research Associate Joseph C. Genereux.
Wiseman said the next step in this area of research, which also is being conducted in collaboration with the Kelly lab, is to identify drug candidates that can activate ATF6.
In addition to Cooley, Ryno, Kelly and Wiseman, other contributors to theProceedings of National Academy of Sciences paper, “Unfolded protein response activation reduces secretion and extracellular aggregation of amyloidogenic immunoglobulin light chain,” are Lars Plate, Gareth J. Morgan and John D. Hulleman, all of TSRI. Support for this study came from Arlene and Arnold Goldstein, the Ellison Medical Foundation, the Skaggs Institute for Chemical Biology at TSRI, the Lita Annenberg Hazen Foundation, TSRI and the National Institutes of Health (AG046495, DK075295, NS079882, F32 AG042259). For more information on this study. http://www.pnas.org/content/111/36/13046.full
In addition to Chen, Genereux and Wiseman, other contributors to theChemistry and Biology paper, “ATF6 Activation Reduces the Secretion and Extracellular Aggregation of Destabilized Variants of an Amyloidogenic Protein,” are Song Qu, John D. Hulleman and Matthew D. Shoulders, all of TSRI. Support for this study came from Arlene and Arnold Goldstein, the Ellison Medical Foundation, TSRI, the American Cancer Society and the National Institutes of Health (AG036634, NS079882, DK075295, DK102635, AG046495, HL099245). For more information on this study, http://www.cell.com/chemistry-biology/abstract/S1074-5521(14)00327-5
An insight to the conserved water mediated dynamics of catalytic His88 and its recognition to thyroxin and RBP binding residues in human transthyretin
http://dx.doi.org:/10.1080/07391102.2014.984632
Avik Banerjeea & Bishnu P. Mukhopadhyaya**
online: 08 Dec 2014
Human transthyretin (hTTR) is a multifunctional protein involved in several amyloidogenic diseases. Besides transportation of thyroxin and vitamin-A, its role towards the catalysis of apolipoprotein-A1 and Aβ-peptide are also drawing interest. The role of water molecules in the catalytic mechanism is still unknown. Extensive analyses of 14 high-resolution X-ray structures of human transthyretin and MD simulation studies have revealed the presence of eight conserved hydrophilic centres near its catalytic zone which may be indispensable for the function, dynamics and stability of the protein. Three water molecules (W1, W2 and W3) form a cluster and play an important role in the recognition of the catalytic and RBP-binding residues. They also induce the reorganisation of the His88 for coupling with other catalytic residues (His90, Glu92). Another water molecule (W5) participate in inter-monomer recognition between the catalytic and thyroxin binding sites. The rest four water molecules (W6, W*, W# and W†) form a distorted tetrahedral cluster and impart stability to the catalytic core of hTTR. The conserved water mediated recognition dynamics of the different functional sites may provide some rational clues towards the understanding of the activity and mechanism of hTTR.
Amyloid Formation by Human Carboxypeptidase D Transthyretin-like Domain under Physiological Conditions*
Javier Garcia-Pardo‡,§1, Ricardo Graña-Montes‡,§2, Marc Fernandez-Mendez‡,§, Angels Ruyra‡3, Nerea Roher‡,¶4, Francesc X. Aviles‡,§, Julia Lorenzo‡,§5 and Salvador Ventura‡,§6
↵6 Recipient of an ICREA academia award. To whom correspondence may be addressed. Tel.: 34-935868956; Fax: 34-935811264; E-mail: salvador.ventura@uab.es.
Capsule
Background: Proteins can form amyloid aggregates from initially folded states.
Results: The transthyretin-like domain of human carboxypeptidase D forms amyloid aggregates without extensive unfolding.
Conclusion: The monomeric transthyretin fold has an inherent propensity to aggregate due to the presence of preformed amyloidogenic structural elements.
Significance: Generic aggregation from initially folded states would have a huge impact on cell proteostasis.
Protein aggregation is linked to a growing list of diseases, but it is also an intrinsic property of polypeptides, because the formation of functional globular proteins comes at the expense of an inherent aggregation propensity. Certain proteins can access aggregation-prone states from native-like conformations without the need to cross the energy barrier for unfolding. This is the case of transthyretin (TTR), a homotetrameric protein whose dissociation into its monomers initiates the aggregation cascade. Domains with structural homology to TTR exist in a number of proteins, including the M14B subfamily carboxypeptidases. We show here that the monomeric transthyretin-like domain of human carboxypeptidase D aggregates under close to physiological conditions into amyloid structures, with the population of folded but aggregation-prone states being controlled by the conformational stability of the domain. We thus confirm that the TTR fold keeps a generic residual aggregation propensity upon folding, resulting from the presence of preformed amyloidogenic β-strands in the native state. These structural elements should serve for functional/structural purposes, because they have not been purged out by evolution, but at the same time they put proteins like carboxypeptidase D at risk of aggregation in biological environments and thus can potentially lead to deposition diseases.
The importance of a gatekeeper residue on the aggregation of transthyretin implications for transthyretin-related amyloidoses (Article)
Journal of Biological Chemistry 10 October 2014; 289(41): 28324-28337
Sant’Anna, R.a, Braga, C.a, Varejão, N.a, Pimenta, K.M.a, Granã-Montes, R.d, Alves, A.a, Cortines, J.c, Cordeiro, Y.b, Ventura, S.d , Foguel, D.a
a Instituto de Bioquímica Médica Leopoldo de Meis, Programa de Biologia Estrutural, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil
b Faculdade de Farmácia, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil
c Instituto de Microbiologia Professor Paulo de Goés, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil
d Institut de Biotecnologia I Biomedicina, Universitat Autònoma de Barcelona, Bellaterra, Spain
Protein aggregation into β-sheet-enriched amyloid fibrils is associated with an increasing number of human disorders. The adoption of such amyloid conformations seems to constitute a generic property of polypeptide chains. Therefore, during evolution, proteins have adopted negative design strategies to diminish their intrinsic propensity to aggregate, including enrichment of gatekeeper charged residues at the flanks of hydrophobic aggregation-prone segments. Wild type transthyretin (TTR) is responsible for senile systemic amyloidosis, and more than 100 mutations in the TTR gene are involved in familial amyloid polyneuropathy. The TTR 26-57 segment bears many of these aggressive amyloidogenic mutations as well as the binding site for heparin. We demonstrate here that Lys-35 acts as a gatekeeper residue in TTR, strongly decreasing its amyloidogenic potential. This protective effect is sequence-specific because Lys-48 does not affect TTR aggregation. Lys-35 is part of the TTR basic heparin-binding motif. This glycosaminoglycan blocks the protective effect of Lys-35, probably by neutralization of its side chain positive charge. A K35L mutation emulates this effect and results in the rapid self-assembly of the TTR 26-57 region into amyloid fibrils. This mutation does not affect the tetrameric protein stability, but it strongly increases its aggregation propensity. Overall, we illustrate how TTR is yet another amyloidogenic protein exploiting negative design to prevent its massive aggregation, and we show how blockage of conserved protective features by endogenous factors or mutations might result in increased disease susceptibility.
Unfolded protein response-induced ERdj3 secretion links ER stress to extracellular proteostasis
Joseph C Genereux1,2,‡, Song Qu1,2,3,‡, Minghai Zhou4, Lisa M Ryno1,2, Shiyu Wang5, Matthew D Shoulders2,†, Randal J Kaufman5, Corinne I Lasmézas4, Jeffery W Kelly1,2,6 and R Luke Wiseman1,3,*
EMBO J, 2 Jan 2015; 34(1): 4–19. published online: 31 OCT 2014 http://dx.doi.org:/10.15252/embj.201488896
The Unfolded Protein Response (UPR) indirectly regulates extracellular proteostasis through transcriptional remodeling of endoplasmic reticulum (ER) proteostasis pathways. This remodeling attenuates secretion of misfolded, aggregation-prone proteins during ER stress. Through these activities, the UPR has a critical role in preventing the extracellular protein aggregation associated with numerous human diseases. Here, we demonstrate that UPR activation also directly influences extracellular proteostasis through the upregulation and secretion of the ER HSP40 ERdj3/DNAJB11. Secreted ERdj3 binds misfolded proteins in the extracellular space, substoichiometrically inhibits protein aggregation, and attenuates proteotoxicity of disease-associated toxic prion protein. Moreover, ERdj3 can co-secrete with destabilized, aggregation-prone proteins in a stable complex under conditions where ER chaperoning capacity is overwhelmed, preemptively providing extracellular chaperoning of proteotoxic misfolded proteins that evade ER quality control. This regulated co-secretion of ERdj3 with misfolded clients directly links ER and extracellular proteostasis during conditions of ER stress. ERdj3 is, to our knowledge, the first metazoan chaperone whose secretion into the extracellular space is regulated by the UPR, revealing a new mechanism by which UPR activation regulates extracellular proteostasis.
Synopsis
The unfolded protein response (UPR) triggers secretion of ER chaperone ERdj3 to prevent formation of toxic aSggregates, providing the first mechanistic link between intracellular stress signaling and extracellular proteostasis.
- ERdj3 is an ER-stress-induced secreted chaperone
- Secretion of ERdj3 is induced by the ATF6 arm of the UPR
- Secretion of ERdj3 enhances extracellular proteostasis capacity
- ERdj3 can co-secrete as a stable complex with misfolding-prone protein clients
- ERdj3 serves as a link between intracellular and extracellular proteostasis capacity during ER stress.

http://onlinelibrary.wiley.com/store/10.15252/embj.201488896/asset/image
Imbalances in extracellular protein homeostasis (or proteostasis) and consequent protein aggregation are inextricably linked to degenerative phenotypes in over 30 human protein misfolding diseases, including Alzheimer’s disease, Creutzfeldt–Jakob disease and the transthyretin (TTR) amyloidoses (Kelly, 1996; Rochet & Lansbury, 2000; Stefani & Dobson, 2003; Buxbaum, 2004; Haass & Selkoe, 2007). Compelling genetic and pharmacologic evidence supports a causal relationship between protein aggregation (including amyloidogenesis) and the degeneration of post-mitotic tissue in these disorders (Tanzi & Bertram, 2005; Aguzzi et al, 2007; Gotz et al, 2011; Coelho et al,2012). Primary determinants of extracellular proteostasis capacity include the spectrum and concentration of secreted proteostasis factors (e.g., chaperones) (Wyatt et al, 2013) and the efficiency of protein folding and quality control in the endoplasmic reticulum (ER) (Wisemanet al, 2007).
Extracellular proteostasis capacity is regulated by secreted proteins that prevent the formation of protein aggregates associated with disease. The best characterized secreted chaperones such as clusterin directly bind misfolded proteins in the extracellular environment and prevent their aggregation through an ATP-independent “holdase” mechanism (Wyatt et al, 2012). Deletion of clusterin predisposes mice to aging-dependent progressive glomerulopathy (Rosenberg et al, 2002) and increases Aβ(1–42) aggregation and deposition in mouse models of Alzheimer’s disease (DeMattos et al, 2004). Furthermore, genome-wide association studies implicate clusterin in the development of Alzheimer’s disease (Harold et al, 2009; Lambert et al, 2009; Wijsman et al, 2011). Small populations of some ER chaperones, such as the HSP70 BiP and the lectin calreticulin, can be trafficked to the plasma membrane, particularly under stress or during apoptosis (Martins et al, 2010; Zhang et al, 2010). Protein disulfide isomerases (PDIs) can also be secreted to promote extracellular disulfide exchange (Jordan & Gibbins, 2006; Hahm et al, 2013). A role for these chaperones in extracellular proteostasis maintenance has not been demonstrated to date, rather their surface expression has been implicated in immunological signaling (Peters & Raghavan, 2011; Lee, 2014).
Extracellular proteostasis is also impacted by proteostasis in the ER, which is responsible for the folding and trafficking of the 1/3 of the human proteome that is targeted to the cellular secretory pathway (Fewell et al, 2001; Braakman & Bulleid, 2011). In the ER, nascent polypeptides interact with components of ER protein folding pathways to facilitate their folding into native three-dimensional conformations (Buck et al, 2007). Folded proteins are then packaged into vesicles at the ER membrane and trafficked to downstream compartments of the secretory pathway or the extracellular space. Proteins unable to attain native three-dimensional conformations in the ER are instead targeted to ER degradation pathways such as ER-associated degradation (ERAD) (Benyair et al, 2011). The partitioning of polypeptides between ER protein folding/trafficking and degradation pathways, also referred to as ER quality control, prevents the secretion of misfolded, aggregation-prone proteins (Powers et al, 2009; Araki & Nagata, 2011).
Despite the typical efficiency of ER quality control, exposure to genetic, environmental or aging-related stresses leads to increased protein misfolding within the ER lumen and imbalances in ER proteostasis. Such stresses can increase secretion of misfolding-prone proteins into the extracellular space, directly challenging extracellular proteostasis capacity and facilitating concentration-dependent protein aggregation into proteotoxic oligomeric conformations. As such, ER stress is pathologically associated with numerous extracellular protein aggregation diseases including the systemic amyloidoses and Alzheimer’s disease (Teixeira et al, 2006; Kim et al, 2008).
To restore ER proteostasis following stress, cells activate the Unfolded Protein Response (UPR). The UPR consists of three integrated stress-responsive signaling pathways activated downstream of the ER stress-sensing proteins IRE1, ATF6, and PERK (Schroder & Kaufman, 2005; Walter & Ron, 2011). These stress sensors are activated by the accumulation of misfolded proteins within the ER lumen (a consequence of ER stress) (Bertolotti et al, 2000; Okamura et al, 2000; Shen et al, 2002). The activation of UPR signaling pathways results in the attenuation of new protein synthesis (Harding et al, 1999) and transcriptional remodeling of ER protein folding, trafficking, and degradation pathways (Lee et al, 2003; Yamamoto et al, 2007; Shoulders et al, 2013), thus enhancing ER proteostasis capacity and quality control. Through these mechanisms, UPR activation reduces accumulation of misfolded proteins in the ER and attenuates the aberrant secretion of aggregation-prone proteins into the extracellular space (Adachi et al, 2008; Shoulders et al, 2013).
In contrast, the functional impact of UPR signaling on extracellular proteostasis capacity remains poorly defined. Not only are the established secreted chaperones not transcriptional targets of the UPR, but clusterin secretion is attenuated during conditions of ER stress, indicating that clusterin secretion is not a protective mechanism to regulate extracellular proteostasis in response to pathologic ER insults (Nizard et al, 2007). Similarly, ER stress reduces secretion of ER proteostasis factors such as protein disulfide isomerase (PDI), suggesting reduced extracellular regulation of disulfide integrity during ER stress (Terada et al, 1995). Thus, we sought to study the functional role for UPR signaling in adapting extracellular proteostasis capacity during conditions of ER stress.
Here, we report that UPR activation regulates extracellular proteostasis directly through the secretion of the ER-targeted HSP40 co-chaperone ERdj3. While ERdj3 is established to function as an HSP40 co-chaperone for BiP within the ER HSP70 chaperoning pathway (Shen & Hendershot, 2005), we show that ERdj3 is also a UPR-induced secreted chaperone, whose extracellular levels increase both in response to ER stress and stress-independent activation of the UPR-associated transcription factor ATF6. Secreted ERdj3 binds to misfolded proteins in the extracellular space, prevents the aggregation of amyloidogenic Aβ1–40 at substoichiometric concentrations, and ameliorates the toxic effects of misfolded prion protein on neuronal cells. Furthermore, we demonstrate that ERdj3 can co-secrete with destabilized, misfolding-prone clients under conditions where the ER HSP70 chaperoning pathway is overwhelmed, preemptively chaperoning these misfolding-prone secreted proteins that evade ER quality control in the extracellular environment. Thus, the capacity for ERdj3 to function in both ER and extracellular proteostasis provides an unanticipated direct link between these two environments that is regulated by the UPR during conditions of ER stress.
ER stress increases ERdj3 secretion
…..
We confirmed the ER stress-dependent increase in ERdj3 mRNA in HEK293T cells treated with the small molecule SERCA inhibitor thapsigargin (Tg)—a potent inducer of ER stress (Fig 1B). Upon Tg-induced ER stress, ERdj3 protein levels increased primarily in conditioned media and not intracellularly for HEK293T-Rex (Fig 1C and D). RNAi depletion of ERdj3 reduced intracellular ERdj3 levels > 90% and completely ablated extracellular ERdj3 upon Tg treatment. Tg treatment also selectively increased extracellular, as opposed to intracellular, ERdj3 from Huh7 cells (Supplementary Fig S1D). In stark contrast to ERdj3, BiP and HYOU1, two abundantly expressed, UPR-induced, ER chaperones, were not detected in the conditioned media (Fig 1C), reflecting the presence of ER retention motifs on these proteins. These results are consistent with previous results showing that negligible BiP is secreted to the extracellular space (Munro & Pelham, 1987; Yamamoto et al, 2003; Kern et al, 2009). Rather, BiP that evades the KDEL receptor is typically still retained at the cellular membrane (Wang et al, 2009; Zhang et al, 2010), as is common for other canonical ER-localized chaperones, particularly under apoptotic conditions (Jordan & Gibbins, 2006; Martins et al, 2010; Lee, 2014). HYOU1, the ER resident Hsp110 that both serves as a nucleotide exchange factor for BiP and displays its own chaperone function (Andreasson et al, 2010; Behnke & Hendershot, 2014), has not been implicated in either secretion or presentation at the cellular membrane. Alternatively, intracellular levels of BiP and HYOU1 were significantly increased upon Tg treatment. These data indicate that increased extracellular ERdj3 levels result from constitutive secretion and not from leakage of ER proteins into the extracellular space, as has been proposed for other ER chaperones (Booth & Koch, 1989).
…..
We next evaluated whether ER stress increases ERdj3 serum levels in mice. In the absence of stress, ERdj3 has a serum concentration of 23 ± 5 nM (Supplementary Fig S1E). Mice subjected to an 18 h fast followed by refeeding on a high-fat diet, which rapidly induces hepatic ER stress (Oyadomari et al, 2008), show a significant twofold increase in serum ERdj3 levels (Fig 1H). ERdj3 serum levels also significantly increased following 7 days on a high-fructose diet (Supplementary Fig S1F and G), which also induces ER stress in hepatic cells (Wanget al, 2012), as indicated by increased BiP, Grp94, and phosphorylated eIF2α in hepatic lysates (Supplementary Fig S1H). These results demonstrate that increased ERdj3 serum levels in mice correlate with hepatic ER stress, strongly indicating that hepatic ER stress increases ERdj3 secretion in vivo.
Stress-independent activation of the UPR-associated transcription factor ATF6 increases ERdj3 secretion
….
Figure 2. ERdj3 is efficiently secreted from cells following stress-independent activation of the UPR-associated transcription factor ATF6
We further characterized the ATF6-dependent increase in ERdj3 secretion using a [35S] metabolic pulse-chase approach. ATF6 preactivation increased the extracellular concentration of newly synthesized ERdj3 fourfold relative to vehicle-treated cells, with nearly 40% of [35S] labeled ERdj3 being secreted following a 4 h incubation in non-radioactive chase media (Fig 2B–D). Despite an increase in ERdj3 synthesis (Supplementary Fig S2B), XBP1s preactivation reduced the fraction of newly synthesized ERdj3 in media, relative to the vehicle treatment (Fig 2D). This decrease in ERdj3 secretion is attributed to an increase in ERdj3 degradation, with ~40% of newly synthesized ERdj3 being degraded after 4 h in cells following XBP1s preactivation (Fig 2E). ATF6 and XBP1s co-activation demonstrated a similar increase in ERdj3 secretion to that observed with ATF6 preactivation alone, demonstrating that ATF6 activation prevents the increased ERdj3 degradation observed upon XBP1s activation (Fig 2B–E). Collectively, these results show that ERdj3 secretion is increased by stress-independent preactivation of the UPR-associated transcription factor ATF6, directly implicating protective UPR signaling in the regulation of the extracellular ERdj3 concentration.
Secreted ERdj3 increases extracellular proteostasis capacity
To evaluate whether ERdj3 secretion is critical for the maintenance of intracellular ER proteostasis, we overexpressed either ERdj3WT or the non-secreted ERdj3KDEL and then measured expression of UPR target genes and cellular viability, both in the absence and the presence of Tg-induced ER stress (Supplementary Fig S3A and B). Despite the known role of ERdj3 in folding and degradation of specific clients (Shen & Hendershot, 2005; Hoshino et al, 2007; Jin et al, 2008; Buck et al, 2010; Tan et al, 2014), ERdj3 retention does not significantly impair global ER proteostasis maintenance in the absence or the presence of ER stress.
…..
Figure 3. Secreted ERdj3 inhibits extracellular protein aggregation
…..
Figure 4. Secreted ERdj3 attenuates vacuole formation induced by toxic prion protein in mammalian cells
…..
ERdj3 is co-secreted in complex with secreted destabilized mutant proteins
…..

http://onlinelibrary.wiley.com/store/10.15252/embj.201488896/asset/image
…..
ERdj3–client co-secretion is regulated by ER proteostasis capacity
…..
Figure 6. ERdj3–client protein complexes are co-secreted when BiP activity is limiting
…..
Discussion
Protein synthesis is necessarily an intracellular process. Hence, extracellular metazoan environments such as the blood rely on cellular protein secretion to define extracellular protein concentrations and regulate the integrity of the secreted proteome. The presence of misfolding-prone proteins in the ER directly threatens this environment, as imbalances in ER proteostasis can impair the capacity for ER quality control pathways to prevent secretion of misfolded, aggregation-prone client proteins that in turn challenge the integrity of the extracellular proteome (Hetz & Mollereau, 2014). The UPR indirectly regulates extracellular proteostasis by attenuating the secretion of misfolded protein conformations that can both accumulate during and induce ER stress. Here, we demonstrate a direct role for the UPR in regulating extracellular proteostasis through the increased transcription and secretion of ERdj3. The capacity for UPR activation to influence extracellular proteostasis through ERdj3 secretion links protein misfolding in the secretory pathway to extracellular proteostasis, revealing a new mechanism by which cells coordinate intra- and extracellular environments in response to pathologic insults that induce ER stress (Fig 7).

http://onlinelibrary.wiley.com/store/10.15252/embj.201488896/asset/image
In response to ER stress, newly synthesized ERdj3 binds misfolded ER client proteins and delivers them to BiP for chaperoning in the Hsp70 cycle. When free BiP becomes limiting, or if repeated BiP cycling cannot productively deplete the levels of the misfolded client, the stable ERdj3–client complex is co-secreted to the extracellular environment, preemptively binding the misfolded protein and preventing the aggregation of the misfolded client protein in the extracellular space. Furthermore, stress-induced ERdj3 can be secreted on its own into the extracellular space where it can bind to misfolded, aggregation-prone client proteins and attenuate pathologic protein aggregation in the extracellular environment.
In its role as a traditional HSP40 co-chaperone to BiP, ERdj3 can influence the trafficking of ER client proteins and is exploited for pathogenic toxin internalization (Yu & Haslam, 2005; Massey et al, 2011) and viral infection (Wen & Damania, 2010; Goodwin et al, 2011) (Fig 7). However, we find that at least half of newly synthesized ERdj3 is secreted. Secreted ERdj3 has the capacity to bind misfolded proteins and prevent their extracellular aggregation and proteotoxicity, suggesting that UPR-dependent increases in ERdj3 secretion offer a new potential mechanism to protect the local extracellular environment from toxic protein conformations that can be secreted during ER stress (Fig 7). Interestingly, ERdj3 also can also influence extracellular proteostasis through its co-secretion with destabilized, misfolding-prone client proteins. This capacity of ERdj3 to be co-secreted with misfolding-prone proteins avoids the problem of diffusion-limited encounter in the extracellular space.
ERdj3–client co-secretion serves as a natural bridge between the dual roles of ERdj3 as an ER HSP40 and in regulating extracellular proteostasis (Fig 7). In the ER, ERdj3 delivers misfolded clients (e.g., FTTTRA25T) into the BiP cycle for chaperoning. If inadequate free BiP is available, ERdj3 remains bound to its client protein throughout the secretory pathway and the client-ERdj3 complex can be secreted into the extracellular space, where lack of BiP prevents ERdj3 release from the substrate. When the ER must deal with high, persistent levels of misfolding-prone proteins, the UPR is activated, increasing the levels of ERdj3, BiP and other chaperones to offer misfolding-prone proteins more ER chaperoning capacity. Simultaneously, increased intracellular ERdj3 is available to bind and co-secrete with misfolding-prone protein clients that evade ER quality control and traverse the secretory pathway, providing preemptive chaperone capacity to the extracellular space. Importantly, clusterin co-secretion, unlike ERdj3 co-secretion, assists destabilized clients in evading ER quality control, providing a potential reason for the reduced secretion of clusterin during conditions of ER stress. Thus, ERdj3 offers a unique link between ER and extracellular proteostasis, employing a mechanism that cannot be achieved with other known secreted chaperones.
The capacity of UPR-dependent activation of ATF6 to influence extracellular proteostasis through ERdj3 secretion indicates a potential role for this pathway in extracellular protein aggregation pathologies. UPR signaling is activated in many extracellular protein aggregation diseases, including Alzheimer’s disease and the systemic amyloidoses (Teixeira et al, 2006; Saxena et al, 2009; Hoozemans et al, 2012; Hetz & Mollereau, 2014). Our results showing that secreted ERdj3 can increase the extracellular chaperoning and prevent extracellular aggregation and/or proteotoxicity of disease-associated proteins such as TPrP, TTRA25T, and Aβ suggest that ATF6-dependent regulation of ERdj3 secretion is a potential mechanism to protect the extracellular space from proteotoxicity. Consistent with this prediction, presenilin mutations causatively associated with Alzheimer’s disease significantly impair ATF6 activation during stress (Katayama et al, 1999, 2001). Thus, a decreased capacity to regulate extracellular proteostasis through ATF6 activation and consequent ERdj3 secretion could be a contributing factor in the disease pathology of patients harboring these mutations.
We have established that UPR activation directly and adaptively regulates the composition of the extracellular proteostasis network through ERdj3 secretion. In particular, UPR-induced secretion of ERdj3 offers a mechanism for organisms to adapt to the presence of destabilized proteins in the secretory pathway and increase extracellular chaperoning capacity through two mechanisms: (1) the secretion of free ERdj3 available to bind misfolding-prone proteins in the extracellular environment and (2) the co-secretion of ERdj3–client complexes, which preemptively protects the extracellular environment from proteotoxic protein conformations. This potentially provides an endogenous mechanism to prevent ER stress-induced increases in extracellular protein aggregation and proteotoxicity that can lead to degenerative phenotypes. Our identification of ERdj3 as a UPR-induced secreted chaperone demonstrates that targeting adaptive stress responses, particularly the ATF6 arm of the UPR, can enhance the maintenance of extracellular proteostasis, offering a promising approach to better understand and intervene in diseases characterized by extracellular protein aggregation.
References
- , , , , , (2008) ATF6 is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum. Cell Struct Funct 33: 75–89
- , , (2007) Insights into prion strains and neurotoxicity. Nat Rev Mol Cell Biol 8: 552–561
- , , , , (2010) The endoplasmic reticulum Grp170 acts as a nucleotide exchange factor of Hsp70 via a mechanism similar to that of the cytosolic Hsp110. J Biol Chem 285: 12445–12453
- , (2011) Protein folding and quality control in the ER. Cold Spring Harb Perspect Biol 3: a007526
- , (2014) The large Hsp70 Grp170 binds to unfolded protein substrates in vivo with a regulation distinct from conventional Hsp70s. J Biol Chem 289: 2899–2907
- , , (2011) Protein quality control, retention, and degradation at the endoplasmic reticulum. Int Rev Cell Mol Biol 292: 197–280
- , , , , (2000) Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol 2: 326–332
- , , , , , , , , , (1999) Endogenous proteins controlling amyloid beta-peptide polymerization. Possible implications for beta-amyloid formation in the central nervous system and in peripheral tissues. J Biol Chem 274: 15990–15995
………..
Late Onset of Alzheimer’s Disease and One-carbon Metabolism
Reporter and Curator: Dr. Sudipta Saha, Ph.D.
http://pharmaceuticalintelligence.com/2013/05/06/alzheimers-disease-and-one-carbon-metabolism/
The two main molecular signs of AD are:
- Extracellular deposits of Amyloid-beta (Aβ) peptides (amyloidogenic pathway) and
- Intracellular deposits of phosphorylated protein TAU (fibrillogenic pathway)
For many years, both these two pathways (amyloidogenic and fibrillogenic) contended the role of “responsible” for AD onset in the researchers’ debates, even originating respectively the two groups of “BAptists” and “TAUists” scientists. In the recent years, however, these absolutist hypotheses were confuted by the emerging data evidencing that late onset AD (LOAD) has the characteristics to be considered a multifactorial disease and by scientific reports demonstrating possible interconnection between (but not limited to) the two above-mentioned “pathogenic” pathways.
For example, it was demonstrated that
- GSK-3β (glycogen synthase kinase 3-beta), a phosphorylase involved in tau phosphorylation, is also responsible for APP (Amyloid Precursor Protein) phosphorylation and that
- Aβ peptides are able to induce GSK-3β.
Among the several possible cocauses and interconnected pathways involved in LOAD onset and progression, a very rapidly emerging topic is related to the role of epigenetics. Moreover, it was hypothesized that methylation impairment could be a common promoter and/or a connection between amyloid and tau pathogenic pathways involving not only DNA methylation but also protein methylation mechanisms. This observation rises from studies on PP2A (protein phosphatase 2A) protein methylation showing that downregulation of neuronal PP2A methylation occurs in affected brain regions from AD patients, causing the accumulation of both phosphorylated tau and APP isoforms and increased secretion of Aβ peptides.
Altered methylation metabolism could represent the connection between B vitamins and LOAD. B vitamins are essential cofactors of homocysteine (HCY) metabolism, also called 1-carbon metabolism. One-carbon metabolism is a complex biochemical pathway regulated by the presence of folate, vitamin B12 and B6 (among other metabolites), and leading to the production of methyl donor molecule S-adenosylmethionine (SAM). High HCY and low B vitamin levels are associated to LOAD, even if a cause-effect relationship is still far to be ascertained; moreover, a clear correlation between HCY and Aβ levels has been found.
In addition, SAM, the principal metabolite in the HCY cycle and the main methyl donor in eukaryotes, appears to be altered in some neurological disorders, including AD. HCY, a thiol containing amino acid produced during the methionine metabolism via the adenosylated compound SAM, once formed is either converted to cysteine by transsulfuration or remethylated to form methionine. In the remethylation pathway HCY is remethylated by the vitamin B12-dependent enzyme methionine synthase (MS) using 5-methyltetrahydrofolate as cosubstrate. Alternatively, mainly in liver, betaine can donate a methyl group in a vitamin B12-independent reaction, catalyzed by betaine-homocysteine methyltransferase (BHMT). In the transsulfuration pathway, HCY can condense with serine to form cystathionine in a reaction catalyzed by the cystathionine beta synthase (CBS), a vitamin B6-dependent enzyme, and the cystathionine is hydrolyzed to cysteine (Cys). Cysteine is used for protein synthesis, metabolized to sulfate, or used for glutathione (GSH) synthesis. The tripeptide GSH is the most abundant intracellular nonprotein thiol, and it is a versatile reductant, serving multiple biological functions, acting, among others, as a quencher of free radicals and a cosubstrate in the enzymatic reduction of peroxides. HCY accumulation causes the accumulation of S-adenosylhomocysteine (SAH) because of the reversibility of the reaction converting SAH to HCY and adenosine (Ado); the equilibrium dynamic favors SAH synthesis. The reaction proceeds in the hydrolytic direction only if HCY and adenosine are efficiently removed. SAH is a strong DNA methyltransferases inhibitor, which reinforces DNA hypomethylation (Chiang et al., 1996). Thus, an alteration of the metabolism through either remethylation or transsulfuration pathways can lead to hyperhomocysteinemia, decrease of SAM/SAH ratio (methylation potential; MP), and alteration of GSH levels, suggesting that hypomethylation is a mechanism through which HCY is involved in vascular disease and AD, together with the oxidative damage. To add insult to injury, oxidative stress also promotes the formation of oxidized derivatives of HCY, like homocysteic acid and homocysteine sulfinic acid. These compounds, through the interaction with glutamate receptors, generate intracellular free radicals.
The first observations about B vitamins or HCY deficiency in neurological disorders were hypothesized in the 80 seconds. Despite this recent acknowledgement, alterations of HCY levels and related compounds were only recently widely recognized as risk factors for LOAD and other forms of dementia. Few mechanisms are suggested as possible protagonists in the toxic pathway of HCY in LOAD onset:
- oxidative stress and neurotoxicity,
These results were obtained by using both transgenic and dietary models of hyperhomocysteinemia or altered 1-carbon metabolism. On the one hand, this variety of experimental models allowed to investigate multiple aspects of the biochemical alterations and their consequences; on the other, the lacking of common methods or goals generated a large body of literature in part overlapping for some aspects but fragmentary or incomplete for others. This aspect represents, together with the scarce interplay between clinical/epidemiological and biomolecular research, one of the reasons for the poor relevance given by the scientific community to the role of 1-carbon metabolism in certain diseases like dementia.
A causal connection between 1-carbon alterations:
- hyperhomocysteinemia,
- low B vitamins,
- low SAM, or
- high SAH
and biological alterations responsible for LOAD onset and progression is still missing. So, it was previously demonstrated that 1-carbon metabolism was related to AD-like hallmarks (increased Aβ production) via PSEN1 (presenilin 1) and BACE (beta-site APP cleaving enzyme 1) upregulation in cellular and animal models. More recently, it was added to the rising literature body dealing with 1-carbon metabolism and GSK-3β and PP2A modulation; it was also demonstrated that PSEN1 promoter is regulated by site-specific DNA methylation in cell cultures and mice and that this modulation of methylation is dependent on the regulation of the DNA methylation machinery. Although all the proposed pathways of HCY toxicity are possibly involved and nonmutually exclusive, as suggested by the multifactorial origin of LOAD, the recent advances in the connection between epigenetics and LOAD (as discussed above) stress a primary role for methylation dishomeostasis dependent on 1-carbon metabolism alterations.
Protein misfolding and prions
Larry H/ Bernstein, MD, FCAP, Curator
Leaders in Pharmaceutical Intelligence
Series E. 2; 4.9
Revised 9/30/2015
Susan L. Lindquist, Stanley B. Prusiner
http://pharmaceuticalintelligence.com/2015/09/11/protein-misfolding-and-prions-3/
Whitehead Member Susan Lindquist is a pioneer in the study of protein folding. She has shown that changes in protein folding can have profound and unexpected influences in fields as wide-ranging as human disease, evolution and nanotechnology.
Protein misfolding has been implicated as a major mechanism in many severe neurological disorders including Parkinson’s and Huntington’s diseases. Lindquist and colleagues have developed yeast strains that serve as living test tubes in which to study these disorders, unraveling how protein folding contributes to them.
Prions are proteins that can change into a self-perpetuating form. They have only been discovered recently, but one of them is already well known as the cause of mad cow disease. The Lindquist lab investigates both how prions form and the diseases they cause. In addition, Lindquist is convinced that other prion proteins play many important and positive roles in biological processes. The first evidence for this was shown in her work with Nobel Laureate Eric Kandel, which demonstrated that prions may be integral to memory storage in the brain.
Heat shock proteins are a group of molecular chaperone proteins that, as their name might suggest, guide other proteins to fold and mature correctly. Lindquist has established that heat shock protein 90 (Hsp90) can reveal hidden genetic variation in fruit flies and in cress plants (Arabidopsis) under certain environmental conditions.
Lindquist is a Member and former Director (2001-2004) of Whitehead Institute, a Professor of Biology at MIT, and a Howard Hughes Medical Institute investigator. Previously she was the Albert D. Lasker Professor of Medical Sciences from 1999-2001, and a Professor in the Department of Molecular Biology, University of Chicago, since 1978. She received a PhD in Biology from Harvard University in 1976, and was elected to the American Academy of Arts and Sciences in 1997, the National Academy of Sciences in 1997 and the Institute of Medicine in 2006.
Dr. Susan Lindquist – “Alzheimer’s Disease: An Entirely New Point of …
http://www.youtube.com/watch%3Fv%3DZ3tK50LQH_c Nov 15, 2011 …
Whitehead Institute Member Susan Lindquist’s keynote from the 2011 Whitehead Colloquium, November 5, 2011.
Susan Lindquist Lab uploaded a video 1 year ago
Sue Lindquist Plenary Lecture at AAAS Annual Meeting 2014
From Yeast Cells to Patient Neurons: A Powerful Discovery Platform for Parkinson’s and Alzheimer’s Disease
Stanley B. Prusiner, MD
Director, Institute for Neurodegenerative Diseases
Professor, Department of Neurology
Prusiner discovered an unprecedented class of pathogens that he named prions. Prions are proteins that acquire an alternative shape that becomes self-propagating. As prions accumulate, they cause neurodegenerative diseases in animals and humans. Prusiner’s discovery lead him to develop a novel disease paradigm: prions cause disorders such as Creutzfeldt-Jakob disease (CJD) in humans that manifest as (1) sporadic, (2) inherited and (3) infectious illnesses. Based on his seminal discovery that prions can assemble into amyloid fibrils, Prusiner proposed that the more common neurodegenerative diseases including Alzheimer’s and Parkinson’s diseases may be caused by prions.
Prusiner’s contributions to scientific research have been internationally recognized: He is a member of the National Academy of Sciences, the Institute of Medicine, the American Academy of Arts and Sciences and the American Philosophical Society, and a foreign member of the Royal Society, London. He is the recipient of numerous prizes, including the Potamkin Prize for Alzheimer’s Disease Research from the American Academy of Neurology (1991); the Richard Lounsbery Award for Extraordinary Scientific Research in Biology and Medicine from the National Academy of Sciences (1993); the Gairdner Foundation International Award (1993); the Albert Lasker Award for Basic Medical Research (1994); the Wolf Prize in Medicine from the State of Israel (1996); the Nobel Prize in Physiology or Medicine (1997); and the United States Presidential National Medal of Science (2009).
Stanley Prusiner – National Medal of Science – YouTube
http://www.youtube.com/watch%3Fv%3DkghMfXrtvAY
Nov 29, 2010 … 2009 Medal of Science Laureate for his discovery of prions — a new class of infectious agents comprised only of proteins. Produced by Evolving ..
2011 Bay Area Council Outlook Conference – Dr. Stanley Prusiner …
http://www.youtube.com/watch%3Fv%3DcSZA8VUXxZ8
Apr 27, 2011 … 2011 Bay Area Council Outlook Conference – Dr. Stanley Prusiner …. President Obama Awards National Medal of Scienceand Medal of …
By Jennifer O’Brien on October 15, 2010
UCSF Nobel laureate Stanley B. Prusiner, MD, UCSF professor of neurology and director of the Institute for Neurodegenerative Diseases, today (Oct. 15, 2010) was named to receive the National Medal of Science, the nation’s highest honor for science and technology.
Prusiner received the medal for his discovery of and ongoing research on a novel infectious agent, which he named the prion (PREE-on). The prion, composed solely of protein, causes bovine spongiform encephalopathy, or “mad cow” disease, and other related fatal neurodegenerative diseases in animals and humans.
Prions
PNAS Nov 10, 1998; 95(23):13363–13383, http://dx.doi.org:/10.1073/pnas.95.23.13363
Prions are unprecedented infectious pathogens that cause a group of invariably fatal neurodegenerative diseases by an entirely novel mechanism. Prions are transmissible particles that are devoid of nucleic acid and seem to be composed exclusively of a modified protein (PrPSc). The normal, cellular PrP (PrPC) is converted into PrPSc through a posttranslational process during which it acquires a high β-sheet content. The species of a particular prion is encoded by the sequence of the chromosomal PrP gene of the mammals in which it last replicated. In contrast to pathogens carrying a nucleic acid genome, prions appear to encipher strain-specific properties in the tertiary structure of PrPSc.
The torturous path of the scientific investigation that led to an understanding of familial Creutzfeldt–Jakob disease (CJD) chronicles a remarkable scientific odyssey. By 1930, the high incidence of familial (f) CJD in some families was known (1, 2). Almost 60 years were to pass before the significance of this finding could be appreciated (3–5). CJD remained a curious, rare neurodegenerative disease of unknown etiology throughout this period of three score years (6)(7).
Once CJD was shown to be an infectious disease, relatively little attention was paid to the familial form of the disease since most cases were not found in families(8–12). Libyan Jews living in Israel developed CJD about 30 times more frequently than other Israelis (13). This finding prompted some investigators to propose that the Libyan Jews had contracted CJD by eating lightly cooked brain from scrapie-infected sheep when they lived in Tripoli prior to emigration. Subsequently, the Libyan Jewish patients were all found to carry a mutation at codon 200 in their prion protein (PrP) gene (14–16).
Slow Viruses.
The term “slow virus” had been coined by Bjorn Sigurdsson in 1954 while he was working in Iceland on scrapie and visna of sheep (17). Five years later, William Hadlow had suggested that kuru, a disease of New Guinea highlanders, was similar to scrapie and thus, it, too, was caused by a slow virus (18). Seven more years were to pass before the transmissibility of kuru was established by passaging the disease to chimpanzees inoculated intracerebrally (19). Just as Hadlow had made the intellectual leap between scrapie and kuru, Igor Klatzo made a similar connection between kuru and CJD (20). Neuropathologists were struck by the similarities in light microscopic pathology of the central nervous system (CNS) that kuru exhibited with scrapie or CJD. In 1968, the transmission of CJD to chimpanzees after intracerebral inoculation was reported (7).
In scrapie, kuru, CJD, and all of the other disorders now referred to as prion diseases (Table 1), spongiform degeneration and astrocytic gliosis is found upon microscopic examination of the CNS (Fig. 1) (21–22).
Table 1
The prion disease
Figure 1
http://www.pnas.org/content/95/23/13363/F1.medium.gif
Neuropathologic changes in Swiss mice after inoculation with RML scrapie prions. (a) Hematoxylin and eosin stain of a serial section of the hippocampus shows spongiform degeneration of the neuropil, with vacuoles 10–30 μm in diameter.
Prions: A Brief Overview.
Prions are unprecedented infectious pathogens that cause a group of invariably fatal neurodegenerative diseases mediated by an entirely novel mechanism. Prion diseases may present as genetic, infectious, or sporadic disorders, all of which involve modification of the prion protein (PrP), a constituent of normal mammalian cells (23). CJD generally presents as progressive dementia, whereas scrapie of sheep and bovine spongiform encephalopathy (BSE) are generally manifest as ataxic illnesses (Table 1) (24).
Prions are devoid of nucleic acid and seem to be composed exclusively of a modified isoform of PrP designated PrPSc.‡ The normal, cellular PrP, denoted PrPC, is converted into PrPSc through a process whereby a portion of its α-helical and coil structure is refolded into β-sheet (25). This structural transition is accompanied by profound changes in the physicochemical properties of the PrP. The amino acid sequence of PrPSc corresponds to that encoded by the PrP gene of the mammalian host in which it last replicated. In contrast to pathogens with a nucleic acid genome that encode strain-specific properties in genes, prions encipher these properties in the tertiary structure of PrPSc (26–28). Transgenetic studies argue that PrPScacts as a template upon which PrPC is refolded into a nascent PrPSc molecule through a process facilitated by another protein.
More than 20 mutations of the PrP gene are now known to cause the inherited human prion diseases, and significant genetic linkage has been established for five of these mutations (4, 16, 29–31). The prion concept readily explains how a disease can be manifest as a heritable as well as an infectious illness.
Families of hypotheses.
Once the requirement for a protein was established, it was possible to revisit the long list of hypothetical structures that had been proposed for the scrapie agent and to eliminate carbohydrates, lipids, and nucleic acids as the infective elements within a scrapie agent devoid of protein (58) (58, 68).
The family of hypotheses that remained after identifying a protein component was still large and required a continued consideration of all possibilities in which a protein was a critical element (49). The prion concept evolved from a family of hypotheses in which an infectious protein was only one of several possibilities. With the accumulation of experimental data on the molecular properties of the prion, it became possible to discard an increasing number of hypothetical structures. (69).
……..
With the discovery of PrP 27–30 and the production of antiserum (87), brains from humans and animals with putative prion diseases were examined for the presence of this protein. In each case, PrP 27–30 was found, and it was absent in other neurodegenerative disorders such as Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis (88–91). The extreme specificity of PrPSc for prion disease is an important feature of the protein and is consistent with the postulated role of PrPSc in both the transmission and pathogenesis of these illnesses (Table 2) (92).
Table 2
Arguments for prions being composed largely, if not entirely, of PrPSc molecules and devoid of nucleic acid
The accumulation of PrPSc contrasts markedly with that of glial fibrillary acidic protein (GFAP) in prion disease. In scrapie, GFAP mRNA and protein levels rise as the disease progresses (93), but the accumulation of GFAP is neither specific nor necessary for either the transmission or the pathogenesis of disease. Mice deficient for GFAP show no alteration in their incubation times (94, 95).
Except for PrPSc, no macromolecule has been found in tissues of patients dying of the prion diseases that is specific for these encephalopathies. In searches for a scrapie-specific nucleic acid, cDNAs have been identified that are complementary to mRNAs encoding other proteins with increased expression in prion disease (96–98). Yet none of the proteins has been found to be specific for prion disease.
more…
Alzheimer’s Outlook 2014
Six leading experts provide the latest thinking on new and emerging approaches to the prevention, diagnosis and treatment of Alzheimer’s disease and other dementias
* * * * * * * * * * * * * * *
If you or a loved one has been diagnosed with Alzheimer’s disease or another memory disorder…
Or if you are caring for someone with Alzheimer’s and are wondering if there’s a new drug or therapy in the pipeline that might help…
Then it’s vitally important to stay on top of developments in the field — so you can ask your doctor the key questions — and discuss the critical issues that affect the management of the disease.
To help you, we have just published Alzheimer’s Outlook 2014 — a valuable new resource that allows you to sit down with a group of preeminent physicians and listen in as they share their insights and ideas about the future course of Alzheimer’s disease — and provide a clear sense of what caregivers and patients can hope for.
Alzheimer’s Outlook 2014 is part of a series of annual research reports written for concerned lay readers. It gives you special access to information you won’t find anywhere else on the future of Alzheimer’s research.
What’s in the Alzheimer’s pipeline?
In the past few years, researchers have made meaningful strides in the understanding of dementia prevention, diagnosis and treatment. Many important breakthroughs have come from the talented physicians and scientists working here at Johns Hopkins Medicine.
In the pages of Alzheimer’s Outlook 2014 you’ll gain unprecedented access to the insights of Hopkins experts, as well as from colleagues at other renowned research centers.
And there’s so much exciting information to report!
Although we don’t yet have a drug to stop the disease progression, new techniques in molecular biology and genetics are providing remarkable insights into how and why Alzheimer’s begins, how it progresses and how it produces symptoms.
Great progress has also been made in brain imaging and other biomarkers that might allow us to diagnose Alzheimer’s when no or minimal symptoms are present. Thanks to two new radiologic compounds researchers can now see the abnormal proteins in the brain and track the disease from one part of the brain to the next.
Here’s a sample of other key highlights in Alzheimer’s Outlook 2014:
- Investigating Causes and Risks by Peter V. Rabins, M.D., M.P.H., Professor of Psychiatry at Johns Hopkins and Medical Editor of the Johns Hopkins Memory Disorders Bulletin. Dr. Rabins takes a close look at the amyloid cascade hypothesis, which predominates Alzheimer’s research and drug development. He also discusses promising new brain tracers, apolipoprotein E as a risk factor for late-onset Alzheimer’s and progress on understanding the genetics of Alzheimer’s.
- New Research Efforts to Prevent or Slow Dementia by Peter V. Rabins, M.D., M.P.H. Do vitamin E supplements have any effect on cognitive impairment or Alzheimer’s disease? In his second chapter, Dr. Rabins reviews recent research on this question. He also reports on the role of statins in Alzheimer’s, research on cocoa and enhanced brain health and the status of funding for dementia research.
- The Ongoing Search for Drugs That Will Affect Alzheimer’s Disease by Paul Rosenberg, M.D., Associate Director of the Memory and Alzheimer’s Treatment Center at the Johns Hopkins Bayview Medical Center. Researchers now believe that to have any significant benefit, a treatment has to stop the disease long before symptoms of Alzheimer’s appear and before damage to the brain becomes widespread. Dr. Rosenberg describes efforts at early diagnosis, including the A4 trial, the DIAN study, the API study and the SNIFF study.
- Noninvasive Brain Stimulation for Aphasia by Argyle Hillis, M.D., Professor of Neurology at the Johns Hopkins University School of Medicine. Primary progressive aphasia causes degeneration of nerve cells in the brain’s left hemisphere, which controls speech and language. It can also be an early symptom of Alzheimer’s. Dr. Hillis describes her work with transcranial direct current stimulation to help aphasia patients recover.
- Reducing Risks for Alzheimer’s by Marilyn Albert, Ph.D., Director of the Johns Hopkins Alzheimer’s Disease Research Center. Aerobic exercise promotes better mental functioning by improving cerebral blood flow. But can exercise improve the outlook for Alzheimer’s patients? Dr. Alpert looks at research on exercise and dementia and also reports good news on increased federal funding for dementia research.
- Assessing Cognitive Impairment Online by Jason Brandt, Ph.D., Professor of Neurology at the Johns Hopkins University School of Medicine. Low-tech cognitive screening tests offer a quick, inexpensive assessment of a person’s cognitive health. Dr. Brandt has been working on an online assessment tool called the “Dementia Risk Assessment,” which will help patients decide if they should pursue in-person evaluation from a doctor.
- Brain Training: The ACTIVE Study by George W. Rebok, Ph.D., Professor of Mental Health at the Johns Hopkins Bloomberg School of Public Health. Dr. Rebok is a principal investigator in the ACTIVE (Advanced Cognitive Training for Independent and Vital Elderly) trial which looks at ways to improve cognitive performance in older adults. In this section, Dr. Rebok explains what he discovered.
Scientists Discover New Disease Caused By Prion Protein
Giles and his colleagues at the University of California, San Francisco took a look at a disease called multiple-system atrophy or MSA.
GILES: Initially, it looks like Parkinson’s disease. People have the tremors and the loads of problems of Parkinson’s disease.
STEIN: But MSA destroys the brain even faster than Parkinson’s. Giles and his colleagues suspected MSA might be caused by a misshapen version of a protein called alpha-synuclein. So they created mice that have the human form of that protein in their brains and injected the mice with alpha-synuclein from the brains of 14 people who had died from MSA.
GILES: And in every case, the mice died about four months later of the disease.
STEIN: Under a microscope, their brains looked exactly like the brains of people who died from MSA. And tissue taken from the brains of the dead mice could do the same thing to the brains of other mice.
GILES: So that really shows that this is a transmissible disease and that the protein involved in it is acting as a prion.
STEIN: That has all kinds of implications. For starters, Giles says it raises the disturbing possibility that MSA could spread from one person to another by surgical instruments that have been used on the brains of MSA patients.
GILES: Protein sticks very tightly to the stainless steel. And when you clean it afterwards, you could potentially still have misfolded clumps of protein on the surgical instruments that, if you then do surgery on a second person, could potentially induce that in the other person.
STEIN: And the new research, which is being published in the proceedings of the National Academy of Sciences, could be really important for other reasons.
CORINNE LASMEZAS: The fact that these proteins behave like prions – it has tremendous implications.
STEIN: Corinne Lasmezas is a neuroscientist at the Scripps Research Institute in Florida. If a prion can cause MSA, Lasmezas says that’s a big boost for the idea that prions could cause other much more common diseases like Alzheimer’s, Parkinson’s and Lou Gehrig’s disease.
Another Fatal Brain Disease May Come from the Spread of ‘Prion’ Proteins

 , of many years, but is ultimately fatal.
, of many years, but is ultimately fatal.A rare and fatal brain disorder called multiple system atrophy (MSA) may be caused by a newly discovered prion, a protein similar to the ones that cause mad cow and Creutzfeldt-Jakob disease (CJD), according to a new study.
The findings may set the stage for new treatments for MSA, a progressive disorder that causes symptoms similar to Parkinson’s disease and has no cure.
What’s more, the researchers say that the prion they believe causes MSA, called alpha-synuclein, is the first new prion to be discovered in half a century. Prions are infectious proteins that fold abnormally and trigger the misfolding of other, similar proteins. Eventually, the buildup of misfolded proteins can cause lesions to form in the brain, leading to disease.
Genomic Promise for Neurodegenerative Diseases, Dementias, Autism Spectrum, Schizophrenia, and Serious Depression
Reporter and writer: Larry H Bernstein, MD, FCAP
There has been an considerable success in the current state of expanding our knowledge in genomics and therapeutic targets in cancer (although clinical remission targets and relapse are a concern), cardiovascular disease, and infectious disease. Our knowledge of prenatal and perinatal events is still at an early stage. The neurology front is by no means unattended. Here there are two prominent drivers of progress –
- genomic control of cellular apoptosis by ubiquitin pathways, and
- epigenetic investigations,
among a complex sea of sequence-changes. I indicate some of the current status in this. However, as much as we have know, there is an incredible barrier to formulate working models because:
- ligand binding between DNA short-sequences is not predictable over time
- binding between proteins and DNA is still largely unknown
- specific regulatory roles between nucleotide-sequences and histone proeins are still unclear
- the relationship between intracellular as well as extracellular cations and the equilibria between cations and anions in intertitial fluid that bathes the cell and between organelles is virgin territory
Consequently, it is quite an accomplishment to have come as far as we have come, and yet, even with the huge compuational power at our disposal, there is insuficient data to unravel the complexity. This may be especially true in the pathway to understanding of neurological and behavioral disorders.
Broad Map of Brain
John Markoff reports in the Feb 18 front-page of New York Times (Project would construct a broad map of the brain) that the Obama administration envisions a decade-long effort to examine the workings of the human brain and construct a map, comparable to what the Human Genome Project did for genetics. It will be a collaboration between universities, the federal government, private foundations, and teams of scientists (neuro-, nano- and whoever else). The goal is to break through the barrier to understanding the brain’s billions of neurons and gain greater insight into
- perception
- actions
- and consciousness.
Essentially, it holds great promise for understanding
Alzheimer’s disease and Parkinson’s, as well as finding therapies for a variety of mental illnesses. An open-ended question is whether it will also advance artificial intelligence research. It is termed the Brain Activity Map project.
http://NYTimes/broad-map-of-brain/
Alzheimer’s Genomic Diagnosis and Treatment
Larry H Bernstein, MD, FCAP
Gene Mutation Protects Against Alzheimer’s
by Greg Miller on 11 July 2012
Brain preserver. A newly discovered gene mutation appears to protect against Alzheimer’s disease. Credit: Alzheimer’s Disease Education and Referral Center/NIA/NIH
http://news.sciencemag.org/sciencenow/2012/07/gene-mutation-protects-against-a.html
A rare mutation that alters a single letter of the genetic code protects people from the
- memory-robbing dementia of Alzheimer’s disease.
The DNA change may inhibit the buildup of β amyloid, the
- protein fragment that forms the hallmark plaques in the brains of Alzheimer’s patients.
- The mutation affects a gene called APP,
- which encodes a protein that gets broken down into pieces,
- including β amyloid.
Researchers previously identified more than 30 mutations to APP, none of them good. Several of these changes increase β amyloid formation and cause
• a devastating inherited form of Alzheimer’s that afflicts people in their 30s and 40s—
• much earlier than the far more common “late-onset” form of Alzheimer’s
- that typically strikes people their 70s and 80s.
The new mutation, discovered from whole-genome data from 1795 Icelanders for variations in APP that protect against Alzheimer’s, appears to do the opposite. The mutation interferes with one of the enzymes that breaks down the APP protein and causes a 40% reduction in β amyloid formation
New pharmacological strategies for treatment of Alzheimer’s disease: focus on disease modifying drugs.
Salomone S, Caraci F, Leggio GM, Fedotova J, Drago F.
University of Catania, Viale Andrea Doria 6, Catania, Italy.
Br J Clin Pharmacol. 2012 Apr;73(4):504-17. doi: 10.1111/j.1365-2125.2011.04134.x.
Current approved drug treatments for Alzheimer disease (AD) include
- cholinesterase inhibitors (donepezil, rivastigmine, galantamine) and the
- NMDA receptor antagonist memantine.
These drugs provide symptomatic relief but poorly affect the progression of the disease. Drug discovery has been directed, in the last 10 years, to develop ‘disease modifying drugs’ hopefully able to counteract the progression of AD. Because in a chronic, slow progressing pathological process, such as AD, an early start of treatment enhances the chance of success,
- it is crucial to have biomarkers for early detection of AD-related brain dysfunction,
- usable before clinical onset.
Reliable early biomarkers need therefore to be prospectively tested for predictive accuracy,
- with specific cut off values validated in clinical practice.
Disease modifying drugs developed so far include drugs to
- reduce β amyloid (Aβ) production,
- drugs to prevent Aβ aggregation,
- drugs to promote Aβ clearance,
- drugs targeting tau phosphorylation and assembly
None of these drugs has demonstrated efficacy in phase 3 studies. The failure of clinical trials with disease modifying drugs raises a number of questions, spanning from
- methodological flaws to
- fundamental understanding of AD pathophysiology and biology.
Diagnostic criteria applicable to presymptomatic stages of AD have now been published.
These new criteria may impact on drug development, such that future trials on disease modifying drugs will include populations susceptible to AD, before clinical onset. http://www.ncbi.nlm.nih.gov/pubmed/22035455
Gene mutation defends against Alzheimer’s disease
Rare genetic variant suggests a cause and treatment for cognitive decline.
Ewen Callaway 11 July 2012
http://www.nature.com/news/gene-mutation-defends-against-alzheimer-s-disease-1.10984
J. NIETH/CORBIS
Almost 30 million people live with Alzheimer’s disease worldwide, a staggering health-care burden that is expected to quadruple by 2050. Yet doctors can offer no effective treatment, and scientists have been unable to pin down the underlying mechanism of the disease.
Research published this week offers some hope on both counts – few people carry a genetic mutation that naturally prevents them from developing the condition – 0.5% of Icelanders have a protective gene, as are 0.2–0.5% of Finns, Swedes and Norwegians. Icelanders who carry it have a 50% better chance of reaching age 85, are more than five times more likely to reach it 85 without Alzheimer’s. The mutation seems to put a brake on the milder mental deterioration that most elderly people experience. Carriers are about 7.5 times more likely than non-carriers to reach the age of 85 without major cognitive decline, and perform better on the cognitive tests that are administered thrice yearly to Icelanders who live in nursing homes.
The discovery not only confirms the principal suspect that is responsible for Alzheimer’s, it also suggests that the disease could be
- an extreme form of the cognitive decline seen in many older people.
The mutation — the first ever found to protect against the disease — lies in a gene that produces
- amyloid-β precursor protein (APP),
- which has an unknown role in the brain
APP was discovered 25 years ago in patients with rare,
- inherited forms of Alzheimer’s that strike in middle age.
- In the brain, APP is broken down into a smaller molecule called amyloid-β.
Visible clumps, or plaques, of amyloid-β found in the autopsied brains of patients are a hallmark of Alzheimer’s.
Scientists have long debated whether the plaques are a cause of the neurodegenerative condition
- or a consequence of other biochemical changes associated with the disease.
The latest finding supports other genetics studies blaming amyloid-β, according to Rudolph Tanzi, a neurologist at the Massachusetts General Hospital in Boston and a member of one of the four teams that discovered APP’s role in the 1980s.
If amyloid-β plaques were confirmed as the cause of Alzheimer’s, it would bolster efforts to develop drugs that block their formation, says Kári Stefánsson, chief executive of deCODE Genetics in Reykjavik, Iceland, who led the latest research. He and his team first discovered the mutation by comparing the complete genome sequences of 1,795 Icelanders with their medical histories. The researchers then studied the variant in nearly 400,000 more Scandinavians.
This suggests that Alzheimer’s disease and cognitive decline are two sides of the same coin, with a common cause — the build-up of amyloid-β plaques in the brain, something seen to a lesser degree in elderly people who do not develop full-blown Alzheimer’s. A drug that mimics the effects of the mutation, might slow cognitive decline as well as prevent Alzheimer’s.
Stefánsson and his team discovered that the mutation introduces a single amino-acid alteration to APP. This amino acid is close to the site where an enzyme called
- β-secretase 1 (BACE1) ordinarily snips APP into smaller amyloid-β chunks —
- and the alteration is enough to reduce the enzyme’s efficiency.
Stefánsson’s study suggests that blocking β-secretase from cleaving APP has the potential to prevent Alzheimer’s, but Philippe Amouyel, an epidemiologist at the Pasteur Institute in Lille, France, says “it is very difficult to identify the
- precise time when this amyloid toxic effect could still be modified”.
“If this effect needs to be blocked as early as possible in life to protect against Alzheimer’s disease, we will need to propose a new design for clinical trials” to identify an effective treatment.
The results demonstrate that whole-genome sequencing can uncover very rare mutations that might offer insight into common diseases.
- disease risk, may be determined by genetic variants that slightly tilt the odds of developing disease
- In this case a rare mutant may provide very key mechanistic insights into Alzheimer’s
Jonsson, T. et al. Nature http://dx.doi.org/10.1038/nature11283 (2012).
Kang, J. et al. Nature 325, 733–736 (1987).
Goldgaber, D., Lerman, M. I., McBride, O. W., Saffiotti, U. & Gajdusek, D. C. Science 235, 877–880 (1987).
BHCE genetic data combined with brain imaging using agent florbetapir connects the BHCE gene to AD plaque buildup. BHCE is an enzyme that breaks down acetylcholine in the brain, which is depleted early in the disease and results in memory loss. http://www.genengnews.com/
New Alzheimer’s Genes Found
Gigantic Scientific Effort Discovers Clues to Treatment, Diagnosis of Alzheimer’s Disease
By Daniel J. DeNoon
WebMD Health News Reviewed by Laura J. Martin, MD
http://www.webmd.com/alzheimers/news/20110403/new-alzheimers-genes-found
A massive scientific effort has found five new gene variants linked to Alzheimer’s disease. The undertaking involved analyzing the genomes of nearly 40,000 people with and without Alzheimer’s. This study was undertaken by two separate research consortiums in the U.S. and in Europe, which collaborated to confirm each other’s results.
Four genes had previously been linked to Alzheimer’s. Three of them affect only the risk of relatively rare forms of Alzheimer’s. The fourth is APOE, until now the only gene known to affect risk of the common, late-onset form of Alzheimer’s. Roughly 27% of Alzheimer’s disease can be attributed to the five new gene variants. Even though Alzheimer’s is a very complex disease, the new findings represent a large chunk of Alzheimer’s risk, according to Margaret A. Pericak-Vance, PhD, of the U.S. consortium –
- 20% of the causal risk of Alzheimer’s disease and
- 32% of the genetic risk.
Alzheimer’s Tied to Mutation Harming Immune Response
By GINA KOLATA Published: November 14, 2012 in NY Times
http://www.nytimes.com/2012/11/15/health/gene-mutation-that-hobbles-immune-response-is-linked-to-alzheimers.html?_r=0
Alzheimer’s researchers and drug companies have for years concentrated on one hallmark of Alzheimer’s disease: the production of toxic shards of a protein that accumulate in plaques on the brain.
Two groups of researchers working from entirely different starting points have converged on a mutated gene involved in another aspect of Alzheimer’s disease:
- the immune system’s role in protecting against the disease.
The mutation is suspected of interfering with
- the brain’s ability to prevent the buildup of plaque.
When the gene is not mutated, white blood cells in the brain spring into action,
- gobbling up and eliminating the plaque-forming toxic protein, beta amyloid.
As a result, Alzheimer’s can be staved off or averted. People with the mutated gene have a threefold to fivefold increase in the likelihood of developing Alzheimer’s disease in old age.
Comparing Differences
Dr. Julie Williams’s, Cardiff, Wales (European team leader) report identified CLU and Picalm. A second study published in Nature Genetics, by Philippe Amouyel from Institut Pasteur de Lille in France, pinpointed CLU and CR1. The greatest inherited risk comes from the APOE gene, discovered in 1993 by a team led by Allen Roses, now director of the Deane Drug Discovery Institute at Duke UMC, in Durham, North Carolina.
The findings “are beginning to give us insight into the biology, but I don’t think you can expect treatments overnight,” Dr. Michael Owen (Cardiff, Wales) said. Instead, the genes will show a mosaic of risk, and “the key issue is what hand of cards you’re dealt,” he said.
Promise for Early Diagnosis
BHCE genetic data combined with brain imaging using agent florbetapir connects the BHCE gene to AD plaque buildup. BHCE is an enzyme that breaks down acetylcholine in the brain, which is depleted early in the disease and results in memory loss.
Dr. Bernstein’s comments:
- There has been a long history of failure of drugs to slow down the progression of Alzheimer’s. Regression of the plaques has not corresponded with retention of cognitive ability, which has been behind the arguments over beta amyloid or tau.
- We now have two particularly interesting mutations –
- ApoE gene mutation that increases risk
- APP mutation that quite dramatically affects retention of cognition
The Alzheimer Scene around the Web
Larry H Bernstein, MD, FCAP, Curator
http://pharmaceuticalintelligence.com/2012/11/02/the-alzheimer-scene-around-the-web/
Neurodegerative Disease
Tumeric-Derived Compound Curcumin May Treat Alzheimer’s
Curry chemical shows promise for treating the memory-robbing disease
By Lauren K. Wolf
Department: Science & Technology
News Channels: Biological SCENE
Keywords: alternative medicine, dietary supplements, curcumin, tumeric, Alzheimer’s disease
CURRY WONDER
Curcumin, derived from the rootstalk of the turmeric plant, not only gives Indian dishes their color but might treat Alzheimer’s.
Credit: Shutterstock
More than 5 million people in the U.S. currently live with Alzheimer’s disease. And according to the Alzheimer’s Association, the situation is only going to get worse.
By 2050, the nonprofit estimates, up to 16 million Americans will have the memory-robbing disease. It will cost the U.S. $1.1 trillion annually to care for them unless a successful therapy is found.
Pharmaceutical companies have invested heavily in developing Alzheimer’s drugs, many of which target amyloid-β, a peptide that misfolds and clumps in the brains of patients. But so far, no amyloid-β-targeted medications have been successful. Expectation for the most advanced drugs—bapineuzumab from Pfizer and Johnson & Johnson and solanezumab from Eli Lilly & Co.—are low on the basis of lackluster data from midstage clinical trials. That sentiment was reinforced last week when bapineuzumab was reported to have failed the first of four Phase III studies.
Even if these late-stage hopefuls do somehow work, they won’t come cheap, says Gregory M. Cole, a neuroscientist at the University of California, Los Angeles. These drugs “would cost patients tens of thousands of dollars per year,” he estimates. That hefty price tag stems from bapineuzumab and solanezumab being costly-to-manufacture monoclonal antibodies against amyloid-β.
“There’s a great need for inexpensive Alzheimer’s treatments,” as well as a backup plan if pharma fails, says Larry W. Baum, a professor in the School of Pharmacy at the Chinese University of Hong Kong. As a result, he says, a great many researchers have turned their attention to less pricy alternatives, such as compounds from plants and other natural sources.
Curcumin, a spice compound derived from the rootstalk of the turmeric plant (Curcuma longa), has stood out among some of the more promising naturally derived candidates.
When administered to mice that develop Alzheimer’s symptoms, curcumin decreases inflammation and reactive oxygen species in the rodents’ brains, researchers have found. The compound also inhibits the aggregation of troublesome amyloid-β strands among the animals’ nerve cells. But the development of curcumin as an Alzheimer’s drug has been stymied, scientists say, both by its low uptake in the body and a lack of funds for effective clinical trials—obstacles researchers are now trying to overcome.
In addition to contributing to curry dishes’ yellow color and pungent flavor, curcumin has been a medicine in India for thousands of years. Doctors practicing traditional Hindu medicine admire turmeric’s active ingredient for its anti-inflammatory properties and have used it to treat patients for ailments including digestive disorders and joint pain.
Only in the 1970s did Western researchers catch up with Eastern practices and confirm curcumin’s anti-inflammatory properties in the laboratory. Scientists also eventually determined that the polyphenolic compound is an antioxidant and has chemotherapeutic activity.
Bharat B. Aggarwal, a professor at the University of Texas M. D. Anderson Cancer Center, says curcumin is an example of a pleiotropic agent: It has a number of different effects and interacts with many targets and biochemical pathways in the body. He and his group have discovered that one important molecule targeted and subsequently suppressed by curcumin is NF-κB, a transcription factor that switches on the body’s inflammatory response when activated (J. Biol. Chem., DOI: 10.1074/jbc.270.42.24995).
Aside from NF-κB, curcumin seems to interact with several other molecules in the inflammatory pathway, a biological activity that Aggarwal thinks is advantageous. “All chronic diseases are caused by dysregulation of multiple targets,” he says. “Chemists don’t yet know how to design a drug that hits multiple targets.” With curcumin, “Mother Nature has already provided a compound that does so.”
Curcumin’s pleiotropy also brought it to the attention of UCLA’s Cole during the early 1990s while he was searching for possible Alzheimer’s therapeutics. “That was before we knew about amyloid-β” and its full role in Alzheimer’s, he says. “We were working on the disease from an oxidative damage and inflammation point of view—two processes implicated in aging.”
When Cole and his wife, Sally A. Frautschy, also at UCLA, searched the literature for compounds that could tackle both of these age-related processes, curcumin jumped out at them. It also didn’t hurt that the incidence of Alzheimer’s in India, where large amounts of curcumin are consumed regularly, is lower than in other parts of the developing world (Lancet Neurol., DOI:10.1016/s1474-4422(08)70169-8).
In 2001, Cole, Frautschy, and colleagues published the first papers that demonstrated curcumin’s potential to treat neurodegenerative disease (Neurobiol. Aging, DOI: 10.1016/s0197-4580(01)00300-1; J. Neurosci.2001, 8370). The researchers studied the effects of curcumin on rats that had amyloid-β injected into their brains, as well as mice engineered to develop amyloid brain plaques. In both cases, curcumin suppressed oxidative tissue damage and reduced amyloid-β deposits.
Those results, Cole says, “turned us into curcuminologists.”
Although the UCLA team observed that curcumin decreased amyloid plaques in animal models, at the time, the researchers weren’t sure of the molecular mechanism involved.
Soon after the team’s first results were published, Cole recalls, a colleague brought to his attention the structural similarity between curcumin and the dyes used to stain amyloid plaques in diseased brain tissue. When Cole and Frautschy tested the spice compound, they saw that it, too, could stick to aggregated amyloid-β. “We thought, ‘Wow, not only is curcumin an antioxidant and an anti-inflammatory, but it also might be an anti-amyloid drug,’ ” he says.
In 2004, a group in Japan demonstrated that submicromolar concentrations of curcumin in solution could inhibit aggregation of amyloid-β and break up preformed fibrils of the stuff (J. Neurosci. Res., DOI: 10.1002/jnr.20025). Shortly after that, the UCLA team demonstrated the same (J. Biol. Chem., DOI: 10.1074/jbc.m404751200).
As an Alzheimer’s drug, however, it’s unclear how important it is that the spice compound inhibits amyloid-β aggregation, Cole says. “When you have something that’s so pleiotropic,” he adds, “it’s hard to know” which of its modes of action is most effective.
Having multiple targets may be what helps curcumin have such beneficial, neuroprotective effects, says David R. Schubert, a neurobiologist at the Salk Institute for Biological Studies, in La Jolla, Calif. But its pleiotropy can also be a detriment, he contends.
The pharmaceutical world, Schubert says, focuses on designing drugs aimed at hitting single-target molecules with high affinity. “But we don’t really know what ‘the’ target for curcumin is,” he says, “and we get knocked for it on grant requests.”
Another problem with curcumin is poor bioavailability. When ingested, UCLA’s Cole says, the compound gets converted into other molecular forms, such as curcumin glucuronide or curcumin sulfate. It also gets hydrolyzed at the alkaline and neutral pHs present in many areas of the body. Not much of the curcumin gets into the bloodstream, let alone past the blood-brain barrier, in its pure, active form, he adds.
Unfortunately, neither Cole nor Baum at the Chinese University of Hong Kong realized the poor bioavailability until they had each launched a clinical trial of curcumin. So the studies showed no significant difference between Alzheimer’s patients taking the spice compound and those taking a placebo (J. Clin. Psychopharmacol., DOI: 10.1097/jcp.0b013e318160862c).
“But we did show curcumin was safe for patients,” Baum says, finding a silver lining to the blunder. “We didn’t see any adverse effects even at high doses.”
Some researchers, such as Salk’s Schubert, are tackling curcumin’s low bioavailability by modifying the compound to improve its properties. Schubert and his group have come up with a molecule, called J147, that’s a hybrid of curcumin and cyclohexyl-bisphenol A. Like Cole and coworkers, they also came upon the compound not by initially screening for the ability to interact with amyloid-β, but by screening for the ability to alleviate age-related symptoms.
The researchers hit upon J147 by exposing cultured Alzheimer’s nerve cells to a library of compounds and then measuring changes to levels of biomarkers for oxidative stress, inflammation, and nerve growth. J147 performed well in all categories. And when given to mice engineered to accumulate amyloid-β clumps in their brains, the hybrid molecule prevented memory loss and reduced formation of amyloid plaques over time (PLoS One, DOI: 10.1371/journal.pone.0027865).
Other researchers have tackled curcumin’s poor bioavailability by reformulating it. Both Baum and Cole have encapsulated curcumin in nanospheres coated with either polymers or lipids to protect the compound from modification after ingestion. Cole tells C&EN that by packaging the curcumin in this way, he and his group have gotten micromolar quantities of it into the bloodstream of humans. The researchers are now preparing for a small clinical trial to test the formulation on patients with mild cognitive impairment, who are at an increased risk of developing Alzheimer’s.
An early-intervention human study such as this one comes with its own set of challenges, Cole says. People with mild cognitive impairment “have good days and bad days,” he says. A large trial over a long period would be the best way to get any meaningful data, he adds. Such a trial can cost up to $100 million, a budget big pharma might be able to scrape together but that is far out of reach for academics funded by grants, Cole says. “If you’re down at the level of what an individual investigator can do, you’re running a small trial,” he says, “and even if the result is positive, it might be inconclusive” because of its small size or short duration. That’s one of the reasons the curcumin work is slow-going, Cole contends.
NIH-Funded Research Provides New Clues on How ApoE4 Affects Alzheimer’s Risk
Published: Tuesday, October 30, 2012
Last Updated: Tuesday, October 30, 2012
Researchers found that ApoE4 triggers an inflammatory reaction that weakens the blood-brain barrier.
Common variants of the ApoE gene are strongly associated with the risk of developing late-onset Alzheimer’s disease, but the gene’s role in the disease has been unclear.
Now, researchers funded by the National Institutes of Health have found that in mice, having the most risky variant of ApoE damages the blood vessels that feed the brain.
The researchers found that the high-risk variant, ApoE4, triggers an inflammatory reaction that weakens the blood-brain barrier, a network of cells and other components that lines brain’s brain vessels.
Normally, this barrier allows nutrients into the brain and keeps harmful substances out.
The study appears in Nature, and was led by Berislav Zlokovic, M.D., Ph.D., director of the Center for Neurodegeneration and Regeneration at the Zilkha Neurogenetic Institute, Keck School of Medicine, University of Southern California, Los Angeles.
“Understanding the role of ApoE4 in Alzheimer’s disease may be one of the most important avenues to a new therapy,” Dr. Zlokovic said. “Our study shows that ApoE4 triggers a cascade of events that damages the brain’s vascular system,” he said, referring to the system of blood vessels that supply the brain.
The ApoE gene encodes a protein that helps regulate the levels and distribution of cholesterol and other lipids in the body. The gene exists in three varieties.
ApoE2 is thought to play a protective role against both Alzheimer’s and heart disease, ApoE3 is believed to be neutral, and ApoE4 confers a higher risk for both conditions.
Outside the brain, the ApoE4 protein appears to be less effective than other versions at clearing away cholesterol; however, inside the brain, exactly how ApoE4 contributes to Alzheimer’s disease has been a mystery.
Dr. Zlokovic and his team studied several lines of genetically engineered mice, including one that lacks the ApoE gene and three other lines that produce only human ApoE2, ApoE3 or ApoE4. Mice normally have only a single version of ApoE.
The researchers found that mice whose bodies made only ApoE4, or made no ApoE at all, had a leaky blood-brain barrier. With the barrier compromised, harmful proteins in the blood made their way into the mice’s brains, and after several weeks, the researchers were able to detect loss of small blood vessels, changes in brain function, and a loss of connections between brain cells.
“The study demonstrates that damage to the brain’s vascular system may play a key role in Alzheimer’s disease, and highlights growing recognition of potential links between stroke and Alzheimer’s-type dementia,” said Roderick Corriveau, Ph.D., a program director at NIH’s National Institute of Neurological Disorders and Stroke (NINDS), which helped fund the research. “It also suggests that we might be able to decrease the risk of Alzheimer’s disease among ApoE4 carriers by improving their vascular health.”
The researchers also found that ApoE2 and ApoE3 help control the levels of an inflammatory molecule called cyclophilin A (CypA), but ApoE4 does not. Levels of CypA were raised about five-fold in blood vessels of mice that produce only ApoE4.
The excess CypA then activated an enzyme, called MMP-9, which destroys protein components of the blood-brain barrier. Treatment with the immunosuppressant drug cyclosporine A, which inhibits CypA, preserved the integrity of the blood-brain barrier and lessened damage to the brain.
An inhibitor of the MMP-9 enzyme had similar beneficial effects. In prior studies, inhibitors of this enzyme have been shown to reduce brain damage after stroke in animal models.
“These findings point to cyclophilin A as a potential new drug target for Alzheimer’s disease,” said Suzana Petanceska, Ph.D., a program director at NIH’s National Institute on Aging (NIA), which also funded Dr. Zlokovic’s study.
“Many population studies have shown an association between vascular risk factors in mid-life, such as high blood pressure and diabetes, and the risk for Alzheimer’s in late-life. We need more research aimed at deepening our understanding of the mechanisms involved and to test whether treatments that reduce vascular risk factors may be helpful against Alzheimer’s.”
Alzheimer’s disease is the most common cause of dementia in older adults, and affects more than 5 million Americans. A hallmark of the disease is a toxic protein fragment called beta-amyloid that accumulates in clumps, or plaques, within the brain.
Gene variations that cause higher levels of beta-amyloid are associated with a rare type of Alzheimer’s that appears early in life, between age 30 and 60.
However, it is the ApoE4 gene variant that is most strongly tied to the more common, late-onset type of Alzheimer’s disease. Inheriting a single copy of ApoE4 from a parent increases the risk of Alzheimer’s disease by about three-fold. Inheriting two copies, one from each parent, increases the risk by about 12-fold.
Dr. Zlokovic’s study and others point to a complex interplay between beta-amyloid and ApoE4. On the one hand, beta-amyloid is known to build up in and damage blood vessels and cause bleeding into the brain.
On the other hand, Dr. Zlokovic’s data suggest that ApoE4 can damage the vascular system independently of beta-amyloid. He theorizes that this damage makes it harder to clear beta-amyloid from the brain.
Some therapies under investigation for Alzheimer’s focus on destroying amyloid plaques, but therapies designed to compensate for ApoE4 might help prevent the plaques from forming, he said.
Compound Could Become Alzheimer’s Treatment
Thu, 10/11/2012 – 1:29pm
A new molecule designed to treat Alzheimer’s disease has significant promise and is potentially the safest to date, according to researchers.
Purdue University professor Arun Ghosh designed the molecule, which is a highly potent beta-secretase inhibitor with unique features that ensure it goes only to its target and does not affect healthy physiological processes, he said.
“This molecule maintains the disease-fighting properties of earlier beta-secretase inhibitors, but is much less likely to cause harmful side effects,” said Ghosh, the Ian P. Rothwell Distinguished Professor of Chemistry and Medicinal Chemistry and Molecular Pharmacology. “The selectivity we achieved is unprecedented, which gives it great promise for the long-term medication required to treat Alzheimer’s. Each time a treatment misses its disease target and instead interacts with a healthy cell or molecule, damage is done that we call toxicity. Even low levels of this toxicity could build up over years and years of treatment, and an Alzheimer’s patient would need to be treated for the rest of his or her life.”
The new molecule shows a 7,000-fold selectivity for its target enzyme, which far surpasses the benchmark of a 1,000-fold selectivity for a viable treatment molecule, and dwarfs the selectivity values in the hundreds for past beta-secretase inhibitors, he said. A paper detailing the work will be published in an upcoming Alzheimer’s research issue of the Journal of Medicinal Chemistry and is currently available online. The National Institutes of Health funded the research.
Beta-secretase inhibitors, which could allow for intervention in the early stages of Alzheimer’s disease, have promise as a potential treatment. Several drugs based on this molecular target have made it to clinical trials, including one based on a molecule Ghosh designed previously. These molecules prevent the first step in a chain of events that leads to the formation of amyloid plaque in the brain, fibrous clumps of toxic proteins that are believed to cause the disease’s devastating symptoms.
The National Institute on Aging estimates that 5.1 million Americans suffer from Alzheimer’s disease, which leads to dementia by affecting parts of the brain that control thought, memory and language.
“Alzheimer’s is a progressive disease that destroys the brain and also destroys the quality of life for those who suffer from it,” Ghosh said. “It eventually robs people of their ability to recognize their own spouse or child and to complete basic tasks necessary for independence, like getting dressed. It is a truly devastating disease for those who suffer from it and for their friends and loved ones.”
Earlier versions of the beta-secretase inhibitor were able to stop and even reverse the progression of amyloid plaques in tests on mice, but potency and selectivity are only two of the three pillars of a viable Alzheimer’s treatment, Ghosh said. It has yet to be shown whether this molecule possesses the third pillar, the ability to be turned into an easily administered drug that passes through the blood-brain barrier.
Ghosh collaborates with Jordan Tang, the J.G. Puterbaugh Chair in Medical Research at the Oklahoma Medical Research Foundation, who in 2000 identified beta-secretase and its role in the progression of Alzheimer’s. Later that year Ghosh designed his first molecule that bound to and inhibited the activity of the enzyme. He has strived to create the needed improvements ever since.
Ghosh bypasses the usual lengthy process of trial and error in finding useful inhibitor molecules by using a structure-based design strategy. He uses the structures of the inhibitor bound to the enzyme as a guide to what molecular features are important for desirable and undesirable characteristics. Then he removes, replaces and adds molecular groups to amplify the desirable and eliminate the undesirable.
“I believe structure-based design is vital to the development of new and improved medicine,” said Ghosh, who also is a member of the Purdue University Center for Cancer Research. “These strategies have the potential to eliminate enormous costs and time needed in traditional random screening protocols for drug development. Structure-based strategies allow us to design molecules that do precisely what we need them to do with fewer undesirable side effects.”
Tang performed the X-ray crystallography and captured the crystal structures to reveal important insights and serve as a guide for Ghosh’s designs.
“Developing inhibitors into clinically useful drugs is an evolutionary process,” Tang said. “We learn what works and what doesn’t along the way, and the knowledge permits us to do better in the next step. The miracles of modern medicine are built on top of excellent scientific findings. We try to do good science and know that the consequence will be a better chance for conquering diseases and improving lives.”
Beta-secretase belongs to a class of enzymes called aspartyl proteases. Research into beta-secretase inhibitors faced setbacks when other aspartyl proteases similar in structure, called memapsin 1 and cathepsin D, were discovered and found to be involved in many important physiological processes. Earlier designed beta-secretase inhibitors were found also to work against the biologically necessary enzymes.
Ghosh’s team focused on developing ways to make the inhibitor more selective so that it would avoid these other, physiologically important enzymes. They compared the structures of beta-secretase and memapsin 1 as they interacted with the inhibitor to find an active area unique only to beta-secretase. Then they added a functional molecular feature that targets and interacts with the unique area, making the inhibitor more attractive to beta-secretase and less attractive to the other enzymes.
“The added feature serves as a bait on the inhibitor molecule that entices beta-secretase and also grabs onto it tightly, greatly enhancing its selectivity,” he said. “This is a fundamental insight into the origins of selectivity and ways to increase it.”
Ghosh said this work highlights an important purpose of academic research.
“Academic research lays out and shares the fundamentals to advance drug discovery,” he said. “Advances in treatment are built upon the basic research happening at universities.”

















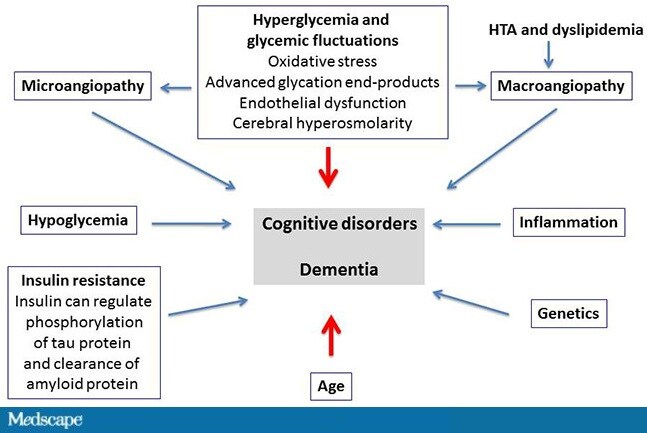{kind=link}
{kind=link}
{kind=link}