Introduction – The Evolution of Cancer Therapy and Cancer Research: How We Got Here?
Author and Curator: Larry H Bernstein, MD, FCAP
The evolution of progress we have achieved in cancer research, diagnosis, and therapeutics has originated from an emergence of scientific disciplines and the focus on cancer has been recent. We can imagine this from a historical perspective with respect to two observations. The first is that the oldest concepts of medicine lie with the anatomic dissection of animals and the repeated recurrence of war, pestilence, and plague throughout the middle ages, and including the renaissance. In the awakening, architecture, arts, music, math, architecture and science that accompanied the invention of printing blossomed, a unique collaboration of individuals working in disparate disciplines occurred, and those who were privileged received an education, which led to exploration, and with it, colonialism. This also led to the need to increasingly, if not without reprisal, questioning long-held church doctrines.
It was in Vienna that Rokitansky developed the discipline of pathology, and his student Semelweis identified an association between then unknown infection and childbirth fever. The extraordinary accomplishments of John Hunter in anatomy and surgery came during the twelve years war, and his student, Edward Jenner, observed the association between cowpox and smallpox resistance. The development of a nursing profession is associated with the work of Florence Nightengale during the Crimean War (at the same time as Leo Tolstoy). These events preceded the work of Pasteur, Metchnikoff, and Koch in developing a germ theory, although Semelweis had committed suicide by infecting himself with syphilis. The first decade of the Nobel Prize was dominated by discoveries in infectious disease and public health (Ronald Ross, Walter Reed) and we know that the Civil War in America saw an epidemic of Yellow Fever, and the Armed Services Medical Museum was endowed with a large repository of osteomyelitis specimens. We also recall that the Russian physician and playwriter, Anton Checkov, wrote about the conditions in prison camps.
But the pharmacopeia was about to open with the discoveries of insulin, antibiotics, vitamins, thyroid action (Mayo brothers pioneered thyroid surgery in the thyroid iodine-deficient midwest), and pitutitary and sex hormones (isolatation, crystal structure, and synthesis years later), and Karl Landsteiner’s discovery of red cell antigenic groups (but he also pioneered in discoveries in meningitis and poliomyelitis, and conceived of the term hapten) with the introduction of transfusion therapy that would lead to transplantation medicine. The next phase would be heralded by the discovery of cancer, which was highlighted by the identification of leukemia by Rudolph Virchow, who cautioned about the limitations of microscopy. This period is highlighted by the classic work – “Microbe Hunters”.

John Hunter

Walter Reed

Robert Koch

goldberger 1916 Pellagra

Louis Pasteur
A multidisciplinary approach has led us to a unique multidisciplinary or systems view of cancer, with different fields of study offering their unique expertise, contributions, and viewpoints on the etiology of cancer. Diverse fields in immunology, biology, biochemistry, toxicology, molecular biology, virology, mathematics, social activism and policy, and engineering have made such important contributions to our understanding of cancer, that without cooperation among these diverse fields our knowledge of cancer would never had evolved as it has. In a series of posts “Heroes in Medical Research:” the work of researchers are highlighted as examples of how disparate scientific disciplines converged to produce seminal discoveries which propelled the cancer field, although, at the time, they seemed like serendipitous findings. In the post Heroes in Medical Research: Barnett Rosenberg and the Discovery of Cisplatin (Translating Basic Research to the Clinic) discusses the seminal yet serendipitous discoveries by bacteriologist Dr. Barnett Rosenberg, which eventually led to the development of cisplatin, a staple of many chemotherapeutic regimens. Molecular biologist Dr. Robert Ting, working with soon-to-be Nobel Laureate virologist Dr. James Gallo on AIDS research and the associated Karposi’s sarcoma identified one of the first retroviral oncogenes, revolutionizing previous held misconceptions of the origins of cancer (described in Heroes in Medical Research: Dr. Robert Ting, Ph.D. and Retrovirus in AIDS and Cancer). Located here will be a MONTAGE of PHOTOS of PEOPLE who made seminal discoveries and contributions in every field to cancer Each of these paths of discovery in cancer research have led to the unique strategies of cancer therapeutics and detection for the purpose of reducing the burden of human cancer. However, we must recall that this work has come at great cost, while it is indeed cause for celebration. The current failure rate of clinical trials at over 70 percent, has been a cause for disappointment, and has led to serious reconsideration of how we can proceed with greater success. The result of the evolution of the cancer field is evident in the many parts and chapters of this ebook. Volume 4 contains chapters that are in a predetermined order:
- The concepts of neoplasm, malignancy, carcinogenesis, and metastatic potential, which encompass:
(a) How cancer cells bathed in an oxygen rich environment rely on anaerobic glycolysis for energy, and the secondary consequences of cachexia and sarcopenia associated with progression

invasion

ARTS protein and cancer

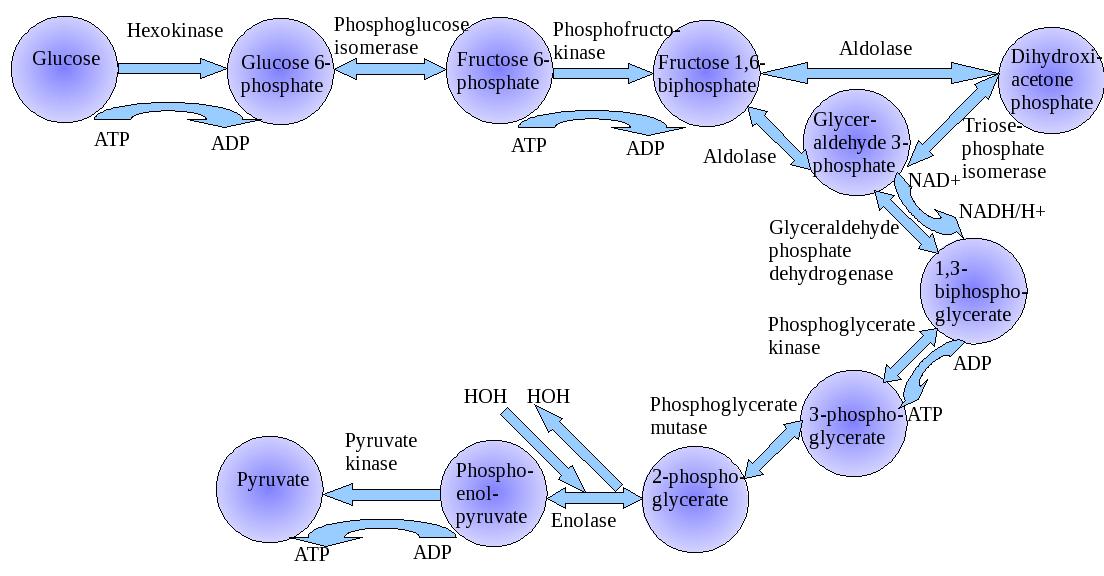
Glycolysis

Krebs cycle

Metabolic control analysis of respiration in human cancer tissue

akip1-expression-modulates-mitochondrial-function
(b) How advances in genetic analysis, molecular and cellular biology, metabolomics have expanded our basic knowledge of the mechanisms which are involved in cellular transformation to the cancerous state.

nucleotides

Methylation of adenine

ampk-and-ampk-related-kinase-ark-family-

ubiquitylation
(c) How molecular techniques continue to advance our understanding of how genetics, epigenetics, and alterations in cellular metabolism contribute to cancer and afford new pathways for therapeutic intervention.

genomic effects

LKB1AMPK pathway

mutation-frequencies-across-12-cancer-types

AMPK-activating drugs metformin or phenformin might provide protection against cancer

pim2-phosphorylates-pkm2-and-promotes-glycolysis-in-cancer-cells
pim2-phosphorylates-pkm2-and-promotes-glycolysis-in-cancer-cells
2. The distinct features of cancers of specific tissue sites of origin
3. The diagnosis of cancer by
(a) Clinical presentation
(b) Age of onset and stage of life
(c) Biomarker features

hairy cell leukemia

lymphoma leukemia
(d) Radiological and ultrasound imaging
- Treatments
- Prognostic differences within and between cancer types
We have introduced the emergence of a disease of great complexity that has been clouded in more questions than answers until the emergence of molecular biology in the mid 20th century, and then had to await further discoveries going into the 21st century. What gave the research impetus was the revelation of
1 the mechanism of transcription of the DNA into amino acid sequences

Proteins in Disease
2 the identification of stresses imposed on cellular function

NO beneficial effects
3 the elucidation of the substructure of the cell – cell membrane, mitochondria, ribosomes, lysosomes – and their functions, respectively
AKIP1 Expression Modulates Mitochondrial Function
4 the elucidation of oligonucleotide sequences
nucleotides

dna-replication-unwinding

dna-replication-ligation

dna-replication-primer-removal

dna-replication-leading-strand

dna-replication-lagging-strand

dna-replication-primer-synthesis

dna-replication-termination
5 the further elucidation of functionally relevant noncoding lncDNA

6 the technology to synthesis mRNA and siRNA sequences

Figure. RNAi and gene silencing
7 the repeated discovery of isoforms of critical enzymes and their pleiotropic properties
8. the regulatory pathways involved in signaling

Figure. Signaling Pathways Map
This is a brief outline of the modern progression of advances in our understanding of cancer. Let us go back to the beginning and check out a sequence of Nobel Prizes awarded and related discoveries that have a historical relationship to what we know. The first discovery was the finding by Louis Pasteur that fungi that grew in an oxygen poor environment did not put down filaments. They did not utilize oxygen and they produced used energy by fermentation. This was the basis for Otto Warburg sixty years later to make the comparison to cancer cells that grew in the presence of oxygen, but relied on anaerobic glycolysis. He used a manometer to measure respiration in tissue one cell layer thick to measure CO2 production in an adiabatic system.
video width=”1280″ height=”720″ caption=”1741-7007-11-65-s1 Macromolecular juggling by ubiquitylation enzymes.” mp4=”http://pharmaceuticalintelligence.com/wp-content/uploads/2014/04/1741-7007-11-65-s1-macromolecular-juggling-by-ubiquitylation-enzymes.mp4“][/video]
An Introduction to the Warburg Apparatus
http://www.youtube.com/watch?v=M-HYbZwN43o
Lavoisier Antoine-Laurent and Laplace Pierre-Simon (1783) Memoir on heat. Mémoirs de l’Académie des sciences. Translated by Guerlac H, Neale Watson Academic Publications, New York, 1982.
Instrumental background 200 years later: Gnaiger E (1983) The twin-flow microrespirometer and simultaneous calorimetry. In Gnaiger E, Forstner H, eds. Polarographic Oxygen Sensors. Springer, Heidelberg, Berlin, New York: 134-166.

otto_heinrich_warburg

The Warburg apparatus is a manometric respirometer which was used for decades in biochemistry for measuring oxygen consumption of tissue homogenates or tissue slices.
The Warburg apparatus has its name from the German biochemist Otto Heinrich Warburg (1883-1970) who was awarded the Nobel Prize in physiology or medicine in 1931 for his “discovery of the nature and mode of action of the respiratory enzyme” [1].
The aqueous phase is vigorously shaken to equilibrate with a gas phase, from which oxygen is consumed while the evolved carbon dioxide is trapped, such that the pressure in the constant-volume gas phase drops proportional to oxygen consumption. The Warburg apparatus was introduced to study cell respiration, i.e. the uptake of molecular oxygen and the production of carbon dioxide by cells or tissues. Its applications were extended to the study of fermentation, when gas exchange takes place in the absence of oxygen. Thus the Warburg apparatus became established as an instrument for both aerobic and anaerobic biochemical studies [2, 3].
The respiration chamber was a detachable glass flask (F) equipped with one or more sidearms (S) for additions of chemicals and an open connection to a manometer (M; pressure gauge). A constant temperature was provided by immersion of the Warburg chamber in a constant temperature water bath. At thermal mass transfer equilibrium, an initial reading is obtained on the manometer, and the volume of gas produced or absorbed is determined at specific time intervals. A limited number of ‘titrations’ can be performed by adding the liquid contained in a side arm into the main reaction chamber. A Warburg apparatus may be equipped with more than 10 respiration chambers shaking in a common water bath. Since temperature has to be controlled very precisely in a manometric approach, the early studies on mammalian tissue respiration were generally carried out at a physiological temperature of 37 °C.
The Warburg apparatus has been replaced by polarographic instruments introduced by Britton Chance in the 1950s. Since Chance and Williams (1955) measured respiration of isolated mitochondria simultaneously with the spectrophotometric determination of cytochrome redox states, a water chacket could not be used, and measurements were carried out at room temperature (or 25 °C). Thus most later studies on isolated mitochondria were shifted to the artifical temperature of 25 °C.
Today, the importance of investigating mitochondrial performance at in vivo temperatures is recognized again in mitochondrial physiology. Incubation times of 1 hour were typical in experiments with the Warburg apparatus, but were reduced to a few or up to 20 min, following Chance and Williams, due to rapid oxygen depletion in closed, aqueous phase oxygraphs with high sample concentrations. Today, incubation times of 1 hour are typical again in high-resolution respirometry, with low sample concentrations and the option of reoxygenations.
“The Nobel Prize in Physiology or Medicine 1931”. Nobelprize.org. 27 Dec 2011 www.nobelprize.org/nobel_prizes/medicine/laureates/1931/
- Oesper P (1964) The history of the Warburg apparatus: Some reminiscences on its use. J Chem Educ 41: 294.
- Koppenol WH, Bounds PL, Dang CV (2011) Otto Warburg’s contributions to current concepts of cancer metabolism. Nature Reviews Cancer 11: 325-337.
- Gnaiger E, Kemp RB (1990) Anaerobic metabolism in aerobic mammalian cells: information from the ratio of calorimetric heat flux and respirometric oxygen flux. Biochim Biophys Acta 1016: 328-332. – “At high fructose concentrations, respiration is inhibited while glycolytic end products accumulate, a phenomenon known as the Crabtree effect. It is commonly believed that this effect is restricted to microbial and tumour cells with uniquely high glycolytic capacities (Sussman et al, 1980). However, inhibition of respiration and increase of lactate production are observed under aerobic conditions in beating rat heart cell cultures (Frelin et al, 1974) and in isolated rat lung cells (Ayuso-Parrilla et al, 1978). Thus, the same general mechanisms responsible for the integration of respiration and glycolysis in tumour cells (Sussman et al, 1980) appear to be operating to some extent in several isolated mammalian cells.”
Mitochondria are sometimes described as “cellular power plants” because they generate most of the cell’s supply of adenosine triphosphate (ATP), used as a source of chemical energy.[2] In addition to supplying cellular energy, mitochondria are involved in other tasks such as signaling, cellular differentiation, cell death, as well as the control of the cell cycle and cell growth.[3] The organelle is composed of compartments that carry out specialized functions. These compartments or regions include the outer membrane, the intermembrane space, the inner membrane, and the cristae and matrix. Mitochondrial proteins vary depending on the tissue and the species. In humans, 615 distinct types of proteins have been identified from cardiac mitochondria,[9 Leonor Michaelis discovered that Janus green can be used as a supravital stain for mitochondria in 1900. Benjamin F. Kingsbury, in 1912, first related them with cell respiration, but almost exclusively based on morphological observations.[13] In 1913 particles from extracts of guinea-pig liver were linked to respiration by Otto Heinrich Warburg, which he called “grana”. Warburg and Heinrich Otto Wieland, who had also postulated a similar particle mechanism, disagreed on the chemical nature of the respiration. It was not until 1925 when David Keilin discovered cytochromes that the respiratory chain was described.[13]
The Clark Oxygen Sensor
Dr. Leland Clark – inventor of the “Clark Oxygen Sensor” (1954); the Clark type polarographic oxygen sensor remains the gold standard for measuring dissolved oxygen in biomedical, environmental and industrial applications . ‘The convenience and simplicity of the polarographic ‘oxygen electrode’ technique for measuring rapid changes in the rate of oxygen utilization by cellular and subcellular systems is now leading to its more general application in many laboratories. The types and design of oxygen electrodes vary, depending on the investigator’s ingenuity and specific requirements of the system under investigation.’Estabrook R (1967) Mitochondrial respiratory control and the polarographic measurement of ADP:O ratios. Methods Enzymol. 10: 41-47. “one approach that is underutilized in whole-cell bioenergetics, and that is accessible as long as cells can be obtained in suspension, is the oxygen electrode, which can obtain more precise information on the bioenergetic status of the in situ mitochondria than more ‘high-tech’ approaches such as fluorescent monitoring of Δψm.” Nicholls DG, Ferguson S (2002) Bioenergetics 3. Academic Press, London.
Great Figures in Cancer

Dr. Elizabeth Blackburn,

j_michael_bishop onogene

Harold Varmus

Potts and Habener (PTH mRNA, Harvard MIT) JCI

JCI Fuller Albright and hPTH AA sequence

Dr. E. Donnall Thomas Bone Marrow Transplants

Dr Haraldzur Hausen EBV HPV

Dr. Craig Mello


Lee Hartwell – Hutchinson Cancer Res Center

Judah Folkman, MD

Gertrude B. Elien (1918-1999)
The Nobel Prize in Physiology or Medicine 1922
Archibald V. Hill, Otto Meyerhof
AV Hill –
“the production of heat in the muscle” Hill started his research work in 1909. It was due to J.N. Langley, Head of the Department of Physiology at that time that Hill took up the study on the nature of muscular contraction. Langley drew his attention to the important (later to become classic) work carried out by Fletcher and Hopkins on the problem of lactic acid in muscle, particularly in relation to the effect of oxygen upon its removal in recovery. In 1919 he took up again his study of the physiology of muscle, and came into close contact with Meyerhof of Kiel who, approaching the problem differently, arrived at results closely analogous to his study. In 1919 Hill’s friend W. Hartree, mathematician and engineer, joined in the myothermic investigations – a cooperation which had rewarding results.
Otto Meyerhof –

otto-fritz-meyerhof
lactic acid production in muscle contraction Under the influence of Otto Warburg, then at Heidelberg, Meyerhof became more and more interested in cell physiology. In 1923 he was offered a Professorship of Biochemistry in the United States, but Germany was unwilling to lose him. In 1929 he was he was placed in charge of the newly founded Kaiser Wilhelm Institute for Medical Research at Heidelberg. From 1938 to 1940 he was Director of Research at the Institut de Biologie physico-chimique at Paris, but in 1940 he moved to the United States, where the post of Research Professor of Physiological Chemistry had been created for him by the University of Pennsylvania and the Rockefeller Foundation. Meyerhof’s own account states that he was occupied chiefly with oxidation mechanisms in cells and with extending methods of gas analysis through the calorimetric measurement of heat production, and especially the respiratory processes of nitrifying bacteria. The physico-chemical analogy between oxygen respiration and alcoholic fermentation caused him to study both these processes in the same subject, namely, yeast extract. By this work he discovered a co-enzyme of respiration, which could be found in all the cells and tissues up till then investigated. At the same time he also found a co-enzyme of alcoholic fermentation. He also discovered the capacity of the SH-group to transfer oxygen; after Hopkins had isolated from cells the SH bodies concerned, Meyerhof showed that the unsaturated fatty acids in the cell are oxidized with the help of the sulfhydryl group. After studying closer the respiration of muscle, Meyerhof investigated the energy changes in muscle. Considerable progress had been achieved by the English scientists Fletcher and Hopkins by their recognition of the fact that lactic acid formation in the muscle is closely connected with the contraction process. These investigations were the first to throw light upon the highly paradoxical fact, already established by the physiologist Hermann, that the muscle can perform a considerable part of its external function in the complete absence of oxygen.
But it was indisputable that in the last resort the energy for muscle activity comes from oxidation, so the connection between activity and combustion must be an indirect one, and observed that in the absence of oxygen in the muscle, lactic acid appears, slowly in the relaxed state and rapidly in the active state, disappearing in the presence of oxygen. Obviously, then, oxygen is involved when muscle is in the relaxed state. http://upload.wikimedia.org/wikipedia/commons/e/e1/Glycolysis.jpg
The Nobel Prize committee had been receiving nominations intermittently for the previous 14 years (for Eijkman, Funk, Goldberger, Grijns, Hopkins and Suzuki but, strangely, not for McCollum in this period). Tthe Committee for the 1929 awards apparently agreed that it was high time to honor the discoverer(s) of vitamins; but who were they? There was a clear case for Grijns, but he had not been re-nominated for that particular year, and it could be said that he was just taking the relatively obvious next steps along the new trail that had been laid down by Eijkman, who was also now an old man in poor health, but there was no doubt that he had taken the first steps in the use of an animal model to investigate the nutritional basis of a clinical disorder affecting millions. Goldberger had been another important contributor, but his recent death put him out of consideration. The clearest evidence for lack of an unknown “something” in a mammalian diet was presented by Gowland Hopkins in 1912. This Cambridge biochemist was already well known for having isolated the amino acid tryptophan from a protein and demonstrated its essential nature. He fed young rats on an experimental diet, half of them receiving a daily milk supplement, and only those receiving milk grew well, Hopkins suggested that this was analogous to human diseases related to diet, as he had suggested already in a lecture published in 1906. Hopkins, the leader of the “dynamic biochemistry” school in Britain and an influential advocate for the importance of vitamins, was awarded the prize jointly with Eijkman. A door was opened. Recognition of work on the fat-soluble vitamins begun by McCollum. The next award related to vitamins was given in 1934 to George Whipple, George Minot and William Murphy “for their discoveries concerning liver therapy in cases of [then incurable pernicious] anemia,” The essential liver factor (cobalamin, or vitamin B12) was isolated in 1948, and Vitamin B12 was absent from plant foods. But William Castle in 1928 had demonstrated that the stomachs of pernicious anemia patients were abnormal in failing to secrete an “intrinsic factor”.
1937 Albert von Szent-Györgyi Nagyrápolt –
” the biological combustion processes, with special reference to vitamin C and the catalysis of fumaric acid”
http://www.biocheminfo.org/klotho/html/fumarate.html
structure of fumarate
Szent-Györgyi was a Hungarian biochemist who had worked with Otto Warburg and had a special interest in oxidation-reduction mechanisms. He was invited to Cambridge in England in 1927 after detecting an antioxidant compound in the adrenal cortex, and there, he isolated a compound that he named hexuronic acid. Charles Glen King of the University of Pittsburgh reported success In isolating the anti-scorbutic factor in 1932, and added that his crystals had all the properties reported by Szent-Györgyi for hexuronic acid. But his work on oxidation reactions was also important. Fumarate is an intermediate in the citric acid cycle used by cells to produce energy in the form of adenosine triphosphate (ATP) from food. It is formed by the oxidation of succinate by the enzyme succinate dehydrogenase. Fumarate is then converted by the enzyme fumarase to malate. An enzyme adds water to the fumarate molecule to form malate. The malate is created by adding one hydrogen atom to a carbon atom and then adding a hydroxyl group to a carbon next to a terminal carbonyl group.
In the same year, Norman Haworth from the University of Birmingham in England received a Nobel prize from the Chemistry Committee for having advanced carbohydrate chemistry and, specifically, for having worked out the structure of Szent-Györgyi’s crystals, and then been able to synthesize the vitamin. This was a considerable achievement. The Nobel Prize in Chemistry was shared with the Swiss organic chemist Paul Karrer, cited for his work on the structures of riboflavin and vitamins A and E as well as other biologically interesting compounds. This was followed in 1938 by a further Chemistry award to the German biochemist Richard Kuhn, who had also worked on carotenoids and B-vitamins, including riboflavin and pyridoxine. But Karrer was not permitted to leave Germany at that time by the Nazi regime. However, the American work with radioisotopes at Lawrence Livermore Laboratory, UC Berkeley, was already ushering in a new era of biochemistry that would enrich our studies of metabolic pathways. The importance of work involving vitamins was acknowledged in at least ten awards in the 20th century.
1. Carpenter, K.J., Beriberi, White Rice and Vitamin B, University of California Press, Berkeley (2000).
2. Weatherall, M.W. and Kamminga, H., The making of a biochemist: the construction of Frederick Gowland Hopkins’ reputation. Medical History vol.40, pp. 415-436 (1996).
3. Becker, S.L., Will milk make them grow? An episode in the discovery of the vitamins. In Chemistry and Modern Society (J. Parascandela, editor) pp. 61-83, American Chemical Society,
Washington, D.C. (1983).
4. Carpenter, K.J., The History of Scurvy and Vitamin C, Cambridge University Press, New York (1986).
Transport and metabolism of exogenous fumarate and 3-phosphoglycerate in vascular smooth muscle.
Molecular and Cellular Biochemistry (Impact Factor: 2.33). 05/1999; 195(1-2):113-21. http://dx.doi.org/10.1023/A:1006976432578
The keto (linear) form of exogenous fructose 1,6-bisphosphate, a highly charged glycolytic intermediate, may utilize a dicarboxylate transporter to cross the cell membrane, support glycolysis, and produce ATP anaerobically. We tested the hypothesis that fumarate, a dicarboxylate, and 3-phosphoglycerate (3-PG), an intermediate structurally similar to a dicarboxylate, can support contraction in vascular smooth muscle during hypoxia. 3-PG improved maintenance of force (p < 0.05) during the 30-80 min period of hypoxia. Fumarate decreased peak isometric force development by 9.5% (p = 0.008) but modestly improved maintenance of force (p < 0.05) throughout the first 80 min of hypoxia. 13C-NMR on tissue extracts and superfusates revealed 1,2,3,4-(13)C-fumarate (5 mM) metabolism to 1,2,3,4-(13)C-malate under oxygenated and hypoxic conditions suggesting uptake and metabolism of fumarate. In conclusion, exogenous fumarate and 3-PG readily enter vascular smooth muscle cells, presumably by a dicarboxylate transporter, and support energetically important pathways.
Comparison of endogenous and exogenous sources of ATP in fueling Ca2+ uptake in smooth muscle plasma membrane vesicles.
C D Hardin, L Raeymaekers, R J Paul
The Journal of General Physiology (Impact Factor: 4.73). 12/1991; 99(1):21-40. http://dx.doi.org:/10.1085/jgp.99.1.21
A smooth muscle plasma membrane vesicular fraction (PMV) purified for the (Ca2+/Mg2+)-ATPase has endogenous glycolytic enzyme activity. In the presence of glycolytic substrate (fructose 1,6-diphosphate) and cofactors, PMV produced ATP and lactate and supported calcium uptake. The endogenous glycolytic cascade supports calcium uptake independent of bath [ATP]. A 10-fold dilution of PMV, with the resultant 10-fold dilution of glycolytically produced bath [ATP] did not change glycolytically fueled calcium uptake (nanomoles per milligram protein). Furthermore, the calcium uptake fueled by the endogenous glycolytic cascade persisted in the presence of a hexokinase-based ATP trap which eliminated calcium uptake fueled by exogenously added ATP. Thus, it appears that the endogenous glycolytic cascade fuels calcium uptake in PMV via a membrane-associated pool of ATP and not via an exchange of ATP with the bulk solution. To determine whether ATP produced endogenously was utilized preferentially by the calcium pump, the ATP production rates of the endogenous creatine kinase and pyruvate kinase were matched to that of glycolysis and the calcium uptake fueled by the endogenous sources was compared with that fueled by exogenous ATP added at the same rate. The rate of calcium uptake fueled by endogenous sources of ATP was approximately twice that supported by exogenously added ATP, indicating that the calcium pump preferentially utilizes ATP produced by membrane-bound enzymes.
Evidence for succinate production by reduction of fumarate during hypoxia in isolated adult rat heart cells.
C Hohl, R Oestreich, P Rösen, R Wiesner, M Grieshaber
Archives of Biochemistry and Biophysics (Impact Factor: 3.37). 01/1988; 259(2):527-35. http://dx.doi.org:/10.1016/0003-9861(87)90519-4 It has been demonstrated that perfusion of myocardium with glutamic acid or tricarboxylic acid cycle intermediates during hypoxia or ischemia, improves cardiac function, increases ATP levels, and stimulates succinate production. In this study isolated adult rat heart cells were used to investigate the mechanism of anaerobic succinate formation and examine beneficial effects attributed to ATP generated by this pathway. Myocytes incubated for 60 min under hypoxic conditions showed a slight loss of ATP from an initial value of 21 +/- 1 nmol/mg protein, a decline of CP from 42 to 17 nmol/mg protein and a fourfold increase in lactic acid production to 1.8 +/- 0.2 mumol/mg protein/h. These metabolite contents were not altered by the addition of malate and 2-oxoglutarate to the incubation medium nor were differences in cell viability observed; however, succinate release was substantially accelerated to 241 +/- 53 nmol/mg protein. Incubation of cells with [U-14C]malate or [2-U-14C]oxoglutarate indicates that succinate is formed directly from malate but not from 2-oxoglutarate. Moreover, anaerobic succinate formation was rotenone sensitive.
We conclude that malate reduction to succinate occurs via the reverse action of succinate dehydrogenase in a coupled reaction where NADH is oxidized (and FAD reduced) and ADP is phosphorylated. Furthermore, by transaminating with aspartate to produce oxaloacetate, 2-oxoglutarate stimulates cytosolic malic dehydrogenase activity, whereby malate is formed and NADH is oxidized.
In the form of malate, reducing equivalents and substrate are transported into the mitochondria where they are utilized for succinate synthesis.
1953 Hans Adolf Krebs –
” discovery of the citric acid cycle” and In the course of the 1920’s and 1930’s great progress was made in the study of the intermediary reactions by which sugar is anaerobically fermented to lactic acid or to ethanol and carbon dioxide. The success was mainly due to the joint efforts of the schools of Meyerhof, Embden, Parnas, von Euler, Warburg and the Coris, who built on the pioneer work of Harden and of Neuberg. This work brought to light the main intermediary steps of anaerobic fermentations.
In contrast, very little was known in the earlier 1930’s about the intermediary stages through which sugar is oxidized in living cells. When, in 1930, I left the laboratory of Otto Warburg (under whose guidance I had worked since 1926 and from whom I have learnt more than from any other single teacher), I was confronted with the question of selecting a major field of study and I felt greatly attracted by the problem of the intermediary pathway of oxidations.
These reactions represent the main energy source in higher organisms, and in view of the importance of energy production to living organisms (whose activities all depend on a continuous supply of energy) the problem seemed well worthwhile studying. http://www.johnkyrk.com/krebs.html
Interactive Krebs cycle
There are different points where metabolites enter the Krebs’ cycle. Most of the products of protein, carbohydrates and fat metabolism are reduced to the molecule acetyl coenzyme A that enters the Krebs’ cycle. Glucose, the primary fuel in the body, is first metabolized into pyruvic acid and then into acetyl coenzyme A. The breakdown of the glucose molecule forms two molecules of ATP for energy in the Embden Meyerhof pathway process of glycolysis.
On the other hand, amino acids and some chained fatty acids can be metabolized into Krebs intermediates and enter the cycle at several points. When oxygen is unavailable or the Krebs’ cycle is inhibited, the body shifts its energy production from the Krebs’ cycle to the Embden Meyerhof pathway of glycolysis, a very inefficient way of making energy.
Fritz Albert Lipmann –
“discovery of co-enzyme A and its importance for intermediary metabolism”.
In my development, the recognition of facts and the rationalization of these facts into a unified picture, have interplayed continuously. After my apprenticeship with Otto Meyerhof, a first interest on my own became the phenomenon we call the Pasteur effect, this peculiar depression of the wasteful fermentation in the respiring cell. By looking for a chemical explanation of this economy measure on the cellular level, I was prompted into a study of the mechanism of pyruvic acid oxidation, since it is at the pyruvic stage where respiration branches off from fermentation.
For this study I chose as a promising system a relatively simple looking pyruvic acid oxidation enzyme in a certain strain of Lactobacillus delbrueckii1. In 1939, experiments using minced muscle cells demonstrated that one oxygen atom can form two adenosine triphosphate molecules, and, in 1941, the concept of phosphate bonds being a form of energy in cellular metabolism was developed by Fritz Albert Lipmann.
In the following years, the mechanism behind cellular respiration was further elaborated, although its link to the mitochondria was not known.[13]The introduction of tissue fractionation by Albert Claude allowed mitochondria to be isolated from other cell fractions and biochemical analysis to be conducted on them alone. In 1946, he concluded that cytochrome oxidase and other enzymes responsible for the respiratory chain were isolated to the mitchondria. Over time, the fractionation method was tweaked, improving the quality of the mitochondria isolated, and other elements of cell respiration were determined to occur in the mitochondria.[13]
The most important event during this whole period, I now feel, was the accidental observation that in the L. delbrueckii system, pyruvic acid oxidation was completely dependent on the presence of inorganic phosphate. This observation was made in the course of attempts to replace oxygen by methylene blue. To measure the methylene blue reduction manometrically, I had to switch to a bicarbonate buffer instead of the otherwise routinely used phosphate. In bicarbonate, pyruvate oxidation was very slow, but the addition of a little phosphate caused a remarkable increase in rate. The phosphate effect was removed by washing with a phosphate free acetate buffer. Then it appeared that the reaction was really fully dependent on phosphate.
A coupling of this pyruvate oxidation with adenylic acid phosphorylation was attempted. Addition of adenylic acid to the pyruvic oxidation system brought out a net disappearance of inorganic phosphate, accounted for as adenosine triphosphate. The acetic acid subunit of acetyl CoA is combined with oxaloacetate to form a molecule of citrate. Acetyl coenzyme A acts only as a transporter of acetic acid from one enzyme to another. After Step 1, the coenzyme is released by hydrolysis to combine with another acetic acid molecule and begin the Krebs’ Cycle again.
Hugo Theorell –
“the nature and effects of oxidation enzymes”
From 1933 until 1935 Theorell held a Rockefeller Fellowship and worked with Otto Warburg at Berlin-Dahlem, and here he became interested in oxidation enzymes. At Berlin-Dahlem he produced, for the first time, the oxidation enzyme called «the yellow ferment» and he succeeded in splitting it reversibly into a coenzyme part, which was found to be flavin mononucleotide, and a colourless protein part. On return to Sweden, he was appointed Head of the newly established Biochemical Department of the Nobel Medical Institute, which was opened in 1937.
Succinate is oxidized by a molecule of FAD (Flavin Adenine Dinucleotide). The FAD removes two hydrogen atoms from the succinate and forms a double bond between the two carbon atoms to create fumarate.
1953

double-stranded-dna

crick-watson-with-their-dna-model.
Watson & Crick double helix model
A landmark in this journey
They followed the path that became clear from intense collaborative work in California by George Beadle, by Avery and McCarthy, Max Delbruck, TH Morgan, Max Delbruck and by Chargaff that indicated that genetics would be important.
1965
François Jacob, André Lwoff and Jacques Monod –
” genetic control of enzyme and virus synthesis”.
In 1958 the remarkable analogy revealed by genetic analysis of lysogeny and that of the induced biosynthesis of ß-galactosidase led François Jacob, with Jacques Monod, to study the mechanisms responsible for the transfer of genetic information as well as the regulatory pathways which, in the bacterial cell, adjust the activity and synthesis of macromolecules. Following this analysis, Jacob and Monod proposed a series of new concepts, those of messenger RNA, regulator genes, operons and allosteric proteins.
Francois Jacob
Having determined the constants of growth in the presence of different carbohydrates, it occurred to me that it would be interesting to determine the same constants in paired mixtures of carbohydrates. From the first experiment on, I noticed that, whereas the growth was kinetically normal in the presence of certain mixtures (that is, it exhibited a single exponential phase), two complete growth cycles could be observed in other carbohydrate mixtures, these cycles consisting of two exponential phases separated by a-complete cessation of growth.
Lwoff, after considering this strange result for a moment, said to me, “That could have something to do with enzyme adaptation.”
“Enzyme adaptation? Never heard of it!” I said.
Lwoff’s only reply was to give me a copy of the then recent work of Marjorie Stephenson, in which a chapter summarized with great insight the still few studies concerning this phenomenon, which had been discovered by Duclaux at the end of the last century. Studied by Dienert and by Went as early as 1901 and then by Euler and Josephson, it was more or less rediscovered by Karström, who should be credited with giving it a name and attracting attention to its existence.
Lwoff’s intuition was correct. The phenomenon of “diauxy” that I had discovered was indeed closely related to enzyme adaptation, as my experiments, included in the second part of my doctoral dissertation, soon convinced me. It was actually a case of the “glucose effect” discovered by Dienert as early as 1900. That agents that uncouple oxidative phosphorylation, such as 2,4-dinitrophenol, completely inhibit adaptation to lactose or other carbohydrates suggested that “adaptation” implied an expenditure of chemical potential and therefore probably involved the true synthesis of an enzyme.
With Alice Audureau, I sought to discover the still quite obscure relations between this phenomenon and the one Massini, Lewis, and others had discovered: the appearance and selection of “spontaneous” mutants. We showed that an apparently spontaneous mutation was allowing these originally “lactose-negative” bacteria to become “lactose-positive”. However, we proved that the original strain (Lac-) and the mutant strain (Lac+) did not differ from each other by the presence of a specific enzyme system, but rather by the ability to produce this system in the presence of lactose. This mutation involved the selective control of an enzyme by a gene, and the conditions necessary for its expression seemed directly linked to the chemical activity of the system.
1974
Albert Claude, Christian de Duve and George E. Palade –
” the structural and functional organization of the cell”.
I returned to Louvain in March 1947 after a period of working with Theorell in Sweden, the Cori’s, and E Southerland in St. Louis, fortunate in the choice of my mentors, all sticklers for technical excellence and intellectual rigor, those prerequisites of good scientific work. Insulin, together with glucagon which I had helped rediscover, was still my main focus of interest, and our first investigations were accordingly directed on certain enzymatic aspects of carbohydrate metabolism in liver, which were expected to throw light on the broader problem of insulin action. But I became distracted by an accidental finding related to acid phosphatase, drawing most of my collaborators along with me. The studies led to the discovery of the lysosome, and later of the peroxisome.
In 1962, I was appointed a professor at the Rockefeller Institute in New York, now the Rockefeller University, the institution where Albert Claude had made his pioneering studies between 1929 and 1949, and where George Palade had been working since 1946. In New York, I was able to develop a second flourishing group, which follows the same general lines of research as the Belgian group, but with a program of its own.
1968
Robert W. Holley, Har Gobind Khorana and Marshall W. Nirenberg –
“interpretation of the genetic code and its function in protein synthesis”.
1969
Max Delbrück, Alfred D. Hershey and Salvador E. Luria –
” the replication mechanism and the genetic structure of viruses”.
1975 David Baltimore, Renato Dulbecco and Howard Martin Temin –
” the interaction between tumor viruses and the genetic material of the cell”.
1976
Baruch S. Blumberg and D. Carleton Gajdusek –
” new mechanisms for the origin and dissemination of infectious diseases” The editors of the Nobelprize.org website of the Nobel Foundation have asked me to provide a supplement to the autobiography that I wrote in 1976 and to recount the events that happened after the award. Much of what I will have to say relates to the scientific developments since the last essay. These are described in greater detail in a scientific memoir first published in 2002 (Blumberg, B. S., Hepatitis B. The Hunt for a Killer Virus, Princeton University Press, 2002, 2004).
1980
Baruj Benacerraf, Jean Dausset and George D. Snell
” genetically determined structures on the cell surface that regulate immunological reactions”.
1992
Edmond H. Fischer and Edwin G. Krebs
“for their discoveries concerning reversible protein phosphorylation as a biological regulatory mechanism”
1994
Alfred G. Gilman and Martin Rodbell –
“G-proteins and the role of these proteins in signal transduction in cells”
2011
Bruce A. Beutler and Jules A. Hoffmann –
” the activation of innate immunity“ and the other half to Ralph M. Steinman – “the dendritic cell and its role in adaptive immunity”.
Renato L. Baserga, M.D.
Kimmel Cancer Center and Keck School of Medicine
Dr. Baserga’s research focuses on the multiple roles of the type 1 insulin-like growth factor receptor (IGF-IR) in the proliferation of mammalian cells. The IGF-IR activated by its ligands is mitogenic, is required for the establishment and the maintenance of the transformed phenotype, and protects tumor cells from apoptosis. It, therefore, serves as an excellent target for therapeutic interventions aimed at inhibiting abnormal growth. In basic investigations, this group is presently studying the effects that the number of IGF-IRs and specific mutations in the receptor itself have on its ability to protect cells from apoptosis.
This investigation is strictly correlated with IGF-IR signaling, and part of this work tries to elucidate the pathways originating from the IGF-IR that are important for its functional effects. Baserga’s group has recently discovered a new signaling pathway used by the IGF-IR to protect cells from apoptosis, a unique pathway that is not used by other growth factor receptors. This pathway depends on the integrity of serines 1280-1283 in the C-terminus of the receptor, which bind 14.3.3 and cause the mitochondrial translocation of Raf-1.
Another recent discovery of this group has been the identification of a mechanism by which the IGF-IR can actually induce differentiation in certain types of cells. When cells have IRS-1 (a major substrate of the IGF-IR), the IGF-IR sends a proliferative signal; in the absence of IRS-1, the receptor induces cell differentiation. The extinction of IRS-1 expression is usually achieved by DNA methylation.
Janardan Reddy, MD
Northwestern University
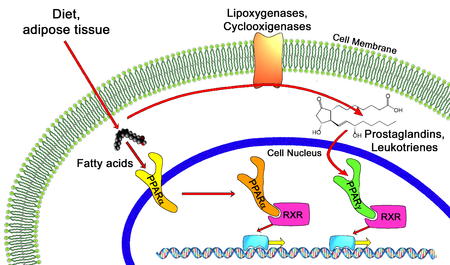
The central effort of our research has been on a detailed analysis at the cellular and molecular levels of the pleiotropic responses in liver induced by structurally diverse classes of chemicals that include fibrate class of hypolipidemic drugs, and phthalate ester plasticizers, which we designated hepatic peroxisome proliferators. Our work has been central to the establishment of several principles, namely that hepatic peroxisome proliferation is associated with increases in fatty acid oxidation systems in liver, and that peroxisome proliferators, as a class, are novel nongenotoxic hepatocarcinogens.
We introduced the concept that sustained generation of reactive oxygen species leads to oxidative stress and serves as the basis for peroxisome proliferator-induced liver cancer development. Furthermore, based on the tissue/cell specificity of pleiotropic responses and the coordinated transcriptional regulation of fatty acid oxidation system genes, we postulated that peroxisome proliferators exert their action by a receptor-mediated mechanism. This receptor concept laid the foundation for the discovery of
- a three member peroxisome proliferator-activated receptor (PPARalpha-, ß-, and gamma) subfamily of nuclear receptors.
- PPARalpha is responsible for peroxisome proliferator-induced pleiotropic responses, including
- hepatocarcinogenesis and energy combustion as it serves as a fatty acid sensor and regulates fatty acid oxidation.
Our current work focuses on the molecular mechanisms responsible for PPAR action and generation of fatty acid oxidation deficient mouse knockout models. Transcription of specific genes by nuclear receptors is a complex process involving the participation of multiprotein complexes composed of transcription coactivators.
Jose Delgado de Salles Roselino, Ph.D.
Leloir Institute, Brazil
Warburg effect, in reality “Pasteur-effect” was the first example of metabolic regulation described. A decrease in the carbon flux originated at the sugar molecule towards the end metabolic products, ethanol and carbon dioxide that was observed when yeast cells were transferred from anaerobic environmental condition to an aerobic one. In Pasteur´s works, sugar metabolism was measured mainly by the decrease of sugar concentration in the yeast growth media observed after a measured period of time. The decrease of the sugar concentration in the media occurs at great speed in yeast grown in anaerobiosis condition and its speed was greatly reduced by the transfer of the yeast culture to an aerobic condition. This finding was very important for the wine industry of France in Pasteur time, since most of the undesirable outcomes in the industrial use of yeast were perceived when yeasts cells took very long time to create a rather selective anaerobic condition. This selective culture media was led by the carbon dioxide higher levels produced by fast growing yeast cells and by a great alcohol content in the yeast culture media. This finding was required to understand Lavoisier’s results indicating that chemical and biological oxidation of sugars produced the same calorimetric results. This observation requires a control mechanism (metabolic regulation) to avoid burning living cells by fast heat released by the sugar biological oxidative processes (metabolism). In addition, Lavoisier´s results were the first indications that both processes happened inside similar thermodynamics limits.
In much resumed form, these observations indicates the major reasons that led Warburg to test failure in control mechanisms in cancer cells in comparison with the ones observed in normal cells. Biology inside classical thermo dynamics poses some challenges to scientists. For instance, all classical thermodynamics must be measured in reversible thermodynamic conditions. In an isolated system, increase in P (pressure) leads to decrease in V (volume) all this in a condition in which infinitesimal changes in one affects in the same way the other, a continuum response. Not even a quantic amount of energy will stand beyond those parameters. In a reversible system, a decrease in V, under same condition, will led to an increase in P.
In biochemistry, reversible usually indicates a reaction that easily goes from A to B or B to A. This observation confirms the important contribution of E Schrodinger in his What´s Life: “This little book arose from a course of public lectures, delivered by a theoretical physicist to an audience of about four hundred which did not substantially dwindle, though warned at the outset that the subject-matter was a difficult one and that the lectures could not be termed popular, even though the physicist’s most dreaded weapon, mathematical deduction, would hardly be utilized. The reason for this was not that the subject was simple enough to be explained without mathematics, but rather that it was much too involved to be fully accessible to mathematics.”
Hans Krebs describes the cyclic nature of the citrate metabolism. Two major research lines search to understand the mechanism of energy transfer that explains how ADP is converted into ATP. One followed the organic chemistry line of reasoning and therefore, searched how the breakdown of carbon-carbon link could have its energy transferred to ATP synthesis. A major leader of this research line was B. Chance who tried to account for two carbon atoms of acetyl released as carbon dioxide in the series of Krebs cycle reactions. The intermediary could store in a phosphorylated amino acid the energy of carbon-carbon bond breakdown. This activated amino acid could transfer its phosphate group to ADP producing ATP. Alternatively, under the possible influence of the excellent results of Hodgkin and Huxley a second line of research appears.
The work of Hodgkin & Huxley indicated the storage of electrical potential energy in transmembrane ionic asymmetries and presented the explanation for the change from resting to action potential in excitable cells. This second line of research, under the leadership of P Mitchell postulated a mechanism for the transfer of oxide/reductive power of organic molecules oxidation through electron transfer as the key for energetic transfer mechanism required for ATP synthesis. Paul Boyer could present how the energy was transduced by a molecular machine that changes in conformation in a series of 3 steps while rotating in one direction in order to produce ATP and in opposite direction in order to produce ADP plus Pi from ATP (reversibility). Nonetheless, a victorious Peter Mitchell obtained the correct result in the conceptual dispute, over the B. Chance point of view, after he used E. Coli mutants to show H gradients in membrane and its use as energy source.
However, this should not detract from the important work of Chance. B. Chance got the simple and rapid polarographic assay method of oxidative phosphorylation and the idea of control of energy metabolism that bring us back to Pasteur. This second result seems to have been neglected in searching for a single molecular mechanism required for the understanding of the buildup of chemical reserve in our body. In respiring mitochondria the rate of electron transport, and thus the rate of ATP production, is determined primarily by the relative concentrations of ADP, ATP and phosphate in the external media (cytosol) and not by the concentration of respiratory substrate as pyruvate. Therefore, when the yield of ATP is high as is in aerobiosis and the cellular use of ATP is not changed, the oxidation of pyruvate and therefore of glycolysis is quickly (without change in gene expression), throttled down to the resting state. The dependence of respiratory rate on ADP concentration is also seen in intact cells. A muscle at rest and using no ATP has very low respiratory rate.
I have had an ongoing discussion with Jose Eduardo de Salles Roselino, inBrazil. He has made important points that need to be noted.
- The constancy of composition which animals maintain in the environment surrounding their cells is one of the dominant features of their physiology. Although this phenomenon, homeostasis, has held the interest of biologists over a long period of time, the elucidation of the molecular basis for complex processes such as temperature control and the maintenance of various substances at constant levels in the blood has not yet been achieved. By comparison, metabolic regulation in microorganisms is much better understood, in part because the microbial physiologist has focused his attention on enzyme-catalyzed reactions and their control, as these are among the few activities of microorganisms amenable to quantitative study. Furthermore, bacteria are characterized by their ability to make rapid and efficient adjustments to extensive variations in most parameters of their environment; therefore, they exhibit a surprising lack of rigid requirements for their environment, and appears to influence it only as an incidental result of their metabolism. Animal cells on the other hand have only a limited capacity for adjustment and therefore require a constant milieu. Maintenance of such constancy appears to be a major goal in their physiology (Regulation of Biosynthetic Pathways H.S. Moyed and H EUmbarger Phys Rev,42 444 (1962)).
- A living cell consists in a large part of a concentrated mixture of hundreds of different enzymes, each a highly effective catalyst for one or more chemical reactions involving other components of the cell. The paradox of intense and highly diverse chemical activity on the one hand and strongly poised chemical stability (biological homeostasis) on the other is one of the most challenging problems of biology (Biological feedback Control at the molecular Level D.E. Atkinson Science vol. 150: 851, 1965). Almost nothing is known concerning the actual molecular basis for modulation of an enzyme`s kinetic behavior by interaction with a small molecule. (Biological feedback Control at the molecular Level D.E. Atkinson Science vol. 150: 851, 1965). In the same article, since the core of Atkinson´s thinking seems to be strongly linked with Adenylates as regulatory effectors, the previous phrases seems to indicate a first step towards the conversion of homeostasis to an intracellular phenomenon and therefore, one that contrary to Umbarger´s consideration could be also studied in microorganisms.
- Most biochemical studies using bacteria, were made before the end of the third upper part of log growth phase. Therefore, they could be considered as time-independent as S Luria presented biochemistry in Life an Unfinished Experiment. The sole ingredient on the missing side of the events that led us into the molecular biology construction was to consider that proteins, a macromolecule, would never be affected by small molecules translational kinetic energy. This, despite the fact that in a catalytic environment and its biological implications S Grisolia incorporated A K Balls observation indicating that the word proteins could be related to Proteus an old sea god that changed its form whenever he was subjected to inquiry (Phys Rev v 4,657 (1964).
- In D.E. Atkinson´s work (Science vol 150 p 851, 1965), changes in protein synthesis acting together with factors that interfere with enzyme activity will lead to “fine-tuned” regulation better than enzymatic activity regulation alone. Comparison of glycemic regulation in granivorous and carnivorous birds indicate that when no important nutritional source of glucose is available, glycemic levels can be kept constant in fasted and fed birds. The same was found in rats and cats fed on high protein diets. Gluconeogenesis is controlled by pyruvate kinase inhibition. Therefore, the fact that it can discriminate between fasting alone and fasting plus exercise (carbachol) requirement of gluconeogenic activity (correspondent level of pyruvate kinase inhibition) the control of enzyme activity can be made fast and efficient without need for changes in genetic expression (20 minute after stimulus) ( Migliorini,R.H. et al Am J. Physiol.257 (Endocrinol. Met. 20): E486, 1989). Regrettably, this was not discussed in the quoted work. So, when the control is not affected by the absorption of nutritional glucose it can be very fast, less energy intensive and very sensitive mechanism of control despite its action being made in the extracellular medium (homeostasis).


{kind=link}