Graft-versus-Host Disease
Writer and Curator: Larry H. Bernstein, MD, FCAP
Introduction
This piece is a follow up to the article on allogeneic transfusion reactions, which extends into transplantation and transplantation outcomes for hematological diseases, both malignant and nonmalignant. The safety of transfusions in Western countries has improved substantially, and the causes for transfusion mishaps has been reduced to unexpected infectious sources, uncommon immune incompatibilities, and errors in processing the blood products. The greatest risk is incurred in platelet transfusions because of the short shelf-life of the product, and the time needed for testing prior to release. This portion of the review is concerned with Graft-versus-Host Disease, which is unique to transfusion and transplanting of blood. In other transplantation, there is graft failure because of host versus graft incompatibility or complications. The reverse order applies to blood. In this case, on the contrary, the transfused or grafted donor tissue becomes a pursuer after the recipients hematopoietic cells.
Peter Brian Medawar: Father of Transplantation
Thomas E. Starzl, M.D., PH.D., F.A.C.S.
J Am Coll Surg. 1995 Mar; 180(3): 332–336
Most of the surgical specialities can be tracked to the creative vision of a surgeon. Transplantation is an exception. Here, the father of the field is succinctly defined in the dictionary as: “Peter Brian Medawar: a Brazilian born British Zoologist who at the age of 45 shared a 1960 Nobel Prize for his work on acquired immunologic tolerance”. Medawar was mysteriously overwhelming to many colleagues and observers, even when he was young. He was the son of a Lebanese father and an English mother—tall, athletic, abnormally handsome, hypnotically articulate in public, and politely cordial in his personal relations. In September 1969, at the age of 54, he had the first of a series of strokes. These crippled him physically but not in spirit. Although I saw Medawar often professionally and privately over a 22 year period, before and after he was disabled, this sporadic exposure was not enough to understand him. My sense is that no one did, except perhaps Jean, his wife for nearly 50 years.
Medawar’s dazzling personality before and great courage after his strokes was inspirational, but his fame was based on the unique achievement in 1953 captured by the terse dictionary mention of “acquired immunologic tolerance.” The roots leading to this accomplishment had fed on the blood of war. More than 12 years earlier, the recently wed zoologist Medawar—24 years of age and fresh from graduate studies at Oxford University—was assigned to
the service of the British surgeon, Dr. Thomas Gibson, to determine if skin allografts could be used to treat casualties from the Battle of Britain. First,
in human studies with Gibson, and then with simple and logical rabbit experiments, Medawar showed that rejection of the skin was an immunologic phenomenon. This later was shown to be analogous to the cell-mediated delayed hypersensitivity that confers immunity to diseases such as tuberculosis. The principal evidence in the early studies was that repetitive grafts from the same donor were rejected more rapidly with each successive attempt —the sensitization and donor specificity confirming an earlier clinical observations by Emil Holman of Stanford in skin-grafted burn victims. Once it was established that rejection was an immune reaction, strategies began to evolve to weaken the recipient immune system. By 1953, total body irradiation and adrenal cortical steroids had been shown to delay skin rejection. However, this immunosuppressive effect was either minor if the animals survived, or lethal to the recipient if the grafts were spared.
Bombshell
In the resulting atmosphere of nihilism about clinical applications, a three and one-half page article by Billingham, Brent, and Medawar in the October 3, 1953 issue of Nature describing acquired tolerance, came as a blinding beacon of hope. The three men had learned that donor splenocytes could be engrafted by their intravenous infusion into immunologically immature mice in utero or perinatally. When these inoculated recipients matured, they could accept skin and other tissues from the donor (but from no other) mouse strain. The immune system of the recipients had been populated by the immunocytes of the donor, meaning that they were now chimeras. The race was on to convert this principle to humans. However, the dark side of their accomplishment soon was revealed by the two younger members of Medawar’s team, Billingham and Brent and by the Dane, Simonsen. The engrafted donor cells could turn the tables and reject the defenseless recipient unless the tissue match was a good one. This was the dreaded graft versus host disease (GVHD) in which transplanted donor cells attacked the recipient skin, gastrointestinal tract, lungs, liver, and the bone marrow itself. Medawar’s dream of 1953 was suddenly a nightmare. Or was it?
On the contrary, the work took a straight line to clinical application, after the demonstration by Prehn and Main that similar tolerance could be induced in adult mice rendered immunologically defenseless by total body irradiation before splenocyte (or later bone marrow) infusion. The recipient conditioning, known as cytoablation, also could be accomplished with myelotoxic drugs. However, as Billingham, Brent, and Medawar had predicted, donor specific tolerance could be induced in humans without GVHD only if there was a good tissue (HLA) match. In 1968, 15 years after the epic Billingham, Brent and Medawar publication, Robert Good and Fritz Bach reported the first two successful human bone marrow transplants. Both recipients of well matched bone marrow from blood relatives are still alive. This was a triumph in which the principal clinicians were internists, as summarized 25 years later in the acceptance speech by the 1990 Nobel Laureate Donnall Thomas.

The growth of bone marrow and whole organ transplantation
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2681237/bin/nihms-87975-f0001.gif
The growth of bone marrow (right) and whole organ transplantation (left) from the seed planted by Peter Medawar during World War II. GVHD, Graft versus host disease.
Immunological Tolerance: Medawar Nobel Acceptance Lecture
“Immunological tolerance” may be described as a state of indifference or non-reactivity towards a substance that would normally be expected to excite an immunological response. The term first came to be used in the context of tissue transplantation immunity, i.e. of the form of immunity that usually prohibits the grafting of tissues between individuals of different genetic make-up; and it was used to refer only to a non-reactivity caused by exposing animals to antigenic stimuli before they were old enough to undertake an immunological response. For example, if living cells from a mouse of strain CBA are injected into an adult mouse of strain A, the CBA cells will be destroyed by an immunological process, and the A-line mouse that received them will destroy any later graft of the same origin with the speed to be expected of an animal immunologically forearmed. But if the CBA cells are injected into a foetal or newborn A-line mouse, they are accepted; more than that, the A-line mouse, when it grows up, will accept any later graft from a CBA donor as if it were its own. I shall begin by using the term “immunological tolerance” in the rather restricted sense that is illustrated by this experiment, and shall discuss its more general usage later on.
The experiment I have just described can be thought of as an artificial reproduction of an astonishing natural curiosity, the phenomenon of red-cell chimerism in certain dizygotic twins. The blood systems of twin cattle before birth are not sharply distinct from each other, as they are in most other twins; instead, the blood systems make anastomoses with each other, with the effect that the twins can indulge in a prolonged exchange of blood before birth. In 1945, R.D. Owen2 made the remarkable discovery that most twin cattle are born with, and may retain throughout life, a stable mixture – not necessarily a fifty-fifty mixture – of each other’s red cells; it followed, then, that the twin cattle must have exchanged red-cell precursors and not merely red cells in their mutual transfusion before birth. This is the first example of the phenomenon we came to call immunological tolerance; the red cells could not have “adapted” themselves to their strange environment, because they were in fact identified as native or foreign by those very antigenie properties which, had an adaptation occurred, must necessarily have been transformed. A few years later R.E. Billingham and I3, with the help of three members of the scientific staff of the Agricultural Research Council, showed that most dizygotic cattle twins would accept skin grafts from each other, and that this mutual tolerance was specific, for skin transplanted from third parties was cast off in the expected fashion.
Some properties of the tolerant state
The main points that emerged from our analysis of the tolerant state were these. In the first place, tolerance must be due to an alteration of the host, not to an antigenic adaptation of the grafted cells, for grafts newly transplanted in adult life have no opportunity to adapt themselves, and the descendants of the cells injected into foetal or newborn animals can be shown by N.A. Mitcbison’s methods to retain their antigenic power10. Once established, the state of tolerance is systemic; if one part of the body will tolerate a foreign graft, so will another; we found no evidence that a tolerated graft builds up a privileged position for itself within its own lymphatic territory. The stimulus that is responsible for instating tolerance is an antigenic stimulus – one which, had it been applied to older animals, would have caused them to become sensitive or immune. A plural stimulus can induce plural tolerance; the donor will usually contain several important antigens that are lacking in the recipient, and long-lasting tolerance must imply tolerance of them all. The state of tolerance is specific in the sense that it will discriminate between one individual and another, for an animal made tolerant of grafts from one individual will not accept grafts from a second individual unrelated to the first; but it will not discriminate between one tissue and another from the same donor.
Tolerance and auto-immunity: 50 years after Burnet.
Martini A1, Burgio GR
Eur J Pediatr. 1999 Oct;158(10):769-75.
Fifty years ago Sir F. Macfarlane Burnet published his first fundamental contribution to the theory of immune tolerance he perfected 10 years later. Since then an impressive amount of new information on the function of the immune system has been gathered. As any original meaningful theory, Burnet’s hypothesis on the development of immune tolerance has undergone extensive modifications to take into account all these new findings. An improved understanding of the mechanisms of tolerance has led to new possibilities for the treatment of auto-immune diseases.
Clonal Selection
http://en.wikipedia.org/wiki/Clonal_selection
Clonal selection theory is a scientific theory in immunology that explains the functions of cells (lymphocytes) of the immune system in response to specific antigens invading the body. The concept was introduced by an Australian doctor Frank Macfarlane Burnet in 1957 in an attempt to explain the formation of a diversity of antibodies during initiation of the immune response. The theory has become a widely accepted model for how the immune system responds to infection and how certain types of B and T lymphocytes are selected for destruction of specific antigens.
The theory states that in a pre-existing group of lymphocytes (specifically B cells), a specific antigen only activates (i.e. selection) its counter-specific cell so that particular cell is induced to multiply (producing its clones) for antibody production. In short the theory is an explanation of the mechanism for the generation of diversity of antibody specificity. The first experimental evidence came in 1958, when Gustav Nossal and Joshua Lederberg showed that one B cell always produces only one antibody. The idea turned out to be the foundation of molecular immunology, especially in adaptive immunity.
The fundamental contribution of Robert A. Good to the discovery of the crucial role of thymus in mammalian immunity
Domenico Ribatti
Immunology. 2006 Nov; 119(3): 291–295.
http://dx.doi.org:/10.1111/j.1365-2567.2006.02484.x
Robert Alan Good was a pioneer in the field of immunodeficiency diseases. He and his colleagues defined the cellular basis and functional consequences of many of the inherited immunodeficiency diseases. His was one of the groups that discovered the pivotal role of the thymus in the immune system development and defined the separate development of the thymus-dependent and bursa-dependent lymphoid cell lineages and their responsibilities in cell-mediated and humoral immunity. Keywords: bursa of Fabricius, history of medicine, immunology, thymus
Robert Alan Good (May 21, 1922 – June 13, 2003) was an American physician who performed the first successful human bone marrow transplant
Robert A. Good began his intellectual and experimental queries related to the thymus in 1952 at the University of Minnesota, initially with pediatric patients. However, his interest in the plasma cell, antibodies and the immune response began in 1944, while still in Medical School at the University of Minnesota in Minneapolis, with his first publication appearing in 1945.
Idiopathic Acquired Agammaglobulinemia Associated with Thymoma (1953)
- a markedly deficient ability to produce antibodies and significant deficits of all or most of the cell-mediated immunities
- in no instance did removal of the thymic tumour restore immunological function or correct the protein deficit
Good syndrome: thymoma with immunodeficiency
- increased susceptibility to bacterial infections by encapsulated organisms and opportunistic viral and fungal infections
- immunodeficiencies, leukopenia, lymphopenia and eosinophylopenia
- severely hypogammaglobulinemic rather than agammaglobulinemic
Good and others found that the patients lacked all of the subsequently described immunoglobulins. These patients were found not to have plasma cells or germinal centers in their hematopoietic and lymphoid tissues. They possessed circulating lymphocytes in normal numbers.
Speculation on the reason for immunological failure following neonatal thymectomy has centered on the thymus as a source of cells or humoral factors essential to normal lymphoid development and immunological maturation.
The bursa of Fabricius and the thymus are ‘central lymphoid organs’ in the chicken, essential to the ontogenetic development of adaptive immunity in that species. Studies by Papermaster and co-workers in Good’s laboratory34,35 indicated that bursectomy in the newly hatched chicks did not completely abolish immunological potential in the adult animal but rather produced a striking quantitative reduction insufficient to eliminate the homograft reaction. The failure of thymectomy in newly hatched chicks to alter the immunological potential of the maturing animal probably only reflected the participation of the bursa of Fabricius in the development of full immunological capacity.
Bursectomized and irradiated birds were completely devoid of germinal centers, plasma cells and the capacity to make antibodies yet they had perfectly normal development of thymocytes and lymphocytes elsewhere in the body that mediated cellular immune reactions. On the other hand, thymectomized and irradiated animals were deficient in lymphocytes that mediated cellular immunity as assessed by skin graft rejection, delayed-type hypersensitivity and graft versus host assays, but they still produced germinal centers, plasma cells and circulating immunoglobulins.
Graft vs Host Disease
Graft-versus-host disease (GVHD) is a complication that can occur after a stem cell or bone marrow transplant. With GVHD, the newly transplanted donor cells attack the transplant recipient’s body.
Graft-versus-host disease (GVHD) is a common complication following an allogeneic tissue transplant. It is commonly associated with stem cell or bone marrow transplant but the term also applies to other forms of tissue graft. Immune cells (white blood cells) in the tissue (the graft) recognize the recipient (the host) as “foreign“. The transplanted immune cells then attack the host’s body cells. GVHD can also occur after a blood transfusion if the blood products used have not been irradiated or treated with an approved pathogen reduction system.
http://en.wikipedia.org/wiki/Graft-versus-host_disease
Causes
GVHD may occur after a bone marrow or stem cell transplant in which someone receives bone marrow tissue or cells from a donor. This type of transplant is called allogeneic. The new, transplanted cells regard the recipient’s body as foreign. When this happens, the newly transplanted cells attack the recipient’s body.
GVHD does not occur when someone receives his or her own cells during a transplant. This type of transplant is called autologous.
Before a transplant, tissue and cells from possible donors are checked to see how closely they match the person having the transplant. GVHD is less likely to occur, or symptoms will be milder, when the match is close. The chance of GVHD is:
- Around 30 – 40% when the donor and recipient are related
- Around 60 – 80% when the donor and recipient are not related
There are two types of GVHD: acute and chronic. Symptoms in both acute and chronic GVHD range from mild to severe.
- Acute GVHD usually happens within the first 6 months after a transplant.
- Chronic GVHD usually starts more than 3 months after a transplant, and can last a lifetime.
Bone marrow transplant
A bone marrow transplant is a procedure to replace damaged or destroyed bone marrow with healthy bone marrow stem cells. Stem cells are immature cells in the bone marrow that give rise to all of your blood cells.
There are three kinds of bone marrow transplants:
- Autologous bone marrow transplant: The term auto means self. Stem cells are removed from you before you receive high-dose chemotherapy or radiation treatment. The stem cells are stored in a freezer (cryopreservation). After high-dose chemotherapy or radiation treatments, your stems cells are put back in your body to make (regenerate) normal blood cells. This is called a rescue transplant.
- Allogeneic bone marrow transplant: The term allo means other. Stem cells are removed from another person, called a donor. Most times, the donor’s genes must at least partly match your genes. Special blood tests are done to see if a donor is a good match for you. A brother or sister is most likely to be a good match. Sometimes parents, children, and other relatives are good matches. Donors who are not related to you may be found through national bone marrow registries.
- Umbilical cord blood transplant: This is a type of allogeneic transplant. Stem cells are removed from a newborn baby’s umbilical cord right after birth. The stem cells are frozen and stored until they are needed for a transplant. Umbilical cord blood cells are very immature so there is less of a need for matching. But blood counts take much longer to recover.
Before the transplant, chemotherapy, radiation, or both may be given. This may be done in two ways:
- Ablative (myeloablative) treatment: High-dose chemotherapy, radiation, or both are given to kill any cancer cells. This also kills all healthy bone marrow that remains, and allows new stem cells to grow in the bone marrow.
- Reduced intensity treatment, also called a mini transplant: Patients receive lower doses of chemotherapy and radiation before a transplant. This allows older patients, and those with other health problems to have a transplant.
Histocompatibility antigen:
- A histocompatibility antigen blood test looks at proteins called human leukocyte antigens (HLAs). These are found on the surface of almost all cells in the human body. HLAs are found in large amounts on the surface of white blood cells. They help the immune system tell the difference between body tissue and substances that are not from your own body.
http://www.nlm.nih.gov/medlineplus/ency/article/001309.htm
Induction of transplantation tolerance in haploidenical transplantation under reduced intensity conditioning: The role of ex-vivo generated donor CD8+ T cells with central memory phenotype
Eran Ophir, Y Eidelstein, E Bachar-Lustig, D Hagin, N Or-Geva, A Lask, , Y Reisner
Best Practice & Research Clinical Haematology 24 (2011) 393–401
http://dx.doi.org:/10.1016/j.beha.2011.05.007
Haploidentical hematopoietic stem cell transplantation (HSCT) offers the advantage of readily available family member donors for nearly all patients. A ‘megadose’ of purified CD34þ hematopoietic stem cells is used to overcome the host’s residual immunity surviving the myeloablative conditioning, while avoiding severe GVHD. However, the number of CD34+ cells that can be harvested is insufficient for overcoming the large numbers of host T cells remaining after reduced intensity conditioning (RIC). Therefore, combining a ‘megadose’ of CD34+ HSCT with other tolerizing cells could potentially support and promote successful engraftment of haploidentical purified stem cell transplantation under a safer RIC. One approach to address this challenge
could be afforded by using Donor CD8 T cells directed against 3rd-party stimulators, bearing an ex-vivo induced central memory phenotype (Tcm). These Tcm cells, depleted of GVH reactivity, were shown to be highly
efficient in overcoming host T cells mediated rejection and in promoting
fully mismatched bone-marrow (BM) engraftment, in HSCT murine models.
This is likely due to the marked lymph node homing of the Tcm, their strong proliferative capacity and prolonged persistence in BM transplant recipients. Thus, combining anti 3rd-party Tcm cell therapy with a ‘megadose’ of purified CD34+ stem cells, could offer a safer RIC protocol for attaining hematopoietic chimerism in patients with hematological diseases and as a platform for organ transplantation or cell therapy in cancer patients.
Induction of tolerance in organ recipients by hematopoietic stem cell transplantation
Eran Ophir, Yair Reisner
International Immunopharmacology 9 (2009) 694–700
http://dx.doi.org:/10.1016/j.intimp.2008.12.009
The use of hematopoietic stem cell transplantation (HSCT) for the establishment of mixed chimerism represents a viable and attractive approach for generating tolerance in transplantation biology, as it generally leads to durable immune tolerance, enabling the subsequent engraftment of organ transplants without the need for a deleterious continuous immunosuppressive therapy. However, in order to apply HSCT to patients in a manner that enables long term survival, transplant-related mortality must be minimized by eliminating the risk for graft-versus-host-disease (GVHD) and by reducing the toxicity of the conditioning protocol. T-cell depleted bone marrow transplants (TDBMT) have been shown to adequately eliminate GVHD. However, even in leukemia patients undergoing supralethal conditioning, mismatched TDBMT are vigorously rejected. This barrier can be overcome through the modulatory activity of CD34 cells, which are endowed with veto activity, by the use of megadose stem cell transplants. In mice, megadoses of Sca+lin– hematopoietic stem cells can induce mixed chimerism following sub-lethal conditioning. Nevertheless, the number of human CD34 cells that can be harvested is not likely to be sufficient to overcome rejection under reduced intensity conditioning (RIC), which might be acceptable in recipients of organ transplantation. To address this challenge, we investigated a novel source of veto cells, namely anti 3rd-party cytotoxic T cells (CTLs) which are depleted of GVH reactivity, combined with megadoses of purified stem cells and a RIC protocol. This approach might provide a safer modality for the induction of durable chimerism.
Intrinsic unresponsiveness of Mertk/B cells to chronic graft-versus-host disease is associated with unmodulated CD1d expression
Wen-Hai Shao, Y Zhen, FD Finkelman, RA Eisenberg, PL Cohen
Journal of Autoimmunity 39 (2012) 412e419
http://dx.doi.org/10.1016/j.jaut.2012.07.001
Activation and migration of marginal zone B (MZB) cells into follicular (FO) regions of the spleen has been proposed as one of the mechanisms that regulate the development of autoreactive B cells. The mer receptor tyrosine kinase (Mertk) mediates apoptotic cell clearance and regulates activation and cytokine secretion. In the well-studied class II chronic GVH model of bm12 cells into B6 hosts, we observed that Mertk deficient B6 mice did not generate autoantibodies in response to this allogeneic stimulus. We posited that Mertk is important in MHC-II-mediated B cell signaling. In the present study, we show that B cells from Mertk-/- mice but not WT B6 mice exhibited decreased calcium mobilization and tyrosine phosphorylation when stimulated by MHC-II cross-linking. The finding that Mertk was important for class II signaling in B cells was further supported by the preponderance of a-allotype autoantibodies in cGVH in RAG-KO mice reconstituted with a mixture of bone marrow from Mertk-/-mice (b-allotype) and C20 mice (a-allotype). MZB cells from Mertk-/- mice were unable to down regulate surface CD1d expression and subsequent inclusion in the MZ, associated with significantly lower germinal center responses compared to MZB cells from WT. Moreover, Mertk-/- mice treated with an anti-CD1d down regulating antibody responded significantly to bm12 cells, while no response was observed in Mertk-/- mice treated with control antibodies. Taken together, these findings extend the role of Mertk to include CD1d down regulation on MZB cells, a potential mechanism limiting B cell activation in cGVH.
Galectin-9 ameliorates acute GVH disease through the induction of T-cell apoptosis
Kazuki Sakai, Eri Kawata, Eishi Ashihara, Yoko Nakagawa, et al.
Eur. J. Immunol. 2011. 41: 67–75 http://dx.doi.org:/10.1002/eji.200939931
Galectins comprise a family of animal lectins that differ in their affinity for β-galactosides. Galectin-9 (Gal-9) is a tandem-repeat-type galectin that was recently shown to function as a ligand for T-cell immunoglobin domain and mucin domain-3 (Tim-3) expressed on terminally differentiated CD41 Th1 cells. Gal-9 modulates immune reactions, including the induction of apoptosis in Th1 cells. In this study, we investigated the effects of Gal-9 in murine models of acute GVH disease (aGVHD). First, we demonstrated that recombinant human Gal-9 inhibit MLR in a dose-dependent manner, involving both Ca21 influx and apoptosis in T cells. Next, we revealed that recombinant human Gal-9 significantly inhibit the progression of aGVHD in murine BM transplantation models. In conclusion, Gal-9 ameliorates aGVHD, possibly by inducing T-cell apoptosis, suggesting that gal-9 may be an attractive candidate for the treatment of aGVHD.
GVHD Prevention: An Ounce Is Better Than a Pound
Pavan Reddy, Gerard Socie, Corey Cutler, Daniel Weisdorf
Biol Blood Marrow Transplant 18:S17-S26, 2012 http://dx.doi.org:/10.1016/j.bbmt.2011.10.034
The pathophysiology of acute graft-versus-host disease (aGVHD) is known to involve donor T cells responding to host histoincompatible allo-antigens presented by the host antigen presenting cells (APCs) and the subsequent induction of pro-inflammatory cytokines and cellular effectors that cause target organ damage. In a more general sense, GVHD can be considered as an immune response against foreign antigens that has gone awry. Similar to all immune responses, GVHD, can be understood as a process that consists of (A) triggers, (B) sensors, (C) mediators, and (D) effectors of GVHD.
Like all immune responses, certain triggers are critical for induction of acute graft-versus-host disease (aGVHD). These include: (1) Disparities between histocompatibility antigens: antigen disparity can be at the level of major histocompatibility complex (MHC), that is, MHC mismatched or at the level of minor histocompatibility antigens (miHA), that is, MHC matched but miHA mismatched. The severity of aGVHD is directly related to the degree of M HC mismatch. In bone marrow transplants (BMT) that are MHC matched but miHA disparate, donor T cells still recognize MHC-peptide derived from the products of recipient polymorphic genes, the miHAs.
Damage induced by conditioning regimens and underlying diseases: under most circumstances, the initiation of an adaptive immune response is triggered by the innate immune response. The innate immune system is triggered by certain exogenous and endogenous molecules. This is likely the case in the induction of aGVHD. Pattern recognition receptors such as Toll-like receptors (TLR), nucleotide-binding oligomerization domain containing 2 (NOD2) play an essential role in innate immunity and in initiating the cellular signaling pathways that activate cytokine secretion, such as NF-kB. Some of their ligands, such as lipopolysaccharide, CpG, and MDP2, which is recognized by TLR-4, TLR-9, and NOD2, respectively, are released by the preparative regimens and contribute to the induction and enhancement of allo-T cell responses. In this way, the conditioning regimens amplify the secretion of proinflammatory cytokines like interleukin (IL)-1, tumor necrosis factor (TNF)-α, IL-6, and other interferon family members in a process described as a ‘‘cytokine storm.’’
The triggers that initiate an immune response have to be sensed and presented. APCs might be considered the sensors for aGVHD. The APCs sense the DAMPs, present the MHC disparate or miHA disparate protein, and provide the critical secondary (costimulatory) and tertiary (cytokine) signals for activation of the alloreactive T cells, the mediators of aGVHD. APCs sense allo-disparity through MHC and peptide complexes. Dendritic cells (DCs) are the most potent APCs and the primary sensors of allo-disparity.
APCs provide the critical costimulation signals for turning on the aGVHD process. The interaction between the MHC/allopeptide complex on APCs and the T cell receptor of donor T cells along with the signal via T cell costimulatory molecules and their ligands on APCs is required to achieve T cell activation, proliferation, differentiation, and survival and the in vivo blockade of positive costimulatory molecules (such as CD28, ICOS, CD40, CD30, etc.), or inhibitory signals (such as PD-1 and CTLA-4) mitigate or exacerbate aGVHD, respectively.
Evidence suggests that alloreactive donor T cells consist of several subsets with different stimuli responsiveness, activation thresholds, and effector functions.
The allo-antigen composition of the host determines which donor T cells subsets differentiate and proliferate. As mentioned previously, in the majority of HLA-matched HCT, aGVHD may be induced by either or both CD41 and CD81 subsets responses to miHAs. The repertoire and immunodominance of the GVHD-associated peptides presented by MHC class I and class II molecules has not been defined. Donor naive CD62L1 T cells are the primary alloreactive T cells that drive the GVHD reaction while the donor effector memory CD62L2 T cells do not. Interestingly, donor regulatory T cells (Tregs) expressing CD62L are also critical to the regulation of GVHD. We now know that it is possible to modulate the alloreactivity of na€ıve T cells by inducing anergy with costimulation blockade, deletion via cytokine modulation, or mixed chimerism. Donor effector memory T cells that are nonalloreactive do not induce GVHD, yet are able to transfer functional memory. In contrast, memory T cells that are alloreactive can cause severe GVHD.
The effector phase that leads to GVHD target organ damage is a complex cascade that involves cytolytic cellular effectors such as CD8 cytotoxic T lymphocytes (CTLs), CD4 T cells, natural killer cells, and inflammatory molecules such as IL-1β, TNF-α, IFN-ϒ, IL-6, and reactive oxygen species. The cellular effectors require cell-cell contact to kill the cells of the target tissues via activation of perforin granzyme, Fas-FasL (CD95-CD95L), or TNFR TRAIL pathways. Other CTLs killing mechanisms such as TWEAK, and LTβ/LIGHT pathways have also been implicated in GVHD. It is important to note that
CTL pathways are essential for GVL effects as well.
All of the above aspects of the biology of aGVHD have been summarized in the mold of a normal immune response. Although this allows for accessing the biology of GVHD, it is important to note that GVHD is a complicated systemic process with as yet still many unknowns and is not a simplified, linear, or cyclical process.
Kinetics of CD4+ and CD8+ T-cell subsets in graft-versus-host reaction (GVHR) in ginbuna crucian carp Carassius auratus langsdorfii
Yasuhiro Shibasakia, H Todaa, Isao kobayashib, T Moritomoa, T Nakanishia
Developmental and Comparative Immunology 34 (2010) 1075–1081
http://dx.doi.org:/10.1016/j.dci.2010.05.009
We have previously demonstrated the presence of graft-versus-host reaction (GVHR) in fish employing a model system of clonal triploid ginbuna and tetraploid ginbuna-goldfish hybrids. To elucidate the role of CD8+ T cells in the induction of GVHR, we investigate the kinetics of CD4+ and CD8+ T-cell subsets in GVHR along with the pathological changes associated with GVH disease (GVHD) in ginbuna. GVHR was not induced with a leukocyte fraction lacking CD8+ T cells separated by magnetic cell sorting. Ploidy and immunofluorescence analysis revealed that CD4+ and CD8+ T cells from sensitized donors greatly
increased in the host trunk kidney, constituting more than 80% of total cells 1–2 weeks after donor cell injection, while those from non-sensitized donors constituted less than 50% of cells present. The increase of CD4+ T cells was greater and more rapid than that of CD8+ T cells. The number of donor CD4+ and CD8+ T cells was highest in trunk kidney followed by spleen. Increases in donor CD4+ and CD8+ T cells were also found in liver and PBL, although the percentages were not as high. Pathologic changes similar to those in human and murine acute GVHD were observed in the lymphoid organs as well as target organs such as skin, liver and intestine, including the destruction of cells and tissues and massive leukocyte infiltration. The pathologic changes became more severe with the increase of CD8+ T cells. These results suggest that donor-derived CD8+ T cells play essential roles for the induction of acute GVHR/D in teleosts as in mammals.
Fludarabine and Exposure-Targeted Busulfan Compares Favorably with Busulfan/Cyclophosphamide-Based Regimens in Pediatric Hematopoietic
Cell Transplantation: Maintaining Efficacy with Less Toxicity
I.H. Bartelink, E.M.L. van Reij, C.E. Gerhardt, E.M. van Maarseveen, et al
Biol Blood Marrow Transplant 20 (2014) 345e353
http://dx.doi.org/10.1016/j.bbmt.2013.11.027
Busulfan (Bu) is used as a myeloablative agent in conditioning regimens before allogeneic hematopoietic cell transplantation (allo-HCT). In line with strategies explored in adults, patient outcomes may be optimized by replacing cyclophosphamide (Cy) with or without melphalan (Mel) with fludarabine (Flu). We compared outcomes in 2 consecutive cohorts of HCT recipients with a nonmalignant HCT indication, a myeloid malignancy, or a lymphoid malignancy with a contraindication for total body irradiation (TBI). Between 2009 and 2012, 64 children received Flu + Bu at a target dose of 80-95 mg-h/L, and between 2005 and 2008, 50 children received Bu targeted to 74-80 mg-h/L þ Cy. In the latter group, Mel was added for patients with myeloid malignancy (n = 12). Possible confounding effects of calendar time were studied in 69 patients receiving a myeloablative dose of TBI between 2005 and 2012. Estimated 2-year survival and event-free survival were 82% and 78%, respectively, in the FluBu arm and 78% and 72%, respectively, in the BuCy (Mel) arm (P, not significant). Compared with the BuCy (Mel) arm, less toxicity was noted in the FluBu arm, with lower rates of acute (noninfectious) lung injury (16% versus 36%; P < .007), veno-occlusive disease (3% versus 28%; P < .003), chronic graft-versus-host disease (9% versus 26%; P < .047), adenovirus infection (3% versus 32%; P < .001), and human herpesvirus 6 infection reactivation (21% versus 44%; P < .005). Furthermore, the median duration of neutropenia was shorter in the FluBu arm (11 days versus 22 days; P < .001), and the patients in this arm required fewer transfusions. Our data indicate that Flu (160 mg/m2) with targeted myeloablative Bu (90 mg-h/L) is less toxic than and equally effective
as BuCy (Mel) in patients with similar indications for allo-HCT.
Fibrotic and Sclerotic Manifestations of Chronic Graft-versus-Host Disease
Carrie L. Kitko, Eric S. White, Kristin Baird
Biol Blood Marrow Transplant 18:S46-S52, 2012
http://dx.doi.org:/10.1016/j.bbmt.2011.10.021
Chronic graft-versus-host disease (cGVHD) is a common cause of morbidity
and mortality following allogeneic stem cell transplantation (HCT), with approximately 50% to 60% of long-term HCT survivors developing one or more manifestations of the disorder. Although acute GVHD is typically limited to skin, liver, and gastrointestinal involvement, virtually every organ is at risk for the development of cGVHD. Although the pathophysiology of cGVHD remains poorly understood, some of the most severe organ manifestations are linked by end-organ fibrosis. In particular, fibrotic cutaneous and bronchiolar changes, resulting in scleroderma-like changes and bronchiolitis obliterans syndrome (BOS), respectively, are two of the most devastating outcomes for these patients. Both sclerotic GVHD (ScGVHD) and BOS have been reported in 5% to 15% of patients with cGVHD.
Many of the manifestations of cGVHD share clinical characteristics seen in nontransplant conditions, including systemic sclerosis or pulmonary fibrosis. Thus, understanding the pathophysiology underlying these related conditions may help identify potential mechanisms and ultimately new therapeutic options for patients with cGVHD.
Tyrosine kinase inhibitors (TKIs) have been shown to inhibit two different profibrotic pathways (transforming growth factor β [TGF-β] and platelet-derived growth factor [PDGF]) in various mouse models of fibrotic disease and offer a possible novel treatment approach for cGVHD patients suffering from severe sclerosis. Likewise, overexpression of TNF-α has been shown to induce fibrogenesis in experimental hepatocellular disease and has been linked with human scleroderma-associated interstitial pulmonary fibrosis and profibrotic responses in human osteoarthritic hip joint fibroblasts. The use of TNF antagonists has been examined in some clinical situations associated with fibrosis, suggesting they may also be of some benefit to patients with cGVHD; however, this must first be prospectively tested.
Table. Proposed Modifications to NIH BOS Clinical Definition
- Absence of infection (no change)
- Another cGVHD manifestation in another organ (no change)
- FEV1 <75% predicted (no change) or >10% decline from pre-HCT value (modification)
- Signs of Obstruction
- FEV1/SVC ratio <0.7 (modification), or
- RV >120% predicted (no change), or
- RV/TLC >120% (modification), and
- HRCT with evidence of air trapping (no change)
SVC indicates slow vital capacity; RV, residual volume; TLC, total lung capacity; HRCT, high-resolution computed tomography
Figure (not shown)
Effect of etanercept on survival in post-HCT patients with subacute lung injury. (A) Overall 5-year survival by pulmonary function testing defect. Patients with an obstructive defect (solid line) had a 5-year survival of 67% compared with 44% in those with a restrictive lung defect (dashed line) (P 5 .19). (B) Overall 5-year survival by response to therapy. Patients who responded to etanercept therapy (solid line) had a 5-year survival of 90% compared with 55% in patients who failed to respond (dashed line) (P 5.07). (Figures reprinted with permission, Biol Blood and Marrow Trans).
Extensive, sclerotic skin changes with superficial or deep subcutaneous or fascial involvement are seen in approximately 4% to 13% of patients with cGVHD and can be a life-threatening manifestation. ScGVHD of the skin includes several cutaneous presentations characterized by inflammation and progressive fibrosis of the dermis and subcutaneous tissues. These changes can resemble morphea, systemic sclerosis, or eosinophilic fasciitis and may or may not occur in the setting of concurrent overlying epidermal GVHD. When severe, ScGVHD can result in contractures, severe wasting, and chest wall restriction.
Development of clinical trials for patients with cGVHD is difficult because of the complexity and heterogeneity of disease, variable approaches to treatment, and the lack of standardized assessments of disease. In particular, the study of ScGVHD lacks universally accepted measures of disease burden and response. Investigators have used several measures to assess ScGVHD involvement including body surface area, magnetic resonance imaging, ultrasound, and range-of-motion measurements. Additionally, investigators have tried to apply the Rodnan score, the standardmeasure for skin involvement in scleroderma. Thus far, none of these measures has proven
to be completely reliable in the setting of ScGVHD, and it is likely that multiple measures will need to be integrated into the assessment of ScGVHD.
Imatinib mesylate (Gleevec in the US; Glivec in Europe, Australia, and Latin America, marketed by Novartis) is a TKI that has biological activity against both PDGF and TGF-β signaling pathways. Both cytokines have been implicated in the pathogenesis of several fibrosing diseases, including hepatic, renal, and lung, as well as in scleroderma, a disease that closely resembles ScGVHD. In addition, stimulatory antibodies specific for the PDGF receptor (PDGFR) were identified in a series of 39 patients with extensive cGVHD with higher levels detected in those patients with skin involvement. Similar stimulatory antibodies targeting PDGFR have been reported in patients with scleroderma, suggesting an important therapeutic target for these fibrosing conditions. Imatinib mesylate has particularly potent activity against PDGF and is FDA approved in the United States for the treatment of several disorders associated with aberrant PDGFR signaling. The side effect profile of the drug is well established in non-HCT patients, which is helpful in the setting of a therapy for allogenic HCT patients, many of whom have multiorgan system symptoms and possible dysfunction and who will require ongoing immunosuppressive therapy.
Through the efforts of the Chronic GVHD Consortium, led by Stephanie Lee at the Fred Hutchinson Cancer Research Center, there is a multicenter, ongoing prospective evaluation of the NIH diagnostic and assessment tools. This effort has already resulted in several publications that have further refined essential criteria for cGVHD evaluation, including organ-specific manifestations such as BOS and ScGVHD. Currently, the Consortium is conducting a multicenter prospective clinical trial of fluticasone propionate, azithromycin, and montelukast for the treatment of BOS (ClinicalTrials.gov NCT01307462); a separate trial of imatinib versus rituximab for treatment of ScGVHD is also enrolling subjects (ClinicalTrials.gov NCT01309997).
Although cGVHD remains a significant problem for many long-term survivors of HCT, critical advances in cGVHD research and treatment can be achieved by cooperative group efforts such as those put forth by the Chronic GVHD Consortium and the Clinical Trials Network.
Hematopoietic stem cell transplantation (HSCT): An approach to autoimmunity
Carmen Alaez, Mariana Loyola, Andrea Murguıa, Hilario Flores, et al.
Autoimmunity Reviews 5 (2006) 167– 179
http://dx.doi.org:/10.1016/j.autrev.2005.06.003
HSCT provides the opportunity to replace a damaged tissue. It is the most important treatment for high risk hematologic malignant and nonmalignant disorders. An important challenge in the identification of matched donors/patients is the HLA diversity. The Mexican Bone Marrow Registry (DONORMO) has nowadays N5000 donors. The prevalent alleles are Amerindian, Mediterranean (Semitic and Spanish genes) and African. In theory, it is possible to find 11% of 6/6 A–B–DR low resolution matches for 70% of patients with Mexican ancestry. We contributed with 39 unrelated, cord blood and autologous HSCT for patients with malignant, genetic and autoimmune disorders. Overall disease survival was 50% (2–7 years) depending on the initial diagnosis, conditioning, disease evolution or other factors. Clinical studies using autologous and unrelated HSC are performed on patients with refractory autoimmune diseases producing mixed results: mainly, T1D, RA, MS, SLE. Improvement has been observed in skin damage and quality of life in SLE and systemic sclerosis. Disease stabilization in 2/3 of MS patients. However, in RA and T1D, initial benefits have been followed by eventual relapse. With growing clinical experience and protocol improvement, treatment-related mortality is decreasing. Proof efficacy will be achieved by comparing HSCT with standard therapy in autoimmunity.
Monoclonal Antibody-Mediated Targeting of CD123, IL-3 Receptor α Chain, Eliminates Human Acute Myeloid Leukemic Stem Cells
Liqing Jin, Erwin M. Lee, Hayley S. Ramshaw, Samantha J. Busfield, et al.
Cell: Stem Cell 5, 31–42, July 2, 2009
http://dx.doi.org:/10.1016/j.stem.2009.04.018
Leukemia stem cells (LSCs) initiate and sustain the acute myeloid leukemia (AML) clonal hierarchy and possess biological properties rendering them resistant to conventional chemotherapy. The poor survival of AML patients raises expectations that LSC-targeted therapies might achieve durable remissions. We report that an anti-interleukin-3 (IL-3) receptor α chain (CD123)-neutralizing antibody (7G3) targeted AML-LSCs, impairing homing
to bone marrow (BM) and activating innate immunity of nonobese diabetic/ severe-combined immunodeficient (NOD/SCID) mice. 7G3 treatment profoundly reduced AML-LSC engraftment and improved mouse survival.
Mice with preestablished disease showed reduced AML burden in the BM
and periphery and impaired secondary transplantation upon treatment, establishing that AMLLSCs were directly targeted. 7G3 inhibited IL-3-mediated intracellular signaling of isolated AML CD34+ CD38[1] cells in vitro and reduced their survival. These results provide clear validation for therapeutic monoclonal antibody (mAb) targeting of AML-LSCs and for translation of in vivo preclinical research findings toward a clinical application.
Many Days at Home during Neutropenia after Allogeneic Hematopoietic Stem Cell Transplantation Correlates with Low Incidence of Acute Graft-versus-Host Disease
Olle Ringdén, Mats Remberger, Katarina Holmberg, Charlotta Edeskog, et al.
Biol Blood Marrow Transplant 19 (2013) 314e320
http://dx.doi.org/10.1016/j.bbmt.2012.10.011
Patients are isolated in the hospital during the neutropenic phase after allogeneic hematopoietic stem cell transplantation. We challenged this by allowing patients to be treated at home. A nurse from the unit visited and checked the patient. One hundred forty-six patients treated at home were compared with matched hospital control subjects. Oral intake was intensified from September 2006 and improved (P < .002). We compared 4 groups: home care and control subjects before and after September 2006. The cumulative incidence of acute graft-versus-host disease (GVHD) of grades II to IV was 15% in the “old” home care group, which was significantly lower than that of 32% to 44% in the other groups (P <.03). Transplantation-related mortality, chronic GVHD, and relapse were similar in the groups. The “new” home care patients spent fewer days at home (P < .002). In multivariate analysis, GVHD of grades 0 to I was associated with home care (hazard ratio [HR], 2.46; P <.02) and with days spent at home (HR, .92; P < .005) but not with oral nutrition (HR, .98; P = .13). Five year survival was 61% in the home care group as compared with 49% in the control subjects (P < .07). Home care is safe. Home care and many days spent at home were correlated with a low risk of acute GVHD.
Impact on Outcomes of Human Leukocyte Antigen Matching by Allele-Level Typing in Adults with Acute Myeloid Leukemia Undergoing Umbilical Cord Blood Transplantation
Jaime Sanz, Francisco J. Jaramillo, Dolores Planelles, Pau Montesinos, et al.
Biol Blood Marrow Transplant 20 (2014) 106e110
http://dx.doi.org/10.1016/j.bbmt.2013.10.016
This retrospective study analyzed the impact of directional donor-recipient human leukocyte antigen (HLA) disparity using allele-level typing at HLA-A, -B, -C, and -DRB1 in 79 adults with acute myeloid leukemia (AML) who received single-unit umbilical cord blood (UCB) transplant at a single institution. With extended high resolution HLA typing, the donor-recipient compatibility ranged from 2/8 to 8/8. HLA disparity showed no negative impact on nonrelapse mortality (NRM), graft-versus-host (GVH) disease or engraftment. Considering disparities in the GVH direction, the 5-year cumulative incidence of relapse was 44% and 22% for patients receiving an UCB unit matched > 6/8 and < 6/8, respectively (P <.04). In multivariable analysis, a higher HLA disparity in the GVH direction using extended high-resolution typing (Risk ratio [RR] 2.8; 95% confidence interval [CI], 1.5 to 5.1; P ¼.0009) and first complete remission at time of transplantation (RR 2.1; 95% CI, 1.2 to 3.8; P < .01) were the only variables significantly associated with an improved disease-free survival. In conclusion, we found that in adults with AML undergoing single-unit UCBT, an increased number of HLA disparities at allele-level typing improved disease-free survival by decreasing the relapse rate without a negative effect on NRM.
HLA mismatch direction in cord blood transplantation: impact on outcome and implications for cord blood unit selection
Cladd E. Stevens, C Carrier, C Carpenter, D Sung, and A Scaradavou
Blood. 2011; 118(14):3969-3978
http://dx.doi.org:/10.1182/blood-2010-11-317271
Donor-recipient human leukocyte antigen mismatch level affects the outcome of unrelated cord blood (CB) transplantation. To identify possible “permissive” mismatches, we examined the relationship between direction of human leukocyte antigen mismatch (“vector”) and transplantation outcomes in 1202 recipients of single CB units from the New York Blood Center National Cord Blood Program treated in United States Centers from 1993-2006. Altogether, 98 donor/patient pairs had only unidirectional mismatches: 58 in the graft-versus-host (GVH) direction only (GVH-O) and 40 in the host-versus-graft or rejection direction only (R-O). Engraftment was faster in patients with GVH-O mismatches compared with those with 1 bidirectional mismatch (hazard ratio [HR] = 1.6, P < .003). In addition, patients with hematologic malignancies given GVH-O grafts had lower transplantation-related mortality (HR = 0.5, P < .062), overall mortality (HR = 0.5, P < .019), and treatment failure (HR = 0.5, P < .016), resulting in outcomes similar to those of matched CB grafts. In contrast, R-O mismatches had slower engraftment, higher graft failure, and higher relapse rates (HR = 2.4, P < .010). Based on our findings, CB search algorithms should be modified to identify unidirectional mismatches. We recommend that transplant centers give priority to GVH-O-mismatched units over other mismatches and avoid selecting R-O mismatches, if possible.
Mutation of the NPM1 gene contributes to the development of donor cell–derived acute myeloid leukemia after unrelated cord blood transplantation
for acute lymphoblastic leukemia
G Rodríguez-Macías, C Martínez-Laperche, J Gayoso, V Noriega, .., Ismael Buño
Human Pathology (2013) 44, 1696–1699
http://dx.doi.org/10.1016/j.humpath.2013.01.001
Donor cell leukemia (DCL) is a rare but severe complication after allogeneic stem cell transplantation. Its true incidence is unknown because of a lack of correct recognition and reporting, although improvements in molecular analysis of donor-host chimerism are contributing to a better diagnosis of this complication. The mechanisms of leukemogenesis are unclear, and multiple factors can contribute to the development of DCL. In recent years, cord blood has emerged as an alternative source of hematopoietic progenitor cells, and at least 12 cases of DCL have been reported after unrelated cord blood transplantation. We report a new case of DCL after unrelated cord blood transplantation in a 44-year-old woman diagnosed as having acute lymphoblastic leukemia with t(1;19) that developed acute myeloid leukemia with normal karyotype and nucleophosmin (NPM1) mutation in donor cells. To our knowledge, this is the first report of NPM1 mutation contributing to DCL development.
Graft-versus-leukemia in the bone marrow
Blood, 23 JAN 2014; 123(4)
http://imagebank.hematology.org.
63-year-old female with relapsed acute myeloid leukemia (AML) after allogeneic stem cell transplantation reached CR2 after re-induction therapy followed by consolidation with donor lymphocyte infusions: 3 x 107/kg and 3 x 108/kg after 1 and 2.5 months, respectively. No signs of graft-versus-host disease were observed at this time. At 5 months follow-up, her blood count deteriorated: hemoglobin: 6.9 mmol/L, thrombocytes: 58 x 109/L and leukocytes: 1.37 x 109/L. Bone marrow aspirate was not evaluable. Bone marrow trephine biopsy showed relapse AML with hypercellularity in the H&E staining (340 objective lens, panel A) and 20% CD341 blast cells without any signs of maturation (panel B). Also, a high number of CD3 positive T cells (panel C) was noted, intermingling with the CD34 positive blasts, both staining positively with CD43 (panel D). Only supportive care was given. However, normalization of the blood count was observed in the following months and she developed graft-versus-host disease of the lung, which was treated with ciclosporin and prednisone. A bone marrow aspirate performed 3 months after relapse showed a third remission with 0.8% myeloid blasts. In retrospect, one could therefore consider the picture of the bone marrow trephine biopsy at the second relapse as graft-versus-leukemia in the bone marrow.

GVL- panel A

GVL – panel B

GVL – panel C

Long-Term Outcomes of Alemtuzumab-Based Reduced-Intensity Conditioned Hematopoietic Stem Cell Transplantation for Myelodysplastic Syndrome and Acute Myelogenous Leukemia Secondary to Myelodysplastic Syndrome
Victoria T. Potter, Pramila Krishnamurthy, Linda D. Barber, ZiYi Lim, et al.
Biol Blood Marrow Transplant 20 (2014) 111e117
http://dx.doi.org/10.1016/j.bbmt.2013.10.021
Allogeneic hematopoietic stem cell transplantation (HSCT) with reduced-intensity conditioning (RIC) offers a potential cure for patients with myelodysplastic syndrome (MDS) who are ineligible for standard-intensity regimens. Previously published data from our institution suggest excellent outcomes at 1 yr using a uniform fludarabine, busulfan, and alemtuzumab-based regimen. Here we report long-term follow-up of 192 patients with MDS and acute myelogenous leukemia (AML) secondary to MDS (MDS-AML) transplanted with this protocol, using sibling (n = 45) or matched unrelated (n = 147) donors. The median age of the cohort was 57 yr (range, 21 to 72 yr), and median follow-up was 4.5 yr (range, 0.1 to 10.6 yr). The 5-yr overall survival (OS), event-free survival, and nonrelapse mortality were 44%, 33%, and 26% respectively. The incidence of de novo chronic graft-versus-host disease (GVHD) was low at 19%, illustrating the efficacy of alemtuzumab for GVHD prophylaxis. Conversely, the 5-yr relapse rate was 51%. For younger patients (age <50 yr), the 5-yr OS and relapse rates were 58% and 39%, respectively. On multivariate analysis, advanced age predicted significantly worse outcomes, with patients age >60 yr having a 5-yr OS of 15% and relapse rate of 66%. Patients receiving preemptive donor lymphocyte infusions had an impressive 5-yr OS of 67%, suggesting that this protocol may lend itself to the incorporation of immunotherapeutic strategies. Overall, these data demonstrate good 5-yr OS for patients with MDS and MDS-AML undergoing alemtuzumab-based RIC-HSCT. The low rate of chronic GVHD is encouraging, and comparative studies with other RIC protocols are warranted.
Natural killer cell activity influences outcome after T cell depleted stem cell transplantation from matched unrelated and haploidentical donors
Peter Lang, Matthias Pfeiffer, Heiko-Manuel Teltschik, Patrick Schlegel, et al.
Best Practice & Research Clinical Haematology 24 (2011) 403–411
http://dx.doi.org:/10.1016/j.beha.2011.04.009
Lytic activity and recovery of natural killer (NK) cells was monitored in pediatric patients with leukemias (ALL, AML, CML, JMML) and myelodysplastic syndromes after transplantation of T cell depleted stem cells from matched unrelated (n = 18) and mismatched related (haploidentical, n = 29) donors. CD34+ selection with magnetic microbeads resulted in 8 x 103/kg residual T cells. No post-transplant immune suppression was given. NK cells recovered rapidly after transplantation (300 CD56+/mL at day 30, median), whereas T cell recovery was delayed (median: 12 CD3+/mL at day 90). NK activity was measured as specific lysis of K 562 targets several times (mean: 3 assays per patient). Four temporal patterns of lytic activity could be differentiated: consistently low, consistently high, decreasing and increasing activity. Patients with consistently high or increasing activity had significantly lower relapse probability than patients with consistently low or decreasing levels (0.18 vs 0.73 at 2 years, p < 0.05). The subgroup of patients with ALL showed similar results (0.75 vs 0.14 at 2 years, p < 0.05). Speed of T cell recovery had no influence. These data suggest that both achieving and maintaining a high level of NK activity may contribute to prevent relapse. Since NK activity could be markedly increased by in vitro stimulation with Interleukin 2 (IL-2), in vivo administration should be considered.
Graft-versus-host disease: Pathogenesis and clinical manifestations of graft-versus-host disease
Sharon R. Hymes, Amin M. Alousi, and Edward W. Cowen
J Am Acad Dermatol 2012; 66: 515.e1-18.
- Graft-versus-host disease is the primary cause of morbidity and nonerelapse related mortality in patients who undergo allogeneic hematopoietic cell transplantation.
- Acute graft-versus-host disease manifests as a skin exanthem, liver dysfunction, and gastrointestinal involvement.
- Chronic graft-versus-host disease of the skin is remarkably variable in its clinical presentation.
- Chronic graft-versus-host disease is a multisystem disorder that may affect nearly any organ; the most common sites are the skin, oral mucosa, and eyes.
Key points
- Allogeneic transplantation is in widespread use for hematologic malignancies, but is also increasingly used for marrow failure syndromes, immunodeficiencies, and other life-threatening conditions
- Graft-versus-host disease is the primary cause of morbidity and nonerelapse related mortality after allogeneic hematopoietic cell transplantation
- Minimizing graft-versus-host disease without losing the graft-versus-tumor effect is an area of active research
- The skin is the most common organ affected in patients with graft-versus-host disease
Outcomes of Thalassemia Patients Undergoing Hematopoietic Stem Cell Transplantation by Using a Standard Myeloablative versus a Novel Reduced-Toxicity Conditioning Regimen According to a New Risk Stratification
Usanarat Anurathapan, S Pakakasama, P Mekjaruskul, N Sirachainan, et al.
Biol Blood Marrow Transplant 20 (2014) 2056e2075
http://dx.doi.org/10.1016/j.bbmt.2014.07.016
Improving outcomes among class 3 thalassemia patients receiving allogeneic hematopoietic stem cell transplantations (HSCT) remains a challenge. Before HSCT, patients who were > 7 years old and had a liver size > 5 cm constitute what the Center for International Blood and Marrow Transplant Research defined as a very high risk subset of a conventional high-risk class 3 group (here referred to as class 3 HR). We performed HSCT in 98 patients with related and unrelated donor stem cells. Seventy-six of the patients with age < 10 years received the more conventional myeloablative conditioning (MAC) regimen (cyclophos-phamide, busulfan, + fludarabine); the remaining 22 patients with age > 10 years and hepatomegaly (class 3 HR), and in several instances additional comorbidity problems, underwent HSCT with a novel reduced-toxicity conditioning (RTC) regimen (fludarabine and busulfan). We then compared the outcomes between these 2 groups (MAC versus RTC). Event-free survival (86% versus 90%) and overall survival (95% versus 90%) were not significantly different between the respective groups; however, there was a higher incidence of serious treatment-related complications in the MAC group, and although we experienced 6 graft failures in the MAC group (8%), there were none in the RTC group. Based on these results, we suggest that (1) class 3HRthalassemia patients can safely receive HSCT with our novel RTC regimen and achieve the same excellent outcome as low/standard-risk thalassemia patients who received the standard MAC regimen, and further, (2) that this novel RTC approach should be tested in the low/standard-risk patient population.
Pharmacological Immunosuppression Reduces But Does Not Eliminate the Need for Total-Body Irradiation in Nonmyeloablative Conditioning Regimens for Hematopoietic Cell Transplantation
Marco Mielcarek, Beverly Torok-Storb, Rainer Storb
Biol Blood Marrow Transplant 17: 1255-1260 (2011)
http://dx.doi.org:/10.1016/j.bbmt.2011.01.003
In the dog leukocyte antigen (DLA)-identical hematopoietic cell transplantation (HCT) model, stable marrow engraftment can be achieved with total-body irradiation (TBI) of 200 cGy when used in combination with postgrafting immunosuppression. The TBI dose can be reduced to 100 cGy without compromising engraftment rates if granulocyte colony-stimulating factor (G-CSF)-mobilized peripheral blood mononuclear cells (G-PBMC) are infused with the marrow. T cell-depleting the G-PBMC product abrogates this effect. These results were interpreted to suggest that the additional T cells provided with G-PBMC facilitated engraftment by overcoming host resistance.We therefore hypothesized that the TBI dose may be further reduced to 50 cGy by augmenting immunosupression either by (1) tolerizing or killing recipient T cells, or (2) enhancing the graft-versus-host (GVH) activity of donor T cells. To test the first hypothesis, recipient T cells were activated before HCT by repetitive donor-specific PBMC infusions followed by administration of methotrexate (MTX) (n 5 5), CTLA4-Ig (n = 4), denileukin diftitox (Ontak; n = 4), CTLA4-Ig 1 MTX (n = 8), or 5c8 antibody (anti-CD154) 1 MTX (n = 3). To test the second hypothesis, recipient dendritic cells were expanded in vivo by infusion of Flt3 ligand given either pre-HCT (n = 4) or pre- and post-HCT (n = 5) to augment GVH reactions. Although all dogs showed initial allogeneic engraftment, sustained engraftment was seen in only 6 of 42 dogs (14% of all dogs treated in 9 experimental groups). Hence, unless more innovative pharmacotherapy can be developed that more forcefully shifts the immunologic balance in favor of the donor, noncytotoxic immunosuppressive drug therapy as the sole component of HCT preparative regimens may not suffice to ensure sustained engraftment.
Pretransplant Immunosuppression followed by Reduced-Toxicity Conditioning and Stem Cell Transplantation in High-Risk Thalassemia: A Safe Approach to Disease Control
Usanarat Anurathapan, S Pakakasama, P Rujkijyanont, N Sirachainan, et al.
Biol Blood Marrow Transplant 19 (2013) 1254e1270
http://dx.doi.org/10.1016/j.bbmt.2013.04.023
Patients with class 3 thalassemia with high-risk features for adverse events after high-dose chemotherapy with hematopoietic stem cell transplantation (HSCT) are difficult to treat, tending to either suffer serious toxicity or fail to establish stable graft function. We performed HSCT in 18 such patients age 7 years and hepatomegaly using a novel approach with pretransplant immunosuppression followed by a myeloablative reduced-toxicity conditioning regimen (fludarabine and i.v. busulfan [Flu-IV Bu]) and then HSCT. The median patient age was 14 years (range, 10 to 18 years). Before the Flu-IV Bu þ antithymocyte globulin conditioning regimen, all patients received 1 to 2 cycles of pretransplant immunosuppression with fludarabine and dexamethasone. Thirteen patients received a related donor graft, and 5 received an unrelated donor graft. An initial prompt engraftment of donor cells with full donor chimerism was observed in all 18 patients, but 2 patients developed secondary mixed chimerism that necessitated withdrawal of immunosuppression to achieve full donor chimerism. Two patients (11%) had acute grade III-IV graft-versus-host disease, and 5 patients had limited chronic graft-versus-host disease. The only treatment-related mortality was from infection, and with a median follow-up of 42 months (range, 4 to 75), the 5-year overall survival and thalassemia-free survival were 89%. We conclude that this novel sequential immunoablative pretransplant-ation conditioning program is safe and effective for patients with high-risk class 3 thalassemia exhibiting additional comorbidities.
Profiling antibodies to class II HLA in transplant patient sera
Curtis McMurtrey, D Lowe, R Buchli, S Daga, D Royer, A Humphrey, et al.
Human Immunology 75 (2014) 261–270
http://dx.doi.org/10.1016/j.humimm.2013.11.015
Immunizing events including pregnancy, transfusions, and transplantation promote strong alloantibody responses to HLA. Such alloantibodies to HLA preclude organ transplantation, foster hyperacute rejection, and contribute to chronic transplant failure. Diagnostic antibody-screening assays detect alloreactive antibodies, yet key attributes including antibody concentration and isotype remain largely unexplored. The goal here was to provide a detailed profile of allogeneic antibodies to class II HLA. Methodologically, alloantibodies were purified from sensitized patient sera using an HLA-DR11 immunoaffinity column and subsequently categorized. Antibodies to DR11 were found to fix complement, exist at a median serum concentration of 2.3 lg/mL, consist of all isotypes, and isotypes IgG2, IgM, and IgE were elevated. Because multimeric isotypes can confound diagnostic determinations of antibody concentration, IgM and IgA isotypes were removed and DR11-IgG tested alone. Despite removal of multimeric isotypes, patient-to patient antibody concentra-tions did not correlate with MFI values. In conclusion, allogeneic antibody responses to DR11 are comprised of all antibody isotypes at differing proportions, these combined isotypes fix complement at nominal serum concentrations, and enhancements other than the removal of IgM and IgA multimeric isotypes may be required if MFI is to be used as a means of determining anti-HLA serum antibody concentrations in diagnostic clinical assays.
Reduced-intensity conditioning and HLA-matched hemopoietic stem-cell transplantation in patients with chronic granulomatous disease: a prospective multicenter study
Tayfun Güngör, P Teira, M Slatter, G Stussi, P Stepensky, D Moshous, et al.
Lancet 2014; 383: 436–48
http://dx.doi.org/10.1016/S0140-6736(13)62069-3
Background In chronic granulomatous disease allogeneic hemopoietic stem-cell transplantation (HSCT) in adolescents and young adults and patients with high-risk disease is complicated by graft-failure, graft-versus-host disease (GVHD), and transplant-related mortality. We examined the effect of a reduced-intensity conditioning regimen designed to enhance myeloid engraftment and reduce organ toxicity in these patients. Methods This prospective study was done at 16 centers in ten countries worldwide. Patients aged 0–40 years with chronic granulomatous disease were assessed and enrolled at the discretion of individual centers. Reduced-intensity conditioning consisted of high-dose fludarabine (30 mg/m² [infants <9 kg 1∙2 mg/kg]; one dose per day on days –8 to –3), serotherapy (anti-thymocyte globulin [10 mg/kg, one dose per day on days –4 to –1; or thymoglobulin 2·5 mg/kg, one dose per day on days –5 to –3]; or low-dose alemtuzumab [<1 mg/kg on days –8 to –6]), and low-dose (50–72% of myeloablative dose) or targeted busulfan administration (recommended cumulative area under the curve: 45–65 mg/L × h). Busulfan was administered mainly intravenously and exceptionally orally from days –5 to –3. Intravenous busulfan was dosed according to weight-based recommendations and was administered in most centers (ten) twice daily over 4 h. Unmanipulated bone marrow or peripheral blood stem cells from HLA-matched related donors or HLA-9/10 or HLA-10/10 matched unrelated-donors were infused. The primary endpoints were overall survival and event-free survival (EFS), probabilities of overall survival and EFS at 2 years, incidence of acute and chronic GVHD, achievement of at least 90% myeloid donor chimerism, and incidence of graft failure after at least 6 months of follow-up. Findings 56 patients (median age 12∙7 years; IQR 6·8–17·3) with chronic granulomatous disease were enrolled from June 15, 2003, to Dec 15, 2012. 42 patients (75%) had high-risk features (ie, intractable infections and autoinflammation), 25 (45%) were adolescents and young adults (age 14–39 years). 21 HLA-matched related-donor and 35 HLA-matched unrelated-donor transplants were done. Median time to engraftment was 19 days (IQR 16–22) for neutrophils and 21 days (IQR 16–25) for platelets. At median follow-up of 21 months (IQR 13–35) overall survival was 93% (52 of 56) and EFS was 89% (50 of 56). The 2-year probability of overall survival was 96% (95% CI 86∙46–99∙09) and of EFS was 91% (79∙78–96∙17). Graft-failure occurred in 5% (three of 56) of patients. The cumulative incidence of acute GVHD of grade III–IV was 4% (two of 56) and of chronic graft-versus-host disease was 7% (four of 56). Stable (≥90%) myeloid donor chimerism was documented in 52 (93%) surviving patients. Interpretation This reduced-intensity conditioning regimen is safe and efficacious in high-risk patients with chronic granulomatous disease.
Refinement of the Definition of Permissible HLA-DPB1 Mismatches with Predicted Indirectly ReCognizable HLA-DPB1 Epitopes
Kirsten A. Thus, MTA Ruizendaal, TA de Hoop, Eric Borst, et al.
Biol Blood Marrow Transplant 20 (2014) 1705e1710
http://dx.doi.org/10.1016/j.bbmt.2014.06.026
Hematopoietic stem cell transplantation with HLA-DPB1emismatched donors leads to an increased risk of acute graft-versus-host disease (GVHD). Studies have indicated a prognostic value for classifying HLA-DPB1 mismatches based on T cell epitope (TCE) groups. The aim of this study was to determine the contribution of indirect recognition of HLA-DPe derived epitopes, as determined with the Predicted Indirectly ReCognizable HLA Epitopes (PIRCHE) method. We therefore conducted a retrospective single-center analysis on 80 patients transplanted with a 10/10 matched unrelated donor that was HLA-DPB1 mismatched. HLADPB1 mismatches that were classified as GVH nonpermissive by the TCE algorithm correlated to higher numbers of HLA class I as well as HLA class II presented PIRCHE (PIRCHE-I and -II) compared with permissive or host-versus-graft nonpermissive mismatches. Patients with acute GVHD grades II to IV presented significantly higher numbers of PIRCHE-I compared with patients without acute GVHD (P < .05). Patients were divided into 2 groups based on the presence or absence of PIRCHE. Patients with PIRCHE-I or -II have an increased hazard of acute GVHD when compared with patients without PIRCHE-I or -II (hazard ratio [HR], 3.19; 95% confidence interval [CI], 1.10 to 9.19; P <.05; and HR, 4.07; 95% CI, .97 to 17.19; P < .06, respectively). Patients classified as having an HLA-DPB1 permissive mismatch by the TCE model had an increased risk of acute GVHD when comparing presence of PIRCHE-I with absence of PIRCHE-I (HR, 2.96; 95% CI, .84 to 10.39; P < .09). We therefore conclude that the data presented in this study describe an attractive and feasible possibility to better select permissible HLA-DPB1 mismatches by including both a direct and an indirect recognition model.
Treosulfan-Thiotepa-FludarabineeBased Conditioning Regimen for
Allogeneic Transplantation in Patients with Thalassemia Major: A
Single-Center Experience from North India
Dharma Choudhary, SK Sharma, N Gupta,…, Satyendra Katewa
Biol Blood Marrow Transplant 19 (2013) 492e503
http://dx.doi.org/10.1016/j.bbmt.2012.11.007
Hematopoietic stem cell transplantation (HSCT) is the definite treatment
for patients with thalassemia major. A busulfan (Bu) and cyclophosphamide
(Cy)ebased regimen has been the standard myeloablative chemotherapy,
but it is associated with higher treatment-related toxicity, particularly in
patients classified as high risk by the Pesaro criteria. Treosulfan-based
conditioning regimens have been found to be equally effective and less
toxic. Consequently, we analyzed the safety and efficacy of treosulfan/
thiotepa/fludarabine (treo/thio/flu)-based conditioning regimens for
allogeneic HSCT in patients with thalassemia major between February
2010 and September 2012. We compared those results retrospectively
with results in patients who underwent previous HSCT with a Bu/Cy/
antithymocyte globulin (ATG)ebased conditioning regimen. A treo/thio/
flu-based conditioning regimen was used in 28 consecutive patients with
thalassemia major. The median patient age was 9.7 years (range, 2-18
years), and the mean CD34+ stem cell dose was 6.18 x 106/kg. Neutrophil
and platelet engraftment occurred at a median of 15 days (range, 12-23
days) and 21 days (range, 14-34 days), respectively. Three patients
developed veno-occlusive disease, 4 patients developed acute graft
versus-host disease (GVHD), and 2 patients had chronic GVHD. Treatment-
related mortality (TRM) was 21.4%. Two patients experienced secondary
graft rejection. We compared these results with results in patients who
underwent previous HSCT using a Bu/Cy/ATG-based conditioning regimen.
Twelve patients were treated with this protocol, at a median age of 7.2
years (range, 2-11 years). One patient had moderate veno-occlusive disease,
2 patients developed acute GVHD, 2 patients had chronic GVHD, and 2
patients experienced graft rejection. There was no TRM in this group. We
found no significant differences between the 2 groups (treo/thio/flu vs Bu/
Cy/ATG) in terms of the incidence of acute GVHD, chronic GVHD, TRM,
and graft failure, although a trend toward higher TRM was seen with the
treo/thio/flu regimen.
Graft-versus-Host Disease
James L.M. Ferrara, John E. Levine, Pavan Reddy, and Ernst Holler
Lancet. 2009 May 2; 373(9674): 1550–1561
http:dx.doi.org:/10.1016/S0140-6736(09)60237-3
The number of allogeneic hematopoietic cell transplantations (HCT)
continues to increase with more than 25,000 allogeneic transplantations
performed annually. The graft-versus leukemia/ tumor (GVL) effect during
allogeneic HCT effectively eradicates many hematological malignancies.
The development of novel strategies that use donor leukocyte infusions,
non-myeloablative conditioning and umbilical cord blood (UCB)
transplantation have helped expand the indications for allogeneic HCT
over the last several years, especially among older patients. Improvements
in infectious prophylaxis, immunosuppressive medications, supportive care
and DNA-based tissue typing have also contributed to improved outcomes
after allogeneic HCT. Yet the major complication of allogeneic HCT, graft-
versus-host disease (GVHD), remains lethal and limits the use of this
important therapy. Given current trends, the number of transplants from
unrelated donors is expected to double within the next five years,
significantly increasing the population of patients with GVHD. In this
seminar we review advances made in identifying the genetic risk
factors and pathophysiology of this major HCT complication, as well
as its prevention, diagnosis and treatment.
Non-HLA Genetics—Despite HLA identity between a patient and donor,
approximately 40% of patients receiving HLA-identical grafts develop
acute GVHD due to genetic differences that lie outside the HLA loci,
or “minor” histocompatibility antigens (HA). Some minor HAs, such as HY
and HA-3, are expressed on all tissues and are targets for both GVHD
and GVL. Other minor HAs, such as HA-1 and HA-2, are expressed most
abundantly on hematopoietic cells (including leukemic cells) and may
therefore induce a greater GVL effect with less GVHD. Polymorphisms
in both donors and recipients for cytokines that are involved in the
classical `cytokine storm’ of GVHD have been implicated as risk factors
for GVHD. Tumor Necrosis Factor (TNF)-α, Interleukin 10 (IL-10),
Interferon-γ (IFNγ) variants have correlated with GVHD in some, but
not all, studies. Genetic polymorphisms of proteins involved in innate
immunity, such as nucleotide oligomerization domain 2 and Keratin 18
receptors, have also been associated with GVHD.
Future strategies to identify the best possible transplant donor will
probably incorporate both HLA and non-HLA genetic factors. Skin is most
commonly affected and is usually the first organ involved, often coinciding
with engraftment of donor cells. The characteristic maculopapular rash is
pruritic and can spread throughout the body, sparing the scalp. In severe
cases the skin may blister and ulcerate. Apoptosis at the base of epidermal
rete pegs is a characteristic pathologic finding. Other features include
dyskeratosis, exocytosis of lymphocytes, satellite lymphocytes adjacent
to dyskeratotic epidermal keratinocytes, and a perivascular lymphocytic
infiltration in the dermis.
Gastrointestinal tract involvement of acute GVHD usually presents as
diarrhea but may also include vomiting, anorexia, and/or abdominal pain
when severe. The diarrhea of GVHD is secretory and often voluminous
(greater than two liters per day). Bleeding, which carries a poor prognosis,
occurs as a result of mucosal ulceration but patchy involvement of the
mucosa often leads to a normal appearance on endoscopy.
The incidence of the severity of acute GVHD is determined by the extent
of involvement of three principal target organs. The overall grades are
classified as I (mild), II (moderate), III (severe) and IV (very severe). Severe
GVHD carries a poor prognosis, with 25% long term survival for grade III and
5% for grade IV. The incidence of acute GVHD is directly related to the
degree of mismatch between HLA proteins and ranges from 35-45% in
recipients of full matched sibling donor grafts to 60-80% in recipients of
one-antigen HLA mismatched unrelated donor grafts. The same degree
of mismatch causes less GVHD using UCB grafts and incidence of acute
GVHD is lower following the transplant of partially matched UCB units
and ranges from 35-65%.
Two important principles are important to consider regarding the
pathophysiology of acute GVHD. First, acute GVHD reflects exaggerated
but normal inflammatory mechanisms mediated by donor lymphocytes infused
into the recipient where they function appropriately, given the foreign
environment they encounter. Second, the recipient tissues that stimulate
donor lymphocytes have usually been damaged by underlying disease,
prior infections, and the transplant conditioning regimen. As
a result, these tissues produce molecules (sometimes referred to as
“danger” signals) that promote the activation and proliferation of donor
immune cells. Based largely on experimental models, the development
of acute GVHD can be conceptualized in three sequential steps or phases:
(1) activation of the APCs; (2) donor T cell activation, proliferation,
differentiation and migration; and (3) target tissue destruction.
Alemtuzumab is a monoclonal antibody that binds CD52, a protein
expressed on a broad spectrum of leukocytes including lymphocytes,
monocytes, and dendritic cells. Its use in GVHD prophylaxis in a
Phase II trial decreased the incidence of acute and chronic GVHD
following reduced intensity transplant.98 In two prospective studies,
patients who received alemtuzumab rather than methotrexate showed
significantly lower rates of acute and chronic GVHD, but experienced
more infectious complications and higher rates of relapse, so that there
was no overall survival benefit. Alemtuzumab may also contribute to
graft failure when used with minimal intensity conditioning regimens.
An alternative strategy to TCD attempted to induce anergy in donor
T cells by ex vivo antibody blockade of co-stimulatory pathways prior
to transplantation. A small study using this approach in haploidentical
HCT recipients was quite encouraging, but has not yet been replicated.
Thus the focus of most prevention strategies remains pharmacological
manipulation of T cells after transplant.
Administration of anti-T cell antibodies in vivo as GVHD prophylaxis
has also been extensively tested. The best studied drugs are anti-
thymocyte globulin (ATG) or antilymphocyte globulin (ALG) preparations.
These sera, which have high titers of polyclonal antibodies, are made
by immunizing animals (horses or rabbits) to thymocytes or lymphocytes,
respectively. A complicating factor in determining the role of these
polyclonal sera in transplantation is the observation that even different
brands of the same class of sera exert different biologic effects. However,
the side effects of ATG/ALG infusions are common across different
preparations and include fever, chills, headache, thrombocytopenia
(from cross-reactivity to platelets), and, infrequently, anaphylaxis. In
retrospective studies, rabbit ATG reduced the incidence of GVHD in
related donor HSCT recipients without appearing to improve survival.
In recipients of unrelated donor HSCT, addition of ALG to standard
GVHD prophylaxis effectively prevented severe GVHD, but did not
result in improved survival because of increased infections. In a long
term follow-up study, however, pretransplant ATG provided significant
protection against extensive chronic GVHD and chronic lung dysfunction.
As allogeneic transplantation becomes an increasingly attractive therapeutic
option, the need for novel approaches to GVHD has accelerated. The
number of patients receiving transplants from unrelated donors is
expected to double in the next five years, significantly increasing
the population of patients with GVHD. The advent of RIC regimens
has reduced transplant-related mortality and lengthened the period
during which acute GVHD may develop (many new cases present up
to day 200) and the need for close monitoring of patients in this period
has increased. Patients have often returned to the care of their primary
hematologists by this time, increasing the need for these physicians to
collaborate with transplant specialists in the management of GVHD and
its complications.
Identification of biomarkers for GVHD with diagnostic (and possibly
prognostic) significance may eventually make the treatment of GVHD
preemptive rather than prophylactic. The use of cellular component therapy,
such as regulatory T cells that have been expanded ex vivo. will also
enter clinical trials in the near future, but the extensive infrastructure
required for such cellular approaches will likely limit their use initially.
Immunomodulatory Effects of Palifermin (Recombinant Human
Keratinocyte Growth Factor) in an SLE-Like Model of Chronic
Graft-Versus-Host Disease
C. A. Ellison, Y. V. Lissitsyn, I. Gheorghiu & J. G. Gartner
Scandinavian Journal of Immunology 2011; 75, 69–76
http://dx.doi.org:/10.1111/j.1365-3083.2011.02628.x
Keratinocyte growth factor (KGF) promotes epithelial cell proliferation
and survival. Recombinant human KGF, also known as palifermin, protects
epithelial cells from injury induced by chemicals, irradiation and acute murine
graft versus-host disease (GVHD). Findings from our studies and others
have shown that palifermin also has immunomodulatory properties. In a
model of acute GVHD, we showed that it shifts the immune response
from one in which Th1 cytokines dominate to mixed Th1 and Th2 cytokine
profile. Using the DBA⁄ 2 fi (C57BL ⁄ 6 · DBA⁄ 2)F1-hybrid model of chronic,
systemic lupus erythematosus-like GVHD, we showed that palifermin
treatment is associated with higher levels of Th2 cytokines, the production
of anti-nuclear antibodies, cryoglobulinemia and the development of more
severe pathological changes in the kidney. The aim of our current study
was to gain a better understanding of the immunobiology of KGF by
further characterizing the palifermin-mediated effects in this model of
chronic GVHD. Because the pathological changes we observed resemble
those seen in thymic stromal lymphopoietin (TSLP) transgenic mice, we
had originally hypothesized that palifermin might augment TSLP levels.
Surprisingly, we did not observe an increase in thymic
TSLP mRNA expression in palifermin-treated recipients. We did, however,
observe some differences in the percentages of CD4+CD25+Foxp3+
regulatory T cells in the spleen at some time points in palifermin-treated
recipients. Most importantly, we found that TGFβ levels were higher in
palifermin-treated recipients early in the GVH reaction, raising the
possibility that KGF might indirectly induce the development of fibrosis
and glomerulonephritis through a pathway involving TGFβ.
Keratinocyte growth factor (KGF) is an epithelial cell growth factor that is
produced by both mesenchymal cells and intraepithelial cdT cells. It is
also known as fibroblast growth factor 7. Its receptor, (KGFR⁄FGF7R), an
alternatively spliced form of FGFR2 ⁄ bek, is found on epithelial cells in
the intestine, mammary glands, ovaries and urinary tract, and on
hepatocytes, keratinocytes and alveolar type II cells. Previously, it
was shown that recombinant human KGF, also known as palifermin,
can protect the lung, bladder or intestine from chemical- or irradiation-
induced injury. This has been attributed to the ability of KGF to reduce
oxidative damage and enhance DNA repair.
Our own studies have provided a better understanding of the immuno-
biological properties of KGF in pathologically distinct models of systemic
disease driven by intense immunological and inflammatory responses.
The acute GVHD that develops in the C57BL ⁄ 6 fi (C57BL ⁄ 6 · DBA⁄ 2)F1-
hybrid model is characterized by the activation of alloreactive donor T cells,
the production of Th1 cytokines and tissue injury in the skin, gastrointestinal
tract, liver, thymus and lung, where epithelia are present. Injury to the
intestinal mucosa permits the translocation of endotoxin into the system,
which, if untreated, leads to the development of endotoxemic shock. We
showed that palifermin treatment protects recipients from epithelial
cell injury, endotoxemia and morbidity in GVH mice. Palifermin also
shifts the immune response away from one that is predominated by Th1
cytokines towards a profile of mixed Th1 and Th2 cytokines, with a
preponderance of Th2 cytokines. The DBA⁄ 2 fi (C57BL ⁄ 6 · DBA⁄ 2)F1-
hybrid model of chronic GVHD is characterized by pathological changes
resembling those seen in systemic lupus erythematosus (SLE). Using this
model, we showed that palifermin treatment augments the production of Th2
cytokines such as IL-4, IL-5 and IL-13 and obviates IFN-c production. Both
untreated and palifermin-treated recipients developed pathological changes
in the kidney, but these changes were more severe in palifermin-treated
recipients. Some of the changes that developed in the palifermin-treated
recipients resemble those seen in thymic stromal lymphopoietin (TSLP)
transgenic mice. These similarities include the presence of ANA in the
sera, the development of cryoglobulinemia and the development of
glomerulonephritis featuring the deposition of immune complexes
consisting of IgG, IgA, IgM and C3 in the mesangium and the glomerular
capillaries. This led us to hypothesize that treating the recipient mice with
palifermin might induce TSLP expression in this model.
In this study, we were interested in determining whether palifermin
treatment was indeed associated with increased TSLP expression.
We were also interested in knowing whether palifermin treatment
changes the percentage of CD4+CD25+FoxP3+ cells in the spleen,
because palifermin treatment has been associated with increased
percentages of CD4+CD25+FoxP3+ cells in other studies including
our own. Lastly, we wished to study the effect of palifermin treatment
on TGFb levels, because this cytokine is known to play a pivotal role
in the development of glomerulonephritis.
We studied the histopathological changes to confirm that the pathological
changes seen in the kidney in this study were the same as those reported
by us previously.We examined kidney sections from both untreated and
palifermin-treated recipients. In these experiments, we were able to
reproduce findings from an earlier study that showed that palifermin-
treated recipients mice in this model of chronic GVHD develop a severe,
extracapillary proliferative glomerular nephritis characterized by epithelial
crescents and hyaline thrombi. These changes were associated with higher
levels of protein in the urine and the development of ascites, presumably
related to the development of nephrotic syndrome, as a consequence
of glomerular injury.

Pathological changes in the kidney. (A) shows a section from a BDF1-hybrid control
mouse that did not receive a graft. (B) shows increased epithelial cellularity within a
glomerulus from an untreated recipient with chronic graft-versus-host disease, on
day 50. No crescents were observed in sections from this group of recipients.
(C and D) show examples of pathological changes observed in kidneys from
palifermin-treated recipients on day 50. Arrows indicate examples of crescentic
glomerulonephritis and the development of protein casts within tubular lumena.
(E and F) show examples of the hyaline thrombi (arrows) seen in the glomeruli
in kidney sections from palifermin-treated recipients on day 50. All sections
were stained with haematoxylin and eosin except for that shown in (F), which
was stained with Masson Trichrome. The concentration of protein measured in
the urine is shown in the lower left corner of each photomicrograph. Original
magnification: ·200 (B–E) and ·400 (A and F).
TGFβ is a highly pleiotropic cytokine with three isoforms, TGFβ1, TGFβ2 and
TGFβ3 . Nearly, all cells have receptors for at least one of these isoforms,
but cells of the immune system primarily express TGFβ1. This cytokine
was implicated in the development of experimental glomerulonephritis in
experiments in which rats were treated with antiserum directed against
TGFβ1. The ability of palifermin to induce TGFβ release and reverse
limited airflow was demonstrated in a mouse model of emphysema. The
authors further showed that palifermin induced the release of TGFβ1
from primary cultures of mouse alveolar type 2 cells. Our results show
that palifermin treatment is associated with a rise in splenic TGFβ levels
during the first month of the GVH reaction. It is possible that by inducing
TGFβ production shortly after transplantation, palifermin treatment is able
to promote the development of the severe, crescentic glomerulonephritis
that we observed at later time points. As such, our findings raise the
possibility that endogenous KGF might play a role in the development
of glomerulonephritis and ⁄ or other autoimmune phenomena associated
with chronic GVHD and ⁄ or SLE.
T cells, murine chronic graft-versus-host disease and autoimmunity
Robert A. Eisenberg, Charles S. Via
Journal of Autoimmunity 39 (2012) 240e247
http://dx.doi.org:/10.1016/j.jaut.2012.05.017
The chronic graft-versus-host disease (cGVHD) in mice is characterized by
the production of autoantibodies and immunopathology characteristic of
systemic lupus erythematosus (lupus). The basic pathogenesis involves
the cognate recognition of foreign MHC class II of host B cells by alloreactive
CD4 T cells from the donor. CD4 T cells of the host are also necessary for
the full maturation of host B cells before the transfer of donor T cells.
CD8 T cells play critical roles as well. Donor CD8 T cells that are highly
cytotoxic can ablate or prevent the lupus syndrome, in part by killing
recipient B cells. Host CD8 T cells can reciprocally downregulate donor
CD8 T cells, and thus prevent them from suppressing the autoimmune
process. Thus, when the donor inoculum contains both CD4 T cells and
CD8 T cells, the resultant syndrome depends on the balance of activities
of these various cell populations. For example, in one cGVHD model
(DBA/2 (C57BL/6xDBA/2)F1, the disease is more severe in females, as
it is in several of the spontaneous mouse models of lupus, as well as in
human disease. The mechanism of this female skewing of disease
appears to depend on the relative inability of CD8 cells of the female host
to downregulate the donor CD4 T cells that drive the autoantibody response.
In general, then, the abnormal CD4 T cell help and the modulating roles
of CD8 T cells seen in cGVHD parallel the participation of T cells in
genetic lupus in mice and human lupus, although these spontaneous
syndromes are presumably not driven by overt alloreactivity.
Systemic lupus erythematosus (SLE) is characterized by a spectrum of auto-
antibodies that targets multiple normal cellular components, particularly
nucleic acids or proteins that are physiologically bound to nucleic acids.
Although SLE is highly diverse in its manifestations, a common theme
is the loss of B cell tolerance to these cellular autoantigens. More than
for any other human condition, several spontaneously arising mouse
models for SLE have been described, beginning with the New Zealand
strains in 1959. These models are largely genetic. In some cases, an
individual gene such as fas or Yaa plays a major role in driving the loss
of tolerance. However, in general the genetic contribution is complex and
involves multiple loci, which are not yet fully defined.
Despite extensive investigations, the failures in immunoregulation that
underlie the genetic SLE models remain poorly understood. It is not known
for sure which B cell tolerance checkpoints are breached in a given model,
and why. The autoantibody response to DNA, Sm, and other autoantigens
resembles the normal response to exogenous antigens: it involves clonal
expansion, somatic mutation, and a pattern of isotype use characteristic of
a T-cell dependent immunization. Thus the cellular dynamics of the response
may be basically normal. Yet the B-cell repertoire is abnormally autoreactive.
In this review we wish to focus more on the role of the T cell in SLE. As
stated above, the loss of B cell tolerance in SLE does appear in general
to require the participation of T cells. Multiple T cells abnormalities have
been described in human and in murine SLE, although in most cases it is
not clear if these are primary or secondary manifestations. Nevertheless,
it is striking how difficult it has been to demonstrate definitively the specificity
of the T cells that provide help for autoantibody production.
The key cellular mechanism in the cGVHD that results in the loss of B cell
tolerance and the production of the autoantibodies typical of SLE is the
cognate interaction of CD4 T cells with an MHC class II determinant on
the B cell surface. A variety of protocols have achieved this interaction.
In general, either the donor/recipient strains are paired in such away
that they only differ at the MHC class II loci, or the CD4 cells are isolated
free of CD8 cells that would recognize MHC class I. If the allorecognition
involves both CD4 T cell interaction with MHC II and CD8 interaction with
MHC I, an acute GVHD occurs, which is immunosuppressive, rather than
immunostimulatory. The DBA/2 (C57BL/6 DBA/2)F1 (B6D2F1) and the
BALB/c (BALB/c A/J)F1 models are exceptions to this rule. The former
has been investigated extensively for a deficiency in CD8 cytotoxic
lymphocytes.
The MHC class II recognition may be at either the I-A or the I-E locus.
However, the autoantibody specificities seen and the degree of immuno-
pathology differ depending on the locus targeted. In one set of experiments,
F1 mice were bred between either B6 or coisogenic bm12 mice and
B10.A(2R) or B10.A(4R) MHC recombinant congenics. The MHC class II
of B6 is I-Ab, while that of bm12 is I-Abm12. These two alleles differ by
only three amino acids, which is sufficient for a full strength MLR (mixed
lymphocyte reaction) between the two strains. Otherwise B6 and bm12
are identical. B10.A(2R) and B10.A(4R) differ only by the expression of
I-E in the former strain, but not in the latter strain. Thus, donor/recipient
combinations could be employed that provided for allogeneic differs only
at I-A, only at I-E, or at both loci.
Results from Busser et al. delineate requirements for this MHC class II
recognition. Utilizing several transgenic mouse strains that express a
more or less constricted CD4 autoreactive repertoire, they showed that
a diverse repertoire was essential to the production of SLE autoantibodies
by MHC II recognition. On the other hand, the non-specific, early polyclonal
B cell activation phase of cGVHD occurred even with a limited CD4 repertoire.
Figure not shown. Chronic GVHD in bm12 C57BL/6 mice. The MHC of the
bm12 donor differs from the MHC of the C57BL/6 recipient just in three
amino acids in the I-A class II molecule. Thus donor CD4 T cells recognize
MHC IIþ B cells as foreign. Donor CD8 T cells see only self MHC I. All T
cells do not express MHC II. Polyclonal activation and specific lupus
autoantibody responses ensue..
Lupus can result from unchecked CD4 T cell cognate help to a polyclonal
population of B cells. CD8 T cells can downregulate this CD4 driven B-cell
hyperactivity through CD8 CTL effectors and can maintain remission,
possibly through memory CD8 T cells. Whether CD8 CTL actually prevent
lupus in normals and fail in lupus prone individuals is not known; however,
data from the P F1 model suggest that therapeutic induction of CD8 CTL
and possibly long term memory cells may be beneficial in preventing or
limiting disease expression. The potential major role played by either
IFNa and IL-21 in both lupus expression and CD8 CTL function remains
to be further defined, but already these cytokines are being targeted in
human or murine lupus.
It is not surprising that the T cells have been shown to have diverse roles in
the autoimmune cGVHD in mice. Donor CD4 T cells drive the host B cell
activation, while host CD4 T cells are required to mature these B cells prior
to their encounter with donor T cells. Donor CD4 T cells also help activate
donor CD8 T cells, which in turn can downregulate or even ablate the
autoimmune response. Donor CD4 T cells license host DC cells, which in
turn can interact with donor CD8 T cells. Host CD8 T cells can suppress
the activity of donor CD8 T cells, and thereby favor the development of
the lupus syndrome. Although the precise mechanisms of T cell participation
in spontaneous lupus are still being defined, it seems reasonable to probe
these syndromes in humans and in mice for T cell mechanism that have
been shown to participate in cGVHD, CD4-B cell interactions almost
certainly are central to the pathogenesis of spontaneous lupus, and they
have been a target of investigation for several decades. If we understood
the peptide specificity of the alloreactive CD4 T cells that drive the formation
of the characteristic lupus autoantibodies, we would have a much clearer
idea where to look for such epitopes in spontaneous disease. Much less
is known about the other T cell activities defined in cGVHD, particularly
those that involve CD8 T cells. This area should invite further detailed
investigation. For example, the striking role of CD8 T cells in the stronger
female disease in the DBA BDF1 model clearly demands that similar
mechanisms be sought for in spontaneous disease.
Understanding Chronic GVHD from Different Angles
Bruce Blazar, Eric S. White, Daniel Couriel
Biol Blood Marrow Transplant 18:S184-S188, 2012
http://dx.doi.org:/10.1016/j.bbmt.2011.10.025
Whereas acute graft-versus-host disease (aGVHD) rates have decreased
with more intensive GVHD preventive agents and use of single and double
umbilical cord blood units as a source of donor cells in adult recipients,
significant chronic GVHD (cGVHD) rates unexpectedly have remained high.
Moreover, granulocyte colony stimulating factor mobilized peripheral blood
stem cell grafts have been associated with an increased overall risk of
cGVHD. As such, cGVHD has emerged as a primary cause of morbidity
and mortality following allogeneic hematopoietic stem cell transplantation.
Progress in developing cGVHD interventional strategies has been hampered
by variable onset and clinical and pathological manifestations of cGVHD, now
better defined by the National Institutes of Health (NIH) consensus conference,
and a dearth of preclinical models that closely mimic the conditions in which
cGVHD is generated and manifested. Although the exact causes of cGVHD
remain unknown, higher antibody levels have been associated with auto-
immunity and implicated in cGVHD. Newly diagnosed patients with
extensive cGVHD had elevated soluble B cell activating factor levels and
anti-double-strand DNA antibodies were found, which was associated with
higher circulating levels of pregerminal center (GC) B cells and post-GC
plasmablasts. B cells from cGVHD patients were hyperresponsive to Toll-like
receptor-9 signaling and have up-regulated CD86 levels.
By using a Cy and low doses of donor T cells, aGVHD was avoided and
cGVHD with BO favored. Histologic changes were similar to the findings in
human cGVHD with peribronchiolar and perivascular cuffing and infiltration
of the airway epithelium. The liver had inflammation and lymphocytic
infiltration, along with collagen deposition. The parotid and submandibular
salivary glands displayed lymphocytic infiltrates in both the bone marrow
and cGVHD groups, likely because of transplantation conditioning.
Treatment of steroid refractory cGVHD patients with rituximab, a B cell–
depleting anti-CD20 monoclonal antibody, has shown a beneficial role in
resolution of the autoimmune disorders such as systemic lupus erythmatosus
and rheumatoid arthritis, andcGVHD, with overall response rates of 29%
to 36% for oral, hepatic, gastrointestinal, and lung cGVHD, and 60% for
cutaneous cGVHD in aggregate data from multiple trials. Thus, we recently
undertook studies to identify the presence of CD41 T helper cells and B2201
B cells in the airways of mice that had BO, tissue-specific antibodies from sera,
and alloantibody deposition in the lung and liver of cGVHD recipients. cGVHD
development was associated with IgG2c deposition in the lung and liver,
abrogated if the donor bone marrow was deficient in mature B cells or
incapable of producing antihost reactive IgG. Robust GC formation was
seen in mice with cGVHD. Alleviation of symptoms in mice that received
B cell–deficient bone marrow confirms the requirement of B cells for lung
dysfunction and inflammation and fibrosis in the lung and liver.
Given a role for IgG antibodies, allo- or auto-Ab binding to the cGVHD organs
could enable tissue destruction or the pathology could be defined by the
specific function of these secreted antibodies. Pathogenic antibody production
therefore is likely to be an important inducer of cGVHD, and targeting this
specific function of the B cells is an attractive strategy for cGVHD. Because
GC B cells display lower susceptibility to rituximab-mediated clearance, probably
because they reside in a nonoptimal environment for antibody-based depletion,
our observation that GC B cells are critical to the development of cGVHD
suggests that agents that are more effective at disrupting the GC might be
more clinically useful. Treatment with LTbR-Ig, a fusion protein that blocks
interactions between LTbR and its ligands, had a direct effect on the
symptoms of cGVHD, at least in part by blocking GC formation and suggest
that LTbR-Ig could be a potential clinical interventional strategy for prevention
and therapy of cGVHD.
Fibrosis is the end result of a number of inflammatory and other injurious events,
resulting in replacement of normal tissue with a dense extracellular matrix (ECM)
scar composed primarily of collagens. While some degree of tissue fibrosis is
considered protective (e.g. in the setting of cutaneous wound healing),
exaggerated or unrelenting ECM deposition with replacement of the normal
tissue architecture is considered pathologic. Fibroproliferative disorders as
a class involving multiple organs (e.g. cGVHD following hematopoietic stem
cell transplant [affecting up to 30% of recipients surviving more than 100 days,
scleroderma [estimated to affect 70,000 in the US], idiopathic pulmonary fibrosis
[estimated to affect 200,000 in the US], hepatic cirrhosis [estimated to affect
up to 400,000 in the US], and renal fibrosis due to diabetic nephropathy and
other causes [estimated to affect over 400,000 in the US]) are a major cause
of morbidity and mortality. Combined, these disorders alone are conservatively
estimated to affect approximately 1 in 300 persons in the United States. When
coupled with a host of other disorders in which tissue fibrosis contributes to
morbidity (e.g. fibroproliferative acute respiratory distress syndrome,
hypersensitivity pneumonitis, solid organ transplant rejection), that estimate
is likely to be much greater.
Wound healing occurs by a highly orchestrated, complex process that has
been well defined. In general, wound repair occurs in 4 stages which overlap
considerably: clotting/coagulation, inflammation, fibroproliferation, and tissue
remodeling. The initial injury leads to a local disruption of epithelial and
endothelial barriers resulting in the elaboration of inflammatory mediators and
extravasation of cells and plasma proteins that serve to achieve hemostasis
and provide a provisional fibrin-rich matrix for the influx of inflammatory and
other reparative cells. Simultaneously, platelet degranulation provides a local
“boost” of vasodilators, growth factors, and ECM proteins that aid in the wound
healing response. Inflammatory cell influx occurs next, with polymorphonuclear
leukocytes (PMNs) arriving first. Following PMN degranulation, mononuclear
cells (macrophages and lymphocytes) arrive next and, along with PMN derived
products, sterilize and remove foreign materials from the wound. This process
also results in the elaboration of cytokines and chemokines designed to
augment the inflammatory response, to promote angiogenesis (allowing for
enhanced nutrient and oxygen delivery to the wound bed), and to recruit
fibroblasts to the wound bed. Fibroblast recruitment and transdifferentiation to
myofibroblasts (or recruitment of already-differentiated myofibroblasts or
fibroblast precursors; this point is still controversial) marks the fibroproliferative
stage, with the result being the elaboration of ECM proteins (collagens,
fibronectins) to repair the tissue defect.
Vorinostat plus tacrolimus and mycophenolate to prevent graft-versus-host
disease after related-donor reduced-intensity conditioning allogeneic
hemopoietic stem-cell transplantation: a phase 1/2 trial
Sung Won Choi, T Braun, L Chang, JLM Ferrara, A Pawarode, et al.
Lancet Oncol 2014; 15: 87–95
http://dx.doi.org/10.1016/S1470-2045(13)70512-6
Background Acute graft-versus-host disease (GVHD) remains a barrier to more
widespread application of allogeneic hemopoietic stem-cell transplantation.
Vorinostat is an inhibitor of histone deacetylases and was shown to attenuate
GVHD in preclinical models. We aimed to study the safety and activity of
vorinostat, in combination with standard immunoprophylaxis, for prevention of
GVHD in patients undergoing related-donor reduced-intensity conditioning
hemopoietic stem-cell transplantation. Methods Between March 31, 2009,
and Feb 8, 2013, we did a prospective, single-arm, phase 1/2 study at two
centers in the USA. We recruited adults (aged ≥18 years) with high-risk
hematological malignant diseases who were candidates for reduced-intensity
conditioning hemopoietic stem-cell transplantation and had an available 8/8
or 7/8 HLA matched related donor. All patients received a conditioning regimen
of fl udarabine (40 mg/m² daily for 4 days) and busulfan (3·2 mg/kg daily for
2 days) and GVHD immunoprophylaxis of mycophenolate mofetil (1 g three
times a day, days 0–28) and tacrolimus (0·03 mg/kg a day, titrated to a goal
level of 8–12 ng/mL, starting day –3 until day 180). Vorinostat (either 100 mg
or 200 mg, twice a day) was initiated 10 days before haemopoietic stem-cell
transplantation until day 100. The primary endpoint was the cumulative
incidence of grade 2–4 acute GVHD by day 100. This trial is registered with
ClinicalTrials.gov, number NCT00810602.
Findings 50 patients were assessable for both toxic effects and response;
eight additional patients were included in the analysis of toxic effects. All
patients engrafted neutrophils and platelets at expected times after
hemopoietic stem-cell transplantation. The cumulative incidence of grade
2–4 acute GVHD by day 100 was 22% (95% CI 13–36). The most common
non-hematological adverse events included electrolyte disturbances (n=15),
hyperglycemia (11), infections (six), mucositis (four), and increased activity
of liver enzymes (three). Non-symptomatic thrombocytopenia after
engraftment was the most common hematological grade 3–4 adverse
event (nine) but was transient and all cases resolved swiftly.
Interpretation Administration of vorinostat in combination with standard
GVHD prophylaxis after related-donor reduced-intensity conditioning
hemopoietic stem-cell transplantation is safe and is associated with a
lower than expected incidence of severe acute GVHD. Future studies
are needed to assess the effect of vorinostat for prevention of GVHD in
broader settings of hemopoietic stem-cell transplantation.
Read Full Post »

 References in https://www.wjnm.org/article.asp?issn=1450-1147;year=2019;volume=18;issue=1;spage=8;epage=12;aulast=Hertz
References in https://www.wjnm.org/article.asp?issn=1450-1147;year=2019;volume=18;issue=1;spage=8;epage=12;aulast=Hertz











 Article Info
Article Info


![Deep imaging of [agr]-catulin-GFP+ HSCs in digitally reconstructed bone marrow.](https://i0.wp.com/www.nature.com/nature/journal/v526/n7571/carousel/nature15250-f1.jpg?resize=210%2C200)
![[agr]-catulin-GFP expression among haematopoietic cells is highly restricted to HSCs.](https://i0.wp.com/www.nature.com/nature/journal/v526/n7571/images/nature15250-sf3.jpg?w=500)




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
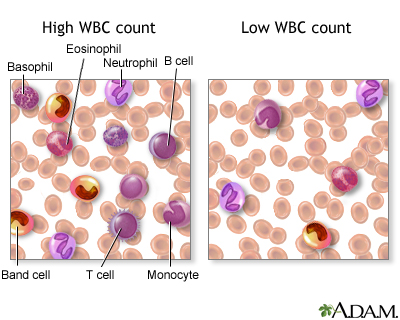{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}