Hematological Cancer Classification
Author and Curator: Larry H. Bernstein, MD, FCAP
Introduction to leukemias and lymphomas
2.4.1 Ontogenesis of the blood elements: hematopoiesis
http://www.britannica.com/EBchecked/topic/69747/blood-cell-formation
Blood cells are divided into three groups: the red blood cells (erythrocytes), the white blood cells (leukocytes), and the blood platelets (thrombocytes). The white blood cells are subdivided into three broad groups: granulocytes, lymphocytes, and monocytes.
Blood cells do not originate in the bloodstream itself but in specific blood-forming organs, notably the marrow of certain bones. In the human adult, the bone marrow produces all of the red blood cells, 60–70 percent of the white cells (i.e., the granulocytes), and all of the platelets. The lymphatic tissues, particularly the thymus, the spleen, and the lymph nodes, produce the lymphocytes (comprising 20–30 percent of the white cells). The reticuloendothelial tissues of the spleen, liver, lymph nodes, and other organs produce the monocytes (4–8 percent of the white cells). The platelets, which are small cellular fragments rather than complete cells, are formed from bits of the cytoplasm of the giant cells (megakaryocytes) of the bone marrow.
In the human embryo, the first site of blood formation is the yolk sac. Later in embryonic life, the liver becomes the most important red blood cell-forming organ, but it is soon succeeded by the bone marrow, which in adult life is the only source of both red blood cells and the granulocytes. Both the red and white blood cells arise through a series of complex, gradual, and successive transformations from primitive stem cells, which have the ability to form any of the precursors of a blood cell. Precursor cells are stem cells that have developed to the stage where they are committed to forming a particular kind of new blood cell.
In a normal adult the red cells of about half a liter (almost one pint) of blood are produced by the bone marrow every week. Almost 1 percent of the body’s red cells are generated each day, and the balance between red cell production and the removal of aging red cells from the circulation is precisely maintained.

http://interactive-biology.com/wp-content/uploads/2012/07/Cells-in-the-Bone-Marrow-1024×747.png
Erythropoiesis
http://www.interactive-biology.com/3969/erythropoiesis-formation-of-red-blood-cells/
Erythropoiesis – Formation of Red Blood Cells
Because of the inability of erythrocytes (red blood cells) to divide to replenish their own numbers, the old ruptured cells must be replaced by totally new cells. They meet their demise because they don’t have the usual specialized intracellular machinery, which controls cell growth and repair, leading to a short life span of 120 days.
This short life span necessitates the process erythropoiesis, which is the formation of red blood cells. All blood cells are formed in the bone marrow. This is the erythrocyte factory, which is soft, highly cellar tissue that fills the internal cavities of bones.
Erythrocyte differentiation takes place in 8 stages. It is the pathway through which an erythrocyte matures from a hemocytoblast into a full-blown erythrocyte. The first seven all take place within the bone marrow. After stage 7 the cell is then released into the bloodstream as a reticulocyte, where it then matures 1-2 days later into an erythrocyte. The stages are as follows:
- Hemocytoblast, which is a pluripotent hematopoietic stem cell
- Common myeloid progenitor, a multipotent stem cell
- Unipotent stem cell
- Pronormoblast
- Basophilic normoblast also called an erythroblast.
- Polychromatophilic normoblast
- Orthochromatic normoblast
- Reticulocyte
These characteristics can be seen during the course of erythrocyte maturation:
- The size of the cell decreases
- The cytoplasm volume increases
- Initially there is a nucleus and as the cell matures the size of the nucleus decreases until it vanishes with the condensation of the chromatin material.
Low oxygen tension stimulates the kidneys to secrete the hormone erythropoietin into the blood, and this hormone stimulates the bone marrow to produce erythrocytes.
Rarely, a malignancy or cancer of erythropoiesis occurs. It is referred to as erythroleukemia. This most likely arises from a common myeloid precursor, and it may occur associated with a myelodysplastic syndrome.
Summary of erythrocyte maturation
White blood cell series: myelopoiesis
http://www.nlm.nih.gov/medlineplus/ency/presentations/100151_3.htm
http://www.nlm.nih.gov/medlineplus/ency/images/ency/fullsize/15220.jpg
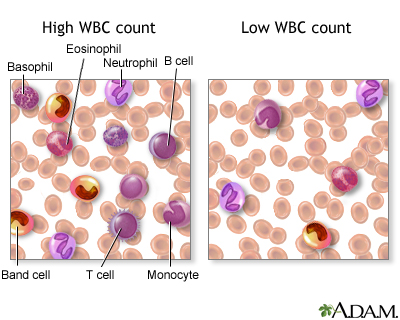
There are various types of white blood cells (WBCs) that normally appear in the blood: neutrophils (polymorphonuclear leukocytes; PMNs), band cells (slightly immature neutrophils), T-type lymphocytes (T cells), B-type lymphocytes (B cells), monocytes, eosinophils, and basophils. T and B-type lymphocytes are indistinguishable from each other in a normal slide preparation. Any infection or acute stress will result in an increased production of WBCs. This usually entails increased numbers of cells and an increase in the percentage of immature cells (mainly band cells) in the blood. This change is referred to as a “shift to the left” People who have had a splenectomy have a persistent mild elevation of WBCs. Drugs that may increase WBC counts include epinephrine, allopurinol, aspirin, chloroform, heparin, quinine, corticosteroids, and triamterene. Drugs that may decrease WBC counts include antibiotics, anticonvulsants, antihistamine, antithyroid drugs, arsenicals, barbiturates, chemotherapeutic agents, diuretics and sulfonamides. (Updated by: David C. Dugdale, III, MD)
https://www.med-ed.virginia.edu/courses/path/innes/nh/wcbmaturation.cfm
Note that the mature forms of the myeloid series (neutrophils, eosinophils, basophils), all have lobed (segmented) nuclei. The degree of lobation increases as the cells mature.
The earliest recognizable myeloid cell is the myeloblast (10-20m dia) with a large round to oval nucleus. There is fine diffuse immature chromatin (without clumping) and a prominant nucleolus.
The cytoplasm is basophilic without granules. Although one may see a small golgi area adjacent to the nucleus, granules are not usually visible by light microscopy. One should not see blast cells in the peripheral blood.

https://www.med-ed.virginia.edu/courses/path/innes/images/nhjpeg/nh%20myeloblast%20x100b.jpeg
The promyelocyte (10-20m) is slightly larger than a blast. Its nucleus, although similar to a myeloblast shows slight chromatin condensation and less prominent nucleoli. The cytoplasm contains striking azurophilic granules or primary granules. These granules contain myeloperoxidase, acid phosphatase, and esterase enzymes. Normally no promyelocytes are seen in the peripheral blood.
At the point in development when secondary granules can be recognized, the cell becomes a myelocyte.

https://www.med-ed.virginia.edu/courses/path/innes/images/nhjpeg/nh%20promyelocyte%20×100%20a.jpeg
Myelocytes (10-18m) are not normally found in the peripheral blood. Nucleoli may not be seen in the late myelocyte. Primary azurophilic granules are still present, but secondary granules predominate. Secondary granules (neut, eos, or baso) first appear adjacent to the nucleus. In neutrophils this is the “dawn” of neutrophilia.
Metamyelocytes (10-18m) have kidney shaped indented nuclei and dense chromatin along the nuclear membrane. The cytoplasm is faintly pink, and they have secondary granules (neutro, eos, or baso). Zero to one percent of the peripheral blood white cells may be metamyelocytes (juveniles).

https://www.med-ed.virginia.edu/courses/path/innes/images/nhjpeg/nh%20metamyelocyte%20×100.jpeg
Bands, slightly smaller than juveniles, are marked by a U-shaped or deeply indented nucleus.

https://www.med-ed.virginia.edu/courses/path/innes/images/nhjpeg/nh%20band%20x100a.jpeg
Segmented (segs) or polymorphonuclear (PMN) leukocytes (average 14 m dia) are distinguished by definite lobation with thin thread-like filaments of chromatin joining the 2-5 lobes. 45-75% of the peripheral blood white cells are segmented neutrophils.
https://www.med-ed.virginia.edu/courses/path/innes/images/nhjpeg/nh%20neutrophil%20×100%20d.jpeg
Thrombocytogenesis
The incredible journey: From megakaryocyte development to platelet formation
Kellie R. Machlus1,2 and Joseph E. Italiano Jr
JCB 2013; 201(6): 785-796
http://dx.doi.org:/10.1083/jcb.201304054
Large progenitor cells in the bone marrow called megakaryocytes (MKs) are the source of platelets. MKs release platelets through a series of fascinating cell biological events. During maturation, they become polyploid and accumulate massive amounts of protein and membrane. Then, in a cytoskeletal-driven process, they extend long branching processes, designated proplatelets, into sinusoidal blood vessels where they undergo fission to release platelets.

http://dm5migu4zj3pb.cloudfront.net/manuscripts/26000/26891/medium/JCI0526891.f4.jpg

http://www.nature.com/nri/journal/v11/n4/images/nri2956-f3.jpg
2.4.2 Classification of hematological malignancies
Practical Diagnosis of Hematologic Disoreders. 4th edition. Vol 2.
Kjeldsberg CR, Ed. ASCP Press. 2006. Chicago, IL.
2.4.2.1 Primary Classification
Acute leukemias
Myelodysplastic syndromes
Acute myeloid leukemia
Acute lymphoblastic leukemia
Myeloproliferative Disorders
Chronic myeloproliferative disorders
Chronic myelogenous leukemia and related disorders
Myelofibrosis, including chronic idiopathic
Polycythemia, including polycythemia rubra vera
Thrombocytosis, including essential thrombocythemia
Chronic lymphoid leukemia and other lymphoid leukemias
Lymphomas
Non-Hodgkin Lymphoma
Hodgkin lymphoma
Lymphoproliferative disorders associated with immunodeficiency
Plasma Cell dyscrasias
Mast cell disease and Histiocytic neoplasms
2.4.2.2 Secondary Classification
2.4.2.3 Nuance – PathologyOutlines
Nat Pernick, Ed.
Leukemia – Acute
Primary references acute leukemia-general AML general AML classification transient abnormal myelopoiesis
Recurrent genetic abnormalities: AML with t(6;9) AML with t(8;21) AML with 11q23 abnormalities AML with inv(16) or t(16;16) AML with Down syndrome AML with FLT3 mutations AML with myelodysplastic related changes AML therapy related APL microgranular variant APL with t(15;17) APL with t(V;17) APL therapy related
AML not otherwise categorized: minimally differentiated (M0) without maturation (M1) with maturation (M2) M3 myelomonocytic monoblastic and monocytic erythroid megakaryoblastic CD13/CD33 negative basophilic myeloid sarcoma acute panmyelosis with myelofibrosis with Philadelphia chromosome with pseudo Chediak-Higashi anomaly hypocellular
ALL: general WHO classification with eosinophilia
PreB ALL: general t(9;22) t(v;11q23) t(1;19) t(5;14) t(12;21) hyperdiploidy hypodiploidy mature B ALL/Burkitt
Other ALL: T ALL ambiguous lineage mixed phenotype
AML and related malignancies
Acute myeloid leukemias with recurrent genetic abnormalities:
- AML with t(8;21)(q22;q22); RUNX1-RUNX1T1
- AML with inv(16)(p13.1;q22) or t(16;16)(p13.1;q22); CBF&beta-MYH11
- Acute promyelocytic leukemia with t(15;17)(q22;q12); PML/RAR&alpha and variants
- AML with t(9;11)(p22;q23); MLLT3-MLL
- AML with t(6;9)(p23;q34); DEK-NUP214
- AML with inv(3)(q21q26.2) or t(3;3)(q21;q26.2); RPN1-EVI1
- AML (megakaryoblastic) with t(1;22)(p13;q13); RBM15-MKL1
- AML with mutated NPM1*
- AML with mutated CEBPA*
* provisional
Acute myeloid leukemia with myelodysplasia related changes
Therapy related acute myeloid leukemia
- Alkylating agent related
- Topoisomerase II inhibitor related (some maybe lymphoid)
Acute myeloid leukemia not otherwise categorized:
- AML minimally differentiated (M0)
- AML without maturation (M1)
- AML with maturation (M2)
- Acute myelomonocytic leukemia (M4)
- Acute monoblastic and monocytic leukemia (M5a, M5b)
- Acute erythroid leukemia (M6)
- Acute megakaryoblastic leukemia (M7)
- Acute basophilic leukemia
- Acute panmyelosis with myelofibrosis
Myeloid Sarcoma
Myeloid proliferations related to Down syndrome:
- Transient abnormal myelopoeisis
- Myeloid leukemia associated with Down syndrome
Blastic plasmacytoid dentritic cell neoplasm:
Acute leukemia of ambiguous lineage:
- Acute undifferentiated leukemia
- Mixed phenotype acute leukemia with t(9;22)(q34;q11.2); BCR-ABL1
- Mixed phenotype acute leukemia with t(v;11q23); MLL rearranged
- Mixed phenotype acute leukemia, B/myeloid, NOS
- Mixed phenotype acute leukemia, T/myeloid, NOS
- Mixed phenotype acute leukemia, NOS, rare types
- Other acute leukemia of ambiguous lineage
- References: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissue (IARC, 2008), Discovery Medicine 2010, eMedicine
Acute lymphocytic leukemia
General
=================================================================
- WHO classification system includes former FAB classifications ALL-L1 and L2
● FAB L3 is now considered Burkitt lymphoma
WHO classification of acute lymphoblastic leukemia
=================================================================
Precursor B lymphoblastic leukemia / lymphoblastic lymphoma:
● ALL with t(9;22)(q34;q11.2); BCR-ABL (Philadelphia chromosome)
● ALL with t(v;11q23) (MLL rearranged)
● ALL with t(1;19)(q23;p13.3); TCF3-PBX1 (E2A-PBX1)
● ALL with t(12;21)(p13;q22); ETV6-RUNX1 (TEL-AML1)
● Hyperdiploid > 50
● Hypodiploid
● t(5;14)(q31;q32); IL3-IGH
Precursor T lymphoblastic leukemia / lymphoma
Additional references
=================================================================
Mixed phenotype acute leukemia (MPAL)
General
=================================================================
- De novo acute leukemia containing separate populations of blasts of more than one lineage (bilineal or bilineage), or a single population of blasts co-expressing antigens of more than one lineage (biphenotypic)Excludes:
● Acute myeloid leukemia (AML) with recurrent translocations t(8;21), t(15;17) or inv(16)
● Leukemias with FGFR1 mutations
● Chronic myelogenous leukemia (CML) in blast crisis
● Myelodysplastic syndrome (MDS)-related AML and therapy-related AML, even if they have MPAL immunophenotypeCriteria for biphenotypic leukemia:
● Score of 2 or more for each of two separate lineages:The European Group for the Immunological Classification of Leukemias (EGIL) scoring system2008 WHO classification of acute leukemias of ambiguous lineage
Prognosis
=================================================================
- Poor, overall survival of 18 months
● Young age, normal karyotype and ALL induction therapy are associated with favorable survival
● Ph+ is a predictor for poor prognosis
● Bone marrow transplantation should be considered in first remission
Major Categories
MPAL with t(9;22)(q34;q11.2); BCR-ABL1
=================================================================
- 20% of all MPAL
● Blasts with t(9;22)(q34;q11.2) translocation or BCR-ABL1 rearrangement (Ph+) without history of CML
● Majority in adults
● High WBC counts● Most of the cases B/myeloid phenotype
● Rare T/myeloid, B and T lineage, or trilineage leukemiasMorphology:
● Many cases show a dimorphic blast population, one resembling myeloblasts and the other lymphoblastsCytogenetic abnormalities:
● Conventional karyotyping for t(9;22), FISH or PCR for BCR-ABL1 translocation
● Additional complex karyotypes
● Ph+ is a poor prognostic factor for MPAL, with a reported median survival of 8 months
● Worse than patients of all other types of MPAL
MPAL with t(v;11q23); MLL rearranged
=================================================================
- Meeting the diagnostic criteria for MPAL with blasts bearing a translocation involving the 11q23 breakpoint (MLL gene)
● MPAL with MLL rearranged rare
● More often seen in children and relatively common in infancy
● High WBC counts
● Poor prognosis
● Dimorphic blast population, with one resembling monoblasts and the other resembling lymphoblasts
● Lymphoblast population often shows a CD19+, CD10- B precursor immunophenotype, frequently CD15+
● Expression of other B markers usually weak
● Translocations involving MLL gene include t(4;11)(q21;q23), t(11;19)(q23;p13), and t(9;11)(p22;q23)
● Cases with chromosome 11q23 deletion should not be classified in this category
B cell acute lymphoblastic leukemia (ALL) / lymphoblastic lymphoma (LBL)
General
=================================================================
- Current 2008 WHO classification: B lymphoblastic leukemia / lymphoma, NOS or B lymphoblastic leukemia / lymphoma with recurrent genetic abnormalities
- See also lymphomas: B cell chapter
- Also called B cell acute lymphoblastic leukemia / lymphoblastic lymphoma, pre B ALL / LBL
- Usually children
- B acute lymphoblastic leukemia presents with pancytopenia due to extensive marrow involvement, stormy onset of symptoms, bone pain due to marrow expansion, hepatosplenomegaly due to neoplastic infiltration, CNS symptoms due to meningeal spread and testicular involvement
- B acute lymphoblastic lymphoma often presents with cutaneous nodules, bone or nodal involvement, < 25% lymphoblasts in bone marrow and peripheral blood; aleukemic cases are usually asymptomatic
- Depending on specific leukemia, arises in either hematopoietic stem cell or B-cell progenitor
- Tumors are derived from pre-germinal center naive B cells with unmutated VH region genes
- Have multiple immunophenotyping aberrancies relative to normal B cell precursors (hematogones); at relapse, 73% show loss of 1+ aberrance and 60% show new aberrancies (Am J Clin Pathol 2007;127:39)
Prognostic features
=================================================================
- Favorable prognosis: age 1-10 years, female, white; preB phenotype, hyperdiploidy>50, t(12,21), WBC count at presentation <50×108/L, non-traumatic tap with no blasts in CNS, rapid response to chemotherapy < 5% blasts on morphology on day 15, remission status after induction <5% blasts on morphology and <0.01% blast on flow or PCR, CD10+
- Intermediate prognosis: hyperdiploidy 47-50, diploid, 6q- and rearrangements of 8q24
- Unfavorable prognosis: under age 1 (usually have 11q23 translocations) or over age 10; t(9;22) (but not if age 59+ years, Am J Clin Pathol 2002;117:716); male, > 50×108/L WBC at presentation, hypodiploidy, near tetraploidy, 17p- and MLL rearrangements t(v;11q23); CD10-; non-traumatic tap with > 5% blasts or traumatic tap (7%); also increased microvessel staining using CD105 in children (Leuk Res 2007;31:1741), MDR1 expression in children (Oncol Rep 2004;12:1201) and adults (Blood 2002;100:974), 25%+ blasts on morphology on day 15, remission status after induction ≥ 5% blasts on morphology and ≥ 0.1% blasts on flow or PCR
Case reports
=================================================================
- 12 month old girl and 13 month old boy with mature phenotype but no translocations (Arch Pathol Lab Med 2003;127:1340)
- 56 year old man with ALL arising from follicular lymphoma (Arch Pathol Lab Med 2002;126:997)
- 76 year old man with basal cell carcinoma (Diagn Pathol 2007;2:32)
- With hemophagocytic lymphohistiocytosis (Pediatr Blood Cancer 2008;50:381)
Treatment
================================================================
- Chemotherapy cures more children than adults; adolescents benefit from intensive regimens (Hematology Am Soc Hematol Educ Program 2005:123)
Micro description
=================================================================
- Bone marrow smears: small to intermediate blast-like cells with scant, variably basophilic cytoplasm, round / oval or convoluted nuclei, fine chromatin and indistinct nucleoli; frequent mitotic figures; may have “starry sky” appearance similar to Burkitt lymphoma; may have large lymphoblasts with 1-4 prominent nucleoli resembling myeloblasts; usually no sclerosis
- Bone marrow biopsy: usually markedly hypercellular with reduction of trilinear maturation; cells have minimal cytoplasm, medium sized nuclei that are often convoluted, moderately dense chromatin and indistinct nucleoli, brisk mitotic activity
- Other tissues: may have “starry sky” appearance similar to Burkitt lymphoma; collagen dissection, periadipocyte growth pattern and single cell linear filing
Chronic Leukemia
Chronic Myeloid Neoplasms
Myelodysplastic syndromes (MDS): general, WHO classification, childhood, refractory anemia, refractory anemia with ringed sideroblasts, refractory cytopenia with multilineage dysplasia, refractory anemia with excess blasts, 5q-syndrome, therapy related, unclassified, arsenic toxicity
Myeloproliferative neoplasms (MPN): general, WHO classification, chronic eosinophilic leukemia, chronic myelogenous leukemia, chronic neutrophilic leukemia, essential thrombocythemia, hypereosinophilic syndrome, mast cell disease, polycythemia vera, primary myelofibrosis, unclassifiable
MDS/MPN: general, WHO classification, atypical CML, chronic myelomonocytic leukemia (CMML), chronic myelomonocytic leukemia with eosinophilia, juvenile myelomonocytic leukemia, unclassifiable
Myeloid neoplasms associated with eosinophilia and abnormalities of PDGFRA, PDGFRB, or FGFR1: PDGFRA, PDGFRB, FGFR1
Miscellaneous: transient myeloproliferative disorder of DownÃs syndrome
Lymphoma and plasma cell neoplasms
Lymph nodes: normal development-general B cells T cells NK cells normal histology grossing lymph nodes features to report
Molecular testing: theory FISH northern blot PCR southern blot
Non-Hodgkin lymphoma: general cytogenetics staging staging-pediatric morphologic clues hemophagocytic syndrome chemotherapeutic atypia
B cell disorders: general post-rituximab bone marrow biopsy classification-historical WHO classification
B cell lymphoma subtypes: age-related EBV-associated ALK positive large cell Burkitt unclassifiable-intermediate between Burkitt and diffuse large B cell lymphoma CLL
diffuse large B cell: diffuse-NOS CD5+ T cell / histiocyte rich primary cutaneous-general primary cutaneous-leg primary sites-other
follicular: general childhood cutaneous GI
hairy cell leukemia HCL variant intravascular large B cell lymphomatoid granulomatosis lymphoplasmacytic mantle cell-classic mantle cell-blastoid marginal zone-general marginal zone-MALT MALT-primary sites marginal zone-nodal mediastinal (thymic) plasmablastic pre B lymphoblastic leukemia/lymphoma primary effusion prolymphocytic leukemia pyothorax associated SLL splenic marginal zone splenic lymphoma with villous lymphocytes
Plasma cell neoplasms: general myeloma plasmacytoma heavy chain disease primary amyloidosis MGUS Osteosclerotic myeloma (POEMS) cryoglobulinemia
T/NK cell disorders: general WHO classification adult T cell aggressive NK cell leukemia anaplastic large cell ALK+ ALK- angioimmunoblastic T cell blastic plasmacytoid chronic lymphoproliferative disorders of NK cells cutaneous CD4+ small/medium sized T cell lymphoma cutaneous CD30 positive T cell lymphoproliferative disorders cutaneous gamma delta T cell lymphoma enteropathy epidermotropic CD8+ T cell lymphoma hepatosplenic indolent T cell proliferations mycosis fungoides NK/T cell lymphoma-nasal type nodal CD8+ cytotoxic T cell nonB nonT lymphoblastic peripheral T cell lymphoma, NOS primary effusion lymphoma Sezary syndrome staging subcutaneous panniculitis-like T cell large granular lymphocytic leukemia T cell lymphoblastic leukemia/lymphoma T cell prolymphocytic leukemia
Hodgkin lymphoma: general/staging classic lymphocyte depleted lymphocyte rich classical mixed cellularity nodular lymphocyte predominant nodular sclerosis
Post-transplantation: general WHO classification plasmacytic hyperplasia/IM-like lesions polymorphic B cell lymphoproliferative disorders monomorphic B cell lymphoproliferative disorders other graft versus host disease
Other: AIDS associated-general AIDS associated-examples EBV+ T cell lymphoproliferative disorders of childhood primary immune disorders related
Myeloproliferative neoplasms (MPN)
WHO 2008 – Myeloproliferative neoplasms (MPN)
General
=================================================================
- Chronic myelogenous leukemia
● Polycythemia vera
● Essential thrombocythemia
● Primary myelofibrosis
● Chronic neutrophilic leukemia
● Chronic eosinophilic leukemia, not otherwise categorized
● Mast cell disease
● MPNs, unclassifiable
WHO 2001 – Chronic myeloproliferative diseases
Definition
=================================================================
- Chronic myelogenous leukemia (Philadelphia chromosome, t(9;22)(q34;q11), BCR-ABL positive)
● Chronic neutrophilic leukemia
● Chronic eosinophilic leukemia (and the hypereosinophilic syndrome)
● Polycythemia vera
● Chronic idiopathic myelofibrosis (with extramedullary hematopoiesis)
● Essential thrombocythemia
● Chronic myeloproliferative disease, unclassifiable
Additional references
=================================================================
The World Health Organization (WHO) classification of the myeloid neoplasms James W. Vardiman, Nancy Lee Harris, and Richard D. Brunning
Blood 2002; 100(7) http://dx.doi.org/10.1182/blood-2002-04-1199
Lymphoma – Non B cell neoplasms
T/NK cell disorders/WHO classification (2008)
Principles of classification
=================================================================
- Based on all available information (morphology, immunophenotype, genetics, clinical)
● No one antigenic marker is specific for any neoplasm (except ALK1)
● Immune profiling less helpful in subclassification of T cell lymphomas then B cell lymphomas
● Certain antigens commonly associated with specific disease entities but not entirely disease specific
● CD30: common in anaplastic large cell lymphoma but also classic Hodgkin lymphoma and other B and T cell lymphomas
● CD56: characteristic for nasal NK/T cell lymphoma, but also other T cell neoplasms and plasma cell disorders
● Variation of immunophenotype within a given disease (hepatosplenic T cell lymphoma: usually γδ but some are αβ)
● Recurrent genetic alterations have been identified for many B cell lymphomas but not for most T cell lymphomas
● No attempt to stratify lymphoid malignancies by grade
● Recognize the existence of grey zone lymphomas
● This multiparameter approach has been validated in international studies as highly reproducible
WHO 2008 classification of tumors of hematopoietic and lymphoid tissues (T/NK)
=================================================================
Precursor T-lymphoid neoplasms
● T lymphoblastic leukemia/lymphoma, 9837/3
Mature T cell and NK cell neoplasms
● T cell prolymphocytic leukemia, 9834/3
● T cell large granular lymphocytic leukemia, 9831/3
● Chronic lymphoproliferative disorder of NK cells, 9831/3
● Aggressive NK cell leukemia, 9948/3
● Systemic EBV-positive T cell lymphoproliferative disease of childhood, 9724/3
● Hydroa vacciniforme-like lymphoma, 9725/3
● Adult T cell leukemia/lymphoma, 9827/3
● Extranodal NK/T cell lymphoma, nasal type, 9719/3
● Enteropathy-associated T cell lymphoma, 9717/3
● Hepatosplenic T cell lymphoma, 9716/3
● Subcutaneous panniculitis-like T cell lymphoma, 9708/3
● Mycosis fungoides, 9700/3
● Sézary syndrome, 9701/3
● Primary cutaneous CD30-positive T cell lymphoproliferative disorders
● Lymphomatoid papulosis, 9718/1
● Primary cutaneous anaplastic large cell lymphoma, 9718/3
● Primary cutaneous gamma-delta T cell lymphoma, 9726/3
● Primary cutaneous CD8-positive aggressive epidermotropic cytotoxic T cell lymphoma, 9709/3
● Primary cutaneous CD4-positive small/medium T cell lymphoma, 9709/3
● Peripheral T cell lymphoma, NOS, 9702/3
● Angioimmunoblastic T cell lymphoma, 9705/3
● Anaplastic large cell lymphoma, ALK-positive, 9714/3
● Anaplastic large cell lymphoma, ALK-negative, 9702/3
Chronic Lymphocytic Leukemia
Chronic Lymphocytic Leukemia Staging
Author: Sandy D Kotiah, MD; Chief Editor: Jules E Harris, MD
Medscape Sep 6, 2013
http://emedicine.medscape.com/article/2006578-overview
General considerations in the staging of chronic lymphocytic leukemia (CLL) and the revised Rai (United States) and Binet (Europe) staging systems for CLL are provided below.[1, 2, 3]
See Chronic Leukemias: 4 Cancers to Differentiate, a Critical Images slideshow, to help detect chronic leukemias and determine the specific type present.
General considerations
- CLL and small lymphocytic lymphoma (SLL) are different manifestations of the same disease; SLL is diagnosed when the disease is mainly nodal, and CLL is diagnosed when the disease is seen in the blood and bone marrow
- CLL is diagnosed by > 5000 monoclonal lymphocytes/mm3 for longer than 3mo; the bone marrow usually has more than 30% monoclonal lymphocytes and is either normocellular or hypercellular
- Monoclonal B lymphocytosis is a precursor form of CLL that is defined by a monoclonal B cell lymphocytosis < 5000 monoclonal lymphocytes/mm3; all lymph nodes smaller than 1.5 cm; no anemia; and no thrombocytopenia
Revised Rai staging system (United States)
Low risk (formerly stage 0)[1] :
- Lymphocytosis, lymphocytes in blood > 15000/mcL, and > 40% lymphocytes in the bone marrow
Intermediate risk (formerly stages I and II):
- Lymphocytosis as in low risk with enlarged node(s) in any site, or splenomegaly or hepatomegaly or both
High risk (formerly stages III and IV):
- Lymphocytosis as in low risk and intermediate risk with disease-related anemia (hemoglobin level < 11.0 g/dL or hematocrit < 33%) or platelets < 100,000/mcL
Binet staging system (Europe)
Stage A:
- Hemoglobin ≥ 10 g/dL, platelets ≥ 100,000/mm3, and < 3 enlarged areas
Stage B:
- Hemoglobin ≥ 10 g/dL, platelets ≥ 100,000/mm3, and ≥ 3 enlarged areas
Stage C:
- Hemoglobin < 10 g/dL, platelets < 100,000/mm3, and any number of enlarged areas
Read Full Post »









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}