Metastatic Disease (4.3)
Writer and Curator: Larry H. Bernstein, MD, FCAP
In the preceding discussions the hematological and nonhematological cancers were elaborated. These were tumors of blood or solid tumors that are malignant. Malignant solid tumors have a loss of normal architecture. Malignant cancers of the blood forming organ also have a disruption of the architecture in the blood forming organs, and they are circulating elements that are either acutely increased in number or chronically increased to very high circulating counts as well as many cells in the marrow. The diagnosis depends on the type of cell elements and the stage of maturation. In the case of blood cell cancers, one might consider an intermediate stage that has a long course that is in the case of the myelogenous series, myeloid dysplasia, which includes myelofibrosis, which in either case is not a benign course. In the case of solid tumors, there is an anatomic structure of the cancer site.
The usual structure for a carcinoma is either adjoining cells surrounding a vascular supply, as in the liver, a parenchymal gland, as in pancreas, a tubular structure, as the gastrointestinal tract and lungs (which are embryologically and outpouching of the gut), or a skin surface. In the case of carcinomas, the cells mature from a basement membrane of small flattened cells that overlie a fibrovascular matrix and an underlying myxoid stroma, perhaps beneath which is a muscular organ, then covered by a flat layer of cells. In the case of all epithelial structures there is an orderly maturation of epithelium from the basal layer to the mature epithelial cells that are elongate, have a brush border, and secrete into the glandular structure. The cell maturation becomes disrupted and disorderly to different degrees in the development of malignancy from a dysplasia to low grade malignancy, to high grade anaplastic cancer.
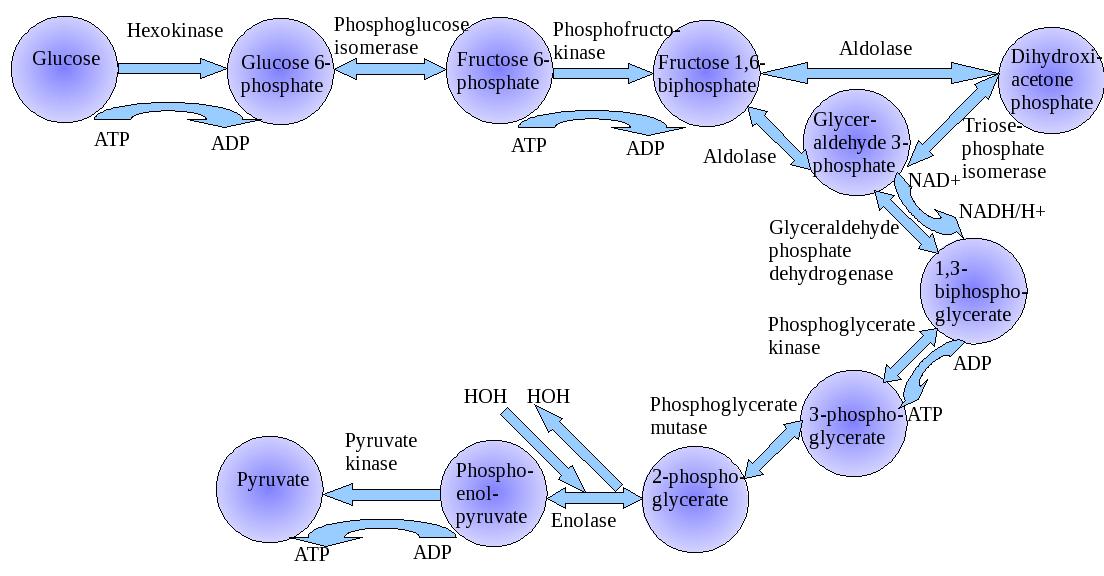
The development of a cancer implies the loss of tissue architecture, the replication of cells, the development of a neoplasms circulation (which is the topic of vascular endothelial growth factor (VEGF)), the overgrowth of the circulation so that the tumor has insufficient blood supply, and vascular invasion. We refer to the Warburg Hypothesis with respect to the malignancy relying on glycolysis in the presence of oxygen (aerobic glycolysis), but it may be questionable to imply that there is sufficient oxygen supply. In some cases a cancer may occur from a longstanding inflammatory focus. This has been seen to occur in osteomyelitis and in gastrointestinal fistulas. The growth of a neoplasm, when it exceeds its blood supply, requires adaptive changes. The most obvious to consider would be a decreased reliance of mitochondrial respiration. Warburg refer to the increase production of lactic acid as analogous to Pasteur observation of fermentation in yeast (Pasteur effect). He measured the lactic acid production by various tissues, and the consumption with the oxygen consumption showed that in many tissues approximately two molecules of lactate are prevented from appearing when one molecule of oxygen is consumed – a relationship that Meyerhof had found in muscle. This he expressed as the “Meyerhof quotient”:
Anaerobic glycolysis – aerobic glycolysis/oxygen consumption
Ref: Otto Warburg: Cell Physiologist, Biochemist, and Eccentric
Hans Krebs in collaboration with Roswitha Schmid
Clarendon Press, Oxford, 1981. Pp 19-25.
The special feature of cancer cells was the high rate of glycolysis in the presence of oxygen, whereas muscle can form lactate from carbohydrate in the absence of oxygen. This led to the discovery that all animal tissues are capable of glycolysis both aerobically and anaerobically. Pasteur had established 60 years earlier that the rates of fermentation are generally hiugh anaerobically, but low aerobically. This led Warburg to the conclusion that cancer cells are distinguished from noncancer cells by their failure to suppress glycolysis in the presence of oxygen. He discovered in 1926 that the link between respiration and fermentation can be severed by a specific inhibitor, ethylcarbylamine. He looked at carbylethylamine as an inhibitor of the ‘Pasteur effect’, and determined that the catalyst was a heavy metal ion. But the proposed mechanism was shown not to be correct by Engelhardt, Lynen, Bucher, Lowry, Racker, and Sols.The activity of the enzyme phospofructokinase is regulated by the concentrations of ATP, ADP and inorganic phosphate (Pi). The “allosteric properties” of PFK could account for the ‘Pasteur effect’. ATP inactivates PFK, while ADP and Pi activate it. Further, etylcarbylamine was found to be an uncoupler of oxidative phosphorylation (OxPhos), but Warburg was right in postulating that a heavy metal was involved since heavy metals are involved in
OxPhos. The explanation for this is now that when malignant transformation occurs, the cells’ energy supply is redirected from their normal function to growth. This change was found to be irreversible upon restoration of oxygen supply.
The topic of discussion is metastasis. What does it have to do with malignancy and respiration? Metastasis is the other key feature of cancer cells. What it has to do with respiration would probably tie in with the change in the cells’ energy supply that is directed toward proliferation. As the cell metabolism is reconfigured, there is also a change in the cell signaling with respect to apoptosis and the events regarding autophagy. This has to extend beyond the mitochondria, mainly because autophagy involves mitophagy, the ER and the entire cytoskeleton. This means that the cytoplasmic relationship to the intercellular matrix and the fibroblast stroma would have to be affected, as the cell breaks away from its close association with adjacent cells. Cells can migrate to adjacent lymphatic structures, and either enter the circulation by way of the lymphatics or by invasion of the venous circulation directly. In any case, entry into the circulation allows for transport to distant sites. With respect to migration to distant sites, we recall the hypothesis of Paget that the cells metastasize directly into the circulating blood, and they may ‘seed’ to favorable organs.
The discussion now turns to the assessment of apoptosis as a means to inhibition of cancer cell lines, which proliferate if unchecked and migrate away from the primary site. I use a few examples from a symposium volume of the Annals of the New York Academy of Sciences:
Apoptosis: From Signaling Pathways to Therapeutic Tools.
Ed, Mark Diederich
ANYAA9 2003; 1010:1-799
The role of β-glucuronidase in induction of apoptosis by Genistein Combined Polysaccharide (GCP) in xenogenetic mice bearing human mammary cancer cells.
Yuan L, Wagatsuma C, Sun B, Kim Jung-Hwan, Surh Young-Joon
Ann NY Acad Sci 2003;1010: 347-349.
http://dx.doi.org:/10.1196/annals.1299.063
- GCP inhibits tumor cell growth through multiple mechanisms, including induction of tumor apoptosis
- The biological activities of genistein (aglycon) are more evident in tumor tissues than in normal tissues.
- Hiugh doses of genistein administration rarely induces toxicity to normal tissues.
- Higher levels of β-glucuronidase expression in tumor tissues results in more genistein aglycon, leading to tumor destruction.
Induction of apoptosis in human pancreatic cancer cells by docosahexanoic acid
Merendino N, Molinari R, Loppi B, Pessina G, D’Aquino M, Tomassi G, Velotti F.
Ibid 361-364. http://dx.doi.org:/10.1196/annals.1299.143
Polyunsaturated fatty acids have been indicated to induce anti-proliferative and/or apoptotic effects in various tumor cells. We showed that, at a 200-μM concentration, both alpha-linoleic (18:2 n-6; LA) or docosahexaenoic (22:6 n-3; DHA) acid inhibited cell growth, while only DHA induced apoptosis in the human Paca-44 pancreatic cancer cell line. Investigating the mechanism underlying DHA-induced apoptosis, we showed that DHA induced a rapid and dramatic (>60%) intracellular depletion of reduced glutathione (GSH), without affecting oxidized glutathione (GSSG). Moreover, using two specific inhibitors of carrier-mediated GSH extrusion, cystathionine or methionine, we observed that GSH depletion occurred via an active GSH extrusion, and that inhibition of GSH efflux completely reversed apoptosis. These results provide the first evidence for a possible causative role of GSH depletion in DHA-induced apoptosis.
Opposite phenotypes of cancer and aging arise from alternative regulation of common signaling pathways.
Ukraintseva SV1, Yashin AI.
Ann N Y Acad Sci. 2003 Dec; 1010:489-92.
http://dx.doi.org:/10.1196/annals.1299.089
Phenotypic features of malignant and senescent cells are in many instances opposite. Cancer cells do not “age”; their metabolic, proliferative, and growth characteristics are opposite to those observed with cellular aging (both replicative and functional). In many such characteristics cancer cells resemble embryonic cells. One can say that cancer manifests itself as a local, uncontrolled “rejuvenation” in an organism. Available evidence from human and animal studies suggests that the opposite phenotypic features of aging and cancer arise from the opposite regulation of genes participating in apoptosis/growth arrest or growth signal transduction pathways in cells. This fact may be applicable in the development of new anti-aging treatments. Genes that are contrarily regulated in cancer and aging cells (e.g., proto-oncogenes or tumor suppressors) could be candidate targets for anti-aging interventions. Their “cancer-like” regulation, if strictly controlled, might help to rejuvenate the human organism.
CUGBP2 Plays a Critical Role in Apoptosis of Breast Cancer Cells in Response to Genotoxic Injury
Mukhopadhyay D, Jung J, Murmu N, Houchen CW, Dieckgraefe BK, Anant A
Ibid 504–509. http://dx.doi.org:/10.1196/annals.1299.093
Posttranscriptional control of gene expression plays a key role in regulating gene expression in cells undergoing apoptosis. Cyclooxygenase-2 (COX-2) is a crucial enzyme in the conversion of arachidonic acid to prostaglandin E2 (PGE2) and is significantly upregulated in many types of adenocarcinomas. COX-2 overexpression leads to increased PGE2 production, resulting in increased cellular proliferation. PGE2 enhances the resistance of cells to ionizing radiation. Accordingly, understanding mechanisms regulating COX-2 expression may lead to important therapeutic advances. Besides transcriptional control, COX-2 expression is significantly regulated by mRNA stability and translation. We have previously demonstrated that RNA binding protein CUGBP2 binds AU-rich sequences to regulate COX-2 mRNA translation. In the current study, we have determined that expression of both COX-2 mRNA and CUGBP2 mRNA are induced in MCF-7 cells, a breast cancer cell line, following exposure to 12 Gy γ-irradiation. However, only CUGBP2 protein is induced, but COX-2 protein levels were not altered. Silencer RNA (siRNA)-mediated inhibition of CUGBP2 reversed the block in COX-2 protein expression. Furthermore, MCF-7 cells underwent apoptosis in response to radiation injury, which was also reversed by CUGBP2 siRNAs. These data suggest that CUGBP2 is a critical regulator of the apoptotic response to genotoxic injury in breast cancer cells.
Multiple and synergistic deregulations of apoptosis-controlling genes in pancreatic carcinoma cells
A Trauzold,1 S Schmiedel,1 C Röder,1 C Tams,1 M Christgen,1 S Oestern,1 A Arlt,2 S Westphal,1 M Kapischke,1 H Ungefroren,1 and H Kalthoff1,
Ibid 510-513. Br J Cancer. 2003 Nov 3; 89(9): 1714–1721.
http://dx.doi.org/10.1038%2Fsj.bjc.6601330
CD95, TRAIL-R1 (tumor necrosis factor-related apoptosis inducing ligand-receptor 1) and TRAIL-R2 are members of the TNF-receptor family of transmembrane proteins that are capable of inducing apoptosis (Wiley et al, 1995; Pitti et al, 1996; Pan et al, 1997; Peter et al, 1998). Following ligand binding, the receptors oligomerize and the pro-apoptotic molecules TRADD, FADD and FLICE/caspase-8 are recruited to their intracellular death domain forming the ‘death-inducing signaling complex’ (DISC) (Krammer, 1999). The subsequent events leading to apoptosis depend on the specific cell type being challenged. In type I cells the bulk induction of caspase-8 at the DISC leads to the direct activation of the effector caspase 3. In type II cells only little amounts of caspase-8 are activated at the DISC requiring the pro-apoptotic mitochondrial amplification loop for efficient caspase-3 activation (Scaffidi et al, 1998).
In Vivo Imaging of Chemotherapy-Induced Apoptosis in Human Cancers
T Belhocine, N Steinmetz, A Green, P Rigo
Ibid 525-529. http://dx.doi.org:/10.1196/annals.1299.097
Rationale. Induction of apoptosis in sensitive tumor cells is the main mechanism of action of chemotherapy agents in human cancers. Also, the assessment of drug-induced apoptosis soon after chemotherapy may be an early predictor of treatment efficacy. Patients and Methods. A phase I/II study was prospectively conducted in 15 patients presenting with proven lung cancers (n= 10), breast cancers (n= 2), and lymphomas (n= 3) to assess the value of the 99mTc-radiolabeled recombinant human (rh) Annexin V for imaging apoptosis immediately after completion of the first course of chemotherapy. Early Annexin V findings post-chemotherapy (day+1, day+2) were also compared to the tumor status at 6 to 12 weeks post-treatment.
Results. All lung and lymphoma patients with an increased tracer uptake post-treatment (n= 8) had either partial or complete tumor response. Five patients with no tracer uptake had progressive disease. However, two breast cancers had a response to treatment, although no significant tracer uptake was observed. Tumor response and survival time were significantly correlated with the 99mTc-labeled Annexin V uptake. No serious events related to tracer administration were noted. Conclusion. Preliminary results of this pilot study demonstrate the feasibility of the 99mTc-labeled Annexin V uptake for the in vivo imaging of apoptosis after one course of chemotherapy. If confirmed on larger series, these promising results may open new perspectives in the management of oncology patients.
In vivo photoacoustic imaging of chemotherapy-induced apoptosis in squamous cell carcinoma using a near-infrared caspase-9 probe.
Yang Q1, Cui H, Cai S, Yang X, Forrest ML.
J Biomed Opt. 2011 Nov; 16(11):116026.
http://dx.doi.org:/10.1117/1.3650240
Anti-cancer drugs typically exert their pharmacological effect on tumors by inducing apoptosis, or programmed cell death, within the cancer cells. However, no tools exist in the clinic for detecting apoptosis in real time. Microscopic examination of surgical biopsies and secondary responses, such as morphological changes, are used to verify efficacy of a treatment. Here, we developed a novel near-infrared dye-based imaging probe to directly detect apoptosis with high specificity in cancer cells by utilizing a noninvasive photoacoustic imaging (PAI) technique. Nude mice bearing head and neck tumors received cisplatin chemotherapy (10 mg/kg) and were imaged by PAI after tail vein injection of the contrast agent. In vivo PAI indicated a strong apoptotic response to chemotherapy on the peripheral margins of tumors, whereas untreated controls showed no contrast enhancement by PAI. The apoptotic status of the mouse tumor tissue was verified by immunohistochemical techniques staining for cleaved caspase-3 p11 subunit. The results demonstrated the potential of this imaging probe to guide the evaluation of chemotherapy treatment.
Noninvasive imaging techniques are necessary for early cancer detection and evaluation of the chemotherapeutic effect on tumors. Current diagnostic imaging techniques generally include γ-scintigraphy, magnetic resonance imaging, computed tomography, and ultrasonography; however, these techniques only give morphological information on the tumor. These techniques do not report the biochemical response of the tumor to treatment and physical changes in the tumor in response to treatment may take days to weeks to fully manifest. Positron emission topography and SPECT can indirectly detect tumor response to treatment due to changes in metabolic activity and blood perfusion, respectively. However, no clinical imaging technique can directly detect the biochemical response, e.g., apoptosis, of tumors to treatment. Since apoptosis often occurs within in the first 18 to 36 h after treatment, direct imaging of apoptosis would rapidly indicate if there is a response in the tumor to chemotherapy.
Photoacoustic imaging (PAI) overcomes the spatial and resolution limitations of conventional imaging techniques at a relatively low cost,1, 2 and it has shown its potential to monitor the growth of melanoma brain tumors3 and melanoma metastasis in sentinel lymph nodes.4 However, ascribed to the fact that PAI utilizes the optical absorption of tissues for contrast, it cannot differentiate normal from cancerous cells unless the cells are overexpressing chromomeric marker (e.g., melanomas) or labeled by reporter moieties as contrast agent to enhance the contrast between normal and pathological tissues. In this case, application of a contrast agent such as fluorochromes is expected to facilitate both the visualization of head and neck squamous cell carcinoma (HNSCC) cancer cells and their response to treatment in vivo by PAI.
We have synthesized a near-infrared fluorescent imaging probe – IR780-linker-Val-Ala-Glu(OMe)-FMK by conjugating a fluorochrome (IR780) to Z-Val-Ala-Glu (OMe), a cell permeable caspase inhibitor. The activation of caspase family of cysteine proteases has been recognized as a critical event of apoptosis, which is a physiological process of type I programmed cell death. Typically anti-cancer agents act on cancer cells to induce apoptosis, so apoptosis is a rapid and definite indicator of tumor response. For this reason, apoptosis is used in screening drug candidates in cell culture. The fluoromethyl ketone of the tripeptides valine, alanine, and O-methyle-glutamic acid [Val-Ala-Glu(OMe)-FMK] can specifically and irreversibly bind to the cysteine residue at the active site of caspase-9.5 Our preliminary in vitro cell-imaging test with prostate cancer DU 145 cells demonstrated the sensitivity of this imaging probe for cell apoptosis.6 In this study, we evaluated the application of IR780-linker-Val-Ala-Glu(OMe)-FMK for PAI to detect procaspase-9 activation caused by anticancer drug treatment in living nude mice bearing HNSCC tumors.

Increase in PA amplitude within the HNSCC tumor after intravenous injection of imaging agent
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3221716/bin/JBOPFO-000016-116026_1-g002.jpg
Fourteen micron (thickness) sections of the tumor tissue were stained with a goat primary polyclonal antibody for cleaved caspase-3 p11 subunit (Asp-175-Ser-176) and a donkey anti-goat secondary antibody with a fluorescein isothiocyanate (FITC) fluorophore (Santa Cruz Biotechnology Inc., Santa Cruz, California). Cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI).
Maximum amplitude projection images obtained from the PAI of the HNSCC tumor region shown in Fig. (Fig.2).2 were converted to grayscale images. The grayscale images at various time points were linearly aligned using the scale-invariant feature transform function of Fiji/ImageJA software (ver. 20110307, http://pacific.mpi-cbg.de/wiki/index.php/Fiji) (Fig. (Fig.3).3). Quantification of PA signal intensity within the tumor region was performed in triplet for each image by measuring the mean gray value (units: gray/pixel) of the circled tumor region. The extent of signal enhancement was calculated by normalizing the tumor signal against a background reading taken immediately before injection of the imaging agent (Fig. (Fig.2).2)
Apoptosis in the tumor tissues was independently verified by immunohistochemical staining for caspase 3, a downstream indicator of apoptosome-activated caspase-mediated apoptosis that would not cross-react with the caspase-9 PA probe. Figure (Figure4a) 4a represents a control section stained with the secondary antibody alone (autofluorescence of the tissue without apparent staining); while, Fig. (Fig.4b).4b shows the immunostaining of the caspase-3 p11 subunit (green) and the DAPI staining of cell nuclei. The intense green fluorescence in these sections suggests the wide spread apoptosis of cells in the tumor tissues after intravenous administration of high-dose cisplatin. In addition, cells on the peripheral of the tumor stained more strongly for caspase 3 (green fluorescence) compared to cells at the tumor interior. This was consistent with the PA imaging of apoptosis that showed strong apoptosis at the tumor peripheral, suggesting chemotherapeutics had penetrated the outer layers of the tumor and induced apoptosis.

Immunostaining for apoptosis in tumor
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3221716/bin/JBOPFO-000016-116026_1-g004.jpg
Immunostaining for apoptosis in tumor. (a) Representative control section stained with secondary antibody alone and (b) tissue section of the HNSCC tumor stained for caspase-3 p11 subunit after cisplatin treatment.
I now consider mechanisms of metastasis as currently viewed.
Metastasis mechanisms.
Geiger TR1, Peeper DS.
Biochim Biophys Acta. 2009 Dec; 1796(2):293-308. http://dx.doi.org:/10.1016/j.bbcan.2009.07.006.
Metastasis, the spread of malignant cells from a primary tumor to distant sites, poses the biggest problem to cancer treatment and is the main cause of death of cancer patients. It occurs in a series of discrete steps, which have been modeled into a “metastatic cascade”. In this review, we comprehensively describe the molecular and cellular mechanisms underlying the different steps, including Epithelial-Mesenchymal Transition (EMT), invasion, anoikis, angiogenesis, transport through vessels and outgrowth of secondary tumors. Furthermore, we implement recent findings that have broadened and challenged the classical view on the metastatic cascade, for example the establishment of a “premetastatic niche”, the requirement of stem cell-like properties, the role of the tumor stroma and paracrine interactions of the tumor with cells in distant anatomical sites. A better understanding of the molecular processes underlying metastasis will conceivably present us with novel targets for therapeutic intervention.
Axis of evil: molecular mechanisms of cancer metastasis Thomas Bogenrieder1 and Meenhard Herlyn1
Oncogene (2003) 22, 6524–6536.
http://dx.doi.org:/doi:10.1038/sj.onc.1206757
Although the genetic basis of tumorigenesis may vary greatly between different cancer types, the cellular and molecular steps required for metastasis are similar for all cancer cells. Not surprisingly, the molecular mechanisms that propel invasive growth and metastasis are also found in embryonic development, and to a less perpetual extent, in adult tissue repair processes. It is increasingly apparent that the stromal microenvironment, in which neoplastic cells develop, profoundly influences many steps of cancer progression, including the ability of tumor cells to metastasize. In carcinomas, the influences of the microenvironment are mediated, in large part, by bidirectional interactions (adhesion, survival, proteolysis, migration, immune escape mechanisms lymph-/angiogenesis, and homing on target organs) between epithelial tumor cells and neighboring stromal cells, such as fibroblasts as well as endothelial and immune cells. In this review, we summarize recent advances in understanding the molecular mechanisms that govern this frequently lethal metastatic progression along an axis from primary tumor to regional lymph nodes to distant organ sites. Affected proteins include growth factor signaling molecules, chemokines, cell–cell adhesion molecules (cadherins, integrins) as well as extracellular proteases (matrix metalloproteinases). We then discuss promising new therapeutic approaches targeting the microenvironment. We note, however, that there is still too little knowledge of how the many events are coordinated and integrated by the cancer cell, with conspiratorial help by the stromal component of the host. Before drug development can proceed with a legitimate chance of success, significant gaps in basic knowledge need to be filled.
Metastases to regional lymph nodes are detected at diagnosis and surgery in approximately one-third of breast, colorectal, uterine cervix, and oral cavity and pharynx cancer patients, and one-quarter of esophageal, lung pancreas, gastric and bladder cancer patients (Greenlee et al., 2001). The high mortality rates associated with cancer are caused by the metastatic spread of tumor cells from the site of their origin. In fact, metastases are the cause of 90% of cancer deaths (Hanahan and Weinberg, 2000). The prognosis for a patient who is diagnosed with advanced invasive or metastatic disease remains little better than it was decades ago (Sporn, 1997). Tumor cells invade either the blood or lymphatic vessels to access the general circulation and then establish themselves in other (visceral) tissues. Ultimately, they become surgically unresectable, with pharmacological or radiological long-term control being uncommon (Stacker et al., 2002).
Although the genetic basis of tumorigenesis may vary greatly between different cancer types, the cellular and molecular steps required for metastasis are generally similar for all solid tumor cells (Woodhouse et al., 1997; Liotta and Kohn, 2003). Not surprisingly, the molecular mechanisms that propel invasive growth and metastasis are also found in embryonic development, and, however to a less perpetual/chronic/aggressive/quantitatively different extent, in adult tissue maintenance (e.g. involving stem cell differentiation) and repair processes (‘tumors are wounds that do not heal’) (Dvorak, 1986). We now view cancer as a complex tissue resulting from disrupted organ homeostasis, rather than focusing on the cancer cell, and the genes within it, alone (Hanahan and Weinberg, 2000;Bissell and Radisky, 2001; Bogenrieder and Herlyn, 2002; Wiseman and Werb, 2002). Normal tissue homeostasis is maintained between epithelial cells and their microenvironment, such as fibroblasts, endothelial and immunocompetent cells, and the extracellular matrix (ECM). Similarly, during malignant transformation and progression, there are (however deregulated) reciprocal and conspirational interactions between the neoplastic cells and the adjacent stromal cells (Hsu et al., 2002). A series of recent investigations have shown that aberrations in the stroma can both precede and stimulate the development of cancers (reviewed in Bissell and Radisky, 2001; Wiseman and Werb, 2002).
The process of metastasis involves an intricate interplay between altered cell adhesion, survival, proteolysis, migration, lymph-/angiogenesis (see articles in this issue by R Kerbel and P Campochiaro, pp. NNN–NNN), immune escape mechanisms, and homing on target organs (Table 1). However, there is still very little knowledge of how these events are coordinated by the cancer cell, with conspirational help by the stromal component (microenvironment) of the host (Clark, 1991; Hsu et al., 2002). This process is usually said to be ‘uncontrolled’. As we shall see, however, it is by no means purely stochastic, but rather a finely tuned molecular machinery with active tumor cell–host collaboration. Thus, all explanations of ‘success’ of the metastatic axis contain a strong element of determinism. Whereas the early steps in the metastastic campaign are completed very efficiently, metastasis is an inefficient process in its later steps, especially the regulation of cancer cell growth at the secondary sites (Luzzi et al., 1998; Cameron et al., 2000; Chambers et al., 2002). Given that spread of the tumor to distant organs is usually lethal, more intense studies of these molecular mechanisms assume general importance to develop more effective anticancer strategies. In the following discussion of specific molecular mechanisms, we have often chosen to draw mainly from examples that pertain to melanoma progression, although similar processes are most likely also operating during oncogenesis of a wide range of cancers.
The classical metastatic cascade encompasses intravasation by tumor cells, their circulation in lymph and blood vascular systems, arrest in distant organs, extravasation, and growth into metastatic foci (Herlyn and Malkowicz, 1991;Woodhouse et al., 1997). Ann Chambers et al. (2001),(2002) have demonstrated in murine models that the limiting factor for metastasis formation is growth after extravasation (Figure 1a). Recently, this extravasation model has been challenged by Ruth Muschel and co-workers (Al-Mehdi et al., 2000; Wong et al., 2002), who showed that tumor cells can readily proliferate after arrest in blood vessels, suggesting that extravasation is not a prerequisite for metastatic growth (Figure 1b). In a separate series of experiments, Mary Hendrix and co-workers described that tumor cells can even have endothelial cell-like functions and form channels that allow fluid flow (Maniotis et al., 1999; Folberg et al., 2000) (Figure 1c). The group has identified some of the players, such as EphA2 and VE-cadherin, on aggressive melanoma cells that are shared with endothelial cells and that are likely involved in ‘vasculogenic mimicry’. Vasculogenic mimicry is the ability of aggressive cancer cells to form de novo vessel-like networks in vitro in the absence of endothelial cells or fibroblasts, concomitant with their expression of vascular-associated cellular marker (Sood et al., 2001,2002). Tumor cell plasticity is demonstrated by the ability of tumor cells to adopt a variety of phenotypes, including an endothelial phenotype (Sood et al., 2001, 2002). These exciting new findings underscore the plasticity of malignant cells from advanced tumor progression stages, and they require from tumor biologists a more dynamic view of the metastatic cascade. If the biological phenotype of metastasis must be portrayed flexibly, then we need a new ‘yardstick,’ a normal cell, to better characterize and understand the many faces of metastasis. We need to understand how the malignant cells exert cooperation from the normal cells. Our central hypothesis is that both normal and malignant cells utilize the same molecules for invasion, but that differences in downstream signaling events allow the tumor cells to dominate over normal cells in the microenvironment. This ‘dominant plasticity’ model of cancer metastasis takes into account the flexible response of malignant cells to microenvironmental pressures while maintaining dominance over the normal parenchymal and stromal cells.

Models of metastasis
http://www.nature.com/onc/journal/v22/n42/images/1206757f1.jpg
Models of metastasis. (a) According to Chambers and co-workers, only a very small population of injected cells (2%) form micrometastases, although over 87% are arrested in the liver. Furthermore, not all of the micrometastases persist, and the progressively growing metastases that kill the mice arise only from a small subset (0.02%) of the injected cells. (b) Muschel and co-workers recently proposed a new model for pulmonary metastasis in which endothelium-attached tumor cells that survived the initial apoptotic stimuli proliferate intravascularly. Thus, a principal tenet of this new model is that the extravasation of tumor cells is not a prerequisite for metastatic colony formation and that the initial proliferation takes place within the blood vessels. (c) The unique ability of aggressive tumor cells to generate patterned networks, similar to the patterned networks during embryonic vasculogenesis, and concomitantly to express vascular markers associated with endothelial cells, their precursors and other vascular cells has been termed ‘vasculogenic mimicry’ by Hendrix and co-workers




















































{kind=link}
{kind=link}
{kind=link}
{kind=link}