Rosa’s to like
Curator & Reporter: Larry H. Bernstein, MD, FCAP
Reality Check: Cancer Experts Discuss Hurdles Facing CAR-T Therapy
September 18th, 2015
There’s a lot of excitement these days about a type of cellular immunotherapy known as CAR-T, a method of modifying peoples’ immune cells to fight cancer. But you could also fill a book listing all the problems its makers will have to solve—how to test, manufacture, and even the define the nature of these cancer-killing cells—before the CAR-T story is a successful one.
These hurdles, not the hype, were the subject of a panel of experts from industry, academia, and the FDA at the Inaugural International Cancer Immunotherapy Conference in New York Thursday afternoon. The panelists included University of Pennsylvania professor Carl June, whose work has led to programs now in clinical testing at Novartis; Adaptimmune executive vice president Gwendolyn Binder-Scholl; and GlaxoSmithKline’s head of immuno-oncology Cedrik Britten, among others.
CAR-T stands for chimeric antigen receptor T cell, which describes an engineered version of the immune system’s attack dogs. CAR-T cells are a patient’s own T cells altered outside the body to be cancer killers, then put back in to go after tumor cells.
CAR-T therapies from Novartis, Juno Therapeutics (NASDAQ: JUNO), and Kite Pharma (NASDAQ: KITE) have produced impressive results so far for certain blood cancers, leading to long-lasting remissions in some patients.
But the field is early in its development. Researchers are trying to figure out how to make these therapies useful for more common cancers, such as lung, breast, and ovarian, and how to mitigate the overactive immune responses they can cause. Biotechs and pharma companies developing autologous therapies—which modify the cells of each individual patient—are wrestling with how to manufacture and distribute them at scale.
But a different, larger question looms, and it gets to the heart of why autologous T cell therapy is truly a new medical frontier. The cells that are delivered back into the patient are not what ends up doing the bulk of the therapeutic work.
The panelists Thursday noted how T cell therapies could throw a wrench into a typical, and crucial, clinical strategy. Early in the clinical testing of a drug, companies usually run what are known as dose escalation studies. Different doses of products are tested, low to high, to establish a trend of responses, see what safety issues pop up, and pick the optimal dose to move forward.
But because CAR-T cell populations expand once they’re put back into a patient’s body, doses are harder to define. What’s more, cranking up a dose for such a powerful therapy could be dangerous. “Are classical trial designs applicable, or do they have to be changed?” asked GSK’s Britten. “You can not have a simple dose escalation [study] with a drug that replicates.”
Adaptimmune’s Binder-Scholl called for more guidance from regulators to help figure out a more standardized scheme for dose escalation studies.
“I think the biology is going to make [that type of guidance] awfully challenging,” says Juno’s chief financial officer Steve Harr, who also wasn’t on the panel. “I would like to think over time we get into something a bit more predictable, and maybe we have some type of a standard, but we’re very early in this process.”
Medscape Cardiology > Black on Cardiology
SPRINT Hypertension Trial: Preliminary Results Discussed
Henry R. Black, MD; William C. Cushman, MD Disclosures | Sept 18, 2015
Stopped Early for Benefit
Henry R. Black, MD: Hi. I’m Dr Henry Black. I’m adjunct professor of medicine at the Langone New York University School of Medicine, and I’m here today with my long-term friend and colleague, Dr Bill Cushman. Bill, thank you very much for doing this.
William C. Cushman, MD: Delighted to be here.
Dr Black: What I want to talk about is the SPRINT study,[1]which you’ve been a primary participant in. The top-line resultswere just released. Tell us a little bit about SPRINT: who was in it, what the hypothesis was, and how it compares to the ACCORD study, which you also participated in.
Dr Cushman: Sure. I’m Dr Bill Cushman. I’m from Memphis, Tennessee. And I’m chief of the preventive medicine section at the VA and professor of preventive medicine at the University of Tennessee.
I was a network principal investigator in SPRINT, which meant that I oversaw about a quarter of the sites. SPRINT was a study sponsored by the National Institutes of Health (NIH), primarily the National Heart, Lung, and Blood Institute (NHLBI). But other institutes—the National Institute of Diabetes and Digestive and Kidney Diseases, the National Institute on Aging, the National Institute of Neurological Disorders and Stroke—were also involved.
SPRINT was a study of 9361 participants who were randomized to either a lower, more intensive goal of less than 120 mm Hg systolic blood pressure (SBP) compared with a goal of less than 140 mm Hg systolic. That was considered standard when we designed the study, and all guidelines recommended at least getting below 140 mm Hg.
We recruited a participant pool of high-risk hypertensive patients with SBPs of ≥130 mm Hg. They could be on medications (the majority were), but they didn’t have to be. Participants not only had to have elevated blood pressure, but they also had to be above age 50 and they had to have some other indices of risk: known cardiovascular disease, chronic kidney disease, or being above age 75, for example, or having a Framingham risk assessment for cardiovascular disease of ≥ 15% over 10 years.
They were randomly allocated to these two groups, with the intent of being followed for about 5 years. The primary outcome in SPRINT was a combined cardiovascular outcome that included myocardial infarction (MI), acute coronary syndrome other than MI, stroke, heart failure, or cardiovascular death.
Now, there are a lot of other outcomes in SPRINT, including whether this lower blood pressure goal would prevent dementia, changes on MRI, or chronic kidney disease. Those outcomes have not been stopped or announced yet, and we’re still collecting data on that.
The cardiovascular outcomes were viewed as so positive in terms of the benefit that the Data and Safety Monitoring Board recommended to Gary Gibbons, the director of the NHLBI, that the cardiovascular part of the trial—and the intensive intervention in particular—should be stopped and that the investigators and the participants should be unblinded. And that was done.
Dr Black: Were the antihypertensive regimens prescribed, or was it whatever the docs wanted to do?
Dr Cushman: Good point. We actually recommended using the major classes that were proven to be of benefit in cardiovascular outcome trials in hypertension: either thiazide-type diuretics, ACE inhibitors, angiotensin receptor blockers, or calcium blockers. It was primarily those four classes, and they could be combined in whatever way the investigators wanted. We did put a lot of emphasis on using thiazide-type diuretics because of the ALLHAT[2] results.
But the way they could be combined was really up to the investigators. Now, if the participants had known coronary disease or some other indication for a beta-blocker, that could certainly be used. And then other drugs could be added. We had a very large formulary representative of all the major classes of drugs— not only those classes, but also beta-blockers, alpha blockers, aldosterone inhibitors (spironolactone or amiloride, for example).
We had a lot of drugs available. They were predominantly purchased for the study, by NIH. There were only two drugs that were donated by the pharmaceutical companies. The study was entirely funded by NIH.
SPRINT vs ACCORD
Dr Black: How is this different from ACCORD?[3]
Dr Cushman: In ACCORD, we had the same two SBP goals: less than 120 mm Hg compared with a SBP of less than 140 mm Hg. However, SPRINT is twice as large as ACCORD.
As you may remember, we did not show a significant benefit for the lower SBP goal for the overall cardiovascular outcome in the ACCORD trial. We did see a significant reduction in stroke of about 40%, but that was a secondary outcome. The primary outcomes in mortality were not reduced in ACCORD.
However, ACCORD was about half the size of SPRINT. And even though the ACCORD blood pressure study was done in patients with diabetes, on average, they were probably a little lower-risk than our SPRINT participants because of their somewhat younger age (average age, 62 years), the absence of real chronic kidney disease, and several other reasons.
Even though ACCORD didn’t show a statistically significant benefit, it did show a 12% reduction in the cardiovascular outcome with a confidence interval that could have included up to a 27% benefit.
In contrast, SPRINT was twice as large, with a higher-risk population with an older average age. We excluded people with diabetes because that was being looked at in ACCORD. And we excluded people who’d had a prior stroke because that was being looked at in the SPS3[4] post-stroke study in terms of blood pressure goals.
Despite that, we had a very high-risk population. And what we found was about a third of a reduction in the primary cardiovascular events. That was significant.
We also saw, quite importantly, about a 25% reduction in all-cause mortality. That was surprising. The results are quite clear that there’s dramatic benefit in terms of both cardiovascular events and total mortality.
Dr Black: You probably can’t tell us this yet, but what was the blood pressure achieved in the less-than-140 group compared with the less-than-120 group?
We also saw, quite importantly, about a 25% reduction in all-cause mortality. That was surprising. The results are quite clear that there’s dramatic benefit in terms of both cardiovascular events and total mortality.
Diabetes Drug Empagliflozin Cuts CV Deaths in Landmark EMPA-REG Trial
STOCKHOLM ( updated with commentary ) — Patients with type 2 diabetes and established cardiovascular disease receiving the glucose-lowering agent empagliflozin (Jardiance, Boehringer Ingelheim/Lilly), a sodium glucose cotransporter-2 (SGLT-2) inhibitor, were less likely to die than those taking placebo in the large, much-anticipated EMPA-REG OUTCOME study, hailed here as a landmark trial.
The benefit on survival was seen regardless of the cause of death — empagliflozin prevented one in three cardiovascular deaths, with a significant 38% relative risk reduction in cardiovascular mortality, as well as a significant 32% relative reduction in all-cause mortality.
CV death was one component of the primary composite outcome, which also included nonfatal myocardial infarction (MI) or nonfatal stroke. It was the CV mortality benefit, however, that primarily drove the reduction in this end point.
“Empagliflozin is reducing death, the ultimate outcome,” senior author of the study, Silvio Inzucchi, MD, of Yale Diabetes Center, New Haven, Connecticut, told Medscape Medical News. “This is a first in my lifetime — a diabetes drug trial that has shown improved outcomes in high-risk cardiovascular patients.”
This is a first in my lifetime — a diabetes drug trial that has shown improved outcomes in high-risk cardiovascular patients.
Dr Inzucchi was given multiple rounds of applause as he presented the findings of EMPA-REG OUTCOME here at the European Association for the Study of Diabetes (EASD) 2015 Meeting, The study was also published simultaneously in the New England Journal of Medicine, by a team led by Bernard Zinman MD, director, Diabetes Centre, Mount Sinai Hospital, Toronto, Ontario.
Sept 17, 2015 http://dx.doi.org:/10.1056/NEJMoa1504720
Type 2 diabetes is a major risk factor for cardiovascular disease,1,2 and the presence of both type 2 diabetes and cardiovascular disease increases the risk of death.3 Evidence that glucose lowering reduces the rates of cardiovascular events and death has not been convincingly shown,4-6although a modest cardiovascular benefit may be observed after a prolonged follow-up period.7Furthermore, there is concern that intensive glucose lowering or the use of specific glucose-lowering drugs may be associated with adverse cardiovascular outcomes.8 Therefore, it is necessary to establish the cardiovascular safety benefits of glucose-lowering agents.9
Cohen’s Brain Bits: Let the Sunshine in?
http://www.medpagetoday.com/Blogs/CohensBrainBits/53630?xid=nl_mpt_DHE_2015-09-19&eun=g337145d0r Published: Sep 18, 2015
By Joshua Cohen MD, MPH
Vitamin D is actually not a vitamin at all — it is a group of fat-soluble steroid hormones responsible for a host of important functions in the body. As it is found in low levels in most foods other than fish and dairy, vitamin D is primarily synthesized from cholesterol in the skin upon exposure to UVB radiation.
While the discovery of vitamin D nearly a century ago stemmed from its role in calcium homeostasis and metabolism, an abundance of studies in the past decade have demonstrated the critical role vitamin D plays in neuronal development and protection. Indeed, in the past few years, researchers have uncovered an association between vitamin D deficiency and an array of important neurologic diseases.
A study in this week’s JAMA Neurology investigated the relationship between vitamin D levels, as measured in the blood as 25-hydroxyvitamin D, and the rate of cognitive decline in a population of 382 multi-ethnic older adults. Both vitamin D insufficient (12-20 ng/mL) and deficient (<12 ng/mL) participants demonstrated accelerated cognitive decline in multiple functional domains, especially episodic memory and executive function, that are the domains most affected in patients with Alzheimer’s dementia.
Previous studies have emphasized the essential role of vitamin D in the brain and have raised concern about the effect of vitamin D deficiency on the brain. Vitamin D’s neuroprotective roles include stimulation of neurotrophin release, neuroimmunomodulation, and interaction with reactive nitrogen and oxygen species. Vitamin D appears to also play a role in neurodevelopment through its regulation of nerve growth factor synthesis. Imaging studies have found increases in white matter hyperintensities and enlarged ventricles in vitamin D deficient study participants.
Channel Molecular Noise to Keep Cells Healthy
Molecular fluctuations or noise within and among cells can be manipulated to control the networks that govern the workings of living cells—promoting cellular health and potentially alleviating diseases such as cancer. [Daniel K. Wells]
Complex networks are noisy, whether they constitute food webs, power grids, or cells. And when networks buzz and crackle beyond normal bounds, bad things can happen: ecosystems can collapse, power grids can leave us in the dark, and cells can tumble into cancerous states.
All these networks are amenable to similar mathematical treatments says a scientific team at Northwestern University. The team, led by physicist Adilson E. Motter, Ph.D., substantiated this claim by focusing on a particularly difficult biophysical problem: the rational control of cellular behavior. To date, attempts to exert such control have been frustrated by the high dimensionality and noise that are inherent properties of large intracellular networks.
Dr. Motter and his colleagues noted that the response of biological systems to noise has been studied extensively. Yet they also realized that little had been done to exploit noise, or to at least channel it. They hoped to find a way to do so and thereby demonstrate the possibility of preserving or inducing desirable cell states.
Using a newly developed computational algorithm, Dr. Motter and colleagues showed that molecular-level noise can be manipulated to control the networks that govern the workings of living cells—promoting cellular health and potentially alleviating diseases such as cancer. They presented their results September 16 in the journal Physical Review X, in an article entitled, “Control of Stochastic and Induced Switching in Biophysical Networks.”
“Here we present a scalable, quantitative method based on the Freidlin-Wentzell action to predict and control noise-induced switching between different states in genetic networks that, conveniently, can also control transitions between stable states in the absence of noise,” wrote the authors. “We apply this methodology to models of cell differentiation and show how predicted manipulations of tunable factors can induce lineage changes, and further utilize it to identify new candidate strategies for cancer therapy in a cell death pathway model.”
Essentially, by leveraging noise, the team found that the high-dimensional gene regulatory dynamics could be controlled instead by controlling a much smaller and simpler network, termed a “network of state transitions.” In this network, cells randomly transition from one phenotypic state to another—sometimes from states representing healthy cell phenotypes to unhealthy states where the conditions are potentially cancerous. The transition paths between these states can be predicted, as cells making the same transition will typically travel along similar paths in their gene expression.
The team began by using noise to define the most-likely transition pathway between different system states, and connecting these paths into the network of state transitions. By doing so, the researchers could then focus on just one path between any two states, distilling a multidimensional system to a series of one-dimensional interconnecting paths.
Then, using their computational approach, the team identified optimal modifications of experimentally adjustable parameters, such as protein activation rates, to encourage desired transitions between different states.
Mitochondrial Protein Finding May Allow Scientists to Control Apoptosis
http://www.genengnews.com/Media/images/GENHighlight/Sep18_2015_NIH_Mitochondria1481397318.jpg
A protein embedded in the surface of mitochondria opens the door to apoptosis, causing cells to experience severe power failures, according to new work by researchers at Temple University School of Medicine. The study, appearing in Molecular Cell, suggests that blocking the door with a small-molecule inhibitor could be key to the treatment of cardiovascular diseases such as heart attack and stroke, where extensive mitochondrial dysfunction and cell death hinder tissue recovery.
The study (“SPG7 Is an Essential and Conserved Component of the Mitochondrial Permeability Transition Pore”), led by Muniswamy Madesh, Ph.D., associate professor in the department of biochemistry, the Cardiovascular Research Center, and the Center for Translational Medicine at Temple University School of Medicine (TUSM), shows that the protein, spastic paraplegia 7 (SPG7), is the central component of the so-called permeability transition pore (PTP), a protein complex in the mitochondrial membrane that mediates necrotic cell death (death caused by cell injury).
The identification of SPG7 marks a major advance in scientists’ understanding of how the PTP affects necrosis. Although first described in 1976, the molecular parts of the pore have eluded discovery. “The only known molecular component of the PTP prior to our discovery of SPG7 was a protein called CypD, which is necessary for pore function,” Dr. Madesh explained.
To identify genes that modulate PTP opening induced by calcium overload or increased levels of reactive oxygen species (ROS), the two primary factors that cause mitochondrial dysfunction and cell death via pore opening, Dr. Madesh’s team devised an RNA interference-based screen in which the activity of each gene under investigation was knocked down, or silenced, to examine its effects on mitochondrial calcium levels.
The researchers began with a panel of 128 different genes but after initial screening narrowed the field to just 14 candidate PTP components. Subsequent experiments showed that the loss of only one of them, SPG7, prevented pore opening.
Much of what is known about the PTP comes from studies of mitochondria in disease. In pathological states, particularly those involving hypoxia, calcium, and ROS accumulate within mitochondria, causing them to swell and prompting the PTP to open. Because pore opening disrupts the flow of electrons and protons across the mitochondrial membranes, which normally sustains energy production, it results in a catastrophic drop in cellular energy levels.
In the absence of disease, precisely how the PTP helps to mediate normal cellular physiology remains unclear. According to Dr. Madesh, “Under physiological conditions, SPG7 may function through transient pore openings to release toxic metabolites that have accumulated in mitochondria.” He plans to explore this possibility with knockout animal models.
“See-Through” Brain Developed by Japanese Researchers
Scientists at the RIKEN Brain Science Institute in Japan have developed a new method for creating transparent tissue that can be used to illuminate 3D brain anatomy at high resolutions. Published in Nature Neuroscience, the work showcases the novel technology and its practical importance in clinical science by showing how it has given new insights into Alzheimer’s disease plaques.
“The usefulness of optical clearing techniques can be measured by their ability to gather accurate 3D structural information that cannot be readily achieved through traditional 2D methods,” explains lead scientist Atsushi Miyawaki, M.D., Ph.D. “Here, we achieved this goal using a new procedure, and collected data that may resolve several current issues regarding the pathology of Alzheimer’s disease. While Superman’s x-ray vision is only the stuff of comics, our method, called ScaleS, is a real and practical way to see through brain and body tissue.”
In recent years, generating see-through tissue—a process called optical clearing—has become a goal for many researchers in life sciences because of its potential to reveal complex structural details of our bodies, organs, and cells—both healthy and diseased—when combined with advanced microscopy imaging techniques. Previous methods were limited because the transparency process itself can damage the structures under study.
The original recipe reported by the Miyawaki team in 2011, termed Scale, was an aqueous solution based on urea that suffered from this same problem. The research team spent five years improving the effectiveness of the original recipe to overcome this critical challenge, and the result is ScaleS, a new technique with many practical applications.
“The key ingredient of our new formula is sorbitol, a common sugar alcohol,” notes Dr. Miyawaki. “By combining sorbitol in the right proportion with urea, we could create transparent brains with minimal tissue damage, that can handle both florescent and immunohistochemical labeling techniques, and is even effective in older animals.”
The team has devised several variations of the Scale technique that can be used together. By combining ScaleS with AbScale—a variation for immunolabeling—and ChemScale—a variation for fluorescent chemical compounds—they generated multicolor high-resolution 3D images of amyloid beta plaques in older mice from a genetic mouse model of Alzheimer’s disease developed at the RIKEN BSI by the Takaomi Saido team.
After showing how ScaleS treatment can preserve tissue, the researchers put the technique to practical use by visualizing in 3D the mysterious “diffuse” plaques seen in the postmortem brains of Alzheimer’s disease patients that are typically undetectable using 2D imaging. Contrary to current assumptions, the diffuse plaques proved not to be isolated, but showed extensive association with microglia —mobile cells that surround and protect neurons.
GEN Roundup on Cell-Based Assays for Biological Relevancy
Cell-Based Assay Platforms are Evolving to Meet Diverse Challenges
- Cell-based assay platforms are evolving to meet diverse challenges—mimicking disease states, preserving signaling pathways, modeling drug responses, and recreating environments conducive to tissue development.
GEN recently interviewed a number of experts on cell-based assay technology to get a sense of the state of the art and to find out where this technology might be most valuable to life sciences research.
- GEN: What are some of the main challenges that are faced when validating cell-based assays?
- Dr. Kelly: Considerable challenges come from using systems involving a living organism in the validation of cell-based assays. The characteristics of such systems will likely affect the criteria for validation suitability. These criteria might be specific for primary cells, immortalized cell lines, cancerous cell lines, or cells generated de novo from multipotent stem cells.
- Chemical reagents are generally well characterized by parameters such as molecular weight, solubility, etc., which are unlikely to change between assays.
- However, characteristics of primary cells or established cell lines, such as viability, growth phase, proliferation rate, level of metabolism, and even cell size are much more vulnerable.
- Mr. Trinquet: Beyond developing the right cell-based assay, the main challenge remains the relevancy of the cell model for the target being investigated. Generally, a single assay must also be compatible with a broad variety of cell technologies/models, from engineered cells to more complex models, such as 2D, 3D, microtissue, primary culture, and induced pluripotent stem cell models.
- This certainly adds some difficulty, given that protein expression levels may differ from one model to another. Also, these assays must generally translate well all along the value chain, from high-throughput screening to late stages of lead op, so that end users do not have to switch between too many assay technologies.
- Dr. Hsu: Cell-based assays provide more biologically relevant information than biochemical assays for high-throughput screening and ADME/Tox. One challenge in developing and validating cell-based assays is to generate cells that reliably express the drug target and give reproducible results with good Z′ over time.
- We developed and launched the industry’s first cell-based assays and profiling services for G-protein-coupled receptors. The expression of G-protein-coupled receptors has been worked out, but ion channels are challenging. Another challenge is to make sure the assays and readouts are target specific and predictive, with a good dynamic range and signal-to-noise ratio to differentiate compounds with different potencies and efficacies.
- Dr. Khimani: Cell-based assays provide a complex and physiologically relevant medium to evaluate the effect of novel therapeutic or modulatory candidates. However, unlike traditional assay formats, cell-based assays introduce a number of challenging factors that must be considered—such as cell type, expression level, stability, and passage viability—when optimizing the assay conditions.
- In addition, with complex cell-based assay systems, data extraction and signal-to-noise optimization can be time-consuming bottlenecks. Other challenges, particularly with high-content screening, include separate investments in instrumentation, training, data analysis, and data management, all leading to a lower throughput.
- Dr. Fan: Cell-based assays are model systems, and the most critical challenge facing such assays is how well they reflect real biology. Cell-based assays offer great advantages over biochemical assays because they are conducted in cellular contexts. That said, most of the current cell-based assays use a homogeneous population of cells grown from immortalized cell lines, many of which express target proteins or reporters in excessive, nonphysiological amounts via transient transfection or randomly integrated stable clones. These cell models are far from the actual cellular context in normal or diseased tissue such as a tumor.
- In addition, phenotypical consequences of an analyte of interest to the cell could reflect a combination of effects that a single cell-based assay would not be able to fully address. These factors impact the validation or correlation of the results of a cell-based assay with a phenotypical consequence, an animal model study, or a clinically relevant finding.
- Dr. Piper: The most formidable challenge in generating and validating cell-based assays is achieving predictability and translatability. Next-generation re-targeting systems (such as the Jump-In™ platform) have made over-expression of genes, even multigene cassettes, fast, reliable, and easy compared to traditional single-cell cloning.
- While simple overexpression of a target may be sufficient to drive a primary screen and identify hits, it often lacks a sufficiently complex pathophysiological context to robustly convert hits to lead candidates that are meaningful in clinical trials. These systems have value at early stages, but they would benefit from improvements or secondary screens that can better translate to clinical results.
- Dr. Payne: The choice of a cell system remains a challenge. Cell lines produce reproducible results, but do not accurately model living systems. Although primary cells are more physiologically relevant, they are inherently variable, making it harder to deliver a robust cell-based assay.
- Choosing appropriate endpoints can be time consuming: measuring one parameter is not enough to accurately determine the functionality of a drug. The ability to analyze several markers in multiplex assays provides greater information on drug efficacy and toxicity, the latter being important for failing flawed drugs earlier. Finally, once validated offline, assays still require revalidation when transferred to automated context.
- GEN: What is more valuable to researchers with respect to cell-based assays miniaturization or ultra-high throughput?
- Dr. Kelly: A single cell contains the complete genome of the species and thousands of expressed genes, implying that one cell could provide the same information as millions. High-throughput efforts should be aimed at our ability to multiplex, multivisualize, and microarray the enormous amount of information that one cell can provide.Mr. Trinquet: Miniaturization may be more important because the cells that are used are more complex and costly to produce massively. It comes to be particularly important when several assays need to be run in parallel using the same sample, such as cell lysate after stimulation.
Dana-Farber Researchers Use Gene Editing to Short-Circuit Sickle Cell Disease
Sep 16, 2015
NEW YORK (GenomeWeb) – Scientists have developed a gene editing strategy that could help treat sickle cell disease by short-circuiting the mutated hemoglobin causing the disease.
“We’ve now targeted the modifier of the modifier of a disease-causing gene,” Stuart Orkin, chairman of pediatric oncology at Dana-Farber Cancer Institute and associate chief of hematology/oncology at Boston Children’s Hospital, said in a statement. “It’s a very different approach to treating disease.”
Using CRISPR/Cas9 gene editing tools to systematically excise stretches of a promoter region of the enhancer gene BCL11A — which selects the type of hemoglobin that blood cells create — the researchers found an edit that inactivated BCL11A in human blood stem cells. The cut leads cells to increase levels of fetal hemoglobin, resulting in a milder form of sickle cell disease.
The scientists, led by Orkin and Daniel Bauer of Dana-Farber and Boston Children’s, and Feng Zhang of the Broad Institute, published their study today in Nature.
The human genome codes for both a fetal version and an adult version of hemoglobin. A mutation in the adult version of the protein causes sickle cell disease. BCL11A became a target of sickle cell disease research after Orkin’s laboratory revealed its direct role in the transition from fetal to adult hemoglobin in a 2009 study published in Nature. In 2013, a study led by Orkin and Bauer found the promoter region which controls expression of BCL11A in red blood cells.
Musical Scales
http://www.the-scientist.com//?articles.view/articleNo/43794/title/Musical-Scales/
The quest to document an ancient sea creature reveals a cyclical chorus of fish songs.
By Kerry Grens | September 1, 2015
http://www.the-scientist.com/Sept2015/notebook2.jpg

fish songs
Several years ago, ichthyologist Eric Parmentier met a French marine biologist and filmmaker, Laurent Ballesta, who was organizing an expedition to South Africa to produce a documentary film on the coelacanth. This ancient fish—one whose fossil record dates back at least 350 million years—has an almost mythical legacy. Although it was widely assumed to have gone extinct 65 million years ago, a live specimen was found in 1938, and scientists have identified two extant species of coelacanth. Both species move in a peculiar way, waggling four lobe-like fins in an alternating pattern, as we do our arms and legs. Their anatomy is also unusual: a tiny brain, a joint at the back of the head that allows the animal to open its jaws widely, and only rudimentary vertebrae. Ballesta’s trip inspired Parmentier, who studies fish acoustics, to collaborate with the team. “I hoped to be the first guy to record [sounds of] the coelacanth.”
In the spring of 2013, divers successfully planted a hydrophone inside the cave and also shot video footage of a coelacanth. (The resulting documentary by Ballesta is available on YouTube. Although it is in French, the footage obviates the need for fluency to enjoy the film.) Day and night, for weeks, the hydrophone dutifully recorded the sounds within the cave. When Parmentier retrieved the files and went to analyze the recordings, there was one big problem: it was filled with dozens of different fish calls. “Maybe the coelacanth is in these sound files, but it’s completely masked by the other sounds,” he says.
Nonetheless, the tape captured ceaseless, never-before-heard chatter among the aquatic organisms within the cave (PNAS, 112:6092-97, 2015). To make some sense of it, Parmentier’s team undertook the laborious task of characterizing the sounds recorded over 19 nonconsecutive days (to make this feasible, the group pared down its analysis to the first nine minutes of every hour). The researchers assigned more than 2,700 sounds to 17 groups, most of which sounded to Parmentier like fish (one group was clearly dolphin, based on its high frequency, he says). These included frog-like croaks, grunts that sounded like a creaking door, a moan, and one that sounded like a whistle blown under water. “It’s fair to say, based on the characteristics of the sounds they were hearing, they are probably fish sounds,” says Erica Staaterman, a postdoc at the Smithsonian who studies fish acoustic communication.
NIH Awards Beth Israel Team $3M to Continue Study of Heart Disease Biomarker Sep 17, 2015
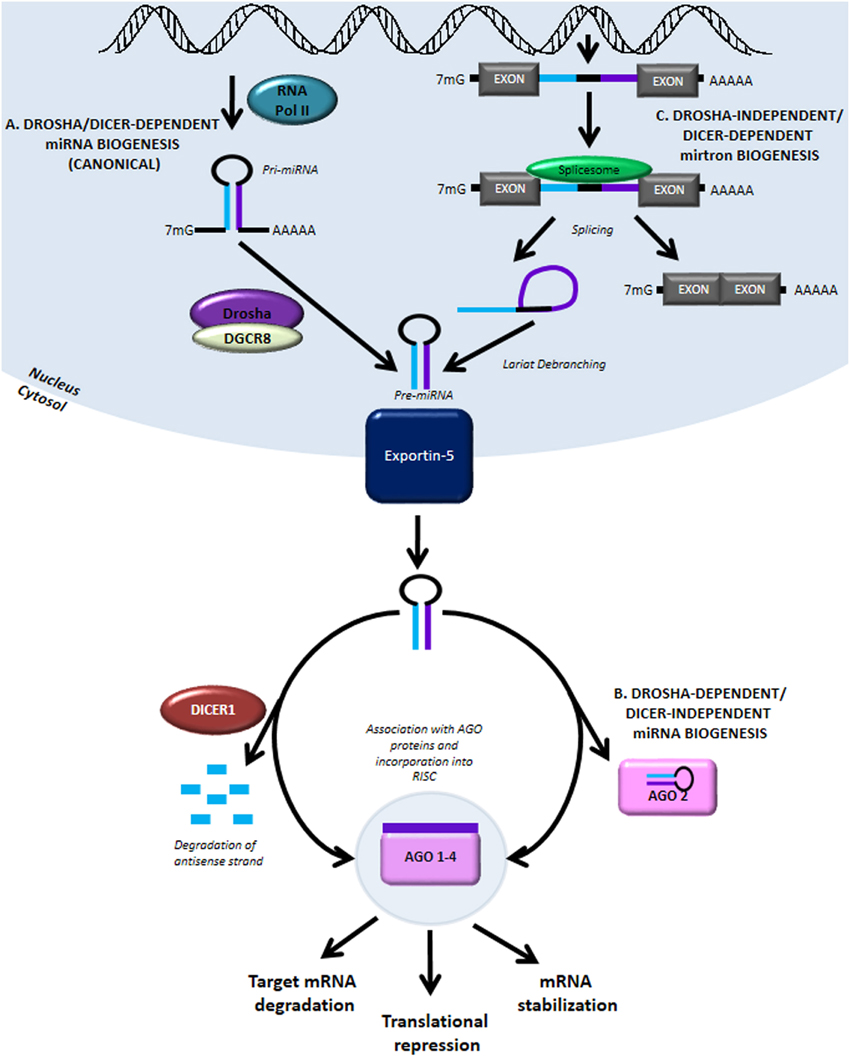
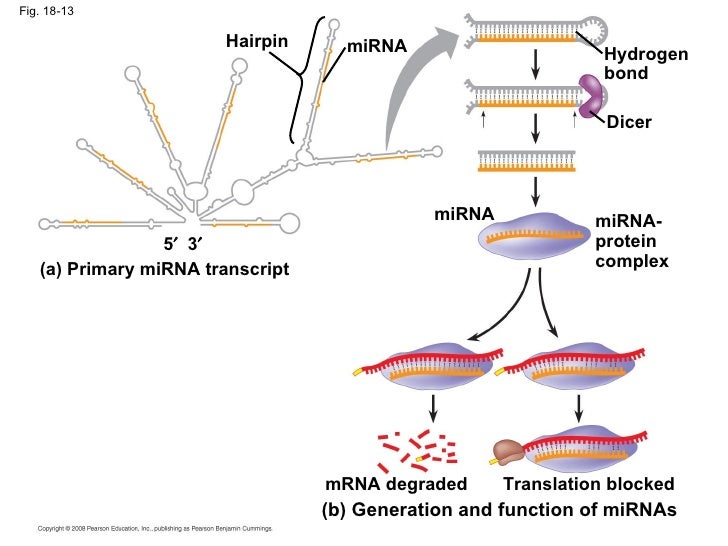
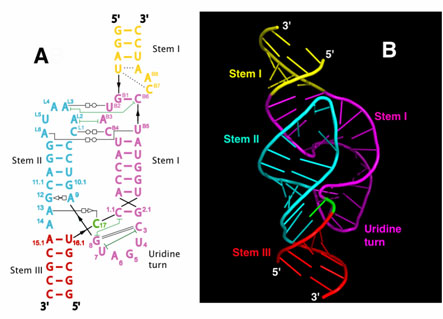
NEW YORK (GenomeWeb) – The National Institutes of Health has awarded a Beth Israel Deaconess Medical Center (BIDMC) research team $3 million in funding to support the second phase of an effort to identify microRNAs that can be used to predict clinical outcomes of heart disease patients.
The grant, which was awarded under the NIH’s Extracellular RNA Communication program, follows a $4 million award the group received to kick off the project in 2013.
To date, the team has identified a number of miRNA biomarker candidates including miR-30d, which the researchers reported earlier this year as a predictor of beneficial cardiac remodeling in patients following a heart attack and a key player in preventing cell death.
With the latest grant, the investigators aim to validate miR-30d and other candidate miRNAs in several large patient cohorts.

microRNA-based tests
https://www.youtube.com/watch?v=oMaiIyGfhQw
Unraveling determinants of transcription factor binding outside the core binding site
Michal Levo, Einat Zalckvar, Eilon Sharon, Ana Carolina Dantas Machado, Yael Kalma, Maya Lotam-Pompan, Adina Weinberger, Zohar Yakhini, Remo Rohs and Eran Segal. “Unraveling determinants of transcription factor binding outside the core binding site”. Genome Res. July 2015 25: 1018-1029.
http://genome.cshlp.org/content/25/7/1018.abstract
Binding of transcription factors (TFs) to regulatory sequences is a pivotal step in the control of gene expression. Despite many advances in the characterization of sequence motifs recognized by TFs, our ability to quantitatively predict TF binding to different regulatory sequences is still limited. Here, we present a novel experimental assay termed BunDLE-seq that provides quantitative measurements of TF binding to thousands of fully designed sequences of 200 bp in length within a single experiment. Applying this binding assay to two yeast TFs, we demonstrate that sequences outside the core TF binding site profoundly affect TF binding. We show that TF-specific models based on the sequence or DNA shape of the regions flanking the core binding site are highly predictive of the measured differential TF binding. We further characterize the dependence of TF binding, accounting for measurements of single and co-occurring binding events, on the number and location of binding sites and on the TF concentration. Finally, by coupling our in vitro TF binding measurements, and another application of our method probing nucleosome formation, to in vivo expression measurements carried out with the same template sequences serving as promoters, we offer insights into mechanisms that may determine the different expression outcomes observed. Our assay thus paves the way to a more comprehensive understanding of TF binding to regulatory sequences and allows the characterization of TF binding determinants within and outside of core binding sites.
Defective Mitochondria Transform Normal Cells into Tumors
An international research team reports that defects in mitochondria, play a key role in the transition from normal cells to cancerous ones. When the scientists disrupted a key component of mitochondria, otherwise normal cells took on characteristics of cancerous tumor cells.
Their study (“Disruption of cytochrome c oxidase function induces the Warburg effect and metabolic reprogramming”) is published Oncogene and was led by members of the lab of Narayan G. Avadhani, Ph.D., the Harriet Ellison Woodward Professor of Biochemistry in the department of biomedical sciences in the school of veterinary medicine at the University of Pennsylvania. Satish Srinivasan, Ph.D., a research investigator in Dr. Avadhani’s lab, was the lead author.
In 1924, German biologist Otto Heinrich Warburg observed that cancerous cells consumed glucose at a higher rate than normal cells and had defects in their grana, the organelles that are now known as mitochondria. He postulated that the mitochondrial defects led to problems in the process by which the cell produces energy, called oxidative phosphorylation, and that these defects contributed to the cells becoming cancerous.
“The first part of the Warburg hypothesis has held up solidly in that most proliferating tumors show high dependence on glucose as an energy source and they release large amounts of lactic acid,” said Dr. Avadhani. “But the second part, about the defective mitochondrial function causing cells to be tumorigenic, has been highly contentious.”




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}