Peroxisome proliferator-activated receptor (PPAR-gamma) Receptors Activation: PPARγ transrepression for Angiogenesis in Cardiovascular Disease and PPARγ transactivation for Treatment of Diabetes
UPDATED on 11/27/2018
A new combination drug therapy for CVD patients with co-morbidity of DM2 is presented in the following article, representing different mechanism of actions, pathways and a novel treatment proposed in 2018:
Cardiovascular (CV) Disease and Diabetes: New ACC Guidelines for use of two major new classes of diabetes drugs — sodium-glucose cotransporter type 2 (SGLT2) inhibitors and glucagon-like peptide 1 receptor agonists (GLP-1RAs) for reduction of adverse outcomes
https://pharmaceuticalintelligence.com/2018/11/27/cardiovascular-cv-disease-and-diabetes-new-acc-guidelines-for-use-of-two-major-new-classes-of-diabetes-drugs-sodium-glucose-cotransporter-type-2-sglt2-inhibitors-and-glucagon-like/
The title of this article
Peroxisome proliferator-activated receptor (PPAR-gamma) Receptors Activation: PPARγ transrepression for Angiogenesis in Cardiovascular Disease and PPARγ transactivation for Treatment of Diabetes
represents an explanation for pathways and mechanism of actions of combination drug therapy novel in its conceptualization in 2013.
The research is presented in the following three parts. References for each part are at the end.
PART I: Genetics and Biochemistry of Peroxisome proliferator-activated receptor
Reporter: Aviva Lev-Ari, PhD, RN
PART II: Peroxisome proliferator-activated receptors as stimulants of angiogenesis in cardiovascular disease and diabetes
Reporter: Aviva Lev-Ari, PhD, RN
PART III: PPAR-gamma Role in Activation of eNOS: The Cardiovascular Benefit
Author and Curator: Aviva Lev-Ari, PhD, RN
PART I:
Genetics and Biochemistry of Peroxisome proliferator-activated receptor
PPAR -alpha and -gamma pathways
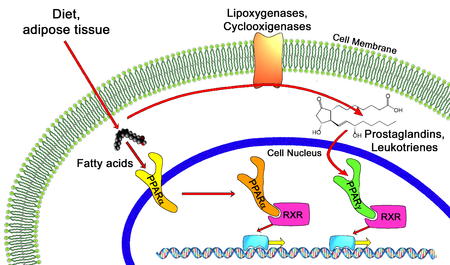
In the field of molecular biology, the peroxisome proliferator-activated receptors (PPARs) are a group of nuclear receptor proteins that function as transcription factors regulating the expression of genes.[1] PPARs play essential roles in the regulation of cellular differentiation, development, and metabolism (carbohydrate, lipid, protein), and tumorigenesis[2] of higher organisms.[3][4]
Three types of PPARs have been identified: alpha, gamma, and delta (beta):[3]
- α (alpha) – expressed in liver, kidney, heart, muscle, adipose tissue, and others[5]
- β/δ (beta/delta) – expressed in many tissues but markedly in brain, adipose tissue, and skin
- γ (gamma) – although transcribed by the same gene, this PPAR through alternative splicing is expressed in three forms:
- γ1 – expressed in virtually all tissues, including heart, muscle, colon, kidney, pancreas, and spleen
- γ2 – expressed mainly in adipose tissue (30 amino acids longer)
- γ3 – expressed in macrophages, large intestine, white adipose tissue
Physiological function
All PPARs heterodimerize with the retinoid X receptor (RXR) and bind to specific regions on the DNA of target genes. These DNA sequences are termed PPREs (peroxisome proliferator hormone response elements). The DNA consensus sequence is AGGTCANAGGTCA, with N being a random nucleotide. In general, this sequence occurs in the promotor region of a gene, and, when the PPAR binds its ligand, transcription of target genes is increased or decreased, depending on the gene. The RXR also forms a heterodimer with a number of other receptors (e.g., vitamin D and thyroid hormone).
The function of PPARs is modified by the precise shape of their ligand-binding domain (see below) induced by ligand binding and by a number of coactivator and corepressor proteins, the presence of which can stimulate or inhibit receptor function, respectively.[9]
Endogenous ligands for the PPARs include free fatty acids and eicosanoids. PPARγ is activated by PGJ2 (a prostaglandin). In contrast, PPARα is activated by leukotriene B4. PPARγ activation by agonist RS5444 may inhibit anaplastic thyroid cancer growth.[10]

Peroxisome proliferator-activated receptor (Photo credit: Wikipedia)
Genetics
The three main forms are transcribed from different genes:
Hereditary disorders of all PPARs have been described, generally leading to a loss in function and concomitant lipodystrophy, insulin resistance, and/or acanthosis nigricans.[11] Of PPARγ, a gain-of-function mutation has been described and studied (Pro12Ala) which decreased the risk of insulin resistance; it is quite prevalent (allele frequency 0.03 – 0.12 in some populations).[12] In contrast, pro115gln is associated with obesity. Some other polymorphisms have high incidence in populations with elevated body mass indexes.
SOURCE:
http://en.wikipedia.org/wiki/Peroxisome_proliferator-activated_receptor
Mechanism of action
Thiazolidinediones or TZDs act by activating PPARs (peroxisome proliferator-activated receptors), a group of nuclear receptors with greatest specificity for PPARγ (gamma). The endogenous ligands for these receptors are free fatty acids (FFAs) and eicosanoids. When activated, the receptor binds to DNA in complex with the retinoid X receptor (RXR), another nuclear receptor, increasing transcription of a number of specific genes and decreasing transcription of others.
PPARγ transactivation

Thiazolidinedione ligand dependent transactivation is responsible for the majority of anti-diabetic effects.
The activated PPAR/RXR dimer binds to peroxisome proliferator hormone response elements upstream of target genes in complex with a number of coactivators such as nuclear receptor coactivator 1 and CREB binding protein, this causes upregulation of genes (for a full list see PPARγ:
TZDs also increase the synthesis of certain proteins involved in fat and glucose metabolism, which reduces levels of certain types of lipids, and circulating free fatty acids. TZDs generally decrease triglycerides and increase high-density lipoprotein cholesterol (HDL-C) and low-density lipoprotein cholesterol (LDL-C). Although the increase in LDL-C may be more focused on the larger LDL particles, which may be less atherogenic, the clinical significance of this is currently unknown. Nonetheless, rosiglitazone, a certain glitazone, was suspended from allowed use by medical authorities in Europe, as it has been linked to an increased risk of heart attack and stroke.[3]
PPARγ transrepression

Thiazolidinedione ligand dependent transrepression mediates the majority of anti-inflammatory effects.
Binding of PPARγ to coactivators appears to reduce the levels of coactivators available for binding to pro-inflammatory transcription factors such as NF-κB, this causes a decrease in transcription of a number of pro inflammatory genes, including various interleukins and tumour necrosis factors.
SOURCE:
http://en.wikipedia.org/wiki/Thiazolidinedione
1. Waki H, Yamauchi T, Kadowaki T (February 2010). “[Regulation of differentiation and hypertrophy of adipocytes and adipokine network by PPARgamma]” (in Japanese). Nippon Rinsho 68 (2): 210–6. PMID 20158086.
2. Panigrahy D, Singer S, Shen LQ, et al. (2002). “PPARγ ligands inhibit primary tumor growth and metastasis by inhibiting angiogenesis”. J. Clin. Invest. 110 (7): 923–32. doi:10.1172/JCI15634. PMC 151148. PMID 12370270.
3. NHS: Avandia diabetes drug suspended, Friday 24th September 2010
Members of the class
The chemical structure of thiazolidinedione
Chemically, the members of this class are derivatives of the parent compound thiazolidinedione, and include:
- Rosiglitazone (Avandia), which was put under selling restrictions in the US and withdrawn from the market in Europe due to an increased risk of cardiovascular events.
- Pioglitazone (Actos), France and Germany have suspended the sale of the diabetes drug Actos after a study suggested the drug, also known as pioglitazone, could raise the risk of bladder cancer.[4]
- Troglitazone (Rezulin), which was withdrawn from the market due to an increased incidence of drug-induced hepatitis.
Experimental agents include netoglitazone, an antidiabetic agent, rivoglitazone, and the early non-marketed thiazolidinedione ciglitazone.
Replacing one oxygen atom in a thiazolidinedione with an atom of sulfur gives a rhodanine.
SOURCE:
http://en.wikipedia.org/wiki/Thiazolidinedione
PART II:
Peroxisome proliferator-activated receptors as Stimulants of Angiogenesis in Cardiovascular Disease and Diabetes
In 2009 in Diabetes Metab Syndr Obes a seminal paper was published on the topic by Desouza, Rentschler and Fonseca. (2009). This work constitutes Part II. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3048019/
Mechanisms by which PPARs may stimulate angiogenesis
PPARs seem to have a protective role in ischemic tissues, including brain, cardiac and skin. A part of this may be by stimulating angiogenesis and improving blood supply. Hypoxia is a trigger for the development of angiogenesis. One of the key mediators in hypoxia-induced angiogenesis is hypoxia inducible factor (HIF-1), which is induced in hypoxic cells and binds to hypoxia response element (HRE). HIF-1 mediates the transcriptional activation of several genes that promote angiogenesis, including VEGF, angiopoeitin (Ang-1, Ang-2), and matrix metalloproteinases (MMP-2, MMP-9).55 15-deoxy-delta(12, 14)-prostaglandin J(2) (15d-PGJ(2)), a PPAR-γ agonist, has been shown to induce HIF-1 expression and thereby angiogenesis (Figure 1).34 However pioglitazone has been shown to suppress the induction of HIF-1.56 Conditions that influence the stimulation or suppression of HIF activation by PPAR-γ are largely unknown.
Several studies suggest that eNOS synthase activation is required for angiogenesis that may be protective under certain conditions.57–59 In one study pioglitazone reduced the myocardial infarct size in part via activation of eNOS.60 PPAR-α activation has also been shown to protect the type 2 diabetic rat myocardium against ischemia-reperfusion injury via the activation of the NO pathway (Table 1, Figure 1).61 However, stimulation of the inducible nitric oxide (iNOS) pathway can lead to undesirable angiogenesis that may be contribute to pathological states such as proliferative retinopathy. PPARs in fact have been shown to suppress iNOS expression, thereby suppressing undesirable angiogenesis.62,63 Here again the factors that allow for activation of eNOS and suppression of iNOS is largely unknown.
The most studied pathway by which PPARs may stimulate angiogenesis is the VEGF pathway. VEGF can stimulate angiogenesis via stimulation of the ERK1/2 pathway. PPAR-β/δ activation has been shown to increase VEGF expression and thereby stimulate angiogenesis (Figure 1).26 In some studies PPAR-α and PPAR-γ have also been shown to increase VEGF expression.47,48 However the majority of studies still show that PPAR activation suppresses VEGF expression. The end result of whether PPAR activation suppresses or stimulates VEGF expression seems to lie in the pathological condition in which its actions are observed (Figure 1). It is likely that PPAR activation results in increased VEGF expression in conditions where new blood vessel formation is required, such as ischemic skin flaps, brain, or cardiac tissue ischemia. On the other hand, pathological angiogenesis such as in the eye or within an atherosclerotic plaque is suppressed by PPAR activation via a suppression of VEGF (Figure 1).

Mechanisms by which PPARs effect angiogenesis.

Effect of PPARs on angiogenesis
Recently some studies indicate that PPARs may increase the expression and activation of the phosphatidylinositol-3-kinase (PI3K/AKT) pathway.61,64 The PI3K/AKT pathway stimulates angiogenesis.59,65 Again the majority of studies show that PPAR activation inhibits PI3K/AKT activation.
It is very likely that a large amount of variation found in different studies is due to the use of agonists and antagonists of the PPAR receptors that exhibit direct PPAR-independent effects. Most study designs do not distinguish between direct effects and indirect effects of various pharmacological agonists/antagonist used. Fibrates and TZDs have both been shown to have direct independent effects on inflammation, proliferation and angiogenesis. Hence it is difficult to conclude that all the pro and antiangiogenic effects seen in various studies are a result of PPAR activation exclusively.
Clinical significance and conclusions
Some compounds such as TZDs and fibrates are routinely used in patients with diabetes, dyslipidemia, and cardiovascular disease. Other compounds such as partial agonists or dual agonists of PPAR-α and PPAR-γ are in development. The effects of these newer compounds, on angiogenesis and cardiovascular disease are yet to be determined. Current evidence from clinical trials suggest a mixed picture. TZD treatment in patients with type 2 diabetes has been shown to be associated with macular edema. On the other hand, the FIELD study using fenofibrate showed a decrease in the need for laser treatments in patients with diabetic retinopathy. The PROACTIVE study showed that pioglitazone trended to decrease certain cardiovascular endpoints. In some studies, rosiglitazone increased the risk of cardiovascular events. In other studies such as ACCORD and VADT, TZD treatment was not associated with increased cardiovascular event risk. Several factors, including the study design, PPAR receptor affinity, and the PPAR-independent actions of these compounds, possibly play a role in the differences in results seen. The duration of the pathological state and the vasculature of the effected organ likely play a role in whether PPARs prove beneficial or harmful. In conclusion it may be prudent to summarize that at this point the evidence suggests that PPARs can either stimulate or inhibit angiogenesis, depending on the biological context and pathological process.
Clinical Trials: Controversial Research Results
Peroxisome proliferator-activated receptors (PPARs) are a group of nuclear hormone receptors that regulate lipid and glucose metabolism. PPAR-α agonists such as fenofibrate and PPAR-γ agonists such as the thiozolidinediones have been used to treat dyslipidemia and insulin resistance in diabetes. Over the past few years research has discovered the role of PPARs in the regulation of inflammation, proliferation, and angiogenesis. Clinical trials looking at the effect of PPAR agonists on cardiovascular outcomes have produced controversial results. Studies looking at angiogenesis and proliferation in various animal models and cell lines have shown a wide variation in results. This may be due to the differential effects of PPARs on proliferation and angiogenesis in various tissues and pathologic states. This review discusses the role of PPARs in stimulating angiogenesis. It also reviews the settings in which stimulation of angiogenesis may be either beneficial or harmful.
affect inflammation, proliferation, immune function and angiogenesis.3 There are three PPAR isotypes, PPAR-α, PPAR-β/δ, and PPAR-γ. They form heterodimers with the retinoid X receptors and bind to specific DNA sequences, called peroxisome proliferator response elements (PPRE), in the promoter regions of their target genes. PPARs exhibit isotype-specific tissue expression patterns. PPAR-α is primarily expressed in organs with significant fatty acid catabolism. PPAR-β/δ is expressed in nearly all cell types and the level of expression seems to depend on the amount of angiogenesis, cell proliferation, and differentiation occurring in that specific tissue.4 PPAR-γ is found in adipose tissue and at lower levels in immune cells vascular tissue and some organs. PPAR-γ exists in two protein isoforms, PPAR-γ1 and PPAR-γ2, with different lengths of the N-terminal. The PPAR-γ2 isoform is predominantly expressed in adipose tissue, whereas PPAR-γ1 is relatively widely expressed.5 Expression of each isoform is driven by a specific promoter that confers the distinct tissue expression patterns. There are also two other mRNA variants of PPAR-γ, proteins identical to PPAR-γ1: PPAR-γ3, which is restricted to macrophages, adipose tissue, and colon, and PPAR-γ4, the tissue distribution of which is unclear at this time.5 Human PPAR-γ plays a critical physiological role as a central transcriptional regulator of both adipogenic and lipogenic programs. Its transcriptional activity is induced by the binding of endogenous and synthetic lipophilic ligands, which has led to the determination of many roles for PPAR-γ in pathological states such as type 2 diabetes, atherosclerosis, inflammation, and cancer.
The role of PPARs has traditionally been recognized as antiproliferative and antiangiogenic in a large number of disease states including cancer and cardiovascular disease.4 These studies have led to clinical trials with PPAR agonists to evaluate their benefits in cancer and cardiovascular disease. The results of some of these trials especially in cardiovascular disease have been mixed and hence controversial.
The results obtained with a PPAR-γ agonist pioglitazone do suggest a better impact on the lipid profile compared to rosiglitazone (the former lowers triglyceride significantly and has less adverse effects on low-density lipoprotein [LDL] cholesterol), and at least a mixed result (the primary composite endpoint was not reduced significantly but myocardial infarction, stroke, and death were reduced by 16%), in an outcome trial – PROspective pioglitAzone Clinical Trial In macroVascular Events (PROACTIVE).6 Rosiglitazone on the other hand was found to increase cardiovascular events in a large restrospective analysis study.7
This has led to a lot of recent research into PPARs that is contrary to the traditional literature in their role as inhibitors of angiogenesis. This review will examine the role and evidence of PPARs as promoters of angiogenesis, the mechanisms involved, and the implications thereof.
SOURCE:
Desouza, Rentschler and Fonseca. (2009).
Angiogenesis is described as the formation of new capillaries from the existing vasculature. This process involves the breakdown of the extracellular matrix and formation of an endothelial tube. Angiogenesis is an important physiologic process in the female reproductive cycle, wound healing, and bone formation. Angiogenesis is also a crucial step in several disease states including cancer, diabetic retinopathy, rheumatoid arthritis, stroke, and ischemic coronary artery disease.8–10 Neoangiogenesis has harmful as well as beneficial effects in the setting of type 2 diabetes and cardiovascular disease.10 In the setting of diabetes, there is abnormal regulation and signaling of vascular endothelial growth factor (VEGF) and its receptor Flk-1.11 This may lead to increased levels of circulating VEGF, resulting in increased permeability of vascular structures throughout the body. In the retina, this results in the formation of protein-rich exudates containing VEGF that induces a local inflammatory response resulting in capillary sprouting. A similar process might take place in the arterial wall, thereby promoting capillary sprouting and plaque destabilization.12 At the same time, the lack of Flk-1 activation in endothelial cells and abnormal VEGF-dependent activation of monocytes impair the arteriogenic response that requires monocyte recruitment, and monocyte and endothelial cell migration and proliferation.11 This could lead to a deficient angiogenic response in ischemic tissue. VEGF/Flk-1 signaling may also be required for bone marrow release of circulating endothelial progenitor cells that play a role in endothelial function and arteriogenesis.13 The abnormal release of endothelial progenitors could further reduce arteriogenic response. This has therapeutic implications in terms of vascularization and survival of skin grafts in patients with diabetes as well as vascularization of the ischemic myocardium. An important mechanism by which PPARs seem to regulate angiogenesis is via VEGF.11,12 It would therefore appear that PPARs have a role in regulating both beneficial and harmful effects of angiogenesis thereby leading to controversial results (Figure 1).
The other factor influencing the results of angiogenesis studies is the use of PPAR agonists that have pleotropic effects. PPAR-α agonists such as fibrates stimulate pathways that do not depend on PPAR-α.14 PPAR-γ agonists such as thiozolidinediones (TZDs) have PPARγ independent actions on proliferative and inflammatory pathways.14 Therefore to conclude that the effects of commonly used PPAR agonists on angiogenesis are specifically due to PPAR activation is at best controversial.15
Although the majority of studies point towards the antiproliferative, antiangiogenic properties of PPAR-α, this may be due to the use of fibrates as agonists in these experiments. A lot more research needs to be done using methods such as spontaneous PPAR-α activation, overexpression, silencing and knockout mice, rather than using chemical agonists and antagonists which might have pleotropic effects unrelated to PPAR-α.
PPAR-γ and angiogenesis
PPAR-γ is probably the most studied PPAR, likely due to the use and development of several PPAR-γ agonists such as thiozolidinediones in the treatment of type 2 diabetes. Endogenous ligands for PPAR-γ include long chain polyunsaturated fatty acids and their derivatives, 15-deoxy-Δ12, 14-prostaglandin J2 (15d-PGJ2).4 Other natural ligands include nitrolinoleic acids. 15d-PGJ2 has been found to upregulate the expression of PPAR-γ and also the DNA binding and transcriptional activity.34 Synthetic ligands include TZDs and various nonsteroidal anti-inflammatory drugs.35
Studies supporting antiproliferative properties of PPAR-γ
PPAR-γ has widespread effects involving, inflammation, atherosclerosis, obesity, diabetes, and cancer.36 PPAR-γ agonists directly inhibit tumor cell growth, induce cell differentiation, and apoptosis in various cancer types (Table 1).37 TZDs have been shown to decrease post angioplasty neointimal hyperplasia in both animals and humans (Table 1).38,39 PPAR-γ ligands have been shown to inhibit and stimulate angiogenesis (Table 1). Inhibition by PPAR-γ ligands can occur through direct effects on the endothelium or through indirect effects on the net balance of proangiogenic and antiangiogenic mediators.37 PPAR-γ expressed in choroidal endothelial cells inhibits the differentiation and proliferation of those cells.38,39 Rosiglitazone inhibited endothelial cell proliferation and migration and decreased VEGF-induced tubule formation in human umbilical vein endothelial cells.40,41 In another study PPAR-γ ligands stimulated endothelial cell caspase-mediated apoptosis.42 15d-PGJ2, an endogenous ligand of PPAR-γ, induces growth inhibition, differentiation, and apoptosis of tumor cells.43 PPAR-γ activation interrupts NF-kβ signaling with subsequent blockade of proinflammatory gene expression.43 Pioglitazone and rosiglitazone inhibit the effects of growth factors such as bFGF and VEGF. Endothelial cell migration is also inhibited by both compounds.44 Thus natural and synthetic ligands of PPAR-γ exhibit antiangiogenic properties under certain conditions.
Studies supporting proangiogenic role of PPAR-γ
However, PPAR- ligands have also been shown to stimulate the angiogenic pathway (Table 1). In bovine aortic endothelial cells, prolonged treatment with troglitazone increased VEGF and endothelial nitric oxide (NO) production with no change in endothelial nitric oxide synthase (eNOS) expression.45 In cultured rat myofibroblasts, activation of PPAR-γ by troglitazone and 15-dPGJ2 induced VEGF expression and augmented tubule formation.46 In mice treated with rosiglitazone, angiogenesis was stimulated in adipose tissue with increased expression of VEGF and angiopoeitin-4 (Ang-4). Ang-4 stimulated endothelial cell growth and tubule formation. 47 In rats with focal cerebral ischemia, rosiglitazone treatment enhanced neurologic improvement and reduced the infarct size by reducing caspase-3 activity, increasing the number of endothelial cells, and increasing eNOS expression.48 In the setting of diabetes, PPAR-γ agonists may promote revascularization of ischemic tissue. Diabetic mice with induced unilateral hind limb ischemia, when treated with pioglitazone showed normalization of VEGF, upregulation of eNOS activity, and partial restoration of blood flow recovery.49 In mice treated with pioglitazone, VEGR-receptor-2 positive EPCs were upregulated and migratory capacity was increased. In vivo angiogenesis was increased 2-fold.50 In an endothelial/interstitial cell co-culture assay, treatment with PPAR-γ agonists stimulated production of VEGF. In the same study, corneas treated with the same PPAR-γ agonists increased phosphorylation of eNOS.20
Few studies have evaluated angiogenesis in humans. Pioglitazone treatment has been shown to increase serum VEGF, IL-8, and angiogenin levels in patients with type 2 diabetes.51 In another study thiozolidinedione use in patients with type 2 diabetes was associated with diabetic macular edema.52
PGC-1α and angiogenesis
Peroxisome proliferator-activated receptor (PPAR)-gamma coactivator 1alpha (PGC-1α) is a nuclear transcriptional coactivator that regulates several important metabolic processes, including mitochondrial biogenesis, adaptive thermogenesis, respiration, insulin secretion and gluconeogenesis. 53 PGC-1α also co-activates PPAR-α, PPAR-β/δ, and PPAR-γ which are important transcription factors of genes regulating lipid and glucose metabolism.53 Recently Arany and colleagues have shown that PGC-1α stimulates angiogenesis in ischemic tissues. Using a combination of muscle cell assays and genetically modified mice that over or underexpess PGC-1α, they showed that PGC-1α is a powerful inducer of VEGF expression. PGC-1α did not involve HIF-1 but activated the nuclear receptor, estrogen-related receptor-α (ERR-α).33 PGC-1α−/− mice are viable, suggesting that PGC-1α is not essential in embryonic vascularization but they show a striking failure to reconstitute blood flow in a normal manner to the limb after an ischaemic insult.54 Transgenic expression of PGC-1α in skeletal muscle is protective against ischemic insults. This suggests that PGC-1α plays a more important role in a disease state rather than a physiologically healthy state.
PART III: PPAR-gamma Role in Activation of eNOS: The Cardiovascular Benefit
Author and Curator: Aviva Lev-Ari, PhD, RN
Mechanism of Action (MOA) for ElectEagle‘s component 3
Treatment Regime with PPAR-gamma agonists (TZDs)
For ElectEagle‘s component 1:
Lev-Ari, A., (2012 X). Clinical Trials Results for Endothelin System: Pathophysiological role in Chronic Heart Failure, Acute Coronary Syndromes and MI – Marker of Disease Severity or Genetic Determination?
http://pharmaceuticalintelligence.com/2012/10/19/clinical-trials-results-for-endothelin-system-pathophysiological-role-in-chronic-heart-failure-acute-coronary-syndromes-and-mi-marker-of-disease-severity-or-genetic-determination/
Lev-Ari, A., (2012W). Endothelin Receptors in Cardiovascular Diseases: The Role of eNOS Stimulation
http://pharmaceuticalintelligence.com/2012/10/04/endothelin-receptors-in-cardiovascular-diseases-the-role-of-enos-stimulation/
Lev-Ari, A., (2012V). Inhibition of ET-1, ETA and ETA-ETB, Induction of NO production, stimulation of eNOS and Treatment Regime with PPAR-gamma agonists (TZD): cEPCs Endogenous Augmentation for Cardiovascular Risk Reduction – A Bibliography
http://pharmaceuticalintelligence.com/2012/10/04/inhibition-of-et-1-eta-and-eta-etb-induction-of-no-production-and-stimulation-of-enos-and-treatment-regime-with-ppar-gamma-agonists-tzd-cepcs-endogenous-augmentation-for-cardiovascular-risk-reduc/
For ElectEagle‘s component 2:
Lev-Ari, A. (2012L).. Cardiovascular Disease (CVD) and the Role of agent alternatives in endothelial Nitric Oxide Synthase (eNOS) Activation and Nitric Oxide Production
http://pharmaceuticalintelligence.com/2012/07/19/cardiovascular-disease-cvd-and-the-role-of-agent-alternatives-in-endothelial-nitric-oxide-synthase-enos-activation-and-nitric-oxide-production/
Lev-Ari, A. (2012i). Bystolic’s generic Nebivolol – positive effect on circulating Endothelial Proginetor Cells endogenous augmentation
http://pharmaceuticalintelligence.com/2012/07/16/bystolics-generic-nebivolol-positive-effect-on-circulating-endothilial-progrnetor-cells-endogenous-augmentation/
Three indications for PPAR-gamma agonist (TZD): Experimental agents include netoglitazone, an antidiabetic agent, rivoglitazone, and the early non-marketed thiazolidinedione ciglitazone
- Antisclerosis, angiogenic progenitor cell differentiation and endogenous augmentation of cEPCs
- Stimulation of eNOS
- Decrease insulin resistance
Classic indication: action to decrease insulin resistance. PPAR-gamma receptors are complex and modulate the expression of the genes involved in lipid and glucose metabolism, insulin signal transduction and adipocyte and other tissue differentiation. TZDs have significant effects on vascular endothilium, the immune system, the ovaries, and tumor cells. Some of these responses may be independent of the PPAR-gamma pathway (Nolte and Karam, 2004). TZDs are ligands of PPAR-gamma receptors part of the steroid (estrogen receptor ligands) and thyroid superfamily of nuclear receptors found in muscle, liver and adipocytes. In the gold standard of all pharmacology books, the cardinal indication for TZDs is action to decrease insulin resistance (Nolte and Karam, 2004 in Katzung). However, the recent research has proposed two new indications for Rosiglitazone in addition to the original insulin sensitivity reduction indication.
As implied in Part I of the ElectEagle Project, TDZs were selected for a new indication in the domain of modulation of atherosclerosis (Verma and Szmitko, 2006), (Li et al., 2004)and facilitation of the differentiation of angiogenic progenitor cells, inhibition of vascular smooth muscle, proliferation and migration to improve endothelial function (Wang et al., 2004).
The following three seminal papers on the function of TDZs in modulation of vascular disease served as an inspiration for our extension of their new indication for TDZs in the anti-atherosclerosis domain into the cEPCs endogenous augmentation proposed treatment area.
Verma S, Szmitko, PE, (2006). The vascular biology of peroxisome proliferator-activated receptors: Modulation of atherosclerosis. Can J Cardiol, 22 (Suppl B):12B-17B.
Wang C-H, Ciliberti N, Li S-H, Szmitko PE, Weisel RD, Fedak PWM, Al-Omran M, Cherng W-J, Li R-K, Stanford WL, Verma S., (2004). Rosiglitazone facilitates angiogenic progenitor cell differentiation toward endothelial lineage: a new paradigm in glitazone pleiotropy. Circulation, 109:1392-1400.
Li AC, Binder CJ, Gutierrez A, Brown KK, Plotkin CR, Pattison JW, Valledor AF, Davis RA, Wilson TM, Wizttum JL, Palinski W, Glass CK., (2004). Differential inhibition of macrophage foam-cell formation and atherosclerosis in mice by PPAR alpha, beta/delta, and gamma. J. Clin. Invest., 114:1564-1576.
Namely, in the ElectEagleProject, a finely tuned interpretation is provided. We assume that TZDs may have a potential therapeutic effect on augmentation of cEPCs in a significant way should a combination drug therapy be designed to include Rosiglitazoneand two other drugsonewhich inhibits receptors ETA and ETA-ETB and the other which induces eNOS. TDZs were selected for a new indication related to anti-atherosclerosis, however, we extend and emphasize TZDs function in cell differentiation and cell migration of EPCs following encouraging results by Wang et al., (2004). Thus, we are shifting the indication from atherosclerosis and peripheral vascular disease to cardiovascular and CAD.
In 2005 a new indication for TZDs emerged from new finding about the PPAR-gamma receptors function in cell nitric oxide (NO) release without increasing the expression of endothelial nitric oxide synthase (eNOS) (Polikandriotis et al., 2005). This is an important finding for the drug combination components selected for ElectEagleProject. This subject is covered in the following section, Role of PPAR-gamma in eNOS stimulation.
Mechanism of action (MOA) for ElectEagle‘s components 2 & 3
Role of PPAR-gamma in eNOS stimulation
Polikandriotis et al. (2005), recently reported that the peroxisome proliferator-activated receptor gamma (PPARgamma) ligands 15-deoxy-Delta(12,14)-prostaglandin J2 (15d-PGJ2) and ciglitazone increased cultured endothelial cell nitric oxide (NO) release without increasing the expression of endothelial nitric oxide synthase (eNOS). Their study was designed to characterize further the molecular mechanisms underlying PPARgamma-ligand-stimulated increases in endothelial cell NO production.
Their methods and Results: Treating human umbilical vein endothelial cells (HUVEC) with PPARgamma ligands (10 micromol/L 15d-PGJ2, ciglitazone, or rosiglitazone) for 24 hours increased NOS activity and NO release. In selected studies, HUVEC were treated with PPARgamma ligands and with the PPARgamma antagonist GW9662 (2 micromol/L), which fully inhibited stimulation of a luciferase reporter gene, or with small interfering RNA to PPARgamma, which reduced HUVEC PPARgamma expression. Treatment with either small interfering RNA to PPARgamma or GW9662 inhibited 15d-PGJ2-, ciglitazone-, and rosiglitazone-induced increases in endothelial cell NO release. Rosiglitazone and 15d-PGJ2, but not ciglitazone, increased heat shock protein 90-eNOS interaction and eNOS ser1177 phosphorylation. The heat shock protein 90 inhibitor geldanamycin attenuated 15d-PGJ2- and rosiglitazone-stimulated NOS activity and NO production. Their Conclusion: These findings further clarify mechanisms involved in PPARgamma-stimulated endothelial cell NO release and emphasize that individual ligands exert their effects through distinct PPARgamma-dependent mechanisms
Originally, Rosiglitazone was indicated as an adjunct to diet and exercise to improve glycemic control in patients with type 2 diabetes mellitus who are already treated with combination rosiglitazone and metformin or who are not adequately controlled on metformin alone. As a result of the FDA drug recall of Rosiglitazone, we suggest here several alternatives: Experimental agents include netoglitazone, an antidiabetic agent, rivoglitazone, and the early non-marketed thiazolidinedione ciglitazone
In the ElectEagle project, Rosiglitazone was identified for a new indication – as a PPAR-gamma agonist implicated with efficacy for endogenous augmentation of cEPCs which serves as a biomarker for CVD risk reduction — an extension of the anti-atherosclerosis indication or the confinement to perileral vascular endothelium (Verma & Szmitko), (Wang et al., 2004), (Li et al., 2004).
Polikandriotis et al. (2005) is a very import publication for ElectEagle project for the following critical five reasons:
- Polikandriotis et al. (2005) clarify the mechanism of action of PPAR-gamma agonists at the protein level in a set of novel experiments, thus contributes to the understanding of the physiological process of the mechanism of action of PPAReceptor-gamma and its relations to L-arginine: NO pathway and its impact in many areas of research, notably vascular biology.
- Polikandriotis et al. (2005) compare two PPAR-gamma agonist agents and confirm Rosiglitazone to be the more potent among the two for the experiments described above
- Polikandriotis et al. (2005) identify Rosiglitazone capability to stimulate endothelial cell NO release, which is a third indication for Rosiglitazone.
- The combination drug therapy selected in May 2006, for the ElectEagle project involved three drugs. Two of which where a PPAR-gamma agonist, specifically, Rosiglitazone. The other drug was an eNOS agonist to stimulate NO production and reuptake. By identifying Rosiglitazone capability to stimulate endothelial cell NO release, Polikandriotis et al. (2005) offer reassurance for the selection of Rosiglitazone in the first place, and further more we became aware that it will exert synergies with the drug chosen as an eNOS agonist.
- In the ElectEagle project, a new experiment is called for following Polikandriotis et al. (2005) findings on Rosiglitazone impact on NO release. It will be needed to measure the incremental induction of NO release resulting from a combination therapy which includes an eNOS agonist and a PPAR-gamma agonist implicated in 2005 with stimulant effects on the release NO.
Moncada & Higgs, (2006) explain that the low concentrations of NO generated by eNOS protect against atherosclerosis by promoting vasodilatation, inhibiting leucocyte and platelet adhesion and/or aggregation and smooth muscle cell proliferation. However, higher concentrations of NO generated by iNOS promote atherosclerosis, either directly or via the formation of NO adducts, such as peroxynitrite. Such a paradox in the action of NO was apparent from their experiments some years ago, in which they found that the acute vascular injury in the ileum and colon following administration of lipopolysaccharide is aggravated by early treatment with a NO synthase inhibitor, whereas delayed administration of such a compound provides protection against the damage to the intestinal vasculature (Laszlo et al., 1994). A prominent example of this comes from experiments in Apo-Emutant mice in which the concomitant knocking out of eNOS leads to an increase in atherosclerosis, while the knocking out of iNOS reduces atherosclerosis (Moncada, 2005).
Research Goals in characterization of ElectEagle Version I
Provided rationale for agent selection for
ElectEagle Version I – Component 3: Treatment Regime with PPAR-gamma agonists (TZD)
agent selection: Rosiglitazone
As a result of the FDA drug recall of Rosiglitazone, we suggest here several alternatives: Experimental agents include netoglitazone, an antidiabetic agent, rivoglitazone, and the early non-marketed thiazolidinedione ciglitazone
Retionale: See discussion on TZDs MOA, above
a-priori postulates presented in Part I for Component 3: PPAR-gamma
- dose concentration dependence on PPAReceptor-gamma – confirmed by a study for Rosiglitazone and a study for Ciglitazone
| PPAReceptor-gamma agonists |
time concentration dependence manner |
dose concentration dependencemanner |
time and dose |
dose |
| Rosiglitazone |
|
Polikandriotis et al., (2005) |
|
maximum recommended daily dose of 8 mg to 2,000 mg. |
| Ciglitazone |
|
Polikandriotis et al., (2005) |
|
|
Proposed integration plan for ElectEagle’s Version I with CVD patients current medication regimen for selective medical diagnoses
Blood Pressure Medicine:
Beta blockers, Verapamil (Calan), Reserpine (Hydropes), Clonidine (Catapres), Methyldopa (Aldomet)
Diuretics:
Thiazides, Spironolactone (Aldactone), Hydralazine
Antidepressants:
Prozac, Lithium, MOA’s, Tricyclics
Stomach Medicine:
Tagamet and Zantac, plus other compounds containing Cimetidine and Ranitidine or associated compounds in Anticholesterol Drugs
Antipsychotics:
Chlorpromazine (Thorazine), Pimozide (Orap), Thiothixine (Navane), Thiordazine (Mellaril), Sulpiride, Haloperidol (haldol), Fluphenazine (Modecate, Prolixin)
Heart Medicine:
Clofibrate (Atromid), Gemfibrozil, Diagoxin
Hormones:
Estrogen, Progesterone, Proscar, Casodex, Eulexin, Corticosteroids Gonadotropin releasing antagonists: Zoladex and Lupron
Cytotoxic agents:
Cyclophosphamide, Methotrexate, Roferon Non-steroidal anti-inflammatories
Others-
Alprazolam, Amoxapine, Chlordiazepoxide, Sertraline, Paroxetine, Clomipramine, Fluvoxamine, Fluoxetine, Imipramine, Doxepine, Desipramine, Clorprothixine, Bethanidine, Naproxen, Nortriptyline, Thioridazine, Tranylcypromine, Venlafaxine, Citalopram.
INTERACTIONS for Nebivolol – Component 2
Calcium Antagonists:
Caution should be exercised when administering beta-blockers with calcium antagonists of the verapamil or diltiazem type because of their negative effect on contractility and atrio-ventricular conduction. Exaggeration of these effects can occur particularly in patients with impaired ventricular function and/or SA or AV conduction abnormalities. Neither medicine should therefore be administered intravenously within 48 hours of discontinuing the other.
Anti-arrhythmics:
Caution should be exercised when administering beta-blockers with Class I anti-arrhythmic drugs and amiodarone as their effect on atrial conduction time and their negative inotropic effect may be potentiated. Such interactions can have life threatening consequences.
Clonidine:
Beta-blockers increase the risk of rebound hypertension after sudden withdrawal of chronic clonidine treatment.
Digitalis:
Digitalis glycosides associated with beta-blockers may increase atrio-ventricular conduction times. Nebivolol does not influence the kinetics of digoxin & clinical trials have not shown any evidence of an interaction.
Special note: Digitalisation of patients receiving long term beta-blocker therapy may be necessary if congestive cardiac failure is likely to develop. The combination can be considered despite the potentiation of the negative chronotropic effect of the two medicines. Careful control of dosages and of individual patient’s response (notably pulse rate) is essential in this situation.
Insulin & Oral Antidiabetic drugs:
Glucose levels are unaffected, however symptoms of hypoglycemia may be masked.
Anaesthetics:
Concomitant use of beta-blockers & anaesthetics e.g. ether, cyclopropane & trichloroethylene may attenuate reflex tachycardia & increase the risk of hypotension
| Medical Diagnoses |
Current medication regiment |
ET-1, ETA and ETA-ETBinhibition |
eNOS agonistsproduction stimulation of NO |
PPAR-gamma agonist (TZD) |
PPAR-gamma agonist (TZD) as eNOS stimulant |
| CAD patients |
Beta blockers, ACEI, ARB, CCB, Diagoxin, Coumadin |
yes |
yes |
yes |
|
| Endothelial Dysfunction in DM patients with or without Erectile Dysfunction |
Insulin |
yes |
yes |
yes |
yes |
| Atherosclerosis patients: Arteries and or veins |
AntihypertensiveCoumadin |
yes |
yes |
yes |
yes |
| pre-stenting treatment phase |
Beta blockers, Verapamil (Calan), Reserpine (Hydropes), Clonidine (Catapres), Methyldopa (Aldomet) |
yes |
yes |
yes |
|
| post-stenting treatment phase |
Antiplatelets |
yes |
|
|
yes |
| if stent is a Bare Mesh stent (BMS) |
CoumadinBeta blockers |
yes |
|
|
yes |
| if stent is Drug Eluting stent (DES) |
antibiotics |
yes |
|
|
|
| if stent is EPC antibody coated |
|
yes |
|
|
yes |
| post CABG patients |
CoumadinBeta blockers, Verapamil(Calan), Reserpine (Hydropes), Clonidine (Catapres), Methyldopa (Aldomet) |
yes |
|
|
yes |
| CVD patients on blood thinner |
Coumadin |
yes |
yes |
yes |
|
Conclusions
- Most favorable and unexpected to us was finding in the literature new indications for TDZs as stimulators of eNOS, in addition to the new indication for atherosclerosis besides the classic indication in pharmacology books, being in the reduction of insulin resistance. Reassuring our selection of Rosiglitazone. As a result of the FDA recoll, the drug substitute will be an Experimental agents include netoglitazone, an antidiabetic agent, rivoglitazone, and the early non-marketed thiazolidinedione ciglitazone
- Most favorable and unexpected to us was finding in the literature new indications for beta blockers as NO stimulant, nebivolol, a case in point, thus, fulfilling two indications in one drug along the direction of the study to identify eNOS agonists.
- The following combination of drugs was selected for ElectEagle Version I
Bosentan (Tracleer), Oral: 62.5 mg tablets
Nebivolol, Oral: 5mg once daily
Experimental agents include netoglitazone, an antidiabetic agent, rivoglitazone, and the early non-marketed thiazolidinedione ciglitazone
- We confirmed time and dose concentrations postulating apriori in most cases. Additional literature searches will benefit the project for the three drugs selected
- We have identified Inhibition of ET-1, ETA and ETA-ETB as one of the agent in the drug combination. The entire literature on cEPCs does not implicate Endothelin with impact on eEPCs while it is known that mechanical stress increase its secretion, this type of stress is implicated with hypertension. To leave out ET-1 from the cEPCs function in CVD risk equates to leaving out Thrombin from the coagulation cascade. ElectEagle Version I corrects that ommission.
REFERENCES for PART I:
1. Michalik L, Auwerx J, Berger JP, Chatterjee VK, Glass CK, Gonzalez FJ, Grimaldi PA, Kadowaki T, Lazar MA, O’Rahilly S, Palmer CN, Plutzky J, Reddy JK, Spiegelman BM, Staels B, Wahli W (2006). “International Union of Pharmacology. LXI. Peroxisome proliferator-activated receptors”. Pharmacol. Rev. 58 (4): 726–41. doi:10.1124/pr.58.4.5. PMID 17132851.
2. Belfiore A, Genua M, Malaguarnera R (2009). “PPAR-gamma Agonists and Their Effects on IGF-I Receptor Signaling: Implications for Cancer”. PPAR Res 2009: 830501. doi:10.1155/2009/830501. PMC 2709717. PMID 19609453.
3.a b Berger J, Moller DE (2002). “The mechanisms of action of PPARs”. Annu. Rev. Med. 53: 409–35. doi:10.1146/annurev.med.53.082901.104018. PMID 11818483.
4 Feige JN, Gelman L, Michalik L, Desvergne B, Wahli W (2006). “From molecular action to physiological outputs: peroxisome proliferator-activated receptors are nuclear receptors at the crossroads of key cellular functions”. Prog. Lipid Res. 45 (2): 120–59. doi:10.1016/j.plipres.2005.12.002. PMID 16476485.
5 Tyagi S, Gupta P, Saini AS, Kaushal C, Sharma S (October 2011). “The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases”. J Adv Pharm Technol Res 2 (4): 236–40. doi:10.4103/2231-4040.90879. PMC 3255347. PMID 22247890.
6 Dreyer C, Krey G, Keller H, Givel F, Helftenbein G, Wahli W (1992). “Control of the peroxisomal beta-oxidation pathway by a novel family of nuclear hormone receptors”. Cell 68 (5): 879–87. doi:10.1016/0092-8674(92)90031-7. PMID 1312391.
7 Issemann I, Green S (1990). “Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators”. Nature 347 (6294): 645–50. doi:10.1038/347645a0. PMID 2129546.
8 Schmidt A, Endo N, Rutledge SJ, Vogel R, Shinar D, Rodan GA (1992). “Identification of a new member of the steroid hormone receptor superfamily that is activated by a peroxisome proliferator and fatty acids”. Mol. Endocrinol. 6 (10): 1634–41. doi:10.1210/me.6.10.1634. PMID 1333051.
9 Yu S, Reddy JK (2007). “Transcription coactivators for peroxisome proliferator-activated receptors”. Biochim. Biophys. Acta 1771 (8): 936–51. doi:10.1016/j.bbalip.2007.01.008. PMID 17306620.
10 Marlow LA, Reynolds LA, Cleland AS, Cooper SJ, Gumz ML, Kurakata S, Fujiwara K, Zhang Y, Sebo T, Grant C, McIver B, Wadsworth JT, Radisky DC, Smallridge RC, Copland JA (February 2009). “Reactivation of suppressed RhoB is a critical step for the inhibition of anaplastic thyroid cancer growth”. Cancer Res. 69 (4): 1536–44. doi:10.1158/0008-5472.CAN-08-3718. PMC 2644344. PMID 19208833.
11 Meirhaeghe A, Amouyel P (2004). “Impact of genetic variation of PPARgamma in humans”. Mol. Genet. Metab. 83 (1-2): 93–102. doi:10.1016/j.ymgme.2004.08.014. PMID 15464424.
12 Buzzetti R, Petrone A, Ribaudo MC, Alemanno I, Zavarella S, Mein CA, Maiani F, Tiberti C, Baroni MG, Vecci E, Arca M, Leonetti F, Di Mario U (2004). “The common PPAR-gamma2 Pro12Ala variant is associated with greater insulin sensitivity”. Eur. J. Hum. Genet. 12 (12): 1050–4. doi:10.1038/sj.ejhg.5201283. PMID 15367918.
13 Zoete V, Grosdidier A, Michielin O (2007). “Peroxisome proliferator-activated receptor structures: ligand specificity, molecular switch and interactions with regulators”. Biochim. Biophys. Acta 1771 (8): 915–25. doi:10.1016/j.bbalip.2007.01.007. PMID 17317294.
REFERENCES for PART II:
Part II is based on the following:
Cyrus V Desouza, Lindsey Rentschler, and Vivian Fonseca
Peroxisome proliferator-activated receptors as stimulants of angiogenesis in cardiovascular disease and diabetes Diabetes Metab Syndr Obes. 2009; 2: 165–172. Published online 2009 September 25 PMCID: PMC3048019
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3048019/
1. Wee CC, Hamel MB, Huang A, Davis RB, Mittleman MA, McCarthy EP. Obesity and undiagnosed diabetes in the US. Diabetes Care. 2008;31:1813–1815. [PMC free article] [PubMed]
2. Westphal SA. Obesity, abdominal obesity, and insulin resistance. Clin Cornerstone. 2008;9:23–31. [PubMed]
3. Calkin AC, Thomas MC. PPAR agonists and cardiovascular disease in diabetes. PPAR Res. 2008;245410
4. Duan SZ, Ivashchenko CY, Usher MG, Mortensen RM. PPAR-gamma in the cardiovascular system. PPAR Res. 2008;745804
5. Knouff C, Auwerx J. Peroxisome proliferator-activated receptor-gamma calls for activation in moderation: lessons from genetics and pharmacology. Endocr Rev. 2004;25:899–918. [PubMed]
6. Erdmann E, Dormandy J, Wilcox R, Massi-Benedetti M, Charbonnel B. PROactive 07: pioglitazone in the treatment of type 2 diabetes: results of the PROactive study. Vasc Health Risk Manag. 2007;3:355–370. [PMC free article] [PubMed]
7. Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457–2471. [PubMed]
8. Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1:27–31. [PubMed]
9. Carmeliet P, Baes M. Metabolism and therapeutic angiogenesis. N Engl J Med. 2008;358:2511–2512. [PubMed]
10. Martin A, Komada MR, Sane DC. Abnormal angiogenesis in diabetes mellitus. Med Res Rev. 2003;23:117–145. [PubMed]
11. Simons M. Angiogenesis, arteriogenesis, and diabetes: paradigm reassessed? J Am Coll Cardiol. 2005;46:835–837. [PubMed]
12. Qaum T, Xu Q, Joussen AM, et al. VEGF-initiated blood-retinal barrier breakdown in early diabetes. Invest Ophthalmol Vis Sci. 2001;42:2408–2413. [PubMed]
13. Pitchford SC, Furze RC, Jones CP, Wengner AM, Rankin SM. Differential mobilization of subsets of progenitor cells from the bone marrow. Cell Stem Cell. 2009;4:62–72. [PubMed]
14. Jandeleit-Dahm KA, Calkin A, Tikellis C, Thomas M. Direct antiatherosclerotic effects of PPAR agonists. Curr Opin Lipidol. 2009;20:24–29. [PubMed]
15. Pozzi A, Ibanez MR, Gatica AE, et al. Peroxisomal proliferator-activated receptor-alpha-dependent inhibition of endothelial cell proliferation and tumorigenesis. J Biol Chem. 2007;282:17685–17695. [PubMed]
16. Grabacka M, Reiss K. Anticancer properties of PPARalpha – effects on cellular metabolism and inflammation. PPAR Res. 2008;930705
17. Scott R, O’Brien R, Fulcher G, et al. Effects of fenofibrate treatment on cardiovascular disease risk in 9,795 individuals with type 2 diabetes and various components of the metabolic syndrome: the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) study. Diabetes Care. 2009;32:493–498. [PMC free article] [PubMed]
18. Kasai T, Miyauchi K, Yokoyama T, Aihara K, Daida H. Efficacy of peroxisome proliferative activated receptor (PPAR)-alpha ligands, fenofibrate, on intimal hyperplasia and constrictive remodeling after coronary angioplasty in porcine models. Atherosclerosis. 2006;188:274–280. [PubMed]
19. Gizard F, Amant C, Barbier O, et al. PPAR alpha inhibits vascular smooth muscle cell proliferation underlying intimal hyperplasia by inducing the tumor suppressor p16INK4a. J Clin Invest. 2005;115:3228–3238. [PMC free article] [PubMed]
20. Biscetti F, Gaetani E, Flex A, et al. Selective activation of peroxisome proliferator-activated receptor (PPAR)alpha and PPAR gamma induces neoangiogenesis through a vascular endothelial growth factor-dependent mechanism. Diabetes. 2008;57:1394–1404. [PubMed]
21. Biscetti F, Gaetani E, Flex A, et al. Peroxisome proliferator-activated receptor alpha is crucial for iloprost-induced in vivo angiogenesis and vascular endothelial growth factor upregulation. J Vasc Res. 2009;46:103–108. [PubMed]
22. Fauconnet S, Lascombe I, Chabannes E, et al. Differential regulation of vascular endothelial growth factor expression by peroxisome proliferator-activated receptors in bladder cancer cells. J Biol Chem. 2002;277:23534–23543. [PubMed]
23. Wang N. PPAR-delta in Vascular Pathophysiology. PPAR Res. 2008;164163
24. Berry DC, Noy N. All-trans-retinoic acid represses obesity and insulin resistance by activating both PPAR{beta}/{delta} and RAR. Mol Cell Biol. 2009;29:3286–3296. [PMC free article] [PubMed]
25. Stephen RL, Gustafsson MC, Jarvis M, et al. Activation of peroxisome proliferator-activated receptor delta stimulates the proliferation of human breast and prostate cancer cell lines. Cancer Res. 2004;64:3162–3170. [PubMed]
26. Piqueras L, Reynolds AR, Hodivala-Dilke KM, et al. Activation of PPARbeta/delta induces endothelial cell proliferation and angiogenesis. Arterioscler Thromb Vasc Biol. 2007;27:63–69. [PubMed]
27. Gaudel C, Schwartz C, Giordano C, Abumrad NA, Grimaldi PA. Pharmacological activation of PPARbeta promotes rapid and calcineurin-dependent fiber remodeling and angiogenesis in mouse skeletal muscle. Am J Physiol Endocrinol Metab. 2008;295:E297–E304. [PubMed]
28. Yoshinaga M, Kitamura Y, Chaen T, et al. The simultaneous expression of peroxisome proliferator-activated receptor delta and cyclooxygenase-2 may enhance angiogenesis and tumor venous invasion in tissues of colorectal cancers. Dig Dis Sci. 2009;54:1108–1114. [PubMed]
29. He T, Lu T, d’Uscio LV, Lam CF, Lee HC, Katusic ZS. Angiogenic function of prostacyclin biosynthesis in human endothelial progenitor cells. Circ Res. 2008;103:80–88. [PMC free article] [PubMed]
30. Muller-Brusselbach S, Komhoff M, Rieck M, et al. Deregulation of tumor angiogenesis and blockade of tumor growth in PPARbeta-deficient mice. Embo J. 2007;26:3686–3698. [PMC free article] [PubMed]
31. Muller R, Komhoff M, Peters JM, Muller-Brusselbach S. A Role for PPARbeta/delta in Tumor Stroma and Tumorigenesis. PPAR Res. 2008;534294
32. Wang D, Wang H, Guo Y, et al. Crosstalk between peroxisome proliferator-activated receptor delta and VEGF stimulates cancer progression. Proc Natl Acad Sci U S A. 2006;103:19069–19074. [PMC free article] [PubMed]
33. Hollingshead HE, Killins RL, Borland MG, et al. Peroxisome proliferator-activated receptor-beta/delta (PPARbeta/delta) ligands do not potentiate growth of human cancer cell lines. Carcinogenesis. 2007;28:2641–2649. [PubMed]
34. Kim EH, Surh YJ. 15-deoxy-Delta12, 14-prostaglandin J2 as a potential endogenous regulator of redox-sensitive transcription factors. Biochem Pharmacol. 2006;72:1516–1528. [PubMed]
35. Lehmann JM, Lenhard JM, Oliver BB, Ringold GM, Kliewer SA. Peroxisome proliferator-activated receptors alpha and gamma are activated by indomethacin and other non-steroidal anti-inflammatory drugs. J Biol Chem. 1997;272:3406–3410. [PubMed]
36. Pershadsingh HA. Peroxisome proliferator-activated receptor-gamma: therapeutic target for diseases beyond diabetes: quo vadis? Expert Opin Investig Drugs. 2004;13:215–228.
37. Giaginis C, Tsantili-Kakoulidou A, Theocharis S. Peroxisome proliferator-activated receptor-gamma ligands: potential pharmacological agents for targeting the angiogenesis signaling cascade in cancer. PPAR Res. 2008;431763
38. Rosmarakis ES, Falagas ME. Effect of thiazolidinedione therapy on restenosis after coronary stent implantation: a meta-analysis of randomized controlled trials. Am Heart J. 2007;154:144–150. [PubMed]
39. Desouza CV, Gerety M, Hamel FG. Long-term effects of a PPAR-gamma agonist, pioglitazone, on neointimal hyperplasia and endothelial regrowth in insulin resistant rats. Vascul Pharmacol. 2007;46:188–194. [PubMed]
40. Panigrahy D, Singer S, Shen LQ, et al. PPARgamma ligands inhibit primary tumor growth and metastasis by inhibiting angiogenesis. J Clin Invest. 2002;110:923–932. [PMC free article] [PubMed]
41. Sheu WH, Ou HC, Chou FP, Lin TM, Yang CH. Rosiglitazone inhibits endothelial proliferation and angiogenesis. Life Sci. 2006;78:1520–1528. [PubMed]
42. Bishop-Bailey D, Hla T. Endothelial cell apoptosis induced by the peroxisome proliferator-activated receptor (PPAR) ligand 15-deoxy-Delta12, 14-prostaglandin J2. J Biol Chem. 1999;274:17042–17048. [PubMed]
43. Giri S, Rattan R, Singh AK, Singh I. The 15-deoxy-delta12,14-prostaglandin J2 inhibits the inflammatory response in primary rat astrocytes via down-regulating multiple steps in phosphatidylinositol 3-kinase-Akt-NF-kappaB- p300 pathway independent of peroxisome proliferator-activated receptor gamma. J Immunol. 2004;173:5196–5208. [PubMed]
44. Aljada A, O’Connor L, Fu YY, Mousa SA. PPAR gamma ligands, rosiglitazone and pioglitazone, inhibit bFGF- and VEGF-mediated angiogenesis. Angiogenesis. 2008;11:361–367. [PubMed]
45. Cho DH, Choi YJ, Jo SA, Jo I. Nitric oxide production and regulation of endothelial nitric-oxide synthase phosphorylation by prolonged treatment with troglitazone: evidence for involvement of peroxisome proliferator-activated receptor (PPAR) gamma-dependent and PPARgamma-independent signaling pathways. J Biol Chem. 2004;279:2499–2506. [PubMed]
46. Chintalgattu V, Harris GS, Akula SM, Katwa LC. PPAR-gamma agonists induce the expression of VEGF and its receptors in cultured cardiac myofibroblasts. Cardiovasc Res. 2007;74:140–150. [PubMed]
47. Gealekman O, Burkart A, Chouinard M, Nicoloro SM, Straubhaar J, Corvera S. Enhanced angiogenesis in obesity and in response to PPAR-gamma activators through adipocyte VEGF and ANGPTL4 production. Am J Physiol Endocrinol Metab. 2008;295:E1056–E1064. [PMC free article] [PubMed]
48. Chu K, Lee ST, Koo JS, et al. Peroxisome proliferator-activated receptor-gamma-agonist, rosiglitazone, promotes angiogenesis after focal cerebral ischemia. Brain Res. 2006;1093:208–218. [PubMed]
49. Huang PH, Sata M, Nishimatsu H, Sumi M, Hirata Y, Nagai R. Pioglitazone ameliorates endothelial dysfunction and restores ischemia-induced angiogenesis in diabetic mice. Biomed Pharmacother. 2008;62:46–52. [PubMed]
50. Gensch C, Clever YP, Werner C, Hanhoun M, Bohm M, Laufs U. The PPAR-gamma agonist pioglitazone increases neoangiogenesis and prevents apoptosis of endothelial progenitor cells. Atherosclerosis. 2007;192:67–74. [PubMed]
51. Vijay SK, Mishra M, Kumar H, Tripathi K. Effect of pioglitazone and rosiglitazone on mediators of endothelial dysfunction, markers of angiogenesis and inflammatory cytokines in type-2 diabetes. Acta Diabetol. 2009;46:27–33. [PubMed]
52. Fong DS, Contreras R. Glitazone use associated with diabetic macular edema. Am J Ophthalmol. 2009;147:583–586. e1. [PubMed]
53. Finck BN, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) regulatory cascade in cardiac physiology and disease. Circulation. 2007;115:2540–2548. [PubMed]
54. Arany Z, Foo SY, Ma Y, et al. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha. Nature. 2008;451:1008–1012. [PubMed]
55. Hickey MM, Simon MC. Regulation of angiogenesis by hypoxia and hypoxia-inducible factors. Curr Top Dev Biol. 2006;76:217–257. [PubMed]
56. Lee KS, Kim SR, Park SJ, et al. Peroxisome proliferator activated receptor-gamma modulates reactive oxygen species generation and activation of nuclear factor-kappaB and hypoxia-inducible factor 1alpha in allergic airway disease of mice. J Allergy Clin Immunol. 2006;118:120–127. [PubMed]
57. Chen J, Cui X, Zacharek A, Roberts C, Chopp M. eNOS mediates TO90317 treatment-induced angiogenesis and functional outcome after stroke in mice. Stroke. 2009;40:2532–2538. [PMC free article] [PubMed]
58. Howell K, Costello CM, Sands M, Dooley I, McLoughlin P. L-arginine promotes angiogenesis in the chronically hypoxic lung: a novel mechanism ameliorating pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2009;296:L1042–L1050. [PubMed]
59. Namkoong S, Kim CK, Cho YL, et al. Forskolin increases angiogenesis through the coordinated cross-talk of PKA-dependent VEGF expression and Epac-mediated PI3K/Akt/eNOS signaling. Cell Signal. 2009;21:906–915. [PubMed]
60. Yasuda S, Kobayashi H, Iwasa M, et al. Antidiabetic drug pioglitazone protects the heart via activation of PPAR-{gamma} receptors, PI3-kinase, Akt, and eNOS pathway in a rabbit model of myocardial infarction. Am J Physiol Heart Circ Physiol. 2009;296:H1558–H1565. [PubMed]
61. Bulhak AA, Jung C, Ostenson CG, Lundberg JO, Sjoquist PO, Pernow J. PPAR-alpha activation protects the type 2 diabetic myocardium against ischemia-reperfusion injury: involvement of the PI3-Kinase/Akt and NO pathway. Am J Physiol Heart Circ Physiol. 2009;296:H719–H727. [PubMed]
62. Cuzzocrea S, Pisano B, Dugo L, et al. Rosiglitazone, a ligand of the peroxisome proliferator-activated receptor-gamma, reduces acute inflammation. Eur J Pharmacol. 2004;483:79–93. [PubMed]
63. Tao L, Liu HR, Gao E, et al. Antioxidative, antinitrative, and vasculoprotective effects of a peroxisome proliferator-activated receptor-gamma agonist in hypercholesterolemia. Circulation. 2003;108:2805–2811. [PubMed]
64. Pedchenko TV, Gonzalez AL, Wang D, DuBois RN, Massion PP. Peroxisome proliferator-activated receptor beta/delta expression and activation in lung cancer. Am J Respir Cell Mol Biol. 2008;39:689–696. [PMC free article] [PubMed]
65. Ma J, Sawai H, Ochi N, et al. PTEN regulate angiogenesis through PI3K/Akt/VEGF signaling pathway in human pancreatic cancer cells. Mol Cell Biochem. 2009 May 13; Epub ahead of print.
66. Panigrahy D, Kaipainen A, Huang S, et al. PPARalpha agonist fenofibrate suppresses tumor growth through direct and indirect angiogenesis inhibition. Proc Natl Acad Sci U S A. 2008;105:985–990. [PMC free article] [PubMed]
67. Minutoli L, Antonuccio P, Polito F, et al. Peroxisome proliferator activated receptor beta/delta activation prevents extracellular regulated kinase 1/2 phosphorylation and protects the testis from ischemia and reperfusion injury. J Urol. 2009;181:1913–1921. [PubMed]
68. Lim HJ, Lee S, Park JH, et al. PPAR delta agonist L-165041 inhibits rat vascular smooth muscle cell proliferation and migration via inhibition of cell cycle. Atherosclerosis. 2009;202:446–454. [PubMed]
69. Borland MG, Foreman JE, Girroir EE, et al. Ligand activation of peroxisome proliferator-activated receptor-beta/delta inhibits cell proliferation in human HaCaT keratinocytes. Mol Pharmacol. 2008;74:1429–1442. [PMC free article] [PubMed]
70. Piqueras L, Sanz MJ, Perretti M, et al. Activation of PPAR{beta}/{delta} inhibits leukocyte recruitment, cell adhesion molecule expression, and chemokine release. J Leukoc Biol. 2009;86:115–122. [PubMed]
REFERENCES for PART III:
Inhibition of ET-1, ETA and ETA-ETB, Induction of NO production, stimulation of eNOS and Treatment Regime with PPAR-gamma agonists (TZD): cEPCs Endogenous Augmentation for Cardiovascular Risk Reduction – A Bibliography
http://pharmaceuticalintelligence.com/2012/10/04/inhibition-of-et-1-eta-and-eta-etb-induction-of-no-production-and-stimulation-of-enos-and-treatment-regime-with-ppar-gamma-agonists-tzd-cepcs-endogenous-augmentation-for-cardiovascular-risk-reduc/
Additional References to Studies on PPAR-Gamma
Repository on BioInfoBank Library on Peroxisome proliferator-activated receptor
http://lib.bioinfo.pl/paper:11030710
Repository on Science.gov on Peroxisome proliferator-activated receptor
http://www.science.gov/topicpages/e/exhibits+ppargamma+ligand.html
On this Open Access OnLine Scientific Journal, Dr. Lev-Ari’s research on Pharmaco-Therapy of Cardiovascular Diseases includes the following:
Lev-Ari, A., (2012 X). Clinical Trials Results for Endothelin System: Pathophysiological role in Chronic Heart Failure, Acute Coronary Syndromes and MI – Marker of Disease Severity or Genetic Determination?
http://pharmaceuticalintelligence.com/2012/10/19/clinical-trials-results-for-endothelin-system-pathophysiological-role-in-chronic-heart-failure-acute-coronary-syndromes-and-mi-marker-of-disease-severity-or-genetic-determination/
Lev-Ari, A., (2012W). Endothelin Receptors in Cardiovascular Diseases: The Role of eNOS Stimulation
http://pharmaceuticalintelligence.com/2012/10/04/endothelin-receptors-in-cardiovascular-diseases-the-role-of-enos-stimulation/
Lev-Ari, A., (2012V). Inhibition of ET-1, ETA and ETA-ETB, Induction of NO production, stimulation of eNOS and Treatment Regime with PPAR-gamma agonists (TZD): cEPCs Endogenous Augmentation for Cardiovascular Risk Reduction – A Bibliography
http://pharmaceuticalintelligence.com/2012/10/04/inhibition-of-et-1-eta-and-eta-etb-induction-of-no-production-and-stimulation-of-enos-and-treatment-regime-with-ppar-gamma-agonists-tzd-cepcs-endogenous-augmentation-for-cardiovascular-risk-reduc/
Lev-Ari, A., (2012U). Cardiovascular Outcomes: Function of circulating Endothelial Progenitor Cells (cEPCs): Exploring Pharmaco-therapy targeted at Endogenous Augmentation of cEPCs
http://pharmaceuticalintelligence.com/2012/08/28/cardiovascular-outcomes-function-of-circulating-endothelial-progenitor-cells-cepcs-exploring-pharmaco-therapy-targeted-at-endogenous-augmentation-of-cepcs/
Lev-Ari, A., (2012T). Endothelial Dysfunction, Diminished Availability of cEPCs, Increasing CVD Risk for Macrovascular Disease – Therapeutic Potential of cEPCs
http://pharmaceuticalintelligence.com/2012/08/27/endothelial-dysfunction-diminished-availability-of-cepcs-increasing-cvd-risk-for-macrovascular-disease-therapeutic-potential-of-cepcs/
Lev-Ari, A., (2012S). Vascular Medicine and Biology: CLASSIFICATION OF FAST ACTING THERAPY FOR PATIENTS AT HIGH RISK FOR MACROVASCULAR EVENTS Macrovascular Disease – Therapeutic Potential of cEPCs
http://pharmaceuticalintelligence.com/2012/08/24/vascular-medicine-and-biology-classification-of-fast-acting-therapy-for-patients-at-high-risk-for-macrovascular-events-macrovascular-disease-therapeutic-potential-of-cepcs/
Lev-Ari, A. (2012L).. Cardiovascular Disease (CVD) and the Role of agent alternatives in endothelial Nitric Oxide Synthase (eNOS) Activation and Nitric Oxide Production
http://pharmaceuticalintelligence.com/2012/07/19/cardiovascular-disease-cvd-and-the-role-of-agent-alternatives-in-endothelial-nitric-oxide-synthase-enos-activation-and-nitric-oxide-production/
Lev-Ari, A. (2012a). Resident-cell-based Therapy in Human Ischaemic Heart Disease: Evolution in the PROMISE of Thymosin beta4 for Cardiac Repair
http://pharmaceuticalintelligence.com/2012/04/30/93/
Lev-Ari, A. (2012b). Triple Antihypertensive Combination Therapy Significantly Lowers Blood Pressure in Hard-to-Treat Patients with Hypertension and Diabetes
http://pharmaceuticalintelligence.com/2012/05/29/445/
Lev-Ari, A. (2012h). Macrovascular Disease – Therapeutic Potential of cEPCs: Reduction Methods for CV Risk
http://pharmaceuticalintelligence.com/2012/07/02/macrovascular-disease-therapeutic-potential-of-cepcs-reduction-methods-for-cv-risk/
Lev-Ari, A. (2012j) Mitochondria Dysfunction and Cardiovascular Disease – Mitochondria: More than just the “powerhouse of the cell”
http://pharmaceuticalintelligence.com/2012/07/09/mitochondria-more-than-just-the-powerhouse-of-the-cell/
Lev-Ari, A. (2012i). Bystolic’s generic Nebivolol – positive effect on circulating Endothelial Proginetor Cells endogenous augmentation
http://pharmaceuticalintelligence.com/2012/07/16/bystolics-generic-nebivolol-positive-effect-on-circulating-endothilial-progrnetor-cells-endogenous-augmentation/
Electronic versions NOT available for:
Lev-Ari, A. & Abourjaily, P. (2006a) “An Investigation of the Potential of circulating Endothelial Progenitor Cells (cEPC) as a Therapeutic Target for Pharmacologic Therapy Design for Cardiovascular Risk Reduction.”Part I: Macrovascular Disease – Therapeutic Potential of cEPCs – Reduction methods for CV risk. Part II: (2006b) Therapeutic Strategy for cEPCs Endogenous Augmentation: A Concept-based Treatment Protocol for a Combined Three Drug Regimen. Part III: (2006c) Biomarker for Therapeutic Targets of Cardiovascular Risk Reduction by cEPCs Endogenous Augmentation using New Combination Drug Therapy of Three Drug Classes and Several Drug Indications. Northeastern University, Boston, MA 02115
Lev-Ari, A. (2007) Heart Vasculature Regeneration and Protection of Coronary Artery Endothelium and Smooth Muscle: A Concept-based Pharmacological Therapy of a Combined Three Drug Regimen. Bouve College of Health Sciences, Northeastern University, Boston, MA 02115
Read Full Post »
