Author and Curator: Larry H Bernstein, MD, FCAP
and
Curator: Aviva-Lev Ari, PhD, RN
This article is a followup of the wonderful study of the effect of oxidation of a methionine residue in calcium dependent-calmodulin kinase Ox-CaMKII on stabilizing the atrial cardiomyocyte, giving protection from atrial fibrillation. It is also not so distant from the work reviewed, mostly on the ventricular myocyte and the calcium signaling by initiation of the ryanodyne receptor (RyR2) in calcium sparks and the CaMKII d isoenzyme.
We refer to the following related articles published in pharmaceutical Intelligence:
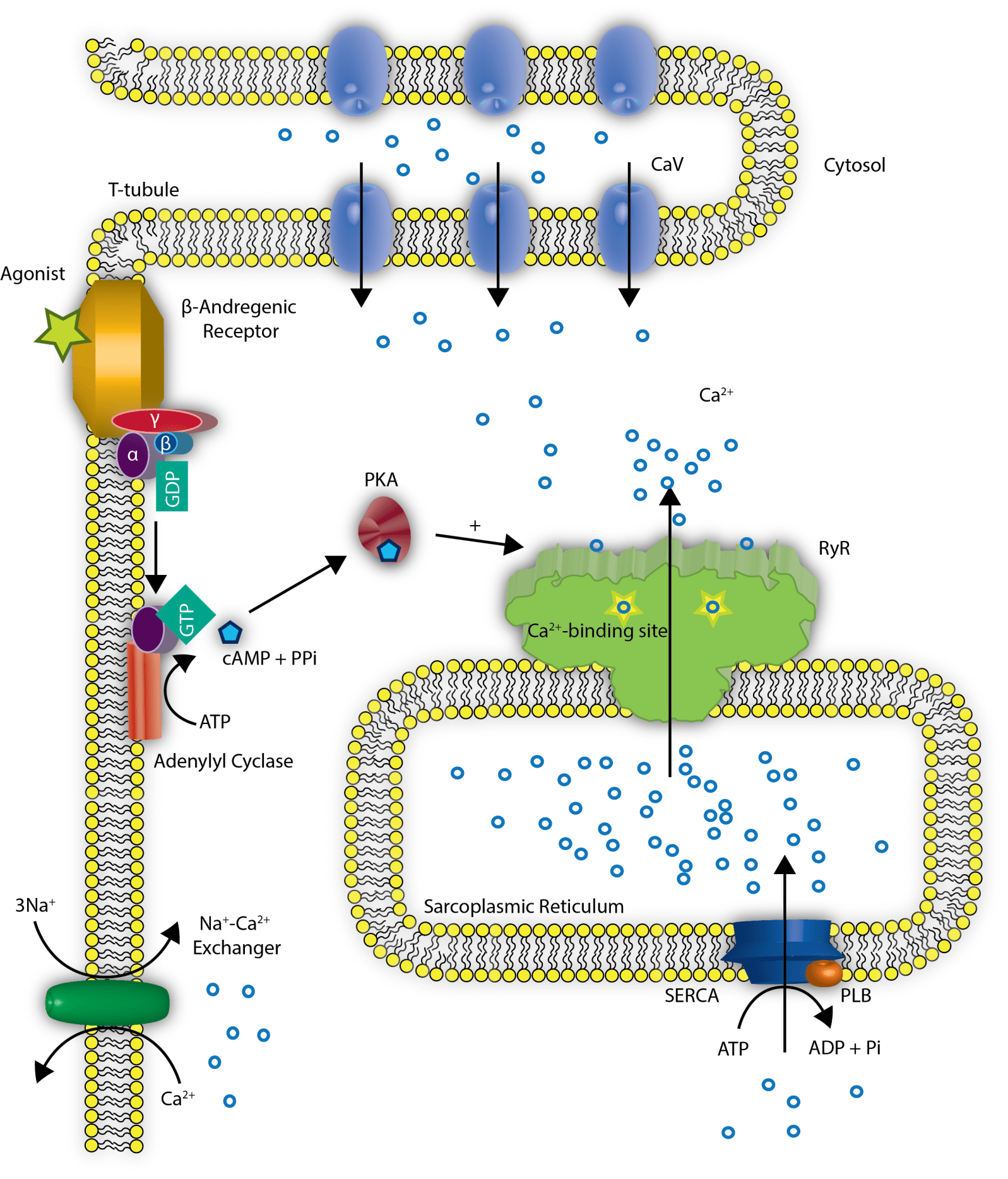
Oxidized Calcium Calmodulin Kinase and Atrial Fibrillation
Author: Larry H. Bernstein, MD, FCAP and Curator: Aviva Lev-Ari, PhD, RN
http://pharmaceuticalintelligence.com/2013/10/26/oxidized-calcium-calmodulin-kinase-and-atrial-fibrillation/
Jmjd3 and Cardiovascular Differentiation of Embryonic Stem Cells
Author: Larry H. Bernstein, MD, FCAP and Curator: Aviva Lev-Ari, PhD, RN
http://pharmaceuticalintelligence.com/2013/10/26/jmjd3-and-cardiovascular-differentiation-of-embryonic-stem-cells/
Contributions to cardiomyocyte interactions and signaling
Author and Curator: Larry H Bernstein, MD, FCAP and Curator: Aviva Lev-Ari, PhD, RN
http://pharmaceuticalintelligence.com/2013/10/21/contributions-to-cardiomyocyte-interactions-and-signaling/
Cardiac Contractility & Myocardium Performance: Therapeutic Implications for Ryanopathy (Calcium Release-related Contractile Dysfunction) and Catecholamine Responses
Editor: Justin Pearlman, MD, PhD, FACC, Author and Curator: Larry H Bernstein, MD, FCAP, and Article Curator: Aviva Lev-Ari, PhD, RN
http://pharmaceuticalintelligence.com/2013/08/28/cardiac-contractility-myocardium-performance-ventricular-arrhythmias-and-non-ischemic-heart-failure-therapeutic-implications-for-cardiomyocyte-ryanopathy-calcium-release-related-contractile/
Part I. Identification of Biomarkers that are Related to the Actin Cytoskeleton
Curator and Writer: Larry H Bernstein, MD, FCAP
http://pharmaceuticalintelligence.com/2012/12/10/identification-of-biomarkers-that-are-related-to-the-actin-cytoskeleton/
Part II: Role of Calcium, the Actin Skeleton, and Lipid Structures in Signaling and Cell Motility
Larry H. Bernstein, MD, FCAP, Stephen Williams, PhD and Aviva Lev-Ari, PhD, RN
http://pharmaceuticalintelligence.com/2013/08/26/role-of-calcium-the-actin-skeleton-and-lipid-structures-in-signaling-and-cell-motility/
Part IV: The Centrality of Ca(2+) Signaling and Cytoskeleton Involving Calmodulin Kinases and Ryanodine Receptors in Cardiac Failure, Arterial Smooth Muscle, Post-ischemic Arrhythmia, Similarities and Differences, and Pharmaceutical Targets
Larry H Bernstein, MD, FCAP, Justin Pearlman, MD, PhD, FACC and Aviva Lev-Ari, PhD, RN
http://pharmaceuticalintelligence.com/2013/09/08/the-centrality-of-ca2-signaling-and-cytoskeleton-involving-calmodulin-kinases-and-ryanodine-receptors-in-cardiac-failure-arterial-smooth-muscle-post-ischemic-arrhythmia-similarities-and-differen/
Part VI: Calcium Cycling (ATPase Pump) in Cardiac Gene Therapy: Inhalable Gene Therapy for Pulmonary Arterial Hypertension and Percutaneous Intra-coronary Artery Infusion for Heart Failure: Contributions by Roger J. Hajjar, MD
Aviva Lev-Ari, PhD, RN
http://pharmaceuticalintelligence.com/2013/08/01/calcium-molecule-in-cardiac-gene-therapy-inhalable-gene-therapy-for-pulmonary-arterial-hypertension-and-percutaneous-intra-coronary-artery-infusion-for-heart-failure-contributions-by-roger-j-hajjar/
Part VII: Cardiac Contractility & Myocardium Performance: Ventricular Arrhythmias and Non-ischemic Heart Failure – Therapeutic Implications for Cardiomyocyte Ryanopathy (Calcium Release-related Contractile Dysfunction) and Catecholamine Responses
Justin Pearlman, MD, PhD, FACC, Larry H Bernstein, MD, FCAP and Aviva Lev-Ari, PhD, RN
http://pharmaceuticalintelligence.com/2013/08/28/cardiac-contractility-myocardium-performance-ventricular-arrhythmias-and-non-ischemic-heart-failure-therapeutic-implications-for-cardiomyocyte-ryanopathy-calcium-release-related-contractile/
Part VIII: Disruption of Calcium Homeostasis: Cardiomyocytes and Vascular Smooth Muscle Cells: The Cardiac and Cardiovascular Calcium Signaling Mechanism
Justin Pearlman, MD, PhD, FACC, Larry H Bernstein, MD, FCAP and Aviva Lev-Ari, PhD, RN
http://pharmaceuticalintelligence.com/2013/09/12/disruption-of-calcium-homeostasis-cardiomyocytes-and-vascular-smooth-muscle-cells-the-cardiac-and-cardiovascular-calcium-signaling-mechanism/
Part IX: Calcium-Channel Blockers, Calcium Release-related Contractile Dysfunction (Ryanopathy) and Calcium as Neurotransmitter Sensor
Justin Pearlman, MD, PhD, FACC, Larry H Bernstein, MD, FCAP and Aviva Lev-Ari, PhD, RN
http://pharmaceuticalintelligence.com/2013/09/16/calcium-channel-blocker-calcium-as-neurotransmitter-sensor-and-calcium-release-related-contractile-dysfunction-ryanopathy/
Part X: Synaptotagmin functions as a Calcium Sensor: How Calcium Ions Regulate the fusion of vesicles with cell membranes during Neurotransmission
Larry H Bernstein, MD, FCAP and Aviva Lev-Ari, PhD, RN
http://pharmaceuticalintelligence.com/2013/09/10/synaptotagmin-functions-as-a-calcium-sensor-how-calcium-ions-regulate-the-fusion-of-vesicles-with-cell-membranes-during-neurotransmission/
The material presented is very focused, and cannot be found elsewhere in Pharmaceutical Intelligence with respedt to genetics and heart disease. However, there are other articles that may be of interest to the reader.
Volume Three: Etiologies of Cardiovascular Diseases – Epigenetics, Genetics & Genomics
Curators: Larry H Bernstein, MD, FCAP and Aviva Lev-Ari, PhD, RN
http://pharmaceuticalintelligence.com/biomed-e-books/series-a-e-books-on-cardiovascular-diseases/volume-three-etiologies-of-cardiovascular-diseases-epigenetics-genetics-genomics/
PART 3. Determinants of Cardiovascular Diseases: Genetics, Heredity and Genomics Discoveries
3.2 Leading DIAGNOSES of Cardiovascular Diseases covered in Circulation: Cardiovascular Genetics, 3/2010 – 3/2013
The Diagnoses covered include the following – relevant to this discussion
- MicroRNA in Serum as Bimarker for Cardiovascular Pathologies: acute myocardial infarction, viral myocarditis, diastolic dysfunction, and acute heart failure
- Genomics of Ventricular arrhythmias, A-Fib, Right Ventricular Dysplasia, Cardiomyopathy
- Heredity of Cardiovascular Disorders Inheritance
3.2.1: Heredity of Cardiovascular Disorders Inheritance
The implications of heredity extend beyond serving as a platform for genetic analysis, influencing diagnosis,
- prognostication, and
- treatment of both index cases and relatives, and
- enabling rational targeting of genotyping resources.
This review covers acquisition of a family history, evaluation of heritability and inheritance patterns, and the impact of inheritance on subsequent components of the clinical pathway.
SOURCE: Circulation: Cardiovascular Genetics.2011; 4: 701-709. http://dx.doi.org/10.1161/CIRCGENETICS.110.959379
3.2.2: Myocardial Damage
3.2.2.1 MicroRNA in Serum as Biomarker for Cardiovascular Pathologies: acute myocardial infarction, viral myocarditis, diastolic dysfunction, and acute heart failure
Increased MicroRNA-1 and MicroRNA-133a Levels in Serum of Patients With Cardiovascular Disease Indicate Myocardial Damage
Y Kuwabara, Koh Ono, T Horie, H Nishi, K Nagao, et al.
SOURCE: Circulation: Cardiovascular Genetics. 2011; 4: 446-454 http://dx.doi.org/10.1161/CIRCGENETICS.110.958975
3.2.2.2 Circulating MicroRNA-208b and MicroRNA-499 Reflect Myocardial Damage in Cardiovascular Disease
MF Corsten, R Dennert, S Jochems, T Kuznetsova, Y Devaux, et al.
SOURCE: Circulation: Cardiovascular Genetics. 2010; 3: 499-506. http://dx.doi.org/10.1161/CIRCGENETICS.110.957415
3.2.4.2 Large-Scale Candidate Gene Analysis in Whites and African Americans Identifies IL6R Polymorphism in Relation to Atrial Fibrillation
The National Heart, Lung, and Blood Institute’s Candidate Gene Association Resource (CARe) Project
RB Schnabel, KF Kerr, SA Lubitz, EL Alkylbekova, et al.
SOURCE: Circulation: Cardiovascular Genetics.2011; 4: 557-564 http://dx.doi.org/10.1161/CIRCGENETICS.110.959197
Weighted Gene Coexpression Network Analysis of Human Left Atrial Tissue Identifies Gene Modules Associated With Atrial Fibrillation
N Tan, MK Chung, JD Smith, J Hsu, D Serre, DW Newton, L Castel, E Soltesz, G Pettersson, AM Gillinov, DR Van Wagoner and J Barnard
From the Cleveland Clinic Lerner College of Medicine (N.T.), Department of Cardiovascular Medicine (M.K.C., D.W.N.), and Department of Thoracic & Cardiovascular Surgery (E.S., G.P., A.M.G.); and Department of Cellular & Molecular Medicine (J.D.S., J.H.), Genomic Medicine Institute (D.S.), Department of Molecular Cardiology (L.C.), and Department of Quantitative Health Sciences (J.B.), Cleveland Clinic Lerner Research Institute, Cleveland, OH
Circ Cardiovasc Genet. 2013;6:362-371; http://dx.doi.org/10.1161/CIRCGENETICS.113.000133
http://circgenetics.ahajournals.org/content/6/4/362 The online-only Data Supplement is available at http://circgenetics.ahajournals.org/lookup/suppl/doi:10.1161/CIRCGENETICS.113.000133/-/DC1
Background—Genetic mechanisms of atrial fibrillation (AF) remain incompletely understood. Previous differential expression studies in AF were limited by small sample size and provided limited understanding of global gene networks, prompting the need for larger-scale, network-based analyses.
Methods and Results—Left atrial tissues from Cleveland Clinic patients who underwent cardiac surgery were assayed using Illumina Human HT-12 mRNA microarrays. The data set included 3 groups based on cardiovascular comorbidities: mitral valve (MV) disease without coronary artery disease (n=64), coronary artery disease without MV disease (n=57), and lone AF (n=35). Weighted gene coexpression network analysis was performed in the MV group to detect modules of correlated genes. Module preservation was assessed in the other 2 groups. Module eigengenes were regressed on AF severity or atrial rhythm at surgery. Modules whose eigengenes correlated with either AF phenotype were analyzed for gene content. A total of 14 modules were detected in the MV group; all were preserved in the other 2 groups. One module (124 genes) was associated with AF severity and atrial rhythm across all groups. Its top hub gene, RCAN1, is implicated in calcineurin-dependent signaling and cardiac hypertrophy. Another module (679 genes) was associated with atrial rhythm in the MV and coronary artery disease groups. It was enriched with cell signaling genes and contained cardiovascular developmental genes including TBX5.
Conclusions—Our network-based approach found 2 modules strongly associated with AF. Further analysis of these modules may yield insight into AF pathogenesis by providing novel targets for functional studies. (Circ Cardiovasc Genet. 2013;6:362-371.)
Key Words: arrhythmias, cardiac • atrial fibrillation • bioinformatics • gene coexpression • gene regulatory networks • genetics • microarrays
Introduction
trial fibrillation (AF) is the most common sustained cardiac arrhythmia, with a prevalence of ≈1% to 2% in the general population.1,2 Although AF may be an isolated condition (lone AF [LAF]), it often occurs concomitantly with other cardiovascular diseases, such as coronary artery disease (CAD) and valvular heart disease.1 In addition, stroke risk is increased 5-fold among patients with AF, and ischemic strokes attributed to AF are more likely to be fatal.1 Current antiarrhythmic drug therapies are limited in terms of efficacy and safety.1,3,4 Thus, there is a need to develop better risk prediction tools as well as mechanistically targeted therapies for AF. Such developments can only come about through a clearer understanding of its pathogenesis.
Family history is an established risk factor for AF. A Danish Twin Registry study estimated AF heritability at 62%, indicating a significant genetic component.5 Substantial progress has been made to elucidate this genetic basis. For example, genome-wide association studies (GWASs) have identified several susceptibility loci and candidate genes linked with AF. Initial studies performed in European populations found 3 AF-associated genomic loci.6–9 Of these, the most significant single-nucleotide polymor-phisms (SNPs) mapped to an intergenic region of chromosome 4q25. The closest gene in this region, PITX2, is crucial in left-right asymmetrical development of the heart and thus seems promising as a major player in initiating AF.10,11 A large-scale GWAS meta-analysis discovered 6 additional susceptibility loci, implicating genes involved in cardiopulmonary development, ion transport, and cellular structural integrity.12
Differential expression studies have also provided insight into the pathogenesis of AF. A study by Barth et al13 found that about two-thirds of the genes expressed in the right atrial appendage were downregulated during permanent AF, and that many of these genes were involved in calcium-dependent signaling pathways. In addition, ventricular-predominant genes were upregulated in right atrial appendages of subjects with AF.13 Another study showed that inflammatory and transcription-related gene expression was increased in right atrial appendages of subjects with AF versus controls.14 These results highlight the adaptive responses to AF-induced stress and ischemia taking place within the atria.
Despite these advances, much remains to be discovered about the genetic mechanisms of AF. The AF-associated SNPs found thus far only explain a fraction of its heritability15; furthermore, the means by which the putative candidate genes cause AF have not been fully established.9,15,16 Additionally, previous differential expression studies in human tissue were limited to the right atrial appendage, had small sample sizes, and provided little understanding of global gene interactions.13,14 Weighted gene coexpression network analysis (WGCNA) is a technique to construct gene modules within a network based on correlations in gene expression (ie, coexpression).17,18 WGCNA has been used to study genetically complex diseases, such as metabolic syndrome,19 schizophrenia,20 and heart failure.21 Here, we obtained mRNA expression profiles from human left atrial appendage tissue and implemented WGCNA to identify gene modules associated with AF phenotypes.
Methods
Subject Recruitment
From 2001 to 2008, patients undergoing cardiac surgery at the Cleveland Clinic were prospectively screened and recruited. Informed consent for research use of discarded atrial tissues was obtained from each patient by a study coordinator during the presurgical visit. Demographic and clinical data were obtained from the Cardiovascular Surgery Information Registry and by chart review. Use of human atrial tissues was approved by the Institutional Review Board of the Cleveland Clinic.
Table S1: Clinical definitions of cardiovascular phenotype groups
| Criterion Type |
Mitral Valve (MV) Disease |
Coronary Artery Disease (CAD) |
Lone Atrial Fibrillation (LAF) |
| Inclusion Criteria |
Surgical indication – |
Surgical indication – |
History of atrial fibrillation |
|
mitral valve repair or replacement |
coronary artery bypass graft |
|
|
|
|
Surgical indication |
|
|
|
– MAZE procedure |
|
|
|
Preserved ejection fraction (≥50%) |
| Exclusion Criteria |
Significant coronary artery disease: |
Significant mitral valve disease: |
Significant |
|
|
|
coronary artery |
|
– Significant (≥50%) stenosis |
– Documented echocardiography |
disease: |
|
in at least |
finding of |
– Significant |
|
one coronary artery |
mitral regurgitation (≥3) or |
(≥50%) stenosis in |
|
via cardiac catheterization |
mitral stenosis |
at least one |
|
– History of revascularization |
– History of mitral valve |
coronary artery via |
|
(percutaneous coronary intervention or coronary artery bypass graft surgery) |
repair or replacement |
cardiac catheterization |
|
|
|
– History of revascularization |
|
|
|
(percutaneous coronary intervention or coronary artery bypass graft surgery) |
|
|
|
Significant valvular heart disease: |
|
|
|
-Documented echocardiography finding of valvular regurgitation (≥3) or stenosis |
|
|
|
-History of valve repair or replacement |
RNA Microarray Isolation and Profiling
Left atria appendage specimens were dissected during cardiac surgery and stored frozen at −80°C. Total RNA was extracted using the Trizol technique. RNA samples were processed by the Cleveland Clinic Genomics Core. For each sample, 250-ng RNA was reverse transcribed into cRNA and biotin-UTP labeled using the TotalPrep RNA Amplification Kit (Ambion, Austin, TX). cRNA was quantified using a Nanodrop spectrophotometer, and cRNA size distribution was assessed on a 1% agarose gel. cRNA was hybridized to Illumina Human HT-12 Expression BeadChip arrays (v.3). Arrays were scanned using a BeadArray reader.
Expression Data Preprocessing
Raw expression data were extracted using the beadarray package in R, and bead-level data were averaged after log base-2 transformation. Background correction was performed by fitting a normal-gamma deconvolution model using the NormalGamma R package.22 Quantile normalization and batch effect adjustment with the ComBat method were performed using R.23 Probes that were not detected (at a P<0.05 threshold) in all samples as well as probes with relatively lower variances (interquartile range ≤log2[1.2]) were excluded.
The WGCNA approach requires that genes be represented as singular nodes in such a network. However, a small proportion of the genes in our data have multiple probe mappings. To facilitate the representation of singular genes within the network, a probe must be selected to represent its associated gene. Hence, for genes that mapped to multiple probes, the probe with the highest mean expression level was selected for analysis (which often selects the splice isoform with the highest expression and signal-to-noise ratio), resulting in a total of 6168 genes.
Defining Training and Test Sets
Currently, no large external mRNA microarray data from human left atrial tissues are publicly available. To facilitate internal validation of results, we divided our data set into 3 groups based on cardiovascular comorbidities: mitral valve (MV) disease without CAD (MV group; n=64), CAD without MV disease (CAD group; n=57), and LAF (LAF group; n=35). LAF was defined as the presence of AF without concomitant structural heart disease, according to the guidelines set by the European Society of Cardiology.1 The MV group, which was the largest and had the most power for detecting significant modules, served as the training set for module derivation, whereas the other 2 groups were designated test sets for module reproducibility. To minimize the effect of population stratification, the data set was limited to white subjects. Differences in clinical characteristics among the groups were assessed using Kruskal–Wallis rank-sum tests for continuous variables and Pearson x2 test for categorical variables.
Weight Gene Coexpression Network Analysis
WGCNA is a systems-biology method to identify and characterize gene modules whose members share strong coexpression. We applied previously validated methodology in this analysis.17 Briefly, pair-wise gene (Pearson) correlations were calculated using the MV group data set. A weighted adjacency matrix was then constructed. I is a soft-thresholding parameter that provides emphasis on stronger correlations over weaker and less meaningful ones while preserving the continuous nature of gene–gene relationships. I=3 was selected in this analysis based on the criterion outlined by Zhang and Horvath17 (see the online-only Data Supplement).
Next, the topological overlap–based dissimilarity matrix was computed from the weighted adjacency matrix. The topological overlap, developed by Ravasz et al,24 reflects the relative interconnectedness (ie, shared neighbors) between 2 genes.17 Hence, construction of the network dendrogram based on this dissimilarity measure allows for the identification of gene modules whose members share strong intercon-nectivity patterns. The WGCNA cutreeDynamic R function was used to identify a suitable cut height for module identification via an adaptive cut height selection approach.18 Gene modules, defined as branches of the network dendrogram, were assigned colors for visualization.
Network Preservation Analysis
Module preservation between the MV and CAD groups as well as the MV and LAF groups was assessed using network preservation statistics as described in Langfelder et al.25 Module density–based statistics (to assess whether genes in each module remain highly connected in the test set) and connectivity-based statistics (to assess whether connectivity patterns between genes in the test set remain similar compared with the training set) were considered in this analysis.25 In each comparison, a Z statistic representing a weighted summary of module density and connectivity measures was computed for every module (Zsummary). The Zsummary score was used to evaluate module preservation, with values ≥8 indicating strong preservation, as proposed by Langfelder et al.25 The WGCNA R function network preservation was used to implement this analysis.25
Table S2: Network preservation analysis between the MV and CAD groups – size and Zsummary scores of gene modules detected.
| Module |
Module Size |
ZSummary
|
| Black |
275 |
15.52 |
| Blue |
964 |
44.79 |
| Brown |
817 |
12.80 |
| Cyan |
119 |
13.42 |
| Green |
349 |
14.27 |
| Green-Yellow |
215 |
19.31 |
| Magenta |
239 |
15.38 |
| Midnight-Blue |
83 |
15.92 |
| Pink |
252 |
23.31 |
| Purple |
224 |
16.96 |
| Red |
278 |
17.30 |
| Salmon |
124 |
13.84 |
| Tan |
679 |
28.48 |
| Turquoise |
1512 |
44.03 |
Table S3: Network preservation analysis between the MV and LAF groups – size and Zsummary scores of gene modules detected
| Module |
Module Size |
ZSummary |
| Black |
275 |
13.14 |
| Blue |
964 |
39.26 |
| Brown |
817 |
14.98 |
| Cyan |
119 |
11.46 |
| Green |
349 |
14.91 |
| Green-Yellow |
215 |
20.99 |
| Magenta |
239 |
18.58 |
| Midnight-Blue |
83 |
13.87 |
| Pink |
252 |
19.10 |
| Purple |
224 |
8.80 |
| Red |
278 |
16.62 |
| Salmon |
124 |
11.57 |
| Tan |
679 |
28.61 |
| Turquoise |
1512 |
42.07 |
Clinical Significance of Preserved Modules
Principal component analysis of the expression data for each gene module was performed. The first principal component of each module, designated the eigengene, was identified for the 3 cardiovascular disease groups; this served as a summary expression measure that explained the largest proportion of the variance of the module.26 Multivariate linear regression was performed with the module ei-gengenes as the outcome variables and AF severity (no AF, paroxysmal AF, persistent AF, permanent AF) as the predictor of interest (adjusting for age and sex). A similar regression analysis was performed with atrial rhythm at surgery (no AF history, AF history in sinus rhythm, AF history in AF rhythm) as the predictor of interest. The false discovery rate method was used to adjust for multiple comparisons. Modules whose eigengenes associated with AF severity and atrial rhythm were identified for further analysis.
In addition, hierarchical clustering of module eigengenes and selected clinical traits (age, sex, hypertension, cholesterol, left atrial size, AF state, and atrial rhythm) was used to identify additional module–trait associations. Clusters of eigengenes/traits were detected based on a dissimilarity measure D, as given by
D=1−cor(Vi,Vj),i≠j (3)
where V=the eigengene or clinical trait.
Enrichment Analysis
Gene modules significantly associated with AF severity and atrial rhythm were submitted to Ingenuity Pathway Analysis (IPA) to determine enrichment for functional/disease categories. IPA is an application of gene set over-representation analysis; for each dis-ease/functional category annotation, a P value is calculated (using Fisher exact test) by comparing the number of genes from the module of interest that participate in the said category against the total number of participating genes in the background set.27 All 6168 genes in the current data set served as the background set for the enrichment analysis.
Hub Gene Analysis
Hub genes are defined as genes that have high intramodular connectivity17,20
Alternatively, they may also be defined as genes with high module membership21,25
Both definitions were used to identify the hub genes of modules associated with AF phenotype.
To confirm that the hub genes identified were themselves associated with AF phenotype, the expression data of the top 10 hub genes (by intramodular connectivity) were regressed on atrial rhythm (adjusting for age and sex). In addition, eigengenes of AF-associated modules were regressed on their respective (top 10) hub gene expression profiles, and the model R2 indices were computed.
Membership of AF-Associated Candidate Genes From Previous Studies
Previous GWAS studies identified multiple AF-associated SNPs.8,9,12,15,28 We selected candidate genes closest to or containing these SNPs and identified their module locations as well as their closest within-module partners (absolute Pearson correlations).
Sensitivity Analysis of Soft-Thresholding Parameter
To verify that the key results obtained from the above analysis were robust with respect to the chosen soft-thresholding parameter (I=3), we repeated the module identification process using I=5. The eigen-genes of the detected modules were computed and regressed on atrial rhythm (adjusting for age and sex). Modules significantly associated with atrial rhythm in ≥2 groups of data set were compared with the AF phenotype–associated modules from the original analysis.
Results
Subject Characteristics
Table 1 describes the clinical characteristics of the cardiac surgery patients who were recruited for the study. Subjects in the LAF group were generally younger and less likely to be a current smoker (P=2.0×10−4 and 0.032, respectively). Subjects in the MV group had lower body mass indices (P=2.7×10−6), and a larger proportion had paroxysmal AF compared with the other 2 groups (P=0.033).
Table 1. Clinical Characteristics of Study Subjects
| Characteristics |
MV Group (n=64)
|
CAD Group (n=57)
|
LAF Group (n=35)
|
P Value*
|
| Age, median y (1st–3rd quartiles) |
60 (51.75–67.25)
|
64 (58.00–70.00)
|
56 (45.50–60.50)
|
2.0×10−4
|
| Sex, female (%) |
19 (29.7) |
6 (10.5) |
7 (20.0)
|
0.033
|
| BMI, median (1st–3rd quartiles) |
25.97 (24.27–28.66)
|
29.01 (27.06–32.11)
|
29.71 (26.72–35.10)
|
2.7×10−6
|
| Current smoker (%) |
29 (45.3) |
35 (61.4) |
12 (21.1)
|
0.032
|
| Hypertension (%) |
21 (32.8) |
39 (68.4) |
16 (45.7)
|
4.4×10−4
|
| AF severity (%) |
|
|
|
|
| No AF |
7 (10.9) |
7 (12.3) |
0 (0.0)
|
0.033
|
| Paroxysmal |
19 (29.7) |
10 (17.5) |
7 (20.0)
|
|
| Persistent |
30 (46.9) |
26 (45.6) |
15 (42.9)
|
|
| Permanent |
8 (12.5) |
14 (24.6) |
13 (37.1)
|
|
| Atrial rhythm at surgery (%) |
|
|
|
|
| No AF history in sinus rhythm |
7 (10.9) |
7 (12.3) |
0 (0)
|
0.065
|
| AF history in sinus rhythm |
28 (43.8) |
16 (28.1) |
11 (31.4)
|
|
| AF History in AF rhythm |
29 (45.3) |
34 (59.6) |
24 (68.6)
|
|
Gene Coexpression Network Construction and Module Identificationsee document at http://circgenetics.ahajournals.org/content/6/4/362
A total of 14 modules were detected using the MV group data set (Figure 1), with module sizes ranging from 83 genes to 1512 genes; 38 genes did not share similar coexpression with the other genes in the network and were therefore not included in any of the identified modules

Figure 1. Network dendrogram (top) and colors of identified modules (bottom). The dendrogram was constructed using the topological overlap matrix as the similarity measure. Modules corresponded to branches of the dendrogram and were assigned colors for visualization.
Network Preservation Analysis Revealed Strong Preservation of All Modules Between the Training and Test Sets
All 14 modules showed strong preservation across the CAD and LAF groups in both comparisons, with Z [summary] scores of >10 in most modules (Figure 2). No major deviations in the Z [summary] score distributions for the 2 comparisons were noted, indicating that modules were preserved to a similar extent across the 2 groups

Figure 2. Preservation of modules between mitral valve (MV) and coronary artery disease (CAD) groups (left), and MV and lone atrial fibrillation (LAF) groups (right). A Zsummary statistic was computed for each module as an overall measure of its preservation relating to density and connectivity. All modules showed strong preservation in both comparisons with Zsummary scores >8 (red dotted line).
Regression Analysis of Module Eigengene Profiles Identified 2 Modules Associated With AF Severity and Atrial Rhythm
Table IV in the online-only Data Supplement summarizes the proportion of variance explained by the first 3 principal components for each module. On average, the first principal component (ie, the eigengene) explained ≈18% of the total variance of its associated module. For each group, the module eigengenes were extracted and regressed on AF severity (with age and sex as covariates). The salmon module (124 genes) eigengene was strongly associated with AF severity in the MV and CAD groups (P=1.7×10−6 and 5.2×10−4, respectively); this association was less significant in the LAF group (P=9.0×10−2). Eigengene levels increased with worsening AF severity across all 3 groups, with the greatest stepwise change taking place between the paroxysmal AF and persistent AF categories (Figure 3A). When the module eigen-genes were regressed on atrial rhythm, the salmon module eigengene showed significant association in all groups (MV: P=1.1×10−14; CAD: P=1.36×10−6; LAF: P=2.1×10−4). Eigen-gene levels were higher in the AF history in AF rhythm category (Figure 3B).
Table S4: Proportion of variance explained by the principal components for each module.
|
Dataset
Group
|
Principal
Component
|
Black
|
Blue
|
Brown
|
Cyan
|
Green
|
Green-
Yellow
|
Magenta
|
|
Mitral
|
1
|
20.5% |
22.2% |
20.1% |
21.8% |
21.4% |
22.8% |
19.6% |
|
2
|
4.1% |
3.6% |
4.8% |
5.7% |
4.5% |
5.9% |
3.9% |
|
3
|
3.4% |
3.1% |
3.8% |
4.4% |
3.9% |
3.7% |
3.7% |
|
CAD
|
1
|
12.5% |
18.6% |
7.1% |
16.8% |
12.2% |
20.3% |
12.8% |
|
2
|
6.0% |
5.5% |
5.0% |
7.0% |
5.5% |
6.1% |
6.4% |
|
3
|
4.9% |
4.1% |
4.4% |
6.5% |
4.8% |
4.4% |
4.8% |
|
LAF
|
1
|
14.0% |
16.6% |
11.7% |
14.3% |
14.7% |
20.8% |
20.2% |
|
2
|
8.9% |
8.5% |
7.6% |
9.3% |
7.3% |
11.1% |
6.9% |
|
3
|
6.5% |
6.3% |
5.5% |
8.2% |
6.1% |
5.3% |
6.2% |
|
Dataset
Group
|
Principal
Component
|
Midnight- Blue |
Pink
|
Purple
|
Red
|
Salmon
|
Tan
|
Turquoise
|
|
Mitral
|
1
|
28.5% |
22.6% |
18.7% |
20.5% |
22.3% |
19.0% |
25.8% |
|
2
|
4.6% |
6.0% |
4.7% |
4.1% |
6.9% |
4.0% |
3.5% |
|
3
|
4.2% |
4.2% |
4.2% |
3.5% |
4.0% |
3.6% |
3.3% |
|
CAD
|
1
|
23.4% |
17.1% |
15.5% |
15.0% |
18.0% |
14.6% |
18.2% |
|
2
|
7.4% |
8.6% |
6.0% |
6.4% |
7.2% |
5.8% |
6.6% |
|
3
|
5.1% |
5.4% |
5.3% |
5.4% |
6.2% |
5.1% |
4.5% |
|
LAF
|
1
|
23.5% |
18.4% |
12.0% |
15.9% |
16.9% |
13.7% |
16.5% |
|
2
|
7.9% |
8.5% |
9.8% |
9.4% |
9.5% |
9.1% |
9.6% |
|
3
|
6.7% |
7.0% |
6.6% |
6.0% |
6.9% |
6.8% |
6.3% |

Figure 3. Boxplots of salmon module eigengene expression levels with respect to atrial fibrillation (AF) severity (A) and atrial rhythm (B).
A, Eigengene expression correlated positively with AF severity, with the largest stepwise increase between the paroxysmal AF and permanent AF categories. B, Eigengene expression was highest in the AF history in AF rhythm category in all 3 groups. CAD indicates coronary artery disease; LAF, lone AF; and MV, mitral valve.
The regression analysis also revealed statistically significant associations between the tan module (679 genes) eigengene and atrial rhythm in the MV and CAD groups (P=5.8×10−4 and 3.4×10−2, respectively). Eigengene levels were lower in the AF history in AF rhythm category compared with the AF history in sinus rhythm category (Figure 4); this trend was also observed in the LAF group, albeit with weaker statistical evidence (P=0.15).

Figure 4. Boxplots of tan module eigengene expression levels with respect to atrial rhythm.
Eigengene expression levels were lower in the atrial fibrillation (AF) history in AF rhythm category compared with the AF history in sinus rhythm category. CAD indicates coronary artery disease; LAF, lone AF; and MV, mitral valve
Hierarchical Clustering of Eigengene Profiles With Clinical Traits
Hierarchical clustering was performed to identify relationships between gene modules and selected clinical traits. The salmon module clustered with AF severity and atrial rhythm; in addition, left atrial size was found in the same cluster, suggesting a possible relationship between salmon module gene expression and atrial remodeling (Figure 5A). Although the tan module was in a separate cluster from the salmon module, it was negatively correlated with both atrial rhythm and AF severity (Figure 5B).

Figure 5. Dendrogram (A) and correlation heatmap (B) of module eigengenes and clinical traits.
A, The salmon module eigengene but not the tan module eigengene clustered with atrial fibrillation (AF) severity, atrial rhythm, and left atrial size. B, AF severity and atrial rhythm at surgery correlated positively with the salmon module eigengene and negatively with the tan module eigengene. Arhythm indicates atrial rhythm at surgery; Chol, cholesterol; HTN, hypertension; and LASize, left atrial size.
IPA Enrichment Analysis of Salmon and Tan Modules
The salmon module was enriched in genes involved in cardiovascular function and development (smallest P=4.4×10−4) and organ morphology (smallest P=4.4×10−4). In addition, the top disease categories identified included endocrine system disorders (smallest P=4.4×10−4) and cardiovascular disease (smallest P=2.59×10−3).
The tan module was enriched in genes involved in cell-to-cell signaling and interaction (smallest P=8.9×10−4) and cell death and survival (smallest P=1.5×10−3). Enriched disease categories included cancer (smallest P=2.2×10−4) and cardiovascular disease (smallest P=4.5×10−4).
Hub Gene Analysis of Salmon and Tan Modules
We identified hub genes in the 2 modules based on intramod-ular connectivity and module membership. For the salmon module, the gene RCAN1 exhibited the highest intramodular connectivity and module membership. The top 10 hub genes (by intramodular connectivity) were significantly associated with atrial rhythm, with false discovery rate–adjusted P values ranging from 1.5×10−5 to 4.2×10−12. These hub genes accounted for 95% of the variation in the salmon module eigengene.
In the tan module, the top hub gene was CPEB3. The top 10 hub genes (by intramodular connectivity) correlated with atrial rhythm as well, although the statistical associations in the lower-ranked hub genes were relatively weaker (false discovery rate–adjusted P values ranging from 1.1×10−1 to 3.4×10−4). These hub genes explained 94% of the total variation in the tan module eigengene.
The names and connectivity measures of the hub genes found in both modules are presented in Table 2.
Table 2. Top 10 Hub Genes in the Salmon (Left) and Tan (Right) Modules as Defined by Intramodular Connectivity and Module Membership
|
|
Salmon Module
|
|
|
|
Tan Module
|
|
| Gene |
IMC
|
Gene
|
MM
|
Gene
|
IMC
|
Gene
|
MM
|
| RCAN1 |
8.2 |
RCAN1
|
0.81 |
CPEB3
|
43.3 |
CPEB3
|
0.85 |
| DNAJA4 |
7.7 |
DNAJA4
|
0.81 |
CPLX3
|
42.4 |
CPLX3
|
0.84 |
| PDE8B |
7.7 |
PDE8B
|
0.80 |
NEDD4L
|
40.8 |
NEDD4L
|
0.83 |
| PRKAR1A |
6.9 |
PRKAR1A
|
0.77 |
SGSM1
|
40.7 |
SGSM1
|
0.82 |
| PTPN4 |
6.7 |
PTPN4
|
0.75 |
UCKL1
|
39.0 |
UCKL1
|
0.81 |
| SORBS2 |
6.0 |
FHL2
|
0.69 |
SOSTDC1
|
37.2 |
SOSTDC1
|
0.79 |
| ADCY6 |
5.7 |
ADCY6
|
0.69 |
PRDX1
|
35.5 |
RCOR2
|
0.78 |
| FHL2 |
5.7 |
SORBS2
|
0.68 |
RCOR2
|
35.4 |
EEF2K
|
0.77 |
| BVES |
5.4 |
DHRS9
|
0.67 |
NPPB
|
35.3 |
PRDX1
|
0.76 |
| TMEM173 |
5.3 |
LAPTM4B
|
0.65 |
LRRN3
|
34.6 |
MMP11
|
0.76 |
A visualization of the salmon module is shown using the Cytoscape tool (Figure 6). A full list of the genes in the salmon and tan modules is provided in the online-only Data Supplement.
Figure 6. Cytoscape visualization of genes in the salmon module.
Nodes representing genes with high intramodu-lar connectivities, such as RCAN1 and DNAJA4, appear larger in the network. Strong connections are visualized with darker lines, whereas weak connections appear more translucent

Membership of AF-Associated Candidate Genes From Previous Studies
The tan module contained MYOZ1, which was identified as a candidate gene from the recent AF meta-analysis. PITX2 was located in the green module (n=349), and ZFHX3 was located in the turquoise module (n=1512). The locations of other candidate genes (and their closest partners) are reported in the online-only Data Supplement.
Sensitivity Analysis of Key Results
We repeated the WGCNA module identification approach using a different soft-thresholding parameter (β=5). One module (n=121) was found to be strongly associated with atrial rhythm at surgery across all 3 groups of data set, whereas another module (n=244) was associated with atrial rhythm at surgery in the MV and CAD groups. The first module overlapped significantly with the salmon module in terms of gene membership, whereas most of the second modules’ genes were contained within the tan module. The top hub genes found in the salmon and tan modules remained present and highly connected in the 2 new modules identified with the different soft-thresholding parameter.
Discussion
To our knowledge, our study is the first implementation of an unbiased, network-based analysis in a large sample of human left atrial appendage gene expression profiles. We found 2 modules associated with AF severity and atrial rhythm in 2 to 3 of our cardiovascular comorbidity groups. Functional analyses revealed significant enrichment of cardiovascular-related categories for both modules. In addition, several of the hub genes identified are implicated in cardiovascular disease and may play a role in AF initiation and progression.
In our study, WGCNA was used to construct modules based on gene coexpression, thereby reducing the net-work’s dimensionality to a smaller set of elements.17,21 Relating modulewise changes to phenotypic traits allowed statistically significant associations to be detected at a lower false discovery rate compared with traditional differential expression studies. Furthermore, shared functions and pathways among genes in the modules could be inferred via enrichment analyses.
We divided our data set into 3 groups to verify the reproducibility of the modules identified by WGCNA; 14 modules were identified in the MV group in our gene network. All were strongly preserved in the CAD and LAF groups, suggesting that gene coexpression patterns are robust and reproducible despite differences in cardiovascular comorbidities.
The use of module eigengene profiles as representative summary measures has been validated in a number of studies.20,26 Additionally, we found that the eigengenes accounted for a significant proportion (average 18%) of gene expression variability in their respective modules. Regression analysis of the module eigengenes found 2 modules associated with AF severity and atrial rhythm in ≥2 groups of data set. The association between the salmon module eigengene and AF severity was statistically weaker in the LAF group (adjusted P=9.0×10−2). This was probably because of its significantly smaller sample size compared with the MV and CAD groups. Despite this weaker association, the relationship between the salmon module eigengene and AF severity remained consistent among the 3 groups (Figure 3A). Similarly, the lack of statistical significance for the association between the tan module eigengene and atrial rhythm at surgery in the LAF group was likely driven by the smaller sample size and (by definition) lack of samples in the no AF category.
A major part of our analysis focused on the identification of module hub genes. Hubs are connected with a large number of nodes; disruption of hubs therefore leads to widespread changes within the network. This concept has powerful applications in the study of biology, genetics, and disease.29,30 Although mutations of peripheral genes can certainly lead to disease, gene network changes are more likely to be motivated by changes in hub genes, making them more biologically interesting targets for further study.17,29,31 Indeed,
- the hub genes of the salmon and tan modules accounted for the vast majority of the variation in their respective module eigengenes, signaling their importance in driving gene module behavior.
The hub genes identified in the salmon and tan modules were significantly associated with AF phenotype overall. It was noted that this association was statistically weaker for the lower-ranked hub genes in the tan module. This highlights an important aspect and strength of WGCNA—to be able to capture module-wide changes with respect to disease despite potentially weaker associations among individual genes.
The implementation of WGCNA necessitated the selection of a soft-thresholding parameter 13. Unlike hard-thresholding (where gene correlations below a certain value are shrunk to zero), the soft-thresholding approach gives greater weight to stronger correlations while maintaining the continuous nature of gene–gene relationships. We selected a 13 value of 3 based on the criteria outlined by Zhang and Horvath.17 His team and other investigators have demonstrated that module identification is robust with respect to the 13 parameter.17,19–21 In our data, we were also able to reproduce the key findings reported with a different, larger 13 value, thereby verifying the stability of our results relating to 13.
The salmon module (124 genes) was associated with both AF phenotypes; furthermore, IPA analysis of its gene contents suggested enrichment in cardiovascular development as well as disease. Its eigengene increased with worsening AF severity, with the largest stepwise change occurring between the paroxysmal AF and persistent AF categories (Figure 3). Hence,
- the gene expression changes within the salmon module may reflect the later stages of AF pathophysiology.
The top hub gene of the salmon module was RCAN1 (regulator of calcineurin 1). Calcineurin is a cytoplasmic Ca2+/ calmodulin-dependent protein phosphatase that stimulates cardiac hypertrophy via its interactions with NFAT and L-type Ca2+ channels.32,33 RCAN1 is known to inhibit calcineurin and its associated pathways.32,34 However, some data suggest that RCAN1 may instead function as a calcineurin activator when highly expressed and consequently potentiate hypertrophic signaling.35 Thus,
- perturbations in RCAN1 levels (attributable to genetic variants or mutations) may cause an aberrant switching in function, which in turn triggers atrial remodeling and arrhythmogenesis.
Other hub genes found in the salmon module are also involved in cardiovascular development and function and may be potential targets for further study.
- DNAJA4 (DnaJ homolog, subfamily A, member 4) regulates the trafficking and maturation of KCNH2 potassium channels, which have a prominent role in cardiac repolarization and are implicated in the long-QT syndromes.36
FHL2 (four-and-a-half LIM domain protein 2) interacts with numerous cellular components, including
- actin cytoskeleton,
- transcription machinery, and
- ion channels.37
FHL2 was shown to enhance the hypertrophic effects of isoproterenol, indicating that
- FHL2 may modulate the effect of environmental stress on cardiomyocyte growth.38
- FHL2 also interacts with several potassium channels in the heart, such as KCNQ1, KCNE1, and KCNA5.37,39
Additionally, blood vessel epicardial substance (BVES) and other members of its family were shown to be highly expressed in cardiac pacemaker cells. BVES knockout mice exhibited sinus nodal dysfunction, suggesting that BVES regulates the development of the cardiac pacemaking and conduction system40 and may therefore be involved in the early phase of AF development.
The tan module (679 genes) eigengene was negatively correlated with atrial rhythm in the MV and CAD groups (Figure 4); this may indicate a general decrease in gene expression of its members in fibrillating atrial tissue. IPA analysis revealed enrichment in genes involved in cell signaling as well as apoptosis. The top-ranked hub gene, cytoplasmic polyade-nylation element binding protein 3 (CPEB3), regulates mRNA translation and has been associated with synaptic plasticity and memory formation.41 The role of CPEB3 in the heart is currently unknown, so further exploration via animal model studies may be warranted.
Natriuretic peptide-precursor B (NPPB), another highly interconnected hub gene, produces a precursor peptide of brain natriuretic peptide, which
- regulates blood pressure through natriuresis and vasodilation.42
(NPPB) gene variants have been linked with diabetes mellitus, although associations with cardiac phenotypes are less clear.42 TBX5 and GATA4, which play important roles in the embryonic heart development,43 were members of the tan module. Although not hub genes, they may also contribute toward developmental susceptibility of AF. In addition, TBX5 was previously reported to be near an SNP associated with PR interval and AF in separate large-scale GWAS studies.12,28 MYOZ1, another candidate gene identified in the recent AF GWAS meta-analysis, was found to be a member as well; it associates with proteins found in the Z-disc of skeletal and cardiac muscle and may suppress calcineurin-dependent hypertrophic signaling.12
Some, but not all, of the candidate genes found in previous GWAS studies were located in the AF-associated modules. One possible explanation for this could be the difference in sample sizes. The meta-analysis involved thousands of individuals, whereas the current study had <100 in each group of data set, which limited the power to detect significant differences between levels of AF phenotype even with the module-wise approach. Additionally, transcription factors like PITX2 are most highly expressed during the fetal phase of development. Perturbations in these genes (attributable to genetic variants or mutations) may therefore initiate the development of AF at this stage and play no significant role in adults (when we obtained their tissue samples).
Limitations in Study
We noted several limitations in this study. First, no human left atrial mRNA data set of adequate size currently exists publicly. Hence, we were unable to validate our results with an external, independent data set. However, the network preservation assessment performed within our data set showed strong preservation in all modules, indicating that our findings are robust and reproducible.
Although the module eigengenes captured a significant proportion of module variance, a large fraction of variability did remain unaccounted for, which may limit their use as representative summary measures.
We extracted RNA from human left atrial appendage tissue, which consists primarily of cardiomyocytes and fibroblasts. Atrial fibrosis is known to occur with AF-associated remodeling.44 As such, the cardiomyocyte to fibroblast ratio is likely to change with different levels of AF severity, which in turn influences the amount of RNA extracted from each cell type. Hence, true differences in gene expression (and coexpression) within cardiomyocytes may be confounded by changes in cellular composition attributable to atrial remodeling. Also, there may be significant regional heterogeneity in the left atrium with respect to structure, cellular composition, and gene expression,45 which may limit the generaliz-ability of our results to other parts of the left atrium.
All subjects in the study were whites to minimize the effects of population stratification. However, it is recognized that the genetic basis of AF may differ among ethnic groups.9 Thus, our results may not be generalizable to other ethnicities.
Finally, it is possible for genes to be involved in multiple processes and functions that require different sets of genes. However, WGCNA does not allow for overlapping modules to be formed. Thus,
- this limits the method’s ability to characterize such gene interactions.
Conclusions
In summary, we constructed a weighted gene coexpression network based on RNA expression data from the largest collection of human left atrial appendage tissue specimens to date. We identified 2 gene modules significantly associated with AF severity or atrial rhythm at surgery. Hub genes within these modules may be involved in the initiation or progression of AF and may therefore be candidates for functional studies.
Refererences
1. European Heart Rhythm Association, European Association for Cardio-Thoracic Surgery, Camm AJ, Kirchhof P, Lip GY, Schotten U, et al. Guidelines for the management of atrial fibrillation: the task force for the management of atrial fibrillation of the European Society of Cardiology (ESC). Eur Heart J. 2010;31:2369–2429.
2. Lemmens R, Hermans S, Nuyens D, Thijs V. Genetics of atrial fibrillation and possible implications for ischemic stroke. Stroke Res Treat. 2011;2011:208694.
3. Wann LS, Curtis AB, January CT, Ellenbogen KA, Lowe JE, Estes NA III, et al; ACCF/AHA/HRS. 2011 ACCF/AHA/HRS focused update on the management of patients with atrial fibrillation (Updating the 2006 Guideline): a report of the American College of Cardiology Foundation/ American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2011;57:223–242.
4. Dobrev D, Carlsson L, Nattel S. Novel molecular targets for atrial fibrillation therapy. Nat Rev Drug Discov. 2012;11:275–291.
5. Christophersen IE, Ravn LS, Budtz-Joergensen E, Skytthe A, Haunsoe S, Svendsen JH, et al. Familial aggregation of atrial fibrillation: a study in Danish twins. Circ Arrhythm Electrophysiol. 2009;2:378–383.
6. Gudbjartsson DF, Arnar DO, Helgadottir A, Gretarsdottir S, Holm H, Sig-urdsson A, et al. Variants conferring risk of atrial fibrillation on chromosome 4q25. Nature. 2007;448:353–357.
7. Ellinor PT, Lunetta KL, Glazer NL, Pfeufer A, Alonso A, Chung MK, et al. Common variants in KCNN3 are associated with lone atrial fibrillation. Nat Genet. 2010;42:240–244.
8. Benjamin EJ, Rice KM, Arking DE, Pfeufer A, van Noord C, Smith AV, et al. Variants in ZFHX3 are associated with atrial fibrillation in individuals of European ancestry. Nat Genet. 2009;41:879–881.
9. Sinner MF, Ellinor PT, Meitinger T, Benjamin EJ, Kääb S. Genome-wide association studies of atrial fibrillation: past, present, and future. Cardio-vasc Res. 2011;89:701–709.
10. Clauss S, Kääb S. Is Pitx2 growing up? Circ Cardiovasc Genet. 2011;4:105–107.
11. Kirchhof P, Kahr PC, Kaese S, Piccini I, Vokshi I, Scheld HH, et al. PITX2c is expressed in the adult left atrium, and reducing Pitx2c expression promotes atrial fibrillation inducibility and complex changes in gene expression. Circ Cardiovasc Genet. 2011;4:123–133.
12. Ellinor PT, Lunetta KL, Albert CM, Glazer NL, Ritchie MD, Smith AV, et al. Meta-analysis identifies six new susceptibility loci for atrial fibrillation. Nat Genet. 2012;44:670–675.
13. Barth AS, Merk S, Arnoldi E, Zwermann L, Kloos P, Gebauer M, et al. Reprogramming of the human atrial transcriptome in permanent atrial fibrillation: expression of a ventricular-like genomic signature. Circ Res. 2005;96:1022–1029.
Continues to 45. see
http://circgenetics.ahajournals.org/content/6/4/362
CLINICAL PERSPECTIVE
Atrial fibrillation is the most common sustained cardiac arrhythmias in the United States. The genetic and molecular mechanisms governing its initiation and progression are complex, and our understanding of these mechanisms remains incomplete despite recent advances via genome-wide association studies, animal model experiments, and differential expression studies. In this study, we used weighted gene coexpression network analysis to identify gene modules significantly associated with atrial fibrillation in a large sample of human left atrial appendage tissues. We further identified highly interconnected genes (ie, hub genes) within these gene modules that may be novel candidates for functional studies. The discovery of the atrial fibrillation-associated gene modules and their corresponding hub genes provide novel insight into the gene network changes that occur with atrial fibrillation, and closer study of these findings can lead to more effective targeted therapies for disease management.
Read Full Post »




![Poster-ProBNP_final[1]](https://i0.wp.com/pharmaceuticalintelligence.com/wp-content/uploads/2013/12/poster-probnp_final1.jpg)









































{kind=link}