The Centrality of Ca(2+) Signaling and Cytoskeleton Involving Calmodulin Kinases and Ryanodine Receptors in Cardiac Failure, Arterial Smooth Muscle, Post-ischemic Arrhythmia, Similarities and Differences, and Pharmaceutical Targets
Author and Curator: Larry H Bernstein, MD, FCAP
Author, and Content Consultant to e-SERIES A: Cardiovascular Diseases: Justin Pearlman, MD, PhD, FACC
and
Curator: Aviva Lev-Ari, PhD, RN

Image generated by Adina Hazan, 06/30/2021
Abbreviations:
TAC – Transverse Aortic Constriction, AP, action potential; ARVD2, arrhythmogenic right ventricular cardiomyopathy type 2; CaMKII, Ca2+/calmodulim-dependent protein kinase II; CICR, Ca2+ induced Ca2+ release;CM, calmodulin; CPVT, catecholaminergic polymorphic ventricular tachycardia; ECC, excitation–contraction coupling; FKBP12/12.6, FK506 binding protein; HF, heart failure; LCC, L-type Ca2+ channel; P-1 or P-2, phosphatase inhibitor type-1 or type-2; PKA, protein kinase A; PLB, phosphoplamban; PP1, protein phosphatase 1; PP2A, protein phosphatase 2A; RyR1/2, ryanodine receptor type-1/type-2; SCD, sudden cardiac death; SERCA, sarcoplasmic reticulum Ca2+ ATPase; SL, sarcolemma; SR, sarcoplasmic reticulum.
This is the Part IV of a series on the cytoskeleton and structural shared thematics in cellular movement and cellular dynamics. The last two are specific to the heart, and the third was renal tubular caicium exchange and the effects of Na+ and hormones.
In Part I, Identification of Biomarkers that are Related to the Actin Cytoskeleton
http://pharmaceuticalintelligence.com/2012/12/10/identification-of-biomarkers-that-are-related-to-the-actin-cytoskeleton/
The prior articles discussed common management motifs across cell-types that are essential for cell division, embryogenesis, cancer metastasis, osteogenesis, musculoskeletal function, vascular compliance, and cardiac contractility. This second article concentrates on specific functionalities for cardiac contractility based on Ca++ signaling in excitation-contraction coupling, addressing modifications specific to cardiac muscle and not to skeletal muscle. In Part I there was discussion of the importance of Ca2+ signaling on innate immune system, and the roles of calcium in immunology will be further expanded in a third article of the series.
The Series consists of the following articles:
Part I: Identification of Biomarkers that are Related to the Actin Cytoskeleton
Larry H Bernstein, MD, FCAP
http://pharmaceuticalintelligence.com/2012/12/10/identification-of-biomarkers-that-are-related-to-the-actin-cytoskeleton/
Part II: Role of Calcium, the Actin Skeleton, and Lipid Structures in Signaling and Cell Motility
Larry H. Bernstein, MD, FCAP, Stephen Williams, PhD and Aviva Lev-Ari, PhD, RN
http://pharmaceuticalintelligence.com/2013/08/26/role-of-calcium-the-actin-skeleton-and-lipid-structures-in-signaling-and-cell-motility/
Part III: Renal Distal Tubular Ca2+ Exchange Mechanism in Health and Disease
Larry H. Bernstein, MD, FCAP, Stephen J. Williams, PhD
and Aviva Lev-Ari, PhD, RN
http://pharmaceuticalintelligence.com/2013/09/02/renal-distal-tubular-ca2-exchange-mechanism-in-health-and-disease/
Part IV: The Centrality of Ca(2+) Signaling and Cytoskeleton Involving Calmodulin Kinases and Ryanodine Receptors in Cardiac Failure, Arterial Smooth Muscle, Post-ischemic Arrhythmia, Similarities and Differences, and Pharmaceutical Targets
Larry H Bernstein, MD, FCAP, Justin Pearlman, MD, PhD, FACC and Aviva Lev-Ari, PhD, RN
http://pharmaceuticalintelligence.com/2013/09/08/the-centrality-of-ca2-signaling-and-cytoskeleton-involving-calmodulin-kinases-and-ryanodine-receptors-in-cardiac-failure-arterial-smooth-muscle-post-ischemic-arrhythmia-similarities-and-differen/
Part V: Ca2+-Stimulated Exocytosis: The Role of Calmodulin and Protein Kinase C in Ca2+ Regulation of Hormone and Neurotransmitter
Larry H Bernstein, MD, FCAP
and
Aviva Lev-Ari, PhD, RN
http://pharmaceuticalintelligence.com/2013/12/23/calmodulin-and-protein-kinase-c-drive-the-ca2-regulation-of-hormone-and-neurotransmitter-release-that-triggers-ca2-stimulated-exocytosis/
Part VI: Calcium Cycling (ATPase Pump) in Cardiac Gene Therapy: Inhalable Gene Therapy for Pulmonary Arterial Hypertension and Percutaneous Intra-coronary Artery Infusion for Heart Failure: Contributions by Roger J. Hajjar, MD
Aviva Lev-Ari, PhD, RN
http://pharmaceuticalintelligence.com/2013/08/01/calcium-molecule-in-cardiac-gene-therapy-inhalable-gene-therapy-for-pulmonary-arterial-hypertension-and-percutaneous-intra-coronary-artery-infusion-for-heart-failure-contributions-by-roger-j-hajjar/
Part VII: Cardiac Contractility & Myocardium Performance: Ventricular Arrhythmias and Non-ischemic Heart Failure – Therapeutic Implications for Cardiomyocyte Ryanopathy (Calcium Release-related Contractile Dysfunction) and Catecholamine Responses
Justin Pearlman, MD, PhD, FACC, Larry H Bernstein, MD, FCAP and Aviva Lev-Ari, PhD, RN
http://pharmaceuticalintelligence.com/2013/08/28/cardiac-contractility-myocardium-performance-ventricular-arrhythmias-and-non-ischemic-heart-failure-therapeutic-implications-for-cardiomyocyte-ryanopathy-calcium-release-related-contractile/
Part VIII: Disruption of Calcium Homeostasis: Cardiomyocytes and Vascular Smooth Muscle Cells: The Cardiac and Cardiovascular Calcium Signaling Mechanism
Justin Pearlman, MD, PhD, FACC, Larry H Bernstein, MD, FCAP and Aviva Lev-Ari, PhD, RN
http://pharmaceuticalintelligence.com/2013/09/12/disruption-of-calcium-homeostasis-cardiomyocytes-and-vascular-smooth-muscle-cells-the-cardiac-and-cardiovascular-calcium-signaling-mechanism/
Part IX: Calcium-Channel Blockers, Calcium Release-related Contractile Dysfunction (Ryanopathy) and Calcium as Neurotransmitter Sensor
Justin Pearlman, MD, PhD, FACC, Larry H Bernstein, MD, FCAP and Aviva Lev-Ari, PhD, RN
Part X: Synaptotagmin functions as a Calcium Sensor: How Calcium Ions Regulate the fusion of vesicles with cell membranes during Neurotransmission
Larry H Bernstein, MD, FCAP and Aviva Lev-Ari, PhD, RN
http://pharmaceuticalintelligence.com/2013/09/10/synaptotagmin-functions-as-a-calcium-sensor-how-calcium-ions-regulate-the-fusion-of-vesicles-with-cell-membranes-during-neurotransmission/
Part XI: Sensors and Signaling in Oxidative Stress
Larry H. Bernstein, MD, FCAP
http://pharmaceuticalintelligence.com/2013/11/01/sensors-and-signaling-in-oxidative-stress/
Part XII: Atherosclerosis Independence: Genetic Polymorphisms of Ion Channels Role in the Pathogenesis of Coronary Microvascular Dysfunction and Myocardial Ischemia (Coronary Artery Disease (CAD))
Larry H Bernstein, MD, FCAP and Aviva Lev-Ari, PhD, RN
http://pharmaceuticalintelligence.com/2013/12/21/genetic-polymorphisms-of-ion-channels-have-a-role-in-the-pathogenesis-of-coronary-microvascular-dysfunction-and-ischemic-heart-disease/
Observations of Tissues Dependent on Electrical Impulses and Differences in Calcium-Efflux Mechanisms
Voice of Justin Pearlman
Skeletal muscles are named for muscle bundles attached to skeleton elements, including head and neck, thorax, and the long bones of limbs, but the same structural and neuronally controlled muscle type is also in the abdomenal wall and the scalp, face, and eyes (for eye motion), each serving the function of movement on demand. The skeletal element these muscles attach to are tendons (fibrous tissue), often anchored to bone before and after an articulation (joint). There are several features that distinguish skeletal muscle from smooth muscle and from myocardium (heart muscle). Skeletal muscles are striated. They have fast-twitch and slow-twitch fibers in various proportions. They are under voluntary neural control, not autonomic (involuntary).
In distinction, smooth muscles line arterial blood vessels, lymphatics, the urinary bladder, the gastrointestinal tract, the respiratory tract, and also the uterus, the pili of the skin (goose bumps), and are in the eyes to control pupil diameter and lens focus. They are controlled by autonomic innervation.
The myocardium, or heart muscle, is distinct in many ways. The heart muscle has a unique architecture with Z-bands. The heart muscle a syncytium of cardiac muscle made of cardiomyocytes, which means instead of a bundle of separate cells each distinctly bounded by a cell membrane, the entire heart muscle can be viewed as a single multinucleated cell (or merger of cells). Skeletal muscle has multinucleated cells also from the merger of multiple blast cells, but unlike the heart there are distinct cell boundaries between skeletal myocytes, known as myofibers. The heart has fiber layers with different orientations (spiral clockwise and counterclockwise arrangement of muscle fibers) that result in multiple types of motion, but technically all of the heart muscle fibers are part of a single conglomerate cell. The motions of the heart include: translation, tilting, shortening, thickening, narrowing, twisting, rotating, lengthening and widening. The heart cell contracts and has innervation to the AV node and the SA node, with both sympathetic and parasymptathetic innervation.
All three types of muscle apply a basic Motif of proteins that change length in response to a calcium signal. The calcium is stored is sacks called the sarcoplasmic reticulum. The calcium is released into the main fluid of the cell (the cytoplasm), where it controls different functions. Even in skeletal muscle there is a difference between thigh and thorax, and we know from comparative ornithology that the enzymology and energy metabolism of the wings of birds that soar, hawks and eagles, differs from the chicken, or the turkey.
Key features are illustrated below.
Figure 1….. skeletal muscle vs heart calcium channels.
receptors voltage gated Ca(2) channel

We see in Figure 1 that both the skeletal muscle and the cardiomyocyte have a Ryanodyne receptor that is the flow device for carrying the Ca(2+) ions from the sarcoplasm into the cytoplasm. In the skeletal muscle there is a dihydropyridine receptor. The heart muscle is voltage gated. The interaction with calmodulin (not shown) via Calcium/calmodulin-dependent Protein Kinase Type II delta = CaMKI, II – IV. CaMKII has isoforms a, b, c, d – and CaMKIId has two splice variants (cytoplasmic and nuclear). These will be discussed fully in the fifth of the series. Take note of the fact the CaMKII isoform is found only in the heart. So we have here molecules with similar structure, but not completely homologous. Structure and function have made small, requiring significant adaptations.
Figure 2. A cardiomycyte structure with the sarcomere and calcium efflux into the cytoplasn, and with the mitochondrion available for Ca(2+) exchange with the cytoplasm, and with Ca(2+), Na(+) and K(+) channels contiguous with the extracellular space.
RyR

The arterial endothelium is functionally protected by eNOS converting arginine to citrulline. This does not occur with adult form of urea cycle (Krebs Henseleit) disorder, as there is no substrate. iNOS, a nitric oxide isoform present in macrophages that invade through intercellular spaces into the underlying matrix. A large study presented at the European Society of Cardiology (ESC) 2013 Congress has indicated that there is not a relationship of tight control of type 2 diabetes and cardiovascular events, even though we know that there is a relationship between diabetes and
- insulin resistance
- endothelial activation
- inflammatory markers
- homocysteine
Adipokines interact in type 2 diabetes with inflammatory cytokines for development of insulin resistance, and these are markers of arterial vascular disease. But the association of diabetes with heart disease, long considered valid, has come into some dispute. Recently, saxagliptin was associated with a significant 27% increased risk of hospitalizations for heart failure in the Saxagliptin Assessment of Vascular Outcomes Recorded in Patients with Diabetes Mellitus (SAVOR-TIMI 53) study, a component of the prespecified secondary end point. In the Examination of Cardiovascular Outcomes with Alogliptin versus Standard of Care in Patients with Type 2 Diabetes Mellitus and Acute Coronary Syndrome (EXAMINE) study, there was no increased risk of heart failure with alogliptin. While saxagliptin and alogliptin significantly reduced glycated hemoglobin levels, there was some debate about the role of the drugs, which are dipeptidyl peptidase-4 (DPP-4) inhibitors, in clinical practice. There is some disappointment with respect to the diabetes issue, but that might be remedied by improvement based on the appropriate combination of biomarkers for prediction asnd monitoring at the earliest onset. Dr William White said alogliptin lowers the glycemic index significantly, and such reductions can reduce the risk of microvascular complications. We know from the prior literature that it might take five years-plus before we determine a microvascular benefit. A serious problem in the validity of the results was that statistically, saxagliptin met the primary end point of noninferiority, with the drug no worse than placebo. Glycated hemoglobin levels were reduced with saxagliptin, down from 8.0% at baseline to 7.7% at the end of the trial (p<0.001 vs placebo). In addition, more patients in the saxagliptin arm had glycated hemoglobin levels reduced to less than 7.0%. The relevant question is what the effect was for patients who achieved a glycated Hb of < 7.7%, which makes the p-value meaningless for an 0.3% change overall.
Implications of ca(2+) handling dysfunction
A. if the dysfuction is in smooth muscle – effect on arterial elasticity
B. if the dysfunction is in cardiomyocytes – Ventricular contractility & arrhythmias
We now review the calcium cycling of smooth muscle based on extracted work at MIT and Harvard Medical School, and at the University of Iowa. The work focuses on the disordered Ca(2+) signaling that plays a large role in the development of “arterial stiffness”, not disregarding the competing roles of endothelial nitric oxide and the inflammatory cell mediated oxidative stress related iNOS in the arterial circulation, and the preference for stress points at the junction of arteries. Disordered Ca(2+) in vascular smooth muscle leads to ischemic arterial disease, vascular rigidity from loss of flexibility, which can lead to ischemic myocardial damage.
Calcium Cycling in Synthetic and Contractile Phasic or Tonic Vascular Smooth Muscle Cells
L Lipskaia, I Limon, R Bobe and R Hajjar.
Chapter 2. Intech Open. @2012. http://dx.doi.org/10.5772/48240
Calcium ions (Ca2+) are present in low concentrations in the cytosol (~100 nM) and in high concentrations (in mM range) in both the extracellular medium and intracellular stores (mainly sarco/endo/plasmic reticulum, SR). This differential allows the calcium ion to be a ubiquitous 2nd messenger that carries information essential for cellular functions as diverse as contraction, metabolism, apoptosis, proliferation and/or hypertrophic growth. The mechanisms responsible for generating a Ca2+ signal greatly differ from one cell type to another. In the different types of vascular smooth muscle cells (VSMC), enormous variations do exist with regard to the mechanisms responsible for generating Ca2+ signal. In each VSMC phenotype (synthetic/proliferating1 and contractile2 [1], tonic or phasic), the Ca2+ signaling system is adapted to its particular function and is due to the specific patterns of expression and regulation of Ca2+ handling molecules (Figure 1).
1Synthetic VSMCs have a fibroblast appearance, proliferate readily, and synthesize increased levels of various extracellular matrix components, particularly fibronectin, collagen types I and III, and tropoelastin [1].
2Contractile VSMCs have a muscle-like or spindle-shaped appearance and well-developed contractile apparatus resulting from the expression and intracellular accumulation of thick and thin muscle filaments [1].
in contractile VSMCs, the initiation of contractile events is driven by membrane depolarization; and the principal entry-point for extracellular Ca2+ is the voltage-operated L-type calcium channel (LTCC). In contrast, in synthetic/proliferating VSMCs, the principal way-in for extracellular Ca2+ is the store-operated calcium (SOC) channel. Whatever the cell type, the calcium signal consists of limited elevations of cytosolic free calcium ions in time and space. The calcium pump, sarco/endoplasmic reticulum Ca2+ ATPase (SERCA), has a critical role in determining the frequency of SR Ca2+ release by controlling the velocity of Ca2+ upload into the sarcoplasmic reticulum (SR) and the Ca2+ sensitivity of SR calcium channels, Ryanodin Receptor, RyR and Inositol tri-Phosphate Receptor, IP3R.
Figure 1. Schematic representation of Calcium Cycling in Contractile and Proliferating VSMCs.

Schematic representation of Calcium Cycling in Contractile and Proliferating VSMCs
Left panel: schematic representation of calcium cycling in quiescent /contractile VSMCs. Contractile response is initiated by extracellular Ca2* influx due to activation of Receptor Operated Ca2* channels (through phosphoinositol-coupled receptor) or to activation of L-Type Calcium channels (through an increase in luminal pressure). Small increase of cytosolic due IP3 binding to IP3R (puff) or RyR activation by LTCC or ROC-dependent Ca2* influx leads to large SR Ca2* release due to the activation of IP3R or RyR clusters (“Ca2*-induced Ca2*release” phenomenon). Cytosolic Ca2* is rapidly reduced by SR calcium pumps (both SERCA2a and SERCA2b are expressed in quiescent VSMCs), maintaining high concentration of cytosolic Ca2* and setting the sensitivity of RyR or IP3R for the next spike. Contraction of VSMCs occurs during oscillatory Ca2* transient. Middle panel: schematic representation of atherosclerotic vessel wall. Contractile VSMC are located in the media layer, synthetic VSMC are located in sub-endothelial intima. Right panel: schematic representation of calcium cycling in quiescent /contractile VSMCs. Agonist binding to phosphoinositol-coupled receptor leads to the activation of IP3R resulting in large increase in cytosolic Ca2*. Calcium is weakly reduced by SR calcium pumps (only SERCA2b, having low turnover and low affinity to Ca2* is expressed). Store depletion leads to translocation of SR Ca2* sensor STIM1 towards PM, resulting in extracellular Ca2* influx though opening of Store Operated Channel (CRAC). Resulted steady state Ca2* transient is critical for activation of proliferation-related transcription factors ‘NFAT). Abbreviations: PLC – phospholipase C; PM – plasma membrane; PP2B – Ca2*/calmodulin-activated protein phosphatase 2B (calcineurin); ROC- receptor activated channel; IP3 – inositol-1,4,5-trisphosphate, IP3R – inositol-1,4,5-trisphosphate receptor; RyR – ryanodine receptor; NFAT – nuclear factor of activated T-lymphocytes; VSMC – vascular smooth muscle cells; SERCA – sarco(endo)plasmic reticulum Ca2* ATPase; SR – sarcoplasmic reticulum.
General aspects of calcium cycling and signaling in vascular smooth muscle cells
Besides maintaining vascular tone in mature vessels, VSMCs also preserve blood vessel integrity. VSMCs are instrumental for vascular remodeling and repair via proliferation and migration. Interestingly, Ca2* plays a central role in both physiological processes. In VSMCs, calcium signaling involves a cross-regulation of Ca2* influx, sarcolemmal membrane signaling molecules and Ca2* release and uptake from the sarco/endo/plasmic reticulum and mitochondria, which plays a central role in both vascular tone and integrity.
Calcium handling by the plasma membrane’s calcium channels and pumps
Membrane depolarization is believed to be a key process for the activation of calcium events in mature VSMCs. Thus, much attention has been given to uncovering the various mechanisms responsible for triggering this depolarization. Increased intra-vascular pressure of resistance arteries stimulates gradual membrane depolarization in VSMCs, increasing the probability of opening L-type high voltage-gated Ca2* channels (Cav1.2) (LTCC). Alternatively, the calcium-dependent contractile response can be induced through the activation of specific membrane receptors coupled to phospholipase C (PLC) isoforms3. The various isoforms of transient receptor potential (TRP) ion channel family, particularly TRPC3, TRPC6 and TRPC7 possibly activated directly by diacyl glycerol (DAG), can also contribute to initial plasma membrane Ca2* influx and subsequent membrane depolarization.
Among voltage-insensitive calcium influx pathways, the store-operated Ca2* channels (SOC), maintain a long-term cellular Ca2* signal. They are activated upon a decrease of internal store Ca2* concentration resulting from a Ca2* release via the opening of SR Ca2* release channels. SOC has two essential regulatory components, the SR/ER located Ca2* sensor STIM1 (stromal interaction molecule) and the Ca2* channels Orai. Upon decrease of [Ca2*] in the reticulum (<500µM), Ca2* dissociates from STIM1; then STIM1 molecules oligomerize and translocate to specialized cortical reticulum compartments adjacent to the plasma membrane. There, the STIM1 cytosolic activating domains bind to and cluster the Orai proteins into an opened archaic Ca2* channel known as Ca2*-release activated Ca2* channel (CRAC).
- All isoforms of PLC, catalyze the hydrolysis of phosphatidylinositol4,5-biphosphate (PIP2) to produce the intracellular messengers IP3 increase and diacylglycerol (DAG); both of which promote cytosolic Ca2* rise through activation of plasma membrane or sarcoplasmic reticulum calcium channels.
- The CRAC is responsible for the “2h cytosolic Ca2* increase” required to induce VSMCs proliferation.
The calcium signal is terminated by membrane hyper-polarization and cytosolic Ca2+ removal. First, calcium sparks resulting from the opening of sub-plasmalemmal clusters of RyR activate large-conductance Ca2+ sensitive K+ (BK) channels. Then, the resulting spontaneous transient outward currents (STOC) hyperpolarize the membrane and decrease the open probability of L-type Ca2+ channels. Cytosolic calcium is extruded at the level of plasma membrane by plasma membrane Ca2+ ATPase (PMCA) and the Na+/Ca2+ exchanger (NCX). The principal amount of cytosolic Ca2+ (> 70%) is re-uploaded to the internal store.
Calcium handling by the sarco/endoplasmic reticulum’s calcium channels and pumps
The initial entry of Ca2+ through plasma membrane channels triggers large Ca2+ release from the internal store via the process of Ca2+-induced Ca2+-release (CICR). The mechanism responsible for initiating Ca2+ release depends on Ca2+ sensitive SR calcium channels, the ryanodin receptor (RyR)5 or the IP3 receptor (IP3R). Indeed, IP3R and RyR are highly sensitive to cytosolic Ca2+ concentrations and when cytosolic Ca2+ concentration ranges from nM to µM, they open up. On the contrary, a higher cytosolic Ca2+ concentration (from µM to mM) closes them. In other words, cytosolic Ca2+ increase first exerts a positive feedback and facilitates SR channels opening whereas a further increase has an opposite effect and actually inhibits the SR channels opening. Importantly enough to be mentioned, RyR phosphorylation by the second messenger cyclic ADP ribose (cADPR) and protein kinase A (PKA) enhances Ca2+ sensitivity, the phosphorylation induced by the protein kinase C (PKC) decreases RyR sensitivity to Ca2+.
Sarco/Endoplasmic Ca2+ATPases (SERCA), the only calcium transporters expressed within sarco/endoplasmic reticulum (SR), serve to actively return calcium into this organelle. In mammals, three SERCA genes ATP2A1, ATP2A2 and ATP2A3 coding for SERCA1, SERCA2 and SERCA3 isoforms respectively have been identified [35]. Each gene gives rise to a different SERCA isoform through alternative splicing (Figure 2); they all have discrete tissue distributions and unique regulatory properties, providing a potential focal point within the cell for the integration of diverse stimuli to adjust and fine-tune calcium homeostasis in the SR/ER. In VSMCs, SERCA2a and the ubiquitous SERCA2b isoforms are expressed; besides vascular smooth muscle, SERCA2a is preferentially expressed in cardiac and skeletal muscles. SERCA2b differs from SERCA2a by an extension of 46 amino acids. Diversity of SERCA isoforms in the same cell suggests that each of them could be responsible for controlling unique cell functions.
- RyR are structurally and functionally analogous to IP3R, although they are approximately twice as large and have twice the conductance of IP3R [27]; RyR channels are sensitive to store loading and IP3R channels are sensitized by the agonist-dependent formation of IP3.
SERCA2’s activity depends on its interaction with phospholamban and is inhibitory in its de-phosphorylated form. PKA phosphorylation of phospholamban results in its dissociation from SERCA2, thus activating the Ca2+ pumps. Cyclic ADP-ribose was also reported to stimulate SERCA pump activity.
As previously mentioned, SR Ca2+ content controls the sensitivity of SR Ca2+ channels, RyR and IP3R, as well as functioning of SOC-mediated Ca2+ entry, thereby determining the type of intracellular calcium transient. Since SOCs opening depends on Ca2+ content of the store, one may suggest that SERCA participates to its regulation. Consistent with this, SOCs open up when the leak of Ca2+ from intracellular stores is not compensated with SERCA activity; SERCA inhibitors such as thapsigargin which prevent Ca2+ uptake are commonly used to chemically induce SOC currents; several works have established that SERCA can cluster with STIM1 and Orai1 in various cellular types.
Mechanisms of cytosolic Ca2+ oscillations in VSMC
Ca2+ oscillations are one of the ways that VSMCs respond to agonists. These Ca2+ oscillations are maintained during receptor occupancy and are driven by an endogenous pacemaker mechanism, called the cellular Ca2+ oscillator. Ca2+ oscillators were classified into two main types, the membrane oscillators and the cytosolic oscillators.
Membrane oscillators are those which generate oscillations at the cell membrane by successive membrane depolarization. In most small resistance arteries, inhibitors of plasma membrane voltage-dependent channels reduce or even abolish the membrane potential oscillations which precede rhythmical contractions. This suggests that rhythmic extracellular Ca2+ influx can be required for calcium oscillatory transient. Besides, membrane oscillators greatly depend on Ca2+ entry in order to provide enough Ca2+ to charge up the intracellular stores for each oscillatory cycle.
Cytosolic oscillators do not depend on the cell membrane to generate oscillations. Instead, they arise from intracellular store membrane instability. The pacemaker mechanism of cytosolic Ca2+ oscillator is based on the velocity of luminal Ca2+ loading and luminal Ca2+ content. The mechanism responsible for initiating Ca2+ release depends either on RyRs or IP3R activation. As soon as stores are sufficiently charged with Ca2+, the SR Ca2+ channels become sensitive to cytosolic Ca2+ and can participate to the process of Ca2+-induced Ca2+-release, which is responsible for orchestrating the regenerative release of Ca2+ from the SR/ER. Importantly, extracellular Ca2+ influx is not required for cytosolic oscillator function. Indeed, the Ca2+ oscillations can be observed in the absence of extracellular Ca2+.
In mature vessels, VSMCs mainly exhibit a tonic or phasic contractile phenotype. In contractile VSMCs extracellular calcium influx predominantly takes place through the voltage-dependent L-type calcium channel, LTCC9 (Figure 3). Extracellular Ca2* influx causes a small increase of cytosolic Ca2* generated by the opening of IP3R clusters, called puff and/or RyR2 clusters, called spark. These local rises of cytosolic Ca2* generate a larger SR Ca2* release through the Ca2*-induced Ca2* release phenomenon. Elevation of free cytosolic calcium triggers VSMC contraction.
- In contractile VSMCs, NFAT can be activated by sustained Ca2* influx (persistent Ca2* sparklets) mediated by clusters of L-type Ca2* channels operating in a high open probability mode
Steady state increase in cytosolic Ca2* triggers tonic contraction; oscillatory type of Ca2* transient triggers phasic contraction. It is worth mentioning that accumulating evidence indicate that SR Ca2*ATPase functioning/location within the cell (which greatly influences the velocity of calcium upload) determines the mode of Ca2* transient in VSMCs. Consistent with this, i) “phasic” VSMCs display a greater number of peripherally located SR than “tonic” VSMCs; indeed “tonic” VSMCs exhibit centrally located SR; (rev in [61, 77]); ii) drugs which interfere with the IP3 pathway or intracellular stores abolish spontaneous vaso-motion; iii) blocking SERCA strongly inhibits the Ca2* oscillations, demonstrating that they are induced by SR Ca2* release; this latter argument is further supported by the fact that oscillations are present even in the absence of extracellular Ca2*
SERCA2a has a higher catalytic turnover when compared to SERCA2b due to a higher rate of de-phosphorylation and a lower affinity for Ca2+; ii) SER-CA2a is absent in synthetic VSMCs, which only exhibit tonic contraction, iii) transferring the SERCA2a gene to synthetic cultured VSMCs modifies the agonist-induced calcium transient from steady-state to oscillatory mode. Therefore, one might suggest that the physiological role of SERCA2a in VSMCs consists of controlling the “cytosolic oscillator”, thereby determining phasic vs tonic type of smooth muscle contraction.
SERCA2a as a potential target for treating vascular proliferative diseases
Abundant proliferation of VSMCs is an important component of the chronic inflammatory response associated to atherosclerosis and related vascular occlusive diseases (intra-stent restenosis, transplant vasculopathy, and vessel bypass graft failure). Great efforts have been made to prevent/reduce trans-differentiation and proliferation of synthetic VSMCs. Anti-proliferative therapies including the use of pharmacological agents and gene therapy approaches are, until now, considered as a suitable approach in the treatment of these disorders. Indeed, coronary stenting is the only procedure that has been proven to reduce the incidence of late restenosis after percutaneous transluminal coronary angioplasty. Nevertheless, post-interventional intra-stent restenosis, characterized by the re-narrowing of the arteries caused by VSMC proliferation, occurs in 10 to 20 % of patients. These disorders remain the major limitation of revascularization by percutaneous transluminal angioplasty and artery bypass surgery. The use of drug-eluting stents (stent eluting anti-proliferative drug) significantly reduces restenosis but impairs the re-endothelialization process and subsequently often induces late thrombosis. In human, trans-differentiation of contractile VSMCs towards a synthetic/proliferating inflammatory/migratory phenotype after percutaneous transluminal angioplasty appears to be a fundamental process of vascularproliferative disease.
Concluding remarks
Over the last decade, great progress has been made in the understanding of the various intracellular molecular mechanisms in VSMCs which control calcium cycling and excitation/contraction or excitation/transcription coupling. VSMCs employ a great variety of Ca2+ signaling systems that are adapted to control their different contractile functions. Alterations in the expressions of Ca2+ handling molecules are closely associated with VSMC phenotype modulation. Furthermore, these changes in expression are inter-connected and each acquired or lost Ca2+ signaling molecule represents a component of signaling module functioning as a single unit.
In non-excitable synthetic VSMCs, calcium cycling results from the protein module ROC/IP3R/STIM1/ORAI1 which controls SOC influx. Agonist stimulation of synthetic VSMCs translates into a sustained increase in cytosolic Ca2+. This increase is required for the activation of NFAT downstream cellular signaling pathways inducing proliferation, migration and possibly an inflammatory response. Calcium cycling in excitable contractile VSMCs is governed by the protein module composed of ROC/LTCC/RyR2/SERCA2a and controls the contractile response.
Author details
Larissa Lipskaia
Mount Sinai School of Medicine, Department of Cardiology, New York, NY, USA
Isabelle Limon
Univ Paris 6, UR4 stress inflammation and aging, Paris, France
Abbreviations
BK – large-conductance Ca2+ sensitive K+ channel; cADPR – cyclic Adenosine Diphosphate Ribose; CICR – Ca2+- Induced Ca2+ Release; CRAC – Ca2+- Release Activated Ca2+ Channels; DAG – Diacyl Glycerol; IP3R – sarco/endoplasmic reticulum Ca2+ channel Inositol tri-Phosphate Receptor; LTCC – voltage-dependent L-type Ca2+ channels; NCX – Na+/Ca2+ exchanger; PKA – Protein Kinase A (activated by cAMP, cyclic adenosine monophosphate); PLC – Phospholipase C; PMCA – Plasmic Membrane Ca2+ ATPase; RyR – sarco/endoplasmic reticulum Ca2+ channel Ryanodin Receptor
B. cardiomyocyte or smooth muscle. Let’s look a little further.
CaM kinase and disordering of intracellular calcium homeostasis , molecular link to arrhythmias
Mark E. Anderson, MD, PhD, Professor of Medicine and Pharmacology, University of Iowa, Iowa City, IADr. Anderson has presented a large body of work done at Vanderbilt University and University of Iowa Medical Schools for over a decade. The major hypothesis is that in the aftermath of a heart attack, the structural and electrical remodeling renders the heart prone to arrhythmias . The signaling molecule called calmodulin (CaM) kinase is a key and the work suggests that drugs that block CaM kinase activity might make good anti-arrhythmic medications. CaM kinase is a molecule that is intricately involved in calcium signaling and regulation. CaM kinase regulates calcium entry into the cell and calcium storage and release inside the cell.
Calcium enters heart cells through proteins called L-type calcium channels, donut-like pores in the cell membrane that open and close. If these channels stay open and let too much calcium into the cell, the risk of arrhythmia increases. Studies have shown that CaM kinase activity is increased in animal models and human heart disease. Dr. Anderson poses the question – does CaM kinase — which we know is elevated in heart disease — drive arrhythmias? The question is driven by their findings that the addition of activated CaM kinase allowed more calcium than normal to flow into isolated heart cells. The investigators measured the opening and closing of single calcium channels using a technique called patch-clamp electrophysiology. Then they added an already-activated form of CaM kinase to the preparation. When we added the activated CaM kinase, the calcium channels opened like crazy,” Anderson said. “In fact, they were more likely to open and stay open for long periods of time.
They also showed that cardiac cells with added CaM kinase had electrical changes called early afterdepolarizations (EADs). EADs are believed to be the triggering cause of arrhythmias in cardiomyopathy, hypertrophy, and long QT syndrome. The investigators implanted tiny telemeters into the mice and recorded electrocardiograms (ECGs) , which revealed not only the electrical changes expected in diseased hearts, Anderson said, but also an increased tendency for arrhythmias. Next, they treated the mice with a drug that blocks CaM kinase activity significantly suppressed the arrhythmias. They also found that cardiac cells isolated from the mice and found spontaneous EADs, which disappeared when the cells were treated with the CaM kinase-blocking drug. The evidence all points to CaM kinase driving arrhythmias.
They have demonstrated that CaM kinase is also important for calcium-activated gene expression and that it may be involved in the changes that occur in association with cardiac hypertrophy and heart failure. Anderson suggests that CaM kinase could be the link to explain why calcium channels open more frequently in heart failure, why people in heart failure have arrhythmias. He postulates that it would good to have a target that addresses both phenotypic disorders — the arrhythmia phenotype and the heart failure phenotype — and CaM kinase may be that target. Further, he observes that with the exception of so-called beta blockers, none of the current anti-arrhythmic drugs have been shown to reduce the mortality rate. More recent work in Iowa has identified a new link – a link between the inflammation in heart muscle following a heart attack and the enzyme calcium/calmodulin-dependent protein kinase II or CaM kinase II.
CaM kinase II, a pivotal enzyme that registers changes in calcium levels and oxidative stress and translates these signals into cellular effects, including changes in heart rate, cell proliferation and cell death. CaM kinase II also regulates gene expression — which genes are turned on or off at any given time. We have seen how Inhibition of CaM kinase II in mice protects the animals’ hearts against some of the damaging effects of a heart attack. A study compared a large number of genes that were expressed in the protected mice compared to the non-protected control mice. A particularly interesting finding was that a cluster of inflammatory genes was differently expressed depending on whether CaM kinase II was active or inhibited. Specifically, the research showed that heart attack triggered increased expression of a set of pro-inflammatory genes, and inhibition of CaM kinase II substantially reduced this effect.
The main research themes pursued by the Anderson laboratory are
- Oxidative activation of CaMKII;
- CaMKII signaling to ion channels;
- The role of CaMKII in inflammation;
- The role of CaMKII in cardiac pacemaker cells;
- The role of CaMKII in cell survival.
Keywords: Calcium-Calmodulin-Dependent Protein Kinase Type 2, Calcium, Calcium-Calmodulin-Dependent Protein Kinases, Calcium Channels, L-Type, Calmodulin, Arrhythmia, Ion channel, Hypertrophy, Cell Signaling, Signal Transduction
Regulation of cardiac ATP-sensitive potassium channel surface expression by calcium/calmodulin-dependent protein kinase II.
Ana Sierra; Asipu Sivaprasadarao; Peter M Snyder; Ekaterina Subbotina; Michel Vivaudou; Zhiyong Zhu; Leonid V Zingman; et al.
Differential regulated interactions of calcium/calmodulin-dependent protein kinase II with isoforms of voltage-gated calcium channel beta subunits.
Grueter, CE, Abiria, SA, Wu, Y, Anderson, ME, Colbran, RJ.
Biochemistry, 47(6), 1760-7, 2008.
Differential effects of phospholamban and Ca2+/calmodulin-dependent kinase II on [Ca2+]i transients in cardiac myocytes at physiological stimulation frequencies.
Werdich, AA, Lima, EA, Dzhura, I, Singh, MV, Li, J, Anderson, ME, Baudenbacher, FJ.
Am J Physiol Heart Circ Physiol, 294(5), H2352-62, 2008.
Conserved Regulation of Cardiac Calcium Uptake by Peptides Encoded in Small Open Reading Frames
Emile G. Magny1, Jose Ignacio Pueyo1, Frances M.G. Pearl1,2, MA Cespedes1, et al.
1 School of Life Sciences, University of Sussex, Falmer, Brighton, East Sussex, UK.
2 Institute of Cancer Research, Sutton, Surrey SM2 5NG, UK Science
http:/dx.doi.org/10.1126/science.1238802
Small Open Reading Frames (smORFs) are short DNA sequences able to encode small peptides of less than 100 amino acids. Study of these elements has been neglected despite thousands existing in our genomes. We and others showed previously that peptides as short as 11 amino acids are translated and provide essential functions during insect development. Here, we describe two peptides of less than 30 amino acids regulating calcium transport in the Drosophila heart influencing regular muscle contraction. These peptides seem conserved for more than 550 million years in a range of species from flies to humans, where they have been implicated in cardiac pathologies. Such conservation suggests that the mechanisms for heart regulation are ancient and that smORFs may be a fundamental genome component that should be studied systematically.
Excitation-contraction coupling in the heart: the state of the question.
MD Stern, EG Lakatta
Laboratory of Cardiovascular Science, National Institute on Aging, NIH, Baltimore, Md.
The FASEB Journal (impact factor: 5.71). 10/1992; 6(12):3092-100.
Source: PubMed
www.researchgate.net/publication/21829642_Excitation-contraction_coupling_in_the_heart_the_state_of_the_question
Recent developments have led to great progress toward determining the mechanism by which calcium is released from the sarcoplasmic reticulum in the heart. The data support the notion of calcium-induced calcium release via a calcium-sensitive release channel. Calcium release channels have been isolated and cloned. This situation creates a paradox, as it has also been found that calcium release is smoothly graded and closely responsive to sarcolemmal membrane potential, properties that would not be expected of calcium-induced calcium release, which has intrinsic positive feedback. There is, therefore, no quantitative understanding of how the properties of the calcium release channel can lead to the macroscopic physiology of the whole cell. This problem could, in principle, be solved by various schemes involving heterogeneity at the ultrastructural level. The simplest of these require only that the sarcolemmal calcium channel be located in close proximity to one or more sarcoplasmic reticulum release channels. Theoretical modeling shows that such arrangements can, in fact, resolve the positive feedback paradox. An agenda is proposed for future studies required in order to reach a specific, quantitative understanding of the functioning of calcium-induced calcium release.
The role of protein kinases and protein phosphatases in the regulation of cardiac sarcoplasmic reticulum function
EG Kranias, RC Gupta, G Jakab, HW Kim, NAE Steenaart, ST Rapundalo
Molecular and Cellular Biochemistry 06/1988; 82(1):37-44. · 2.06 Impact Factor
www.researchgate.net/publication/6420466_Protein_phosphatases_decrease_sarcoplasmic_reticulum_calcium_content_by_stimulating_calcium_release_in_cardiac_myocytes
Canine cardiac sarcoplasmic reticulum is phosphorylated by adenosine 3,5-monophosphate (cAMP)-dependent and by calcium calmodulin-dependent protein kinases on a 27 000 proteolipid, called phospholamban. Both types of phosphorylation are associated with an increase in the initial rates of Ca(2+) transport by SR vesicles which reflects an increased turnover of elementary steps of the calcium ATPase reaction sequence. The stimulatory effects of the protein kinases on the calcium pump may be reversed by an endogenous protein phosphatase, which can dephosphorylate both the CAMP-dependent and the calcium calmodulin-dependent sites on phospholamban. Thus, the calcium pump in cardiac sarcoplasmic reticulum appears to be under reversible regulation mediated by protein kinases and protein phosphatases.
Regulation of the cardiac ryanodine receptor channel by luminal Ca2+ involves luminal Ca2+ sensing sites
I Györke, S Györke
Biophysical Journal 01/1999; 75(6):2801-10. · 3.65 Impact Factor
www.researchgate.net/publication/13459335_Regulation_of_the_cardiac_ryanodine_receptor_channel_by_luminal_Ca2_involves_luminal_Ca2_sensing_sites
The mechanism of activation of the cardiac calcium release channel/ryanodine receptor (RyR) by luminal Ca(2+) was investigated in native canine cardiac RyRs incorporated into lipid bilayers in the presence of 0.01 microM to 2 mM Ca(2+) (free) and 3 mM ATP (total) on the cytosolic (cis) side and 20 microM to 20 mM Ca(2+) on the luminal (trans) side of the channel and with Cs+ as the charge carrier. Under conditions of low [trans Ca(2+)] (20 microM), increasing [cis Ca(2+)] from 0.1 to 10 microM caused a gradual increase in channel open probability (Po). Elevating [cis Ca(2+)] above 100 microM resulted in a gradual decrease in Po. Elevating trans [Ca(2+)] enhanced channel activity (EC50 approximately 2.5 mM at 1 microM cis Ca2+) primarily by increasing the frequency of channel openings. The dependency of Po on trans [Ca2+] was similar at negative and positive holding potentials and was not influenced by high cytosolic concentrations of the fast Ca(2+) chelator, 1,2-bis(2-aminophenoxy)ethane-N,N,N, N-tetraacetic acid. Elevated luminal Ca(2+) enhanced the sensitivity of the channel to activating cytosolic Ca(2+), and it essentially reversed the inhibition of the channel by high cytosolic Ca(2+). Potentiation of Po by increased luminal Ca(2+) occurred irrespective of whether the electrochemical gradient for Ca(2+) supported a cytosolic-to-luminal or a luminal-to-cytosolic flow of Ca(2+) through the channel. These results rule out the possibility that under our experimental conditions, luminal Ca(2+) acts by interacting with the cytosolic activation site of the channel and suggest that the effects of luminal Ca2+ are mediated by distinct Ca(2+)-sensitive site(s) at the luminal face of the channel or associated protein.
Contemporary Definitions and Classification of the Cardiomyopathies
AHA Scientific Statement: Council on Clin. Cardiol.; HF and Transplant. Committee; Quality of Care and Outcomes Res. and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention
BJ Maron, Chair; JA Towbin; G Thiene; C Antzelevitch; D Corrado; D Arnett; AJ Moss; et al.
Circulation. 2006; 113: 1807-1816 http://dx.doi.org/10.1161/CIRCULATIONAHA.106.174287
Classifications of heart muscle diseases have proved to be exceedingly complex and in many respects contradictory. Indeed, the precise language used to describe these diseases is profoundly important. A new contemporary and rigorous classification of cardiomyopathies (with definitions) is proposed here. This reference document affords an important framework and measure of clarity to this heterogeneous group of diseases. Of particular note, the present classification scheme recognizes the rapid evolution of molecular genetics in cardiology, as well as the introduction of several recently described diseases, and is unique in that it incorporates ion channelopathies as a primary cardiomyopathy.
Ryanopathy: causes and manifestations of RyR2 dysfunction in heart failure
Belevych AE, Radwański PB, Carnes CA, Györke S.
College of Medicine, The Ohio State University, Columbus, OH.
Cardiovasc Res. 2013; 98(2):240-7. http://dx.doi.org/10.1093/cvr/cvt024.
Epub 2013 Feb 12. PMID: 23408344 PMCID: PMC3633158 [Available on 2014/5/1]
The cardiac ryanodine receptor (RyR2), a Ca(2+) release channel on the membrane of the sarcoplasmic reticulum (SR), plays a key role in determining the strength of the heartbeat by supplying Ca(2+) required for contractile activation. Abnormal RyR2 function is recognized as an important part of the pathophysiology of heart failure (HF). While in the normal heart, the balance between the cytosolic and intra-SR Ca(2+) regulation of RyR2 function maintains the contraction-relaxation cycle, in HF, this behaviour is compromised by excessive post-translational modifications of the RyR2. Such modification of the Ca(2+) release channel impairs the ability of the RyR2 to properly deactivate leading to a spectrum of Ca(2+)-dependent pathologies that include cardiac systolic and diastolic dysfunction, arrhythmias, and structural remodeling. In this article, we present an overview of recent advances in our understanding of the underlying causes and pathological consequences of abnormal RyR2 function in the failing heart. We also discuss the implications of these findings for HF therapy.
Up-regulation of Sarcoplasmic Reticulum Ca(2+) Uptake Leads to Cardiac Hypertrophy, Contractile Dysfunction and Early Mortality in mice deficient in CASQ2
Kalyanasundaram A, Lacombe VA, Belevych AE, Brunello L, Carnes CA, Janssen PM, … Gyørke S.
Department of Physiology and Cell Biology, College of Medicine, Ohio State University, Columbus, OH.
Cardiovasc Res. May 2013; 98(2):297-306. http://dx.doi.org/10.1093/cvr/cvs334. Epub 2012 Nov 6.
Aberrant Ca(2+) release (i.e. Ca(2+) ‘leak’) from the sarcoplasmic reticulum (SR) through cardiac ryanodine receptors (RyR2) is linked to heart failure (HF). Does SR-derived Ca(2+) can actually cause HF? We ask whether and by what mechanism combining dysregulated RyR2 function with facilitated Ca(2+) uptake into SR exacerbates abnormal SR Ca(2+) release and induces HF.
We crossbred mice deficient in expression of cardiac calsequestrin (CASQ2) with mice overexpressing the skeletal muscle isoform of SR Ca(2+)ATPase (SERCA1a). The new double-mutant strains displayed early mortality, congestive HF with left ventricular dilated hypertrophy, and decreased ejection fraction. Intact right ventricular muscle preparations from double-mutant mice preserved normal systolic contractile force but were susceptible to spontaneous contractions. Double-mutant cardiomyocytes while preserving normal amplitude of systolic Ca(2+) transients displayed marked disturbances in diastolic Ca(2+) handling in the form of multiple, periodic Ca(2+) waves and wavelets. Dysregulated myocyte Ca(2+) handling and structural and functional cardiac pathology in double-mutant mice were associated with increased rate of apoptotic cell death. Qualitatively similar results were obtained in a hybrid strain created by crossing CASQ2 knockout mice with mice deficient in phospholamban.
We demonstrate that enhanced SR Ca(2+) uptake combined with dysregulated RyR2s results in sustained diastolic Ca(2+) release causing apoptosis, dilated cardiomyopathy, and early mortality. Further, up-regulation of SERCA activity must be advocated with caution as a therapy for HF in the context of abnormal RyR2 function.
Comment in
Mind the store: modulating Ca(2+) reuptake with a leaky sarcoplasmic reticulum. [Cardiovasc Res. 2013] PMID: 23135969 [PubMed – in process] PMCID: PMC3633154 [Available on 2014/5/1]
Myocardial Delivery of Stromal Cell-Derived Factor 1 in Patients With Ischemic Heart Disease: Safe and Promising Circ. Res.. 2013;112:746-747
Circulation Research Thematic Synopsis: Cardiovascular Genetics Circ. Res.2013;112:e34-e50,
Ryanodine Receptor Phosphorylation and Heart Failure: Phasing Out S2808 and ³Criminalizing² S2814 ,
Héctor H Valdivia
Center for Arrhythmia Research, University of Michigan, Ann Arbor, MI.
Circ. Res.. 2012;110:1398-1402 http://dx.doi.org/10.1161/CIRCRESAHA.112.270876 (IF: 9.49).
By the time the heart reaches the pathological state clinically recognized as heart failure (HF), it has undergone profound and often irreversible alterations in structure and function at the molecular, cellular and organ level. Although the etiologies of HF are diverse:
- hypertension,
- myocardial infarction,
- atherosclerosis,
- valvular insufficiency,
- mutations in genes encoding sarcomeric proteins
Some alterations are commonly found in most forms of HF, and they may account for the maladaptive structural remodeling and systolic dysfunction that characterize this syndrome.
At the cellular level, there are well documented changes in
- ionic channel density and function (electrical remodeling),
- increased ROS production,
- mitochondrial dysfunction,
- imbalanced energy intake and consumption,
- genetic reprogramming,
- altered excitation-contraction coupling,
and in general, dysregulation of a multitude of other processes and pathways that are essential for proper cardiac function. Combined, this myriad of alterations leads to
- loss in contractility and
- loss ejection fraction,
- ventricular wall remodeling,
- increased vascular resistance, and
- dysregulated fluid homeostasis.
In this issue of Circulation Research, Respress et al.2 report that preventing phosphorylation of cardiac ryanodine receptors (RyR2) at a single residue, S2814, is sufficient to avert many of these alterations and improve cardiac function in HF. The results presented here follow a string of papers that touch on the delicate and controversial subject of ryanodine receptor phosphorylation and HF. They offer a new twist to a contentious story and attempt to reconcile many apparently contradicting results, but key issues remain.
Calcium “Leak” in HF
It appears that suppressing the dysfunction of a select group of biological and molecular signaling pathways may substantially improve or even reverse the cardiac deterioration observed in HF. For example, correcting the characteristically depressed sarcoplasmic reticulum (SR) calcium content of failing cardiomyocytes is a target of HF gene therapy. SR calcium “leak”, an operational term that indicates increased and untimely calcium release by RyR2s, also appears common to several models of HF. Therefore, stemming off calcium “leak” may prevent the progression of cardiac malfunction in HF patients. However, a rationalized therapy towards this aim must be founded on the precise knowledge of the mechanisms leading to calcium leak. Marks group, in a landmark publication in 2000 (ref. 6) and later in multiple other high-impact factor papers (many of them co-authored by Wehrens 7-10) postulated that RyR2 “hyperphosphorylation” at S2808 by PKA was the primary mechanism leading to increased calcium “leak” in HF. This idea was initially appealing and fueled intensive research in the subject, but many groups failed to reproduce central tenets of this hypothesis. (11 and 12) The controversies surrounding the Marks-Wehrens hypothesis of increased calcium leak by hyperphosphorylation of RyR2-S2808 have been recently and comprehensibly reviewed by Bers.13 Here I will focus on the modifications to this hypothesis as derived from the new findings of Respress et al.2 Emerging points from these new findings will be the demotion of S2808, to intervene not as universal player in HF but only in selective forms of this syndrome, and the role of S2814 as pre-eminent generator of calcium leak that leads to arrhythmias and exacerbates other forms of HF. The “criminalization” of S2814 has begun in earnest.
CaMKII Effect on Calcium Leak and the Role of S2808 and S2814
Many studies have provided evidence that persistent CaMKII activity can lead to cardiac arrhythmias and promote HF.14-16 Animals and patients with congestive HF display increased levels of CaMKII,17,18 and overexpression of AC3-I, a peptide inhibitor of CaMKII, delays the onset of HF in mice.19 There is also good agreement4,20 (although not universal21) that CaMKII, and not PKA, increases calcium leak, and therefore, it is likely that the arrhythmogenic and deleterious activity of CaMKII in HF may be associated with this effect. Obviously, if PKA does not cause calcium leak directly, this by itself imposes insurmountable constraints on the Marks-Wehrens hypothesis that posits that PKA phosphorylation of RyR2-S2808 is responsible for the high calcium leak of HF. With the focus now on CaMKII, the obligated question is then, by what mechanisms CaMKII increases calcium leak from the SR? To increase calcium leak, the cell must either increase SR calcium content, and/or increase the activity of the RyR2 (albeit the latter alone would have only transient effects due to autoregulatory mechanisms22). Since both PKA and CaMKII increase SR calcium load by phosphorylating phospholamban (but at different residues) and relieving the inhibition it exerts on SERCA2a, the differential effect of these kinases must result from the regulation they exert on RyR2s. Wehrens group offers here2 at least a partial explanation of this complex mechanism and, along with previous papers co- authored with Marks, these groups set specific roles for S2808 and S2814 on regulation of RyR2 activity and their protective effect (or lack thereof) in HF. In their view, PKA exclusively phosphorylates S2808 and dissociates FKBP12.6, which destabilizes the closed state of the channel and increases RyR2 activity, whereas CaMKII (almost) exclusively phosphorylates S2814, has no effect on FKBP12.6 binding, and equally activates RyR2s. In this issue, Respress et al.2 report that preventing phosphorylation of S2814 (by genetic substitution of Ser by Ala, S2814A) protects against non-ischemic (pressure overload) HF but has no effect on ischemic HF; conversely, and against other data by the same groups, S2808 phosphorylation was not significantly different in non-ischemic HF, implying that it is relevant only in ischemic HF. This clean targeting of RyR2 phospho-epitopes by PKA and CaMKII and their nice “division of labor” for pathogenicity in distinct forms of HF would really simplify phosphorylation schemes and reconcile apparent contradictions. However, as is generally the case, the proposal appears oversimplified and almost too good to be true. Let’s discuss each of the premises on which the Respress et al.2 results have been interpreted and the problems associated with these premises.
One kinase = one site = one effect. Is it really that simple?
The RyR2 is a huge protein. It is assembled as a tetrameric complex of ~2 million Da, with each subunit composed of ~5,000 amino acids.
Using canonical phosphorylation consensus and high confidence values, the RyR2 may be phosphorylated in silico at more than 100 sites by the combined action of PKA,
- CaMKII,
- PKG, and
- PKC, to name a few.11
Granted, a “potential” phosphorylation site is very different than a demonstrated, physiologically-relevant phosphorylation site and it is possible that many of the predicted residues are not phosphorylated in vivo. Even then, several groups have demonstrated that CaMKII phosphorylates RyR2 with stoichiometry of at least 3 or 4 to 1 with respect to PKA.23-26 This fact is by itself compelling evidence that there are multiple phosphorylation sites in RyR2. Now, let’s make the optimistic assumption that all the PKA sites have already been mapped, and that S2808 and S2030 (ref. 27) are the only PKA sites. Taking into account the CaMKII:PKA phosphorylation ratio (3:1 or 4:1), this would then yield a minimum of ~6 – 8 CaMKII phosphorylation sites (per channel subunit!). In this perspective, it is almost disingenuous to label S2808 as “the” PKA site, and we may purposely deceive ourselves when we label S2814 “the” CaMKII site. Against this sense of pessimism and intractability, let’s not forget that S2808 was actually discovered as a CaMKII site.24 It is possible then that the number of CaMKII sites is smaller if only S2030 remains as a bona fide PKA site. Still, neither scheme supports one CaMKII site per channel subunit.
But let’s go along for a moment with the possibility, however unlikely, that PKA phosphorylates S2808 only, and CaMKII phosphorylates S2814 only. When calling these sites by their distinctive numbers, it is easy to forget that these phospho-sites are only 6 residues apart, that is, a minuscule proportion (~0.000003%) in the context of the whole channel protein. How can the same reaction (phosphorylation) that occurs at sites so close to one another be differentially transmitted to the very distant gating domains of the channel? If these residues were lining the pore of the channel, where critical differences emerge by substituting one residue but not the neighboring one, then it would be easier to understand how S2808 and S2814 could transmit distinct signals. But both are part of a “phosphorylation hot spot”, a cytoplasmic loop that contains additional potential phospho-sites11 and that has been mapped to the external surface of the channel.28 Marks and Wehrens groups have shown that phosphorylation of S2808A by CaMKII or of S2814A by PKA fully activate the channel.7,9 At face value, this means that knocking out one phospho-residue does not cripple this “hot spot” and that phosphorylation of at least one residue in this external loop enables it to transmit conformational changes to the gating domains of the channel. Seen in this structural context in which the “hot spot” works in unison upon phosphorylation of at least one residue, it is very difficult (but not impossible) to accommodate the notion that phosphorylation of S2808 or S2814 alone dictates the differential response of the RyR2 to PKA and CaMKII.
An Alternative Model to explain Differential PKA and CaMKII Effects
An alternative model to explain the differential effect of PKA and CaMKII to elicit calcium leak from RyR2 that takes into account other phospho-sites is needed. Before formulating it, let’s consider some important points. First, it is not difficult to assume that the role of the “phosphorylation hot spot” is to readily pick up signals from different kinases. The multi-valence of this “hot spot” is demonstrated so far by the fact that S2808 may be phosphorylated by CaMKII24,25,26 and by PKA,6,25,26 and its eagerness to undergo phosphorylation by the fact that S2808 is at least ~50% phosphorylated even at basal state25-27,29,30 and phospho-signals from these sites may be readily detected upon β-adrenergic stimulation of the heart.30,31Second, if we accept the Shannon and Bers results that CaMKII, and not PKA, elicits calcium leak from the SR,4,20 this obligatorily means that PKA phosphorylation of S2808 is not responsible for eliciting calcium leak (in direct conflict with the Marks-Wehrens hypothesis). In support of this notion, studies by the Houser and Valdivia groups have provided evidence that preventing S2808 phosphorylation has negligible impact on the β-adrenergic response of the heart and on the progression of non-ischemic and ischemic HF.30-32 Third, another PKA site, S2030, largely ignored in the Marks-Wehrens scheme, has been mapped and shown to activate channel openings27 and although its place in the larger context of RyR2 phosphorylation has not been determined yet, I think it is illogical to assume that its existence is futile and that it contributes nothing to regulation of the channel. Thus, according to the preceding discussion, it is almost unsustainable to postulate that the differential effects of CaMKII and PKA to elicit calcium leak stems from their effects on the RyR2 “phosphorylation hot spot” alone. Instead, I would like to posit an alternative model that integrates findings by many of the above-referenced groups (Fig. 1). In this model, the surface domain of the RyR2 comprising residues 2804-2814 (mouse nomenclature) is an eager target for phosphorylation by PKA, CaMKII and probably other kinases (4 Ser/Thr).11,24-26,29 Phosphorylation of this “hot spot” by either PKA or CaMKII (or both) “primes” the RyR2 for subsequent signals and is probably responsible for the coordinated openings in response to fast calcium stimuli detected in single channel recordings33 and in cellular settings34 (but this has yet to be demonstrated). The differential effect of PKA and CaMKII on RyR2 activity would then depend on the integrated response of the phosphorylated “hot spot” and of additional phosphorylation sites. For example, phosphorylation of S2808 and S2030 by PKA could coordinate channel openings in response to fast calcium stimuli, and phosphorylation of S2814 and other CaMKII site(s) could open RyR2s at diastolic [Ca2+], which would translate in calcium leak. Examples of proteins acting as molecular switchboards in response to various degrees of phosphorylation are not unprecedented.35 In fact, RyR2s are activated by phosphorylation and dephosphorylation as well36,37 and their relative degree of phosphorylation determines a final functional output.38 It is therefore conceivable that the complex response of RyR2s to any type of phosphorylation and the variable results obtained by investigators apparently using the same experimental conditions may be due to the variable degree of phosphorylation in which the RyR2s were found. Of course, until the 3D structure of the RyR2 is solved and we understand the mechanism by which the “phosphorylation hot spot” and other phospho-sites “talk” to the channel’s gating domains this structurally-based model will remain speculative, but it at least takes into consideration compelling evidence on the existence of various phosphorylation sites and departs substantially from the simplified notion of one kinase = one site = one effect.
Fig. 1 Models of RyR2 modulation by phosphorylation

See – Ryanodine Receptor Phosphorylation and Heart Failure – Phasing Out S2808 and “Criminalizing” S2814. Héctor H. Valdivia. http://circres.ahajournals.org/content/110/11/1398.full www.ncbi.nlm.nih.gov/pmc/articles/PMC3386797
Models of RyR2 modulation by phosphorylation. In the Marks-Wehrens model (A), S2808 is the only site phosphorylated by PKA, and S2814 by CaMKII. PKA phosphorylation of S2808 dissociates FKBP12.6, which destabilizes the closed state of the channel and induces subconductance states, eliciting calcium leak. Calcium leak from the SR then causes deleterious effects such as arrhythmias and worsening of (ischemic) HF. CaMKII phosphorylation of S2814 does not dissociate FKBP12.6 but also causes calcium leak. This leak is also arrhythmogenic but is not relevant in ischemic HF, only in nonischemic HF. In the multiphosphorylation site model (B), S2808 and S2814 are part of a “phosphorylation hot spot” that is located in a protruding part of the channel, is targeted by several kinases, and may contain other phospho-epitopes not yet characterized. Phosphorylation of individual residues within this “hot spot” may be undistinguishable by the channel’s gating domains; instead, the differential regulation of PKA and CaMKII on channel gating may come about by the combined effect of each kinase on phospho-residues of the “hot spot” and other phosphorylation sites.
see- Is ryanodine receptor phosphorylation key to the fight or flight response and heart failure? Thomas Eschenhagen. JCI 210; 120(12): 4197-4203. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2994341/
In situations of stress the heart beats faster and stronger. According to Marks and colleagues, this response is, to a large extent, the consequence of facilitated Ca2+ release from intracellular Ca2+ stores via ryanodine receptor 2 (RyR2), thought to be due to catecholamine-induced increases in RyR2 phosphorylation at serine 2808 (S2808). If catecholamine stimulation is sustained (for example, as occurs in heart failure), RyR2 becomes hyperphosphorylated and “leaky,” leading to arrhythmias and other pathology. This “leaky RyR2 hypothesis” is highly controversial. In this issue of the JCI, Marks and colleagues report on two new mouse lines with mutations in S2808 that provide strong evidence supporting their theory.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2994341/bin/JCI45251.f1.jpg

In the signalling scheme outlined in Figure1 of this commentary, which prevailed until the end of the last century, the two major determinants of intracellular Ca2+ transients and thereby the contractile force of the heart were (a) the size of the Ca2+ current entering via the LTCC (well exemplified by the negative inotropic effects of LTCC blockers) and (b) the activity of SERCA and thus the Ca2+ load of the SR. The critical role of the latter was convincingly demonstrated by the fact that Plb–/– mice, which have maximal SERCA activity, exhibit higher basal force and reduced inotropic response to isoprenaline (1).
See also Table 1
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2994341/table/T1/?report=thumb

In the Marks-Wehrens model, S2808 is the only site phosphorylated by PKA, and S2814 by CaMKII. PKA phosphorylation of S2808 dissociates FKBP12.6, which destabilizes the closed state of the channel and induces subconductance states, eliciting calcium leak. Calcium leak from the SR then causes deleterious effects such as arrhythmias and worsening of (ischemic) HF. CaMKII phosphorylation of S2814 does not dissociate FKBP12.6 but also causes calcium leak. This leak is also arrhythmogenic, but is not relevant in ischemic HF, only in non-ischemic HF. In the multi-phosphorylation site model, S2808 and S2814 are part of a “phosphorylation hot spot” that is located in a protruding part of the channel, is targeted by several kinases, and may contain other phospho-epitopes not yet characterized. Phosphorylation of individual residues within this “hot spot” may be undistinguishable by the channel’s gating domains; instead, the differential regulation of PKA and CaMKII on channel gating may come about by the combined effect of each kinase on phospho-residues of the “hot spot” and other phosphorylation sites.



Appealing as Marks’ theory is, the concept has been challenged and remains controversial (Tables1 and 2). On the one hand, some theoretical considerations argue against it. For example, it seems counterintuitive that phosphorylation at a single residue in a protein of more than 5,000 amino acids could profoundly affect channel open probability. Second, S2808, the proposed site of phosphorylation by PKA, is located in an area distant from the FKBP12.6/RyR2 interaction site (3), making it somewhat unlikely that phosphorylation affects FKPB12.6 binding. Third, it seems unlikely and to contradict experimental results (4) that an isolated increase in RyR2 open probability has more than a transient consequence on Ca2+ handling, because an isolated increase in Ca2+release from the RyR2 will automatically lead to reduced Ca2+ load in the SR and therefore fast normalization of Ca2+ transients (autoregulation).
More concerning than theoretical considerations are numerous reports that failed to reproduce important aspects of the data that support the leaky RyR2 hypothesis and the critical importance of S2808 (Tables (Tables11and and2).2). (a) Phosphorylation of RyR2 at S2808 has been found by others to be either not altered in heart failure at all or to be only moderately increased (5–8). Others have reported that 75% of the available RyR2 S2808 sites are phosphorylated under normal conditions, making a 9-fold change in chronic heart failure somewhat unlikely (9). (b) Whereas general consensus exists that β-adrenergic stimulation increases spontaneous Ca2+ release (the “Ca2+ leak”) from the SR, the role of RyR2 phosphorylation and FKBP12.6 dissociation remains controversial. Importantly, PKA had no effect on Ca2+release in permeabilized Plb–/– mouse myocytes, i.e., cells in which the SR is maximally loaded with Ca2+ and one would have expected a particularly strong effect of increasing RyR2 open probability.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2994341
Now, let’s go back to the results of Respress et al.2 and consider them in this light. They found that preventing phosphorylation of S2814 alone mitigates non-ischemic HF induced by transverse aortic constriction (TAC) in mice. This implies that other CaMKII sites are not necessary to mitigate the CaMKII-induced calcium leak that they propose is responsible for the deleterious effect in WT mice subjected to TAC. If phosphorylation of the “hot spot” is compulsory to prime the RyR2 to process and discriminate other phosphorylation signals, then other residues in that “hot spot” must have been phosphorylated to fulfill this need. Surprisingly, S2808 was not significantly phosphorylated in this setting. This leaves a very difficult conundrum: if S2808 was not phosphorylated significantly and the other CaMKII sites are not necessary to stop calcium leak, how then can we explain the results of Respress et al.2? Of course there are always alternatives, and we would be inconsistent if we rigidly adhere to one model and fell into the dogmatism we are criticizing. The conclusions of Respress et al.2 are in line with their findings, but at this point the numbers do not add up and it’s obvious that the great complexity of this process (RyR2 phosphorylation) precludes simplified and neatly organized schemes. As a clear example of this, in the landmark paper by Marks group,6 S2808 was found substantially hyperphosphorylated in tachypacing-induced failing dogs, also a non-ischemic model of HF. This does not fit well in the current scheme of Wehrens where S2808A protects against ischemic HF, but has no prominent role in non-ischemic HF.
In summary, CaMKII and PKA may have specific roles in calcium leak and, since they both increase SR calcium load, their differential effect likely resides on their effect on RyR2s. However, the effect of PKA- or CaMKII-phosphorylation of RyR2s does not appear solved yet. Starting in 2000 and up to the present day, Marks and Wehrens have provided high-quality data in prominent journals aggressively pursuing the notion that PKA phosphorylates S2808 only, that CaMKII phosphorylates S2814 only, and that these sites alone integrate multiple signals to open RyR2s. Many key aspects of their general hypothesis including dissociation of FKBP12.6 by PKA phosphorylation of S2808, subconductance states as hallmarks of phosphorylation, and the prominent role of S2808 as promoter of arrhythmias and HF have not been confirmed by several groups. The present paper by the Wehrens group modifies slightly the original claim that S2808 was involved in ischemic and non-ischemic forms of HF and continues to shift the lion’s share of pathogenicity to S2814. However, as discussed above, the Marks-Wehrens model largely ignores compelling data on the presence of multiple phosphorylation sites and the complexity they add to the finely graded response of RyR2s to phosphorylation.
2. Respress JL, van Oort RJ, Li N, Rolim N, Dixit S, Dealmeida A, Voigt N, Lawrence WS, Skapura DG, Skårdal K, Wisloff U, Wieland T, Ai X, Pogwizd SM, Dobrev D, Wehrens XH. Role of RyR2 Phosphorylation at S2814 During Heart Failure Progression. Circ Res. 2012;xx:xx–xx. [in the issue; printer, please update] [PMC free article] [PubMed]
6. Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101(4):365–376. [PubMed]
7. Wehrens XH, Lehnart SE, Reiken S, Vest JA, Wronska A, Marks AR. Ryanodine receptor/calcium release channel PKA phosphorylation: a critical mediator of heart failure progression. Proc Natl Acad Sci U S A. 2006;103:511–518. [PMC free article] [PubMed]
36. Lokuta AJ, Rogers TB, Lederer WJ, Valdivia HH. Modulation of cardiac ryanodine receptors of swine and rabbit by a phosphorylation-dephosphorylation mechanism. J Physiol. 1995;487:609–622. [PMC free article] [PubMed]
37. Terentyev D, Viatchenko-Karpinski S, Gyorke I, Terentyeva R, Gyorke S. Protein phosphatases decrease sarcoplasmic reticulum calcium content by stimulating calcium release in cardiac myocytes. J Physiol. 2003;552(Pt 1):109–118. [PMC free article] [PubMed]
38. Carter S, Colyer J, Sitsapesan R. Maximum phosphorylation of the cardiac ryanodine receptor at Ser-2809 by protein kinase A produces unique modifications to channel gating and conductance not observed at lower levels of phosphorylation. Circ Res. 2006; 98:1506–1513. [PubMed]
The Cardiac Ryanodine Receptor (calcium release channel) – Emerging role in Heart Failure and Arrhythmia Pathogenesis
Cardiovasc Res (2002) 56 (3): 359-372. http://dx.doi.org/10.1016/S0008-6363(02)00574-6
The cardiac sarcoplasmic reticulum calcium release channel, commonly referred to as the ryanodine receptor, is a key component in cardiac excitation–contraction coupling, where it is responsible for the release of calcium from the sarcoplasmic reticulum. As our knowledge of the ryanodine receptor has advanced an appreciation that this key E–C coupling component may have a role in the pathogenesis of human cardiac disease has emerged. Heart failure and arrhythmia generation are both pathophysiological states that can result from deranged excitation–contraction coupling. Evidence is now emerging that hyperphosphorylation of the cardiac ryanodine receptor is an important event in chronic heart failure, contributing to impaired contraction and the generation of triggered ventricular arrhythmias.
Furthermore the therapeutic benefits of β blockers in heart failure appear to be partly explained through a reversal of this phenomenon. Two rare inherited arrhythmogenic conditions, which can cause sudden death in children, have also been shown to result from mutations in the cardiac ryanodine receptor. These conditions,
- catecholaminergic polymorphic ventricular tachycardia and
- arrhythmogenic right ventricular cardiomyopathy (subtype 2),
further implicate the ryanodine receptor as a potentially arrhythmogenic substrate and suggest this channel may offer a new therapeutic target in the treatment of both cardiac arrhythmias and heart failure.
Protein phosphatases decrease sarcoplasmic reticulum calcium content by stimulating calcium release in cardiac myocytes
D Terentyev, S Viatchenko-Karpinski, I Gyorke, R Terentyeva and S Gyorke
Texas Tech University Health Sciences Center, Lubbock, TX
J Physiol 2003; 552(1), pp. 109–118. http:/dx.doi.org/10.1113/jphysiol.2003.046367
Phosphorylation/dephosphorylation of Ca2+ transport proteins by cellular kinases and phosphatases plays an important role in regulation of cardiac excitation–contraction coupling; furthermore, abnormal protein kinase and phosphatase activities have been implicated in heart failure. However, the precise mechanisms of action of these enzymes on intracellular Ca2+ handling in normal and diseased hearts remains poorly understood. We have investigated the effects of protein phosphatases PP1 and PP2A on spontaneous Ca(2+) sparks and SR Ca(2+) load in myocytes permeabilized with saponin. Exposure of myocytes to PP1 or PP2A caused a dramatic increase in frequency of Ca(2+) sparks followed by a nearly complete disappearance of events. These effects were accompanied by depletion of the SR Ca(2+) stores, as determined by application of caffeine. These changes in Ca(2+) release and SR Ca(2+) load could be prevented by the inhibitors of PP1 and PP2A phosphatase activities okadaic acid and calyculin A. At the single channel level, PP1 increased the open probability of RyRs incorporated into lipid bilayers. PP1-medited RyR dephosphorylation in our permeabilized myocytes preparations was confirmed biochemically by quantitative immunoblotting using a phosphospecific anti-RyR antibody. Our results suggest that increased intracellular phosphatase activity stimulates RyR mediated SR Ca(2+) release leading to depleted SR Ca(2+) stores in cardiac myocytes.
In heart muscle cells, the process of excitation–contraction (EC) coupling is mediated by Ca(2+) influx through sarcolemmal L-type Ca(2+) channels activating Ca(2+) release channels (ryanodine receptors, RyRs) in the sarcoplasmicreticulum (SR). Once activated, the RyR channels allow Ca(2+) to be released from the SR into the cytosol to induce contraction. This mechanism is known as Ca(2+)-induced calcium release (CICR) (Fabiato, 1985; Bers, 2002).
During relaxation, most of the Ca(2+) is resequestered into the SR by the Ca(2+)-ATPase. The amount of Ca(2+) released and the force of contraction depend on the magnitude of the Ca(2+) trigger signal, the functional state of the RyRs and the amount of Ca(2+) stored in the SR. Reversible phosphorylation of proteins composing the EC coupling machinery plays an important role in regulation of cardiac contractility (Bers, 2002). Thus, during stimulation of the b-adrenergic pathway, phosphorylation of several target proteins, including the L-type Ca(2+) channels, RyRs and phospholamban, by protein kinase A (PKA) leads to an overall increase in SR Ca2+ release and contractile force in heart cells (Callewaert et al. 1988, Spurgeon et al. 1990; Hussain & Orchard, 1997; Zhou et al. 1999; Song et al. 2001; Viatchenko-Karpinski & Gyorke, 2001). PKA-dependent phosphorylation of the L-type Ca(2+) channels increases the Ca2+ current (ICa), increasing both the Ca2+ trigger for SR Ca2+ release and the SR Ca(2+) content (Callewaert et al. 1988; Hussain & Orchard, 1997; Del Principe et al. 2001). Phosphorylation of phospholamban (PLB) relieves the tonic inhibition dephosphorylated PLB exerts on the SR Ca(2+)-ATPase (SERCA) resulting in enhanced SR Ca(2+) accumulation and enlarged Ca(2+) release (Kranias et al. 1985; Simmermann & Jones, 1998). With regard to the RyR, despite clear demonstration of phosphorylation of the channel in biochemical studies (Takasago et al. 1989; Yoshida et al. 1992), the consequences of this reaction to channel function have not been clearly defined. RyR phosphorylation by PKA and Ca(2+)–calmodulin dependent protein kinase (CaMKII) has been reported to increase RyR activity in lipid bilayers (Hain et al. 1995; Marx et al. 2000; Uehara et al. 2002). Moreover, it has been reported that in heart failure (HF), hyperphosphorylation of RyR causes the release of FK-506 binding protein (FKBP12.6) from the RyR, rendering the channel excessively leaky for Ca(2+) (Marx et al. 2000). However, other studies have reported no functional effects (Li et al. 2002) or even found phosphorylation to reduce RyR channel steady-state open probability (Valdivia et al. 1995; Lokuta et al. 1995).
The Action of Protein Kinases is Opposed by Dephosphorylating Phosphatases.
Three types of protein: phosphatases (PPs), referred to as
- PP1,
- PP2A and
- PP2B (calcineurin),
have been shown to influence cardiac performance (Neumann et al. 1993; Rusnak & Mertz, 2000). Overall, according to most studies phosphatases appear to downregulate SR Ca(2+) release and contractile performance (Neumann et al. 1993; duBell et al. 1996, 2002; Carr et al. 2002; Santana et al. 2002). Furthermore, PP1 and PP2A activities appear to be increased in heart failure (Neumann, 2002; Carr et al. 2002). However, again the precise mode of action of these enzymes on intracellular Ca(2+) handling in normal and diseased hearts remains poorly understood. In the present study, we have investigated the effects of protein phosphatases PP1 and PP2A on local Ca(2+) release events, Ca(2+) sparks, in cardiac cells. Our results show that phosphatases activate RyR mediated SR Ca(2+) release leading to depletion of SR Ca(2+) stores. These results provide novel insights into the mechanisms and potential role of protein phosphorylation/dephosphorylation in regulation of Ca(2+) signaling in normal and diseased hearts.
RESULTS
Effects of PP1 and PP2A on Ca2+ Sparks and SR Ca(2+) Content.
- PP1 caused an early transient potentiation of Ca2+ spark frequency followed by a delayed inhibition of event occurrence.
- PP1 produced similar biphasic effects on the magnitude and spatio-temporal characteristics of Ca(2+) sparks
Specifically, during the potentiatory phase (1 min after addition of the enzyme), PP1 significantly increased the amplitude, rise-time, duration and width of Ca(2+) sparks; during the inhibitory phase (5 min after addition of the enzyme), all these parameters were significantly suppressed by PP1.
- The SR Ca(2+) content decreased by 35 % or 69 % following the exposure of myocytes to either 0.5 or 2Uml_1 PP1, respectively (Fig. 1C).
Qualitatively similar results were obtained with phosphatase PP2A. Similar to the effects of PP1, PP2A (5Uml_1) produced a transient increase in Ca(2+) spark frequency (~4-fold) followed by a depression of event occurrence and decreased SR Ca(2+) content (by 82 % and 65 %, respectively). Also similar to the action of PP1, PP2A increased the amplitude and spatio-temporal spread (i.e. rise-time, duration and width) of Ca(2+) sparks at 1 min and suppressed the same parameters at 5 min of exposure to the enzyme (Table 1). Together, these results suggest that phosphatases enhance spark-mediated SR Ca2+ release, leading to decreased SR Ca(2+) content.
- Preventive effects of calyculin A and okadaic acid
- Preventive effects of ryanodine
PP1-mediated RyR dephosphorylation
The cardiac RyR is phosphorylated at Ser-2809 (in the rabbit sequence) by both PKA and CAMKII (Witcher et al. 1991; Marx et al. 2000). Although additional phosphorylation sites may exist on the RyR (Rodriguez et al. 2003), Ser-2809 is believed to be the only site that is phosphorylated by PKA, and RyR hyperphosphorylation at this site has been reported in heart failure (Marx et al. 2000). To test whether indeed phosphatases dephosphorylated the RyR in our permeabilized myocyte experiments we performed quantitative immunoblotting using an antibody that specifically recognizes the phosphorylated form of the RyR at Ser-2809 (Rodriguez et al. 2003). Myocytes exhibited a significant level of phosphorylation under baseline conditions. Maximal phosphorylation was 201 % of control. When exposed to 2Uml_1 PP1, RyR phosphorylation was 58 % of the control basal condition. Exposing to a higher PP1 concentration (10Uml_1) further reduced RyR phosphorylation to 22% of control. Thus, consistent with the results of our functional measurements, PP1 decreased RyR phosphorylation in cardiac myocytes.
Figure 1. Effects of PP1 on properties of Ca(2+) sparks and SR Ca(2+) content in rat permeabilized myocytes
http://dx.doi.org/10.1113/jphysiol.2003.046367
A, spontaneous Ca(2+) spark images recorded under reference conditions, and 1 or 5 min after exposure of the cell to 2Uml_1 PP1. Traces below the images are Ca(2+) transients induced by application of 10 mM caffeine immediately following the acquisition of sparks before (3 min) and after (5 min) application of PP1 in the same cell. The Ca(2+) transients were elicited by a whole bath application of 10 mM caffeine. B, averaged spark frequency at early (1 min) and late (5 min) times following the addition of either 0.5 or 2Uml_1 of PP1 to the bathing solution. C, averaged SR Ca(2+) content for 0.5 or 2Uml_1 of PP1 measured before and 5 min after exposure to the enzyme. Data are presented as means ± S.E.M. of 6 experiments in different cells.
Figure 2. Effects of PP2A on properties of Ca2+ sparks and SR Ca2+ content in rat permeabilized myocytes
http://dx.doi.org/10.1113/jphysiol.2003.046367
A, spontaneous Ca(2+) spark images recorded under reference conditions, and 1 or 5 min after exposure of the cell to 5Uml_1 PP2A. Traces below the images are Ca(2+) transients induced by application of 10 mM caffeine immediately following the acquisition of sparks before (3 min) and after (5 min) application of PP2A in the same cell. B and C, averaged spark frequency (B) and SR Ca(2+) content (C) for the same conditions as in A. Data are presented as means ± S.E.M. of 6 experiments in different cells.

DISCUSSION
In the present study, we have investigated the impact of physiologically relevant exogenous protein phosphatases PP1 and PP2A on RyR-mediated SR Ca(2+) release (measured as Ca(2+) sparks) in permeabilized heart cells. Our principal finding is that phosphatases stimulated RyR channels leading to depleted SR Ca(2+) stores. These results have important ramifications for understanding the mechanisms and role of protein phosphorylation/dephosphorylation in modulation of Ca(2+) handling in normal and diseased heart.

Modulation of SR Ca2+ release by Protein Phosphorylation/Dephophorylation
Since protein dephosphorylation clearly resulted in increased functional activity of the Ca(+)release channel, our results imply that a reverse, phosphorylation reaction should reduce RyR activity. If indeed such effects take place, why do they not manifest in inhibition of Ca(+)sparks? One possibility is that enhanced Ca(+) uptake by SERCA masks or overcomes the effects phosphorylation may have on RyRs. In
addition, the potential inhibitory influence of protein phosphorylation on RyR activity in myocytes could be countered by feedback mechanisms involving changes in luminal Ca(+)(Trafford et al. 2002; Gyorke et al. 2002). In particular, reduced open probability of RyRs would be expected to lead to increased Ca2+ accumulation in the SR; increased intra-SR [Ca(2+)] in turn would increase activity of RyRs at their luminal Ca(2+) regulatory sites as demonstrated for the RyR channel inhibitor tetracaine (Gyorke et al. 1997; Overend et al. 1997). Thus potentiation of SERCA combined with the intrinsic capacity of the release mechanism to self-regulate could explain at least in part why PKA-mediated protein phoshorylation results in maintained potentiation of Ca(2+) sparks despite a potential initial decrease in RyR activity.

Role of altered RyR Phosphorylation in Heart Failure
Marx et al. (2000) have proposed that enhanced levels of circulating catecholamines lead to increased phosphorylation of RyR in heart failure. Based on biochemical observations as well as on studying properties of single RyRs incorporated into artificial lipid bilayers, these investigators have hypothesized that hyperphosphorylation of RyRs contributes to pathogenesis of heart failure by making the channel excessively leaky due to dissociation of FKBP12.6 from the channel. We show that the mode of modulation of RyRs by phosphatases does not support this hypothesis as dephosphorylation caused activation instead of inhibition of activity of RyR channels in a relatively intact setting. Interestingly, our results provide the basis for a different possibility in which dephophosphorylation of RyR rather than its phosphorylation causes depletion of SR Ca(2+) stores by stimulating RyRs in failing hearts. It has been reported that PP1 and PP2 activities are increased in heart failure (Huang et al. 1999; Neumann et al. 1997; Neuman, 2002). Furthermore, overexpression of PP1 or ablation of the endogenous PP1 inhibitor, l-1, results in depressed contractile performance and heart failure (Carr et al. 2002). Our finding that PP1 causes depletion of SR Ca(2+) stores by activating RyRs could account for, or contribute to, these results.
DelPrincipe F, Egger M, Pignier C & Niggli E (2001). Enhanced E-C coupling efficiency after beta-stimulation of cardiac myocytes. Biophys J 80, 64a.
Gyorke I & Gyorke S (1998). Regulation of the cardiac ryanodine receptor channel by luminal Ca2+ involves luminal Ca2+ sensing sites. Biophys J 75, 2801–2810.
Gyorke S, Gyorke I, Lukyanenko V, Terentyev D, Viatchenko-Karpinski S & Wiesner TF (2002). Regulation of sarcoplasmic reticulum calcium release by luminal calcium in cardiac muscle. Front Biosci 7, d1454–d1463.
Gyorke I, Lukyanenko V & Gyorke S (1997). Dual effects of tetracaine on spontaneous calcium release in rat ventricular myocytes. J Physiol 500, 297–309.
MacDougall LK, Jones LR & Cohen P (1991). Identification of the major protein phosphatases in mammalian cardiac muscle which dephosphorylate phospholamban. Eur J Biochem 196, 725–734.
Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N & Marks AR (2000). PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell 101, 365–376.
Rodriguez P, Bhogal MS & Colyer J (2003). Stoichiometric phosphorylation of cardiac ryanodine receptor on serine-2809 by calmodulin-dependent kinase II and protein kinase A. J Biol Chem (in press).
Proc Natl Acad Sci U S A. 2010 August 3; 107(31): E124.
Reply to Eisner et al.: CaMKII phosphorylation of RyR2 increases cardiac contractility
The ryanodine receptor/calcium-release channel (RyR2) on the sarcoplasmic reticulum (SR) is the source of Ca2+ required for myocardial excitation–contraction (EC) coupling. During stress (i.e., exercise), contractility of the cardiac muscle is increased largely because of phosphorylation and activation of key proteins that regulate SR Ca2+ release. These include the voltage-gated calcium channel (Cav1.2) on the plasma membrane through which Ca2+ enters the cardiomyocyte, the sarco/endoplasmic reticulum calcium ATPase (SERCA2a)/phospholamban complex that pumps Ca2+ into the SR, and the RyR2 channel that releases Ca2+ from the SR, all of which are activated by phosphorylation.
For the past 10 y, Eisner et al. (1) have advanced the idea that activation of the RyR2 channel (e.g., by phosphorylation) cannot play a role in regulating systolic Ca2+ release and cardiac contractility. They base their position on an experiment in which they used caffeine to activate the RyR2 channel and showed that Ca2+ release was increased but after a few beats, returned to baseline (1). However, their experiment is not a good model for the physiological response to stress in which the three key regulators of EC coupling are all activated by the same signal (i.e., phosphorylation) such that there is increased Ca2+ influx, increased SR Ca2+ uptake, and increased SR Ca2+ release.
In the Eisner caffeine experiment, RyR2 was activated, but the Cav1.2 and SERCA2a were not. Selective activation of RyR2 is not physiological, and the outcome of their experiment was predictable. Caffeine-induced activation of RyR2 resulted in a transient increase in SR Ca2+ release, but because there was no concomitant increase in Ca2+ influx or SR Ca2+ uptake, the increase in SR Ca2+ release could not be sustained. However, on the basis of this experiment, Eisner et al. (1) concluded that activation of RyR2 plays no role in stress-induced increased cardiac contractility.
We have shown that, during stress, the increased heart rate results in a rate-dependent activation of CaMKII that phosphorylates and activates RyR2. We showed the essential role of this rate-dependent activation of RyR2 by CaMKII by showing that genetically engineered mice, lacking the CaMKII phosphorylation site on RyR2 (RyR2-S2814A), exhibit blunted increases in systolic Ca2+-transient amplitudes and contractile responses as heart rate increases (2). We also showed that a reduction in the amount of CaMKII in the RyR2 complex in failing hearts results in defective regulation of the channel, which could explain the loss of the rate-dependent increase in contractility in heart failure.
Eisner et al. (3) challenge all of our findings based on their caffeine experiment. However, our experiments have been conducted under physiological conditions in which all three components involved in Ca2+signaling during muscle contraction are activated, not just one. The only perturbation that we have introduced is to ablate the CaMKII phosphorylation site on RyR2 using a single amino acid substitution. This results in a blunted contractile response, leading us to conclude that CaMKII phosphorylation of RyR2 does indeed play a key role in enhancing contractility as the heart rate increases.
Cardiac Ryanodine Receptor Function and Regulation in Heart Disease
SE LEHNART, AHT WEHRENS, A KUSHNIR, AR MARKS*
Annals NY Acad Sci JAN 2006 http://dx.doi.org/10.1196/annals.1302.012
Cardiac Engineering: From Genes and Cells to Structure and Function 2004; 1015(1), pp 144–159
The cardiac ryanodine receptor (RyR2) located on the sarcoplasmic reticulum (SR) controls intracellular Ca2+ release and muscle contraction in the heart. Ca2+ release via RyR2 is regulated by several physiological mediators. Protein kinase (PKA) phosphorylation dissociates the stabilizing FKBP12.6 subunit (calstabin2) from the RyR2 complex, resulting in increased contractility and cardiac output. Congestive heart failure is associated with
- elevated plasma catecholamine levels, and
- chronic stimulation of β-adrenergic receptors
- leads to PKA hyperphosphorylation of RyR2 in failing hearts.
- PKA hyperphosphorylation results in calstabin2-depleted RyR2 that displays altered channel gating and
- may cause aberrant SR Ca2+ release,
- depletion of SR Ca2+ stores, and
- reduced myocardial contractility in heart failure.
Calstabin2-depleted RyR2 may also trigger cardiac arrhythmias that cause sudden cardiac death. In patients with catecholaminergic polymorphic ventricular tachycardia (CPVT), RyR2 missense mutations cause reduced calstabin2 binding to RyR2. Increased RyR2 phosphorylation and pathologically increased calstabin2 dissociation during exercise results in aberrant diastolic calcium release, which may trigger ventricular arrhythmias and sudden cardiac death. In conclusion, heart failure and exercise-induced sudden cardiac death have been linked to defects in RyR2-calstabin2 regulation, and this may represent a novel target for the prevention and treatment of these forms of heart disease

The δC Isoform of CaMKII Is Activated in Cardiac Hypertrophy and Induces Dilated Cardiomyopathy and Heart Failure
T Zhang, LS Maier, ND Dalton, S Miyamoto, J Ross, DM Bers, JH Brown
University of California, San Diego, La Jolla, Calif; and Loyola University, Chicago, Ill.
Circ Res. 2003;92:912-919. http://dx.doi.org/10.1161/01.RES.0000069686.31472.C5
Recent studies have demonstrated that transgenic (TG) expression of either Ca(2+)/calmodulin-dependent protein kinase IV (CaMKIV) or CaMKIIδB, both of which localize to the nucleus, induces cardiac hypertrophy. However, CaMKIV is not present in heart, and cardiomyocytes express not only the nuclear CaMKIIδB but also a cytoplasmic isoform, CaMKII δC. In the present study, we demonstrate that expression of the δC isoform of CaMKII is selectively increased and its phosphorylation elevated as early as 2 days and continuously for up to 7 days after pressure overload. To determine whether enhanced activity of this cytoplasmic δC isoform of CaMKII can lead to phosphorylation of Ca(2+) regulatory proteins and induce hypertrophy, we generated TG mice that expressed the δC isoform of CaMKII. Immunocytochemical staining demonstrated that the expressed transgene is confined to the cytoplasm of cardiomyocytes isolated from these mice. These mice develop a dilated cardiomyopathy with up to a 65% decrease in fractional shortening and die prematurely. Isolated myocytes are enlarged and exhibit reduced contractility and altered Ca2(2+) handling. Phosphorylation of the ryanodine receptor (RyR) at a CaMKII site is increased even before development of heart failure, and CaMKII is found associated with the RyR in immunoprecipitates from the CaMKII TG mice. Phosphorylation of phospholamban is also increased specifically at the CaMKII but not at the PKA phosphorylation site. These findings are the first to demonstrate that CaMKIIδC can mediate phosphorylation of Ca(2+) regulatory proteins in vivo and provide evidence for the involvement of CaMKIIδC activation in the pathogenesis of dilated cardiomyopathy and heart failure.
Multifunctional Ca(2+)/calmodulin-dependent protein kinases (CaM kinases or CaMKs) are transducers of Ca2+ signals that phosphorylate a wide range of substrates and thereby affect Ca(2+)-mediated cellular responses.1 The family includes CaMKI and CaMKIV, monomeric enzymes activated by CaM kinase kinase,2,3 and CaMKII, a multimer of 6 to 12 subunits activated by autophosphorylation.1 The CaMKII subunits α, β, γ, and δ show different tissue distributions,1 with the δisoform predominating in the heart.4–7 Splice variants of the δisoform, characterized by the presence of a second variable domain,4,7 include δB, which contains a nuclear localization signal (NLS), and δC, which does not. CaMKII composed of δB subunits localizes to the nucleus, whereas CaMKIIδC localizes to the cytoplasm.4,8,9
CaMKII has been implicated in several key aspects of acute cellular Ca(2+) regulation related to cardiac excitation-contraction (E-C) coupling. CaMKII phosphorylates sarcoplasmic reticulum (SR) proteins including the ryanodine receptors (RyR2) and phospholamban (PLB).10–14 Phosphorylation of RyR has been suggested to alter the channel open probability,14,15 whereas phosphorylation of PLB has been suggested to regulate SR Ca(2+) uptake.14 It is also likely that CaMKII phosphorylates the L-type Ca2 channel complex or an associated regulatory protein and thus mediates Ca2 current (ICa) facilitation.16-18 and the development of early after-depolarizations and arrhythmias.19 Thus, CaMKII has significant effects on E-C coupling and cellular Ca2 regulation. Nothing is known about the CaMKII isoforms regulating these responses.
Contractile dysfunction develops with hypertrophy, characterizes heart failure, and is associated with changes in cardiomyocyte Ca2homeostasis.20 CaMKII expression and activity are altered in the myocardium of rat models of hypertensive cardiac hypertrophy21,22 and heart failure,23 and in cardiac tissue from patients with dilated cardiomyopathy.24,25
Several transgenic mouse models have confirmed a role for CaMK in the development of cardiac hypertrophy, as originally suggested by studies in isolated neonatal rat ventricular myocytes.9,26–28 Hypertrophy develops in transgenic mice that overexpress CaMKIV,27 but this isoform is not detectable in the heart,4,29 and CaMKIV knockout mice still develop hypertrophy after transverse aortic constriction (TAC).29
Transgenic mice overexpressing calmodulin developed severe cardiac hypertrophy,30 later shown to be associated with an increase in activated CaMKII31; the isoform of CaMKII involved in hypertrophy could not be determined from these studies. We recently reported that transgenic mice that overexpress CaMKIIδB, which is highly concentrated in cardiomyocyte nuclei, develop hypertrophy and dilated cardiomyopathy.32 To determine whether in vivo expression of the cytoplasmic CaMKIIδC can phosphorylate cytoplasmic Ca2regulatory proteins and induce hypertrophy or heart failure, we generated transgenic (TG) mice that expressed the δC isoform of CaMKII under the control of the cardiac specific α-myosin heavy chain (MHC) promoter. Our findings implicate CaMKIIδC in the pathogenesis of dilated cardiomyopathy and heart failure and suggest that this occurs at least in part via alterations in Ca2handling proteins.33
Results
Expression and Activation of CaMKIIδC Isoform After TAC
To determine whether CaMKII was regulated in pressureoverload–induced hypertrophy, CaMKIIδ expression and phosphorylation were examined by Western blot analysis using left ventricular samples obtained at various times after TAC. A selective increase (1.6-fold) in the lower band of CaMKIIδwas observed as early as 1 day and continuously for 4 days (2.3-fold) and 7 days (2-fold) after TAC (Figure 1A). To confirm that CaMKIIδC was increased and determine whether this occurred at the transcriptional level, we performed semiquantitative RT-PCR using primers specific for the CaMKIIδC isoform. These experiments revealed that mRNA levels for CaMKIIδC were increased 1 to 7 days after TAC (Figure 1B). In addition to examining CaMKII expression, the activation state of CaMKII was monitored by its autophosphorylation, which confers Ca2-independent activity.
Figure 1. Expression and activation of CaMKII δC isoform after TAC.
see http://dx.doi.org/10.1161/01.RES.0000069686.31472.C5
A, Western blot analysis of total CaMKII in left ventricular (LV) homogenates obtained at indicated times after TAC. Cardiomyocytes transfected with CaMKIIδB and δC (right) served as positive controls and molecular markers. Top band (58 kDa) represents CaMKIIδB plus δ9, and the bottom band (56 kDa) corresponds to CaMKIIδC. *P0.05 vs control. B, Semiquantitative RT-PCR using primers specific for CaMKIIδC isoform (24 cycles) and GAPDH (19 cycles) using total RNA isolated from the same LV samples. C, Western blot analysis of phospho-CaMKII in LV homogenates obtained at various times after TAC. Three bands seen for each sample represent CaMKIIγ subunit (uppermost), CaMKIIδB plus δ9 (58 kDa), and CaMKIIδC (56 kDa). Quantitation is based on the sum of all of the bands. *P0.05 vs control.
Figure 2. Expression and activation of CaMKII in CaMKIIδC transgenic mice.
see http://dx.doi.org/10.1161/01.RES.0000069686.31472.C5
A, Transgene copy number based on Southern blots using genomic DNA isolated from mouse tails (digested with EcoRI). Probe (a 32P-labeled 1.7-kb EcoRI-SalI -MHC fragment) was hybridized to a 2.3-kb endogenous fragment (En) and a 3.9-kb transgenic fragment (TG). Transgene copy number was determined from the ratio of the 3.9-kb/2.3-kb multiplied by 2. B, Immunocytochemical staining of ventricular myocytes isolated from WT and CaMKIIδTG mice. Myocytes were cultured on laminin-coated slides overnight. Transgene was detected by indirect immunofluorescence staining using rabbit anti-HA antibody (1:100 dilution) followed by FITC-conjugated goat antirabbit IgG antibody (1:100 dilution). CaMKIIδB localization to the nucleus in CaMKIIδB TG mice (see Reference 32) is shown here for comparative purpose. C, Quantitation of the fold increase in CaMKIIδprotein expression in TGL and TGM lines. Different amounts of ventricular protein (numbers) from WT control, TG () and their littermates () were immunoblotted with an anti-CaMKIIδ antibody. Standard curve from the WT control was used to calculate fold increases in protein expression in TGL and TGM lines. D, Phosphorylated CaMKII in ventricular homogenates was measured by Western blot analysis (n5 for each group). **P0.01 vs WT.
Generation and Identification of CaMKIIδC Transgenic Mice
TG mice expressing HA-tagged rat wild-type CaMKIIδC under the control of the cardiac-specific α-MHC promoter were generated as described in Materials and Methods. By Southern blot analysis, 3 independent TG founder lines carrying 3, 5, and 15 copies of the transgene were identified. They were designated as TGL (low copy number), TGM (medium copy number), and TGH (high copy number),
The founder mice from the TGH line died at 5 weeks of age with marked cardiac enlargement. The other two lines showed germline transmission of the transgene. The transgene was expressed only in the heart.
Although CaMKII protein levels in TGL and TGM hearts were increased 12- and 17-fold over wild-type (WT) controls
(Figure 2C), the amount of activated CaMKII was only increased 1.7- and 3-fold in TGL and TGM hearts (Figure 2D). The relatively small increase in CaMKII activity in the TG lines probably reflects the fact that the enzyme is not constitutively activated and that the availability of Ca2/CaM, necessary for activation of the overexpressed CaMKII, is limited. Importantly, the extent of increase in active CaMKII in the TG lines was similar to that elicited by TAC.
Cardiac Overexpression of CaMKIIδC Induces Cardiac Hypertrophy and Dilated Cardiomyopathy
There was significant enlargement of hearts from CaMKIIδC TGM mice by 8 to 10 weeks (Figure 3A) and from TGL mice by 12 to 16 weeks. Histological analysis showed ventricular dilation (Figure 3B), cardiomyocyte enlargement (Figure 3C), and mild fibrosis (Figure 3D) in CaMKIIδC TG mice. Quantitative analysis of cardiomyocyte cell volume from 12-week-old TGM mice gave values of 54.7 + 0.1 pL for TGM (n = 96) versus 28.6 + 0.1 pL for WT littermates (n=94; P0.001).
Ventricular dilation and cardiac dysfunction developed over time in proportion to the extent of transgene expression. Left ventricular end diastolic diameter (LVEDD) was increased by 35% to 45%, left ventricular posterior wall thickness (LVPW) decreased by 26% to 29% and fractional shortening decreased by 50% to 60% at 8 weeks for TGM and at 16 weeks for TGL. None of these parameters were significantly altered at 4 weeks in TGM or up to 11 weeks in TGL mice, indicating that heart failure had not yet developed. Contractile function was significantly decreased.
Figure 6. Dilated cardiomyopathy and dysfunction in CaMKIIδC TG mice at both whole heart and single cell levels.
see Fig 6 http://dx.doi.org/10.1161/01.RES.0000069686.31472.C5
C, Decreased contractile function in ventricular myocytes isolated from 12-week old TGM and WT controls presented as percent change of resting cell length (RCL) stimulated at 0.5 Hz. Representative trace and mean values are shown. *P0.05 vs WT.
Figure 7. Phosphorylation of PLB in CaMKIIδC TG mice.
see Fig 7: http://dx.doi.org/10.1161/01.RES.0000069686.31472.C5
(Figures 7A and 7B). (see http://dx.doi.org/10.1161/01.RES.0000069686.31472.C6
(Figure 8C). (http://dx.doi.org/10.1161/01.RES.0000069686.31472.C5
Thr17 and Ser16 phosphorylated PLB was measured by Western blots using specific anti-phospho antibodies. Ventricular homogenates were from 12- to 14-week-old WT and TGM mice (A) or 4 to 5-week-old WT and TGM mice (B). Data were normalized to total PLB examined by Western blots (data not shown here). n = 6 to 8 mice per group; *P0.05 vs WT.
Cardiac Overexpression of CaMKIIδC Results in Changes in the Phosphorylation of Ca2 Handling Proteins
To assess the possible involvement of phosphorylation of Ca2cycling proteins in the phenotypic changes observed in the CaMKIIC TG mice, we first compared PLB phosphorylation state in homogenates from 12- to 14-week-old TGM and WT littermates. Western blots using antibodies specific for phosphorylated PLB showed a 2.3-fold increase in phosphorylation of Thr17 (the CaMKII site) in hearts from TGM versus WT (Figure 7A). Phosphorylation of PLB at the CaMKII site was also increased 2-fold in 4- to 5-week-old TGM mice (Figure 7B). Significantly, phosphorylation of the PKA site (Ser16) was unchanged in either the older or the younger TGM mice (Figures 7A and 7B).
To demonstrate that the RyR2 phosphorylation changes observed in the CaMKII transgenic mice are not secondary to development of heart failure, we performed biochemical studies examining RyR2 phosphorylation in 4- to 5-week-old TGM mice. At this age, most mice showed no signs of hypertrophy or heart failure (see Figure 6B) and there was no significant increase in myocyte size (21.3 + 1.3 versus 27.7 + 4.6 pL; P0.14). Also, twitch Ca2 transient amplitude was not yet significantly depressed, and mean δ [Ca2+]i (1 Hz) was only 20% lower (192 + 36 versus 156 + 13 nmol/L; P0.47) versus 50% lower in TGM at 13 weeks.33
The in vivo phosphorylation of RyR2, determined by back phosphorylation, was significantly (2.10.3-fold; P0.05) increased in these 4- to 5-week-old TGM animals (Figure 8C), an increase equivalent to that seen in 12- to 14-week-old mice. We also performed the RyR2 back-phosphorylation assay using purified CaMKII rather than PKA. RyR2 phosphorylation at the CaMKII site was also significantly increased (2.2 + 0.3-fold; P0.05) in 4- to 5-week-old TGM mice (Figure 8C).
The association of CaMKII with the RyR2 is consistent with a physical interaction between this protein kinase and its substrate. The catalytic subunit of PKA and the phosphatases PP1 and PP2A were also present in the RyR2 immunoprecipitates, but not different in WT versus TG mouse hearts (Figure 8D). These data provide further evidence that the increase in RyR2 phosphorylation, which precedes development of failure in the 4- to 5-week-old CaMKIIδC TG hearts, can be attributed to the increased activity of CaMKII.
Discussion
CaMKII is involved in the dynamic modulation of cellular Ca2 regulation and has been implicated in the development of cardiac hypertrophy and heart failure.14 Published data from CaMK-expressing TG mice demonstrate that forced expression of CaMK can induce cardiac hypertrophy and lead to heart failure.27,32 However, the CaMK genes expressed in these mice are neither the endogenous isoforms of the enzyme nor the isoforms likely to regulate cytoplasmic Ca(2+) handling, because they localize to the nucleus.
First, we demonstrate that the cytoplasmic cardiac isoform of CaMKII is upregulated at the expression level and is in the active state (based on autophosphorylation) after pressure overload induced by TAC. Second, we demonstrate that two cytoplasmic CaMKII substrates (PLB and RyR) are phosphorylated in vivo when CaMKII is overexpressed and its activity increased to an extent seen under pathophysiological conditions. Moreover, CaMKIIδis found to associate physically with the RyR in the heart. Finally, our data indicate that heart failure can result from activation of the cytoplasmic form of CaMKII and this may be due to altered Ca(2+) handling.
Differential Regulation of CaMKIIδ Isoforms in Cardiac Hypertrophy
The isoform of CaMKII that predominates in the heart is the δ isoform.4–7 Neither the α nor the β isoforms are expressed and there is only a low level of expression of the γ isoforms.39 Both δB and δC splice variants of CaMKIIδ are present in the adult mammalian myocardium36,40 and expressed in distinct cellular compartments.4,8,9
We suggest that the CaMKIIδisoforms are differentially regulated in pressure-overload–induced hypertrophy, because the expression of CaMKIIδC is selectively increased as early as 1 day after TAC. Studies using RT-PCR confirm that CaMKIIδC is regulated at the transcriptional level in response to
TAC. In addition, activation of both CaMKIIδB and CaMKIIδC, as indexed by autophosphorylation, increases as early as 2 days after TAC. Activation of CaMKIIδB by TAC is relevant to our previous work indicating its role in hypertrophy.9,32 The increased expression, as well as activation of the CaMKIIδC isoform, suggests that it could also play a critical role in both the acute and longer responses to pressure overload.
In conclusion, we demonstrate here that CaMKIIδC can phosphorylate RyR2 and PLB when expressed in vivo at levels leading to 2- to 3-fold increases in its activity. Similar increases in CaMKII activity occur with TAC or in heart failure. Data presented in this study and in the accompanying article33 suggest that altered phosphorylation of Ca(2+) cycling proteins is a major component of the observed decrease in contractile function in CaMKIIδC TG mice. The early occurrence of increased CaMKII activity after TAC, and of RyR and PLB phosphorylation in the CaMKIIδC TG mice suggest that CaMKIIδC plays an important role in the pathogenesis of dilated cardiomyopathy and heart failure. These results have major implications for considering CaMKII and its isoforms in exploring new treatment strategies for heart failure.
Unique phosphorylation site on the cardiac ryanodine receptor regulates calcium channel activity.
DR Witcher, RJ Kovacs, H Schulman, DC Cefali, LR Jones
Krannert Institute of Cardiology and the Indiana University School of Medicine, Indianapolis,
Stanford University School of Medicine, Stanford.
Journal of Biological Chemistry 07/1991; 266(17):11144-52. · 4.77 Impact
http://www.jbc.org/content/266/17/11144.full.pdf
Ryanodine receptors have recently been shown to be the Ca2+ release channels of sarcoplasmic reticulum in both cardiac muscle and skeletal muscle. Several regulatory sites are postulated to exist on these receptors, but to date, none have been definitively identified. In the work described here, we localize one of these sites by showing that the cardiac isoform of the ryanodine receptor is a preferred substrate for multifunctional Ca2+/calmodulin-dependent protein kinase (CaM kinase). Phosphorylation by CaM kinase occurs at a single site encompassing serine 2809. Antibodies generated to this site react only with the cardiac isoform of the ryanodine receptor, and immunoprecipitate only cardiac [3H]ryanodine-binding sites. When cardiac junctional sarcoplasmic reticulum vesicles or partially purified ryanodine receptors are fused with planar bilayers, phosphorylation at this site activates the Ca2+ channel. In tissues expressing the cardiac isoform of the ryanodine receptor, such as heart and brain, phosphorylation of the Ca(2+) release channel by CaM kinase may provide a unique mechanism for regulating intracellular (Ca2+) release.
The Ca(2+) release from the SR causes an increase in Ca(2+) concentration which leads to muscle contraction (1). Recently, the sites of Ca(2+) release have been identified and purified from both cardiac (2-4) and skeletal muscle SR (5- 7) and shown to be the same as the ryanodine receptors or high molecular weight proteins. The structures attach the transverse tubules to the junctional SR both in intact tissues and isolated membrane fractions (1, 8-10). Although the Ca(2+) release channels from cardiac and skeletal muscle show many similarities such as nearly identical
- myoplasmic 3- EGTA,
- Ca2+ conductances (2-7),
- protease sensitivities (11, E ) ,
- calmodulin-binding capabilities (ll), and
- modulation by allosteric regulators such as Ca2+, Mg2+, ATP, and calmodulin (13-15),
they also exhibit several differences in protein structure and function. Quantitative differences have been noted on the effects of modulators on ryanodine binding to the two proteins (16-18), as well as on Ca(2+) channel kinetics. In addition, the cardiac ryanodine smaller apparent molecular weight than the skeletal muscle receptor on SDS-PAGE (ll), and monoclonal antibodies can be made which react with the cardiac receptor but not the skeletal receptor (16).
Recent work on characterization receptors has culminated in elucidation of structures of the proteins by sequencing of their cDNAs (19-21). Consistent with the differences between the two protein iso- forms noted above, the cardiac and skeletal muscle receptors have been found to be the products of different genes, with overall amino acid identities of 66% (21). Both protein isoforms are very large, containing approximately 5,000 amino acids and exhibiting predicted molecular weights of 564,711 for the cardiac protein (21) and 565,223 (19) or 563,584 (20) for the skeletal muscle protein. In the native state, ryanodine receptors are arranged as tetramers (1-7). In an earlier study (22), we demonstrated that the canine cardiac high molecular weight protein (or ryanodine receptor; Ref. 3) was an excellent substrate CaM kinase (23,24) endogenous to junctional SR membranes. In the work described here, we show that phosphorylation of the cardiac receptor by CaM kinase occurs at a single site, which is not substantially phosphorylated in the skeletal muscle receptor, and that phosphorylation ryanodine receptor at this site activates the Ca2+ channel.
Our data are the first to support the hypothesis (21), that the modulator-binding sites of the cardiac ryanodine receptor are contained within residues 2619-3016. (13, 14). The ryanodine receptor is compared with the primary structure for the multifunctional of the cardiac model of Otsu et al. (21).
Experimental Procedures.
See Figs 1-6 http://www.jbc.org/content/266/17/11144.full.pdf
RESULTS AND DISCUSSION.
Preferential Phosphorylation Receptor-(Fig. 1, arrowheads) is phosphorylated in junctional vesicles by an endogenous calmodulin-requiring proteinase and this phosphorylation is stimulated several fold when exogenous CaM kinase is added. In contrast, the ryanodine receptor in canine fast and vesicles, which migrates with weight on SDS-PAGE (2, 11, 16), is not significantly phosphorylated by either endogenous or exogenous protein kinase (Fig. 1, small arrows).
Similar results were obtained with rabbit skeletal muscle SR vesicles. The identity of the skeletal muscle ryanodine receptor in these studies (Fig. 1, small arrow) was confirmed by immunoblotting with a skeletal muscle isoform-specific antibody (supplied by K. Campbell, University of Iowa). We did detect a low level of phosphorylation of a protein in slow skeletal muscle samples migrating slightly faster than the cardiac receptor, but this protein did not cross-react with skeletal muscle (or cardiac, see below) antibodies, suggesting that it is unrelated to the ryanodine receptor. CaM kinase-catalyzed phosphorylation of the cardiac ryanodine receptor was always at least 10-fold greater than skeletal receptor phosphorylation. These results demonstrate that the skeletal muscle ryanodine receptor phosphorylation is insignificant compared to cardiac protein phosphorylation. Consistent with our results, Otsu et al. (21) have recently shown that, the cardiac isoform receptor is absent from fast and slow skeletal muscle. Phosphorylation of the cardiac ryanodine receptor by cAMP kinase also occurs, but phosphorylation by added cAMP kinase is no greater than that achieved with endogenous CaM kinase. (Fig. 2). In contrast, the amount of exogenous CaM kinase increases receptor phosphorylation 4-fold, to a maximal level of 26 pmol of P/mg of SR protein (Fig. 2). We observed no significant phosphorylation of canine fast and slow or rabbit skeletal muscle ryanodine. Maximal ryanodine binding (3) in these preparations ranged between 5 and 6 pmol/mg of protein, a value nearly identical to the level of receptor phosphorylation achieved with exogenous cAMP kinase (see CaM kinase), but one-fourth the value achieved with added CaM kinase. Since the functional unit release channel contains only one high affinity ryanodine- binding site/tetramer (4), our results suggest that the endogenous CaM kinase is capable of phosphorylating only one-fourth of the available sites, whereas the exogenous kinase can fully phosphorylate the receptor (below) of the Cardiac Ryanodine. The canine Slow skeletal muscle SR receptor of the ryanodine it was recently reported is phosphorylated 1/20th by the of the CaM kinase.
TABLE 1
Immunoprecipitation of Ryanodine receptors from CHAPS-solubilized canine SR membranes. Values are expressed for aliquots of the following fractions: S, solubilized receptors after treatment of membranes with 2% CHAPS; B, bound fraction, containing ryanodine receptors immunoprecipitated from CHAPS superna- tant; F, free fraction, containing ryanodine receptors not immunoprecipitated. Total binding was measured using 20 nM [3H]ryanodine. For nonspecific binding, 10 PM cold ryanodine was added. FIG. 7.
Effect of ATP and calmodulin on the cardiac Ca(2+) release channel. Holding potential was 0 mV, with upward current deflections representing movement of Ba(2+) from the trans to the cis chamber. Gaussian distributions were fit to the peaks of activity in the histograms. Signals were filtered at 300 Hz (low pass Bessel) and digitized at 1 KHz (Axotape, Axon Instruments) for * off-line analysis. In the control (A), p(open) was 0.26. Addition of 1 mM ATP (B) produced prolonged openings of the channel, increasing p(0pen) to 0.81. Subsequent addition of calmodulin (C) decreased p(open) to 0.12, producing long closures and brief aborted openings.
Sequencing of the Cardiac Phosphorylation Site. In order to sequence the phosphorylation site of the cardiac ryanodine receptor, we phosphorylated junctional SR membranes on large scale with added CaM kinase and purified the phosphorylated denatured ryanodine receptor to homogeneity in one step using SDS-gel filtration chromatography (Fig. 3). The purified cardiac ryanodine receptor was digested with trypsin, and the radioactive peptides recovered using Fe(3+) affinity chromatography (30,37). 90% of the loaded radioactivity was recovered in the pH 8.6 and 10 eluates from the Fe column (Fig. 4). These fractions were then combined and subjected to reverse-phase chromatography, yielding a single major radioactive peptide peak eluting at approximately 24% acetonitrile (Fig. 4, inset).
http://www.jbc.org/content/266/17/11144.full.pdf
Gas-phase sequencing of the radioactive tryptic peptide gave a single sequence of 18 consecutive residues, which corresponded exactly to residues 2807-2824 reported for the rabbit cardiac ryanodine receptor from cDNA cloning (Fig. 5) (21). When CNBr and endoproteinase Lys-C were used to cleave the receptor, another “P-labeled peptide was isolated and sequenced, which matched with residues 2800-2811 of the rabbit cardiac ryanodine receptor (Fig. 5).
Serine 2809 within the phosphorylated tryptic peptide is situated on the carboxyl-terminal side of 2 arginine residues. The fact that R-R-X-S and R-X-X-S/T are minimal consensus phosphorylation sequences (38,39) for CAMP kinase and CaM kinase, respectively, makes this residue the likely phosphorylation site. Consistent with this, the ratio threitol-serine to phenylthiohydantoin-serine recovered dur- ing cycle 3 of sequencing of this peptide was 10 times greater than that recovered during cycles 6 and 9. It is known that dithiothreitol-serine is the predominant breakdown product of phosphoserine (40, 41). Phosphoamino acid analysis revealed that this peptide contained only phosphoserine; more- over, >90% of the 3’Pi was released from the peptide by cycle 10 (40, 42), demonstrating that no serine residue downstream of this region was significantly labeled.
Based on these results, we conclude that serine 2809 is the amino acid phosphorylated by CaM kinase. When only endogenous CaM kinase was used to phosphorylate the cardiac ryanodine receptor, the same labeled tryptic peptide was recovered and sequenced in four separate runs. Thus, although exogenously added kinase gives a 4-fold stimulation of receptor phosphorylation (Fig. 2), no new sites are phosphorylated. The reason for the low level of phosphorylation obtained with endogenous CaM kinase remains undefined.
Cardiac Electrophysiological Dynamics From the Cellular Level to the Organ Level
Daisuke Sato and Colleen E. Clancy
Department of Pharmacology, University of California – Davis, Davis, CA.
Biomedical Engineering and Computational Biology 2013:5: 69–75
http://www.la-press.com. http://dx.doi.org/10.4137/BECB.S10960
Figure 1. (Top): APD and DI. (Bottom): The physiological mechanism of APD alternans involves recovery from inactivation of ICaL. [see http://dx.doi.org/10.4137/BECB.S10960]
Figure 2. APD restitution and dynamical mechanism of APD alternans. [see http://dx.doi.org/10.4137/BECB.S10960]
Review Series. Genetic Causes of Human Heart Failure
Hiroyuki Morita, Jonathan Seidman and Christine E. Seidman
Harvard Medical School, Brigham and Women’s Hospital, Howard Hughes Medical Institute, Boston, MA
J Clin Invest. 2005;115(3):518–526. http://dx.doi.org/10.1172/JCI24351.
Correspondence to: Christine E. Seidman, Department of Genetics, Harvard Medical School, Boston, MA. Ph: (617) 432-7871; E-mail: cseidman@genetics.med.harvard.edu
Factors that render patients with cardiovascular disease at high risk for heart failure remain incompletely defined. Recent insights into molecular genetic causes of myocardial diseases have highlighted the importance of single-gene defects in the pathogenesis of heart failure. Through analyses of the mechanisms by which a mutation selectively perturbs one component of cardiac physiology and triggers cell and molecular responses, studies of human gene mutations provide a window into the complex processes of cardiac remodeling and heart failure. Knowledge gleaned from these studies shows promise for defining novel therapeutic targets for genetic and acquired causes of heart failure.
Introduction
Heart failure currently affects 4.8 million Americans, and each year over 500,000 new cases are diagnosed. In 2003 heart failure contributed to over 280,000 deaths and accounted for 17.8 billion health care dollars (1).
Heart failure almost universally arises in the context of antecedent cardiovascular disease:
- atherosclerosis,
- cardiomyopathy,
- myocarditis,
- congenital malformations, or
- valvular disease.
The study of single-gene mutations that trigger heart failure provides an opportunity for defining important molecules involved in these processes. Although these monogenic disorders account for only a small subset of overall heart failure cases, insights into the responses triggered by gene mutations are likely to also be relevant to more common etiologies of heart failure.
Early Manifestation – Heart Failure – Ventricular Remodeling.
One of 2 distinct morphologies occurs: left ventricular hypertrophy (increased wall thickness without chamber expansion) or dilation (normal or thinned walls with enlarged chamber volumes).
Each is associated with specific hemodynamic changes. Systolic function is normal, but diastolic relaxation is impaired in hypertrophic remodeling; diminished systolic function characterizes dilated remodeling. Clinical recognition of these cardiac findings usually prompts diagnosis of hypertrophic cardiomyopathy (HCM) or dilated cardiomyopathy (DCM). There is now considerable evidence that many different gene mutations can cause these pathologies (Figure 1), and with these discoveries has come recognition of distinct histopathologic features that further delineate several subtypes of remodeling. The current compendia of genes that remodel the heart already suggest a multiplicity of pathways by which the human heart can fail.
To facilitate a discussion, we have grouped known cardiomyopathy genes according to the probable functional consequences of mutations on
- force generation and transmission,
- metabolism,
- calcium homeostasis, or
- transcriptional control.
Gene mutations in one functional category inevitably have an impact on multiple myocyte processes, and, the eventual delineation of signals between functional groups may be critical to understanding cardiac decompensation and heart failure development.
Figure 1. see http:/dx.doi.org/10.1172/JCI24351
Human gene mutations can cause cardiac hypertrophy (blue), dilation (yellow), or both (green). In addition to these two patterns of remodeling, particular gene defects produce hypertrophic remodeling with glycogen accumulation (pink) or dilated remodeling with fibrofatty degeneration of the myocardium (orange). Sarcomere proteins denote β-myosin heavy chain, cardiac troponin T, cardiac troponin I, α-tropomyosin, cardiac actin, and titin. Metabolic/storage proteins denote AMP-activated protein kinase γ subunit, LAMP2, lysosomal acid α 1,4–glucosidase, and lysosomal hydrolase α-galactosidase A. Z-disc proteins denote MLP and telethonin. Dystrophin-complex proteins denote δ-sarcoglycan, β-sarcoglycan, and dystrophin. Ca2+ cycling proteins denote PLN and RyR2. Desmosome proteins denote plakoglobin, desmoplakin, and plakophilin-2.
Force generation and propagation. Generation of contractile force by the sarcomere and its transmission to the extracellular matrix are the fundamental functions of heart cells. Inadequate performance in either component prompts cardiac remodeling (hypertrophy or dilation), produces symptoms, and leads to heart failure. Given the importance of these processes for normal heart function and overt clinical manifestations of deficits in either force generation or transmission, it is not surprising that more single-gene mutations have been identified in molecules involved in these critical processes than in those of other functional classes.
Figure 2 see http:/dx.doi.org/10.1172/JCI24351
Human mutations affecting contractile and Z-disc proteins. The schematic depicts one sarcomere,
the fundamental unit of contraction encompassing the protein segment between flanking Z discs. Sarcomere thin filament proteins are composed of actin and troponins C, T, and I. Sarcomere thick filament proteins include myosin heavy chain, myosin essential and regulatory light chains, myosin-binding protein-C and titin. The sarcomere is anchored through titin and actin interactions with Z disc proteins α-actinin, calsarcin-1, MLP, telethonin (T-cap), and ZASP. Human mutations (orange text) in contractile proteins and Z-disc proteins can cause HCM or DCM.
Sarcomere protein mutations. Human mutations in the genes encoding protein components of the sarcomere cause either HCM or DCM. While progression to heart failure occurs with both patterns of remodeling, the histopathology, hemodynamic profiles, and biophysical consequences of HCM or DCM mutations suggest that distinct molecular processes are involved.
Over 300 dominant mutations in genes encoding β-cardiac myosin heavy chain (MYH7), cardiac myosin-binding protein-C (MYBPC3), cardiac troponin T (TNNT2), cardiac troponin I (TNNI3), essential myosin light chain (MYL3), regulatory myosin light chain (MYL2), α-tropomyosin (TPM1), cardiac actin (ACTC), and titin (TTN) have been reported to cause HCM (Figure 2) (2, 3). Recent reports of comprehensive sequencing of sarcomere protein genes in diverse patient populations indicate that MYBPC3 and MYH7 mutations are most frequent (4, 5). Sarcomere gene mutations that cause HCM produce a shared histopathology with enlarged myocytes that are disorganized and die prematurely, which results in increased cardiac fibrosis.
The severity and pattern of ventricular hypertrophy,
- age at onset of clinical manifestations, and
- progression to heart failure
are, in part, dependent on the precise sarcomere protein gene mutation. For example, TNNT2 mutations are generally associated with a high incidence of sudden death despite only mild left ventricular hypertrophy (6, 7). While only a small subset (10–15%) of HCM patients develop heart failure, this end-stage phenotype has a markedly poor prognosis and often necessitates cardiac transplantation. Accelerated clinical deterioration has been observed with MYH7 Arg719Trp, TNNT2 Lys273Glu, TNNI3 Lys183del, and TPM1 Glu180Val mutations (8–11).
Most HCM mutations encode defective polypeptides containing missense residues or small deletions; these are likely to be stably incorporated into cardiac myofilaments and to produce hypertrophy because normal sarcomere function is disturbed. Many HCM mutations in MYBPC3 fall within carboxyl domains that interact with titin and myosin; however, the exact biophysical properties altered by these defects remain unknown (Figure 2). HCM mutations in myosin are found in virtually every functional domain, which suggests that the biophysical consequences of these defects may vary. Genetic engineering of some human myosin mutations into mice has indicated more consistent sequelae. Isolated single-mutant myosin molecules containing different HCM mutations
- had increased actin-activated ATPase activity and
- showed greater force production and
- faster actin-filament sliding,
biophysical properties that may account for hyperdynamic contractile performance observed in HCM hearts and that suggest a mechanism for premature myocyte death in HCM (12–14). Uncoordinated contraction due to
- heterogeneity of mutant and normal sarcomere proteins,
- increased energy consumption, and
- changes in Ca2+ homeostasis
could diminish myocyte survival and trigger replacement fibrosis. With insidious myocyte loss and increased fibrosis, the HCM heart transitions from hypertrophy to failure.
Mice that are engineered to carry a sarcomere mutation replicate the genetics of human disease; heterozygous mutations cause HCM. One exception is a deletion of proximal myosin-binding protein-C sequences; heterozygous mutant mice exhibited normal heart structure while homozygous mutant mice developed hypertrophy (15). Remarkably, while most heterozygous mouse models with a mutation in myosin heavy chain, myosin-binding protein-C, or troponin T developed HCM (16–18), homozygous mutant mice (19, 20) developed DCM with fulminant heart failure and, in some cases, premature death. These mouse studies might indicate that HCM, DCM, and heart failure reflect gradations of a single molecular pathway. Alternatively, significant myocyte death caused by homozygous sarcomere mutations may result in heart failure. Human data suggest a more complicated scenario. The clinical phenotype of rare individuals who carry homozygous sarcomere mutations in either MYH7 (21) or in TNNT2 (22) is severe hypertrophy, not DCM. Furthermore, individuals with compound heterozygous sarcomere mutations exhibit HCM, not DCM. The absence of ventricular dilation in human hearts with 2 copies of mutant sarcomere proteins is consistent with distinct cellular signaling programs that remodel the heart into hypertrophic or dilated morphologies.
DCM sarcomere protein gene mutations affect distinct amino acids from HCM-causing mutations, although the proximity of altered residues is remarkable. The histopathology of sarcomere DCM mutations is quite different from those causing HCM, and is remarkably nonspecific. Degenerating myocytes with increased interstitial fibrosis are present, but myocyte disarray is notably absent. There are 2 mechanisms by which sarcomere mutations may cause DCM and heart failure: deficits of force production and deficits of force transmission. Diminished force may occur in myosin mutations (e.g., MYH7 Ser532Pro) that alter actin-binding residues involved in initiating the power stroke of contraction. Impaired contractile force may also occur in DCM troponin mutations (TNNT2 ΔLys210, ref. 23; and TNNI3 Ala2Val, ref. 24) that alter residues implicated in tight binary troponin interactions. Because troponin molecules modulate calcium-stimulated actomyosin ATPase activity, these defects may cause inefficient ATP hydrolysis and therein decrease contractile power.
Other DCM sarcomere mutations are more likely to impair force transmission (Figure 2). For example, a myosin mutation (at residue 764) located within the flexible fulcrum that transmits movement from the head of myosin to the thick filament is likely to render ineffectual the force generated by actomyosin interactions (23). DCM TPM1 mutations (25) are predicted to destabilize actin interactions and compromise force transmission to neighboring sarcomere. Likewise, ACTC mutations (26) that impair binding of actin to Z-disc may compromise force propagation. TTN mutations provide quintessential evidence that deficits in force transmission cause DCM and heart failure. By spanning the sarcomere from Z-disc to M-line, this giant muscle protein assembles contractile filaments and provides elasticity through serial spring elements. Titin interacts with α-actinin and telethonin (T-cap) at the Z-disc, with calpain3 and obscurin at the I-band (the extensible thin filament regions flanking Z-discs), and with myosin-binding protein-C, calmodulin, and calpain3 at the M-line region. Human mutations identified in
- the Z-disc–I-band transition zone (27),
- in the telethonin and α-actinin–binding domain, and
- in the cardiac-specific N2B domain (an I-band subregion; ref. 28) each cause DCM and heart failure.
Intermediate filaments and dystrophin-associated glycoprotein mutations. Intermediate filaments function as cytoskeletal proteins linking the Z-disc to the sarcolemma. Desmin is a type III intermediate filament protein, which, when mutated, causes skeletal and cardiac muscle disease (Figure 3). The hearts of mice deficient in desmin (29) are more susceptible to mechanical stress, which is consistent with the function of intermediate proteins in force transmission.
Figure 3
Human mutations (orange text) in components of myocyte cytoarchitecture cause DCM and heart failure. Force produced by sarcomeric actin-myosin interactions is propagated through the actin cytoskeleton and dystrophin to the dystrophin-associated glycoprotein complex (composed of α- and β-dystroglycans, α-, β-, γ- and δ-sarcoglycans, caveolin-3, syntrophin, and dystrobrevin). Desmosome proteins plakoglobin, desmoplakin, and plakophilin-2, provide functional and structural contacts between adjacent cells and are linked through intermediate filament proteins, including desmin, to the nuclear membrane, where lamin A/C is localized. (Adapted from ref. 96.)
Through dystrophin and actin interactions, the dystrophin-associated glycoprotein complex (composed of α- and β-dystroglycans, α-, β-, γ- and δ-sarcoglycans, caveolin-3, syntrophin, and dystrobrevin) provides stability to the sarcomere and transmits force to the extracellular matrix. Human mutations in these proteins cause muscular dystrophy with associated DCM and heart failure (Figure 3). Skeletal muscle manifestations can be minimal in female carriers of X-linked dystrophin defects, and some individuals present primarily with heart failure (30). In the mouse experiment, coxsackievirus B3–encoded protease2A, which can cleave dystrophin, was shown to produce sarcolemmal disruption and cause DCM, which suggests that dystrophin is also involved in the pathologic mechanism of DCM and heart failure that follow viral myocarditis (31).
While deficiencies of proteins that link the sarcomere to the extracellular matrix are likely to impair force transmission, recent studies of mice engineered to carry mutations in these molecules indicate other mechanisms for heart failure. A model of desmin-related cardiomyopathies (32) uncovered striking intracellular aggresomes, electron dense accumulations of heat shock and chaperone protein, α-B-crystalline, desmin, and amyloid in association with sarcomeres. While particularly abundant in the amyloid heart, aggresomes were also found in some DCM and HCM specimens, which suggests that excessive degenerative processing induced by myocyte stress or gene mutation may be toxic to sarcomere function.
Analyses of δ-sarcoglycan null mice (33) also yielded unexpected disease mechanisms, primary coronary vasospasm and myocardial ischemia. Selective restoration of δ-sarcoglycan to the cardiac myocytes extinguished this pathology, thereby implicating chronic ischemia as a contributing factor to heart failure development in patients with sarcoglycan mutations.
Mutations in intercalated and Z-disc proteins. To generate contraction, one end of each actin thin filament must be immobilized. The Z-disc defines the lateral boundary of the sarcomere, where actin filaments, titin, and nebulette filaments are anchored. Metavinculin provides attachment of thin filaments to the plasma membrane and plays a key role in productive force transmission. Two metavinculin gene mutations cause DCM by disruption of disc structure and actin-filament organization (34).
Other Z-disc protein constituents may also function as mechano-stretch receptors (35). Critical components include α-actinin, which aligns actin and titin from neighboring sarcomeres and interacts with muscle LIM protein (MLP encoded by CSRP3), telethonin (encoded by TCAP), which interacts with titin and MLP to subserve overall sarcomere function, and Cypher/Z-band alternatively spliced PDZ-motif protein (Cypher/ZASP), a striated muscle-restricted protein that interacts with α-actinin–2 through a PDZ domain and couples to PKC-mediated signaling via its LIM domains (Figure 2). Mutations in these molecules cause either DCM (35, 36) or HCM (37, 38) and predispose the affected individuals to heart failure. Genetically engineered mice with MLP deficiency (39) help to model the mechanism by which mutations in distinct proteins cause disease. Without MLP, telethonin is destabilized and gradually lost from the Z-disc; as a consequence, MLP-deficient cardiac papillary muscle shows an impairment in tension generation following the delivery of a 10% increase in passive stretch of the muscle and a loss of stretch-dependent induction of molecular markers (e.g., brain natriuretic peptide), which suggests that an MLP-telethonin–titin complex is an essential component of the cardiac muscle mechanical stretch sensor machinery. An important question is how signaling proteins (e.g., Cyper/ZASP) within the Z-disc translate mechanosensing into activation of survival or cell death pathways.
Lamin A/C mutations. The inner nuclear-membrane protein complex contains emerin and lamin A/C. Defects in emerin cause X-linked Emery-Dreifuss muscular dystrophy, joint contractures, conduction system disease, and DCM. Dominant lamin A/C mutations exhibit a more cardiac-restricted phenotype with fibrofatty degeneration of the myocardium and conducting cells, although subclinical involvement of skeletal muscles and contractures are sometimes apparent. The remarkable electrophysiologic deficits (progressive atrioventricular block and atrial arrhythmias) observed in mutations of lamin A/C and emerin indicate the particular importance of these proteins in electrophysiologic cells. A recent study of lamin A/C mutant mice showed evidence of marked nuclear deformation, fragmentation of heterochromatin, and defects in mechanotransduction (40, 41), all of which likely contribute to reduced myocyte viability. The similarities of cardiac histopathology (fibrofatty degeneration) observed in mutations of the nuclear envelope and desmosomes raise the possibility that these structures may both function as important mechanosensors in myocytes (Figure 3).
Desmosome protein mutations. Arrhythmogenic right ventricular cardiomyopathy (ARVD) identifies an unusual group of cardiomyopathies characterized by progressive fibrofatty degeneration of the myocardium, electrical instability, and sudden death (42). While right ventricular dysplasia predominates, involvement of the left ventricle also occurs. Progressive myocardial dysfunction is seen late in the course of disease, often with right-sided heart failure. ARVD occurs in isolation or in the context of Naxos syndrome, an inherited syndrome characterized by prominent skin (palmar-plantar keratosis), hair, and cardiac manifestations. Mutations in protein components of the desmosomes (Figure 3) (plakoglobin, ref. 43; desmoplakin, refs. 44, 45; and plakophilin-2, ref. 46) and in the cardiac ryanodine receptor (RyR2) (ref. 47; discussed below) cause syndromic and nonsydromic ARVD. Desmosomes are organized cell membrane structures that provide functional and structural contacts between adjacent cells and that may be involved in signaling processes. Whether mutations in the desmosomal proteins render cells of the heart (and skin) inappropriately sensitive to normal mechanical stress or cause dysplasia via another mechanism is unknown.
Energy production and regulation
Mitochondrial mutations. Five critical multiprotein complexes, located within the mitochondria, synthesize ATP by oxidative phosphorylation. While many of the protein components of these complexes are encoded by the nuclear genome, 13 are encoded by the mitochondrial genome. Unlike nuclear gene mutations, mitochondrial gene mutations exhibit matrilineal inheritance. In addition, the mitochondrial genome is present in multiple copies, and mutations are often heteroplasmic, affecting some but not all copies. These complexities, coupled with the dependence of virtually all tissues on mitochondrial-derived energy supplies, account for the considerable clinical diversity of mitochondrial gene mutations (Figure 4). While most defects cause either dilated or hypertrophic cardiac remodeling in the context of mitochondrial syndromes such as Kearns-Sayre syndrome, ocular myopathy, mitochondrial encephalomyopathy with lactic-acidosis and stroke-like episodes (MELAS), and myoclonus epilepsy with ragged-red fibers (MERFF) (48), there is some evidence that particular mitochondrial mutations can produce predominant or exclusive cardiac disease (49, 50). An association between heteroplasmic mitochondrial mutations and DCM has been recognized (51).
Figure 4
Human gene mutations affecting cardiac energetics and metabolism. Energy substrate utilization is directed by critical metabolic sensors in myocytes, including AMP-activated protein kinase (AMPK), which, in response to increased AMP/ATP levels, phosphorylates target proteins and thereby regulates glycogen and fatty acid metabolism, critical energy sources for the heart. Glycogen metabolism involves a large number of proteins including α-galactosidase A (mutated in Fabry disease) and LAMP2 (mutated in Danon disease). Glycogen and fatty acids are substrates for multiprotein complexes located within the mitochondria for the synthesis of ATP. KATP channels composed of an enzyme complex and a potassium pore participate in decoding metabolic signals to maximize cellular functions during stress adaptation. Human mutations (orange text) that cause cardiomyopathies have been identified in the regulatory SUR2A subunit of KATP, the γ2 subunit of AMPK, mitochondrial proteins, α-galactosidase A, and LAMP2.
Nuclear-encoded metabolic mutations. Nuclear gene mutations affecting key regulators of cardiac metabolism are emerging as recognized causes of hypertrophic cardiac remodeling and heart failure (Figure 4). Mutations in genes encoding the γ2 subunit of AMP-activated protein kinase (PRKAG2), α-galactosidase A (GLA), and lysosome-associated membrane protein-2 (LAMP2) can cause profound myocardial hypertrophy in association with electrophysiologic defects (52). AMP-activated protein kinase functions as a metabolic-stress sensor in all cells. This heterotrimeric enzyme complex becomes activated during energy-deficiency states (low ATP, high ADP) and modulates (by phosphorylation) a large number of proteins involved in cell metabolism and energy (53). Most GLA mutations can cause multisystem classic Fabry disease (angiokeratoma, corneal dystrophy, renal insufficiency, acroparesthesia, and cardiac hypertrophy), but some defects produce primarily cardiomyopathy. LAMP2 mutations can also produce either multisystem Danon disease (with skeletal muscle, neurologic, and hepatic manifestations) or a more restricted cardiac phenotype.
Cardiac histopathology reveals that, unlike sarcomere gene mutations, which cause hypertrophic remodeling, the mutations in PRKAG2, LAMP2, and GLA accumulate glycogen in complexes with protein and/or lipids, thereby defining these pathologies as storage cardiomyopathies. Progression from hypertrophy to heart failure is particularly common and occurs earlier with LAMP2 mutations than with other gene mutations that cause metabolic cardiomyopathies. Since both GLA and LAMP2 are encoded on chromosome X, disease expression is more severe in men, but heterozygous mutations in women are not entirely benign, perhaps due to X-inactivation that equally extinguishes a normal or mutant allele. The cellular and molecular pathways that produce either profound hypertrophy or progression to heart failure from PRKAG2, GLA, or LAMP2 mutations are incompletely understood. While accumulated byproducts are likely to produce toxicity, animal models indicate that mutant proteins cause far more profound consequences by changing cardiac metabolism and altering cell signaling. This is particularly evident in PRKAG2 mutations that increase glucose uptake by stimulating translocation of the glucose transporter GLUT-4 to the plasma membrane, increase hexokinase activity, and alter expression of signaling cascades (54).
The cooccurrence of electrophysiologic defects in metabolic mutations raises the possibility that pathologic cardiac conduction and arrhythmias contribute to cardiac remodeling and heart failure in these gene mutations. One mechanism for electrophysiologic defects appears to be the direct consequence of storage: transgenic mice that express a human PRKAG2 mutation (55) developed ventricular pre-excitation due to pathologic atrioventricular connections by glycogen-filled myocytes that ruptured the annulus fibrosis (the normal anatomic insulator which separates atrial and ventricular myocytes). A second and unknown mechanism may be that these gene defects are particularly deleterious to specialized cells of the conduction system. Little is known about the metabolism of these cells, although historical histopathologic data indicate glycogen to be particularly more abundant in the conduction system than in the working myocardium (56–58).
Ca2+ Cycling
Considerable evidence indicates the presence of abnormalities in myocyte calcium homeostasis to be a prevalent and important mechanism for heart failure. Protein and RNA levels of key calcium modulators are altered in acquired and inherited forms of heart failure, and human mutations in molecules directly involved in calcium cycling have been found in several cardiomyopathies (Figure 5).
Figure 5
Human mutations affecting Ca2+ cycling proteins. Intracellular Ca2+ handling is the central coordinator of cardiac contraction and relaxation. Ca2+ entering through L-type channels (LTCC) triggers Ca2+ release (CICR) from the SR via the RyR2, and sarcomere contraction is initiated. Relaxation occurs with SR Ca2+ reuptake through the SERCA2a. Calstabin2 coordinates excitation and contraction by modulating RyR2 release of Ca2+. PLN, an SR transmembrane inhibitor of SERCA2a modulates Ca2+ reuptake. Dynamic regulation of these molecules is effected by PKA-mediated phosphorylation. Ca2+ may further function as a universal signaling molecule, stimulating Ca2+-calmodulin and other molecular cascades. Human mutations (orange text) in molecules involved in calcium cycling cause cardiac remodeling and heart failure. NCX, sodium/calcium exchanger.
Calcium enters the myocyte through voltage-gated L-type Ca2+ channels; this triggers release of calcium from the sarcoplasmic reticulum (SR) via the RyR2. Emerging data define FK506-binding protein (FKBP12.6; calstabin2) as a critical stabilizer of RyR2 function (59), preventing aberrant calcium release during the relaxation phase of the cardiac cycle (Figure 5). Stimuli that phosphorylate RyR2 (such as exercise) by protein kinase A (PKA) dissociate calstabin2 from the receptor, thereby increasing calcium release and enhancing contractility. At low concentrations of intracellular calcium, troponin I and actin interactions block actomyosin ATPase activity; increasing levels foster calcium binding to troponin C, which releases troponin I inhibition and stimulates contraction. Cardiac relaxation occurs when calcium dissociates from troponin C, and intracellular concentrations decline as calcium reuptake into the SR occurs through the cardiac sarcoplasmic reticulum Ca2+-ATPase pump (SERCA2a). Calcium reuptake into SR is regulated by phospholamban (PLN), an inhibitor of SERCA2a activity that when phosphorylated dissociates from SERCA2a and accelerates ventricular relaxation.
RyR2 mutations. While some mutations in the RyR2 are reported to cause ARVD (47) (see discussion of desmosome mutations), defects in this calcium channel are more often associated with catecholaminergic polymorphic ventricular tachycardia (60, 61), a rare inherited arrhythmic disorder characterized by normal heart structure and sudden cardiac death during physical or emotional stress. Mutations in calsequestrin2, an SR calcium-binding protein that interacts with RyR2, also cause catecholaminergic polymorphic ventricular tachycardia (62, 63). Whether the effect of calsequestrin2 mutations directly or indirectly alters RyR2 function is unknown (Figure 5).
While RyR2 mutations affect residues in multiple functional domains of the calcium channel, those affecting residues involved in calstabin2-binding provide mechanistic insights into the substantial arrhythmias found in affected individuals. Mutations that impair calstabin2-binding may foster calcium leak from the SR and trigger depolarization. Diastolic calcium leak can also affect excitation-contraction coupling and impair systolic contractility.
Studies of mice deficient in FKBP12.6 (64) confirmed the relevance of SR calcium leak from RyR2 to clinically important arrhythmias. RyR2 channel activity in FKBP12.6-null mice was significantly increased compared with that of wild-type mice, consistent with a diastolic Ca2+ leak. Mutant myocytes demonstrated delayed after-depolarizations, and exercise-induced syncope, ventricular arrhythmias, and sudden death were observed in FKBP12.6-null mice.
Calcium dysregulation is also a component of hypertrophic remodeling that occurs in sarcomere gene mutations. Calcium cycling is abnormal early in the pathogenesis of murine HCM (65, 66): SR calcium stores are decreased and calcium-binding proteins and RyR2 levels are diminished. Whether calcium changes contribute to ventricular arrhythmias in mouse and human HCM remains an intriguing question.
Related mechanisms may contribute to ventricular dysfunction and arrhythmias in acquired forms of heart failure, in which chronic phosphorylation of RyR2 reduces calstabin2 levels in the channel macromolecular complex and increases calcium loss from SR stores. These data indicate the potential benefit of therapeutics that improve calstabin2-mediated stabilization of RyR2 (67, 68); such agents may both improve ventricular contractility and suppress arrhythmias in heart failure.
PLN mutations. Rare human PLN mutations cause familial DCM and heart failure (69, 70). The pathogenetic mechanism of one mutation (PLN Arg9Cys) was elucidated through biochemical studies, which indicated unusual PKA interactions that inhibited phosphorylation of mutant and wild-type PLN. The functional consequence of the mutation was predicted to be constitutive inhibition of SERCA2a, a result confirmed in transgenic mice expressing mutant, but not wild-type, PLN protein. In mutant transgenic mice, calcium transients were markedly prolonged, myocyte relaxation was delayed, and these abnormalities were unresponsive to β-adrenergic stimulation. Profound biventricular cardiac dilation and heart failure developed in mutant mice, providing clear evidence of the detrimental effects of protracted SERCA2a inhibition due to excess PLN activity.
The biophysical consequences accounting for DCM in humans who are homozygous for a PLN null mutation (Leu39stop; ref. 70) are less clear. PLN-deficient mice show increased calcium reuptake into the SR and enhanced basal contractility (71). Indeed, these effects on calcium cycling appear to account for the mechanism by which PLN ablation rescues DCM in MLP-null mice (72). However, normal responsiveness to β-adrenergic stimulation is blunted in PLN-deficient myocytes, and cells are less able to recover from acidosis that accompanies vigorous contraction or pathologic states, such as ischemia (73). The collective lesson from human PLN mutations appears to be that too little or too much PLN activity is bad for long-term heart function.
Acquired causes of heart failure are also characterized by a relative decrease in SERCA2a function due to excessive PLN inhibition. Downregulation of β-adrenergic responsiveness attenuates PLN phosphorylation, which compromises calcium reuptake and depletes SR calcium levels, which may impair contractile force and enhance arrhythmias. Heterozygote SERCA2 null mice are a good model of this phenotype and exhibit impaired restoration of SR calcium with deficits in systolic and diastolic function (74).
Cardiac ATP-sensitive potassium channel mutations. In response to stress such as hypoxia and ischemia, myocardial cells undergo considerable changes in metabolism and membrane excitability. Cardiac ATP-sensitive potassium channels (KATP channels) contain a potassium pore and an enzyme complex that participate in decoding metabolic signals to maximize cellular functions during stress adaptation (Figure 4) (75). KATP channels are multimeric proteins containing the inwardly rectifying potassium channel pore (Kir6.2) and the regulatory SUR2A subunit, an ATPase-harboring, ATP-binding cassette protein. Recently, human mutations in the regulatory SUR2A subunit (encoded by ABCC9) were identified as a cause of DCM and heart failure (76). These mutations reduced ATP hydrolytic activities, rendered the channels insensitive to ADP-induced conformations, and affected channel opening and closure. Since KATP-null mouse hearts have impaired response to stress and are susceptible to calcium overload (75), some of the pathophysiology of human KATP mutations (DCM and arrhythmias) may reflect calcium increases triggered by myocyte stress.
Transcriptional Regulators
Investigation of the molecular controls of cardiac gene transcription has led to the identification of many key molecules that orchestrate physiologic expression of proteins involved in force production and transmission, metabolism, and calcium cycling. Given that mutation in the structural proteins involved in these complex processes is sufficient to cause cardiac remodeling, it is surprising that defects in transcriptional regulation of these same proteins have not also been identified as primary causes of heart failure. Several possible explanations may account for this. Transcription factor gene mutations may be lethal or may at least substantially impair reproductive fitness so as to be rapidly lost. The consequences of transcription factor gene mutations may be so pleiotropic that these cause systemic rather than single-organ disease. Changes in protein function (produced by a structural protein mutation) may be more potent for remodeling than changes in levels of structural protein (produced by transcription factor mutation). While many other explanations may be relevant, the few human defects discovered in transcriptional regulators that cause heart failure provide an important opportunity to understand molecular mechanisms for heart failure.
Nkx2.5 mutations. The homeodomain-containing transcription factor Nkx2.5, a vertebrate homolog of the Drosophila homeobox gene tinman, is one of the earliest markers of mesoderm. When Nkx2.5 is deleted in the fly, cardiac development is lost (77). Targeted disruption of Nkx2.5 in mice (Nkx2.5–/–) causes embryonic lethality due to the arrested looping morphogenesis of the heart tube and growth retardation (78, 79). Multiple human dominant Nkx2.5 mutations have been identified as causing primarily structural malformations (atrial and ventricular septation defects) accompanied by atrioventricular conduction delay, although cardiac hypertrophic remodeling has also been observed (80). Although the mechanism for ventricular hypertrophy in humans with Nkx2.5 mutations is not fully understood, the pathology is unlike that found in HCM, which perhaps indicates that cardiac hypertrophy is a compensatory event. Several human Nkx2.5 mutations have been shown to abrogate DNA binding (81), which suggests that the level of functional transcription factor is the principle determinant of structural phenotypes. Heterozygous Nkx2.5+/– mice exhibit only congenital malformations with atrioventricular conduction delay (82, 83). Remarkably, however, transgenic mice expressing Nkx2.5 mutations develop profound cardiac conduction disease and heart failure (84) and exhibit increased sensitivity to doxorubicin-induced apoptosis (85), which suggests that this transcription factor plays an important role in postnatal heart function and stress response.
Insights into transcriptional regulation from mouse genetics. Dissection of the combinatorial mechanisms that activate or repress cardiac gene transcription has led to the identification of several key molecules that directly or indirectly lead to cardiac remodeling. While human mutations in these genes have not been identified, these molecules are excellent candidates for triggering cell responses to structural protein gene mutations.
Hypertrophic remodeling is associated with reexpression of cardiac fetal genes. Molecules that activate this program may also regulate genes that directly cause hypertrophy. Activation of calcineurin (Ca2+/calmodulin-dependent serine/threonine phosphatase) results in dephosphorylation and nuclear translocation of nuclear factor of activated T cells 3 (NFAT3), which, in association with the zinc finger transcription factor GATA4, induces cardiac fetal gene expression. Transgenic mice that express activated calcineurin or NFAT3 in the heart develop profound hypertrophy and progressive decompensation to heart failure (86), responses that were prevented by pharmacologic inhibition of calcineurin. Although these data implicated NFAT signaling in hypertrophic heart failure, pharmacologic inhibition of this pathway fails to prevent hypertrophy caused by sarcomere gene mutations in mice and even accelerates disease progression to heart failure (65). Mice lacking calsarcin-1, which is localized with calcineurin to the Z-disc, showed an increase in Z-disc width, marked activation of the fetal gene program, and exaggerated hypertrophy in response to calcineurin activation or mechanical stress, which suggests that calsarcin-1 plays a critical role in linking mechanical stretch sensor machinery to the calcineurin-dependent hypertrophic pathway (87).
Histone deacetylases (HDACs) are emerging as important regulators of cardiac gene transcription. Class II HDACs (4/5/7/9) bind to the cardiac gene transcription factor MEF2 and inhibit MEF2-target gene expression. Stress-responsive HDAC kinases continue to be identified but may include an important calcium-responsive cardiac protein, calmodulin kinase. Kinase-induced phosphorylation of class II HDACs causes nuclear exit, thereby releasing MEF2 for association with histone acetyltransferase proteins (p300/CBP) and activation of hypertrophic genes. Mice deficient in HDAC9 are sensitized to hypertrophic signals and exhibit stress-dependent cardiac hypertrophy. The discovery that HDAC kinase is stimulated by calcineurin (88) implicates crosstalk between these hypertrophic signaling pathways.
Recent attention has also been focused on Hop, an atypical homeodomain-only protein that lacks DNA-binding activity. Hop is expressed in the developing heart, downstream of Nkx2-5. While its functions are not fully elucidated, Hop can repress serum response factor–mediated (SRF-mediated) transcription. Mice with Hop gene ablation have complex phenotypes. Approximately half of Hop-null embryos succumb during mid-gestation with poorly developed myocardium; some have myocardial rupture and pericardial effusion. Other Hop-null embryos survive to adulthood with apparently normal heart structure and function. Cardiac transgenic overexpression of epitope-tagged Hop causes hypertrophy, possibly by recruitment of class I HDACs that may inhibit anti-hypertrophic gene expression (89–92).
PPARα plays important roles in transcriptional control of metabolic genes, particularly those involved in cardiac fatty acid uptake and oxidation. Mice with cardiac-restricted overexpression of PPARα replicate the phenotype of diabetic cardiomyopathy: hypertrophy, fetal gene activation, and systolic ventricular dysfunction (93). Heterozygous PPARγ-deficient mice, when subjected to pressure overload, developed greater hypertrophic remodeling than wild-type controls, implicating the PPARγ-pathway as a protective mechanism for hypertrophy and heart failure (94).
Retinoid X receptor α (RXRα) is a retinoid-dependent transcriptional regulator that binds DNA as an RXR/retinoic acid receptor (RXR/RAR) heterodimer. RXRα-null mice die during embryogenesis with hypoplasia of the ventricular myocardium. In contrast, overexpression of RXRα in the heart does not rescue myocardial hypoplasia but causes DCM (95).
Integrating Functional and Molecular Signals
Study of human gene mutations that cause HCM and DCM provides information about functional triggers of cardiac remodeling. In parallel with evolving information about molecular-signaling cascades that influence cardiac gene expression, there is considerable opportunity to define precise pathways that cause the heart to fail. To understand the integration of functional triggers with molecular responses, a comprehensive data set of the transcriptional and proteomic profiles associated with precise gene mutations is needed. Despite the plethora of information associated with such studies, bioinformatic assembly of data and deduction of pathways should be feasible and productive for defining shared or distinct responses to signals that cause cardiac remodeling and heart failure. Accrual of this data set in humans is a desirable goal, although confounding clinical variables and tissue acquisition pose considerable difficulties that can be more readily addressed by study of animal models with heart disease. With more knowledge about the pathways involved in HCM and DCM, strategies may emerge to attenuate hypertrophy, reduce myocyte death, and diminish myocardial fibrosis, processes that ultimately cause the heart to fail.
CardioGenomics. Genomics of Cardiovascular Development, Adaptation, and Remodeling.
NHLBI program for genomic applications. Harvard Medical School. http://cardiogenomics.med.harvard.edu
Morita, H, et al. Molecular epidemiology of hypertrophic cardiomyopathy. Cold Spring Harb. Symp. Quant. Biol. 2002. 67:383-388.
Richard, P, et al. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation. 2003. 107:2227-2232
Palmer, BM, et al. Effect of cardiac myosin binding protein-C on mechanoenergetics in mouse myocardium. Circ. Res. 2004. 94:1615-1622.
Harris, SP, et al. Hypertrophic cardiomyopathy in cardiac myosin binding protein-C knockout mice. Circ. Res. 2002. 90:594-601.
Kamisago, M, et al. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N. Engl. J. Med. 2000. 343:1688-1696.
Itoh-Satoh, M, et al. Titin mutations as the molecular basis for dilated cardiomyopathy. Biochem. Biophys. Res. Commun. 2002. 291:385-393.
Gerull, B, et al. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat. Genet. 2004. 36:1162-1164.
Tiso, N, et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum. Mol. Genet. 2001. 10:189-194.
Anan, R, et al. Cardiac involvement in mitochondrial diseases. A study on 17 patients with documented mitochondrial DNA defects. Circulation. 1995. 91:955-961.
Cardiovascular Autonomic Dysfunction and Predicting Outcomes in Diabetes
Marlene Busko Aug 27, 2013 http://www.medscape.com/viewarticle/810063?src=wnl_edit_tpal&uac=62859DN
Autonomic Dysfunction and Risk of a CV Event In patients with CAD and type 2 diabetes, autonomic dysfunction is common, but its prognostic value is unknown.
A.
data a substudy of patients enrolled in the ARTEMIS trial
,530 patients with CAD and diabetes matched with 530 patients with CAD without diabetes. The patients had a mean age of 67, and 69% were males
patients performed a test on an exercise bicycle, which allowed the researchers to determine their heart-rate recovery, defined as the drop in heart rate from the rate at maximal exercise to the rate one minute after stopping the exercise In univariate analysis, among patients with CAD and type 2 diabetes, those who had a blunted heart-rate recovery after exercise–defined as a drop in heart rate of less than 21 beats per minute–had a 1.69-fold greater risk having a cardiovascular event than their peers. Similarly, those with blunted heart-rate turbulence (<3.4 ms/R-R interval) had a 2.08-fold increased risk of an event, and those with low heart-rate variability (<110 ms) had a 1.96-fold greater risk of having a cardiovascular event. After multivariate analysis, C-reactive protein (CRP), but none of the three measures of autonomic function, still predicted an increased risk of having a cardiovascular event during this short follow-up.
During a two-year follow-up, 127 patients (13%) reached the composite end point of a cardiovascular event, which included
- cardiovascular death (2%),
- acute coronary event (8%),
- stroke (3%), or
- hospitalization for heart failure (2%).
B. Autonomic Dysfunction and Risk of Severe Hypoglycemia
Dr Seung-Hyun Ko (Catholic University of Korea, Gyeonggi-do, South Korea
data 894 consecutive patients with type 2 diabetes, aged 25 to 75
heart-rate variability measured at three times: during a Valsalva maneuver, deep breathing, and going from lying down to standing. During close to 10 years of follow-up, 77 episodes of severe hypoglycemia occurred among 62 patients (9.9%). About 16% of patients were diagnosed with early autonomic dysfunction and another 15% were diagnosed with definite autonomic dysfunction. Patients with type 2 diabetes and definite autonomic dysfunction were more than twice as likely to have an episode of severe hypoglycemia as those with normal autonomic function (HR 2.43).
patient education concerning hypoglycemia is essential for patients with definite [cardiovascular autonomic neuropathy] to prevent [severe hypoglycemia] and related mortality
Measurement of heart-rate turbulence (HRT), an ECG phenomenon that reflects hemodynamic responses to premature ventricular contractions (PVCs), can risk-stratify patients in the post-MI setting and may be similarly useful in heart failure or other heart disease, according to a state-of-the-art review in the October 21, 2008 issue of the Journal of the American College of Cardiology [1]. “Several large-scale retrospective and prospective studies have established beyond any doubt that HRT is one of the strongest independent risk predictors after MI. It thus appears that the stage has now been reached when HRT might be used in large prospective intervention studies,” according to the authors, led by Dr Axel Bauer (Deutsches Herzzentrum, Munich, Germany). The group had been asked to write the review by the International Society for Holter and Noninvasive Electrophysiology (ISHNE), it states. HRT, first published as a potential CV risk stratifier in 1999 [2], and other measures of autonomic function aren’t as well established or even studied as much as some other prognostic markers based on electrocardiography, such as T-wave alternans. As the authors note, it’s usually measured from an average of multiple PVCs on 24-hour Holter monitoring.
The strongest support for the parameter’s risk-stratification role comes from “six large-scale studies and from two prospective studies, both of which have been specifically designed to validate the prognostic value of HRT in post-MI patients receiving state-of-the-art treatment,” the report states.
Other evidence suggests a role for HRT evaluation after PCI to assess the strength of perfusion from the treated coronary artery. “Persistent impairment of HRT after PCI in patients with incomplete reperfusion implies prolonged baroreflex impairment and is consistent with poor prognosis,” write Bauer et al. “Thus, early assessment of HRT may be detecting pathological loss of reflex autonomic response due to incomplete reperfusion or severe microvascular dysfunction after PCI. In heart failure, according to the authors, patients “are known to have significantly impaired baroreflex sensitivity as well as reduced heart-rate variability. . . . This may suggest the possibility of guiding pharmacological therapy [according to HRT responses] in heart-failure patients.” They also note that the prognostic power of HRT in heart failure appears limited to patients with ischemic cardiomyopathy.
Bauer A, Malik M, Schmidt G, et al. Heart rate turbulence: Standards of measurement, physiological interpretation, and clinical use. International Society for Holter and Noninvasive Electrophysiology consensus. J Am Coll Cardiol 2008; 52:1353–1365.
http://dx.doi.org/10.1016/j.jacc.2008.07.041
Schmidt G, Malik M, Barthel P, et al. Heart-rate turbulence after ventricular premature beats as a predictor of mortality after acute myocardial infarction. Lancet 1999; 353:1390–1396. Abstract
http://www.medscape.com/viewarticle/582091
Read Full Post »












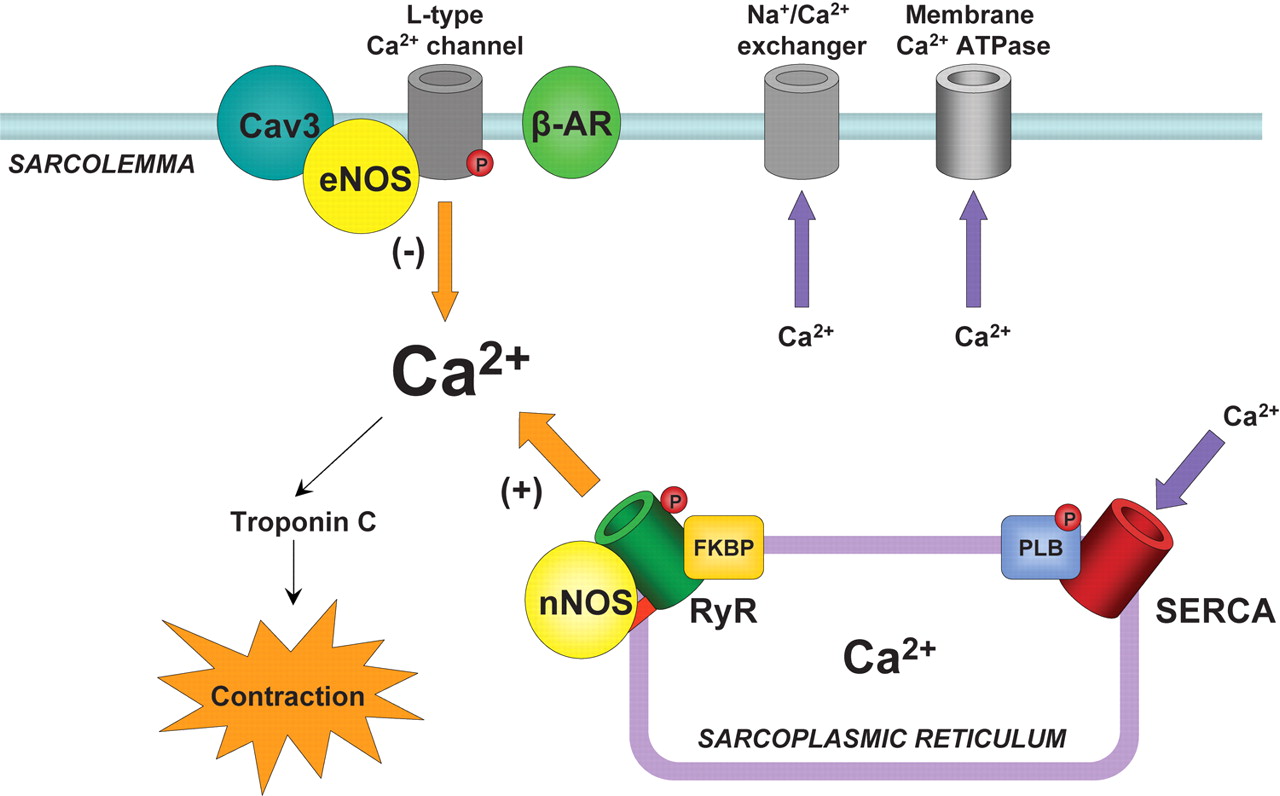


















![[low asterisk]](https://i0.wp.com/www.ncbi.nlm.nih.gov/corehtml/pmc/pmcents/x204E.gif?w=500) and
and 







 Carmat’s bioprosthetic device is designed to replace the real heart for as much as five years, mimicking nature’s work using biological materials and sensors. (Benoit Tessier/Reuters)
Carmat’s bioprosthetic device is designed to replace the real heart for as much as five years, mimicking nature’s work using biological materials and sensors. (Benoit Tessier/Reuters) PARIS (Reuters) – France’s Carmat said on Friday it had carried out the first human implantation of its artificial heart.The operation, performed on Wednesday at the Georges Pompidou European Hospital in Paris, went smoothly, Carmat said in a statement, adding that the patient was being monitored in the intensive care unit but was awake and talking.(Reporting by Natalie Huet; editing by Mark John)
PARIS (Reuters) – France’s Carmat said on Friday it had carried out the first human implantation of its artificial heart.The operation, performed on Wednesday at the Georges Pompidou European Hospital in Paris, went smoothly, Carmat said in a statement, adding that the patient was being monitored in the intensive care unit but was awake and talking.(Reporting by Natalie Huet; editing by Mark John)































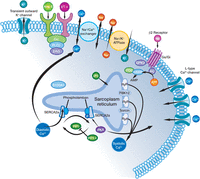
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}