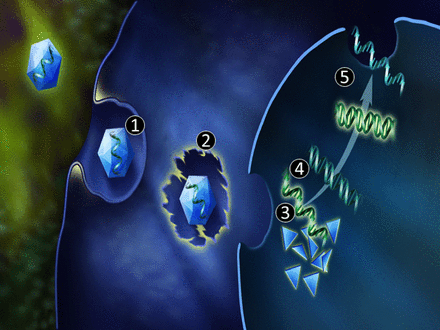
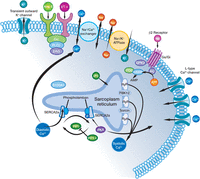
Although many pathways are associated with cardiomyopathy, up-regulation of transcription and induction of apoptosis are major mediators of pathogenic responses in the heart. The GPCR-associated pathway (dark red) can be activated by ET-1 and AngII, which are released in response to reduced contractility, and mediates contractile adaptation through increased calcium release from the sarcoplasmic reticulum. Increased intracellular calcium activates calmodulin and induces activation of the transcription factor MEF2. Incorporation into the sarcomere of mutant proteins that exhibit reduced ATP efficiency inhibits the sequestration of calcium from the cytosol and further enhances increases in intracellular calcium concentration. GPCR signaling is also associated with activation of the Akt signaling pathway (light green) that induces fetal gene expression and the cardiac hypertrophic response through inhibition of GSK3β. Apoptotic pathways (light blue) are induced by cytochrome c (CytC) release from mitochondria and activation of death receptors (like FasR) by cytokines such as TNF. Calcium overload and myocyte loss significantly contribute to reduced contractility in many forms of cardiomyopathy. ET-1, endothelin-1; HDAC, histone deacetylase; NFAT, nuclear factor of activated T cells; MEF-2, myocyte enhancer factor 2; SERCA, sarco/endoplasmic reticulum calcium-ATPase; cFLIP, cellular FLICE-inhibitory protein; AngII, angiotensin II; FasR, Fas receptor.
http://jcb.rupress.org/content/194/3/355.full
For Disruption of Calcium Homeostasis in Cardiomyocyte Cells, see
Part VI: Calcium Cycling (ATPase Pump) in Cardiac Gene Therapy: Inhalable Gene Therapy for Pulmonary Arterial Hypertension and Percutaneous Intra-coronary Artery Infusion for Heart Failure: Contributions by Roger J. Hajjar, MD
Aviva Lev-Ari, PhD, RN
http://pharmaceuticalintelligence.com/2013/08/01/calcium-molecule-in-cardiac-gene-therapy-inhalable-gene-therapy-for-pulmonary-arterial-hypertension-and-percutaneous-intra-coronary-artery-infusion-for-heart-failure-contributions-by-roger-j-hajjar/
Part VII: Cardiac Contractility & Myocardium Performance: Ventricular Arrhythmias and Non-ischemic Heart Failure – Therapeutic Implications for Cardiomyocyte Ryanopathy (Calcium Release-related Contractile Dysfunction) and Catecholamine Responses
Justin Pearlman, MD, PhD, FACC, Larry H Bernstein, MD, FCAP and Aviva Lev-Ari, PhD, RN
http://pharmaceuticalintelligence.com/2013/08/28/cardiac-contractility-myocardium-performance-ventricular-arrhythmias-and-non-ischemic-heart-failure-therapeutic-implications-for-cardiomyocyte-ryanopathy-calcium-release-related-contractile/
Section Three
The Calcium Sensor: How Calcium Ions Regulate the fusion of vesicles with cell membranes during Neurotransmission
This topic is covered in
Synaptotagmin functions as a Calcium Sensor: How Calcium Ions Regulate the fusion of vesicles with cell membranes during Neurotransmission
Larry H Bernstein, MD, FCAP and Aviva Lev-Ari, PhD, RN
http://pharmaceuticalintelligence.com/2013/09/10/synaptotagmin-functions-as-a-calcium-sensor-how-calcium-ions-regulate-the-fusion-of-vesicles-with-cell-membranes-during-neurotransmission/
Justin D Pearlman, MD, PhD, FACC PENDING
REFERENCES
Vascular Smooth Muscle Cells
Amberg GC, Navedo MF, Nieves-Cintr ´on M, Molkentin JD &
Santana LF (2007). Calcium sparklets regulate local and
global calcium in murine arterial smooth muscle. J Physiol
579, 187–201.
Aoyama M, Yamada A,Wang J, Ohya S, Furuzono S, Goto T,
Hotta S, Ito Y, Matsubara T, Shimokata K, Chen SRW,
Imaizumi Y & Nakayama S (2004). Requirement of
ryanodine receptors for pacemaker Ca2+ activity in ICC and
HEK293 cells. J Cell Sci 117, 2813–2825.
Berridge MJ (2007). Inositol trisphosphate and calcium
oscillations. Biochem Soc Symp 74, 1–7.
Berridge MJ (2008). Cell Signalling Biology. Portland Press
Limited (www.cellsignallingbiology.org).
Berridge MJ, Bootman MD & Roderick HL (2003). Calcium
signalling: Dynamics, homeostasis and remodelling. Nat Rev
Mol Cell Biol 4, 517–529.
Berridge MJ & Dupont G (1994). Spatial and temporal
signalling by calcium. Curr Opin Cell Biol 6, 267–274.
Blanks AM, Zhao Z-H, Shmygol A, Bru-Mercier G, Astle S &
Thornton S (2007). Characterization of the molecular and
electrophysiological properties of the T-type calcium
channel in human myometrium. J Physiol 581, 915–926.
Bradley KN, Currie S, MacMillan D, Muir TC & McCarron JG
(2003). Cyclic ADP-ribose inceases Ca2+ removal in smooth
muscle. J Cell Sci 116, 4291–4306.
Bradley E, HollywoodMA, Johnston L, Large RJ, Matsuda T,
Baba A, McHale NG, Thornbury KD & Sergeant GP (2006).
Contribution of reverse Na+–Ca2+ exchange to spontaneous
activity in interstitial cells of Cajal in the rabbit urethra.
J Physiol 574, 651–661.
Brain KL, Cuprian AM,Williams DJ & Cunnane TC (2003).
The sources and sequestration of Ca2+ transients in the
mouse vas deferens. J Physiol 553, 627–635.
Brain KL, Jackson MJ, Trout SJ & Cunnane TC (2002).
Intermittent ATP release from nerve terminals elicits focal
smooth muscle Ca2+ transients in mouse vas deferens.
J Physiol 541, 849–862.
Brown A, Cornwell T, Korniyenko I, Solodushko V, Bond CT,
Adelman JP & Taylor MS (2007). Myometrial expression of
small conductance Ca2+-activated K+ channels depresses
phasic uterine contractions. Am J Physiol Cell Physiol 292,
C832–C840.
Burdyga T &Wray S (2005). Action potential refractory period
in ureter smooth muscle is set by Ca sparks and BK channels.
Nature 436, 559–562.
Collier ML, Ji G,Wang Y-X & Kotlikoff MI (2000).
Calcium-induced calcium release in smooth muscle. Loose
coupling between the action potential and calcium release.
J Gen Physiol 115, 653–662.
Craven M, Sergeant GP, Hollywood MA, McHale NG &
Thornbury KD (2004). Modulation of spontaneous
Ca2+-activated Cl− currents in the rabbit corpus cavernosum
by the nitric oxide-cGMP pathway. J Physiol 556, 495–506.
Dai JM, Kuo K-H, Leo JM, van Breemen C & Lee C-H (2006).
Mechanism of ACh-induced asynchronous calcium waves
and tonic contraction in porcine tracheal muscle bundle.
Am J Physiol Lung Cell Mol Physiol 290, L459–L469.
Deshpande DA,White TA, Dogan S,Walseth TF, Panettieri RA
& Kanna MS (2005). CD38/cyclic ADP-ribose signalling: role
in the regulation of calcium homeostasis in airway smooth
muscle. Am J Physiol Lung Cell Mol Physiol 288, L733–L788.
Essin K,Welling A, Hofmann F, Luft FC, Gollasch M &
Moosmang S (2007). Indirect coupling between CaV1.2
channels and ryanodine receptors to generate Ca2+ sparks in
murine arterial smooth muscle cells. J Physiol 584, 205–219.
Haddock RE & Hill CE (2002). Differential activation of ion
channels by inositol 1,4,5-trisphosphate (IP3)- and
ryanodine-sensitive calcium stores in rat basilar artery
vasomotion. J Physiol 545, 615–627.
C2008 The Author. Journal compilation C 2008 The Physiological Society
Downloaded from J Physiol (jp.physoc.org) by guest on September 12, 2013
5060 M. J. Berridge J Physiol 586.21
Haddock RE & Hill CE (2005). Rhythmicity in arterial smooth
muscle. J Physiol 566, 645–656.
Hashitani H, Fukuta H, Takano H, Klemm MF & Suzuki H
(2001). Origin and propagation of spontaneous excitation in
smooth muscle of the guinea-pig urinary bladder. J Physiol
530, 273–286.
Hashitani H (2006). Interaction between interstitial cells and
smooth muscles in the lower urinary tract and penis.
J Physiol 576, 707–714.
Hashitani H & Brading AF (2003a). Electrical properties of
detrusor smooth muscles from the pig and human urinary
bladder. Br J Pharmacol 140, 146–158.
Hashitani H & Brading AF (2003b). Ionic basis for the
regulation of spontaneous excitation in detrusor smooth
muscle cells of the guinea-pig urinary bladder.
Br J Pharmacol 140, 159–169.
Hashitani H, Brading AF & Suzuki H (2004). Correlation
between spontaneous electrical, calcium and mechanical
activity of detrusor smooth muscle of the guinea-pig
bladder. Br J Pharmacol 141, 183–193.
Hashitani H, Bramich NJ & Hirst GDS (2000). Mechanisms of
excitatory neuromuscular transmission in the guinea-pig
urinary bladder. J Physiol 524, 565–579.
Hashitani H & Suzuki H (2007). Properties of spontaneous
Ca2+ transients recorded from interstitial cells of Cajal-like
cells of the rabbit urethra in situ. J Physiol 583, 505–519.
Hashitani H, Yanai Y, Shirasawa N, Soji T, Tomita A, Kohri K &
Suzuki H (2005). Interaction between spontaneous and
neurally mediated regulation of smooth muscle cell tone in
the rabbit corpus cavernosum. J Physiol 569, 723–735.
Heppner TJ, Bonev AD & Nelson MT (2005). Elementary
purinergic Ca2+ transients evoked by nerve stimulation in
rat urinary bladder smooth muscle. J Physiol 564, 201–212.
Hirst GDS &Ward SM (2003). Interstitial cells: involvement in
rhythmicity and neural control of gut smooth muscle.
J Physiol 550, 337–346.
Hotta S, Morimura K, Ohya S, Muraki K, Takeshima H &
Imaizumi Y (2007). Ryanodine receptor type 2 deficiency
changes excitation–contraction coupling and membrane
potential in urinary bladder smooth muscle. J Physiol 582,
489–506.
Iino M, Kasai H & Yamazawa T (1994). Visualization of neural
control of intracellular Ca2+ concentration in single vascular
smooth muscle cells in situ. EMBO J 13, 5026–5031.
Imtiaz MS, Katnik CP, Smith DW& van Helden DF (2006).
Role of voltage-dependent modulation of store Ca2+ release
in synchronization of Ca2+ oscillations. Biophys J 90, 1–23.
Imtiaz MS, Zhao J, Hosaka K, von derWeid P-Y, Crowe M &
van Helden DF (2007). Pacemaking through Ca2+ stores
interacting as coupled oscillators via membrane
depolarization. Biophys J 92, 3843–3861.
Johnstone L, Sergeant GP, Hollywood MA, Thornbury KD &
McHale NG (2005). Calcium oscillations in interstitial cells
of the rabbit urethra. J Physiol 565, 449–461.
Kim YC, Koh SD & Sanders KM (2002). Voltage-dependent
inward currents of intestinal cells of Cajal from murine colon
and small intestine. J Physiol 541, 797–810.
Kito Y & Suzuki H (2003). Properties of pacemaker potentials
recorded from myenteric interstitial cells of Cajal distributed
in the mouse small intestine. J Physiol 553, 803–818.
Kito Y,Ward SM & Sanders KM (2005). Pacemaker potentials
generated by interstitial cells of Cajal in the murine intestine.
Am J Physiol Cell Physiol 288, C710–C720.
Komuro T (2006). Structure and organization of interstitial
cells of Cajal in the gastrointestinal tract. J Physiol 576,
653–658.
Kuo K-H, Dai J, Seow CY, Lee C-H & van Breemen C (2003).
Relationship between asynchronous Ca2+ waves and force
development in intact smooth muscle bundles of the porcine
trachea. Am J Physiol Lung Cell Mol Physiol 285,
L1345–L1353.
Kupittayanant S, Luckas MJM &Wray S (2002). Effect of
inhibiting the sarcoplasmic reticulum on spontaneous and
oxytocin-induced contractions of human myometrium.
Br J Obstet Gynaec 109, 289–296.
Lamboley M, Schuster A, B´eny J-L & Meister J-J (2003).
Recruitment of smooth muscle cells and arterial vasomotion.
Am J Physiol Heart Circ Physiol 285, H562–H569.
Lamont C, Vainorius E &WierWG (2003). Purinergic and
adrenergic Ca2+ transients during neurogenic contractions
of rat mesenteric small arteries. J Physiol 549, 801–808.
Lamont C &WierWG (2002). Evoked and spontaneous
purinergic junctional Ca2+ transients (jCaTs) in rat small
arteries. Circ Res 91, 454–456.
Lang RJ, Hashitani H, Tonta MA, Parkington HC & Suzuki H
(2007). Spontaneous electrical and Ca2+ signals in typical
and atypical smooth muscle cells and interstitial cell of
Cajal-like cells of mouse renal pelvis. J Physiol 583,
1049–1068.
Lee C-H, Poburko D, Sahota P, Sandhu J, Ruehlmann DO &
van Breemen C (2001). The mechanism of
phenylephrine-mediated [Ca2+]i oscillations underlying
tonic contraction in the rabbit inferior vena cava. J Physiol
534, 641–650.
Liu X & Farley JM (1996). Acetylcholine-induced chloride
current oscillations in swine tracheal smooth muscle cells.
J Pharmacol Exp Ther 276, 178–186.
McCarron JG, MacMillan D, Bradley KN, Chalmers S & Muir
TC (2004). Origin and mechanisms of Ca2+ waves in smooth
muscle as revealed by localized photolysis of caged inositol
1,4,5-trisphosphate. J Biol Chem 279, 8417–8427.
McHale NG, Hollywood MA, Sergeant GP, Shafei M,
Thornbury KT &Ward SM (2006). Organization and
function of ICC in the urinary tract. J Physiol 576, 689–694.
Mauban JRH, Lamont C, Balke CW&WierWG (2001).
Adrenergic stimulation of rat resistance arteries affects Ca2+
sparks, Ca2+ waves, and Ca2+ oscillations. Am J Physiol Heart
Circ Physiol 280, H2399–H2405.
Meredith AL, Thorneloe KS,WernerME, Nelson MT & Aldrich
RW(2004). Overactive bladder and incontinence in the
absence of the BK large conductance Ca2+-activated K+
channel. J Biol Chem 279, 36746–36752.
Morimura K, Ohi Y, Yamamura H, Ohya S, Muraki K &
Imaizumi Y (2005). Two-step Ca2+ intracellular release
underlies excitation-contraction coupling in mouse urinary
bladder myocytes. Am J Physiol Cell Physiol 290, C388–C403.
Mulryan K, Gitterman DP, Lewis CJ, Vial C, Leckie BJ, Cobb
AL, Brown JE, Conley EC, Buell G, Pritchard CA & Evans RJ
(2000). Reduced vas deferens contraction and male
infertility in mice lacking P2X1 receptors. Nature 403, 86–89.
C2008 The Author. Journal compilation C 2008 The Physiological Society
Downloaded from J Physiol (jp.physoc.org) by guest on September 12, 2013
J Physiol 586.21 Smooth muscle cell calcium activation mechanisms 5061
Nakao K, Inoue Y, Okabe K, Kawarabayashi T & Kitamura K
(1997). Oxytocin enhances action potentials in pregnant
human myometrium – a study with microelectrodes.
Am J Obstet Gynecol 177, 222–228.
Ohi Y, Yamamura H, Nagano N, Ohya S, Muraki K,Watanabe
M & Imaizumi Y (2001). Local Ca2+ transients and
distribution of BK channels and ryanodine receptors in
smooth muscle cells of guinea-pig vas deferens and urinary
bladder. J Physiol 534, 313–326.
Park KJ, Hennig GW, Lee H-T, Spencer NJ,Ward SM, Sith TK
& Sanders KM (2006). Spatial and temporal mapping of
pacemaker activity in interstitial cells of Cajal in mouse
ileum in situ. Am J Physiol Cell Physiol 290,
C1411–C1427.
Peng H, Matchkov V, Ivarsen A, Aalkjaer C & Nilsson H
(2001). Hypothesis for the initiation of vasomotion. Circ Res
88, 810–815.
Peppiatt-Wildman CM, Albert AP, Saleh SN & LargeWA
(2007). Endothelin-1 activates a Ca2+-permeable cation
channel with TRPC3 and TRPC7 properties in rabbit
coronary artery myocytes. J Physiol 580,
755–764.
Perez JF & Sanderson MJ (2005a). The frequency of calcium
oscillations induced by 5-HT, ACH, and KCl determine the
contraction of smooth muscle cells of intrapulmonary
bronchioles. J Gen Physiol 125, 535–553.
Perez JF & Sanderson MJ (2005b). The contraction of smooth
muscle cells of intrapulmonary arterioles is determined by
the frequency of Ca2+ oscillations induced by 5-HT and KCl.
J Gen Physiol 125, 555–567.
Rebolledo A, Speroni F, Raingo J, Salemme SV, Tanzi F, Munin
V, A˜n´on MC & Milesi V (2006). The Na+/Ca2+ exchanger is
active and working in the reverse mode in human umbilical
artery smooth muscle. Biochem Biophys Res Commun 339,
840–845.
Saleh SN, Albert AP, Peppiatt-Wildman CM & LargeWA
(2008). Diverse properties of store-operated TRPC channels
activated by protein kinase C in vascular myocytes. J Physiol
586, 2463–2476.
Sanders KM, Koy SD &Ward SM (2006). Interstitial cells of
Cajal as pacemakers in the gastrointestinal tract. Annu Rev
Physiol 68, 307–343.
Sanderson MJ, Delmotte P, Bai Y & Perez-Zogbhi JF (2008).
Regulation of airway SMC contractility by Ca2+ signaling
and sensitivity. Proc Am Thorac Soc 5, 23–31.
Sergeant GP, Hollywood MA, McCloskey KD, Thornbury KD
& McHale NG (2000). Specialised pacemaking cells in the
rabbit urethra. J Physiol 526, 359–366.
Sergeant GP, Hollywood MA, McHale NG & Thornbury KD
(2006a). Ca2+ signalling in urethral interstitial cells of Cajal.
J Physiol 576, 715–720.
Sergeant GP, Johnston L, McHale NG, Thornbury KD &
Hollywood MA (2006b). Activation of the cGMP/PKG
pathway inhibits electrical activity in rabbit urethral
interstitial cells of Cajal by reducing the spatial spread of
Ca2+ waves. J Physiol 574, 167–181.
Shaw L, O’Neill S, Jones CJP, Austin C & Taggart MJ (2004).
Comparison of U46619-, endothelin-1- or
phenylephrine-induced changes in cellular Ca2+ profiles and
Ca2+ sensitization of constriction of pressurised rat
resistance arteries. Br J Pharm 141, 678–688.
Shmygol A, Blanks AM, Bru-Mercier G, Gullam JE & Thornton
S (2007). Control of uterine Ca2+ by membrane voltage:
Toward understanding the excitation-contraction coupling
in human myometrium. Ann N Y Acad Sci 1101, 97–109.
Somlyo AP & Somlyo AV (2003). Ca2+ sensitivity of smooth
muscle and nonmuscle myosin II: modulated by G proteins,
kinases, and myosin phosphatase. Physiol Rev 83, 1325–1358.
van Helden DF & Imtiaz MS (2003). Ca2+ phase waves: a basis
for cellular pacemaking and long-range synchronicity in the
guinea-pig gastric pylorus. J Physiol 548, 271–296.
Wang H, Eto M, SteersWD, Somlyo AP & Somlyo AV (2002).
RhoA-mediated Ca2+ sensitization in erectile function. J Biol
Chem 277, 30614–30621.
Ward SM & Sanders KM (2006). Involvement of intramuscular
interstitial cells of Cajal in neuroeffector transmission in the
gastrointestinal tract. J Physiol 576, 675–682.
White C & McGeown JG (2003). Inositol 1,4,5-trisphosphate
receptors modulate Ca2+ sparks and Ca2+ store content in
vas deferens myocytes. Am J Physiol Cell Physiol 285,
C195–C204.
Wray S (2007). Insights into the uterus. Exp Physiol 92,
621–631.
Wray S, Burdyga T & Noble K (2005). Calcium signalling in
smooth muscle. Cell Calcium 38, 397–407.
Wray S & Noble K (2008). Sex hormones and excitationcontraction
coupling in the uterus: The effects of oestrus and
hormones. J Neuroendocrinol 20, 451–461.
Wray S & Shmygol A (2007). Role of the calcium store in
uterine contractility. Semin Cell Dev Biol 18, 315–320.
Yamazawa T & Iino M (2002). Simultaneous imaging of Ca2+
signals in interstitial cells of Cajal and longitudinal smooth
muscle cells during rhythmic activity in mouse ileum.
J Physiol 538, 823–835.
Young RC (2007). Myocytes, myometrium, and uterine
contraction. Ann N Y Acad Sci 1101, 72–84.
C2008 The Author. Journal compilation C 2008 The Physiological Society
Downloaded from JCB
Cardiomyocytes Cells
. ↵ Adams, J.W., D.S. Migita, M.K. Yu, R. Young, M.S. Hellickson, F.E. Castro-Vargas, J.D. Domingo, P.H. Lee, J.S. Bui, S.A. Henderson. 1996. Prostaglandin F2 alpha stimulates hypertrophic growth of cultured neonatal rat ventricular myocytes. J. Biol. Chem. 271:1179–1186. doi:10.1074/jbc.271.2.1179 Abstract/FREE Full Text
. ↵ Akyürek, O., N. Akyürek, T. Sayin, I. Dinçer, B. Berkalp, G. Akyol, M. Ozenci, D. Oral. 2001. Association between the severity of heart failure and the susceptibility of myocytes to apoptosis in patients with idiopathic dilated cardiomyopathy. Int. J. Cardiol. 80:29–36. doi:10.1016/S0167-5273(01)00451-X CrossRefMedline
. ↵ Ashrafian, H., M.P. Frenneaux. 2007. Metabolic modulation in heart failure: the coming of age. Cardiovasc. Drugs Ther. 21:5–7. doi:10.1007/s10557-007-6000-z CrossRefMedline
. ↵ Basso, C., D. Corrado, F.I. Marcus, A. Nava, G. Thiene. 2009. Arrhythmogenic right ventricular cardiomyopathy. Lancet. 373:1289–1300. doi:10.1016/S0140-6736(09)60256-7 CrossRefMedline
. ↵ Berko, B.A., M. Swift. 1987. X-linked dilated cardiomyopathy. N. Engl. J. Med. 316:1186–1191. doi:10.1056/NEJM198705073161904 Medline
. ↵ Buvoli, M., M. Hamady, L.A. Leinwand, R. Knight. 2008. Bioinformatics assessment of beta-myosin mutations reveals myosin’s high sensitivity to mutations. Trends Cardiovasc. Med. 18:141–149. doi:10.1016/j.tcm.2008.04.001 CrossRefMedline
. ↵ Bybee, K.A., T. Kara, A. Prasad, A. Lerman, G.W. Barsness, R.S. Wright, C.S. Rihal. 2004. Systematic review: transient left ventricular apical ballooning: a syndrome that mimics ST-segment elevation myocardial infarction. Ann. Intern. Med. 141:858–865. Abstract/FREE Full Text
. ↵ Chin, T.K., J.K. Perloff, R.G. Williams, K. Jue, R. Mohrmann. 1990. Isolated noncompaction of left ventricular myocardium. A study of eight cases. Circulation. 82:507–513. doi:10.1161/01.CIR.82.2.507 Abstract/FREE Full Text
. ↵ Communal, C., K. Singh, D.R. Pimentel, W.S. Colucci. 1998. Norepinephrine stimulates apoptosis in adult rat ventricular myocytes by activation of the beta-adrenergic pathway. Circulation. 98:1329–1334. Abstract/FREE Full Text
. ↵ Corrado, D., C. Basso, G. Thiene, W.J. McKenna, M.J. Davies, F. Fontaliran, A. Nava, F. Silvestri, C. Blomstrom-Lundqvist, E.K. Wlodarska, et al. 1997. Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: a multicenter study. J. Am. Coll. Cardiol. 30:1512–1520. doi:10.1016/S0735-1097(97)00332-X Abstract
. ↵ Cregler, L.L. 1989. Progression from hypertrophic cardiomyopathy to dilated cardiomyopathy. J. Natl. Med. Assoc. 81:820: 824–826. Search Google Scholar
. ↵ D’Angelo, D.D., Y. Sakata, J.N. Lorenz, G.P. Boivin, R.A. Walsh, S.B. Liggett, G.W. Dorn II. 1997. Transgenic Galphaq overexpression induces cardiac contractile failure in mice. Proc. Natl. Acad. Sci. USA. 94:8121–8126. doi:10.1073/pnas.94.15.8121 Abstract/FREE Full Text
. ↵ Dávila-Román, V.G., G. Vedala, P. Herrero, L. de las Fuentes, J.G. Rogers, D.P. Kelly, R.J. Gropler. 2002. Altered myocardial fatty acid and glucose metabolism in idiopathic dilated cardiomyopathy. J. Am. Coll. Cardiol. 40:271–277. doi:10.1016/S0735-1097(02)01967-8 Abstract/FREE Full Text
. ↵ Davis, J., H. Wen, T. Edwards, J.M. Metzger. 2007. Thin filament disinhibition by restrictive cardiomyopathy mutant R193H troponin I induces Ca2+-independent mechanical tone and acute myocyte remodeling. Circ. Res. 100:1494–1502. doi:10.1161/01.RES.0000268412.34364.50 Abstract/FREE Full Text
. ↵ de Jonge, H.W., D.H. Dekkers, B.C. Tilly, J.M. Lamers. 2002. Cyclic stretch and endothelin-1 mediated activation of chloride channels in cultured neonatal rat ventricular myocytes. Clin. Sci. 103(Suppl 48):148S–151S. Medline
. ↵ Deinum, J., J.M. van Gool, M.J. Kofflard, F.J. ten Cate, A.H. Danser. 2001. Angiotensin II type 2 receptors and cardiac hypertrophy in women with hypertrophic cardiomyopathy. Hypertension. 38:1278–1281. doi:10.1161/hy1101.096114 Abstract/FREE Full Text
. ↵ Dobrin, J.S., D. Lebeche. 2010. Diabetic cardiomyopathy: signaling defects and therapeutic approaches. Expert Rev. Cardiovasc. Ther. 8:373–391. doi:10.1586/erc.10.17 CrossRefMedline
. ↵ Dolci, A., R. Dominici, D. Cardinale, M.T. Sandri, M. Panteghini. 2008. Biochemical markers for prediction of chemotherapy-induced cardiotoxicity: systematic review of the literature and recommendations for use. Am. J. Clin. Pathol. 130:688–695. doi:10.1309/AJCPB66LRIIVMQDR Abstract/FREE Full Text
. ↵ Edwards, B.S., R.S. Zimmerman, T.R. Schwab, D.M. Heublein, J.C. Burnett Jr. 1988. Atrial stretch, not pressure, is the principal determinant controlling the acute release of atrial natriuretic factor. Circ. Res. 62:191–195. Abstract/FREE Full Text
. ↵ Esposito, G., S.V. Prasad, A. Rapacciuolo, L. Mao, W.J. Koch, H.A. Rockman. 2001. Cardiac overexpression of a G(q) inhibitor blocks induction of extracellular signal-regulated kinase and c-Jun NH(2)-terminal kinase activity in in vivo pressure overload. Circulation. 103:1453–1458. Abstract/FREE Full Text
. ↵ Flavigny, J., M. Souchet, P. Sébillon, I. Berrebi-Bertrand, B. Hainque, A. Mallet, A. Bril, K. Schwartz, L. Carrier. 1999. COOH-terminal truncated cardiac myosin-binding protein C mutants resulting from familial hypertrophic cardiomyopathy mutations exhibit altered expression and/or incorporation in fetal rat cardiomyocytes. J. Mol. Biol. 294:443–456. doi:10.1006/jmbi.1999.3276 CrossRefMedline
. ↵ Force, T., R. Hajjar, F. Del Monte, A. Rosenzweig, G. Choukroun. 1999. Signaling pathways mediating the response to hypertrophic stress in the heart. Gene Expr. 7:337–348. Medline
. ↵ Freedom, R.M., S.J. Yoo, D. Perrin, G. Taylor, S. Petersen, R.H. Anderson. 2005. The morphological spectrum of ventricular noncompaction. Cardiol. Young. 15:345–364. doi:10.1017/S1047951105000752 CrossRefMedline
. ↵ Frey, N., E.N. Olson. 2003. Cardiac hypertrophy: the good, the bad, and the ugly. Annu. Rev. Physiol. 65:45–79. doi:10.1146/annurev.physiol.65.092101.142243 CrossRefMedline
. ↵ Geng, Y.J., Y. Ishikawa, D.E. Vatner, T.E. Wagner, S.P. Bishop, S.F. Vatner, C.J. Homcy. 1999. Apoptosis of cardiac myocytes in Gsalpha transgenic mice. Circ. Res. 84:34–42. Abstract/FREE Full Text
. ↵ Gill, C., R. Mestril, A. Samali. 2002. Losing heart: the role of apoptosis in heart disease—a novel therapeutic target? FASEB J. 16:135–146. doi:10.1096/fj.01-0629com Abstract/FREE Full Text
. ↵ Grogan, M., M.M. Redfield, K.R. Bailey, G.S. Reeder, B.J. Gersh, W.D. Edwards, R.J. Rodeheffer. 1995. Long-term outcome of patients with biopsy-proved myocarditis: comparison with idiopathic dilated cardiomyopathy. J. Am. Coll. Cardiol. 26:80–84. doi:10.1016/0735-1097(95)00148-S Abstract
. ↵ Gupta, A., N.S. Aberle II, J. Ren, A.C. Sharma. 2005. Endothelin-converting enzyme-1-mediated signaling in adult rat ventricular myocyte contractility and apoptosis during sepsis. J. Mol. Cell. Cardiol. 38:527–537. doi:10.1016/j.yjmcc.2005.01.002 CrossRefMedline
. ↵ Haq, S., G. Choukroun, Z.B. Kang, H. Ranu, T. Matsui, A. Rosenzweig, J.D. Molkentin, A. Alessandrini, J. Woodgett, R. Hajjar, et al. 2000. Glycogen synthase kinase-3beta is a negative regulator of cardiomyocyte hypertrophy. J. Cell Biol. 151:117–130. doi:10.1083/jcb.151.1.117 Abstract/FREE Full Text
. ↵ Heineke, J., J.D. Molkentin. 2006. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat. Rev. Mol. Cell Biol. 7:589–600. doi:10.1038/nrm1983 CrossRefMedline
. ↵ Herron, T.J., K.S. McDonald. 2002. Small amounts of alpha-myosin heavy chain isoform expression significantly increase power output of rat cardiac myocyte fragments. Circ. Res. 90:1150–1152. doi:10.1161/01.RES.0000022879.57270.11 Abstract/FREE Full Text
. ↵ Herron, T.J., R. Vandenboom, E. Fomicheva, L. Mundada, T. Edwards, J.M. Metzger. 2007. Calcium-independent negative inotropy by beta-myosin heavy chain gene transfer in cardiac myocytes. Circ. Res. 100:1182–1190. doi:10.1161/01.RES.0000264102.00706.4e Abstract/FREE Full Text
. ↵ Hoogerwaard, E.M., P.A. van der Wouw, A.A. Wilde, E. Bakker, P.F. Ippel, J.C. Oosterwijk, D.F. Majoor-Krakauer, A.J. van Essen, N.J. Leschot, M. de Visser. 1999. Cardiac involvement in carriers of Duchenne and Becker muscular dystrophy. Neuromuscul. Disord. 9:347–351. doi:10.1016/S0960-8966(99)00018-8 CrossRefMedline
. ↵ Huang, X.P., J.F. Du. 2004. Troponin I, cardiac diastolic dysfunction and restrictive cardiomyopathy. Acta Pharmacol. Sin. 25:1569–1575. Medline
. ↵ Huang, Y., R.P. Hickey, J.L. Yeh, D. Liu, A. Dadak, L.H. Young, R.S. Johnson, F.J. Giordano. 2004. Cardiac myocyte-specific HIF-1alpha deletion alters vascularization, energy availability, calcium flux, and contractility in the normoxic heart. FASEB J. 18:1138–1140. doi:10.1096/fj.03-1377com Abstract/FREE Full Text
. ↵ Iacovoni, A., R. De Maria, A. Gavazzi. 2010. Alcoholic cardiomyopathy. J. Cardiovasc. Med. (Hagerstown). 11:884–892. doi:10.2459/JCM.0b013e32833833a3 CrossRefMedline
. ↵ Ingwall, J.S., R.G. Weiss. 2004. Is the failing heart energy starved? On using chemical energy to support cardiac function. Circ. Res. 95:135–145. doi:10.1161/01.RES.0000137170.41939.d9 Abstract/FREE Full Text
. ↵ Iwai-Kanai, E., K. Hasegawa, M. Araki, T. Kakita, T. Morimoto, S. Sasayama. 1999. alpha- and beta-adrenergic pathways differentially regulate cell type-specific apoptosis in rat cardiac myocytes. Circulation. 100:305–311. Abstract/FREE Full Text
. ↵ Kalsi, K.K., R.T. Smolenski, R.D. Pritchard, A. Khaghani, A.M. Seymour, M.H. Yacoub. 1999. Energetics and function of the failing human heart with dilated or hypertrophic cardiomyopathy. Eur. J. Clin. Invest. 29:469–477. doi:10.1046/j.1365-2362.1999.00468.x CrossRefMedline
. ↵ Kamisago, M., S.D. Sharma, S.R. DePalma, S. Solomon, P. Sharma, B. McDonough, L. Smoot, M.P. Mullen, P.K. Woolf, E.D. Wigle, et al. 2000. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N. Engl. J. Med. 343:1688–1696. doi:10.1056/NEJM200012073432304 CrossRefMedline
. ↵ Kaneda, T., C. Naruse, A. Kawashima, N. Fujino, T. Oshima, M. Namura, S. Nunoda, S. Mori, T. Konno, H. Ino, et al. 2008. A novel beta-myosin heavy chain gene mutation, p.Met531Arg, identified in isolated left ventricular non-compaction in humans, results in left ventricular hypertrophy that progresses to dilation in a mouse model. Clin. Sci. 114:431–440. doi:10.1042/CS20070179 CrossRefMedline
. ↵ Kantor, P.F., M.A. Robertson, J.Y. Coe, G.D. Lopaschuk. 1999. Volume overload hypertrophy of the newborn heart slows the maturation of enzymes involved in the regulation of fatty acid metabolism. J. Am. Coll. Cardiol. 33:1724–1734. doi:10.1016/S0735-1097(99)00063-7 Abstract/FREE Full Text
. ↵ Karam, S., M.J. Raboisson, C. Ducreux, L. Chalabreysse, G. Millat, A. Bozio, P. Bouvagnet. 2008. A de novo mutation of the beta cardiac myosin heavy chain gene in an infantile restrictive cardiomyopathy. Congenit. Heart Dis. 3:138–143. doi:10.1111/j.1747-0803.2008.00165.x CrossRefMedline
. ↵ Katritsis, D., P.T. Wilmshurst, J.A. Wendon, M.J. Davies, M.M. Webb-Peploe. 1991. Primary restrictive cardiomyopathy: clinical and pathologic characteristics. J. Am. Coll. Cardiol. 18:1230–1235. doi:10.1016/0735-1097(91)90540-P Abstract
. ↵ Keeling, P.J., Y. Gang, G. Smith, H. Seo, S.E. Bent, V. Murday, A.L. Caforio, W.J. McKenna. 1995. Familial dilated cardiomyopathy in the United Kingdom. Br. Heart J. 73:417–421. doi:10.1136/hrt.73.5.417 Abstract/FREE Full Text
. ↵ Kinnunen, P., O. Vuolteenaho, H. Ruskoaho. 1993. Mechanisms of atrial and brain natriuretic peptide release from rat ventricular myocardium: effect of stretching. Endocrinology. 132:1961–1970. doi:10.1210/en.132.5.1961 Abstract/FREE Full Text
. ↵ Knowlton, K.U., M.C. Michel, M. Itani, H.E. Shubeita, K. Ishihara, J.H. Brown, K.R. Chien. 1993. The alpha 1A-adrenergic receptor subtype mediates biochemical, molecular, and morphologic features of cultured myocardial cell hypertrophy. J. Biol. Chem. 268:15374–15380. Abstract/FREE Full Text
. ↵ Kojima, M., I. Shiojima, T. Yamazaki, I. Komuro, Z. Zou, Y. Wang, T. Mizuno, K. Ueki, K. Tobe, T. Kadowaki, et al. 1994. Angiotensin II receptor antagonist TCV-116 induces regression of hypertensive left ventricular hypertrophy in vivo and inhibits the intracellular signaling pathway of stretch-mediated cardiomyocyte hypertrophy in vitro. Circulation. 89:2204–2211. Abstract/FREE Full Text
. ↵ Krown, K.A., M.T. Page, C. Nguyen, D. Zechner, V. Gutierrez, K.L. Comstock, C.C. Glembotski, P.J. Quintana, R.A. Sabbadini. 1996. Tumor necrosis factor alpha-induced apoptosis in cardiac myocytes. Involvement of the sphingolipid signaling cascade in cardiac cell death. J. Clin. Invest. 98:2854–2865. doi:10.1172/JCI119114 Medline
. ↵ Kuwahara, K., Y. Saito, M. Takano, Y. Arai, S. Yasuno, Y. Nakagawa, N. Takahashi, Y. Adachi, G. Takemura, M. Horie, et al. 2003. NRSF regulates the fetal cardiac gene program and maintains normal cardiac structure and function. EMBO J. 22:6310–6321. doi:10.1093/emboj/cdg601 CrossRefMedline
. ↵ Le Guennec, J.Y., N. Peineau, J.A. Argibay, K.G. Mongo, D. Garnier. 1990. A new method of attachment of isolated mammalian ventricular myocytes for tension recording: length dependence of passive and active tension. J. Mol. Cell. Cardiol. 22:1083–1093. doi:10.1016/0022-2828(90)90072-A CrossRefMedline
. ↵ Lopaschuk, G.D., M.A. Spafford, D.R. Marsh. 1991. Glycolysis is predominant source of myocardial ATP production immediately after birth. Am. J. Physiol. 261:H1698–H1705. Medline
. ↵ Lowes, B.D., W. Minobe, W.T. Abraham, M.N. Rizeq, T.J. Bohlmeyer, R.A. Quaife, R.L. Roden, D.L. Dutcher, A.D. Robertson, N.F. Voelkel, et al. 1997. Changes in gene expression in the intact human heart. Downregulation of alpha-myosin heavy chain in hypertrophied, failing ventricular myocardium. J. Clin. Invest. 100:2315–2324. doi:10.1172/JCI119770 Medline
. ↵ Luckey, S.W., L.A. Walker, T. Smyth, J. Mansoori, A. Messmer-Kratzsch, A. Rosenzweig, E.N. Olson, L.A. Leinwand. 2009. The role of Akt/GSK-3beta signaling in familial hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 46:739–747. doi:10.1016/j.yjmcc.2009.02.010 CrossRefMedline
. ↵ Maass, A.H., M. Buvoli. 2007. Cardiomyocyte preparation, culture, and gene transfer. Methods Mol. Biol. 366:321–330. doi:10.1007/978-1-59745-030-0_18 CrossRefMedline
. ↵ Marian, A.J. 2000. Pathogenesis of diverse clinical and pathological phenotypes in hypertrophic cardiomyopathy. Lancet. 355:58–60. doi:10.1016/S0140-6736(99)06187-5 CrossRefMedline
. ↵ Maron, B.J., A. Pelliccia. 2006. The heart of trained athletes: cardiac remodeling and the risks of sports, including sudden death. Circulation. 114:1633–1644. doi:10.1161/CIRCULATIONAHA.106.613562 FREE Full Text
. ↵ Maron, B.J., P.F. Nichols III, L.W. Pickle, Y.E. Wesley, J.J. Mulvihill. 1984. Patterns of inheritance in hypertrophic cardiomyopathy: assessment by M-mode and two-dimensional echocardiography. Am. J. Cardiol. 53:1087–1094. doi:10.1016/0002-9149(84)90643-X CrossRefMedline
. ↵ Maron, B.J., J.M. Gardin, J.M. Flack, S.S. Gidding, T.T. Kurosaki, D.E. Bild. 1995. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation. 92:785–789. Abstract/FREE Full Text
. ↵ Matsui, T., L. Li, J.C. Wu, S.A. Cook, T. Nagoshi, M.H. Picard, R. Liao, A. Rosenzweig. 2002. Phenotypic spectrum caused by transgenic overexpression of activated Akt in the heart. J. Biol. Chem. 277:22896–22901. doi:10.1074/jbc.M200347200 Abstract/FREE Full Text
. ↵ Menon, S.C., B.W. Eidem, J.A. Dearani, S.R. Ommen, M.J. Ackerman, D. Miller. 2009. Diastolic dysfunction and its histopathological correlation in obstructive hypertrophic cardiomyopathy in children and adolescents. J. Am. Soc. Echocardiogr. 22:1327–1334. doi:10.1016/j.echo.2009.08.014 CrossRefMedline
. ↵ Mestroni, L., C. Rocco, D. Gregori, G. Sinagra, A. Di Lenarda, S. Miocic, M. Vatta, B. Pinamonti, F. Muntoni, A.L. Caforio, et al.; Heart Muscle Disease Study Group. 1999. Familial dilated cardiomyopathy: evidence for genetic and phenotypic heterogeneity. J. Am. Coll. Cardiol. 34:181–190. doi:10.1016/S0735-1097(99)00172-2 Abstract/FREE Full Text
. ↵ Michael, A., S. Haq, X. Chen, E. Hsich, L. Cui, B. Walters, Z. Shao, K. Bhattacharya, H. Kilter, G. Huggins, et al. 2004. Glycogen synthase kinase-3beta regulates growth, calcium homeostasis, and diastolic function in the heart. J. Biol. Chem. 279:21383–21393. doi:10.1074/jbc.M401413200 Abstract/FREE Full Text
. ↵ Michels, V.V., P.P. Moll, F.A. Miller, A.J. Tajik, J.S. Chu, D.J. Driscoll, J.C. Burnett, R.J. Rodeheffer, J.H. Chesebro, H.D. Tazelaar. 1992. The frequency of familial dilated cardiomyopathy in a series of patients with idiopathic dilated cardiomyopathy. N. Engl. J. Med. 326:77–82. doi:10.1056/NEJM199201093260201 Medline
. ↵ Minamisawa, S., M. Hoshijima, G. Chu, C.A. Ward, K. Frank, Y. Gu, M.E. Martone, Y. Wang, J. Ross Jr, E.G. Kranias, et al. 1999. Chronic phospholamban-sarcoplasmic reticulum calcium ATPase interaction is the critical calcium cycling defect in dilated cardiomyopathy. Cell. 99:313–322. doi:10.1016/S0092-8674(00)81662-1 CrossRefMedline
. ↵ Miyata, S., W. Minobe, M.R. Bristow, L.A. Leinwand. 2000. Myosin heavy chain isoform expression in the failing and nonfailing human heart. Circ. Res. 86:386–390. Abstract/FREE Full Text
. ↵ Mogensen, J., T. Kubo, M. Duque, W. Uribe, A. Shaw, R. Murphy, J.R. Gimeno, P. Elliott, W.J. McKenna. 2003. Idiopathic restrictive cardiomyopathy is part of the clinical expression of cardiac troponin I mutations. J. Clin. Invest. 111:209–216. CrossRefMedline
. ↵ Molkentin, J.D., G.W. Dorn II. 2001. Cytoplasmic signaling pathways that regulate cardiac hypertrophy. Annu. Rev. Physiol. 63:391–426. doi:10.1146/annurev.physiol.63.1.391 CrossRefMedline
. ↵ Molkentin, J.D., J.R. Lu, C.L. Antos, B. Markham, J. Richardson, J. Robbins, S.R. Grant, E.N. Olson. 1998. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 93:215–228. doi:10.1016/S0092-8674(00)81573-1 CrossRefMedline
. ↵ Nagata, K., R. Liao, F.R. Eberli, N. Satoh, B. Chevalier, C.S. Apstein, T.M. Suter. 1998. Early changes in excitation-contraction coupling: transition from compensated hypertrophy to failure in Dahl salt-sensitive rat myocytes. Cardiovasc. Res. 37:467–477. doi:10.1016/S0008-6363(97)00278-2 Abstract/FREE Full Text
. ↵ Narula, J., N. Haider, R. Virmani, T.G. DiSalvo, F.D. Kolodgie, R.J. Hajjar, U. Schmidt, M.J. Semigran, G.W. Dec, B.A. Khaw. 1996. Apoptosis in myocytes in end-stage heart failure. N. Engl. J. Med. 335:1182–1189. doi:10.1056/NEJM199610173351603 CrossRefMedline
. ↵ Narula, J., P. Pandey, E. Arbustini, N. Haider, N. Narula, F.D. Kolodgie, B. Dal Bello, M.J. Semigran, A. Bielsa-Masdeu, G.W. Dec, et al. 1999. Apoptosis in heart failure: release of cytochrome c from mitochondria and activation of caspase-3 in human cardiomyopathy. Proc. Natl. Acad. Sci. USA. 96:8144–8149. doi:10.1073/pnas.96.14.8144 Abstract/FREE Full Text
. ↵ Neubauer, S. 2007. The failing heart—an engine out of fuel. N. Engl. J. Med. 356:1140–1151. doi:10.1056/NEJMra063052 CrossRefMedline
. ↵ O’Neill, B.T., E.D. Abel. 2005. Akt1 in the cardiovascular system: friend or foe? J. Clin. Invest. 115:2059–2064. doi:10.1172/JCI25900 CrossRefMedline
. ↵ Ohler, A., J. Weisser-Thomas, V. Piacentino, S.R. Houser, G.F. Tomaselli, B. O’Rourke. 2009. Two-photon laser scanning microscopy of the transverse-axial tubule system in ventricular cardiomyocytes from failing and non-failing human hearts. Cardiol. Res. Pract. 2009:802373. Medline
. ↵ Olivetti, G., R. Abbi, F. Quaini, J. Kajstura, W. Cheng, J.A. Nitahara, E. Quaini, C. Di Loreto, C.A. Beltrami, S. Krajewski, et al. 1997. Apoptosis in the failing human heart. N. Engl. J. Med. 336:1131–1141. doi:10.1056/NEJM199704173361603 CrossRefMedline
. ↵ Palmiter, K.A., M.J. Tyska, D.E. Dupuis, N.R. Alpert, D.M. Warshaw. 1999. Kinetic differences at the single molecule level account for the functional diversity of rabbit cardiac myosin isoforms. J. Physiol. 519:669–678. doi:10.1111/j.1469-7793.1999.0669n.x Abstract/FREE Full Text
. ↵ Parvatiyar, M.S., J.R. Pinto, D. Dweck, J.D. Potter. 2010. Cardiac troponin mutations and restrictive cardiomyopathy. J. Biomed. Biotechnol. 2010:350706. doi:10.1155/2010/350706 Medline
. ↵ Peddy, S.B., L.A. Vricella, J.E. Crosson, G.L. Oswald, R.D. Cohn, D.E. Cameron, D. Valle, B.L. Loeys. 2006. Infantile restrictive cardiomyopathy resulting from a mutation in the cardiac troponin T gene. Pediatrics. 117:1830–1833. doi:10.1542/peds.2005-2301 Abstract/FREE Full Text
. ↵ Ritter, M., E. Oechslin, G. Sütsch, C. Attenhofer, J. Schneider, R. Jenni. 1997. Isolated noncompaction of the myocardium in adults. Mayo Clin. Proc. 72:26–31. doi:10.4065/72.1.26 Abstract/FREE Full Text
. ↵ Ro, A., W.H. Frishman. 2006. Peripartum cardiomyopathy. Cardiol. Rev. 14:35–42. doi:10.1097/01.crd.0000174805.68081.f7 CrossRefMedline
. ↵ Rodeheffer, R.J., I. Tanaka, T. Imada, A.S. Hollister, D. Robertson, T. Inagami. 1986. Atrial pressure and secretion of atrial natriuretic factor into the human central circulation. J. Am. Coll. Cardiol. 8:18–26. doi:10.1016/S0735-1097(86)80086-9 Abstract
. ↵ Rose, E.A., A.C. Gelijns, A.J. Moskowitz, D.F. Heitjan, L.W. Stevenson, W. Dembitsky, J.W. Long, D.D. Ascheim, A.R. Tierney, R.G. Levitan, et al.; Randomized Evaluation of Mechanical Assistance for the Treatment of Congestive Heart Failure (REMATCH) Study Group. 2001. Long-term use of a left ventricular assist device for end-stage heart failure. N. Engl. J. Med. 345:1435–1443. doi:10.1056/NEJMoa012175 CrossRefMedline
. ↵ Sack, M.N., T.A. Rader, S. Park, J. Bastin, S.A. McCune, D.P. Kelly. 1996. Fatty acid oxidation enzyme gene expression is downregulated in the failing heart. Circulation. 94:2837–2842. Abstract/FREE Full Text
. ↵ Sadoshima, J., S. Izumo. 1993. Mechanical stretch rapidly activates multiple signal transduction pathways in cardiac myocytes: potential involvement of an autocrine/paracrine mechanism. EMBO J. 12:1681–1692. Medline
. ↵ Sakata, Y., B.D. Hoit, S.B. Liggett, R.A. Walsh, G.W. Dorn II. 1998. Decompensation of pressure-overload hypertrophy in G alpha q-overexpressing mice. Circulation. 97:1488–1495. Abstract/FREE Full Text
. ↵ Sasse-Klaassen, S., B. Gerull, E. Oechslin, R. Jenni, L. Thierfelder. 2003. Isolated noncompaction of the left ventricular myocardium in the adult is an autosomal dominant disorder in the majority of patients. Am. J. Med. Genet. A. 119A:162–167. doi:10.1002/ajmg.a.20075 Medline
. ↵ Schram, K., S. De Girolamo, S. Madani, D. Munoz, F. Thong, G. Sweeney. 2010. Leptin regulates MMP-2, TIMP-1 and collagen synthesis via p38 MAPK in HL-1 murine cardiomyocytes. Cell. Mol. Biol. Lett. 15:551–563. doi:10.2478/s11658-010-0027-z CrossRefMedline
. ↵ Schwartz, K., Y. Lecarpentier, J.L. Martin, A.M. Lompré, J.J. Mercadier, B. Swynghedauw. 1981. Myosin isoenzymic distribution correlates with speed of myocardial contraction. J. Mol. Cell. Cardiol. 13:1071–1075. doi:10.1016/0022-2828(81)90297-2 CrossRefMedline
. ↵ Seidman, J.G., C. Seidman. 2001. The genetic basis for cardiomyopathy: from mutation identification to mechanistic paradigms. Cell. 104:557–567. doi:10.1016/S0092-8674(01)00242-2 CrossRefMedline
. ↵ Semsarian, C., M.J. Healey, D. Fatkin, M. Giewat, C. Duffy, C.E. Seidman, J.G. Seidman. 2001. A polymorphic modifier gene alters the hypertrophic response in a murine model of familial hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 33:2055–2060. doi:10.1006/jmcc.2001.1466 CrossRefMedline
. ↵ Shen, W.K., W.D. Edwards, S.C. Hammill, K.R. Bailey, D.J. Ballard, B.J. Gersh. 1995. Sudden unexpected nontraumatic death in 54 young adults: a 30-year population-based study. Am. J. Cardiol. 76:148–152. doi:10.1016/S0002-9149(99)80047-2 CrossRefMedline
. ↵ Shiojima, I., K. Sato, Y. Izumiya, S. Schiekofer, M. Ito, R. Liao, W.S. Colucci, K. Walsh. 2005. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J. Clin. Invest. 115:2108–2118. doi:10.1172/JCI24682 CrossRefMedline
. ↵ Silberbach, M., T. Gorenc, R.E. Hershberger, P.J. Stork, P.S. Steyger, C.T. Roberts Jr. 1999. Extracellular signal-regulated protein kinase activation is required for the anti-hypertrophic effect of atrial natriuretic factor in neonatal rat ventricular myocytes. J. Biol. Chem. 274:24858–24864. doi:10.1074/jbc.274.35.24858 Abstract/FREE Full Text
. ↵ Simpson, P., A. McGrath, S. Savion. 1982. Myocyte hypertrophy in neonatal rat heart cultures and its regulation by serum and by catecholamines. Circ. Res. 51:787–801. Abstract/FREE Full Text
. ↵ Smith, C.S., P.A. Bottomley, S.P. Schulman, G. Gerstenblith, R.G. Weiss. 2006. Altered creatine kinase adenosine triphosphate kinetics in failing hypertrophied human myocardium. Circulation. 114:1151–1158. doi:10.1161/CIRCULATIONAHA.106.613646 Abstract/FREE Full Text
. ↵ Stanley, W.C., G.D. Lopaschuk, J.G. McCormack. 1997. Regulation of energy substrate metabolism in the diabetic heart. Cardiovasc. Res. 34:25–33. doi:10.1016/S0008-6363(97)00047-3 FREE Full Text
. ↵ Stauffer, B.L., J.P. Konhilas, E.D. Luczak, L.A. Leinwand. 2006. Soy diet worsens heart disease in mice. J. Clin. Invest. 116:209–216. doi:10.1172/JCI24676 CrossRefMedline
. ↵ Taigen, T., L.J. De Windt, H.W. Lim, J.D. Molkentin. 2000. Targeted inhibition of calcineurin prevents agonist-induced cardiomyocyte hypertrophy. Proc. Natl. Acad. Sci. USA. 97:1196–1201. doi:10.1073/pnas.97.3.1196 Abstract/FREE Full Text
. ↵ Teekakirikul, P., S. Eminaga, O. Toka, R. Alcalai, L. Wang, H. Wakimoto, M. Nayor, T. Konno, J.M. Gorham, C.M. Wolf, et al. 2010. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-β. J. Clin. Invest. 120:3520–3529. doi:10.1172/JCI42028 CrossRefMedline
. ↵ Torre-Amione, G., S. Kapadia, J. Lee, J.B. Durand, R.D. Bies, J.B. Young, D.L. Mann. 1996. Tumor necrosis factor-alpha and tumor necrosis factor receptors in the failing human heart. Circulation. 93:704–711. Abstract/FREE Full Text
. ↵ Vikstrom, K.L., S.M. Factor, L.A. Leinwand. 1996. Mice expressing mutant myosin heavy chains are a model for familial hypertrophic cardiomyopathy. Mol. Med. 2:556–567. Medline
. ↵ Wang, L., J.G. Seidman, C.E. Seidman. 2010. Narrative review: harnessing molecular genetics for the diagnosis and management of hypertrophic cardiomyopathy. Ann. Intern. Med. 152:513–520: W181. Abstract/FREE Full Text
. ↵ Weiford, B.C., V.D. Subbarao, K.M. Mulhern. 2004. Noncompaction of the ventricular myocardium. Circulation. 109:2965–2971. doi:10.1161/01.CIR.0000132478.60674.D0 FREE Full Text
. ↵ Yamaji, K., S. Fujimoto, Y. Ikeda, K. Masuda, S. Nakamura, Y. Saito, C. Yutani. 2005. Apoptotic myocardial cell death in the setting of arrhythmogenic right ventricular cardiomyopathy. Acta Cardiol. 60:465–470. doi:10.2143/AC.60.5.2004965 CrossRefMedline
. ↵ Yang, Z., N.E. Bowles, S.E. Scherer, M.D. Taylor, D.L. Kearney, S. Ge, V.V. Nadvoretskiy, G. DeFreitas, B. Carabello, L.I. Brandon, et al. 2006. Desmosomal dysfunction due to mutations in desmoplakin causes arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ. Res. 99:646–655. doi:10.1161/01.RES.0000241482.19382.c6 Abstract/FREE Full Text
. ↵ Yousef, Z.R., P.W. Foley, K. Khadjooi, S. Chalil, H. Sandman, N.U. Mohammed, F. Leyva. 2009. Left ventricular non-compaction: clinical features and cardiovascular magnetic resonance imaging. BMC Cardiovasc. Disord. 9:37. doi:10.1186/1471-2261-9-37 CrossRefMedline
. ↵ Zhang, Y.H., J.B. Youm, H.K. Sung, S.H. Lee, S.Y. Ryu, W.K. Ho, Y.E. Earm. 2000. Stretch-activated and background non-selective cation channels in rat atrial myocytes. J. Physiol. 523:607–619. doi:10.1111/j.1469-7793.2000.00607.x Abstract/FREE Full Text



























{kind=link}
{kind=link}
{kind=link}