Heart, Vascular Smooth Muscle, Excitation-Contraction Coupling (E-CC), Cytoskeleton, Cellular Dynamics and Ca2 Signaling
Author and Curator: Larry H Bernstein, MD, FCAP
Author and Cardiovascular Three-volume Series, Editor: Justin Pearlman, MD, PhD, FACC, and
Curator: Aviva Lev-Ari, PhD, RN

Image created by Adina Hazan 06/30/2021
Abbreviations
AP, action potential; ARVD2, arrhythmogenic right ventricular cardiomyopathy type 2; CaMKII, Ca2+/calmodulim-dependent protein kinase II; CICR, Ca2+ induced Ca2+ release;CM, calmodulin; CPVT, catecholaminergic polymorphic ventricular tachycardia; ECC, excitation–contraction coupling; FKBP12/12.6, FK506 binding protein; HF, heart failure; LCC, L-type Ca2+ channel; P-1 or P-2, phosphatase inhibitor type-1 or type-2; PKA, protein kinase A; PLB, phosphoplamban; PP1, protein phosphatase 1; PP2A, protein phosphatase 2A; RyR1/2, ryanodine receptor type-1/type-2; SCD, sudden cardiac death; SERCA, sarcoplasmic reticulum Ca2+ ATPase; SL, sarcolemma; SR, sarcoplasmic reticulum.
This is Part V of a series on the cytoskeleton and structural shared thematics in cellular movement and cellular dynamics.
The Series consists of the following articles:
Part I: Identification of Biomarkers that are Related to the Actin Cytoskeleton
Larry H Bernstein, MD, FCAP
Part II: Role of Calcium, the Actin Skeleton, and Lipid Structures in Signaling and Cell Motility
Larry H. Bernstein, MD, FCAP, Stephen Williams, PhD and Aviva Lev-Ari, PhD, RN
Part III: Renal Distal Tubular Ca2+ Exchange Mechanism in Health and Disease
Larry H. Bernstein, MD, FCAP, Stephen J. Williams, PhD and Aviva Lev-Ari, PhD, RN
Part IV: The Centrality of Ca(2+) Signaling and Cytoskeleton Involving Calmodulin Kinases and Ryanodine Receptors in Cardiac Failure, Arterial Smooth Muscle, Post-ischemic Arrhythmia, Similarities and Differences, and Pharmaceutical Targets
Larry H Bernstein, MD, FCAP, Justin Pearlman, MD, PhD, FACC and Aviva Lev-Ari, PhD, RN
Part V: Heart, Vascular Smooth Muscle, Excitation-Contraction Coupling (E-CC), Cytoskeleton, Cellular Dynamics and Ca2 Signaling
Larry H Bernstein, MD, FCAP, Justin Pearlman, MD, PhD, FACC and Aviva Lev-Ari, PhD, RN
Part VI: Calcium Cycling (ATPase Pump) in Cardiac Gene Therapy: Inhalable Gene Therapy for Pulmonary Arterial Hypertension and Percutaneous Intra-coronary Artery Infusion for Heart Failure: Contributions by Roger J. Hajjar, MD
Aviva Lev-Ari, PhD, RN
Part VII: Cardiac Contractility & Myocardium Performance: Ventricular Arrhythmias and Non-ischemic Heart Failure – Therapeutic Implications for Cardiomyocyte Ryanopathy (Calcium Release-related Contractile Dysfunction) and Catecholamine Responses
Justin Pearlman, MD, PhD, FACC, Larry H Bernstein, MD, FCAP and Aviva Lev-Ari, PhD, RN
In the first part, we discussed common MOTIFs across cell-types that are essential for cell division, embryogenesis, cancer metastasis, osteogenesis, musculoskeletal function, vascular compliance, and cardiac contractility. This second article concentrates on specific functionalities for cardiac contractility based on Ca++ signaling in excitation-contraction coupling. The modifications discussed apply specifically to cardiac muscle and not to skeletal muscle. Considering the observations described might raise additional questions specifically address to the unique requirements of smooth muscle, abundant in the GI tract and responsible for motility in organ function, and in blood vessel compliance or rigidity. Due to the distinctly different aspects of the cardiac contractility and contraction force, and the interactions with potential pharmaceutical targets, there are two separate articles on calcium signaling and cardiac arrhythmias or heart failure (Part 2 and Part 3). Part 2 focuses on the RYANODINE role in cardiac Ca(2+) signaling and its effect in heart failure. Part 3 takes up other aspects of heart failure and calcium signaling with respect to phosporylation/dephosphorylation. I add a single review and classification of genetic cardiac disorders of the same cardiac Ca(2+) signaling and the initiation and force of contraction. Keep in mind that the heart is a syncytium, and this makes a huge difference compared with skeletal muscle dynamics. In Part 1 there was some discussion of the importance of Ca2+ signaling on innate immune system, and the immunology will be further expanded in a fourth of the series.
SUMMARY:
This second article on the cardiomyocyte and the Ca(2+) cycling between the sarcomere and the cytoplasm, takes a little distance from the discussion of the ryanodine that precedes it. In this discussion we found that there is a critical phosphorylation/dephosphorylation balance that exists between Ca(+) ion displacement and it occurs at a specific amino acid residue on the CaMKIId, specific for myocardium, and there is a 4-fold increase in contraction and calcium release associated with this CAM kinase (ser 2809) dependent exchange. These events are discussed in depth, and the research holds promise for therapeutic application. We also learn that Ca(2+) ion channels are critically involved in the generation of arrhythmia as well as dilated and hypertrophic cardiomyopathy. In the case of arrhythmiagenesis, there are two possible manners by which this occurs. One trigger is Ca(2+) efflux instability. The other is based on the finding that when the cellular instability is voltage driven, the steady-state wavelength (separation of nodes in space) depends on electrotonic coupling between cells and the steepness of APD and CV restitution. The last article is an in depth review of the genetic mutations that occur in cardiac diseases. It is an attempt at classifying them into reasonable groupings. What are the therapeutic implications of this? We see that the molecular mechanism of cardiac function has been substantially elucidated, although there are contradictions in experimental findings that are unexplained. However, for the first time, it appears that personalized medicine is on a course that will improve health in the population, and the findings will allow specific targets designed for the individual with a treatable impairment in cardiac function that is identifiable early in the course of illness. This article is a continuation to the following articles on tightly related topics: Part I: Identification of Biomarkers that are Related to the Actin Cytoskeleton Larry H Bernstein, MD, FCAP http://pharmaceuticalintelligence.com/2012/12/10/identification-of-biomarkers-that-are-related-to-the-actin-cytoskeleton/ Part II: Role of Calcium, the Actin Skeleton, and Lipid Structures in Signaling and Cell Motility Larry H. Bernstein, MD, FCAP, Stephen Williams, PhD and Aviva Lev-Ari, PhD, RN http://pharmaceuticalintelligence.com/2013/08/26/role-of-calcium-the-actin-skeleton-and-lipid-structures-in-signaling-and-cell-motility/ Part III: Renal Distal Tubular Ca2+ Exchange Mechanism in Health and Disease Larry H. Bernstein, MD, FCAP, Stephen J. Williams, PhD and Aviva Lev-Ari, PhD, RN http://pharmaceuticalintelligence.com/2013/09/02/renal-distal-tubular-ca2-exchange-mechanism-in-health-and-disease/ Part IV: The Centrality of Ca(2+) Signaling and Cytoskeleton Involving Calmodulin Kinases and Ryanodine Receptors in Cardiac Failure, Arterial Smooth Muscle, Post-ischemic Arrhythmia, Similarities and Differences, and Pharmaceutical Targets Larry H Bernstein, MD, FCAP, Justin Pearlman, MD, PhD, FACC and Aviva Lev-Ari, PhD, RN http:/pharmaceuticalintelligence.com/2013.09.089/lhbern/The Centrality of Ca(2+) Signaling and Cytoskeleton Involving Calmodulin Kinases and Ryanodine Receptors in Cardiac Failure, Arterial Smooth Muscle, Post-ischemic Arrhythmia, Similarities and Differences, and Pharmaceutical Targets
Part V: Heart Smooth Muscle and Cardiomyocyte Cells: Excitation-Contraction Coupling & Ryanodine Receptor (RyR) type-1/type-2 in Cytoskeleton Cellular Dynamics and Ca2+ Signaling
Larry H Bernstein, MD, FCAP, Justin Pearlman, MD, PhD, FACC and Aviva Lev-Ari, PhD, RN http://pharmaceuticalintelligence.com/2013/08/26/heart-smooth-muscle-excitation-contraction-coupling-cytoskeleton-cellular-dynamics-and-ca2-signaling/ Part VI: Calcium Cycling (ATPase Pump) in Cardiac Gene Therapy: Inhalable Gene Therapy for Pulmonary Arterial Hypertension and Percutaneous Intra-coronary Artery Infusion for Heart Failure: Contributions by Roger J. Hajjar, MD Curator: Aviva Lev-Ari, PhD, RN http://pharmaceuticalintelligence.com/2013/08/01/calcium-molecule-in-cardiac-gene-therapy-inhalable-gene-therapy-for-pulmonary-arterial-hypertension-and-percutaneous-intra-coronary-artery-infusion-for-heart-failure-contributions-by-roger-j-hajjar/ and Advanced Topics in Sepsis and the Cardiovascular System at its End Stage Larry H Bernstein, MD, FCAP http://pharmaceuticalintelligence.com/2013/08/18/advanced-topics-in-sepsis-and-the-cardiovascular-system-at-its-end-stage/
The Role of Protein Kinases and Protein Phosphatases in the Regulation of Cardiac Sarcoplasmic Reticulum Function
EG Kranias, RC Gupta, G Jakab, HW Kim, NAE Steenaart, ST Rapundalo Molecular and Cellular Biochemistry 06/1988; 82(1):37-44. · 2.06 Impact Factor http://www.researchgate.net/publication/6420466_Protein_phosphatases_decrease_sarcoplasmic_reticulum_calcium_content_by_stimulating_calcium_release_in_cardiac_myocytes Canine cardiac sarcoplasmic reticulum is phosphorylated by
- adenosine 3,5-monophosphate (cAMP)-dependent and
- calcium calmodulin-dependent protein kinases on
- a proteolipid, called phospholamban.
Both types of phosphorylation are associated with
- an increase in the initial rates of Ca(2+) transport by SR vesicles
- which reflects an increased turnover of elementary steps of the calcium ATPase reaction sequence.
The stimulatory effects of the protein kinases on the calcium pump may be reversed by an endogenous protein phosphatase, which
- can dephosphorylate both the CAMP-dependent and the calcium calmodulin-dependent sites on phospholamban.
Thus, the calcium pump in cardiac sarcoplasmic reticulum appears to be under reversible regulation mediated by protein kinases and protein phosphatases. 
Regulation of the Cardiac Ryanodine Receptor Channel by Luminal Ca2+ involves Luminal Ca2+ Sensing Sites
I Györke, S Györke. Biophysical Journal 01/1999; 75(6):2801-10. · 3.65 Impact factor http:// www.researchgate.net/publication/13459335/Regulation_of_the_cardiac_ryanodine_receptor_channel_by_luminal_Ca2_involves_luminal_Ca2_sensing_sites The mechanism of activation of the cardiac calcium release channel/ryanodine receptor (RyR) by luminal Ca(2+) was investigated in native canine cardiac RyRs incorporated into lipid bilayers in the presence of 0.01 microM to 2 mM Ca(2+) (free) and 3 mM ATP (total) on the cytosolic (cis) side and 20 microM to 20 mM Ca(2+) on the luminal (trans) side of the channel and with Cs+ as the charge carrier. Under conditions of low [trans Ca(2+)] (20 microM), increasing [cis Ca(2+)] from 0.1 to 10 microM caused a gradual increase in channel open probability (Po). Elevating [cis Ca(2+)] [cytosolic] above 100 microM resulted in a gradual decrease in Po. Elevating trans [Ca(2+)] [luminal] enhanced channel activity (EC50 approximately 2.5 mM at 1 microM cis Ca2+) primarily by increasing the frequency of channel openings. The dependency of Po on trans [Ca2+] [luminal] was similar at negative and positive holding potentials and was not influenced by high cytosolic concentrations of the fast Ca(2+) chelator, 1,2-bis(2-aminophenoxy)ethane-N,N,N, N-tetraacetic acid. Elevated luminal Ca(2+)
- enhanced the sensitivity of the channel to activating cytosolic Ca(2+), and it
- essentially reversed the inhibition of the channel by high cytosolic Ca(2+).
Potentiation of Po by increased luminal Ca(2+) occurred irrespective of whether the electrochemical gradient for Ca(2+) supported a cytosolic-to-luminal or a luminal-to-cytosolic flow of Ca(2+) through the channel. These results rule out the possibility that under our experimental conditions, luminal Ca(2+) acts by interacting with the cytosolic activation site of the channel and suggest that the effects of luminal Ca2+ are mediated by distinct Ca(2+)-sensitive site(s) at the luminal face of the channel or associated protein. 
Protein phosphatases Decrease Sarcoplasmic Reticulum Calcium Content by Stimulating Calcium Release in Cardiac Myocytes
D Terentyev, S Viatchenko-Karpinski, I Gyorke, R Terentyeva and S Gyorke Texas Tech University Health Sciences Center, Lubbock, TX J Physiol 2003; 552(1), pp. 109–118. http://dx.doi.org/10.1113/jphysiol.2003.046367 Phosphorylation/dephosphorylation of Ca2+ transport proteins by cellular kinases and phosphatases plays an important role in regulation of cardiac excitation–contraction coupling; furthermore,
- abnormal protein kinase and phosphatase activities have been implicated in heart failure.
However, the precise mechanisms of action of these enzymes on intracellular Ca2+ handling in normal and diseased hearts remains poorly understood. We have investigated
- the effects of protein phosphatases PP1 and PP2A on spontaneous Ca(2+) sparks and SR Ca(2+) load in myocytes permeabilized with saponin.
Exposure of myocytes to PP1 or PP2A caused a dramatic increase in frequency of Ca(2+) sparks followed by a nearly complete disappearance of events, which were accompanied by depletion of the SR Ca(2+) stores, as determined by application of caffeine. These changes in
- Ca(2+) release and
- SR Ca(2+) load
could be prevented by the inhibitors of PP1 and PP2A phosphatase activities okadaic acid and calyculin A. At the single channel level, PP1 increased the open probability of RyRs incorporated into lipid bilayers. PP1-medited RyR dephosphorylation in our permeabilized myocytes preparations was confirmed biochemically by quantitative immunoblotting using a phosphospecific anti-RyR antibody. Our results suggest that
- increased intracellular phosphatase activity stimulates
- RyR mediated SR Ca(2+) release
- leading to depleted SR Ca(2+) stores in cardiac myocytes.
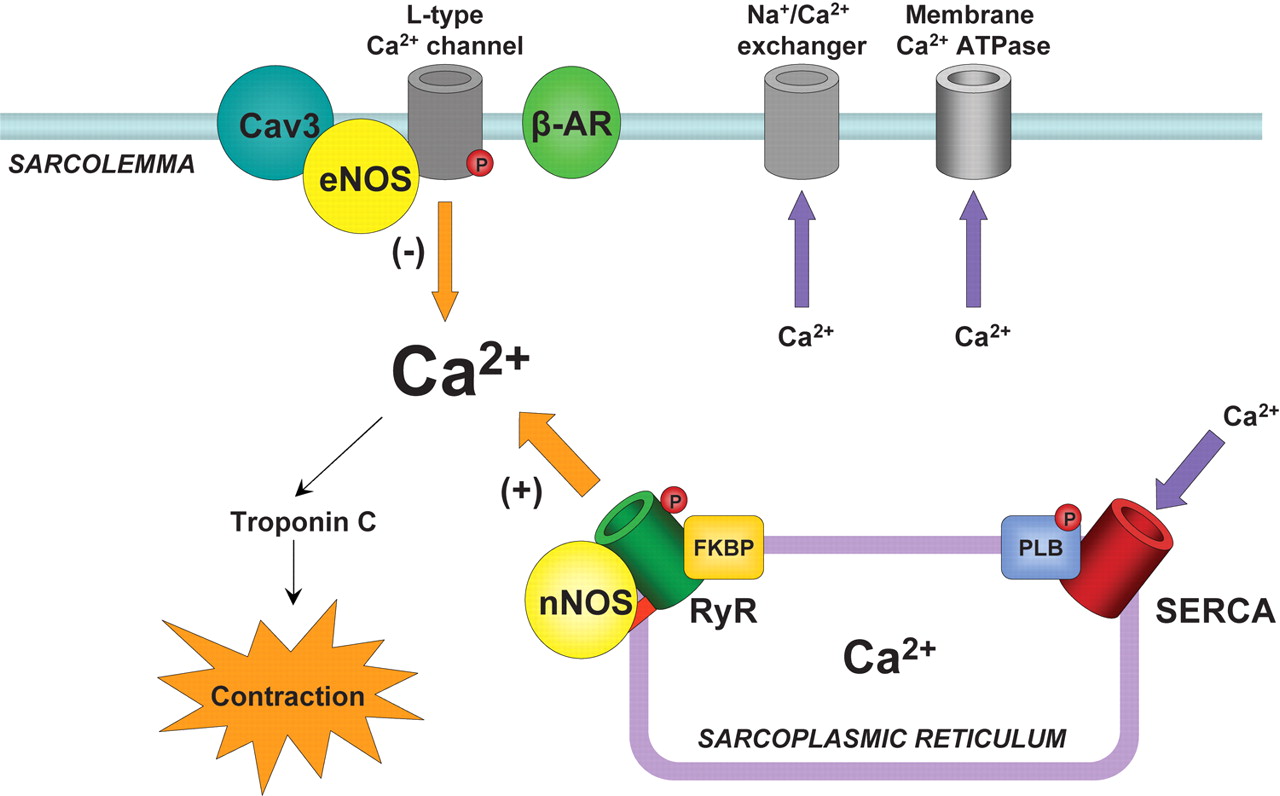
In heart muscle cells, the process of excitation–contraction (EC) coupling is mediated by
- Ca(2+) influx through sarcolemmal L-type Ca(2+) channels
- activating Ca(2+) release channels (ryanodine receptors, RyRs) in the sarcoplasmic reticulum (SR).
Once activated, the RyR channels allow Ca(2+) to be released from the SR into the cytosol to induce contraction. This mechanism is known as Ca(2+)-induced calcium release (CICR) (Fabiato, 1985; Bers, 2002). During relaxation, most of the Ca(2+) is resequestered into the SR by the Ca(2+)-ATPase. The amount of Ca(2+) released and the force of contraction depend on
- the magnitude of the Ca(2+) trigger signal,
- the functional state of the RyRs and
- the amount of Ca(2+) stored in the SR.
F1.large calcium movement and RyR2 receptor  calcium release calmodulin + ER Reversible phosphorylation of proteins composing the EC coupling machinery plays an important role in regulation of cardiac contractility (Bers, 2002). Thus, during stimulation of the b-adrenergic pathway, phosphorylation of several target proteins, including
calcium release calmodulin + ER Reversible phosphorylation of proteins composing the EC coupling machinery plays an important role in regulation of cardiac contractility (Bers, 2002). Thus, during stimulation of the b-adrenergic pathway, phosphorylation of several target proteins, including
- the L-type Ca(2+) channels,
- RyRs and
- phospholamban,
by protein kinase A (PKA) leads to an overall increase in SR Ca2+ release and contractile force in heart cells (Callewaert et al. 1988, Spurgeon et al. 1990; Hussain & Orchard, 1997; Zhou et al. 1999; Song et al. 2001; Viatchenko-Karpinski & Gyorke, 2001). PKA-dependent phosphorylation of the L-type Ca(2+) channels increases the Ca2+ current (ICa), increasing both
- the Ca2+ trigger for SR Ca2+ release and
- the SR Ca(2+) content
(Callewaert et al. 1988; Hussain & Orchard, 1997; Del Principe et al. 2001). Phosphorylation of phospholamban (PLB) relieves the tonic inhibition dephosphorylated PLB exerts on the SR Ca(2+)-ATPase (SERCA) resulting in enhanced SR Ca(2+) accumulation and enlarged Ca(2+) release (Kranias et al. 1985; Simmermann & Jones, 1998). With regard to the RyR, despite clear demonstration of phosphorylation of the channel in biochemical studies (Takasago et al. 1989; Yoshida et al. 1992), the consequences of this reaction to channel function have not been clearly defined. RyR phosphorylation by PKA and Ca(2+)–calmodulin dependent protein kinase (CaMKII) has been reported to increase RyR activity in lipid bilayers (Hain et al. 1995; Marx et al. 2000; Uehara et al. 2002). Moreover, it has been reported that in heart failure (HF), hyperphosphorylation of RyR causes
- the release of FK-506 binding protein (FKBP12.6) from the RyR,
- rendering the channel excessively leaky for Ca(2+) (Marx et al. 2000).
However, other studies have reported no functional effects (Li et al. 2002) or even found phosphorylation to reduce RyR channel steady-state open probability (Valdivia et al. 1995; Lokuta et al. 1995). The action of protein kinases is opposed by dephosphorylating phosphatases. Three types of protein phosphatases (PPs), referred to as PP1, PP2A and PP2B (calcineurin), have been shown to influence cardiac performance (Neumann et al. 1993; Rusnak & Mertz, 2000). Overall, according to most studies phosphatases appear to downregulate SR Ca(2+) release and contractile performance (Neumann et al. 1993; duBell et al. 1996, 2002; Carr et al. 2002; Santana et al. 2002). Furthermore, PP1 and PP2A activities appear to be increased in heart failure (Neumann, 2002; Carr et al. 2002). However, again the precise mode of action of these enzymes on intracellular Ca(2+) handling in normal and diseased hearts remains poorly understood. In the present study, we have investigated the effects of protein phosphatases PP1 and PP2A on local Ca(2+) release events, Ca(2+) sparks, in cardiac cells. Our results show that
- phosphatases activate RyR mediated SR Ca(2+) release
- leading to depletion of SR Ca(2+) stores.
These results provide novel insights into the mechanisms and potential role of protein phosphorylation/dephosphorylation in regulation of Ca(2+) signaling in normal and diseased hearts. 
RESULTS
Effects of PP1 and PP2A on Ca2+ sparks and SR Ca(2+) content.
[1] PP1 caused an early transient potentiation of Ca2+ spark frequency followed by a delayed inhibition of event occurrence. [2] PP1 produced similar biphasic effects on the magnitude and spatio-temporal characteristics of Ca(2+) sparks Specifically, during the potentiatory phase (1 min after addition of the enzyme), PP1 significantly increased
- the amplitude,
- rise-time,
- duration and
- width of Ca(2+) sparks;
during the inhibitory phase (5 min after addition of the enzyme),
- all these parameters were significantly suppressed by PP1.
The SR Ca(2+) content decreased by 35 % or 69 % following the exposure of myocytes to either 0.5 or 2Uml_1 PP1, respectively (Fig. 1C). Qualitatively similar results were obtained with phosphatase PP2A. Similar to the effects of PP1, PP2A (5Uml_1) produced a transient increase in Ca(2+) spark frequency (~4-fold) followed by a depression of event occurrence and decreased SR Ca(2+) content (by 82 % and 65 %, respectively). Also similar to the action of PP1, PP2A increased
- the amplitude and
- spatio-temporal spread (i.e. rise-time, duration and width) of Ca(2+) sparks at 1 min
- and suppressed the same parameters at 5 min of exposure to the enzyme (Table 1).
Together, these results suggest that phosphatases enhance spark-mediated SR Ca2+ release, leading to decreased SR Ca(2+) content. Preventive effects of calyculin A and okadaic acid Preventive effects of ryanodine
PP1-mediated RyR dephosphorylation
F3.large cardiomyocyte SR  F2.large RyR and calcium
F2.large RyR and calcium  coupled receptors The cardiac RyR is phosphorylated at Ser-2809 (in the rabbit sequence) by both PKA and CAMKII (Witcher et al. 1991; Marx et al. 2000). Although additional phosphorylation sites may exist on the RyR (Rodriguez et al. 2003), but Ser-2809 is believed to be the only site that is phosphorylated by PKA, and RyR hyperphosphorylation at this site has been reported in heart failure (Marx et al. 2000). To test whether indeed phosphatases dephosphorylated the RyR in our permeabilized myocyte experiments we performed quantitative immunoblotting using an antibody that specifically recognizes the phosphorylated form of the RyR at Ser-2809 (Rodriguez et al. 2003). Myocytes exhibited a significant level of phosphorylation under baseline conditions. Maximal phosphorylation was 201 % of control. When exposed to 2Uml_1 PP1, RyR phosphorylation was 58 % of the control basal condition. Exposing to a higher PP1 concentration (10Uml_1) further reduced RyR phosphorylation to 22% of control. Thus, consistent with the results of our functional measurements,
coupled receptors The cardiac RyR is phosphorylated at Ser-2809 (in the rabbit sequence) by both PKA and CAMKII (Witcher et al. 1991; Marx et al. 2000). Although additional phosphorylation sites may exist on the RyR (Rodriguez et al. 2003), but Ser-2809 is believed to be the only site that is phosphorylated by PKA, and RyR hyperphosphorylation at this site has been reported in heart failure (Marx et al. 2000). To test whether indeed phosphatases dephosphorylated the RyR in our permeabilized myocyte experiments we performed quantitative immunoblotting using an antibody that specifically recognizes the phosphorylated form of the RyR at Ser-2809 (Rodriguez et al. 2003). Myocytes exhibited a significant level of phosphorylation under baseline conditions. Maximal phosphorylation was 201 % of control. When exposed to 2Uml_1 PP1, RyR phosphorylation was 58 % of the control basal condition. Exposing to a higher PP1 concentration (10Uml_1) further reduced RyR phosphorylation to 22% of control. Thus, consistent with the results of our functional measurements,
- PP1 decreased RyR phosphorylation in cardiac myocytes.
Figure 1. Effects of PP1 on properties of Ca(2+) sparks and SR Ca(2+) content in rat permeabilized myocytes see . http://dx.doi.org/10.1113/jphysiol.2003.046367 A, spontaneous Ca(2+) spark images recorded under reference conditions, and 1 or 5 min after exposure of the cell to 2Uml_1 PP1. Traces below the images are Ca(2+) transients induced by application of 10 mM caffeine immediately following the acquisition of sparks before (3 min) and after (5 min) application of PP1 in the same cell. The Ca(2+) transients were elicited by a whole bath application of 10 mM caffeine. B, averaged spark frequency at early (1 min) and late (5 min) times following the addition of either 0.5 or 2Uml_1 of PP1 to the bathing solution. C, averaged SR Ca(2+) content for 0.5 or 2Uml_1 of PP1 measured before and 5 min after exposure to the enzyme. Data are presented as means ± S.E.M. of 6 experiments in different cells. Figure 2. Effects of PP2A on properties of Ca2+ sparks and SR Ca2+ content in rat permeabilized myocytes see . http://dx.doi.org/10.1113/jphysiol.2003.046367 A, spontaneous Ca(2+) spark images recorded under reference conditions, and 1 or 5 min after exposure of the cell to 5Uml_1 PP2A. Traces below the images are Ca(2+) transients induced by application of 10 mM caffeine immediately following the acquisition of sparks before (3 min) and after (5 min) application of PP2A in the same cell. B and C, averaged spark frequency (B) and SR Ca(2+) content (C) for the same conditions as in A. Data are presented as means ± S.E.M. of 6 experiments in different cells.
DISCUSSION
In the present study, we have investigated the impact of physiologically relevant exogenous protein phosphatases PP1 and PP2A on RyR-mediated SR Ca(2+) release (measured as Ca(2+) sparks) in permeabilized heart cells. Our principal finding is that
- phosphatases stimulated RyR channels lead to depleted SR Ca(2+) stores.
These results have important ramifications for understanding the mechanisms and role of protein phosphorylation/dephosphorylation in
- modulation of Ca(2+) handling in normal and diseased heart.
Modulation of SR Ca2+ release by protein phosphorylation/dephophorylation
Since protein dephosphorylation clearly resulted in increased functional activity of the Ca(+)release channel, our results imply that a reverse, phosphorylation reaction should reduce RyR activity. If indeed such effects take place, why do they not manifest in inhibition of Ca(+)sparks? One possibility is that enhanced Ca(+) uptake by SERCA
- masks or overcomes the effects phosphorylation may have on RyRs.
In addition, the potential inhibitory influence of protein phosphorylation on RyR activity in myocytes could be countered by feedback mechanisms involving changes in luminal Ca(2+)(Trafford et al. 2002; Gyorke et al. 2002). In particular, reduced open probability of RyRs would be expected to lead to
- increased Ca2+ accumulation in the SR;
- and increased intra-SR [Ca(2+)], in turn would
- increase activity of RyRs at their luminal Ca(2+) regulatory sites
as demonstrated for the RyR channel inhibitor tetracaine (Gyorke et al. 1997; Overend et al. 1997). Thus
- potentiation of SERCA
- combined with the intrinsic capacity of the release mechanism to self-regulate
could explain at least in part why PKA-mediated protein phoshorylation results in maintained potentiation of Ca(2+) sparks despite a potential initial decrease in RyR activity.
Role of altered RyR Phosphorylation in Heart Failure
Marx et al. (2000) have proposed that enhanced levels of circulating catecholamines lead to increased phosphorylation of RyR in heart failure. Based on biochemical observations as well as on studying properties of single RyRs incorporated into artificial lipid bilayers, these investigators have hypothesized that
- hyperphosphorylation of RyRs contributes to pathogenesis of heart failure
- by making the channel excessively leaky due to dissociation of FKBP12.6 from the channel.
We show that the mode of modulation of RyRs by phosphatases does not support this hypothesis as
- dephosphorylation caused activation instead of
Interestingly, our results provide the basis for a different possibility in which
- dephophosphorylation of RyR rather than its phosphorylation causes depletion of SR Ca(2+) stores by stimulating RyRs in failing hearts.
It has been reported that PP1 and PP2 activities are increased in heart failure (Huang et al. 1999; Neumann et al. 1997; Neuman, 2002). Furthermore, overexpression of PP1 or ablation of the endogenous PP1 inhibitor, l-1, results in
- depressed contractile performance and heart failure (Carr et al. 2002).
Our finding that PP1 causes depletion of SR Ca(2+) stores by activating RyRs could account for, or contribute to, these results.
References
1 DelPrincipe F, Egger M, Pignier C & Niggli E (2001). Enhanced E-C coupling efficiency after beta-stimulation of cardiac myocytes. Biophys J 80, 64a. 2 Gyorke I & Gyorke S (1998). Regulation of the cardiac ryanodine receptor channel by luminal Ca2+ involves luminal Ca2+ sensing sites. Biophys J 75, 2801–2810. 3 Gyorke S, Gyorke I, Lukyanenko V, Terentyev D, Viatchenko-Karpinski S & Wiesner TF (2002). Regulation of sarcoplasmic reticulum calcium release by luminal calcium in cardiac muscle. Front Biosci 7, d1454–d1463. 4 Gyorke I, Lukyanenko V & Gyorke S (1997). Dual effects of tetracaine on spontaneous calcium release in rat ventricular myocytes. J Physiol 500, 297–309. 5 MacDougall LK, Jones LR & Cohen P (1991). Identification of the major protein phosphatases in mammalian cardiac muscle which dephosphorylate phospholamban. Eur J Biochem 196, 725–734. 6 Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N & Marks AR (2000). PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell 101, 365–376. 7 Rodriguez P, Bhogal MS & Colyer J (2003). Stoichiometric phosphorylation of cardiac ryanodine receptor on serine-2809 by calmodulin-dependent kinase II and protein kinase A. J Biol Chem (in press).
The δC Isoform of CaMKII Is Activated in Cardiac Hypertrophy and Induces Dilated Cardiomyopathy and Heart Failure
T Zhang, LS Maier, ND Dalton, S Miyamoto, J Ross, DM Bers, JH Brown. University of California, San Diego, La Jolla, Calif; and Loyola University, Chicago, Ill. Circ Res. 2003;92:912-919. http://dx.doi.org/10.1161/01.RES.0000069686.31472.C5 Recent studies have demonstrated that transgenic (TG) expression of either Ca(2+)/calmodulin-dependent protein kinase IV (CaMKIV) or CaMKIIδB, both of which localize to the nucleus, induces cardiac hypertrophy. However,
- CaMKIV is not present in heart, and
- cardiomyocytes express not only the nuclear CaMKIIδB
- but also a cytoplasmic isoform, CaMKII δC.
In the present study, we demonstrate that
- expression of the δC isoform of CaMKII is selectively increased and
- its phosphorylation elevated as early as 2 days and continuously for up to 7 days after pressure overload.
To determine whether enhanced activity of this cytoplasmic δC isoform of CaMKII can lead to phosphorylation of Ca(2+) regulatory proteins and induce hypertrophy, we generated TG mice that expressed the δC isoform of CaMKII. Immunocytochemical staining demonstrated that the expressed transgene is confined to the cytoplasm of cardiomyocytes isolated from these mice. These mice develop a dilated cardiomyopathy with up to a 65% decrease in fractional shortening and die prematurely. Isolated myocytes are enlarged and exhibit reduced contractility and altered Ca2(2+) handling. Phosphorylation of the ryanodine receptor (RyR) at a CaMKII site is increased even before development of heart failure, and
- CaMKII is found associated with the RyR from the CaMKII TG mice.
- Phosphorylation of phospholamban is increased specifically at the CaMKII but not at the PKA phosphorylation site.
These findings are the first to demonstrate that CaMKIIδC can mediate phosphorylation of Ca(2+) regulatory proteins in vivo and provide evidence for the involvement of CaMKIIδC activation in the pathogenesis of dilated cardiomyopathy and heart failure. Multifunctional Ca(2+)/calmodulin-dependent protein kinases (CaM kinases or CaMKs) are transducers of Ca2+ signals that phosphorylate a wide range of substrates and thereby affect Ca(2+)-mediated cellular responses.1 The family includes CaMKI and CaMKIV, monomeric enzymes activated by CaM kinase kinase,2,3 and CaMKII, a multimer of 6 to 12 subunits activated by autophosphorylation.1 The CaMKII subunits α, β, γ, and δ show different tissue distributions,1 with
- the δ isoform predominating in the heart.4–7
- Splice variants of the δ isoform, characterized by the presence of a second variable domain,4,7 include δB, which contains a nuclear localization signal (NLS), and
- δC, which does not. CaMKII composed of δB subunits localizes to the nucleus, whereas CaMKIIδC localizes to the cytoplasm.4,8,9
CaMKII has been implicated in several key aspects of acute cellular Ca(2+) regulation related to cardiac excitation-contraction (E-C) coupling. CaMKII
- phosphorylates sarcoplasmic reticulum (SR) proteins including the ryanodine receptors (RyR2) and
- phospholamban (PLB).10–14
Phosphorylation of RyR has been suggested to alter the channel open probability,14,15 whereas phosphorylation of PLB has been suggested to regulate SR Ca(2+) uptake.14 It is also likely that CaMKII phosphorylates the L-type Ca(2+) channel complex or an associated regulatory protein and thus
- mediates Ca(2+) current (ICa) facilitation.16-18 and
- the development of early after-depolarizations and arrhythmias.19
Thus, CaMKII has significant effects on E-C coupling and cellular Ca(2 +) regulation. Nothing is known about the CaMKII isoforms regulating these responses. Contractile dysfunction develops with hypertrophy, characterizes heart failure, and is associated with changes in cardiomyocyte (Ca2+) homeostasis.20 CaMKII expression and activity are altered in the myocardium of rat models of hypertensive cardiac hypertrophy21,22 and heart failure,23 and
- in cardiac tissue from patients with dilated cardiomyopathy.24,25
Several transgenic mouse models have confirmed a role for CaMK in the development of cardiac hypertrophy, as originally suggested by studies in isolated neonatal rat ventricular myocytes.9,26–28 Hypertrophy develops in transgenic mice that overexpress CaMKIV,27 but this isoform is not detectable in the heart,4,29 and CaMKIV knockout mice still develop hypertrophy after transverse aortic constriction (TAC).29 Transgenic mice overexpressing calmodulin developed severe cardiac hypertrophy,30 later shown to be associated with an increase in activated CaMKII31; the isoform of CaMKII involved in hypertrophy could not be determined from these studies. We recently reported that transgenic mice that overexpress CaMKIIδB, which is highly concentrated in cardiomyocyte nuclei, develop hypertrophy and dilated cardiomyopathy.32 To determine whether
- in vivo expression of the cytoplasmic CaMKIIδC can phosphorylate cytoplasmic Ca(2+) regulatory proteins and
- induce hypertrophy or heart failure,
we generated transgenic (TG) mice that expressed the δC isoform of CaMKII under the control of the cardiac specific α-myosin heavy chain (MHC) promoter. Our findings implicate CaMKIIδC in the pathogenesis of dilated cardiomyopathy and heart failure and suggest that
- this occurs at least in part via alterations in Ca(2+) handling proteins.33
Ca(2+) and contraction RyR  yuan_image3 Ca++ exchange
yuan_image3 Ca++ exchange
Results
Expression and Activation of CaMKIIδC Isoform After TAC
To determine whether CaMKII was regulated in pressure overload–induced hypertrophy, CaMKIIδ expression and phosphorylation were examined by Western blot analysis using left ventricular samples obtained at various times after TAC. A selective increase (1.6-fold) in the lower band of CaMKIIδwas observed as early as 1 day and continuously for 4 days (2.3-fold) and 7 days (2-fold) after TAC (Figure 1A). To confirm that CaMKIIδC was increased and determine whether this occurred at the transcriptional level, we performed semiquantitative RT-PCR using primers specific for the CaMKIIδC isoform. These experiments revealed that
- mRNA levels for CaMKIIδC were increased 1 to 7 days after TAC (Figure 1B).
In addition to examining CaMKII expression, the activation state of CaMKII was monitored by its autophosphorylation, which confers Ca2-independent activity.
Figure 1. Expression and activation of CaMKII δC isoform after TAC.
see http://dx.doi.org/10.1161/01.RES.0000069686.31472.C5 A, Western blot analysis of total CaMKII in left ventricular (LV) homogenates obtained at indicated times after TAC. Cardiomyocytes transfected with CaMKIIδB and δC (right) served as positive controls and molecular markers. Top band (58 kDa) represents CaMKIIδB plus δ9, and the bottom band (56 kDa) corresponds to CaMKIIδC. *P0.05 vs control. B, Semiquantitative RT-PCR using primers specific for CaMKIIδC isoform (24 cycles) and GAPDH (19 cycles) using total RNA isolated from the same LV samples. C, Western blot analysis of phospho-CaMKII in LV homogenates obtained at various times after TAC. Three bands seen for each sample represent CaMKIIγ subunit (uppermost), CaMKIIδB plus δ9 (58 kDa), and CaMKIIδC (56 kDa). Quantitation is based on the sum of all of the bands. *P0.05 vs control.
Figure 2. Expression and activation of CaMKII in CaMKIIδC transgenic mice.
see http://dx.doi.org/10.1161/01.RES.0000069686.31472.C5 A, Transgene copy number based on Southern blots using genomic DNA isolated from mouse tails (digested with EcoRI). Probe (a 32P-labeled 1.7-kb EcoRI-SalI -MHC fragment) was hybridized to a 2.3-kb endogenous fragment (En) and a 3.9-kb transgenic fragment (TG). Transgene copy number was determined from the ratio of the 3.9-kb/2.3-kb multiplied by 2. B, Immunocytochemical staining of ventricular myocytes isolated from WT and CaMKIIδTG mice. Myocytes were cultured on laminin-coated slides overnight. Transgene was detected by indirect immunofluorescence staining using rabbit anti-HA antibody (1:100 dilution) followed by FITC-conjugated goat antirabbit IgG antibody (1:100 dilution). CaMKIIδB localization to the nucleus in CaMKIIδB TG mice (see Reference 32) is shown here for comparative purpose. C, Quantitation of the fold increase in CaMKIIδprotein expression in TGL and TGM lines. Different amounts of ventricular protein (numbers) from WT control, TG () and their littermates () were immunoblotted with an anti-CaMKIIδ antibody. Standard curve from the WT control was used to calculate fold increases in protein expression in TGL and TGM lines. D, Phosphorylated CaMKII in ventricular homogenates was measured by Western blot analysis (n5 for each group). **P0.01 vs WT.
Generation and Identification of CaMKIIδC Transgenic Mice
TG mice expressing HA-tagged rat wild-type CaMKIIδC under the control of the cardiac-specific α-MHC promoter were generated as described in Materials and Methods. By Southern blot analysis, 3 independent TG founder lines carrying 3, 5, and 15 copies of the transgene were identified. They were designated as TGL (low copy number), TGM (medium copy number), and TGH (high copy number), The founder mice from the TGH line died at 5 weeks of age with marked cardiac enlargement. The other two lines showed germline transmission of the transgene. The transgene was expressed only in the heart. Although CaMKII protein levels in TGL and TGM hearts were increased 12- and 17-fold over wild-type (WT) controls (Figure 2C), the amount of activated CaMKII was only increased 1.7- and 3-fold in TGL and TGM hearts (Figure 2D). The relatively small increase in CaMKII activity in the TG lines probably reflects the fact that the enzyme is not constitutively activated and that the availability of Ca2/CaM, necessary for activation of the overexpressed CaMKII, is limited. Importantly,
- the extent of increase in active CaMKII in the TG lines was similar to that elicited by TAC.
Cardiac Overexpression of CaMKIIδC Induces Cardiac Hypertrophy and Dilated Cardiomyopathy
There was significant enlargement of hearts from CaMKIIδC TGM mice by 8 to 10 weeks [see http://dx.doi.org/10.1161/01.RES.0000069686.31472.C5%5D (Figure 3A) and from TGL mice by 12 to 16 weeks. Histological analysis showed ventricular dilation (Figure 3B), cardiomyocyte enlargement (Figure 3C), and mild fibrosis (Figure 3D) in CaMKIIδC TG mice. Quantitative analysis of cardiomyocyte cell volume from 12-week-old TGM mice gave values of 54.7 + 0.1 pL for TGM (n = 96) versus 28.6 + 0.1 pL for WT littermates (n=94; P0.001). Ventricular dilation and cardiac dysfunction developed over time in proportion to the extent of transgene expression. Left ventricular end diastolic diameter (LVEDD) was increased by 35% to 45%, left ventricular posterior wall thickness (LVPW) decreased by 26% to 29% and fractional shortening decreased by 50% to 60% at 8 weeks for TGM and at 16 weeks for TGL. None of these parameters were significantly altered at 4 weeks in TGM or up to 11 weeks in TGL mice, indicating that heart failure had not yet developed. Contractile function was significantly decreased. Figure 6. Dilated cardiomyopathy and dysfunction in CaMKIIδC TG mice at both whole heart and single cell levels. [see Fig 6: http://dx.doi.org/10.1161/01.RES.0000069686.31472.C5] C, Decreased contractile function in ventricular myocytes isolated from 12-week old TGM and WT controls presented as percent change of resting cell length (RCL) stimulated at 0.5 Hz. Representative trace and mean values are shown. *P0.05 vs WT. Figure 7. Phosphorylation of PLB in CaMKIIδC TG mice. [see Fig 7: http://dx.doi.org/10.1161/01.RES.0000069686.31472.C5] Thr17 and Ser16 phosphorylated PLB was measured by Western blots using specific anti-phospho antibodies. Ventricular homogenates were from 12- to 14-week-old WT and TGM mice (A) or 4 to 5-week-old WT and TGM mice (B). Data were normalized to total PLB examined by Western blots (data not shown here). n = 6 to 8 mice per group; *P0.05 vs WT.
Cardiac Overexpression of CaMKIIδC Results in Changes in the Phosphorylation of Ca2 Handling Proteins
To assess the possible involvement of phosphorylation of Ca2cycling proteins in the phenotypic changes observed in the CaMKIIC TG mice, we first compared PLB phosphorylation state in homogenates from 12- to 14-week-old TGM and WT littermates. Western blots using antibodies specific for phosphorylated PLB showed a 2.3-fold increase in phosphorylation of Thr17 (the CaMKII site) in hearts from TGM versus WT (Figure 7A). Phosphorylation of PLB at the CaMKII site was also increased 2-fold in 4- to 5-week-old TGM mice (Figure 7B). Significantly, phosphorylation of the PKA site (Ser16) was unchanged in either the older or the younger TGM mice (Figures 7A and 7B). (see http://dx.doi.org/10.1161/01.RES.0000069686.31472.C5) To demonstrate that the RyR2 phosphorylation changes observed in the CaMKII transgenic mice are not secondary to development of heart failure, we performed biochemical studies examining RyR2 phosphorylation in 4- to 5-week-old TGM mice. At this age, most mice showed no signs of hypertrophy or heart failure (see Figure 6B) and there was no significant increase in myocyte size (21.3 + 1.3 versus 27.7 + 4.6 pL; P0.14). Also, twitch Ca2 transient amplitude was not yet significantly depressed, and mean δ [Ca2+]i (1 Hz) was only 20% lower (192 + 36 versus 156 + 13 nmol/L; P0.47) versus 50% lower in TGM at 13 weeks.33 The in vivo phosphorylation of RyR2, determined by back phosphorylation, was significantly (2.10.3-fold; P0.05) increased in these 4- to 5-week-old TGM animals (Figure 8C), an increase equivalent to that seen in 12- to 14-week-old mice. We also performed the RyR2 back-phosphorylation assay using purified CaMKII rather than PKA. RyR2 phosphorylation at the CaMKII site was also significantly increased (2.2 + 0.3-fold; P0.05) in 4- to 5-week-old TGM mice (Figure 8C). (http://dx.doi.org/10.1161/01.RES.0000069686.31472.C5) The association of CaMKII with the RyR2 is consistent with a physical interaction between this protein kinase and its substrate. The catalytic subunit of PKA and the phosphatases PP1 and PP2A were also present in the RyR2 immunoprecipitates, but not different in WT versus TG mouse hearts (Figure 8D). These data provide further evidence that
- the increase in RyR2 phosphorylation, which precedes development of failure in the 4- to 5-week-old CaMKIIδC TG hearts, can be attributed to the increased activity of CaMKII.
Discussion
- CaMKII is involved in the dynamic modulation of cellular
- Ca2 regulation and has been implicated in the development of cardiac hypertrophy and heart failure.14
- Published data from CaMK-expressing TG mice demonstrate that forced expression of CaMK can induce cardiac hypertrophy and lead to heart failure.27,32
However, the CaMK genes expressed in these mice are neither the endogenous isoforms of the enzyme nor the isoforms likely to regulate cytoplasmic Ca(2+) handling, because they localize to the nucleus.
- the cytoplasmic cardiac isoform of CaMKII is upregulated at the expression level and is in the active state (based on autophosphorylation) after pressure overload induced by TAC.
- two cytoplasmic CaMKII substrates (PLB and RyR) are phosphorylated in vivo when CaMKII is overexpressed and its activity increased to an extent seen under pathophysiological conditions.
- CaMKIIδ is found to associate physically with the RyR in the heart.
- heart failure can result from activation of the cytoplasmic form of CaMKII and this may be due to altered Ca(2+) handling.
Differential Regulation of CaMKIIδ Isoforms in Cardiac Hypertrophy
- The isoform of CaMKII that predominates in the heart is the δ isoform.4–7 Neither the α nor the β isoforms are expressed and there is only a low level of expression of the γ isoforms.39
- Both δB and δC splice variants of CaMKIIδ are present in the adult mammalian myocardium36,40 and expressed in distinct cellular compartments.4,8,9
We suggest that the CaMKIIδ isoforms are differentially regulated in pressure-overload–induced hypertrophy, because the expression of CaMKIIδC is selectively increased as early as 1 day after TAC. Studies using RT-PCR confirm that
- CaMKIIδC is regulated at the transcriptional level in response to TAC. In addition,
- activation of both CaMKIIδB and CaMKIIδC, as indexed by autophosphorylation, increases as early as 2 days after TAC.
- Activation of CaMKIIδB by TAC is relevant to our previous work indicating its role in hypertrophy.9,32
- The increased expression, as well as activation of the CaMKIIδC isoform, suggests that it could also play a critical role in both the acute and longer responses to pressure overload.
In conclusion, we demonstrate here that CaMKIIδC can phosphorylate RyR2 and PLB when expressed in vivo at levels leading to 2- to 3-fold increases in its activity. Similar increases in CaMKII activity occur with TAC or in heart failure. Data presented in this study and in the accompanying article33 suggest that altered phosphorylation of Ca(2+) cycling proteins is a major component of the observed decrease in contractile function in CaMKIIδC TG mice. The occurrence of increased CaMKII activity after TAC, and of RyR and PLB phosphorylation in the CaMKIIδC TG mice suggest that
- CaMKIIδC plays an important role in the pathogenesis of dilated cardiomyopathy and heart failure.
These results have major implications for considering CaMKII and its isoforms in exploring new treatment strategies for heart failure.
Cardiac Electrophysiological Dynamics From the Cellular Level to the Organ Level
Daisuke Sato and Colleen E. Clancy Department of Pharmacology, University of California – Davis, Davis, CA. Biomedical Engineering and Computational Biology 2013:5: 69–75 http://www.la-press.com. http://dx.doi.org/10.4137/BECB.S10960 Abstract: Cardiac alternans describes contraction of the ventricles in a strong-weak-strong-weak sequence at a constant pacing frequency. Clinically, alternans manifests as alternation of the T-wave on the ECG and predisposes individuals to arrhythmia and sudden cardiac death. In this review, we focus on the fundamental dynamical mechanisms of alternans and show how alternans at the cellular level underlies alternans in the tissue and on the ECG. A clear picture of dynamical mechanisms underlying alternans is important to allow development of effective anti-arrhythmic strategies. The cardiac action potential is the single cellular level electrical signal that triggers contraction of the heart.1 Under normal conditions, the originating activation signal comes from a small bundle of tissue in the right atrium called the sinoatrial node (SAN). The action potentials generated by the SAN initiate an excitatory wave that, in healthy tissue, propagates smoothly through a well-defined path and causes excitation and contraction in the ventricles. In disease states, the normal excitation pathway is disrupted and a variety of abnormal rhythms can occur, including cardiac alternans, a well-known precursor to sudden cardiac death. Cardiac alternans was initially documented in 1872 by a German physician, Ludwig Traube.2 He observed contraction of the ventricles in a strong-weak-strong-weak sequence even though the pacing frequency was constant. Clinically, alternans manifests as alternation of the T-wave on the ECG, typically in the microvolt range. It is well established that individuals with microvolt T-wave alternans are at much higher risk for arrhythmia and sudden cardiac death. A clear picture of physiological mechanisms underlying alternans is important to allow development of effective anti-arrhythmic drugs. It is also important to understand dynamical mechanisms because while the cardiac action potential is composed of multiple currents, each of which confers specific properties, revelation of dynamical mechanisms provides a unified fundamental view of the emergent phenomena that holds independently of specific current interactions. The ventricular myocyte is an excitable cell providing the cellular level electrical activity that underlies cardiac contraction. Under resting conditions, the membrane potential is about -80 mV. When the cell is stimulated, sodium (Na) channels open and the membrane potential goes above 0 mV. Then, a few ms later, the inward current L-type calcium (Ca) current activates and maintains depolarization of the membrane potential. During this action potential plateau, several types of outward current potassium (K) channels also activate. Depending on the balance between inward and outward currents, the action potential duration (APD) is determined.The diastolic interval (DI) that follows cellular repolarization describes the duration the cell resides in the resting state until the next excitation. During the DI, channels recover with kinetics determined by intrinsic time constants. APD restitution defines the relationship between the APD and the previous DI (Fig. 1 top panel). In most cases1, the APD becomes longer as the previous DI becomes longer due to recovery of the L-type Ca channel (Fig. 1, bottom panel), and thus the APD restitution curve has a positive slope. Figure 1. (Top): APD and DI. (Bottom): The physiological mechanism of APD alternans involves recovery from inactivation of ICaL. [see http://dx.doi.org/10.4137/BECB.S10960]
Action Potential Duration Restitution
In 1968 Nolasco and Dahlen showed graphically that APD alternans occurs when the slope of the APD restitution curve exceeds unity. Why is the steepness of the slope important? As shown graphically in Figure 2, APD alternans amplitude is multiplied by the slope of the APD restitution curve in each cycle. When the slope is larger than one, then the alternans amplitude will be amplified until the average slope reaches 1 or the cell shows a 2:1 stimulus to response ratio. The one-dimensional mapping between APD and DI fails to explain quasi-periodic oscillation of the APD. Figure 2. APD restitution and dynamical mechanism of APD alternans. [see http://dx.doi.org/10.4137/BECB.S10960]
Calcium Driven Alternans
A strong-weak-strong-weak oscillation in contraction implies that the Ca transient (CaT) is alternating. Until 1999 it was assumed that if the APD is alternating then the CaT alternates because the CaT follows APD changes. However, Chudin et al showed that CaT can alternate even when APD is kept constant during pacing with a periodic AP clamp waveform.14 This implies that the intracellular Ca cycling has intrinsic nonlinear dynamics. A critical component in this process is the sarcoplasmic reticulum (SR), a subcellular organelle that stores Ca inside the cell. When Ca enters a cell through the L-type Ca channel (or reverse mode Na-Ca exchanger (NCX) ryanodine receptors open and large Ca releases occur from the SR (Ca induced Ca release). The amount of Ca release steeply depends on SR Ca load. This steep relation between Ca release and SR Ca load is the key to induce CaT alternans. A one-dimensional map between Ca release and SR calcium load can be constructed to describe the relationship21 similar to the map used in APD restitution.
Subcellular Alternans
A number of experimental and computational studies have been undertaken to identify molecular mechanisms of CaT alternans by identifying the specific components in the calcium cycling process critical to formation of CaT alternans. These components include SR Ca leak and load, Ca spark frequency and amplitude, and rate of SR refilling. For example, experiments have shown that alternation in diastolic SR Ca is not required for CaT alternans.24 In addition, stochastic openings of ryanodine receptors (RyR) lead to Ca sparks that occur randomly, not in an alternating sequence that would be expected to underlie Ca altern-ans. So, how do local random sparks and constant diastolic SR calcium load lead to global CaT alternans? Mathematical models with detailed representations of subcellular Ca cycling have been developed in order to elucidate the underlying mechanisms. Modeling studies have shown that even when SR Ca load is not changing, RyRs, which are analogous to ICaL in APD alternans, recover gradually from refractoriness. As RyR availability increases (for example during a long diastolic interval) a single Ca spark from a RyR will be larger in amplitude and recruit neighboring Ca release units to generate more sparks. The large resultant CaT causes depletion of the SR and when complete recovery of RyRs does not occur prior to the arrival of the next stimulus, the subsequent CaT will be small. This process results in an alternans of CaT amplitude from beat-to-beat.
Coupling Between the Membrane Potential and Subcellular Calcium Dynamics
Importantly, the membrane voltage and intracellular Ca cycling are coupled via Ca sensitive channels such as the L-type Ca channel and the sodium-calcium exchanger (NCX). The membrane voltage dynamics and the intracellular Ca dynamics are bi-directionally coupled. One direction is from voltage to Ca. As the DI becomes longer, the CaT usually becomes larger since the recovery time for the L-type Ca channel in increased and the SR Ca release becomes larger. The other direction is from Ca to voltage. Here we consider two major currents, NCX and ICaL. As the CaT becomes larger, forward mode NCX becomes larger and prolongs APD. On the other hand, as the CaT becomes larger, ICaL becomes smaller due to Ca-induced inactivation, and thus, larger CaT shortens the APD. Therefore, depending on which current dominates, larger CaT can prolong or shorten APD. If a larger CaT prolongs (shortens) the APD, then the coupling is positive (negative). The coupled dynamics of the membrane voltage and the intracellular Ca cycling can be categorized by the instability of membrane voltage (steep APD restitution), instability of the intracellular Ca cycling (steep relation between Ca release versus SR Ca load), and the coupling (positive or negative). If the coupling is positive, alternans is electromechanically concordant (long-short-long-short APD corresponds to large-small-large-small CaT sequence) regardless of the underlying instability mechanism. On the other hand, if the coupling is negative, alternans is electromechanically concordant in a voltage-driven regime. However, if alternans is Ca driven, alternans becomes electromechanically discordant (long-short-long-short APD corresponds to small-large-small-large CaT sequence). It is also possible to induce quasi- periodic oscillation of APD and CaT when voltage and Ca instabilities contribute equally.
Alternans in Higher Dimensions
Tissue level alternans in APD and CaT also occur and here we describe how the dynamical mechanism of alternans at the single cell level determines the phenomena in tissue. Spatially discordant alternans (SDA) where APDs in different regions of tissue alternate out-of-phase, is more arrhythmogenic since it causes large gradients of refractoriness and wave-break, which can initiate ventricular tachycardia and ventricular fibrillation. How is SDA induced? As the APD is a function of the previous DI, conduction velocity (CV) is also function of the previous DI (CV restitution) since the action potential propagation speed depends on the availability of the sodium channel. As the DI becomes shorter, sodium channels have less time to recover. Therefore, in general, as the DI becomes shorter, the CV becomes slower. When tissue is paced rapidly, action potentials propagate slowly near the stimulus, and thenac-celerate downstream as the DI becomes longer. This causes heterogeneity in APD (APD is shorter near the stimulus). During the following tissue excitation, APD becomes longer and the CV becomes faster at the pacing site then gradually APD becomes shorter and the CV becomes slower. The interaction between steep APD restitution and steep CV restitution creates SDA. This mechanism applies only when the cellular instability is voltage driven. When the cellular instability is Ca driven, the mechanism of SDA formation is different. If the voltage-Ca coupling is negative, SDA can form without steep APD and CV restitution. The mechanism can be understood as follows. First, when cells are uncoupled, alternans of APD and Ca are electromechanically discordant. If two cells are alternating in opposite phases, once these cells are coupled by voltage, due to electrotonic coupling, the membrane voltage of both cells is synchronized and thus APD becomes the same. This synchronization of APD amplifies the difference of CaT between two cells (Fig. 5 in). In other words it desynchronizes CaT. This instability mechanism is also found in subcellular SDA. In the case where the instability is Ca driven and the coupling is positive, there are several interesting distinctive phenomena that can occur. First, the profile of SDA of Ca contains a much steeper gradient at the node (point in space where no alternans occurs–cells downstream of the node are alternating out of phase with those upstream of the node) compared to the case of voltage driven SDA. Thus, the cellular mechanism of instability can be identified by evaluating the steepness of the alternans amplitude gradient in space around the node. When the cellular instability is voltage driven, the steady-state wavelength (separation of nodes in space) depends on electrotonic coupling between cells and the steepness of APD and CV restitution, regardless of the initial conditions. However, if the cellular instability is Ca driven, the location of nodes depends on the pacing history, which includes pacing cycle length and other parameters affected by pacing frequency. In this case, once the node is formed, the location of the node may be fixed, especially when Ca instability is strong. Such an explanation may apply to recent experimental results. Summary In this review, we described how the origin of alternans at the cellular level (voltage driven, Ca drive, coupling between voltage and Ca) affects the formation of spatially discordant alternans at the tissue level. Cardiac alternans is a multi-scale emergent phenomenon. Channel properties determine the instability mechanism at the cellular level. Alternans mechanisms at cellular level determine SDA patterns at the tissue level. In order to understand alternans and develop anti-arrhythmic drug and therapy, multi-scale modeling of the heart is useful, which is increasingly enabled by emerging technologies such as general-purpose computing on graphics processing units (GPGPU) and cloud computing.
Related articles
- The Centrality of Ca(2+) Signaling and Cytoskeleton Involving Calmodulin Kinases and Ryanodine Receptors in Cardiac Failure, Arterial Smooth Muscle, Post-ischemic Arrhythmia, Similarities and Differences, and Pharmaceutical Targets (pharmaceuticalintelligence.com)
- Renal Distal Tubular Ca2+ Exchange Mechanism in Health and Disease (pharmaceuticalintelligence.com)
- Role of Calcium, the Actin Skeleton, and Lipid Structures in Signaling and Cell Motility (pharmaceuticalintelligence.com)
- Cardiac Contractility & Myocardium Performance: Ventricular Arrhythmias and Non-ischemic Heart Failure – Therapeutic Implications for Cardiomyocyte Ryanopathy (Calcium Release-related Contractile Dysfunction) and Catecholamine Responses in the Human Heart (pharmaceuticalintelligence.com)
- New coating may reduce blood clot risk inside stents (medicalnewstoday.com)
- Alternative Designs for the Human Artificial Heart: The Patients in Heart Failure – Outcomes of Transplant (donor)/Implantation (artificial) and Monitoring Technologies for the Transplant/Implant Patient in the Community (pharmaceuticalintelligence.com)
- Enzyme in airway lining cells could hold key for asthma sufferers (medicalnewstoday.com)
- Scientists Use Human Stem Cells to Engineer a Beating Mouse Heart (news.softpedia.com)
- New pathway in blood vessel inflammation and disease discovered – Kruppel-like factors as master regulators of vascular health (medicalnewstoday.com)

English: Diagram of contraction of smooth muscle fiber (Photo credit: Wikipedia)







