Nobel Prize in Physiology or Medicine 2013 for Cell Transport: James E. Rothman of Yale University; Randy W. Schekman of the University of California, Berkeley; and Dr. Thomas C. Südhof of Stanford University
Reporter: Aviva Lev-Ari, PhD, RN
Comments by Graduate Students of the nobel Prize Recipients and other in NYT, 10/7/2013:
- Metastasis, Chapel Hill, NC Oct. 7, 2013 at 11:48 a.m
I had the privilege of meeting Randy Schekman a few times when I was a postdoc at Berkeley. In addition to pioneering the understand of cellular trafficking, he was also a great colleague and educator (of undergrads, grad students, postdocs). Hats off to a wonderful scientist who also pays it forward to future generations as a mentor!
- Paul Knoepfler, Davis, CA Oct. 7, 2013 at 12:16 p.m
Last couple years, including this year, the Nobel for Physiology or Medicine Award has been dominated by Cell Biologists. I think this highlights how understanding cells is really the key to most medicine.
Paul Knoepfler
http://www.ipscell.com
- Raj, Long Island, NY, Oct. 7, 2013 at 8:20 a.m
I guess UC Berkeley will have to add a few more Nobel Laureate Parking Spots on their campus now!
Yes, in parking-challenged Berkeley campus, some of the best parking spots are reserved for the Nobel Laureate Faculty. They have so many winners, and rather spotty on-campus parking, so they don’t want such brains to go hunt for parking. They reason that the Laureates should be doing better things, like more research, or assisting newer researchers and students. A most elegant solution!
I don’t think there is any other institution anywhere in the world that has dedicated parking for their Nobel-winning employees. Or has so many Nobels on the payroll. But then, there is just one Cal.
This prize is another testament to UC Berkeley’s standing.
Congratulations to the scientists, and a big thank you to their institutions that allowed them the freedom and resources to pursue their ideas.
Randy Schekman awarded 2013 Nobel Prize in Physiology or Medicine
By Robert Sanders, Media Relations | October 7, 2013
 Randy Schekman, who will share the 2013 Nobel Prize in Physiology or Medicine (Peg Skorpinski photo)
Randy Schekman, who will share the 2013 Nobel Prize in Physiology or Medicine (Peg Skorpinski photo)Randy W. Schekman, professor of molecular and cell biology at the University of California, Berkeley, has won the 2013 Nobel Prize in Physiology or Medicine for his role in revealing the machinery that regulates the transport and secretion of proteins in our cells. He shares the prize with James E. Rothman of Yale University and Thomas C. Südhof of Stanford University.
Discoveries by Schekman about how yeast secrete proteins led directly to the success of the biotechnology industry, which was able to coax yeast to release useful protein drugs, such as insulin and human growth hormone. The three scientists’ research on protein transport in cells, and how cells control this trafficking to secrete hormones and enzymes, illuminated the workings of a fundamental process in cell physiology.
Schekman is UC Berkeley’s 22nd Nobel Laureate, and the first to receive the prize in the area of physiology or medicine.
In a statement, the 50-member Nobel Assembly lauded Rothman, Schekman and Südhof for making known “the exquisitely precise control system for the transport and delivery of cellular cargo. Disturbances in this system have deleterious effects and contribute to conditions such as neurological diseases, diabetes, and immunological disorders.”
“My first reaction was, ‘Oh, my god!’ said Schekman, 64, who was awakened at his El Cerrito home with the good news at 1:30 a.m. “That was also my second reaction.”
Be part of our developing story on Storify and Twitter: Tweet your congratulations to Professor Schekman, using hashtag #BerkeleyNobel.
Also see:
Happy ending for Berkeley’s newest Nobel winner
Schekman and Rothman separately mapped out one of the body’s critical networks, the system in all cells that shuttles hormones and enzymes out and adds to the cell surface so it can grow and divide. This system, which utilizes little membrane bubbles to ferry molecules around the cell interior, is so critical that errors in the machinery inevitably lead to death.
“Ten percent of the proteins that cells make are secreted, including growth factors and hormones, neurotransmitters by nerve cells and insulin from pancreas cells,” said Schekman, a Howard Hughes Medical Institute Investigator and a faculty member in the Li Ka Shing Center for Biomedical and Health Sciences.
 Schekman takes a call at home after getting the news. (Carol Ness photo)
Schekman takes a call at home after getting the news. (Carol Ness photo)In what some thought was a foolish decision, Schekman decided in 1976, when he first joined the College of Letters and Science at UC Berkeley, to explore this system in yeast. In the ensuing years, he mapped out the machinery by which yeast cells sort, package and deliver proteins via membrane bubbles to the cell surface, secreting proteins important in yeast communication and mating. Yeast also use the process to deliver receptors to the surface, the cells’ main way of controlling activities such as the intake of nutrients like glucose.
In the 1980s and ’90s, these findings enabled the biotechnology industry to exploit the secretion system in yeast to create and release pharmaceutical products and industrial enzymes. Today, diabetics worldwide use insulin produced and discharged by yeast, and most of the hepatitis B vaccine used around the world is secreted by yeast. Both systems were developed by Chiron Corp. of Emeryville, Calif., now part of Novartis International AG, during the 20 years Schekman consulted for the company.
Various diseases, including some forms of diabetes and a form of hemophilia, involve a hitch in the secretion system of cells, and Schekman is now investigating a possible link to Alzheimer’s disease.
“Our findings have aided people in understanding these diseases,” said Schekman.
Based on the machinery discovered by Schekman and Rothman, Südhof subsequently discovered how nerve cells release signaling molecules, called neurotransmitters, which they use to communicate.
For his scientific contributions, Schekman was elected to the National Academy of Sciences in 1992, received the Gairdner International Award in 1996 and the Lasker Award for basic and clinical research in 2002. He was elected president of the American Society for Cell Biology in 1999. On Oct. 3, Schekman received the Otto Warburg Medal of the German Society for Biochemistry and Molecular Biology, which is considered the highest German award in the fields of biochemistry and molecular biology.
Schekman, formerly editor of the journal Proceedings of the National Academy of Sciences, currently is editor-in-chief of the new open access journal eLife.
Schekman and his wife, Nancy Walls, have two adult children.
MORE INFORMATION
- Nobel Prize summary
- Randy Schekman: A passion for yeast (2002)
- HHMI research summary (2012)
- UC Berkeley cell biologist receives Lasker Award for cell secretion research important to biotech industry (2002)
SOURCE
Thomas Südhof wins Nobel Prize in Physiology or Medicine
Neuroscientist Thomas Südhof, MD, professor of molecular and cellular physiology at the Stanford School of Medicine, won the 2013 Nobel Prize in Physiology or Medicine.
BY KRISTA CONGER
 Thomas Sudhof won the 2013 Nobel Prize in Physiology or Medicine.
Thomas Sudhof won the 2013 Nobel Prize in Physiology or Medicine.Neuroscientist Thomas Südhof, MD, professor of molecular and cellular physiology at the Stanford University School of Medicine, won the 2013 Nobel Prize in Physiology or Medicine.
He shared the prize with James Rothman, PhD, a former Stanford professor of biochemistry, andRandy Schekman, PhD, who earned his doctorate at Stanford under the late Arthur Kornberg, MD, another winner of the Nobel Prize in Physiology or Medicine.
The three were awarded the prize “for their discoveries of machinery regulating vesicle traffic, a major transport system in our cells.” Rothman is now a professor at Yale University, and Schekman is a professor at UC-Berkeley.
“I’m absolutely surprised,” said Südhof, who was in the remote town of Baeza in Spain to attend a conference and give a lecture. “Every scientist dreams of this. I didn’t realize there was chance I would be awarded the prize. I am stunned and really happy to share the prize with James Rothman and Randy Schekman.”
The three winners will share a prize that totals roughly $1.2 million, with about $413,600 going to each.
Robert Malenka, MD, Stanford’s Nancy Friend Pritzker Professor in Psychiatry and Behavioral Sciences, is at the conference with Südhof, a close collaborator. “He’s dazed, tired and happy,” Malenka said by phone. “The only time I’ve seen him happier was when his children were born.”
Südhof, the Avram Goldstein Professor in the School of Medicine, received the award for his work in exploring how neurons in the brain communicate with one another across gaps called synapses. Although his work has focused on the minutiae of how molecules interact on the cell membranes, the fundamental questions he’s pursuing are large.
“The brain works by neurons communicating via synapses,” Südhof said in a phone conversation this morning. “We’d like to understand how synapse communication leads to learning on a larger scale. How are the specific connections established? How do they form? And what happens in schizophrenia and autism when these connections are compromised?” In 2009, he published research describing how a gene implicated in autism and schizophrenia alters mice’s synapses and produces behavioral changes in the mice, such as excessive grooming and impaired nest building, that are reminiscent of these human neuropsychiatric disorders.
Lloyd Minor, MD, dean of the School of Medicine, said, “Thomas Südhof is a consummate citizen of science. His unrelenting curiosity, his collaborative spirit, his drive to ascertain the minute details of cellular workings, and his skill to carefully uncover these truths — taken together it’s truly awe-inspiring.
“He has patiently but relentlessly probed one of the fundamental questions of medical science — perhaps the fundamental question in neuroscience: How nerve cells communicate with each other. The answer is at the crux of human biology and of monumental importance to human health. Dr. Südhof’s receipt of this prize is inordinately well-deserved, and I offer him my heartfelt congratulations. His accomplishment represents what Stanford Medicine and the biomedical revolution are all about.”
The Nobel committee called Südhof on his cell phone after trying his home in Menlo Park, Calif. His wife, Lu Chen, PhD, associate professor of neurosurgery and of psychiatry and behavioral sciences, then gave the committee his cell phone number to reach him in Spain.
“The phone rang three times before I decided to go downstairs and pick it up,” Chen said. “I thought it was one of my Chinese relatives who couldn’t figure out the time zone.”
Chen and Südhof have two young children, and Südhof has four adult children from a previous marriage. “I was very surprised,” Chen said, “but he’s more concerned about how I’ll get the kids up this morning in time for school.”
“I was expecting a call from a colleague about the conference I’m here to attend, so I pulled off in a parking lot,” said Südhof, who was driving from Madrid to Baeza at the time he received the announcement. “I hadn’t slept at all the previous night, and I certainly wasn’t expecting a call from the Nobel committee.”
On the day he got the call from the Nobel committee, he was scheduled to give a talk at a conference, Membrane Traffic at the Synapse: The Cell Biology of Synaptic Plasticity, held in a 17th-century building that now serves as a conference center.
“Professor Sudhof’s contributions to the understanding of how cells operate have been of enormous importance to medicine, and to his own work in understanding how connections form within the human brain,” said Stanford President John Hennessy. “The recognition by the Nobel committee is a remarkable achievement.”
Südhof, who is also a Howard Hughes Medical Institute investigator, has spent the past 30 years prying loose the secrets of the synapse, the all-important junction where information, in the form of chemical messengers called neurotransmitters, is passed from one neuron to another. The firing patterns of our synapses underwrite our consciousness, emotions and behavior. The simple act of taking a step forward, experiencing a fleeting twinge of regret, recalling an incident from the morning commute or tasting a doughnut requires millions of simultaneous and precise synaptic firing events throughout the brain and peripheral nervous system.
Even a moment’s consideration of the total number of synapses in the typical human brain adds up to instant regard for that organ’s complexity. Coupling neuroscientists’ ballpark estimate of 200 billion neurons in a healthy adult brain with the fact that any single neuron may share synaptic contacts with as few as one or as many as 1 million other neurons (the median is somewhere in the vicinity of 10,000) suggests that your brain holds perhaps 2 quadrillion synapses — 10,000 times the number of stars in the Milky Way.
“The computing power of a human or animal brain is much, much higher than that of any computer,” said Südhof. “A synapse is not just a relay station. It is not even like a computer chip, which is an immutable element. Every synapse is like a nanocomputer all by itself. The amount of neurotransmitter released, or even whether that release occurs at all, depends on that particular synapse’s previous experience.”
Much of a neuron can be visualized as a long, hollow cord whose outer surface conducts electrical impulses in one direction. At various points along this cordlike extension are bulbous nozzles known as presynaptic terminals, each one housing myriad tiny, balloon-like vesicles containing neurotransmitters and each one abutting a downstream (or postsynaptic) neuron.
When an electrical impulse traveling along a neuron reaches one of these presynaptic terminals, calcium from outside the neuron floods in through channels that open temporarily, and a portion of the neurotransmitter-containing vesicles fuse with the surrounding bulb’s outer membrane and spill their contents into the narrow gap separating the presynaptic terminal from the postsynaptic neuron’s receiving end.
Südhof, along with other researchers worldwide, has identified integral protein components critical to the membrane fusion process. Südhof purified key protein constituents sticking out of the surfaces of neurotransmitter-containing vesicles, protruding from nearby presynaptic-terminal membranes, or bridging them. Then, using biochemical, genetic and physiological techniques, he elucidated the ways in which the interactions among these proteins contribute to carefully orchestrated membrane fusion: As a result, synaptic transmission is today one of the best-understood phenomena in neuroscience.
Südhof, who was born in Germany in 1955, received an MD in 1982 from Georg-August-Universität in Göttingen. He came to Stanford in 2008 after 25 years at the University of Texas Southwestern Medical Center at Dallas, where he first worked as a postdoctoral fellow at the laboratories of Michael Brown, MD, and Joseph Goldstein, MD.. Brown and Goldstein were awarded the Nobel Prize in Physiology or Medicine in 1985 for their work in understanding the regulation of cholesterol metabolism. In 1986, Südhof established his own laboratory at the university.
Südhof became an HHMI investigator in 1991, and moved to Stanford as a professor in molecular and cellular physiology in 2008.
The proteins Südhof has focused on for close to three decades are disciplined specialists. They recruit vesicles, bring them into “docked” positions near the terminals, herd calcium channels to the terminal membrane, and, cued by calcium, interweave like two sides of a zipper and force the vesicles into such close contact with terminal membranes that they fuse with them and release neurotransmitters into the synaptic gap. Although these specialists perform defined roles at the synapses, similar proteins, discovered later by Südhof and others, play comparable roles in other biological processes ranging from hormone secretion to fertilization of an egg during conception to immune cells’ defense against foreign invaders.
“We’ve made so many major advances during the past 50 years in this field, but there’s still much more to learn,” said Südhof, who in a 2010 interview with The Lancet credited his bassoon instructor as his most influential teacher for helping him to learn the discipline to practice for hours on end. “Understanding how the brain works is one of the most fundamental problems in neuroscience.”
Südhof’s accomplishments also earned him the 2013 Lasker Basic Medical Research Award. He is a member of the National Academy of Sciences, the Institute of Medicine and the American Academy of Arts & Sciences. He also is a recipient of the 2010 Kavli Prize in neuroscience.
In the Lancet interview, Südhof defined basic research as an approach often neglected in the pursuit of medicine. “This ‘solid descriptive science,’ like neuroanatomy or biochemistry, [are] disciplines that cannot claim to immediately understand functions or provide cures, but which form the basis for everything we do.”
Südhof said this morning he is excited to speak with his family about the prize, although it may be too much for his youngest children, ages 3 and 4, to grasp. “I will try to explain it to them,” he said. “It will be a wonderful occasion.” He noted that he has already received congratulatory calls from two of his four adult children. For them, the news may have come as less of a surprise.
“The Nobel prize became an inevitable topic of conversation when Tom won the Lasker award,” Chen said. “But the two of us share a feeling that one should never work for prizes.”
“Everyone has pegged him as a potential Nobel prize winner for many years,” said Malenka, who described the scene at the conference during the lunch hour. “It was just a matter of time. The attendees were clapping and cheering for him.”
Although he plans to return to the United States as soon as possible, Südhof has no plans to let the award slow his research — or even his plans for the day. He responded to an inquiry with a characteristically low-key reply. “Well, I think I’ll go ahead and give my talk.”
Rothman Lab
Membrane fusion is a fundamental biological process for organelle formation, nutrient uptake, and the secretion of hormones and neurotransmitters.
It is central to vesicular transport, storage, and release in many areas of endocrine and exocrine physiology, and imbalances in these processes give rise to important diseases, such as diabetes.
We employ diverse biophysical, biochemical, and cell biological approaches to characterize the fundamental participants in intracellular transport processes.

Time lapse images of fusing flipped-SNARE cells.
SNARE Overview
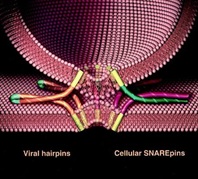
Over 30 years ago, we observed what we interpreted to be vesicular transport in crude extracts of tissue culture cells. In subsequent years we found that we had reconstituted vesicle trafficking in the Golgi, including the process of membrane fusion. Using this assay as a guide, we purified as a required factor the NEM-Sensitive Fusion protein (NSF). This led to the purification of the Soluble NSF Attachment Factor (SNAP), which bound NSF to Golgi membranes, and then with these tools discovered that the receptors for SNAP in membranes were actually complexes of proteins (which we called SNAREs) which we envisioned could potentially partner as a bridge between membranes to contribute to the process of membrane fusion and provide specificity to it (as captured in the ‘SNARE hypothesis’ proposed at the time).
We now know that organisms have a large family of SNARE proteins that indeed form cognate partnerships in just this way, and that NSF is an ATPase that (using SNAP as an adaptor protein) disrupts the SNARE complex after fusion is complete so its subunits can be recycled for repeated use. Recombinant cognate SNAREs introduced into artificial bilayers or expressed ectopically on the outside of cells ( “flipped SNAREs”) spontaneously and efficiently result in membrane (or cell) fusion, demonstrating that the SNARE complex is not only necessary but is sufficient for fusion. There are many proteins known and rapidly being discovered which closely regulate this vital process, but the muscle – if not always the brains – is in the SNAREs. Compartmental specificity is encoded to a remarkable degree in the functional partnering of SNARE proteins, a fact which is in no way inconsistent with the emerging contribution of upstream regulatory components (like rabGTPases and tethering complexes) to domain/compartment specificity.
Current Research & Projects
Our lab is working to elucidate the underlying mechanisms of vesicular transport within cells and the secretion of proteins and neurotransmitters.
Projects include:
- The biochemical and biophysical mechanisms of vesicle budding and fusion;
- Cellular regulation of vesicle fusion in exocytosis and synaptic transmission;
- Structural and functional organization of the Golgi apparatus from a cellular systems view.
We take an interdisciplinary approach which includes cell-free biochemistry, single molecule biophysics, high resolution optical imaging of single events/single molecules in the cell and in cell-free formats.
The overall goal is to understand transport pathways form structural mechanism to cellular physiology. The latter is facilitated by high throughput functional genomics at the cellular level (see Yale Center for High Throughput Cell Biology).
We have a strong interest in new lab members who bring backgrounds in chemistry, physics, and engineering.
SOURCE
http://medicine.yale.edu/cellbio/rothman/index.aspx
3 Americans Win Joint Nobel Prize in Medicine

From left: Randy W. Schekman, Thomas C. Südhof and James E. Rothman.
<nyt_byline>
By LAWRENCE K. ALTMAN
Published: October 7, 2013 151 Comments
Three Americans won the Nobel Prize in Physiology or Medicine Monday for discovering the machinery that regulates how cells transport major molecules in a cargo system that delivers them to the right place at the right time in cells.
The Karolinska Institute in Stockholmannounced the winners: James E. Rothman of Yale University; Randy W. Schekman of the University of California, Berkeley; and Dr. Thomas C. Südhof of Stanford University.
The molecules are moved around cells in small packages called vesicles, and each scientist discovered different facets that are needed to ensure that the right cargo is shipped to the correct destination at precisely the right time.
Their research solved the mystery of how cells organize their transport system, the Karolinska committee said. Dr. Schekman discovered a set of genes that were required for vesicle traffic. Dr. Rothman unraveled protein machinery that allows vesicles to fuse with their targets to permit transfer of cargo. Dr. Südhof revealed how signals instruct vesicles to release their cargo with precision.
The tiny vesicles, which have a covering known as membranes, shuttle the cargo between different compartments or fuse with the membrane. The transport system activates nerves. It also controls the release of hormones.
Disturbances in this exquisitely precise control system cause serious damage that, in turn, can contribute to conditions like neurological diseases, diabetes and immunological disorders.
Dr. Schekman, 64, who was born in St. Paul, used yeast cells as a model system when he began his research in the 1970s. He found that vesicles piled up in parts of the cell and that the cause was genetic. He went on to identify three classes of genes that control different facets of the cell’s transport system. Dr. Schekman studied at the University of California in Los Angeles and at Stanford University, where he obtained his Ph.D. in 1974.
In 1976, he joined the faculty of the University of California, Berkeley, where he is currently professor in the Department of Molecular and Cell Biology. Dr. Schekman is also an investigator at the Howard Hughes Medical Institute.
Dr. Rothman, 63, who was born in Haverhill, Mass., studied vesicle transport in mammalian cells in the 1980s and 1990s. He discovered that a protein complex allows vesicles to dock and fuse with their target membranes. In the fusion process, proteins on the vesicles and target membranes bind to each other like the two sides of a zipper. The fact that there are many such proteins and that they bind only in specific combinations ensures that cargo is delivered to a precise location.
The same principle operates inside the cell and when a vesicle binds to the cell’s outer membrane to release its contents. Dr. Rothman received a Ph.D. from Harvard Medical School in 1976, was a postdoctoral fellow at Massachusetts Institute of Technology, and moved in 1978 to Stanford University, where he started his research on the vesicles of the cell. Dr. Rothman has also worked at Princeton University, Memorial Sloan-Kettering Cancer Institute and Columbia University.
In 2008, he joined the faculty of Yale University where he is currently professor and chairman in the Department of Cell Biology. Some of the genes Dr. Schekman discovered in yeast coded for proteins correspond to those Dr. Rothman identified in mammals. Collectively, they mapped critical components of the cell´s transport machinery.
Dr. Südhof, 57, who was born in Göttingen, Germany, studied neurotransmission, the process by which nerve cells communicate with other cells in the brain. At the time he set out to explore the field 25 years ago, much of it was virgin scientific territory. Researchers had not identified a single protein in the neurotransmission process.
Dr. Südhof helped transform what had been a rough outline into a number of molecular activities to provide insights into the elaborate mechanisms at the crux of neurological activities, from the simplest to the most sophisticated. He did so by systematically identifying, purifying and analyzing proteins that can rapidly release chemicals that underlie the brain’s activities. The transmission process can take less than a thousandth of a second.
Dr. Südhof studied at the Georg-August-Universität in Göttingen, where he received a medical degree in 1982 and a doctorate in neurochemistry the same year. In 1983, he moved to the University of Texas Southwestern Medical Center in Dallas. Dr. Südhof, who has American citizenship, became an investigator at the Howard Hughes Medical Institute in 1991 and was appointed professor of molecular and cellular physiology at Stanford University in 2008.
All three scientists have won other awards, including the Lasker Prize, for their research.
<nyt_correction_bottom>
This article has been revised to reflect the following correction:
Correction: October 7, 2013
An earlier version of this article misstated Randy W. Schekman’s age. He is 64, not 65.
SOURCE
http://www.nytimes.com/2013/10/08/health/3-win-joint-nobel-prize-in-medicine.html?_r=0
Nobel for Cell Transport
This year’s Nobel Prize in Physiology or Medicine is going jointly to three scientists for their work figuring out how cells transport their cargo, according to the Karolinska Institute. They will share the $1.25 million prize.
“Imagine hundreds of thousands of people who are traveling around hundreds of miles of streets; how are they going to find the right way? Where will the bus stop and open its doors so that people can get out?” says Nobel committee secretary Goran Hansson, according to the Associated Press. “There are similar problems in the cell.”
By studying yeast cells with defective vesicles, Randy Schekman from the University of California, Berkeley, uncovered three classes of genes that control transportation within the cell, the New York Times adds. Schekman was awakened in California by the call from Stockholm. “I wasn’t thinking too straight. I didn’t have anything elegant to say,” he tells the AP. “All I could say was ‘Oh my God,’ and that was that.” Schekman adds that he called his lab manager to arrange a celebration in the lab.
Meanwhile, Yale University’s James Rothman discovered a protein complex that allows vesicles to bind to their intended membrane targets, getting the vesicle contents to a specific location. Rothman notes that he recently lost funding for work building on his discovery, and says that he hopes that having won the Nobel will help him when he reapplies.
And Thomas Südhof at Stanford University systematically studied how nerve cells communicate, finding that vesicles full of neurotransmitters bind to cell membranes to release their contents through a molecular mechanism that responds to the presence of calcium ions. He was on his way to a give a talk when he got his call. “I got the call while I was driving and like a good citizen I pulled over and picked up the phone,” Südhof says to the AP. “To be honest, I thought at first it was a joke. I have a lot of friends who might play these kinds of tricks.”
Other related articles published on these Open Access Online Scientific Journal include the following:
The Series on Cardiovascular Disease and the role of Calcium Signaling consists of the following articles:
Part I: Identification of Biomarkers that are Related to the Actin Cytoskeleton
Larry H Bernstein, MD, FCAP
Part II: Role of Calcium, the Actin Skeleton, and Lipid Structures in Signaling and Cell Motility
Larry H. Bernstein, MD, FCAP, Stephen Williams, PhD and Aviva Lev-Ari, PhD, RN
Part III: Renal Distal Tubular Ca2+ Exchange Mechanism in Health and Disease
Larry H. Bernstein, MD, FCAP, Stephen J. Williams, PhD and Aviva Lev-Ari, PhD, RN
Part IV: The Centrality of Ca(2+) Signaling and Cytoskeleton Involving Calmodulin Kinases and Ryanodine Receptors in Cardiac Failure, Arterial Smooth Muscle, Post-ischemic Arrhythmia, Similarities and Differences, and Pharmaceutical Targets
Larry H Bernstein, MD, FCAP, Justin Pearlman, MD, PhD, FACC and Aviva Lev-Ari, PhD, RN
Part V: Heart, Vascular Smooth Muscle, Excitation-Contraction Coupling (E-CC), Cytoskeleton, Cellular Dynamics and Ca2 Signaling
Larry H Bernstein, MD, FCAP, Justin Pearlman, MD, PhD, FACC and Aviva Lev-Ari, PhD, RN
Part VI: Calcium Cycling (ATPase Pump) in Cardiac Gene Therapy: Inhalable Gene Therapy for Pulmonary Arterial Hypertension and Percutaneous Intra-coronary Artery Infusion for Heart Failure: Contributions by Roger J. Hajjar, MD
Aviva Lev-Ari, PhD, RN
Part VII: Cardiac Contractility & Myocardium Performance: Ventricular Arrhythmiasand Non-ischemic Heart Failure – Therapeutic Implications for Cardiomyocyte Ryanopathy (Calcium Release-related Contractile Dysfunction) and Catecholamine Responses
Justin Pearlman, MD, PhD, FACC, Larry H Bernstein, MD, FCAP and Aviva Lev-Ari, PhD, RN
Part VIII: Disruption of Calcium Homeostasis: Cardiomyocytes and Vascular Smooth Muscle Cells: The Cardiac and Cardiovascular Calcium Signaling Mechanism
Justin Pearlman, MD, PhD, FACC, Larry H Bernstein, MD, FCAP and Aviva Lev-Ari, PhD, RN
Part IX: Calcium-Channel Blockers, Calcium Release-related Contractile Dysfunction (Ryanopathy) and Calcium as Neurotransmitter Sensor
Justin Pearlman, MD, PhD, FACC, Larry H Bernstein, MD, FCAP and Aviva Lev-Ari, PhD, RN
Part X: Synaptotagmin functions as a Calcium Sensor: How Calcium Ions Regulate the fusion of vesicles with cell membranes during Neurotransmission
Larry H Bernstein, MD, FCAP and Aviva Lev-Ari, PhD, RN
