Prime Editing as a New CRISPR Tool to Enhance Precision and Versatility
Curator: Stephen J. Williams, Ph.D.
The following articles is structured as follows
1: Recent update of financial statements from Editas Medicine, with a brief history of the company from formation to present
2: Main article which describes Editas Medicine’s CRISPR/Cas9 gene editing system and advantages of their system over competitors. This includes the early seminal publications showing utility of the system in humans
UPDATED 05/31/2026
The following press release announces a major public offering of shares of Editas Medicine, a biotechnology company founded in 2013 by Nobel Leaureates Dr. Jennifer Doudna, and Dr. George Church, as well as other prominent scientists to use the gene editing capabilities of the CRISPR/Cas9 system to cure genetic diseases. After initial funding of $43 million from Third Rock Ventures and Polaris Partners, Dr. Doudna left in 2014 as a result of patent disagreements. The company has had early successes with ‘in-vivo’ CRISPR in a clinical trial with EDIT-101 for Leber congenital amaurosis 10 (LCA10), however this was not able to reach market. Programs for correcting defects ex-vivo for sickle cell disease and beta-thalassemia (renicel) was terminated in 2024. Due to stiff competition from Veritas and CRSPR Therapeutics, as well as inability to secure a commercial partner, Editas Medicine pivoted their strategy in late 2024. The need for capital is reflecting in their public offering however they still maintain a strong IP portfoliol
A
Source: https://ir.editasmedicine.com/news-releases/news-release-details/editas-medicine-announces-pricing-3194-million-public-offering
Press Release Editas Medicine Announces Pricing of Up to $319.4 Million Public Offering
May 26, 2026 at 9:05 AM EDT
CAMBRIDGE, Mass., May 26, 2026 (GLOBE NEWSWIRE) — Editas Medicine, Inc. (Nasdaq: EDIT), a pioneering gene editing company developing transformative medicines for serious diseases, today announced the pricing of an underwritten public offering of 55,555,556 shares of its common stock and accompanying common stock warrants to purchase an aggregate of 55,555,556 shares of common stock (or pre-funded warrants in lieu thereof). Each share of common stock and accompanying common stock warrant are being sold together at a combined public offering price of $2.25. The aggregate gross proceeds from the offering are expected to be approximately $125.0 million (assuming no exercise of the common stock warrants), before deducting underwriting discounts and commissions and offering expenses. If all of the common stock warrants are exercised at their exercise price, the Company would receive additional gross proceeds from the offering of approximately $194.4 million before deducting underwriting discounts and commissions and offering expenses.
Each common stock warrant will be exercisable for shares of common stock (or pre-funded warrants in lieu thereof), will have an exercise price of $3.50 per share (or $3.4999 per share if exercised for pre-funded warrants), will be exercisable immediately and will expire on the earlier of (i) the date that is thirty (30) days following the first public announcement by the Company of Phase 1 clinical data for the Company’s product candidate, EDIT-401, that discloses at least three patients in the trial that each demonstrated greater than 80% reduction in LDL-cholesterol as compared to baseline with at least one (1) month of follow-up and (ii) three years from the date of issuance. Any pre-funded warrants issued upon the exercise of common stock warrants will have an exercise price of $0.0001 per share of common stock, will be immediately exercisable and will expire on the date the pre-funded warrant is exercised in full.
All of the securities in the offering are being sold by Editas Medicine. The offering is expected to close on or about May 27, 2026, subject to satisfaction of customary closing conditions.Cantor and Wells Fargo Securities are acting as joint book-running managers for the offering.The securities are being offered pursuant to an effective shelf registration statement on Form S-3 (File No. 333-277471) that was filed with the Securities and Exchange Commission (SEC) on February 28, 2024, as amended by Post-Effective Amendment No. 1 to Form S-3 Registration Statement and Post-Effective Amendment No. 2 to Form S-3 Registration Statement, each filed with the SEC on March 5, 2025, and declared effective on March 21, 2025. The offering is being made only by means of a prospectus supplement and accompanying prospectus that form a part of the registration statement. A preliminary prospectus supplement and accompanying prospectus relating to and describing the terms of the offering have been filed with the SEC and are available at www.sec.gov. A final prospectus supplement relating to the offering will be filed with the SEC and will be available for free on the SEC’s website at www.sec.gov. Copies of the final prospectus supplement may be obtained, when available, by contacting Cantor Fitzgerald & Co., Attention: Capital Markets, 110 East 59th Street, 6th Floor New York, New York 10022, Email: prospectus@cantor.com; or Wells Fargo Securities, LLC, Attention: Equity Syndicate Department, 90 South 7th Street, 5th Floor, Minneapolis, Minnesota 55402, at (800) 645-3751 (option #5) or email a request to WFScustomerservice@wellsfargo.com. This press release does not constitute an offer to sell or the solicitation of an offer to buy these securities, nor shall there be any sale of these securities in any state or jurisdiction in which such offer, solicitation or sale would be unlawful prior to registration or qualification under the securities laws of any such state or jurisdiction.
About Editas Medicine As a pioneering gene editing company, Editas Medicine is focused on translating the power and potential of CRISPR genome editing systems into a robust pipeline of transformative in vivo medicines for people living with serious diseases around the world. Editas Medicine aims to discover, develop, manufacture, and commercialize durable, precision in vivo gene editing medicines for a broad class of diseases. Editas Medicine is the exclusive licensee of Broad Institute’s Cas12a patent estate and Broad Institute and Harvard University’s Cas9 patent estates for human medicines.
Investor and Media Contacts:
ir@editasmed.com
media@editasmed.com

Source: Editas Medicine, Inc.
ORIGINAL ARTICLE : 08/29/2020
Curator: Stephen J. Williams, PhD
The following curation describes the technology developed by Editas Medicine which reduces off target effects and is the basis for the in-vivo and ex-vivo therapeutic CRSPR/Cas9 technology. Seminal publication are included,

CRISPR has become a powerful molecular for the editing of genomes tool in research, drug discovery, and the clinic
(see posts and ebook on this site below)
however, as discussed on this site
(see posts below)
there have been many instances of off-target effects where genes, other than the selected target, are edited out. This ‘off-target’ issue has hampered much of the utility of CRISPR in gene-therapy and CART therapy
see posts
However, an article in Science by Jon Cohen explains a Nature paper’s finding of a new tool in the CRISPR arsenal called prime editing, meant to increase CRISPR specificity and precision editing capabilities.
By Jon Cohen | Oct 25th, 2019
Prime editing promises to be a cut above CRISPR Jon Cohen CRISPR, an extraordinarily powerful genome-editing tool invented in 2012, can still be clumsy. … Prime editing steers around shortcomings of both techniques by heavily modifying the Cas9 protein and the guide RNA. … ” Prime editing “well may become the way that disease-causing mutations are repaired,” he says.
Science Vol. 366, No. 6464; DOI: 10.1126/science.366.6464.406
The effort, led by Drs. David Liu and Andrew Anzalone at the Broad Institute (Cambridge, MA), relies on the modification of the Cas9 protein and guide RNA, so that there is only a nick in a single strand of the double helix. The canonical Cas9 cuts both strands of DNA, and so relies on an efficient gap repair activity of the cell. The second part, a new type of guide RNA called a pegRNA, contains an RNA template for a new DNA sequence to be added at the target location. This pegRNA-directed synthesis of the new template requires the attachment of a reverse transcriptase enzymes to the Cas9. So far Liu and his colleagues have tested the technology on over 175 human and rodent cell lines with great success. In addition, they had also corrected mutations which cause Tay Sachs disease, which previous CRISPR systems could not do. Liu claims that this technology could correct over 89% of pathogenic variants in human diseases.
A company Prime Medicine has been formed out of this effort.
Source: https://science.sciencemag.org/content/366/6464/406.abstract
Read an article on Dr. Liu, prime editing, and the companies that Dr. Liu has initiated including Editas Medicine, Beam Therapeutics, and Prime Medicine at https://www.statnews.com/2019/11/06/questions-david-liu-crispr-prime-editing-answers/
(interview by StatNews SHARON BEGLEY @sxbegle)
As was announced, prime editing for human therapeutics will be jointly developed by both Prime Medicine and Beam Therapeutics, each focusing on different types of edits and distinct disease targets, which will help avoid redundancy and allow us to cover more disease territory overall. The companies will also share knowledge in prime editing as well as in accompanying technologies, such as delivery and manufacturing.
Reader of StatNews.: Can you please compare the pros and cons of prime editing versus base editing?
The first difference between base editing and prime editing is that base editing has been widely used for the past 3 1/2 years in organisms ranging from bacteria to plants to mice to primates. Addgene tells me that the DNA blueprints for base editors from our laboratory have been distributed more than 7,500 times to more than 1,000 researchers around the world, and more than 100 research papers from many different laboratories have been published using base editors to achieve desired gene edits for a wide variety of applications. While we are very excited about prime editing, it’s brand-new and there has only been one paper published thus far. So there’s much to do before we can know if prime editing will prove to be as general and robust as base editing has proven to be.
We directly compared prime editors and base editors in our study, and found that current base editors can offer higher editing efficiency and fewer indel byproducts than prime editors, while prime editors offer more targeting flexibility and greater editing precision. So when the desired edit is a transition point mutation (C to T, T to C, A to G, or G to A), and the target base is well-positioned for base editing (that is, a PAM sequence exists approximately 15 bases from the target site), then base editing can result in higher editing efficiencies and fewer byproducts. When the target base is not well-positioned for base editing, or when other “bystander” C or A bases are nearby that must not be edited, then prime editing offers major advantages since it does not require a precisely positioned PAM sequence and is a true “search-and-replace” editing capability, with no possibility of unwanted bystander editing at neighboring bases.
Of course, for classes of mutations other than the four types of point mutations that base editors can make, such as insertions, deletions, and the eight other kinds of point mutations, to our knowledge prime editing is currently the only approach that can make these mutations in human cells without requiring double-stranded DNA cuts or separate DNA templates.
Nucleases (such as the zinc-finger nucleases, TALE nucleases, and the original CRISPR-Cas9), base editors, and prime editors each have complementary strengths and weaknesses, just as scissors, pencils, and word processors each have unique and useful roles. All three classes of editing agents already have or will have roles in basic research and in applications such as human therapeutics and agriculture.

Nature Paper on Prime Editing CRISPR
Andrew V. Anzalone, Peyton B. Randolph, Jessie R. Davis, Alexander A. Sousa,
Luke W. Koblan, Jonathan M. Levy, Peter J. Chen, Christopher Wilson,
Gregory A. Newby, Aditya Raguram & David R. Liu
Nature volume 576, pages149–157(2019)
Abstract
Most genetic variants that contribute to disease1 are challenging to correct efficiently and without excess byproducts2,3,4,5. Here we describe prime editing, a versatile and precise genome editing method that directly writes new genetic information into a specified DNA site using a catalytically impaired Cas9 endonuclease fused to an engineered reverse transcriptase, programmed with a prime editing guide RNA (pegRNA) that both specifies the target site and encodes the desired edit. We performed more than 175 edits in human cells, including targeted insertions, deletions, and all 12 types of point mutation, without requiring double-strand breaks or donor DNA templates. We used prime editing in human cells to correct, efficiently and with few byproducts, the primary genetic causes of sickle cell disease (requiring a transversion in HBB) and Tay–Sachs disease (requiring a deletion in HEXA); to install a protective transversion in PRNP; and to insert various tags and epitopes precisely into target loci. Four human cell lines and primary post-mitotic mouse cortical neurons support prime editing with varying efficiencies. Prime editing shows higher or similar efficiency and fewer byproducts than homology-directed repair, has complementary strengths and weaknesses compared to base editing, and induces much lower off-target editing than Cas9 nuclease at known Cas9 off-target sites. Prime editing substantially expands the scope and capabilities of genome editing, and in principle could correct up to 89% of known genetic variants associated with human diseases.
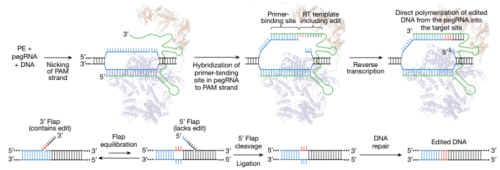
From Anzolone et al. Nature 2019 Figure 1.
Prime editing strategy
Cas9 targets DNA using a guide RNA containing a spacer sequence that hybridizes to the target DNA site. We envisioned the generation of guide RNAs that both specify the DNA target and contain new genetic information that replaces target DNA nucleotides. To transfer information from these engineered guide RNAs to target DNA, we proposed that genomic DNA, nicked at the target site to expose a 3′-hydroxyl group, could be used to prime the reverse transcription of an edit-encoding extension on the engineered guide RNA (the pegRNA) directly into the target site (Fig. 1b, c, Supplementary Discussion).. These initial steps result in a branched intermediate with two redundant single-stranded DNA flaps: a 5′ flap that contains the unedited DNA sequence and a 3′ flap that contains the edited sequence copied from the pegRNA (Fig. 1c). Although hybridization of the perfectly complementary 5′ flap to the unedited strand is likely to be thermodynamically favoured, 5′ flaps are the preferred substrate for structure-specific endonucleases such as FEN122, which excises 5′ flaps generated during lagging-strand DNA synthesis and long-patch base excision repair. The redundant unedited DNA may also be removed by 5′ exonucleases such as EXO123.
- The authors reasoned that preferential 5′ flap excision and 3′ flap ligation could drive the incorporation of the edited DNA strand, creating heteroduplex DNA containing one edited strand and one unedited strand (Fig. 1c).
- DNA repair to resolve the heteroduplex by copying the information in the edited strand to the complementary strand would permanently install the edit (Fig. 1c).
- They had hypothesized that nicking the non-edited DNA strand might bias DNA repair to preferentially replace the non-edited strand.
Results
- The authors evaluated the eukaryotic cell DNA repair outcomes of 3′ flaps produced by pegRNA-programmed reverse transcription in vitro, and performed in vitro prime editing on reporter plasmids, then transformed the reaction products into yeast cells (Extended Data Fig. 2).
- Reporter plasmids encoding EGFP and mCherry separated by a linker containing an in-frame stop codon, +1 frameshift, or −1 frameshift were constructed and when plasmids were edited in vitro with Cas9 nickase, RT, and 3′-extended pegRNAs encoding a transversion that corrects the premature stop codon, 37% of yeast transformants expressed both GFP and mCherry (Fig. 1f, Extended Data Fig. 2).
- They fused a variant of M—MLV-RT (reverse transcriptase) to Cas9 with an extended linker and this M-MLV RT fused to the C terminus of Cas9(H840A) nickase was designated as PE1. This strategy allowed the authors to generate a cell line containing all the required components of the primer editing system. They constructed 19 variants of PE1 containing a variety of RT mutations to evaluate their editing efficiency in human cells
- Generated a pentamutant RT incorporated into PE1 (Cas9(H840A)–M-MLV RT(D200N/L603W/T330P/T306K/W313F)) is hereafter referred to as prime editor 2 (PE2). These were more thermostable versions of RT with higher efficiency.
- Optimized the guide (pegRNA) using a series of permutations and recommend starting with about 10–16 nt and testing shorter and longer RT templates during pegRNA optimization.
- In the previous attempts (PE1 and PE2 systems), mismatch repair resolves the heteroduplex to give either edited or non-edited products. So they next developed an optimal editing system (PE3) to produce optimal nickase activity and found nicks positioned 3′ of the edit about 40–90 bp from the pegRNA-induced nick generally increased editing efficiency (averaging 41%) without excess indel formation (6.8% average indels for the sgRNA with the highest editing efficiency) (Fig. 3b).
- The cell line used to finalize and validate the system was predominantly HEK293T immortalized cell line
- Together, their findings establish that PE3 systems improve editing efficiencies about threefold compared with PE2, albeit with a higher range of indels than PE2. When it is possible to nick the non-edited strand with an sgRNA that requires editing before nicking, the PE3b system offers PE3-like editing levels while greatly reducing indel formation.
- Off Target Effects: Strikingly, PE3 or PE2 with the same 16 pegRNAs containing these four target spacers resulted in detectable off-target editing at only 3 out of 16 off-target sites, with only 1 of 16 showing an off-target editing efficiency of 1% or more (Extended Data Fig. 6h). Average off-target prime editing for pegRNAs targeting HEK3, HEK4, EMX1, and FANCFat the top four known Cas9 off-target sites for each protospacer was <0.1%, <2.2 ± 5.2%, <0.1%, and <0.13 ± 0.11%, respectively (Extended Data Fig. 6h).
- The PE3 system was very efficient at editing the most common mutation that causes Tay-Sachs disease, a 4-bp insertion in HEXA(HEXA1278+TATC).
References
- Landrum, M. J. et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 44, D862–D868 (2016).
- Jinek, M. et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science337, 816–821 (2012).
- Cong, L. et al. Multiplex genome engineering using CRISPR/Cas systems. Science339, 819–823 (2013).
- Mali, P. et al. RNA-guided human genome engineering via Cas9. Science339, 823–826 (2013).
- Kosicki, M., Tomberg, K. & Bradley, A. Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Biotechnol. 36, 765–771 (2018).
- Anzalone, A.V., Randolph, P.B., Davis, J.R. et al.Search-and-replace genome editing without double-strand breaks or donor DNA. Nature576, 149–157 (2019). https://doi.org/10.1038/s41586-019-1711-4
Read Full Post »










{kind=link}
{kind=link}