Perspectives on Anti-metastatic Effects in Cancer Research 2015
Curator: Larry H. Bernstein, MD, FCAP
Combining Kinetic Ligand Binding and 3D Tumor Invasion Technologies to Assess Drug Residence Time and Anti-metastatic Effects of CXCR4 Inhibitors
Application Note 3D Cell Culture, ADME/Tox, Cell Imaging, Cell-Based Assays
BioTek Instruments, Inc. P.O. Box 998, Highland Park, Winooski, Vermont 05404-0998
Brad Larson and Leonie Rieger, BioTek Instruments, Inc., Winooski, VT
Nicolas Pierre, Cisbio US, Inc., Bedford, MA
Hilary Sherman, Corning Incorporated, Life Sciences, Kennebunk, ME
Metastasis, the spread of cancer cells from the original tumor to secondary locations within the body, is linked to approximately 90% of cancer deaths1 . The expression of chemokine receptors, such as CXCR4 and CCR7, is tightly correlated with the metastatic properties of breast cancer cells. In vivo, neutralizing the interaction of CXCR4 and its known ligand, SDF1-α (CXCL12), significantly impaired the metastasis of breast cancer cells and cell migration2 . Traditionally, the discovery of novel agents has been guided by the affinity of the ligand for the receptor under equilibrium conditions, largely ignoring the kinetic aspects of the ligandreceptor interaction. However, awareness of the importance of binding kinetics has started to increase due to accumulating evidence3, 4, 5, 6 suggesting that the in vivo effectiveness of ligands may be attributed to the time a particular ligand binds to its receptor (drug-target residence time).
Similarly, appropriate in vitro cell models have also been lacking to accurately assess the ability of novel therapies to inhibit tumor invasion. Tumors in vivo exist as a three-dimensional (3D) mass of multiple cell types, including cancer and stromal cells7 . Therefore, incorporating a 3D spheroid-type cellular structure that includes co-cultured cell types forming a tumoroid, provides a more predictive model than the use of individual cancer cells cultured on the bottom of a well in traditional two-dimensional (2D) format.
Here we examine the drug-target residence time of various CXCR4 inhibitors using a direct, homogeneous ligand binding assay and CXCR4 expressing cell line in a kinetic format. This inhibitor panel was further tested in a 3D tumor invasion assay to determine whether there is a correlation between the molecule’s CXCR4 residence time and inhibition of the phenotypic effect of tumor invasion. MDA-MB-231 breast adenocarcinoma cells, known to be invasive, and metastasize to lung from primary mammary fat pad tumors8 , were included, in addition to primary human dermal fibroblasts. Cellular analysis algorithms provided accurate quantification of changes to the original tumoroid structure, as well as invadopodia development. The combination presents an accurate, yet easy-to-use method to assess target-based and phenotypic effects of new, potential anti-metastatic drugs.
……
Cytation™ 5 Cell Imaging Multi-Mode Reader Cytation 5 is a modular multi-mode microplate reader that combines automated digital microscopy and microplate detection. Cytation 5 includes filter- and monochromator-based microplate reading; the microscopy module provides high resolution microscopy in fluorescence, brightfield, color brightfield and phase contrast. With special emphasis on live-cell assays, Cytation 5 features temperature control to 65 °C, CO2 / O2 gas control and dual injectors for kinetic assays. Shaking and Gen5 software are also standard. The instrument was used to image spheroids, as well as individual cell invasion through the Matrigel matrix.
Tag-lite® Receptor Ligand Binding Assay
Figure 1. Tag-lite® Receptor Ligand Binding Assay Procedure. The Tag-lite CXCR4 assay relies on a fully functional SNAP-tag fused CXCR4 receptor and fluorescently labeled ligand SDF1-α. Being homogeneous, the binding assay allows for binding events to be precisely recorded in time. The assay can be used to derive the kinetic binding parameters of unlabeled compounds by application of the Motulsky and Mahan equations.
……
Results and Discussion
Drug-Target Residence Time
Determination Association Kinetics of SDF1-α-d2 Labeled Ligand
The final Drug-Target Residence Time value takes into account the observed on and off rates of the unlabeled inhibitors as well as the labeled SDF1-α-d2 ligand, and is computed by incorporation of the Motulsky and Mahan equation9 . The first step to calculate the final value was to perform an associative binding experiment using a concentration range of 0-100 nM of the d2 acceptor fluor labeled ligand. Binding was monitored kinetically over a period of 40 minutes.
Figure 2. Association binding graph of SDF1-α-d2. Observed associative binding curves calculated from HTRF ratios of wells containing SDF1-α-d2 ligand concentrations ranging from 0-100 nM. Non-specific binding values subtracted from total ratios to determine observed specific binding.
Binding increases over time until it plateaus after several minutes (Figure 2). The plateau in an association experiment depends on the concentration of labeled SDF1-α used. Higher plateaus will be obtained with higher concentrations. Fitting of the curves with Graph Pad Prism yields the observed association rate values for all concentrations tested or kobs.
The Kd value of the labeled ligand was also determined by plotting the HTRF ratios generated after a binding equilibrium was reached with the different concentrations of ligand tested.
Figure 3. SDF1-α-d2 saturation binding curve. HTRF ratios generated upon the achievement of binding equilibrium of tested [SDF1-α-d2].
In a saturation binding experiment, increasing concentrations of labeled SDF1-α result in increased binding. Saturation is obtained when no further binding can be recorded. The ligand concentration that binds to half the receptor sites at equilibrium or Kd was 29 nM.
An assessment of whether the labeled SDF1-α ligand follows the Law of Mass action can also be carried out. If the system does follow the Law of Mass action then kobs increases linearly with increasing concentrations of SDF1-α.
Due to the linear shape of the curve, and an R2 value >0.9, Law of Mass Action was proven for the labeled SDF1-α ligand. This allowed for the use of Graph Pad Prism software to derive association and dissociation rate constants from the linear regression line. The rate constant values experimentally found or mathematically derived are summarized in Table 1. kon,SDF1-α-d2 and koff ,SDF1-α-d2 were 0.001 nM-1.s-1 and 0.04 s-1, respectively
Table SDF1-α-d2 Kinetic Binding Characterization
Association Kinetics of SDF1-α-d2 Labeled Ligand In the theory developed by Motulsky and Mahan, an unlabeled competitor is co-incubated with a labeled ligand during a kinetic association experiment. Here, a single concentration of the SDF1-α-d2 ligand, 25 nM, was co-incubated with multiple concentrations of the unlabeled SDF1-α competitors in the presence of the CXCR4 expressing cells. Kinetic binding of the labeled ligand was then monitored over time.
Figure 5. Kinetics of Competitive Binding. Plot of specific binding HTRF ratios over time for the SDF1-α-d2 ligand when in the presence of 100, 10, or 1 nM concentrations of (A.) AMD 3100, (B.) AMD 3465, or (C.) IT1t.
From the curve fitting of the observed SDF1-α-d2 kinetic binding, and incorporation of the Law of Mass Action linear regression line, k(off) (Min-1) values were then calculated. Final residence time (R) values could then be determined using the following formula:
R = 1/k(off)
Therefore, molecules having a lower k(off) rate reside at the target receptor for longer periods of time.
Table 2. SDF1-α Competitor Dissociation Rate and Residence Time Values.
From the shape of the curves in Figure 5, and a comparison of the residence time values generated for the labeled ligand and unlabeled competitors (Table 2), qualitative and quantitative assumptions regarding the various competitors can then be made. First, if the competitor dissociates faster from its target than the ligand (smaller R value), such as is seen with AMD 3100 (Figure 5A), the specific binding of the ligand will slowly and monotonically approach its equilibrium in time. However, when the competitor dissociates slower (larger R value), the association curve of the ligand consists of two phases, starting with a typical “overshoot” and then a decline until a new equilibrium is reached. Competitors whose residence times are greater than that of the SDF1-α-d2 ligand, such as AMD 3465 and IT1t (Figure 5B and C), may then exhibit a stronger inhibitory response when used in the confirmatory phenotypic 3D tumor invasion assay.
Interruption of Invasion via SDF1-α Ligand Binding Inhibition As stated previously, interruption of the interaction between CXCR4 and its known ligand, SDF1-α, impairs metastasis of breast cancer and cell migration2 . Therefore, a phenotypic assessment of the CXCR4 inhibitor panel was then performed to determine whether changes in the level of tumor migration could be detected, and more importantly, if compounds exhibiting longer residence times compared to SDF1-α-d2 exhibited a higher inhibitory effect on migration through the 3D matrix. MDA-MB-231 breast adenocarcinoma cells, co-cultured with human dermal fibroblasts, were used as the in vitro tumor model. This breast cancer cell line has been previously shown to express the CXCR4 receptor10.
Figure 6. Image-based Monitoring of MDA-MB-231/Fibroblast Tumor Invasion. Overlaid brightfield and fluorescent images captured using a 4x objective, after a 0 and 5 day incubation period with AMD 3465, IT1t, and CTCE 9908. Imaging channel representation: Brightfield – Total cells and invadopodia; GFP – MDA-MB-231 cells; RFP – Fibroblasts.
Figure 7. Quantification of Invasive Tumor Area. 4x overlaid images captured following 5 day (A.) 100 and (B.) 0 μM IT1t incubation with tumoroids. Object masks automatically drawn by Gen5 using the following criteria: Threshold: 5000 RFU; Min. Object Size: 400 μm; Max. Object Size: 1500 μm; Image Smoothing Strength: 0; Background Flattening Size: Auto.
Cellular analysis is performed with the Cytation 5 using the brightfield signal to quantify the extent of invasion. Minimum and maximum object sizes, as well as brightfield threshold values are set such that a precise object mask is automatically drawn around each tumoroid in its entirety (Figure 7A and B). The same criteria are used for all images evaluated during the experiment. This allows for a quantitative comparison of the area covered within each object mask to be completed.
Figure 8. Tumor Invasion Inhibition Determination. Graphs of individual tumoroid areas on day 0, and subsequent to five day invasion period in the presence of inhibitor concentrations.
The 4x images displayed (Figure 6), as well as the graphs in Figure 8, demonstrating total tumoroid area coverage before and after the incubation period illustrate the ability of CXCR4 inhibitors to interrupt tumor invasion consistent with the previously determined residence time. AMD 3465 and IT1t, which exhibit a residence time longer than SDF1-α-d2, effectively minimize tumor invasion in a dose dependent manner. The decrease in MDAMB-231 GFP and fibroblast RFP expression exhibited after a 5 day 100 μM IT1t incubation, also seen after a 7 day AMD 3465 incubation of the same concentration (data not shown), may also indicate the chronic cytotoxic effects that elevated dosing of these compounds can have on both cancer and stromal cells. All other compounds show little to no effect on the ability of the tumoroid to migrate through the 3D matrix. While AMD 3465 and ITt1 display the same sub-nanomolar potency, AMD3465 prevails as a CXCR4 inhibitor due to its greater residence time.
Conclusions The Tag-lite CXCR4 ligand binding assay provides a simple, yet robust cell-based approach to determine kinetic binding of known receptor ligands, as well as competitive binding of test molecules. The simultaneous dual emission capture and injection capabilities of the Synergy Neo allow accurate calculations of kinetic association and dissociation rates to be made when used in conjunction with the Tag-lite® assay. Corning Spheroid Microplates then provide an easy-to-use, consistent method to perform spheroid aggregation and confirmatory 3D tumor invasion assays. Imaging of spheroid formation, as well as invading structures can be performed by the Cytation™ 5 using brightfield or fluorescent channels to easily track tumoroid invasion. The flexible cellular analysis capacity of the Gen5™ Data Analysis Software also allows for accurate assessment of 3D tumor invasion during the entire incubation period. The combination of assay chemistry, cell model, kinetic microplate and image-based monitoring, in addition to cellular analysis provide an ideal method to better understand the target-based and phenotypic effects of potential inhibitors of tumor invasion and metastasis.
References
- Saxe, Charles. ‘Unlocking The Mysteries Of Metastasis’. ExpertVoices 2013. http://www.cancer.org/ cancer/news/expertvoices/post/2013/01/23/unlockingthe-mysteries-of-metastasis.aspx. Accessed 16 Mar. 2015.
- Müller, A., Homey, B., Soto, H., Ge, N., Catron, D., Buchanan, M., McClanahan, T., Mruphy, E., Yuan, W., Wagner, S., Barrera, J., Mohar, A., Verástegui, E., Zlotnik, A. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001, 410, 50-56.
- Swinney, D. Biochemical mechanisms of drug action: what does it take for success? Nat Rev Drug Discov. 2004, 3, 801-808.
- Copeland, R., Pompliano, D., Meek, T. Drugtarget residence time and its implications for lead optimization. Nat Rev Drug Discov. 2006,5, 730-739.
- Tummino, P., Copeland, R. Residence time of receptor-ligand complexes and its effect on biological function. Biochemistry. 2008, 47, 5481-5492.
- Zhang, R., Monsma, F. The importance of drug-target residence time. Curr Opin Drug Discov Devel. 2009, 12, 488-496.
- Mao, Y., Keller, E., Garfield, D., Shen, K., Wang, J. Stromal cells in tumor microenvironment and breast cancer. Cancer Metast Rev. 2013, 32, 303-315.
- Kamath, L., Meydani, A., Foss, F., Kuliopulos, A. Signaling from protease-activated receptor-1 inhibits migration and invasion of breast cancer cells. Cancer Res. 2001, 61, 5933-5940.
- Motulsky, H., Mahan, L. The kinetics of competitive radioligand binding predicted by the law of mass action. Mol Pharmacol. 1984, 25, 1-9.
- Sun, Y., Mao, X, Fan, C, Liu, C., Guo, A., Guan, S., Jin, Q., Li, B., Yao, F., Jin, F. CXCL12-CXCR4 axis promotes the natural selection of breast cancer cell metastasis. Tumor Biol. 2014, 35, 7765-7773.
Inspired by Nature
Researchers are borrowing designs from the natural world to advance biomedicine.
By Daniel Cossins | August 1, 2015
http://mobile.the-scientist.com/article/43625/inspired-by-nature
When biomedical engineer Jeff Karp has questions, he looks to animals for answers. In 2009, Karp gathered his team at the Brigham and Women’s Hospital in Boston to brainstorm novel ways to capture circulating tumor cells (CTCs) in the bloodstream. They mulled over the latest microfluidic devices. Then the conversation turned to the New England Aquarium, and to jellyfish.
Scientists have tried to grab cancer cells from blood ever since they discovered that tumors shed malignant cells that migrate throughout the vasculature—a process known as metastasis. “If you pluck out these cells, you have a direct indicator of what the cancer looks like,” says Karp. “Then you can screen drugs to get those that will have the greatest impact.” Doctors might also be able to detect such cells during the earliest stages of metastatic cancer, when it’s more readily treatable.
CANCER-CELL CAPTURE DEVICE: Jellyfish’s long, sticky tentacles grab prey and other food particles from water. Researchers have copied this design by coating the channels of a microfluidic chip with long, tentacle-like strands of DNA that bind a protein on the surface of leukemia cells. The device can process 10 times more blood than existing chips in the same amount of time.
See full infographic: JPG SANDCASTLE WORM: PHEBE LI FOR THE SCIENTIST. DIAGRAM: KIMBERLY BATTISTA
The problem is, CTCs make up a tiny fraction of cells in the bloodstream of a person with cancer, meaning an effective diagnostic must process relatively large volumes of blood. However, an existing test, which uses magnetic particles to isolate CTCs, processes just 7.5 milliliters of blood, only a fraction of one percent of the 5 liters of blood in an adult human. Dialysis-like microfluidic devices promise to handle larger volumes and improve efficiency, but the best current prototypes still feature extremely narrow microchannels to ensure CTCs pass within reach of CTC-binding antibodies along the perimeter. “Channel height is extremely low in a lot of the proposed devices, meaning you can barely flow any blood through,” says Karp. (See “Capturing Cancer Cells on the Move,” The Scientist, April 2014.)
Karp wanted to change that. “We asked ourselves, ‘What creatures can capture things at a distance?’” he recalls. One of his graduate students suggested jellyfish, whose long, sticky tentacles grab prey and other food particles from water. Within a year, Karp and his colleagues had designed a microfluidic chip on which 800-micron-wide microchannels are lined with long, tentacle-like strands of DNA that bind a protein on the surface of leukemia cells as they pass through the channels. (See illustration below.) In 2012, Karp showed that the jellyfish-inspired device could process 10 times more blood than existing chips in the same amount of time and trap an average of 50 percent of circulating leukemia cells.1 Karp estimates that a device the size of the standard microscope slide could collect hundreds or thousands of tumor cells in minutes. Encouraged by such results, Karp’s team is now improving the platform, designing chips that can catch any CTC of interest.
The jellyfish is far from the only intriguing organism to have served as a blueprint for scientists in the field of bioinspired medicine. Researchers have taken cues from the adhesive chemistry perfected by mussels and marine worms to create tissue glues that stick in wet and turbulent conditions; from red blood cell membranes to help drug-carrying nanoparticles avoid immune attack; and from the slippery slides that help carnivorous pitcher plants catch prey to produce novel antibacterial surfaces. (See “Bioinspired Antibacterial Surfaces.”) Nature, it seems, provides a compendium of biomedical solutions.
“Nature has used the power of evolution by natural selection to develop the most efficient ways to solve all kinds of problems,” says Donald Ingber, founding director of the Wyss Institute for Biologically Inspired Engineering in Boston. “We’ve uncovered so much about how nature works, builds, controls, and manufactures from the nanoscale up. Now we’re starting to leverage those biological principles.”
Sticking points
Looking to nature is not a new concept, and bioinspiration is just one of several approaches bioengineers employ to devise new medical treatments and devices. But in the last few years, the approach has come to the fore with several promising new products, even if most of them remain a few years away from human trials. “Almost every research institute now has a center for biomimicry or biologically inspired engineering,” says Ingber. “It’s just reaching that tipping point where it’s going to begin to have an impact.”
TISSUE GLUE: The sandcastle worm (Phragmatopoma californica) builds reef-like shelters by gluing together grains of sand with two separate secretions: one containing negatively charged polyphosphate proteins and the other positively charged polyamine proteins. Researchers mimicked this idea with synthetic polyelectrolytes to create an injectible fluid that can patch fetal membrane ruptures in an in vitro model.
See full infographic: JPG SANDCASTLE WORM: PHEBE LI FOR THE SCIENTIST. DIAGRAM: KIMBERLY BATTISTA
Medical adhesion is one area where bioinspiration promises to make an impression. Stitches and staples are still the standard for suturing wounds and closing up surgical incisions, but these technologies can damage tissue, leave gaps for bacteria to infiltrate, and increase the risk of inflammation. For years, surgeons have been in need of new medical adhesives that can bond tissue strongly inside the body without provoking inflammation.
Heeding the call, bioengineers have again turned to the sea. Phillip Messersmith of the University of California, Berkeley, for example, is focused on the protein-filled secretions marine mussels use to fasten themselves to wave-battered rocks. The proteins in these liquid secretions are rich in an amino acid called dihydroxyphenylalanine (DOPA), which features reactive catechol chains. These catechol chains bond tightly with each other in a mussel’s own secretions but also bond with metal atoms present on the surface of rocks. Using this strategy as a blueprint, Messersmith and colleagues chemically synthesized a variant of DOPA to crosslink biocompatible polymers.
Their glue has successfully fastened transplanted insulin-producing islet cells to the outer surface of the liver and nearby tissues in mice.2 The technique could potentially provide an alternative to standard methods of islet transplantation in which islets are infused into the liver vasculature, where they trigger an inflammatory response that quickly kills off about half of the transplanted cells—and impairs the surviving cells’ ability to produce therapeutic insulin. The researchers are also testing the bioinspired adhesive’s ability to repair ruptured fetal membranes, which can lead to premature birth and other serious complications. (See “Mimicking Mussels,” The Scientist, April 2013.)
Cancer Invasion and Metastasis: Molecular and Cellular Perspective
Tracey A. Martin, Lin Ye, Andrew J. Sanders, Jane Lane, and Wen G. Jiang*.
* Metastasis and Angiogenesis Research Group, Institute of Cancer and Genetics, Cardiff University School of Medicine, Department of Surgery, University Hospital of Wales, Cardiff, UK.
Metastatic Cancer: Clinical and Biological Perspectives edited by Rahul Jandial.
Read this chapter in the Madame Curie Bioscience Database here.
Metastasis is the leading reason for the resultant mortality of patients with cancer. The past few decades have witnessed remarkable progress in understanding the molecular and cellular basis of this lethal process in cancer. The current article summarizes some of the key progress in this area and discusses the role of cell junctions, cell adhesions, epithelial-mesenchymal transition, angio and lymphangiogenesis and organ specific metastasis.
Of primary importance in the prognosis of cancer patients is the sequence of events leading to the development of tumor cell invasion and metastasis. The course of tumor metastasis entails a series of stages that lead to the formation of secondary tumors in distant organs and is, largely, responsible for the mortality and morbidity of cancer.
Once tumor cells acquire the ability to penetrate the surrounding tissues, the process of invasion is instigated as these motile cells pass through the basement membrane and extracellular matrix, progressing to intravasation as they penetrate the lymphatic or vascular circulation. The metastatic cells then journey through the circulatory system invading the vascular basement membrane and extracellular matrix in the process of extravasation. Ultimately, these cells will attach at a new location and proliferate to produce the secondary tumor. Concentrating research efforts on identifying and understanding the mechanisms concerned in tumor cell invasion may lead to limiting tumor progression and, as a result, to a reduction in mortality for many cancer patients. In the following, we have summarized some of the recent progress in the area of cell adhesion, epithelial to mesenchymal transition, angiogenesis, lymphangiogenesis and organ specific metastasis in cancer.
Cancer Invasion and Metastasis: The Role of Cell Adhesion Molecules
Cancer metastasis is the spread of cancer cells to tissues and organs beyond where the tumor originated and the formation of new tumors (secondary and tertiary foci) is the single event that results in the death of most patients with cancer. At the time of cancer diagnosis, at least half of the patients already present clinically detectable metastatic disease.1 A higher number of patients will also have micrometastases that would be beyond conventional detection techniques. Thus, metastasis is the most life threatening event in patients with cancer. The process is composed of a number of sequential events which must be completed in order for the tumor cell to successfully metastasize, the so called metastatic cascade. This process contributes to the complexity of cancer as a multiplex disease. During the metastatic cascade, changes in cell-cell and cell-matrix adhesion are of paramount importance.2
The metastatic cascade can be broadly separated into three main processes: invasion, intravasation and extravasation. The loss of cell-cell adhesion capacity allows malignant tumor cells to dissociate from the primary tumor mass and changes in cell-matrix interaction enable the cells to invade the surrounding stroma; the process of invasion. This involves the secretion of substances to degrade the basement membrane and extracellular matrix and also the expression/ suppression of proteins involved in the control of motility and migration. The tumor must also initialize angiogenesis, without which the tumor would fail to develop, as local diffusion for transport of nutrients to and removal of waste products from the tumor site would suffice for tumors up to 2 mm in diameter.3 The blood vessel within the tumor’s vicinity can then provide a route for the detached cells to enter the circulatory system and metastasize to distant sites; the process of intravasation.4,5 Interaction between the tumor cell and the surrounding stroma is extremely important in the development of tumor angiogenesis.6 Once the tumor cell has arrived at a likely point of intravasation, it interacts with the endothelial cells by undergoing biochemical interactions (mediated by carbohydratecarbohydrate locking reactions, which occur weakly but quickly) develops adhesion to the endothelial cells to form stronger bonds, and thus penetrates the endothelium and the basement membrane; the process of extravasation. The new tumor can then proliferate at this secondary focus.
The metastatic cascade is therefore dependent on the loss of adhesion between cells, which results in the dissociation of the cell from the primary tumor, and subsequently the ability of the cell to attain a motile phenotype via changes in cell to matrix interaction.
Cellular Junctions
Epithelial cells are characterized by a remarkable polarization of their plasma membrane, evidenced by the appearance of structurally, compositionally, and functionally distinct surface domains. The cell to cell adhesion complex runs from the apical to the basal membranes and is composed of Tight Junctions (TJ), Adherens Junctions (AJ), Gap Junctions (GJ), Desmosomes and integrins (Fig. 1).
Schematics showing the arrangement of cell-cell junctions and cell-matrix interactions.
Tight Junctions (TJ)
The permeability of epithelial and endothelial cells is governed by the TJ and they are located at the apical membrane of the cell,7–9 (Fig. 1). The TJ is a region where the plasma membrane of adjacent cells forms a series of contacts that appear to completely occlude the extracellular space thus creating an intercellular barrier and intramembrane diffusion fence.10 In epithelial cells the TJ functions in an adhesive manner and can prevent cell dissociation.11 TJ in endothelial cells function as a barrier through which molecules and inflammatory cells can pass. Interaction with and penetration of the vascular endothelium by dissociated cancer cells is an important step in the formation of cancer metastases. TJ are the first barrier that cancer cells must overcome in order to metastasize. We have previously demonstrated that TJ of vascular endothelium in vivo function as a barrier between blood and tissues against metastatic cancer cells.12 Early studies demonstrated a correlation between the reduction of TJ and tumor differentiation and experimental evidence has emerged to place TJ in the frontline as the structure that cancer cells must overcome in order to metastasize.12–15Although a considerable body of work exists on TJ and their role in a number of diseases, following the early work of Martinez-Paloma16 and others,17,18 it is only in recent years that there has been an upsurge in studies investigating their possible role in tumorigenesis and metastasis.
There have now been numerous studies on colorectal cancer,19–21 pancreatic cancers22–24 and an increasing number of studies performed on breast cancer.25–27 Changes in both tumor and endothelial cells are necessary for successful growth and spread of cancer cells and these changes are somewhat similar. A change in cancer cells by upregulation or downregulation of relevant TJ proteins results in loss of cellcell association, cell contact inhibition, leading to uncontrolled growth, loss of adhesion to and degradation of the basement. These must be a concurrent loss of cellcell association in the endothelium and modulation of TJ proteins involved in facilitating the passage of the cancer cells through this barrier.
HGF/SF (hepatocyte growth factor), a cytokine secreted by stromal cells and key to the development and progression of cancer, particularly during metastasis has been shown to be capable of modulating expression and function of TJ molecules in human breast cancer cell lines.28 HGF decreased trans-epithelial resistance and increased paracellular permeability of human breast cancer cell lines, MDA-MB-231 and MCF-7. Q-PCR showed that HGF modulated the levels of several TJ molecule (occludin, claudin-1 and -5, JAM-1 and -2) mRNA transcripts in MDA-MB-231 and MCF-7 cells. Such data shows that HGF disrupts TJ function in human breast cancer cells by effecting changes in the expression of TJ molecules at both the mRNA and protein levels and that regulation of TJ could be of fundamental importance in the prevention of metastasis of breast cancer cells. Regulation of vascular permeability is one of the most important functions of endothelial cells, and endothelial cells from different organ sites show different degrees of permeability.29 Tumor blood vessels are more permeable on macro-molecular diffusion than normal tissue vessels. However, the cause and mechanism of hyperpermeability of human vessels had not been clear. Tumor cells release a number of factors that can assist their transmigration through the endothelium after treating endothelial cells with conditioned media from a highly invasive and metastatic melanoma cell line,29 with TJ being irreversibly damaged (as assessed using TER-trans-epithelial resistance). In fact, HGF has been shown to decrease TER and increase PCP (paracellular permeability) in human endothelial cells.8
An increasing number of studies have shown that numerous TJ components are directly or indirectly involved in cancer progression including ZO-1, ZO-2, claudin-7, claudin-1 and occludin.25 When human tissues and breast cancer cell lines were amplified for functional regions of occludin, tumor tissues showed truncated and/or variant signals. There was also considerable variation in the expression of occludin in the 10 human breast cancer cell lines investigated. Western blotting demonstrated that variants in the MDA-MB-231 and MCF-7 human breast cancer cell lines did not fit the expected occludin signals for changes in phosphorylation status. Immunostaining showed similarly disparate levels of expression. Ribozyme knockdown resulted in increased invasion, reduced adhesion and significantly reduced TJ functions. Q-RT-PCR analysis of 124 tumor and 33 background human breast tissues showed occludin to be significantly decreased in patients with metastatic disease. Immunohistochemical staining showed a decreased expression of occludin in the tumor sections. This study demonstrated for the first time that occludin is differentially expressed in human breast tumor tissues and cell lines. This loss of or aberrant expression has clear repercussions as to the importance of occludin in maintaining TJ integrity in breast tissues,25 (Fig. 2). Highly differentiated adenocarcinomas with well developed TJ provide an important insight into the usefulness of TJ molecules and are possible prognostic indicators and future targets for therapy. In breast cancer, ZO-1 has been demonstrated to be decreased in poorly differentiated tumors and correlated with increasing Grade and TNM (tumor-nodal) status.30 There are a respectable number of reports describing the dysregulation of transmembrane proteins in human cancers and in cell lines. This dysregulation can be the result of both upregulation and downregulation of expression, epigenetic changes and changes in activation and location of the proteins.
Adherens Junctions (AJ)
AJ are cellcell microdomains that provide adherent strength and localize to the basal side of the TJ31 (Fig. 1). The integral membrane proteins of the AJ are of the cadherin family, with E-cadherin being most abundant in epithelia and VE-cadherin in endothelia (Fig. 1). Nectins are also found in AJ of epithelia. In polarized epithelia of vertebrates, the AJ is part of the tripartite junctional complex localized at the juxtaluminal region, which comprises the TJ, AJ, and desmosome aligned in this order from the apical end of the junction.32 In this type of epithelia, the AJ is specifically termed the zonula adherens or adhesion belt, as it completely encloses the cells along with the F-actin lining, called the circumferential actin belt.33 The AJs in other cell types assume different morphologies with the AJ in fibroblastic cells being spotty and discontinuous34 while those in neurons are organized into tiny puncta as a constituent of the synaptic junctions.35 A major function of AJs is to maintain the physical association between cells, as disruption of them causes loosening of cellcell contacts, leading to disorganization of tissue architecture.33
Classical or type I cadherins mediate adhesion at the adherens, cellcell or cellmatrix adhesive junctions that are linked to microfilaments. Type I classical cadherins are composed of five tandem extracellular cadherin domains (EC1-EC5), a single segment transmembrane domain and a distinct, highly conserved cytoplasmic tail that specifically binds catenins.36 In addition to cadherin homophilic binding, it has been reported that cadherin is also capable of heterophilic interactions with numerous extracellular and intracellular proteins. The key to their adhesive activity is the interaction between the catenin-binding sequence and submembrane plaque proteins β-catenin or plakoglobin (γ-catenin), which form the link to the actin cytoskeleton. α-catenin binds to a short region close to the N terminus of β-catenin forming a stable bond between the complex and the actin cytoskeleton.36 In addition to α-, β-, and γ-catenin, a fourth catenin-like protein capable of binding cadherin, p120ctn, has emerged as a key regulator of cadherin function.37 p120ctn was originally identified as a substrate for receptor tyrosine kinases and like the other catenin molecules, binds directly to the cytoplasmic domain of cadherin.37
Nectins are transmembrane proteins that are found in both TJ and AJ. In AJ, during the process of early cellcell contacts, nectins first accumulate at the contacts, and then cadherins follow them, suggesting that the former may guide the latter in their junctional localization. Nectin interaction serves for recruiting cadherins to heterotypic cellcell borders, which are otherwise distributed throughout cellcell borders.33 Thus, nectins recruit cadherins to the synaptic contacts formed between two distinct domains of hippocampal neurons, i.e., axons and dendrites, which express nectin-1 and nectin-3, respectively.38 Thus, nectins show important cooperation with classic cadherins in generating heterotypic cellcell contacts.33
Evidence has long accumulated to point toward a pivitol role for E-cadherin and the catenin complex in the control of cancer cell dissociation and spread. Tumor invasion and metastasis, both hallmarks of tumor malignancy, frequently coincide with the loss of E-cadherin-mediated cell-cell adhesion. Expression of E-cadherin, the most abundant adhesion molecule in adherens junctions of epithelia, is downregulated in most, if not all, epithelial cancers.39 Several studies have shown that reconstitution of a functional E-cadherin adhesion complex suppresses the invasive phenotype of many different tumor cell types.40–42 In the context of cancer, E-cadherin has been categorized as a tumor suppressor, given its essential role in the formation of proper intercellular junctions, and its downregulation in the process of epithelial-mesenchymal transition (EMT) in epithelial tumor progression.
Recent studies in triple-negative breast cancer (TNBC), which is characterized by negativity for estrogen receptor, progesterone receptor and human epidermal growth factor receptor 2 (HER2), have shown there is a high risk breast cancer that lacks specific targets for treatment selection. Chemotherapy is, therefore, the primary systemic modality used in the treatment of this disease, but reliable parameters to predict the chemosensitivity of TNBC have not been clinically available.43 Patients with E-cadherin-negative and Ki67-positive expression showed significantly worse overall survival time than those with either E-cadherin-positive or Ki67-negative expression. Multivariate analysis showed that the combination of E-cadherin-negative and Ki67-positive expression was strongly predictive of poor overall survival in TNBC patients receiving adjuvant chemotherapy. The authors demonstrated that adjuvant therapy is beneficial for Stage II TNBC patients and that the combination of E-cadherin and Ki67 status might be a useful prognostic marker indicating the need for adjuvant chemotherapy in Stage II TNBC patients.43
E-cadherin inactivation with loss of cell adhesion is the hallmark of lesions of the lobular phenotype and E-cadherin is typically absent, as seen by immunohistochemistry in both lobular carcinoma in situ and invasive lobular lesions, suggesting it occurs early in the neoplastic process. In invasive lobular lesions, the cadherin-catenin complex was examined; complete complex dissociation was defined as negative membranous E-cadherin, α- and β-catenin expression.44 E-cadherin was found to be absent in all lesions and positive in all normal tissues. Membranous a and β-catenin expressions decreased with the transition from lobular lesions to invasive lesions, while TWIST expression increased. Gene expression paralleled IHC-staining patterns with a stepwise downregulation of E-cadherin, α and β-catenins from normal to lobular to invasive lesions, and increasing expression of TWIST from normal to lobular to invasive lesions. The decreasing membranous catenin expression in tandem with increasing levels of TWIST across the spectrum of lobular lesions suggests that cadherin-catenin complex dissociation is a progressive process in human breast cancer.44
Desmosomes
In cell-cell junctions, desmosomes form adherent points in the form of a continuum of cells within tissues by linkage of their integral membrane proteins (desmocollin and desmoglein) via desmoplakins (plakophilin and plakoglobin) to intermediate filaments31,45 (Fig. 1). Desmosomes are crucial for tissue integrity by their very strong adherence that resists calcium-depletion in developed tissue, but can be regulated by protein kinase C when dynamic remodelling of cellcell adhesion is required.45 Desmosomes not only provide mechanical stability but also facilitate cellcell communication through signal transmission.46 The desmosome is divided into three parallel identifiable zones, arranged symmetrically on the cytoplasmic faces of the plasma membranes of bordering cells and separated by the extracellular domain, which in mature desmosomes is bisected by a dense midline. Each desmosomal plaque consists of a thick outer dense plaque and a translucent inner dense plaque. The five major desmosomal components are the desmosomal cadherins, represented by desmogleins (14) and desmocollins (13), the armadillo family members, plakoglobin and the plakophilins (13), and the plakin linker protein desmoplakin, which anchors the intermediate keratin filaments.46
Recent studies using mouse genetic approaches have uncovered a role for desmosomes in tumor suppression, demonstrating that desmosome downregulation occurs before that of adherens junctions to drive tumor development and early invasion, suggesting a two-step model of adhesion dysfunction in cancer progression.47 Studies have shown that an increased expression of desmosome proteins, such as Desmoglein 2 and 3 and PKP3, can be observed in certain cancers of the skin, head and neck, prostate and lung compared with normal tissue, and that this overexpression is associated with enhanced tumor progression.46,48–50
Reduced expression of Desmocollin 2 has been reported in colorectal carcinomas, suggesting that it may play a role in the development and/or progression of colorectal cancer. Kolegraff et al.51 reported that the loss of Desmocollin-2 promotes cell proliferation and enables tumor growth in vivo through the activation of Akt/β-catenin signaling. Inhibition of Akt prevented the increase in β-catenin-dependent transcription and proliferation following Desmocollin-2 knockdown and attenuated the in vivo growth of Desmocollin-2 -deficient cells. This provides evidence that loss of Desmocollin-2 contributes to the growth of colorectal cancer cells and highlights a novel mechanism by which the desmosomal cadherins regulate β-catenin signaling.51
Oral squamous cell carcinomas and pre-malignant dysplasia can be suβ-classified according to their in vitro replicative lifespan, where the immortal dysplasia and carcinoma subsets have p16(ink4a) and p53 dysfunction, telomerase deregulation and genetic instability and the mortal subset do not. It has been demonstrated that desmosomal proteins exhibit a distinct expression pattern in oral mucosa when compared with epidermis in vivo. Microarray data from a large panel of lines shows that the transcript levels of Desmoglein 2 and Desmocollin2/3 are reduced in immortal dysplasia and carcinoma cells.52 Interestingly, Desmoglein 2 was upregulated. Reduction of Desmoglein 3 and upregulation of Desmoglein 2 were found in two independent microarray data sets. Significantly, we demonstrated that reduction of Desmoglein 3 and upregulation of Desmoglein 2 was reversible in vitro by using RNAi-mediated knockdown of Desmoglein 2 in carcinoma cells. The remaining desmosomal proteins were largely disrupted or internalized and associated with retraction of keratin intermediate filaments in oral squamous cell carcinomas lines. These findings suggest dysfunction and loss of desmosomal components are common events in the immortal class of oral squamous cell carcinomas and that these events may precede overt malignancy.52
There are numerous links between the desmosome and the adherens junction. A decrease in the levels of the desmosomal plaque protein, plakophilin3, leads to a decrease in desmosome size and cell-cell adhesion. Gosavi et al.53investigated whether plakophilin3 is required for desmosome formation. Plakophilin3 knockdown clones showed decreased cell border staining for multiple desmosomal proteins, when compared with vector controls, and did not form desmosomes in a calcium switch assay. Further analysis demonstrated that plakophilin3, plakoglobin and E-cadherin are present at the cell border at low concentrations of calcium. Loss of either plakoglobin or E-cadherin led to a decrease in the levels of plakophilin3 and other desmosomal proteins at the cell border. The results reported here are consistent with the model that plakoglobin and E-cadherin recruit plakophilin 3 to the cell border to initiate desmosome formation.53
Gap Junctions (GJ)
GJ are unique cell-to-cell channels that allow diffusion of small metabolites, second messengers, ions and other molecules between neighboring cells31 (Fig. 1). GJ communication is essential for electrical transduction, signaling and nutrition. The channels can be open or closed, a highly dynamic process regulated at multiple levels, with the integral membrane proteins forming these channels in vertebrates being the connexins of which over 20 family members have now been identified in humans; connexin43 the most abundantly expressed connexin.31 ZO-1 acts as a scaffold in GJ and recruits signaling proteins. Connexins are also known to interact with Occludin and also form complexes with CAR and β-catenin.54
For decades, cancer was associated with GJ defects. However, more recently it appeared that connexins can be re-expressed and participate in cancer cell dissemination during the late stages of tumor progression. Since primary tumors of prostate cancer are known to be connexin deficient, Lamiche et al.55 investigated whether their bone-targeted metastatic behavior could be influenced by the re-expression of the connexin type (connexin43) which is originally present in prostate tissue and highly expressed in bone where it participates in the differentiation of osteoblastic cells. It appeared that Cx43 behaved differently in those cell lines and induced different phenotypes. In LNCaP, connexin43 was functional, localized at the plasma membrane and its high expression was correlated with a more aggressive phenotype both in vitro and in vivo. In particular, those connexin43-expressing LNCaP cells exhibited a high incidence of osteolytic metastases generated by bone xenografts in mice. Interestingly, LNCaP cells were also able to decrease the proliferation of cocultured osteoblastic cells. In contrast, the increased expression of connexin43 in PC-3 cells led to an unfunctional, cytoplasmic localization of the protein and was correlated with a reduction of proliferation, adhesion and invasion of the cells. In conclusion, the localization and the functionality of connexin43 may govern the ability of prostate cancer cells to metastasize in bones.55
In colorectal tumors, loss of connexin43 expression is correlated with significantly shorter relapse-free and overall survival. Connexin43 was further found to negatively regulate growth of colon cancer cells, in part by enhancing apoptosis and was found to colocalize with β-catenin and reduce Wnt signaling.56 This study represents the first evidence that Cx43 acts as a colorectal cancer tumor suppressor and that loss of Cx43 expression during colorectal cancer development is associated with reduced patient survival. Connexin43 was downregulated or aberrantly localized in colon cancer cell lines and colorectal carcinomas, which is associated with loss of gap junction intercellular communication. Such data indicate that Cx43 is a colorectal cancer tumor suppressor protein that predicts clinical outcome.56
Integrins and Selectins
There is accumulating evidence for the role of integrins and selectins in cancer progression of various cancer types, including colon and lung carcinomas and melanomas.57 While selectin-mediated tumor cells arrest and adhesion contribute to metastasis, integrin-mediated interaction from both tumor cells and the surrounding environment further contribute to cancer progression.
Integrins
Integrins are large and complex transmembrane glycoproteins that consist of two distinct chains, α and β-subunits, which form a non-covalent heterodimer and combine to form 24 unique canonical α/β receptors.57 Integrins mediate cell adhesion and directly bind components of the extracellular matrix, such as fibronectin, vitronectin, laminin, or collagen and provide anchorage for cell motility and invasion. Integrins mediate bidirectional signaling where intracellular signals induce alterations in the conformation.57 Integrins participate in multiple cellular processes, including cell adhesion, migration, proliferation, survival, and the activation of growth factor receptors. As many human tumors originate from epithelial cells, integrins expressed on epithelial cells are generally also present in tumor cells and therefore, integrins have become linked with patient survival and metastatic status. Recent studies have shown that expression of αv integrins is elevated in the prostate cancer stem/progenitor cell subpopulation compared with more differentiated, committed precursors. Van den Hoogen et al.58 examined the functional role of αv integrin receptor expression in the acquisition of a metastatic stem/ progenitor phenotype in human prostate cancer. Stable knockdown of αv integrin expression in PC-3M-Pro4 prostate cancer cells coincided with a significant decrease of prostate cancer stem/ progenitor cell characteristics (α2 integrin, CD44, and ALDH(hi)) and decreased expression of invasion-associated genes Snail, Snail2, and Twist. Consistent with these observations, αv-knockdown strongly inhibited the clonogenic and migratory potentials of human prostate cancer cells in vitro and significantly decreased tumorigenicity and metastatic ability in preclinical models of orthotopic growth and bone metastasis. This indicates that integrin αv expression is functionally involved in the maintenance of a highly migratory, mesenchymal cellular phenotype as well as the acquisition of a stem/progenitor phenotype in human prostate cancer cells with metastasis-initiating capacity.58,59
Lu et al.59 investigated the expression of osteopontin and integrin αv (ITGAV, main receptor of the osteopontin) in laryngeal and hypopharyngeal squamous cell carcinoma and any correlation of the expression quantity with tumor biological behavior. The expression quantity of osteopontin and integrin αv in primary and metastatic carcinomas is significantly higher than in normal tissues. The expression of osteopontin and integrin αv in the well-differentiated group was significantly lower than in moderately and poorly differentiated groups; the expression quantity of osteopontin and integrin αv in groups with lymph node metastasis was significantly higher than in groups without lymph node metastasis. The authors conclude that the expression of osteopontin and integrin αv significantly influenced the differentiation and metastasis of the laryngeal and hypopharyngeal squamous cell carcinoma. Overexpression of both proteins may have contributed to invasion and metastasis of the laryngeal and hypopharyngeal squamous cell carcinoma, and therefore, they both may have value as a target for chemotherapy in laryngeal and hypopharyngeal squamous cell carcinoma treatment.59
Selectins
The selectins: E-selectin, P-selectin, and L-selectin are adhesion molecules that are crucial for binding of circulating leukocytes to vascular endothelium during the inflammatory response to injury or infection. Accumulated evidence indicates that selectins regulate adhesion of circulating cancer cells to the walls of blood vessels.60 Selectin ligands are transmembrane glycoproteins expressed on leukocytes and cancer cells that promote bond formations with selectins to mediate inflammatory processes and selectins and their ligands also participate in signal transduction to regulate diverse cellular functions.60
Haematogenous metastasis of small cell lung cancer is still a poorly understood process and represents the life threatening event in this malignancy.61 In particular, the rate-limiting step within the metastatic cascade is not yet clearly defined although, many findings indicate that extravasation of circulating tumor cells is crucially important as most tumor cells within the circulation undergo apoptosis. If extravasation of small cell lung cancer tumor cells mimics leukocyte-endothelial interactions, small cell lung cancer cells should adhere to E- and P-selectins expressed on the luminal surface of activated endothelium. The adhesion to E- and P-selectin under physiological shear stress with regard to adhesive events, rolling behavior and rolling velocity was determined in the human small cell lung cancer cell lines SW2, H69, H82, OH1 and OH3. OH1 SCLC cells adhered best to recombinant human (rh) E-selectin FC-chimeras and human lung endothelial cells (HPMEC), H82 small cell lung cancer cells adhered best to activated human umbilical vein endothelial cells (HUVEC) under physiological shear stress. As OH1 cells had also produced by far the highest number of spontaneous lung metastases when xenografted into pfp/rag2 mice in previous experiments the findings implicate that adhesion of small cell lung cancer cells to E-selectin is of paramount importance in small cell lung cancer metastasis formation.61
Cell-Matrix Interactions
Controlled interaction between the cells and the extracellular matrix is essential for many processes, including normal development, migration and proliferation.31 Interaction between the cell and the matrix can occur through a number of routes; cell adhesion molecules (CAM) including integrins, selectins, cadherins, the Ig superfamily, CD44 and focal adhesions.
Integrins
Integrin-mediated adhesions to the extracellular matrix are among the first adhesion junctions where bidirectional signaling occurs.31 At the extracellular side integrins bind directly to the extracellular matrix which includes collagen, fibronectin and laminins etc. Cytoplasmic partners include talins, paxillin, focal adhesion kinase and linkage to α-actinin and actin-stress fibers. These focal adhesion complexes control a variety of signaling pathways regulated by the interplay with the extracellular partners. Substantial cross-talk between the diverse cellcell and cellextracellular matrix junctions has been found, and the architecture of the epithelial monolayer is highly regulated by their concerted actions.31
Cell Adhesion Molecules (CAM)
Cell adhesion molecules (CAM) facilitate cellular processes such as cell proliferation, migration, and differentiation and are essential during development and for maintaining the integrity of tissue architecture in adults.62 CAMs include cadherins, integrins, selectins, and the immunoglobulin superfamily (IgSF). In normal tissue, CAM expression is tightly regulated. However, aberrant expression of CAMs disrupts normal cell-cell and cell-matrix interactions and can facilitate tumor formation and metastasis. A number of IgSF members have been identified as biomarkers for cancer progression and have also been associated with metastatic progression in a range of huma tumors.62
CD44
CD44 is a multifunctional cell surface adhesion molecule that is involved in cell-cell and cell-matrix interaction and has been implicated in tumor cell invasion and metastasis. In humans, the CD44 family is encoded by a single gene located on chromosome 11p13 and comprises at least 20 exons. Exons 15, 1618 and 20, are spliced together to form a CD44 transcript that has become known as the standard isoform (CD44s). At least ten exons can be alternatively spliced and inserted into the standard isoform at an insertion site between exons 5 and 16 to give rise to variant isoforms of CD44. Thus, exons 615 are variant exons and are typically identified as v1v10.63 CD44 is the principal ligand for hyaluronic acid (HA), a major component of the extracellular matrix. However CD44 can also bind to other ECM components including collagen, fibronectin, laminin and non-ECM component such as osteopontin and serglycin. CD44 is expressed on a variety of cells and tissues including T- lymphocytes, B-cells, monocytes, granulocytes, erythrocytes, many epithelial cell types; Keratinocytes, chondrocytes, mesothelial and some endothelial cells. It is also expressed in many cancer cell types and their metastases in particular; high molecular weight forms of CD44 show restricted expression in tumors and may correlate with tumor development and metastasis and have potential diagnostic and prognostic value in some cancers. Additionally, it has been shown in experimental models that CD44 can inhibit tumor growth and metastatic spread. Further investigation is still needed but CD44 may yet prove to be a potential target for cancer therapy.63
The importance of non-coding RNA transcripts in regulating microRNA (miRNA) functions, especially the 3′ untranslated region (UTR), has been revealed in recent years. Genes encoding the extracellular matrix normally produce large mRNA transcripts including the 3UTR. How these large transcripts affect miRNA functions and how miRNAs modulate the extracellular matrix protein expression are largely unknown. Jeyapalan and Yang64 demonstrated that the overexpression of the CD44 3UTR results in enhanced cell motility, invasion and cell adhesion in human breast carcinoma cell line MDA-MB-231. They also found that expression of the CD44 3UTR enhances metastasis in vivo. Computational analysis indicated that miRNAs that interact with the CD44 3UTR also have binding sites in other matrix encoding mRNA 3UTRs, including collagen type 1α1 (Col1α1) repressed by miR-328 and fibronectin type 1 (FN1) repressed by miR-5123p, miR-491 and miR-671. Protein analysis demonstrated that expression of CD44, Col1a1, and FN1 were synergistically upregulated in vitro and in vivo upon transfection of the CD44 3UTR. The non-coding 3UTR of CD44 interacts with multiple miRNAs that target extracellular matrix properties and thus can be used to antagonize miRNA activities.64
CD44 is also a causal factor for tumor invasion, metastasis and acquisition of resistance to apoptosis. CD44 knockdown using inducible short hairpin RNA (shRNA) significantly reduces cell growth and invasion. Short hairpin RNA against CD44 and pGFP-V-RS-vector was used for knockdown of CD44 expression in SW620 colon cancer cells. Short hairpin RNA against CD44 reduced the expression of CD44. Cell proliferation, migration and invasion were markedly inhibited and apoptosis was increased in shRNA CD44-transfected cells. Knockdown of CD44 decreased the phosphorylation of PDK1, Akt and GSK3β, and β-catenin levels. Decreased phosphorylated Akt led to an increase in phosphorylated FoxO1 and induced cell cycle arrest in the G0-G1 phase and a decrease in the S phase. The levels of Bcl-2 and Bcl-xL expression were downregulated, while the levels of BAX expression and cleaved caspase-3, -8 and -9 were increased. CD44 knockdown by way of shRNA inhibited cell proliferation and induced cell apoptosis which suggests that it could be used as a therapeutic intervention with the anti-survival/pro-apoptotic machinery in human colon cancer.65
Focal Adhesions
Focal adhesion kinase (FAK), a crucial mediator of integrin and growth factor signaling, is a novel and promising target in cancer therapy. FAK resides within focal adhesions which are contact points between extracellular matrix (ECM) and cytoskeleton, and increased expression of the kinase has been linked with cancer cell migration, proliferation and survival.66 Migration is a coordinated process that involves dynamic changes in the actin cytoskeleton and its interplay with focal adhesions. At the leading edge of a migrating cell, it is the re-arrangement of actin and its attachment to focal adhesions that generates the driving force necessary for movement.67 Signaling by the FAK-Src complex plays a crucial role in regulating the formation of protein complexes at focal adhesions to which the actin filaments are attached. Cortactin, an F-actin associated protein and a substrate of Src kinase interacts with FAK through its SH3 domain and the C-terminal proline-rich regions of FAK. Wang et al.67 showed that the autophosphorylation of Tyr(397) in FAK, which is necessary for FAK activation, was not required for the interaction with cortactin, but was essential for the tyrosine phosphorylation of the associated cortactin. At focal adhesions, cortactin was phosphorylated at tyrosine residues known to be phosphorylated by Src. The tyrosine phosphorylation of cortactin and its ability to associate with the actin cytoskeleton were required in tandem for the regulation of cell motility. Cell motility could be inhibited by truncating the N-terminal F-actin binding domains of cortactin or by blocking tyrosine phosphorylation (Y421/466/475/482F mutation). In addition, the mutant cortactin phosphorylation mimic (Y421/466/475/482E) had a reduced ability to interact with FAK and promoted cell motility. The promotion of cell motility by the cortactin phosphorylation mimic could also be inhibited by truncating its N-terminal F-actin binding domains. This suggests that cortactin acts as a bridging molecule between actin filaments and focal adhesions. The cortactin N-terminus associates with F-actin, while its C-terminus interacts with focal adhesions. The tyrosine phosphorylation of cortactin by the FAK-Src complex modulates its interaction with FAK and increases its turnover at focal adhesions to promote cell motility.67
Clinical Considerations
A number of cell adhesion molecules have now become classed as clinical indicators and there is a clear trend toward using them for prognosis or diagnosis. The number of studies identifying these molecules as biomarkers are legion and cannot be thoroughly reviewed here. Some timely examples are as follows: The TJ transmembrane protein claudin-7 has achieved status as a prognostic indicator in invasive ductal carcinoma of the breast68 and is a candidate expression marker for distinguishing chromophobe renal cell carcinoma from other renal tumor subtypes, including the morphologically similar oncocytoma.69 Moreover, decreased claudin-7 correlated with high tumor grade in prostate cancer70 and is able to regulate the expression of prostate specific antigen.71 When considering potential targets for therapy, claudin-1 has been found to act as a cancer invasion/metastasis suppressor in addition to its use as a prognostic predictor and potential drug treatment target for patients with lung adenocarcinoma.72 E-Cadherin and vimentin have now been described predictive markers of outcome among patients with non-small cell lung cancer treated with erlotinib.73
Epithelial-Mesenchymal Transition
Cell Motility
A major factor shaping the metastatic character of cancer cells lies in their motility. Cell motility and migration is crucial to normal development and is a major component of organogenesis, inflammation and wound healing. However, changes in the signaling pathways directing its regulation can lead to the pathological processes of tumor cell invasion and metastasis.
The development and progression of cell motility is orchestrated by a sequence of specific biophysical, interdependent processes involving cytoskeletal modifications, changes in cell-substrate adhesive properties and alterations in the extracellular matrix. Reacting to a stimulus, a cell will commence polarization and extend protrusions in the direction of migration74 which originates with extension of the leading edge by protrusion of lamellipodia and/or filopodia, driven by actin polymerisation and filament elongation, with frequently associated membrane ruffling,75 which extends the cell body to then produce new, distal adhesion sites. Following protrusion, adhesion is instigated between the cell and substratum at the leading edge accomplished largely by integrin and non-integrin receptors binding to specific extracellular matrix protein domains.74,76 Subsequently, actomyosin-mediated contraction of the cell occurs with resultant forward motion of the cell body, initiated by contractile forces being generated at or near the leading edge, coupled with detachment of the trailing edge from the substratum. In addition, the migrating cell secretes the proteases required to break down the extracellular matrix proteins thus providing a pathway for the advancing cell.
Several molecules have been identified as having important roles to play in the signaling processes leading to cell motility/migration, with the associated loss of epithelial characteristics and gain of a migratory and mesenchymal phenotype. Thus, the acquisition of a mesenchymal-like cell phenotype provides one of the major characteristics of metastatic progression of most carcinomas.
Mechanisms of EMT
There is growing acknowledgment that the detachment and escape of cells from the primary tumor mimics the developmental process known as epithelialmesenchymal transition (EMT) (Fig. 3), a dynamic process permitting polarized epithelial cells to go through multiple biochemical and morphological changes enabling them to assume a mesenchymal phenotype with enhanced migratory and invasive capabilities.77–80
Schematic description of EMT/MET showing effectors of these processes; dissociation/ association of cell to cell adhesions together with characteristic markers of either epithelial or mesenchymal cells.
Initiation of the process of EMT entails the loss of cell-cell adhesions; activation of transcription factors; alterations in expression of specific cell-surface proteins; reorganization and expression of cytoskeletal proteins; and production of ECM degrading enzymes. Consequently, the course of EMT involves a shift in the characteristic morphology and gene expression pattern of epithelial cells resulting in the acquisition of a characteristic mesenchymal, migratory phenotype.81,82
EMT Progression
Epithelial cells present a highly polarized morphology, intimately linked by cell-cell junctions in the form of TJ, AJ, desmosomes and GJ. Loss of these intercellular connections provides a critical step during EMT allowing for physical detachment of cancer cells from the primary tumor. Thus, EMT is characterized by the combined loss of epithelial cell junction proteins, including E-cadherin, α-catenin, claudins, occludin and ZO-1, an increased expression of mesenchymal markers, such as N-cadherin, vimentin and fibronectin, as well as reorganization of the cytoskeleton, which collectively results in the loss of apical-basal cell polarity and the attainment of a spindle-shaped morphology.77,83
Loss of expression of the cellcell adhesion molecule E-cadherin is a characteristic trait of EMT in development and in the progression of epithelial tumors to invasive, metastatic cancers. The loss of E-cadherin is generally seen to coincide with a gain of expression of the mesenchymal cadherin, N-cadherin in many cancer types; this ‘cadherin switch’ is thought to be necessary for tumor cells to gain invasive properties and is also a characteristic of EMT.39
It is evident from recent studies that EMT-inducing signals are, in part, initiated by growth factors, including hepatocyte growth factor (HGF), epidermal growth factor (EGF) and transforming growth factor β (TGFβ). These induce downstream activation of a number of EMT-inducing transcription factors including Snail, Slug, Twist and zinc finger E-box binding homeobox 1 (ZEB1).81,84–86
EMT Biomarkers
A number of biomarkers have been found to be useful indicators for EMT (Table 1.).
Biomarkers of EMT .
E-Cadherin
It is essential that weakening of cell-cell adhesion occurs to allow cells to become motile and metastasise and a modification in the adhesive properties of cells is a necessary element of the metastatic process. Cell adhesion molecules (CAMs) regulate cell-cell and cell-matrix adhesion and are implicated in almost all stages of metastasis, therefore alterations in normal levels of CAMs such as E-cadherin will be significant in tumor progression. E-cadherin is a member of a family of Ca2+ dependent CAMs made up of intracellular, extracellular and transmembrane domains. These domains play vital roles in cellular recognition during morphogenesis and development and are responsible for cell-cell adhesion87 thus holding a central role in the maintenance of tissue integrity. E-cadherin and its adhesion complex play an essential function in the adhesion of breast cancer cells, being involved in the control of tumor progression and metastasis. Members of the complex, such as β-catenin, act as regulators of cell adhesion, and also of cell signaling and transcription regulation.88 Studies exploring the expression of E-cadherin and α-catenin in tumor tissues have shown that loss of both molecules is linked to an increased invasiveness of tumor cells.89 Evidence for this comes from in vitro and in vivo studies which demonstrate that E-cadherin expression is inversely correlated with the motile and invasive behavior of tumor cells and also with metastasis in cancer patients.90 Further studies have revealed that the relocalization of β-catenin to the nucleus correlates with the acquisition of the mesenchymal phenotype,91,92 and is associated with the loss of E-cadherin. This reduction of cell surface E-cadherin causes the cells to be receptive to initiation of EMT.93 Numerous reports have indicated that E-cadherin plays a role in meningiomas, tumors of the central nervous system; with upregulation and nuclear localization of β-catenin in 60% of anaplastic memingiomas.94
Transcription Factors in EMT
Important transcription factors shown to be significant in EMT, as they affect the regulation of E-cadherin expression, are Slug and Snail (SNAI1),95 Zeb-185 and Twist.96,97 Importantly, Snail has been identified as having a significant role in the differentiation of epithelial cells into mesenchymal cells during embryonic development98,99 with Slug and Snail effecting the downregulation of E-cadherin expression by binding directly to two proximal E2-boxes of the E-cadherin promoter.84,100 It has been shown that Snail and E-cadherin expression are inversely correlated in squamous cell carcinoma101 and cancer of the breast.102 Snail also represses expression of genes encoding tight junction components, such as claudins and occludins.103
The basic helix-loop-helix protein Twist is also a key transcription factor in EMT and is known to trigger EMT mechanisms possibly by the regulation of the E-cadherin to N-cadherin switch. It is not known if E-cadherin expression can be repressed directly by Twist however, forced N-cadherin expression exerts a dominant effect over E-cadherin in breast cancer cells.104,105 Similarly, expression of N-cadherin in normal epithelial cells results in downregulation of E-cadherin expression.104 Work on glioblastoma (GBM) by Mikheeva et al.106 has shown that TWIST1 promotes GBM invasion through instigation of mesenchymal molecular and cellular changes. This study showed, however, that this effect was not reliant on a cadherin switch as a reduction in levels of E-cadherin and consequent increase in N-cadherin did not occur with TWIST1 overexpression.
Nevertheless many of the genes regulated by TWIST1 in GBM cell lines mirror those which it regulates in cancer metastasis which suggests some overlap with that of TWIST1-mediated EMT in carcinomas.106 In work on medulloblastoma, evidence for a significant role for EMT has been seen with intermittent hypoxic conditions in the tumor microenvironment.107 Hypoxia is recognized as a factor involved in overexpression of the urokinase plasminogen activator (uPA) and its receptor (uPAR) with overexpression promoting uPAR-mediated survival signaling in various cancers.108 Likewise, hypoxia/overexpression of uPAR in cancer cells promotes EMT and thus invasiveness and metastasis. The study by Gupta also showed that when medulloblastoma cells are exposed to intermittent hypoxia this initiates various molecular and phenotypic changes consistent with EMT, as the cell signaling molecules vimentin, N-cadherin, Snail are overexpressed in these medulloblastoma cells with a reduction in the epithelial markers ZO-1 and E-cadherin.
EMT-Related Factors
Bone Morphogenetic Protein (BMP7)
Numerous signaling pathways have been implicated in the initiation of EMT, in particular, TGF-β1 has been identified as a potent initiator of EMT in renal tubular epithelial cells,109 and also in cancer cells, stimulating cell invasion and metastasis.110 However, it has been reported that a member of the TGF-β superfamily, bone morphogenetic protein 7 (BMP-7) reverses TGF-β induced EMT by induction of E-cadherin.111 Indeed, BMP-7 has been shown to regulate epithelial homeostasis in the human mammary gland by preserving the epithelial phenotype.79 Similarly, a decrease in BMP-7 expression in human breast cancer leads to the acquisition of a bone metastatic phenotype,79 with loss of BMP-7 being associated with a more invasive and motile mesenchymal phenotype, in PC-3 prostate cancer cells.112Furthermore, systemic administration of recombinant BMP-7 to mice with severe renal fibrosis has resulted in reversal of EMT with repair of damaged epithelial structures111 as BMP-7 acts to reverse TGF-β1 induced EMT by upregulating E-cadherin in renal cells. Linked with this, BMP member growth and differentiation factor 9 (GDF-9) has been shown to promote the invasiveness of PC-3 cells together with an induction in the expression of genes including SNAI1, RhoC, ROCK-1 and N-cadherin, while reducing levels of E-cadherin. Thus in PC-3 cells, GDF-9 signaling via ALK-5, promotes cell invasiveness via a complex signaling network working collectively to trigger EMT, thus aiding in the aggressiveness and progression of prostate cancer cells.113
Matrix Metalloproteinases (MMPs)
The matrix metalloproteinases (MMPs) are an important component of cell invasion capable of degrading a range of extracellular matrix proteins allowing cancer cells to migrate and invade. In epithelial ovarian cancer TGFβ and EGF act as inducers of MMP2 production and enhance cell motility,114 while in breast cancer there is an upregulation of MMP9.115
In oral squamous cell carcinoma Snail and Slug are seen to act as regulators of TGFβ triggered EMT, with Snail upregulating MMP2 and MMP9 initiating EMT; while Slug and Snail maintain longer term EMT by stimulating MMP9 expression.116 The MMPs not only function in membrane/ matrix degradation but are also involved in cell adhesion. Treatment of MCF-7 cells with MMP7 results in E-cadherin cleavage producing an 80kDa fraction which is detectable in the serum and urine of cancer patients and has been proposed as a biomarker.117 Similarly, MMP9 appears to cleave the TJ molecule Occludin (personal communication).
Epithelial Protein Lost in Neoplasm (EPLIN)
The cytoskeletal protein EPLIN has been identified as a key molecule linking the cadherin-catenin complex to F-actin and stabilizing the Zona Adherens in MDCK and DLD-1 cells.118 It is an actin cross linking protein that bundles actin in the cells and stabilizes the cytoskeletal filaments. By doing so, EPLIN protein inhibits cell motility, and has been found to be downregulated in a number of oral, breast and prostate cancer cell lines. Forced expression of EPLIN in the EPLIN-α negative breast cancer cell line, MDA MB-231 has been shown to reduce migration and invasion in these cells so reducing their aggressiveness.119 Similarly, overexpression of EPLIN in the PC-3 cell line results in a reduction in both in vivo and in vitro growth potential together with a reduction in cell invasiveness and ability to adhere to extracellular matrix.120
Thus, EPLIN could be seen to be acting as a tumor suppressor. Recently, biochemical and functional evidence has exposed EPLIN as a negative regulator of EMT and invasiveness in prostate cancer cells. Evidence has emerged to show that a downregulation of EPLIN significantly disrupts epithelial structures, initiates actin cytoskeleton remodelling via the EPLIN link between actin filaments and β-catenin, affects explicit gene expression profiles and triggers a pro-EMT program.121
A great deal of energy has been focused, over the last four decades, on the elucidation of the molecular mechanisms governing EMT/MET since the concepts were first defined by Hay (1968).122 Evidence has emerged that the process of EMT can be classified into three different subtypes; type 1 associated with implantation, embryo formation, and organ development; type 2 EMT associated with wound healing, tissue regeneration, and organ fibrosis and type 3 EMT which arises in neoplastic cells in relation to tumor growth and cancer progression, occurring in cells that have gone through epigenetic changes in genes that support the instigation of localized tumors. Many investigators have found that applying the principles of carcinoma EMT to their studies has aided in the understanding of tumor cell invasion in various cancer types and pinpointed many of the genes specifically associated with EMT in relation to tumor growth and metastasis. Continued studies will hopefully provide significantly more information concerning the molecular mechanisms that drive EMT, in relation to the effects of EMT on the progression of carcinomas and will possibly offer new approaches and targets to prevent the most fatal characteristic of tumorigenesis-metastasis.
Angiogenesis and Lymphangiogenesis in Cancer Metastasis
Introduction to Angiogenesis and Lymphangiogenesis
The growth of new blood or lymphatic vessels from pre-existing vessels (the process of angiogenesis or lymphangiogenesis) is essential in physiological events such as reproduction, development, wound-healing and immunity. However, imbalance or manipulation of these essential processes is seen in a number of disease states and these processes are frequently involved in cancer progression and metastasis.123,124
Angiogenic and Lymphangiogenic Cascade
The angiogenic process is made up of a complex multi-step cascade, which is tightly regulated through the balance of a number of pro- and anti- angiogenic factors. Tumor cells frequently tip this balance in favor of blood vessel production through the secretion of pro-angiogenic factors as summarized in Fig. 4. The production of angiogenic factors from a source tissue or tumor bind to and activate endothelial cells of a neighboring blood vessel. Following activation, the endothelial cells begin to produce enzymes that break down the basement membrane of the blood vessel creating tiny pores. Endothelial cells then proliferate and migrate through these pores, toward the angiogenic source, a mechanism that involves a variety of adhesion molecules to aid movement of the new blood vessel toward the source and also the production of various enzymes, such as matrix metalloproteinases, at the sprouting tip, to facilitate this movement through the extrα-cellular matrix. Endothelial cells of the new vessel then undergo a tubule formation phase, where these cells roll to form a tube like structure before establishment of a blood vessel loop between the source and the existing vessel. Finally, structural stabilization of this loop is obtained through recruitment of additional cell types, such as smooth muscle cells, providing support to the vessel and allowing blood flow to the angiogenic source.125
Summary of key steps involved in the angiogenic cascade
While the vasculature system and lymphatic system are structurally different, the process of lymphangiogenesis shares similarities with the angiogenesis process. New lymphatic vessel growth can be stimulated by a variety of factors such as members of the vascular endothelial growth factor (VEGF) family (e.g., VEGF-C and VEGF-D), which induce sprouting of new vessels and proliferation of lymphatic endothelial cells (LEC),126,127 a process which, similar to angiogenesis, is utilized by metastasising tumor cells. Key angiogenic and lymphangiogenic factors are summaried inTable 2.
Key angiogenic and lymphangiogenic factors .
Therapeutic Potential of Angiogenesis and Lymphangiogenesis in Targeting Cancer Metastasis
While lymphangiogenesis and angiogenesis are essential in numerous physiological processes they are also commonly involved in disease states, in particular the progression of cancer and metastasis.
Angiogenesis and Anti-Angiogenesis Strategies in Cancer
The importance of angiogenesis in advanced tumor development has been known for many years. Without their own vasculature, tumors are unable to grow beyond a size of approximately 23 mm and are limited by their reliance on simple diffusion to obtain required resources.3,128 To overcome this, cancer cells often secrete certain factors to encourage new blood vessel growth to the tumor (tumor angiogenesis). These new blood vessels provide the required resources for advanced and rapid development of the tumor and also provide direct links with the vascular system to the tumor, facilitating metastatic invasion into this system and dissemination around the body.
There are a number of factors that have been demonstrated to enhance angiogenesis such as vascular endothelial growth factor (VEGF) and hepatocyte growth factor (HGF) and, given the importance of tumor angiogenesis in facilitating advanced tumor growth and metastatic spread, research into effective targeting of tumor angiogenesis has been a key area of interest in the scientific community, employing various strategies to disrupt or block new blood vessel growth to the developing tumor.
VEGF is perhaps one of the best known and established angiogenesis regulators to date and given its major role in angiogenesis, it has been subjected to vast scientific study. The VEGF family itself consists of several members, which signal through a number of VEGF receptors, however, the main angiogenesis regulator in normal physiology and cancer appears to be VEGF (also known as VEGF-A) and the VEGF receptor-2 (VEGFR-2 or FLK).129,130 Early research established the importance of VEGF in regulating endothelial proliferation and survival and its ability to promote angiogenesis using in vitro models.131 Given its vital role in tumor angiogenesis, specific targeting of VEGF signaling has been one of the key avenues in developing anti-angiogenic therapies. One such strategy has employed the development and use of a VEGF neutralising antibody termed Bevacizumab (also known as Avastin). This therapy has been approved for use in a variety of cancer types, such as non-squamous non-small-cell lung cancer and colorectal cancer.130 Scientific research into the benefits of Bevacizumab is ongoing, with studies examining and demonstrating the potential of Bevacizumab in additional cancer types such as epithelial ovarian cancer, where previous trials have yielded promising results.132
HGF represents another potential target for the treatment of cancer progression and angiogenesis. The role of HGF in contributing to cancer progression has been well demonstrated within the literature. This is largely due to the ability of HGF to promote pro-metastatic traits such as motogenesis, morphogenesis, mitogenesis and angiogenesis.133 HGF has the capacity to enhance angiogenesis both directly and in-directly, either through its motogenic or morphogenic effects on endothelial cells or through its capacity to enhance other pro-angiogenic factors such as VEGF and its receptor.133,134 Several earlier studies conducted in our labs have highlighted the potential anti-angiogenic application for targeting HGF. HGF treatment in vivo was found to enhance the expression of tumor endothelial markers (TEMs) in tumors obtained from the inoculation of PC-3 prostate cancer cells into CD1 athymic nude mice. However, the addition of NK4, a HGF antagonist, to the treatment was able to prevent the elevation of these TEMs in the tumors.135 Similarly, in a breast cancer in vivo model HGF treatment was found to enhance vessel formation in tumors arising from MDA-MB-231 inoculation into CD1 athymic nude mice using immunohistochemical staining (IHC) analysis of resulting tumor tissues. In keeping with its role, addition of NK4 again prevented the enhanced angiogenesis seen in HGF treatment groups.136 In both studies HGF treatment caused enhanced tumor development, whereas co-treatment could suppress these increases in tumor growth.135,136
Given its involvement in the processes of angiogenesis and tumor progression, inhibitors to the cMET tyrosine kinase receptor of HGF have been developed as treatment regimes. Strategies such as Foretinib, an oral multikinase inhibitor targeting a variety of proteins including cMET and the VEGF receptor have been developed and are being assessed for their efficacy.137
Lymphangiogenesis and Anti-Lymphangiogenesis in Cancer
The area of lymphangiogenesis and the potential of anti-lymphangiogenic therapies in the treatment of cancer has been somewhat over-shadowed by research into anti-angiogenic strategies and the relative lack of pro-lymphangiogenic markers. However, the last 15 -20 years has seen the identification of lymphangiogenic markers and markers of lymphatic endothelial cells, such as lymphatic vessel endothelial hyaluronan receptor-1 LYVE-1138 and vascular endothelial growth factor receptor-3 (VEFGR-3).139 Studies such as these have aided in the progression of this field of research and demonstrated its importance in cancer metastasis.
Lymphatic metastases are common, with a number of cancers first metastasising to regional lymph nodes. The determination of lymph node involvement is an important factor in determining the aggressive nature of a particular cancer, with lymphatic metastasis commonly being associated with a poorer patient outlook.140 Scientific research, examining the role of VEGF-C and D in mouse models has demonstrated the potential of these factors to enhance tumor lymphatics and promote metastatic spread of tumor cells.141,142 In keeping with this, a number of recent studies have reported the association of lymphatic factors such as VEGF-C and D and the VEGFR3 receptor with lymph node metastasis and patient survival.143–145 Taken together, these studies highlight the importance of tumor lymphangiogenesis in cancer spread and survival and demonstrate the potential for anti-lymphatic therapies, targeting factors such as VEGF-C, -D or the VEGFR3 receptor, to limit cancer spread and enhance survival rates.
In summary, anti-angiogenesis and anti-lympangiogenesis therapies hold great potential in combating the ongoing problem of cancer metastasis and the poor survival rates associated with cancer spread. Research and development of drugs in this area have so far begun to yield positive results with therapies such as Bevacizumab being implemented in the treatment of several cancer types. However, resistance to these anti-angiogenic strategies are possible and thus further research into new and multi target inhibitors of angiogenesis and lymphangiogenesis is essential in the ongoing fight against cancer spread.
Organ Specific Metastasis
Cancer metastases are responsible for the majority of cancer-related deaths. From a primary tumor to a distant site and eventually developing a secondary tumor, cancerous cells need to proceed along a series of interrelated and sequential steps, including invasion through extracellular matrix, intravasation, survival in the circulation, extravasation into a distant site, and progressive growth at that site. The metastatic procedure is an inefficient process whereby the vast majority of circulating tumor cells are not able to progressively grow at distant sites. A latent period may exist between infiltration of cancer cells at a distant site and colonization leading progressively to the growth of a secondary tumor. Such a period can be as long as a couple of years seen in some metastases of breast cancer after initial management, and it can also be as short as a few months in lung cancer which may develop a metastasis rapidly within a few months of diagnosis. The cellular origin, intrinsic properties of the tumor, tissue affinities and circulation patterns determine not only the sites of tumor spread, but also the temporal course and severity of metastasis to vital organs. In addition to the above aspects of metastases, certain metastatic cells exhibit tissue tropism, preferring to grow in certain organs (Table 3). In breast cancer, for example, metastasis affects the bone and the lung, and less frequently the liver, brain, and adrenal medulla. Although the genetic and epigenetic basis of these metastatic properties is yet to be fully established, acquisition of the ability to complete each step involved in metastasis is thought to be driven by the accumulation of genetic mutations and epigenetic events that may result in a cells acquisition of metastatic traits during the process of developing a secondary tumor.
Common metastatic sites of certain solid tumors .
The organs mostly assaulted by metastases are lung, liver, brain and bone146 (Fig. 5). The lungs are the commonest site of metastases for many primary tumors. However, there is a great difference in propensity between the malignancies. It is just as high as 90% in melanomas at autopsy. The lungs serve as first filter for tumor cells spreading through blood circulation in malignancies whose venous drainage flows directly into the lungs. The tumors of testis, melanoma, osteosarcoma, and head and neck tumors have the highest incidence of pulmonary metastases.146 The liver is one of the most common sites for metastatic disease, accounting for 25% of all metastases to solid organs.147 In the United States and Europe, secondary liver neoplasms are far more common than primary hepatic neoplasms. In the adult oncology patient, most are metastatic carcinomas, of which adenocarcinomas are the predominant subtype, followed by squamous cell carcinomas and neuroendocrine carcinomas. Other tumor types that metastasize to the liver include melanomas, lymphomas, and rarely sarcomas. The most frequent metastasis to the brain occurs in patients with lung, breast, melanoma, renal, and colorectal tumors.148 In 2700 cases from the Memorial Sloan-Kettering Cancer Center in New York, the distribution of primary cancers was as follows: 48% lung, 15% breast, 9% melanoma, 1% lymphoma (mainly non-Hodgkin), 3% GI (3% colon and 2% pancreatic), 11% genitourinary (21% kidney, 46% testes, 5% cervix, 5% ovary), 10% osteosarcoma, 5% neuroblastoma, and 6% head and neck tumor . Once metastasis to the brain is diagnosed, the median survival of untreated patients is 12 mo. Bone metastases are most commonly seen in prostate, breast and lung cancer, which are leading malignancies in female and/or male having the highest incidence and mortality rates.149–151 Bone metastasis usually leads to severe morbidities, which always persist until the death of patients, including bone pain, hypercalcemia, pathological fracture, spinal cord compression and consequent paralysis. In the following part, we generally reviewed the process and molecular mechanisms of organ specific metastases with a focus on bone metastasis.
Organ specific metastases from primary tumors.
Metastatic Course, Routes and Steps
At an early stage, cancerous cells are confined to the primary site within the boundary of certain surrounding tissues. As the disease progresses, some cancer cells, as the result of genetic/ epigenetic predisposition, environmental interaction/stimulation, and indeed the combination of these elements, become more aggressive and begin to breach the surrounding structure. These cells would either directly invade the surrounding tissue, or disseminate via lymphatic and hematogenous routes. Direct invasion may result in the spreading of cancer cells to surrounding tissues and neighboring organs. For example, the local invasion of prostate cancer, can affect the erectile nerves, seminal vesicles, bladder and rectum nearby the prostate. The lymphatic and vascular routes differ from cancer to cancer according to their primary sites, however, frequently result in the systemic spread of cancer cells to distant organs, including bones, lung, and liver. For example, the primary lymphatic drainage of the prostate is via the internal iliac, perivesical, external iliac, obturator, and presacral nodes. The secondary lymphatic drainage includes the inguinal, common iliac and parα-aortic nodes. These nodes are therefore prime locations when one searches for the involved positive lymph nodes. Since the end of last century, a new technique, sentinel lymph node dissection has been developed and introduced in the detection, staging and management of lymph node involvement in cancer. The detection of a positive sentinel node indicates the need for a wide dissection of lymph nodes during surgery.
Both lymphatic and hematogenous dissemination frequently occur, even during early stages of the disease, and are seen in a vast majority of the patients who have an advanced cancer. To determine if systemic spread ‘occurred’ or not is a highly controversial topic, a conclusion of which is dependent on a wide variety of factors, from the type of samples to test, location and timing of sampling, techniques to detect cancer cells, to the interpretation of the presence of cancer cells or a cancer cell in a sample. Nonetheless, brain, bone, lung and liver are the most leading hematogenous sites from certain solid tumors.152–155
The process of metastasis is complex and arduous, which incorporates multiple cells, factors and stages. During the development and progression of primary tumors, certain clones of tumor cells will have the required genotypic and phenotypic characteristics to enable themselves to interact with the local microenvironment. For example, tumor cells release VEGF to initiate angiogenesis, thus enhancing the blood supply to the tumor. The stromal cells are rich sources of protein factors that directly act on cancer cells thus driving the growth of tumors and dissemination of cancer cells. On the other hand, some of the stromal cell derived factors will directly induce angiogenesis thus supporting the growth and spread of an aggressive tumor. A good example of these stromα-derived protein factors is hepatocyte growth factor (HGF), a cytokine secreted by the stroma cells, which has been implicated in the angiogenesis and the dissemination of tumor cells.133 The disruption of intercellular adhesion in the tumor causes some tumor cells to detach from the tumor mass (detachment), followed by these cells invading through the extracellular matrix, a process so-called invasion which incorporates the motility, migration of tumor cells and breakdown of extracellular matrix. Some tumor cells will penetrate the blood vessels, thus entering the circulation (intravasation). From this point, these tumor cells move away from the primary site and circulate in the blood circulation where, they would encounter resistance by the immune system and the mechanical stresses of blood flow. Some tumor cells will eventually survive and adopt a process to leave the blood circulation, known as extravasation, in which cells adhere and penetrate the blood vessel again (a virtual reversal of the intravasation process). Once the tumor cells escape from the circulation, they will have to survive and finally develop a secondary tumor at the other site, in this case in bone. This complex process also needs the integration of multiple factors and events, such as invasion of tumor, angiogenesis and the interaction between tumor cells and the local microenvironment at a distant site/organ.
Metastasis Regulators
The interrelated and sequential multi-steps of metastasis require certain transformations of cancer cells at each step, from primary site to metastatic site. Numerous genes and molecules have been implicated into this dynamic and adaptable evolution of metastatic cancer cells, including suppressors and promoters of metastasis which may be altered genetically or epigenetically in accordance with the requirements at each step. Initiating factors for tumor progression and metastasis are critical and essential, particularly for dissociation and invasion which allow cancer cells to leave primary sites. The genes that determine these activities have been defined as metastasis initiation genes.156,157 These genes could promote cell motility, epithelial mesenchymal transition (EMT), extracellular matrix degradation, angiogenesis or evasion of the immune system. For example, EMT is mediated by developmental programmes that are under the control of aberrantly regulated transcription factors, such as Twist1, Snai1 and Snai2 (also known as Slug). Other determinants of invasion are components and modulators of certain pathways which include hepatocyte growth factor (HGF), VEGF and ERK pathways. Metastatic growth is also initiated by the suppression of non-coding RNAs, such as miR-126 and miR-335 in breast and gastric carcinomas.158,159 Some of the initiating factors that allow transformed cells to invade the surrounding tissue and attract a supportive stroma facilitate the dissemination of cancer cells and probably continue to do so after cancer cells infiltrate distant tissues. This is why some prognosis signatures of a malignancy can also be utilized as a signature to predict metastases.153
Metastasis suppressor genes are defined by their ability to inhibit metastasis at any step of the metastatic cascade. These metastasis suppressor genes inhibit metastasis of cancer cells, in vivo, without blocking tumorigenicity. To date, some metastasis suppressor genes have been identified, such as nonmetastatic gene 23 (NM23), Kangai 1 (KAI1), KISS1, mitogen-activated protein kinase 4 (MKK4), breast cancer metastasis suppressor 1 (BRMS1), Rho GDP dissociation inhibitor 2 (RhoGDI2), cofactor required for Sp1 transcriptional activation subunit 3 (CRSP3) and Vitamin D3 upregulated protein 1 (VDUP1). Deregulation of these metastasis suppressor genes has been indicated in certain solid tumors.160–162
‘Seeds’ and ‘Soil’ Crosstalk between Cancer Cells and the Microenvironment during Bone Metastasis
Bone metastasis has been characterized as either osteolytic or osteoblastic. This classification actually represents two extremes of a continuum in which dysregulation of the normal bone remodelling process occurs. Patients can have both osteolytic and osteoblastic metastases or mixed lesions containing both elements. Most metastatic bone tumors from breast cancer have predominantly osteolytic lesions. In contrast, the metastatic lesions from prostate cancer are predominantly osteoblastic. During osteoblastic bone metastases, the balance between bone resorption and bone formation is tipped in favor of the latter. Patients suffer severe bone pain and the poor quality of bone produced in osteoblastic bone metastases frequently leads to bone fractures. Models to investigate osteoblastic metastases are rather rare, compared with models of osteolytic metastasis. Mechanisms, by which a metastatic lesion becomes osteoblastic or osteolytic remain unclear. However, a number of factors produced by cancer cells, such as platelet-derived growth factor (PDGF), insulin-like growth factors (IGFs), fibroblast growth factors (FGFs), VEGF, Wingless and NT-1 (WNT1), parathyroid hormone related protein (PTHrP), urokinase-type plasminogen activator (uPA), prostate specific antigen (PSA), endothelin-1 (ET-1) and BMPs, have been implicated in osteoblastic lesions.
The question of why the bone is the most preferred metastatic site of some solid tumors (breast, prostate and lung cancer) has aroused intense interest. One would first contemplate the anatomical characteristics of the organs at primary sites. The blood supply to the organs may provide a shortcut for the hematogenous dissemination of tumor cells from primary tumor to certain bones. For example, a rich venous plexus surrounds the prostate and connects to the venous drainage of the spine: this collection of veins (Batson’s plexus) is potentially one of the reasons why the lumbosacral spinal metastases are common in advanced prostate cancer.163 However, the anatomical explanation is not able to explain why the other axial skeleton, skull and ribs may also be involved in the bone metastasis from prostate cancer.
The ‘seed and soil’ theory proposed by Paget may provide some clue from a different standpoint.164 Osteotropic ‘seeds’ (tumor cells) may be developed during the progression of prostate cancer. These tumor cells may have acquired specific genetic phenotype, or activation of specific cytokine and proteases. These features direct the metastasis to bone. For example, elevated expression of BMPs and TGF-β in prostate cancer cells have been implicated in bone metastasis.165–168 The “seeds” may also attach to the bone endothelium more effectively than to the endothelia of other organs.169 It has been suggested that the protease-activated receptor (PAR1, thrombin receptor) and integrin αVβ3 which are highly expressed in primary prostate cancer cell lines and metastatic prostate cancer cells derived from bone metastasis, may contribute to the bone metastases through facilitating the attachment of tumor cells to blood vessel walls and the process of extravasation.170–173 The vascular endothelial growth factor (VEGF) secreted by the tumor cells may also contribute to the bone metastasis due to both the promotion of angiogenesis and the activation of osteoblasts.174–176
On the other hand, bone also provides a fertile “soil” for the “seeds”. The bone matrix synthesized by osteoblasts has a particular abundance of cytokines and non-collagen proteins, which may attract prostate cancer cells and allow them to survive and proliferate in the bone matrix. For example, BMPs and TGF-β enriched in bone matrix can facilitate the development of bone metatstasis. Osteonectin, osteopontin, osteocalcin, and bone sialoprotein can also modulate the properties of prostate cancer cells and facilitate the spreading and growth, including promoting their migration, invasion and proliferation.177–182 Bone turnover, as a characteristic of the adult bone, occurs most often in the bones rich in trabecular bone, such as the vertebrae, proximal femur, calcaneous, and ultradistal radius. During the bone turnover, cytokines and NCPs released or synthesized through bone resorption and bone formation thus generate a fertile ‘soil’. This may supplement the explanation of the favorite locations in bone metastases.
During the development of bone metastasis from prostate cancer, the interactions among tumor cells, bone cells and bone matrix constitute a “vicious cycle” of osteoblast/ osteoclast-mediated bone metastasis. For example, during the osteoblastic bone metastases of prostate cancer, cancer cells produce osteogenic factors such as ET-1, BMPs and PDGF, to activate osteoblasts. The osteoblasts differentiated from their progenitor cells deposit new matrix for bone formation. However, this unmineralised new matrix provides a more fertile soil to tumor cells, which is enriched with growth factors and NCPs. These factors help prostate cancer cells survive and proliferate in the bone microenvironment. The prostate cancer cells then further activate osteoblasts. In addition to this vicious cycle, at certain stages, both tumor-derived factors and osteoblasts expressing RANKL can activate osteoclasts, leading to some level of bone resorption, and subsequently generate bigger space for dominant osteoblastic lesion. The cytokines and NCPs released from bone matrix during bone resorption can also enhance this “vicious cycle” through facilitating proliferation of both prostate cancer cells and osteoblasts.
Conclusion
Metastasis, the leading cause of mortality in patients with cancer, is receiving increasing attention in both scientfic and clinical research. Yet the mechanisms remain poorly understood and methods in combatting metastasis remain limited. It is however pleasing to observe some of the major progresses in this vital area of cancer research. With the increasing knowledge in gene expression, cellular behavior, biological events in the spread paths of cancer cells, there are now new prospects of taking some of the observations into the diagnosis, prognosis and treatment in the metastatic disease. For example, new knowledge on barrier function and paracelluar permeability may allow one to devise new direction in controlling the trepassing cancer cells and their entry into the destination tissues and organs. New biomarkers in areas such as epithelial to mesenchymal transtion offer new opportunities in predictive methods of metastatic potential of a primary tumor and new target for therapy. Angiogenesis has already been a fruitful area in new therapies and the organ specific spread of a solid tumor may allow new method of detection and a new way of targeting metastatic tumor cells. Although enormous challenges remain, it is anticipated that these lines of research will steadily find their into clinical practice.
Acknowledgment
The authors wish to thank Cancer Research Wales, the Albert Hung Foundation, the Breast Cancer Hope Foundation, and the Welsh Assembly Government for supporting their work.
References
1.
DeVita VT Jr., Young RC, Canellos GP. Combination versus single agent chemotherapy: a review of the basis for selection of drug treatment of cancer. Cancer. 1975;35:98–110. http://dx.doi.org/10.1002/1097-0142(197501)35:1<98::AID-CNCR2820350115>3.0.CO;2-B . [PubMed]
2.
Martin TA, Jiang WG. Loss of tight junction barrier function and its role in cancer metastasis. Biochim Biophys Acta. 2009;1788:872–91. http://dx.doi.org/10.1016/j.bbamem.2008.11.005 . [PubMed]
3.
Brooks PC. Cell adhesion molecules in angiogenesis. Cancer Metastasis Rev. 1996;15:187–94.http://dx.doi.org/10.1007/BF00437471 . [PubMed]
4.
Folkman J, Shing Y. Angiogenesis. J Biol Chem. 1992;267:10931–4. [PubMed]
5.
Folkman J. Fighting cancer by attacking its blood supply. Sci Am. 1996;275:150–4.http://dx.doi.org/10.1038/scientificamerican0996-150 . [PubMed]
6.
Ono M, Torisu H, Fukushi J, Nishie A, Kuwano M. Biological implications of macrophage infiltration in human tumor angiogenesis. Cancer Chemother Pharmacol. 1999;43(Suppl):S69–71.http://dx.doi.org/10.1007/s002800051101 . [PubMed]
7.
Jiang WG, Bryce RP, Horrobin DF, Mansel RE. Regulation of tight junction permeability and occludin expression by polyunsaturated fatty acids. Biochem Biophys Res Commun. 1998;244:414–20.http://dx.doi.org/10.1006/bbrc.1998.8288 . [PubMed]
8.
Jiang WG, Martin TA, Matsumoto K, Nakamura T, Mansel RE. Hepatocyte growth factor/scatter factor decreases the expression of occludin and transendothelial resistance (TER) and increases paracellular permeability in human vascular endothelial cells. J Cell Physiol. 1999;181:319–29. http://dx.doi.org/10.1002/(SICI)1097-4652(199911)181:2<319::AID-JCP14>3.0.CO;2-S . [PubMed]
9.
Tsukita S, Furuse M. Occludin and claudins in tight-junction strands: leading or supporting players? Trends Cell Biol. 1999;9:268–73. http://dx.doi.org/10.1016/S0962-8924(99)01578-0 . [PubMed]
10.
Wong V, Gumbiner BM. A synthetic peptide corresponding to the extracellular domain of occludin perturbs the tight junction permeability barrier. J Cell Biol. 1997;136:399–409. http://dx.doi.org/10.1083/jcb.136.2.399 . [PMC free article] [PubMed]
11.
Hollande F, Blanc EM, Bali JP, Whitehead RH, Pelegrin A, Baldwin GS, et al. HGF regulates tight junctions in new nontumorigenic gastric epithelial cell line. Am J Physiol Gastrointest Liver Physiol. 2001;280:G910–21.[PubMed]
12.
Martin TA, Mansel RE, Jiang WG. Antagonistic effect of NK4 on HGF/SF induced changes in the transendothelial resistance (TER) and paracellular permeability of human vascular endothelial cells. J Cell Physiol.2002;192:268–75. http://dx.doi.org/10.1002/jcp.10133 . [PubMed]
13.
Ren J, Hamada J, Takeichi N, Fujikawa S, Kobayashi H. Ultrastructural differences in junctional intercellular communication between highly and weakly metastatic clones derived from rat mammary carcinoma. Cancer Res.1990;50:358–62. [PubMed]
14.
Satoh H, Zhong Y, Isomura H, Saitoh M, Enomoto K, Sawada N, et al. Localization of 7H6 tight junction-associated antigen along the cell border of vascular endothelial cells correlates with paracellular barrier function against ions, large molecules, and cancer cells. Exp Cell Res. 1996;222:269–74.http://dx.doi.org/10.1006/excr.1996.0034 . [PubMed]
15.
Hoevel T, Macek R, Mundigl O, Swisshelm K, Kubbies M. Expression and targeting of the tight junction protein CLDN1 in CLDN1-negative human breast tumor cells. J Cell Physiol. 2002;191:60–8.http://dx.doi.org/10.1002/jcp.10076 . [PubMed]
16.
Martinez-Paloma A. Ultrastructural modifications of intercellular junctions in some epithelial tumors Lab Invest1970. 22 605 14. [PubMed]
17.
Inoue T, Shimono M, Yamamura T, Saito I, Watanabe O, Kawahara H. Acinic cell carcinoma arising in the glossopalatine glands: a report of two cases with electron microscopic observations. Oral Surg Oral Med Oral Pathol. 1984;57:398–407. http://dx.doi.org/10.1016/0030-4220(84)90159-2 . [PubMed]
18.
Mullin JM, O’Brien TG. Effects of tumor promoters on LLC-PK1 renal epithelial tight junctions and transepithelial fluxes. Am J Physiol. 1986;251:C597–602. [PubMed]
19.
Bornholdt J, Friis S, Godiksen S, Poulsen SS, Santoni-Rugiu E, Bisgaard HC, et al. The level of claudin-7 is reduced as an early event in colorectal carcinogenesis. BMC Cancer. 2011;11:65. http://dx.doi.org/10.1186/1471-2407-11-65 . [PMC free article] [PubMed]
20.
Nakagawa S, Miyoshi N, Ishii H, Mimori K, Tanaka F, Sekimoto M, et al. Expression of CLDN1 in colorectal cancer: a novel marker for prognosis. Int J Oncol. 2011;39:791–6. [PubMed]
21.
Wang X, Tully O, Ngo B, Zitin M, Mullin JM. Epithelial tight junctional changes in colorectal cancer tissues.ScientificWorldJournal. 2011;11:826–41. http://dx.doi.org/10.1100/ tsw.2011.86 . [PubMed]
22.
Takai E, Tan X, Tamori Y, Hirota M, Egami H, Ogawa M. Correlation of translocation of tight junction protein Zonula occludens-1 and activation of epidermal growth factor receptor in the regulation of invasion of pancreatic cancer cells. Int J Oncol. 2005;27:645–51. [PubMed]
23.
Zhou L, Tan X, Wang W, Wang B, Dai X, Liu J. Analysis of invasion-metastasis in pancreatic cancer: Correlation between the expression and arrangement of tight junction protein-2 and cell dissociation in pancreatic cancer cells.Mol Med Report. 2010;3:149–53. [PubMed]
24.
Kojima T, Takasawa A, Kyuno D, Ito T, Yamaguchi H, Hirata K, et al. Downregulation of tight junction-associated MARVEL protein marvelD3 during epithelial-mesenchymal transition in human pancreatic cancer cells.Exp Cell Res. 2011;317:2288–98. http://dx.doi.org/10.1016/j. yexcr.2011.06.020 . [PubMed]
25.
Martin TA, Mansel RE, Jiang WG. Loss of occludin leads to the progression of human breast cancer. Int J Mol Med. 2010;26:723–34. http://dx.doi.org/10.3892/ijmm_00000519 . [PubMed]
26.
Myal Y, Leygue E, Blanchard AA. Claudin 1 in breast tumorigenesis: revelation of a possible novel “claudin high” subset of breast cancers. J Biomed Biotechnol. 2010;2010:956897. http://dx.doi.org/10.1155/2010/956897 . [PMC free article] [PubMed]
27.
Wu Q, Liu Y, Ren Y, Xu X, Yu L, Li Y, et al. Tight junction protein, claudin-6, downregulates the malignant phenotype of breast carcinoma. Eur J Cancer Prev. 2010;19:186–94.http://dx.doi.org/10.1097/CEJ.0b013e328337210e . [PubMed]
28.
Martin TA, Watkins G, Mansel RE, Jiang WG. Hepatocyte growth factor disrupts tight junctions in human breast cancer cells. Cell Biol Int. 2004a;28:361–71. http://dx.doi. org/10.1016/j.cellbi.2004.03.003 . [PubMed]
29.
Utoguchi N, Mizuguchi H, Dantakean A, Makimoto H, Wakai Y, Tsutsumi Y, et al. Effect of tumour cell-conditioned medium on endothelial macromolecular permeability and its correlation with collagen. Br J Cancer.1996;73:24–8. http://dx.doi.org/10.1038/bjc.1996.5 . [PMC free article] [PubMed]
30.
Martin TA, Watkins G, Mansel RE, Jiang WG. Loss of tight junction plaque molecules in breast cancer tissues is associated with a poor prognosis in patients with breast cancer. Eur J Cancer. 2004b;40:271725.http://dx.doi.org/10.1016/j.ejca.2004.08.008 . [PubMed]
31.
Giepmans BN, van Ijzendoorn SC. Epithelial cell-cell junctions and plasma membrane domains. Biochim Biophys Acta. 2009;1788:820–31. http://dx.doi.org/10.1016/j.bbamem.2008.07.015 . [PubMed]
32.
Farquhar MG, Palade GE. Junctional complexes in various epithelia. J Cell Biol. 1963;17:375–412.http://dx.doi.org/10.1083/jcb.17.2.375 . [PMC free article] [PubMed]
33.
Meng W, Takeichi M. Adherens junction: molecular architecture and regulation. Cold Spring Harb Perspect Biol.2009;1:a002899. http://dx.doi.org/10.1101/cshperspect.a002899 . [PMC free article] [PubMed]
34.
Yonemura S, Itoh M, Nagafuchi A, Tsukita S. Cell-to-cell adherens junction formation and actin filament organization: similarities and differences between non-polarized fibroblasts and polarized epithelial cells. J Cell Sci.1995;108:127–42. [PubMed]
35.
Uchida N, Honjo Y, Johnson KR, Wheelock MJ, Takeichi M. The catenin/cadherin adhesion system is localized in synaptic junctions bordering transmitter release zones. J Cell Biol. 1996;135:767–79.http://dx.doi.org/10.1083/jcb.135.3.767 . [PMC free article] [PubMed]
36.
Gooding JM, Yap KL, Ikura M. The cadherin-catenin complex as a focal point of cell adhesion and signalling: new insights from three-dimensional structures. Bioessays. 2004;26:497–511. http://dx.doi.org/10.1002/bies.20033 . [PubMed]
37.
Reynolds AB, Daniel J, McCrea PD, Wheelock MJ, Wu J, Zhang Z. Identification of a new catenin: the tyrosine kinase substrate p120cas associates with E-cadherin complexes. Mol Cell Biol. 1994;14:8333–42. [PMC free article] [PubMed]
38.
Togashi H, Miyoshi J, Honda T, Sakisaka T, Takai Y, Takeichi M. Interneurite affinity is regulated by heterophilic nectin interactions in concert with the cadherin machinery. J Cell Biol. 2006;174:141–51.http://dx.doi.org/10.1083/jcb.200601089 . [PMC free article] [PubMed]
39.
Cavallaro U, Christofori G. Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat Rev Cancer.2004;4:118–32. http://dx.doi.org/10.1038/nrc1276 . [PubMed]
40.
Vleminckx K, Vakaet L Jr., Mareel M, Fiers W, van Roy F. Genetic manipulation of E-cadherin expression by epithelial tumor cells reveals an invasion suppressor role. Cell. 1991;66:107–19. http://dx.doi.org/10.1016/0092-8674(91)90143-M . [PubMed]
41.
Luo J, Lubaroff DM, Hendrix MJ. Suppression of prostate cancer invasive potential and matrix metalloproteinase activity by E-cadherin transfection. Cancer Res. 1999;59:3552–6. [PubMed]
42.
Hsu MY, Meier FE, Nesbit M, Hsu JY, Van Belle P, Elder DE, et al. E-cadherin expression in melanoma cells restores keratinocyte-mediated growth control and down-regulates expression of invasion-related adhesion receptors. Am J Pathol. 2000;156:1515–25. http://dx.doi.org/10.1016/ S0002-9440(10)65023-7 . [PMC free article] [PubMed]
43.
Kashiwagi S, Yashiro M, Takashima T, Aomatsu N, Ikeda K, Ogawa Y, et al. Advantages of adjuvant chemotherapy for patients with triple-negative breast cancer at Stage II: usefulness of prognostic markers E-cadherin and Ki67. Breast Cancer Res. 2011;13:R122. http://dx.doi.org/10.1186/ bcr3068 . [PMC free article] [PubMed]
44.
Morrogh M, Andrade VP, Giri D, Sakr RA, Paik W, Qin LX, et al. Cadherin-catenin complex dissociation in lobular neoplasia of the breast. Breast Cancer Res Treat. 2012;132:641–52. http://dx.doi.org/10.1007/s10549-011-1860-0 . [PMC free article] [PubMed]
45.
Garrod D, Chidgey M. Desmosome structure, composition and function. Biochim Biophys Acta. 2008;1778:572–87. http://dx.doi.org/10.1016/j.bbamem.2007.07.014 . [PubMed]
46.
Brooke MA, Nitoiu D, Kelsell DP. Cell-cell connectivity: desmosomes and disease. J Pathol. 2012;226:158–71.http://dx.doi.org/10.1002/path.3027 . [PubMed]
47.
Dusek RL, Attardi LD. Desmosomes: new perpetrators in tumour suppression. Nat Rev Cancer. 2011;11:317–23.http://dx.doi.org/10.1038/nrc3051 . [PMC free article] [PubMed]
48.
Furukawa C, Daigo Y, Ishikawa N, Kato T, Ito T, Tsuchiya E, et al. Plakophilin 3 oncogene as prognostic marker and therapeutic target for lung cancer. Cancer Res. 2005;65:7102–10. http://dx.doi.org/10.1158/0008-5472.CAN-04-1877 . [PubMed]
49.
Brennan D, Mahoney MG. Increased expression of Dsg2 in malignant skin carcinomas: A tissuemicroarray based study. Cell Adh Migr. 2009;3:148–54. http://dx.doi.org/10.4161/ cam.3.2.7539 . [PMC free article] [PubMed]
50.
Breuninger S, Reidenbach S, Sauer CG, Strbel P, Pfitzenmaier J, Trojan L, et al. Desmosomal plakophilins in the prostate and prostatic adenocarcinomas: implications for diagnosis and tumor progression. Am J Pathol.2010;176:2509–19. http://dx.doi.org/10.2353/ajpath.2010.090737 . [PMC free article] [PubMed]
51.
Kolegraff K, Nava P, Helms MN, Parkos CA, Nusrat A. Loss of desmocollin-2 confers a tumorigenic phenotype to colonic epithelial cells through activation of Akt/b-catenin signaling. Mol Biol Cell. 2011;22:1121–34.http://dx.doi.org/10.1091/mbc.E10-10-0845 . [PMC free article] [PubMed]
52.
Teh MT, Parkinson EK, Thurlow JK, Liu F, Fortune F, Wan H. A molecular study of desmosomes identifies a desmoglein isoform switch in head and neck squamous cell carcinoma. J Oral Pathol Med. 2011;40:67–76.http://dx.doi.org/10.1111/j.1600-0714.2010.00951.x . [PubMed]
53.
Gosavi P, Kundu ST, Khapare N, Sehgal L, Karkhanis MS, Dalal SN. E-cadherin and plakoglobin recruit plakophilin3 to the cell border to initiate desmosome assembly. Cell Mol Life Sci. 2011;68:1439–54.http://dx.doi.org/10.1007/s00018-010-0531-3 . [PubMed]
54.
Derangeon M, Spray DC, Bourmeyster N, Sarrouilhe D, Herv JC. Reciprocal influence of connexins and apical junction proteins on their expressions and functions. Biochim Biophys Acta. 2009;1788:76878.http://dx.doi.org/10.1016/j.bbamem.2008.10.023 . [PMC free article] [PubMed]
55.
Lamiche C, Clarhaut J, Strale PO, Crespin S, Pedretti N, Bernard FX, et al. The gap junction protein Cx43 is involved in the bone-targeted metastatic behaviour of human prostate cancer cells. Clin Exp Metastasis.2012;29:111–22. http://dx.doi.org/10.1007/s10585-011-9434-4 . [PubMed]
56.
Sirnes S, Bruun J, Kolberg M, Kjenseth A, Lind GE, Svindland A, et al. Connexin43 acts as a colorectal cancer tumor suppressor and predicts disease outcome. Int J Cancer. 2011. http://dx.doi.org/10.1002/ijc.26392 . [PubMed]
57.
Bendas G, Borsig L. Cancer cell adhesion and metastasis: selectins, integrins, and the inhibitory potential of heparins. Int J Cell Biol. 2012;2012:676731. http://dx.doi. org/10.1155/2012/676731 . [PMC free article] [PubMed]
58.
van den Hoogen C, van der Horst G, Cheung H, Buijs JT, Pelger RC, van der Pluijm G. Integrin αv expression is required for the acquisition of a metastatic stem/progenitor cell phenotype in human prostate cancer. Am J Pathol.2011;179:2559–68. http://dx.doi.org/10.1016/j. ajpath.2011.07.011 . [PMC free article] [PubMed]
59.
Lu JG, Li Y, Li L, Kan X. Overexpression of osteopontin and integrin αv in laryngeal and hypopharyngeal carcinomas associated with differentiation and metastasis. J Cancer Res Clin Oncol. 2011;137:1613–8.http://dx.doi.org/10.1007/s00432-011-1024-y . [PubMed]
60.
St Hill CA. Interactions between endothelial selectins and cancer cells regulate metastasis. [Elite Ed] Front Biosci.2012;17:3233–51. [PubMed]
61.
Richter U, Schrder C, Wicklein D, Lange T, Geleff S, Dippel V, et al. Adhesion of small cell lung cancer cells to E- and P-selectin under physiological flow conditions: implications for metastasis formation. Histochem Cell Biol.2011;135:499–512. http://dx.doi.org/10.1007/s00418-0110804-4 . [PubMed]
62.
Wai Wong C, Dye DE, Coombe DR. The role of immunoglobulin superfamily cell adhesion molecules in cancer metastasis. Int J Cell Biol. 2012;2012:340296. http://dx.doi. org/10.1155/2012/340296 . [PMC free article] [PubMed]
63.
Martin TA, Harrison G, Mansel RE, Jiang WG. The role of the CD44/ezrin complex in cancer metastasis. Crit Rev Oncol Hematol. 2003;46:165–86. http://dx.doi.org/10.1016/ S1040-8428(02)00172-5 . [PubMed]
64.
Jeyapalan Z., Yang B. B. The non-coding 3’UTR of CD44 induces metastasis by regulating extracellular matrix functions. J Cell Sci. 2012 [PubMed]
65.
Park YS, Huh JW, Lee JH, Kim HR. shRNA against CD44 inhibits cell proliferation, invasion and migration, and promotes apoptosis of colon carcinoma cells. Oncol Rep. 2012;27:339–46. [PubMed]
66.
Schwock J, Dhani N, Hedley DW. Targeting focal adhesion kinase signaling in tumor growth and metastasis.Expert Opin Ther Targets. 2010;14:77–94. http://dx.doi. org/10.1517/14728220903460340 . [PubMed]
67.
Wang W, Liu Y, Liao K. Tyrosine phosphorylation of cortactin by the FAK-Src complex at focal adhesions regulates cell motility. BMC Cell Biol. 2011;12:49. http://dx.doi.org/10.1186/14712121-12-49 . [PMC free article] [PubMed]
68.
Bernardi MA, Logullo AF, Pasini FS, Nonogaki S, Blumke C, Soares FA, et al. Prognostic significance of CD24 and claudin-7 immunoexpression in ductal invasive breast cancer. Oncol Rep. 2012;27:28–38. [PubMed]
69.
Hornsby CD, Cohen C, Amin MB, Picken MM, Lawson D, Yin-Goen Q, et al. Claudin-7 immunohistochemistry in renal tumors: a candidate marker for chromophobe renal cell carcinoma identified by gene expression profiling.Arch Pathol Lab Med. 2007;131:1541–6. [PubMed]
70.
Sheehan GM, Kallakury BV, Sheehan CE, Fisher HA, Kaufman RP Jr., Ross JS. Loss of claudins-1 and -7 and expression of claudins-3 and -4 correlate with prognostic variables in prostatic adenocarcinomas. Hum Pathol.2007;38:564–9. http://dx.doi.org/10.1016/j.humpath.2006.11.007 . [PubMed]
71.
Zheng JY, Yu D, Foroohar M, Ko E, Chan J, Kim N, et al. Regulation of the expression of the prostate-specific antigen by claudin-7. J Membr Biol. 2003;194:187–97. http://dx.doi. org/10.1007/s00232-003-2038-4 . [PubMed]
72.
Chao YC, Pan SH, Yang SC, Yu SL, Che TF, Lin CW, et al. Claudin-1 is a metastasis suppressor and correlates with clinical outcome in lung adenocarcinoma. Am J Respir Crit Care Med. 2009;179:12333.http://dx.doi.org/10.1164/rccm.200803-456OC . [PubMed]
73.
Richardson F, Young GD, Sennello R, Wolf J, Argast GM, Mercado P, et al. The evaluation of E-Cadherin and vimentin as biomarkers of clinical outcomes among patients with non-small cell lung cancer treated with erlotinib as second- or third-line therapy. Anticancer Res. 2012;32:537–52. [PubMed]
74.
Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, et al. Cell migration: integrating signals from front to back. Science. 2003;302:1704–9. http://dx.doi.org/10.1126/science.1092053 . [PubMed]
75.
Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–10. http://dx.doi.org/10.1016/0092-8674(92)90164-8 . [PubMed]
76.
Faassen AE, Schrager JA, Klein DJ, Oegema TR, Couchman JR, McCarthy JB. A cell surface chondroitin sulfate proteoglycan, immunologically related to CD44, is involved in type I collagen-mediated melanoma cell motility and invasion. J Cell Biol. 1992;116:521–31. http://dx.doi.org/10.1083/ jcb.116.2.521 . [PMC free article] [PubMed]
77.
Huber MA, Kraut N, Beug H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr Opin Cell Biol. 2005;17:548–58. http://dx.doi.org/10.1016/j.ceb.2005.08.001 . [PubMed]
78.
Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol.2006;7:131–42. http://dx.doi.org/10.1038/nrm1835 . [PubMed]
79.
Buijs JT, Henriquez NV, van Overveld PG, van der Horst G, ten Dijke P, van der Pluijm G. TGF-beta and BMP7 interactions in tumour progression and bone metastasis. Clin Exp Metastasis. 2007;24:60917.http://dx.doi.org/10.1007/s10585-007-9118-2 . [PubMed]
80.
Guarino M. Epithelial-mesenchymal transition and tumour invasion. Int J Biochem Cell Biol. 2007;39:2153–60.http://dx.doi.org/10.1016/j.biocel.2007.07.011 . [PubMed]
81.
Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–54.http://dx.doi.org/10.1038/nrc822 . [PubMed]
82.
Thiery JP. Epithelial-mesenchymal transitions in development and pathologies. Curr Opin Cell Biol. 2003;15:740–6. http://dx.doi.org/10.1016/j.ceb.2003.10.006 . [PubMed]
83.
Trimboli AJ, Fukino K, de Bruin A, Wei G, Shen L, Tanner SM, et al. Direct evidence for epithelial-mesenchymal transitions in breast cancer. Cancer Res. 2008;68:937–45. http://dx.doi.org/10.1158/0008-5472.CAN-07-2148 . [PubMed]
84.
Cano A, Prez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, et al. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2:76–83.http://dx.doi.org/10.1038/35000025 . [PubMed]
85.
Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7:415–28. http://dx.doi. org/10.1038/nrc2131 . [PubMed]
86.
Medici D, Hay ED, Olsen BR. Snail and Slug promote epithelial-mesenchymal transition through betacatenin-T-cell factor-4-dependent expression of transforming growth factor-beta3. Mol Biol Cell. 2008;19:4875–87.http://dx.doi.org/10.1091/mbc.E08-05-0506 . [PMC free article] [PubMed]
87.
Takeichi M. Cadherin cell adhesion receptors as a morphogenetic regulator. Science. 1991;251:1451–5.http://dx.doi.org/10.1126/science.2006419 . [PubMed]
88.
Jiang WG, Mansel RE. E-cadherin complex and its abnormalities in human breast cancer. Surg Oncol.2000;9:151–71. http://dx.doi.org/10.1016/S0960-7404(01)00010-X . [PubMed]
89.
Zschiesche W, Schnborn I, Behrens J, Herrenknecht K, Hartveit F, Lilleng P, et al. Expression of E-cadherin and catenins in invasive mammary carcinomas. Anticancer Res. 1997;17(1B):561–7. [PubMed]
90.
Jiang WG. E-cadherin and its associated protein catenins, cancer invasion and metastasis. Br J Surg. 1996;83:437–46. http://dx.doi.org/10.1002/bjs.1800830404 . [PubMed]
91.
Gottardi CJ, Wong E, Gumbiner BM. E-cadherin suppresses cellular transformation by inhibiting betacatenin signaling in an adhesion-independent manner. J Cell Biol. 2001;153:1049–60.http://dx.doi.org/10.1083/jcb.153.5.1049 . [PMC free article] [PubMed]
92.
Stockinger A, Eger A, Wolf J, Beug H, Foisner R. E-cadherin regulates cell growth by modulating proliferation-dependent beta-catenin transcriptional activity. J Cell Biol. 2001;154:1185–96.http://dx.doi.org/10.1083/jcb.200104036 . [PMC free article] [PubMed]
93.
Kim K, Lu Z, Hay ED. Direct evidence for a role of beta-catenin/LEF-1 signaling pathway in induction of EMT.Cell Biol Int. 2002;26:463–76. http://dx.doi.org/10.1006/cbir.2002.0901 . [PubMed]
94.
Pecina-Slaus N, Cicvara-Pecina T, Kafka A. Epithelial-to-mesenchymal transition: possible role in meningiomas [Elite Ed] Front Biosci (Elite Ed) 2012;4:889–96. http://dx.doi. org/10.2741/427 . [PubMed]
95.
Martin TA, Goyal A, Watkins G, Jiang WG. Expression of the transcription factors snail, slug, and twist and their clinical significance in human breast cancer. Ann Surg Oncol. 2005;12:488–96.http://dx.doi.org/10.1245/ASO.2005.04.010 . [PubMed]
96.
Rosivatz E, Becker I, Specht K, Fricke E, Luber B, Busch R, et al. Differential expression of the epithelial-mesenchymal transition regulators snail, SIP1, and twist in gastric cancer. Am J Pathol. 2002;161:1881–91.http://dx.doi.org/10.1016/S0002-9440(10)64464-1 . [PMC free article] [PubMed]
97.
Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927–39.http://dx.doi.org/10.1016/j.cell.2004.06.006 . [PubMed]
98.
Leptin M, Grunewald B. Cell shape changes during gastrulation in Drosophila. Development. 1990;110:73–84.[PubMed]
99.
Smith DE, Franco del Amo F, Gridley T. Isolation of Sna, a mouse gene homologous to the Drosophila genes snail and escargot: its expression pattern suggests multiple roles during postimplantation development.Development. 1992;116:1033–9. [PubMed]
100.
Batlle E, Sancho E, Franc C, Domnguez D, Monfar M, Baulida J, et al. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol. 2000;2:84–9.http://dx.doi.org/10.1038/35000034 . [PubMed]
101.
Yokoyama K, Kamata N, Hayashi E, Hoteiya T, Ueda N, Fujimoto R, et al. Reverse correlation of E-cadherin and snail expression in oral squamous cell carcinoma cells in vitro. Oral Oncol. 2001;37:6571.http://dx.doi.org/10.1016/S1368-8375(00)00059-2 . [PubMed]
102.
Blanco MJ, Moreno-Bueno G, Sarrio D, Locascio A, Cano A, Palacios J, et al. Correlation of Snail expression with histological grade and lymph node status in breast carcinomas. Oncogene. 2002;21:32416.http://dx.doi.org/10.1038/sj.onc.1205416 . [PubMed]
103.
Ikenouchi J, Matsuda M, Furuse M, Tsukita S. Regulation of tight junctions during the epithelium-mesenchyme transition: direct repression of the gene expression of claudins/occludin by Snail. J Cell Sci. 2003;116:1959–67.http://dx.doi.org/10.1242/jcs.00389 . [PubMed]
104.
Islam S, Carey TE, Wolf GT, Wheelock MJ, Johnson KR. Expression of N-cadherin by human squamous carcinoma cells induces a scattered fibroblastic phenotype with disrupted cell-cell adhesion. J Cell Biol.1996;135:1643–54. http://dx.doi.org/10.1083/jcb.135.6.1643 . [PMC free article] [PubMed]
105.
Nieman MT, Prudoff RS, Johnson KR, Wheelock MJ. N-cadherin promotes motility in human breast cancer cells regardless of their E-cadherin expression. J Cell Biol. 1999;147:631–44. http://dx.doi.org/10.1083/jcb.147.3.631 . [PMC free article] [PubMed]
106.
Mikheeva SA, Mikheev AM, Petit A, Beyer R, Oxford RG, Khorasani L, et al. TWIST1 promotes invasion through mesenchymal change in human glioblastoma. Mol Cancer. 2010;9:194. http://dx.doi.org/10.1186/1476-4598-9-194 . [PMC free article] [PubMed]
107.
Gupta R, Chetty C, Bhoopathi P, Lakka S, Mohanam S, Rao JS, et al. Downregulation of uPA/uPAR inhibits intermittent hypoxia-induced epithelial-mesenchymal transition (EMT) in DAOY and D283 medulloblastoma cells. Int J Oncol. 2011;38:733–44. [PubMed]
108.
Lester RD, Jo M, Montel V, Takimoto S, Gonias SL. uPAR induces epithelial-mesenchymal transition in hypoxic breast cancer cells. J Cell Biol. 2007;178:425–36. http://dx.doi.org/10.1083/ jcb.200701092 . [PMC free article] [PubMed]
109.
Strutz F, Zeisberg M, Ziyadeh FN, Yang CQ, Kalluri R, Mller GA, et al. Role of basic fibroblast growth factor-2 in epithelial-mesenchymal transformation. Kidney Int. 2002;61:1714–28. http://dx.doi.org/10.1046/j.1523-1755.2002.00333.x . [PubMed]
110.
Akhurst RJ, Derynck R. TGF-beta signaling in cancer–a double-edged sword. Trends Cell Biol. 2001;11:S44–51. [PubMed]
111.
Zeisberg M, Hanai J, Sugimoto H, Mammoto T, Charytan D, Strutz F, et al. BMP-7 counteracts TGFbeta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med. 2003;9:964–8.http://dx.doi.org/10.1038/nm888 . [PubMed]
112.
Ye L, Lewis-Russell JM, Kynaston H, Jiang WG. Endogenous bone morphogenetic protein-7 controls the motility of prostate cancer cells through regulation of bone morphogenetic protein antagonists. J Urol.2007;178:1086–91. http://dx.doi.org/10.1016/j.juro.2007.05.003 . [PubMed]
113.
Bokobza SM, Ye L, Kynaston H, Jiang WG. Growth and differentiation factor 9 (GDF-9) induces epithelial-mesenchymal transition in prostate cancer cells. Mol Cell Biochem. 2011;349:33–40.http://dx.doi.org/10.1007/s11010-010-0657-5 . [PubMed]
114.
Xu Z, Jiang Y, Steed H, Davidge S, Fu Y. TGFβ and EGF synergistically induce a more invasive phenotype of epithelial ovarian cancer cells. Biochem Biophys Res Commun. 2010;401:376–81.http://dx.doi.org/10.1016/j.bbrc.2010.09.059 . [PubMed]
115.
Wang X, Lu H, Urvalek AM, Li T, Yu L, Lamar J, et al. KLF8 promotes human breast cancer cell invasion and metastasis by transcriptional activation of MMP9. Oncogene. 2011;30:1901–11.http://dx.doi.org/10.1038/onc.2010.563 . [PMC free article] [PubMed]
116.
Qiao B, Johnson NW, Gao J. Epithelial-mesenchymal transition in oral squamous cell carcinoma triggered by transforming growth factor-beta1 is Snail family-dependent and correlates with matrix metalloproteinase-2 and -9 expressions. Int J Oncol. 2010;37:663–8. [PubMed]
117.
Lynch CC, Vargo-Gogola T, Matrisian LM, Fingleton B. Cleavage of E-Cadherin by Matrix Metalloproteinase-7 Promotes Cellular Proliferation in Nontransformed Cell Lines via Activation of RhoA. J Oncol.2010;2010:530745. http://dx.doi.org/10.1155/2010/530745 . [PMC free article] [PubMed]
118.
Abe K, Takeichi M. EPLIN mediates linkage of the cadherin catenin complex to F-actin and stabilizes the circumferential actin belt. Proc Natl Acad Sci U S A. 2008;105:13–9. http://dx.doi.org/10.1073/pnas.0710504105 . [PMC free article] [PubMed]
119.
Jiang WG, Martin TA, Lewis-Russell JM, Douglas-Jones A, Ye L, Mansel RE. Eplin-alpha expression in human breast cancer, the impact on cellular migration and clinical outcome. Mol Cancer. 2008;7:71.http://dx.doi.org/10.1186/1476-4598-7-71 . [PMC free article] [PubMed]
120.
Sanders AJ, Martin TA, Ye L, Mason MD, Jiang WG. EPLIN is a negative regulator of prostate cancer growth and invasion. J Urol. 2011;186:295–301. http://dx.doi.org/10.1016/j.juro.2011.03.038 . [PubMed]
121.
Zhang S, Wang X, Osunkoya AO, Iqbal S, Wang Y, Chen Z, et al. EPLIN downregulation promotes epithelial-mesenchymal transition in prostate cancer cells and correlates with clinical lymph node metastasis. Oncogene.2011;30:4941–52. http://dx.doi.org/10.1038/onc.2011.199 . [PMC free article] [PubMed]
122.
Hay ED. Organization and fine structure of epithelium and mesenchyme in the developing chick embryo. In: Epithelial-mesenchymal interactions. In: Fleischmajer R, Billingham RE, editors. Baltimore: Williams & Wilkins; 1968. pp. 31–55.
123.
Folkman J. Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov. 2007;6:273–86.http://dx.doi.org/10.1038/nrd2115 . [PubMed]
124.
Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. 2011;146:87387.http://dx.doi.org/10.1016/j.cell.2011.08.039 . [PubMed]
125.
Pandya NM, Dhalla NS, Santani DD. Angiogenesis–a new target for future therapy. Vascul Pharmacol.2006;44:265–74. http://dx.doi.org/10.1016/j.vph.2006.01.005 . [PubMed]
126.
Al-Rawi MA, Jiang WG. Lymphangiogenesis and cancer metastasis. Front Biosci. 2011;16:723–39.http://dx.doi.org/10.2741/3715 . [PubMed]
127.
Albrecht I, Christofori G. Molecular mechanisms of lymphangiogenesis in development and cancer. Int J Dev Biol. 2011;55:483–94. http://dx.doi.org/10.1387/ijdb.103226ia . [PubMed]
128.
Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–6.http://dx.doi.org/10.1056/NEJM197111182852108 . [PubMed]
129.
Klagsbrun M, D’Amore PA. Vascular endothelial growth factor and its receptors. Cytokine Growth Factor Rev.1996;7:259–70. http://dx.doi.org/10.1016/S1359-6101(96)00027-5 . [PubMed]
130.
Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. http://dx.doi.org/10.1038/nature10144 . [PMC free article] [PubMed]
131.
Ferrara N, Davis-Smyth T. The biology of vascular endothelial growth factor. Endocr Rev. 1997;18:425.http://dx.doi.org/10.1210/er.18.1.4 . [PubMed]
132.
Sato S, Itamochi H. Bevacizumab and ovarian cancer. Curr Opin Obstet Gynecol. 2012;24:8–13.http://dx.doi.org/10.1097/GCO.0b013e32834daeed . [PubMed]
133.
Jiang WG, Martin TA, Parr C, Davies G, Matsumoto K, Nakamura T. Hepatocyte growth factor, its receptor, and their potential value in cancer therapies. Crit Rev Oncol Hematol. 2005;53:35–69.http://dx.doi.org/10.1016/j.critrevonc.2004.09.004 . [PubMed]
134.
Wojta J, Kaun C, Breuss JM, Koshelnick Y, Beckmann R, Hattey E, et al. Hepatocyte growth factor increases expression of vascular endothelial growth factor and plasminogen activator inhibitor-1 in human keratinocytes and the vascular endothelial growth factor receptor flk-1 in human endothelial cells. Lab Invest. 1999;79:427–38.[PubMed]
135.
Davies G, Mason MD, Martin TA, Parr C, Watkins G, Lane J, et al. The HGF/SF antagonist NK4 reverses fibroblast- and HGF-induced prostate tumor growth and angiogenesis in vivo. Int J Cancer. 2003;106:348–54.http://dx.doi.org/10.1002/ijc.11220 . [PubMed]
136.
Martin TA, Parr C, Davies G, Watkins G, Lane J, Matsumoto K, et al. Growth and angiogenesis of human breast cancer in a nude mouse tumour model is reduced by NK4, a HGF/SF antagonist. Carcinogenesis. 2003;24:1317–23. http://dx.doi.org/10.1093/carcin/bgg072 . [PubMed]
137.
Eder JP, Shapiro GI, Appleman LJ, Zhu AX, Miles D, Keer H, et al. A phase I study of foretinib, a multi-targeted inhibitor of c-Met and vascular endothelial growth factor receptor 2. Clin Cancer Res. 2010;16:3507–16.http://dx.doi.org/10.1158/1078-0432.CCR-10-0574 . [PubMed]
138.
Banerji S, Ni J, Wang SX, Clasper S, Su J, Tammi R, et al. LYVE-1, a new homologue of the CD44 glycoprotein, is a lymph-specific receptor for hyaluronan. J Cell Biol. 1999;144:789–801.http://dx.doi.org/10.1083/jcb.144.4.789 . [PMC free article] [PubMed]
139.
Kaipainen A, Korhonen J, Mustonen T, van Hinsbergh VW, Fang GH, Dumont D, et al. Expression of the fms-like tyrosine kinase 4 gene becomes restricted to lymphatic endothelium during development. Proc Natl Acad Sci U S A. 1995;92:3566–70. http://dx.doi.org/10.1073/pnas.92.8.3566 . [PMC free article] [PubMed]
140.
McAllaster JD, Cohen MS. Role of the lymphatics in cancer metastasis and chemotherapy applications. Adv Drug Deliv Rev. 2011;63:867–75. http://dx.doi.org/10.1016/j.addr.2011.05.014 . [PubMed]
141.
Skobe M, Hawighorst T, Jackson DG, Prevo R, Janes L, Velasco P, et al. Induction of tumor lymphangiogenesis by VEGF-C promotes breast cancer metastasis. Nat Med. 2001;7:192–8. http://dx.doi.org/10.1038/84643 . [PubMed]
142.
Stacker SA, Caesar C, Baldwin ME, Thornton GE, Williams RA, Prevo R, et al. VEGF-D promotes the metastatic spread of tumor cells via the lymphatics. Nat Med. 2001;7:186–91. http://dx.doi.org/10.1038/84635 . [PubMed]
143.
Feng Y, Wang W, Hu J, Ma J, Zhang Y, Zhang J. Expression of VEGF-C and VEGF-D as significant markers for assessment of lymphangiogenesis and lymph node metastasis in non-small cell lung cancer. Anat Rec (Hoboken) 2010;293:802–12. http://dx.doi.org/10.1002/ar.21096 . [PubMed]
144.
Kilvaer TK, Valkov A, Sorbye S, Smeland E, Bremnes RM, Busund LT, et al. Profiling of VEGFs and VEGFRs as prognostic factors in soft tissue sarcoma: VEGFR-3 is an independent predictor of poor prognosis.PLoS One. 2010;5:e15368. http://dx.doi.org/10.1371/journal. pone.0015368 . [PMC free article] [PubMed]
145.
Raica M, Cimpean AM, Ceausu R, Ribatti D. Lymphatic microvessel density, VEGF-C, and VEGFR-3 expression in different molecular types of breast cancer. Anticancer Res. 2011;31:1757–64. [PubMed]
146.
Debois JM. 2002. TxNxM1: The Anatomy and Clinics of Metastatic Cancer, Kluwer Academic Publisher.
147.
Abbruzzese JL, Abbruzzese MC, Lenzi R, Hess KR, Raber MN. Analysis of a diagnostic strategy for patients with suspected tumors of unknown origin. J Clin Oncol. 1995;13:2094–103. [PubMed]
148.
Schouten LJ, Rutten J, Huveneers HA, Twijnstra A. Incidence of brain metastases in a cohort of patients with carcinoma of the breast, colon, kidney, and lung and melanoma. Cancer. 2002;94:2698–705.http://dx.doi.org/10.1002/cncr.10541 . [PubMed]
149.
Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer.2002;2:584–93. http://dx.doi.org/10.1038/nrc867 . [PubMed]
150.
Hess KR, Varadhachary GR, Taylor SH, Wei W, Raber MN, Lenzi R, et al. Metastatic patterns in adenocarcinoma. Cancer. 2006;106:1624–33. http://dx.doi.org/10.1002/cncr.21778 . [PubMed]
151.
Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–49.http://dx.doi.org/10.3322/caac.20006 . [PubMed]
152.
Bubendorf L, Schpfer A, Wagner U, Sauter G, Moch H, Willi N, et al. Metastatic patterns of prostate cancer: an autopsy study of 1,589 patients. Hum Pathol. 2000;31:578–83. http://dx.doi.org/10.1053/hp.2000.6698 . [PubMed]
153.
Minn AJ, Kang Y, Serganova I, Gupta GP, Giri DD, Doubrovin M, et al. Distinct organ-specific metastatic potential of individual breast cancer cells and primary tumors. J Clin Invest. 2005;115:44–55. [PMC free article] [PubMed]
154.
Schlter K, Gassmann P, Enns A, Korb T, Hemping-Bovenkerk A, Hlzen J, et al. Organ-specific metastatic tumor cell adhesion and extravasation of colon carcinoma cells with different metastatic potential. Am J Pathol.2006;169:1064–73. http://dx.doi.org/10.2353/ ajpath.2006.050566 . [PMC free article] [PubMed]
155.
Nguyen DX, Bos PD, Massagu J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer.2009;9:274–84. http://dx.doi.org/10.1038/nrc2622 . [PubMed]
156.
Nguyen DX, Massagu J. Genetic determinants of cancer metastasis. Nat Rev Genet. 2007;8:341–52.http://dx.doi.org/10.1038/nrg2101 . [PubMed]
157.
Chiang AC, Massagu J. Molecular basis of metastasis. N Engl J Med. 2008;359:2814–23.http://dx.doi.org/10.1056/NEJMra0805239 . [PMC free article] [PubMed]
158.
Feng R, Chen X, Yu Y, Su L, Yu B, Li J, et al. miR-126 functions as a tumour suppressor in human gastric cancer. Cancer Lett. 2010;298:50–63. http://dx.doi.org/10.1016/j.canlet.2010.06.004 . [PubMed]
159.
Yang R, Dick M, Marme F, Schneeweiss A, Langheinz A, Hemminki K, et al. Genetic variants within miR-126 and miR-335 are not associated with breast cancer risk. Breast Cancer Res Treat. 2011;127:54954.http://dx.doi.org/10.1007/s10549-010-1244-x . [PubMed]
160.
Steeg PS. Metastasis suppressors alter the signal transduction of cancer cells. Nat Rev Cancer. 2003;3:55–63.http://dx.doi.org/10.1038/nrc967 . [PubMed]
161.
Malik FA, Sanders AJ, Jones AD, Mansel RE, Jiang WG. Transcriptional and translational modulation of KAI1 expression in ductal carcinoma of the breast and the prognostic significance. Int J Mol Med. 2009;23:273–8.[PubMed]
162.
Malik FA, Sanders AJ, Kayani MA, Jiang WG. Effect of expressional alteration of KAI1 on breast cancer cell growth, adhesion, migration and invasion. Cancer Genomics Proteomics. 2009;6:205–13. [PubMed]
163.
Batson OV. The function of the vertebral veins and their role in the spread of metastases. 1940. Clin Orthop Relat Res. 1995:4–9. [PubMed]
164.
Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989;8:98–101. [PubMed]
165.
Autzen P, Robson CN, Bjartell A, Malcolm AJ, Johnson MI, Neal DE, et al. Bone morphogenetic protein 6 in skeletal metastases from prostate cancer and other common human malignancies. Br J Cancer. 1998;78:1219–23.http://dx.doi.org/10.1038/bjc.1998.658 . [PMC free article] [PubMed]
166.
De Pinieux G, Flam T, Zerbib M, Taupin P, Bellahcne A, Waltregny D, et al. Bone sialoprotein, bone morphogenetic protein 6 and thymidine phosphorylase expression in localized human prostatic adenocarcinoma as predictors of clinical outcome: a clinicopathological and immunohistochemical study of 43 cases. J Urol.2001;166:1924–30. http://dx.doi.org/10.1016/S00225347(05)65722-9 . [PubMed]
167.
Shariat SF, Shalev M, Menesses-Diaz A, Kim IY, Kattan MW, Wheeler TM, et al. Preoperative plasma levels of transforming growth factor beta(1) (TGF-beta(1)) strongly predict progression in patients undergoing radical prostatectomy. J Clin Oncol. 2001;19:2856–64. [PubMed]
168.
Masuda H, Fukabori Y, Nakano K, Takezawa Y, CSuzuki T, Yamanaka H. Increased expression of bone morphogenetic protein-7 in bone metastatic prostate cancer. Prostate. 2003;54:268–74.http://dx.doi.org/10.1002/pros.10193 . [PubMed]
169.
Lehr JE, Pienta KJ. Preferential adhesion of prostate cancer cells to a human bone marrow endothelial cell line. J Natl Cancer Inst. 1998;90:118–23. http://dx.doi.org/10.1093/jnci/90.2.118 . [PubMed]
170.
Chay CH, Cooper CR, Gendernalik JD, Dhanasekaran SM, Chinnaiyan AM, Rubin MA, et al. A functional thrombin receptor (PAR1) is expressed on bone-derived prostate cancer cell lines. Urology. 2002;60:760–5.http://dx.doi.org/10.1016/S0090-4295(02)01969-6 . [PubMed]
171.
Cooper CR, Chay CH, Pienta KJ. The role of alpha(v)beta(3) in prostate cancer progression. Neoplasia.2002;4:191–4. http://dx.doi.org/10.1038/sj.neo.7900224 . [PMC free article] [PubMed]
172.
Cooper CR, Chay CH, Gendernalik JD, Lee HL, Bhatia J, Taichman RS, et al. Stromal factors involved in prostate carcinoma metastasis to bone. Cancer. 2003;97(Suppl):739–47. http://dx.doi.org/10.1002/cncr.11181 . [PubMed]
173.
Kumar CC. Integrin alpha v beta 3 as a therapeutic target for blocking tumor-induced angiogenesis. Curr Drug Targets. 2003;4:123–31. http://dx.doi.org/10.2174/1389450033346830 . [PubMed]
174.
Krupski T, Harding MA, Herce ME, Gulding KM, Stoler MH, Theodorescu D. The role of vascular endothelial growth factor in the tissue specific in vivo growth of prostate cancer cells. Growth Factors. 2001;18:287–302.http://dx.doi.org/10.3109/08977190109029117 . [PubMed]
175.
Chen J, De S, Brainard J, Byzova TV. Metastatic properties of prostate cancer cells are controlled by VEGF. Cell Commun Adhes. 2004;11:1–11. http://dx.doi.org/10.1080/15419060490471739 . [PubMed]
176.
Dai J, Kitagawa Y, Zhang J, Yao Z, Mizokami A, Cheng S, et al. Vascular endothelial growth factor contributes to the prostate cancer-induced osteoblast differentiation mediated by bone morphogenetic protein. Cancer Res.2004;64:994–9. http://dx.doi.org/10.1158/0008-5472.CAN03-1382 . [PubMed]
177.
Jacob K, Webber M, Benayahu D, Kleinman HK. Osteonectin promotes prostate cancer cell migration and invasion: a possible mechanism for metastasis to bone. Cancer Res. 1999;59:4453–7. [PubMed]
178.
Rosol TJ. Pathogenesis of bone metastases: role of tumor-related proteins. J Bone Miner Res. 2000;15:844–50.http://dx.doi.org/10.1359/jbmr.2000.15.5.844 . [PubMed]
179.
Denhardt DT, Giachelli CM, Rittling SR. Role of osteopontin in cellular signaling and toxicant injury. Annu Rev Pharmacol Toxicol. 2001;41:723–49. http://dx.doi.org/10.1146/annurev. pharmtox.41.1.723 . [PubMed]
180.
Fizazi K, Yang J, Peleg S, Sikes CR, Kreimann EL, Daliani D, et al. Prostate cancer cells-osteoblast interaction shifts expression of growth/survival-related genes in prostate cancer and reduces expression of osteoprotegerin in osteoblasts. Clin Cancer Res. 2003;9:2587–97. [PubMed]
181.
Zhang JH, Tang J, Wang J, Ma W, Zheng W, Yoneda T, et al. Over-expression of bone sialoprotein enhances bone metastasis of human breast cancer cells in a mouse model. Int J Oncol. 2003;23:1043–8. [PubMed]
182.
Khodavirdi AC, Song Z, Yang S, Zhong C, Wang S, Wu H, et al. Increased expression of osteopontin contributes to the progression of prostate cancer. Cancer Res. 2006;66:883–8. http://dx.doi.org/10.1158/0008-5472.CAN-05-2816 . [PubMed]
183.
Resnick MB, Konkin T, Routhier J, Sabo E, Pricolo VE. Claudin-1 is a strong prognostic indicator in stage II colonic cancer: a tissue microarray study. Mod Pathol. 2005;18:511–8.http://dx.doi.org/10.1038/modpathol.3800301 . [PubMed]
184.
Montgomery E, Mamelak AJ, Gibson M, Maitra A, Sheikh S, Amr SS, et al. Overexpression of claudin proteins in esophageal adenocarcinoma and its precursor lesions. Appl Immunohistochem Mol Morphol. 2006;14:24–30.http://dx.doi.org/10.1097/01.pai.0000151933.04800.1c . [PubMed]
185.
Sommers CL, Heckford SE, Skerker JM, Worland P, Torri JA, Thompson EW, et al. Loss of epithelial markers and acquisition of vimentin expression in adriamycin- and vinblastine-resistant human breast cancer cell lines.Cancer Res. 1992;52:5190–7. [PubMed]
186.
Zajchowski DA, Bartholdi MF, Gong Y, Webster L, Liu HL, Munishkin A, et al. Identification of gene expression profiles that predict the aggressive behavior of breast cancer cells. Cancer Res. 2001;61:5168–78.[PubMed]
187.
Ponnusamy MP, Lakshmanan I, Jain M, Das S, Chakraborty S, Dey P, et al. MUC4 mucin-induced epithelial to mesenchymal transition: a novel mechanism for metastasis of human ovarian cancer cells. Oncogene.2010;29:5741–54. http://dx.doi.org/10.1038/onc.2010.309 . [PMC free article] [PubMed]
188.
Wang J, Chen L, Li Y, Guan XY. Overexpression of cathepsin Z contributes to tumor metastasis by inducing epithelial-mesenchymal transition in hepatocellular carcinoma. PLoS One. 2011;6:e24967.http://dx.doi.org/10.1371/journal.pone.0024967 . [PMC free article] [PubMed]
189.
Sarri D, Rodriguez-Pinilla SM, Hardisson D, Cano A, Moreno-Bueno G, Palacios J. Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res. 2008;68:989–97.http://dx.doi.org/10.1158/0008-5472.CAN-07-2017 . [PubMed]
190.
Davies BR, Barraclough R, Davies MP, Rudland PS. Production of the metastatic phenotype by DNA transfection in a rat mammary model. Cell Biol Int. 1993;17:871–9. http://dx.doi. org/10.1006/cbir.1993.1150 . [PubMed]
191.
Andersen K, Mori H, Fata J, Bascom J, Oyjord T, Mlandsmo GM, et al. The metastasis-promoting protein S100A4 regulates mammary branching morphogenesis. Dev Biol. 2011;352:181–90.http://dx.doi.org/10.1016/j.ydbio.2010.12.033 . [PMC free article] [PubMed]
192.
Cavallaro U. N-cadherin as an invasion promoter: a novel target for antitumor therapy? Curr Opin Investig Drugs.2004;5:1274–8. [PubMed]
193.
Cme C, Magnino F, Bibeau F, De Santa Barbara P, Becker KF, Theillet C, et al. Snail and slug play distinct roles during breast carcinoma progression. Clin Cancer Res. 2006;12:5395–402. http://dx.doi.org/10.1158/1078-0432.CCR-06-0478 . [PubMed]
194.
Comijn J, Berx G, Vermassen P, Verschueren K, van Grunsven L, Bruyneel E, et al. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell. 2001;7:1267–78.http://dx.doi.org/10.1016/S1097-2765(01)00260-X . [PubMed]
195.
Blissett AR, Garbellini D, Calomeni EP, Mihai C, Elton TS, Agarwal G. Regulation of collagen fibrillogenesis by cell-surface expression of kinase dead DDR2. J Mol Biol. 2009;385:902–11.http://dx.doi.org/10.1016/j.jmb.2008.10.060 . [PMC free article] [PubMed]
196.
Pea C, Garca JM, Silva J, Garca V, Rodrguez R, Alonso I, et al. E-cadherin and vitamin D receptor regulation by SNAIL and ZEB1 in colon cancer: clinicopathological correlations. Hum Mol Genet. 2005;14:3361–70.http://dx.doi.org/10.1093/hmg/ddi366 . [PubMed]
197.
Spoelstra NS, Manning NG, Higashi Y, Darling D, Singh M, Shroyer KR, et al. The transcription factor ZEB1 is aberrantly expressed in aggressive uterine cancers. Cancer Res. 2006;66:3893–902.http://dx.doi.org/10.1158/0008-5472.CAN-05-2881 . [PubMed]
198.
Conn G, Soderman DD, Schaeffer MT, Wile M, Hatcher VB, Thomas KA. Purification of a glycoprotein vascular endothelial cell mitogen from a rat glioma-derived cell line. Proc Natl Acad Sci U S A. 1990;87:1323–7.http://dx.doi.org/10.1073/pnas.87.4.1323 . [PMC free article] [PubMed]
199.
Gerber HP, Dixit V, Ferrara N. Vascular endothelial growth factor induces expression of the antiapoptotic proteins Bcl-2 and A1 in vascular endothelial cells. J Biol Chem. 1998;273:13313–6.http://dx.doi.org/10.1074/jbc.273.21.13313 . [PubMed]
200.
Dimmeler S, Dernbach E, Zeiher AM. Phosphorylation of the endothelial nitric oxide synthase at ser-1177 is required for VEGF-induced endothelial cell migration. FEBS Lett. 2000;477:258–62.http://dx.doi.org/10.1016/S0014-5793(00)01657-4 . [PubMed]
201.
Khurana R, Simons M. Insights from angiogenesis trials using fibroblast growth factor for advanced arteriosclerotic disease. Trends Cardiovasc Med. 2003;13:116–22. http://dx.doi. org/10.1016/S1050-1738(02)00259-1 . [PubMed]
202.
DAmore PA, Smith SR. Growth factor effects on cells of the vascular wall: a survey. Growth Factors.1993;8:61–75. http://dx.doi.org/10.3109/08977199309029135 . [PubMed]
203.
Otrock ZK, Mahfouz RA, Makarem JA, Shamseddine AI. Understanding the biology of angiogenesis: review of the most important molecular mechanisms. Blood Cells Mol Dis. 2007;39:212–20.http://dx.doi.org/10.1016/j.bcmd.2007.04.001 . [PubMed]
204.
Falcone DJ, McCaffrey TA, Haimovitz-Friedman A, Garcia M. Transforming growth factor-beta 1 stimulates macrophage urokinase expression and release of matrix-bound basic fibroblast growth factor. J Cell Physiol.1993;155:595–605. http://dx.doi.org/10.1002/jcp.1041550317 . [PubMed]
205.
Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;9:653–60. http://dx.doi.org/10.1038/nm0603-653 . [PubMed]
206.
Thurston G. Role of Angiopoietins and Tie receptor tyrosine kinases in angiogenesis and lymphangiogenesis.Cell Tissue Res. 2003;314:61–8. http://dx.doi.org/10.1007/ s00441-003-0749-6 . [PubMed]
207.
Breiteneder-Geleff S, Soleiman A, Kowalski H, Horvat R, Amann G, Kriehuber E, et al. Angiosarcomas express mixed endothelial phenotypes of blood and lymphatic capillaries: podoplanin as a specific marker for lymphatic endothelium. Am J Pathol. 1999;154:385–94. http://dx.doi. org/10.1016/S0002-9440(10)65285-6 . [PMC free article] [PubMed]
208.
Wigle JT, Harvey N, Detmar M, Lagutina I, Grosveld G, Gunn MD, et al. An essential role for Prox1 in the induction of the lymphatic endothelial cell phenotype. EMBO J. 2002;21:1505–13.http://dx.doi.org/10.1093/emboj/21.7.1505 . [PMC free article] [PubMed]
209.
He Y, Karpanen T, Alitalo K. Role of lymphangiogenic factors in tumor metastasis. Biochim Biophys Acta.2004;1654:3–12. [PubMed]
Copyright © 2013 Landes Bioscience.
In this Page
- Introduction
- Cancer Invasion and Metastasis: The Role of Cell Adhesion Molecules
- Epithelial-Mesenchymal Transition
- Angiogenesis and Lymphangiogenesis in Cancer Metastasis
- Organ Specific Metastasis
- Conclusion
- Acknowledgment
- References
Yirong Li 30.31
NYU Langone Medical Center
How do cancer cells survive in blood circulation?
I am wondering how cancer cells escape from immunosystem and survive during blood circulation. Is there some ways to isolate cancer cells during their blood circulation?
Constantine Kaniklidis · 88.51 · 7.14 · No Surrender Breast Cancer Foundation (NSBCF)
This excellent question is essentially about two related subprocesses that constitute the “early game” of the metastatic process, namely (1) intravasation which is the endothelial transmigration of tumor cells into blood vessels in the vasculature, and (2) hematogenous survival (tumor cell survival in the circulatory system), which together are called hematogenous dissemination. I will give, below, a reasonably brief sketch of these subprocesses here (distilled from a lengthier 40+ page review of the metastatic process and cascade I recently completed [Kaniklidis, C. The Early Game of Metastasis: Tumor Cell Intravasation and Hematogenous Survival. (pending)].
The Metastatic Process: Brief summary
The multi-step process of metastasis is a complex and coordinated choreography encompassing:
(1) local infiltration of tumor cells into the surrounding/adjacent tissue (tumor cell penetration through the ECM / the basement membrane),
(2) intravasation (endothelial transmigration of tumor cells into vessels),
(3) hematogenous survival and translocation, that is, the tumor cell survival in the circulatory system and its translocation through the bloodstream to microvessels of distant tissues
(4) extravasation (exit from bloodstream, and
(5) adaption to the foreign microenvironment of distant site tissue and subsequent colonization (cell proliferation and the formation of a macroscopic secondary tumor) in competent organs.
Subprocesses (2) and (3) together, that is, the combination of intravasation + hematogenous survival constitute collectively what is known as hematogenous dissemination.
Intravasation
Tumor cells intravasation, which is the endothelial transmigration of cancer cells into vessels, involves two different types of motility: tumor cells can intravasate the blood, or the lymph vasculature, although dissemination via the hematogenous route seems to represents the major mechanism for dispersal of metastatic carcinoma cells [1], and for both routes, the process is mechanistically via interaction of tumor cells with the vascular endothelium. Note however that although the primary main route of the metastatic spread has generally until recently been the blood / circulation system, mounting evidence suggests that the lymphatic system is also a key player in cancer cell dissemination. But as to the central matter of endothelial transmigration of tumor cells, there remains indeterminacy and continued debate on the question of active versus passive dissemination, that is, as to whether (1) tumor cells actively migrate through blood and lymph vasculature as a response to phenomenon like growth factor gradients, or (2) do so passively by “crawling” into the vasculature even in the absence an active cell migration machinery, leading to a neatly phrased article title from Lance Munn and colleagues, namely “Do cancer cells crawl into vessels, or are they pushed?” [2].
There are a number of molecular phenomena that facilitate endothelial transmigration, that is, the crossing by tumor cells of the pericyte and endothelial cell barriers that constitute the microvessel walls:
(1) Twist:
Jing Yang et al. have shown in a murine breast cancer model that the transcription factor, Twist, appears to allow the step of intravasation and hence functions as an EMT-inducing transcription factor and thus a key regulator of metastasis [3], both augmenting EMT (epithelial-to-mesenchymal) transitions and promoting the rate of hematogenous intravasation.
(2) Chemoattractive Gradients and the Role of EGF / CSF-1:
In addition, a second mechanism is at play, as documented in the breast cancer context, that involves what is called chemoattractive gradients, confirmed by direct visualization using multiphoton microscopy by researchers at the Albert Einstein College of Medicine [4,5]. These direct observations demonstrated how perivascular macrophages in mammary tumors are critically involved in intravasation and hematogenous survival, and that these perivascular macrophages synergistically induce tumor cell intravasation even in the absence of local angiogenesis. These perivascular macrophages are recruited by the tumor cells to the injured site (Condeelis), inducing intravasation into the blood system via chemoattractive gradients generated by these same perivascular macrophages, with crosstalk and collaboration between the tumor microenvironment and the tumor cells at the intravasation site is enabled thorough a positive-feedback loop constituted by the reciprocal secretion of epidermal growth factor (EGF) created by the macrophages and colony stimulating factor-1 (CSF-1) by the tumor cells, jointly augmenting chemotaxis and the intravasation process, with EGF promoting tumor cell migration into the hematogenous vasculature through interaction with the EGF receptor, and CSF-1 expressed on the tumor cells functioning as a potent chemoattractant for CSF-1 receptor positive macrophages [6,7].
(3) Transforming Growth Factor-beta (TGF-beta):
In mammary carcinoma, the cytokine TGF-beta (transforming growth factor-beta) enhances intravasation via increased penetration of microvessel walls, suggesting that transient TGF-beta signalling is critical for blood-born metastasis [8].
(4) VEGF and Neoangiogenesis:
Via VEGF and neoangiogenesis, tumor cells stimulate formation of new blood vessels within the local microenvironment, with the neovasculature created by tumor cells being prone to leakiness, and ultimately facilitate intravasation [9].
Hematogenous Survival: Survival in Vasculature
But after successful invasion of the hematogenous vasculature, tumor cells must survive a perilous microenvironment of challenging hurdles that include hemodynamic shear forces turbulence, surveillance from and attack by immune cells especially natural killer (NK) cells, and lack of substratum, and entrapment-by-size in early-encountered capillary beds, which occurs usually even in the first capillary bed encountered by the tumor cells consequent to the fact that the diameter of most tumor cells is too large for passage through small capillaries [10].
A main defense for hematogenous survival used by tumor cells is using platelets as a shield, by binding coagulation factors on the platelets, forming an embolus aggregate that protects the tumor cells from immune-cell-mediated lysis / destruction, as well as decreases the level and impact of the circulation system’s hemodynamic shear forces and turbulence, to enhance survival [11-14].
In addition, tumor cells are physically shielded from the stress of blood flow, shear forces and turbulence, as well as from lysis by NK cells by two related processes:
(1) activation of the coagulation cascade and
(2) formation of platelet-rich thrombi around tumor cells in the vasculature [15-18]. The process,
in essence, is that tumor cell tissue factor triggers thrombin formation that initiates both coagulation and platelet activation, which in turn enhance metastatic spread. And Fibrin can be bound by integrins on tumor cells and on activated platelets, triggering the formation of tumor-cell–fibrin–platelet aggregates. These large aggregates and emboli then have the strength and resiliency to survive hematogenous shear forces and turbulence [17,19-22]
And it appears that the normal anti-tumor reactivity of NK immune cells can be subverted by a platelet-derived coating (called MHC Class I) which disguises the tumor cell with a pseudo-normal phenotype, exempting it from immune response and attack. [23,24].
References
1. Gupta GP, Massagué J. Cancer metastasis: building a framework. Cell 2006 Nov 17; 127(4):679-95.
2. Bockhorn M, Jain RK, Munn LL. Active versus passive mechanisms in metastasis: do cancer cells crawl into vessels, or are they pushed? Lancet Oncol 2007; 8(5):444-8.
3. Yang J, Mani SA, Donaher JL, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004 Jun 25; 117(7):927-39.
4. Condeelis J, Segall JE. “Intravital imaging of cell movement in tumours,” Nat Rev Cancer. 2003 Dec;3(12):921-30;
5. Wyckoff JB, Wang Y, Lin EY, et al. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res 2007 Mar 15; 67(6):2649-56.
6. Wyckoff J, Wang W, Lin EY, et al. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res 2004 Oct 1; 64(19):7022-9.
7. Goswami S, Sahai E, Wyckoff JB, et al. Macrophages promote the invasion of breast carcinoma cells via a colony-stimulating factor-1/epidermal growth factor paracrine loop. Cancer Res. 2005 Jun 15;65(12):5278-83.
8. Giampieri S, Manning C, Hooper S, Jones L, Hill CS, Sahai E. Localized and reversible TGFbeta signalling switches breast cancer cells from cohesive to single cell motility. Nat Cell Biol. 2009 Nov;11(11):1287-96.
9. Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov 2011; 10(6):417-27.
10. Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med 2006; 12(8):895-904.
11. Palumbo JS. Mechanisms linking tumor cell-associated procoagulant function to tumor dissemination. Semin Thromb Hemost 2008; 34(2):154-60.
12. Im JH, Fu W, Wang H, et al. Coagulation facilitates tumor cell spreading in the pulmonary vasculature during early metastatic colony formation. Cancer Res 2004; 64(23): 8613–8619.
13. Palumbo JS, Talmage KE, Massari JV, et al. Tumor cell-associated tissue factor and circulating hemostatic factors cooperate to increase metastatic potential through natural killer cell-dependent and -independent mechanisms. Blood 2007 110(1):133–141.
14. Khamis ZI, Sahab ZJ, Sang QX. Active roles of tumor stroma in breast cancer metastasis. Int J Breast Cancer 2012; 2012:574025.
15. Palumbo JS, Talmage KE, Massari JV, et al. Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell-mediated elimination of tumor cells. Blood 2005; 105:178–85.
16. [Erpenbeck L, Schon MP. Deadly allies: the fatal interplay between platelets and metastasizing cancer cells. Blood 2010; 115:3427–36.
17. Gay LJ, Felding-Habermann B. Contribution of platelets to tumour metastasis. Nat Rev Cancer 2011; 11:123–34.
18. Degen JL , Palumbo JS . Hemostatic factors, innate immunity and malignancy. Thromb Res 2012; 129 Suppl 1:S1–5.
19. Liu Y, Jiang P, Capkova K, et al. Tissue factor activated coagulation cascade in the tumor microenvironment is critical for tumor progression and an effective target for therapy. Cancer Res 2011; 71:6492–502.]
20. Labelle M, Begum S, Hynes RO. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 2011; 20:576–90.
21. Camerer E, Qazi AA, Duong DN, Cornelissen I, Advincula R, Coughlin SR. Platelets, protease-activated receptors, and fibrinogen in hematogenous metastasis. Blood 2004;104:397–401.
22. [Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell 2011 Oct 14; 147(2):275-92.
23. Placke T, Oergel M, Schaller M, J, et al. Platelet-derived MHC Class I confers a pseudo-normal phenotype to cancer cells that subverts the anti-tumor reactivity of natural killer immune cells. Cancer Res 2012; 72:440–8.
24. Nieswandt B, Hafner M, Echtenacher B, Mannel DN. Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Res 1999 59:1295–1300.
Constantine Kaniklidis, Director of Medical Research,
No Surrender Breast Cancer Foundation (NSBCF)
European Association for Cancer Research (EACR)
Christopher Daniel Duntsch · 79.15 · 110.86 · Hybrid Bioscience, Inc., Synthetic Investments, Inc., The University of Tennessee Health Science Center
This is simple to model. Epigenetic and genetic changes result in cells having a gain of function, developing the ability to migrate, to express extracellular MMPs and related proteins, to migrate from a primary focus and get into the lymphatic system and hemotopoietic system, to up regulate cell surface proteins that allow binding a distant sites, to survive immunosurveillance (rarely) by changing HLA/B expression and other immunoprotiens and / or cell surface antigens, or to express anti-immune proteins such as FAS Ligan, to express proteins to digest and create pathways into distant organs. Much research as of late suggest the most difficult thing for a metastatic cancer cell to do is to survive in a foreign tissue, because it is a hostile environment. Other research suggests cancer stem cells adapt by creating pseudo environments around said metastatic cells that create an environment similar to the primary foci. Regardless, most research demonstrates that for every cancer stell attempting to metastasize, a small fraction succeed.
Salwa Hassan Teama · 11.71 · 2.35 · Ain Shams University
Cancer is a prominent cause of death worldwide. When cancer disseminates from the primary lesion to other vital organs, it becomes a devastating disease. In fact, it is the metastatic process that leads to 90% of cancer-related deaths Metastatic tumors are spread over the entire human body and are more difficult to remove or treat than the primary tumor. In a patient with metastatic disease, circulating tumor cells (CTCs) can be found in venous blood. These circulating tumor cells are part of the metastatic cascade.
METASTATIC PROCESS
A complex multi-step event, this biological process requires tumor cells to break free of the primary solid tumor, penetrate into the blood or lymphatic circulation, and ultimately extravasate out of the circulation and into an organ or tissue distant from the primary lesion.
Cancer occurs after a cell is progressively genetically damaged and turns into a cell bearing a malignant phenotype. These cells are able to undergo uncontrolled abnormal mitosis, which leads to an increase of these cancerous cells at that location. In absence of regular control mechanisms a heterogeneous population of cells is created and these cancerous cells together form the primary tumor. A tumor is considered benign if it lacks the ability to invade other tissue. When cells acquire the ability to penetrate and infiltrate surrounding normal tissues, the cancer is considered malignant and has the potential to metastasize.
Before tumor cells can start to metastasize, they need to succeed in stimulating angiogenesis. In this way tumor cells gain direct access to the blood circulation. This leads to improved access to the nutrients and oxygen carried by the blood, but also an opportunity for the tumor cells to enter the blood stream. An alternative route for tumor cells to end up in the blood circulation is through the lymphatic system. Tumor cells circulating in the blood can reach in principle most sites of the body, but different kinds of cancer create metastasis at different sites. For example breast cancer generally creates metastases in liver, lung and bone while prostate cancer most often metastasizes in bone. This preference is driven by two processes. The first is mechanical of nature, a large amount of CTC arrests in the first capillary bed they encounter. The second is more biological, the CTCs will form a metastasis in tissue only if they are able to extravasate out of the blood stream and the local environment is suitable for them to grow. This preference has been noted for the first time by
Stephen Paget and is known as the seed and soil hypothesis. Tumor cells thus have a preference for a certain site, and this opens an interesting research field to identify the cell surface molecules on the tumor cells and the endothelial cells aligning the capillaries at the specific sites.
More:http://www.ifcc.org/media/209935/eJIFCC%20n%203_2012_07%20Van%20Dalum.pdf
http://www.aacc.org/publications/cln/2008/november/Pages/Series1108.aspx
What are CTC?
Epithelial tumors or carcinomas represent about 80% of all cancers. CTC originate from the epithelium and are not present in the circulating peripheral blood of individuals free of neoplastic disease. Derived from clones of the primary tumor, these cells can be detected before the primary tumor is identified and often persist even after the primary tumor has been removed.
More:http://www.aacc.org/publications/cln/2008/november/Pages/Series1108.aspx
Cancer cell heterogeneity
Heterogeneity among cancer cells (Pleiomorphism) was a common and predominant features of most common solid tumors.
Heterogeneity and Clonality
Cancer cells are genetically unstable and as the population expands the probability of mutation increases. This is in turn lead to possibilities that epigenetic mechanisms could also exert selective differential selective pressure on heterogeneous cancer cell population.
Wolman (1982, 1986) considered that genetic and chromosomal instability were the potential source of genetic heterogeneity among all the tumors and that variation in local environment selective pressure and differential survival may contribute to cellular heterogeneity within expanding tumors also heterogeneity itself might permit selection and increase the number of aberrant cells responsible for tumor progression and metastasis
More:http://link.springer.com/article/10.1023%2FA%3A1010614909387?LI=true#page-2
Developing tumors must acquire nutrients to ensure their rapid growth. Second, they must escape the attack from the host immune system.
The vast majority of tumor cells that enter the circulation are rapidly eliminated by factors such as blood turbulence,natural killer cells, and macrophages.
Nitric oxide secretion by activated macrophages and endothelial cells is a major cytotoxic mediator responsible for the destruction of tumor cells passing through capillary beds. In addition, activation of apoptosis also contributes to eliminate metastatic cells.
In contrast, fibrin deposits, platelet aggregation, and adhesion around the tumor emboli may protect circulating cells from mechanical trauma, facilitate their arrest in capillary beds, and protect tumor emboli from destruction by host immunity .
More: http://www.molmed.org/content/1999/5_99/5_99_Fournier.PDF
Recent studies suggest that these phenomena could be related and that tumor cell metabolism may propel tumor immune escape. Tumor cell metabolism tends to avoid mitochondrial activity and oxidative phosphorylation (OXPHOS), and largely relies on glycolysis to produce energy. This specific metabolism helps tumor cells to avoid the immune attack from the host by blocking or avoiding the immune attack. By changing their metabolism, tumor cells produce or sequester a variety of amino acids, lipids and chemical compounds that directly alter immune function therefore promoting immune evasion. A second group of metabolism-related modification targets the major histocompatibility complex-I (MHC-I) and related molecules. Tumor MHC-I presents tumor-associated antigens (TAAs) to cytotoxic Tcells (CTLs) and hence, sensitizes cancer cells to the cytolytic actions of the anti-tumor adaptive immune response. Blocking tumor mitochondrial activity decreases expression of MHC-I molecules at the tumor cell surface. And peroxynitrite (PNT), produced by tumor-infiltrating myeloid cells, chemically modifies MHC-I avoiding TAA expression in the plasma membrane.
These evidences on the role of tumor cell metabolism on tumor immune escape open the possibility of combining drugs designed to control tumor cell metabolism with new procedures of anti-tumor immunotherapy.
From tumor cell metabolism to tumor immune escape
More: http://hal.archives-ouvertes.fr/docs/00/72/67/17/PDF/villalba_et_al-IJBCB.pdf
Andrew Sunters · 52.96 · 160.24 · Royal Veterinary College
An interesting question, and is at the heart of one of the “hallmarks of cancer” and the “enabling characteristics” of cancer in Hanahan and Weinbergs theory of the hallmarks of cancer, which is a good starting place. One of the hallmarks is the ability to locally invade tissue and form distant metastases. Whilst there is debate about how well these cells survive, it is obvious that some do survive. Two strategies are for the cells to coat them selves with something to impare immune recognition, and this has been shown to be platelets or other cell types, as Naresh and others point out. Another is related to mutations-in tumours such as colon tumours which commonly lacking mismatch repair genes frameshift mutations generate nonsensical proteins which when expressed on the MHC can attract the interest of the immune system. Loss of the MHC or the protein machinery which process antigen peptides for MHC presentation means that these mutant peptides are not seen by the immune system, and the cancer cells can avoid detection to some degree. I would urge you to read the paper and updates and discussion:
http://www.sciencedirect.com/science/article/pii/S0092867400816839
Primitive Human Leukemia Cells Grown in Lab
Rogue stem cells are at the root of all leukemias
http://www.technologynetworks.com/HTS/news.aspx?ID=185776
Chronic myeloid leukemia (CML), a family of cancers that affect blood and bone marrow, is treatable in many instances. But the therapies used to keep the cancer in check have no effect on the primitive stem cells, also known as leukemia stem cells, that cause the disease in the first place – leaving patients susceptible to a relapse if they go off their meds.
Now, using cells from a patient with CML, researchers at the University of Wisconsin-Madison have found the recipe to generate cells with properties of primitive human leukemia cells in the lab. The work establishes a potent model for studying CML stem cells and identifying new drugs that could potentially provide better treatment options for leukemia.
“Treatment doesn’t eliminate the stem cells that cause chronic myeloid leukemia,” explains Igor Slukvin, a UW-Madison professor of pathology and laboratory medicine and an expert on stem cells and human blood. “We know we can treat CML, but we can’t cure it. The stem cells persist.”
Slukvin and his collaborators report their work online. By genetically reprogramming the patient’s bone marrow cells, the Wisconsin group was able to turn back the developmental clock and make all-purpose induced pluripotent stem cells (iPSCs), which capture the underlying genetic alterations driving the leukemia. Directing the induced primitive CML cells to become early blood cells, researchers were able to generate cells that share many properties of leukemia stem cells, including increased long-term survival and proliferation as well as innate resistance to drugs.
The drugs now used to treat CML are known as tyrosine kinase inhibitors; they work by stopping the progression and proliferation of the cancer cells emanating from the bone marrow. Though these drugs are very effective, patients risk relapse if they stop taking the medication. Moreover, in some patients the leukemia cells develop resistance to the drug, making it less effective.
The ability to make leukemia stem cells in the lab using reprogrammed adult bone marrow cells from patients will give scientists a new way to explore the development and progression of the disease in a laboratory dish. Currently, only mouse models of leukemia stem cells are available in the lab.
The advent of a human cell model opens the door to exploring the differences in how the disease manifests itself within different people. “The induced cells capture the genetic abnormality of the individual patient,” says Slukvin, creating the potential for more personalized treatment of the disease.
In addition, the new cell model creates a path to capture the genetic variations of the disease as it manifests itself in individual patients. Because iPSCs arise from a single cell, the selection of individual cloned iPSC cells makes it possible to capture the diversity of genetic alterations within individual stem cells and study their effects.
Because the disease transitions through chronic, accelerated and acute phases in patients, the new model may also let scientists study the progression of leukemia in the lab.
“If we make iPSCs from stored patient samples collected at different stages of diseases,” notes Slukvin, “we can produce from these iPSCs primitive leukemia cells that capture different stages of leukemia progression.”
An important potential application is that the lab-created human stem cells, like other types of synthesized stem cells or their derivatives, can also be used to learn more about leukemia stem cell survival factors and develop high-throughput drug screens where chemical compounds can be tested for efficacy and safety as potential drug candidates. Slukvin and his team, in fact, used the new system to discover a novel primitive leukemia cell survival factor, a protein known as olfactomedin 4.
Using drugs or antibodies to target the protein, which helps the primitive CML cells survive, may open an avenue to clear the leukemia and potentially cure the disease, although much work remains to be done to achieve such an outcome.
Neoplastic blood cells become pluripotent
Igor I. Slukvin
In this issue of Blood, Ye and colleagues show that CD34+ cells obtained from patients with JAK2-V617F MPDs could be reprogrammed to iPSCs and be differentiated back into hematopoietic progenitors.1
Myeloproliferative disorders (MPDs) represent a group of clonal hematopoietic progenitor/stem cell disorders associated with excess production of cells of myeloid lineages, resulting in an increase in one or more mature peripheral blood elements. This group of myeloproliferative neoplasms includes polycythemia vera (PV), essential thrombocythemia (ET), primary myelofibrosis (PMF), chronic myeloid leukemia (CML), and other rarer disorders. Whereas CML was the first blood cancer known to be linked to a chromosomal translocation, the JAK2-V617F mutation associated with PV was discovered only several years ago.2 However, the mechanisms of transformation by JAK2-V617F mutation are not well understood, particularly why the same mutation causes different phenotypes including PV, ET, or PMF. It has been hypothesized that disease manifestation depends on the cell affected by the original mutation, the genetic background of the patient, or the level of JAK2-V617F activity. The work by Ye et al provides a novel approach to ask and answer important questions about MPD pathogenesis, by modeling development of myeloproliferative neoplasia in vitro using patient-specific induced pluripotent stem cells (iPSCs).1
In 2006, the Yamanaka group revealed that mouse skin fibroblasts could be reprogrammed to pluripotency via ectopic expression of 4 transcription factors.3 A year later, iPSCs were obtained from human fibroblasts.4,5 These discoveries opened opportunities to generate disease-specific iPSCs carrying a particular genetic trait at the cellular level. As proof of this concept, iPSCs have been generated from fibroblasts obtained from patients with several genetic diseases including the inherited bone marrow failure syndrome Fanconi anemia.6 However, a fibroblast-based approach would not work for acquired blood diseases such as MPDs or leukemia, because cytogenetic abnormalities defining such diseases are limited to bone marrow cells in most of the cases. Several months ago, Loh et al demonstrated that iPSCs could be generated by reprogramming mobilized peripheral blood CD34+cells.7 The work published in this issue of Blood by Ye et al is the first description of successful reprogramming of CD34+ cells from patients with acquired blood diseases.1 Using retroviral vectors encoding Oct4, Sox2, Klf4, and c-Myc genes, Ye et al generated iPSCs from CD34+ cells obtained from healthy controls and MPD patients carrying the JAK2-V617F mutation. While MPD-derived iPSCs retained the JAK2-V617F mutation, they had a normal karyotype, embryonic stem cell–like phenotype, and pluripotent differentiation potential. When control and diseased iPSCs were differentiated back into CD34+CD45+ hematopoietic progenitors, the progenitors derived from MPD-iPSCs recapitulated the features of somatic CD34+ cells from which the iPSCs were originally derived. Similar to somatic MPD CD34+ cells, iPSC-derived CD34+CD45+ cells demonstrated enhanced erythropoiesis and up-regulation of genes known to be increased in PV.
This study clearly demonstrates how iPSC technology could be used to model acquired blood diseases. This technology would be of particular value for the study of blood disorders such as myelodysplastic syndromes, paroxysmal nocturnal hemoglobinuria, and others for which animal models are not available or difficult to create. In addition, iPSCs carrying leukemia-specific cytogenetic translocation could be used to analyze how cancer stem cells develop. Importantly, the iPSC-based approach would be helpful in addressing the role of genetic background in manifestation of neoplastic blood disorders. Because iPSCs are capable of indefinite self-renewal, diseased blood cells can be generated continuously in the laboratory, eliminating the need for a constant supply of hematopoietic progenitors from the patients. In particular, a continuous supply of genetically diverse diseased blood cells for drug screening and discovery could be created. Because multiple types of cells can be generated from iPSCs, interaction of diseased blood cells with endothelial or stromal cells could be modeled in vitro. However, several important issues related to iPSC models of blood diseases remain to be addressed. It is known that the hematopoietic differentiation potential of iPSC lines generated from the same starting material varies significantly.8 If several clones were generated from iPSCs, which clones should be selected to make an appropriate conclusion regarding differences in differentiation potential? What would be an appropriate control for diseased versus nondiseased iPSCs? For studies of acquired blood diseases, iPSC lines can be generated from hematopoietic cells and fibroblasts or bone marrow mesenchymal stem cells (see figure). In this way, iPSCs with the same genetic background, but different in terms of presence or absence of acquired mutations, will be available for comparative analysis. The majority of disease-specific iPSCs have been made using retroviral vectors. Although the impact of exogenous expression is unclear, the possibility remains that retroviral integration and background expression of pluripotency genes may affect the behavior of iPSC-derived hematopoietic progenitors. Recently developed new reprogramming methods allowing for the generation of transgene-free iPSCs will be helpful to overcome this limitation.
http://d3md5dngttnvbj.cloudfront.net/content/bloodjournal/114/27/5409/F1.medium.gif
The use of iPSCs in modeling for acquired blood disease. Bone marrow samples from patients with acquired blood diseases can be used to obtain mutation-free mesenchymal stem cells (MSCs) and CD34+ cells or other types of hematopoietic progenitors (HPs) carrying disease-associated mutation. Alternatively, diseased peripheral blood CD34+ cells and fibroblasts or other types of cells lacking mutation from the same patient can be used. By reprogramming cells with or without genetic abnormality from the same patient, iPSCs with the same genetic background but different in expression of mutation can be generated. Using an in vitro differentiation system, hematopoietic precursors at different stages of maturation and terminally differentiated cells can be obtained for studies of disease pathogenesis. Transplantation of de novo generated cells with neoplasia-specific mutation into immunocompromised mice can be used to address emergence of blood cancer stem cells. Drug screening and discovery is another obvious and immediate benefit of iPSC technology for development of new therapies for blood diseases.
REFERENCES
- ↵
- Ye Z,
- Zhan H,
- Mali P,
- et al
. Human-induced pluripotent stem cells from blood cells of healthy donors and patients with acquired blood disorders. Blood2009;114(27):5473-5480.
- ↵
- James C,
- Ugo V,
- Le Couedic JP,
- et al
. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005;434(7037):1144-1148.
- ↵
- Takahashi K,
- Yamanaka S
. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006;126(4):663-676.
- ↵
- Yu J,
- Vodyanik MA,
- Smuga-Otto K,
- et al
. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007;318(5858):1917-1920.
- ↵
- Takahashi K,
- Tanabe K,
- Ohnuki M,
- et al
. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007;131(5):861-872.
- ↵
- Raya A,
- Rodriguez-Piza I,
- Guenechea G,
- et al
. Disease-corrected haematopoietic progenitors from Fanconi anaemia induced pluripotent stem cells. Nature2009;460(7251):53-59.
- ↵
- Loh YH,
- Agarwal S,
- Park IH,
- et al
. Generation of induced pluripotent stem cells from human blood. Blood 2009;113(22):5476-5479.
- ↵
- Choi K,
- Yu J,
- Smuga-Otto K,
- et al
. Hematopoietic and endothelial differentiation of human induced pluripotent stem cells. Stem Cells 2009;27(3):559-567.
Gene Test Finds Which Breast Cancer Patients Can Skip Chemo
9/28/2015 Marilynn Marchione, AP Chief Medical Writer
In this Sept. 5, 2013 file photo, chemotherapy is administered to a cancer patient via intravenous drip in Durham, N.C. In a study sponsored by the National Cancer Institute and results published online Monday, Sept. 28, 2015, by the New England Journal of Medicine, a gene-activity test that was used to gauge early-stage breast cancer patient’s risk accurately identified a group of women whose cancers are so likely to respond to hormone-blocking drugs that adding chemo would do little if any good while exposing them to side effects and other health risks. (Gerry Broome, Associated Press) Many women with early-stage breast cancer can skip chemotherapy without hurting their odds of beating the disease – good news from a major study that shows the value of a gene-activity test to gauge each patient’s risk.
The test accurately identified a group of women whose cancers are so likely to respond to hormone-blocking drugs that adding chemo would do little if any good while exposing them to side effects and other health risks. In the study, women who skipped chemo based on the test had less than a 1 percent chance of cancer recurring far away, such as the liver or lungs, within the next five years.
“You can’t do better than that,” said the study leader, Dr. Joseph Sparano of Montefiore Medical Center in New York.
An independent expert, Dr. Clifford Hudis of New York’s Memorial Sloan Kettering Cancer Center, agreed.
“There is really no chance that chemotherapy could make that number better,” he said. Using the gene test “lets us focus our chemotherapy more on the higher risk patients who do benefit” and spare others the ordeal.
The study was sponsored by the National Cancer Institute. Results were published online Monday by the New England Journal of Medicine and discussed at the European Cancer Congress in Vienna.
The study involved the most common type of breast cancer – early stage, without spread to lymph nodes; hormone-positive, meaning the tumor’s growth is fueled by estrogen or progesterone; and not the type that the drug Herceptin targets. Each year, more than 100,000 women in the United States alone are diagnosed with this.
The usual treatment is surgery followed by years of a hormone-blocking drug. But many women also are urged to have chemo, to help kill any stray cancer cells that may have spread beyond the breast and could seed a new cancer later. Doctors know that most of these women don’t need chemo but there are no great ways to tell who can safely skip it.
A California company, Genomic Health Inc., has sold a test called Oncotype DX since 2004 to help gauge this risk. The test measures the activity of genes that control cell growth, and others that indicate a likely response to hormone therapy treatment.
Past studies have looked at how women classified as low, intermediate or high risk by the test have fared. The new study is the first to assign women treatments based on their scores and track recurrence rates.
Of the 10,253 women in the study, 16 percent were classified as low risk, 67 percent as intermediate and 17 percent as high risk for recurrence by the test. The high-risk group was given chemotherapy and hormone-blocking drugs. Women in the middle group were randomly assigned to get hormone therapy alone or to add chemo. Results on these groups are not yet ready – the study is continuing.
But independent monitors recommended the results on the low-risk group be released, because it was clear that adding chemo would not improve their fate.
After five years, about 99 percent had not relapsed, and 98 percent were alive. About 94 percent were free of any invasive cancer, including new cancers at other sites or in the opposite breast.
“These patients who had low risk scores by Oncotype did extraordinarily well at five years,” said Dr. Hope Rugo, a breast cancer specialist at the University of California, San Francisco, with no role in the study. “There is no chance that for these patients, that chemotherapy would have any benefit.”
Dr. Karen Beckerman, a New York City obstetrician diagnosed with breast cancer in 2011, said she was advised to have chemo but feared complications. A doctor suggested the gene test and she scored very low for recurrence risk.
“I was convinced that there was no indication for chemotherapy. I was thrilled not to have to have it,” and has been fine since then, she said.
Mary Lou Smith, a breast cancer survivor and advocate who helped design the trial for ECOG, the Eastern Cooperative Oncology Group, which ran it, said she thought women “would be thrilled” to skip chemo.
“Patients love the idea of a test” to help reduce uncertainty about treatment, she said. “I’ve had chemotherapy. It’s not pretty.”
The test costs $4,175, which Medicare and many insurers cover. Others besides Oncotype DX also are on the market, and Hudis said he hopes the new study will encourage more, to compete on price and accuracy.
“The future is bright” for gene tests to more precisely guide treatment, he said.
Source: Associated Press
Sequencing Metastatic Cancers Could Lead to Improved Therapies
- Unravelling the genetic sequences of cancer that has spread to the brain could offer unexpected targets for effective treatment, according to a study (“Genomic characterization of brain metastases and paired primary tumors reveals branched evolution and potential therapeutic targets”) published in Cancer Discovery.
Scientists say they found that the original, or primary, cancer in a patient’s body may have important differences at a genetic level from cancer that has spread to the patient’s brain. This insight could suggest new lines of treatment.
Priscilla Brastianos, M.D., a neurooncologist and director of the Brain Metastasis program at Massachusetts General Hospital, points out that “brain metastases are a devastating complication of cancer. Approximately eight to ten percent of cancer patients will develop brain metastases, and treatment options are limited. Even where treatment is successfully controlling cancer elsewhere in the body, brain metastases often grow rapidly.”
She and her colleagues studied tissue samples from 104 adults with cancer. In collaboration with researchers at the Broad Institute, they analyzed the genetics of biopsies taken from the primary tumor, brain metastases, and normal tissues in each adult. For 20 patients, they also had access to metastases elsewhere in the body.
The team discovered that, in every patient, the brain metastasis and primary tumor shared some of their genetics, but there were also key differences. In 56% of patients, genetic alterations that potentially could be targeted with drugs were found in the brain metastasis but not in the primary tumor.
“We found genetic alterations in brain metastases that could affect treatment decisions in more than half of the patients in our study,” notes Dr. Brastianos. “We could not detect these genetic alterations in the biopsy of the primary tumor. This means that when we rely on analysis of a primary tumor we may miss mutations in the brain metastases that we could potentially target and treat effectively with drugs.”
This study also found that if a patient had more than one brain metastasis, each was genetically similar. The researchers used their findings to map the evolution of a cancer through a patient’s body, and draw up a phylogenetic tree for each patient to demonstrate how the cancer had spread and where each metastasis had come from.
They concluded that brain metastases and the primary tumor share a common genetic ancestor. Once a cancer cell, or clone, has moved from the primary site to the brain, it continues to develop and amass genetic mutations. The genetic similarity of the brain metastases in individual patients suggests that each brain metastasis has developed from a single clone entering the brain.
The genetic changes in brain metastases are independent of any occurring at the same time in the primary tumor, and in metastases elsewhere in the body, the researchers said. Characterization of the genetics of a patient’s primary cancer can be used to optimize treatment decisions, so that drugs that target specific mutations in the cancer can be chosen. However, brain metastases are not routinely biopsied and analyzed.
“When brain metastasis tissue is available as part of clinical care, we are suggesting sequencing and analysis of that sample,” continues Dr. Brastianos. “It may offer more therapeutic opportunities for the patient. Genetic characterization of even a single brain metastasis may be superior to that of the primary tumor or a lymph node biopsy for selection of a targeted treatment.”
Efficient generation of transgene-free induced pluripotent stem cells from normal and neoplastic bone marrow and cord blood mononuclear cells
Kejin Hu,1Junying Yu,1Kran Suknuntha,2Shulan Tian,3Karen Montgomery,4Kyung-Dal Choi,1Ron Stewart,3James A. Thomson,3 and Igor I. Slukvin corresponding author 1,2
Blood. 2011 Apr 7; 117(14): e109–e119. http://dx.doi.org:/10.1182/blood-2010-07-298331
Reprogramming blood cells to induced pluripotent stem cells (iPSCs) provides a novel tool for modeling blood diseases in vitro. However, the well-known limitations of current reprogramming technologies include low efficiency, slow kinetics, and transgene integration and residual expression. In the present study, we have demonstrated that iPSCs free of transgene and vector sequences could be generated from human BM and CB mononuclear cells using nonintegrating episomal vectors. The reprogramming described here is up to 100 times more efficient, occurs 1-3 weeks faster compared with the reprogramming of fibroblasts, and does not require isolation of progenitors or multiple rounds of transfection. Blood-derived iPSC lines lacked rearrangements of IGH and TCR, indicating that their origin is non–B- or non–T-lymphoid cells. When cocultured on OP9, blood-derived iPSCs could be differentiated back to the blood cells, albeit with lower efficiency compared to fibroblast-derived iPSCs. We also generated transgene-free iPSCs from the BM of a patient with chronic myeloid leukemia (CML). CML iPSCs showed a unique complex chromosomal translocation identified in marrow sample while displaying typical embryonic stem cell phenotype and pluripotent differentiation potential. This approach provides an opportunity to explore banked normal and diseased CB and BM samples without the limitations associated with virus-based methods
The advent of reprogramming technology has opened up the possibility of obtaining patient-specific induced pluripotent stem cells (iPSCs) for the study of blood diseases and for potential therapeutic applications. Although skin fibroblasts initially were used to obtain human iPSCs,1,2 several studies demonstrated successful reprogramming of CD34+ cells from CB or mobilized peripheral blood.3,4 Recently, T cells and peripheral blood mononuclear cells have also been successfully reprogrammed to iPSCs.5–7 Because genetic abnormalities are limited to hematopoietic cells in many blood diseases, successful reprogramming of blood cells represents a major advance in establishing iPSC-based models for hematologic diseases. However, because the current reprogramming methods use virus-based delivery of reprogramming factors, permanent integration of transgene and/or vector sequences into the genome, residual transgene expression, low efficiency, and slow kinetics remain the major problems surrounding this technology. To overcome these problems, several approaches have been used, including transient transfection, RNA transfection, the “PiggyBac” system, protein transduction, the Cre-LoxP excision system, minicircle vectors, and episomal plasmids.8–13 Nevertheless, limitations related to low reprogramming efficiency and/or genomic integration and complexity of genetic manipulations are still not completely resolved, and the suitability of these newest techniques for blood reprogramming remains unknown.
We recently developed a method for obtaining human iPSCs free of vector and transgene sequences from human fibroblasts using nonintegrating episomal vectors.14 In the present study, we have demonstrated that this technology could be applied to efficiently reprogram mononuclear cells from human BM and CB to pluripotency with up to 100 times more reprogramming efficiency compared with fibroblasts. The iPSCs generated by this method were free of transgene and vector sequences and were able to differentiate back to the blood, albeit with lower efficiency compared with fibroblast-derived iPSCs. Using the same protocol, we also efficiently reprogrammed a BM sample from a patient with chronic myeloid leukemia (CML), and were able to obtain transgene-free iPSCs with unique, patient-specific complex chromosomal translocation, which would be impossible to generate using currently available genetic-engineering methods. The elimination of genomic integration and background transgene expression, some of which are oncogenes, is a critical step toward advancing iPSC technology for the modeling of blood diseases and therapeutic applications.
Generation of iPSCs from mononuclear cells
Frozen CB mononuclear cells were obtained from AllCells. BM mononuclear cells from normal donors and from a patient with CML in the chronic phase were purchased from AllCells. Total BM cells intended for final disposition were also obtained from the University of Wisconsin Hospital and Clinics. Whole BM was cultured overnight in expansion medium consisting of StemSpan SFEM (StemCell Technologies) supplemented with Ex-Cyte (0.2%; Celliance) and recombinant human IL-3 (10 ng/mL), IL-6 (100 ng/mL), SCF (100 ng/mL), and FMS-related tyrosine kinase-3 ligand (Flt3L;100 ng/mL; all from PeproTech). The next day, Histopaque (Sigma-Aldrich) separation was performed to obtain the mononuclear cells. For reprogramming, BM mononuclear cells were cultured in expansion medium for 2 days (Figure 1A). After removing the dead cells by spinning over a 20% Percoll gradient (Sigma-Aldrich), 1 × 105 to 3.7 × 106viable cells were transfected with combination 19 of reprogramming factors (9 μg of pEP4EO2SET2K and pEP4EO2SEN2K and 6 μg of pCEP4M2L)14 using the CD34+ Nucleofector kit (Lonza). After an additional 2 days of culturing in expansion medium and removing the dead cells by Percoll density centrifugation, cells were transferred onto MEFs and cultured in iPSC medium. Starting from day 10, MEF-conditioned medium was used, and this was changed every day. The individual iPSC colonies were picked up for expansion from days 17-21. CB mononuclear cells were reprogrammed using the same conditions with or without the addition of 1μM thiazovivin (Stemgent).
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3083304/figure/F1/
Figure 1
Efficient generation of transgene-free iPSCs from BM mononuclear cells. (A) Schematic diagram of reprogramming protocol. (B) Kinetics of morphologic changes after blood reprogramming. (C-D) Comparison of reprogramming efficiency between blood cells and …
High efficiency of reprogramming of mononuclear cells from human BM and CB
For the production of iPSCs, BM mononuclear cells were cultured in serum-free expansion medium supplemented with human SCF, IL-3, IL-6, and Flt3L for 2 days to expand hematopoietic progenitors, and transfected with episomal vectors (combination 19)14 by nucleofection. After an additional 2 days of culture in hematopoietic medium, floating cells were transferred onto MEF feeders (Figure 1A). Cells in coculture underwent a series of changes, including morphologic transformation from round to cuboidal shape, with eventual formation of ALP+ colonies with typical ESC morphology at approximately day 17-21 of culture (Figure 1B-C). By picking up 50 of 88 high-quality iPSC colonies, we were able to obtain 47 iPSC lines in a single reprogramming experiment, representing 352 iPSC lines per 106 transfected cells. This high reprogramming efficiency of blood cells was reproduced in another experiment (Figure 1D). In contrast, we obtained only a few iPSC lines by transfection of 106 fibroblasts with episomal plasmids expressing the same set of reprogramming factors.14 To confirm superior efficiency of BM-cell reprogramming, we performed side-by-side reprogramming experiments with BM mononuclear cells and neonatal fibroblasts and evaluated the number of ALP+ colonies after the first passage. As shown in Figure 1C, reprogrammed BM mononuclear cells generated a much higher number of ALP+ colonies compared with fibroblasts in 2 independent experiments. BM iPSCs expressed the typical ESC markers OCT4, SOX2, NANOG, LIN28, SSEA3, SSEA4, TRA-1-60, TRA-1-81, and ALP as determined by RT-PCR and flow cytometry (Figure 1E,J). We also observed up-regulation of other ESC signature genes REX1 (ZFP42), GDF3, DNMT3B, andTDGF1, which were not present in our reprogramming cocktails (Figure 1F,J). As expected, BM iPSCs lost expression of the pan-hematopoietic markers CD45 and CD43 (data not shown) and genes typically found in the BM hematopoietic cells (Figure 2C). To characterize the molecular properties of BM iPSCs, we performed a global analysis of the gene expression of blood-derived iPSCs and compared them with 5 hESC lines and 3 iPSCs derived from fibroblasts using plasmid combination 19 (DF19 iPSC lines).14 In this analysis, we also included 2 iPSC lines derived from fibroblasts using the same set of reprogramming factors but using expression vectors with different transgene arrangements (combination 6, DF6 iPSC lines).14Global analysis of gene expression confirmed the similarity of BM iPSCs to 5 hESC and 5 fibroblast iPSC lines. As shown in Figure 2A, BM iPSCs clustered together with hESCs and fibroblast-derived iPSCs, but were distant from the parental BM cells. Similarly, analysis of scatter plots shows a much tighter correlation of reprogrammed BM cells with hESCs than with parental cells (Figure 2B). The pluripotency of iPSC-derived cell lines was confirmed using a teratoma-formation assay with demonstration of derivatives of all 3 germ layers (Figure 1G). Whereas we detected an abnormal karyotype in one BM iPSC line, the majority of them maintained the normal karyotype (Figure 1H).
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3083304/bin/zh89991168710002.gif
Figure 2
Global analysis of gene expression in hESCs and iPSCs generated from BM, CB, and fibroblasts and their parental cells. (A) Pearson correlation analysis of global gene expression. (B) Scatter plots comparing the global gene-expression profiles of BM9 iPSC …
Although we used single-cell subcloning to isolate cells that had lost episomal plasmids in our previous reprogramming studies,14 our initial subcloning experiments with BM iPSCs demonstrated that all clones obtained at passage 15 were transgene-free (Figure 1I). Based on these experiments, we concluded that episomal plasmids were cured from BM iPSCs faster than we had previously thought. To analyze the kinetics of episomal plasmid loss, we extracted episomal DNA at different passage from 10 random BM iPSC lines. We found that episomal DNA was lost progressively, and was absent in some samples as early as passage 3. By passage 7, we did not detect any transgene in 7 of 10 lines checked with multiple pairs of primers (Figure 1K).
We applied a similar approach to the reprogramming of mononuclear cells of CB. Although the efficiency of reprogramming was much lower, we were able to obtain 6 CB iPSCs from approximately 3 × 106transfected CB mononuclear cells. By adding small-molecule thiazovivin21 to reprogramming cultures, we were able to increase the reprogramming efficiency of CB cells by more than 10 times (Figure 3B). We obtained a total of 22 CB iPSC lines from 2 reprogramming experiments. All CB iPSCs displayed the typical hESC phenotype and gene-expression profile (Figure 3A,G). Six selected CB iPSC lines showed pluripotency in the teratoma assay and were free of episomal vectors and genomic integration CB iPSCs (Figure 3E-F).
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3083304/bin/zh89991168710003.gif
Figure 3
Reprogramming of CB mononuclear cells with nonintegrating constructs. (A) All 22 CB iPSC lines express hESC-specific surface markers as indicated, and express OCT4, NANOG, and SOX2. iPSC lines checked are: CB iPSC1 to CB iPSC6, CB iPSCT1 to CB iPSCT10, …
Hematopoietic differentiation potential of blood-derived iPSCs
To test hematopoietic differentiation potential of blood-derived iPSCs, we used iPSC cocultured with OP9.22As we showed previously, hematopoietic differentiation from hESCs proceeds through the formation of a population of CD34+ cells, which includes CD34+CD43+ hematopoietic progenitors, CD34+CD31+CD43−endothelial cells, and CD34+CD31−CD43− mesenchymal cells. The 3 major populations of CD43+hematopoietic cells include CD235a/CD41a+ erythro-megakaryocytic progenitors and lin−CD43+CD45−and CD45+ multipotent progenitors.18 Earlier, we found that fibroblast-derived iPSCs and hESCs follow a very similar pattern of hematopoietic differentiation, although significant variation in blood-forming potential was observed between different iPSC clones. In addition, we noted that the generation of 4 iPSC clones was sufficient to ensure that at least one clone showed good hematopoietic differentiation potential.23 Testing of 4 BM iPSC lines revealed a similar differentiation pattern of BM iPSCs (Figure 4A). However, opposite our expectations, all 4 BM iPSCs produced fewer CD43+ hematopoietic progenitors than H1 hESCs or transgene-free fibroblast-derived iPSCs obtained using a similar method. Screening 5 additional BM iPSCs and 6 CB iPSCs failed to reveal a clone with higher differentiation potential, indicating that our blood-derived iPSCs were somewhat resistant to differentiating back to the blood in coculture with OP9 (Figure 4B). Because recent studies have suggested that lymphoid cell–derived iPSCs differentiate into blood less efficiently than CD34+ cell–derived iPSCs,7 we evaluated the rearrangement of TCR and IGH genes in our cells to determine whether our iPSCs originated from lymphoid cells. As shown in Figure 5, all 9 tested iPSC lines lacked rearrangements of TCR and IGH, indicating that their origin was non–B- or non–T-lymphoid cells.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3083304/bin/zh89991168710004.gif
Figure 4
Hematopoietic differentiation potential of BM- and CB-derived iPSCs. (A) In coculture with OP9, blood-derived iPSCs generate a CD34+ population of cells with typical subsets including CD43+hematopoietic progenitors, CD31+CD43− endothelial cells, …
Figure 5
Analyses of TCR and IGH rearrangement in BM and CB iPSC lines. (A) PCR analyses of TCRB rearrangements. (B) PCR analyses of TCRG rearrangement. (C) PCR analyses of IGH rearrangements. FR indicates framework. (D) Specimen controls. M indicates the 50-bp…
Reprogramming of BM samples with CML
Reprogramming of neoplastic BM cells provides an opportunity to address the effect of oncogenes and patient-specific chromosomal abnormalities on the development of the leukemia phenotype in vitro. However the virus-based approach for reprogramming leukemic cells is highly undesirable because of genomic integration and background expression of reprogramming factors, some of which are oncogenes. Therefore, we applied episomal vectors to generate transgene-free iPSCs from a patient with CML in the chronic phase. We picked, expanded, and froze 50 CML iPSC lines from a single reprogramming. As with normal BM, we were able to generate multiple transgene-free CML iPSC lines with typical features of pluripotent stem cells. Two transgene-free CML iPSC lines were selected and characterized (Figure 6). RT-PCR analysis revealed that both CML iPSCs retained typical BCR-ABL fusion (Figure 6H). Moreover, the CML iPSCs were found to have a complex karyotype with a 4-way translocation between chromosomes 1, 9, 22, and 11 that was present in the patient BM (Figure 7). CML iPSC lines lacked rearrangement of TCR or IGH, indicating derivation from nonlymphoid cells (Figure 5). After hematopoietic differentiation, these cell lines generated CD43+ hematopoietic progenitors, which included typical subsets of CD235a/CD41a+ erythro-megakaryocytic and lin−CD34+CD43+CD45+/− multipotent progenitors (Figure 6E). In a colony-forming assay, these differentiated CML iPSCs formed all types of hematopoietic colonies, including granulocyte, erythrocyte, monocyte, megakaryocyte and giant granulocyte-macrophage colonies (Figure 6F).
Figure 6
Generation of iPSCs from BM samples from a patient in the chronic phase of CML. (A) Flow cytometric analysis of hESC-specific marker expression in CML iPSC15 and CML iPSC17. (B) Bright-field image demonstrating typical hESC morphology of CML iPSCs growing …
Figure 7
Karyograms of BM cells from a patient with CML and the 2 iPSCs derived from these cells. Top left panel shows spectral karyogram of CML iPSC15. SKY analysis demonstrates the 4-way translocation between chromosomes 1, 9, 11, and 22, shown here by classification-colored …
Current methods for blood reprogramming rely on use of genome-integrating viruses and require several rounds of viral infection. Our data show that iPSC lines free of any transgene or vector sequence could be obtained using EBV-based episomal vectors. The efficiency of reprogramming blood cells by this method was at least 100 times higher than that of fibroblasts and was similar or higher to reported reprogramming efficiency using virus-based methods. Although previous studies have demonstrated the generation of iPSCs from blood using CD34+ cells3,4 or T cells,5–7 these methods require the isolation of progenitors or mature blood cells before reprogramming. We demonstrated that successful reprogramming could be achieved using just 106-107 mononuclear cells from CB or BM without any additional purification steps. Moreover, iPSCs with rearranged TCR or IGH may be undesirable for potential therapeutic applications and modeling of lymphoid development, because prearranged antigen-receptor genes are expressed precociously in early hematopoietic progenitors, leading to abnormal hematopoietic and lymphoid development and predisposition for lymphomas.24 A selective reprogramming of nonlymphoid cells using our method makes it possible to obtain iPSCs lacking TCR and IGH rearrangements using nonseparated mononuclear cells. Reprogramming of blood cells with episomal vectors occurs more rapidly than fibroblasts and is associated with a loss of episomal DNA in the majority of iPSC lines after 7 passages, thus eliminating the requirement for extensive additional subcloning steps. Human BM and CB represent the most accessible sources of somatic cells, with extensive and diverse archived samples available. Successful reprogramming of frozen blood samples containing less than 107 mononuclear cells in the present study clearly demonstrates the applicability of the described method for the generation of transgene-free iPSCs without rearranged antigen-receptor genes from archived samples of normal and diseased blood cells for studies of hematopoietic development, blood disease pathogenesis, and drug screening, and potentially for therapeutic purposes.
397 Induced Pluripotent Stem Cell Model of Chronic Myeloid Leukemia Revealed Olfactomedin 4 As a Novel Survival Factor for Primitive Leukemia Cells
Program: Oral and Poster Abstracts
Type: Oral
Session: 631. Chronic Myeloid Leukemia: Biology and Pathophysiology, excluding Therapy: Strategies to Circumvent Therapy Resistance
Kran Suknuntha, MD, PhD1*, Yuki Ishii, PhD2*, Kejin Hu, PhD3*, Mcintosch Brayan, PhD4*, David T. Yang, MD5,…, Jean YJ Wang, PhD2*, James Thomson, PhD, DVM6* and Igor Slukvin, MD, PhD
56th ASH Meeting 2014 https://ash.confex.com/ash/2014/webprogram/Paper70688.html
CML is a myeloproliferative disorder characterized by unregulated growth of predominantly myeloid cells, and their subsequent accumulation in the bone marrow and peripheral blood. CML originates in hematopoietic stem cells (HSCs) with t(9;22)(q34;q11.2) translocation, which causes the constitutively expression of the BCR-ABL kinase driving the expansion of leukemic progeny. Ex vivo cultures of CML-derived cell lines and primary CML cells, ectopic expression ofBCR-ABL in CD34+ cells and mouse models have provided important insights into CML pathogenesis and led to the development of targeted therapy for this neoplastic disease with BCR-ABL thyrosine kinase inhibitor (TKI), imatinib. Despite these achievements, in many cases CML remains incurable because of innate resistance of CML leukemia stem cells (LSCs) to TKI. Thus, a definitive cure for leukemia requires identifying novel therapeutic targets to eradicate LSCs. However, the rarity of LSCs within the pool of malignant cells remains a major limiting factor for their study in humans. Recently we generated transgene-free iPSCs from the bone marrow mononuclear cells of a patient in the chronic phase of CML (CML15 iPSCs and CML17 iPSCs) and showed that these iPSCs capture the entire genome of neoplastic cells, including the unique 4-way translocation between chromosomes 1, 9, 22, and 11 that was present in the patient bone marrow (BM) (Hu et al., Blood 2011). By differentiating CML iPSCs back to the blood we were able to generate iCD34+primitive hematopoietic cells with typical LSC properties, including HSC phenotype (lin-CD34+CD45+CD90+CD117+CD45RA-RholowALDHhigh), adhesion defect, increased long-term survival and proliferation, and innate resistance to TKI imatinib. By analyzing transcriptome of CML and normal BM iCD34+ cells treated or non-treated with imatinib we discovered OLFM4 as top-ranking gene, which is selectively upregulated by imatinib in CML, but not normal BM iCD34+ cells. Using siRNA, we demonstrated that OLFM4 knockdown potentiate imatinib-induced apoptosis and suppression of CFCs in iCD34+ cells, thereby indicating that OLFM4 is involved in regulation of imatinib resistance and survival of de novo generated primitive CML cells. To find out whether findings obtained using iCD34+ cells can be translated to somatic cells, we evaluated the expression and functional role of OLFM4 in CD34+ cells obtained from parental bone marrow and bone marrow from the several other CML patients in the chronic phase. Using immunohistochemistry and RT-PCR we confirmed OLFM4 expression in lin-CD34+ and CD34- bone marrow cells from patients. Knockdown OLFM4 with siRNA in somatic CML lin-CD34+ potentiated imatininb-induced CFC suppression, abrogated LTC-ICs and engraftment of lin-CD34+ cells in NSGW41 mice, thereby indicating that OLFM4 is critical for survival of CML LSCs. In summary, we showed that reprogramming leukemia cells to pluripotency and then differentiating them back into blood cells can be used as a novel approach to produce an unlimited number of primitive hematopoietic cells with LSC properties and identify of novel LSC survival factors and drug targets. We validated this approach by demonstrating the successful application of the iPSC-based platform to discover OLFM4 as a novel LSC survival factor in patients in the chronic phase of CML.
Scientists Discover How Cancer Cells Escape Blood Vessels
12/16/2015 – Anne Trafton, MIT News Office http://www.biosciencetechnology.com/news/2015/12/scientists-discover-how-cancer-cells-escape-blood-vessels
A rounded cancer cell (top left) sends out nanotubes connecting with endothelial cells. Genetic material can be injected via these nanotubes, transforming the endothelial cells and making them more hospitable to additional cancer cells. (Image: Sengupta Lab)
Scientists at MIT and Massachusetts General Hospital have discovered how cancer cells latch onto blood vessels and invade tissues to form new tumors — a finding that could help them develop drugs that inhibit this process and prevent cancers from metastasizing.
Cancer cells circulating in the bloodstream can stick to blood vessel walls and construct tiny “bridges” through which they inject genetic material that transforms the endothelial cells lining the blood vessels, making them much more hospitable to additional cancer cells, according to the new study.
The researchers also found that they could greatly reduce metastasis in mice by inhibiting the formation of these nanobridges.
“Endothelial cells line every blood vessel and are the first cells in contact with any blood-borne element. They serve as the gateway into and out of tumors and have been the focus of intense research in vascular and cancer biology. These findings bring these two fields together to add greater insight into control of cancer and metastases,” said Elazer Edelman, the Thomas D. and Virginia W. Cabot Professor of Health Sciences and Technology, a member of MIT’s Institute for Medical Engineering and Science, and one of the leaders of the research team.
The lead author of the paper, which appears in the Dec. 16 issue of Nature Communications, is Yamicia Connor, a graduate student in the Harvard-MIT Division of Health Sciences and Technology (HST). The paper’s senior author is Shiladitya Sengupta, an assistant professor at HST and at Harvard Medical School.
Building bridges
Metastasis is a multistep process that allows cancer to spread from its original site and form new tumors elsewhere in the body. Certain cancers tend to metastasize to specific locations; for example, lung tumors tend to spread to the brain, and breast tumors to the liver and bone.
To metastasize, tumor cells must first become mobile so they can detach from the initial tumor. Then they break into nearby blood vessels so they can flow through the body, where they become circulating tumor cells (CTCs). These CTCs must then find a spot where they can latch onto the blood vessel walls and penetrate into adjacent tissue to form a new tumor.
Blood vessels are lined with endothelial cells, which are typically resistant to intruders.
“Normal endothelial cells should not enable a cancer cell to invade, but if a cancer cell can connect with an endothelial cell, and inject signals that enable this endothelial cell to be controlled and completely transformed, then it facilitates metastasis,” Sengupta said.
The researchers first spotted tiny bridges between cancer cells and endothelial cells while using electron microscopy to study the interactions between those cell types. They speculated that the cancer cells might be sending some kind of signal to the endothelial cells.
“Once we saw that these structures allowed for a ubiquitous transfer of a lot of different materials, microRNAs were an obvious interesting molecule because they’re able to very broadly control the genome of a cell in ways that we don’t really understand,” Connor said. “That became our focus.”
MicroRNA, discovered in the early 1990s, helps a cell to fine-tune its gene expression. These strands of RNA, about 22 base pairs long, can interfere with messenger RNA, preventing it from being translated into proteins.
In this case, the researchers found, the injected microRNA makes the endothelial cells “sticky.” That is, the cells begin to express proteins on their surfaces that attract other cells to adhere to them. This allows additional CTCs to bind to the same site and penetrate through the vessels into the adjacent tissue, forming a new tumor.
“It’s almost like the cancer cells are cooperating with each other to facilitate migration,” Sengupta said. “You just need maybe 1 percent of the endothelial cells to become sticky, and that’s good enough to facilitate metastasis.”
Non-metastatic cancer cells did not produce these invasive nanobridges when grown on endothelial cells.
Erkki Ruoslahti, a professor of cell, molecular, and developmental biology at the University of California at Santa Barbara, said that the discovery is an important advance in understanding tumor metastasis.
“I found it particularly interesting that the transfer of regulatory macromolecules from tumor cells to endothelial cells via intercellular nanotubes appears to be more effective (at least over relatively short distances) than exosome-mediated transfer, which has received a lot of attention lately,” said Ruoslahti, who was not part of the research team.
Shutting down metastasis
The nanobridges are made from the proteins actin and tubulin, which also form the cytoskeleton that gives cells their structure. The researchers found that they could inhibit the formation of these nanobridges, which are about 300 microns long, by giving low doses of drugs that interfere with actin.
When the researchers gave these drugs to mice with tumors that normally metastasize, the tumors did not spread.
Sengupta’s lab is now trying to figure out the mechanism of nanobridge formation in more detail, with an eye toward developing drugs that act more specifically to inhibit the process.
“If we can first understand how these structures are formed, then we can try to design targeted therapies to inhibit their formation, which could be a promising new area for developing drugs that specifically target metastasis,” Connor said.
Source: Massachusetts Institute of Technology
Back-to-the-Future with Tumor Cell-Based Avatars
Researchers Looking for Alternatives to Individual Avatars Have Found Reason to Be Hopeful in Tumor-Cell Based Predictive Models
Formidable barriers, including time and expense required to breed and maintain mice engrafted with human tumor tissue, impede the widespread use of mouse avatars.
- Mice grafted with human tumors, known as patient-derived xenograft (PDX) mice, have migrated from cancer research labs to the clinic. But as limitations to modeling patient individual tumors in mice emerge, some investigators are turning to cell-based models and applying new methodologies to support and grow cells in culture.
Conceived by Heinz-Herbert Fiebig and colleagues at the University of Freiburg in the early 1980s, it was hoped that PDX mice would more accurately reflect an individual patient’s tumor in a model system and predict tumor responses to drug therapies. Dr. Fiebig is the founder and CEO of Oncotest, a company that specializes in preclinical pharmacological contract research.
Since their introduction, commercial labs, including Oncotest, the Jackson Laboratory, and Discovery Group plc Horizon (Horizon), have provided access to a wide range of PDX mice made from donated tumor tissue. The tissue, cryopreserved for future use after biopsy, serves as the basis for offering drug-testing services to researchers and pharmaceutical companies. Oncotest, for example, says it provides drug-testing services to 16 of the 20 largest pharmaceutical companies, using a library of more than 350 PDX mouse models.
And beyond PDX mouse model libraries for pharma companies, companies now offer individualized avatar mice directly to patients developed using their own tumors. Champions Oncology provides mouse avatars directly to patients, at a cost of $10,000 to $12,000. Proponents of these mouse models say they can facilitate the identification of a personalized therapeutic regimen, may prove more useful than genomic analysis, and eliminate the cost and toxicity associated with nontargeted chemotherapeutics.
But formidable barriers impede the widespread use of mouse avatars, scientists say, including the time and expense required to breed and maintain mice engrafted with human tumor tissue. Development of an individualized avatar takes anywhere from three to six months, more time than some critically ill patients can survive and, in about 30% of cases, Champions points out it hasn’t been able to grow the patient’s tumor in mice.
In a study published in Cancer in April 2014, Justin Stebbing, M.D., Ph.D., and colleagues at Imperial College, London, reported that they worked with Champions to develop avatars with the company’s TumorGraft system for 22 patients with advanced sarcoma. But nine patients died before the results were ready. “Within a couple of months after their surgery or biopsy, they get chemotherapy and they pass away,” says Champions CEO Ronnie Morris. “We build the avatar, but the patient can’t use it.”
In this study, the scientists said that of implanted tumors, 22 (76%) successfully engrafted, permitting the identification of treatment regimens for these patients. Although several patients died before completion of TumorGraft testing, a correlation between TumorGraft results and clinical outcome was observed in 13 of the 16 (81%) remaining individuals. No patients died during the TumorGraft-predicted therapy.
On the other hand the authors noted that a primary advantage of Champions’ TumorGraft is “that it allows discrimination between the different standard-of-care therapies that may be available, as well as other potential treatments not normally indicated for that tumor.
“Our increased understanding of tumor heterogeneity, even within a single subtype, means that knowing how patients with the same tumor previously responded to a particular drug is no guarantee that the current patient will respond similarly. TumorGraft overcomes this problem by helping guide oncologists to those treatments that are most likely to provide a positive clinical outcome.”
- Search for Alternatives
Given the obstacles to using individual avatars to guide patient therapy, researchers in several laboratories are currently looking for alternatives, turning in some cases to tumor-cell based predictive models in a back to the future approach utilizing up-to-date pharmacogenomics and novel cell culture technologies to improve the longstanding odds against success culture of tumor cells from biopsied material.
The team of Jeffrey Engelman, M.D., Ph.D., director of thoracic oncology and molecular therapeutics at Massachusetts General Hospital Cancer Center, has successfully established cell culture models from biopsy samples of lung cancer patients for functional pharmacologic studies. Dr. Engelman noted that while “Genetics has been extremely useful to guiding treatment, in many cases tumor genetics are ambiguous or do not reveal a mutation that informs a therapeutic strategy. These functional pharmacologic studies can identify effective therapeutic choices even when the genetics fail to do so.”
Dr. Engelman and colleagues described in Science a pharmacogenomic platform that facilitates rapid discovery of drug combinations that can overcome drug resistance. Their cell culture models were derived from patients whose disease had progressed while on treatment with epidermal growth factor receptor (EGFR) or anaplastic lymphoma kinase (ALK) tyrosine kinase inhibitors and then subjected to genetic analyses and a pharmacological screen.
With the system they could identify multiple effective drug combinations, they said. These included the combination of ALK and mitogen-activated protein kinases (MAPK) inhibitors active in an ALK-positive resistant tumor that had developed a MAP2K1 activating mutation. A combination of EGFR and fibroblast growth factor receptor (FGFR) inhibitors was active in an EGFR mutant-resistant cancer with a mutation in FGFR3. Combined ALK and SRC (pp60c-src) inhibition was effective in several ALK-driven patient-derived models, a result not predicted by genetic analysis alone. With further refinements, the authors said their strategy could help direct therapeutic choices for individual patients.
- Several Approaches
Noting the historical difficulty of coaxing tumor cells obtained from tumor biopsies to grow in culture, Dr. Engelman told GEN that his team typically tries three or four different approaches to optimize the growth of cells from a single biopsy, including 3D culture, organoids, and feeder layers to support the best cancer cell growth. “We want to get the biopsy to the high-throughput screening phase as quickly as possible and get the results to inform patient therapy as quickly as possible,” he said.
While the application described in their publication involved lung cancer, he notes that his lab is trying the approach on breast cancer, colorectal tumors, and melanoma. “What’s interesting for us is that there are cancers for which no work has ever been done before,” he noted.
To date, the investigators are “not applying the cell culture technology to the clinic, but are inching closer to doing so,” Dr. Engleman said. “We are confident in the results we get from the screen and believe the data is quite valuable, but we want to make sure there is clinical outcome with therapeutics prior to having a patient enroll in a clinical trial or embark on a specific therapy.”
Dr. Engelman also believes that the technology can be commercialized, but that he is “focused on making it work.” These initial studies demonstrated success in developing NSCLC models NSCLC models in 50% of collected specimens. However, the team believes that success rates could be further improved by using biopsies acquired for specifically for cell line generation.
The authors noted that with their pharmacologic platform, they discovered several previously undescribed combinations in EGFR mutant and ALK-positive lung cancers that were validated in follow-up studies and in vivo. They speculate that a similar approach could be explored in the future as a diagnostic test to identify therapeutic strategies for individual patients (under the auspices of an IRB-approved protocol).
In their study, they screened the cells after they became fully established cell lines, often requiring two to six months, a time frame that would make this approach less than ideal as a routine diagnostic test. But they say, their results of the program provides the groundwork for performing screens on viable cells obtained within weeks of a biopsy using newer technologies that would permit screening of the cancer cells while still in the presence of the stroma present in the biopsy.
In a proof of concept study in Nature Methods, investigators working at MGH, Harvard Medical School, the Karolinska Institute, and other institutions showed that circulating tumor cells (CTCs) can be captured in viable form and used to establish cell cultures, potentially bypassing the need for a biopsy as a source of tumor cells to culture.
The investigators captured the CTCs using microchip technology (the Cluster-Chip) developed to capture CTC clusters independently of tumor-specific markers from unprocessed blood. The device isolates the CTC clusters through specialized bifurcating traps under low-shear stress conditions that preserve their integrity, and, the investigators said, even two-cell clusters can be efficiently captured.
Maheswaran et al., in Cancer Research, used the device to show that the culture of CTCs in the blood of patients with breast cancer enabled them to study patterns of drug susceptibility linked to the genetic context that is unique to an individual tumor.
The investigators established CTC cultures from six patients with estrogen receptor–positive breast cancer. Three of five CTC lines tested were tumorigenic in mice. Genome sequencing of the CTC lines revealed preexisting mutations in the PIK3CA gene and newly acquired mutations in the estrogen receptor gene (ESR1), PIK3CA gene, and fibroblast growth factor receptor gene (FGFR2), among others. Drug sensitivity testing of CTC lines with multiple mutations revealed potential new therapeutic targets.
The authors noted that with optimization of CTC culture conditions, this strategy could help identify the best therapies for individual cancer patients over the course of their disease.
These and other investigators believe, that cell-based methods, once optimized, could bypass the need for whole animal cancer avatars, providing another resource to help inform the choice of therapies likely to be effective in a given patient.
Linking Phenotypes and Modes of Action Through High-Content Screen Fingerprints
The Use of High-Content Screening as a Powerful Technique for Monitoring Phenotypic Responses
Felix Reisen, Amelie Sauty de Chalon, Martin Pfeifer, Xian Zhang, Daniela Gabriel, Paul Selzer
Fig. 2. Phenotypes of snuclei are colored purple, the cytoplasm redix tool compounds targeting different cellular compartments. In all figures nuclei are colored purple, the cytoplasm red.
- In today’s drug discovery campaigns we observe a clear trend toward more complex assay environments. While target-based high-throughput screening (HTS) still plays an important role, phenotypic screening techniques are gaining importance. Phenotypic screening assays are believed to be more closely linked to a given disease state than target-based approaches where the molecular hypothesis might not be relevant for disease pathogenesis.
One approach to phenotypic drug discovery is high-content screening (HCS), an HTS technique based on automated microscopy. HCS allows for highly multiplexed assay readouts that can be used to simultaneously assay several modes of action or toxicity. Additionally, HCS enables screening in a controlled and disease-relevant environment by even using patient-derived cell cultures.
While there are many advantages to phenotypic screening, additional knowledge about the targets being modulated to bring about the desired phenotype can be highly beneficial, for example, in lead optimization, by helping interpretation of structure activity relationships. In addition, knowledge of the target can also help to identify related targets that may bring about challenges in designing selective lead molecules.
Various techniques have been developed to support target identification for compounds active in phenotypic assays. These include approaches such as affinity chromatography, biochemical fractionation, radioactive ligand binding assays, drug affinity responsive target stability. Alternative approaches are based on in vivo chemical genomic assays developed in yeast Saccharomyces cerevisiae or in silico approaches using historic knowledge about compound target associations. In silico methods predict possible targets for a compound by comparing the similarity of the compound’s profile (using chemical similarity, gene expression profile, or HCS experiments) to those of previously characterized compounds with known target.
For the rest of the story, click here.
ASSAY & Drug Development Technologies, published by Mary Ann Liebert, Inc., offers a unique combination of original research and reports on the techniques and tools being used in cutting-edge drug development. GEN presents here one article “Linking Phenotypes and Modes of Action Through High-Content Screen Fingerprints.” Authors of the paper are Felix Reisen, Amelie Sauty de Chalon, Martin Pfeifer, Xian Zhang, Daniela Gabriel, and Paul Selzer.
Immuno-Oncology Landscape Expands
New Techniques Enable Closer Look into Genetic & Cellular Alterations in Tumor Microenvironment
- For years, researchers and physicians have suspected, and have worked to demonstrate, how the immune system affects susceptibility to, defense against, and progression of certain cancers. It is now understood that the immune system has the ability to influence the fate of developing cancers by not only functioning as a tumor promoter that facilitates cellular transformation, promotes tumor growth, and sculpts tumor cell immunogenicity, but also as an extrinsic tumor suppressor that either destroys developing tumors or restrains their expansion.
In the last few decades, drugs, biologicals, and vaccines targeting certain attributes of the immune system, known as immunotherapeutics, have become available, and emerging clinical data suggest that cancer immunotherapy is likely to become a key part of the clinical management of cancer for years to come.
Although immunotherapies represent a major step forward in cancer care, providing in some cases unprecedented response rates, there is still much work to do to discover new druggable targets, find biomarkers to predict response, as well as gain deeper understanding of why some cancer types are incredibly responsive to immunotherapeutic treatments while others are not.
- How Immunotherapies Work
Figure 1. Inhibitory costimulatory checkpoints are a natural immune mechanism for self-tolerance and minimization of collateral tissue damage. Inhibitory checkpoint receptors such as PD-1, LAG-3, TIM-3, and CTLA-4 are expressed by T cells, and their ligands are expressed by macrophage and dendritic cells. Tumor cells can express multiple inhibitory ligands to repress T-cell function and thereby evade clearance by the immune system.
- A deeper understanding of cancer as a disease requires the acknowledgement of its inherent heterogeneity. As with the cancer cells within a tumor, the immunological microenvironments in which they grow are similarly heterogeneous. Emerging and well-established scientific tools and techniques for the analysis of cancer cells, immune cells and their microenvironment can be combined to yield new insights into the nature of tumorigenesis, immune system recruitment, and treatment optimization.In general, immunotherapies direct an individual’s immune system to fight cancer by either stimulating it to attack cancer cells or by introducing manufactured immune system components to augment immune function. Immunotherapy treatments work in different ways. Some boost the body’s immune system in a very general way. Others help train the immune system to attack cancer cells specifically.
- On an immuno-oncological level, the genetic and cellular alterations that define a cancer cell provide the immune system with the means to be recruited to the tumor and generate T-cell responses to recognize and eradicate those cells. Elimination of cancer by T cells is only one step in the cancer immunity cycle. T-cell activation is controlled by both stimulatory and inhibitory checkpoints. Tumors use the expression of inhibitory ligands as a mechanism of suppressing cytotoxic T-cell response and inducing an immunosuppressive environment.
- Identification of specific cancer T-cell inhibitory signals, such as PD-L1, has prompted the development of a new class of cancer immunotherapy that specifically hinders immune effector inhibition, reinvigorating and potentially expanding preexisting anticancer immune responses (Figure 1).
- The presence of environment-altering immunosuppressive innate myeloid lineages in the tumor microenvironment may further explain the limited activity observed with previous immune-based therapies and why these therapies may be more effective in combination with agents that target other steps of the cycle.
- Understanding the Tumor and Its Microenvironment
In addition, the presence and quantity of various immune cell types in the tumor microenvironment may have prognostic value. Many scientists believe that a deepening appreciation of oncology genomics and the quantity and type of antigens expressed by the tumor cells, when coupled with an analysis of the patient’s immune system, will greatly progress the field and unlock the next generation of immunotherapies.
Flow cytometry and immunohistochemistry are established tools for the labeling and analysis of immunological and oncology cellular components. New techniques are likewise becoming more widely used that enable simultaneous detection of proteins and nucleic acids at single-cell resolution.
New Cellular Analysis Tools
- eBioscience, a business unit of Affymetrix, has recently expanded commercialization of two such novel assays that provide exciting new technologies in the armament of cellular analysis techniques for immuno-oncology research. The first is PrimeFlow™ RNA Assay, which is the only commercially available assay for the simultaneous detection of RNA and protein expression within millions of cells at single-cell resolution using a standard flow cytometer. The assay is compatible with cell surface and intracellular antibody staining, using traditional fluorochromes for multiparameter cellular analysis.
- With this technology an immune-oncology researcher could explore gene expression heterogeneity among different rare tumor-infiltrating immune cell subsets with single-cell resolution and without laborious cell sorts, as well as compare kinetics of both RNA and protein in the same cell.
http://www.genengnews.com/Media/images/Article/thumb_eBioscience_Fig21361229223.jpg
Figure 2. The PrimeFlow RNA Assay workflow contains several steps: antibody staining, fixation and permeabilization including intracellular staining if desired, followed by target hybridization with a target-specific probe set containing 20 to 40 oligonucleotide pairs. Next, branched DNA signal amplification is achieved through a series of sequential hybridization steps consisting of pre-amplifiers, amplifiers, and labeled probes, followed by detection by flow cytometric analysis. This results in excellent specificity, low background, and a high signal-to-noise ratio. For simplicity, two RNA targets are shown in the schematic above (red and green), and only 3 of the 20 to 40 oligonucleotide target probe pairs per target RNA are shown.
http://www.genengnews.com/gen-articles/immuno-oncology-landscape-expands/5577/
- S. Shalapour et al. recently published a study in the journal Nature (April 29, 2015) applying these techniques to mouse models of castrate-resistant prostate cancer demonstrating that the presence of a very specific and rare (0.04–3% of total) B cell population in the tumor microenvironment correlates to a immunotherapeutic response allowing a CTL-dependent eradication of oxaliplatin-treated tumors.
- ViewRNA® In Situ Hybridization (ISH) Cell and Tissue Assays comprise the second new technique from eBioscience. Similar to the PrimeFlow RNA assay, but compatible with microscopy, these assays enable the visualization of single-copy RNA transcripts within adherent and suspended single cells or single cells in tissue sections, and in the case of ViewRNA ISH Tissue Assays, the spatial separation of tumor subclones by phenotypic RNA expression. Similarly, this technique can be used to visualize and quantitate cellular and molecular attributes of tumor-infiltrating immune cells to elucidate biomarkers of resistance and response. Leveraging these novel cell analysis approaches, immuno-oncology researchers can analyze cellular diversity in the tumor microenvironment as well as the diversity of immune cell responses at a single-cell level.
- Breakthrough responses to new immunotherapies are stimulating a renewed interest in basic immune biology. With our quest to develop strategies to harness the human immune response against cancer to achieve durable responses and/or complete eradication of cancer in patients safely, we must explore multiple approaches simultaneously. Which immune checkpoints can be manipulated? Are there dual therapies that can be applied to improve responses? Are there biomarkers inherent to the immune system in general, the specific tumor and the tumor microenvironment that can be used to stratify responders?
- Multiple approaches to cancer therapy exist, and few are as complicated as immune-based therapy. That being said, few therapies in recent history have demonstrated such extraordinary and durable responses for the patients who do respond. As such, many believe that this will be an intensifying area of research and clinical focus for years to come.
New Research for Prostate Cancer Therapies
Dr. Glenn Bubley has been treating patients with prostate cancer for more than 25 years.
“When a patient’s diagnosis is latter-stage prostate cancer, the standard treatment is androgen deprivation therapy [ADT],” says Bubley, Director of the Genitourinary Cancer Program in the Cancer Center at Beth Israel Deaconess Medical Center. “ADT works by lowering testosterone production and thereby depriving prostate tumors of the ‘fuel’ that helps them grow.”
But, he adds, although this hormone therapy is almost always effective, all tumors eventually grow resistant to ADT — and cancer recurs. Over the past two years, Bubley has been part of a BIDMC scientific team that has been testing a targeted treatment alternative for late-stage prostate cancer using a unique type of study known as a “Co-Clinical Trial.”
This new approach to clinical research — in which specially-created mouse models with genetic mutations are matched with tumor tissue from human cancer patients in order to test new therapies — was developed by BIDMC Cancer Center Director Pier Paolo Pandolfi, MD, PhD.
“Targeted therapies are designed to attack cancers by pinpointing the genes and genetic mutations that underlie diseases,” says Pandolfi (right). “The problem is that cancer cells are genetically complex, sometimes containing hundreds of genetic mutations. We needed to develop a way to cut down on all this ‘genetic noise’ to get at the root of the disease. The Co-Clinical Trial enables us to streamline and expedite the process in order to more quickly test a variety of new cancer drugs.”
Here’s how it works: In the Co-Clinical Trial, human participants are matched with animal models that have been genetically engineered to carry different combinations of just a few major human prostate cancer genes.
“When the animals develop tumors — just as the human patients did — they will receive the same therapies as the patients receive,” says Bubley (right). But, he adds, because each animal has only a few mutations, the researchers will be able to quickly assess which treatments are effective and which are not — and will be able to go back and adjust treatment accordingly for the human patients.
A particular advantage to this approach, say Bubley and Pandolfi, will be the ability to test combinations of different drugs to treat prostate cancer and overcome ADT resistance.
“Going forward, we think that combinations of targeted and conventional therapies may prove to be effective, particularly for drug-resistant disease,” says Bubley. “And the only realistic way to be able to quickly test numerous different drug combinations will be through the Co-Clinical Trial process.”
Nanocarriers May Carry New Hope for Brain Cancer Therapy
Fri, 11/20/2015 – DOE/Lawrence Berkeley National Laboratory
http://www.dddmag.com/news/2015/11/nanocarriers-may-carry-new-hope-brain-cancer-therapy
At only 20 nanometers in size and featuring a unique hierarchical structure, 3HM nanocarriers meet all the size and stability requirements for effectively delivering therapeutic drugs to brain cancer tumors. Credit: Ting Xu, Berkeley Lab
Glioblastoma multiforme, a cancer of the brain also known as “octopus tumors” because of the manner in which the cancer cells extend their tendrils into surrounding tissue, is virtually inoperable, resistant to therapies, and always fatal, usually within 15 months of onset. Each year, glioblastoma multiforme (GBM) kills approximately 15,000 people in the United States. One of the major obstacles to treatment is the blood brain barrier, the network of blood vessels that allows essential nutrients to enter the brain but blocks the passage of other substances. What is desperately needed is a means of effectively transporting therapeutic drugs through this barrier. A nanoscience expert at Lawrence Berkeley National Laboratory (Berkeley Lab) may have the solution.
Ting Xu, a polymer scientist with Berkeley Lab’s Materials Sciences Division who specializes in self-assembling bio/nano hybrid materials, has developed a new family of nanocarriers formed from the self-assembly of amphiphilic peptides and polymers. Called “3HM” for coiled-coil 3-helix micelles, these new nanocarriers meet all the size and stability requirements for effectively delivering a therapeutic drug to GBM tumors. Amphiphiles are chemical compounds that feature both hydrophilic (water-loving) and lipophilic (fat-loving) properties. Micelles are spherical aggregates of amphiphiles.
In a recent collaboration between Xu, Katherine Ferrara at the University of California (UC) Davis, and John Forsayeth and Krystof Bankiewicz of UC San Francisco, 3HM nanocarriers were tested on GBM tumors in rats. Using the radioactive form of copper (copper-64) in combination with positron emission tomography (PET) and magnetic resonance imaging (MRI), the collaboration demonstrated that 3HM can cross the blood brain barrier and accumulate inside GBM tumors at nearly double the concentration rate of current FDA-approved nanocarriers.
“Our 3HM nanocarriers show very good attributes for the treatment of brain cancers in terms of long circulation, deep tumor penetration and low accumulation in off-target organs such as the liver and spleen,” says Xu, who also holds a joint appointment with the UC Berkeley’s Departments of Materials Sciences and Engineering, and Chemistry. “The fact that 3HM is able to cross the blood brain barrier of GBM-bearing rats and selectively accumulate within tumor tissue, opens the possibility of treating GBM via intravenous drug administration rather than invasive measures. While there is still a lot to learn about why 3HM is able to do what it does, so far all the results have been very positive.”
Glial cells provide physical and chemical support for neurons. Approximately 90-percent of all the cells in the brain are glial cells which, unlike neurons, undergo a cycle of birth, differentiation, and mitosis. Undergoing this cycle makes glial cells vulnerable to becoming cancerous. When they do, as the name “multiforme” suggests, they can take on different shapes, which often makes detection difficult until the tumors are dangerously large. The multiple shapes of a cancerous glial cell also make it difficult to identify and locate all of the cell’s tendrils. Removal or destruction of the main tumor mass while leaving these tendrils intact is ineffective therapy: like the mythical Hydra, the tendrils will sprout new tumors.
Although there are FDA approved therapeutic drugs for the treatment of GBM, these treatments have had little impact on patient survival rate because the blood brain barrier has limited the accumulation of therapeutics within the brain. Typically, GBM therapeutics are ferried across the blood brain barrier in special liposomes that are approximately 110 nanometers in size. The 3HM nanocarriers developed by Xu and her group are only about 20 nanometers in size. Their smaller size and unique hierarchical structure afforded the 3HM nanocarriers much greater access to rat GBM tumors than 110-nanometer liposomes in the tests carried out by Xu and her colleagues.
“3HM is a product of basic research at the interface of materials science and biology,” Xu says. “When I first started at Berkeley, I explored hybrid nanomaterials based on proteins, peptides and polymers as a new family of biomaterials. During the process of understanding the hierarchical assembly of amphiphilic peptide-polymer conjugates, my group and I noticed some unusual behavior of these micelles, especially their unusual kinetic stability in the 20 nanometer size range. We looked into critical needs for nanocarriers with these attributes and identified the treatment of GBM cancer as a potential application.”
Copper-64 was used to label both 3HM and liposome nanocarriers for systematic PET and MRI studies to find out how a nanocarrier’s size might affect the pharmacokinetics and biodistribution in rats with GBM tumors. The results not only confirmed the effectiveness of 3HM as GBM delivery vessels, they also suggest that PET and MRI imaging of nanoparticle distribution and tumor kinetics can be used to improve the future design of nanoparticles for GBM treatment.
“I thought our 3HM hybrid materials could bring new therapeutic opportunities for GBM but I did not expect it to happen so quickly,” says Xu, who has been awarded a patent for the 3HM technology.








 and Protein C Pat...")
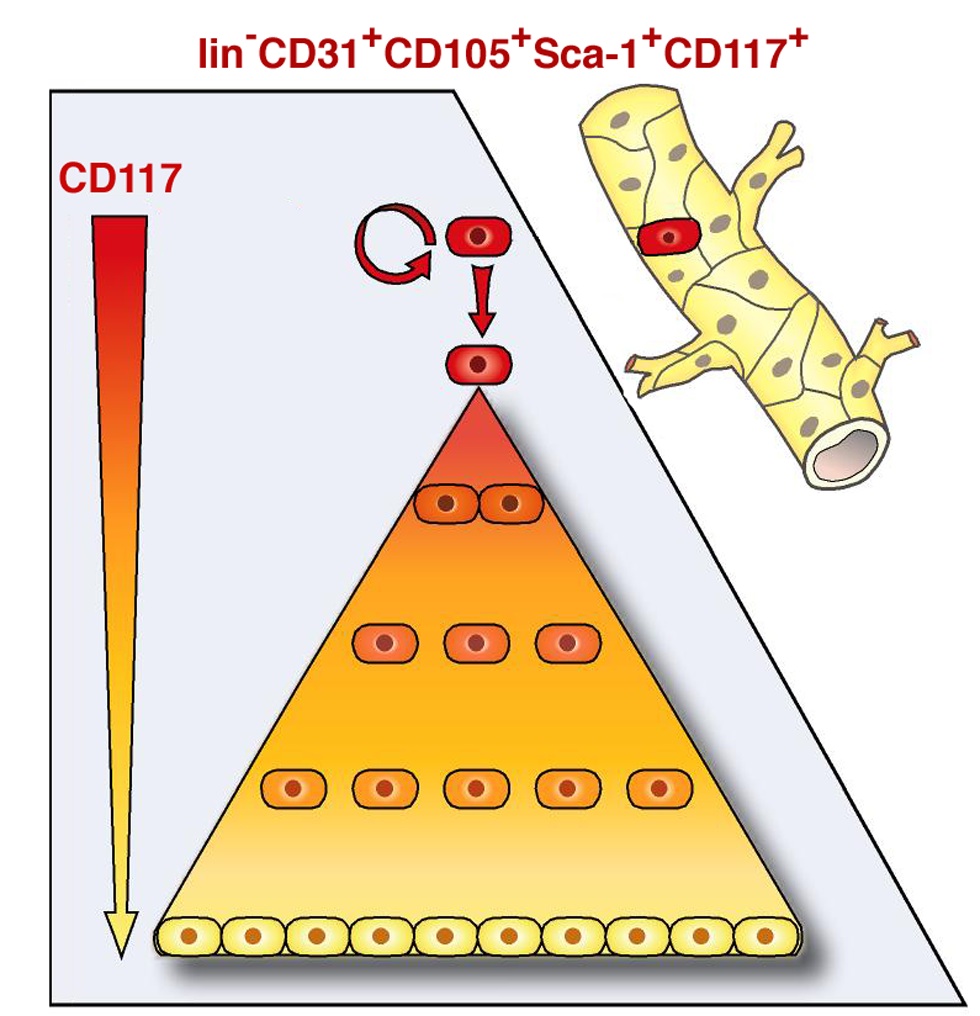
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}