Refined Warburg hypothesis -2.1.2
Writer and Curator: Larry H. Bernstein, MD, FCAP
Refined Warburg Hypothesis -2.1.2
The Warburg discoveries from 1922 on, and the influence on metabolic studies for the next 50 years was immense, and then the revelations of the genetic code took precedence. Throughout this period, however, the brilliant work of Briton Chance, a giant of biochemistry at the University of Pennsylvania, opened new avenues of exploration that led to a recent resurgence in this vital need for answers in cancer research. The next two series of presentations will open up this resurgence of fundamental metabolic research in cancer and even neurodegenerative diseases.
2.1.2.1 Cancer Cell Metabolism. Warburg and Beyond
Hsu PP, Sabatini DM
Cell, Sep 5, 2008; 134:703-707
http://dx.doi.org:/10.016/j.cell.2008.08.021
Described decades ago, the Warburg effect of aerobic glycolysis is a key metabolic hallmark of cancer, yet its significance remains unclear. In this Essay, we re-examine the Warburg effect and establish a framework for understanding its contribution to the altered metabolism of cancer cells.
It is hard to begin a discussion of cancer cell metabolism without first mentioning Otto Warburg. A pioneer in the study of respiration, Warburg made a striking discovery in the 1920s. He found that, even in the presence of ample oxygen, cancer cells prefer to metabolize glucose by glycolysis, a seeming paradox as glycolysis, when compared to oxidative phosphorylation, is a less efficient pathway for producing ATP (Warburg, 1956). The Warburg effect has since been demonstrated in different types of tumors and the concomitant increase in glucose uptake has been exploited clinically for the detection of tumors by fluorodeoxyglucose positron emission tomography (FDG-PET). Although aerobic glycolysis has now been generally accepted as a metabolic hallmark of cancer, its causal relationship with cancer progression is still unclear. In this Essay, we discuss the possible drivers, advantages, and potential liabilities of the altered metabolism of cancer cells (Figure 1). Although our emphasis on the Warburg effect reflects the focus of the field, we would also like to encourage a broader approach to the study of cancer metabolism that takes into account the contributions of all interconnected small molecule pathways of the cell.
Figure 1. The Altered Metabolism of Cancer Cells
Drivers (A and B). The metabolic derangements in cancer cells may arise either from the selection of cells that have adapted to the tumor microenvironment or from aberrant signaling due to oncogene activation. The tumor microenvironment is spatially and temporally heterogeneous, containing regions of low oxygen and low pH (purple). Moreover, many canonical cancer-associated signaling pathways induce metabolic reprogramming. Target genes activated by hypoxia inducible factor (HIF) decrease the dependence of the cell on oxygen, whereas Ras, Myc, and Akt can also upregulate glucose consumption and glycolysis. Loss of p53 may also recapitulate the features of the Warburg effect, that is, the uncoupling of glycolysis from oxygen levels. Advantages (C–E). The altered metabolism of cancer cells is likely to imbue them with several proliferative and survival advantages, such as enabling cancer cells to execute the biosynthesis of macromolecules (C), to avoid apoptosis (D), and to engage in local metabolite-based paracrine and autocrine signaling (E). Potential Liabilities (F and G). This altered metabolism, however, may also confer several vulnerabilities on cancer cells. For example, an upregulated metabolism may result in the build up of toxic metabolites, including lactate and noncanonical nucleotides, which must be disposed of (F). Moreover, cancer cells may also exhibit a high energetic demand, for which they must either increase flux through normal ATP-generating processes, or else rely on an increased diversity of fuel sources (G).
The Tumor Microenvironment Selects for Altered Metabolism
One compelling idea to explain the Warburg effect is that the altered metabolism of cancer cells confers a selective advantage for survival and proliferation in the unique tumor microenvironment. As the early tumor expands, it outgrows the diffusion limits of its local blood supply, leading to hypoxia and stabilization of the hypoxia-inducible transcription factor, HIF. HIF initiates a transcriptional program that provides multiple solutions to hypoxic stress (reviewed in Kaelin and Ratcliffe, 2008). Because a decreased dependence on aerobic respiration becomes advantageous, cell metabolism is shifted toward glycolysis by the increased expression of glycolytic enzymes, glucose transporters, and inhibitors of mitochondrial metabolism. In addition, HIF stimulates angiogenesis (the formation of new blood vessels) by upregulating several factors, including most prominently vascular endothelial growth factor (VEGF).
The oxygen levels within a tumor vary both spatially and temporally, and the resulting rounds of fluctuating oxygen levels potentially select for tumors that constitutively upregulate glycolysis. Interestingly, with the possible exception of tumors that have lost the von Hippel-Lindau protein (VHL), which normally mediates degradation of HIF, HIF is still coupled to oxygen levels, as evident from the heterogeneity of HIF expression within the tumor microenvironment (Wiesener et al., 2001; Zhong et al., 1999). Therefore, the Warburg effect—that is, an uncoupling of glycolysis from oxygen levels—cannot be explained solely by upregulation of HIF.
Recent work has demonstrated that the key components of the Warburg effect—increased glucose consumption, decreased oxidative phosphorylation, and accompanying lactate production—are also distinguishing features of oncogene activation. The signaling molecule Ras, a powerful oncogene when mutated, promotes glycolysis (reviewed in Dang and Semenza, 1999; Samanathan et al., 2005). Akt kinase, a well-characterized downstream effector of insulin signaling, reprises its role in glucose uptake and utilization in the cancer setting (reviewed in Manning and Cantley, 2007), whereas the Myc transcription factor upregulates the expression of various metabolic genes (reviewed in Gordan et al., 2007). The most parsimonious route to tumorigenesis may be activation of key oncogenic nodes that execute a proliferative program, of which metabolism may be one important arm. Moreover, regulation of metabolism is not exclusive to oncogenes. Loss of the tumor suppressor protein p53 prevents expression of the gene encoding SCO2 (the synthesis of cytochrome c oxidase protein), which interferes with the function of the mitochondrial respiratory chain (Matoba et al., 2006). A second p53 effector, TIGAR (TP53-induced glycolysis and apoptosis regulator), inhibits glycolysis by decreasing levels of fructose-2,6-bisphosphate, a potent stimulator of glycolysis and inhibitor of gluconeogenesis (Bensaad et al., 2006). Other work also suggests that p53-mediated regulation of glucose metabolism may be dependent on the transcription factor NF-κB (Kawauchi et al., 2008).
It has been shown that inhibition of lactate dehydrogenase A (LDH-A) prevents the Warburg effect and forces cancer cells to revert to oxidative phosphorylation in order to reoxidize NADH and produce ATP (Fantin et al., 2006; Shim et al., 1997). While the cells are respiratory competent, they exhibit attenuated tumor growth, suggesting that aerobic glycolysis might be essential for cancer progression. In a primary fibroblast cell culture model of stepwise malignant transformation through overexpression of telomerase, large and small T antigen, and the H-Ras oncogene, increasing tumorigenicity correlates with sensitivity to glycolytic inhibition. This finding suggests that the Warburg effect might be inherent to the molecular events of transformation (Ramanathan et al., 2005). However, the introduction of similar defined factors into human mesenchymal stem cells (MSCs) revealed that transformation can be associated with increased dependence on oxidative phosphorylation (Funes et al., 2007). Interestingly, when introduced in vivo these transformed MSCs do upregulate glycolytic genes, an effect that is reversed when the cells are explanted and cultured under normoxic conditions. These contrasting models suggest that the Warburg effect may be context dependent, in some cases driven by genetic changes and in others by the demands of the microenvironment. Regardless of whether the tumor microenvironment or oncogene activation plays a more important role in driving the development of a distinct cancer metabolism, it is likely that the resulting alterations confer adaptive, proliferative, and survival advantages on the cancer cell.
Altered Metabolism Provides Substrates for Biosynthetic Pathways
Although studies in cancer metabolism have largely been energy-centric, rapidly dividing cells have diverse requirements. Proliferating cells require not only ATP but also nucleotides, fatty acids, membrane lipids, and proteins, and a reprogrammed metabolism may serve to support synthesis of macromolecules. Recent studies have shown that several steps in lipid synthesis are required for and may even actively promote tumorigenesis. Inhibition of ATP citrate lyase, the distal enzyme that converts mitochondrial-derived citrate into cytosolic acetyl coenzyme A, the precursor for many lipid species, prevents cancer cell proliferation and tumor growth (Hatzivassiliou et al., 2005). Fatty acid synthase, expressed at low levels in normal tissues, is upregulated in cancer and may also be required for tumorigenesis (reviewed in Menendez and Lupu, 2007). Furthermore, cancer cells may also enhance their biosynthetic capabilities by expressing a tumor-specific form of pyruvate kinase (PK), M2-PK. Pyruvate kinase catalyzes the third irreversible reaction of glycolysis, the conversion of phosphoenolpyruvate (PEP) to pyruvate. Surprisingly, the M2-PK of cancer cells is thought to be less active in the conversion of PEP to pyruvate and thus less efficient at ATP production (reviewed in Mazurek et al., 2005). A major advantage to the cancer cell, however, is that the glycolytic intermediates upstream of PEP might be shunted into synthetic processes.
Biosynthesis, in addition to causing an inherent increase in ATP demand in order to execute synthetic reactions, should also cause a decrease in ATP supply as various glycolytic and Krebs cycle intermediates are diverted. Lipid synthesis, for example, requires the cooperation of glycolysis, the Krebs cycle, and the pentose phosphate shunt. As pyruvate must enter the mitochondria in this case, it avoids conversion to lactate and therefore cannot contribute to glycolysis-derived ATP. Moreover, whereas increased biosynthesis may explain the glucose hunger of cancer cells, it cannot explain the increase in lactic acid production originally described by Warburg, suggesting that lactate must also result from the metabolism of non-glucose substrates. Recently, it has been demonstrated that glutamine may be metabolized by the citric acid cycle in cancer cells and converted into lactate, producing NADPH for lipid biosynthesis and oxaloacetate for replenishment of Krebs cycle intermediates (DeBerardinis et al., 2007).
Metabolic Pathways Regulate Apoptosis
In addition to involvement in proliferation, altered metabolism may promote another cancer-essential function: the avoidance of apoptosis. Loss of the p53 target TIGAR sensitizes cancer cells to apoptosis, most likely by causing an increase in reactive oxygen species (Bensaad et al., 2006). On the other hand, overexpression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) prevents caspase-independent cell death, presumably by stimulating glycolysis, increasing cellular ATP levels, and promoting autophagy (Colell et al., 2007). Whether or not GAPDH plays a physiological role in the regulation of cell death remains to be determined. Intriguingly, Bonnet et al. (2007) have reported that treating cancer cells with dichloroacetate (DCA), a small molecule inhibitor of pyruvate dehydrogenase kinase, has striking effects on their survival and on xenograft tumor growth.
DCA, a currently approved treatment for congenital lactic acidosis, activates oxidative phosphorylation and promotes apoptosis by two mechanisms. First, increased flux through the electron transport chain causes depolarization of the mitochondrial membrane potential (which the authors found to be hyperpolarized specifically in cancer cells) and release of the apoptotic effector cytochrome c. Second, an increase in reactive oxygen species generated by oxidative phosphorylation upregulates the voltage-gated K+ channel, leading to potassium ion efflux and caspase activation. Their work suggests that cancer cells may shift their metabolism to glycolysis in order to prevent cell death and that forcing cancer cells to respire aerobically can counteract this adaptation.
Cancer Cells May Signal Locally in the Tumor Microenvironment
Cancer cells may rewire metabolic pathways to exploit the tumor microenvironment and to support cancer-specific signaling. Without access to the central circulation, it is possible that metabolites can be concentrated locally and reach suprasystemic levels, allowing cancer cells to engage in metabolite-mediated autocrine and paracrine signaling that does not occur in normal tissues. So called androgen-independent prostate cancers may only be independent from exogenous, adrenal-synthesized androgens. Androgen-independent prostate cancer cells still express the androgen receptor and may be capable of autonomously synthesizing their own androgens (Stanbrough et al., 2006).
Metabolism as an Upstream Modulator of Signaling Pathways
Not only is metabolism downstream of oncogenic pathways, but an altered upstream metabolism may affect the activity of signaling pathways that normally sense the state of the cell. Individuals with inherited mutations in succinate dehydrogenase and fumarate hydratase develop highly angiogenic tumors, not unlike those exhibiting loss of the VHL tumor suppressor protein that acts upstream of HIF (reviewed in Kaelin and Ratcliffe, 2008). The mechanism of tumorigenesis in these cancer syndromes is still contentious. However, it has been proposed that loss of succinate dehydrogenase and fumarate hydratase causes an accumulation of succinate or fumarate, respectively, leading to inhibition of the prolyl hydroxylases that mark HIF for VHL-mediated degradation (Isaacs et al., 2005; Pollard et al., 2005; Selak et al., 2005). In this rare case, succinate dehydrogenase and fumarate hydratase are acting as bona fide tumor suppressors.
There are many complex questions to be answered: Is it possible that cancer cells exhibit “metabolite addiction”? Are there unique cancer-specific metabolic pathways, or combinations of pathways, utilized by the cancer cell but not by normal cells? Are different stages of metabolic adaptations required for the cancer cell to progress from the primary tumor stage to invasion to metastasis? How malleable is cancer metabolism?
2.1.2.2 Cancer metabolism. The Warburg effect today
Ferreira LMR
Exp Molec Pathol 2010; 89:372-383.
http://dx.doi.org/10.1016/j.yexmp.2010.08.006
One of the first studies on the energy metabolism of a tumor was carried out, in 1922, in the laboratory of Otto Warburg. He established that cancer cells exhibited a specific metabolic pattern, characterized by a shift from respiration to fermentation, which has been later named the Warburg effect. Considerable work has been done since then, deepening our understanding of the process, with consequences for diagnosis and therapy. This review presents facts and perspectives on the Warburg effect for the 21st century.
Research highlights
► Warburg first established a tumor metabolic pattern in the 1920s. ► Tumors’ increased glucose uptake has been studied since then. ► Cancer bioenergetics’ study provides insights in all its hallmarks. ► New cancer diagnostic and therapeutic techniques focus on cancer metabolism.
Introduction
Contestation to Warburg’s ideas
Glucose’s uptake and intracellular fates
Lactate production and induced acidosis
Hypoxia
Impairment of mitochondrial function
Tumour microenvironment
Proliferating versus cancer cells
More on cancer bioenergetics – integration of metabolism
Perspectives
2.1.2.3 New aspects of the Warburg effect in cancer cell biology
Bensinger SJ, Cristofk HR
Sem Cell Dev Biol 2012; 23:352-361
http://dx.doi.org:/10.1016/j.semcdb.2012.02.003
Altered cellular metabolism is a defining feature of cancer [1]. The best studied metabolic phenotype of cancer is aerobic glycolysis–also known as the Warburg effect–characterized by increased metabolism of glucose to lactate in the presence of sufficient oxygen. Interest in the Warburg effect has escalated in recent years due to the proven utility of FDG-PET for imaging tumors in cancer patients and growing evidence that mutations in oncogenes and tumor suppressor genes directly impact metabolism. The goals of this review are to provide an organized snapshot of the current understanding of regulatory mechanisms important for Warburg effect and its role in tumor biology. Since several reviews have covered aspects of this topic in recent years, we focus on newest contributions to the field and reference other reviews where appropriate.
Highlights
► This review discusses regulatory mechanisms that contribute to the Warburg effect in cancer. ► We list cancers for which FDG-PET has established applications as well as those cancers for which FDG-PET has not been established. ► PKM2 is highlighted as an important integrator of diverse cellular stimuli to modulate metabolic flux and cancer cell proliferation. ► We discuss how cancer metabolism can directly influence gene expression programs. ► Contribution of aerobic glycolysis to the cancer microenvironment and chemotherapeutic resistance/susceptibility is also discussed.
Regulation of the Warburg effect

PKM2 integrates diverse signals to modulate metabolic flux and cell proliferation
Fig. 1. PKM2 integrates diverse signals to modulate metabolic flux and cell proliferation

Metabolism can directly influence gene expression programs
Fig. 2. Metabolism can directly influence gene expression programs. A schematic representation of how metabolism can intrinsically influence epigenetics resulting in durable and heritable gene expression programs in progeny.
2.1.2.4 Choosing between glycolysis and oxidative phosphorylation. A tumor’s dilemma
Jose C, Ballance N, Rossignal R
Biochim Biophys Acta 201; 1807(6): 552-561.
http://dx.doi.org/10.1016/j.bbabio.2010.10.012
A considerable amount of knowledge has been produced during the last five years on the bioenergetics of cancer cells, leading to a better understanding of the regulation of energy metabolism during oncogenesis, or in adverse conditions of energy substrate intermittent deprivation. The general enhancement of the glycolytic machinery in various cancer cell lines is well described and recent analyses give a better view of the changes in mitochondrial oxidative phosphorylation during oncogenesis. While some studies demonstrate a reduction of oxidative phosphorylation (OXPHOS) capacity in different types of cancer cells, other investigations revealed contradictory modifications with the upregulation of OXPHOS components and a larger dependency of cancer cells on oxidative energy substrates for anabolism and energy production. This apparent conflictual picture is explained by differences in tumor size, hypoxia, and the sequence of oncogenes activated. The role of p53, C-MYC, Oct and RAS on the control of mitochondrial respiration and glutamine utilization has been explained recently on artificial models of tumorigenesis. Likewise, the generation of induced pluripotent stem cells from oncogene activation also showed the role of C-MYC and Oct in the regulation of mitochondrial biogenesis and ROS generation. In this review article we put emphasis on the description of various bioenergetic types of tumors, from exclusively glycolytic to mainly OXPHOS, and the modulation of both the metabolic apparatus and the modalities of energy substrate utilization according to tumor stage, serial oncogene activation and associated or not fluctuating microenvironmental substrate conditions. We conclude on the importance of a dynamic view of tumor bioenergetics.
Research Highlights
►The bioenergetics of cancer cells differs from normals. ►Warburg hypothesis is not verified in tumors using mitochondria to synthesize ATP. ►Different oncogenes can either switch on or switch off OXPHOS. ►Bioenergetic profiling is a prerequisite to metabolic therapy. ►Aerobic glycolysis and OXPHOS cooperate during cancer progression.
- Cancer cell variable bioenergetics
Cancer cells exhibit profound genetic, bioenergetic and histological differences as compared to their non-transformed counterpart. All these modifications are associated with unlimited cell growth, inhibition of apoptosis and intense anabolism. Transformation from a normal cell to a malignant cancer cell is a multi-step pathogenic process which includes a permanent interaction between cancer gene activation (oncogenes and/or tumor-suppressor genes), metabolic reprogramming and tumor-induced changes in microenvironment. As for the individual genetic mapping of human tumors, their metabolic characterization (metabolic–bioenergetic profiling) has evidenced a cancer cell-type bioenergetic signature which depends on the history of the tumor, as composed by the sequence of oncogenes activated and the confrontation to intermittent changes in oxygen, glucose and amino-acid delivery.
In the last decade, bioenergetic studies have highlighted the variability among cancer types and even inside a cancer type as regards to the mechanisms and the substrates preferentially used for deriving the vital energy. The more popular metabolic remodeling described in tumor cells is an increase in glucose uptake, the enhancement of glycolytic capacity and a high lactate production, along with the absence of respiration despite the presence of high oxygen concentration (Warburg effect) [1]. To explain this abnormal bioenergetic phenotype pioneering hypotheses proposed the impairment of mitochondrial function in rapidly growing cancer cells [2].
Although the increased consumption of glucose by tumor cells was confirmed in vivo by positron emission tomography (PET) using the glucose analog 2-(18F)-fluoro-2-deoxy-d-glucose (FDG), the actual utilization of glycolysis and oxidative phosphorylation (OXPHOS) cannot be evaluated with this technique. Nowadays, Warburg’s “aerobic-glycolysis” hypothesis has been challenged by a growing number of studies showing that mitochondria in tumor cells are not inactive per se but operate at low capacity [3] or, in striking contrast, supply most of the ATP to the cancer cells [4]. Intense glycolysis is effectively not observed in all tumor types. Indeed not all cancer cells grow fast and intense anabolism is not mandatory for all cancer cells. Rapidly growing tumor cells rely more on glycolysis than slowly growing tumor cells. This is why a treatment with bromopyruvate, for example is very efficient only on rapidly growing cells and barely useful to decrease the growth rate of tumor cells when their normal proliferation is slow. Already in 1979, Reitzer and colleagues published an article entitled “Evidence that glutamine, not sugar, is the major energy source for cultured Hela cells”, which demonstrated that oxidative phosphorylation was used preferentially to produce ATP in cervical carcinoma cells [5]. Griguer et al. also identified several glioma cell lines that were highly dependent on mitochondrial OXPHOS pathway to produce ATP [6]. Furthermore, a subclass of glioma cells which utilize glycolysis preferentially (i.e., glycolytic gliomas) can also switch from aerobic glycolysis to OXPHOS under limiting glucose conditions [7] and [8], as observed in cervical cancer cells, breast carcinoma cells, hepatoma cells and pancreatic cancer cells [9], [10] and [11]. This flexibility shows the interplay between glycolysis and OXPHOS to adapt the mechanisms of energy production to microenvironmental changes as well as differences in tumor energy needs or biosynthetic activity. Herst and Berridge also demonstrated that a variety of human and mouse leukemic and tumor cell lines (HL60, HeLa, 143B, and U937) utilize mitochondrial respiration to support their growth [12]. Recently, the measurement of OXPHOS contribution to the cellular ATP supply revealed that mitochondria generate 79% of the cellular ATP in HeLa cells, and that upon hypoxia this contribution is reduced to 30% [4]. Again, metabolic flexibility is used to survive under hypoxia. All these studies demonstrate that mitochondria are efficient to synthesize ATP in a large variety of cancer cells, as reviewed by Moreno-Sanchez [13]. Despite the observed reduction of the mitochondrial content in tumors [3], [14], [15], [16], [17], [18] and [19], cancer cells maintain a significant level of OXPHOS capacity to rapidly switch from glycolysis to OXPHOS during carcinogenesis. This switch is also observed at the level of glutamine oxidation which can occur through two modes, “OXPHOS-linked” or “anoxic”, allowing to derive energy from glutamine or serine regardless of hypoxia or respiratory chain reduced activity [20].
While glutamine, glycine, alanine, glutamate, and proline are typically oxidized in normal and tumor mitochondria, alternative substrate oxidations may also contribute to ATP supply by OXPHOS. Those include for instance the oxidation of fatty-acids, ketone bodies, short-chain carboxylic acids, propionate, acetate and butyrate (as recently reviewed in [21]).
- Varying degree of mitochondrial utilization during tumorigenesis
In vivo metabolomic analyses suggest the existence of a continuum of bioenergetic remodeling in rat tumors according to tumor size and its rate of growth [22]. Peter Vaupel’s group showed that small tumors were characterized by a low conversion of glucose to lactate whereas the conversion of glutamine to lactate was high. In medium sized tumors the flow of glucose to lactate as well as oxygen utilization was increased whereas glutamine and serine consumption were reduced. At this stage tumor cells started with glutamate and alanine production. Large tumors were characterized by a low oxygen and glucose supply but a high glucose and oxygen utilization rate. The conversion of glucose to glycine, alanine, glutamate, glutamine, and proline reached high values and the amino acids were released [22]. Certainly, in the inner layers constituting solid tumors, substrate and oxygen limitation is frequently observed. Experimental studies tried to reproduce these conditions in vitro and revealed that nutrients and oxygen limitation does not affect OXPHOS and cellular ATP levels in human cervix tumor [23]. Furthermore, the growth of HeLa cells, HepG2 cells and HTB126 (breast cancer) in aglycemia and/or hypoxia even triggered a compensatory increase in OXPHOS capacity, as discussed above. Yet, the impact of hypoxia might be variable depending on cell type and both the extent and the duration of oxygen limitation.
In two models of sequential oncogenesis, the successive activation of specific oncogenes in non-cancer cells evidenced the need for active OXPHOS to pursue tumorigenesis. Funes et al. showed that the transformation of human mesenchymal stem cells increases their dependency on OXPHOS for energy production [24], while Ferbeyre et al. showed that cells expressing oncogenic RAS display an increase in mitochondrial mass, mitochondrial DNA, and mitochondrial production of reactive oxygen species (ROS) prior to the senescent cell cycle arrest [25]. Such observations suggest that waves of gene regulation could suppress and then restore OXPHOS in cancer cells during tumorigenesis [20]. Therefore, the definition of cancer by Hanahan and Weinberg [26] restricted to six hallmarks (1—self-sufficiency in growth signals, 2—insensitivity to growth-inhibitory (antigrowth) signals, 3—evasion of programmed cell death (apoptosis), 4—limitless replicative potential, 5—sustained angiogenesis, and 6—tissue invasion and metastases) should also include metabolic reprogramming, as the seventh hallmark of cancer. This amendment was already proposed by Tennant et al. in 2009 [27]. In 2006, the review Science published a debate on the controversial views of Warburg theory [28], in support of a more realistic description of cancer cell’s variable bioenergetic profile. The pros think that high glycolysis is an obligatory feature of human tumors, while the cons propose that high glycolysis is not exclusive and that tumors can use OXPHOS to derive energy. A unifying theory closer to reality might consider that OXPHOS and glycolysis cooperate to sustain energy needs along tumorigenesis [20]. The concept of oxidative tumors, against Warburg’s proposal, was introduced by Guppy and colleagues, based on the observation that breast cancer cells can generate 80% of their ATP by the mitochondrion [29]. The comparison of different cancer cell lines and excised tumors revealed a variety of cancer cell’s bioenergetic signatures which raised the question of the mechanisms underlying tumor cell metabolic reprogramming, and the relative contribution of oncogenesis and microenvironment in this process. It is now widely accepted that rapidly growing cancer cells within solid tumors suffer from a lack of oxygen and nutrients as tumor grows. In such situation of compromised energy substrate delivery, cancer cell’s metabolic reprogramming is further used to sustain anabolism (Fig. 1), through the deviation of glycolysis, Krebs cycle truncation and OXPHOS redirection toward lipid and protein synthesis, as needed to support uncontrolled tumor growth and survival [30] and [31]. Again, these features are not exclusive to all tumors, as Krebs cycle truncation was only observed in some cancer cells, while other studies indicated that tumor cells can maintain a complete Krebs cycle [13] in parallel with an active citrate efflux. Likewise, generalizations should be avoided to prevent over-interpretations.
Fig. 1. Energy metabolism at the crossroad between catabolism and anabolism.

Energy metabolism at the crossroad between catabolism and anabolism.
The oncogene C-MYC participate to these changes via the stimulation of glutamine utilization through the coordinate expression of genes necessary for cells to engage in glutamine catabolism [30]. According to Newsholme EA and Board M [32] both glycolysis and glutaminolysis not only serve for ATP production, but also provide precious metabolic intermediates such as glucose-6-phosphate, ammonia and aspartate required for the synthesis of purine and pyrimidine nucleotides (Fig. 1). In this manner, the observed apparent excess in the rates of glycolysis and glutaminolysis as compared to the requirement for energy production could be explained by the need for biosynthetic processes. Yet, one should not reduce the shift from glycolysis to OXPHOS utilization to the sole activation of glutaminolysis, as several other energy substrates can be used by tumor mitochondria to generate ATP [21]. The contribution of these different fuels to ATP synthesis remains poorly investigated in human tumors.
- The metabolism of pre-cancer cells and its ongoing modulation by carcinogenesis
At the beginning of cancer, there might have been a cancer stem cell hit by an oncogenic event, such as alterations in mitogen signaling to extracellular growth factor receptors (EGFR), oncogenic activation of these receptors, or oncogenic alterations of downstream targets in the pathways that leads to cell proliferation (RAS–Raf–ERK and PI3K–AKT, both leading to m-TOR activation stimulating cell growth). Alterations of checkpoint genes controlling the cell cycle progression like Rb also participate in cell proliferation (Fig. 2) and this re-entry in the cell cycle implies three major needs to fill in: 1) supplying enough energy to grow and 2) synthesize building blocks de novo and 3) keep vital oxygen and nutrients available. However, the bioenergetic status of the pre-cancer cell could determine in part the evolution of carcinogenesis, as shown on mouse embryonic stem cells. In this study, Schieke et al. showed that mitochondrial energy metabolism modulates both the differentiation and tumor formation capacity of mouse embryonic stem cells [37]. The idea that cancer derives from a single cell, known as the cancer stem cell hypothesis, was introduced by observations performed on leukemia which appeared to be organized as origination from a primitive hematopoietic cell [38]. Nowadays cancer stem cells were discovered for all types of tumors [39], [40], [41] and [42], but little is known of their bioenergetic properties and their metabolic adaptation to the microenvironment. This question is crucial as regards the understanding of what determines the wide variety of cancer cell’s metabolic profile.

Impact of different oncogenes on tumor progression and energy metabolism remodeling.
Fig. 2. Impact of different oncogenes on tumor progression and energy metabolism remodeling.
The analysis of the metabolic changes that occur during the transformation of adult mesenchymal stem cells revealed that these cells did not switch to aerobic glycolysis, but their dependency on OXPHOS was even increased [24]. Hence, mitochondrial energy metabolism could be critical for tumorigenesis, in contrast with Warburg’s hypothesis. As discussed above, the oncogene C-MYC also stimulates OXPHOS [30]. Furthermore, it was recently demonstrated that cells chronically treated with oligomycin repress OXPHOS and produce larger tumors with higher malignancy [19]. Likewise, alteration of OXPHOS by mutations in mtDNA increases tumorigenicity in different types of cancer cells [43], [44] and [45].
Recently, it was proposed that mitochondrial energy metabolism is required to generate reactive oxygen species used for the carcinogenetic process induced by the K-RAS mutation [46]. This could explain the large number of mitochondrial DNA mutations found in several tumors. The analysis of mitochondria in human embryonic cells which derive energy exclusively from anaerobic glycolysis have demonstrated an immature mitochondrial network characterized by few organelles with poorly developed cristae and peri-nuclear distribution [47] and [48]. The generation of human induced pluripotent stem cell by the introduction of different oncogenes as C-MYC and Oct4 reproduced this reduction of mitochondrial OXPHOS capacity[49] and [50]. This indicates again the impact of oncogenes on the control of OXPHOS and might explain the existence of pre-cancer stem cells with different bioenergetic backgrounds, as modeled by variable sequences of oncogene activation. Accordingly, the inhibition of mitochondrial respiratory chain has been recently found associated with enhancement of hESC pluripotency [51].
Based on the experimental evidence discussed above, one can argue that 1) glycolysis is indeed a feature of several tumors and associates with faster growth in high glucose environment, but 2) active OXPHOS is also an important feature of (other) tumors taken at a particular stage of carcinogenesis which might be more advantageous than a “glycolysis-only” type of metabolism in conditions of intermittent shortage in glucose delivery. The metabolic apparatus of cancer cells is not fixed during carcinogenesis and might depend both on the nature of the oncogenes activated and the microenvironment. It was indeed shown that cancer cells with predominant glycolytic metabolism present a higher malignancy when submitted to carcinogenetic induction and analysed under fixed experimental conditions of high glucose [19]. Yet, if one grows these cells in a glucose-deprived medium they shift their metabolism toward predominant OXPHOS, as shown in HeLa cells and other cell types [9]. Therefore, one might conclude that glycolytic cells have a higher propensity to generate aggressive tumors when glucose availability is high. However, these cells can become OXPHOS during tumor progression [24] and [52]. All these observations indicate again the importance of maintaining an active OXPHOS metabolism to permit evolution of both embryogenesis and carcinogenesis, which emphasizes the importance of targeting mitochondria to alter this malignant process.
- Oncogenes and the modulation of energy metabolism
Several oncogenes and associated proteins such as HIF-1α, RAS, C-MYC, SRC, and p53 can influence energy substrate utilization by affecting cellular targets, leading to metabolic changes that favor cancer cell survival, independently of the control of cell proliferation. These oncogenes stimulate the enhancement of aerobic glycolysis, and an increasing number of studies demonstrate that at least some of them can also target directly the OXPHOS machinery, as discussed in this article (Fig. 2). For instance, C-MYC can concurrently drive aerobic glycolysis and/or OXPHOS according to the tumor cell microenvironment, via the expression of glycolytic genes or the activation of mitochondrial oxidation of glutamine [53]. The oncogene RAS has been shown to increase OXPHOS activity in early transformed cells [24], [52] and [54] and p53 modulates OXPHOS capacity via the regulation of cytochrome c oxidase assembly [55]. Hence, carcinogenic p53 deficiency results in a decreased level of COX2 and triggers a shift toward anaerobic metabolism. In this case, lactate synthesis is increased, but cellular ATP levels remain stable [56]. The p53-inducible isoform of phosphofructokinase, termed TP53-induced glycolysis and apoptotic regulator, TIGAR, a predominant phosphatase activity isoform of PFK-2, has also been identified as an important regulator of energy metabolism in tumors [57].
- Tumor specific isoforms (or mutated forms) of energy genes
Tumors are generally characterized by a modification of the glycolytic system where the level of some glycolytic enzymes is increased, some fetal-like isozymes with different kinetic and regulatory properties are produced, and the reverse and back-reactions of the glycolysis are strongly reduced [60]. The GAPDH marker of the glycolytic pathway is also increased in breast, gastric, lung, kidney and colon tumors [18], and the expression of glucose transporter GLUT1 is elevated in most cancer cells. The group of Cuezva J.M. developed the concept of cancer bioenergetic signature and of bioenergetic index to describe the metabolic profile of cancer cells and tumors [18], [61], [64], [65]. This signature describes the changes in the expression level of proteins involved in glycolysis and OXPHOS, while the BEC index gives a ratio of OXPHOS protein content to glycolytic protein content, in good correlation with cancer prognostic[61]. Recently, this group showed that the beta-subunit of the mitochondrial F1F0-ATP synthase is downregulated in a large number of tumors, thus contributing to the Warburg effect [64] and [65]. It was also shown that IF1 expression levels were increased in hepatocellular carcinomas, possibly to prevent the hydrolysis of glytolytic ATP [66]. Numerous changes occur at the level of OXPHOS and mitochondrial biogenesis in human tumors, as we reviewed previously [67]. Yet the actual impact of these changes in OXPHOS protein expression level or catalytic activities remains to be evaluated on the overall fluxes of respiration and ATP synthesis. Indeed, the metabolic control analysis and its extension indicate that it is often required to inhibit activity beyond a threshold of 70–85% to affect the metabolic fluxes [68] and [69]. Another important feature of cancer cells is the higher level of hexokinase II bound to mitochondrial membrane (50% in tumor cells). A study performed on human gliomas (brain) estimated the mitochondrial bound HK fraction (mHK) at 69% of total, as compared to 9% for normal brain [70]. This is consistent with the 5-fold amplification of the type II HK gene observed by Rempel et al. in the rapidly growing rat AS-30D hepatoma cell line, relative to normal hepatocytes [71]. HKII subcellular fractionation in cancer cells was described in several studies [72], [73] and [74]. The group led by Pete Pedersen explained that mHK contributes to (i) the high glycolytic capacity by utilizing mitochondrially regenerated ATP rather than cytosolic ATP (nucleotide channelling) and (ii) the lowering of OXPHOS capacity by limiting Pi and ADP delivery to the organelle [75] and [76].
All these observations are consistent with the increased rate of FDG uptake observed by PET in living tumors which could result from both an increase in glucose transport, and/or an increase in hexokinase activity. However, FDG is not a complete substrate for glycolysis (it is only transformed into FDG-6P by hexokinase before to be eliminated) and cannot be used to evidence a general increase in the glycolytic flux. Moreover, FDG-PET scan also gives false positive and false negative results, indicating that some tumors do not depend on, or do not have, an increased glycolytic capacity. The fast glycolytic system described above is further accommodated in cancer cells by an increase in the lactate dehydrogenase isoform A (LDH-A) expression level. This isoform presents a higher Vmax useful to prevent the inhibition of high glycolysis by its end product (pyruvate) accumulation. Recently, Fantin et al. showed that inhibition of LDH-A in tumors diminishes tumorigenicity and was associated with the stimulation of mitochondrial respiration [79]. The preferential expression of the glycolytic pyruvate kinase isoenzyme M2 (PKM2) in tumor cells, determines whether glucose is converted to lactate for regeneration of energy (active tetrameric form, Warburg effect) or used for the synthesis of cell building blocks (nearly inactive dimeric form) [80]. In the last five years, mutations in proteins of the respiratory system (SDH, FH) and of the TCA cycle (IDH1,2) leading to the accumulation of metabolite and the subsequent activation of HIF-1α were reported in a variety of human tumors [81], [82] and [83].
- Tumor microenvironment modulates cancer cell’s bioenergetics
It was extensively described how hypoxia activates HIF-1α which stimulates in turn the expression of several glycolytic enzymes such as HK2, PFK, PGM, enolase, PK, LDH-A, MCT4 and glucose transporters Glut 1 and Glut 3. It was also shown that HIF-1α can reduce OXPHOS capacity by inhibiting mitochondrial biogenesis [14] and [15], PDH activity [87] and respiratory chain activity [88]. The low efficiency and uneven distribution of the vascular system surrounding solid tumors can lead to abrupt changes in oxygen (intermittent hypoxia) but also energy substrate delivery. .. The removal of glucose, or the inhibition of glycolysis by iodoacetate led to a switch toward glutamine utilization without delay followed by a rapid decrease in acid release. This illustrates once again how tumors and human cancer cell lines can utilize alternative energy pathway such as glutaminolysis to deal with glucose limitation, provided the presence of oxygen. It was also observed that in situations of glucose limitation, tumor derived-cells can adapt to survive by using exclusively an oxidative energy substrate [9] and [10]. This is typically associated with an enhancement of the OXPHOS system. … In summary, cancer cells can survive by using exclusively OXPHOS for ATP production, by altering significantly mitochondrial composition and form to facilitate optimal use of the available substrate (Fig. 3). Yet, glucose is needed to feed the pentose phosphate pathway and generate ribose essential for nucleotide biosynthesis. This raises the question of how cancer cells can survive in the growth medium which do not contain glucose (so-called “galactose medium” with dialysed serum [9]). In the OXPHOS mode, pyruvate, glutamate and aspartate can be derived from glutamine, as glutaminolysis can replenish Krebs cycle metabolic pool and support the synthesis of alanine and NADPH [31]. Glutamine is a major source for oxaloacetate (OAA) essential for citrate synthesis. Moreover, the conversion of glutamine to pyruvate is associated with the reduction of NADP+ to NADPH by malic enzyme. Such NADPH is a required electron donor for reductive steps in lipid synthesis, nucleotide metabolism and GSH reduction. In glioblastoma cells the malic enzyme flux was estimated to be high enough to supply all of the reductive power needed for lipid synthesis [31].
Fig. 3. Interplay between energy metabolism, oncogenes and tumor microenvironment during tumorigenesis (the “metabolic wave model”).

Interplay between energy metabolism, oncogenes and tumor microenvironment
While the mechanisms leading to the enhancement of glycolytic capacity in tumors are well documented, less is known about the parallel OXPHOS changes. Both phenomena could result from a selection of pre-malignant cells forced to survive under hypoxia and limited glucose delivery, followed by an adaptation to intermittent hypoxia, pseudo-hypoxia, substrate limitation and acidic environment. This hypothesis was first proposed by Gatenby and Gillies to explain the high glycolytic phenotype of tumors [91], [92] and [93], but several lines of evidence suggest that it could also be used to explain the mitochondrial modifications observed in cancer cells.
- Aerobic glycolysis and mitochondria cooperate during cancer progression
Metabolic flexibility considers the possibility for a given cell to alternate between glycolysis and OXPHOS in response to physiological needs. Louis Pasteur found that in most mammalian cells the rate of glycolysis decreases significantly in the presence of oxygen (Pasteur effect). Moreover, energy metabolism of normal cell can vary widely according to the tissue of origin, as we showed with the comparison of five rat tissues[94]. During stem cell differentiation, cell proliferation induces a switch from OXPHOS to aerobic glycolysis which might generate ATP more rapidly, as demonstrated in HepG2 cells [95] or in non-cancer cells[96] and [97]. Thus, normal cellular energy metabolism can adapt widely according to the activity of the cell and its surrounding microenvironment (energy substrate availability and diversity). Support for this view came from numerous studies showing that in vitro growth conditions can alter energy metabolism contributing to a dependency on glycolysis for ATP production [98].
Yet, Zu and Guppy analysed numerous studies and showed that aerobic glycolysis is not inherent to cancer but more a consequence of hypoxia[99].
Table 1. Impact of different oncogenes on energy metabolism

Impact of different oncogenes on energy metabolism.
2.1.2.5 Mitohormesis
Yun J, Finkel T
Cell Metab May 2014; 19(5):757–766
http://dx.doi.org/10.1016/j.cmet.2014.01.011
For many years, mitochondria were viewed as semiautonomous organelles, required only for cellular energetics. This view has been largely supplanted by the concept that mitochondria are fully integrated into the cell and that mitochondrial stresses rapidly activate cytosolic signaling pathways that ultimately alter nuclear gene expression. Remarkably, this coordinated response to mild mitochondrial stress appears to leave the cell less susceptible to subsequent perturbations. This response, termed mitohormesis, is being rapidly dissected in many model organisms. A fuller understanding of mitohormesis promises to provide insight into our susceptibility for disease and potentially provide a unifying hypothesis for why we age.
Figure 1. The Basis of Mitohormesis. Any of a number of endogenous or exogenous stresses can perturb mitochondrial function. These perturbations are relayed to the cytosol through, at present, poorly understood mechanisms that may involve mitochondrial ROS as well as other mediators. These cytoplasmic signaling pathways and subsequent nuclear transcriptional changes induce various long-lasting cytoprotective pathways. This augmented stress resistance allows for protection from a wide array of subsequent stresses.
Figure 2. Potential Parallels between the Mitochondrial Unfolded Protein Response and Quorum Sensing in Gram-Positive Bacteria. In the C. elegans UPRmt response, mitochondrial proteins (indicated by blue swirls) are degraded by matrix proteases, and the oligopeptides that are generated are then exported through the ABC transporter family member HAF-1. Once in the cytosol, these peptides can influence the subcellular localization of the transcription factor ATFS-1. Nuclear ATFS-1 is capable of orchestrating a broad transcriptional response to mitochondrial stress. As such, this pathway establishes a method for mitochondrial and nuclear genomes to communicate. In some gram-positive bacteria, intracellularly generated peptides can be similarly exported through an ABC transporter protein. These peptides can be detected in the environment by a membrane-bound histidine kinases (HK) sensor. The activation of the HK sensor leads to phosphorylation of a response regulator (RR) protein that, in turn, can alter gene expression. This program allows communication between dispersed gram-positive bacteria and thus coordinated behavior of widely dispersed bacterial genomes.
Figure 3. The Complexity of Mitochondrial Stresses and Responses. A wide array of extrinsic and intrinsic mitochondrial perturbations can elicit cellular responses. As detailed in the text, genetic or pharmacological disruption of electron transport, incorrect folding of mitochondrial proteins, stalled mitochondrial ribosomes, alterations in signaling pathways, or exposure to toxins all appear to elicit specific cytoprotective programs within the cell. These adaptive responses include increased mitochondrial number (biogenesis), alterations in metabolism, increased antioxidant defenses, and augmented protein chaperone expression. The cumulative effect of these adaptive mechanisms might be an extension of lifespan and a decreased incidence of age-related pathologies.
2.1.2.6 Mitochondrial function and energy metabolism in cancer cells. Past overview and future perspectives
Mayevsky A
Mitochondrion. 2009 Jun; 9(3):165-79
http://dx.doi.org:/10.1016/j.mito.2009.01.009
The involvements of energy metabolism aspects of mitochondrial dysfunction in cancer development, proliferation and possible therapy, have been investigated since Otto Warburg published his hypothesis. The main published material on cancer cell energy metabolism is overviewed and a new unique in vivo experimental approach that may have significant impact in this important field is suggested. The monitoring system provides real time data, reflecting mitochondrial NADH redox state and microcirculation function. This approach of in vivo monitoring of tissue viability could be used to test the efficacy and side effects of new anticancer drugs in animal models. Also, the same technology may enable differentiation between normal and tumor tissues in experimental animals and maybe also in patients.
Energy metabolism in mammalian cells
Fig. 1. Schematic representation of cellular energy metabolism and its relationship to microcirculatory blood flow and hemoglobin oxygenation.
Fig. 2. Schematic representation of the central role of the mitochondrion in the various processes involved in the pathology of cancer cells and tumors. Six issues marked as 1–6 are discussed in details in the text.
In vivo monitoring of tissue energy metabolism in mammalian cells
Fig. 3. Schematic presentation of the six parameters that could be monitored for the evaluation of tissue energy metabolism (see text for details).
Optical spectroscopy of tissue energy metabolism in vivo
Multiparametric monitoring system
Fig. 4. (A) Schematic representation of the Time Sharing Fluorometer Reflectometer (TSFR) combined with the laser Doppler flowmeter (D) for blood flow monitoring. The time sharing system includes a wheel that rotates at a speed of3000 rpm wit height filters: four for the measurements of mitochondrial NADH(366 nm and 450 nm)and four for oxy-hemoglobin measurements (585 nm and 577 nm) as seen in (C). The source of light is a mercury lamp. The probe includes optical fibers for NADH excitation (Ex) and emission (Em), laser Doppler excitation (LD in), laser Doppler emission (LD out) as seen in part E The absorption spectrum of Oxy- and Deoxy- Hemoglobin indicating the two wave length used (C).
Fig. 7. Comparison between mitochondrial metabolic states in vitro and the typical tissue metabolic states in vivo evaluated by NADH redox state, tissue blood flow and hemoglobin oxygenation as could be measured by the suggested monitoring system.
(very important)
2.1.2.7 Metabolic Reprogramming. Cancer Hallmark Even Warburg Did Not Anticipate
Ward PS, Thompson CB.
Cancer Cell 2012; 21(3):297-308
http://dx.doi.org/10.1016/j.ccr.2012.02.014
Cancer metabolism has long been equated with aerobic glycolysis, seen by early biochemists as primitive and inefficient. Despite these early beliefs, the metabolic signatures of cancer cells are not passive responses to damaged mitochondria but result from oncogene-directed metabolic reprogramming required to support anabolic growth. Recent evidence suggests that metabolites themselves can be oncogenic by altering cell signaling and blocking cellular differentiation. No longer can cancer-associated alterations in metabolism be viewed as an indirect response to cell proliferation and survival signals. We contend that altered metabolism has attained the status of a core hallmark of cancer.
The propensity for proliferating cells to secrete a significant fraction of glucose carbon through fermentation was first elucidated in yeast. Otto Warburg extended these observations to mammalian cells, finding that proliferating ascites tumor cells converted the majority of their glucose carbon to lactate, even in oxygen-rich conditions. Warburg hypothesized that this altered metabolism was specific to cancer cells, and that it arose from mitochondrial defects that inhibited their ability to effectively oxidize glucose carbon to CO2. An extension of this hypothesis was that dysfunctional mitochondria caused cancer (Koppenol et al., 2011). Warburg’s seminal finding has been observed in a wide variety of cancers. These observations have been exploited clinically using 18F-deoxyglucose positron emission tomography (FDG-PET). However, in contrast to Warburg’s original hypothesis, damaged mitochondria are not at the root of the aerobic glycolysis exhibited by most tumor cells. Most tumor mitochondria are not defective in their ability to carry out oxidative phosphorylation. Instead, in proliferating cells mitochondrial metabolism is reprogrammed to meet the challenges of macromolecular synthesis. This possibility was never considered by Warburg and his contemporaries.
Advances in cancer metabolism research over the last decade have enhanced our understanding of how aerobic glycolysis and other metabolic alterations observed in cancer cells support the anabolic requirements associated with cell growth and proliferation. It has become clear that anabolic metabolism is under complex regulatory control directed by growth factor signal transduction in non-transformed cells. Yet despite these advances, the repeated refrain from traditional biochemists is that altered metabolism is merely an indirect phenomenon in cancer, a secondary effect that pales in importance to the activation of primary proliferation and survival signals (Hanahan and Weinberg, 2011). Most proto-oncogenes and tumor suppressor genes encode components of signal transduction pathways. Their roles in carcinogenesis have traditionally been attributed to their ability to regulate the cell cycle and sustain proliferative signaling while also helping cells evade growth suppression and/or cell death (Hanahan and Weinberg, 2011). But evidence for an alternative concept, that the primary functions of activated oncogenes and inactivated tumor suppressors are to reprogram cellular metabolism, has continued to build over the past several years. Evidence is also developing for the proposal that proto-oncogenes and tumor suppressors primarily evolved to regulate metabolism.
We begin this review by discussing how proliferative cell metabolism differs from quiescent cell metabolism on the basis of active metabolic reprogramming by oncogenes and tumor suppressors. Much of this reprogramming depends on utilizing mitochondria as functional biosynthetic organelles. We then further develop the idea that altered metabolism is a primary feature selected for during tumorigenesis. Recent advances have demonstrated that altered metabolism in cancer extends beyond adaptations to meet the increased anabolic requirements of a growing and dividing cell. Changes in cancer cell metabolism can also influence cellular differentiation status, and in some cases these changes arise from oncogenic alterations in metabolic enzymes themselves.
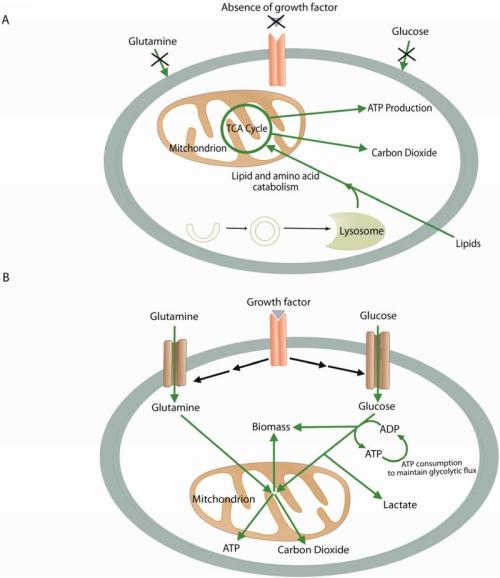
Metabolism in quiescent vs. proliferating cells: both use mitochondria.
(A) In the absence of instructional growth factor signaling, cells in multicellular organisms lack the ability to take up sufficient nutrients to maintain themselves. Neglected cells will undergo autophagy and catabolize amino acids and lipids through the TCA cycle, assuming sufficient oxygen is available. This oxidative metabolism maximizes ATP production. (B) Cells that receive instructional growth factor signaling are directed to increase their uptake of nutrients, most notably glucose and glutamine. The increased nutrient uptake can then support the anabolic requirements of cell growth: mainly lipid, protein, and nucleotide synthesis (biomass). Excess carbon is secreted as lactate. Proliferating cells may also use strategies to decrease their ATP production while increasing their ATP consumption. These strategies maintain the ADP:ATP ratio necessary to maintain glycolytic flux. Green arrows represent metabolic pathways, while black arrows represent signaling.

Metabolism is a direct, not indirect, response to growth factor signaling nihms-360138-f0002
Metabolism is a direct, not indirect, response to growth factor signaling.
(A) The traditional demand-based model of how metabolism is altered in proliferating cells. In response to growth factor signaling, increased transcription and translation consume free energy and decrease the ADP:ATP ratio. This leads to enhanced flux of glucose carbon through glycolysis and the TCA cycle for the purpose of producing more ATP. (B) Supply-based model of how metabolism changes in proliferating cells. Growth factor signaling directly reprograms nutrient uptake and metabolism. Increased nutrient flux through glycolysis and the mitochondria in response to growth factor signaling is used for biomass production. Metabolism also impacts transcription and translation through mechanisms independent of ATP availability.
Alterations in classic oncogenes directly reprogram cell metabolism to increase nutrient uptake and biosynthesis. PI3K/Akt signaling downstream of receptor tyrosine kinase (RTK) activation increases glucose uptake through the transporter GLUT1, and increases flux through glycolysis. Branches of glycolytic metabolism contribute to nucleotide and amino acid synthesis. Akt also activates ATP-citrate lyase (ACL), promoting the conversion of mitochondria-derived citrate to acetyl-CoA for lipid synthesis. Mitochondrial citrate can be synthesized when glucose-derived acetyl-CoA, generated by pyruvate dehydrogenase (PDH), condenses with glutamine-derived oxaloacetate (OAA) via the activity of citrate synthase (CS). mTORC1 promotes protein synthesis and mitochondrial metabolism. Myc increases glutamine uptake and the conversion of glutamine into a mitochondrial carbon source by promoting the expression of the enzyme glutaminase (GLS). Myc also promotes mitochondrial biogenesis. In addition, Myc promotes nucleotide and amino acid synthesis, both through direct transcriptional regulation and through increasing the synthesis of mitochondrial metabolite precursors.
Pyruvate kinase M2 (PKM2) expression in proliferating cells is regulated by signaling and mitochondrial metabolism to facilitate macromolecular synthesis. PKM2 is a less active isoform of the terminal glycolytic enzyme pyruvate kinase. It is also uniquely inhibited downstream of tyrosine kinase signaling. The decreased enzymatic activity of PKM2 in the cytoplasm promotes the accumulation of upstream glycolytic intermediates and their shunting into anabolic pathways. These pathways include the serine synthetic pathway that contributes to nucleotide and amino acid production. When mitochondrial metabolism is excessive, reactive oxygen species (ROS) from the mitochondria can feedback to inhibit PKM2 activity. Acetylation of PKM2, dependent on acetyl-CoA availability, may also promote PKM2 degradation and further contribute to increased flux through anabolic synthesis pathways branching off glycolysis.
IDH1 and IDH2 mutants convert glutamine carbon to the oncometabolite 2-hydroxyglutarate to dysregulate epigenetics and cell differentiation. (A) α-ketoglutarate, produced in part by wild-type isocitrate dehydrogenase (IDH), can enter the nucleus and be used as a substrate for dioxygenase enzymes that modify epigenetic marks. These enzymes include the TET2 DNA hydroxylase enzyme which converts 5-methylcytosine to 5-hydroxymethylcytosine, typically at CpG dinucleotides. 5-hydroxymethylcytosine may be an intermediate in either active or passive DNA demethylation. α-ketoglutarate is also a substrate for JmjC domain histone demethylase enzymes that demethylate lysine residues on histone tails. (B) The common feature of cancer-associated mutations in cytosolic IDH1 and mitochondrial IDH2 is the acquisition of a neomorphic enzymatic activity. This activity converts glutamine-derived α-ketoglutarate to the oncometabolite 2HG. 2HG can competitively inhibit α-ketoglutarate-dependent enzymes like TET2 and the JmjC histone demethylases, thereby impairing normal epigenetic regulation. This results in altered histone methylation marks, in some cases DNA hypermethylation at CpG islands, and dysregulated cellular differentiation.
Hypoxia and HIF-1 activation promote an alternative pathway for citrate synthesis through reductive metabolism of glutamine. (A) In proliferating cells under normoxic conditions, citrate is synthesized from both glucose and glutamine. Glucose carbon provides acetyl-CoA through the activity of PDH. Glutamine carbon provides oxaloacetate through oxidative mitochondrial metabolism dependent on NAD+. Glucose-derived acetyl-CoA and glutamine-derived oxaloacetate condense to form citrate via the activity of citrate synthase (CS). Citrate can be exported to the cytosol for lipid synthesis. (B) In cells proliferating in hypoxia and/or with HIF-1 activation, glucose is diverted away from mitochondrial acetyl-CoA and citrate production. Citrate can be maintained through an alternative pathway of reductive carboxylation, which we propose to rely on reverse flux of glutamine-derived α-ketoglutarate through IDH2. This reverse flux in the mitochondria would promote electron export from the mitochondria when the activity of the electron transport chain is inhibited because of the lack of oxygen as an electron acceptor. Mitochondrial reverse flux can be accomplished by NADH conversion to NADPH by mitochondrial transhydrogenase and the resulting NADPH use in α-ketoglutarate carboxylation. When citrate/isocitrate is exported to the cytosol, some may be metabolized in the oxidative direction by IDH1 and contribute to a shuttle that produces cytosolic NADPH.
A major paradox remaining with PKM2 is that cells expressing PKM2 produce more glucose-derived pyruvate than PKM1-expressing cells, despite having a form of the pyruvate kinase enzyme that is less active and more sensitive to inhibition. One way to get around the PKM2 bottleneck and maintain/enhance pyruvate production may be through an proposed alternative glycolytic pathway, involving an enzymatic activity not yet purified, that dephosphorylates PEP to pyruvate without the generation of ATP (Vander Heiden et al., 2010). Another answer to this paradox may emanate from the serine synthetic pathway. The decreased enzymatic activity of PKM2 can promote the accumulation of the 3-phosphoglycerate glycolytic intermediate that serves as the entry point for the serine synthetic pathway branch off glycolysis. The little studied enzyme serine dehydratase can then directly convert serine to pyruvate. A third explanation may lie in the oscillatory activity of PKM2 from the inactive dimer to active tetramer form. Regulatory inputs into PKM2 like tyrosine phosphorylation and ROS destabilize the tetrameric form of PKM2 (Anastasiou et al., 2011; Christofk et al., 2008b; Hitosugi et al., 2009), but other inputs present in glycolytic cancer cells like fructose-1,6-bisphosphate and serine can continually allosterically activate and/or promote reformation of the PKM2 tetramer (Ashizawa et al., 1991; Eigenbrodt et al., 1983). Thus, PKM2 may be continually switching from inactive to active forms in cells, resulting in an apparent upregulation of flux through anabolic glycolytic branching pathways while also maintaining reasonable net flux of glucose carbon through PEP to pyruvate. With such an oscillatory system, small changes in the levels of any of the above-mentioned PKM2 regulatory inputs can cause exquisite, rapid, adjustments to glycolytic flux. This would be predicted to be advantageous for proliferating cells in the setting of variable extracellular nutrient availability. The capability for oscillatory regulation of PKM2 could also provide an explanation for why tumor cells do not select for altered glycolytic metabolism upstream of PKM2 through deletions and/or loss of function mutations of other glycolytic enzymes.
IDH1 mutations at R132 are not simply loss-of-function for isocitrate and α-ketoglutarate interconversion, but also acquire a novel reductive activity to convert α-ketoglutarate to 2-hydroxyglutarate (2HG), a rare metabolite found at only trace amounts in mammalian cells under normal conditions (Dang et al., 2009). However, it still remained unclear if 2HG was truly a pathogenic “oncometabolite” resulting from IDH1 mutation, or if it was just the byproduct of a loss of function mutation. Whether 2HG production or the loss of IDH1 normal function played a more important role in tumorigenesis remained uncertain.
A potential answer to whether 2HG production was relevant to tumorigenesis arrived with the study of mutations in IDH2, the mitochondrial homolog of IDH1. Up to this point a small fraction of gliomas lacking IDH1 mutations were known to harbor mutations at IDH2 R172, the analogous residue to IDH1 R132 (Yan et al., 2009). However, given the rarity of these IDH2 mutations, they had not been characterized for 2HG production. The discovery of IDH2 R172 mutations in AML as well as glioma samples prompted the study of whether these mutations also conferred the reductive enzymatic activity to produce 2HG. Enzymatic assays and measurement of 2HG levels in primary AML samples confirmed that these IDH2 R172 mutations result in 2HG elevation (Gross et al., 2010; Ward et al., 2010).
It was then investigated if the measurement of 2HG levels in primary tumor samples with unknown IDH mutation status could serve as a metabolite screening test for both cytosolic IDH1 and mitochondrial IDH2 mutations. AML samples with low to undetectable 2HG were subsequently sequenced and determined to be IDH1 and IDH2 wild-type, and several samples with elevated 2HG were found to have neomorphic mutations at either IDH1 R132 or IDH2 R172 (Gross et al., 2010). However, some 2HG-elevated AML samples lacked IDH1 R132 or IDH2 R172 mutations. When more comprehensive sequencing of IDH1 and IDH2 was performed, it was found that the common feature of this remaining subset of 2HG-elevated AMLs was another mutation in IDH2, occurring at R140 (Ward et al., 2010). This discovery provided additional evidence that 2HG production was the primary feature being selected for in tumors.
In addition to intensifying efforts to find the cellular targets of 2HG, the discovery of the 2HG-producing IDH1 and IDH2 mutations suggested that 2HG measurement might have clinical utility in diagnosis and disease monitoring. While much work is still needed in this area, serum 2HG levels have successfully correlated with IDH1 R132 mutations in AML, and recent data have suggested that 1H magnetic resonance spectroscopy can be applied for 2HG detection in vivo for glioma (Andronesi et al., 2012; Choi et al., 2012; Gross et al., 2010; Pope et al., 2012). These methods may have advantages over relying on invasive solid tumor biopsies or isolating leukemic blast cells to obtain material for sequencing of IDH1 and IDH2. Screening tumors and body fluids by 2HG status also has potentially increased applicability given the recent report that additional IDH mutations can produce 2HG (Ward et al., 2011). These additional alleles may account for the recently described subset of 2HG-elevated chondrosarcoma samples that lacked the most common IDH1 or IDH2 mutations but were not examined for other IDH alterations (Amary et al., 2011). Metabolite screening approaches can also distinguish neomorphic IDH mutations from SNPs and sequencing artifacts with no effect on IDH enzyme activity, as well as from an apparently rare subset of loss-of-function, non 2HG-producing IDH mutations that may play a secondary tumorigenic role in altering cellular redox (Ward et al., 2011).
Will we find other novel oncometabolites like 2HG? We should consider basing the search for new oncometabolites on those metabolites already known to cause disease in pediatric inborn errors of metabolism (IEMs). 2HG exemplifies how advances in research on IEMs can inform research on cancer metabolism, and vice versa. Methods developed by those studying 2HG aciduria were used to demonstrate that R(-)-2HG (also known as D-2HG) is the exclusive 2HG stereoisomer produced by IDH1 and IDH2 mutants (Dang et al., 2009; Ward et al., 2010). Likewise, following the discovery of 2HG-producing IDH2 R140 mutations in leukemia, researchers looked for and successfully found germline IDH2 R140 mutations in D-2HG aciduria. IDH2 R140 mutations now account for nearly half of all cases of this devastating disease (Kranendijk et al., 2010). While interest has surrounded 2HG due to its apparent novelty as a metabolite not found in normal non-diseased cells, there are situations where 2HG appears in the absence of metabolic enzyme mutations. For example, in human cells proliferating in hypoxia, α-ketoglutarate can accumulate and be metabolized through an enhanced reductive activity of wild-type IDH2 in the mitochondria, leading to 2HG accumulation in the absence of IDH mutation (Wise et al., 2011). The ability of 2HG to alter epigenetics may reflect its evolutionary ancient status as a signal for elevated glutamine/glutamate metabolism and/or oxygen deficiency.
With this broadened view of what constitutes an oncometabolite, one could argue that the discoveries of two other oncometabolites, succinate and fumarate, preceded that of 2HG. Loss of function mutations in the TCA cycle enzymes succinate dehydrogenase (SDH) and fumarate hydratase (FH) have been known for several years to occur in pheochromocytoma, paraganglioma, leiomoyoma, and renal carcinoma. It was initially hypothesized that these mutations contribute to cancer through mitochondrial damage producing elevated ROS (Eng et al., 2003). However, potential tumorigenic effects were soon linked to the elevated levels of succinate and fumarate arising from loss of SDH and FH function, respectively. Succinate was initially found to impair PHD2, the α-ketoglutarate-dependent enzyme regulating HIF stability, through product inhibition (Selak et al., 2005). Subsequent work confirmed that fumarate could inhibit PHD2 (Isaacs et al., 2005), and that succinate could also inhibit the related enzyme PHD3 (Lee et al., 2005). These observations linked the elevated HIF levels observed in SDH and FH deficient tumors to the activity of the succinate and fumarate metabolites. Recent work has suggested that fumarate may have other important roles that predominate in FH deficiency. For example, fumarate can modify cysteine residues to inhibit a negative regulator of the Nrf2 transcription factor. This post-translational modification leads to the upregulation of antioxidant response genes (Adam et al., 2011; Ooi et al., 2011).
There are still many unanswered questions regarding the biology of SDH and FH deficient tumors. In light of the emerging epigenetic effects of 2HG, it is intriguing that succinate has been shown to alter histone demethylase activity in yeast (Smith et al., 2007). Perhaps elevated succinate and fumarate resulting from SDH and FH mutations can promote tumorigenesis in part through epigenetic modulation.
Despite rapid technological advances in studying cell metabolism, we remain unable to reliably distinguish cytosolic metabolites from those in the mitochondria and other compartments. Current fractionation methods often lead to metabolite leakage. Even within one subcellular compartment, there may be distinct pools of metabolites resulting from channeling between metabolic enzymes. A related challenge lies in the quantitative measurement of metabolic flux; i.e., measuring the movement of carbon, nitrogen, and other atoms through metabolic pathways rather than simply measuring the steady-state levels of individual metabolites. While critical fluxes have been quantified in cultured cancer cells and methods for these analyses continue to improve (DeBerardinis et al., 2007; Mancuso et al., 2004; Yuan et al., 2008), many obstacles remain such as cellular compartmentalization and the reliance of most cell culture on complex, incompletely defined media.
Over the past decade, the study of metabolism has returned to its rightful place at the forefront of cancer research. Although Warburg was wrong about mitochondria, he was prescient in his focus on metabolism. Data now support the concepts that altered metabolism results from active reprogramming by altered oncogenes and tumor suppressors, and that metabolic adaptations can be clonally selected during tumorigenesis. Altered metabolism should now be considered a core hallmark of cancer. There is much work to be done.
2.1.2.8 A Role for the Mitochondrial Pyruvate Carrier as a Repressor of the Warburg Effect and Colon Cancer Cell Growth
Schell JC, Olson KA, …, Xie J, Egnatchik RA, Earl EG, DeBerardinis RJ, Rutter J.
Mol Cell. 2014 Nov 6; 56(3):400-13
http://dx.doi.org:/10.1016/j.molcel.2014.09.026
Cancer cells are typically subject to profound metabolic alterations, including the Warburg effect wherein cancer cells oxidize a decreased fraction of the pyruvate generated from glycolysis. We show herein that the mitochondrial pyruvate carrier (MPC), composed of the products of the MPC1 and MPC2 genes, modulates fractional pyruvate oxidation. MPC1 is deleted or underexpressed in multiple cancers and correlates with poor prognosis. Cancer cells re-expressing MPC1 and MPC2 display increased mitochondrial pyruvate oxidation, with no changes in cell growth in adherent culture. MPC re-expression exerted profound effects in anchorage-independent growth conditions, however, including impaired colony formation in soft agar, spheroid formation, and xenograft growth. We also observed a decrease in markers of stemness and traced the growth effects of MPC expression to the stem cell compartment. We propose that reduced MPC activity is an important aspect of cancer metabolism, perhaps through altering the maintenance and fate of stem cells.
Figure 2. Re-Expressed MPC1 and MPC2 Form a Mitochondrial Complex (A and B) (A) Western blot and (B) qRT-PCR analysis of the indicated colon cancer cell lines with retroviral expression of MPC1 (or MPC1-R97W) and/or MPC2. (C) Western blots of human heart tissue, hematologic cancer cells, and colon cancer cell lines with and without MPC1 and MPC2 re-expression. (D) Fluorescence microscopy of MPC1-GFP and MPC2-GFP overlaid with Mitotracker Red in HCT15 cells. Scale bar: 10 mm. (E) Blue-native PAGE analysis of mitochondria from control and MPC1/2-expressing cells. (F) Western blots of metabolic and mitochondrial proteins across four colon cancer cell lines with or without MPC1/2 expression
Figure 3. MPC Re-Expression Alters Mitochondrial Pyruvate Metabolism (A) OCR at baseline and maximal respiration in HCT15 (n = 7) and HT29 (n = 13) with pyruvate as the sole carbon source (mean ± SEM). (B and C) Schematic and citrate mass isotopomer quantification in cells cultured with D-[U-13C]glucose and unlabeled glutamine for 6 hr (mean ± SD, n = 2). (D) Glucose uptake and lactate secretion normalized to protein concentration (mean ± SD, n = 3). (E–G) (E) Western blots of PDH, phospho-PDH, and PDK1; (F) PDH activity assay and (G) CS activity assay with or without MPC1 and MPC2 expression (mean ± SD, n = 4). (H and I) Effects of MPC1/2 re-expression on mitochondrial membrane potential and ROS production (mean ± SD, n = 3). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Figure 4. MPC Re-Expression Alters Growth under Low-Attachment Conditions (A) Cell number of control and MPC1/2 re-expressing cell lines in adherent culture (mean ± SD, n = 7). (B) Cell viability determined by trypan blue exclusion and Annexin V/PI staining (mean ± SD, n = 3). (C–F) (C) EdU incorporation of MPC re-expressing cell lines at 3 hr post EdU pulse. Growth in 3D culture evaluated by (D) soft agar colony formation (mean ± SD, n = 12, see also Table S1) and by ([E] and [F]) spheroid formation ± MPC inhibitor UK5099 (mean ± SEM, n = 12). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Figure 7. MPC Re-Expression Alters the Cancer Initiating Cell Population (A) Western blot quantification of ALDHA and Lin28A from control or MPC re-expressing HT29 xenografts (mean ± SEM, n = 10). (B and C) Percentage of ALDHhi (n = 3) and CD44hi (n = 5) cells as determined by flow cytometry (mean ± SEM). (D) Western blot analysis of stem cell markers in control and MPC re-expressing cell lines. (E) Relative MPC1 and MPC2 mRNA levels in ALDH sorted HCT15 cells (n = 4,mean ± SEM). 2D growth of (F) whole-population HCT15 cells and (G) ALDH sorted cells. Area determined by ImageJ after crystal violet staining (mean ± SD, n = 6). (H and I) (H) Adherent and (I) spheroid growth of main population (MP) versus side population (SP) HCT15 cells. (mean ± SD, n = 6). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001
Our demonstration that the MPC is lost or underexpressed in many cancers might provide clarifying context for earlier attempts to exploit metabolic regulation for cancer therapeutics. The PDH kinase inhibitor dichloroacetate, which impairs PDH phosphorylation and increases pyruvate oxidation, has been explored extensively as a cancer therapy (Bonnet et al., 2007; Olszewski et al., 2010). It has met with mixed results, however, and has typically failed to dramatically decrease tumor burden as a monotherapy (Garon et al., 2014;
Sanchez-Arago et al., 2010; Shahrzadetal.,2010). Is one possible reason for these failures that the MPC has been lost or inactivated, thereby limiting the metabolic effects of PDH activity? The inclusion of the MPC adds additional complexity to targeting cancer metabolism for therapy but has the potential to explain why treatments may be more effective in some studies than in others (Fulda et al., 2010; Hamanaka and Chandel, 2012; Tennant et al., 2010; Vander Heiden, 2011). The redundant measures to limit pyruvate oxidation make it easy to understand why expression of the MPC leads to relatively modest metabolic changes in cells grown in adherent culture conditions. While subtle, we observed a number of changes in metabolic parameters, all of which are consistent with enhanced mitochondrial pyruvate entry and oxidation. There are at least two possible explanations for the discrepancy that we observed between the impact on adherent and nonadherent cell proliferation. One hypothesis is that the stress of nutrient deprivation and detachment combines with these subtle metabolic effects to impair survival and proliferation.
2.1.2.9 ECM1 promotes the Warburg effect through EGF-mediated activation of PKM2
Lee KM, Nam K, Oh S, Lim J, Lee T, Shin I.
Cell Signal. 2015 Feb; 27(2):228-35
http://dx.doi.org:/10.1016/j.cellsig.2014.11.004
The Warburg effect is an oncogenic metabolic switch that allows cancer cells to take up more glucose than normal cells and favors anaerobic glycolysis. Extracellular matrix protein 1 (ECM1) is a secreted glycoprotein that is overexpressed in various types of carcinoma. Using two-dimensional digest-liquid chromatography-mass spectrometry (LC-MS)/MS, we showed that the expression of proteins associated with the Warburg effect was upregulated in trastuzumab-resistant BT-474 cells that overexpressed ECM1 compared to control cells. We further demonstrated that ECM1 induced the expression of genes that promote the Warburg effect, such as glucose transporter 1 (GLUT1), lactate dehydrogenase A (LDHA), and hypoxia-inducible factor 1 α (HIF-1α). The phosphorylation status of pyruvate kinase M2 (PKM-2) at Ser37, which is responsible for the expression of genes that promote the Warburg effect, was affected by the modulation of ECM1 expression. Moreover, EGF-dependent ERK activation that was regulated by ECM1 induced not only PKM2 phosphorylation but also gene expression of GLUT1 and LDHA. These findings provide evidence that ECM1 plays an important role in promoting the Warburg effect mediated by PKM2.
Fig. 1.ECM1 induces a metabolic shift toward promoting Warburg effect. (A) The levels of glucose uptake were examined with a cell-based assay. (B) Levels of lactate production were measured using a lactate assay kit. (C) Cellular ATP content was determined with a Cell Titer-Glo luminescent cell viability assay. Error bars represent mean ± SD of triplicate experiments (*p b 0.05, ***p b 0.0005).
Fig.2. ECM1 up-regulates expression of gene sassociated with the Warburg effect. (A) Cell lysates were analyzed by western blotting using antibodies specific for ECM1, LDHA, GLUT1,and actin (as a loading control). The intensities of the bands were quantified using 1D Scan software and plotted. (BandC) mRNA levels of each gene were determined by real-time PCR using specific primers. (D) HIF-1α-dependent transcriptional activities were examined using a hypoxia response element (HRE) reporter indual luciferase assays. Error bars represent mean ± SD of triplicate experiments (*p b 0.05, **p b 0.005, ***p b 0.0005).
Fig.3. ECM1-dependent upregulation of gene expression is not mediated byEgr-1.
Fig.4. ECM1 activates PKM2 via EGF-mediated ERK activation
Fig. 5. TheWarburg effect is attenuated by silencing of PKM2 in breast cancer cells
Recently, a non-glycolytic function of PKM2 was reported. Phosphorylated PKM2 at Ser37 is translocated into the nucleus after EGFR and ERK activation and regulates the expression of cyclin D1, c-Myc, LDHA, and GLUT1[19,37]. Here, we showed that ECM1 regulates the phosphorylation level and translocation of PKM2 via the EGFR/ ERK pathway. As we previously showed that ECM1 enhances the EGF response and increases EGFR expression through MUC1-dependent stabilization [17], it seemed likely that activation of the EGFR/ERK pathway by ECM1 is linked to PKM2 phosphorylation. Indeed, we show here that ECM1 regulates the phosphorylation of PKM2 at Ser37 and enhances the Warburg effect through the EGFR/ERK pathway. HIF-1α is known to be responsible for alterations in cancer cell metabolism [38] and our current studies showed that the expression level of HIF-1α is up-regulated by ECM1 (Fig. 2C and D). To determine the mechanism by which ECM1 upregulated HIF-1α expression, we focused on the induction of Egr-1 by EGFR/ERK signaling [39]. However, although Egr-1 expression was regulated by ECM1 we failed to find evidence that Egr-1 affected the expression of genes involved in the Warburg effect (Fig. 3C). Moreover, ERK-dependent PKM2 activation did not regulate HIF-1α expression in BT-474 cells (Fig. 4D and5B). These results suggested that the upregulation of HIF-1α by ECM1 is not mediated by the EGFR/ERK pathway.
Conclusions
In the current study we showed that ECM1 altered metabolic phenotypes of breast cancer cells toward promoting the Warburg effect.
Phosphorylation and nuclear translocation of PKM2 were induced by ECM1 through the EGFR/ERK pathway. Moreover, phosphorylated PKM2 increased the expression of metabolic genes such as LDHA and GLUT1, and promoted glucose uptake and lactate production. These findings provide a new perspective on the distinct functions of ECM1 in cancer cell metabolism. Supplementary data to this article can be found online at
http://dx.doi.org/10.1016/j.cellsig.2014.11.004
References
[1] R.A. Cairns, I.S. Harris, T.W. Mak, Cancer 11 (2011) 85–95.
[2] O. Warburg, Science 123 (1956) 309–314.
[3] G.L. Semenza, D.Artemov, A.Bedi, …, J. Simons, P. Taghavi, H. Zhong, Novartis Found. Symp. 240 (2001) 251–260 (discussion 260–254).
[4] N.C. Denko, Cancer 8 (2008) 705–713.
[5] C. Chen, N. Pore, A. Behrooz, F. Ismail-Beigi, A. Maity, J. Biol. Chem. 276 (2001) 9519–9525.
[6] J.Lum, T.Bui, M.Gruber, J.D.Gordan, R.J.DeBerardinis,.. ,C.B. Thompson, Genes Dev. 21 (2007) 1037–1049.
[7] J.T. Chi, Z. Wang, D.S. Nuyten, E.H. Rodriguez, .., P.O. Brown, PLoS Med.
3 (2006) e47.
[8] G.L. Semenza, Cancer 3 (2003) 721–732.
2.1.2.10 Glutamine Oxidation Maintains the TCA Cycle and Cell Survival during impaired Mitochondrial Pyruvate Transport
Chendong Yang, B Ko, CT. Hensley,…, J Rutter, ME. Merritt, RJ. DeBerardinis
Molec Cell 6 Nov 2014; 56(3):414–424
http://dx.doi.org/10.1016/j.molcel.2014.09.025
Highlights
- Mitochondria produce acetyl-CoA from glutamine during MPC inhibition
•Alanine synthesis is suppressed during MPC inhibition
•MPC inhibition activates GDH to supply pools of TCA cycle intermediates
•GDH supports cell survival during periods of MPC inhibition
Summary
Alternative modes of metabolism enable cells to resist metabolic stress. Inhibiting these compensatory pathways may produce synthetic lethality. We previously demonstrated that glucose deprivation stimulated a pathway in which acetyl-CoA was formed from glutamine downstream of glutamate dehydrogenase (GDH). Here we show that import of pyruvate into the mitochondria suppresses GDH and glutamine-dependent acetyl-CoA formation. Inhibiting the mitochondrial pyruvate carrier (MPC) activates GDH and reroutes glutamine metabolism to generate both oxaloacetate and acetyl-CoA, enabling persistent tricarboxylic acid (TCA) cycle function. Pharmacological blockade of GDH elicited largely cytostatic effects in culture, but these effects became cytotoxic when combined with MPC inhibition. Concomitant administration of MPC and GDH inhibitors significantly impaired tumor growth compared to either inhibitor used as a single agent. Together, the data define a mechanism to induce glutaminolysis and uncover a survival pathway engaged during compromised supply of pyruvate to the mitochondria.

Yang et al, Graphical Abstract
Graphical abstract
Figure 1. Pyruvate Depletion Redirects Glutamine Metabolism to Produce AcetylCoA and Citrate (A) Top: Anaplerosis supplied by [U-13C]glutamine. Glutamine supplies OAA via a-KG, while acetylCoA is predominantly supplied by other nutrients, particularly glucose. Bottom: Glutamine is converted to acetyl-CoA in the absence of glucosederived pyruvate. Red circles represent carbons arising from [U-13C]glutamine, and gray circles are unlabeled. Reductive carboxylation is indicated by the green dashed line. (B) Fraction of succinate, fumarate, malate, and aspartate containing four 13C carbons after culture of SFxL cells for 6 hr with [U-13C]glutamine in the presence or absence of 10 mM unlabeled glucose (Glc). (C) Mass isotopologues of citrate after culture of SFxL cells for 6 hr with [U-13C]glutamine and 10 mM unlabeled glucose, no glucose, or no glucose plus 6 mM unlabeled pyruvate (Pyr). (D) Citrate m+5 and m+6 after culture of HeLa or Huh-7 cells for 6 hr with [U-13C]glutamine and 10 mM unlabeled glucose, no glucose, or no glucose plus 6 mM unlabeled pyruvate. Data are the average and SD of three independent cultures. *p < 0.05; **p < 0.01; ***p < 0.001.
Figure 2. Isolated Mitochondria Convert Glutamine to Citrate (A) Western blot of whole-cell lysates (Cell) and preparations of isolated mitochondria (Mito) or cytosol from SFxL cells. (B) Oxygen consumption in a representative mitochondrial sample. Rates before and after addition of ADP/GDP are indicated. (C) Mass isotopologues of citrate produced by mitochondria cultured for 30 min with [U-13C] glutamine and with or without pyruvate.
Figure 3. Blockade of Mitochondrial Pyruvate Transport Activates Glutamine-Dependent Citrate Formation (A) Dose-dependent effects of UK5099 on citrate labeling from [U-13C]glucose and [U-13C]glutamine in SFxL cells. (B) Time course of citrate labeling from [U-13C] glutamine with or without 200 mM UK5099. (C) Abundance of total citrate and citrate m+6 in cells cultured in [U-13C]glutamine with or without 200 mM UK5099. (D) Mass isotopologues of citrate in cells cultured for 6 hr in [U-13C]glutamine with or without 10 mM CHC or 200 mM UK5099. (E) Effect of silencing ME2 on citrate m+6 after 6 hr of culture in [U-13C]glutamine. Relative abundances of citrate isotopologues were determined by normalizing total citrate abundance measured by mass spectrometry against cellular protein for each sample then multiplying by the fractional abundance of each isotopologue. (F) Effect of silencing MPC1 or MPC2 on formation of citrate m+6 after 6 hr of culture in [U-13C]glutamine. (G) Citrate isotopologues in primary human fibroblasts of varying MPC1 genotypes after culture in [U-13C]glutamine. Data are the average and SD of three independent cultures. *p < 0.05; **p < 0.01; ***p < 0.001. See also Figure S1.
Figure 4. Kinetic Analysis of the Metabolic Effects of Blocking Mitochondrial Pyruvate Transport (A) Summation of 13C spectra acquired over 2 min of exposure of SFxL cells to hyperpolarized [1-13C] pyruvate. Resonances are indicated for [1-13C] pyruvate (Pyr1), the hydrate of [1-13C]pyruvate (Pyr1-Hydr), [1-13C]lactate (Lac1), [1-13C]alanine (Ala1), and H[13C]O3 (Bicarbonate). (B) Time evolution of appearance of Lac1, Ala1, and bicarbonate in control and UK5099-treated cells. (C) Relative 13C NMR signals for Lac1, Ala1, and bicarbonate. Each signal is summed over the entire acquisition and expressed as a fraction of total 13C signal. (D) Quantity of intracellular and secreted alanine in control and UK5099-treated cells. Data are the average and SD of three independent cultures. *p < 0.05; ***p < 0.001. See also Figure S2.
Figure 5. Inhibiting Mitochondrial Pyruvate Transport Enhances the Contribution of Glutamine to Fatty Acid Synthesis (A) Mass isotopologues of palmitate extracted from cells cultured with [U-13C] glucose or [U-13C]glutamine, with or without 200 mM UK5099. For simplicity, only even-labeled isotopologues (m+2, m+4, etc.) are shown. (B) Fraction of lipogenic acetyl-CoA derived from glucose or glutamine with or without 200 mM UK5099. Data are the average and SD of three independent cultures. ***p < 0.001. See also Figure S3.
Figure 6. Blockade of Mitochondrial Pyruvate Transport Induces GDH (A) Two routes by which glutamate can be converted to AKG. Blue and green symbols are the amide (g) and amino (a) nitrogens of glutamine, respectively. (B) Utilization and secretion of glutamine (Gln), glutamate (Glu), and ammonia (NH4+) by SFxL cells with and without 200 mM UK5099. (C) Secretion of 15N-alanine and 15NH4+ derived from [a-15N]glutamine in SFxL cells expressing a control shRNA (shCtrl) or either of two shRNAs directed against GLUD1 (shGLUD1-A and shGLUD1-B). (D) Left: Phosphorylation of AMPK (T172) and acetyl-CoA carboxylase (ACC, S79) during treatment with 200 mM UK5099. Right: Steady-state levels of ATP 24 hr after addition of vehicle or 200 mM UK5099. (E) Fractional contribution of the m+6 isotopologue to total citrate in shCtrl, shGLUD1-A, and shGLUD1-B SFxL cells cultured in [U-13C]glutamine with or without 200 mM UK5099. Data are the average and SD of three independent cultures. *p < 0.05; **p < 0.01; ***p < 0.001. See also Figure S4.
Figure 7. GDH Sustains Growth and Viability during Suppression of Mitochondrial Pyruvate Transport (A) Relative growth inhibition of shCtrl, shGLUD1A, and shGLUD1-B SFxL cells treated with 50 mM UK5099 for 3 days. (B) Relative growth inhibition of SFxL cells treated with combinations of 50 mM of the GDH inhibitor EGCG, 10 mM of the GLS inhibitor BPTES, and 200 mM UK5099 for 3 days. (C) Relative cell death assessed by trypan blue staining in SFxL cells treated as in (B). (D) Relative cell death assessed by trypan blue staining in SF188 cells treated as in (B) for 2 days. (E) (Left) Growth of A549-derived subcutaneous xenografts treated with vehicle (saline), EGCG, CHC, or EGCG plus CHC (n = 4 for each group). Data are the average and SEM. Right: Lactate abundance in extracts of each tumor harvested at the end of the experiment. Data in (A)–(D) are the average and SD of three independent cultures. NS, not significant; *p < 0.05; **p < 0.01; ***p < 0.001. See also Figure S5.
Mitochondrial metabolism complements glycolysis as a source of energy and biosynthetic precursors. Precursors for lipids, proteins, and nucleic acids are derived from the TCA cycle. Maintaining pools of these intermediates is essential, even under circumstances of nutrient limitation or impaired supply of glucose-derived pyruvate to the mitochondria. Glutamine’s ability to produce both acetyl-CoA and OAA allows it to support TCA cycle activity as a sole carbon source and imposes a greater cellular dependence on glutamine metabolism when MPC function or pyruvate supply is impaired. Other anaplerotic amino acids could also supply both OAA and acetyl-CoA, providing flexible support for the TCA cycle when glucose is limiting. Although fatty acids are an important fuel in some cancer cells (Caro et al., 2012), and fatty acid oxidation is induced upon MPC inhibition, this pathway produces acetyl-CoA but not OAA. Thus, fatty acids would need to be oxidized along with an anaplerotic nutrient in order to enable the cycle to function as a biosynthetic hub. Notably, enforced MPC overexpression also impairs growth of some tumors (Schell et al., 2014), suggesting that maximal growth may require MPC activity to be maintained within a narrow window. After decades of research on mitochondrial pyruvate transport, molecular components of the MPC were recently reported (Halestrap, 2012; Schell and Rutter, 2013). MPC1 and MPC2 form a heterocomplex in the inner mitochondrial membrane, and loss of either component impairs pyruvate import, leading to citrate depletion (Bricker et al., 2012; Herzig et al., 2012). Mammalian cells lacking functional MPC1 display normal glutamine-supported respiration (Bricker et al., 2012), consistent with our observation that glutamine supplies the TCA cycle in absence of pyruvate import. We also observed that isolated mitochondria produce fully labeled citrate from glutamine, indicating that this pathway operates as a self-contained mechanism to maintain TCA cycle function. Recently, two well-known classes of drugs have unexpectedly been shown to inhibit MPC. First, thiazolidinediones, commonly used as insulin sensitizers, impair MPC function in myoblasts (Divakaruni et al.,2013). Second, the phosphodiesterase inhibitor Zaprinast inhibits MPC in the retina and brain (Du et al., 2013b). Zaprinast also induced accumulation of aspartate, suggesting that depletion of acetyl-CoA impaired the ability of a new turn of the TCA cycle to be initiated from OAA; as a consequence, OAA was transaminated to aspartate. We noted a similar phenomenon in cancer cells, suggesting that UK5099 elicits a state in which acetyl-CoA supply is insufficient to avoid OAA accumulation. Unlike UK5099, Zaprinast did not induce glutamine-dependent acetyl-CoA formation. This may be related to the reliance of isolated retinas on glucose rather than glutamine to supply TCA cycle intermediates or the exquisite system used by retinas to protect glutamate from oxidation (Du et al., 2013a). Zaprinast was also recently shown to inhibit glutaminase (Elhammali et al., 2014), which would further reduce the contribution of glutamine to the acetyl-CoA pool.
Comment by reader –
The results from these studies served as a good
reason to attempt the vaccination of patients using p53-
derived peptides, and a several clinical trials are currently
in progress. The most advanced work used a long
synthetic peptide mixture derived from p53 (p53-SLP; ISA
Pharmaceuticals, Bilthoven, the Netherlands) (Speetjens
et al., 2009; Shangary et al., 2008; Van der Burg et al.,
2001). The vaccine is delivered in the adjuvant setting
and induces T helper type cells.
Like this:
Like Loading...
Read Full Post »





























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Larry,
I think you have made a great effort in order to connect basic ideas of metabolic regulation with those of gene expression control “modern” mechanisms.
Yet, I do not think that at this stage it will be clear for all readers. At least, for the great majority of the readers. The most important factor I my opinion, is derived from the fact that modern readers considers that metabolic regulation deals with so called “housekeeping activities” of the cell. Something that is of secondary, tertiary or even less level of relevance.
My idea, that you have mentioned in the text when you write at the beginning, the word biochemistry, in order to resume it, derives from the reading of What is life together with Prof. Leloir . For me and also, for him, biochemistry comprises a set of techniques and also a framework of reasoning about scientific results. As a set of techniques, Schrodinger has considered that it will lead to better understanding of genetics and of physiology as a two legs structure supporting the future progress related to his time (mid-forties). For Leloir, the key was the understanding of chemical reactivity and I agree with him. However, as I was able to talk and discuss it with him in detail, we should also take into account levels of stabilities of macromolecules and above all, regulation of activities and function (this is where) Pasteur effect that I was studying in Leloir´s lab at that time, 1970-72, gets into the general picture.
Regulation for complex living beings , that also have cancer cell as a great topic of research problem can be understood through the understanding of two quite different results when opposition with lack of regulation is taken into account or experimentally elicited. The most clearly line of experiments can follow the Pasteur Effect as the intracellular result best seen when aerobiosis is compared with anaerobiosis as conditions in which maintenance of ATP levels and required metabolic regulation (Energy charge D.E, Atkinson etc) is studied. Another line of experiments is one that takes into account the extracellular result or for instance the homeostatic regulation of blood glucose levels. The blood glucose level is the most conspicuous and related to Pasteur Effect regulatory event that can be studied in the liver taking into account both final results tested or compared regarding its regulation, ATP levels maintenance (intracellular) and blood glucose maintenance (extracellular).
My key idea is to consider that the same factors that elicits fast regulatory responses also elicits the slow energetic expensive regulatory responses. The biologic logic behind this common root is the ATP economy. In case, the regulatory stimulus fades out quickly the fast regulatory responses are good enough to maintain life and the time requiring, energetic costly responses will soon be stopped cutting short the ATP expenditure. In case, the stimulus last for long periods of time the fast responses are replaced by adaptive responses that in general will follow the line of cell differentiation mechanisms with changes in gene expression etc.
The change from fast response mechanisms to long lasting developmentally linked ones is not sharp. Therefore, somehow, cancer cells becomes trapped into a metastable regulatory mechanism that prevents cell differentiation and reinforces those mechanisms linked to its internal regulatory goals. This metastable mechanism takes advantage from the fact that other cells, tissues and organs will take good care of homeostatic mechanisms that provide for their nutritional needs. In the case of my Hepatology work you will see a Piruvate kinase that does not responds to homeostatic signals .