Recent progress in neurodegenerative diseases and gliomas
Curator: Larry H. Bernstein, MD, FCAP
LPBI
Alzheimer’s Protein Not All Bad, Says MassGen Study
A controversial idea—that amyloid-beta (Aβ) protein fights bacterial infections in the brain—has gained additional support from a new study. Previously, the idea seemed worthy of investigation, if a bit of a stretch, on the basis of cell culture results. Now, thanks to the efforts of a scientific team lead by researchers based at Massachusetts General Hospital, it has been reinforced by observations of how the Aβ protein functions in animals’ brains.
Details of the new study appeared May 25 in the journal Science Translational Medicine, in an article entitled, “Amyloid-β Peptide Protects against Microbial Infection in Mouse and Worm Models of Alzheimer’s Disease.” The article suggests that the tendency of Aβ protein to form insoluble aggregates is not, as has been widely assumed, intrinsically abnormal, even though the aggregates are recognized as a hallmark of Alzheimer’s disease. Rather, Aβ protein appears to be a natural antibiotic that can trap and imprison bacterial pathogens that manage to pass the blood–brain barrier, which becomes increasingly “leaky” with age.
“We present in vivo data showing that Aβ expression protects against fungal and bacterial infections in mouse, nematode, and cell culture models of AD,” wrote the article’s authors. “We show that Aβ oligomerization, a behavior traditionally viewed as intrinsically pathological, may be necessary for the antimicrobial activities of the peptide.”
The MassGen scientists and their colleagues found that transgenic mice expressing human Aβ survived significantly longer after the induction of Salmonella infection in their brains than did mice with no genetic alteration. Mice lacking the amyloid precursor protein died even more rapidly. Transgenic Aβ expression also appeared to protect C. elegans roundworms from either Candida orSalmonella infection. Similarly, human Aβ expression protected cultured neuronal cells from Candida. In fact, human Aβ expressed by living cells appears to be 1000 times more potent against infection than does the synthetic Aβ used in previous studies.
That superiority appears to relate to properties of Aβ that have been considered part of Alzheimer’s disease pathology—the propensity of small molecules to form oligomers and then aggregate into Aβ plaques. This propensity, suggests the MassGen-led team, may indicate that Aβ acts like an antimicrobial peptide (AMP).
While AMPs fight infection through several mechanisms, a fundamental process involves forming oligomers that bind to microbial surfaces and then clump together into aggregates that both prevent the pathogens from attaching to host cells and allow the AMPs to kill microbes by disrupting their cellular membranes. The synthetic Aβ preparations used in earlier studies did not include oligomers. In the current study, however, oligomeric human Aβ not only showed an even stronger antimicrobial activity, its aggregation into the sorts of fibrils that form Aβ plaques was also seen to entrap microbes in both mouse and roundworm models.
“Our findings raise the intriguing possibility that β-amyloid may play a protective role in innate immunity and infectious or sterile inflammatory stimuli may drive amyloidosis,” the study’s authors concluded. “These data suggest a dual protective/damaging role for Aβ, as has been described for other antimicrobial peptides.”
One of the study’s co-corresponding authors, Rudolph Tanzi, Ph.D., director of the Genetics and Aging Research Unit in the MassGeneral Institute for Neurodegenerative Disease (MGH-MIND), pointed out that AMPs are known to play a role in the pathologies of a broad range of major and minor inflammatory disease. “For example, LL-37, which has been our model for Aβ’s antimicrobial activities, has been implicated in several late-life diseases, including rheumatoid arthritis, lupus, and atherosclerosis,” he elaborated. “The sort of dysregulation of AMP activity that can cause sustained inflammation in those conditions could contribute to the neurodegenerative actions of Aβ in Alzheimer’s disease.”
The study’s other co-corresponding author, Robert Moir, M.D., also of the MGH-MIND Genetics and Aging unit, noted that the study’s findings may lead to potential new therapeutic strategies. He also indicated that therapies designed to eliminate amyloid plaques from patient’s brains may have their limitations.
“It does appear likely that the inflammatory pathways of the innate immune system could be potential treatment targets, Dr. Moir explained. “If validated, our data also warrant the need for caution with therapies aimed at totally removing Aβ plaques. Amyloid-based therapies aimed at dialing down but not wiping out Aβ in the brain might be a better strategy.”
It remains to be determined, however, whether Aβ typically fights real infections or is apt to behave errantly, forming aggregates as though microbes are present, even if they are, in fact, not. “Our findings raise the intriguing possibility that Alzheimer’s pathology may arise when the brain perceives itself to be under attack from invading pathogens,” said Dr. Moir. “Further study will be required to determine whether or not a bona fide infection is involved.”Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease
Deepak Kumar, Vijaya Kumar, Se Hoon Choi, Kevin J. Washicosky, et al.
Science Translational Medicine 25 May 2016; 8 (340): 340ra72
http://dx.doi.org:/10.1126/scitranslmed.aaf1059
Rehabilitation of a β-amyloid bad boy
A protein called Aβ is thought to cause neuronal death in Alzheimer’s disease (AD). Aβ forms insoluble aggregates in the brains of patients with AD, which are a hallmark of the disease. Aβ and its propensity for aggregation are widely viewed as intrinsically abnormal. However, in new work, Kumar et al. show that Aβ is a natural antibiotic that protects the brain from infection. Most surprisingly, Aβ aggregates trap and imprison bacterial pathogens. It remains unclear whether Aβ is fighting a real or falsely perceived infection in AD. However, in any case, these findings identify inflammatory pathways as potential new drug targets for treating AD.
Abstract
The amyloid-β peptide (Aβ) is a key protein in Alzheimer’s disease (AD) pathology. We previously reported in vitro evidence suggesting that Aβ is an antimicrobial peptide. We present in vivo data showing that Aβ expression protects against fungal and bacterial infections in mouse, nematode, and cell culture models of AD. We show that Aβ oligomerization, a behavior traditionally viewed as intrinsically pathological, may be necessary for the antimicrobial activities of the peptide. Collectively, our data are consistent with a model in which soluble Aβ oligomers first bind to microbial cell wall carbohydrates via a heparin-binding domain. Developing protofibrils inhibited pathogen adhesion to host cells. Propagating β-amyloid fibrils mediate agglutination and eventual entrapment of unatttached microbes. Consistent with our model, Salmonella Typhimurium bacterial infection of the brains of transgenic 5XFAD mice resulted in rapid seeding and accelerated β-amyloid deposition, which closely colocalized with the invading bacteria. Our findings raise the intriguing possibility that β-amyloid may play a protective role in innate immunity and infectious or sterile inflammatory stimuli may drive amyloidosis. These data suggest a dual protective/damaging role for Aβ, as has been described for other antimicrobial peptides.
CRISPR Crossing New Barriers
Researchers Are Developing Ways to Edit Some of the Most Difficult-to-Edit DNA-Neuronal DNA
http://www.genengnews.com/insight-and-intelligence/crispr-crossing-new-barriers/77900666/
Confocal microscopic image of the hippocampus showing immunoreactivities for mEGFP (magenta) and the HA tag (green) fused to ß-Actin.
Ryohei Yasuda, Ph.D., scientific director, and his team at the Max Planck Florida Institute of Neuroscience (MPFI) are working to understand the way individual cells in our brains change as we learn and form memories. One of their main goals is to understand how different proteins behave and impact the structure and function of an individual cell, but, much like the field of genetics was once limited by the inability to visualize the structure of DNA, their research has been limited by their ability to locate and visualize the many different types of proteins within a single cell. Current imaging methods do not provide contrast and specificity high enough to see distinct proteins. Plus, the best methods are time-consuming and expensive; it can take a year or more to develop engineered models.
Over the past few years, the development of CRISPR technology has helped scientists overcome countless genetic engineering challenges, and allowed them to edit genes with unmatched precision and speed, massively increasing clarity and cutting the cost of research requiring genetic engineering. The technique has been used in myriad ways to increase understanding and treatment of diseases and disorders, but some cells are more difficult to edit than others. Brain cells have proven especially difficult to manipulate using CRISPR.
Recently, MPFI researchers Takayasu Mikuni, Ph.D., M.D., and Jun Nishiyama, Ph.D., M.D., and Dr. Yasuda were able to harness the power of the CRISPR/Cas9 system in order to create a quick, scalable, and high-resolution technique to edit neuronal DNA, which they called “SLENDR,” (single-cell labeling of endogenous proteins by CRISPR/Cas9-mediated homology-directed repair.) Using the technique, the researchers labeled several distinct proteins with fluorescence, and were able to observe protein localization in the brain that was previously invisible. That’s just the start of what researchers may be able to accomplish using this reliable, new technique for inserting genes into neurons.
CRISPR/Cas9 and Neurons
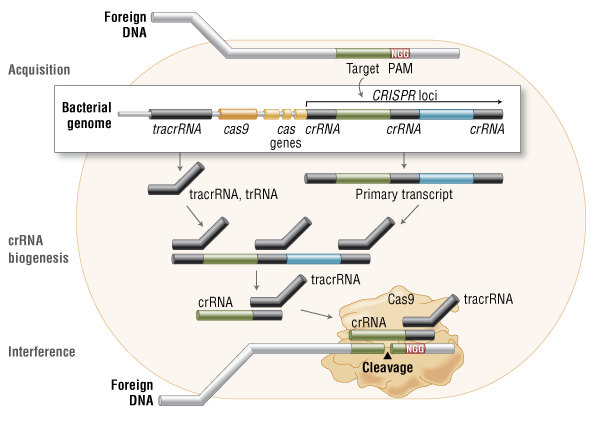
CRISPR is a tool built into bacterial DNA that the organisms use to fight infections. When a virus invades and attempts to insert its infectious DNA into that of a bacterial cell, a special section of the bacterial DNA, called CRISPR, cuts the viral DNA and renders it unable to wreak havoc on the bacteria. The organism then inserts a copy of the viral DNA into its own DNA to work as a type of adaptive immune system, to better recognize and defeat the invader in the future. As scientists have begun to understand how this system works, they have manipulated it to target and damage specific, functional genes in a variety of organisms, and in some cases, insert a new gene in its place.
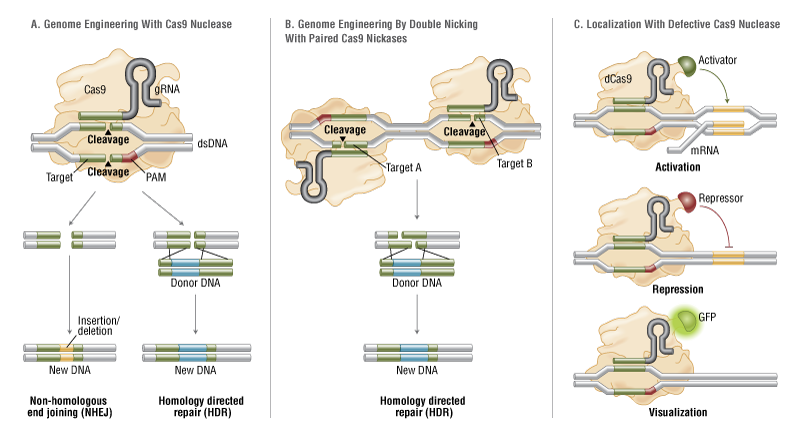
Once the section of DNA is damaged, the technique relies on the cell to naturally repair its own DNA. There are two methods that the cell might use to accomplish this. One is homology-directed repair (HDR), the other is non-homologous end joining (NHEJ). HDR rebuilds or replaces the damaged locus of the genome, whereas NHEJ reattaches the damaged ends. When the reattachment occurs following the degradation of the ends, it often leads to the deletion of function of the gene (“knock-out” the gene). If a cell uses HDR to repair itself, scientists can include a desired gene in the CRISPR system that will be inserted into the DNA to replace the damaged gene.
Despite the impressive power of CRISPR system, its use in brain cells has been limited because by the time the brain has developed, its cells are no longer dividing. Most mature brain cells will repair themselves using NHEJ. The researcher can’t give the cell a gene to insert if it’s not going to insert one to begin with. While scientists can use CRISPR relatively easily to damage and knock out certain genes through NHEJ in the brain, the lack of cell division has made it very difficult for them to knock indesired sequences to genes, through HDR, with reliable precision. That’s where the SLENDR technique comes in.
SLENDR combines the power of the CRISPR/Cas9 system with the specificity and timing of in utero electroporation. Electroporation is a well-known technique used for introducing new material into cells and creating genetic knock-outs and knock-ins. Using in utero electroporation allows researchers to insert the CRISPR/CAS9 system into prenatal models, where brain cells are still developing and dividing. Thus, the broken DNA is still being repaired via HDR, giving researchers the opportunity to precisely modify a gene. This is a big deal. “I believe that SLENDR will be a standard tool for molecular and cellular neurobiology,” said Dr. Yasuda. “SLENDR provides a valuable means to determine subcellular localization of proteins, and will help researchers to determine the function of the proteins.”
In the recent study, the researchers at MPFI inserted a gene that made proteins of interest fluoresce under the microscope. They were even able to reliably label two different proteins with distinct colors at the same time in the same cell. The researchers were able to use the technique to visualize the proteins both in vivo and in vitro. And they were able to do it in a matter of days rather than years.
With existing knowledge of how brains develop, researchers can adjust the timing and position of the electroporation in utero to accurately target cells that will go on to populate particular cortical layers of the brain, even if they haven’t differentiated and moved to that layer yet.
The recent study used the technique primarily to tag certain proteins within brain cells and observe their behavior. But, with continued optimization, the method has the potential to elucidate immeasurable brain activities in both normal and diseased brains, and lead to a deeper understanding of brain function. “The most important part is that precise genome editing is possible in the brain. That’s what’s important,” said Dr. Nishiyama, post-doctoral researcher who worked on the study. “That’s the biggest thing.” Neuroscientists would be remiss to ignore its worth and not explore its potential.
Emma Yasinski is a scientific writer at Max Planck Florida Institute for Neuroscience. Correspondence should be directed to Ryohei Yasuda, Ph.D. (ryohei.yasuda@mpfi.org), scientific director, Max Planck Florida Institute for Neuroscience.
Altered Metabolism of Four Compounds Drives Glioblastoma Growth
Findings suggest new ways to treat the malignancy, slow its progression and reveal its extent more precisely.
http://www.technologynetworks.com/Metabolomics/news.aspx?ID=190732
The altered metabolism of two essential amino acids helps drive the development of the most common and lethal form of brain cancer, according to a new study led by researchers at The Ohio State University Comprehensive Cancer Center – Arthur G. James Cancer Hospital and Richard J. Solove Research Institute (OSUCCC – James).
The study shows that in glioblastoma (GBM), the essential amino acids methionine and tryptophan are abnormally metabolized due to the loss of key enzymes in GBM cells.
The altered methionine metabolism leads to activation of oncogenes, while the changes in tryptophan metabolism shield GBM cells from detection by immune cells. Together, the changes promote tumor progress and cancer-cell survival.
“Our findings suggest that restricting dietary intake of methionine and tryptophan might help slow tumor progression and improve treatment outcomes,” says first author and OSUCCC – James researcher Kamalakannan Palanichamy, PhD, research assistant professor in Radiation Oncology.
“While we need to better understand how these abnormally regulated metabolites activate oncogenic proteins, our intriguing discovery suggests novel therapeutic targets for this disease,” says principal investigator and study leader Arnab Chakravarti, MD, chair and professor of Radiation Oncology and co-director of the Brain Tumor Program.
“For example, restoring the lost enzymes in the two metabolic pathways might slow tumor progression and reduce aggressiveness by inactivating oncogenic kinases and activating immune responses,” says Chakravarti, who holds the Max Morehouse Chair in Cancer Research.
Chakravarti further notes that because GBM cells take up methionine much faster than normal glioma cells, positron emission tomography that uses methionine as a tracer (MET-PET) might help map GBM tumors more accurately, allowing more precise surgical removal and radiation therapy planning. (MET-PET is currently an experimental imaging method.)
More than 11,880 new cases of GBM were estimated to occur in 2015, with overall survival averaging 12 to 15 months, so there is an urgent need for more effective therapies.
Amino acids are the building blocks of proteins. Tryptophan and methionine are essential amino acids – the diet must provide them because cells cannot make them. Normally, the lack of an essential amino acid in the diet can lead to serious diseases and even death. Foods rich in tryptophan and methionine include cheese, lamb, beef, pork, chicken, turkey, fish, eggs, nuts and soybeans.
Palanichamy, Chakravarti and their colleagues conducted this study using 13 primary GBM cell lines derived from patient tumors, four commercially available GBM cell lines and normal human astrocyte cells. Metabolite analyses were done using liquid chromatography coupled with mass spectrometry.
http://www.oncology-central.com/2016/04/01/study-highlights-altered-amino-acid-metabolism-in-glioblastoma/
AUTHORS: EMILY BROWN, FUTURE SCIENCE GROUP
An investigation carried out at The Ohio State University Comprehensive Cancer Center (OH, USA) has uncovered abnormal metabolism of the essential amino acids methionine and tryptophan in glioblastoma.
The study suggests that this abnormal amino acid metabolism aids in the development of the disease. Furthermore, the findings, published recently in Clinical Cancer Research, hint at novel methods to potentially treat the malignancy, slow its progression and reveal its extent more precisely.
According to the study, it is the loss of key enzymes within glioblastoma cells that results in this abnormal metabolism. Modified methionine metabolism is described as promoting the activation of oncogenes, and the changes in tryptophan aid in masking the malignant cells from the immune system.
“While we need to better understand how these abnormally regulated metabolites activate oncogenic proteins, our intriguing discovery suggests novel therapeutic targets for this disease,” commented principal investigator and study leader Arnab Chakravarti (The Ohio State University Comprehensive Cancer Center).
Rapid eye movement sleep (dreaming) shown necessary for memory formation
Rapid eye movement sleep (dreaming) shown necessary for memory formationA study published in the journal Science by researchers at the Douglas Mental Health University Institute at McGill University and the University of Bern provides the first evidence that rapid eye movement (REM) sleep — the phase where dreams appear — is directly involved in memory formation (at least in mice). “We already knew that … more…
May 16, 2016
Inhibition of media septum GABA neurons during rapid eye movement (REM) sleep reduces theta rhythm (a characteristic of REM sleep). Schematic of the in vivo recording configuration: an optic fiber delivered orange laser light to the media septum part of the brain, allowing for optogenetic inhibition of media septum GABA neurons while recording the local field potential signal from electrodes implanted in hippocampus area CA1. (credit: Richard Boyce et al./Science)
A study published in the journal Science by researchers at the Douglas Mental Health University Institute at McGill University and the University of Bern provides the first evidence that rapid eye movement (REM) sleep — the phase where dreams appear — is directly involved in memory formation (at least in mice).
“We already knew that newly acquired information is stored into different types of memories, spatial or emotional, before being consolidated or integrated,” says Sylvain Williams, a researcher and professor of psychiatry at McGill*. “How the brain performs this process has remained unclear until now. We were able to prove for the first time that REM sleep (dreaming) is indeed critical for normal spatial memory formation in mice,” said Williams.
Dream quest
Hundreds of previous studies have tried unsuccessfully to isolate neural activity during REM sleep using traditional experimental methods. In this new study, the researchers instead used optogenetics, which enables scientists to precisely target a population of neurons and control its activity by light.
“We chose to target [GABA neurons in the media septum] that regulate the activity of the hippocampus, a structure that is critical for memory formation during wakefulness and is known as the ‘GPS system’ of the brain,” Williams says.
To test the long-term spatial memory of mice, the scientists trained the rodents to spot a new object placed in a controlled environment where two objects of similar shape and volume stand. Spontaneously, mice spend more time exploring a novel object than a familiar one, showing their use of learning and recall.
Shining orange laser light on media septum (MS) GABA neurons during REM sleep reduces frequency and power (purple section) of neuron signals in dorsal CA1 area of hippocampus (credit: Richard Boyce et al./Science)
When these mice were in REM sleep, however, the researchers used light pulses to turn off their memory-associated neurons to determine if it affects their memory consolidation. The next day, the same rodents did not succeed the spatial memory task learned on the previous day. Compared to the control group, their memory seemed erased, or at least impaired.
“Silencing the same neurons for similar durations outside of REM episodes had no effect on memory. This indicates that neuronal activity specifically during REM sleep is required for normal memory consolidation,” says the study’s lead author, Richard Boyce, a PhD student.
Implications for brain disease
REM sleep is understood to be a critical component of sleep in all mammals, including humans. Poor sleep quality is increasingly associated with the onset of various brain disorders such as Alzheimer’s and Parkinson’s disease.
In particular, REM sleep is often significantly perturbed in Alzheimer’s diseases (AD), and results from this study suggest that disruption of REM sleep may contribute directly to memory impairments observed in AD, the researchers say.
This work was partly funded by the Canadian Institutes of Health Research (CIHR), the Natural Science and Engineering Research Council of Canada (NSERC), a postdoctoral fellowship from Fonds de la recherche en Santé du Québec (FRSQ) and an Alexander Graham Bell Canada Graduate scholarship (NSERC).
* Williams’ team is also part of the CIUSSS de l’Ouest-de-l’Île-de-Montréal research network. Williams co-authored the study with Antoine Adamantidis, a researcher at the University of Bern’s Department of Clinical Research and at the Sleep Wake Epilepsy Center of the Bern University Hospital.
Abstract of Causal evidence for the role of REM sleep theta rhythm in contextual memory consolidation
Rapid eye movement sleep (REMS) has been linked with spatial and emotional memory consolidation. However, establishing direct causality between neural activity during REMS and memory consolidation has proven difficult because of the transient nature of REMS and significant caveats associated with REMS deprivation techniques. In mice, we optogenetically silenced medial septum γ-aminobutyric acid–releasing (MSGABA) neurons, allowing for temporally precise attenuation of the memory-associated theta rhythm during REMS without disturbing sleeping behavior. REMS-specific optogenetic silencing of MSGABA neurons selectively during a REMS critical window after learning erased subsequent novel object place recognition and impaired fear-conditioned contextual memory. Silencing MSGABA neurons for similar durations outside REMS episodes had no effect on memory. These results demonstrate that MSGABA neuronal activity specifically during REMS is required for normal memory consolidation.
Quantifying Consciousness
By Tanya Lewis
Overall brain metabolic rate can distinguish between pathological states of human consciousness, a study shows.
Time-resolved studies define the nature of toxic IAPP intermediates, providing insight for anti-amyloidosis therapeutics.
Abedini A, Plesner A, Cao P, Ridgway Z, et al.
eLife May 23, 2016; 10.7554/eLife.12977. http://dx.doi.org/10.7554/eLife.12977
Islet amyloidosis by IAPP contributes to pancreatic β-cell death in diabetes, but the nature of toxic IAPP species remains elusive. Using concurrent time-resolved biophysical and biological measurements, we define the toxic species produced during IAPP amyloid formation and link their properties to induction of rat INS-1 β-cell and murine islet toxicity. These globally flexible, low order oligomers upregulate pro-inflammatory markers and induce reactive oxygen species. They do not bind 1-anilnonaphthalene-8-sulphonic acid and lack extensive β-sheet structure. Aromatic interactions modulate, but are not required for toxicity. Not all IAPP oligomers are toxic; toxicity depends on their partially structured conformational states. Some anti-amyloid agents paradoxically prolong cytotoxicity by prolonging the lifetime of the toxic species. The data highlight the distinguishing properties of toxic IAPP oligomers and the common features that they share with toxic species reported for other amyloidogenic polypeptides, providing information for rational drug design to treat IAPP induced β-cell death.
NIH study visualizes proteins involved in cancer cell metabolism
Cryo-EM methods can determine structures of small proteins bound to potential drug candidates.
https://www.nih.gov/news-events/news-releases/nih-study-visualizes-proteins-involved-cancer-cell-metabolism
Scientists using a technology called cryo-EM (cryo-electron microscopy) have broken through a technological barrier in visualizing proteins with an approach that may have an impact on drug discovery and development. They were able to capture images of glutamate dehydrogenase, an enzyme found in cells, at a resolution of 1.8 angstroms, a level of detail at which the structure of the central parts of the enzyme could be visualized in atomic detail. The scientists from the National Cancer Institute (NCI), part of the National Institutes of Health, and their colleagues also reported achieving another major milestone, by showing that the shapes of cancer target proteins too small to be considered within the reach of current cryo-EM capabilities can now be determined at high resolution.
The research team was led by NCI’s Sriram Subramaniam, Ph.D., with contributions from scientists at the National Center for Advancing Translational Sciences (NCATS), also part of NIH. The findings appeared online May 26, 2016, in Cell.
“These advances demonstrate a real-life scenario in which drug developers now could potentially use cryo-EM to tweak drugs by actually observing the effects of varying drug structure — much like an explorer mapping the shoreline to find the best place to dock a boat — and alter its activity for a therapeutic effect,” said Doug Lowy, M.D., acting director, NCI.
Both discoveries have the potential to have an impact on drug discovery and development. Cryo-EM imaging enables analysis of structures of target proteins bound to drug candidates without first needing a step to coax the proteins to form ordered arrays. These arrays were needed for the traditional method of structure determination using X-ray crystallography, a powerful technique that has served researchers well for more than a half century. However, not all proteins can be crystallized easily, and those that do crystallize may not display the same shape that is present in their natural environment, either since the protein shape can be modified by crystallization additives or by the contacts that form between neighboring proteins within the crystal lattice.
“It is exciting to be able to use cryo-EM to visualize structures of complexes of potential drug candidates at such a high level of detail.”
—Sriram Subramaniam, Ph.D.,National Caner Institute
“It is exciting to be able to use cryo-EM to visualize structures of complexes of potential drug candidates at such a high level of detail,” said Subramaniam. “The fact that we can obtain structures of small cancer target proteins bound to drug candidates without needing to form 3D crystals could revolutionize and accelerate the drug discovery process.”
Two of the small proteins the researchers imaged in this new study, isocitrate dehydrogenase (IDH1) and lactate dehydrogenase (LDH), are active targets for cancer drug development. Mutations in the genes that code for these proteins are common in several types of cancer. Thus, imaging the surfaces of these proteins in detail can help scientists identify molecules that will bind to them and aid in turning the protein activity off.
In publications in the journal Science last year and this year, Subramaniam and his team reported resolutions of 2.2 angstroms and 2.3 angstroms in cryo-EM with larger proteins, including a complex of a cancer target protein with a small molecule inhibitor. Of note, the journal Nature Methods deemed cryo-EM as the “Method of the Year” in January 2016. “Our earlier work showed what was technically possible,” Subramaniam said. “This latest advance is a delivery of that promise for small cancer target proteins.” For more information on cryo-EM, go to http://electron.nci.nih.gov.
Time-resolved studies define the nature of toxic IAPP intermediates, providing insight for anti-amyloidosis therapeutics.
Abedini A, Plesner A, Cao P, Ridgway Z, et al.
eLife May 23, 2016; 10.7554/eLife.12977. http://dx.doi.org/10.7554/eLife.12977
Islet amyloidosis by IAPP contributes to pancreatic β-cell death in diabetes, but the nature of toxic IAPP species remains elusive. Using concurrent time-resolved biophysical and biological measurements, we define the toxic species produced during IAPP amyloid formation and link their properties to induction of rat INS-1 β-cell and murine islet toxicity. These globally flexible, low order oligomers upregulate pro-inflammatory markers and induce reactive oxygen species. They do not bind 1-anilnonaphthalene-8-sulphonic acid and lack extensive β-sheet structure. Aromatic interactions modulate, but are not required for toxicity. Not all IAPP oligomers are toxic; toxicity depends on their partially structured conformational states. Some anti-amyloid agents paradoxically prolong cytotoxicity by prolonging the lifetime of the toxic species. The data highlight the distinguishing properties of toxic IAPP oligomers and the common features that they share with toxic species reported for other amyloidogenic polypeptides, providing information for rational drug design to treat IAPP induced β-cell death.
Single domain antibodies (sdAbs) aid in x-ray crystallography of mammalian serotonin 5-HT3 receptor
Serotonin 5-HT3 is part of the cys-loop receptor family, the mechanism of this family is not well understood due to difficulties in obtaining high resolution crystal structures. Serotonin 5-HT3 receptor is an important druggable target in alleviating nausea and vomiting induced by chemotherapy or anesthesia, as well as psychiatric disorders. It’s structure is critical in discovering new drugs to modulate its activity.
Previously, electron microscopy imaging of non-mammalian homologs of Cys-loop receptors provided basic understanding of extracellular ligand binding sites and pore forming domains. Little was known about intracellular domains and the way they interact with cellular scaffolding proteins, as they are absent in non-mammalian homologs. A recent publication in Nature extends our understanding behind the mechanism of serotonin 5-HT3 receptors, by resolving a 3.5A crystal structure.
Mouse 5-HT3 exists as a homopentamer and is difficult to express, purify and crystallize. To overcome this challenge, researchers split the receptor by proteolyzing each subunit into two fragments. In addition, an sdAb chaperone, which acts as an inhibitor locking the channel into a non-conducting conformation, was used to stabilized the pentameric structure, enabling resolution of a 3.5A crystal structure. Most importantly the split receptor displays an intracellular domain that is tightly coupled to the membrane domain, which provides important structural information that will lead to further understanding of the physiological conformation of 5-HT3 and Cys-loop receptors.
Hassaine G. et al. X-ray structure of the mouse serotonin 5-HT3 receptor Nature. Aug 2014. 512(7514):276-281
UCLA animal study shows how brain connects memories across time
Wednesday, May 25, 2016
Using a miniature microscope that opens a window into the brain, UCLA neuroscientists have identified in mice how the brain links different memories over time–and this may help develop new drugs in the future for memory-robbing diseases such as Alzheimer’s.
FDA approves new antibody drug for treating pediatric neuroblastoma
Pediatric neuroblastoma is a rare and difficult to treat cancer that forms from immature nerve cells. This form of cancer occurs in 1 in 100,000 children, with 650 new cases each year in the United States. Current therapies, which are non-specific, only provide 40-50% long term survival rate to patients suffering from high-risk neuroblastoma, making this form of cancer an area of high medical unmet need.
A new drug, called dinutuxumab was granted priority review and orphan drug designation by the FDA. It is the first drug of its kind to be approved that specifically treats pediatric neuroblastoma. In addition to the approval, the FDA also issued a rare pediatric review priority voucher to the makers of the drug, for future groundbreaking therapies in pediatric neuroblastoma.
Dinutuxumab (formerly called ch14.18) is a disialoganglioside (GD2) binding chimeric monoclonal antibody that works in combination with granulocyte-macrophage colony-stimulating factor (GM-CSF), interleukin-2 (IL-2), and 13-cis-retinoic acid (RA) for treating high-risk pediatric neuroblastoma.
Antibody therapeutics are highly efficacious and specific towards rare and difficult-to-treat cancers and discovery of new antibody therapeutics will help address critical needs. Antibody drug discovery may be challenging, but working with an experienced partner can help.
FDA approves first therapy for high-risk neuroblastoma
Electronic Biosensor Detects Molecules Linked to Cancer, Alzheimer’s, and Parkinson’s
5/20/2016 by Fundação de Amparo À Pesquisa Do Estado de São Paulo
A biosensor developed by researchers at the National Nanotechnology Laboratory (LNNano) in Campinas, São Paulo State, Brazil, has been proven capable of detecting molecules associated with neurodegenerative diseases and some types of cancer.
The device is basically a single-layer organic nanometer-scale transistor on a glass slide. It contains the reduced form of the peptide glutathione (GSH), which reacts in a specific way when it comes into contact with the enzyme glutathione S-transferase (GST), linked to Parkinson’s, Alzheimer’s and breast cancer, among other diseases. The GSH-GST reaction is detected by the transistor, which can be used for diagnostic purposes.
An inexpensive portable biosensor has been developed by researchers at Brazil’s National Nanotechnology Laboratory with FAPESP’s support. (Credit: LNNano)
The project focuses on the development of point-of-care devices by researchers in a range of knowledge areas, using functional materials to produce simple sensors and microfluidic systems for rapid diagnosis.
“Platforms like this one can be deployed to diagnose complex diseases quickly, safely and relatively cheaply, using nanometer-scale systems to identify molecules of interest in the material analyzed,” explained Carlos Cesar Bof Bufon, Head of LNNano’s Functional Devices & Systems Lab (DSF) and a member of the research team for the project, whose principal investigator is Lauro Kubota, a professor at the University of Campinas’s Chemistry Institute (IQ-UNICAMP).
In addition to portability and low cost, the advantages of the nanometric biosensor include its sensitivity in detecting molecules, according to Bufon.
“This is the first time organic transistor technology has been used in detecting the pair GSH-GST, which is important in diagnosing degenerative diseases, for example,” he explained. “The device can detect such molecules even when they’re present at very low levels in the examined material, thanks to its nanometric sensitivity.” A nanometer (nm) is one billionth of a meter (10-9 meter), or one millionth of a millimeter.
The system can be adapted to detect other substances, such as molecules linked to different diseases and elements present in contaminated material, among other applications. This requires replacing the molecules in the sensor with others that react with the chemicals targeted by the test, which are known as analytes.
The team is working on paper-based biosensors to lower the cost even further and to improve portability and facilitate fabrication as well as disposal.
The challenge is that paper is an insulator in its usual form. Bufon has developed a technique to make paper conductive and capable of transporting sensing data by impregnating cellulose fibers with polymers that have conductive properties.
The technique is based on in situ synthesis of conductive polymers. For the polymers not to remain trapped on the surface of the paper, they have to be synthesized inside and between the pores of the cellulose fibers. This is done by gas-phase chemical polymerization: a liquid oxidant is infiltrated into the paper, which is then exposed to monomers in the gas phase. A monomer is a molecule of low molecular weight capable of reacting with identical or different molecules of low molecular weight to form a polymer.
The monomers evaporate under the paper and penetrate the pores of the fibers at the submicrometer scale. Inside the pores, they blend with the oxidant and begin the polymerization process right there, impregnating the entire material.
The polymerized paper acquires the conductive properties of the polymers. This conductivity can be adjusted by manipulating the element embedded in the cellulose fibers, depending on the application for which the paper is designed. Thus, the device can be electrically conductive, allowing current to flow without significant losses, or semiconductive, interacting with specific molecules and functioning as a physical, chemical or electrochemical sensor.
Protein Oxidation in Aging: Not All Proteins Are Created Equal
Cancer, Alzheimer’s disease and other age-related diseases develop over the course of aging, and certain proteins are shown to play critical roles this process. Those proteins are subject to destabilization as a result of oxidation, which further leads to features of aging cells. It is estimated that almost 50% of proteins are damaged due to oxidation for people at their 80s. The oxidative damage mediated by free radicals occurs when converting food to energy in the presence of oxygen. Cellular structures, such as proteins, DNA, and lipids, are prone to these oxidation damages, which further contribute to the development of age-related diseases.
Using computational models with physics principles incorporated, de Graff el al. from Stony Brook University unfolded the molecular mechanism that how natural chemical process affects the aging of proteins. First, the authors revealed the major factor to explain stability loss in aging cells and organisms is likely to be random modification of the protein sidechains. Furthermore, through the evaluation and analysis on the protein electrostatics, the authors suggested that highly charged proteins are in particular subject to the oxidation induced destabilization. Even one single oxidation could lead to unfold the whole structure for these highly charged proteins. Old cells are enriched in those highly charged proteins, thus the destabilization effects are elevated in the aging cells. In addition, 20 proteins associated with aging are further identified to be at high risk of oxidation. The list includes telomerase proteins and histones, both of which play critical roles in the aging of cells and cancer development. The team is currently working on analyzing more proteins, with the hope to provide key information to aid targeted treatments against age-related diseases.
Further Reading: Emerging Opportunity for Treating Alzheimer Disease by Immunotherapy
Adam M.R. de Graff, Michael J. Hazoglou, Ken A. Dill. Highly Charged Proteins: The Achilles’ Heel of Aging Proteomes.Structure, 24, 285-292 (2016)
Baruch, K. et al. PD-1 Immune Checkpoint Blockade Reduces Pathology and Improves Memory in Mouse Models of Alzheimer’s Disease. Nat. Med. 22, 135-137 (2016)
Single domain antibodies shown to cross blood brain barrier and offers enhanced delivery of therapeutics to CNS targets
A major challenge in developing both small molecule and antibody therapeutics for CNS disorders including brain cancer and neurodegenerative diseases, is penetrating the blood brain barrier (BBB). A study published in FASEB demonstrated that monomeric variable heavy-chain domain of camel homodimeric antibodies (mVHH), can cross the BBB in-vivo, and recognize its intracellular target: glial fibrillary acidic protein (GFAP). The ability of mVHH to cross the BBB of normal animals and those undergoing pathological stress makes it a promising modality for treating CNS diseases as well as for brain imaging.
The investigators of this study expressed a recombinant fusion protein, VHH-GFP, which was able to cross the BBB in-vivo and specifically label astrocytes. GenScript is fully engaged in single-domain antibody lead generation and optimization. With our one-stop services, we are determined to be your best partner in antibody drug discovery from gene synthesis to in-vivo characterization of candidate antibodies. All you need to provide is the Genbank accession number of the antigen protein!
Li T. et al. Cell-penetrating anti-GFAP VHH and corresponding fluorescent fusion protein VHH-GFP spontaneously cross the blood-brain barrier and specifically recognize astrocytes: application to brain imaging. FASEB. Oct 2012. 26:3969-79
New insight behind the success of fighting cancer by targeting immune checkpoint proteins
Immune checkpoint blockade has proven to be highly successful in the clinic at treating aggressive and difficult-to-treat forms of cancer. The mechanism of the blockade, targeting CTLA-4 and PD-1 receptors which act as on/off switches in T cell-mediated tumor rejection, is well understood. However, little is known about the tumor antigen recognition profile of these affected T-cells, once the checkpoint blockade is initiated.
In a recent published study, the authors used genomics and bioinformatics approaches to identify critical epitopes on 3-methylcholanthrene induced sarcoma cell lines, d42m1-T3 and F244. CD8+ T cells in anti-PD-1 treated tumor bearing mice were isolated and fluorescently labeled with tetramers loaded with predicted mutant epitopes. Out of 66 predicted mutants, mLama4 and mAlg8 were among the highest in tetramer-positive infiltrating T-cells. To determine whether targeting these epitopes alone would yield similar results as anti-PD-1 treatment, vaccines against these two epitopes were developed and tested in mice. Prophylactic administration of the combined vaccine against mLama4 and mAlg8 yielded an 88% survival in tumor bearing mice, thus demonstrating that these two epitopes are the major antigenic targets from checkpoint-blockade and therapies against these two targets are similarly efficacious.
In addition to understanding the mechanism, identification of these tumor-specific mutant antigens is the first step in discovering the next wave of cancer immunotherapies via vaccines or antibody therapeutics. Choosing the right antibody platform can speed the discovery of a new therapeutics against these new targets. Single domain antibodies have the advantage of expedited optimization, flexibility of incorporating multiple specificity and functions, superior stability, and low COG over standard antibody approaches.
Gubin MM. et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. Nov 2014. 515:577-584
Anti-PD-1 is poised to be a blockbuster, which other immune-checkpoint targeting drugs are on the horizon?
Clinical studies of anti-immune-checkpoint protein therapeutics have shown not only an improved overall survival, but also a long-term durable response, compared to chemotherapy and genomically-targeted therapy. To expand the success of immune-checkpoint therapeutics into more tumor types and improving efficacy in difficult-to-treat tumors, additional targets involved in checkpoint-blockade need to be explored, as well as testing the synergy between combining approaches.
Currently, CTLA-4 and PD-1/PD-L1 are furthest along in development, and have shown very promising results in metastatic melanoma patients. This is just a fraction of targets involved in the checkpoint-blockade pathway. Several notable targets include:
- LAG-3 – Furthest along in clinical development with both a fusion protein and antibody approach, antibody apporach being tested in combination with anti-PD-1
- TIM-3 – Also in clinical development. Pre-clinical studies indicate that it co-expresses with PD-1 on tumor-infiltrating lymphocytes. Combination with anti-PD-improves anti-tumor response
- VISTA – Antibody targeting VISTA was shown to improve anti-tumor immune response in mice
In addition, there are also co-stimulatory factors that are also being explored as viable therapeutic targets
- OX40 – Both OX40 and 4-1BB are part of the TNF-receptor superfamily. Phase I data shows acceptable safety profile, and evidence of anti-tumor response in some patients
- 4-1BB – Phase I/II data on an antibody therapeutic targeting OX40 shows promising clinical response for melanoma, renal cell carcinoma and ovarian cancer.
- Inducible co-stimulator (ICOS) – Member of the CD28/B7 family. Its expression was found to increase upon T-cell activation. Anti-CTLA-4 therapy increases ICOS-positive effector T-cells, indicating that it may work in synergy with anti-CTLA-4. Clinical trials of anti-ICOS antibody are planned for 2015.
Sharma P and Allison JP. Immune Checkpoint Targeting in Cancer Therapy: Toward Combination Strategies with Curative Potential. Cell. April 2015;161:205-214
CTLA-4 found in dendritic cells suggests New cancer treatment possibilities
Both dendritic cells and T cells are important in triggering the immune response, whereas antigen presenting dendritic cells act as the “general” leading T cells “soldiers” to chase and eliminate enemies in the battle against cancer. The well-known immune checkpoint break, CTLA-4, is believed to be present only in T cells (and cells of the same lineage). However, a new study published in Stem Cells and Development suggests that CTLA-4 also presents in dendritic cells. It further explores the mechanism on how turning off the dendritic cells in the immune response against tumors.
Matthew Halpert, et al. Dendritic Cell Secreted CTLA-4 Regulates the T-cell Response by Downmodulating Bystander Surface B7. Stem Cells and Development, 2016; DOI: 10.1089/scd.2016.0009
With a wide range of animal models to choose from, what are the crucial factors to consider?
A recent perspective published in Nature Medicine addresses these gaps by comparing the strengths and limitations of different tumor models, as well as best models to use for answering different biological questions and best practices for preclinical modeling.
Below is a summary of the authors’ key considerations:
- It is important to choose a model based on the biology of the target. Several diverse tumor models may be required to address complex biology
- If the biology of the target includes signaling between the tumor and the stroma, then it is crucial to understand drug efficacy in the presence of an appropriate tumor microenvironment with orthotopic models
- Avoid overuse of models that are highly sensitive to the drug, unless there is clinically relevant biomarker data to support the findings
- For studying agents that reduce pre-existing tumors, make sure that the tumors are established in the model prior to treatment
- Understanding the pharmacokinetics of a drug in the model prior to studies is important to ensure that the dosing is within range, and that off-target and toxic side effects are not skewing anti-tumor activity.
Gould SE, Junttila MR and de Sauvage FJ. Translational value of mouse models in oncology drug development. Nat Med. May 2015. 21(5):431-439
Revolutionary Impact of Nanodrug Delivery on Neuroscience
Reza Khanbabaie1,2,3 and Mohsen Jahanshahi
Curr Neuropharmacol. 2012 Dec; 10(4): 370–392. doi: 10.2174/157015912804143513
Brain research is the most expanding interdisciplinary research that is using the state of the art techniques to overcome limitations in order to conduct more accurate and effective experiments. Drug delivery to the target site in the central nervous system (CNS) is one of the most difficult steps in neuroscience researches and therapies. Taking advantage of the nanoscale structure of neural cells (both neurons and glia); nanodrug delivery (second generation of biotechnological products) has a potential revolutionary impact into the basic understanding, visualization and therapeutic applications of neuroscience. Current review article firstly provides an overview of preparation and characterization, purification and separation, loading and delivering of nanodrugs. Different types of nanoparticle bioproducts and a number of methods for their fabrication and delivery systems including (carbon) nanotubes are explained. In the second part, neuroscience and nervous system drugs are deeply investigated. Different mechanisms in which nanoparticles enhance the uptake and clearance of molecules form cerebrospinal fluid (CSF) are discussed. The focus is on nanodrugs that are being used or have potential to improve neural researches, diagnosis and therapy of neurodegenerative disorders.
Keywords: Nanodrug, Nanofabrication and purification, Neuroscience, Nervous system, Nano-nervous drugs.
1. INTRODUCTION
The delivery of drugs to the nervous system is mainly limited by the presence of two anatomical and biochemical dynamic barriers: the blood–brain barrier (BBB) and blood–cerebrospinal fluid barrier (BCSFB) separating the blood from the cerebral parenchyma [1]. These barriers tightly seal the central nervous system (CNS) from the changeable milieu of blood. With the advancement of electron microscopy it is found that the ultrastructural localization of the blood–brain barrier is correlated with the capillary endothelial cells within the brain [2]. The BBB inhibits the free paracellular diffusion of water-soluble molecules by an elaborate network of complex tight junctions (TJs) that interconnects the endothelial cells. Similar to the endothelial barrier, the morphological correlate of the BCSFB is found at the level of unique apical tight junctions between the choroid plexus epithelial cells inhibiting paracellular diffusion of water-soluble molecules across this barrier [1, 3]. Beside its barrier function, it allows the directed transport of ions and nutrients into the cerebrospinal fluid (CSF) and removal of toxic agents out of the CSF using numerous transport systems.
One of the most challenging steps in neuroscience researches and therapy is the availability of techniques to penetrate these permeability barriers and delivering drugs to the CNS. Several strategies have been used to circumvent the barriers inhibiting CNS penetration. These strategies generally fall into one or more of the following three categories: manipulating drugs, disrupting the BBB (BBBD) and finding alternative routes for drug delivery. Drug manipulation methods include: Lipophilic Analogs, prodrugs, chemical drug delivery systems (CDDS), Carrier-mediated transport (CMT) and Receptor-mediated drug delivery. The drug manipulating strategy has been frequently employed, but the results have often been disappointing [4–6]. All of these methods have major limitations: they are invasive procedures, have toxic side effects and low efficiency, and are not sufficiently safe [7]. Two methods for disrupting the BBB have been reported: osmotic blood-brain barrier disruption and biochemical blood-brain barrier disruption. However, these procedures also break down the self-defense mechanism of the brain and make it vulnerable to damage or infection from all circulating chemicals or toxins. Since the above techniques aim to enhance the penetration of drugs to the CNS via circulatory system, they will increase the penetration of drugs throughout the entire body. This will frequently result in unwanted systemic side effects. In the other hand, systemically administered agents must penetrate the BBB to enter the CNS, which is a difficult task. Despite advances in rational CNS drug design and BBBD, many potentially efficacious drug molecules still cannot penetrate into the brain parenchyma at therapeutic concentrations. The alternative strategy to enhance CNS penetration of drug molecules is based on methodology that does not rely on the cardiovascular system. These strategies circumvent the BBB altogether and do not need drug manipulation to enhance BBB permeability and/or BBBD. Using alternative routes to deliver drugs to the CNS, e.g. intraventricular/intrathecal route and olfactory pathway, is one of these strategies.
One strategy for bypassing the BBB that has been studied extensively both in laboratory and in clinical trials is the intralumbar injection or intreventricular infusion of drugs directly into the CSF. Compared to vascular drug delivery, intra-CSF drug administration theoretically has several advantages. Intra-CSF administration bypasses the BCB and results in immediate high CSF drug concentrations. Due to the fact that the drug is somewhat contained within the CNS, a smaller dose can be used, potentially minimizing systemic toxicity. Furthermore, drugs in the CSF encounter minimize protein binding and decrease enzymatic activity relative to drugs in plasma, leading to longer drug half-life in the CSF. Finally, since the CSF freely exchanges molecules with the extracellular fluid of the brain parenchyma, delivering drugs into the CSF could theoretically result in therapeutic CNS drug concentrations [7, 8]. However, for several reasons this delivery was not as successful as predicted. These include a slow rate of drug distribution within the CSF and increase in intracranial pressure associated with fluid injection or infusion into small ventricular volumes.
Another CNS drug delivery route is the intranasal route. In this method drugs are transported intranasally along olfactory sensory neurons to yield significant concentrations in the CSF and olfactory bulb. An obvious advantage of this method is that it is noninvasive relative to other strategies. This method has received relatively little attention, since there are difficulties that have to be overcome. Among these obstacles is an enzymatically active, low pH nasal epithelium, the possibility of mucosal irritation or the possibility of large variability caused by nasal pathology, such as common cold.
Based on the advantages and disadvantages of aforementioned strategies, researchers are still looking for novel and better methods of CNS drug deliveries. The most direct way of circumventing the BBB is to deliver drugs directly to the brain interstitium which mainly includes the use of small colloidal particles like liposomes and nanoparticles [8]. By directing agents uniquely to an intracranial target, interstitial drug delivery can theoretically yield high CNS drug concentrations with minimal systemic exposure and toxicity. Furthermore, with this strategy, intracranial drug concentrations can be sustained, which is crucial in treatment with many chemotherapeutic agents. The basic reason of common acceptance of these carriers is due to their controlled profile or drug release nature as well as due to their selected targeting mechanism. Targeting action maybe due to the steric hindrance created by nano-vectors for achieving targeting ability. These carriers are usually administered through parenteral route and due to their steric phenomenon they conceal themselves from opsonisation event induced by tissue macrophages. By this way they achieve targeting ability to brain and other reticuloendothelial system (RES) organs like liver, spleen, etc.
Several approaches have been developed for delivering drugs directly to the brain interstitium like injections, catheters, and pumps. One such methodology is the Ommaya reservoir or implantable pump which achieves truly continuous drug delivery. Though interstitial drug delivery to the CNS has had only modest clinical impact, its therapeutic potential may soon be realized using new advances in polymer technologies to modify the aforementioned techniques. Polymeric or lipidbased devices that can deliver drug molecules at defined rates for specific periods of time are now making a tremendous impact in clinical medicine.
Among the strategies of direct drug delivery to the CNS, nanoparticles have attracted considerable interest from the last few decades. It has been shown that nano delivery systems have great potential to facilitate the movement of drugs across barriers (e.g., BBB). Nanosystems employed for the development of nano drug delivery systems in the treatment of CNS disorders include polymeric nanoparticles, nanospheres, nanosuspensions, nanoemulsions, nanogels, nano-micelles and nano-liposomes, carbon nanotubes, nanofibers and nanorobots, solid lipid nanoparticles (SLN), nanostructured lipid carriers (NLC) and lipid drug conjugates (LDC). Although the exact mechanism of barrier opening by nanoparticles is not known, the novel properties such as tiny size, tailored surface, better solubility, and multi-functionality of nanoparticles present the capability to interact with composite cellular functions in new ways. In fact, nanotechnology has now emerged as an area of research for invention of newer approaches for the CNS drug delivery and a revolutionary method to improve diagnosis and therapy of neurodegenerative disorders.
In this line, an overview of preparation and characterization, purification and separation, loading and delivering of nanodrugs is the first subject of this review. Different types of nanoparticle bioproducts including carbon nanotubes as a drug delivery system and also as a novel tool in neuroscience research are explored. For instance, nanodrug delivery systems like human serum albumin (HSA) nanoparticles, bovine serum albumin (BSA)-Gum Arabic (Acacia) nanoparticles and α-lactalbumin nanoparticles are explained.
The impact of nanotechnology on neuroscience and drug delivery to the central nervous system (CNS) is the subject of the second part of this review. Different mechanisms in which nanoparticles enhance the uptake of molecules both hydrophilic and hydrophobic across the BBB and the impact of various physiochemical parameters of nanoparticles on its uptake and clearance form CSF are discussed. Also nanodrugs that are being used or have potential to improve neural researches, diagnosis and therapy of neurodegenerative disorders are investigated.
2. FROM NANOTECHNOLOGY TO NEUROPHARMACOLOGY
Nanotechnology started by the suggestion of a famous physicist, Richard Feynman, that it should be possible, in principle, to make nanoscale machines that “arrange the atoms the way we want”, and do chemical synthesis by mechanical manipulation [9, 10]. Nanotechnologies exploit materials and devices with a functional organization that has been engineered at the nanometer scale. In a broad sense, they can be defined as the science and engineering involved in the design, syntheses, characterization, and application of materials and devices whose smallest functional organization in at least one dimension is on the nanometer scale, ranging from a few to several hundred nanometers. A nanometer is roughly the size of a molecule itself (e.g., a DNA molecule is about 2.5 nm long while a sodium atom is about 0.2 nm) [10]. Nanotechnology is not in itself a single emerging scientific discipline but rather a meeting of traditional sciences such as chemistry, physics, materials science, and biology to bring together the required collective expertise needed to develop these novel technologies.
The application of nanotechnology in cell biology and physiology enables targeted interactions at a fundamental molecular level. Nanotechnology, in the context of nanomedicine, can be defined as the technologies for making nanocarriers of therapeutics and imaging agents, nanoelectronic biosensors, nanodevices, and microdevices with nanostructures. It also covers possible future applications of molecular nanotechnology (MNT) and nanovaccinology. Unlike the definition in core nanotechnology field, which restricts the “nano” to at least 1–100 nm in one dimension, nanocarriers in the biomedical field are often referred to as particles with a dimension a few nanometers to 1000 nm [8, 11]. Although, the initial properties of nanomaterials studied were for its physical, mechanical, electrical, magnetic, chemical and biological applications, recently, attention has been geared towards its pharmaceutical application, especially in the area of drug delivery [8]. There are a few challenges in use of large size materials in drug deliveries. Some of these challenges are poor bioavailability, in vivo stability, solubility, intestinal absorption, sustained and targeted delivery to site of action, therapeutic effectiveness, generalized side effects, and plasma fluctuations of drugs (see Table 11).
The most important innovations are taking place in nanopharmocology and drug delivery which involves developing nanoscale particles or molecules to improve bioavailability. These pharmacological applications of nanotechnology include: the formation of novel nanoscopic entities [11, 27], exploring and matching specific compounds to particular patients for maximum effectiveness; and advanced pharmaceutical delivery systems and discovery of new pharmacological molecular entities; selection of pharmaceuticals for specific individuals to maximize effectiveness and minimize side effects, and delivery of pharmaceuticals to targeted locations or tissues within the body. Examples of nanomaterials include nanotubes and nanofibers, liposomes, nanoparticles, polymeric micelles, block ionomer complexes, nanogels, and dendrimers.
Nanotubes [28, 29] and nanofibers mimic tubular structures that appear in nature, such as rod shaped bacteria or viruses, microtubules, ion channels, as well as axons and dendrites. They are low-dimensional nanostructures, having a very large axial ratio. Properties of a molecule in a nanotube or nanofiber structure can be different from those in the bulk or in other nanomaterials, such as spherical nanoparticles. These materials have a large surface–volume ratio, which results in a high exposure of the material components to the surrounding environment [30]. This makes nanotubes and nanofibers promising structures for biosensing and molecular recognition [31]. However, it provides a way to control drug release through the nanotubes wall, while the large hollow area inside nanotubes provides an excellent storage for drugs and other agents [32]. Furthermore, nanotubes can be synthesized to be open-ended, which can be exploited for certain biological applications.
Carbon nanotubes (CNTs) was discovered by Iijima [33] which are composed of carbon atoms arranged in hexagonal ring structures similar to graphite, with some five-membered or seven-membered rings providing the structure curvature [29, 34,35]. CNTs are compatible with biological tissues for scaffolding purposes and the charge carried by the nanotubes can be manipulated to control neurite outgrowth [36, 37]. It has also been suggested that CNTs functionalized with growth factors, such as nerve growth factor or brain-derived neurotrophic factor, can stimulate growth of neurons on the nanotube scaffold [38–40]. In such application the toxicity of CNTs remains an issue that must be overcome [41, 42]. It has been reported that conductive polymer coatings for living neural cells has been generated using poly (3,4-ethylenedioxythiophene) PEDOT nanotubes [43]. The electric conductivity of PEDOT was used to enhance the electrical activity of the tissue with a long range aim of treating CNS disorders, which show sensory and motor impairments. These observations suggested that nanotube and nanofiber scaffolds have potential for neuroregeneration as well as treatment of CNS trauma [27, 44]. Nanomaterials suggest a promising strategy for neuroprotection [45]. Neuroprotection is an effect that may result in salvage, recovery, or regeneration of the nervous system.
The role of nanotechnology in targeted drug delivery and imaging was discussed in many reviews and papers [46, 47]. As a step towards a realistic system, a brief overview of preparation, characterization, delivery, loading, purification and separation of nanoparticles and nanodrugs are presented herein. In next two sections the fabrication methods of nanoparticle bioproducts and also the delivery systems of nanodrugs are explained. Subsequently we go back to the CNS nanodrugs for research and therapy and the delivery systems of nanodrugs for nervous system.
……
3. NANODRUG DELIVERY SYSTEMS
The major goals in designing nanoparticles as a delivery system are to control particle size, surface properties [85] and release of pharmacologically active agents in order to achieve the site-specific action of the drug at the therapeutically optimal rate and dose regimen [86]. If nanoparticles are considered to be used as drug delivery vehicles, it depends on many factors including: (a) size of nanoparticles required; (b) inherent properties of the drug, e.g., aqueous solubility; (c) surface characteristics such as charge and permeability; (d) degree of biodegradability, biocompatibility and toxicity; (e) drug release profile desired; and (f) antigenicity of the final product. The advantages of using nanoparticles as a drug delivery system might be summarized as follow [87]:
- Particle size and surface characteristics of nanoparticles can be easily manipulated to achieve both passive and active drug targeting after parenteral administration.
- They control and sustain release of the drug during the transportation and at the site of localization, altering organ distribution of the drug and subsequent clearance of the drug so as to achieve increase in drug therapeutic efficacy and reduction in side effects.
- Controlled release and particle degradation characteristics can be readily modulated by the choice of matrix constituents. Drug loading is relatively high and drugs can be incorporated into the systems without any chemical reaction; this is an important factor for preserving the drug activity.
- Site-specific targeting can be achieved by attaching targeting ligands to surface of particles or use of magnetic guidance.
- The system can be used for various routes of administration including oral, nasal, parenteral, intraocular etc.
NANODRUG DELIVERY SYSTEMS
The major goals in designing nanoparticles as a delivery system are to control particle size, surface properties [85] and release of pharmacologically active agents in order to achieve the site-specific action of the drug at the therapeutically optimal rate and dose regimen [86]. If nanoparticles are considered to be used as drug delivery vehicles, it depends on many factors including: (a) size of nanoparticles required; (b) inherent properties of the drug, e.g., aqueous solubility; (c) surface characteristics such as charge and permeability; (d) degree of biodegradability, biocompatibility and toxicity; (e) drug release profile desired; and (f) antigenicity of the final product. The advantages of using nanoparticles as a drug delivery system might be summarized as follow [87]:
- Particle size and surface characteristics of nanoparticles can be easily manipulated to achieve both passive and active drug targeting after parenteral administration.
- They control and sustain release of the drug during the transportation and at the site of localization, altering organ distribution of the drug and subsequent clearance of the drug so as to achieve increase in drug therapeutic efficacy and reduction in side effects.
- Controlled release and particle degradation characteristics can be readily modulated by the choice of matrix constituents. Drug loading is relatively high and drugs can be incorporated into the systems without any chemical reaction; this is an important factor for preserving the drug activity.
- Site-specific targeting can be achieved by attaching targeting ligands to surface of particles or use of magnetic guidance.
- The system can be used for various routes of administration including oral, nasal, parenteral, intraocular etc.
NERVOUS SYSTEM NANODRUGS
Nanomaterials and nanoparticles can interact with biological systems at fundamental and molecular levels [100, 101]. In neuroscience, the application of nanotechnologies entails specific interactions with neurons and glial cells. Nanodevices can target the cells and glia with a high degree of specificity. This unique molecular specificity enables the nanodrugs to stimulate and interact with tissues and neurons in controlled ways, while minimizing undesirable effects. There are two main types of nervous system drugs (neurodrugs): behavioural and molecular. Behavioural neurodrugs are for the study of how different drugs affect human behaviour and human brain. These drugs are usually used for diagnosis and therapy of neurodegeneration disorders [47, 102]. Molecular neurodrugs are used for the study of neurons and their neurochemical interactions. Since for the most part, neurons in the human brain communicate with one another by releasing chemical messengers called neurotransmitters, these drugs have to target specific transmitters and receptors to have beneficial effect on neurological functions. The preparation of these two types of drugs is closely connected. Researchers are studying the interactions of different neurotransmitters [103], neurohormones [104], neuromodulators [105], enzymes [106], second messengers [107], co-transporters [108, 109], ion channels [110], and receptor proteins [111] in the central and peripheral nervous systems to develop drugs to treat many different neurological disorders, including pain [112], neurodegenerative diseases such as Parkinson’s disease [113] and Alzheimer’s disease [114], psychological disorders [115], addiction [116], and many others.
The blood–brain barrier significantly hinders the passage of systemically delivered therapeutics and the brain extracellular matrix limits the distribution and longevity of locally delivered agents. Nanoparticles represent a promising solution to these problems. They can cross blood-brain barrier and enter the CNS. Although the applications of nanotechnology in basic and clinical neuroscience are only in the early stages of development, partly because of the complexities associated with interacting with neural cells and the mammalian nervous system, however the early results show an impressive potential of nanotechnologies to contribute to neuroscience research [117]. One area in which nanotechnology may have a significant clinical impact in neuroscience is the selective transport and delivery of drugs and other small molecules across the blood brain barrier that cannot cross otherwise.
Examples of current research include technologies that are designed to better interact with neural cells, advanced molecular imaging technologies [118, 119], materials and hybrid molecules used in neural regeneration [120], neuroprotection [121], and targeted delivery of drugs and small molecules across the blood–brain barrier [122, 123]. Among all these modern methods of drug delivery to the central nervous system (CNS), the design and application of bionanotechnologies aimed at the CNS provide revolutionary new approaches for studying cell and molecular biology and physiology. The successful and meaningful development of bionanotechnologies designed to interact with the CNS as research or clinical tools require an understanding of the relevant neurophysiology and neuropathology, an appreciation of the inherent ‘nanoscale’ structure of the CNS, and an understanding of the relevant chemistry and materials science and engineering. At nanoscale, consideration of individual molecules and interacting groups of molecules in relation to the bulk macroscopic properties of the material or device becomes important, since it is control over the fundamental molecular structure that allows control over the macroscopic chemical and physical properties [124]. Applications to neuroscience and physiology imply materials and devices designed to interact with the body at subcellular (i.e., molecular) scales with a high degree of specificity. This can potentially translate into targeted cellular and tissue-specific clinical applications designed to achieve maximal therapeutic affects with minimal side effects.
It started with controlled release strategy and the development of miniaturized delivery systems [125] and continued by the application of albumin nanoparticles for the first time in the Johns Hopkins Medical Institution in Baltimore [126]. Other nanoconstructs such as drug-polymer conjugates were first proposed in the 1970s [127] and developed pre-clinically in the 1980s [128]. Prof. Kreuter [129] proposed a definition of polymeric nanoparticles for pharmaceutical purposes for the first time that later was adopted by the Encyclopaedia of Pharmaceutical Technology [130] and the Encyclopedia of nanotechnology [131]. Today, more than 25 nanomedicines have already been approved for human use [102]. Usually the application of nanodrugs to neuroscience is divided into two parts: application in basic neuroscience [124], and in clinical neuroscience [27].
The development of nanotechnology products may play an important role in adding a new group of therapeutics to the products of pharmaceutical companies [132]. Nanotechnology enhances (1) delivery of poorly water-soluble drugs; (2) delivery of large macromolecule drugs to intracellular sites of action; (3) targeted delivery of drugs in a cell- or tissue-specific manner; (4) transcytosis of drugs across tight epithelial and endothelial barriers; (5) co-delivery of two or more drugs or therapeutic modality for combination therapy; (6) visualization of sites of drug delivery by combining therapeutic agents with imaging modalities; and (7) real-time read on the in vivo efficacy of a therapeutic agent [133]. Additionally, the manufacturing complexity of nanotechnology therapeutics may also create a significant hurdle for generic drug companies to develop equivalent therapeutics readily [132].
…….
Safe, site-specific, and efficient delivery of compounds to CNS disease sites remains a singular goal in achieving optimal therapeutic outcomes to combat neurodegenerative diseases. Treatment of CNS disorders by systemic administration or local delivery of drugs is currently inefficient in many cases. Furthermore, clinical neuroscience faces great challenges due to the extremely heterogeneous cellular and molecular environment and the complexities of the brain’s anatomical and functional “wiring” and associated information processing [224]. However, the emergence of nanotechnology provides hope that it will revolutionize diagnosis and treatment of CNS disorders. Neurodegenerative diseases are usually linked to a loss of brain and spinal cord cells. For example, the neuronal damage in AD and PD is associated with abnormal protein processing and accumulation and results in gradual cognitive and motor deterioration [225].
Like this:
Like Loading...
Read Full Post »





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}