Posts Tagged ‘Nitric oxide synthase’
Protected: Flywheel iNO, Three Novel Adult Patient Inhaled Nitric Oxide Product Concepts by Justin D. Pearlman MD ME PhD FACC
Posted in Medical Devices R&D Investment, Medical Imaging Technology, Image Processing/Computing, MRI, CT, Nuclear Medicine, Ultra Sound, Nitric Oxide in Health and Disease, Technology Transfer: Biotech and Pharmaceutical, tagged Heart Failure, Hypertension, nitric oxide, Nitric oxide synthase, Pulmonary hypertension, Torr on June 3, 2013|
Telling NO to Cardiac Risk
Posted in Bio Instrumentation in Experimental Life Sciences Research, Biological Networks, Gene Regulation and Evolution, Biomarkers & Medical Diagnostics, Cardiac & Vascular Repair Tools Subsegment, Cardiovascular Pharmacogenomics, Cell Biology, Signaling & Cell Circuits, Cerebrovascular and Neurodegenerative Diseases, Chemical Biology and its relations to Metabolic Disease, Chemical Genetics, Disease Biology, Small Molecules in Development of Therapeutic Drugs, Frontiers in Cardiology and Cardiovascular Disorders, Genome Biology, Health Economics and Outcomes Research, HTN, Molecular Genetics & Pharmaceutical, Nitric Oxide in Health and Disease, Origins of Cardiovascular Disease, Personalized and Precision Medicine & Genomic Research, Pharmaceutical R&D Investment, Pharmacotherapy of Cardiovascular Disease, Population Health Management, Genetics & Pharmaceutical, Regulated Clinical Trials: Design, Methods, Components and IRB related issues, Technology Transfer: Biotech and Pharmaceutical, tagged ADMA, Asymmetric dimethylarginine, Aviva Lev-Ari, Biology, Cardiovascular disease, Cardiovascular Disorders, Cell Biology, Conditions and Diseases, DDAH, Endothelium, health, Heart disease, medicine, nitric oxide, Nitric oxide synthase, NO, PubChem, research, ROS, science, Vascular smooth muscle on December 10, 2012| 9 Comments »
Telling NO to Cardiac Risk
DDAH Says NO to ADMA(1); The DDAH/ADMA/NOS Pathway(2)
Author-Writer-Reporter: Stephen J. Williams, PhD
Endothelium-derived nitric oxide (NO) has been shown to be vasoprotective. Nitric oxide enhances endothelial cell survival, inhibits excessive proliferation of vascular smooth muscle cells, regulates vascular smooth muscle tone, and prevents platelets from sticking to the endothelial wall. Together with evidence from preclinical and human studies, it is clear that impairment of the NOS pathway increases risk of cardiovascular disease (3-5).
This post contains two articles on the physiological regulation of nitric oxide (NO) by an endogenous NO synthase inhibitor asymmetrical dimethylarginine (ADMA) and ADMA metabolism by the enzyme DDAH(1,2). Previous posts on nitric oxide, referenced at the bottom of the page, provides excellent background and further insight for this posting. In summary plasma ADMA levels are elevated in patients with cardiovascular disease and several large studies have shown that plasma ADMA is an independent biomarker for cardiovascular-related morbidity and mortality(6-8).
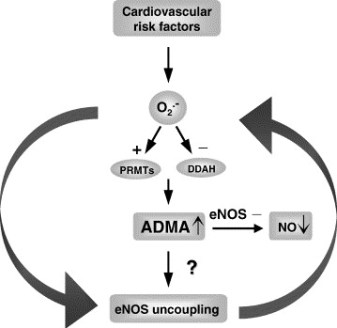

Figure 1 A. Cardiac risks of ADMA B. Effects of ADMA (Photo credit: Wikipedia)
ADMA Production and Metabolism
Nuclear proteins such as histones can be methylated on arginine residues by protein-arginine methyltransferases, enzymes which use S-adenosylmethionine as methyl groups. This methylation event is thought to regulate protein function, much in the way of protein acetylation and phosphorylation (9). And much like phosphorylation, these modifications are reversible through methylesterases. The proteolysis of these arginine-methyl modifications lead to the liberation of free guanidine-methylated arginine residues such as L-NMMA, asymmetric dimethylarginine (ADMA) and symmetrical methylarginine (SDMA).
The first two, L-NMMA and ADMA, have been shown to inhibit the activity of the endothelial NOS. This protein turnover is substantial: for instance the authors note that each day 40% of constitutive protein in adult liver is newly synthesized protein. And in several diseases, such as muscular dystrophy, ischemic heart disease, and diabetes, it has been known since the 1970’s that protein catabolism rates are very high, with corresponding increased urinary excretion of ADMA(10-13). Methylarginines are excreted in the urine by cationic transport. However, the majority of ADMA and L-NMMA are degraded within the cell by dimethylaminohydrolase (DDAH), first cloned and purified in rat(14).

Figure 2. Endogenous inhibitors of NO synthase. Chemical structures generated from PubChem.
DDAH
DDAH specifically hydrolyzes ADMA and L-NMMA to yield citruline and demethylamine and usually shows co-localization with NOS. Pharmacologic inhibition of DDAH activity causes accumulation of ADMA and can reverse the NO-mediated bradykinin-induced relaxation of human saphenous vein.
Two isoforms have been found in human:
- DDAH1 (found in brain and kidney and associated with nNOS) and
- DDAH2 (highly expressed in heart, placenta, and kidney and associated with eNOS).
DDAH2 can be upregulated by all-trans retinoic acid (atRA can increase NO production). Increased reactive oxygen species and possibly homocysteine, a risk factor for cardiovascular disease, can decrease DDAH activity(15,16).
- The importance of DDAH activity can also be seen in transgenic mice which overexpress DDAH, exhibiting increased NO production, increased insulin sensitivity, and reduced vascular resistance (17). Likewise,
- Transgenic mice, null for the DDAH1, showed increase in blood pressure, decreased NO production, and significant increase in tissue and plasma ADMA and L-NMMA.

Figure 3. The DDAH/ADMA/NOS cycle. Figure adapted from Cooke and Ghebremarian (1).
As mentioned in the article by Cooke and Ghebremariam, the authors state: the weight of the evidence indicates that DDAH is a worthy therapeutic target. Agents that increase DDAH expression are known, and 1 of these, a farnesoid X receptor agonist, is in clinical trials
http://www.interceptpharma.com
An alternate approach is to
- develop an allosteric activator of the enzyme. Although
- development of an allosteric activator is not a typical pharmaceutical approach, recent studies indicate that this may be achievable aim(18).
References:
1. Cooke, J. P., and Ghebremariam, Y. T. : DDAH says NO to ADMA.(2011) Arteriosclerosis, thrombosis, and vascular biology 31, 1462-1464
2. Tran, C. T., Leiper, J. M., and Vallance, P. : The DDAH/ADMA/NOS pathway.(2003) Atherosclerosis. Supplements 4, 33-40
3. Niebauer, J., Maxwell, A. J., Lin, P. S., Wang, D., Tsao, P. S., and Cooke, J. P.: NOS inhibition accelerates atherogenesis: reversal by exercise. (2003) American journal of physiology. Heart and circulatory physiology 285, H535-540
4. Miyazaki, H., Matsuoka, H., Cooke, J. P., Usui, M., Ueda, S., Okuda, S., and Imaizumi, T. : Endogenous nitric oxide synthase inhibitor: a novel marker of atherosclerosis.(1999) Circulation 99, 1141-1146
5. Wilson, A. M., Shin, D. S., Weatherby, C., Harada, R. K., Ng, M. K., Nair, N., Kielstein, J., and Cooke, J. P. (2010): Asymmetric dimethylarginine correlates with measures of disease severity, major adverse cardiovascular events and all-cause mortality in patients with peripheral arterial disease. Vasc Med 15, 267-274
6. Kielstein, J. T., Impraim, B., Simmel, S., Bode-Boger, S. M., Tsikas, D., Frolich, J. C., Hoeper, M. M., Haller, H., and Fliser, D. : Cardiovascular effects of systemic nitric oxide synthase inhibition with asymmetrical dimethylarginine in humans.(2004) Circulation 109, 172-177
7. Kielstein, J. T., Donnerstag, F., Gasper, S., Menne, J., Kielstein, A., Martens-Lobenhoffer, J., Scalera, F., Cooke, J. P., Fliser, D., and Bode-Boger, S. M. : ADMA increases arterial stiffness and decreases cerebral blood flow in humans.(2006) Stroke; a journal of cerebral circulation 37, 2024-2029
8. Mittermayer, F., Krzyzanowska, K., Exner, M., Mlekusch, W., Amighi, J., Sabeti, S., Minar, E., Muller, M., Wolzt, M., and Schillinger, M. : Asymmetric dimethylarginine predicts major adverse cardiovascular events in patients with advanced peripheral artery disease.(2006) Arteriosclerosis, thrombosis, and vascular biology 26, 2536-2540
9. Kakimoto, Y., and Akazawa, S.: Isolation and identification of N-G,N-G- and N-G,N’-G-dimethyl-arginine, N-epsilon-mono-, di-, and trimethyllysine, and glucosylgalactosyl- and galactosyl-delta-hydroxylysine from human urine. (1970) The Journal of biological chemistry 245, 5751-5758
10. Inoue, R., Miyake, M., Kanazawa, A., Sato, M., and Kakimoto, Y.: Decrease of 3-methylhistidine and increase of NG,NG-dimethylarginine in the urine of patients with muscular dystrophy. (1979) Metabolism: clinical and experimental 28, 801-804
11. Millward, D. J.: Protein turnover in skeletal muscle. II. The effect of starvation and a protein-free diet on the synthesis and catabolism of skeletal muscle proteins in comparison to liver. (1970) Clinical science 39, 591-603
12. Goldberg, A. L., and St John, A. C.: Intracellular protein degradation in mammalian and bacterial cells: Part 2. (1976) Annual review of biochemistry 45, 747-803
13. Dice, J. F., and Walker, C. D.: Protein degradation in metabolic and nutritional disorders. (1979) Ciba Foundation symposium, 331-350
14. Ogawa, T., Kimoto, M., and Sasaoka, K.: Purification and properties of a new enzyme, NG,NG-dimethylarginine dimethylaminohydrolase, from rat kidney. (1989) The Journal of biological chemistry 264, 10205-10209
15. Ito, A., Tsao, P. S., Adimoolam, S., Kimoto, M., Ogawa, T., and Cooke, J. P.: Novel mechanism for endothelial dysfunction: dysregulation of dimethylarginine dimethylaminohydrolase. (1999) Circulation 99, 3092-3095
16. Stuhlinger, M. C., Tsao, P. S., Her, J. H., Kimoto, M., Balint, R. F., and Cooke, J. P. : Homocysteine impairs the nitric oxide synthase pathway: role of asymmetric dimethylarginine.(2001) Circulation 104, 2569-2575
17. Sydow, K., Mondon, C. E., Schrader, J., Konishi, H., and Cooke, J. P.: Dimethylarginine dimethylaminohydrolase overexpression enhances insulin sensitivity. (2008) Arteriosclerosis, thrombosis, and vascular biology 28, 692-697
18. Zorn, J. A., and Wells, J. A.: Turning enzymes ON with small molecules. (2010) Nature chemical biology 6, 179-188
Other research papers on Nitric Oxide and Cardiac Risk were published on this Scientific Web site as follows:
The Nitric Oxide and Renal is presented in FOUR parts:
Part I: The Amazing Structure and Adaptive Functioning of the Kidneys: Nitric Oxide
Part II: Nitric Oxide and iNOS have Key Roles in Kidney Diseases
Part III: The Molecular Biology of Renal Disorders: Nitric Oxide
Part IV: New Insights on Nitric Oxide donors
Cardiac Arrhythmias: A Risk for Extreme Performance Athletes
What is the role of plasma viscosity in hemostasis and vascular disease risk?
Biochemistry of the Coagulation Cascade and Platelet Aggregation – Part I
Nitric Oxide and iNOS have Key Roles in Kidney Diseases – Part II
Posted in Cell Biology, Signaling & Cell Circuits, Disease Biology, Small Molecules in Development of Therapeutic Drugs, HTN, Human Immune System in Health and in Disease, International Global Work in Pharmaceutical, Nitric Oxide in Health and Disease, Pharmaceutical Industry Competitive Intelligence, Systemic Inflammatory Response Related Disorders, tagged cytokine, Larry H. Bernstein, nephron, nitric oxide, Nitric oxide synthase, oxidative stress, Peroxinitrite, Renal function on November 26, 2012| 10 Comments »
Subtitle: Nitric Oxide, Peroxinitrite, and NO donors in Renal Function Loss
Curator and Author: Larry H. Bernstein, MD, FCAP
The Nitric Oxide and Renal is presented in FOUR parts:
Part I: The Amazing Structure and Adaptive Functioning of the Kidneys: Nitric Oxide
Part II: Nitric Oxide and iNOS have Key Roles in Kidney Diseases
Part III: The Molecular Biology of Renal Disorders: Nitric Oxide
Part IV: New Insights on Nitric Oxide donors
Conclusion to this series is presented in
The Essential Role of Nitric Oxide and Therapeutic NO Donor Targets in Renal Pharmacotherapy
Part II. Oxidative Stress and Regulating a Balance of Redox Potential is Central to Disordered Kidney Function
We have already described the key role that nitric oxide and the NO synthases play in reduction of oxidative stress. The balance that has to be regulated between pro- and anti-oxidative as well as inflammatory elements necessary for renal function, critically involves the circulation of the kidney. It poses an inherent risk in the kidney, where the existence of a rich circulatory and high energy cortical outer region surrounds a medullary inner portion that is engaged in the retention of water, the active transport of glucose, urea and uric acid nitrogenous waste, mineral balance and pH. In this discussion we shall look at kidney function, NO, and the large energy fluxes in the medullary tubules and interstitium. This is a continuation of of a series of posts on NO and NO related disorders, and the kidney in particular.
Part IIa. Nitric Oxide role in renal tubular epithelial cell function
Tubulointerstitial Nephritides
As part of the exponential growth in our understanding of nitric oxide (NO) in health and disease over the past 2 decades, the kidney has become appreciated as a major site where NO may play a number of important roles. Although earlier work on the kidney focused more on effects of NO at the level of larger blood vessels and glomeruli, there has been a rapidly growing body of work showing critical roles for NO in tubulointerstitial disease. In this review we discuss some of the recent contributions to this important field.
Mattana J, Adamidis A, Singhal PC. Nitric oxide and tubulointerstitial nephritides. Seminars in Nephrology 2004; 24(4):345-353.
Nitric oxide donors and renal tubular (subepithelial) matrix
Nitric oxide (NO) and its metabolite, peroxynitrite (ONOO-), are involved in renal tubular cell injury. If NO/ONOO- has an effect to reduce cell adhesion to the basement membrane, does this effect contribute to tubular obstruction and would it be partially responsible for the harmful effect of NO on the tubular epithelium during acute renal failure (ARF)?
Wangsiripaisan A, et al. examined the effect of the NO donors
- (z)-1-[2-(2-aminoethyl)-N-(2-ammonioethyl)amino]diazen-1- ium-1, 2-diolate (DETA/NO),
- spermine NONOate (SpNO), and
- the ONOO- donor 3-morpholinosydnonimine (SIN-1)
on cell-matrix adhesion to collagen types I and IV, and also fibronectin
using three renal tubular epithelial cell lines:
- LLC-PK1,
- BSC-1,
- OK.
It was only the exposure to SIN-1 that caused a dose-dependent impairment in cell-matrix adhesion. Similar results were obtained in the different cell types and matrix proteins. The effect of SIN-1 (500 microM) on LLC-PK1 cell adhesion was not associated with either cell death or alteration of matrix protein and was attenuated by either
- the NO scavenger 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide,
- the superoxide scavenger superoxide dismutase, or
- the ONOO- scavenger uric acid in a dose-dependent manner.
These investigators concluded in this seminal paper that ONOO- generated in the tubular epithelium during ischemia/reperfusion has the potential to impair the adhesion properties of tubular cells, which then may contribute to the tubular obstruction in ARF.
Wangsiripaisan A, Gengaro PE, Nemenoff RA, Ling H, et al. Effect of nitric oxide donors on renal tubular epithelial cell-matrix adhesion. Kidney Int 1999; 55(6):2281-8.

The reaction mechanism of Nitric oxide synthase (Photo credit: Wikipedia)

Nitric Oxide Synthase (Photo credit: Wikipedia)

English: Reactions leading to generation of Nitric Oxide and Reactive Nitrogen Species. Novo and Parola Fibrogenesis & Tissue Repair 2008 1:5 doi:10.1186/1755-1536-1-5 (Photo credit: Wikipedia)
Coexpressed Nitric Oxide Synthase and Apical β1 Integrins
In sepsis-induced acute renal failure, actin cytoskeletal alterations result in shedding of proximal tubule epithelial cells (PTEC) and tubular obstruction. This study examined the hypothesis that inflammatory cytokines, released early in sepsis, cause PTEC cytoskeletal damage and alter integrin-dependent cell-matrix adhesion. The question of whether the intermediate nitric oxide (NO) modulates these cytokine effects was also examined.
After exposure of human PTEC to
- tumor necrosis factor-α,
- interleukin-1α, and
- interferon-γ,
the actin cytoskeleton was disrupted and cells became elongated, with extension of long filopodial processes.
Cytokines induced shedding of
- viable,
- apoptotic, and
- necrotic PTEC,
which was dependent on NO synthesized by inducible NO synthase (iNOS) produced as a result of cytokine actions on PTEC.
Basolateral exposure of polarized PTEC monolayers to cytokines induced maximal NO-dependent cell shedding, mediated in part through NO effects on cGMP. Cell shedding was accompanied by dispersal of
- basolateral β1 integrins and
- E-cadherin,
with corresponding upregulation of integrin expression in clusters of cells elevated above the epithelial monolayer.
These cells demonstrated coexpression of iNOS and apically redistributed β1 integrins. These authors point out that the major ligand involved in cell anchorage was laminin, probably through interactions with the integrin α3β1. This interaction was downregulated by cytokines but was not dependent on NO. They posulate a mechanism by which inflammatory cytokines induce PTEC damage in sepsis, in the absence of hypotension and ischemia.
Glynne PA, Picot J and Evans TJ. Coexpressed Nitric Oxide Synthase and Apical β1 Integrins Influence Tubule Cell Adhesion after Cytokine-Induced Injury. JASN 2001; 12(11): 2370-2383.
Potentiation by Nitric Oxide of Apoptosis in Renal Proximal Tubule Cells
Proximal tubular epithelial cells (PTEC) exhibit a high sensitivity to undergo apoptosis in response to proinflammatory stimuli and immunosuppressors and participate in the onset of several renal diseases. This study examined the expression of inducible nitric oxide (NO) synthase after challenge of PTEC with bacterial cell wall molecules and inflammatory cytokines and analyzed the pathways that lead to apoptosis in these cells by measuring changes in the mitochondrial transmembrane potential and caspase activation.
The data show that the apoptotic effects of proinflammatory stimuli mainly were due to the expression of inducible NO synthase. Cyclosporin A and FK506 inhibited partially NO synthesis. However, both NO and immunosuppressors induced apoptosis, probably through a common mechanism that involved the irreversible opening of the mitochondrial permeability transition pore. Activation of caspases 3 and 7 was observed in cells treated with high doses of NO and with moderate concentrations of immunosuppressors. The conclusion is that the cooperation between NO and immunosuppressors that induce apoptosis in PTEC might contribute to the renal toxicity observed in the course of immunosuppressive therapy.
HORTELANO S, CASTILLA M, TORRES AM, TEJEDOR A, and BOSCÁ L. Potentiation by Nitric Oxide of Cyclosporin A and FK506- Induced Apoptosis in Renal Proximal Tubule Cells. J Am Soc Nephrol 2000; 11: 2315–2323.
Part IIb. Related studies with ROS and/or RNS on nonrenal epithelial cells
Reactive nitrogen species block cell cycle re-entry
Endogenous sources of reactive nitrogen species (RNS) act as second messengers in a variety of cell signaling events, whereas environmental sources of RNS like nitrogen dioxide (NO2) inhibit cell survival and growth through covalent modification of cellular macromolecules.
Murine type II alveolar cells arrested in G0 by serum deprivation were exposed to either NO2 or SIN-1, a generator of RNS, during cell cycle re-entry. In serum-stimulated cells, RNS blocked cyclin D1 gene expression, resulting in cell cycle arrest at the boundary between G0 and G1. Dichlorofluorescin diacetate (DCF) fluorescence indicated that RNS induced sustained production of intracellular hydrogen peroxide (H2O2), which normally is produced only transiently in response to serum growth factors.
Loading cells with catalase prevented enhanced DCF fluorescence and rescued cyclin D1 expression and S phase entry.
These studies indicate environmental RNS interfere with cell cycle re-entry through an H2O2-dependent mechanism that influences expression of cyclin D1 and progression from G0 to the G1 phase of the cell cycle.
Yuan Z, Schellekens H, Warner L, Janssen-Heininger Y, Burch P, Heintz NH. Reactive nitrogen species block cell cycle re-entry through sustained production of hydrogen peroxide. Am J Respir Cell Mol Biol. 2003;28(6):705-12. Epub 2003 Jan 10.
Peroxynitrite modulates MnSOD gene expression
Peroxynitrite (ONOO-) is a strong oxidant derived from nitric oxide (‘NO) and superoxide (O2.-), reactive nitrogen (RNS) and oxygen species (ROS) present in inflamed tissue. Other oxidant stresses, e.g., TNF-alpha and hyperoxia, induce mitochondrial, manganese-containing superoxide dismutase (MnSOD) gene expression.
3-morpholinosydnonimine HCI (SIN-1) (10 or 1000 microM) increased MnSOD mRNA, but did not change hypoxanthine guanine phosphoribosyl transferase (HPRT) mRNA.
Authentic peroxynitrite (ONOO ) (100-500 microM) also increased MnSOD mRNA but did not change constitutive HPRT mRNA expression. ONOO stimulated luciferase gene expression driven by a 2.5 kb fragment of the rat MnSOD gene 5′ promoter region.
MnSOD gene induction due to ONOO- was inhibited effectively by L-cysteine (10 mM) and partially inhibited by N-acetyl cysteine (50 mM) or pyrrole dithiocarbamate (10 mM).
.NO from 1-propanamine, 3-(2-hydroxy-2-nitroso-1-propylhydrazine) (PAPA NONOate) (100 or 1000 microM) did not change MnSOD or HPRT mRNA, nor did either H202 or NO2-, breakdown products of SIN-1 and ONOO, have any effect on MnSOD mRNA expression; ONOO- and SIN-1 also did not increase detectable MnSOD protein content or increase MnSOD enzymatic activity.
Nevertheless, increased steady state [O2.-] in the presence of .NO yields ONOO , and ONOO has direct, stimulatory effects on MnSOD transcript expression driven at the MnSOD gene 5′ promoter region inhibited completely by L-cysteine and partly by N-acetyl cysteine in lung epithelial cells. This raises a question of whether the same effect is seen in renal tubular epithelium.
Jackson RM, Parish G, Helton ES. Peroxynitrite modulates MnSOD gene expression in lung epithelial cells. Free Radic Biol Med. 1998; 25(4-5):463-72.
Comparative impacts of glutathione peroxidase-1 gene knockout on oxidative stress
Selenium-dependent glutathione peroxidase-1 (GPX1) protects against reactive-oxygen-species (ROS)-induced oxidative stress in vivo, but its role in coping with reactive nitrogen species (RNS) is unclear. Primary hepatocytes were isolated from GPX1-knockout (KO) and wild-type (WT) mice to test protection of GPX1 against cytotoxicity of
- superoxide generator diquat (DQ),
- NO donor S-nitroso-N-acetyl-penicillamine (SNAP) and
- peroxynitrite generator 3-morpholinosydnonimine (SIN-1).
Treating cells with SNAP (0.1 or 0.25 mM) in addition to DQ produced synergistic cytotoxicity that minimized differences in apoptotic cell death and oxidative injuries between the KO and WT cells. Less protein nitrotyrosine was induced by 0.05-0.5 mM DQ+0.25 mM SNAP in the KO than in the WT cells.
Total GPX activity in the WT cells was reduced by 65 and 25% by 0.5 mM DQ+0.1 mM SNAP and 0.5 mM DQ, respectively.
Decreases in Cu,Zn-superoxide dismutase (SOD) activity and increases in Mn-SOD activity in response to DQ or DQ+SNAP were greater in the KO cells than in the WT cells.
The study indicates GPX1 was more effective in protecting hepatocytes against oxidative injuries mediated by ROS alone than by ROS and RNS together, and knockout of GPX1 did not enhance cell susceptibility to RNS-associated cytotoxicity. Instead, it attenuated protein nitration induced by DQ+SNAP.
To better understand the mechanism(s) underlying nitric oxide (. NO)-mediated toxicity, in the presence and absence of concomitant oxidant exposure, postmitotic terminally differentiated NT2N cells, which are incapable of producing . NO, were exposed to PAPA-NONOate (PAPA/NO) and 3-morpholinosydnonimine (SIN-1).
Exposure to SIN-1, which generated peroxynitrite (ONOO) in the range of 25-750 nM/min, produced a concentration- and time-dependent delayed cell death.
In contrast, a critical threshold concentration (>440 nM/min) was required for . NO to produce significant cell injury.
There is a largely necrotic lesion after ONOO exposure and an apoptotic-like morphology after . NO exposure. Cellular levels of reduced thiols correlated with cell death, and pretreatment with N-acetylcysteine (NAC) fully protected from cell death in either PAPA/NO or SIN-1 exposure.
NAC given within the first 3 h posttreatment further delayed cell death and increased the intracellular thiol level in SIN-1 but not . NO-exposed cells.
Cell injury from . NO was independent of cGMP, caspases, and superoxide or peroxynitrite formation.
Overall, exposure of non-. NO-producing cells to . NO or peroxynitrite results in delayed cell death, which, although occurring by different mechanisms,
appears to be mediated by the loss of intracellular redox balance.
Gow AJ, Chen Q, Gole M, Themistocleous M, Lee VM, Ischiropoulos H. Two distinct mechanisms of nitric oxide-mediated neuronal cell death show thiol dependency. Am J Physiol Cell Physiol. 2000; 278(6):C1099-107.

Oxidative stress (Photo credit: Wikipedia)

English: Binding of CAPON results in a reduction of NMDA receptor/nitric oxide synthase (NOS) complexes, leading to decreased NMDA receptor–gated calcium influx and a catalytically inactive nitric oxide synthase. Overexpression of either the full-length or the novel shortened CAPON isoform as reported by Brzustowicz and colleagues is, therefore, predicted to lead to impaired NMDA receptor–mediated glutamate neurotransmission. (Photo credit: Wikipedia)
NO2 effect on phosphatidyl choline
Nitrogen dioxide (NO2) inhalation affects the extracellular surfactant as well as the structure and function of type II pneumocytes. The studies had differences in oxidant concentration, duration of exposure, and mode of NO2 application.
This study evaluated the influence of the NO2 application mode on the phospholipid metabolism of type II pneumocytes . Rats were exposed to identical NO2 body doses (720 ppm x h), which were applied continuously (10 ppm for 3 d), intermittently (10 ppm for 8 h per day, for 9 d), and repeatedly (10 ppm for 3 d, 28 d rest, and then 10 ppm for 3 d). Immediately after exposure, type II cells were isolated and evaluated for
- cell yield,
- vitality,
- phosphatidylcholine (PC) synthesis, and
- secretion.
Type II pneumocyte cell yield was only increased from animals that had been continuously exposed to NO2, but vitality of the isolated type II pneumocytes was not affected by the NO2 exposure modes. Continuous application of 720 ppm x h NO2 resulted in increased activity of the cytidine-5-diphosphate (CDP)-choline pathway.
- After continuous NO2 application,
- specific activity of choline kinase,
- cytidine triphosphate (CTP):cholinephosphate cytidylyltransferase,
- uptake of choline, and
- pool sizes of CDP-choline and PC
were significantly increased over those of controls.
Intermittent application of this NO2 body dose provoked less increase in PC synthesis and the synthesis parameters were comparable to those for cells from control animals after repeated exposure. Whereas PC synthesis in type II cells was stimulated by NO2, their secretory activity was reduced. Continuous exposure reduced the secretory activity most, whereas intermittent exposure nonsignificantly reduced this activity as compared with that of controls. The repeated application of NO2 produced no differences.
The authors conclude that type II pneumocytes adapt to NO2 atmospheres depending on the mode of its application, at least for the metabolism of PC and its secretion from isolated type II pneumocytes. Further studies are necessary to determine whether additional metabolic activities will also adapt to NO2 atmospheres, and if these observations are specific for NO2 or represent effects generally due to oxidants. The reader, however, asks whether this effect could also be found in renal epithelial cells, for which PC is not considered vital as for type II pneumocytes and possibly related to surfactant activity in the lung.
Müller B, Seifart C, von Wichert P, Barth PJ. Adaptation of rat type II pneumocytes to NO2: effects of NO2 application mode on phosphatidylcholine metabolism. Am J Respir Cell Mol Biol. 1998; 18(5): 712-20.
iNOS involved in immediate response to anaphylaxis
The generation of large quantities of nitric oxide (NO) is implicated in the pathogenesis of anaphylactic shock. The source of NO, however, has not been established and conflicting results have been obtained when investigators have tried to inhibit its production in anaphylaxis.
This study analyzed the expression of inducible nitric oxide synthase (iNOS) and endothelial nitric oxide synthase (eNOS) in a mouse model of anaphylaxis.
BALB/c mice were sensitized and challenged with ovalbumin to induce anaphylaxis. Tissues were removed from the heart and lungs, and blood was drawn at different time points during the first 48 hours after induction of anaphylaxis. The Griess assay was used to measure nitric oxide generation. Nitric oxide synthase expression was examined by reverse transcriptase polymerase chain reaction and immunohistochemistry.
A significant increase in iNOS mRNA expression and nitric oxide production was evident as early as 10 to 30 minutes after allergen challenge in both heart and lungs. In contrast, expression of eNOS mRNA was not altered during the course of the experiment.
The results support involvement of iNOS in the immediate physiological response of anaphylaxis.
Sade K, Schwartz IF, Etkin S, Schwartzenberg S, et al. Expression of Inducible Nitric Oxide
Synthase in a Mouse Model of Anaphylaxis. J Investig Allergol Clin Immunol 2007; 17(6):379-385.
Part IIc. Additional Nonrenal Related NO References
Nitrogen dioxide induces death in lung epithelial cells in a density-dependent manner.
Persinger RL, Blay WM, Heintz NH, Hemenway DR, Janssen-Heininger YM.
Am J Respir Cell Mol Biol. 2001 May;24(5):583-90.
PMID: 11350828 [PubMed – indexed for MEDLINE] Free Article
2.
Molecular mechanisms of nitrogen dioxide induced epithelial injury in the lung.
Persinger RL, Poynter ME, Ckless K, Janssen-Heininger YM.
Mol Cell Biochem. 2002 May-Jun;234-235(1-2):71-80. Review.
PMID: 12162462 [PubMed – indexed for MEDLINE]
3.
Nitric oxide and peroxynitrite-mediated pulmonary cell death.
Gow AJ, Thom SR, Ischiropoulos H.
Am J Physiol. 1998 Jan;274(1 Pt 1):L112-8.
PMID: 9458808 [PubMed – indexed for MEDLINE] Free Article
4.
Mitogen-activated protein kinases mediate peroxynitrite-induced cell death in human bronchial epithelial cells.
Nabeyrat E, Jones GE, Fenwick PS, Barnes PJ, Donnelly LE.
Am J Physiol Lung Cell Mol Physiol. 2003 Jun;284(6):L1112-20. Epub 2003 Feb 21.
PMID: 12598225 [PubMed – indexed for MEDLINE] Free Article
5.
Peroxynitrite inhibits inducible (type 2) nitric oxide synthase in murine lung epithelial cells in vitro.
Robinson VK, Sato E, Nelson DK, Camhi SL, Robbins RA, Hoyt JC.
Free Radic Biol Med. 2001 May 1;30(9):986-91.
PMID: 11316578 [PubMed – indexed for MEDLINE]
6.
Nitric oxide-mediated chondrocyte cell death requires the generation of additional reactive oxygen species.
Del Carlo M Jr, Loeser RF.
Arthritis Rheum. 2002 Feb;46(2):394-403.
PMID: 11840442 [PubMed – indexed for MEDLINE]
7.
Colon epithelial cell death in 2,4,6-trinitrobenzenesulfonic acid-induced colitis is associated with increased inducible nitric-oxide synthase expression and peroxynitrite production.
Yue G, Lai PS, Yin K, Sun FF, Nagele RG, Liu X, Linask KK, Wang C, Lin KT, Wong PY.
J Pharmacol Exp Ther. 2001 Jun;297(3):915-25.
PMID: 11356911 [PubMed – indexed for MEDLINE] Free Article
Summary
In this piece I have covered the conflicting roles of endogenous end inducible nitric oxide (eNOS and iNOS) in the reaction to reactive oxygen and nitrogen stress (ROS, RNS), and many experiments directed at sorting out these effects using continuous and intermittent delivery of NO2, production of ONOO- from .NO, and several agents that are used to upregulate and downregulate the underlying mechanism of response. These investigations are not only carried out in experiments on renal function and apoptosis, but also there are similar examples taken from studies of lung and liver. This forms a backdrop for the assessment of renal diseases:
- immune related
- acute traumatic injury
- chronic
The continuation of the discussion will be in essays that follow.

A scheme of the shear stress-induced EDRF-NO mechanism (Photo credit: Wikipedia)
Related articles
- Differential Distribution of Nitric Oxide – A 3-D Mathematical Model (pharmaceuticalintelligence.com)
- Low Bioavailability of Nitric Oxide due to Misbalance in Cell Free Hemoglobin in Sickle Cell Disease – A Computational Model (pharmaceuticalintelligence.com)
- Crucial role of Nitric Oxide in Cancer (pharmaceuticalintelligence.com)
- Nitric Oxide and Immune responses: part 1 (pharmaceuticalintelligence.com)
- Nitric Oxide and Sepsis, Hemodynamic Collapse, and the Search for Therapeutic Options (pharmaceuticalintelligence.com)
- Statins’ nonlipid effects on vascular endothelium through eNOS activation (pharmaceuticalintelligence.com)
- Nitric Oxide and Immune Responses: Part 2 (pharmaceuticalintelligence.com)
New Insights on Nitric Oxide donors – Part IV
Posted in Biological Networks, Gene Regulation and Evolution, Biomarkers & Medical Diagnostics, Cell Biology, Signaling & Cell Circuits, Chemical Biology and its relations to Metabolic Disease, Disease Biology, Small Molecules in Development of Therapeutic Drugs, Genome Biology, HTN, Human Immune System in Health and in Disease, Metabolomics, Nitric Oxide in Health and Disease, Pharmaceutical Industry Competitive Intelligence, Proteomics, Stem Cells for Regenerative Medicine, Systemic Inflammatory Response Related Disorders, tagged Cell Biology, Larry H. Bernstein, nitric oxide, Nitric oxide synthase, Peroxinitrite, Reactive oxygen species, Renal function on November 26, 2012| 9 Comments »
Subtitle: Nitric Oxide, Peroxinitrite, and NO donors in Renal Function Loss
Curator and Author: Larry H. Bernstein, MD, FCAP
The Nitric Oxide and Renal is presented in FOUR parts:
Part I: The Amazing Structure and Adaptive Functioning of the Kidneys: Nitric Oxide
Part II: Nitric Oxide and iNOS have Key Roles in Kidney Diseases
Part III: The Molecular Biology of Renal Disorders: Nitric Oxide
Part IV: New Insights on Nitric Oxide donors
Conclusion to this series is presented in
The Essential Role of Nitric Oxide and Therapeutic NO Donor Targets in Renal Pharmacotherapy
The criticality of renal function is easily overlooked until significant loss of nephron mass is overtly seen. The kidneys become acutely and/or chronically dysfunctional in metabolic, systemic inflammatory and immunological diseases of man. We have described how the key role that nitric oxide and the NO synthases (eNOS and iNOS) play competing roles in reduction or in the genesis of reactive oxygen species. There is a balance to be struck between pro- and anti-oxidative as well as inflammatory elements for avoidance of diseases, specifically involving the circulation, but effectively not limited to any organ system. In this discussion we shall look at kidney function, NO and NO donors. This is an extension of a series of posts on NO and NO related disorders.
Part IV. New Insights on NO donors
This study investigated the involvement of nitric oxide (NO) into the irradiation-induced increase of cell attachment. These experiments explored the cellular mechanisms of low-power laser therapy.
HeLa cells were irradiated with a monochromatic visible-tonear infrared radiation (600–860 nm, 52 J/m2) or with a diode laser (820 nm, 8–120 J/m2) and the number of cells attached to a glass matrix was counted after 30 minute incubation at 37oC. The NO donors
- sodium nitroprusside (SNP),
- glyceryl trinitrate (GTN), or
- sodium nitrite (NaNO2)
were added to the cellular suspension before or after irradiation. The action spectra and the concentration and fluence dependencies obtained were compared and analyzed. The well-structured action spectrum for the increase of the adhesion of the cells, with maxima at 619, 657, 675, 740, 760, and 820 nm, points to the existence of a photoacceptor responsible for the enhancement of this property (supposedly cytochrome c oxidase, the terminal respiratory chain enzyme), as well as signaling pathways between the cell mitochondria, plasma membrane, and nucleus. Treating the cellular suspension with SNP before irradiation significantly modifies the action spectrum for the enhancement of the cell attachment property (band maxima at 642, 685, 700, 742, 842, and 856 nm).
The action of SNP, GTN, andNaNO2 added before or after irradiation depends on their concentration and radiation fluence.
The NO donors added to the cellular suspension before irradiation eliminate the radiation induced increase in the number of cells attached to the glass matrix, supposedly by way of binding NO to cytochrome c oxidase. NO added to the suspension after irradiation can also inhibit the light-induced signal downstream. Both effects of NO depend on the concentration of the NO donors added. The results indicate that NO can control the irradiation-activated reactions that increase the attachment of cells.
Karu TI, Pyatibrat LV, and Afanasyeva NI. Cellular Effects of Low Power Laser Therapy Can be Mediated by Nitric Oxide. Lasers Surg. Med 2005; 36:307–314.
Interferon a-2b (IFN-a) effect on barrier function of renal tubular epithelium
IFNa treatment can be accompanied by impaired renal function and capillary leak. This study shows IFNa produced dose-dependent and time-dependent decrease in transepithelial resistance (TER) ameliorated by tyrphostin, an inhibitor of phosphotyrosine kinase with increased expression of occludin and E-cadherin. In conclusion, IFNa can directly affect barrier function in renal epithelial cells via overexpression or missorting of the junctional proteins occludin and E-cadherin.
Lechner J, Krall M, Netzer A, Radmayr C, et al. Effects of interferon a-2b on barrier function and junctional complexes of renal proximal tubular LLC-pK1 cells. Kidney Int 1999; 55:2178-2191.
Ischemia-reperfusion injury
The pathophysiology of acute renal failure (ARF) is complex and not well understood. Numerous models of ARF suggest that oxygen-derived reactive species are important in renal ischemia-reperfusion (I-R) injury, but the nature of the mediators is still controversial. Treatment with
- oxygen radical scavengers,
- antioxidants, and
- iron chelators such as
- superoxide dismutase,
- dimethylthiourea,
- allopurinol, and
- deferoxamine
are protective in some models, and suggest a role for the hydroxyl radical formation. However, these compounds are not protective in all models of I-R injury, and direct evidence for the generation of hydroxyl radical is absent. Furthermore, these inhibitors have another property in common. They all directly scavenge or inhibit the formation of peroxynitrite (ONOO−), a highly toxic species derived from nitric oxide (NO) and superoxide. Thus, the protective effects seen with these inhibitors may be due in part to their ability to inhibit ONOO− formation.
Even though reactive oxygen species are thought to participate in ischemia-reperfusion (I-R) injury, induction of inducible nitric oxide synthase (iNOS) and production of high levels of nitric oxide (NO) also contribute to this injury. NO can combine with superoxide to form the potent oxidant peroxynitrite (ONOO−). NO and ONOO− were investigated in a rat model of renal I-R injury using the selective iNOS inhibitor L-N6-(1-iminoethyl)lysine (L-NIL).
I-R surgery significantly increased plasma creatinine levels to 1.9 ± 0.3 mg/dl (P < .05) and caused renal cortical necrosis. L-NIL administration (3 mg/kg) in animals subjected to I-R significantly decreased plasma creatinine levels to 1.2 ± 0.10 mg/dl (P < .05 compared with I-R) and reduced tubular damage.
ONOO− formation was evaluated by detecting 3-nitrotyrosine-protein adducts, a stable biomarker of ONOO− formation.
- The kidneys from I-R animals had increased levels of 3-nitrotyrosine-protein adducts compared with control animals
- L-NIL-treated rats (3 mg/kg) subjected to I-R showed decreased levels of 3-nitrotyrosine-protein adducts.
These results support the hypothesis that iNOS-generated NO mediates damage in I-R injury possibly through ONOO− formation.
In summary, 3-nitrotyrosine-protein adducts were detected in renal tubules after I-R injury. Selective inhibition of iNOS by L-NIL
- decreased injury,
- improved renal function, and
- decreased apparent ONOO− formation.
Reactive nitrogen species should be considered potential therapeutic targets in the prevention and treatment of renal I-R injury.
Walker LM, Walker PD, Imam SZ, et al. Evidence for Peroxynitrite Formation in Renal Ischemia-Reperfusion Injury: Studies with the Inducible Nitric Oxide Synthase InhibitorL-N6-(1-Iminoethyl)-lysine1. 2000.
Role of TNFa independent of iNOS
Renal failure is a frequent complication of sepsis, mediated by renal vasoconstrictors and vasodilators. Endotoxin induces several proinflammatory cytokines, among which tumor necrosis factor (TNF) is thought to be of major importance. Tumor necrosis factor (TNF) has been suggested to be a factor in the acute renal failure in sepsis or endotoxemia. Passive immunization by anti-TNFa prevented development of septic shock in animal experiments. The development of ARF involves excessive intrarenal vasoconstriction.
Recent studies also suggest involvement of nitric oxide (NO), generated by inducible NO synthase (iNOS), in the pathogenesis of endotoxin-induced renal failure. TNF-a leads to a decrease in glomerular filtration rate (GFR). The present study tested the hypothesis that the role of TNF-a in endotoxic shock related ARF is mediated by iNOS-derived NO.
An injection of lipopolysaccharide (LPS) constituent of gram-negative bacteria to wild-type mice resulted in a 70% decrease in glomerular filtration rate (GFR) and in a 40% reduction in renal plasma flow (RPF) 16 hours after the injection.
The results occurred independent of
- hypotension,
- morphological changes,
- apoptosis, and
- leukocyte accumulation.
In mice pretreated with TNFsRp55, only a 30% decrease in GFR was observed without a significant change in RPF as compared with controls.
Effect of TNFsRp55 (10 mg/kg IP) on renal function in wild-type mice.
Mice were pretreated with TNFsRp55 for one hour before the administration of 5 mg/kg intraperitoneal endotoxin. GFR (A) and RPF (B) were determined 16 hours thereafter. Data are expressed as mean 6, SEM, N 5 6. *P , 0.05 vs. Control; §P , 0.05 vs. LPS, by ANOVA.
The serum NO concentration was significantly lower in endotoxemic wild-type mice pretreated with TNFsRp55, as compared with untreated endotoxemic wild-type mice. In LPS-injected iNOS knockout mice and wild-type mice treated with a selective iNOS inhibitor, 1400W, the development of renal failure was similar to that in wild-type mice. As in wild-type mice,TNFsRp55 significantly attenuated the decrease in GFR (a 33% decline, as compared with 75% without TNFsRp55) without a significant change in RPF in iNOS knockout mice given LPS.
These results demonstrate a role of TNF in the early renal dysfunction (16 h) in a septic mouse model independent of
- iNOS,
- hypotension,
- apoptosis,
- leukocyte accumulation,and
- morphological alterations,
thus suggesting renal hypoperfusion secondary to an imbalance between, as yet to be defined renal vasoconstrictors and vasodilators.
Knotek M, Rogachev B, Wang W,….., Edelstein CL, Dinarello CA, and Schrier RW. Endotoxemic renal failure in mice: Role of tumor necrosis factor independent of inducible nitric oxide synthase. Kidney International 2001; 59:2243–2249
Ischemic acute renal failure
Inflammation plays a major role in the pathophysiology of acute renal failure resulting from ischemia. In this review, we discuss the contribution of endothelial and epithelial cells and leukocytes to this inflammatory response. The roles of cytokines/chemokines in the injury and recovery phase are reviewed. The ability of the mouse kidney to be protected by prior exposure to ischemia or urinary tract obstruction is discussed as a potential model to emulate as we search for pharmacologic agents that will serve to protect the kidney against injury.
the inflammatory mediators produced by tubular epithelial cells and activated leukocytes in renal ischemia/reperfusion (I/R) injury.
Tubular epithelia produce TNF-a, IL-1, IL-6, IL-8, TGF-b, MCP-1, ENA-78, RANTES, and fractalkines, whereas leukocytes produce TNF-a, IL-1, IL-8, MCP-1, ROS, and eicosanoids. The release of these chemokines and cytokines serve as effectors for a positive feedback pathway enhancing inflammation and cell injury
the cycle of tubular epithelial cell injury and repair following renal ischemia/reperfusion.
Tubular epithelia are typically cuboidal in shape and apically-basally polarized; the Na+/K+-ATPase localizes to basolateral plasma membranes, whereas cell adhesion molecules, such as integrins localize basally. In response to ischemia reperfusion, the Na+/K+-ATPase appears apically, and integrins are detected on lateral and basal plasma membranes.
Some of the injured epithelial cells undergo necrosis and/or apoptosis detaching from the underlying basement membrane into the tubular space where they contribute to tubular occlusion. Viable cells that remain attached,
- dedifferentiate,
- spread, and
- migrate to
- repopulate the denuded basement membrane.
With cell proliferation, cell-cell and cell-matrix contacts are restored, and the epithelium redifferentiates and repolarizes, forming a functional, normal epithelium
Inflammation is a significant component of renal I/R injury, playing a considerable role in its pathophysiology. Although significant progress has been made in defining the major components of this process, the complex cross-talk between endothelial cells, inflammatory cells, and the injured epithelium with each generating and often responding to cytokines and chemokines is not well understood. In addition, we have not yet taken full advantage of the large body of data on inflammation in other organ systems.
Furthermore, preconditioning the kidney to afford protection to subsequent bouts of ischemia may serve as a useful model challenging us to therapeutically mimic endogenous mechanisms of protection. Understanding the inflammatory response prevalent in ischemic kidney injury will facilitate identification of molecular targets for therapeutic intervention.
Bonventre JV and Zuk A. Ischemic acute renal failure: An inflammatory disease? Forefronts in Nephrology 2002;.. :480-485
Gene expression profiles in renal proximal tubules
In kidney disease renal proximal tubular epithelial cells (RPTEC) actively contribute to the progression of tubulointerstitial fibrosis by mediating both an inflammatory response and via epithelial-to-mesenchymal transition. Using laser capture microdissection we specifically isolated RPTEC from cryosections of the healthy parts of kidneys removed owing to renal cell carcinoma and from kidney biopsies from patients with proteinuric nephropathies. RNA was extracted and hybridized to complementary DNA microarrays after linear RNA amplification. Statistical analysis identified 168 unique genes with known gene ontology association, which separated patients from controls.
Besides distinct alterations in signal-transduction pathways (e.g. Wnt signalling), functional annotation revealed a significant upregulation of genes involved in
- cell proliferation and cell cycle control (like insulin-like growth factor 1 or cell division cycle 34),
- cell differentiation (e.g. bone morphogenetic protein 7),
- immune response,
- intracellular transport and
- metabolism
in RPTEC from patients.
On the contrary we found differential expression of a number of genes responsible for cell adhesion (like BH-protocadherin) with a marked downregulation of most of these transcripts. In summary, our results obtained from RPTEC revealed a differential regulation of genes, which are likely to be involved in
- either pro-fibrotic or
- tubulo-protective mechanisms
in proteinuric patients at an early stage of kidney disease.
Rudnicki M, Eder S, Perco P, Enrich J, et al. Gene expression profiles of human proximal tubular epithelial cells in proteinuric nephropathies. Kidney International 2006; xx:1-11.
Kidney International advance online publication, 20 December 2006; doi:10.1038/sj.ki.5002043. http://www.kidney-international.org
Oxidative stress involved in diabetic nephropathy
Diabetic Nephropathy (DN) poses a major health problem. There is strong evidence for a potential role of the eNOS gene. The aim of this case control study was to investigate the possible role of genetic variants of the endothelial Nitric Oxide Synthase (eNOS) gene and oxidative stress in the pathogenesis of nephropathy in patients with diabetes mellitus.
The study included 124 diabetic patients;
- 68 of these patients had no diabetic nephropathy (group 1) while
- 56 patients exhibited symptoms of diabetic nephropathy (group 2).
- Sixty two healthy non-diabetic individuals were also included as a control group.
Blood samples from subjects and controls were analyzed to investigate the eNOS genotypes and to estimate the lipid profile and markers of oxidative stress such as malondialdehyde (MDA) and nitric oxide (NO). No significant differences were found in the frequency of eNOS genotypes between diabetic patients (either in group 1 or group 2) and controls (p >0.05).
Also, no significant differences were found in the frequency of eNOS genotypes between group 1 and group 2 (p >0.05).
Both group 1 and group 2 had significantly higher levels of nitrite and MDA when compared with controls (all p = 0.0001). Also group 2 patients had significantly higher levels of nitrite and MDA when compared with group 1 (p = 0.02, p = 0.001 respectively).
The higher serum level of the markers of oxidative stress in diabetic patients particularly those with diabetic nephropathy suggest that oxidative stress and not the eNOS gene polymorphism is involved in the pathogenesis of the diabetic nephropathy in this subset of patients
Badawy A, Elbaz R, Abbas AM, Ahmed Elgendy A, et al. Oxidative stress and not endothelial Nitric Oxide Synthase gene polymorphism involved in diabetic nephropathy. Journal of Diabetes and Endocrinology 2011; 2(3): 29-35.
Metformin in renal ischemia reperfusion
Renal ischemia plays an important role in renal impairment and transplantation.
Metformin is a biguanide used in type 2 diabetes, it inhibits hepatic glucose production and increases peripheral insulin sensitivity. While the mode of action of metformin is incompletely understood, it appears to have anti-inflammatory and antioxidant effects involved in its beneficial effects on insulin resistance.
Control, Sham, ischemia/reperfusion (I/R) and Metformin treated I /R groups
A renal I/R injury was done by a left renal pedicle occlusion to induce ischemia for 45 min followed by 60 min of reperfusion with contralateral nephrectomy. Metformin pretreated I/R rats in a dose of 200 mg/kg/day for three weeks before ischemia induction.
Nitric oxide (NO), tumor necrosis factor alpha (TNF α) , catalase (CAT) and reduced glutathione (GSH) activities were determined in renal tissue, while creatinine clearance (CrCl) , blood urea nitrogen (BUN) were measured and 5 hour urinary volume and electrolytes were estimated .
BUN and CrCl levels in the I/R group were significantly higher than in control rats (p<0.05) table (1).
Table 1: Creatinine clearance (Cr Cl) and blood urea nitrogen( BUN) levels in control and test groups. Mean ± SD.
| Groups | CrCl (ml/min) | BUN mg/dl |
| Control group | 1.30 ±0.11 | 14.30±0.25 |
| Sham group+metformin | 1.27±0.09 | 15.70±0.19 |
| I/R group P1 | 1.85±0.25<0.001*** | 28.00±0.62<0.001*** |
| I/R+metformin group P2 P3 | 1.55±0.220.001**0.028* | 18.10±1.00<0.001***<0.001*** |
P1: Statistical significance between control group and saline treated I/R group.
P2 Statistical significance between control group and Metformin treated I/R group.
P3 Statistical significance between saline treated I/R group and Metformin treated I/R group.
When metformin was administered before I/R, BUN and CrCl levels were still significantly higher than control group but their elevation were significantly lower in comparison to I/R group alone (P<0.05).
TNF α and NO levels were significantly higher in the I/R group than those of the control group (Table 2).
Pre-treatment with metformin significantly lowered their levels in comparison to I/R group (P<0.05).
Table 2: Tumour necrosis factior α (TNF α)and inducible nitric oxide (iNO)levels in control and test groups. (Mean ± SD).
| Groups | TNF α (pmol/mg tissue) | iNO nmol/ mg tissue |
| Control group | 17.60 ±5.98 | 2.54 ± 0.82 |
| Sham group+ metformin | 16.70 ±5.50 | 2.35 ±0.80 |
| I/R group P1 | 54. 00±6.02<0.001*** | 4.50±0.89<0.001** |
| I/R+ metformin group P2 P3 | 39 ± 14.01<0.001***0.006** | 3.53±0.950.02*0.03* |
P1: Statistical significance between control group and saline treated I/R group.
P2 Statistical significance between control group and Metformin treated I/R group.
P3 Statistical significance between saline treated I/R group and Metformin treated I/R group
These results showed significant increase in
- NO,
- TNF α,
- BUN ,
- CrCl and
significant decrease in
- urinary volume ,
- electrolytes,
- CAT and
- GSH activities
in the I/R group than those in the control group.
Metformin
- decreased significantly NO, TNF α, BUN and CrCl while
- increased urinary volume, electrolytes, CAT and GSH activities.
Lipid peroxidation is related to I/R induced tissue injury. Production of inducible NO synthase (NOS) under lipid peroxidation and inflammatory conditions results in the induction of NO
which react with
- O2
- liberating peroxynitrite (OONO–).
NO itself inactivates the antioxidant enzyme system CAT and GSH.
Alteration in NO synthesis have been observed in other kidney injuries as nephrotoxicity and acute renal failure induced by endotoxins. Treatment with iNOS inhibitors improved renal function and decreased peroxynitrite radical which is believed to be responsible for the shedding of proximal convoluted tubules in I/R.
Metformin produced anti-inflammatory renoprotective effect on CrCl and diuresis in renal I/R injury.
Malek HA. The possible mechanism of action of metformin in renal ischemia reperfusion in rats. The Pharma Research Journal 2011; 6(1):42-49.
Possible role of NO donors in ARF
The L-arginine-nitric oxide (NO) pathway has been implicated in many physiological functions in the kidney, including
- regulation of glomerular hemodynamics,
- mediation of pressure-natriuresis,
- maintenance of medullary perfusion,
- blunting of tubuloglomerular feedback (TGF),
- inhibition of tubular sodium reabsorption and
- modulation of renal sympathetic nerve activity.
Its net effect in the kidney is to promote natriuresis and diuresis, contributing to adaptation to variations of dietary salt intake and maintenance of normal blood pressure.
| RAS | Renal hemodynamics | Sodium balance |
| Medullary perfusion | ||
| Pressure-natriuresis | ||
| Salt intake | Tubulo-glomerular feedback | Blood pressure |
| Tubular sodium reabsorption | ||
| Blood pressure | Renal sympathetic activity | Regulation |
| Extrarenal factors | Intrarenal functions | Physiological roles |
Role of nitric oxide in renal physiology. RAS, renin-angiotensin system
Nitric oxide has been implicated in many physiologic processes that influence both acute and long-term control of kidney function. Its net effect in the kidney is to promote natriuresis and diuresis, contributing to adaptation to variations of dietary salt intake and maintenance of normal blood pressure. A pretreatment with nitric oxide donors or L-arginine may prevent the ischemic acute renal injury. In chronic kidney diseases, the systolic blood pressure is correlated with the plasma level of asymmetric dimethylarginine, an endogenous inhibitor of nitric oxide synthase.
A reduced production and biological action of nitric oxide is associated with an elevation of arterial pressure, and conversely, an exaggerated activity may represent a compensatory mechanism to mitigate the hypertension.
JongUn Lee. Nitric Oxide in the Kidney : Its Physiological Role and Pathophysiological Implications. Electrolyte & Blood Pressure 2008; 6:27-34.
Renal Hypoxia and Dysoxia following Reperfusion
Acute renal failure (ARF) is a common condition which develops in 5% of hospitalized patients. Of the patients who develop ARF, ~10% eventually require renal replacement therapy. Among critical care patients who have acute renal failure and survive, 2%-10% develop terminal renal failure and require long-term dialysis.
The kidneys are particularly susceptible to ischemic injury in many clinical conditions such as renal transplantation, treatment of suprarenal aneurysms, renal artery reconstructions, contrast-agent induced nephropathy, cardiac arrest, and shock. One reason for renal sensitivity to ischemia is that the kidney microvasculature is highly complex and must meet a high energy demand. Under normal, steady state conditions, the oxygen (O2) supply to the renal tissues is well in excess of oxygen demand.
Under pathological conditions, the delicate balance of oxygen supply versus demand is easily disturbed due to the unique arrangement of the renal microvasculature and its increasing numbers of diffusive shunting pathways.
The renal microvasculature is serially organized, with almost all descending vasa recta emerging from the efferent arterioles of the juxtamedullary glomeruli. Adequate tissue oxygenation is thus partially dependent on the maintenance of medullary perfusion by adequate cortical perfusion. This, combined with the low amount of medullary blood flow (~10% of total renal blood flow) in the U-shaped microvasculature of the medulla allows O2 shunting between the descending and ascending vasa recta and contributes to the high sensitivity of the medulla and cortico-medullary junction to decreased O2 supply.
Whereas past investigations have focused mainly on tubular injury as the main cause of ischemia-related acute renal failure, increasing evidence implicates alterations in the intra-renal microcirculation pathway and in the O2 handling. Indeed, although acute tubular necrosis (ATN) has classically been believed to be the leading cause of ARF, data from biopsies in patients with ATN have shown few or no changes consistent with tubular necrosis. The role played by microvascular dysfunction, however, has generated increasing interest. The complex pathophysiology of ischemic ARF includes the inevitable reperfusion phase associated with oxidative stress, cellular dysfunction and altered signal transduction.
During this process, alterations in oxygen transport pathways can result in cellular hypoxia and/or dysoxia. In this context, the distinction between hypoxia and dysoxia is that cellular hypoxia refers to the condition of decreased availability of oxygen due to inadequate convective delivery from the microcirculation. Cellular dysoxia, in contrast, refers to a pathological condition where the ability of mitochondria to perform oxidative phosphorylation is limited, regardless of the amount of available oxygen. The latter condition is associated with mitochondrial failure and/or activation of alternative pathways for oxygen consumption. Thus, we would expect that an optimal balance between oxygen supply and demand is essential to reducing damage from renal ischemia-reperfusion (I/R) injury (Figure 1).
Complex interactions exist between tubular injury, microvascular injury, and inflammation after renal I/R. On the one hand, insults to the tubule cells promotes the liberation of a number of inflammatory mediators, such as TNF-á, IL-6, TGF-â, and chemotactic cytokines (RANTES, monocyte chemotactic protein-1, ENA-78, Gro-á, and IL-8). On the other hand, chemokine production can promote leukocyte-endothelium interactions and leukocyte activation, resulting in renal blood flow impairment and the expansion of tubular damage.
renal hemodynamics and electrolyte reabsorption
Adequate medullary tissue oxygenation, in terms of balanced oxygen supply and demand, is dependent on the maintenance of medullary perfusion by adequate cortical perfusion and also on the high rate of O2 consumption required for active electrolyte transport. Furthermore, renal blood flow is closely associated with renal sodium transport.
In addition to having a limited O2 supply due to the anatomy of the microcirculation anatomy, the sensitivity of the medulla to hypoxic conditions results from this high O2 consumption. Renal sodium transport is the main O2-consuming function of the kidney and is closely linked to renal blood flow for sodium transport, particularly in the thick ascending limbs of the loop of Henle and the S3 segments of the proximal tubules.
Medullary renal blood flow is also highly dependent on cortical perfusion, with almost all descending vasa recta emerging from the efferent arteriole of juxta medullary glomeruli. A profound reduction in cortical perfusion can disrupt medullary blood flow and lead to an imbalance between O2 supply and O2 consumption. On theother hand, inhibition of tubular reabsorption by diuretics increases medullary pO2 by decreasing the activity of Na+/K+-ATPases and local O2 consumption.
Mitochondrial activity and NO-mediated O2 consumption
The medulla has been found to be the main site of production of NO in the kidney. In addition to the actions described above, NO appears to be a key regulator of renal tubule cell metabolism by inhibiting the activity of the Na+-K+-2Cl– cotransporter and reducing Na+/H+ exchange. Since superoxide (O2–) is required to inhibit solute transport activity, it was assumed that these effects were mediated by peroxynitrite (OONO–). Indeed, mitochondrial nNOS upregulation, together with an increase in NO production, has been shown to increase mitochondrial peroxynitrite generation, which in turn, can induce cytochrome c release and promote apoptosis. NO has also been shown to directly compete with O2 at the mitochondrial level. These findings support the idea that NO acts as an endogenous regulator to match O2 supply to O2 consumption, especially in the renal medulla.
NO reversibly binds to the O2 binding site of cytochrome oxidase, and acts as a potent, rapid, and reversible inhibitor of cytochrome oxidase in competition with molecular O2. This inhibition could be dependent on the O2 level, since the IC50 (the concentration of NO that reduces the specified response by half) decreases with reduction in O2 concentration. The inhibition of electron flux at the cytochrome oxidase level switches the electron transport chain to a reduced state, and consequently leads to depolarization of the mitochondrial membrane potential and electron leakage.
To summarize, while the NO/O2 ratio can act as a regulator of cellular O2 consumption by matching decreases in O2 delivery to decreases in cellular O2 cellular, the inhibitory effect of NO on mitochondrial respiration under hypoxic conditions further impairs cellular aerobic metabolism This leads to a state of “cytopathic hypoxia,” as described in the sepsis literature.
Only cell-secreted NO competes with O2 and to regulate mitochondrial respiration. In addition to the 3 isoforms (eNOS, iNOS, cnNOS), an α-isoform of neuronal NOS, the mitochondrial isoform (mNOS) located in the inner mitochondrial membrane, has also been shown to regulate mitochondrial respiration.
These data support a role for NO in the balanced regulation of renal O2 supply and O2 consumption after renal I/R However, the relationships between the determinants of O2 supply, O2 consumption, and renal function, and their relation to renal damage remain largely unknown.
Sustained endothelial activation
Ischemic renal failure leads to persistent endothelial activation, mainly in the form of endothelium-leukocyte interactions and the activation of adhesion molecules.
This persistent activation can
- compromise renal blood flow,
- prevent the recovery of adequate tissue oxygenation, and
- jeopardize tubular cell survival despite the initial recovery of renal tubular function.
A 30-50% reduction in microvascular density was seen 40 weeks after renal ischemic injury in a rat model. Vascular rarefaction has been proposed to induce chronic hypoxia resulting in tubulointerstitial fibrosis via the molecular activation of fibrogenic factors such as
- transforming growth factor (TGF)-β,
- collagen, and
- fibronectin,
all of which may play an important role in the progression of chronic renal disease.
Adaptation to hypoxia
Over the last decade, the role of hypoxia-inducible factors (HIFs) in O2 supply and adaptation to hypoxic conditions has found increasing support. HIFs are O2-sensitive transcription factors involved in O2-dependent gene regulation that mediate cellular adaptation to O2 deprivation and tissue protection under hypoxic conditions in the kidney.
NO generation can promote HIF-1α accumulation in a cGMP-independent manner. However, Hagen et al. (2003) showed that NO may reduce the activation of HIF in hypoxia via the inhibitory effect of NO on cytochrome oxidase. Therefore, it seems that NO has pleiotropic effects on HIF expression, with various responses related to different pathways.
HIF-1α upregulates a number of factors implicated in cytoprotection, including angiogenic growth factors, such as
- vascular endothelial growth factors (VEGF),
- endothelial progenitor cell recruitment via the endothelial expression of SDF-1,
- heme-oxygenase-1 (HO-1), and
- erythropoietin (EPO), and
- vasomotor regulation.
HO-1 produces carbon monoxide (a potent vasodilator) while degrading heme, which may preserve tissue blood flow during reperfusion. Thus, it has been suggested that
- the induction of HO-1 can protect the kidney from ischemic damage by decreasing oxidative damage and NO generation.
- in addition to its anti-apoptotic properties, EPO may protect the kidney from ischemic damage by restoring the renal microcirculation
(by stimulating the mobilization and differentiation of progenitor cells toward an endothelial phenotype and by inducing NO release from eNOS).
Pharmacological interventions
Use of pharmacological interventions which act at the microcirculatory level may be a successful strategy to overcome ischemia-induced vascular damage and prevent ARF.
Activated protein C (APC), an endogenous vitamin K-dependent serine protease with multiple biological activities, may meet these criteria. Along with antithrombotic and profibrinolytic properties, APC can reduce the chemotaxis and interactions of leukocytes with activated endothelium. However, renal dysfunction was not improved in the largest study published so far. In addition, APC has been discontinued by Lilly for the use intended in severe sepsis.
- neither drugs with renal vasodilatory effects (i.e., dopamine, fenoldopam, endothelin receptors blockers, adenosine antagonists) or
- agents that decrease renal oxygen consumption (i.e., loop diuretics) have been shown to protect the kidney from ischemic damage.
We have to bear in mind that a magic bullet to treat the highly complex condition of which is renal I/R is not in sight. We can expect that understanding the balance between O2 delivery and O2 consumption, as well as the function of O2-consuming pathways (i.e., mitochondrial function, reactive oxygen species generation) will be central to this treatment strategy.
Take home point
The deleterious effects of NO are thought to be associated with the NO generated by the induction of iNOS and its contribution to oxidative stress both resulting in vascular dysfunction and tissue damage. Ischemic injury also leads to structural damage to the endothelium and leukocyte infiltration. Consequently, renal tissue hypoxia is proposed to promote the initial tubular damage, leading to acute organ dysfunction.
Comment: I express great appreciation for refeering to this work, which does provide enormous new insights into hypoxia-induced acute renal failure, and ties together the anatomy, physiology, and gene regulation through signaling pathways.
Ince C, Legrand M, Mik E , Johannes T, Payen D. Renal Hypoxia and Dysoxia following Reperfusion of the Ischemic Kidney. Molecular Medicine (Proof) 2008; pp36. www.molmed.org

English: Major cellular sources of ROS in living cells. Novo and Parola Fibrogenesis & Tissue Repair 2008 1:5 doi:10.1186/1755-1536-1-5 (Photo credit: Wikipedia)

Figure 1 (Photo credit: Libertas Academica)
The reaction mechanism of Nitric oxide synthase (Photo credit: Wikipedia)
Nitric oxide and non-hemodynamic functions of the kidney
One of the major scientific advances in the past decade in understanding of the renal function and disease is the prolific growth of literature incriminating nitric oxide (NO) in renal physiology and pathophysiology. NO was first shown to be identical with endothelial derived relaxing factor (EDRF) in 1987 and this was followed by a rapid flurry of information defining the significance of NO in not only vascular physiology and hemodynamics but also in neurotransmission, inflammation and immune defense systems.
Although most actions of NO are mediated by cyclic guanosine monophosphate (cGMP) signaling, S-nitrosylation of cysteine residues in target proteins constitutes another well defined non-cGMP dependent mechanism of NO effects.
Recent years have witnessed a phenomenal scientific interest in the vascular biology, particularly the relevance of nitric oxide (NO) in cardiovascular and renal physiology and pathophysiology. Although hemodynamic actions of NO received initial attention, a variety of non-hemodynamic actions are now known to be mediated by NO in the normal kidney,which include
- tubular transport of electrolyte and water,
- maintenance of acid-base homeostasis,
- modulation of glomerular and interstitial functions,
- renin-angiotensin activation and
- regulation of immune defense mechanism in the kidney.
Table 1 : Functions of NO in the kidney
1. Renal macrovascular and microvascular dilatation (afferent > efferent)
2. Regulation of mitochondrial respiration.
3. Modulation renal medullary blood flow
4. Stimulation of fluid, sodium and HCO3 – reabsorption in the proximal tubule
5. Stimulation of renal acidification in proximal tubule by stimulation of NHE activity
6. Inhibition of Na+, Cl- and HCO3 – reabsorption in the mTALH
7. Inhibition of Na+ conductance in the CCD
8. Inhibition of H+-ATPase in CCD
One of the renal regulatory mechanisms related to maintenance of arterial blood pressure involves the phenomenon of pressure-natriuresis in response to elevation of arterial pressure. This effect implies inhibition of tubular sodium reabsorption resulting in natriuresis, in an effort to lower arterial pressure. Experimental evidence indicates that intra-renal NO modulates pressure natriuresis.
Furthermore many studies have confirmed the role of intra renal NO in mediating tubulo-glomerular feedback (TGF). In vivo micropuncture studies have shown that NO derived from nNOS in macula densa specifically inhibits the TGF responses leading to renal afferent arteriolar vasoconstriction in response to sodium reabsorption in the distal tubule. Other recent studies support the inhibitory role of NO from eNOS and iNOS in mTALH segment on TGF effects.
Recent observations in vascular biology have yielded new information that endothelial dysfunction early in the course might contribute to the pathophysiology of acute renal failure. Structural and functional changes in the vascular endothelium are demonstrable in early ischemic renal failure. Altered NO production and /or decreased bioavailability of NO comprise the endothelial dysfunction in acute renal failure.
Several studies have indicated imbalance of NOS activity with enhanced expression and activity of iNOS and decreased eNOS in ischemic kidneys. The imbalance results from enhanced iNOS activity and attenuated eNOS activity in the kidney.
Many experimental studies support a contributory role for NO in glomerulonephritis (GN). Evidence from recent studies pointed out that NO may be involved in peroxynitrite formation, pro-inflammatory chemokines and signaling pathways in addition to direct glomerular effects that promote albumin permeability in GN.
Although originally macrophages and other leukocytes were first considered as the source renal NO production in GN, it is now clear iNOS derived NO from glomerular mesangial cells are the primary source of NO in GN.
In most pathological states, the role of NO is
- dependent on the stage of the disease,
- the nitric oxide synthase (NOS) isoform involved and
- the presence or absence of other modifying intrarenal factors.
Additionally NO may have a dual role in several disease states of the kidney such as
- acute renal failure,
- inflammatory nephritides,
- diabetic nephropathy and
- transplant rejection.
A rapidly growing body of evidence supports a critical role for NO in tubulointerstitial nephritis (TIN). In the rat model of autoimmune TIN, Gabbai et al. demonstrated increased iNOS expression in the kidney and NO metabolites in urine and plasma. However the effects of iNOS on renal damage in TIN seem to have a biphasic effect- since iNOS specific inhibitors (eg. L-Nil) are renoprotective in the acute phase while they actually accelerated the renal damage in the chronic phase. Thus chronic NOS inhibition is used to induce chronic tubulointerstitial injury and fibrosis along with mild glomerulosclerosis and hypertension.
Major pathways of L-arginine metabolism.
- L-arginine may be metabolized by the urea cycle enzyme arginase to L-ornithine and urea
- by arginine decarboxylase to agmatine and CO2 or
- by NOS to nitric oxide (NO) and L-citrulline.
Adapted from Klahr S: Can L-arginine manipulation reduce renal disease? Semin Nephrol 1999; 61:304-309.
It is obvious that kidney is not only a major source of arginine and nitric oxide but NO plays an important role in the water and electrolyte balance and acid-base physiology and many other homeostatic functions in the kidney. Unfortunately we are far from a precise understanding of the significance of NO alterations in various disease states primarily due to conflicting data from the existing literature.
Therapeutic potential for manipulation of L-arginine- nitric oxide axis in renal disease states has been discussed. More studies are required to elucidate the abnormalities in NO
metabolism in renal diseases and to confirm the therapeutic potential of L-arginine.
Sharma SP. Nitric oxide and the kidney. Indian J Nephrol 2004;14: 77-84
Inhibition of Constitutive Nitric Oxide Synthase
Excess NO generation plays a major role in the hypotension and systemic vasodilatation characteristic of sepsis. Yet the kidney response to sepsis is characterized by vasoconstriction resulting in renal dysfunction.
We have examined the roles of inducible nitric oxide synthase (iNOS) and endothelial NOS (eNOS) on the renal effects of lipopolysaccharide administration by comparing the effects of specific iNOS inhibition, L-N6-(1-iminoethyl)lysine (L-NIL), and 2,4-diamino-6-hydroxy-pyrimidine vs. nonspecific NOS inhibitors (nitro-L-arginine-methylester). cGMP responses to carbamylcholine (CCh) (stimulated, basal) and sodium nitroprusside in isolated glomeruli were used as indices of eNOS and guanylate cyclase (GC) activity, respectively.
LPS significantly decreased blood pressure and GFR (P =0.05) and inhibited the cGMP response to CCh. GC activity was reciprocally increased. L-NIL and 2,4-diamino-6-hydroxy-pyrimidine administration prevented the decrease in GFR, restored the normal response to CCh, and GC activity was normalized. In vitro application of L-NIL also restored CCh responses in LPS glomeruli.
Neuronal NOS inhibitors verified that CCh responses reflected eNOS activity. L-NAME, a nonspecific inhibitor, worsened GFR, a reduction that was functional and not related to glomerular thrombosis, and eliminated the CCh response. No differences were observed in eNOS mRNA expression among the experimental groups.
Selective iNOS inhibition prevents reductions in GFR, whereas nonselective inhibition of NOS further decreases GFR. These findings suggest that the decrease in GFR after LPS is due to local inhibition of eNOS by iNOS, possibly via NO autoinhibition.
Schwartz D, Mendonca M, Schwartz I, Xia Y, et al. Inhibition of Constitutive Nitric Oxide Synthase (NOS) by Nitric Oxide Generated by Inducible NOS after Lipopolysaccharide Administration Provokes Renal Dysfunction in Rats. J. Clin. Invest. 1997; 100:439–448.
Salt-Sensitivity and Hypertension
Renin-angiotensin system (RAS) plays a key role in
- the regulation of renal function,
- volume of extracellular fluid and
- blood pressure.
The activation of RAS also induces oxidative stress, particularly superoxide anion (O2-) formation. Although the involvement of O2 – production in the pathology of many diseases has been long known, recent studies also strongly suggest its physiological regulatory function of many organs including the kidney.
However, a marked accumulation of O2- in the kidney alters normal regulation of renal function and may contribute to the development of salt-sensitivity and hypertension. In the kidney, O2- acts as vasoconstrictor and enhances tubular sodium reabsoption.
Nitric oxide (NO), another important radical that exhibits opposite effects than O2 -, is also involved in the regulation of kidney function. O2- rapidly interacts with NO and thus, when O2- production increases, it diminishes the bioavailability of NO leading to the impairment of organ function.
As the activation of RAS, particularly the enhanced production of angiotensin II, can induce both O2- and NO generation, it has been suggested that physiological interactions of RAS, NO and O2- provide a coordinated regulation of kidney function.
The imbalance of these interactions is critically linked to the pathophysiology of salt-sensitivity and hypertension.
Kopkan L, Červenka L. Renal Interactions of Renin-Angiotensin System, Nitric Oxide and Superoxide Anion: Implications in the Pathophysiology of Salt-Sensitivity and Hypertension. Physiol. Res. 2009; 58 (Suppl. 2): S55-S67.
Epicrisis
In this review I attempted to evaulate complex and still incomplete and conflicting conclusions from many studies. I thus broke the report into three major portions:
1 The kidney and its anatomy, physiology, and ontogeny.
2 The pathological disease variation affecting the kidney
a a tie in to eNOS and iNos, nitric oxide, cGMP and glutaminase – in acute renal failure, hypertension, chronic renal failure, dialysis
the pathology of acute tubular necrosis, glomerular function, efferent arteriolar and kidney medullary circulatory impairment, and cast formation related to Tamm Horsfall protein
b The role of NO, eNOS and iNOS in disorders of the lund alveolar cell and subendothelial matrix, and of liver disease also affecting the kidney, and the heart.
c Additional references
3. a Acute renal failure, oxidate stress, ischemia-reperfusion injury, tubulointerstitial chronic inflammation
b Additional references
4. Nitric oxide donors – opportunities for therapeutic targeting?
As we see this in as full a context as possible, it is hard to distinguish the cart from the horse. We know that there is an unquestionable role of NO, and a competing balance to be achieved between eNOS, iNOS, an effect on tubular water and ion-cation reabsorptrion, a role of TNFa, and consequently an impofrtant role in essential/malignant hypertension, with the size of the effect related to the stage of disorder, the amount of interstitial fibrosis, the remaining nephron population, the hypertonicity of the medulla, the vasodilation of the medularry circullation, and the renin-angiotensin-aldosterone system. Substantial data and multiple patientys with many factors per patient would be need to extract the best model using a supercomputer.
Related articles
- Low Bioavailability of Nitric Oxide due to Misbalance in Cell Free Hemoglobin in Sickle Cell Disease – A Computational Model (pharmaceuticalintelligence.com)
- Nitric Oxide and Immune responses: part 1 (pharmaceuticalintelligence.com)
- Differential Distribution of Nitric Oxide – A 3-D Mathematical Model (pharmaceuticalintelligence.com)
- Nitric Oxide and Immune Responses: Part 2 (pharmaceuticalintelligence.com)
- Mitochondrial Damage and Repair under Oxidative Stress (pharmaceuticalintelligence.com)
- Endothelial Function and Cardiovascular Disease (pharmaceuticalintelligence.com)
The Essential Role of Nitric Oxide and Therapeutic NO Donor Targets in Renal Pharmacotherapy
Posted in Biological Networks, Gene Regulation and Evolution, Biomarkers & Medical Diagnostics, Cell Biology, Signaling & Cell Circuits, Chemical Biology and its relations to Metabolic Disease, Disease Biology, Small Molecules in Development of Therapeutic Drugs, HTN, International Global Work in Pharmaceutical, Nitric Oxide in Health and Disease, Pharmaceutical Industry Competitive Intelligence, Proteomics, Systemic Inflammatory Response Related Disorders, tagged Larry H. Bernstein, nephron, nitric oxide, Nitric oxide synthase, NO, Peroxinitrite, Renal function, Tamm-Horsfall protein on November 26, 2012| 14 Comments »
Subtitle: Nitric Oxide, Peroxinitrite, and NO donors in Renal Function Loss
Curator and Author: Larry H. Bernstein, MD, FCAP
This is the last of a series of articles on renal function, the nephron, the vasculature of the kidney, the importance of nitric oxide, the multiple and opposing roles of the NOS isoforms in kidney function, and the impairment of renal function in acute and chronic disease.
The Nitric Oxide and Renal is presented in FOUR parts:
Part I: The Amazing Structure and Adaptive Functioning of the Kidneys: Nitric Oxide
Part II: Nitric Oxide and iNOS have Key Roles in Kidney Diseases
Part III: The Molecular Biology of Renal Disorders: Nitric Oxide
Part IV: New Insights on Nitric Oxide donors
Conclusion to this series is presented in
The Essential Role of Nitric Oxide and Therapeutic NO Donor Targets in Renal Pharmacotherapy
The Essential Role of Nitric Oxide and Therapeutic NO Donor Targets in Renal Pharmacotherapy
")
Loop of Henle (Grey’s Anatomy book) (Photo credit: Wikipedia)
The first section notes the embryonic recapitulation of the evolution of the kidney in the emergence of oxygen dependent species from the deep oceans and swampland to a lung breathing and terrestrial existence. The excretory kidney that all mammals rely on for elimination of nitrogenous wastes, mainly composed of urea, is a metanephric kidney that develops in the fifth week of human embryonic life. The kidneys are paired organs capped by the adrenal glands that lie in the lower posterior abdominal wall supplied by the renal arteries that branch from the aorta.
The kidney has grossly and outer cortex that surrounds the medulla. The cortex is highly vascular containing all of the glomerular structures that receive circulation from an afferent arteriole and return circulation by way of an efferent arteriole. The efferent arteriole can become thickened by sustained high pressure. The glomerulus has a globular structure with a double membrane through which no cells pass, but the plasma is filtered, gaining entry into the tubular nephron. With aging there can be a loss of half of the original nephrons, but the measurement of significant loss is not necessarily measured as decreased glomerular filtration rate (GFR)(120 ml/min) until a loss of 1/3 to 1/2 of kidney mass. The metanephric kidney requires passage of large amounts of fluid volume. The cortex is high-energy and high mitochondrial content because of its concentrated circulation and the pressure need to drive filtration (passive). The next phase of urine formation begins in the cortex, in the proximal convoluted tubule, with the filtrate passing into the descending limb, in which there is active reabsorption of glucose by a transporter, exchange of Na/K by Na/K ATPase, excretion of urea and secretion of uric acid, and regulation of pH by carbonic anhydrase. This is not in itself sufficient to manage the formation of urine that is concentrated, contains nitrogenous waste, and returns key analytes to the circulation.
The medulla contains interstitium, the tubular nephron that consists of descending limb, the Loop of Henley, ascending limb, and the collecting ducts that empty into the medullary pyramids.
The interstitium develops a high pressure bacause of the concentration of Na that is pumped in proximally, and a volume of water that is removed from the adjacent distal end and the collecting ducts under the influence of antidiuretic hormone (ADH). When the circulation and the interstitial pressures are hypoxemic in the medulla, acute medullary necrosis ensues, largely the hallmark of acute tubular necrosis. When the kidney cortex becomes ischemic, partly as a result microthrombi in the arterioles and/or the glomerular capillaries, acute cortical necrosis ensues.
The second section is a description of the role of nitric oxide (NO) in renal function. The earliest concern was focused on the NO and eNOS in the cortex, with the rich vascular bed. The generation of NO by eNOS is essential for vasodilatation. Then, far more interest lie in the role of NO in tubular and interstitial function. The tubules are exposed to large changes in fluid flow pressures and to ionic stresses of the adjacent interstitial tissue. The interest was catalyzed by the wartime crush injuries that led to acute tubular necrosis with an initial 80% mortality, tht has been improved by fluid resuscitation, with caveats.
Nitric oxide (NO) and its metabolite, peroxynitrite (ONOO-), are involved in renal tubular cell injury. However, while eNOS generated NO is beneficial, inducible iNOS generated NO is a large player in ATM. We learn that both forms have an important counter-balancing effect. NO/ONOO- has an effect of reducing cell adhesion to the basement membrane. It is not the NO, but the converted ONOO- peroxynitrite that has the most damaging effect. The damage to tubular epithelium does signal repair, but the damaged cells feed into the tubule and are precursor of Tamm Horsfall protein (TFP), which obstructs flow. The exposure to NO donor SIN-1 caused a dose-dependent impairment in cell-matrix adhesion, that was obtained in different cell types and matrix proteins. In a seminal paper, the authors concluded that ONOO- generated in the tubular epithelium during ischemia/reperfusion has the potential to impair the adhesion properties of tubular cells, which then may contribute to the tubular obstruction in ARF.
In addition, inflammatory cytokines, released early in sepsis, cause Proximal Tubular Epithelial Cells (PTEC) cytoskeletal damage and alter integrin-dependent cell-matrix adhesion. After exposure of human PTEC to tumor necrosis factor-α, interleukin-1α, and interferon-γ, the actin cytoskeleton was disrupted , and the cytokines induced shedding of viable, apoptotic, and necrotic PTEC. The shedding was dependent on NO synthesized by inducible NO synthase (iNOS) produced as a result of cytokine actions on PTEC. The major ligand involved in cell anchorage was laminin, probably through interactions with the integrin α3β1, which was downregulated by the cytokines, but this was independent of NO, so hypotension and ischemia is not involved. Furthermore, the apoptotic effects of proinflammatory stimuli mainly are largely due to the expression of inducible NO synthase. Other expereiments indicated:
- Exposure to SIN-1, which generated peroxynitrite (ONOO) produced a concentration- and time-dependent delayed cell death
- a critical threshold concentration (>440 nM/min) was required for . NO to produce significant cell injury
N acetyl cysteine (NAC) given within the first 3 h posttreatment further delayed cell death and increased the intracellular thiol level in SIN-1 but not . NO-exposed cells but cell injury from . NO was independent of cGMP, caspases, and superoxide or peroxynitrite formation. Exposure of non-. NO-producing cells to . NO or peroxynitrite results in delayed cell death, which, although occurring by different mechanisms, is mediated by the loss of intracellular redox balance.
The third section attends to renal diseases. The best studied type is ARF. The mechanisms of ARF involve both vascular and tubular factors. An ischemic insult to the kidney will in general be the cause of the ARF. While a decrease in renal blood flow with diminished oxygen and substrate delivery to the tubule cells is an important ischemic factor, it must be remembered that a relative increase in oxygen demand by the tubule is also a factor in renal ischemia. In vitro studies using chemical anoxia have revealed abnormalities in the proximal tubule cytoskeleton that are associated with translocation of Na+/K+-ATPase from the basolateral to the apical membrane. A comparison of cadaveric transplanted kidneys with delayed versus prompt graft function has also provided important results regarding the role of Na+/K+-ATPase in ischemic renal injury. A translocation of Na+/K+-ATPase from the basolateral membrane to the cytoplasm may explain the decrease in tubular sodium reabsorption that occurs with ARF. Calpain-mediated breakdown products of the actin-binding protein spectrin have been shown to occur with renal ischemia. Calpain activity was also demonstrated to be increased during hypoxia in isolated proximal tubules. The existence of proteolytic pathways involving cysteine proteases, namely calpain and caspases, may explain the decrease in proximal tubule sodium reabsorption and increased FENa secondary to proteolytic uncoupling of Na+/K+-ATPase from its basolateral membrane anchoring proteins, but not the fall in GFR. An antisense oligonucleotide was shown to block the upregulation of iNOS and afford functional protection against acute renal ischemia. Thus, when isolated proximal tubules from iNOS, eNOS, and neuronal NO synthase (nNOS) knockout mice were exposed to hypoxia, only the tubules from the iNOS knockout mice were protected against hypoxia. This suggests that the protective role of vascular eNOS may be more important than the deleterious effect of iNOS at the tubule level during renal ischemia
The last section is the NO donors as therapeutic targets. IFNa produced dose-dependent and time-dependent decrease in transepithelial resistance (TER) ameliorated by tyrphostin, an inhibitor of phosphotyrosine kinase with increased expression of occludin and E-cadherin. Therefore, IFNa can directly affect barrier function in renal epithelial cells via overexpression or missorting of the junctional proteins occludin and E-cadherin.
There is agreement that oxygen-derived reactive species are important in renal ischemia-reperfusion (I-R) injury. Treatment with oxygen radical scavengers, antioxidants, and iron chelators such as superoxide dismutase, dimethylthiourea, allopurinol, and deferoxamine are protective. They all directly scavenge or inhibit the formation of peroxynitrite (ONOO−), a highly toxic species derived from nitric oxide (NO) and superoxide. Thus, the protective effects seen with these inhibitors may be due in part to their ability to inhibit ONOO− formation. However, induction of inducible nitric oxide synthase (iNOS) and production of high levels of nitric oxide (NO) also contribute to this injury. NO can combine with superoxide to form the potent oxidant peroxynitrite (ONOO−). L-NIL administered to animals subjected to I-R significantly decreased plasma creatinine levels to 1.2 ± 0.10 mg/dl and reduced tubular damage. 3-nitrotyrosine-protein adducts were detected in renal tubules after I-R injury. Selective inhibition of iNOS by L-NIL decreased injury, improved renal function, and decreased apparent ONOO− formation. Therefore, reactive nitrogen species should be considered potential therapeutic targets in the prevention and treatment of renal I-R injury.
NO functions to promote natriuresis and diuresis, contributing to adaptation to variations of dietary salt intake and maintenance of normal blood pressure. A pretreatment with nitric oxide donors or L-arginine may prevent the ischemic acute renal injury. In chronic kidney diseases, the systolic blood pressure is correlated with the plasma level of asymmetric dimethylarginine, an endogenous inhibitor of nitric oxide synthase. A reduced production and biological action of nitric oxide is associated with an elevation of arterial pressure, and conversely, an exaggerated activity may represent a compensatory mechanism to mitigate the hypertension.
Adequate medullary tissue oxygenation, in terms of balanced oxygen supply and demand, is dependent on the maintenance of medullary perfusion by adequate cortical perfusion and also on the high rate of O2 consumption required for active electrolyte transport. The sensitivity of the medulla to hypoxic conditions results from high O2 consumption. Renal sodium transport is the main O2-consuming function of the kidney and is closely linked to renal blood flow for sodium transport, particularly in the thick ascending limbs of the loop of Henle and the S3 segments of the proximal tubules. The medulla has been found to be the main site of production of NO in the kidney. In addition to the actions described above, NO appears to be a key regulator of renal tubule cell metabolism by inhibiting the activity of the Na+-K+-2Cl- cotransporter and reducing Na+/H+ exchange.
NO reversibly binds to the O2 binding site of cytochrome oxidase, and acts as a potent, rapid, and reversible inhibitor of cytochrome oxidase in competition with molecular O2. This inhibition could be dependent on the O2 level, since the IC50 (the concentration of NO that reduces the specified response by half) decreases with reduction in O2 concentration. The inhibition of electron flux at the cytochrome oxidase level switches the electron transport chain to a reduced state, and consequently leads to depolarization of the mitochondrial membrane potential. While the NO/O2 ratio can act as a regulator of cellular O2 consumption by matching decreases in O2 delivery to decreases in cellular O2 cellular, the inhibitory effect of NO on mitochondrial respiration under hypoxic conditions further impairs cellular aerobic metabolism.
HIFs are O2-sensitive transcription factors involved in O2-dependent gene regulation that mediate cellular adaptation to O2 deprivation and tissue protection under hypoxic conditions in the kidney. The induction of HO-1 can protect the kidney from ischemic damage by decreasing oxidative damage and NO generation. Finally, in addition to its anti-apoptotic properties, EPO may protect the kidney from ischemic damage by restoring the renal microcirculation by stimulating the mobilization and differentiation of progenitor cells toward an endothelial phenotype and by inducing NO release from eNOS.
The kidney is not only a major source of arginine and nitric oxide but NO plays an important role in the
- water and electrolyte balance and
- acid-base physiology and
- many other homeostatic functions in the kidney.
We know that there is an unquestionable role of NO, and a competing balance to be achieved between eNOS, iNOS, an effect on tubular water and ion-cation reabsorption, a role of TNFa, and consequently an important role in essential/malignant hypertension, with the size of the effect related to the stage of disorder, the amount of interstitial fibrosis, the remaining nephron population, the hypertonicity of the medulla, the vasodilation of the medullary circulation, and the renin-angiotensin-aldosterone system.

(Photo credit: Wikipedia)

English: Nephron, Diagram of the urine formation. The number inside tubular urin concentration in mOsm/l – when ADH acts Polski: Nefron, Schemat tworzenia moczu. Cyfry wewnątrz kanalików oznaczają lokalne stężenie w mOsm/l – gdy działa ADH (dochodzi do zagęszczania moczu). (Photo credit: Wikipedia)
Related articles
- Low Bioavailability of Nitric Oxide due to Misbalance in Cell Free Hemoglobin in Sickle Cell Disease – A Computational Model (pharmaceuticalintelligence.com)
- Nitric Oxide and Immune responses: part 1 (pharmaceuticalintelligence.com)
- Differential Distribution of Nitric Oxide – A 3-D Mathematical Model (pharmaceuticalintelligence.com)
- Nitric Oxide and Immune Responses: Part 2 (pharmaceuticalintelligence.com)
- MitoQ Blunts Mitochondrial and Renal Damage during Cold Preservation of Porcine Kidneys (plosone.org)
- Nitric Oxide and Sepsis, Hemodynamic Collapse, and the Search for Therapeutic Options (pharmaceuticalintelligence.com)
- Discovery of nitric oxide delivery mechanism may point to new avenue for treating high blood pressure (medicalxpress.com)
- Statins’ nonlipid effects on vascular endothelium through eNOS activation (pharmaceuticalintelligence.com)
Nitric Oxide Function in Coagulation – Part II
Posted in Biological Networks, Gene Regulation and Evolution, Biomarkers & Medical Diagnostics, Cell Biology, Signaling & Cell Circuits, Chemical Biology and its relations to Metabolic Disease, Coagulation Therapy and Internal Bleeding, Disease Biology, Small Molecules in Development of Therapeutic Drugs, Human Immune System in Health and in Disease, International Global Work in Pharmaceutical, Metabolomics, Molecular Genetics & Pharmaceutical, Nitric Oxide in Health and Disease, Personalized and Precision Medicine & Genomic Research, Pharmaceutical Industry Competitive Intelligence, Population Health Management, Genetics & Pharmaceutical, Systemic Inflammatory Response Related Disorders, tagged Coagulation, Endothelial Dysfunction, endothelial progenitor cells, Endothelin, Endothelin Receptors, Endothelium, Factor IX, Factor VII, Factor VIII, Factor XI, nitric oxide, Nitric oxide synthase, Partial thromboplastin time, Platelet, tissue factor on November 26, 2012| 5 Comments »
Nitric Oxide Function in Coagulation – Part II
Curator and Author: Larry H. Bernstein, MD, FCAP
Subtitle: Nitric oxide in hemostatic and bleeding mechanisms. Part II.
Summary: This is the second of a coagulation series on PharmaceuticalIntelligence(wordpress.com) treating the diverse effects of NO on platelets, the coagulation cascade, and protein-membrane interactions with low flow states, local and systemic inflammatory disease, oxidative stress, and hematologic disorders. It is highly complex as the distinction between intrinsic and extrinsic pathways become blurred as a result of endothelial shear stress, distinctly different than penetrating or traumatic injury. In addition, other factors that come into play are also considered.
Please refer to Part I.
Coagulation Pathway
The workhorse tests of the modern coagulation laboratory, the prothrombin time (PT) and the activated partial thromboplastin time (aPTT), are the basis for the published extrinsic and intrinsic coagulation pathways. This is, however, a much simpler model than one encounters delving into the mechanism and interactions involved in hemostasis and thrombosis, or in hemorrhagic disorders.
We first note that there are three components of the hemostatic system in all vertebrates:
- Platelets,
- vascular endothelium, and
- plasma proteins.
The liver is the largest synthetic organ, which synthesizes
- albumin,
- acute phase proteins,
- hormonal and metal binding proteins,
- albumin,
- IGF-1, and
- prothrombin, mainly responsible for the distinction between plasma and serum (defibrinated plasma).
Role of vascular endothelium.
I have identified the importance of prothrombin, thrombin, and the divalent cation Ca 2+ (1% of the total body pool), mention of heparin action, and of vitamin K (inhibited by warfarin). Endothelial functions are inherently related to procoagulation and anticoagulation. The subendothelial matrix is a complex of many materials, most important related to coagulation being collagen and von Willebrand factor.
What about extrinsic and intrinsic pathways? Tissue factor, when bound to factor VIIa, is the major activator of the extrinsic pathway of coagulation. Classically, tissue factor is not present in the plasma but only presented on cell surfaces at a wound site, which is “extrinsic” to the circulation. Or is it that simple?
Endothelium is the major synthetic and storage site for von Willebrand factor (vWF). vWF is…
- secreted from the endothelial cell both into the plasma and also
- abluminally into the subendothelial matrix, and
- acts as the intercellular glue binding platelets to one another and also to the subendothelial matrix at an injury site.
- acts as a carrier protein for factor VIII (antihemophilic factor).
- It binds to the platelet glycoprotein Ib/IX/V receptor and
- mediates platelet adhesion to the vascular wall under shear. [Lefkowitz JB. Coagulation Pathway and Physiology. Chapter I. in Hemostasis Physiology. In ( ???), pp1-12].
Ca++ and phospholipids are necessary for all of the reactions that result in the activation of prothrombin to thrombin. Coagulation is initiated by an extrinsic mechanism that
- generates small amounts of factor Xa, which in turn
- activates small amounts of thrombin.
The tissue factor/factorVIIa proteolysis of factor X is quickly inhibited by tissue factor pathway inhibitor (TFPI).The small amounts of thrombin generated from the initial activation feedback
- to create activated cofactors, factors Va and VIIIa, which in turn help to
- generate more thrombin.
- Tissue factor/factor VIIa is also capable of indirectly activating factor X through the activation of factor IX to factor IXa.
- Finally, as more thrombin is created, it activates factor XI to factor XIa, thereby enhancing the ability to ultimately make more thrombin.
The reconceptualization of hemostasis
The common theme in activation and regulation of plasma coagulation is the reduction in dimensionality. Most reactions take place in a 2D world that will increase the efficiency of the reactions dramatically. The localization and timing of the coagulation processes are also dependent on the formation of protein complexes on the surface of membranes. The coagulation processes can also be controlled by certain drugs that destroy the membrane binding ability of some coagulation proteins – these proteins will be lost in the 3D world and not able to form procoagulant complexes on surfaces.
Assembly of proteins on membranes – making a 3D world flat
• The timing and efficiency of coagulation processes are handled by reduction in dimensionality
– Make 3 dimensions to 2 dimensions
• Coagulation proteins have membrane binding capacity
• Membranes provide non-coagulant and procoagulant surfaces
– Intact cells/activated cells
• Membrane binding is a target for anticoagulant drugs
– Anti-vitamin K (e.g. warfarin)
Modern View
It can be divided into the phases of initiation, amplification and propagation.
- In the initiation phase, small amounts of thrombin can be formed after exposure of tissue factor to blood.
- In the amplification phase, the traces of thrombin will be inactivated or used for amplification of the coagulation process.
At this stage there is not enough thrombin to form insoluble fibrin. In order to proceed further thrombin activates platelets, which provide a procoagulant surface for the coagulation factors. Thrombin will also activate the vital cofactors V and VIII that will assemble on the surface of activated platelets. Thrombin can also activate factor XI, which is important in a feedback mechanism.
In the final step, the propagation phase, the highly efficient tenase and prothrombinase complexes have been assembled on the membrane surface. This yields large amounts of thrombin at the site of injury that can cleave fibrinogen to insoluble fibrin. Factor XI activation by thrombin then activates factor IX, which leads to the formation of more tenase complexes. This ensures enough thrombin is formed, despite regulation of the initiating TF-FVIIa complex, thus ensuring formation of a stable fibrin clot. Factor XIII stabilizes the fibrin clot through crosslinking when activated by thrombin.
Platelet Aggregation
The activities of adenylate and guanylate cyclase and cyclic nucleotide 3′:5′-phosphodiesterase were determined during the aggregation of human blood platelets with
- thrombin, ADP,
- arachidonic acid and
- epinephrine.
[Aggregation is dependent on an intact release mechanism since inhibition of aggregation occurred with adenosine, colchicine, or EDTA. (Herman GE, Seegers WH, Henry RL. Autoprothrombin ii-a, thrombin, and epinephrine: interrelated effects on platelet aggregation. Bibl Haematol 1977;44:21-7)].
- The platelet guanylate cyclase activity during aggregation depends on the nature and mode of action of the inducing agent.
- The membrane adenylate cyclase activity during aggregation is independent of the aggregating agent and is associated with a reduction of activity and
- Cyclic nucleotide phosphodiesterase remains unchanged during the process of platelet aggregation and release.
The role of platelets in arterial thrombosis
Formation of a thrombus on a ruptured plaque is the product of a complex interaction between coagulation factors in the plasma and platelets.
- Tissue factor (TF) released from the subendothelial tissue after endothelial damage induces a cascade of activation of coagulation factors ultimately leading to the formation of thrombin.
- Thrombin cleaves fibrinogen to fibrin, which assembles into a mesh that supports the platelet aggregates.
The Platelet
The platelets are …
- anucleated,
- discoid shaped cell fragments
- originating from megakaryocytes
- fragmented as they are released from the bone marrow
Whether they can in circumstances be developed at extramedullary sites (liver sinusoid) is another matter. They have a lifespan of 7-10 days. Of special interest is:
- They have a network of internal membranes forming a dense tubular system and the open canalicular system (OCS).
- The plasma membrane is an extension of the OCS, thereby greatly increasing the surface area of the platelet.
- The dense tubular system is comparable to the endoplasmatic reticulum in other cell types and is the main storage place of the majority of the platelet’s Ca2+.
Three types of secretory granules exist in platelets:
- the dense granules
- In the dense granules serotonin
- adenosine diphosphate (ADP) and
- Ca2+ are stored.
- a-granules contain
- P-selectin,
- fibrinogen,
- thrombospondin,
- Von Willebrand Factor,
- platelet factor 4 and
- platelet derived growth factor
- lysosomes.
Circulating platelets are kept in a resting state by endothelial cell derived
- prostacyclin (PGI2) and
- nitric oxide (NO).
PGI2 increases cyclic adenosine monophosphate (cAMP), the most potent platelet inhibitor.
Contact activation
The major regulator of the activation of the contact system is the plasma protease inhibitor, C1-INH, which inhibits activated fXII, kallikrein and fXIa. In addition, α2-macroglobulin is an important inhibitor of kallikrein and α1-antitrypsin for fXIa. Factor XII also converts the fXI to an active enzyme, fXIa, which, in turn, converts fIX to fIXa, thereby activating the intrinsic pathway of coagulation.
Activation
Several agonists can activate platelets;
- ADP,
- collagen,
- thromboxane A2 (TxA2),
- epinephrin,
- serotonine and
- thrombin,
which lead to activation previously referred to:
- platelet shape change is
- followed by aggregation and
- granule secretion.
Upon activation the discoid shape changes into a spherical form.
Activation of platelets is increased by two positive feedback loops
- arachidonic acid is cleaved from phospholipids and transformed by cyclooxygenase
(COX) to prostaglandin G2 and H2,
- followed by the formation of TxA2, a potent platelet agonist.
2. the secretion of ADP by the dense granules,
- resulting in activation of the ADP receptor P2Y12.
This causes inhibition of cyclic AMP and sustained aggregation.
Aggregation
The integrin receptor αIIbβ3 plays a vital role in platelet aggregation. The platelet agonists
- induce a conformational change of the αIIbβ3 receptor and
- exposition of binding domains for fibrinogen and von Willebrand Factor.
This allows cross-linking of platelets and the formation of aggregates.
In addition to shape change and aggregation, the membranes of the α- and dense granules fuse with the membranes of the OCS. This causes the release of their contents and the transportation of proteins embedded in their membrane to the plasma membrane.
This complex interaction between
- endothelial cells
- clotting factors
- platelets and
- other factors and cells
can be studied in both in vitro and in vivo model systems. The disadvantage of in vitro assays is that it studies the role of a certain protein or cell in isolation. Given the large number of participants and the complex interactions of thrombus formation there is need to study thrombosis and hemostasis in intact living animals, with all the components important for thrombus formation – a vessel wall and flowing blood – present.
Endothelial Damage and Role as “Primer”
- Endothelial injury changes the permeability of the arterial wall.
- This is followed by an influx of low-density lipoprotein (LDL).
- This elicits an inflammatory response in the vascular wall.
- Monocytes and T-cells bind to the endothelial cells promoting increased migration of the cells into the intima layer
- The monocytes differentiate into macrophages, which take up modified lipoproteins and transform them into foam cells.
- Concurrent with this process macrophages produce cytokines and proteases.
This is a vicious circle of lipid driven inflammation that leads to narrowing of the vessel’s lumen without early clinical consequences. Clinical manifestations of more advanced atherosclerotic disease are caused by destabilization of an atherosclerotic plaque formed as described.
- The first recognizable lesion of the stable atherosclerotic plaque is the fatty streak, which consists of the above described foam cells and T-lymphocytes in the intima.
- Further development of the lesion leads to the intermediate lesion, composed
- of layers of macrophages and smooth muscle cells.
- A more advanced stage is called the vulnerable plaque.
- It has a large lipid core that is covered by a thin fibrous cap.
- This cap separates the lipid contents of the plaque from the circulating blood.
- The vulnerable plaque is prone to rupture, resulting in the formation of a thrombus on the site of disruption or the thrombus can be superimposed on plaque erosion without signs of plaque rupture.
The formation of a superimposed thrombus on a disrupted atherosclerotic plaque in the lumen of the artery leads to
- an acute occlusion of the vessel
- hypoxia of the downstream tissue.
Depending on the location of the atherosclerotic plaque this will cause a myocardial infarction, stroke or peripheral vascular disease.
Endothelial regulation of coagulation
The endothelium attenuates platelet activity by releasing
- nitric oxide and
- prostacyclin.
Several coagulation inhibitors are produced by endothelial cells.
Endothelium-derived TFPI (on its surface) is rapidly released into circulation after heparin administration, reducing the pro-coagulant activities of TF-fVIIa. Endothelial cells also secrete heparin-sulphate, a glycosaminoglycan which catalyzes anti-coagulant activity of AT. Plasma AT binds to heparin-sulphate located on the luminal surface and in the basement membrane of the endothelium. Thrombomodulin is another endothelium-bound protein with anti-coagulant and anti-inflammatory functions. In response to systemic pro-coagulant stimuli, tissue-type plasminogen activator (tPA) is transiently released from the Weibel-Palade bodies of endothelial cells to promote fibrinolysis. Downstream of the vascular injury, the complex of TF-fVIIa/fXa is inhibited by TFPI. Plasma (free) fXa and thrombin are rapidly neutralized by heparan-bound AT. Thrombin is also taken up by endothelial surface-bound thrombomodulin.
The protein C pathway works in hemostasis to control thrombin formation in the area surrounding the clot. Thrombin, generated via the coagulation pathway, is localized to the endothelium by binding to the integral membrane protein, thrombomodulin (TM). TM by occupying exosite I on thrombin, which is required for fibrinogen binding and cleavage, reduces thrombin’s pro-coagulant activities. TM bound thrombin on the endothelial cell surface is able to cleave PC producing activated protein C (APC), a serine protease. In the presence of protein S, APC inactivates FVa and FVIIIa. The proteolytic activity of APC is regulated predominantly by a protein C inhibitor.
Fibrinolytic pathway
Fibrinolysis is the physiological breakdown of fibrin to limit and resolve blood clots. Fibrin is degraded primarily by the serine protease, plasmin, which circulates as plasminogen. In an auto-regulatory manner, fibrin serves as both the co-factor for the activation of plasminogen and the substrate for plasmin. In the presence of fibrin, tissue plasminogen activator (tPA) cleaves plasminogen producing plasmin, which proteolyzes the fibrin. This reaction produces the protein fragment D-dimer, which is a useful marker of fibrinolysis, and a marker of thrombin activity because fibrin is cleaved from fibrinogen to fibrin.
Nitric Oxide and Platelet Energy Production
Nitric oxide (NO) has been increasingly recognized as an important intra- and intercellular messenger molecule with a physiological role in
- vascular relaxation
- platelet physiology
- neurotransmission and
- immune responses.
In vitro NO is a strong inhibitor of platelet adhesion and aggregation. In the blood stream, platelets remain in contact with NO that is permanently released from the endothelial cells and from activated macrophages. It has been suggested that the activated platelet itself is able to produce NO. It has been proposed that the main intracellular target for NO in platelets is soluble cytosolic guanylate cyclase. NO activates the enzyme. When activated, intracellular cGMP elevation inhibits platelet activation. Further, elevated cGMP may not be the sole factor directly involved in the inhibition of platelet activation.

- The reaction mechanism of Nitric oxide synthase (Photo credit: Wikipedia)
Platelets are fairly active metabolically and have a total ATP turnover rate of about 3–8 times that of resting mammalian muscle. Platelets contain mitochondria which enable these cells to produce energy both in the oxidative and anaerobic pathways.
- Under aerobic conditions, ATP is produced by aerobic glycolysis which can account for 30–50% of total ATP production,
- by oxidative metabolism using glucose and glycogen (6–11%), amino-acids (7%) or free fatty acids (20–40%).
The inhibition of mitochondrial respiration by removing oxygen or by respiratory chain blockers (antimycin A, cyanide, rotenone) results in the stimulation of glycolytic flux. This phenomenon indicates that in platelets glycolysis and mitochondrial respiration are tightly functionally connected. It has been reported that the activation of human platelets by high concentration of thrombin is accompanied by an acceleration of lactate production and an increase in oxygen consumption.
The results (in porcine platelets) indicate that:
- NO is able to diminish mitochondrial energy production through the inhibition of cytochrome oxidase
- The inhibitory effect of NO on platelet secretion (but not aggregation) can be attributed to the reduction of mitochondrial energy production.
Porcine blood platelets stimulated by collagen produce more lactate. This indicates that both glycolytic and oxidative ATP production supports platelet responses, and blocking of energy production in platelets may decrease their responses. It is well established that platelet responses have different metabolic energy (ATP) requirements increasing in the order:
- Aggregation
- < dense and alfa granule secretion
- < acid hydrolase secretion.
In addition, exogenously added NO (in the form of NO donors) stimulates glycolysis in intact porcine platelets. Since in platelets glycolysis and mitochondrial respiration are tightly functionally connected, this indicates the stimulatory effect of NO on glycolysis in intact platelets may be produced by non-functional mitochondria.
Can this be the case?
- NO donors are able to inhibit both mitochondrial respiration and platelet cytochrome oxidase.
- Interestingly, the concentrations of NO donors inhibiting mitochondrial respiration and cytochrome oxidase were similar to those stimulating glycolysis in intact platelets.
Studies have shown that mitochondrial complex I is inhibited only after a prolonged (6–18 h) exposure to NO and
- This inhibition appears to result from S-nitrosylation of critical thiols in the enzyme complex.
- Further studies are needed to establish whether long term exposure of platelets to NO affects Mitochondrial complexes I and II.
Comparison of the concentrations of SNAP and SNP affecting cytochrome oxidase activity and mitochondrial respiration with those reducing the platelet responses indicates that NO does not reduce platelet aggregation through the inhibition of oxidative energy production. The concentrations of the NO donors inhibiting platelet secretion, mitochondrial respiration and cytochrome oxidase were similar. Thus, the platelet release reaction strongly depends on the oxidative energy production, and in porcine platelets NO inhibits mitochondrial energy production at the step of cytochrome oxidase.
Taking into account that platelets may contain NO synthase and are able to produce significant amounts of NO it seems possible that nitric oxide can function in these cells as a physiological regulator of mitochondrial energy production.
Key words: glycolysis, mitochondrial energy production, nitric oxide, porcine platelets.
Abbreviations: NO, nitric oxide; SNAP, S-nitroso-N-acetylpenicyllamine; SNP, sodium nitroprusside.
[M Tomasiak, H Stelmach, T Rusak and J Wysocka. Nitric oxide and platelet energy metabolism. Acta Biochimica Polonica 2004; 51(3):789–803.]
Nitric Oxide and Platelet Adhesion
The adhesion of human platelets to monolayers of bovine endothelial cells in culture was studied to determine the role of endothelium-derived nitric oxide in the regulation of platelet adhesion. The adhesion of unstimulated and thrombin-stimulated platelets, washed and labelled with indium-111, was lower in the presence than in the absence of bradykinin or exogenous nitric oxide. The inhibitory action of both bradykinin and nitric oxide was abolished by hemoglobin, but not by aspirin, and was potentiated by superoxide dismutase to a similar degree. It appears that the effect of bradykinin is mediated by the release of nitric oxide from the endothelial cells, and that nitric oxide release contributes to the non-adhesive properties of vascular endothelium.
(Radomski MW, Palmer RMJ, Moncada S. Endogenous Nitric Oxide Inhibits Human Platelet Adhesion to Vascular Endothelium. The Lancet 1987 330; 8567(2): 1057–1058.
http://dx.doi.org/10.1016/S0140-6736(87)91481-4)
1 The interactions between endothelium-derived nitric oxide (NO) and prostacyclin as inhibitors of platelet aggregation were examined to determine whether release of NO accounts for the inhibition of platelet aggregation attributed to EDRF.
2 Porcine aortic endothelial cells treated with indomethacin and stimulated with bradykinin (10-100 nM) released NO in quantities sufficient to account for the inhibition of platelet aggregation attributed to endothelium-derived relaxing factor (EDRF).
3 In the absence of indomethacin, stimulation of the cells with bradykinin (1-3 nM) released small amounts of prostacyclin and EDRF which synergistically inhibited platelet aggregation.
4 EDRF and authentic NO also caused disaggregation of platelets aggregated either with collagen or with U46619.
5 A reciprocal potentiation of both the anti- and the disaggregating activity was also observed between low concentrations of prostacyclin and authentic NO or EDRF released from endothelial cells.
6 It is likely that interactions between prostacyclin and NO released by the endothelium play a role in the homeostatic regulation of platelet-vessel wall interactions.
(Radomski MW, Palmer RMJ & Moncada S. The anti-aggregating properties of vascular endothelium: interactions between prostacyclin and nitric oxide. Br J Pharmac 1987; 92: 639-646.
Factor Xa–Nitric Oxide Signaling
Although primarily recognized for maintaining the hemostatic balance, blood proteases of the coagulation and fibrinolytic cascades elicit rapid cellular responses in
- vascular
- mesenchymal
- inflammatory cell types.
Considerable effort has been devoted to elucidate the molecular interface between protease-dependent signaling and pleiotropic cellular responses. This led to the identification of several membrane protease receptors, initiating intracellular signal transduction and effector functions in vascular cells. In this context, thrombin receptor activation
- generated second messengers in endothelium and smooth muscle cells,
- released inflammatory cytokines from monocytes, fibroblasts, and endothelium, and
- increased the expression of leukocyte-endothelial cell adhesion molecules.
Similarly, binding of factor Xa to effector cell protease receptor-1 (EPR-1) participated in
- in vivo acute inflammatory responses,
- platelet and brain pericyte prothrombinase activity, and
- endothelial cell and smooth muscle cell signaling and proliferation.
Factor Xa stimulated a 5- to 10-fold increased release of nitric oxide (NO) in a dose-dependent reaction (0.1–2.5 mgyml) unaffected by the thrombin inhibitor hirudin but abolished by active site inhibitors, tick anticoagulant peptide, or Glu-Gly-Arg-chloromethyl ketone. In contrast, the homologous clotting protease factor IXa or another endothelial cell ligand, fibrinogen, was ineffective.
A factor Xa inter-epidermal growth factor synthetic peptide L83FTRKL88(G) blocking ligand binding to effector cell protease receptor-1 inhibited NO release by factor Xa in a dose-dependent manner, whereas a control scrambled peptide KFTGRLL was ineffective.
Catalytically active factor Xa induced hypotension in rats and vasorelaxation in the isolated rat mesentery, which was blocked by the NO synthase inhibitor L-NG-nitroarginine methyl ester (LNAME) but not by D-NAME. Factor Xa/NO signaling also produced a dose-dependent endothelial cell release of interleukin 6 (range 0.55–3.1 ngyml) in a reaction
- inhibited by L-NAME and by the
- inter-epidermal growth factor peptide Leu83–Leu88 but
- unaffected by hirudin.
| We observe that incubation of HUVEC monolayers with factor Xa which resulted in a concentration-dependent release of NO, as determined by cGMP accumulation in these cells, was inhibited by the nitric oxide synthase antagonist L-NAME. |
Catalytically inactive DEGR-factor Xa or TAP-treated factor Xa failed to stimulate NO release by HUVEC.
To determine whether factor Xa-induced NO release could also modulate acute phase/inflammatory cytokine gene expression we examined potential changes in IL-6 release following HUVEC stimulation with factor Xa. HUVEC stimulation with factor Xa resulted in a concentration-dependent release of IL-6.
The specificity of factor Xa-induced cytokine release was investigated. Factor Xa-induced IL-6 release from HUVEC was quantitatively indistinguishable from that obtained with tumor necrosis factor-a or thrombin stimulation. This response was abolished by heat denaturation of factor Xa.
Maximal induction of interleukin 6 mRNA required a brief, 30-min stimulation with factor Xa, and was unaffected by subsequent addition of tissue factor pathway inhibitor (TFPI). These data suggest that factor Xa-induced NO release modulates endothelial cell-dependent vasorelaxation and IL-6 cytokine gene expression.
Here, we find that factor Xa induces the release of endothelial cell NO
This pathway requires a dual step cascade, involving
This pathway requiring factor Xa binding to effector cell protease receptor-1 and a secondary step of ligand-dependent proteolysis may preserve an anti-thrombotic phenotype of endothelium but also trigger acute phase responses during activation of coagulation in vivo. |
In summary, these investigators have identified a signaling pathway centered on the ability of factor Xa to rapidly stimulate endothelial cell NO release. This involves a two-step cascade initiated by catalytic active site-independent binding of factor Xa to its receptor, EPR-1, followed by a second step of ligand dependent proteolysis.
(Papapetropoulos A, Piccardoni P, Cirino G, Bucci M, et al. Hypotension and inflammatory cytokine gene expression triggered by factor Xa–nitric oxide signaling. Proc. Natl. Acad. Sci. USA. Pharmacology. 1998; 95:4738–4742.)
Platelets and liver disease
Thrombocytopenia is a marked feature of chronic liver disease and cirrhosis. Traditionally, this thrombocytopenia was attributed to passive platelet sequestration in the spleen. More recent insights suggest an increased platelet breakdown and to a lesser extent decreased platelet production plays a more important role. Besides the reduction in number, other studies suggest functional platelet defects. This platelet dysfunction is probably both intrinsic to the platelets and secondary to soluble plasma factors. It reflects not only a decrease in aggregability, but also an activation of the intrinsic inhibitory pathways. (Witters P, Freson K, Verslype C, Peerlinck K, et al. Review article: blood platelet number and function in chronic liver disease and cirrhosis. Aliment Pharmacol Ther 2008; 27: 1017–1029).
The shortcomings of the old Y-shaped model of normal coagulation are nowhere more apparent than in its clinical application to the complex coagulation disorders of acute and chronic liver disease. In this condition, the clotting cascade is heavily influenced by numerous currents and counter-currents resulting in a mixture of pro- and anticoagulant forces that are themselves further subject to change with altered physiological stress such as super-imposed infection or renal failure.
Multiple mechanisms exist for thrombocytopenia common in patients with cirrhosis besides hypersplenism and expected altered thrombopoietin metabolism. Increased production of two important endothelial derived platelet inhibitors
- nitric oxide and
- prostacyclin
may contribute to defective platelet activation in vivo. On the other hand, high plasma levels of vWF in cirrhosis appear to support platelet adhesion.
Reduced levels of coagulation factors V, VII, IX, X, XI, and prothrombin are also commonly observed in liver failure. Vitamin K–dependent clotting factors (II, VII, IX, X) may be defective in function as a result of decreased y-carboxylation (from vitamin K deficiency or intrinsically impaired carboxylase activity). Fibrinogen levels are decreased with advanced cirrhosis and in patients with acute liver failure.
A hyperfibrinolytic state may develop when plasminogen activation by tPA is accelerated on the fibrin surface. Physiologic stress including infection may be key in tipping this process off through increased release of tPA. Not uncommonly, laboratory abnormalities in decompensated cirrhosis come to resemble disseminated intravascular coagulation (DIC). Relatively stable platelet levels and characteristically high factor VIII levels distinguish this process from DIC as does the absence of uncompensated thrombin generation. The features of both hyperfibrinolysis and DIC are often evident in the decompensated liver disease patient, and the term “accelerated intravascular coagulation and fibrinolysis” (AICF) has been proposed as a way to encapsulate the process under a single heading. The essence of AICF can be postulated to be the result of formation of a fibrin clot that is more susceptible to plasmin degradation due to elevated levels of tPA coupled with inadequate release of PAI to control tPA and lack of a-2 plasmin inhibitor to quench plasmin activity and the maintenance of high local concentrations of plasminogen on clot surfaces despite lower total plasminogen production. These normally balanced processes become pronounced when disturbed by additional stress such as infection.
Normal hemostasis and coagulation is now viewed as primarily a cell-based process wherein key steps in the classical clotting cascade
- occur on the phospholipid membrane surface of cells (especially platelets)
- beginning with activation of tissue factor and factor VII at the site of vascular breach
- which produces an initial “priming” amount of thrombin and a
- subsequent thrombin burst.
Coagulation and hemostasis in the liver failure patient is influenced by multiple, often opposing, and sometimes changing variables. A bleeding diathesis is usually predominant, but the assessment of bleeding risk based on conventional laboratory tests is inherently deficient.
(Caldwell SH, Hoffman M, Lisman T, Gail Macik B, et al. Coagulation Disorders and Hemostasis in Liver Disease: Pathophysiology and Critical Assessment of Current Management. Hepatology 2006;44:1039-1046.)
Bleeding after Coronary Artery bypass Graft
Cardiac surgery with concomitant CPB can profoundly alter haemostasis, predisposing patients to major haemorrhagic complications and possibly early bypass conduit-related thrombotic events as well. Five to seven percent of patients lose more than 2 litres of blood within the first 24 hours after surgery, between 1% and 5% require re-operation for bleeding. Re-operation for bleeding increases hospital mortality 3 to 4 fold, substantially increases post-operative hospital stay and has a sizeable effect on health care costs. Nevertheless, re-exploration is a strong risk factor associated with increased operative mortality and morbidity, including sepsis, renal failure, respiratory failure and arrhythmias.
(Gábor Veres. New Drug Therapies Reduce Bleeding in Cardiac Surgery. Ph.D. Doctoral Dissertation. 2010. Semmelweis University)
Hypercoagulable State in Thalassemia
As the life expectancy of β-thalassemia patients has increased in the last decade, several new complications are being recognized. The presence of a high incidence of thromboembolic events, mainly in thalassemia intermedia patients, has led to the identification of a hypercoagulable state in thalassemia. Patients with thalassemia intermedia (TI) have, in general, a milder clinical phenotype than those with TM and remain largely transfusion independent. The pathophysiology of TI is characterized by extravascular hemolysis, with the release into the peripheral circulation of damaged red blood cells (RBCs) and erythroid precursors because of a high degree of ineffective erythropoiesis. This has also been recently attributed to severe complications such as pulmonary hypertension (PHT) and thromboembolic phenomena.
Many investigators have reported changes in the levels of coagulation factors and inhibitors in thalassemic patients. Prothrombin fragment 1.2 (F1.2), a marker of thrombin generation, is elevated in TI patients. The status of protein C and protein S was investigated in thalassemia in many studies and generally they were found to be decreased; this might be responsible for the occurrence of thromboembolic events in thalassemic patients.
The pathophysiological roles of hemolysis and the dysregulation of nitric oxide homeostasis are correlated with pulmonary hypertension in sickle cell disease and in thalassemia. Nitric oxide binds soluble guanylate cyclase, which converts GTP to cGMP, relaxing vascular smooth muscle and causing vasodilatation. When plasma hemoglobin liberated from intravascularly hemolyzed sickle erythrocytes consumes nitric oxide, the balance is shifted toward vasoconstriction. Pulmonary hypertension is aggravated and in sickle cell disease, it is linked to the intensity of hemolysis. Whether the same mechanism contributes to hypercoagulability in thalassemia is not yet known.
While there are diverse factors contributing to the hypercoagulable state observed in patients with thalassemia. In most cases, a combination of these abnormalities leads to clinical thrombosis. An argument has been made for the a higher incidence of thrombotic events in TI compared to TM patients attributed to transfusion for TM. The higher rate of thrombosis in transfusion-independent TI compared to polytransused TM patients suggests a potential role for transfusions in decreasing the rate of thromboembolic events (TEE). The reduction of TEE in adequately transfused patients may be the result of decreased numbers of pathological RBCs.
(Cappellini MD, Musallam KM, Marcon A, and Taher AT. Coagulopathy in Beta-Thalassemia: Current Understanding and Future Prospects. Medit J Hemat Infect Dis 2009; 1(1):22009029.
DOI 10.4084/MJHID.2009.0292.0), www.mjhid.org/article/view/5250. ISSN 2035-3006.)
Microvascular Endothelial Dysfunction
Severe sepsis, defined as sepsis associated with acute organ dysfunction, results from a generalized inflammatory and procoagulant host response to infection. Coagulopathy in severe sepsis is commonly associated with multiple organ dysfunction, and often results in death. The molecule that is central to these effects is thrombin, although it may also have anticoagulant and antithrombotic effects through the activation of Protein C and induction of prostacyclin. In recent years, it has been recognized that chemicals produced by endothelial cells play a key role in the pathogenesis of sepsis. Thrombomodulin on endothelial cells coverts Protein C to Activated Protein C, which has important antithrombotic, profibrinolytic and anti-inflammatory properties. A number of studies have shown that Protein C levels are reduced in patients with severe infection, or even in inflammatory states without infection. Because coagulopathy is associated with high mortality rates, and animal studies have indicated that therapeutic intervention may result in improved outcomes, it was rational to initiate clinical studies.
Considering the coagulation cascade as a whole, it is the extrinsic pathway (via TF and thrombin activation) rather than the intrinsic pathway that is of primary importance in sepsis. Once coagulation has been triggered by TF activation, leading to thrombin formation, this can have further procoagulant effects, because thrombin itself can activate factors VIII, IX and X. This is normally balanced by the production of anticoagulant factors, such as TF pathway inhibitor, antithrombin and Activated Protein C.
It has been recognized that endothelial cells play a key role in the pathogenesis of sepsis, and that they produce important regulators of both coagulation and inflammation. They can express or release a number of substances, such as TF, endothelin-1 and PAI-1, which promote the coagulation process, as well as other substances, such as antithrombin, thrombomodulin, nitric oxide and prostacyclin, which inhibit it.
Protein C is the source of Activated Protein C. Although Protein C is a biomarker or indicator of sepsis, it has no known specific biological activity. Protein C is converted to Activated Protein C in the presence of normal endothelium. In patients with severe sepsis, the vascular endothelium becomes damaged. The level of thrombomodulin is significantly decreased, and the body’s ability to convert Protein C to Activated Protein C diminishes. Only when activated does Protein C have antithrombotic, profibrinolytic and anti-inflammatory properties.
 and Protein C Pat...")
Blood Coagulation (Thrombin) and Protein C Pathways (Blood_Coagulation_and_Protein_C_Pathways.jpg) (Photo credit: Wikipedia)
Coagulation abnormalities can occur in all types of infection, including both Gram-positive and Gram-negative bacterial infections, or even in the absence of infection, such as in inflammatory states secondary to trauma or neurosurgery. Interestingly, they can also occur in patients with localized disease, such as those with respiratory infection. In a study by Günther et al., procoagulant activity in bronchial lavage fluid from patients with pneumonia or acute respiratory distress syndrome was found to be increased compared with that from control individuals, with a correlation between the severity of respiratory failure and level of coagulant activity.
Severe sepsis, defined as sepsis associated with acute organ dysfunction, results from a generalized inflammatory and procoagulant host response to infection. Once the endothelium becomes damaged, levels of endothelial thrombomodulin significantly decrease, and the body’s ability to convert Protein C to Activated Protein C diminishes. The ultimate cause of acute organ dysfunction in sepsis is injury to the vascular endothelium, which can result in microvascular coagulopathy.
(Vincent JL. Microvascular endothelial dysfunction: a renewed appreciation of sepsis pathophysiology.
Critical Care 2001; 5:S1–S5. http://ccforum.com/content/5/S2/S1)
Endothelial Cell Dysfunction in Severe Sepsis
During the past decade a unifying hypothesis has been developed to explain the vascular changes that occur in septic shock on the basis of the effect of inflammatory mediators on the vascular endothelium. The vascular endothelium plays a central role in the control of microvascular flow, and it has been proposed that widespread vascular endothelial activation, dysfunction and eventually injury occurs in septic shock, ultimately resulting in multiorgan failure. This has been characterized in various models of experimental septic shock. Now, direct and indirect evidence for endothelial cell alteration in humans during septic shock is emerging.
The vascular endothelium regulates the flow of nutrient substances, diverse biologically active molecules and the blood cells themselves. This role of endothelium is achieved through the presence of membrane-bound receptors for numerous molecules, including proteins, lipid transporting particles, metabolites and hormones, as well as through specific junction proteins and receptors that govern cell–cell and cell–matrix interactions. Endothelial dysfunction and/or injury with subendothelium exposure facilitates leucocyte and platelet aggregation, and aggravation of coagulopathy. Therefore, endothelial dysfunction and/or injury should favour impaired perfusion, tissue hypoxia and subsequent organ dysfunction.
Anatomical damage to the endothelium during septic shock has been assessed in several studies. A single injection of bacterial lipopolysaccharide (LPS) has long been demonstrated to be a nonmechanical technique for removing endothelium. In endotoxic rabbits, observations tend to demonstrate that EC surface modification occurs easily and rapidly, with ECs being detached from the internal elastic lamina with an indication of subendothelial oedema. Proinflammatory cytokines increase permeability of the ECs, and this is manifested approximately 6 hours after inflammation is triggered and becomes maximal over 12–24 hours as the combination of cytokines exert potentiating effects. Endothelial physical disruption allows inflammatory fluid and cells to shift from the blood into the interstitial space.
In sepsis
- ECs become injured, prothrombotic and antifibrinolytic
- They promote platelet adhesion
- They promote leucocyte adhesion and inhibit vasodilation
An important point is that EC injury is sustained over time. In an endotoxic rabbit model, we demonstrated that endothelium denudation is present at the level of the abdominal aorta as early as after several hours following injury and persisted for at least 5 days afterward. After 21 days we observed that the endothelial surface had recovered. The de-endothelialized surface accounted for approximately 25% of the total surface.
Thrombomodulin and protein C activation at the microcirculatory level.
The endothelial cell surface thrombin (Th)-binding protein thrombomodulin (TM) is responsible for inhibition of thrombin activity. TM, when bound to Th, forms a potent protein C activator complex. Loss of TM and/or internalization results in Th–thrombin receptor (TR) interaction. Loss of TM and associated protein C activation represents the key event of decreased endothelial coagulation modulation ability and increased inflammation pathways.
( Iba T, Kidokoro A, Yagi Y: The role of the endothelium in changes in procoagulant activity in sepsis. J Am Coll Surg 1998; 187:321-329. Keywords: ATIII, antithrombin III; NF-κ, nuclear factor-κB; PAI,plasminogen activator inhibitor).
In order to test the role of the endothelial-derived relaxing factors NO and PGI2, we investigated, in dogs, the influence of a combination of NG-nitro-L-arginine methyl ester (an inhibitor of NO synthesis) and indomethacin (an inhibitor of PGI2 synthesis). In these dogs treated with indomethacin plus NG-nitro-L-arginine methyl ester, the severity of the oxygen extraction defect was lower than that observed in the deoxycholate-treated dogs, suggesting that other mediators and/or mechanisms may be involved in microcirculatory control during hypoxia. One of these mediators or mechanisms could be related to hyperpolarization. Membrane potential is an important determinant of vascular smooth muscle tone through its influence on calcium influx via voltage-gated calcium channels. Hyperpolarization (as well as depolarization) has been shown to be a means of cell–cell communication in upstream vasodilatation and microcirculatory coordination. It is important to emphasize that intercell coupling exclusively involves ECs.
Interestingly, it was recently shown that sepsis, a situation that is characterized by impaired tissue perfusion and abnormal oxygen extraction, is associated with abnormal inter-EC coupling and reduction in the arteriolar conducted response. An intra-organ defect in blood flow related to abnormal vascular reactivity, cell adhesion and coagulopathy may account for impaired organ oxygen regulation and function. If specific classes of microvessels must or must not be perfused to achieve efficient oxygen extraction during limitation in oxygen delivery, then impaired vascular reactivity and vessel injury might produce a pathological limitation in supply. In sepsis, the inflammatory response profoundly alters circulatory homeostasis, and this has been referred to as a ‘malignant intravascular inflammation’ that alters vasomotor tone and the distribution of blood flow among and within organs. These mechanisms might coexist with other types of sepsis associated cell dysfunction. For example, data suggest that endotoxin directly impairs oxygen uptake in ECs and indicate the importance of endothelium respiration in maintaining vascular homeostasis under conditions of sepsis.
Consistent with the hypothesis that alteration in endothelium plays a major in the pathophysiology of sepsis, it was observed that chronic ecNOS overexpression in the endothelium of mice resulted in resistance to LPS-induced hypotension, lung injury and death . This observation was confirmed by another group of investigators, who used transgenic mice overexpressing adrenomedullin – a vasodilating peptide that acts at least in part via an NO-dependent pathway. They demonstrated resistance of these animals to LPS-induced shock, and lesser declines in blood pressure and less severe organ damage than occurred in the control animals. It might therefore be of importance to favour ecNOS expression and function during sepsis. The recent negative results obtained with therapeutic strategies aimed at blocking inducible NOS with the nonselective NOS inhibitor NG-monomethyl-L-arginine in human septic shock further confirm the overall importance of favoring vessel dilatation.
(Vallet B. Bench-to-bedside review: Endothelial cell dysfunction in severe sepsis: a role in organ dysfunction? Critical Care 2003; 7(2):130-138 (DOI 10.1186/cc1864). (Print ISSN 1364-8535; Online ISSN 1466-609X). http://ccforum.com/content/7/2/130
Thrombosis in Inflammatory Bowel Disease
An association between IBD and thrombosis has been recognized for more than 60 years. Not only are patients with IBD more likely to have thromboembolic complications, but it has also been suggested that thrombosis might be pathogenic in IBD.
Coagulation Described. See Part I. (Cascade)
Endothelial injury exposes TF, which forms a complex with factor VII. This complex activates factors X and, to a lesser extent, IX. TFPI prevents this activation progressing further; for coagulation to progress, factor Xa must be produced via factors IX and VIII. Thrombin, generated by the initial production of factor Xa, activates factor VIII and, through factor XI, factor IX, resulting in further activation of factor X. This positive feedback loop allows coagulation to proceed. Fibrin polymers are stabilized by factor XIIIa. Activated proteins CS (APCS) together inhibit factors VIIIa and Va, whereas antithrombin (AT) inhibits factors VIIa, IXa, Xa, and XIa. Fibrinolysis balances this system through the action of plasmin on fibrin. Plasminogen activator inhibitor controls the plasminogen activator-induced conversion of plasminogen to plasmin.
Inflammation and Thrombotic Processes Linked
Although interest has recently moved away from the proposal that ischemia is a primary cause of IBD, it has become increasingly clear that inflammatory and thrombotic processes are linked. A vascular component to the pathogenesis of CD was first proposed only a year after Crohn et al. described the condition. Subsequently, in 1989, a series of changes comprising vascular injury, focal arteritis, fibrin deposition, arterial occlusion, and then microinfarction or neovascularization was proposed as a possible pathogenetic sequence in CD. In this study, resin casts of the intestinal vasculature showed changes ranging from intravascular fibrin deposition to complete thrombotic occlusion. Furthermore, the early vascular changes appeared to precede mucosal changes, suggesting that they were more likely to cause rather than result from the pathologic features of CD. Subsequent studies showed that intravascular fibrin deposition occurred at the site of granulomatous destruction of mesenteric blood vessels, and positive immunostaining for platelet glycoprotein IIIa occurred in fibrinoid plugs of mucosal capillaries in CD. In addition, intracapillary thrombus has been identified in biopsies from inflamed rectal mucosa from patients with CD. When combined with evidence of ongoing intravascular coagulation in both active and quiescent CD, the above data point toward a thrombotic element contributing to the pathogenesis of CD.
Not only are many different prothrombotic changes described in association with IBD, but they can also have multiple causes. Hyperhomocysteinemia, for example, is known to predispose to thrombosis, and patients with IBD are more likely to have hyperhomocysteinemia than control subjects. Hyperhomocysteinemia in IBD might be due to multiple possible causes, such as deficiencies of vitamin B12 as a result of terminal ileal disease or resection; B6, which is commonly reduced in IBD. A vegan diet can’t be discarded either because of seriously deficient methyl donors (S-adenosyl methionine).
The realization that platelets are not only prothrombotic but also proinflammatory has stimulated interest in their role in both the pathogenesis and complications of IBD. The association between thrombocytosis and active IBD was first described more than 30 years ago. More recent observations link decreased or normal platelet survival to IBD-related thrombocytosis, possibly due to increased thrombopoiesis. This in turn could be driven by an interleukin-6 –induced increase in thrombopoietin synthesis in the liver. Spontaneous in vitro platelet aggregation occurs in platelets isolated from 30% of patients with IBD but not in platelets from control subjects. Moreover, collagen, arachidonic acid, ristocetin, and ADP-induced platelet activation are more marked in platelets from patients with active IBD than in those from healthy volunteers.
The roles of activated platelets and PLAs in mucosal inflammation. Activated platelets can interact with other cells involved in the inflammatory response either through direct contact or through the release of soluble mediators. Activated platelets interact directly with activated vascular endothelium, causing the latter to express adhesion molecules and release inflammatory and chemotactic cytokines.
Platelet activation might be pathogenic in IBD in several ways. Platelet activation might increase platelet aggregation, hence increasing the likelihood of thrombus formation at sites of vascular injury, for example, within the mesenteric circulation. P-selectin is the major ligand for leukocyte-endothelial interaction and is responsible for the rolling of platelets, leukocytes, and PLAs on vascular endothelium. Moreover, platelets adherent to injured vascular endothelium support leukocyte adhesion via P-selectin, an effect that could contribute to leukocyte emigration from the vasculature into the lamina propria in patients with IBD. In addition, P-selectin is the major platelet ligand for platelet-leukocyte interaction, which in turn causes both leukocyte activation and further platelet activation.
Platelet-Leukocyte Aggregation
Recently, studies showing that platelets and leukocytes that circulate together in aggregates (PLA) are more activated than those that circulate alone have generated interest in the role of PLA in various inflammatory and thrombotic conditions. PLA numbers are increased in patients with ischemic heart disease, systemic lupus erythematosus and rheumatoid arthritis, myeloproliferative disorders, and sepsis and are increased by smoking.
We have recently shown that patients with IBD have more PLAs than both healthy and inflammatory control subjects (patients with inflammatory arthritides). As with platelet activation, there was no correlation with disease activity, suggesting that increased PLA formation might be an underlying abnormality. PLAs could contribute to the pathogenesis of IBD in a number of ways. As previously mentioned, TF is key to the initiation of thrombus formation. TF has recently been demonstrated on the surface of activated platelets and in platelet-derived microvesicles. Interaction between neutrophils and activated platelets or microvesicles vastly increases the activity of “intravascular” TF.
Conclusion
It is becoming increasingly apparent that thrombosis and inflammation are intrinsically linked. Hence the involvement of thrombotic processes in the pathogenesis of IBD, although perhaps not as the primary event, seems likely. Indeed, with the recently mounting evidence of the role of activated platelets and of their interaction with leukocytes in the pathogenesis of IBD, it seems even more probable that thrombosis plays some role in the pathogenic process.
(Irving PM, Pasi KJ, and Rampton DS. Thrombosis and Inflammatory Bowel Disease. Clinical Gastroenterology and Hepatology 2005;3:617–628. PII: 10.1053/S1542-3565(05)00154-0.)
Bleeding in Patients with Renal Insufficiency
Approximately 20–40% of critically ill patients will have renal insufficiency at the time of admission or will develop it during their ICU stay, depending on the definition of renal insufficiency and the case mix of the ICU. Such patients are also predisposed to bleeding because of uremic platelet dysfunction, typically multiple comorbidities, coagulopathies and frequent concomitant treatment with antiplatelet or anticoagulant agents.
The impairment in hemostasis in uremic patients is multifactorial and includes physiological defects in platelet hemostasis, an imbalance of mediators of normal endothelial function and frequent comorbidities such as vascular disease, anemia and the frequent need for medical interventions required to treat such comorbidities. Physiologic alterations in uremia include:
- decreased platelet glycoprotein IIb–IIIa binding to both von Willebrand factor (vWf) and fibrinogen, causing an impairment in platelet aggregation;
- increased prostacyclin and nitric oxide production, both potent inhibitors of platelet activation and vasoconstriction; and
- decreased levels of platelet adenosine diphosphate (ADP) and serotonin, causing an impairment in platelet secretion.
In addition to other factors, small peptides containing the RGD (Arg-Gly-Asp) sequence of amino acids have been shown to be inhibitors of platelet aggregation that act by competing with vWf and fibrinogen for binding to the glycoprotein IIb–IIIa receptor.
Conclusion
ICU patients have dynamic risks of thrombosis and bleeding. Invasive procedures may require temporary interruption of anticoagulants. Consequently, approaches to thromboprophylaxis require daily reevaluation.
(Cook DJ, Douketis J, Arnold D, and Crowther MA. Bleeding and venous thromboembolism in the critically ill with emphasis on patients with renal insufficiency. Curr Opin Pulm Med 2009;15:455–462.)
Epicrisis
I have covered a large amount of material on one of the most complex systems in medicine, and still not comprehensive, with a sufficient dash of repetition. The task is to have some grasp of the cell-mediated imbalances inherent if coagulation and bleeding disorders. The key points are:
- inflammation and oxidative stress invariably lurk in the background
- the Y-shaped model with an extrinsic, intrinsic, and common pathway has no basis in understanding
- the current model is based on a cell-mediated concept of endothelial damage and platelet-endothelial interaction
- the model has 3 components: Initiation, Amplification, Propagation
- NO and prostacyclin have key roles in the process
- The plasma proteins involved are in the serine-protease class of enzymes
- The conversion of Protein C to APC has a central role as anti-coagulant
Part II has explored organ system abnormalities that are all related to impairment of the Nitric Oxide balance and dual platelet-endothelial roles.
- interaction of Nitric Oxide and Prostacyclin in Vascular Endothelium (pharmaceuticalintelligence.com)
- Nitric oxide: role in Cardiovascular health and disease (pharmaceuticalintelligence.com)
- Nitric Oxide has a ubiquitous role in the regulation of glycolysis -with a concomitant influence on mitochondrial function (pharmaceuticalintelligence.com)
- Differential Distribution of Nitric Oxide – A 3-D Mathematical Model (pharmaceuticalintelligence.com)
- Nitric Oxide and Sepsis, Hemodynamic Collapse, and the Search for Therapeutic Options (pharmaceuticalintelligence.com)
- Potentiation of Thrombin Generation in Hemophilia A Plasma by Coagulation Factor VIII and Characterization of Antibody-Specific Inhibition (plosone.org)
- Nitric Oxide Covalent Modifications: A Putative Therapeutic Target? (pharmaceuticalintelligence.com)
- Nitric Oxide and Immune responses: part 1 (pharmaceuticalintelligence.com)
- Nitric Oxide and Immune Responses: Part 2 (pharmaceuticalintelligence.com)
- Statins’ nonlipid effects on vascular endothelium through eNOS activation (pharmaceuticalintelligence.com)
Nov 9, 2012 by anamikasarkar
Nitric Oxide (NO) is highly regulated in the blood such that it can be released as vasodilator when needed. The importance and pathway of Nitric Oxide has been nicely reviewed by. “Discovery of NO and its effects of vascular biology”. Other articles which are good readings for the importance of NO are – a) regulation of glycolysis b) NO in cardiovascular disease c) NO and Immune responses Part I and Part II d) NO signaling pathways. The effects of NO in diseased states have been reviewed by the articles – “Crucial role of Nitric Oxide in Cancer”, “Nitric Oxide and Sepsis, Hemodynamic Collapse, and the Search for Therapeutic Options”.. (Also, please see Source for more articles on NO and its significance).
Computational models are very efficient tools to understand complex reactions like NO towards physiological conditions. Among them wall shear stress is one of the major factors which is reviewed in the article – “Differential Distribution of Nitric Oxide – A 3-D Mathematical Model”.
Moreover, decrease in availability of NO can lead to many complications like pulmonary hypertension. Some of the causes of decrease in NO have been identified as clinical hypertension,right ventricular overload which can lead to cardiac heart failure,low levels of zinc and high levels of cardiac necrosis.
Sickle Cell disease patients, a hereditary disease, are also known to have decreased levels of NO which can become physiologically challenging. In USA alone, there are 90,000 people who are affected by Sickle cell disease.
Sickle cell disease is breakage of red blood cells (RBC) membrane and resulting release of the hemoglobin (Hb) into blood plasma. This process is also known as Hemolysis. Sickle cell disease is caused by single mutation of Hb which changes RBC from round shape to sickle or crescent shapes (Figure 1).

Figure 1 (A) shows normal red blood cells flowing freely through veins. The inset shows a cross section of a normal red blood cell with normal hemoglobin.
Figure 1 (B) shows abnormal, sickled red blood cells The inset image shows a cross-section of a sickle cell with long polymerized HbS strands stretching and distorting the cell shape.
Image Source: http://en.wikipedia.org/wiki/Sickle-cell_disease
Figure 1 (B) shows abnormal, sickled red blood cells The inset image shows a cross-section of a sickle cell with long polymerized HbS strands stretching and distorting the cell shape.
Image Source: http://en.wikipedia.org/wiki/Sickle-cell_disease
RBCs generally breakdown and release Hbs in blood plasma after they reach their end of life span. Thus, in case of Sickle cell disease, there is more cell free Hb than normal. Furthermore, it is known that NO has a very high affinity towards Hbs, which is one of the ways free NO is regulated in blood. As a result presence of larger amounts of cell free Hb in Sickle cell disease lead to less availability of NO.
As to “a quantitative relationship between cell free Hb and depletion of NO”, Deonikar and Kavdia (J. Appl. Physiol., 2012) addressed this question by developing a model of a single idealized arteriole, with different layers of blood vessels diffusing nutrients to tissue layers (Figure 2: Deonikar and Kavdia Figure 1).
The model and its parameters are explained in the previously published paper by same authors Deonikar and Kavdia, Annals of Biomed., 2010, who reported that the reaction rate between NO and RBC is 0.2 x 105, M-1 s-1 than 1.4 x 105, M-1 s-1 as previously reported by Butler et.al., Biochim. Biophys. Acta, 1998. Their results show that even small increase in cell free Hb, 0.5uM, can decrease NO concentrations by 3-7 fold. Their mathematical analysis shows that the increase in diffusion resistance of NO from vascular lumen to cell free zone has no effect on NO distribution and concentration with available levels of cell free Hb.
Deonikar and Kavdia’s model, a simple representation shows that for SC disease patients, decrease in levels of bioavailable NO is attributed to cell free Hb, which is in abundant for these patients. Their results show that small increase by 0.5 uM in cell free Hb can cause a large decrease in NO concentrations.
Sources:
Deonikar and Kavdia (2012):http://www.ncbi.nlm.nih.gov/pubmed/22223452
Previous model explaining mathematical representation and parameters used in the model :Deonikar and Kavdia,Annals of Biomed., 2010.
Previous paper stating reaction rate of Hb and NO: Butleret.al., Biochim. Biophys. Acta, 1998.
Causes of decrease in NO
Clinical Hypertension :http://www.ncbi.nlm.nih.gov/pubmed/11311074
Right ventricular overload :http://www.ncbi.nlm.nih.gov/pubmed/9559613
Low levels of zinc and high levels of cardiac necrosis :http://www.ncbi.nlm.nih.gov/pubmed/11243421
Sickle Cell Source:
http://en.wikipedia.org/wiki/Sickle-cell_disease
http://www.nhlbi.nih.gov/health/health-topics/topics/sca/
NO Sources:
Differential Distribution of Nitric Oxide – A 3-D Mathematical Model:
Discovery of NO and its effects of vascular biology
Nitric oxide: role in Cardiovascular health and disease
The rationale and use of inhaled NO in Pulmonary Artery Hypertension and Right Sided Heart Failure
Endothelial Function and Cardiovascular Disease
Interaction of Nitric Oxide and Prostacyclin in Vascular Endothelium
Endothelial Dysfunction, Diminished Availability of cEPCs, Increasing CVD Risk – Macrovascular Disease – Therapeutic Potential of cEPCs
Low Bioavailability of Nitric Oxide due to Misbalance in Cell Free Hemoglobin in Sickle Cell Disease – A Computational Model
Posted in Biological Networks, Gene Regulation and Evolution, Biomarkers & Medical Diagnostics, BioSimilars, CANCER BIOLOGY & Innovations in Cancer Therapy, Cardiovascular Pharmacogenomics, Cell Biology, Signaling & Cell Circuits, Chemical Biology and its relations to Metabolic Disease, Computational Biology/Systems and Bioinformatics, Disease Biology, Small Molecules in Development of Therapeutic Drugs, Nitric Oxide in Health and Disease, Uncategorized, tagged Bioinformatics, Cardiovascular disease, Cardiovascular Disorders, computational biology, health, Hemoglobin, Hemolysis, Hypertension, nitric oxide, Nitric oxide synthase, NO, Pulmonary hypertension, Sickle Cell disease, systems biology on November 9, 2012| 15 Comments »
Author and Reporter: Anamika Sarkar, Ph.D.
Nitric Oxide (NO) is highly regulated in the blood such that it can be released as vasodilator when needed. The importance and pathway of Nitric Oxide has been nicely reviewed by. “Discovery of NO and its effects of vascular biology”. Other articles which are good readings for the importance of NO are – a) regulation of glycolysis b) NO in cardiovascular disease c) NO and Immune responses Part I and Part II d) NO signaling pathways. The effects of NO in diseased states have been reviewed by the articles – “Crucial role of Nitric Oxide in Cancer”, “Nitric Oxide and Sepsis, Hemodynamic Collapse, and the Search for Therapeutic Options”.. (Also, please see Source for more articles on NO and its significance).
Computational models are very efficient tools to understand complex reactions like NO towards physiological conditions. Among them wall shear stress is one of the major factors which is reviewed in the article – “Differential Distribution of Nitric Oxide – A 3-D Mathematical Model”.
Moreover, decrease in availability of NO can lead to many complications like pulmonary hypertension. Some of the causes of decrease in NO have been identified as clinical hypertension, right ventricular overload which can lead to cardiac heart failure, low levels of zinc and high levels of cardiac necrosis.
Sickle Cell disease patients, a hereditary disease, are also known to have decreased levels of NO which can become physiologically challenging. In USA alone, there are 90,000 people who are affected by Sickle cell disease.
Sickle cell disease is breakage of red blood cells (RBC) membrane and resulting release of the hemoglobin (Hb) into blood plasma. This process is also known as Hemolysis. Sickle cell disease is caused by single mutation of Hb which changes RBC from round shape to sickle or crescent shapes (Figure 1).
Figure 1 (A) shows normal red blood cells flowing freely through veins. The inset shows a cross section of a normal red blood cell with normal hemoglobin. Figure 1 (B) shows abnormal, sickled red blood cells The inset image shows a cross-section of a sickle cell with long polymerized HbS strands stretching and distorting the cell shape. Image Source: http://en.wikipedia.org/wiki/Sickle-cell_disease
Sickle Cell RBCs has much shorter life span of 10-20 days when compared with normal RBCs 100-120 days lifespan. Shorter life span of Sickle cell disease RBC’s are compensated by bone marrow generation of new RBCs. However, many times new blood generation cannot cope with the small life span of Sickle cell RBCs and causes pathological condition of Anemia.
RBCs generally breakdown and release Hbs in blood plasma after they reach their end of life span. Thus, in case of Sickle cell disease, there is more cell free Hb than normal. Furthermore, it is known that NO has a very high affinity towards Hbs, which is one of the ways free NO is regulated in blood. As a result presence of larger amounts of cell free Hb in Sickle cell disease lead to less availability of NO.
However, the question remained “what is the quantitative relationship between cell free Hb and depletion of NO”. Deonikar and Kavdia (J. Appl. Physiol., 2012) addressed this question by developing a 2 dimensional Mathematical Model of a single idealized arteriole, with different layers of blood vessels diffusing nutrients to tissue layers (Figure 2: Deonikar and Kavdia Figure 1).

cell free Hb in 2 dimensional representations of blood vessels.
The authors used steady state partial differential equation of circular geometry to represent diffusion of NO in blood and in tissues. They used first and second order biochemical reactions to represent the reactions between NO and RBC and NO autooxidation processes. Some of their reaction model parameters were obtained from literature, rest of them were fitted to experimental results from literature. The model and its parameters are explained in the previously published paper by same authors Deonikar and Kavdia, Annals of Biomed., 2010. The authors found that the reaction rate between NO and RBC is 0.2 x 105, M-1 s-1 than 1.4 x 105, M-1 s-1 as reported before by Butler et.al., Biochim. Biophys. Acta, 1998.
Their results show that even small increase in cell free Hb, 0.5uM, can decrease NO concentrations by 3-7 folds approximately (comparing Fig1(b) and 1(d) of Deonikar and Kavdia, 2012, as shown in Figure 2 of this article). Moreover, their mathematical analysis shows that the increase in diffusion resistance of NO from vascular lumen to cell free zone has no effect on NO distribution and concentration with available levels of cell free Hb.
Deonikar and Kavdia’s mathematical model is a simple representation of actual physiological scenario. However, their model results show that for Sickle cell disease patients, decrease in levels of bioavailable NO is an attribute to cell free Hb, which is in abundant for these patients. Their results show that small increase by 0.5 uM in cell free Hb can cause large decrease in NO concentrations.
These interesting insights from the model can help in further understanding in the context of physiological conditions, by replicating experiments in-vivo and then relating them to other known diseases of Sickle cell disease patients like Anemia, Pulmonary Hypertension. Further, drugs can be targeted towards decreasing free cell Hbs to keep balance in availability of NO, which in turn may help in other related disease like Pulmonary Hypertension of Sickle Cell disease patients.
Sources:
Deonikar and Kavdia (2012) :http://www.ncbi.nlm.nih.gov/pubmed/22223452
Previous model explaining mathematical representation and parameters used in the model :Deonikar and Kavdia, Annals of Biomed., 2010.
Previous paper stating reaction rate of Hb and NO: Butler et.al., Biochim. Biophys. Acta, 1998.
Causes of decrease in NO
Clinical Hypertension : http://www.ncbi.nlm.nih.gov/pubmed/11311074
Right ventricular overload : http://www.ncbi.nlm.nih.gov/pubmed/9559613
Low levels of zinc and high levels of cardiac necrosis : http://www.ncbi.nlm.nih.gov/pubmed/11243421
Sickle Cell Source:
http://en.wikipedia.org/wiki/Sickle-cell_disease
http://www.nhlbi.nih.gov/health/health-topics/topics/sca/
NO Source:
Differential Distribution of Nitric Oxide – A 3-D Mathematical Model:
Discovery of NO and its effects of vascular biology
Nitric oxide: role in Cardiovascular health and disease
Nitric Oxide and Immune Responses: Part 1
Nitric Oxide and Immune Responses: Part 2
Statins’ Nonlipid Effects on Vascular Endothelium through eNOS Activation
Nitric Oxide, Platelets, Endothelium and Hemostasis
Crucial role of Nitric Oxide in Cancer
The rationale and use of inhaled NO in Pulmonary Artery Hypertension and Right Sided Heart Failure
Nitric Oxide and Sepsis, Hemodynamic Collapse, and the Search for Therapeutic Options
Endothelial Function and Cardiovascular Disease
Interaction of Nitric Oxide and Prostacyclin in Vascular Endothelium
Endothelial Dysfunction, Diminished Availability of cEPCs, Increasing CVD Risk – Macrovascular Disease – Therapeutic Potential of cEPCs
Differential Distribution of Nitric Oxide – A 3-D Mathematical Model
Posted in Biological Networks, Gene Regulation and Evolution, Cell Biology, Signaling & Cell Circuits, Computational Biology/Systems and Bioinformatics, Disease Biology, Small Molecules in Development of Therapeutic Drugs, Frontiers in Cardiology and Cardiovascular Disorders, HTN, Human Immune System in Health and in Disease, Nitric Oxide in Health and Disease, Origins of Cardiovascular Disease, Stents & Tools, Uncategorized, Valves & Tools, tagged 3-D Model, Acute coronary syndrome, Aviral Vatsa, Blood flow, Blood vessel, branching in blood vessels, Cardiovascular disease, Cardiovascular Disorders, Circulatory system, Conditions and Diseases, Endothelium, Finite Elements, health, Heart Failure, mathematical model, nitric oxide, Nitric oxide synthase, NO, RBC, Wall Shear Stress on October 28, 2012| 12 Comments »
Author and Reporter: Anamika Sarkar, Ph.D.
Among many important roles of Nitric oxide (NO), one of the key actions is to act as a vasodilator and maintain cardiovascular health. Induction of NO is regulated by signals in tissue as well as endothelium.
Importance of NO has been nicely reviewed in the article “Discovery of NO and its effects of vascular biology”. Other articles which are good readings for the importance of NO are – a) regulation of glycolysis b) NO in cardiovascular disease c) NO and Immune responses Part I and Part II d) NO signaling pathways (Also, please see Source for more articles on NO and its significance).
The rate of production of NO has been established to be dependent on Wall Shear Stress (WSS) (Mashour and Broock, Brain Res., 1999) . Many mathematical models have been developed as 2D diffusion models to predict distribution of NO transport in single vessels, eg. arterioles (Please see Sources for references ).
Chen et. al. (Med. Biol. Eng. Comp., 2011) developed a 3-D model consisting of two branched arterioles and nine capillaries surrounding the vessels. Their model not only takes into account of the 3-D volume, but also branching effects on blood flow (Please see Fig 1 and Fig 2 from Chen et. al. 2011 ).
")
Fig. 1 Blood phase separation with vascular branching. RBC
fractional flow in daughter branch alpha is not necessarily equal
to that in branch beta
")
The mathematical model considers dynamic characteristics related to blood flow, blood vessel structures and transport mechanism in the wall. The authors have considered effects of branching and ratio of diameters between blood vessels of parent and children to determine the fractional blood flow which gets distributed in the network. These branching effects of the vessels will also affect the blood volume or RBC (Red Blood Cell), hence NO consumption in the blood. Parameters in the model are either obtained or fitted with experimental results from literature. Their model assumes a linear relationship of NO production with wall shear stress which in turn will be regulated by blood flow determined by branching characteristics of blood vessels. Moreover, the mathematical model includes transport of NO through the blood vessels in the tissue (in the defined volume of the model) as diffusion model,. The model was solved using Finite Elements method using the software COMSOL.
Their model results show that wall shear stress changes depending upon the distribution of RBC in the microcirculations of blood vessels, which leads to differential production of NO along the vascular network. Levels of NO at vascular walls can be less in branches which receive more blood flow, due to the balance between higher consumption of NO by RBC and production of NO due to high wall stress. Their 3-D simulations showed the importance of capillaries such that NO can be concentrated in tissues far away in distance from arterioles facilitating much controlled NO regulation.
Though, the 3-D model developed by Chen et. al., (2011) is an idealized mathematical model of blood flow with production and consumption of NO, depending upon WSS, yet it shows importance of structure of blood vessels in distributions of NO in vessels and tissues. Such a model with proper extension to larger network can give more insights into differential distributions of NO as a function of blood flow and wall shear stress. As nano-medicine become sophisticated in years to come, information of distribution of NO in tissues and blood vessels can help the medicine to be more targeted.
Sources:
Chen et.al. (2011) : http://www.ncbi.nlm.nih.gov/pubmed/21431938
Mashour and Broock, Brain Res., 1999: http://www.ncbi.nlm.nih.gov/pubmed?term=10526117
Mathematical Modes of NO Distribution in 2-D
- http://www.ncbi.nlm.nih.gov/pubmed/16210436
- http://www.ncbi.nlm.nih.gov/pubmed?term=12826074
- http://www.ncbi.nlm.nih.gov/pubmed/16155098
- http://www.ncbi.nlm.nih.gov/pubmed/9612383
- http://www.ncbi.nlm.nih.gov/pubmed/9841542
Other research on Nitric Oxide and Vascular Biology on this Scientific Web Site include the following:
Nitric Oxide and Immune Responses: Part 1
Curator and Reporter: Aviral Vatsa, 10/18/2012
http://pharmaceuticalintelligence.com/2012/10/18/nitric-oxide-and-immune-responses-part-1/
Clinical Trials Results for Endothelin System: Pathophysiological role in Chronic Heart Failure, Acute Coronary Syndromes and MI – Marker of Disease Severity or Genetic Determination?
Curator: Aviva Lev-Ari, 10/19/2012
Nitric Oxide and Sepsis, Hemodynamic Collapse, and the Search for Therapeutic Options
Curator and Reporter: Larry Bernstein, MD, 10/20/2012
Mitochondrial Damage and Repair under Oxidative Stress
Curator: Larry H Bernstein, MD, FCAP, 10/28/2012
Nitric Oxide and Immune Responses: Part 2
Curator: Aviral Vatsa, PhD, MBBS, 10/28/2012
http://pharmaceuticalintelligence.com/2012/10/28/nitric-oxide-and-immune-responses-part-2/
Differential Distribution of Nitric Oxide – A 3-D Mathematical Model
Author: Anamika Sarkar, PhD, 10/28/2012
Statins’ Nonlipid Effects on Vascular Endothelium through eNOS Activation
Curator, EAW: Larry Bernstein, 10/8/2012
Nitric Oxide Nutritional remedies for hypertension and atherosclerosis. It’s 12 am: do you know where your electrons are?
Author and Reporter: Meg Baker, 10/7/2012.
Inhibition of ET-1, ETA and ETA-ETB, Induction of NO production, stimulation of eNOS and Treatment Regime with PPAR-gamma agonists (TZD): cEPCs Endogenous Augmentation for Cardiovascular Risk Reduction – A Bibliography
Curator: Aviva Lev-Ari, 10/4/2012.
Coronary Artery Disease – Medical Devices Solutions: From First-In-Man Stent Implantation, via Medical Ethical Dilemmas to Drug Eluting Stents August 13, 2012
Author: Aviva Lev-Ari, PhD, RN, 8/13/2012
Vascular Medicine and Biology: CLASSIFICATION OF FAST ACTING THERAPY FOR PATIENTS AT HIGH RISK FOR MACROVASCULAR EVENTS Macrovascular Disease – Therapeutic Potential of cEPCs
Curator; Aviva Lev-Ari, PhD, RN, 8/24/2012
Nitric Oxide and Immune Responses: Part 2
Posted in Biological Networks, Gene Regulation and Evolution, Cell Biology, Signaling & Cell Circuits, Disease Biology, Small Molecules in Development of Therapeutic Drugs, Nitric Oxide in Health and Disease, tagged autoimmune, bacteria, Chemistry, disease, immune modulation, immune system, infection, Infectious disease, iNOS, macrophages, nitric oxide, Nitric oxide synthase, NOS, NOX, parasite, pathways, Reactive oxygen species, redox, RNS, ROS, virus on October 28, 2012| 13 Comments »
Curator/Reporter Aviral Vatsa PhD, MBBS
Based on: A review by (Wink et al., 2011)
This post is in continuation to Part 1 by the same title.
In part one I covered the basics of role of redox chemistry in immune reactions, the phagosome cauldron, and how bacteria bacteria, virus and parasites trigger the complex pathway of NO production and its downstream effects. While we move further in this post, the previous post can be accessed here.
REDOX REGULATION OF IMMUNE FUNCTION
Regulation of the redox immunomodulators—NO/RNS and ROS
In addition to eradicating pathogens, NO/RNS and ROS and their chemical interactions act as effective immunomodulators that regulate many cellular metabolic pathways and tissue repair and proinflammatory pathways. Figure 3 shows these pathways.

Figure 3. Schematic overview of interactive connections between NO and ROS-mediated metabolic pathways. Credit: (Wink et al., 2011)
Regulation of iNOS enzyme activity is critical to NO production. Factors such as the availability of arginine, BH4, NADPH, and superoxide affect iNOS activity and thus NO production. In the absence of arginine and BH4 iNOS becomes a O2_/H2O2 generator (Vásquez-Vivar et al., 1999). Hence metabolic pathways that control arginine and BH4 play a role in determining the NO/superoxide balance. Arginine levels in cells depend on various factors such as type of uptake mechanisms that determine its spatial presence in various compartments and enzymatic systems. As shown in Fig3 Arginine is the sole substrate for iNOS and arginase. Arginase is another key enzyme in immunemodulation. AG is also regulated by NOS and NOX activities. NOHA, a product of NOS, inhibits AG, and O2–increases AG activity. Importantly, high AG activity is associated with elevated ROS and low NO fluxes. NO antagonises NOX2 assembly that in turn leads to reduction in O2_ production. NO also inhibits COX2 activity thus reducing ROS production. Thus, as NO levels decline, oxidative mechanisms increase. Oxidative and nitrosative stress can also decrease intracellular GSH (reduced form) levels, resulting in a reduced antioxidant capability of the cell.
Immune-associated redox pathways regulate other important metabolic cell functions that have the potential for widespread impact on cells, organs, and organisms. These pathways, such as mediated via methionine and polyamines, are critical for DNA stabilization, cell proliferation, and membrane channel activity, all of which are also involved in immune-mediated repair processes.
NO levels dictate the immune signaling pathway
NO/RNS and ROS actively control innate and adaptive immune signaling by participating in induction, maintenance, and/or termination of proinflammatory and anti-inflammatory signaling. As in pathogen eradication, the temporal and spatial concentration profiles of NO are key factors in determining immune-mediated processes.
Brune and coworkers (Messmer et al., 1994) first demonstrated that p53 expression was associated with the concentrations of NO that led to apoptosis in macrophages. Subsequent studies linked NO concentration profiles with expression of other key signaling proteins such as HIF-1α and Akt-P (Ridnour et al., 2008; Thomas et al., 2008). Various levels of NO concentrations trigger different pathways and expectedly this concentration-dependent profile varies with distance from the NO source.NO is highly diffucible and this characteristic can result in 1000 fold reduction in concentration within one cell length distance travelled from the source of production. Time course studies have also shown alteration in effects of same levels of NO over time e.g. NO-mediated ERK-P levels initially increased rapidly on exposure to NO donors and then decreased with continued NO exposure (Thomas et al., 2004), however HIF-1α levels remained high as long as NO levels were elevated. Thus some of the important factors that play critical role in NO effects are: distance from source, NO concentrations, duration of exposure, bioavailability of NO, and production/absence of other redox molecules.

Figure and legend credits: (Wink et al., 2011)
Fig 4: The effect of steady-state flux of NO on signal transduction mechanisms.
This diagram represents the level of sustained NO that is required to activate specific pathways in tumor cells. Similar effects have been seen on endothelial cells. These data were generated by treating tumor or endothelial cells with the NO donor DETANO (NOC-18) for 24 h and then measuring the appropriate outcome measures (for example, p53 activation). Various concentrations of DETANO that correspond to cellular levels of NO are: 40–60 μM DETANO = 50 nM NO; 80–120 μM DETANO = 100 nM NO; 500 μM DETANO = 400 nM NO; and 1 mM DETANO = 1 μM NO. The diagram represents the effect of diffusion of NO with distance from the point source (an activated murine macrophage producing iNOS) in vitro (Petri dish) generating 1 μM NO or more. Thus, reactants or cells located at a specific distance from the point source (i.e., iNOS, represented by star) would be exposed to a level of NO that governs a specific subset of physiological or pathophysiological reactions. The x-axis represents the different zone of NO-mediated events that is experienced at a specific distance from a source iNOS producing >1 μM. Note: Akt activation is regulated by NO at two different sites and by two different concentration levels of NO.
Species-specific NO production
The relationship of NO and immunoregulation has been established on the basis of studies on tumor cell lines or rodent macrophages, which are readily available sources of NO. However in humans the levels of protein expression for NOS enzymes and the immune induction required for such levels of expression are quite different than in rodents (Weinberg, 1998). This difference is most likely due to the human iNOS promotor rather than the activity of iNOS itself. There is a significant mismatch between the promoters of humans and rodents and that is likely to account for the notable differences in the regulation of gene induction between them. The combined data on rodent versus human NO and O2– production strongly suggest that in general, ROS production is a predominant feature of activated human macrophages, neutrophils, and monocytes, and the equivalent murine immune cells generate a combination of O2– and NO and in some cases, favor NO production. These differences may be crucial to understanding how immune responses are regulated in a species-specific manner. This is particularly useful, as pathogen challenges change constantly.
The next post in this series will cover the following topics:
The impact of NO signaling on an innate immune response—classical activation
NO and proinflammatory genes
NO and regulation of anti-inflammatory pathways
NO impact on adaptive immunity—immunosuppression and tissue-restoration response
NO and revascularization
Acute versus chronic inflammatory disease
Bibliography
Further reading on NO:
Nitric Oxide in bone metabolism July 16, 2012
Author: Aviral Vatsa PhD, MBBS
Nitric Oxide production in Systemic sclerosis July 25, 2012
Curator: Aviral Vatsa, PhD, MBBS
Nitric Oxide Signalling Pathways August 22, 2012 by
Curator/ Author: Aviral Vatsa, PhD, MBBS
Nitric Oxide: a short historic perspective August 5, 2012
Author/Curator: Aviral Vatsa PhD, MBBS
http://pharmaceuticalintelligence.com/2012/08/05/nitric-oxide-a-short-historic-perspective-7/
Nitric Oxide: Chemistry and function August 10, 2012
Curator/Author: Aviral Vatsa PhD, MBBS
Nitric Oxide and Platelet Aggregation August 16, 2012 by
Author: Dr. Venkat S. Karra, Ph.D.
The rationale and use of inhaled NO in Pulmonary Artery Hypertension and Right Sided Heart Failure August 20, 2012
Author: Larry Bernstein, MD
Nitric Oxide: The Nobel Prize in Physiology or Medicine 1998 Robert F. Furchgott, Louis J. Ignarro, Ferid Murad August 16, 2012
Reporter: Aviva Lev-Ari, PhD, RN
Coronary Artery Disease – Medical Devices Solutions: From First-In-Man Stent Implantation, via Medical Ethical Dilemmas to Drug Eluting Stents August 13, 2012
Author: Aviva Lev-Ari, PhD, RN
Nano-particles as Synthetic Platelets to Stop Internal Bleeding Resulting from Trauma
August 22, 2012
Reported by: Dr. V. S. Karra, Ph.D.
Cardiovascular Disease (CVD) and the Role of agent alternatives in endothelial Nitric Oxide Synthase (eNOS) Activation and Nitric Oxide Production July 19, 2012
Curator and Research Study Originator: Aviva Lev-Ari, PhD, RN
Macrovascular Disease – Therapeutic Potential of cEPCs: Reduction Methods for CV Risk
July 2, 2012
An Investigation of the Potential of circulating Endothelial Progenitor Cells (cEPCs) as a Therapeutic Target for Pharmacological Therapy Design for Cardiovascular Risk Reduction: A New Multimarker Biomarker Discovery
Curator: Aviva Lev-Ari, PhD, RN
Bone remodelling in a nutshell June 22, 2012
Author: Aviral Vatsa, Ph.D., MBBS
http://pharmaceuticalintelligence.com/2012/06/22/bone-remodelling-in-a-nutshell/
Targeted delivery of therapeutics to bone and connective tissues: current status and challenges- Part, September
Author: Aviral Vatsa, PhD, September 23, 2012
Calcium dependent NOS induction by sex hormones: Estrogen
Curator: S. Saha, PhD, October 3, 2012
http://pharmaceuticalintelligence.com/2012/10/03/calcium-dependent-nos-induction-by-sex-hormones/
Nitric Oxide and Platelet Aggregation,
Author V. Karra, PhD, August 16, 2012
http://pharmaceuticalintelligence.com/2012/08/16/no-and-platelet-aggregation/
Curator: Aviva Lev-Ari, PhD, July 16, 2012
http://pharmaceuticalintelligence.com/?s=Nebivolol
Endothelin Receptors in Cardiovascular Diseases: The Role of eNOS Stimulation
Author: Aviva Lev-Ari, PhD, 10/4/2012
Inhibition of ET-1, ETA and ETA-ETB, Induction of NO production, stimulation of eNOS and Treatment Regime with PPAR-gamma agonists (TZD): cEPCs Endogenous Augmentation for Cardiovascular Risk Reduction – A Bibliography
Curator: Aviva Lev-Ari, 10/4/2012.
Nitric Oxide Nutritional remedies for hypertension and atherosclerosis. It’s 12 am: do you know where your electrons are?
Author and Reporter: Meg Baker, 10/7/2012.
Endothelial Function and Cardiovascular Disease
Posted in Biological Networks, Gene Regulation and Evolution, Biomarkers & Medical Diagnostics, Cardiovascular Pharmacogenomics, Cell Biology, Signaling & Cell Circuits, Chemical Biology and its relations to Metabolic Disease, Chemical Genetics, Computational Biology/Systems and Bioinformatics, Disease Biology, Small Molecules in Development of Therapeutic Drugs, Genome Biology, Health Economics and Outcomes Research, HTN, International Global Work in Pharmaceutical, Metabolomics, Molecular Genetics & Pharmaceutical, Nitric Oxide in Health and Disease, Nutrigenomics, Nutrition, Origins of Cardiovascular Disease, Pharmaceutical Analytics, Pharmaceutical Industry Competitive Intelligence, Pharmaceutical R&D Investment, Pharmacotherapy of Cardiovascular Disease, Population Health Management, Nutrition and Phytochemistry, Scientist: Career considerations, Statistical Methods for Research Evaluation, Systemic Inflammatory Response Related Disorders, Technology Transfer: Biotech and Pharmaceutical, tagged ADMA, alternative medicine, Artery, Atherosclerosis, Cardiovascular disease, CHF, DDAH, DDAH gene, Endothelin ETA/ETB, endothelin inhibitors, Endothelin Receptors, eNOS, health, Heart disease, Heart Failure, nitric oxide, Nitric oxide synthase, Physical exercise, sauna, shear stress, therapeutic targets for CVD, Traditional Chinese Medicine, type 2 diabetes, Vascular resistance on October 25, 2012| 11 Comments »
Endothelial Function and Cardiovascular Disease
Pathologist and Author: Larry H Bernstein, MD, FCAP
This discussion is a continuation of a series on Nitric Oxide, vascular relaxation, vascular integrity, and systemic organ dysfunctions related to inflammatory and circulatory disorders. In some of these, the relationships are more clear than others, and in other cases the vascular disorders are aligned with serious metabolic disturbances. This article, in particular centers on the regulation of NO production, NO synthase, and elaborates more on the assymetrical dimethylarginine (ADMA) inhibition brought up in a previous comment, and cardiovascular disease, including:
- Acute myocardial infarction
- Congestive heart failure
- Chronic renal disease and dialysis
- Hypertension
Recall, though, that in SIRS leading to septic shock, that there is a difference between the pulmonary circulation, the systemic circulation and the portal circulation in these events. The comment calls attention to:
Böger RH. Asymmetric dimethylarginine, an endogenous inhibitor of nitric oxide synthase, explains the ‘L-arginine paradox’ and acts as a novel cardiovascular risk factor. J Nutr 2004; 134: 2842S–7S.
This observer points out that ADMA inhibits vascular NO production at concentrations found in pathophysiological conditions (i.e., 3–15 μmol/l); ADMA also causes local vasoconstriction when it is infused intra-arterially. ADMA is increased in the plasma of humans with hypercholesterolemia, atherosclerosis, hypertension, chronic renal failure, and chronic heart failure.
Increased ADMA levels are associated with reduced NO synthesis as assessed by impaired endothelium-dependent vasodilation. We’ll go into that more with respect to therapeutic targets – including exercise, sauna, and possibly diet, as well as medical drugs.
It is remarkable how far we have come since the epic discovery of 17th century physician, William Harvey, by observing the action of the heart in small animals and fishes, proved that heart receives and expels blood during each cycle, and argued for the circulation in man. This was a huge lead into renaissance medicine. What would he think now?
Key Words: eNOS, NO, endothelin, ROS, oxidative stress, blood flow, vascular resistance, cardiovascular disease, chronic renal disease, hypertension, diabetes, atherosclerosis, MI, exercise, nutrition, traditional chinese medicine, statistical modeling for targeted therapy.
Endothelial Function
The endothelium plays a crucial role in the maintenance of vascular tone and structure by means of eNOS, producing the endothelium-derived vasoactive mediator nitric oxide (NO), an endogenous messenger molecule formed in healthy vascular endothelium from the amino acid precursor L-arginine. Nitric oxide synthases (NOS) are the enzymes responsible for nitric oxide (NO) generation. The generation and actions of NO under physiological and pathophysiological conditions are exquisitely regulated and extend to almost every cell type and function within the circulation. While the molecule mediates many physiological functions, an excessive presence of NO is toxic to cells.
The enzyme NOS, constitutively or inductively, catalyses the production of NO in several biological systems. NO is derived not only from NOS isoforms but also from NOS-independent sources. In mammals, to date, three distinct NOS isoforms have been identified:
- neuronal NOS (nNOS),
- inducible NOS (iNOS), and
- endothelial NOS (eNOS).
The molecular structure, enzymology and pharmacology of these enzymes have been well defined, and reveal critical roles for the NOS system in a variety of important physiological processes. The role of NO and NOS in regulating vascular physiology, through neuro-hormonal, renal and other non-vascular pathways, as well as direct effects on arterial smooth muscle, appear to be more intricate than was originally thought.
Vallance et al. described the presence of asymmetric dimethylarginine (ADMA) as an endogenous inhibitor of eNOS in 1992. Since then, the role of this molecule in the regulation of eNOS has attracted increasing attention.
Endothelins are 21-amino acid peptides, which are active in almost all tissues in the body. They are potent vasoconstrictors, mediators of cardiac, renal, endocrine and immune functions and play a role in bronchoconstriction, neurotransmitter regulation, activation of inflammatory cells, cell proliferation and differentiation.
Endothelins were first characterised by Yanagisawa et al. (1988). The three known endothelins ET-1, -2 and -3 are structurally similar to sarafotoxins from snake venoms. ET-1 is the major isoform generated in blood vessels and appears to be the isoform of most importance in the cardiovascular system with a major role in the maintenance of vascular tone.
The systemic vascular response to hypoxia is vasodilation. However, reports suggest that the potent vasoconstrictor endothelin-1 (ET-1) is released from the vasculature during hypoxia. ET-1 is reported to augment superoxide anion generation and may counteract nitric oxide (NO) vasodilation. Moreover, ET-1 was proposed to contribute to increased vascular resistance in heart failure by increasing the production of asymmetric dimethylarginine (ADMA).
A study investigated the role of ET-1, the NO pathway, the potassium channels and radical oxygen species in hypoxia-induced vasodilation of large coronary arteries and found NO contributes to hypoxic vasodilation, probably through K channel opening, which is reversed by addition of ET-1 and enhanced by endothelin receptor antagonism. These latter findings suggest that endothelin receptor activation counteracts hypoxic vasodilation.
Endothelial dysfunction
Patients with Raynaud’s Phemonenon had abnormal vasoconstrictor responses to cold pressor tests (CPT) that were similar in primary and secondary RP. There were no differences in median flow-mediated and nitroglycerin mediated dilation or CPT of the brachial artery in the 2 populations. Patients with secondary RP were characterized by abnormalities in microvascular responses to reactive hyperemia, with a reduction in area under the curve adjusted for baseline perfusion, but not in time to peak response or peak perfusion ratio.
Plasma ET-1, ADMA, VCAM-1, and MCP-1 levels were significantly elevated in secondary RP compared with primary RP. There was a significant negative correlation between ET-1 and ADMA values and measures of microvascular perfusion but not macrovascular endothelial function. Secondary RP is characterized by elevations in plasma ET-1 and ADMA levels that may contribute to alterations in cutaneous microvascular function.
ADMA inhibits vascular NO production within the concentration range found in patients with vascular disease. ADMA also causes local vasoconstriction when infused intra-arterially, and increases systemic vascular resistance and impairs renal function when infused systemically. Several recent studies have supplied evidence to support a pathophysiological role of ADMA in the pathogenesis of vascular dysfunction and cardiovascular disease. High ADMA levels were found to be associated with carotid artery intima-media-thickness in a study with 116 clinically healthy human subjects. Taking this observation further, another study performed with hemodialysis patients reported that ADMA prospectively predicted the progression of intimal thickening during one year of follow-up.
In a nested, case-control study involving 150 middle-aged, non-smoking men, high ADMA levels were associated with a 3.9-fold elevated risk for acute coronary events. Clinical and experimental evidence suggests elevation of ADMA can cause a relative L-arginine deficiency, even in the presence of “normal” L-arginine levels. As ADMA is a competitive inhibitor of eNOS, its inhibitory action can be overcome by increasing the concentration of the substrate, L-arginine. Elevated ADMA concentration is one possible explanation for endothelial dysfunction and decreased NO production in these diseases.
Metabolic Regulation of L-arginine and NO Synthesis
Methylation of arginine residues within proteins or polypeptides occurs through N-methyltransferases, which utilize S-adenosylmethionine as a methyl donor. After proteolysis of these proteins or polypeptides, free ADMA is present in the cytoplasm. ADMA can also be detected in circulating blood plasma. ADMA acts as an inhibitor of eNOS by competing with the substrate of this enzyme, L-arginine. The ensuing reduction in nitric oxide synthesis causes vascular endothelial dysfunction and, subsequently, atherosclerosis. ADMA is eliminated from the body via urinary excretion and via metabolism by the enzyme DDAH to citrulline and dimethylamine.
Supplementation with L-arginine in animals with experimentally-induced vascular dysfunction atherosclerosis improves endothelium-dependent vasodilation. Moreover, L-arginine supplementation results in enhanced endothelium-dependent inhibition of platelet aggregation, inhibition of monocyte adhesion, and reduced vascular smooth muscle proliferation. One mechanism that explains the occurrence of endothelial dysfunction is the presence of elevated blood levels of asymmetric dimethylarginine (ADMA) – an L-arginine analogue that inhibits NO formation and thereby can impair vascular function. Supplementation with L-arginine has been shown to restore vascular function and to improve the clinical symptoms of various diseases associated with vascular dysfunction.
Beneficial Effects of L-Arginine
- Angina
- Congestive Heart Failure
- Hypertension
- Erectile dysfunction
- Sickle Cell Disease and Pulmonary Hypertension
The ratio of L-arginine to ADMA is considered to be the most accurate measure of eNOS substrate availability. This ratio will increase during L-arginine supplementation, regardless of initial ADMA concentration. Due to the pharmacokinetics of oral L-arginine and the positive results from preliminary studies, it appears supplementation with a sustained-release L-arginine preparation will achieve positive therapeutic results at lower dosing levels.
Many prospective clinical trials have shown that the association between elevated ADMA levels and major cardiovascular events and total mortality is robust and extends to diverse patient populations. However, we need to define more clearly in the future who will profit from ADMA determination, in order to use this novel risk marker as a more specific diagnostic tool.
Elimination of ADMA by way of DDAH
Asymmetric dimethylarginine (ADMA) and monomethyl arginine (L-NMMA) are endogenously produced amino acids that inhibit all three isoforms of nitric oxide synthase (NOS). ADMA accumulates in various disease states, including renal failure, diabetes and pulmonary hypertension, and its concentration in plasma is strongly predictive of premature cardiovascular disease and death. Both LNMMA and ADMA are eliminated largely through active metabolism by dimethylarginine dimethylaminohydrolase (DDAH) and thus DDAH dysfunction may be a crucial unifying feature of increased cardiovascular risk. These investigators ask whether ADMA is the underlying issue related to the pathogenesis of the vascular disorder.
They identified the structure of human DDAH-1 and probed the function of DDAH-1 both by deleting the Ddah1 gene in mice and by using DDAH-specific inhibitors that is shown by crystallography, bind to the active site of human DDAH-1. The loss of DDAH-1 activity leads to accumulation of ADMA and reduction in NO signaling. This in turn causes vascular pathophysiology, including endothelial dysfunction, increased systemic vascular resistance and elevated systemic and pulmonary blood pressure. The results suggest that DDAH inhibition could be harnessed therapeutically to reduce the vascular collapse associated with sepsis.
Methylarginines are formed when arginine residues in proteins are methylated by the action of protein arginine methyltransferases (PRMTs), and free methylarginines are liberated following proteolysis. Clear demonstration of an effect of endogenous ADMA and L-NMMA on cardiovascular physiology would be of importance, not only because of the implications for disease, but also because it would expose a link between post-translational modification of proteins and signaling through a proteolytic product of these modified proteins.
Which is it? ADMA or DDHA: Intrusion of a Genetic alteration.
The study showed that loss of DDAH expression or activity causes endothelial dysfunction, we believe that DDAH inhibition could potentially be used therapeutically to limit excessive NO production, which can have pathological effects. They then showed treated cultured isolated blood vessels with lipopolysaccharide (LPS) induced expression of the inducible isoform of NO synthase (iNOS) and generated high levels of NO, which were blocked by the iNOS-selective inhibitor 1400W and by DDAH inhibitors. Treatment of isolated blood vessels with DDAH inhibitors significantly increased ADMA accumulation in the culture medium. Treatment of isolated blood vessels with bacterial LPS led to the expected hyporeactivity to the contractile effects of phenylephrine, which was reversed by treatment with a DDAH inhibitor. The effect of the DDAH inhibitor was large and stereospecific, and was reversed by the addition of L-arginine.
In conclusion, genetic and chemical-biology approaches provide compelling evidence that loss of DDAH-1 function results in increased ADMA concentrations and thereby disrupts vascular NO signaling. A broader implication of this study is that post-translational methylation of arginine residues in proteins may have downstream effects by affecting NO signaling upon hydrolysis and release of the free methylated amino acid. This signaling pathway seems to have been highly conserved through evolution.
The crucial role of nitric oxide (NO) for normal endothelial function is well known. In many conditions associated with increased risk of cardiovascular diseases such as hypercholesterolemia, hypertension, abdominal obesity, diabetes and smoking, NO biosynthesis is dysregulated, leading to endothelial dysfunction. The growing evidence from animal and human studies indicates that endogenous inhibitors of endothelial NO synthase such as asymmetric dimethylarginine (ADMA) and NG-monomethyl-L-arginine (L-NMMA) are associated with the endothelial dysfunction and potentially regulate NO synthase.
Asymmetric dimethylarginine (ADMA) is one of three known endogenously produced circulating methylarginines (i.e. ADMA, NG-monomethyl-L-arginine (L-NMMA) and symmetrically methylated NG, NG-dimethyl-L-arginine). ADMA is formed by the action of protein arginine methyltransferases that methylate arginine residues in proteins and after which free ADMA is released. ADMA and L-NMMA can competitively inhibit NO elaboration by displacing L-arginine from NO synthase (NOS). The amount of methylarginines is related to overall metabolic activity and the protein turnover rate of cells. Although methylarginines are excreted partly by the kidneys, the major route of elimination of ADMA in humans is metabolism by the dimethylarginine dimethylaminohydrolase enzymes[ dimethylarginine dimethylaminohydrolase-1 and -2 (DDAH)] enzymes. Inhibition of DDAH leads to the accumulation of ADMA and consequently to inhibition of NO-mediated endothelium dependent relaxation of blood vessels.
The potential role of ADMA in angina pectoris has been evaluated by Piatti and co-workers, who reported ADMA levels to be higher in patients with cardiac syndrome X (angina pectoris with normal coronary arteriograms) than in controls. According to preliminary results from the CARDIAC (Coronary Artery Risk Determination investigating the influence of ADMA Concentration) study, patients with coronary heart disease (n 816) had a higher median ADMA plasma concentration than age and sex matched controls (median 0.91 vs. 0.70 mol/l; p 0.0001). Further, in a prospective Chinese study, a high plasma ADMA level independently predicted subsequent cardiovascular adverse events (cardiovascular death, myocardial infarction, and repeated revascularization of a target vessel).

Protein detoxification pathway. (Photo credit: Wikipedia)
There are only few published findings concerning variations in human DDAH. However, polymorphisms in other genes potentially related to risk factors for endothelial dysfunction and cardiovascular events have been studied. Reduced NO synthesis has been implicated in the development of atherosclerosis. For example, there are some functionally important variants of the NOS that could affect individual vulnerability to atherosclerosis by changing the amount of NO generated by the endothelium.
There are probably several functional variations in genes coding DDAH enzymes in different populations. Some of them could confer protection against the harmful effects of elevated ADMA and others impair enzyme function causing accumulation of ADMA in cytosol and/or blood.
In a study of 16 men with either low or high plasma ADMA concentrations were screened to identify DDAH polymorphisms that could potentially be associated with increased susceptibility to cardiovascular diseases. In that study a novel functional mutation of DDAH-1 was identified; the mutation carriers had a significantly elevated risk for cardiovascular disease and a tendency to develop hypertension. These results confirmed the clinical role of DDAH enzymes in ADMA metabolism. Furthermore, it is possible that more common variants of DDAH genes contribute more widely to increased cardiovascular risk.
We found a rare variation in the DDAH-1 gene, which is associated with elevated plasma concentrations of ADMA in heterozygous mutation carriers. There was also an increased prevalence of CHD and a tendency to hypertension among individuals with this DDAH-1 mutation. These observations highlight the importance of ADMA as a possible risk factor and emphasize the essential role of DDAH in regulating ADMA levels.
ADMA Elevation and Coronary Artery Disease
Endothelial dysfunction may be considered as a systemic disorder and involves different vascular beds. Coronary endothelial dysfunction (CED) precedes the development of coronary. Endothelial dysfunction is characterized by a reduction in endogenous nitric oxide (NO) activity, which may be accompanied by elevated plasma asymmetric dimethylarginine (ADMA) levels. ADMA is a novel endogenous competitive inhibitor of NO synthase (NOS), an independent marker for cardiovascular risk.

English: Structure of asymmetric dimethylarginine; ADMA; N,N-Dimethylarginine Deutsch: Asymmetrisches Dimethylarginin; N,N-Dimethyl-L-arginin; Guanidin-N,N-dimethylarginin (Photo credit: Wikipedia)
In a small study fifty-six men without obstructive coronary artery disease (CAD) who underwent coronary endothelial function testing were studied. Men with CED had significant impairment of erectile function (P ¼ 0.008) and significantly higher ADMA levels (0.50+0.06 vs. 0.45+0.07 ng/mL, P ¼ 0.017) compared with men with normal endothelial function. Erectile function positively correlated with coronary endothelial function. This correlation was independent of age, body mass index, high-density lipoprotein, C-reactive protein, homeostasis model assessment of insulin resistance index, and smoking status, suggesting that CED is independently associated with ED and plasma ADMA concentration in men with early coronary atherosclerosis.
ADMA and Chronic Renal Failure in Hepatorenal Syndrome
The concentration of SDMA was significantly higher in the patients with HRS compared to the patients without HRS and it was also higher than the values obtained from the healthy participants (1.76 ± 0.3 μmol/L; 1.01 ± 0.32 and 0.520 ± 0.18 μmol/L, respectively; p < 0.01). The concentrations of ADMA were higher in the cirrhotic patients with HRS than in those without this serious complication of cirrhosis. The concentration of ADMA in all the examined cirrhotic patients was higher than those obtained from healthy volunteers (1.35 ± 0.27 μmol/L, 1.05 ± 0.35 μmol/L and 0.76 ± 0.21 μmol/L, respectively). In the patients with terminal alcoholic liver cirrhosis, the concentrations
of ADMA and SDMA correlated with the progress of cirrhosis as well as with the development of cirrhosis complications. In the patients with HRS there was a positive correlation between creatinine and SDMA in plasma (r2 = 0.0756, p < 0.001) which was not found between creatinine and ADMA. The results demonstrate that the increase in SDMA concentration is proportionate to the progression of chronic damage of the liver and kidneys. Increased ADMA concentration can be a causative agent of renal insufficiency in patients with cirrhosis.
In patients with cirrhosis, ADMA, as well as SDMA could be markers for kidney insufficiency development. Accumulation of ADMA in plasma causes kidney
vasoconstriction and thereby retention of SDMA. Considering that ADMA has several damaging effects, it can be concluded that modulation of the activity of enzyme which participates in ADMA catabolism may represent a new therapeutic goal which is intended to reduce the progress of liver and kidney damage and thus the development of HRS.
ADMA Therapeutic Targets
Elevated plasma concentrations of the endogenous nitric oxide synthase
inhibitor asymmetric dimethylarginine (ADMA) are found in various clinical settings, including
- renal failure,
- coronary heart disease,
- hypertension,
- diabetes and
- preeclampsia.
In healthy people acute infusion of ADMA promotes vascular dysfunction,
and in mice chronic infusion of ADMA promotes progression of atherosclerosis.
Thus, ADMA may not only be a marker but also an active player in cardiovascular disease, which makes it a potential target for therapeutic interventions.
This review provides a summary and critical discussion of the presently available data concerning the effects on plasma ADMA levels of cardiovascular drugs, hypoglycemic agents, hormone replacement therapy, antioxidants, and vitamin supplementation.
We assess the evidence that the beneficial effects of drug therapies on vascular function can be attributed to modification of ADMA levels. To develop more specific ADMA-lowering therapies, mechanisms leading to elevation of plasma ADMA concentrations in cardiovascular disease need to be better understood.
ADMA is formed endogenously by degradation of proteins containing arginine residues that have been methylated by S-adenosylmethionine-dependent methyltransferases (PRMTs). There are two major routes of elimination: renal excretion and enzymatic degradation by the dimethylarginine dimethylaminohydrolases (DDAH-1 and -2).
Oxidative stress causing upregulation of PRMT expression and/or attenuation of DDAH activity has been suggested as a mechanism and possible drug target in clinical conditions associated with elevation of ADMA. As impairment of DDAH activity or capacity is associated with substantial increases in plasma ADMA concentrations, DDAH is likely to emerge as a prime target for specific therapeutic interventions.
Cardiovascular diseases (CVD) in diabetic patients have endothelial dysfunction as a key pathogenetic event. Asymmetric dimethylarginine (ADMA), an endogenous inhibitor of nitric oxide synthase (NOS), plays a pivotal role in endothelial dysfunction. Different natural polyphenols have been shown to preserve endothelial function and prevent CVD. Another study assessed the effect of silibinin, a widely used flavonolignan from milk thistle, on ADMA levels and endothelial dysfunction in db/db mice.
Plasma and aorta ADMA levels were higher in db/db than in control lean mice. Silibinin administration markedly decreased plasma ADMA; consistently, aorta ADMA was reduced in silibinin-treated animals. Plasma and aorta ADMA levels exhibited a positive correlation, whereas liver ADMA was inversely correlated with both plasma and aorta ADMA concentrations. Endothelium-(NO)-dependent vasodilatation to ACh was impaired in db/db mice and was restored in the silibinin group, in accordance with the observed reduction of plasma and vascular levels of ADMA. Endothelium-independent vasodilatation to SNP was not modified by silibinin administration.
Endothelin Inhibitors
Endothelins are potent vasoconstrictors and pressor peptides and are important mediators of cardiac, renal andendocrine functions. Increased ET-1 levels in disease states such as congestive heart failure, pulmonary hypertension, acute myocardial infarction, and renal failure suggest the endothelin system as an attractive target for pharmacotherapy. A non-peptidic, selective, competitive endothelin receptor antagonist with an affinity for the ETA receptor in the subnanomolar range was administered by continuous intravenous infusion to beagle dogs, rats, and Goettingen minipigs. It caused mild arteriopathy characterised by segmental degeneration in the media of mid- to large-size coronary arteries in the heart of dog, but not rat or minipig.
The lesions only occurred in the atrium and ventricle. Frequency and severity of the vascular lesions was not sex or dose related. No effects were noted in blood vessels in other organs or tissue. Plasma concentrations at steady state, and overall exposure in terms of AUC(0–24h) were higher in minipig and rat than the dog but did not cause cardiac arteriopathy. These findings concur with those caused by other endothelin anatagonists, vasodilators and positive inotropic: vasodilating drugs such as potassium channel openers, phosphodiesterase inhibitors and peripheral vasodilators.
Results by echocardiography indicate treatment-related local vasodilatation in the coronary arteries. These data suggest that the coronary arteriopathy may be the result of exaggerated pharmacology. Sustained vasodilatation in the coronary vascular bed may alter flow dynamics and lead to increased shear stress and tension on the coronary wall with subsequent microscopic trauma. In our experience with a number of endothelin receptor antagonists, the cardiac arteriopathy was only noted in studies with multiple daily or continuous intravenous infusion inviting speculation that sustained high plasma levels are needed for development of the lesions.
Up-regulation of vascular endothelin type B (ETB) receptors is implicated in the
pathogenesis of cardiovascular disease. Culture of intact arteries has been shown to induce similar receptor alterations and has therefore been suggested as a suitable method for, ex vivo, in detail delineation of the regulation of endothelin receptors. We hypothesize that mitogen-activated kinases (MAPK) and protein kinase C (PKC) are involved in the regulation of endothelin ETB receptors in human internal mammary arteries.
The endothelin-1-induced contraction (after endothelin ETB receptor desensitization) and the endothelin ETA receptor mRNA expression levels were not altered by culture. The sarafotoxin 6c contraction, endothelin ETB receptor protein and mRNA expression levels were increased. This increase was antagonized by;
PKC inhibitors (10 μM bisindolylmaleimide I and 10 μM Ro-32-0432), and
inhibitors of the p38, extracellular signal related kinases 1 and 2 (ERK1/2) and C-jun terminal kinase (JNK) MAPK pathways
Endothelin Receptor Antagonist Tezosentan
The effects of changes in the mean (Sm) and pulsatile (Sp) components of arterial wall shear stress on arterial dilatation of the iliac artery of the anaesthetized dog were examined in the absence and presence of the endothelin receptor antagonist tezosentan (10 mg kg_1 I.V.; Ro 61-0612; [5-isopropylpyridine-2-sulphonic acid 6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-2-(2-1H-tetrazol-5-ylpyridin-4-yl)-pyrimidin-4-ylamide]).
Changes in shear stress were brought about by varying local peripheral resistance and stroke volume using a distal infusion of acetylcholine and stimulation of the left ansa subclavia. An increase in Sm from 1.81 ± 0.3 to 7.29 ± 0.7 N m_2 (means ± S.E.M.) before tezosentan caused an endothelium-dependent arterial dilatation which was unaffected by administration of tezosentan for a similar increase in Sm from 1.34 ± 0.6 to 5.76 ± 1.4 N m_2 (means ± S.E.M.).
In contrast, increasing the Sp from 7.1 ± 0.8 to a maximum of 11.5 ± 1.1 N m_2 (means ± S.E.M.) before tezosentan reduced arterial diameter significantly. Importantly, after administration of tezosentan subsequent increases in Sp caused arterial dilatation for the same increase in Sp achieved prior to tezosentan, increasing from a baseline of 4.23 ± 0.4 to a maximum of 9.03 ± 0.9 N m_2 (means ± S.E.M.; P < 0.001). The results of this study provide the first in vivo evidence that pulsatile shear stress is a stimulus for the release of endothelin from the vascular endothelium.
Exercise and Diet
Vascular endotheliumis affected by plasma asymmetric dimethylarginine (ADMA), and it is induced by inflammatory cytokines of tumour necrosis factor (TNF)-a in vitro. Would a tight glycemic control restore endothelial function in patients with type-2 diabetes mellitus (DM) with modulation of TNF-a and/or reduction of ADMA level? In 24 patients with type-2 DM, the flow-mediated, endothelium-dependent dilation (FMD: %) of brachial arteries during reactive hyperaemia was determined by a high-resolution ultrasound method. Blood samples for glucose, cholesterol, TNF-a, and ADMA analyses were also collected from these patients after fasting. No significant glycemic or FMD changes were observed in 10 patients receiving the conventional therapy.
In 14 patients who were hospitalized and intensively treated, there was a significant decrease in glucose level after the treatment [from 190+55 to 117+21 (mean+SD) mg/dL, P , 0.01]. After the intensive control of glucose level, FMD increased significantly (from 2.5+0.9 to 7.2+3.0%), accompanied by a significant (P , 0.01) decrease in TNF-a (from 29+16 to 11+9 pg/dL) and ADMA (from 4.8+1.5 to 3.5+1.1 mM/L) levels. The changes in FMD after treatment correlated inversely with those in TNF-a (R ¼ 20.711, P , 0.01) and ADMA (R ¼ 20.717, P , 0.01) levels.
The exaggerated blood pressure response to exercise (EBPR) is an independent predictor of hypertension. Asymmetric dimethylarginine (ADMA) is an endogenous nitric oxide inhibitor and higher plasma levels of ADMA are related to increased cardiovascular risk. The aim of this study is to identify the relationship between ADMA and EBPR.
A total of 66 patients (36 with EBPR and 30 as controls) were enrolled in the study. EBPR is defined as blood pressure (BP) measurements ≥200/100 mmHg during the treadmill test. All the subjects underwent 24-h ambulatory BP monitoring. L-arginine and ADMA levels were measured using a high performance lipid chromatography technique.
The serum ADMA levels were increased in the EBPR group compared to the healthy controls (4.0±1.4 vs 2.6±1.1 μmol/L respectively, P=0.001), but L-arginine levels were similar in the 2 groups (P=0.19). The serum ADMA levels were detected as an independent predictor of EBPR (odds ratio 2.28; 95% confidence interval 1.22–4.24; P=0.002). Serum ADMA levels might play a role in EBPR to exercise.
Endothelial dysfunction occurs early in atherosclerosis in response to cardiovascular risk factors. The occurrence of endothelial dysfunction is primarily the result of reduced nitric oxide (NO) bioavailabilty. It represents an independent predictor of cardiovascular events and predicts the prognosis of the patient. Therefore, endothelial function has been identified as a target for therapeutic intervention. Regular exercise training is a nonpharmacological option to improve endothelial dysfunction in patients with cardiovascular disease by increasing NO bioavailability.
Peripheral Arterial Disease (PAD) is a cause of significant morbidity and mortality in the Western world. risk factor modification and endovascular and surgical revascularisation are the main treatment options at present. However, a significant number of patients still require major amputation. There is evidence that nitric oxide (NO) and its endogenous inhibitor asymmetric dimethylarginine (ADMA) play significant roles in the pathophysiology of PAD.
This paper reviews experimental work implicating the ADMA-DDAH-NO pathway in PAD, focusing on both the vascular dysfunction and both the vascular dysfunction and effects within the ischaemic muscle, and examines the potential of manipulating this pathway as a novel adjunct therapy in PAD.
In patients with CHF, the peripheral vascular resistance is increased via activation of the neurohormonal system, namely by autonomous sympathetic nervous system, rennin -angiotensin- aldosterone system (RAAS), and endothelin system. The vascular endothelial function in patients with CHF, mainly represented by the endothelium-dependent vasodilation, is altered.
Such alteration leads to increased vascular tone and remodeling of the blood vessels, reducing the peripheral blood flow. Hence, the amount of oxygen for the skeletal muscles is compromised, with progressive exercise intolerance. The vascular endothelial dysfunction in the CHF is mainly due to the decrease of the nitric oxide production induced by the reduced gene expression of eNOS and increased oxidative stress.
The endothelium-dependent vasodilation alteration has been virtually reported in all cardiovascular diseases. Using sauna bath as therapeutic option for CHF is not very recent, since in the 1950’s the first studies with CHF patients were conducted and the potential beneficial effect of sauna was suggested. However, some time later the studies emphasized especially its risks and recommended caution in its use for cardiac patients.
Frequently, sports medicine physicians are invited to evaluate the impact of the sauna on diseases and on health in general. Sauna can be beneficial or dangerous depending on its use. In the past few years the sauna is considered beneficial for the cardiovascular diseases’ patients, as the heart failure and lifestyle-related diseases, mainly by improving the peripheral endothelial function through the increase in cardiac output and peripheral vasodilation.
It is widely known that the vasodilators, such as angiotensin converting enzyme inhibitors, improve the CHF and increase the peripheral perfusion. Since the endothelial function is altered in CHF, the endothelium is considered as a new therapeutic target in heart failure. Hence, the angiotensin converting enzyme inhibitors and physical training improve the endothelial function in CHF patients. One of the proposed mechanisms for the alteration of the endothelium-dependent vasodilation would be through the decrease of the NO production in the peripheral vessels in CHF patients. The decrease of peripheral perfusion would decrease the shear stress. The shear stress is an important stimulus for NO production and eNOS expression. On the other hand, the heat increases the cardiac output and improves the peripheral perfusion in CHF patients. Consequently, with the cardiac output improvement in CHF patients, an increase of the shear stress, NO production and eNOS expression are expected.
Sauna bath
The sauna bath represents a heat load of 300-600 W/m2 of body surface area. The skin temperature rapidly increases to ± 40o-41oC and the thermoregulatory mechanisms are triggered. Evaporative heat transfer by sweating is the only effective body heat loss channel in dry sauna. The sweating begins rapidly and reaches its maximum level in ± 15 min. The total sweat secretion represents a heat loss of about 200 W/m2 of the body surface area. The body cannot compensate for the heat load and causing elevation of internal temperature. The skin circulation increases substantially. The skin blood flow, in the thermo-neutral condition (± 20oC) and in rest corresponding to ± 5-10% of the cardiac output, can reach ± 50-70% of the cardiac output.
Thermal therapy in 60oC produced systemic arterial, pulmonary arterial and venous vasodilation, reduced the preload and afterload and improved the cardiac output and the peripheral perfusion, clinical symptoms, life quality, and cardiac arrhythmias in CHF patients. In infants with severe CHF secondary to ventricular septal defect, the sauna therapy decreased the systemic vascular resistance and increased the cardiac output. The sauna benefits in CHF patients are possibly caused by the improvement of the vascular endothelial function and normalization of the neurohormonal system .
Ikeda et al. discovered that the observed improvements in the sauna therapy are due to the eNOS expression increase in the arterial endothelium. They later showed that the thermal therapy with sauna improves the survival of the TO-2 cardiomyopathic hamsters with CHF and, more recently, showed that the repetitive therapy with sauna increases the eNOS expression and the nitric oxide production in artery endothelium of TO-2 cardiomyopathic hamsters with CHF.
Whether n-3 polyunsaturated fatty acid (PUFA) supplementation and/or diet intervention might have beneficial influence on endothelial function was assessed using plasma levels of ADMA and L-arginine. A male population (n = 563, age 70 ± 6 yrs) with long-standing hyperlipidemia, characterized as high risk individuals in 1970–72, was included, randomly allocated to receive placebo n-3 PUFA capsules (corn oil) and no dietary advice (control group), dietary advice (Mediterranean type), n-3 PUFA capsules, or dietary advice and n-3 PUFA combined and followed for 3 years. Fasting blood samples were drawn at baseline and the end of the study.
Compliance with both intervention regimens were demonstrated by changes in serum fatty acids and by recordings from a food frequency questionnaire. No influence of either regimens on ADMA levels were obtained. However, n-3 PUFA supplementation was accompanied by a significant increase in L-arginine levels, different from the decrease observed in the placebo group (p < 0.05). In individuals with low body mass index (<26 kg/m2), the decrease in L-arginine on placebo was strengthened (p = 0.01), and the L-arginine/ADMA ratio was also significantly reduced (p = 0.04). In this rather large randomized intervention study, ADMA levels were not influenced by n-3 PUFA supplementation or dietary counselling. n-3 PUFA did, however, counteract the age related reduction in L-arginine seen on placebo, especially in lean individuals, which might be considered as an improvement of endothelial function.
Traditional Chinese Medicine
Traditional Chinese Medicine (TCM) involves a broad range of empirical testing and refinement and plays an important role in the health maintenance for people all over the world. However, due to the complexity of Chinese herbs, a full understanding of TCM’s action mechanisms is still unavailable despite plenty of successful applications of TCM in the treatment of various diseases, including especially cardiovascular diseases (CVD), one of the leading causes of death.
An integrated system of TCM has been constructed to uncover the underlying action mechanisms of TCM by incorporating the chemical predictors, target predictors and network construction approaches from three representative Chinese herbs, i.e., Ligusticum chuanxiong Hort., Dalbergia odorifera T. Chen and Corydalis yanhusuo WT Wang widely used in CVD treatment, by combined use of drug absorption, distribution, metabolism and excretion (ADME) screening and network pharmacology techniques. These studies have generated 64 bioactive ingredients and identified 54 protein targets closely associated with CVD, to clarify some of the common conceptions in TCM, and provide clues to modernize such specific herbal medicines.
Ligusticum chuanxiong Hort., Dalbergia odorifera T. Chen and Corydalis yanhusuo WT Wang
Twenty-two of 194 ingredients in Ligusticum chuanxiong demonstrate good bioavailability (60%) after oral administration. Interestingly, as the most abundant bioactive compound of Chuanxiong, Ligustilide (M120) only has an adequate OB of 50.10%, although it significantly inhibits the vasoconstrictions induced by norepinephrine bitartrate (NE) and calcium chloride (CaCl2). Indeed, this compound can be metabolized to butylidenephthalide, senkyunolide I (M156), and senkyunolide H (M155) in vivo.
The three natural ingredients produce various pharmacological activities in cerebral blood vessels, the general circulatory system and immune system including spasmolysis contraction effects, inhibitory effects of platelet aggregation and anti-proliferative activity, and thus improve the therapeutic effect on patients. Cnidilide (M93, OB = 77.55%) and spathulenol (M169, OB = 82.37%) also closely correlate with the smooth muscle relaxant action, and thereby have the strongest spasmolytic activity. Carotol (M8) and Ferulic acid (M105) with an OB of 149.03% and 86.56%, respectively, demonstrate better bioavailability compared with cnidilide and spathulenol, which show strong antifungal, antioxidant and anti-inflammatory activity.
The pharmacological activity of ferulic acid results in the improvement of blood fluidity and the inhibition of platelet aggregation, which may offer beneficial effects against cancer, CVD, diabetes and Alzheimer’s disease. As for 3-n-butylphthalide (M85, OB = 71.28%), this compound is not only able to inhibit platelet aggregation, but also decreases the brain infarct volume and enhances microcirculation, thus benefiting patients with ischemic stroke. Platelet aggregation represents a multistep adhesion process involving distinct receptors and adhesive ligands, with the contribution of individual receptor-ligand interactions to the aggregation process depending on the prevailing blood flow conditions, implying that the rheological (blood flow) conditions are an important impact factor for platelet aggregation. Moreover, thrombosis, the pathological formation of platelet aggregates and one of the biggest risk factors for CVD, occludes blood flow causing stroke and heart attack. This explains why the traditional Chinese herb Ligusticum chuanxiong that inhibits platelet aggregates forming and promotes blood circulation can be used in treatment of CVD.
Twenty-six percent (24 of 93) of the ingredients in Dalbergia odorifera meet the OB > 60% criterion irrespective of the pharmacological activity. Relatively high bioavailability values were predicted for the mainly basic compounds odoriflavene (M275, OB = 84.49%), dalbergin (M247, OB = 78.57%), sativanone (M281, OB = 73.01%), liquiritigenin (M262, OB = 67.19%), isoliquiritigenin (M259, OB = 61.38%) and butein (M241, OB = 78.38%). Interestingly, all of the six ingredients show obvious anti-inflammatory property. Butein, liquiritigenin and isoliquiritigenin inhibit cell inflammatory responses by suppressing the NF-κB activation induced by various inflammatory agents and carcinogens, and by decreasing the NF-κB reporter activity. Inflammation occurs with CVD, and Dalbergia odorifera, one of the most potent anti-cardiovascular and anti-cerebrovascular agents, exerts great anti-inflammatory activity.
Corydalis yanhusuo has gained ever-increasing popularity in today’s world because of its therapeutic effects for the treatment of cardiac arrhythmia disease, gastric and duodenal ulcer and menorrhalgia. In our work, 21% (15 of 73) of chemicals in this Chinese herb display good OB (60% or even high), and the four main effective ingredients are natural alkaloid agents.
Dehydrocorydaline blocks the release of noradrenaline from the adrenergic nerve terminals in both the Taenia caecum and pulmonary artery, and thereby inhibits the relaxation or contraction of adrenergic neurons. As for dehydrocavidine with an OB of 47.59%, this alkaloid exhibits a significant spasmolytic effect, which acts via relaxing smooth muscle.
In recent years, CVD has been at the top list of the most serious health problems. Many different types of therapeutic targets have already been identified for the management and prevention of CVD, such as endothelin and others. The key question asked is
- what the interactions of the active ingredients of the Chinese herbs are with their protein targets in a systematic manner and
- how do the corresponding targets change under differential perturbation of the chemicals?
The study used an unbiased approach to probe the proteins that bind to the small molecules of interest in CVD on the basis of the Random Forest (RF) and Support Vector Machine (SVM) methods combining the chemical, genomic and pharmacological information for drug targeting and discovery on a large scale. Applied to 64 ingredients derived from the three traditional Chinese medicines Dalbergia odorifera, Ligusticum chuanxiong and Corydalis yanhusuo, which show good OB, 261 ligand-target interactions have been constructed, 221 of which are enzymes, receptors, and ion channels. This indicates that chemicals with multiple relative targets are responsible for the high interconnectedness of the ligand-target interactions. The promiscuity of drugs has restrained the advance in recent TCM, because they were thought to be undesirable in favor of more target-specific drugs.
Target Identification and Validation
To validate the reliability of these target proteins, the researchers performed a docking analysis to select the ligand-protein interactions with a binding free energies of ≤−5.0 kcal/mol, which leads to the sharp reduction of the interaction number from 5982 to 760. These drug target candidates were subsequently subject to PharmGkb (available online: http://www.pharmgkb.org; accessed on 1 December 2011), a comprehensive disease-target database, to investigate whether they were related to CVD or not, and finally, 54 proteins were collected and retained.
Fourty-two proteins (76%) were identified as the targets of Ligusticum chuanxiong, such as dihydrofolate reductase (P150), an androgen receptor (P210) and angiotensin-converting enzyme (P209) that were involved in the development of CVD. Of the proteins, seven and two were recognized as those of Dalbergia odorifera and Corydalis yanhusuo, respectively. For Dalbergia odorifera, this Chinese herb has 48 potential protein targets, 13 of which have at least one link to other drugs.
The three herbs share 29 common targets, accounting for 52.7% of the total number. Indeed, as one of the most important doctrines of TCM
abstracted from direct experience and perception, “multiple herbal drugs for one disease” has played an undeniable role. These studies explored the targets of the three Chinese herbs, indicating that these drugs target the same targets simultaneously and exhibit similar pharmacological effects on CVD. This is consistent with the theory of “multiple herbal drugs for one disease”.
The three Chinese herbs possess specific targets. The therapeutic efficacy of a TCM depends on multiple components, targets and pathways. The complexity becomes a huge obstacle for the development and innovation of TCM. For example, the Chinese herb Ligusticum chuanxiong identifies the protein caspase-3 (P184), a cysteinyl aspartate-specific protease, as one of its specific targets, and exhibits inhibitory effects on the activity of this protease. In fact, connective tissue growth factor enables the activation of caspase-3 to induce apoptosis in human aortic vascular smooth muscle cells.
Thus, modulation of the activity of caspase-3 with Ligusticum chuanxiong suggests an efficient therapeutic approach to CVD. The Chinese herb Dalbergia odorifera has the α-2A adrenergic receptor (P216) as its specific target and probably blocks the release of this receptor, and thus influences its action. As for Corydalisyanhusuo, the protein tyrosine-protein kinase JAK2 (P9) is the only specific target of this Chinese herb. The results indicate different specific targets possessed by the three Chinese herbs.
Ligand-Candidate Target and Ligand-Potential Target Networks
Previous studies have already reported the relationships of the small molecules with CVD, which indicates the reliability of our results [45,46]. Regarding the candidate targets, we have found that prostaglandin G/H synthase 2 (P46) and prostaglandin G/H synthase 1 (P47) possess the largest number of connected ingredients. Following are nitric-oxide synthase, endothelial (P66) and tyrosine-protein phosphatase non-receptor type 1 (P8), which have 62 and 61 linked chemicals, respectively.
The 29 targets shared by the three traditional Chinese herbs exhibit a high degree of correlations with CVD, which further verifies their effectiveness for the treatment of CVD. These results provide a clear view of the relationships of the target proteins with CVD and other related diseases, which actually link the Chinese herbs and the diseases via the protein targets. This result further explains the theory of “multiple herbal drugs for one disease” based on molecular pharmacology.
Target-Pathway Network
Cells communicate with each other using a “language” of chemical signals. The cell grows, divides,or dies according to the signals it receives. Signals are generally transferred from the outside of the cell. Specialized proteins are used to pass the signal—a process known as signal transduction. Cells have a number of overlapping pathways to transmit signals to multiple targets. Ligand binding in many of the signaling proteins in the pathway can change the cellular communication and finally affect cell growth and proliferation. The authors extracted nine signal pathways closely associated with CVD in PharmGkb (available online: http://www.pharmgkb.org; accessed on 1 December 2011).
As the main components in the VEGF system, proto-oncogene tyrosine-protein kinase Src, eNOS, and hsp90-α is also recognized as common targets of Dalbergia odorifera, Ligusticum chuanxiong and Corydalis yanhusuo, which are efficient for the treatment of CVD. This implies that the candidate drugs can target different target proteins involved in the same or different signal pathways, and thereby have potential effects on the whole signal system.
Target Prediction
In search of the candidate targets, the model that efficiently integrates the chemical, genomic and pharmacological information for drug targeting and discovery on a large scale is based on the two powerful methods Random Forest (RF) and Support Vector Machine (SVM). The model is supported by a large pharmacological database of 6511 drugs and 3999 targets extracted from the DrugBank database (available online: http://drugbank.ca/; accessed on 1 June 2011), and shows an impressive performance of prediction for drug-target interaction, with a concordance of 85.83%, a sensitivity of 79.62% and a specificity of 92.76%. the candidate targets were selected according to the criteria that the possibility of interacting with potential candidate targets was higher than 0.6 for the RF model and 0.7 for the SVM model. The obtained candidate targets were finally reserved and were further predicted for their targets.
Target Validation
Molecular docking analysis was carried out using the AutoDock software (available online: http://autodock.scripps.edu/; accessed on 1 February 2012). This approach performs the docking of the small, flexible ligand to a set of grids describing the target protein. During the docking process, the protein was considered as rigid and the molecules as flexible. The crystal structures of the candidate targets were downloaded from the RCSB Protein Data Bank (available online: http://www.pdb.org/; accessed on 1 December 2011), and the proteins without crystal structures were performed based on homology modeling using the Swiss-Model Automated Protein Modelling Server (available online: http://swissmodel.expasy.org/; accessed on 1 February 2012).
TCM is a heritage that is thousands of years old and is still used by millions of people all over the world—even after the development of modern scientific medicine. Chinese herbal combinations generally include one or more plants and even animal products.
The study identified 54 protein targets, which are closely associated with CVD for the three Chinese herbs, of which 29 are common targets (52.7%), which clarifies the mechanism of efficiency of the herbs for the treatment of CVD.
Activation of NFkB
Extracellular stimuli for NFkB activation and NFkB regulated genes
Extracellular stimuli Regulated genes
TNFa Growth factors (G/M-CSF)
Interleukin 1 G/M CSF, M CSF, G CSF
ROS Cell adhesion molecules
UV light ICAM-1, VCAM, E-Selectin, P-selectin
Ischaemia Cytokines
Lipopolysaccharide TNFa, IL-1, IL-2, IL-6, interferon
Bacteria Transcription regulators
Viruses P53, IkB, c-rel, c-myc
Amyloid Antiapoptotic proteins
Glutamate TRAF-1, TRAF-2, c-IAP1, c-IAP2
Pathophysiology
Reactive oxygen species (ROS) are toxic and in conditions of a dysbalance between their overproduction and the diminished activity of various antioxidant enzymes and other molecules induce cellular injury termed oxidative stress. ROS are often related to a number of diseases like atherosclerosis. However, the mechanism is not clear at all. Latest years of research have brought the idea of connection between ROS and NFkB. And indeed, in vitro studies showed a rapid activation of NFkB after exposure of certain cell types to ROS. Today, no specific receptor for ROS has been found, thus, the details of the ROS induced activation of NFkB are missing.
Natural occurring agents which actions are still a matter of debate in the theory and nouvelle small molecular derivates activate or inhibit the transcriptional factor. Synthetic oligo and polypeptide inhibitors of NFkB can penetrate the cell membrane and directly act on the Rel proteins. The most sophisticated approaches towards inhibiting the activation and translocation of NFkB into the nucleus represent gene deliveries, using plasmids or adenoviruses containing genes for various super repressors—modified IkB proteins, or so called NFkB decoys, which interact with activated NFkB and thus, inhibit the interaction between the transcription factor and nuclear DNA enhancers.
A simplified scheme of the activation of NFkB by the degradation of IkB. IkB is phosphorylated by IKK and ubiquinatated by the ubiquitine ligase system (ULS). IkB is further degradated by the 26S proteasome (26S).Activated NFkB can pass the nuclear membrane and interact with kB binding sequences in enhancers of NFkB regulated genes. LPS, lipopolysaccharide; ROS, reactive oxygen species; FasL, Fas ligand; TRAF, TNFa receptor associated factor; NIK, NFkB inducing kinase; MEKK, mitogen activated protein kinase/extracellular signal regulated kinases kinases.
The medicine of this century is a medicine of molecules, the diagnostic procedure and the therapy moves further from the “clinical picture” to the use of achievements in molecular biology and genetics. However, sober scepticism and awareness are indicated. Especially the role of NFkB in multiple signal transducing pathways and the tissue dependent variability of responses to alternations in NFkB pathway may be the reasons for unwanted side effects of the therapy that are after in vitro or in vivo experiments hardly to expect in the clinical use.
Therapeutic Targets
Modern drug discovery is primarily based on the search and subsequent testing of drug candidates acting on a preselected therapeutic target. Progress in genomics, protein structure, proteomics, and disease mechanisms has led to a growing interest in an effort for finding new targets and more effective exploration of existing targets. The number of reported targets of marketed and investigational drugs has significantly increased in the past 8 years. There are 1535 targets collected in the therapeutic target database.
Knowledge of these targets is helpful for molecular dissection of the mechanism of action of drugs and for predicting features that guide new drug design and the
search for new targets. This article summarizes the progress of target exploration and investigates the characteristics of the currently explored targets to analyze their sequence, structure, family representation, pathway association, tissue distribution, and genome location features for finding clues useful for searching for new targets. Possible “rules” to guide the search for druggable proteins and the feasibility of using a statistical learning method for predicting druggable proteins directly from their sequences are discussed.
Current Trends in Exploration of Therapeutic Targets
There are 395 identifiable targets described in 1606 patents. Of these targets, 264 have been found in more than one patent and 50 appear in more than 10 patents. The number of patents associated with a target can be considered to partly correlate with the level of effort and intensity of interest currently being directed to it. Approximately one third of the patents with an identifiable target were approved in the past year. This suggests that the effort for the exploration of these targets is ongoing, and there has been steady progress in the discovery of new investigational agents directed to these targets.
Various degrees of progress have been made toward discovery and testing of agents directed at these targets. However, for some of these targets, many difficulties remain to be resolved before viable drugs can be derived. The appearance of a high number of patents associated with these targets partly reflects the intensity of efforts for finding effective drug candidates against these targets.
There are 62 targets being explored for the design of subtype-specific drugs, which represents 15.7% of the 395 identifiable targets in U.S. patents approved in 2000 through 2004. Compared with the 11 targets of FDA approved subtype-specific drugs during the same period, a significantly larger number of targets are being explored for the design of subtype-specific drugs.
What Constitutes a Therapeutic Target?
The majority of clinical drugs achieve their effect by binding to a cavity and regulating the activity, of its protein target. Specific structural and physicochemical properties, such as the “rule of five” (Lipinski et al., 2001), are required for these drugs to have sufficient levels of efficacy, bioavailability, and safety, which define target sites to which drug-like molecules can bind. In most cases, these sites exist out of functional necessity, and their structural architectures accommodate target-specific drugs that minimally interact with other functionally important but structurally similar sites.
These constraints limit the types of proteins that can be bound by drug-like molecules, leading to the introduction of the concept of druggable proteins (Hopkins and Groom, 2002; Hardy and Peet, 2004). Druggable proteins do not necessarily become therapeutic targets (Hopkins and Groom, 2002); only those that play key roles in diseases can be explored as potential targets.
Prediction of Druggable Proteins by a Statistical Learning Method
Currently, the support vector machine (SVM) method seems to be the most accurate statistical learning method for protein predictions. SVM is based on the structural risk minimization principle from statistical learning theory. Known proteins are divided into druggable and nondruggable classes; each of these proteins is represented by their sequence-derived physicochemical features.
These features are then used by the SVM to construct a hyperplane in a higher dimensional hyperspace that maximally separates druggable proteins and nondruggable ones. By projecting the sequence of a new protein onto this hyperspace, it can be determined whether this protein is druggable from its location with respect to the hyperplane. It is a druggable protein if it is located on the side of druggable class.
References
Böger RH. Asymmetric dimethylarginine, an endogenous inhibitor of nitric oxide synthase, explains the ‘L-arginine paradox’ and acts as a novel cardiovascular risk factor. J Nutr 2004; 134: 2842S–7S.
B Dobutovi, K Smiljani, S Soski, HD Düngen and ER Isenovi. Nitric Oxide and its Role in Cardiovascular Diseases. The Open Nitric Oxide Journal, 2011; 3: 65-71. 1875-0427/11.
Vallance P, Leone A, Calver A, Collier J, Moncada S. Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet 1992; 339(8793): 572−5.
ER Hedegaard, E Stankevicius, U Simonsen and O Fröbert. Non-endothelial endothelin counteracts hypoxic vasodilation in porcine large coronary arteries. BMC Physiology 2011; 11:8-20. http://www.biomedcentral.com/1472-6793/11/8
S Rajagopalan, D Pfenninger, C Kehrer, A Chakrabarti. Increased Asymmetric Dimethylarginine and Endothelin 1 Levels in Secondary Raynaud’s Phenomenon. Arthritis & Rheumatism 2003; 48(7): 1992–2000. DOI: 10.1002/art.11060
Boger RH. The emerging role of asymmetric dimethylarginine as a novel cardiovascular risk factor. Cardiovasc Res 2003;59:824-833.
Böger RH, Ron ES. L-Arginine Improves Vascular Function by Overcoming the Deleterious Effects of ADMA, a Novel Cardiovascular Risk Factor. Altern Med Rev 2005;10(1):14-23.
Böger RH. Asymmetric dimethylarginine (ADMA) and cardiovascular disease: insights from prospective clinical trials. Vascular Medicine 2005; 10(2): S19-S25. DOI: 10.1191/ 1358863x 05vm602oa.
J Leiper, M Nandi, B Torondel, J Murray-Rust, et al. Disruption of methylarginine metabolism impairs vascular homeostasis.
Murray-Rust, J. et al. Structural insights into the hydrolysis of cellular nitric oxide synthase inhibitors by dimethylarginine dimethylaminohydrolase. Nat. Struct. Biol. 2001; 8:679–683.
D Nilsson, LGustafsson, A Wackenfors, B Gesslein, et al. Up-regulation of endothelin type B receptors in the human internal mammary artery in culture is dependent on protein kinase C and mitogen-activated kinase signaling pathways. MC Cardiovascular Disorders 2008; 8:21-31. doi:10.1186/1471-2261-8-21. http://www.biomedcentral.com/1471-2261/8/21
GL Volti, S Salomone, V Sorrenti, A Mangiameli, et al. Effect of silibinin on endothelial dysfunction and ADMA levels in obese diabetic mice. Cardiovascular Diabetology 2011, 10:62. http://www.cardiab.com/content/10/1/62
Leiper, J., Murray-Rust, J., McDonald, N. & Vallance, P. S-nitrosylation of dimethylarginine dimethylaminohydrolase regulates enzyme activity: further interactions between nitric oxide synthase and DDAH. Proc. Natl. Acad. Sci. USA 2002; 99: 13527–13532.
R Maas. Pharmacotherapies and their influence on asymmetric dimethylargine (ADMA). Vascular Medicine 2005; 10(2): S49-S57. DOI : 10.1191/ 1358863x05vm605oa
Veli-Pekka Valkonen, Tomi-Pekka Tuomainen, R Laaksonen. DDAH gene and cardiovascular risk. Vascular Medicine 2005; 10: S45–48.
AA Elesber, H Solomon, RJ Lennon, V Mathew, et al. Coronary endothelial dysfunction is associated with erectile dysfunction and elevated asymmetric dimethylarginine in patients with early atherosclerosis. European Heart Journal 2006; 27: 824–831. doi:10.1093/eurheartj/ehi749.
S Yasuda, S Miyazaki, M Kanda, Y Goto, et al. Intensive treatment of risk factors in patients with type-2 diabetes mellitus is associated with improvement of endothelial function coupled with a reduction in the levels of plasma asymmetric dimethylarginine an endogenous inhibitor of nitric oxide synthase. European Heart Journal 2006; 27: 1159–1165. doi:10.1093/ eurheartj/ehi876.
F Markos, BA Hennessy, M Fitzpatrick, J O’Sullivan and HM Snow. The effect of tezosentan, a non-selective endothelin receptor antagonist, on shear stress-induced changes in arterial diameter of the anaesthetized dog. Journal of Physiology 2002; 544(3): 913–918. DOI: 10.1113/jphysiol.2002.030478. http://www.jphysiol.org
M Kayrak; A Bacaksiz; MA Vatankulu, SS Ayhan, et al. Association Between Exaggerated Blood Pressure Response to Exercise and Serum Asymmetric Dimethylarginine Levels. Hypertension and Circulatory Control. Circ J 2010; 74: 1135 – 1141.
C Walther, S Gielen, and R Hambrecht. The effect of exercise training on endothelial function in cardiovascular disease in humans. Exerc Sport Sci Rev 2004; 32(4): 129–134 .
D Abraham, S Selvakumar, DM Baker, and JCS Tsui. Nitric Oxide Manipulation: A Therapeutic Target for Peripheral Arterial Disease? Williams, Xu Shi-Wen, Hindawi Publishing Corporation, Cardiology Research and Practice 2012; Article ID 656247, 7 pages doi:10.1155/2012/656247G . Sidney G. Shaw, Ed.
M Stephan-Gueldner, A Inomata. Coronary arterial lesions induced by endothelin antagonists. Toxicology Letters 2000; 112–113: 531–535.
V Ničković, J Nikolić, N Djindjić, М Ilić, et al. Diagnostic significance of dimethylarginine in the development of hepatorenal syndrome in patients with alcoholic liver cirrhosis. Vojnosanit Pregl 2012; 1-6 .UDC: 616.89-008.441.3-06:[616.36-004-07:616.61-008.6-07DOI: 10.2298/ VSP110728009N.
HMA Eid, H Arnesen, EM Hjerkinn, T Lyberg, et al. Effect of diet and omega-3 fatty acid intervention on asymmetric dimethylarginine. Nutrition & Metabolism 2006; 3:4-14. doi:10.1186/1743-7075-3-4.
B Li, X Xu, X Wang, H Yu, X Li, et al. A Systems Biology Approach to Understanding the Mechanisms of Action of Chinese Herbs for Treatment of Cardiovascular Disease. Int. J. Mol. Sci. 2012; 13: 13501-13520; doi:10.3390/ ijms131013501. ISSN 1422-0067. http://www.mdpi.com/journal/ijms
P Celec. Nuclear factor kappa B—molecular biomedicine: the next generation. Biomedicine & Pharmacotherapy 2004; 58:365–371. http://www.elsevier.com/locate/biopha
C. J. ZHENG, L. Y. HAN, C. W. YAP, Z. L. JI, et al. Therapeutic Targets: Progress of Their Exploration and Investigation of Their Characteristics. Pharmacol Rev 2006; 58:259–279. 0031-6997/06/5802-259–279. http://pharmrev.aspetjournals.org/content/suppl/2006/05/26/58.2.259.D
Lev-Ari, A. Stem cells create new heart cells in baby mice, but not in adults, study shows
Lev-Ari, A. Cardiovascular Disease (CVD) and the Role of agent alternatives in endothelial Nitric Oxide Synthase (eNOS) Activation and Nitric Oxide Production
Lev-Ari, A. Bystolic’s generic Nebivolol – positive effect on circulating Endothelial Progenitor Cells endogenous augmentation
Lev-Ari, A. Macrovascular Disease – Therapeutic Potential of cEPCs: Reduction Methods for CV Risk
Lev-Ari, A. Heart patients’ skin cells turned into healthy heart muscle cells
Lev-Ari, A. Resident-cell-based Therapy in Human Ischaemic Heart Disease: Evolution in the PROMISE of Thymosin beta4 for Cardiac Repair
http://pharmaceuticalintelligence.com/2012/04/30/93/
Nitric Oxide and Sepsis, Hemodynamic Collapse, and the Search for Therapeutic Options
Mediterranean Diet is BEST for patients with established Heart Disorders
Endothelin Receptors in Cardiovascular Diseases: The Role of eNOS Stimulation
Inhibition of ET-1, ETA and ETA-ETB, Induction of NO production, stimulation of eNOS and Treatment Regime with PPAR-gamma agonists (TZD): cEPCs
Endogenous Augmentation for Cardiovascular Risk Reduction – A Bibliography
Reveals from ENCODE project will invite high synergistic collaborations to discover specific targets
Related articles
- Nitric Oxide has a ubiquitous role in the regulation of glycolysis -with a concomitant influence on mitochondrial function (pharmaceuticalintelligence.com)
- Clinical Trials Results for Endothelin System: Pathophysiological role in Chronic Heart Failure, Acute Coronary Syndromes and MI – Marker of Disease Severity or Genetic Determination? (pharmaceuticalintelligence.com)
- Latest Study Provides Evidence Confirming the Mechanism of Action of Vasomedical’s EECP Therapy in Treating Cardiovascular Disease (prweb.com)
- Statins’ nonlipid effects on vascular endothelium through eNOS activation (pharmaceuticalintelligence.com)
- Interaction of Nitric Oxide and Prostacyclin in Vascular Endothelium (pharmaceuticalintelligence.com)
- Nitric oxide: role in Cardiovascular health and disease (pharmaceuticalintelligence.com)
Follow Blog via Email
Join 2,056 other subscribers-
Recent Posts
- Grok thinks like PhDs – Hybrid Model for Autonomous Update of Doctoral Dissertations with Original PhD Student Author editing and acceptance of Grok’s updated output. 3rd Joint article, a pilot study for Validation of an Autonomous Journal Articles Updating System (AJAUS) tested on Autonomous Update of Aviva Lev-Ari, PhD, RN Doctoral Thesis, UB, Berkeley, 1983. January 31, 2026
- 2026 World Economic Forum, 1/19 – 1/23/2026, Davos, Switzerland, LPBI Group’s KOLs interpretation of AI in Health Videos January 24, 2026
- Evolving LPBI Group’s Portfolio of Intellectual Properties (IP): From 2021 Vision to 2026 Reality January 12, 2026
- Aviva Lev-Ari, PhD, RN – Biography ->> Grokepedia Entry January 11, 2026
- List of Articles included in the Article SELECTION from Collection of Aviva Lev-Ari, PhD, RN Scientific Articles on PULSE on LinkedIn.com for Training Small Language Models (SLMs) in Domain-aware Content of Medical, Pharmaceutical, Life Sciences and Healthcare by 15 Subjects Matter January 10, 2026
- Article SELECTION from Collection of Aviva Lev-Ari, PhD, RN Scientific Articles on PULSE on LinkedIn.com for Training Small Language Models (SLMs) in Domain-aware Content of Medical, Pharmaceutical, Life Sciences and Healthcare by 15 Subjects Matter January 10, 2026
- Collection of Aviva Lev-Ari, PhD, RN Scientific Articles on PULSE on LinkedIn.com January 10, 2026
- The youngest leader in AI in Health: Tanishq Mathew Abraham, Ph.D. (@iScienceLuvr) January 8, 2026
- 2026 Grok Multimodal Causal Reasoning on Proprietary Cariovascular Corpus: From 2021 Wolfram NLP Baseline to Thousands of Novel Relationships – A Second Head-to-Head Validation of LPBI’s Domain-Aware Training Advantage January 6, 2026
- Workflow for Dynamic Linkage and Transition between two Authoring Systems: LPBI Group’s WordPress.com Multi-Authors Authoring System and Microsoft PowerPoint product for Slide show presentation – Part 10.1 in Composition of Methods January 6, 2026
Archives
Categories
Meta