Healthcare analytics, AI solutions for biological big data, providing an AI platform for the biotech, life sciences, medical and pharmaceutical industries, as well as for related technological approaches, i.e., curation and text analysis with machine learning and other activities related to AI applications to these industries.
Article 11.2.10 Ultrasound based Screening for Ovarian Cancer
Occasionally, I check for news on ovarian cancer screening. I do that for sentimental reasons; I started the HistoScanning project aiming to develop an effective ultrasound-based screening solution for this cancer.
As awareness for ovarian cancer is highest in the USA, I checked for the latest news on the NCI web-site. I found that to-date: “There is no standard or routine screening test for ovarian cancer. Screening for ovarian cancer has not been proven to decrease the death rate from the disease.
Screening for ovarian cancer is under study and there are screening clinical trials taking place in many parts of the country. Information about ongoing clinical trials is available from the NCI Web site.”
I also found that:
Estimated new cases and deaths from ovarian cancer in the United States in 2013:
New cases: 22,240
Deaths: 14,030
To get an idea on the significance of these numbers, lets compare them to the numbers related to breast cancer:
Estimated new cases and deaths from breast cancer in the United States in 2013:
New cases: 232,340 (female); 2,240 (male)
Deaths: 39,620 (female); 410 (male)
Death rate of ovarian cancer patients is almost 4 times higher than the rate in breast cancer patients!
Therefore, I decided to raise awareness to the results achieved for ovarian HistoScanning in a double-blind multicenter European study that was published in European Radiology three years ago. The gynecologists who recruited patients to this study used standard ultrasound machines of GE-Medical. I would like as well to disclose that I am one of the authors of this paper:
To prospectively assess an innovative computer-aided diagnostic technology that quantifies characteristic features of backscattered ultrasound and theoretically allows transvaginal sonography (TVS) to discriminate benign from malignant adnexal masses.
Methods
Women (n = 264) scheduled for surgical removal of at least one ovary in five centres were included. Preoperative three-dimensional (3D)-TVS was performed and the voxel data were analysed by the new technology. The findings at 3D-TVS, serum CA125 levels and the TVS-based diagnosis were compared with histology. Cancer was deemed present when invasive or borderline cancerous processes were observed histologically.
Results
Among 375 removed ovaries, 141 cancers (83 adenocarcinomas, 24 borderline, 16 cases of carcinomatosis, nine of metastases and nine others) and 234 non-cancerous ovaries (107 normal, 127 benign tumours) were histologically diagnosed. The new computer-aided technology correctly identified 138/141 malignant lesions and 206/234 non-malignant tissues (98% sensitivity, 88% specificity). There were no false-negative results among the 47 FIGO stage I/II ovarian lesions. Standard TVS and CA125 had sensitivities/specificities of 94%/66% and 89%/75%, respectively. Combining standard TVS and the new technology in parallel significantly improved TVS specificity from 66% to 92% (p < 0.0001).
An example of an ovary considered to be normal with TVS.
The same TVS false-negative ovary with OVHS-detected foci of malignancy. The presence of an adenocarcinoma was confirmed histologically.
Conclusions
Computer-aided quantification of backscattered ultrasound is highly sensitive for the diagnosis of malignant ovarian masses.
Personal note:
Based on this study a promising offer for ultrasound-based screening method for ovarian cancer was published in: Int J Gynecol Cancer. 2011 Jan;21(1):35-43. doi: 10.1097/IGC.0b013e3182000528.: Mathematical models to discriminate between benign and malignant adnexal masses: potential diagnostic improvement using ovarian HistoScanning. Vaes E, Manchanda R, Nir R, Nir D, Bleiberg H, Autier P, Menon U, Robert A.
Regrettably, the results of these studies were never transformed into routine clinical products due to financial reasons.
Other research papers related to the management of Prostate cancer were published on this Scientific Web site:
Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing[1]
Curator and Reporter: Stephen J. Williams, Ph.D.
UPDATED 3/11/2023
The updates to this article are an expansion of the original article on 4/10/2013 on tumor heterogeneity in renal cancer, extended to other solid tumor types.
Evaluating Tumor Evolution via Genomic Profiling of Individual Tumor Spheroids in a Malignant Ascites
Epithelial ovarian cancer (EOC) is a silent but mostly lethal gynecologic malignancy. Most patients present with malignant ascites and peritoneal seeding at diagnosis. In the present study, we used a laser-aided isolation technique to investigate the clonal relationship between the primary tumor and tumor spheroids found in the malignant ascites of an EOC patient. Somatic alteration profiles of ovarian cancer-related genes were determined for eight spatially separated samples from primary ovarian tumor tissues and ten tumor spheroids from the malignant ascites using next-generation sequencing. We observed high levels of intra-tumor heterogeneity (ITH) in copy number alterations (CNAs) and single-nucleotide variants (SNVs) in the primary tumor and the tumor spheroids. As a result, we discovered that tumor cells in the primary tissues and the ascites were genetically different lineages. We categorized the CNAs and SNVs into clonal and subclonal alterations according to their distribution among the samples. Also, we identified focal amplifications and deletions in the analyzed samples. For SNVs, a total of 171 somatic mutations were observed, among which 66 were clonal mutations present in both the primary tumor and the ascites, and 61 and 44 of the SNVs were subclonal mutations present in only the primary tumor or the ascites, respectively. Based on the somatic alteration profiles, we constructed phylogenetic trees and inferred the evolutionary history of tumor cells in the patient. The phylogenetic trees constructed using the CNAs and SNVs showed that two branches of the tumor cells diverged early from an ancestral tumor clone during an early metastasis step in the peritoneal cavity. Our data support the monophyletic spread of tumor spheroids in malignant ascites.
Conclusion
In this study, we performed genome-wide sequence analysis of the primary tumor and the associated tumor spheroids in the malignant ascites of an EOC patient. We analyzed genetic heterogeneity in the primary tumor and tumor spheroids through multi-region sequencing and the laser-aided cell isolation technique11. From the sequencing data, we discovered clonal or subclonal somatic CNAs and SNVs, based on which we constructed phylogenetic trees and inferred the evolutionary history of tumor cells in the patient. As a result, we found that the tumor cells in the malignant ascites were an independent lineage from the primary tumor. The phylogenetic analysis showed that the lineage branched before the evolution of the cancer cells at the primary tissues, which suggests that analyzing malignant ascites might be used to detect ovarian cancer or metastasis in the early stage. In summary, the genetic plasticity and similarity between a primary tumor and associated tumor spheroids are still not clear, and yet, the nature of the similarity may have profound implications for both tumor progression and therapeutic outcomes in ovarian cancer. Therefore, future prospective studies profiling the genomic information of primary ovarian tumors, distant metastatic tumors, and tumor spheroids to determine the direction of tumor evolution and metastasis of ovarian cancer are warranted.
Tumor evolution and intratumor heterogeneity of an epithelial ovarian cancer investigated using next-generation sequencing
The extent to which metastatic tumors further evolve by accumulating additional mutations is unclear and has yet to be addressed extensively using next-generation sequencing of high-grade serous ovarian cancer.
Methods
Eleven spatially separated tumor samples from the primary tumor and associated metastatic sites and two normal samples were obtained from a Stage IIIC ovarian cancer patient during cytoreductive surgery prior to chemotherapy. Whole exome sequencing and copy number analysis were performed. Omental exomes were sequenced with a high depth of coverage to thoroughly explore the variants in metastatic lesions. Somatic mutations were further validated by ultra-deep targeted sequencing to sort out false positives and false negatives. Based on the somatic mutations and copy number variation profiles, a phylogenetic tree was generated to explore the evolutionary relationship among tumor samples.
Results
Only 6% of the somatic mutations were present in every sample of a given case with TP53 as the only known mutant gene consistently present in all samples. Two non-spatial clusters of primary tumors (cluster P1 and P2), and a cluster of metastatic regions (cluster M) were identified. The patterns of mutations indicate that cluster P1 and P2 diverged in the early phase of tumorigenesis, and that metastatic cluster M originated from the common ancestral clone of cluster P1 with few somatic mutations and copy number variations.
Conclusions
Although a high level of intratumor heterogeneity was evident in high-grade serous ovarian cancer, our results suggest that transcoelomic metastasis arises with little accumulation of somatic mutations and copy number alterations in this patient.
Discussion
We have shown the range of genomic diversity at nucleotide, copy number and gene expression scales present in six patients with HGSC. These samples were resected prior to treatment, revealing intratumoural diversity in the tumours’ natural evolutionary state. Our results were consistent with recent reports 39 that considerable genomic diversity is measurable in HGSCs at the level of copy number. However, in some cases (cases 3 and 5) we observed large-scale copy number differences in spatially separated samples that were not evident at the level of mutational profiling, indicative of different mutational mechanisms operating independently in different parts of a tumour. Together, copy number and mutational profiling identified well-characterized mutations in genes such as PIK3CA, CTNNB1 and PDGFRB, as well as homozygous deletions in NF1, that were not present in all samples of the same case. With the exception of TP53, our data suggest that well-known, actionable driver mutations may only be partially represented if only a single sample is considered per patient. Our results establish that embedded within the context of extreme intra- and intertumour heterogeneity, TP53 mutation remains the most stable genomic feature in HGSC. However, as efforts towards precision medicine informed by mutational landscapes of tumours reach maturity, it will be imprudent to ignore the degree to which regional genomic variation is present in primary samples prior to any treatment-related selective pressures. Moreover, studies of evolution in cancer through analysis of serial biopsies (eg through pre- and post-treatment samples, or comparisons of primary and metastatic samples), will need to account for evidence of divergent profiles simply due to regional sampling bias.
Mutational profiling revealed for the first time that mutations beyond TP53 are present in FT lesions. In case 4, we used the full spectrum of mutations to establish that evolutionary trajectories of two histologically and karyotypically distinct tumours within a single patient arose from a common aetiology, with 26.2% of all mutations (including TP53) conserved in the tubal lesion. Our results indicate how histologically distinct cell populations in the same patient can be linked by common aetiology in the FT. In contrast to HGSC, endometrioid tumours are thought to develop from atypical ovarian endometriosis, with the FT merely acting as a conduit for endometrial epithelium to spread to the ovary (reviewed in 40). The possibility of the FT playing a causative role in rare cases of endometrioid carcinoma, or mixed endometrioid–serous carcinoma, has not yet been explored. Mutations in case 5 revealed the FT lesion to be a metastatic implant more closely related to a lymph node metastasis than to the ovarian samples. Thus, future investigations into aetiological underpinnings of HGSC will likely benefit from mutational comparison to extra-tubal sites to definitively establish the evolutionary origin of FT lesions.
We noted an extreme case of unexpected genomic conservation. In case 6, samples were obtained at primary surgery and after 42 months, with 21 cycles of multi-agent chemotherapy in between. The two samples exhibited near-identical genomic landscapes. Coupled with the long survivorship of this patient, this observation represents an intriguing anecdote. Larger studies will be needed to establish if stable clonal population structure is a feature of long survivorship.
Finally, as proof of concept, examination of plasma ctDNA suggested that representative mutations in the ancestral clone and mutations present in subclones could be detected in ctDNA, although sensitivity to detect mutations varied from patient to patient. This implies that clinical sensitivity analyses will need to be undertaken to establish the generalizability of ctDNA analysis to clonally diverse tumours.
In summary, our study unveils the extensive genomic diversity in primary, untreated, high-grade serous cancers of the ovary prior to treatment-related selection pressures, and illuminates highly individualized evolutionary trajectories that will require detailed consideration if curative therapeutic strategies are to be achieved.
Updated 3/04/2023 articles on specific tumor type intratumoral heterogeneity
Complex Patterns of Genomic Heterogeneity Identified in 42 Tumor Samples and ctDNA of a Pulmonary Atypical Carcinoid Patient
Source: Tamsin J. Robb, Peter Tsai, Sandra Fitzgerald, Paula Shields, Pascalene S. Houseman, Rachna Patel, Vicky Fan, Ben Curran, Rexson Tse, Jacklyn Ting, Nicole Kramer, Braden J. Woodhouse, Esther Coats, Polona Le Quesne Stabej, Jane Reeve, Kate Parker, Ben Lawrence, Cherie Blenkiron, Cristin G. Print; Complex Patterns of Genomic Heterogeneity Identified in 42 Tumor Samples and ctDNA of a Pulmonary Atypical Carcinoid Patient. Cancer Research Communications 3 January 2023; 3 (1): 31–42. https://doi.org/10.1158/2767-9764.CRC-22-0101
Tumor evolution underlies many challenges facing precision oncology, and improving our understanding has the potential to improve clinical care. This study represents a rare opportunity to study tumor heterogeneity and evolution in a patient with an understudied cancer type. A patient with pulmonary atypical carcinoid, a neuroendocrine tumor, metastatic to 90 sites, requested and consented to donate tissues for research. 42 tumor samples collected at rapid autopsy from 14 anatomically distinct sites were analyzed through DNA whole-exome sequencing and RNA sequencing, and five analyzed through linked-read sequencing. Targeted DNA sequencing was completed on two clinical tissue biopsies and one blood plasma sample. Chromosomal alterations and gene variants accumulated over time, and specific chromosomal alterations preceded the single predicted gene driver variant (ARID1A). At the time of autopsy, all sites shared the gain of one copy of Chr 5, loss of one copy of Chr 6 and 21, chromothripsis of one copy of Chr 11, and 39 small variants. Two tumor clones (carrying additional variants) were detected at metastatic sites, and occasionally in different regions of the same organ (e.g., within the pancreas). Circulating tumor DNA (ctDNA) sequencing detected shared tumor variants in the blood plasma and captured marked genomic heterogeneity, including all metastatic clones but few private tumor variants. This study describes genomic tumor evolution and dissemination of a pulmonary atypical carcinoid donated by a single generous patient. It highlights the critical role of chromosomal alterations in tumor initiation and explores the potential of ctDNA analysis to represent genomically heterogeneous disease. Significance: DNA sequencing data from tumor samples and blood plasma from a single patient highlighted the critical early role of chromosomal alterations in atypical carcinoid tumor development. Common tumor variants were readily detected in the blood plasma, unlike emerging tumor variants, which has implications for using ctDNA to capture cancer evolution.
In this article Robb et. al analyzes the tumor evolution of variants and mutations over time in a single patient. Tumor samples were obtained from liquid biopsy as ctDNA, which is unlike the previous study in renal cancer, where multiple fine needle aspirates needed to be conducted to determine intratumoral heterogeneity with respect to spatial distribution within a solid tumor. In this current study the authors were able to study the evolution of tumor heterogeneity in a single patient.
Introduction
Tumor evolution underlies many of the most pressing challenges facing clinical precision oncology today, and a better understanding of this process has the potential to improve clinical care. We present a unique example of tumor evolution in an uncommon tumor type not previously featured in such studies, a pulmonary atypical carcinoid [an intermediate grade neuroendocrine tumor (NET)].
NETs arise from hormone-producing cells of the neuroendocrine system located throughout the body, and are highly heterogeneous, both genetically and pathologically. While once considered rare tumors (1) and thought to be indolent in nature (2), we now recognize that NETs have an age-adjusted incidence of 6.2 cases per 100,000 in the country where this study was undertaken, New Zealand (3), similar to the incidence of ovarian and cervical cancers (4). Pulmonary atypical carcinoids are mitotically active well-differentiated NETs, with one in five presenting with distant metastatic disease at diagnosis (5). They feature relatively few DNA variants, with recurrent variants occurring in chromatin remodeling genes such as MEN1 and ARID1A [reported in 25% and 10% of studied atypical carcinoids, respectively (6)]. In general, NETs feature few small DNA variants and often instead have large-scale chromosomal changes; for example, around 25% of pancreatic NETs lose a suite of 10 chromosomes, and a further 40% feature the loss of chromosome (Chr) 11 (7). Few genomic studies have been completed on pulmonary atypical carcinoids; however, comparative genomic hybridization studies identified recurrent deletions in Chr 11q (harboring MEN1) in around 60% of cases (8–10).
Tumor development is an evolutionary process with similarities to Darwinian natural selection, where tumor cells may be under multiple simultaneous “selective pressures” (11), including immune attack (12) and drug treatment (13). There are multiple debated models for the generation, propagation, and selection of variants throughout tumor development, including linear, branching, punctuated, and neutral evolution (11, 14), each of which may occur in different tumors or at different timepoints within the same patient. Tumor evolution has been associated with key clinical challenges, including cancer metastasis (15), immune evasion (12, 16), and drug resistance (13, 17, 18).
Sequencing the cell-free DNA (cfDNA) in a patient’s blood plasma to identify ctDNA variants derived from tumor cells has been postulated to better represent the total disease burden (including potential tumor heterogeneity) than single tumor biopsies or resections (19, 20). However, we do not yet fully understand the detectability of ctDNA shed from different anatomic sites around a patient’s body—and early results suggest that some tumor sites may be more easily detected than others (21).
In this study, we completed multimodal genomic analysis on samples collected at autopsy from 41 metastatic tumor sites from a single patient with an uncommon pulmonary atypical carcinoid, alongside a blood plasma sample and clinical biopsies, to catalog driver genomic alterations and hypothesize their order of accumulation. We compared the genomic variants identified in the tumors with those detected in the patient’s blood plasma to consider how well the ctDNA analysis represented the genomic tumor heterogeneity, and in turn, infer metastatic sites that clinical ctDNA assays may poorly detect. A potential limitation, though, as stated by authors
“studies that analyze many tumors from a single patient, while revealing detailed and valuable insights about that patient’s cancer, cannot necessarily be generalized to other patients. Furthermore, all samples except for two clinical biopsies were collected at a single timepoint (at autopsy), limiting the extent of computational tumor evolution inferences that could be made, and the small size of these biopsies limited the scope of genomic assays that could be applied
In their conclusions they allude to the branched evolution that was seen with the renal cancer study:
Models of Tumor Evolution and Metastatic Dissemination
We can hypothesize a putative genomic evolution process (Fig. 7) that is consistent with the genomic heterogeneity observed at autopsy and in the two clinical biopsies. The early lung tumor represented by the diagnostic biopsy featured gain of Chr 5, loss of Chr 21 as well as nine small variants, none of which are known or predicted cancer drivers. Genomic features of the tumors collected at autopsy are consistent with further mutation after the lung biopsy was undertaken, followed by an evolutionary event such as a selective sweep, that fixed into the genomes of subsequent tumors a loss of function variant in ARID1A as well as 29 small non-driver variants and additional chromosomal alterations including Chr 11 chromothripsis and Chr 6 loss. There are plausible selective advantages to the tumor of each, including Chr 6 LOH of the HLA genes responsible for presenting peptides to T cells, hypothetically reducing the immune system’s ability to recognize neoantigens presented by the tumor cells (47). The data are consistent with subsequent branching events, including the accumulation of a second somatic “hit” to EPS8L2 (where the first was somatic LOH on Chr 11), before two parallel lineages developed, metastatic group 1 and 2 (Fig. 7). In metastatic group 1, some variants of unknown significance deserve further study. For instance, truncation of tumor suppressor ETV6 [a common fusion gene partner in breast and thyroid cancer (48)], following the LOH of a region of Chr 12 may be functionally significant given this gene’s role in development (49). The missense SLIT1 variant was the only additional protein-affecting variant carried by tumors in metastatic group 2 and may play a role in angiogenesis and migration (50). Interestingly, the two metastatic groups in the pancreas appear to have arisen by two independent seeding events. However, in the literature there is not overwhelming evidence for consistent genetic drivers of metastatic progression (51, 52) and we have no evidence in our data to suggest the additional genomic variants accumulated by each of the two metastatic groups (e.g., in EPS8L2, ETV6, and SLIT1) were necessary to drive their metastasis. Given a general propensity of cancers to metastasize to lung (53), it is possible that the presence of multiple metastatic groups in the lung may be the result of tertiary metastatic seeding events returning tumor cells that have evolved elsewhere to the site of the lung primary tumor.
4/10/2013
Genomic instability is considered a hallmark and necessary for generating the mutations which drive tumorigenesis. Multiple studies had suggested that there may be multiple driver mutations and a plethora of passenger mutations driving a single tumor. This diversity of mutational spectrum is even noticed in cultured tumor cells (refer to earlier post Genome-Wide Detection of Single-Nucleotide and Copy-Number Variation of a Single Human Cell). Certainly, intratumor heterogeneity has been a concern to clinicians in determining the proper personalized therapy for a given cancer patient, and has been debated if multiple biopsies of a tumor is required to acquire a more complete picture of a tumor’s mutations. In the New England Journal of Medicine, lead author Dr. Marco Gerlinger in the laboratory of Dr. Charles Swanton of the Cancer Research UKLondon Research Institute, and colleagues reported the results of a study to determine if intratumoral differences exist in the mutational spectrum of primary and metastatic renal carcinomas, pre- and post-treatment with the mTOR (mammalian target of rapamycin) inhibitor, everolimus (Afinitor®)[1].
The authors compared exome sequencing of multiregion biopsies from four patients with metastatic renal-cell carcinoma who had been enrolled in the Personalized RNA Interference to Enhance the Delivery of Individualized Cytotoxic and Targeted Therapeutics clinical trial of everolimus (E-PREDICT) before and after cytoreductive surgery.
Biopsies taken:
Multiregion spatial biopsy of primary tumor (representing 9 regions of the tumor)
Chest-wall metastases
Perinephric metastases
Germline DNA as control
Multiple platforms were used to determine aberrations as follows:
A phylogenetic reconstruction of all somatic mutations occurring in primary disease and associated metastases was performed to determine the clonal evolution of the metastatic disease given the underlying heterogeneity of the tumor. Basically the authors wanted to know if the mutational spectra of one metastasis could be found in biopsies taken from the underlying primary tumor or if the mutational landscape of metastases had drastically changed.
Results
Multiregion exon-capture sequencing of DNA from pretreatment biopsy samples of the primary tumor, chest wall metastases, and perinephrous metastasis revealed 128 mutations classified as follows:
40 ubiquitous mutations
59 mutations shared by several but not all regions
29 mutations unique to specific regions
31 mutations shared by most primary tumor regions
28 mutations shared by most metastatic regions
The authors mapped these mutations out with respect to their location, in order to determine how the metastatic lesions evolved from the primary tumor, given the massive heterogeneity in the primary tumor. Construction of this “phylogenetic tree” (see Merlo et. al[2]) showed that the disease evolves in a branched not linear pattern, with one branch of clones evolving into a metastatic disease while another branch of clones and mutations evolve into the primary disease.
One of the major themes of the study is shown by results that an average of 70 somatic mutations were found in a single biopsy (a little more than just half of all tumor mutations) yet only 34% of the mutations in multiregion biopsies were detected in all tumor regions.
This indicated to the authors that “a single biopsy was not representative of the mutational landscape of the entire bulk tumor”. In addition, microarray studies concluded that gene-expression signatures from a single biopsy would not be able to predict outcome.
Everolimus therapy did not change the mutational landscape. Interestingly, allelic composition and ploidy analyses revealed an extensive intratumor heterogeneity, with ploidy heterogeneity in two of four tumors and 26 of 30 tumor samples containing divergent allelic-imbalances. This strengthens the notion that multiple clones with diverse genomic instability exist in various regions of the tumor.
The intratumor heterogeneity reveals a convergent tumor evolution with associated heterogeneity in target function
Genes commonly mutated in clear cell carcinoma[3, 4] (and therefore considered the prevalent driver mutations for renal cancer) include:
Only VHL mutations were found in all regions of a given tumor, however there were three distinct SETD2 mutations (frameshift, splice site, missense) which were located in different regions of the tumor.
SETD2 trimethylates histones at various lysine residues, such as lysine residue 36 (H3K36). The trimethylation of H3K36 is found on many actively transcribed genes. Immunohistochemistry showed trimethylated H3K36 was reduced in cancer cells but positive in most stromal cells and in SETD2 wild-type clear-cell carcinomas.
Interestingly most regions of the primary tumor, except one, contained a kinase-domain activating mutation in mTOR. Immunohistochemistry analysis of downstream target genes of mTOR revealed that mTOR activity was enhanced in regions containing this mutation. Therefore the intratumoral heterogeneity corresponded to therapeutic activity, leading to the impression that a single biopsy may result in inappropriate targeted therapy. Additional downstream biomarkers of activity confirmed both the intratumoral heterogeneity of mutational spectrum as well as an intratumoral heterogeneity of therapeutic-target function.
The authors conclude that “intratumor heterogeneity can lead to underestimation of the tumor genomics landscape from single tumor biopsies and may present major challenges to personalized-medicine and biomarker development”.
In an informal interview with Dr. Swanton, he had stressed the importance of performing these multi-region biopsies and the complications that intratumoral heterogeneity would present for personalized medicine, biomarker development, and chemotherapy resistance.
Q: Your data clearly demonstrates that multiple biopsies must be done to get a more complete picture of the tumor’s mutational landscape. In your study, what percentage of the tumor would be represented by the biopsies you had performed?
Dr. Swanton: Realistically this is a very difficult question to answer, the more biopsies we sequence, the more we find, in the near term it may be very difficult to ever formally address this in large metastatic tumours
Q: You have very nice data which suggest that genetic intratumor heterogeneity complicates the tumor biomarker field? do you feel then that quests for prognostic biomarkers may be impossible to attain?
Dr. Swanton: Not necessarily although heterogeneity is likely to complicate matters
Identifying clonally dominant lesions may provide better drug targets
Predicting resistance events may be difficult given the potential impact of tumour sampling bias and the concern that in some tumours a single biopsy may miss a relevant subclonal mutation that may result in resistance
Q: Were you able to establish the degree of genomic instability among the various biopsies?
Dr. Swanton: Yes, we did this by allelic imbalance analysis and found that the metastases were more genomically unstable than the primary region from which the metastasis derived
Q: I was actually amazed that there was a heterogeneity of mTOR mutations and SETD2 after everolimus therapy? Is it possible these clones obtained a growth advantage?
Dr. Swanton: We think so yes, otherwise we wouldn’t identify recurrent mutations in these “driver genes”
Multiple biopsies of primary tumor and metastases are required to determine the full mutational landscape of a patients tumor
The intratumor heterogeneity will have an impact on the personalized therapy strategy for the clinician
Metastases arising from primary tumor clones will have a greater genomic instability and mutational spectrum than the tumor from which it originates
Tumors and their metastases do NOT evolve in a linear path but have a branched evolution and would complicate biomarker development and the prognostic and resistance outlook for the patient
A great video of Dr. Swanton discussing his research can be viewed here
Generic Name: Everolimus Brand Name: Afinitor Other Designation: RAD001, RAD001C
RAD001, an ester of the macrocytic immunosuppressive agent sirolimus (rapamycin), is an inhibitor of mammalian target of rapamycin (mTOR) kinase.Administration Route: intravenous (IV) • PO
• solid organ transplant
• renal cell carcinoma (RCC), metastatic after failure of treatment with sunitinib, sorafenib, or sunitinib plus sorafenib
• renal cell carcinoma, advanced, refractory to treatment with vascular endothelial growth factor (VEGF)-targeted therapy
• treatment of progressive neuroendocrine tumors (NET) of pancreatic origin (PNET) in patients with inoperable, locally advanced or metastatic disease
AMPK is a member of a metabolite-sensing protein kinase family found in all eukaryotes. It functions as a cellular fuel sensor and its activation strongly suppresses cell proliferation in non-malignant cells and cancer cells. AMPK regulates the cell cycle by upregulating the p53-p21 axis and modulating the TSC2-mTOR (mammalian target of rapamycin) pathway. The AMPK signaling network contains a number of tumor suppressor genes including LKB1, p53, TSC1 and TSC2, and modulates growth factor signaling involving proto-oncogenes including PI3K, Akt and ERK. AMPK activation is therefore therapeutic target for cancer (Motoshima H, etal, J Physiol, 1 Jul 2006; 574(Pt 1): 63–71).AMPK is a protein serine/threonine kinase consisting of a heterotrimeric complex of a catalytic alpha subunit and regulatory ß and gamma subunits. AMPK is activated by increased AMP/ATP ratio, under conditions such as glucose deprivation, hypoxia, ischemia and heat shock. It is also activated by several hormones and cytokines. AMPK inhibits ATP-consuming cellular events, protein synthesis, de novo fatty acid synthesis, and generation of mevalonate and the downstream products in the cholesterol synthesis pathway (Motoshima H, etal, J Physiol, 1 Jul 2006; 574(Pt 1): 63–71).
– ovarian cancer
– brain cancer
– liver cancer
– leukemia
– colon cancer
CREB regulated transcription coactivator 2 (CRTC2)TOR complex 2 (TORC2, mTORC2) • RP11-422P24.6 • transducer of regulated cAMP response element-binding protein (CREB)2 • transducer of CREB protein 2 • TOR1Location: 1q21.3
The mammalian target of rapamycin (mTOR) exists in two complexes, TORC1 and TORC2, which are differentially sensitive to rapamycin. cAMP response element-binding protein (CREB) regulated transcription coactivator 2 (CRTC2) or TORC2 is a multimeric kinase composed of mTOR, mLST8, mSin1, and rictor. The complex is insensitive to acute rapamycin exposure and functions in controlling cell growth and actin cytoskeletal assembly.TORC2 controls gene silencing, telomere length maintenance, and survival under DNA-damaging conditions. It is primaily located in the cytoplasm but also shuttles into the nucleus (Schonbrun M, etal, Mol Cell Biol, Aug 2009;29(16):4584-94).
The alpha subunit of the hypoxia inducible factor 1 (HIF-1alpha) is a 826 amino acid antigen consisting of a basic helix-loop-helix (bHLH)-PAS domain at its N-terminus. HIF-1alpha is rapidly degraded by the proteasome under normal conditions, but is stabilized by hypoxia resulting in the transactivation of several proangiogenic genes. HIF-1alpha is responsible for inducing production of new blood vessels as needed when tumors outgrow existing blood supplies. HIF-1alpha serves as a transcriptional factor that regulates gene expression involved in response to hypoxia and promotes angiogenesis.HIF-1alpha is a proangiogenic transcription factor induced primarily by tumor hypoxia that is critically involved in tumor progression, metastasis and overall tumor survival. HIF-1alpha functions as a survival factor that is required for tumorigenesis in many types of malignancies, and is expressed in a majority of metastases and late-stage tumors. HIF-1alpha is overexpressed in brain, breast, colon, endometrial, head and neck, lung, ovarian, and pancreatic cancer, and is associated with increased microvessel density and/or VEGF expression
– prostate cancer
– bladder cancer
– nasopharyngeal cancer
– head and neck cancer
– kidney cancer
– pancreatic cancer
– endometrial cancer
– breast cancer
Mammalian target of rapamycin (mTOR)FK506 binding protein 12-rapamycin associated protein 1 • RAFT1 • FK506 binding protein 12-rapamycin associated protein 2 • FRAP • FRAP1 • FRAP2 • RAPT1 • FKBP-rapamycin associated protein • FKBP12-rapamycin complex-associated protein 1 • rapamycin target protein • TOR • FLJ44809 • MTORC1 • MTORC2 • RPTOR • RAPTOR • KIAA1303 • mammalian target of rapamycin complex 1Location: 1p36.22
The mammalian target of rapamycin (mTOR) is a large serine/threonine protein (Mr 300,000) having heat repeats, and protein-protein interaction domains at its amino terminus, and a protein kinase domain at its carboxy terminus. mTOR is a member of the phosphoinositide 3-kinase (PI3K)-related kinase (PIKK) family and a central modulator of cell growth. It regulates cell growth, proliferation and survival by impacting on protein synthesis and transcription. mTOR is present in two multi-protein complexes, a rapamycin-sensitive complex, TOR complex 1 (TORC1), defined by the presence of Raptor and a rapamycin insensitive complex, TOR complex 2 (TORC2), with Rictor, Protor and Sin1. Rapamycin selectively inhibits mTORC1 by binding indirectly to the mTOR/Raptor complex via FKBP12, resulting in inhibition of p70S6kinase but not the mTORC2 substrate AKTSer473. Selective inhibition of p70S6K attenuates negative feedback loops to IRS1 and TORC2 resulting in an increase in pAKT which may limit the activity of rapamycin.In a hypoxic environment the increase in mass of solid tumors is dependent on the recruitment of mitogens and nutrients. As a function of nutrient levels, particularly essential amino acids, mTOR acts as a checkpoint for ribosome biogenesis and cell growth. Ribosome biogenesis has long been recognized in the clinics as a predictor of cancer progression; increase in size and number of nucleoli is known to be associated with the most aggressive tumors and a poor prognosis. In bacteria, ribosome biogenesis is independently regulated by amino acids and energy charge. The mTOR pathway is controlled by intracellular ATP levels, independent of amino acids, and mTOR itself is an ATP sensor (Kozma SC, etal, AACR02, Abs. 5628).
– breast cancer
– pancreatic cancer
– multiple myeloma
– liver cancer
– brain cancer
– prostate cancer
– kidney cancer
– lymphoma
Signal transducer and activator of transcription 3 (STAT3) is a member of the STAT protein family. STAT3, plays a critical role in hematopoiesis. STAT3 is located in the cytoplasm and translocated to the nucleus after tyrosine phosphorylation. In response to cytokines and growth and other activation factors, STAT family members are phosphorylated by the receptor associated kinases and then form homo- or heterodimers, which translocate to the cell nucleus where they act as transcription activators.
Sonic hedgehog, a secreted hedgehog ligand, is a human homolog of the Drosophila segment polarity gene hedgehog, cloned by investigators at Harvard University (Marigo V, etal, Genomics, 1 Jul 1995;28 (1):44-51).The mammalian sonic hedgehog (Shh) pathway controls proliferation of granule cell precursors in the cerebellum and is essential for normal embryonic development. Shh signaling is disrupted in a variety of malignancies.
– pancreatic cancer
– CNS cancer
References:
1. Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P et al: Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. The New England journal of medicine 2012, 366(10):883-892.
3. Varela I, Tarpey P, Raine K, Huang D, Ong CK, Stephens P, Davies H, Jones D, Lin ML, Teague J et al: Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma.Nature 2011, 469(7331):539-542.
4. Dalgliesh GL, Furge K, Greenman C, Chen L, Bignell G, Butler A, Davies H, Edkins S, Hardy C, Latimer C et al: Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature 2010, 463(7279):360-363.
Other Articles related to this topic appeared on this Open Access Online Scientific Journal, including the following:
To those of you who did not know, 2013 is the year of the ultrasound: http://www.ultrasound2013.org/. This initiative was launched by AIUM and its objectives:
Raise awareness of the value and benefits of ultrasound among patients, health care providers, and insurers
Provide ultrasound education and evidence-based guidelines for health care providers
Educate insurers about the cost savings and patient benefits associated with performing an ultrasound study when scientific evidence supports its potential effectiveness compared to other imaging modalities
Educate patients about the benefits of ultrasound as the appropriate imaging modality for their care
Encourage the incorporation of ultrasound into medical education
The initiative is designed to call attention to the safe, effective, and affordable advantages of ultrasound as an alternative to other imaging modalities that are more costly and/or emit radiation. For a growing number of clinical conditions, ultrasound has been shown to be equally effective in its diagnostic capability, with a distinct advantage in safety and cost over computed tomography and magnetic resonance imaging. Despite this advantage, evidence suggests that ultrasound is vastly underutilized. Ultrasound First focuses on educating health care workers, medical educators, insurers, and patients of the benefits of ultrasound in medical care. “There is growing support and public awareness for the need to reduce and carefully monitor patients’ exposure to radiation during medical imaging. The use of ultrasound as an alternative imaging modality will help achieve that goal while reducing cost,” states AIUM President Alfred Abuhamad, MD. “Many health care workers and insurers are unacquainted with the range of conditions for which ultrasound has been shown to have superior diagnostic capabilities. Disseminating this knowledge to health care workers and incorporating ultrasound in medical protocols where scientific evidence has shown its diagnostic efficacy will undoubtedly improve patient safety and reduce cost. The time to act is now.”
A primary component of Ultrasound First is providing clinical evidence for the use of ultrasound. To that aim, the Journal of Ultrasound in Medicine has launched a special feature, the Sound Judgment Series, consisting of invited articles highlighting the clinical value of using ultrasound first in specific clinical diagnoses where ultrasound has shown comparative or superior value. Clinical conditions that will be addressed in the series include postmenopausal bleeding, right lower quadrant pain, pelvic pain, right upper quadrant pain, and shoulder pain, among others. This series will serve as an important educational resource for health care workers and educators. On the clinical evidence page one can find reasoning for why ultrasound first. Not much related to cancer diagnosis and management. The only interesting claim is: “Ultrasound-guided surgery: Its use to remove tumors from women who have palpable breast cancer is much more successful than standard surgery in excising all the cancerous tissue while sparing as much healthy tissue as possible.”
The article gives clear presentation of the problem and includes demonstrative pictures:
Figure: Basal cell carcinoma with dermal involvement (transverse view, nasal tip). Grayscale sonography (A) and 3-dimensional reconstruction (B, 5- to 8-second sweep) show a 10.1-mm (wide) × 1.4-mm (deep) well-defined hypoechoic oval lesion (between markers in A and outlined in B) that affects the dermis (d) of the left nasal wing. Notice the hyperechoic spots (arrowheads) within the lesion. The nasal cartilage (c) is unremarkable; asterisk indicates basal cell carcinoma.
Basal cell carcinoma with dermal and subcutaneous involvement (transverse view, frontal region). A, Grayscale sonography shows a 11.4-mm (wide) × 6.6-mm (deep) well-defined oval hypoechoic lesion that involves the dermis (d) and subcutaneous tissue (st). There are hyperechoic spots (arrowheads) within the tumor. B, Color Doppler sonography shows increased vascularity within the tumor (asterisk). C, Three-dimensional sonographic reconstruction (5- to 8-second sweep) highlights the lesion (asterisk, outlined); b indicates bony margin of the skull.
Figure: Pleomorphic presentations of basal cell carcinoma lesions on grayscale sonography (transverse views). Notice the variable shapes of the tumors.
Figure: Frequently, blood flow can be detected within the tumor and its periphery, with slow-flow arteries or veins. The latter vascular data can orient the clinician about the distribution and amount of blood flow that he or she will face during surgery. Despite the fact that basal cell carcinomas usually do not present high vascularity, it should be kept in mind that many of basal cell carcinoma operations are performed in the offices of clinicians and not in the main operating rooms of large hospitals. Nevertheless, the finding of high vascularity within a clinically diagnosed basal cell carcinoma may suggest another type of skin cancer that could occasionally mimic basal cell carcinoma, such as squamous cell carcinoma, Merkel cell carcinoma, or a metastatic tumor. The above figure presents variable degrees of vascularity in basal cell carcinoma lesions going from hypovascular to hypervascular on color and power Doppler sonography (transverse views).
Figure: The depth correlation between sonography (variable frequency) and histologic analysis in facial basal cell carcinoma has been reported to be excellent. Thus, the intraclass correlation coefficient for comparing thickness for the two methods (sonography and histologic analysis) that has been described in literature is 0.9 (intraclass correlation coefficient values ≥0.9 are very good; 0.70–0.89 are good; 0.50–0.69 are moderate; 0.30–049 are mediocre; and ≤0.29 are bad). Two rare sonographic artifacts have been described in basal cell carcinoma. One is the “angled border” that is produced by an inflammatory giant cell reaction underlying the tumor, which may falsely increase the apparent size of the tumor. The other is the “blurry border,” which is produced by large hypertrophy of the sebaceous glands surrounding the lesion. According to the literature, both artifacts can be recognized by a well-trained operator. The figure above presents the sonographic involvement of deeper layers such as the nasal cartilage and orbicularis muscles in the face is of critical importance and may change the decision about the type of surgery. Basal cell carcinoma with nasal cartilage involvement (3-dimensional reconstruction, 5- to 8-second sweep, transverse view, left nasal wing). Notice the extension of the tumor (asterisk, outlined) to the nasal cartilage region (c); d indicates dermis.
Basal cell carcinoma with involvement of the orbicularis muscle of the eyelid (m). Grayscale sonography (transverse view, right lower eyelid) shows that the tumor (asterisk) affects the muscle layer (arrows).
Patients with advanced ovarian cancer had significantly better survival if they took beta-blockers, particularly the noncardioselective agents. Overall, beta-blocker users lived about 6 months longer than did nonusers. However, difference more than doubled to a median survival of almost 8 years in the subgroup of patients treated with nonselective beta-blockers. The findings add to evidence from other types of cancer supporting a beneficial effect of beta-blockers on survival and other outcomes. The improved survival with nonselective beta-blockers suggests a potential for novel therapeutic approaches for epithelial ovarian cancer.”
Recently, studies in several types of cancer have demonstrated improved outcomes, including survival, in patients who used beta-blockers. Several lines of evidence support a biologic rationale for a beneficial effect beta-blockers in cancer. Physiologic changes associated with social isolation, depression, and stress includes increased production of the stress-related hormones norepinephrine and epinephrine, which are targeted by beta-blockers. Increased production of stress hormones has been shown to promote cancer-cell growth, progression and spread in several types of cancer, including ovarian cancer, the investigators noted. With respect to specific effects in ovarian cancer, in vitro studies have shown that epithelial ovarian cancer cells express beta-1 and beta-2 adrenergic receptors. Norepinephrine stimulation of ovarian cancer cells induces vascular endothelial growth factor, matrix metalloproteinases, and cancer-cell growth and invasion. Propranolol, a nonselective beta-blocker, has been shown to inhibit the stimulatory effects of norepinephrine on epithelial ovarian cancer cells.
The effects of beta-blockers was examined on survival in patients with epithelial ovarian cancer treated with chemotherapy. Investigators at five medical centers retrospectively identified patients (median age 61) with stage III or IV ovarian cancer and compared records of patients who had been treated with beta-blockers and those who had not. The analysis included 1,425 patients, including 269 patients whose records documented use of beta-blockers. Nonselective agents accounted for 195 (72%) of the beta-blocker users. More than 90% of beta-blocker users had hypertension, compared with 30% of the 1,158 patients who did not receive the drugs. Demographics, disease stage, and surgical outcomes did not differ significantly between beta-blocker users and nonusers.
Patients who did not use beta-blockers had a median overall survival (OS) of 3.5 years, whereas beta-blocker users had a median OS of 3.98 years (P=0.0365). Subanalyses by type of beta-blocker showed that use of cardioselective agents was associated with a median OS of 3.17 years, whereas users of nonselective agents had a median OS of 7.91 years (P<0.0001 versus nonusers). Beta-blocker users also had superior disease-specific survival (DFS), a median of 48.4 months versus 42.4 months for nonusers (P=0.02). Patients who used nonselective beta-blockers had a median DFS of 90 months versus 38.2 months for patients taking cardioselective agents (P<0.001).
The study provided “provocative information” regarding potential novel therapeutic applications of beta-blockers in the treatment of ovarian cancer. In particular, the findings pertaining to nonselective beta-blockers warrant further study. However, the investigators did not perform a multivariate analysis to identify factors that might have explained the results. There remains a significant risk of selection bias and other confounders that may have accounted for some of the survival differences observed.
Clinical Application of miRNAs Remains a Ways Off When its time comes, prognostic tests will be first.
Patricia Fitzpatrick Dimond, Ph.D GEN Insight & Intelligence
It’s still early to tell how well microRNAs (miRNAs) will prove clinically useful. Preclinical research findings indicate their central role in controlling cellular pathways.
This novel class of nucleotides, about 20–25 nucleotides in length, affects gene expression by interacting with messenger RNAs. But unlike Small Interfering RNA, siRNAs, miRNAs are encoded in the human genome and function as natural regulators of global gene expression.
Each of the more than 1,500 encoded miRNAs appears to regulate the expression of tens to hundreds of different genes, on-off switches, regulating multiple cellular functions including
growth and
proliferation.
miRNAs regulate the translation of genes through
sequence-specific binding to mRNA.
Depending on the degree of sequence complimentarity, they can inhibit
the translation and/or degradation of their target mRNAs.
Because of their role in controlling “suites” of genes and, ultimately, pathway function, these molecules have attracted considerable scientific and investor interest in the control of diseases ranging from cardiovascular diseases to cancer.
miRNAs target numerous biomolecules that play a role in carcinogenesis,
functioning as both tumor promoters or suppressors.
Aberrant expression of miRNAs
correlates with the development and progression of tumors;
inhibition of their expression can
modulate the cancer phenotype,
suggesting their potential as anticancer drug targets.
Further supporting their potential use as drug targets, miRNA expression profiling in a variety of tissue, cell, and disease types has revealed
a “miRNA signature” specific to those cell types or disease states.
they identified a 9-miRNA signature that differentiated invasive (IDC) from in situ carcinoma (DCIS).
In studying the global changes of the miRNA repertoire along the transitions defining breast cancer progression, the scientists found that
let-7d, miR-210, and miR-221 were downregulated in the in situ and
upregulated in the invasive transition, thus
featuring an expression reversal along the cancer progression path.
in addition, miRNAs for overall survival and time to metastasis.
Dr. Croce posed that targeted prognostic tests using miRNA will be available within the next two years.
the problem he suggests is validating the signature in a large enough cohort of patients.
They used deep sequencing, an extremely sensitive approach to the determination of miRNAs because you count the molecules. Studies have used microarrays and RT-PCR, and his group used general microarrays and validated RT-PCR. Their method avoided the possibility of artifacts (by counting). Sequencing permits counts of molecules to provide good data.
MicroRNAs (miRs) are small noncoding RNAs (≈23 nucleotides) that regulate gene expression at a posttranscriptional level by degradation or translational inhibition of target mRNAs. Initially discovered as regulators of development in plants, worms, and fruitflies,
miRs are emerging as
pivotal modulators of cardiovascular biology and disease in mice and men.
Besides a cell-specific transcription factor profile,
cell-specific miR-regulated gene expression is integral to cell fate and activation decisions.
Thus, the cell types involved in
atherosclerosis,
vascular disease, and
its myocardial sequelae may be
differentially regulated by distinct miRs, thereby
controlling highly complex processes
smooth muscle cell phenotype and
inflammatory responses of endothelial cells or macrophages.
The generation of mature miR strands requires several steps of processing of the primary miR gene transcript, including
cleavage of the terminal loop of miR-precursors by the RNase III enzyme,Dicer, to produce miR duplexes.
Although either strand of the miR duplex can be stably associated with an Argonaute (Ago) family protein,
preferential loading of a specific strand (ie, the guide strand) onto the miR-induced silencing complex (RISC) is common.
The strand that is not loaded into the RISC (ie, the passenger strand or miR*) is typically degraded.3 Strand selection may be tissue-specific, and an accumulation observed for both strands implies that
each strand can separately enter the silencing complex.4
Because of the often imperfect complementary binding of the miR seed sequence to the mRNA recognition element,
an individual miR can affect the expression of hundreds of target mRNAs.
If a genome is the blueprint for life, then the chief architects are
tiny slices of genetic material that orchestrate how we are assembled and function.
The study pinpoints the molecular regulators of epigenetics — the process by which unchanging genes along our DNA are switched on and off at precisely right time and place.
“Our genome is like a landscape with lakes, mountains, and rivers, but it is not yet a community or a city full of buildings,” said Haifan Lin, director of the Yale Stem Cell Center and senior author of the study. “What this system does is decide where and when to send out the masons, carpenters, and electricians to build a city or a community.”
In the past 20 years, scientists have discovered that some proteins, called epigenetic factors, traverse the static genome and turn the genes on or off. The staggering number of potential combinations of active and inactive genes explains why a relatively small number of genes can carry out such a wide range of functions.
What guides these epigenetic factors to their target? The answer:
specialized RNAs called piRNAs.
In the latest study, the Yale team discovered that
piRNAs guide epigenetic factors to numerous sites throughout the genome of the fruit fly Drosophila, where
these switches work to turn genes on or off.
The dramatic change in gene expression patterns found illustrated
piRNAs key role in coordinating biological activity.
“This is the first major mechanism discovered that controls where epigenetic factors —the gene switches — are to be placed in the genome,” Lin said.
Several types of cancers appeared to be
triggered when the wrong kinds of piRNAs guide epigenetic factors to activate the wrong genes.
Blocking the action of these piRNAs should become a new opportunity to treat cancers, Lin said.
Xiao A. Huang and Hang Yin of Yale are co-lead authors of the paper.
The research was funded by a National Institutes of Health Pioneer Award to Haifan Lin and a grant from Connecticut Stem Cell Research Fund to
Lin and former Yale professor and co-author Michael Snyder, now of Stanford University.
English: A diagram showing at which stages in the DNA-mRNA-protein pathway expression can be controlled. (Photo credit: Wikipedia)
CRACKING THE CODE OF HUMAN LIFE: Recent Advances in Genomic Analysis and Disease – Part IIC
Author: Larry H. Bernstein, MD, FCAP, Triplex Medical Science
Article 1.4 CRACKING THE CODE OF HUMAN LIFE: Recent Advances in Genomics Analysis and Disease – Part IIC
Part I: The Initiation and Growth of Molecular Biology and Genomics – Part I From Molecular Biology to Translational Medicine: How Far Have We Come, and Where Does It Lead Us?
Part IIB. “CRACKING THE CODE OF HUMAN LIFE: The Birth of BioInformatics & Computational Genomics” lays the manifold multivariate systems analytical tools that has moved the science forward to a groung that ensures clinical application.
Part IIC. “CRACKING THE CODE OF HUMAN LIFE: Recent Advances in Genomic Analysis and Disease “ will extend the discussion to advances in the management of patients as well as providing a roadmap for pharmaceutical drug targeting.
This final paper of Part II concludes a thorough review of the scientific events leading to the discovery of the human genome, the purification and identification of the components of the chromosome and the DNA structure and role in regulation of embryogenesis, and potential targets for cancer.
The first two articles, Part IIA, Part IIB, go into some depth to elucidate the problems and breakthoughs encountered in the Human Genome Project, and the construction of a 3-D model necessary to explain interactions at a distance.
Part IIC, the final article, is entirely concerned with clinical application of this treasure trove of knowledge to resolving diseases of epigenetic nature in the young and the old, chronic inflammatory diseases, autoimmune diseases, infectious disease, gastrointestinal disorders, neurological and neurodegenerative diseases, and cancer.
Recently, large studies have identified some of the genetic basis for important common diseases such as heart disease and diabetes, but most of the genetic contribution to them remains undiscovered. Now researchers at the University of Massachusetts Amherst led by biostatistician Andrea Foulkes have applied sophisticated statistical tools to existing large databases to reveal substantial new information about genes that cause such conditions as high cholesterol linked to heart disease.
Foulkes says, “This new approach to data analysis provides opportunities for developing new treatments.” It also advances approaches
to identifying people at greatest risk for heart disease. Another important point is that our method is straightforward to use with freely
available computer software and can be applied broadly to advance genetic knowledge of many diseases.
The new analytical approach she developed with cardiologist Dr. Muredach Reilly at the University of Pennsylvania and others is called “Mixed modeling of Meta-Analysis P-values” or MixMAP. Because it makes use of existing public databases, the powerful new method
represents a low-cost tool for investigators.
MixMAP draws on a principled statistical modeling framework and the vast array of summary data now available from genetic association
studies to formally test at a new, locus-level, association.
While that traditional statistical method looks for one unusual “needle in a haystack” as a possible disease signal, Foulkes and colleagues’
new method uses knowledge of DNA regions in the genome that are likely to
contain several genetic signals for disease variation clumped together in one region.
Thus, it is able to detect groups of unusual variants rather than just single SNPs, offering a way to “call out” gene
regions that have a consistent signal above normal variation.
The LPA gene codes for apolipoprotein(a), which, when linked with low-density lipoprotein particles, forms lipoprotein(a) [Lp(a)] —
a well-studied molecule associated with coronary artery disease (CAD). The Lp(a) molecule has both atherogenic and thrombogenic effects in vitro , but the extent to which these translate to differences in how atherothrombotic disease presents is unknown.
LPA contains many single-nucleotide polymorphisms, and 2 have been identified by previous groups as being strongly associated with
levels of Lp(a) and, as a consequence, strongly associated with CAD.
However, because atherosclerosis is thought to be a systemic disease, it is unclear to what extent Lp(a) leads to atherosclerosis in other arterial beds (eg, carotid, abdominal aorta, and lower extremity),
as well as to other thrombotic disorders (eg, ischemic/cardioembolic stroke and venous thromboembolism).
Such distinctions are important, because therapies that might lower Lp(a) could potentially reduce forms of atherosclerosis beyond the coronary tree.
To answer this question, Helgadottir and colleagues compiled clinical and genetic data on the LPA gene from thousands of previous
participants in genetic research studies from across the world. They did not have access to Lp(a) levels, but by knowing the genotypes for
2 LPA variants, they inferred the levels of Lp(a) on the basis of prior associations between these variants and Lp(a) levels. [1]
Their studies included not only individuals of white European descent but also a significant proportion of black persons, in order to
widen the generalizability of their results.
Their main findings are that LPA variants (and, by proxy, Lp(a) levels) are associated with
CAD,
peripheral arterial disease,
abdominal aortic aneurysm,
number of CAD vessels,
age at onset of CAD diagnosis, and
large-artery atherosclerosis-type stroke.
They did not find an association with
cardioembolic or small-vessel disease-type stroke;
intracranial aneurysm;
venous thrombosis;
carotid intima thickness; or,
in a small subset of individuals, myocardial infarction.
English: Structure of the LPA protein. Based on PyMOL rendering of PDB 1i71. (Photo credit: Wikipedia)
Micrograph of an artery that supplies the heart with significant atherosclerosis and marked luminal narrowing. Tissue has been stained using Masson’s trichrome. (Photo credit: Wikipedia)
Scientists at the Gladstone Institutes have revealed the precise order and timing of hundreds of genetic “switches” required to construct a fully
functional heart from embryonic heart cells — providing new clues into the genetic basis for some forms of congenital heart disease.
In a study being published online today in the journal Cell, researchers in the laboratory of Gladstone Senior Investigator Benoit Bruneau, PhD,
employed stem cell technology, next-generation DNA sequencing and computing tools to piece together the instruction manual, or “genomic
blueprint” for how a heart becomes a heart. These findings offer renewed hope for combating life-threatening heart defects such as arrhythmias (irregular heart beat) and ventricular septal defects (“holes in the heart”).
They approach heart formation with a wide-angle lens by
looking at the entirety of the genetic material that gives heart cells their unique identity.
The news comes at a time of emerging importance for the biological process called “epigenetics,” in which a non-genetic factor impacts a cell’s genetic
makeup early during development — but sometimes with longer-term consequences. All of the cells in an organism contain the same DNA, but the
epigenetic instructions encoded in specific DNA sequences give the cell its identity. Epigenetics is of particular interest in heart formation, as the
incorrect on-and-off switching of genes during fetal development can lead to congenital heart disease — some forms of which may not be apparent until adulthood.
the scientists took embryonic stem cells from mice and reprogrammed them into beating heart cells by mimicking embryonic development in a petri dish. Next, they extracted the DNA from developing and mature heart cells, using an advanced gene-sequencing technique called ChIP-seq that lets scientists “see” the epigenetic signatures written in the DNA.
Map of Heart Disease Death Rates in US White Males from 2000-2004 (Photo credit: Wikipedia)
Estimated propability of death or non-fatal myocardial-infarction over one year corresponding ti selectet values of the individual scores. Ordinate: individual score, abscissa: Propability of death or non-fatal myocardial infarction in 1 year (in %) (Photo credit: Wikipedia)
simply finding these signatures was only half the battle — we next had to decipher which aspects of heart formation they encoded
To do that, we harnessed the computing power of the Gladstone Bioinformatics Core. This allowed us to take the mountains of data collected from
gene sequencing and organize it into a readable, meaningful blueprint for how a heart becomes a heart.”
For each of the above datasets, an upstream analysis from the identified transcription factors correctly identified the stimulus. IPA’s tools were very
easy to use and the
analysis time for the above experiments was less than one minute.
The performance, speed, and ease of use can only be characterized as very good, perhaps leading to breakthroughs when extended and used creatively. Ingenuity’s new transcription factor analysis tool in IPA, coupled with Ingenuity’s established upstream grow tools, should be strongly considered for every lab analyzing differential expression data.
NF-E2-related factor 2 (Nrf2) is an important transcription factor that
activates the expression of cellular detoxifying enzymes.
Nrf2 expression is largely regulated through the association of Nrf2 with Kelch-like ECH-associated protein 1 (Keap1), which
results in cytoplasmic Nrf2 degradation.
Conversely, little is known concerning the regulation of Keap1 expression. Until now, a regulatory role for microRNAs (miRs) in controlling Keap1 gene expression had not been characterized. By using miR array-
based screening, we observed miR-200a silencing in breast cancer cells and
demonstrated that upon re-expression, miR-200a
targets the Keap1 3′-untranslated region (3′-UTR), leading to Keap1 mRNA degradation. Loss of this regulatory mechanism may
contribute to the dysregulation of Nrf2 activity in breast cancer. Previously, we have identified epigenetic repression of miR-200a
in breast cancer cells. Here, we find that treatment with epigenetic therapy, the histone deacetylase inhibitor suberoylanilide hydroxamic acid, restored miR-200a expression and reduced Keap1 levels. This reduction in Keap1 levels corresponded with
Nrf2 nuclear translocation
and activation of Nrf2-dependent NAD(P)H-quinone oxidoreductase 1 (NQO1) gene transcription.
Moreover, we found that Nrf2 activation inhibited the anchorage-independent growth of breast cancer cells. Finally, our in vitro observations were confirmed in a model of carcinogen-induced mammary hyperplasia in vivo. In conclusion, our study demonstrates
that miR-200a regulates the Keap1/Nrf2 pathway in mammary epithelium, and we find that epigenetic therapy can restore miR-200a
regulation of Keap1 expression,
reactivating the Nrf2-dependent antioxidant pathway in breast cancer.
Nuclear factor-like 2 (erythroid-derived 2, also known as NFE2L2 or Nrf2, is a transcription factor that in humans is encoded by the NFE2L2 gene.[1]) NFE2L2 induces the expression of various genes including those that encode for several antioxidant enzymes, and it may play a physiological role in the regulation of oxidative stress. Investigational drugs that target NFE2L2 are of interest as potential therapeutic interventions for
oxidative-stress related pathologies.
4. Highly active zinc finger nucleases by extended modular assembly
Zinc finger nucleases (ZFNs) are important tools for genome engineering. Despite intense interest by many academic groups,
the lack of robust non-commercial methods has hindered their widespread use. The modular assembly (MA) of ZFNs from
publicly-available one-finger archives provides a rapid method to create proteins that can recognize a very broad spectrum of DNA sequences.
However, three- and four-finger arrays often fail to produce active nucleases. Efforts to improve the specificity of the one-finger archives have not increased the success rate above 25%, suggesting that the MA method might
be inherently inefficient due to its insensitivity to context-dependent effects.
Here we present the first systematic study on the effect of array length on ZFN activity. ZFNs composed of six-finger MA arrays produced mutations at 15 of 21 (71%) targeted
loci in human and mouse cells. A novel Drop-Out Linker scheme was used to rapidly assess three- to six-finger combinations,
demonstrating that shorter arrays could improve activity in some cases. Analysis of 268 array variants revealed that half of
MA ZFNs of any array composition that exceed an ab initio
B-score cut-off of 15 were active.
MA ZFNs are able to target more DNA sequences with higher success rates than other methods.
These insightful reviews are based on the strategic data and insights from Thomson Reuters Cortellis™ for Competitive Intelligence. (A Review of April-June 2012).
The majority of diseases are complex and multi-factorial, involving multiple genes interacting with environmental factors. At the genetic level,
information from genome-wide association studies that elucidate common patterns of genetic variation across various human populations,
in addition to profiling, technologies can be utilized in discovery research to provide snapshots of genes and expression profiles that are controlled
by the same regulatory mechanism and are altered between healthy and diseased states.
The characterization of genes that are abnormally expressed in disease tissues could further be employed as
diagnostic markers,
prognostic indicators of efficacy and/or toxicity, or as
targets for therapeutic intervention.
As the defining catalyst that exponentially paved the way for personalized medicine, information from the published genome sequence revealed that much of the genetic variations in humans are concentrated in about 0.1 percent of the over 3 billion base pairs in the haploid DNA. Most of these variations involve substitution of a single nucleotide for another at a given location in the genetic sequence, known as single nucleotide polymorphism (SNP).
Combinations of linked SNPs aggregate together to form haplotypes and
together these serve as markers for locating genetic variations in DNA sequences.
SNPs located within the protein-coding region of a gene or within the control regions of DNA that regulate a gene’s activity could
have a substantial effect on the encoded protein and thus influence phenotypic outcomes.
Analyzing SNPs between patient population cohorts could highlight specific genotypic variations which can be correlated with specific phenotypic variations in disease predisposition and drug responses.
Prior to the genomic revolution, many of the established therapies were directed against less than 500 drug targets, with many of the top selling drugs acting on well defined protein pathways. However, the sequencing of the human genome has massively expanded the pool of molecular targets that could be exploited in unmet medical needs and currently, of the approximately 22,300 protein-coding genes in the human code, it has been estimated that up to 3000 are druggable. Furthermore, genomic technologies such as
high-throughput sequencing
and transcription profiling,
can be used to identify and validate biologically relevant target molecules, or can be applied to cell-based and mice disease models or directly to in vivo human tissues,
helping to correlate gene targets with phenotypic traits of complex diseases.
This is particularly important, as
insufficient validation of target gene/proteins in complex diseases may be a contributing factor in the decline in R&D productivity.
Personalized medicine no doubt is already having a tremendous impact on drug development pipelines. According to a study conducted by the Tufts Center for the Study of Drug Development, more than 90 percent of biopharmaceutical companies now utilize at least some
genomics-derived targets in their drug discovery programs.
However, pipeline analysis from Cortellis for Competitive Intelligence suggests that there is still a scientific gap that has resulted in difficulty optimizing these novel genomic targets into the clinical R&D portfolios of major pharmaceutical companies, particularly outside the oncology field. Selected examples of personalized medicine product candidates in clinical development include (see TABLE 4).
Mutations in Melanomaare in regions that control genes, not in the genes themselves. The mutations are exactly the type caused by exposure to ultraviolet light. The findings are reported in two papers in http://Science.com/ScienceExpress/
The findings do not suggest new treatments, but they help explain how melanomas – and possibly – other cancers – develop and what drives their growth. This is a modification found in the “dark matter”, according to Dr. Levi A. Garraway, the 99 percent of DNA in a region that regulates genes. A small control region was mutated in 7 out of 10 of the tumors, commonly of one or two tiny changes. A German Team led by Rajiv Kumar (Heidelberg) and Dirk Schadendorf (Essen) looked at a family whose members tended to get melanomas. Their findings indicate that those inherited with the mutations might be born with cells that have taken the first step toward cancer.
The mutations spur cells to make telomerase, that keeps the cells immortal by preventing them from losing the ends of their chromosome, the telomere. Abundant telomerase occurs in 90 percent of cancers, according to Immaculata De Vivo at Harvard Medical School.
The importance of the findings is that the mechanism of telomerase involvement in cancer is now within view. But it is not clear how to block the telomerase production in cancer cells.
A slight mutation in the matched nucleotides can lead to chromosomal aberrations and unintentional genetic rearrangement. (Photo credit: Wikipedia)
Comment
This discussion addresses the issues raised about the direction to follow in personalized medicine. Despite the amount of work necessary to bring the clarity that is sought after, the experiments and experimental design is most essential.
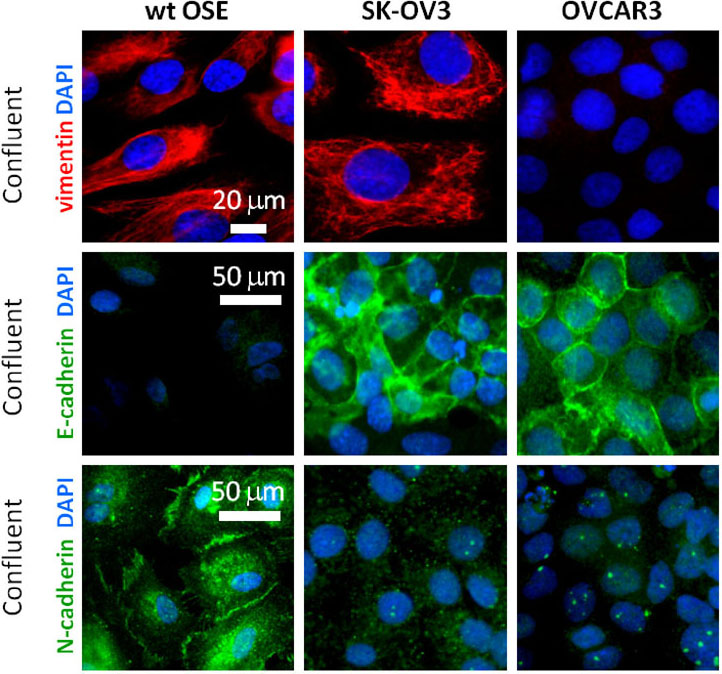
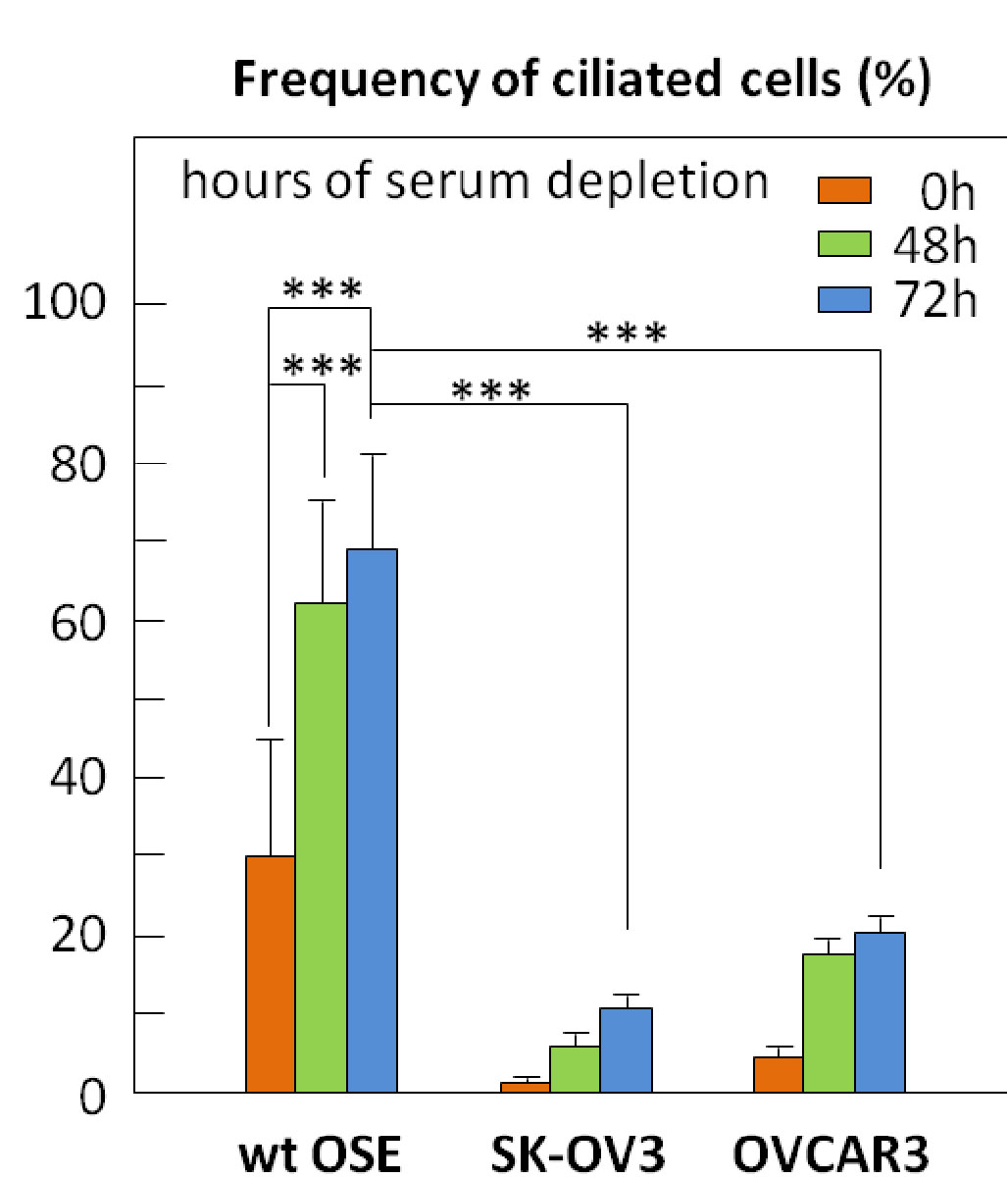
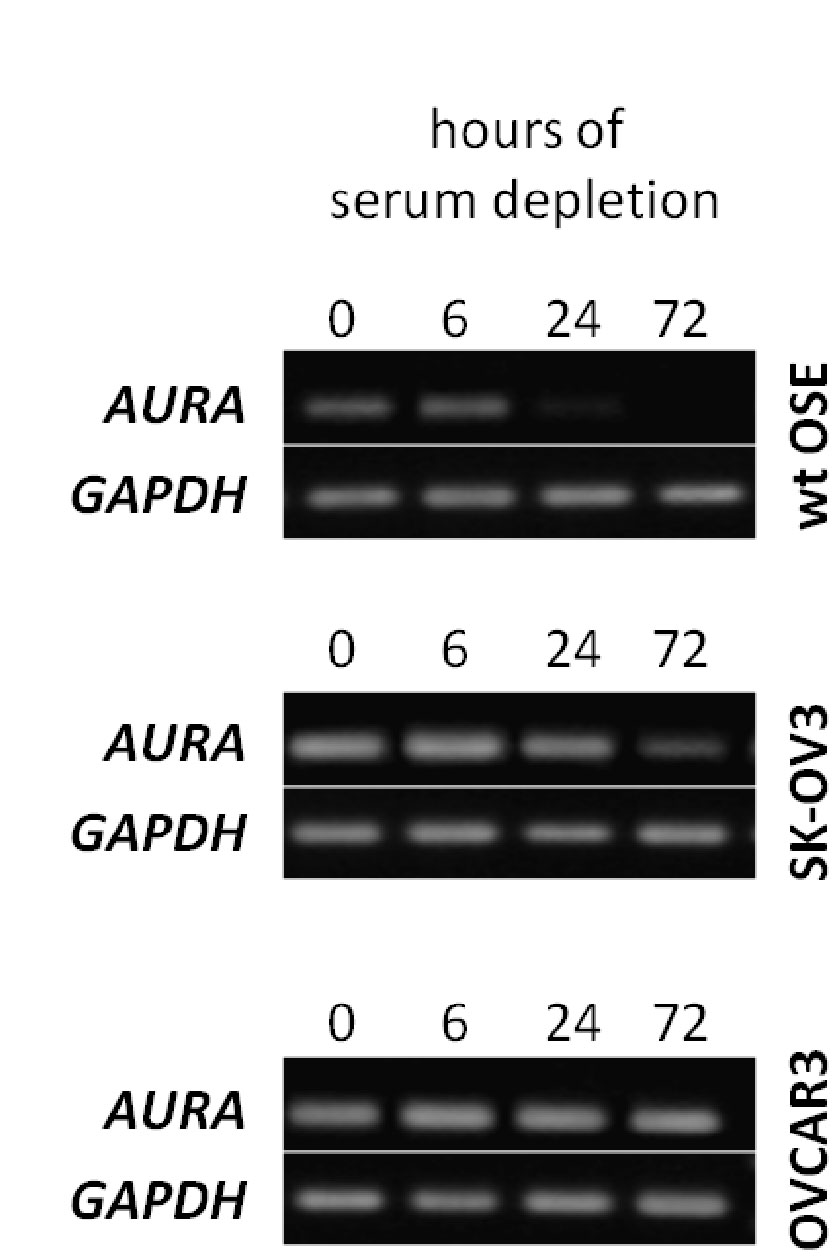
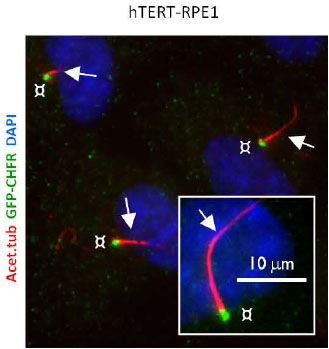
The arrest of ciliogenesis in ovarian cancer cell lines compared to wild type (WT) ovarian epithelial cells, and
The link to suppressing ciliogenesis by AURA protein and CHFR at the base of the cilium, which disappears at mitosis or with proliferation.
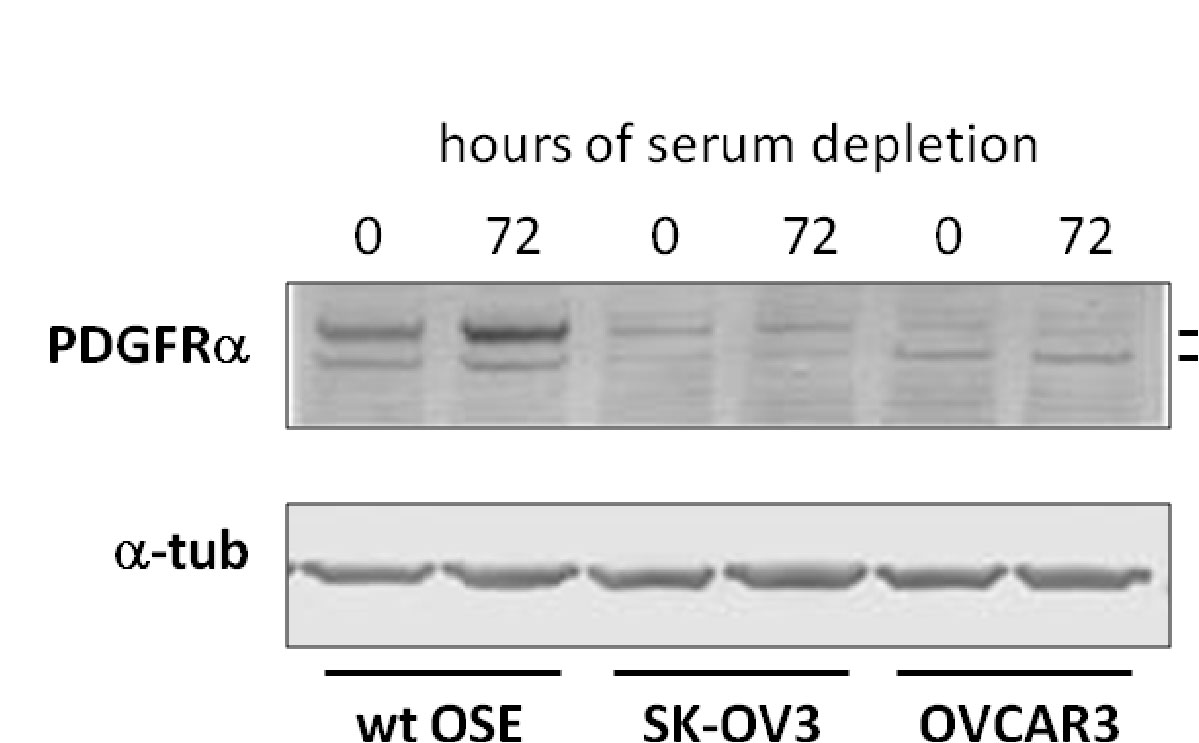
There is no accumulation by upregulation of PDGF under starvation by the cancer cells compared to the effect in WT OSE.
Here we have a systematic combination of signaling events tied to changes in putative biomarkers that occur synchronously in Ov cancer cell lines.
These changes are identified with changes in
proliferation,
loss of ciliary structure, and
proliferation.
In this described scenario,
WT OSE cells would be arrested, and
it appears that they would take the path to apoptosis (under starvation).
Even without more information, this cluster is what one wants to have in a “syndromic classification”. The information used to form the classification entails the identification of strong‘signaling-related’ biomarkers. The Gli2 peptide has to be part of this.
In principle, a syndromic classification would be ideally expected to have no less than 64 classes. If the classification is “weak”, then the class frequencies would be close to what one would expect in the WT OSE. In this case, in reality,
several combinatorial classes would have low frequency, and
others would be quite high.
This obeys the classification rules established by feature identification, and the information gain described by Solomon Kullback and extended by Akaike.
Does this have to be the case for all different cancer types? I don’t think so. The cells are different in ontogenesis. In this case, even the WT OSE have mesenchymal features and so, are not fully directed to epithelial expression. This happens to be the case in actual anatomic expression of the ovary. On the other hand, one would expect shared features of the
ovary,
testes,
thyroid,
adrenals, and
pituitary.
There is biochemical expression in terms of their synthetic function – TPN organs. I would have to put the liver into that broad class. Other organs – skeletal muscle & heart – transform substrate into energy or work. (Where you might also put intestinal smooth muscle).
They have to have different biomarker expressions, even though they much less often don’t form neoplasms. (Bone is not just a bioenergetic force. It is maintained by muscle action. It forms sarcomas. But there has to be a balance between bone removal by osteoclasts and refill by osteoblasts.)
Viewpoint: What we have learned
The Watson-Crick model proposed in 1953 is limited for explaining fully genome effects
The Pauling triplex model may have been prescient because of a more full anticipation of molecular bonding variants
A more adequate triple-helix model has been proposed and is consistent with a compact genome in the nucleus
The structure of the genome is not as we assumed – based on the application of Fractal Geometry. Current body of evidence is building that can reveal a more complete view of genome function.
transcription
cell regulation
mutations
Summary
I have just completed a most comprehensive review of the Human Genome Project. There are key research collaborations, problems in deciphering the underlying structure of the genome, and there are also both obstacles and insights to elucidating the complexity of the final model.
This is because of frequent observations of molecular problems in folding and other interactions between nucleotides that challenge the sufficiency of the original DNA model proposed by Watson and Crick. This has come about because of breakthrough innovation in technology and in computational methods.
Radoslav Bozov •
Molecular biology and growth was primarily initiated on biochemical structural paradigms aiming to define functional spatial dynamics of molecules via assignation of various types of bondings – covalent and non-covalent – hydrogen, ionic , dipole-dipole, hydrophobic interactions.
Lab techniques based on z/m paradigm allowed separation, isolation and identification of bio substances with a general marker identity finding correlation between physiological/cellular states.
The development of electronic/x-ray technologies allowed zooming in nano space without capturing time.
NMR technology identified the existence of space topology of initial and final atomic states giving a highly limited light on time – energy axis of atomic interactions.
Sequence technology and genomic perturbations shed light on uncertainty of genomic dynamics and regulators of functional ever expanding networks.
Transition state theory coupled to structural complexity identification and enzymatic mechanisms ran up parallel to work on various phenomena of strings of nucleotides (oligomers and polymers) – illusion/observation of constructing models on the dynamics of protein-dna-rna interference.
The physical energetic constrains of biochemistry were inapplicable in open biological systems. Biologists have accepted observation as a sole driver towards re-evaluating models.
The separation of matter and time constrains emerged as deviation of energy and space constrains transforming into the full acceptance of code theory of life. One simple thing was left unnoticed over time –
the amount of information of quantum matter within a single codon is larger than that of a single amino acid. This violated all physical laws/principles known to work with a limited degree of certainty.
The limited amount of information analyzed by conventional sequence identity led to the notion of applicability of statistical measures of and PCR technology. Mutations were identified over larger scale of data.
Quantum chemistry itself is being limited due discrete space/energy constrains, thus it transformed into concepts/principles in biology that possess highly limited physical values whatsoever.
The central dogma is partially broken as a result of
regulatory constrains
epigenetic phenomena and
iRNA.
Large scale code computational data run into uncertainty of the processes of evolution and its consequence of signaling transformation. All drugs were ‘lucky based’ applicability and/or discovery with largely unpredictable side effect over time.
Other Related articles on this Open Access Online Sceintific Journal include the following:
Article 2.3 Genome-Wide Detection of Single-Nucleotide and Copy-Number Variation of a Single Human Cell
Most tumors exhibit a level of diversity, at the cellular, histologic, and even genetic level (2). This genetic heterogeneity within a tumor has been a focus of recent research efforts to analyze the characteristics, expression patterns, and genetic differences between individual tumor cells. This genetic diversity is usually manifested as single nucleotide variations (SNV) and copy number variations (CNV), both of which provide selection pressures in both cancer and evolution.
As cancer research and personalized medicine is focused on analyzing this tumor heterogeneity it has become pertinent view the tumor as a heterogeneous population of cells instead of as a homogenous mass. In, fact, studies have suggested that cancer cell lines growing on plastic in culture, even though thought of as clonogenic, can actually display a varied degree of expression differences between neighboring cells growing on the same dish. Indeed, cancer stem cells show an asynchronous cell division, for example a parent CD133-positive cell will divide into a CD133-positive and a CD133-negative cell(3). In addition, the discovery that circulating tumor cells (a rare population of circulating cells in the blood) can be prognostic of outcome in cancer such as inflammatory breast cancer(4), it is ever more important to develop methods to analyze single cell populations.
Harvard University researchers, Dr. Chenghang Zong, Sijia Lu, Alec Chapman and Sunney Xie developed a new amplification method utilizing multiple annealing and looping-based amplification cycles (MALBAC)(1). A quasilinear preamplification process is used on pictograms of DNA genomic fragments (form 10 to 100 kb) isolated from a single cell. This is performed to reduce the bias associated with nonlinear DNA amplification. A series of random primers (which the authors termed MALBAC primers, constructed with a common sequence tags) are annealed at low temperature (0 °C). PCR rounds produce semiamplicons. Further rounds of amplification, after a step of looping the amplicons, result in full amplicons with complementary ends. When the two ends hybridize to form the looped DNA, this prevents use of this loop structure as a template, therefore leading to a close-to–linear amplification. The process allows for a higher fidelity of DNA replication and the ability to amplify a whole genome. The amplicons are then sequenced either by whole-genome sequencing methods using Sanger-sequencing to verify any single nucleotide polymorphisms. This procedure of MALBAC-amplification resulted in coverage of 85-93% of the genome of a single cell.
As proof of principle, the authors used MALBAC to amplify the DNA of single SW480 cancer cells (picked from a clonally expanded population of a heterogeneous population (the bulk DNA). Comparison of the MALBAC method versus the MDA method revealed copy number variations (CNV) between three individual cells, which had been picked from the clonally expanded pool. Their results were in agreement with karyotyping studies on the SW480 cell line. Meticulous quality controls were performed to limit contamination, high false positive rates of SNV detection due to amplification bias, and false positives due to amplification or sequencing errors.
Interestingly, the authors found 35 unique single nucleotide variations which h had occurred from 20 cell divisions from a single SW480 cancer cell. This resulted in an estimated 49 mutations which occurred in 20 generations, yielding a mutation rate of 2.5 nucleotides per generation. In addition, the authors were able to map some of these mutations on various chromosomes and perform next-gen sequencing (deep sequencing) to verify the nucleotide mutations and found an unusually high purine-pyrimidine exchange rate.
In a subsequent paper, investigators from the same group at Harvard used this technology to sequence 99 sperm cells from a single individual to study genetic diversity created during meiotic recombination, a mechanism involved in evolution and development(5).
2. Cooke, S. L., Temple, J., Macarthur, S., Zahra, M. A., Tan, L. T., Crawford, R. A., Ng, C. K., Jimenez-Linan, M., Sala, E., and Brenton, J. D. (2011) British journal of cancer104, 361-368
4. Giuliano, M., Giordano, A., Jackson, S., Hess, K. R., De Giorgi, U., Mego, M., Handy, B. C., Ueno, N. T., Alvarez, R. H., De Laurentiis, M., De Placido, S., Valero, V., Hortobagyi, G. N., Reuben, J. M., and Cristofanilli, M. (2011) Breast cancer research : BCR13, R67
5. Lu, S., Zong, C., Fan, W., Yang, M., Li, J., Chapman, A. R., Zhu, P., Hu, X., Xu, L., Yan, L., Bai, F., Qiao, J., Tang, F., Li, R., and Xie, X. S. (2012) Science338, 1627-1630
Other related posts on this website regarding Cancer and Genomics include:
Ovarian Cancer and fluorescence-guided surgery: A report
Author, Editor: Tilda Barliya PhD
Article 11.4.6 Ovarian Cancer and fluorescence guided surgery A report
Surgery is being commonly used to diagnose, treat and even help prevent cancer. In which the surgeon will cut into the body to remove the cancer along with some surrounding healthy tissue to ensure that all of the cancer is removed. However distinguishing cancer cells from healthy ones during surgery can prove difficult, if not impossible. Sometimes lesions are detected only postoperatively, leading to more surgery down the line. Currently, surgeons rely on vision and touch to detect tumors during surgery but in many cases there is still no good way to determine a tumor’s margins.
In recent years, major progression has been made in imaging-guided surgery and doctors believe that use of fluorescent dye could boost survival rates by guiding them to tiny clusters of malignant cells.
The first fluorescence-guided surgery in ovarian cancer patients have yielded great results and are summarized herein.
Dr. Phillip Low, a Ralph C. Corely Distinguished Professor of Chemistry from Purdue University has invented a fluorescent imaging agent to a modified form of the vitamin folic acid, which acts as a “homing device” to seek out and attach to ovarian cancer cells (1)
” Of all gynecologic malignancies, epithelial ovarian cancer (EOC) is the most frequent cause of death, both in the United States and in Europe. The relative absence of a clear, distinctive clinical presentation in early stages, combined with the lack of a screening tool, often results in the disease being diagnosed only at more advanced stages. The overall 5-year survival rate is 45%, and for stages III and IV it is only 20–25%.” Cytoreduction surgery followed by chemotherapy is considereed the most effective treatment. Radiologic approaches such as X-ray, CT, MRI and ultrasound have been considered for use in assisting surgical procedures, but these are not tumor specific and generally are not useful for intraoperative applications. Therefore, a better tumor-specific detection strategy may drastically improve the patient survival.
The overexpression of folate receptor-α (FR-α) in 90–95% of epithelial ovarian cancers prompted the investigation of intraoperative tumor-specific fluorescence imaging in ovarian cancer surgery using an FR-α–targeted fluorescent agent. Moreover, the absence of FR-α on healthy cells leads to high tumor-to-normal ratios.
As a ligand of FR-α, folate has already been conjugated to DTPA for SPECT/CT imaging and to several PET tracers. It has also been linked to fluorescein for use in imaging metastatic disease in murine tumor models, although this was never tested in humans.
In this article, the authors have conjugated the folate to fluorescein isothiocyanate (FITC) for the use in surgery together with a real-time multispectral intraoperative fluorescence imaging system.
The authors have conducted the first clinical trial using the fluorescence-guided surgery in ovarian cancer patient. Described herein:
Tumor-specific fluorescent agent:
Targeting of the FR-α in ovarian cancer in patients, the imaging agent was produced at clinical grade according to GMP conditions by Endocyte Inc. Folate hapten (vitamin B9) was conjugated with fluorescein isothiocyanate (FITC), yielding folate-FITC (See Fitgure). Folate-FITC has an excitation wavelength of 495 nm and emits light at 520 nm. The conjugate has a very high sensitivity and clusters of cancer cells as small as one-tenth of a millimeter can be detected, as opposed to the earlier average minimal cluster size of 3 millimeters in diameter based on current methods of visual and tactile detection.
Folate-FITC was dissolved in 10 ml sterile normal saline and injected at a dose of 0.3 mg per kg body weight over a period of 10 min and was injected 2 hrs prior to the surgery.
Patients:
10 patients with different stages of the over cancer were recruited, The mean age of all patients was 61.2 ± 11.4 (mean ± s.d.). Four patients were diagnosed with a malignant epithelial ovarian tumor (two serous carcinomas, one undifferentiated carcinoma and one mucinous carcinoma) and one patient with a serous borderline tumor. Five patients were diagnosed with a benign ovarian tumor, as confirmed by histopathology: two fibrothecomas, one cellular fibroma, one cystic teratoma and one benign multicystic ischemic ovary.
Multispectral fluorescence camera system:
The camera system (developed by the Technical University Munich/Helmholtz Center) consists of a charge-coupled digital (EM-CCD) camera (Andor Technology) for sensitive fluorescence detection and two separate cameras for detection of intrinsic fluorescence and color (PCO AG). The system is controlled by a synchronized multi-CPU computer system (Dell Computer) for simultaneous processing of raw data and image registration and rendering. The system allows color imaging and simultaneous sensitive fluorescence detection in the visible light spectrum, as appropriate for FITC imaging. Surgery and imaging procedure are described in detail in the article (1). Shortly, a live imaging during surgery enabled the surgeon to locate the tumor and remove it, biopsy was taken for further histopathology.
Results:
Fluorescence was detectable intraoperatively in all patients with a malignant tumor and FR-αexpression but was absent in the patient with a malignant tumor but no FR-α expression and in those with benign tumors (Table 1)
Table 1: Demographics an individual data for patients
Study no.
Age (years)
Histopathology
FIGO stage
In vivofluorescence
IHC FR-α expression
FM FITC
n = 10 patients. ++, strong; +, moderate; 0, weak; −, absent; FIGO, International Federation of Gynecology and Obstetrics; IHC FR-α, immunohistochemistry folate-receptor alpha; FM FITC, fluorescence microscopy for folate-FITC; n.a., not applicable.
Malignant tumor
1
72
Serous ovarian carcinoma
III
++
++
++
7
76
Serous ovarian carcinoma
III
+
+
+
9
64
Undifferentiated carcinoma
III
–
–
–
10
61
Mucineus ovarian carcinoma
III
+
+
+
Borderline tumor
5
48
Serous borderline tumor
I
0
+
0/+
Benign tumor
2
59
Fibrothecoma
n.a.
–
–
–
3
74
Fibrothecoma
n.a.
–
–
–
4
53
Mature cystic teratoma
n.a.
–
–
–
6
64
Benign multicystic ischemic ovary
n.a.
–
–
–
8
41
Fibroma
n.a.
–
–
–
Healthy tissue did not show any fluorescence signal either in vivo, ex vivo or on histopathological validation. In two separate still images of patients with ovarian cancer, the mean tumor-to-background ratio (as compared to healthy peritoneal surface) for ten demarcated fluorescent tumor deposits in each still image was 3.1 (± 0.8 s.d.). In the patient with a high-grade serous carcinoma and extensive peritoneal disseminated disease (stage III, FR-α positive), widespread tumor-specific fluorescence (white spots) was present throughout the abdominal cavity, as confirmed by ex vivo histopathology. Real-time image-guided excision of fluorescent tumor deposits of size <1 mm was feasible.
Five surgeons independently identified tumor deposits on three separate color images (shown on a representative image in (Left) and on their corresponding fluorescence image of precisely the same area (Right).
The number of tumor deposits detected by surgeons when guided by tumor-specific fluorescence (median 34, range 8–81) was significantly higher than with visual observation alone (median 7, range 4–22, P < 0.001).
Summary:
In this limited series, the authored showed that the use of intraoperative tumor-specific fluorescence imaging of the systemically administered FR-α–targeted agent folate-FITC offers specific and sensitive real-time identification of tumor tissue during surgery in patients with ovarian cancer and the presence of FR-α–positive tumors. Nevertheless, one patient presented with a malignant tumor that did not express FR-α, and consequently, no fluorescence was detected.
A major advantage over current imaging modalities is that an intraoperative fluorescence imaging system offers a large field of view for inspection and staging. This, in turn, may permit future patient-tailored surgical interventions and may decrease the number of needless extensive surgical procedures and the associated morbidity.
The second major advantage of intraoperative imaging as compared to current standard techniques is that it may guide the surgeon in debulking efforts, thus contributing to more efficient cytoreduction and ultimately improving the effect of adjuvant chemotherapy in patients with reduced tumor load
Improving the detection of cancer deposits to submillimeter size might ultimately improve survival rates, but whether this is the case needs to established by additional clinical studies.
Advantages:
In ovarian cancer, the FR-α appears to constitute a good target because it is overexpressed in 90–95% of malignant tumors, especially serous carcinomas.
Targeting ligand, folate, is attractive as it is nontoxic, inexpensive and relatively easily conjugated to a fluorescent dye to create a tumor-specific fluorescent contrast agent.
Disadvantages:
Overexpression of FR-α varies strongly between different solid tumors originating from different organs, a characteristic that reduces the general applicability of folate-FITC in cancer.
Many organs have autofluorescence in the excitation and emission parameters of the FITC dye.
Development of new fluorescent agents in the near-infrared spectrum will allow for identification of more deeply seated tumors, based on the stronger penetration properties of near-infrared dyes with an excitation wavelength >700 nm compared to FITC.
This is the first in-human proof-of-principle and the potential benefit of intraoperative tumor-specific fluorescence imaging in staging and debulking surgery for ovarian cancer using the systemically administered targeted fluorescent agent folate-FITC. Larger international multicenter studies using standardized, uniformly calibrated multispectral fluorescence camera systems combined with folate-FITC are needed to confirm our data and further elucidate the diagnostic (accuracy, sensitivity and specificity) and therapeutic value of the reported approach in larger series of ovarian cancer patients.
Note: Other similar approaches have been explored for brain tumors (3a, 3b) in human clinical trials using 5-aminolevulinic acid (5-ALA). We will not address this trial in this discussion.
Ref:
1. Gooitzen M van Dam, George Themelis, Lucia M A Crane, Niels J Harlaar, Rick G Pleijhuis, Wendy Kelder, Athanasios Sarantopoulos, Johannes S de Jong, Henriette J G Arts, Ate G J van der Zee, Joost Bart, Philip S Low & Vasilis Ntziachristos. Intraoperative tumor-specific fluorescence imaging in ovarian cancer by folate receptor-αtargeting: first in-human results. Nature Medicine 17, 1315–1319 (2011). http://www.nature.com.rproxy.tau.ac.il/nm/journal/v17/n10/full/nm.2472.html
3a. Stummer W, Pichlmeier U, Meinel T, Wiestler OD, Zanella F, Reulen HJ; ALA-Glioma Study group. Fluorescence-guided surgery with 5-aminolevulinic acid for resection of malignant glioma: a randomised controlled multicentre phase III trial. Lancet Oncol 2006 May;7(5):392-401.
Each year, over 20 000 women in the US are diagnosed with ovarian cancer while there are nearly 15 000 deaths (http://www.cdc.gov/uscs). The 5-year (post-diagnosis) mortality rate is less than 46% partly due to the fact that early detection still remains difficult (Jemal et al., 2010). So, it is not surprising to see biomedical research putting a much needed focus on this deadly killer.
As such, a joint collaboration between Dr. Søren T Christensen’s lab (University of Copenhagen, Denmark) and the Laboratory of Reproductive Biology (LRB, RigsHospital, Copenhagen) decided to investigate into the specific molecular mechanisms behind ovarian cancer. This resulted in the recent publication linking aberrant hedgehog/PDGF signaling and ovarian cancer (Egeberg et al., 2012). Specifically, the paper shows that there are specific defects associated with the primary cilium of these cancer cell lines which then lead to a perturbation of two critical signaling systems that were examined in the paper. The starting materials included ovarian cancer lines OVCAR3 and SK-OV3. And, the control WT cells are ovarian surface epithelium (OSE which are thought to be the cells of origin for ovarian cancer) cells that were obtained at LRB where young girls that are diagnosed with cancer can deposit and cryopreserve their ovaries for future implantation after being deemed cancer-free.
Biomarkers evaluated in the study included
1. Gli proteins – glioma transcription factors specific to hedgehog signaling (regulating cell proliferation/differentiation). In the off stage, they are thought to act as repressors while in the on state, the proteins are modified at the primary cilium to become activators (Satir el al., 2010).
2. CHFR (checkpoint with forkhead-associated and ring finger domains) – a tumor suppressor gene with multiple functions in checkpoints during mitosis (Kang et al., 2002)
3. AURA (Aurora Kinase A) mark centrosome maturation, mitotic entry an spindle assembly (Fu et al., 2007).
4. PDGF (platelet derived growth factor) is a signaling pathway involved in cell growth/survival, differentiation, migration and wound healing (Schneider et al., 2010).
The paper begins with characterizing the epithelial nature of OSE to confirm the validity of the primary OSE cell cultures. Of note is the fact that OSE lack E-cadherins present in other epithelial cells while expressing N-cadherin and vimentin which together suggest OSE have a more mesenchymal characteristic in comparison to other epithelial tissues. Meawhile, OVCAR3 was negative for vimentin while both cancer cell lines expressed N-cadherin and E-cadherin (more so) indicating these cancer cell lines retained more of a classic epithelial characteristic in comparison to WT OSE which might imply lack of plasticity (Wong et al., 1999).
Next, ciliogenesis (ability to form a primary cilium) was evaluated in the cancer cell lines vs WT OSE. Under serum starvation (used to induce cilia formation), far fewer cancer cells are able to form a primary cilium. Since primary cilium are formed in mitotically inactive cells (usually at G0), it was speculated that the lack of cilia in the cancer lines implied greater proliferation rates. However, Ki67, phosphorylated retinoblastoma protein (p-RB) and PCNA (cell proliferation markers) stainings showed that under serum starvation conditions, the cancer cells still have the ability to enter growth arrest.
So, there had be another explanation accounting for the difference between WT OSE and the cancer cell lines such as a interruption of signal between the primary cilium and the growth arrest apparatus. Thus, the hedgehog signaling was then examined. Full-length Gli2 acts as a repressor while cleavage at the primary cilia results in the activator form. WT OSE had very little full-length Gli2 whereas the cleaved repressor form is present. This indicates that the hedgehog pathway is mostly inactive in WT OSE. On the other hand, the two cancer lines had higher levels of the full length activator form and little of the cleaved repressor form. Serum starvation decreased this activation of the hedgehog signal as expected (due to the presence of the primary cilium cleaving the Gli2) but not down to the same levels as in WT OSE. This was confirmed by RT-PCR results which in summary stated that there is a higher basal level of hedgehog signaling in the cancer lines vs WT OSE.
PDGFa had previously been reported to be upregulated during serum starvation and consequently is seen accumulating at the primary cilium (Schneider et al., 2005). While this phenomenon is observed In WT OSE, not only are there lower levels of PDGF overall, but there is no accumulation of this protein upon serum starvation. Therefore, PDGF signaling is perturbed in the cancer cell lines.
Next, the expression of AURA was examined. This kinase is responsible for ciliary disassembly which is a necessary step before entering mitosis. The cancer OSE cells were shown to have higher AURA levels as compared to WT OSE. Given that these cancer cell lines have fewer primary cilia, one can deduce that the higher AURA protein levels suppress ciliogenesis while in WT OSE, they have a role in ciliary disassembly. This was further confirmed by the fact that knockdown of AURA in the cancer cell lines resulted in increased ciliogenesis.
Then, the paper looks upstream of AURA at CHFR which is responsible for the ubiquitination/ proteosomal degradation of AURA. During serum starvation (i.e. primary cilia is formed), CHFR is found at the base of the cilium in WT OSE and this localization disappears during mitosis. In the transformed hTERT-RPE1 cells, CHFR is found both in growth-arrested as well as cells in mitosis. Therefore, this result hints at a AURA/CHFR interaction at the centrosome regulating ciliary dynamics.
Thus, an axis linking the primary cilium to different signal transduction systems and eventually leading to cancer is shown below:
Cilia defect Hedgehog/PDGF uncontrolled proliferation cancer
The cilia defect is likely due to a mutation in the AURA/CHFR axis or even further upstream. While this mechanistic explanation summarizes the findings of this paper,its logic can be applied to other cancer types also (Veland et al., 2009).








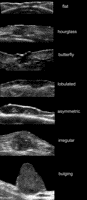

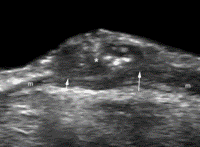












 Intraoperative tumor-specific fluorescence imaging in ovarian cancer by folate receptor-α targeting (
Intraoperative tumor-specific fluorescence imaging in ovarian cancer by folate receptor-α targeting (