The SCID Pig: How Pigs are becoming a Great Alternate Model for Cancer Research[1]
Author/Writer: Stephen J. Williams, Phd

Article ID #80: The SCID Pig: How Pigs are becoming a Great Alternate Model for Cancer Research. Published on 10/10/2013
WordCloud Image Produced by Adam Tubman
UPDATED 3/14/2020
The need for alternate models of human cancer
Many worldwide regulatory bodies are in agreement that proper choice of animal model is necessary for adequate extrapolation of toxicity and efficacy data from animal to human, considering the varied classes of therapeutics now being developed for oncology. The inability of screens, reliant on human xenografts grown in immunocompromised mice to evaluate host-immune and species-dependent effects, has made development of alternative animal-models a priority. This is evident in the fact that ninety percent of new anticancer drugs which showed anti-tumor efficacy in mouse preclinical models failed in human clinical studies. A recently developed “humanized” mouse model may assist in testing the metabolism of cancer drugs but still relies on older “immunosuppression” mouse models (http://stehlin.org/mouse-model-development/). This inadequacy of older, accepted models is clearly evident when evaluating safety and efficacy of adenoviral based gene therapies such as oncolytic conditionally-replicative adenovirus (CRAd). Although new-generation CRAds present with a relative safe profile[2, 3], adenoviral particles, especially the Ad5 based virus used for most CRAds, have the tendency to replicate in non-tumor tissue, such as liver and lung, resulting in tissue-specific toxicities[4-7]. The manifestation of these toxicities is only evident in species permissive for viral replication, such as the pig. Indeed, one of the first clinical trials with older adenovirus gene therapy, resulting in severe hepatic toxicity and fatality, may have been prevented if more appropriate preclinical screens were conducted. Thereafter, strict regulatory guidelines for adenoviral-based clinical trials have been issued, with particular emphasis on vector dosage, safety and toxicity[8]. Indeed, at Schering-Plough, a toxicology program was initiated to evaluate SCH 58500, and adenoviral gene therapy directed against p53, which involved use of non-immunogenic rats compared with testing in Yorkshire pigs made immunoreactive to the vector[9, 10]. In fact, data from the pig study revealed a faster clearance of virus as well as toxicities not seen in non-immunogenic, non-permissive hosts such as rat and mouse.
Therefore, development of a porcine model of cancer would permit both testing of both the efficacy and safety of these therapies in the same animal.
Development of large animal models of cancer
To date, large animal tumor models have been used for studying spontaneously formed tumors in dogs and cats [11](Vail, 2000, Cancer Invest), the most common being canine [12] and feline non-Hodgkin’s lymphoma [13]. The advantages of these companion models are the outbred nature of the animals, comparable size and kinetics to human tumors [14-18], and high incidence rates. Allografts of the outbred-canine transplanted venereal tumor have been used to test the ability to detect tumors using X-ray computed tomography and MRI with the ultimate goal of imaging-guided intervention. Researchers have recently utilized the spontaneously arising canine and feline soft tissue sarcomas to study effects of hyperthermia on chemotherapy pharmocokinetics, development of hypoxic cell markers, and cancer imaging techniques [15, 19-26]
Although it appears that, for a select number of tumor types, spontaneously arising tumors in large outbred animals can be useful to model the human disease, it is disappointing these spontaneous arising tumors are limited to discrete tumor types. However, due to recent advances in sequencing of several domestic animal genomes and the development of new cloning strategies, it is now very feasible to utilize other animal models more relevant to human disease, notably the miniature pig.
 The Gottingen mini-pig
The Gottingen mini-pig
Large animals in medical research: Advantages of the minipig
Due to recent advances in sequencing of several domestic animal genomes [27, 28] and the development of new organism cloning technologies [29-31], it is now very feasible to utilize other species to model human disease, notably the pig. The development of porcine models of human disease has gained much interest lately. Advantages include the resemblance in anatomy, physiology, and genetic makeup with the human, as well as new methods to manipulate the pig genome [32, 33]. To date, porcine models of human metabolic syndrome [34] and diabetes [35], aortic aneurism [36], infectious disease resistance [32, 37], seizure [38], neurologic syndromes [33], and pancreatitis [39] have been developed. Recently, a genetically-engineered porcine model of cystic fibrosis was produced in collaboration with investigators at University of Iowa and Exemplar Genetics [40-42]. Additionally, Cho et al. successfully transplanted spontaneously transformed leukemic and lymphatic tumor cells in a major histocompatibility complex (MHC)-defined inbred miniature swine model [43], suggesting feasibility of an ex vivo strategy to develop a porcine tumor model. Porcine models have, also, been used to develop, test and refine surgical [44, 45] and laparoscopic techniques [46, 47], radio- and cryoablation protocols of tissues [48-52] and robotic surgery using the da Vinci Surgical SystemÒ [53, 54]. In addition, because of the size of porcine organs and their resemblance to the human (in genetics) the minipig is very useful and abundant of a source to isolate specific cell types for in vitro studies. Below is a figure showing the comparable size of human and porcine ovaries to the mouse and ability to purify porcine ovarian epithelial cells and their similarity to human and mouse ovarian epithelial cells.

Figure 1. The human and pig ovary have similar size and can yield a greater number of isolated cells than one can get from a mouse ovary.

Figure 2. Isolation and morphology of ovarian epithelial cells from three sources:
A) Devonshire/Yorkshire pig
B) normal human ovary
c) SV129/BL6 mouse
note cobblestone epithelial morphology from all three sources©
To date, there has been no allograft or xenograft model of cancer in pigs. The consensus amongst many surgeons suggests development of a minipig tumor model would be an invaluable tool for developing surgical skills.
A recent advancement in porcine tumor modeling was made by collaboration between researchers from the laboratories of Dr. Stefan Bossmann and Deryl Troyer at Kansas State and Iowa State, respectively[1]. The joint collaboration resulted in the development of the first severe combined immunodeficient pig line (SCID pig) which was shown to be able to accept human tumor xenografts. The line of immunodeficient pig was discovered when Yorkshire pigs were bred for increased feed efficiency and a line of pigs exhibited SCID-like symptoms including:
- Decreased levels of circulating lymphocytes
- Atrophied thymus and lymph nodes
The SCID phenotype in mice have been ascribed to defects in a DNA-dependent protein kinase gene which prevents variable-diversity-joining [V(D)J] gene region recombination[55]. There have been multiple genetic defects found in humans resulting in SCID, including defects in adenylate kinase2, Janus kinase 3, the IL2 receptor, and the IL-7 receptor[56]. The SCID phenotype in this pig line has a simple autosomal recessive inheritance pattern which, as described below in an interview with the authors, allows for the propagation of this porcine line.
An important feature of SCID models is the ability of these animals to act as a recipient of human tumorigenic cell lines. In fact, growth of cell lines in SCID mice is a common test for tumorigenicity. Therefore, to test if these pigs could act as recipients for human cancer cell lines, the authors inoculated the SCID Yorkshire pigs with 4 million A3755M human melanoma cells or PANC1 human pancreatic carcinoma cells subcutaneously in the left and right ears respectively of three pigs. Some features of the results include:
- All injection sites showed evidence (either histologic or palpable) of tumor growth
- Tumors showed characteristic histologic features of malignant neoplasm including
- Bizarre and atypical mitotic figures
- Anisocytosis (different cell sizes and shapes; feature of malignancy)
- Anisokaryosis (different size and shape of nucleus)
- tumors stained with anti-human mitochondrial antibody (a marker of epithelial cancer cells) showed strong cytoplasmic staining of neoplastic cells
- interestingly no necrotic regions in the tumor
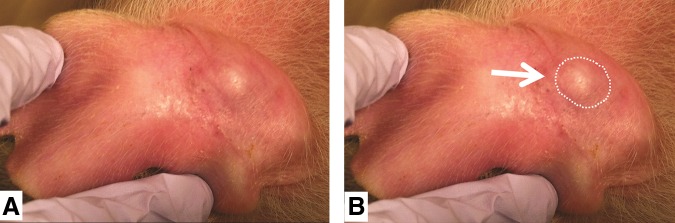
 Figure 3. Visual evidence of human tumor cells growing in SCID pig ear (day 20). B) Same picture as A) but circle outlines growth. From reference 1. Basel et al., used with permission from Mary Liebert.
Figure 3. Visual evidence of human tumor cells growing in SCID pig ear (day 20). B) Same picture as A) but circle outlines growth. From reference 1. Basel et al., used with permission from Mary Liebert.
It is interesting to note that these tumors only grew roughly 10 x 5.5 mm, which is genrally large enough to do preclinical studies but may be too expensive to be of use for xenograft studies. However it would be very feasible to conduct allograft studies in these SCID pigs.
Dr. Jack Dekkers, C.F. Curtiss Distinguished Professor and Section Leader of Animal Breeding and Genetics at Iowa State University, was kind to answer a few questions about the SCID pig model.
Question: You had mentioned this line was identified after breeding Yorkshire pigs for increased feed efficiency. Have you identified or hypothesize which altered pathway or molecular defect which results in a SCID phenotype? Is this SCID phenotype a result of a metabolic syndrome these pigs could have?
Dr. Dekkers: We indeed identified the SCID phenotype in a line of pigs that we had selected for increased feed efficiency. However, I don’t think this phenotype has anything to do with the selection we practiced; it was either already present in the founders of the line or it was a random mutation that occurred in the line, independent of the selection for feed efficiency. We have narrowed the mutation that causes the SCID in our pigs down to a chromosomal region and have a very strong candidate gene in that region that we are currently pursuing.
Question: In your opinion, is it possible to produce a highly inbred immunocompromised strain of pig such as a Gottingen minipig?
Dr. Dekkers: We are working on breeding the SCID mutation into mini pigs. But in the meantime, we have used bone marrow transfer to create a male that is homozygous SCID (it’s an autosomal recessive) and reproducing. This allows us to produce litters that are 50% SCID and 50% normal (carriers) by mating him to carrier females.
REFERENCES
1. Basel MT, Balivada S, Beck AP, Kerrigan MA, Pyle MM, Dekkers JC, Wyatt CR, Rowland RR, Anderson DE, Bossmann SH et al: Human xenografts are not rejected in a naturally occurring immunodeficient porcine line: a human tumor model in pigs. BioResearch open access 2012, 1(2):63-68.
2. Dobbelstein M: Replicating adenoviruses in cancer therapy. Curr Top Microbiol Immunol 2004, 273:291-334.
3. Lichtenstein DL, Wold WS: Experimental infections of humans with wild-type adenoviruses and with replication-competent adenovirus vectors: replication, safety, and transmission. Cancer Gene Ther 2004, 11(12):819-829.
4. Volpers C, Kochanek S: Adenoviral vectors for gene transfer and therapy. J Gene Med 2004, 6 Suppl 1:S164-171.
5. Brand K, Arnold W, Bartels T, Lieber A, Kay MA, Strauss M, Dorken B: Liver-associated toxicity of the HSV-tk/GCV approach and adenoviral vectors. Cancer Gene Ther 1997, 4(1):9-16.
6. Lieber A, He CY, Meuse L, Schowalter D, Kirillova I, Winther B, Kay MA: The role of Kupffer cell activation and viral gene expression in early liver toxicity after infusion of recombinant adenovirus vectors. J Virol 1997, 71(11):8798-8807.
7. Keedy V, Wang W, Schiller J, Chada S, Slovis B, Coffee K, Worrell J, Thet LA, Johnson DH, Carbone DP: Phase I study of adenovirus p53 administered by bronchoalveolar lavage in patients with bronchioloalveolar cell lung carcinoma: ECOG 6597. J Clin Oncol 2008, 26(25):4166-4171.
8. Assessment of adenoviral vector safety and toxicity: report of the National Institutes of Health Recombinant DNA Advisory Committee. Hum Gene Ther 2002, 13(1):3-13.
9. Morrissey RE, Horvath C, Snyder EA, Patrick J, Collins N, Evans E, MacDonald JS: Porcine toxicology studies of SCH 58500, an adenoviral vector for the p53 gene. Toxicol Sci 2002, 65(2):256-265.
10. Morrissey RE, Horvath C, Snyder EA, Patrick J, MacDonald JS: Rodent nonclinical safety evaluation studies of SCH 58500, an adenoviral vector for the p53 gene. Toxicol Sci 2002, 65(2):266-275.
11. Vail DM, MacEwen EG: Spontaneously occurring tumors of companion animals as models for human cancer. Cancer Invest 2000, 18(8):781-792.
12. Leifer CE, Matus RE: Canine lymphoma: clinical considerations. Semin Vet Med Surg (Small Anim) 1986, 1(1):43-50.
13. MacEwen EG: Spontaneous tumors in dogs and cats: models for the study of cancer biology and treatment. Cancer Metastasis Rev 1990, 9(2):125-136.
14. Schwyn U, Crompton NE, Blattmann H, Hauser B, Klink B, Parvis A, Ruslander D, Kaser-Hotz B: Potential tumour doubling time: determination of Tpot for various canine and feline tumours. Vet Res Commun 1998, 22(4):233-247.
15. Zeman EM, Calkins DP, Cline JM, Thrall DE, Raleigh JA: The relationship between proliferative and oxygenation status in spontaneous canine tumors. Int J Radiat Oncol Biol Phys 1993, 27(4):891-898.
16. LaRue SM, Fox MH, Withrow SJ, Powers BE, Straw RC, Cote IM, Gillette EL: Impact of heterogeneity in the predictive value of kinetic parameters in canine osteosarcoma. Cancer Res 1994, 54(14):3916-3921.
17. Vail DM, Kisseberth WC, Obradovich JE, Moore FM, London CA, MacEwen EG, Ritter MA: Assessment of potential doubling time (Tpot), argyrophilic nucleolar organizer regions (AgNOR), and proliferating cell nuclear antigen (PCNA) as predictors of therapy response in canine non-Hodgkin’s lymphoma. Exp Hematol 1996, 24(7):807-815.
18. Guglielmino R, Canese MG, Miniscalco B, Geuna M: Comparison of clinical, morphological, immunophenotypical and cytochemical characteristics of LGL leukemia/lymphoma in dog, cat and human. Eur J Histochem 1997, 41 Suppl 2:23-24.
19. Cline JM, Thrall DE, Rosner GL, Raleigh JA: Distribution of the hypoxia marker CCI-103F in canine tumors. Int J Radiat Oncol Biol Phys 1994, 28(4):921-933.
20. Thrall DE, McEntee MC, Cline JM, Raleigh JA: ELISA quantification of CCI-103F binding in canine tumors prior to and during irradiation. Int J Radiat Oncol Biol Phys 1994, 28(3):649-659.
21. Raleigh JA, La Dine JK, Cline JM, Thrall DE: An enzyme-linked immunosorbent assay for hypoxia marker binding in tumours. Br J Cancer 1994, 69(1):66-71.
22. Thrall DE, Larue SM, Pruitt AF, Case B, Dewhirst MW: Changes in tumour oxygenation during fractionated hyperthermia and radiation therapy in spontaneous canine sarcomas. Int J Hyperthermia 2006, 22(5):365-373.
23. Siddiqui F, Li CY, Larue SM, Poulson JM, Avery PR, Pruitt AF, Zhang X, Ullrich RL, Thrall DE, Dewhirst MW et al: A phase I trial of hyperthermia-induced interleukin-12 gene therapy in spontaneously arising feline soft tissue sarcomas. Mol Cancer Ther 2007, 6(1):380-389.
24. Sostman HD, Prescott DM, Dewhirst MW, Dodge RK, Thrall DE, Page RL, Tucker JA, Harrelson JM, Reece G, Leopold KA et al: MR imaging and spectroscopy for prognostic evaluation in soft-tissue sarcomas. Radiology 1994, 190(1):269-275.
25. Dennis R: Imaging features of orbital myxosarcoma in dogs. Vet Radiol Ultrasound 2008, 49(3):256-263.
26. Mueller F, Fuchs B, Kaser-Hotz B: Comparative biology of human and canine osteosarcoma. Anticancer Res 2007, 27(1A):155-164.
27. Cockett NE: Current status of the ovine genome map. Cytogenet Genome Res 2003, 102(1-4):76-78.
28. Schook LB, Beever JE, Rogers J, Humphray S, Archibald A, Chardon P, Milan D, Rohrer G, Eversole K: Swine Genome Sequencing Consortium (SGSC): A Strategic Roadmap for Sequencing The Pig Genome. Comp Funct Genomics 2005, 6(4):251-255.
29. Wilmut I, Schnieke AE, McWhir J, Kind AJ, Campbell KH: Viable offspring derived from fetal and adult mammalian cells. Nature 1997, 385(6619):810-813.
30. Cibelli JB, Stice SL, Golueke PJ, Kane JJ, Jerry J, Blackwell C, Ponce de Leon FA, Robl JM: Cloned transgenic calves produced from nonquiescent fetal fibroblasts. Science 1998, 280(5367):1256-1258.
31. Polejaeva IA, Chen SH, Vaught TD, Page RL, Mullins J, Ball S, Dai Y, Boone J, Walker S, Ayares DL et al: Cloned pigs produced by nuclear transfer from adult somatic cells. Nature 2000, 407(6800):86-90.
32. Lunney JK: Advances in swine biomedical model genomics. Int J Biol Sci 2007, 3(3):179-184.
33. Schook LB, Kuzmuk K, Adam S, Rund L, Chen K, Rogatcheva M, Mazur M, Pollock C, Counter C: DNA-based animal models of human disease: from genotype to phenotype. Dev Biol (Basel) 2008, 132:15-25.
34. Spurlock ME, Gabler NK: The development of porcine models of obesity and the metabolic syndrome. J Nutr 2008, 138(2):397-402.
35. Palin MF, Labrecque B, Beaudry D, Mayhue M, Bordignon V, Murphy BD: Visfatin expression is not associated with adipose tissue abundance in the porcine model. Domest Anim Endocrinol 2008, 35(1):58-73.
36. Sadek M, Hynecek RL, Goldenberg S, Kent KC, Marin ML, Faries PL: Gene expression analysis of a porcine native abdominal aortic aneurysm model. Surgery 2008, 144(2):252-258.
37. Saetre T, Hoiby EA, Aspelin T, Lermark G, Lyberg T: Acute serogroup A streptococcal shock: A porcine model. J Infect Dis 2000, 182(1):133-141.
38. Marchi N, Angelov L, Masaryk T, Fazio V, Granata T, Hernandez N, Hallene K, Diglaw T, Franic L, Najm I et al: Seizure-promoting effect of blood-brain barrier disruption. Epilepsia 2007, 48(4):732-742.
39. Tao J, Gong D, Ji D, Xu B, Liu Z, Li L: Improvement of monocyte secretion function in a porcine pancreatitis model by continuous dose dependent veno-venous hemofiltration. Int J Artif Organs 2008, 31(8):716-721.
40. Rogers CS, Abraham WM, Brogden KA, Engelhardt JF, Fisher JT, McCray PB, Jr., McLennan G, Meyerholz DK, Namati E, Ostedgaard LS et al: The porcine lung as a potential model for cystic fibrosis. Am J Physiol Lung Cell Mol Physiol 2008, 295(2):L240-263.
41. Rogers CS, Stoltz DA, Meyerholz DK, Ostedgaard LS, Rokhlina T, Taft PJ, Rogan MP, Pezzulo AA, Karp PH, Itani OA et al: Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science 2008, 321(5897):1837-1841.
42. Rogers CS, Hao Y, Rokhlina T, Samuel M, Stoltz DA, Li Y, Petroff E, Vermeer DW, Kabel AC, Yan Z et al: Production of CFTR-null and CFTR-DeltaF508 heterozygous pigs by adeno-associated virus-mediated gene targeting and somatic cell nuclear transfer. J Clin Invest 2008, 118(4):1571-1577.
43. Cho PS, Lo DP, Wikiel KJ, Rowland HC, Coburn RC, McMorrow IM, Goodrich JG, Arn JS, Billiter RA, Houser SL et al: Establishment of transplantable porcine tumor cell lines derived from MHC-inbred miniature swine. Blood 2007, 110(12):3996-4004.
44. Hammond I, Taylor J, Obermair A, McMenamin P: The anatomy of complications workshop: an educational strategy to improve the training and performance of fellows in gynecologic oncology. Gynecol Oncol 2004, 94(3):769-773.
45. Schneider C, Jung A, Reymond MA, Tannapfel A, Balli J, Franklin ME, Hohenberger W, Kockerling F: Efficacy of surgical measures in preventing port-site recurrences in a porcine model. Surg Endosc 2001, 15(2):121-125.
46. Rassweiler JJ, Henkel TO, Potempa DM, Frede T, Stock C, Gunther M, Alken P: [Laparoscopic training in urology. An essential principle of laparoscopic interventions in the retroperitoneum]. Urologe A 1993, 32(5):393-402.
47. Orvieto MA, Zorn KC, Lyon MB, Tolhurst SR, Rapp DE, Seip R, Sanghvi N, Shalhav A: High intensity focused ultrasound renal tissue ablation: a laparoscopic porcine model. J Urol 2009, 181(2):861-866.
48. Ng KK, Lam CM, Poon RT, Shek TW, To JY, Wo YH, Ho DW, Fan ST: Comparison of systemic responses of radiofrequency ablation, cryotherapy, and surgical resection in a porcine liver model. Ann Surg Oncol 2004, 11(7):650-657.
49. Alemany R, Balague C, Curiel DT: Replicative adenoviruses for cancer therapy. Nat Biotechnol 2000, 18(7):723-727.
50. Kahlenberg MS, Volpe C, Klippenstein DL, Penetrante RB, Petrelli NJ, Rodriguez-Bigas MA: Clinicopathologic effects of cryotherapy on hepatic vessels and bile ducts in a porcine model. Ann Surg Oncol 1998, 5(8):713-718.
51. Long JP, Faller GT: Percutaneous cryoablation of the kidney in a porcine model. Cryobiology 1999, 38(1):89-93.
52. Scott DM, Young WN, Watumull LM, Lindberg G, Fleming JB, Rege RV, Brown RJ, Jones DB: Development of an in vivo tumor-mimic model for learning radiofrequency ablation. J Gastrointest Surg 2000, 4(6):620-625.
53. Molpus KL, Wedergren JS, Carlson MA: Robotically assisted endoscopic ovarian transposition. Jsls 2003, 7(1):59-62.
54. Hanly EJ, Marohn MR, Bachman SL, Talamini MA, Hacker SO, Howard RS, Schenkman NS: Multiservice laparoscopic surgical training using the daVinci surgical system. Am J Surg 2004, 187(2):309-315.
55. Finnie NJ, Gottlieb TM, Blunt T, Jeggo PA, Jackson SP: DNA-dependent protein kinase defects are linked to deficiencies in DNA repair and V(D)J recombination. Philosophical transactions of the Royal Society of London Series B, Biological sciences 1996, 351(1336):173-179.
56. Notarangelo LD: Primary immunodeficiencies. The Journal of allergy and clinical immunology 2010, 125(2 Suppl 2):S182-194.
UPDATED 3/14/2020
A recent Research Article and Research Article Summary in Science discusses, by the primary author of her study that describes the utility of the pig as an excellent surrogate model of the human brain and human brain function. The study, by Dr. Evelina Sjostdedt et al., was an integrative analysis of porcine, mouse, and human transcriptomic, genomic, and proteomic data from discrete anatomical regions of the brain. The global analysis suggested that there is similar regional organization and expression patterns among the three mammalian species. The authors found interspecies variability with respect for many neurotransmitter receptors.
However, for some regions of the brain, such as the cerebellum and hypothalamus, the human global expression profile is closer to that of the pig than of the mouse, suggesting that the pig might be considered a preferred animal model to study many brain processes.
In addition, interestingly, the authors found that many signature genes canonically thought to only be expressed in certain brain cells (astrocytes, microglia, oligodendrocytes) are expressed in higher levels in peripheral organs as well as immune cells.
Please go to the full article in Science,
An atlas of the protein-coding genes in the human, pig, and mouse brain,
Science 06 Mar 2020:
Vol. 367, Issue 6482, eaay5947
DOI: 10.1126/science.aay5947
to access the data used in this study, which includes high resolution images and metadata have also been made publicly available in the open-access Human Protein Atlas (HPA) Brain Atlas. at www.proteinatlas.org.
Other articles on this site pertaining to Alternate Animal Models and Cancer and Disease include:
Read Full Post »



















{kind=link}