A Synthesis of the Beauty and Complexity of How We View Cancer
Author: Larry H. Bernstein, MD, FCAP
Cancer Volume One – Summary
A Synthesis of the Beauty and Complexity of How We View Cancer
This document has covered a broad spectrum of the research, translational biology, diagnostics (both laboratory and imaging methodologies), and treatments for a variety of cancers, mainly by organs, and selectively by the most common cancers seen in human populations. A number of observations stand out on review of all the material presented. 1. The most common cancers affecting humans is spread worldwide, with some variation by region. 2. Cancers within geographic regions may be expressed differently in relationship to population migrations, the incidence of specific environmental pollutants, occurrence of insect transmitted and sexually transmitted diseases (HIV, HCV, HPV), and possibly according to age, or relationship to ultraviolet or high dose radiation exposure. 3. Cancers are expressed within generally recognized age timelines. For example, acute lymphocytic leukemia and neuroblastoma in children under 10 years age; malignant giant cell tumor and osteosarcoma in the third and fourth decade; prostate cancer and breast cancer over age 40, and are more aggressive at an earlier age, both having a strong sex hormone dependence. 4. There is dispute about the effectiveness of screening for cancer with respect to what age, excessive risk in treatment modality, and the duration of progression free survival. Despite the evidence of several years potential life extension, a long term survival of 10 years is not the expected outcome. However, the quality of life in the remaining years is a valid point in favor of progress. 5. There has been a significant reduction in toxicity of treatment, but attention has been focused on a patient-centric decision process. 6. There has been a dramatic improvement in surgical approaches, post-surgical surveillance, and in diagnosis by invasive and noninvasive methods, especially in the combination of needle biopsy and imaging techniques. 7. There is significant variation within cancer cell types with respect to disease-free survival.
The work presented has several main components: First, there is the biology and mechanisms involved in carcinogenesis related to (1) mutations; (2) carcinogenesis; (3) cell regulatory mechanisms; (4) cell signaling pathways; (5) apoptosis (6) ubitination (7) mitochondrial dysfunction; (8) cell-cell interactions; (9) cell migration; (10) metastasis. Then there are large portions covering (1) imaging; (2) specific targeted therapy; (3) nanotechology-based therapy; (4) specific organ-type cancers; (5) genomics-based testing; (6) circulating cancer cells; (7) miRNAs; (8) siRNAs; (9) cancer immunology and (10) immunotherapy.
Classically, we refer to cancer development in terms of the germ cell layers – ectoderm, mesoderm, and endoderm. These are formative in embryonic development. The most active development occurs during embryonic development, with a high growth rate of cells and also a high utilization of energy. The cells utilize oxidation for energy in this period characterized by movement of cells in differentiation and organogenesis. This was observed to be unlike the cell metabolism in carcinogenesis, which is characterized by impaired mitochondrial function and reliance on lactate production for energy – termed anaerobic glycolysis, as investigated by Meyerhof, Embden, Warburg, Szent-Gyorgy, H. Krebs, Theorell, AV Hill, B Chance, P Mitchell, P Boyer, F Lippman, and others.
In addition, the body economy has been divided into two major metabolic compartments: fat and lean body mass (LBM), which is further denoted as visceral and structural. This denotes the gut, kidneys, liver, lung, pancreas, sexual organs, endocrines, brain and fat cells in one compartment, and skeletal muscle, bone and cardiovascular in another. LBM is calculated as fat free mass. Further, brown fat is distinguished from white fat. But this was a first layer of construction of the human body. One peels away this layer to find a second layer. For example, the gut viscera have an inner (outer) epithelial layer, a muscularis, and a deep epithelium, which has circulation and fat. There is also an interstitium between the gut epithelium and muscularis. The lung has an epithelium exposed to the airspaces, then capillaries, and then epithelium, designed for exchange of O2 and CO2, the source of heat generation. The pancreas has an endocrine portion in the islets that are embedded in an exocrine secretory organ. The sexual organs have a combination of glandular structures embedded in a mesothelium.
The structural compartment is entirely accounted for by the force of contraction. If this is purely anatomical, that is not really the case when one goes into the functioning substructures of these tissues – cytoplasm, endoplasmic reticulum (ribosomal), mitochondria, liposomes, chromatin apparatus, cell membrane and vesicles. Within and between these structures are the working and interacting mechanisms of the cell in its unique role. What ties these together was first thought to be found in the dogma following the discovery of the genetic code in 1953 that begat DNA to RNA to protein.
This led to many other discoveries that made it clear that it was only a first approximation. It did not account for noncoding DNA, which became unmasked with the culmination of the Human Genome Project and concurrent advances in genomics (mtDNA, mtRNA, siRNA, exosomes, proteomics, synthetic biology, predictive analytics, and regulatory pathways directed by signaling molecules. Here is a list of signaling pathways: 1. JAK-STAT 2. GPCR 3. Endocrine 4. Cytochemical 5. RTK 6. P13K 7. NF-KB 8. MAPK 9. Ubiquitin 10. TGF-beta 11. Stem cell These signaling pathways have become the basis for the discovery of inhibitors of signaling pathways (suppressors), as well as activators, as these have been considered as specific targets for selective therapy. (.See Figure below) Of course, extensive examination of these pathways has required that all such findings are validated based on the STRENGTH of their effect on the target and in the impact of suppression.

Let us continue this discussion elucidating several major points. While the early observations that drove the interest in biochemical behavior of cancer cells has been displaced, it has not faded from view.
Bioenergetics of Cancer cells
Michael J. Gonzalez (Bioenergetic_Theory_of_Carcinigenesis. http://www.academia.edu/2224071/ Bioenergetic_Theory_of_Carcinigenesis) maintains that the altered energy metabolism of tumor cells provides a viable target for a non-toxic chemotherapeutic approach. An increased glucose consumption rate has been observed in malignant cells. Warburg (NobelLaureate in medicine) postulated that the respiratory process of malignant cells was impaired in the malignant transformation. Szent-Györgyi (Nobel in medicine) also viewed cancer as originating from insufficient oxygen utilization. Oxygen inhibits anaerobic metabolism (fermentation and lactic acid production). Interestingly, during cell differentiation (where cell energy level is high) there is an increased cellular production of oxidation products that appear to provide physiological stimulation for changes in gene expression that may lead to a terminal differentiated state. The failure to maintain high ATP production (high cell energy levels) may be a consequence of inactivation of key enzymes, especially those related to the Krebs cycle and the electron transport system. A distorted mitochondrial function (transmembrane potential) may result. This aspect could be suggestive of an important mitochondrial involvement in the carcinogenic process in addition to presenting it as a possible therapeutic target for cancer. Intermediate metabolic correction of the mitochondria is postulated as a possible non-toxic therapeutic approach for cancer.
Fermentation is the anaerobic metabolic breakdown of glucose without net oxidation. Fermentation does not release all the available energy of glucose or need oxygen as part of its biochemical reactions ; it merely allows glycolysis (a process that yields two ATP per mole of glucose) to continue by replenishing reduced coenzymes and yields lactate as its final product. The first step in aerobic and anaerobic energy producing pathways, it occurs in the cytoplasm of cells, not in specialized organelles, and is found in all living organisms. Cancer cells have a fundamentally different energy metabolism compared to normal cells, that are obligate aerobes (oxygen-requiring cells) meeting their energy needs with oxidative metabolic processes., while cancer cells do not require oxygen for their survival. This increase in glycolytic flux is a metabolic strategy of tumor cells to ensure growth and survival in environments with low oxygen concentrations.
Radoslav Bozov has commented that the process of genomic evolution cannot be fully revealed through comparative genomicsHe states that DNA would be entropic- favorable stable state going towards absolute ZERO temp. Themodynamics measurement in subnano discrete space would go negative towards negativity. DNA is like a cold melting/growing crystal, quite stable as it appears not due to hydrogen bonding , but due to interference of C-N-O. That force is contradicted via proteins onto which we now know large amount of negative quantum redox state carbon attaches. The more locally one attempts to observe, the more hidden variables would emerge as a consequence of discrete energy spaces opposing continuity of matter/time. But stability emerges out of non-stable states, and never reaches absolute stability, for there would be neither feelings nor freedom.
Membrane potential(Vm)
Membrane potential (Vm), the voltage across the plasma membrane, arises because of the presence of differention channels/transporters with specific ion selectivity and permeability. Vm is a key biophysical signal in non-excitable cells, modulating important cellular activities, such as proliferation and differentiation. Therefore, the multiplicities of various ion channels/transporters expressed on different cells are finely tuned in order to regulate the Vm. (M Yang and WJ Brackenbury.
Membrane potential and cancer progression. Frontiers in Physiol. 2013(4); 185: 1. http://dx.doi.org/10.3389/fphys.2013.00185)
It is well-established that cancer cells possess distinct bioelectrical properties. Notably, electrophysiological analyses in many cancer cell types have revealed a depolarized Vm that favors cell proliferation. Ion channels/transporters control cell volume and migration, and emerging data also suggest that the level of Vm has functional roles in cancer cell migration. In addition, yperpolarization is necessary for stem cell differentiation. For example, both osteogenesis and adipogenesis are hindered in human mesenchymal stem cells (hMSCs) under depolarizing conditions. Therefore, in the context of cancer, membrane depolarization might be important for the emergence and maintenance of cancer stem cells (CSCs), giving rise to sustained tumor growth. This review aims to provide a broad understanding of the Vm as a bioelectrical signal in cancer cells by examining several key types of ion channels that contribute to its regulation. The mechanisms by which Vm regulates cancer cell proliferation, migration, and differentiation will be discussed. In the long term, Vm might be avaluable clinical marker for tumor detection with prognostic value, and could even be artificially modified in order to inhibit tumor growth and metastasis.
Perspective beyond Cancer Genomics: Bioenergetics of Cancer Stem Cells
Hideshi Ishii, Yuichiro Doki, and Masaki Mori
Yonsei Med J 2010; 51(5):617-621. http://dx.doi.org/10.3349/ymj.2010.51.5.617 pISSN: 0513-5796, eISSN: 1976-2437
Although the notion that cancer is a disease caused by genetic and epigenetic alterations is now widely accepted, perhaps more emphasis has been given to the fact that cancr is a genetic disease. It should be noted that in the post-genome sequencing project period of the 21st century, the underlined phenomenon nevertheless could not be discarded towards the complete control of cancer disaster as the whole strategy, and in depth investigation of the factors associated with tumorigenesis is required for achieving it. Otto Warburg has won a Nobel Prize in 1931 for the discovery of tumor bioenergetics, which is now commonly used as the basis of positron emission tomography (PET), a highly sensitive noninvasive technique used in cancer diagnosis. Furthermore, the importance of the cancer stem cell (CSC) hypothesis in therapy-related resistance and metastasis has been recognized during the past 2 decades. Accumulating evidence suggests that tumor bioenergetics plays a critical role in CSC regulation; this finding has opened up a new era of cancer medicine, which goes beyond cancer genomics.
Efficient execution of cell death in non-glycolytic cells requires the generation of ROS controlled by the activity of mitochondrial H+-ATP synthase.
Gema Santamaría1,#, Marta Martínez-Diez1,#, Isabel Fabregat2 and José M. Cuezva1,*
Carcinogenesis 2006 27(5):925-935 http://dx.doi.org/10.1093/carcin/bgi315
There is a large body of clinical data documenting that most human carcinomas contain reduced levels of the catalytic subunit of the mitochondrial H+-ATP synthase. In colon and lung cancer this alteration correlates with a poor patient prognosis. Furthermore, recent findings in colon cancer cells indicate that down-regulation of the H+-ATP synthase is linked to the resistance of the cells to chemotherapy. However, the mechanism by which the H+-ATP synthase participates in cancer progression is unknown. In this work, we show that inhibitors of the H+-ATP synthase delay
staurosporine-induced cell death in liver cells that are dependent on oxidative phosphorylation for energy provision whereas it has no effect on glycolytic cells. Efficient execution of cell death requires the generation of reactive oxygen species (ROS) controlled by the activity of the H+-ATP synthase in a process that is concurrent with the rapid disorganization of the cellular mitochondrial network. The generation of ROS after staurosporine treatment is highly dependent on the mitochondrial membrane potential and most likely caused by reverse electron flow to Complex I. The generated ROS promote the carbonylation and covalent modification of cellular and mitochondrial proteins. Inhibition of the activity of the H+-ATP synthase blunted ROS production, prevented the oxidation of cellular proteins and the modification of mitochondrial proteins, delaying the release of cyt c and the execution of cell death. The results in this work establish the down-regulation of the H+-ATP synthase, and thus of oxidative phosphorylation, as part of the molecular strategy adapted by cancer cells to avoid reactive oxygen species-mediated cell death. Furthermore, the results provide a mechanistic explanation to understand chemotherapeutic resistance of cancer cells that rely on glycolysis as main energy provision pathway.
see also –
The tumor suppressor function of mitochondria: Translation into the clinics
José M. Cuezva, Álvaro D. Ortega, Imke Willers, et al.
Biochimica et Biophysica Acta (BBA) – Molecular Basis of Disease Dec 2009; 1792(12): 1145–1158 http://dx.doi.org/10.1016/j.bbadis.2009.01.006
Recently, the inevitable metabolic reprogramming experienced by cancer cells as a result of the onset of cellular proliferation has been added to the list of hallmarks of the cancer cell phenotype. Proliferation is bound to the synchronous fluctuation of cycles of an increased glycolysis concurrent with a restrained oxidative phosphorylation. Mitochondria are key players in the metabolic cycling experienced during proliferation because of their essential roles in the transduction of biological energy and in defining the life–death fate of the cell. These two activities are molecularly and functionally integrated and are both targets of commonly altered cancer genes. Moreover, energetic metabolism of the cancer cell also affords a target to develop new therapies because the activity of mitochondria has an unquestionable tumor suppressor function. In this review, we summarize most of these findings paying special attention to the opportunity that translation of energetic metabolism into the clinics could afford for the management of cancer patients. More specifically, we emphasize the role that mitochondrial β-F1-ATPase has as a marker for the prognosis of different cancer patients as well as in predicting the tumor response to therapy.
Self-Destructive Behavior in Cells May Hold Key to a Longer Life
Carl Zimmer, MY Times October 5, 2009
In recent years, scientists have found evidence of autophagy in preventing a much wider range of diseases. Many disorders, like Alzheimer’s disease, are the result of certain kinds of proteins forming clumps. Lysosomes can devour these clumps before they cause damage, slowing the onset of diseases.
Lysosomes may also protect against cancer. As mitochondria get old, they cast off charged molecules that can wreak havoc in a cell and lead to potentially cancerous mutations. By gobbling up defective mitochondria, lysosomes may make cells less likely to damage their DNA. Many scientists suspect it is no coincidence that breast cancer cells are often missing autophagy-related genes. The genes may have been deleted by mistake as a breast cell divided. Unable to clear away defective mitochondria, the cell’s descendants become more vulnerable to mutations.
Unfortunately, as we get older, our cells lose their cannibalistic prowess. The decline of autophagy may be an important factor in the rise of cancer, Alzheimer’s disease and other disorders that become common in old age. Unable to clear away the cellular garbage, our bodies start to fail.
If this hypothesis turns out to be right, then it may be possible to slow the aging process by raising autophagy. It has long been known, for example, that animals that are put on a strict low-calorie diet can live much longer than animals that eat all they can. Recent research has shown that caloric restriction raises autophagy in animals and keeps it high. The animals seem to be responding to their low-calorie diet by feeding on their own cells, as they do during famines. In the process, their cells may also be clearing away more defective molecules, so that the animals age more slowly.
Some scientists are investigating how to manipulate autophagy directly. Dr. Cuervo and her colleagues, for example, have observed that in the livers of old mice, lysosomes produce fewer portals on their surface for taking in defective proteins. So they engineered mice to produce lysosomes with more portals. They found that the altered lysosomes of the old experimental mice could clear away more defective proteins. This change allowed the livers to work better.
Essentiality of pyruvate kinase, oxidation, and phosphorylation
We can move to the next level with greater clarity. Yu et al. reported an important relationship between Pyruvate kinase M2 (PKM2) and the Warburg effect of cancer cells ( M Yu, et al. PIM2 phosphorylates PKM2 and promotes Glycolysis in Cancer Cells. J Biol Chem (PMID: 24142698) http://dx.doi.org10.1074/jbc.M113.508226 ). They found that PIM2 could directly phosphorylate PKM2 on the Thr454 residue, which resulted in an increase of PKM2 protein levels. PKM2 with a phosphorylation-defective mutation displayed a reduced effect on glycolysis compared to the wild-type, thereby co-activating HIF-1α and β-catenin, and enhanced mitochondria respiration and chemotherapeutic sensitivity of cancer cells. This indicated that PIM2-dependent phosphorylation of PKM2 is critical for regulating the Warburg effect in cancer, highlighting PIM2 as a potential therapeutic target.
In another study of the effect of 3 homoplastic mtDNA mutations on oxidative metabolism of osteosarcoma cells, there was a difference proportional to the magnitude of the defect. (Iommarini L, et al. Different mtDNA mutations modify tumor progression in dependence of the degree of respiratory complex I impairment. Hum Mol Genet. 2013 Nov 11. [Epub ahead of print]; PMID: 24163135 ). Osteosarcoma cells carrying the most marked impairment of the gene encoding mitochondrial complex I (CI) of oxidative phosphorylation displayed a reduced tumorigenic potential both in vitro and in vivo, when compared with cells with mild CI dysfunction. The severe CI dysfunction was an energetic defect associated with a compensatory increase in glycolytic metabolism and AMP-activated protein kinase activation. The result suggested that mtDNA mutations may display diverse impact on tumorigenic potential depending on the type and severity of the resulting oxidative phosphorylation dysfunction. The modulation of tumor growth was independent from reactive oxygen species production but correlated with hypoxia-inducible factor 1α stabilization, indicating that structural and functional integrity of CI and oxidative phosphorylation are required for hypoxic adaptation and tumor progression.
An unrelated finding shares some agreement with what has been identified (Systematic isolation of context-dependent vulnerabilities in NSCLC. Cell, 24 Oct 2013; 155 (3): 552-566, http://dx.doi.org/10.1016/ j.cell.2013.09.041). They report three distinct target/response-indicator pairings that are represented with significant frequencies (6%–16%) in the patient population. These include NLRP3 mutation/inflammasome activation-dependent FLIP addiction, co-occurring KRAS and LKB1 mutation-driven COPI addiction, and selective sensitivity to a synthetic indolotriazine that is specified by a seven-gene expression signature. This is depicted in the Figure below. The authors noted a frequency and diversity of somatic lesions detected among lung tumors can confound efforts to identify these targets.

The forging of a cancer-metabolism link and twists in the chain (Biome 19th April 2013)
Ten years ago, Grahame Hardie and Dario Alessi discovered that the elusive upstream kinase required for the activation of AMP-activated protein kinase (AMPK) by metabolic stress that the Hardie lab had been pursuing in their research on the metabolic regulator AMPK was the tumor suppressor, LKB1, that the neighbouring Alessi lab was working on at the time. This finding represented the first clear link between AMPK and cancer.
The resulting paper [1], published in 2003 in what was then Journal of Biology (now BMC Biology), was one [1] of three [2, 3] connecting these two kinases and that helped to swell of a surge of interest in the metabolism of tumor cells that was just beginning at about that time and is still growing. (LKB1 and AMPK and the cancer-metabolism link – ten years after. D Grahame Hardie, and Dario R Alessi. BMC Biology 2013, 11:36. http://dx doi.org.10.1186/1741-7007-11-36.)
In September 2003, both groups published a joint paper [1] in Journal of Biology (now BMC Biology) that identified the long-sought and elusive upstream kinase acting on AMP-activated protein kinase (AMPK) as a complex containing LKB1, a known tumor suppressor. Similar findings were reported at about the same time by David Carling and Marian Carlson [2] and by Reuben Shaw and Lew Cantley [3]; at the time of writing these three papers have received between them a total of over 2,000 citations. These findings provided a direct link between a protein kinase, AMPK, which at the time was mainly associated with regulation of metabolism, and another protein kinase, LKB1, which was known from genetic studies to be a tumor suppressor. While the idea that cancer is in part a metabolic disorder (first suggested by Warburg in the 1920s [4]) is well recognized today [5], this was not the case in 2003, and our paper perhaps contributed towards its renaissance.
The distinctive metabolic feature of tumor cells that enables them to meet the demands of unrestrained growth is the switch from oxidative generation of ATP to aerobic glycolysis – a phenomenon now well known as the Warburg effect. Operating this switch is one of the central functions of the AMP-activated protein kinase (AMPK) that has long been the focus of research in the Hardie lab. AMPK is an energy sensor that is allosterically tuned by competitive binding of ATP, ADP and AMP to sites on its g regulatory subunit (its portrait here, with AMP bound at two sites, was kindly provided by Bing Xiao and Stephen Gamblin). When phosphorylated by LKB1, AMPK responds to depletion of ATP by turning off anabolic reactions required for growth, and turning on catabolic reactions and oxidative phosphorylation – the reverse of the Warburg effect. In this light, it is not surprising that LKB1 is inactivated in some proportion of many different types of tumors.
AMPK as an energy sensor and metabolic switch
AMPK was discovered as a protein kinase activity that phosphorylated and inactivated two key enzymes of fatty acid and sterol biosynthesis: acetyl-CoA carboxylase (ACC) and 3-hydroxy-3-methylglutaryl-CoA reductase (HMGR). The ACC kinase activity was reported to be activated by 5’-AMP, and the HMGR kinase activity by reversible phosphorylation, but for many years the two activities were thought to be due to distinct enzymes. However, in 1987 the DGH laboratory showed that both were functions of a single protein kinase, which we renamed AMPK after its allosteric activator, 5’-AMP. It was subsequently found that AMPK regulated not only lipid biosynthesis, but also many other metabolic pathways, both by direct phosphorylation of metabolic enzymes, and through longer-term effects mediated by phosphorylation of transcription factors and co-activators. In general, AMPK switches off anabolic pathways that consume ATP and NADPH, while switching on catabolic pathways that generate ATP (Figure 1).

Summary of a selection of target proteins and metabolic pathways regulated by AMPK. Anabolic pathways switched off by AMPK are shown in the top half of the ‘wheel’ and catabolic pathways switched on by AMPK in the bottom half. Where a protein target for AMPK responsible for the effect is known, it is shown in the inner wheel; a question mark indicates that it is not yet certain that the protein is directly phosphorylated. For original references see [54].
Key to acronyms: ACC1/ACC2, acetyl-CoA carboxylases-1/-2; HMGR, HMG-CoA reductase; SREBP1c, sterol response element binding protein-1c; CHREBP, carbohydrate response element binding protein; TIF-1A, transcription initiation factor-1A; mTORC1, mechanistic target-of-rapamycin complex-1; PFKFB2/3, 6-phosphofructo-2-kinase, cardiac and inducible isoforms; TBC1D1, TBC1 domain protein-1; SIRT1, sirtuin-1; PGC-1α, PPAR-γ coactivator-1α; ULK1, Unc51-like kinase-1.

Regulation of AMPK. AMPK can be activated by increases in cellular AMP:ATP or ADP:ATP ratio, or Ca2+ concentration. AMPK is activated >100-fold on conversion from a dephosphorylated form (AMPK) to a form phosphorylated at Thr172 (AMPK-P) catalyzed by at least two upstream kinases: LKB1, which appears to be constitutively active, and CaMKKβ, which is only active when intracellular Ca2+ increases. Increases in AMP or ADP activate AMPK by three mechanisms: (1) binding of AMP or ADP to AMPK, causing a conformational change that promotes phosphorylation by upstream kinases (usually this will be LKB1, unless [Ca2+] is elevated); (2) binding of AMP or ADP, causing a conformational change that inhibits dephosphorylation by protein phosphatases; (3) binding of AMP (and not ADP), causing allosteric activation of AMPK-P. All three effects are antagonized by ATP, allowing AMPK to act as an energy sensor.

Members of the AMPK and AMPK-related kinase (ARK) family. All the kinases named in the figure are phosphorylated and activated by LKB1, although what regulates this phosphorylation is known only for AMPK. Alternative names are shown, where applicable.

Three possible mechanisms to explain how the AMPK-activating drugs metformin or phenformin might provide protection against cancer. (a) Metformin acts on the liver and other insulin target tissues by activating AMPK (and probably via other targets), normalizing blood glucose; this reduces insulin secretion from pancreatic β cells, reducing the growth-promoting effects of insulin (and high glucose) on tumor cells. Since metformin does not reduce glucose levels in normoglycemic individuals, this mechanism would only operate in insulin-resistant subjects. (b) Metformin or phenformin activates AMPK in pre-neoplastic cells, restraining their growth and proliferation and thus delaying the onset of tumorigenesis; this mechanism would only operate in cells where the LKB1-AMPK pathway was intact. (c) Metformin or phenformin inhibits mitochondrial ATP synthesis in tumor cells, promoting cell death. If the LKB1-AMPK pathway was down-regulated in the tumor cells, they would be more sensitive to cell death induced by the biguanides than surrounding normal cells.
Metformin and phenformin are biguanides that inhibit mitochondrial function and so deplete ATP by inhibiting its production . AMPK is activated by any metabolic stress that depletes ATP, either by inhibiting its production (as do hypoxia, glucose deprivation, and treatment with biguanides) or by accelerating its consumption (as does muscle contraction). By switching off anabolism and other ATP-consuming processes and switching on alternative ATP-producing catabolic pathways, AMPK acts to restore cellular energy homeostasis.
Findings that AMPK is activated in skeletal muscle during exercise and that it increases muscle glucose uptake and fatty acid oxidation led to the suggestion that AMPK-activating drugs might be useful for treating type 2 diabetes. Indeed, it turned out that AMPK is activated by metformin, a drug that had at that time been used to treat type 2 diabetes for over 40 years, and by phenformin , a closely related drug that had been withdrawn for treatment of diabetes due to side effects of lactic acidosis.
If only it were so simple. Effects of metformin on cancer in type 2 diabetics could be secondary to reduction in insulin levels, and although there is evidence for direct effects of AMPK activation on the development of tumors in mice, there is also recent evidence that tumors that become established without down-regulating LKB1 survive metformin better than those that have lost it – probably because metformin poisons the mitochondrial respiratory chain, depressing ATP levels, and cells in which AMPK can still be activated in response to the challenge do better than those in which it can’t.
In their review, Hardie and Alessi chart these twists and turns, and point to the explosion of further possibilities opened up by the discovery, since their 2003 publication, of at least one other class of kinase upstream of AMPK (the CaM kinases), and at least a dozen other downstream targets of LKB1 (AMPK-related kinases, or ARKs) – not to mention the innumerable downstream targets of AMPK; all which make half their schematic illustrations look like hedgehogs.
Analysis of respiration in human cancer
Bioenergetic profiling of cancer cells is of great potential because it can bring forward new and effective
Therapeutic strategies along with early diagnosis. Metabolic Control Analysis (MCA) is a methodology that enables quantification of the flux control exerted by different enzymatic steps in a metabolic network thus assessing their contribution to the system‘s function.
(T Kaambre,V Chekulayev, I Shevchuk, et al. Metabolic control analysis of respiration in human cancer tissue. Frontiers Physiol 2013 (4); 151: 1. http://dx.doi.org/10.3389/fphys.2013.00151)
Our main goal is to demonstrate the applicability of MCA for in situ studies of energy
Metabolism in human breast and colorectal cancer cells as well as in normal tissues .We seek to determine the metabolic conditions leading to energy flux redirection in cancer cells. A main result obtained is that the adenine nucleotide translocator exhibits the highest control of respiration in human breast cancer thus becoming a prospective therapeutic target. Additionally, we present evidence suggesting the existence of mitochondrial respiratory supercomplexes that may represent a way by which cancer cells avoid apoptosis. The data obtained show that MCA applied in situ can be insightful in cancer cell energetic research.

Metabolic control analysis of respiration in human cancer tissue.
Representative traces of change in the rate of oxygen consumption by permeabilized human colorectal cancer (HCC) fibers after their titration with increasing concentrations of mersalyl, an inhibitor of inorganic phosphate carrier (panel A). The values of respiration rate obtained were plotted vs. mersalyl concentration (panel B) and from the plot the corresponding flux control coefficient was calculated. Bars are ±SEM.
Oncologic diseases such as breast and colorectal cancers are still one of the main causes of premature death. The low efficiency of contemporary medicine in the treatment of these malignancies is largely mediated by a poor understanding of the processes involved in metastatic dissemination of cancer cells as well as the unique energetic properties of mitochondria from tumors. Current knowledge supports the idea that human breast and colorectal cancer cells exhibit increased rates of glucose consumption displaying Warburg phenotype,i.e.,elevated glycolysis even in the presence of oxygen (Warburg and Dickens, 1930; Warburg, 1956 ;Izuishietal., 2012). Notwithstanding, there are some evidences that in these malignancies mitochondrial oxidative phosphorylation (OXPHOS) is the main source of ATP rather than glycolysis. Cancer cells have been classified according to their pattern of metabolic remodeling depending of the relative balance between aerobic glycolysis and OXPHOS (Bellanceetal.,2012). The first type of tumor cells is highly glycolytic, the second OXPHOS deficient and the third type of tumors dislay enhanced OXPHOS. Recent studies strongly sug gest that cancer cells can utilize lactate, free fatty acids, ketone bodies, butyrate and glutamine as key respiratory substrate selic iting metabolic remodeling of normal surrounding cells toward aerobic glycolysis—“reverse Warburg”effect (Whitaker-Menezes et al.,2011;Salem et al.,2012;Sotgia et al.,2012;Witkiewicz et al., 2012).
In normal cells,the OXPHOS system is usually closely linked to phosphotransfer systems, including various creatine kinase(CK) isotypes,which ensure a safe operation of energetics over a broad functional range of cellular activities (Dzejaand Terzic,2003). However, our current knowledge about the function of CK/creatine (Cr) system in human breast and colorectal cancer is insufficient. In some malignancies, for example sarcomas the CK/Cr system was shown to be strongly downregulated (Beraetal.,2008;Patraetal.,2008). Our previous studies showed that the mitochondrial-bound CK (MtCK) activity was significantly decreased in HL-1 tumor cells (Mongeetal.,2009), as compared to normal parent cardiac cells where the OXPHOS is the main ATP source of and the CK system is a main energy carrier. In the present study,we estimated the role of MtCK in maintaining energy homeostasis in human colorectal cancer cells. Understanding the control and regulation of energy metabolism requires analytical tools that take into account the existing interactions between individual network components and their impact on systemic network function. Metabolic Control Analysis(MCA) is a theoretical framework relating the properties of metabolic systems to the kinetic characteristics of their individual enzymatic components (Fell,2005). An experimental approach of MCA has been already successfully applied to the studies of OXPHOS in isolated mitochondria (Tageretal.,1983; Kunzetal.,1999; Rossignoletal.,2000) and in skinned muscle fibers (Kuznetsovetal.,1997;Teppetal.,2010).

Values of basal (Vo) and maximal respiration rate (Vmax, in the presence of 2 mM ADP) and apparent Michaelis Menten constant (Km) for ADP in permeabilized human breast and colorectal cancer samples as well as health tissue. – See more at: http://journal.frontiersin.org/Journal/10.3389/fphys.2013.00151/full#sthash.VBXPdodj.dpuf
Role of Uncoupling Proteins in Cancer
Adamo Valle, Jordi Oliver and Pilar Roca *
Cancers 2010; 2: 567-591; http://dx.doi.org/10.3390/cancers2020567
Since Otto Warburg discovered that most cancer cells predominantly produce energy by glycolysis rather than by oxidative phosphorylation in mitochondria, much interest has been focused on the alterations of these organelles in cancer cells. Mitochondria have been shown to be key players in numerous cellular events tightly related with the biology of cancer. Although energy production relies on the glycolytic pathway in cancer cells, these organelles also participate in many other processes essential for cell survival and proliferation such as ROS production, apoptotic and necrotic cell death, modulation of oxygen concentration, calcium and iron homeostasis, and certain metabolic and biosynthetic pathways. Many of these mitochondrial-dependent processes are altered in cancer cells, leading to a phenotype characterized, among others, by higher oxidative stress, inhibition of apoptosis, enhanced cell proliferation, chemoresistance, induction of angiogenic genes and aggressive fatty acid oxidation. Uncoupling proteins, a family of inner mitochondrial membrane proteins specialized in energy-dissipation, has aroused enormous interest in cancer due to their relevant impact on such processes and their potential for the development of novel therapeutic strategies.
Uncoupling proteins (UCPs) are a family of inner mitochondrial membrane proteins whose function is to allow the re-entry of protons to the mitochondrial matrix, by dissipating the proton gradient and, subsequently, decreasing membrane potential and production of reactive oxygen species (ROS). Due to their pivotal role in the intersection between energy efficiency and oxidative stress UCPs are being investigated for a potential role in cancer. In this review we compile the latest evidence showing a link between uncoupling and the carcinogenic process, paying special attention to their involvement in cancer initiation, progression and drug chemoresistance.
The Warburg Effect
Uncoupling the Warburg effect from cancer
A Najafov and DR Alessi
Proc Nat Acad Sci www.pnas.org/cgi/doi/10.1073/pnas.1014047107
A remarkable trademark of most tumors is their ability to break down glucose by glycolysis at a vastly higher rate than in normal tissues, even when oxygen is copious. This phenomenon, known as the Warburg effect, enables rapidly dividing tumor cells to generate essential biosynthetic building blocks such as nucleic acids, amino acids, and lipids from glycolytic intermediates to permit growth and duplication of cellular components during division (1). An assumption dominating research in this area is that the Warburg effect is specific to cancer. Thus, much of the focus has been on uncovering mechanisms by which cancer-causing mutations influence metabolism to stimulate glycolysis.
This has lead to many exciting discoveries. For example, the p53 tumor suppressor can suppress glycolysis through its ability to control expression of key metabolic genes, such as phosphoglycerate mutase (2), synthesis of cytochrome C oxidase-2 (3), and TP53-induced glycolysis and apoptosis regulator (TIGAR) (4). Many cancer-causing mutations lead to activation of the Akt and mammalian target of rapamycin (mTOR) pathway that profoundly influences metabolism and expression of metabolic enzymes to promoteglycolysis (5).
Strikingly, all cancer cells but not nontransformed cells express a specific splice variant of pyruvate kinase, termed M2-PK, that is less active, leading to the build up of phosphoenolpyruvate (6). Recent work has revealed that reduced activity of M2-PK promotes a unique glycolytic pathway in which phosphoenolpyruvate is converted to pyruvate by a histidine-dependent phosphorylation of phosphoglycerate mutase, promoting assimilation of glycolytic products into biomass (7). However, despite these observations, one might imagine that the Warburg effect need not be specific for cancer and that any normal cell would need to stimulate glycolysis to generate sufficient biosynthetic materials to fuel expansion and division.
Recent work by Salvador Moncada’s group published in PNAS (8) and other recent work from the same group (9, 10) provides exciting evidence supporting the idea that the Warburg effect is also required for the proliferation of noncancer cells.
The key discovery was that the anaphase promoting complex/cyclosome-Cdh1(APC/C-Cdh1), a master regulator of the transition of G1 to S phase of the cell cycle, inhibits glycolysis in proliferating noncancer cells by mediating the degradation of two key metabolic enzymes, namely 6-phosphofructo-2-kinase/ fructose-2,6-bisphosphatase isoform3 (PFKFB3) (9, 10) and glutaminase-(Fig. 1) (8).

Fig. Mechanism by which APC/C-Cdh1 inhibits glycolysis and glutaminolysis to suppress cell proliferation.
APC/C-Cdh1 E3 ligase recognizes KEN-box–containing metabolic enzymes, such as PFKFB3 and glutaminase-1 (GLS1), and ubiquitinates and targets them for proteasomal degradation. This inhibits glycolysis and glutaminolysis, leading to decrease in metabolites that can be assimilated into biomass, thereby suppressing proliferation.
PFKFB3 potently stimulates glycolysis by catalyzing the formation of fructose-2,6-bisphosphate, the allosteric activatorof 6-phosphofructo-1-kinase (11). Glutaminase-1 is the first enzyme in glutaminolysis, converting glutamine to lactate, yielding biosyntheticintermediates required for cell proliferation (12).
APC/C is a cell cycle-regulated E3 ubiquitin ligase that promotes ubiquitination of a distinct set of cell cycle proteins containing either a D-box (destruction box) or a KEN-box, named after the essential Lys-Glu-Asn motif required for APC recognition (13). Among its well-known substrates are crucial cell cycle proteins, such as cyclin B1, securin, and Plk1. By ubiquitinating and targeting its substrates to 26S proteasome-mediated degradation, APC/C regulates processes in late mitotic stage, exit from mitosis, and several events in G1 (14). The Cdh1 subunit is the KENbox binding adaptor of the APC/C ligase and is essential for G1/S transition.
Importantly, APC/C-Cdh1 is inactivated at the initiation of the S-phase of the cell cycle when DNA and cellular organelles are replicated at the time of the greatest need for generation of biosynthetic materials. APC/C-Cdh1 is reactivated later at the mitosis/G1 phase of the cell cycle when there is a lower requirement for biomassgeneration.
Both PFKFB3 (9, 10) and glutaminase-1 (8) possess a KEN-box and are rapidly degraded in nonneoplastic lymphocytes during the cell cycle when APC/C-Cdh1 is active. Consistent with destruction being mediated by APC-C-Cdh1, ablation of the KEN-box prevents degradation of PFKFB3 (9, 10) and glutaminase-1 (8). Inhibiting the proteasomal-dependent degradation with the MG132 inhibitor
markedly increases levels of ubiquitinated PFKFB3 and glutaminase-1 (8). Moreover, overexpression of Cdh1 to activate APC/C-Cdh1 decreases levels of PFKFB3 as well as glutmaninase-1 and concomitantly inhibited glycolysis, as judged by decrease in lactate production. This effect is also observed when cells were treated with a glutaminase-1 inhibitor (6-diazo-5- oxo-L-norleucine) (8). The final evidence supporting the authors’ hypothesis is that proliferation and glycolysis is inhibited after shRNA-mediated silencing of either PFKFB3 or glutaminase-1 (8).
These results are interesting, because unlike most recent work in this area, Colombo et al. (8) link the Warburg effect to the machinery of the cell cycle that is present in all cells rather than to cancer driving mutations. Further work is required to properly define the overall importance of this pathway, which has thus far only been studied in a limited number of cells. It would also be of value to undertake a more detailed analysis of how the rate of glycolysis and other metabolic pathways vary during the cell cycle of normal and cancer cells…(see full 2 page article) at PNAS.
The Warburg Effect Suppresses Oxidative Stress Induced Apoptosis in a Yeast Model for Cancer
C Ruckenstuhl, S Buttner, D Carmona-Gutierre, et al.
PLoS ONE 2009; 4(2): e4592. http://dx.doi.org/10.1371/journal.pone.0004592
Colonies of Saccharomyces cerevisiae, suitable for manipulation of mitochondrial respiration and shows mitochondria-mediated cell death, were used as a model. Repression of respiration as well as ROS-scavenging via glutathione inhibited apoptosis, conferred a survival advantage during seeding and early development of this fast proliferating solid cell population. In contrast, enhancement of respiration triggered cell death.
Conclusion/Significance: The Warburg effect might directly contribute to the initiation of cancer formation – not only by enhanced glycolysis – but also via decreased respiration in the presence of oxygen, which suppresses apoptosis.
PIM2 phosphorylates PKM2 and promotes Glycolysis in Cancer Cells
Z Yu, L Huang, T Zhang, et al.
J Biol Chem 2013; http://dx.doi.org/10.1074/jbc.M113.508226
http://www.jbc.org/cgi/doi/10.1074/jbc.M113.508226
Serine/threonine protein kinase PIM2, a known oncogene is a binding partner of pyruvate kinase M2 (PKM2), a key player in the Warburg effect of cancer cells. PIM2 interacts with PKM2 and phosphorylates PKM2 on the Thr454 residue.
The phosphorylation of PKM2 increases glycolysis and proliferation in cancer cells.
The PIM2-dependent phosphoirylation of ZPKM2 is critical for regulating the Warburg effect in cancer.
Genome-Scale Metabolic Modeling Elucidates the Role of Proliferative Adaptation in Causing the Warburg Effect
Shlomi T, Benyamini T, Gottlieb E, Sharan R, Ruppin E
PLoS Comput Biol 2011; 7(3): e1002018. http://dx.doi.org/10.1371/journal.pcbi.1002018
The Warburg effect – a classical hallmark of cancer metabolism – is a counter-intuitive phenomenon in which rapidly proliferating cancer cells resort to inefficient ATP production via glycolysis leading to lactate secretion, instead of relying primarily on more efficient energy production through mitochondrial oxidative phosphorylation, as most normal cells do.
The causes for the Warburg effect have remained a subject of considerable controversy since its discovery over 80 years ago, with several competing hypotheses. Here, utilizing a genome-scale human metabolic network model accounting for stoichiometric and enzyme solvent capacity considerations, we show that the Warburg effect is a direct consequence of the metabolic adaptation of cancer cells to increase biomass production rate. The analysis is shown to accurately capture a three phase metabolic behavior that is observed experimentally during oncogenic progression, as well as a prominent characteristic of cancer cells involving their preference for glutamine uptake over other amino acids.
The metabolic advantage of tumor cells
Maurice Israël and Laurent Schwartz
Abstract
1- Oncogenes express proteins of “Tyrosine kinase receptor pathways”, a receptor family including insulin or IGF-Growth Hormone receptors. Other oncogenes alter the PP2A phosphatase brake over these kinases.
2- Experiments on pancreatectomized animals; treated with pure insulin or total pancreatic extracts, showed that choline in the extract, preserved them from hepatomas.
Since choline is a methyle donor, and since methylation regulates PP2A, the choline protection may result from PP2A methylation, which then attenuates kinases.
3- Moreover, kinases activated by the boosted signaling pathway inactivate pyruvate kinase and pyruvate dehydrogenase. In addition, demethylated PP2A would no longer dephosphorylate these enzymes. A “bottleneck” between glycolysis and the oxidative-citrate cycle interrupts the glycolytic pyruvate supply now provided via proteolysis and alanine transamination. This pyruvate forms lactate (Warburg effect) and NAD+ for glycolysis. Lipolysis and fatty acids provide acetyl CoA; the citrate condensation increases, unusual oxaloacetate sources are available. ATP citrate lyase follows, supporting aberrant transaminations with glutaminolysis and tumor lipogenesis. Truncated urea cycles, increased polyamine synthesis, consume the methyl donor SAM favoring carcinogenesis.
4- The decrease of butyrate, a histone deacetylase inhibitor, elicits epigenic changes (PETEN, P53, IGFBP decrease; hexokinase, fetal-genes-M2, increase)
5- IGFBP stops binding the IGF – IGFR complex, it is perhaps no longer inherited by a single mitotic daughter cell; leading to two daughter cells with a mitotic capability.
6- An excess of IGF induces a decrease of the major histocompatibility complex MHC1, Natural killer lymphocytes should eliminate such cells that start the tumor, unless the fever prostaglandin PGE2 or inflammation, inhibit them…
Introduction
The metabolic network of biochemical pathways forms a system controlled by a few switches, changing the finality of this system. Specific substrates and hormones control such switches. If for example, glycemia is elevated, the pancreas releases insulin, activating anabolism and oxidative glycolysis, energy being required to form new substance or refill stores. If starvation decreases glycemia, glucagon and epinephrine activate gluconeogenesis and ketogenesis to form nutriments, mobilizing body stores. The different finalities of the system are or oriented by switches sensing the NADH/NAD+, the ATP/AMP, the cAMP/AMP ratios or the O2 supply… We will not describe here these metabolic finalities and their controls found in biochemistry books.
Many of the switches depend of the phosphorylation of key enzymes that are active or not. Evidently, there is some coordination closing or opening the different pathways. Take for example gluconeogenesis, the citrate condensation slows down, sparing OAA, which starts the gluconeogenic pathway. In parallel, one also has to close pyruvate kinase (PK); if not, phosphoenolpyruvate would give back pyruvate, interrupting the pathway. Hence, the properties of key enzymes acting like switches on the pathway specify the finality of the system. Our aim is to show that tumor cells invent a new specific finality, with mixed glycolysis and gluconeogenesis features. This very special metabolism gives to tumor cells a selective advantage over normal cells, helping the tumor to develop at the detriment of the rest of the body.
I Abnormal metabolism of tumors, a selective advantage
The initial observation of Warburg 1956 on tumor glycolysis with lactate production is still a crucial observation [1]. Two fundamental findings complete the metabolic picture: the discovery of the M2 pyruvate kinase (PK) typical of tumors [2] and the implication of tyrosine kinase signals and subsequent phosphorylations in the M2 PK blockade [3–5].
A typical feature of tumor cells is a glycolysis associated to an inhibition of apoptosis. Tumors over-express the high affinity hexokinase 2, which strongly interacts with the mitochondrial ANT-VDAC-PTP complex. In this position, close to the ATP/ADP exchanger (ANT), the hexokinase receives efficiently its ATP substrate [6,7]. As long as hexokinase occupies this mitochondria site, glycolysis is efficient. However, this has another consequence, hexokinase pushes away from the mitochondria site the permeability transition pore (PTP), which inhibits the release of cytochrome C, the apoptotic trigger [8]. The site also contains a voltage dependent anion channel (VDAC) and other proteins. The repulsion of PTP by hexokinase would reduce the pore size and the release of cytochrome C. Thus, the apoptosome-caspase proteolytic structure does not assemble in the cytoplasm. The liver hexokinase or glucokinase, is different it has less interaction with the site, has a lower affinity for glucose; because of this difference, glucose goes preferentially to the brain.
Further, phosphofructokinase gives fructose 1-6 bis phosphate; glycolysis is stimulated if an allosteric analogue, fructose 2-6 bis phosphate increases in response to a decrease of cAMP. The activation of insulin receptors in tumors has multiple effects, among them; a decrease of cAMP, which will stimulate glycolysis.
Another control point is glyceraldehyde P dehydrogenase that requires NAD+ in the glycolytic direction. If the oxygen supply is normal, the mitochondria malate/aspartate (MAL/ASP) shuttle forms the required NAD+ in the cytosol and NADH in the mitochondria. In hypoxic conditions, the NAD+ will essentially come via lactate dehydrogenase converting pyruvate into lactate. This reaction is prominent in tumor cells; it is the first discovery of Warburg on cancer.
At the last step of glycolysis, pyruvate kinase (PK) converts phosphoenolpyruvate (PEP) into pyruvate, which enters in the mitochondria as acetyl CoA, starting the citric acid cycle and oxidative metabolism. To explain the PK situation in tumors we must recall that PK only works in the glycolytic direction, from PEP to pyruvate, which implies that gluconeogenesis uses other enzymes for converting pyruvate into PEP. In starvation, when cells need glucose, one switches from glycolysis to gluconeogenesis and ketogenesis; PK and pyruvate dehydrogenase (PDH) are off, in a phosphorylated form, presumably following a cAMP-glucagon-adrenergic signal. In parallel, pyruvate carboxylase (Pcarb) becomes active. Moreover, in starvation, much alanine comes from muscle protein proteolysis, and is transaminated into pyruvate. Pyruvate carboxylase first converts pyruvate to OAA and then, PEP carboxykinase converts OAA to PEP etc…, until glucose. The inhibition of PK is necessary, if not one would go back to pyruvate. Phosphorylation of PK, and alanine, inhibit the enzyme.
Well, tumors have a PK and a PDH inhibited by phosphorylation and alanine, like for gluconeogenesis, in spite of an increased glycolysis! Moreover, in tumors, one finds a particular PK, the M2 embryonic enzyme [2,9,10] the dimeric, phosphorylated form is inactive, leading to a “bottleneck “. The M2 PK has to be activated by fructose 1-6 bis P its allosteric activator, whereas the M1 adult enzyme is a constitutive active form. The M2 PK bottleneck between glycolysis and the citric acid cycle is a typical feature of tumor cell glycolysis.
We also know that starvation mobilizes lipid stores from adipocyte to form ketone bodies, they are like glucose, nutriments for cells. Growth hormone, cAMP, AMP, activate a lipase, which provides fatty acids; their β oxidation cuts them into acetyl CoA in mitochondria and in peroxisomes for very long fatty acids; forming ketone bodies. Normally, citrate synthase slows down, to spare acetyl CoA for the ketogenic route, and OAA for the gluconeogenic pathway. Like for starvation, tumors mobilize lipid stores. But here, citrate synthase activity is elevated, condensing acetyl CoA and OAA [11–13]; citrate increases, ketone bodies decrease. Consequently, ketone bodies will stop stimulating Pcarb. In tumors, the OAA needed for citrate synthase will presumably come from PEP, via reversible PEP carboxykinase or other sources. The quiescent Pcarb will not process the pyruvate produced by alanine transamination after proteolysis, leaving even more pyruvate to lactate dehydrogenase, increasing the lactate released by the tumor, and the NAD+ required for glycolysis.
Above the bottleneck, the massive entry of glucose accumulates PEP, which converts to OAA via mitochondria PEP carboxykinase, an enzyme requiring biotine-CO2-GDP. This source of OAA is abnormal, since Pcarb, another biotin-requiring enzyme, should have provided OAA. Tumors may indeed contain “morule inclusions” of biotin-enzyme [14] suggesting an inhibition of Pcarb, presumably a consequence of the maintained citrate synthase activity, and decrease of ketone bodies that normally stimulate Pcarb. The OAA coming via PEP carboxykinase and OAA coming from aspartate transamination or via malate dehydrogenase condenses with acetyl CoA, feeding the elevated tumoral citric acid condensation starting the Krebs cycle. Thus, tumors have to find large amounts of acetyl CoA for their condensation reaction; it comes essentially from lipolysis and β oxidation of fatty acids, and enters in the mitochondria via the carnitine transporter. This is the major source of acetyl CoA; since PDH that might have provided acetyl CoA remains in tumors, like PK, in the inactive phosphorylated form. The blockade of PDH [15] was recently reversed by inhibiting its kinase [16,17].
The key question is then to find out why NADH, a natural citrate synthase inhibitor did not switch off the enzyme in tumor cells. Probably, the synthesis of NADH by the dehydrogenases of the Krebs cycle and malate/aspartate shuttle, was too low, or the oxidation of NADH via the respiratory electron transport chain and mitochondrial complex1 (NADH dehydrogenase) was abnormally elevated. Another important point concerns PDH and α ketoglutarate dehydrogenase that are homologous enzymes, they might be regulated in a concerted way; when PDH is off, α ketoglutarate dehydrogenase might be also be slowed. Moreover, this could be associated to an upstream inhibition of aconinase by NO, or more probably to a blockade of isocitrate dehydrogenase, which favors in tumor cells, the citrate efflux from mitochondria, and the ATP citrate lyase route.
Normally, an increase of NADH inhibits the citrate condensation, favoring the ketogenic route associated to gluconeogenesis, which turns off glycolysis. Apparently, this regulation does not occur in tumors, since citrate synthase remains active. Moreover, in tumor cells, the α ketoglutarate not processed by
α ketoglutarate dehydrogenase converts to glutamate, via glutamate dehydrogenase, in this direction the reaction forms NAD+, backing up the LDH production. Other sources of glutamate are glutaminolysis, which increases in tumors [2].
The Figure Figure11 shows how tumors bypass the PK and PDH bottlenecks and evidently, the increase of glucose influx above the bottleneck, favors the supply of substrates to the pentose shunt, as pentose is needed for synthesizing ribonucleotides, RNA and DNA. The Figure Figure11 represents the stop below the citrate condensation. Hence, citrate quits the mitochondria to give via ATP citrate lyase, acetyl CoA and OAA in the cytosol of tumor cells. Acetyl CoA supports the synthesis of fatty acids and the formation of triglycerides. The other product of the ATP citrate lyase reaction, OAA, drives the transaminase cascade (ALAT and GOT transaminases) in a direction that consumes GLU and glutamine and converts in fine alanine into pyruvate and lactate plus NAD+. This consumes protein body stores that provide amino acids and much alanine (like in starvation).
The Figure Figure11 indicates that malate dehydrogenase is a source of NAD+ converting OAA into malate, which backs-up LDH. Part of the malate converts to pyruvate (malic enzyme) and processed by LDH. Moreover, malate enters in mitochondria via the shuttle and gives back OAA to feed the citrate condensation. Glutamine will also provide amino groups for the “de novo” synthesis of purine and pyrimidine bases particularly needed by tumor cells. The Figure Figure11 indicates that ASP shuttled out of the mitochondrial, joins the ASP formed by cytosolic transaminases, to feed the synthesis of pyrimidine bases via ASP transcarbamylase, a process also enhanced in tumor cells. In tumors, this silences the argininosuccinate synthetase step of the urea cycle [18–20].
This blockade also limits the supply of fumarate to the Krebs cycle. The latter, utilizes the α ketoglutarate provided by the transaminase reaction, since α ketoglutarate coming via aconitase slows down. Indeed, NO and peroxynitrite increase in tumors and probably block aconitase. The Figure Figure11 indicates the cleavage of arginine into urea and ornithine. In tumors, the ornithine production increases, following the polyamine pathway. Ornithine is decarboxylated into putrescine by ornithine decarboxylase, then it captures the backbone of S adenosyl methionine (SAM) to form polyamines spermine then spermidine, the enzyme controlling the process is SAM decarboxylase. The other reaction product, 5-methlthioribose is then decomposed into methylthioribose and adenine, providing purine bases to the tumor. We shall analyze below the role of SAM in the carcinogenic mechanism, its destruction aggravates the process.

In summary, it is like if the mechanism switching from gluconeogenesis to glycolysis was jammed in tumors, PK and PDH are at rest, like for gluconeogenesis, but citrate synthase is on. Thus, citric acid condensation pulls the glucose flux in the glycolytic direction, which needs NAD+; it will come from the pyruvate to lactate conversion by lactate dehydrogenase (LDH) no longer in competition with a quiescent Pcarb. Since the citrate condensation consumes acetyl CoA, ketone bodies do not form; while citrate will support the synthesis of triglycerides via ATP citrate lyase and fatty acid synthesis… The cytosolic OAA drives the transaminases in a direction consuming amino acid. The result of these metabolic changes is that tumors burn glucose while consuming muscle protein and lipid stores of the organism. In a normal physiological situation, one mobilizes stores for making glucose or ketone bodies, but not while burning glucose! Tumor cell metabolism gives them a selective advantage over normal cells. However, one may attack some vulnerable points.
II Starters for cancer metabolic anomaly
1. Lessons from oncogenes
Following the discovery of Rous sarcoma virus transmitting cancer [21], we have to wait the work of Stehelin [22] to realize that this retrovirus only transmitted a gene captured from a previous host. When one finds that the transmitted gene encodes the Src tyrosine kinase, we are back again to the tyrosine kinase signals, similar to those activated by insulin or IGF, which control carbohydrate metabolism, anabolism and mitosis.
An up regulation of the gene product, now under viral control causes tumors. However, the captured viral oncogene (v-oncogene) derives from a normal host gene the proto-oncogene. The virus only perturbs the expression of a cellular gene the proto-oncogene. It may modify its expression, or its regulation, or transmit a mutated form of the proto-oncogene. Independently of any viral infection, a similar tumorigenic process takes place, if the proto-oncogene is translocated in another chromosome; and transcribed under the control of stronger promoters. In this case, the proto-oncogene becomes an oncogene of cellular origin (c-oncogene). The third mode for converting a prot-oncogene into an oncogene occurs if a retrovirus simply inserts its strong promoters in front of the proto-oncogene enhancing its expression.
It is impressive to find that retroviral oncogenes and cellular oncogenes disturb this major signaling pathway: the MAP kinases mitogenic pathways. At the ligand level we find tumors such Wilm’s kidney cancer, resulting from an increased expression of insulin like growth factor; we have also the erbB or V-int-2 oncogenes expressing respectively NGF and FGF growth factor receptors. The receptors for these ligands activate tyrosine kinase signals, similarly to insulin receptors. The Rous sarcoma virus transmits the src tyrosine kinase, which activates these signals, leading to a chicken leukemia. Similarly, in murine leukemia, a virus captures and retransmits the tyrosine kinase abl. Moreover, abl is also stimulated if translocated and expressed with the bcr gene of chromosome 22, as a fusion protein (Philadelphia chromosome). Further, ahead Ras exchanging protein for GTP/GDP, and then the Raf serine-threonine kinases proto-oncogenes are known targets for oncogenes. Finally, at the level of transcription factors activated by MAP kinases, one finds cjun, cfos or cmyc. An avian leucosis virus stimulates cmyc, by inserting its strong viral promoter. The retroviral attacks boost the mitogenic MAP kinases similarly to inflammatory cytokins, or to insulin signals, that control glucose transport and gycolysis.
In addition to the MAP kinase mitogenic pathway, tyrosine kinase receptors activate PI3 kinase pathways; PTEN phosphatase counteracts this effect, thus acting as a tumor suppressor. Recall that a DNA virus, the Epstein-Barr virus of infectious mononucleose, gives also the Burkitt lymphoma; the effect of the virus is to enhance PI3 kinase. Down stream, we find mTOR (the target of rapamycine, an immune-suppressor) mTOR, inhibits PP2A phosphatase, which is also a target for the simian SV40 and Polyoma viruses. Schematically, one may consider that the different steps of MAP kinase pathways are targets for retroviruses, while the different steps of PI3 kinase pathway are targets for DNA viruses. The viral-driven enhanced function of these pathways mimics the effects of their prolonged activation by their usual triggers, such as insulin or IGF; one then expects to find an associated increase of glycolysis. The insulin or IGF actions boost the cellular influx of glucose and glycolysis. However, if the signaling pathway gets out of control, the tyrosine kinase phosphorylations may lead to a parallel PK blockade [3–5] explaining the tumor bottleneck at the end of glycolysis. Since an activation of enyme kinases may indeed block essential enzymes (PK, PDH and others); in principle, the inactivation of phosphatases may also keep these enzymes in a phosphorylated form and lead to a similar bottleneck and we do know that oncogenes bind and affect PP2A phosphatase. In sum, a perturbed MAP kinase pathway, elicits metabolic features that would give to tumor cells their metabolic advantage.
2. The methylation hypothesis and the role of PP2A phosphatase
In a remarkable comment, Newberne [23] highlights interesting observations on the carcinogenicity of diethanolamine [24] showing that diethanolamine decreased choline derivatives and methyl donors in the liver, like does a choline deficient diet. Such conditions trigger tumors in mice, particularly in the B6C3F1 strain. Again, the historical perspective recalled by Newberne’s comment brings us back to insulin. Indeed, after the discovery of insulin in 1922, Banting and Best were able to keep alive for several months depancreatized dogs, treated with pure insulin. However, these dogs developed a fatty liver and died. Unlike pure insulin, the total pancreatic extract contained a substance that prevented fatty liver: a lipotropic substance identified later as being choline [25]. Like other lipotropes, (methionine, folate, B12) choline supports transmethylation reactions, of a variety of substrates, that would change their cellular fate, or action, after methylation. In the particular case concerned here, the removal of triglycerides from the liver, as very low-density lipoprotein particles (VLDL), requires the synthesis of lecithin, which might decrease if choline and S-adenosyl methionine (SAM) are missing. Hence, a choline deficient diet decreases the removal of triglycerides from the liver; a fatty liver and tumors may then form. In sum, we have seen that pathways exemplified by the insulin-tyrosine kinase signaling pathway, which control anabolic processes, mitosis, growth and cell death, are at each step targets for oncogenes; we now find that insulin may also provoke fatty liver and cancer, when choline is not associated to insulin.
We must now find how the lipotropic methyl donor controls the signaling pathway. We know that after the tyrosine kinase reaction, serine-threonine kinases take over along the signaling route. It is thus highly probable that serine-threonine phosphatases will counteract the kinases and limit the intensity of the insulin or insulin like signals. One of the phosphatases involved is PP2A, itself the target of DNA viral oncogenes (Polyoma or SV40 antigens react with PP2A subunits and cause tumors). We found a possible link between the PP2A phosphatase brake and choline in works on Alzheimer’s disease [26]. Indeed, the catalytic C subunit of PP2A is associated to a structural subunit A. When C receives a methyle, the dimer recruits a regulatory subunit B. The trimer then targets specific proteins that are dephosphorylated [27].
In Alzheimer’s disease, the poor methylation of PP2A is associated to an increase of homocysteine in the blood [26]. The result of the PP2A methylation failure is a hyperphosphorylation of Tau protein and the formation of tangles in the brain. Tau protein is involved in tubulin polymerization, controlling axonal flow but also the mitotic spindle. It is thus possible that choline, via SAM, methylates PP2A, which is targeted toward the serine-threonine kinases that are counteracted along the insulin-signaling pathway. The choline dependent methylation of PP2A is the brake, the “antidote”, which limits “the poison” resulting from an excess of insulin signaling. Moreover, it seems that choline deficiency is involved in the L to M2 transition of PK isoenzymes [28].
3. Cellular distribution of PP2A
In fact, the negative regulation of Ras/MAP kinase signals mediated by PP2A phosphatase seems to be complex. The serine-threonine phosphatase does more than simply counteracting kinases; it binds to the intermediate Shc protein on the signaling cascade, which is inhibited [29]. The targeting of PP2A towards proteins of the signaling pathway depends of the assembly of the different holoenzymes. The carboxyl methylation of C-terminal leucine 309 of the catalytic C unit, permits to a dimeric form made of C and a structural unit A, to recruit one of the many regulatory units B, giving a great diversity of possible enzymes and effects. The different methylated ABC trimers would then find specific targets. It is consequently essential to have more information on methyl transferases and methyl esterases that control the assembly or disassembly of PP2A trimeric forms.
A specific carboxyl methyltransferase for PP2A [30] was purified and shown to be essential for normal progression through mitosis [31]. In addition, a specific methylesterase that demethylates PP2A has been purified [32]. Is seems that the methyl esterase cancels the action of PP2A, on signaling kinases that increase in glioma [33]. Evidently, the cellular localization of the methyl transferase (LCMT-1) and the phosphatase methyl esterase (PME-1) are crucial for controlling PP2A methylation and targeting. Apparently, LCMT-1 mainly localizes to the cytoplasm and not in the nucleus, where PME-1 is present, and the latter harbors a nuclear localization signal [34]. From these observations, one may suggest that PP2A gets its methyles in the cytoplasm and regulates the tyrosine kinase-signaling pathway, attenuating its effects.
A methylation deficit should then decrease the methylation of PP2A and boost the mitotic insulin signals as discussed above for choline deficiency, steatosis and hepatoma. At the nucleus, where PME-1 is present, it will remove the methyl, from PP2A, favoring the formation of dimeric AC species that have different targets, presumably proteins involved in the cell cycle. It is interesting to quote here the structural mechanism associated to the demethylation of PP2A. The crystal structures of PME-1 alone or in complex with PP2A dimeric core was reported [35] PME-1 binds directly to the active site of PP2A and this rearranges the catalytic triad of PME-1 into an active conformation that should demethylate PP2A, but this also seems to evict a manganese required for the phosphatase activity. Hence, demethylation and inactivation would take place in parallel, blocking mitotic actions.
However, another player is here involved, the so-called PTPA protein, which is a PP2A phosphatase activator. Apparently, this activator is a new type of cis/trans of prolyl isomerase, acting on Pro190 of the catalytic C unit isomerized in presence of Mg-ATP [36], which would then cancel the inactivation mediated by PME-1. Following the PTPA action, the demethylated phosphatase would become active again in the nucleus, and stimulate cell cycle proteins [37,38] inducing mitosis. Unfortunately, the ligand of this new prolyl isomerase is still unknown. Moreover, we have to consider that other enzymes such as cytochrome P450 have also demethylation properties.
In spite of deficient methylations and choline dehydrogenase pathway, tumor cells display an enhanced choline kinase activity, associated to a parallel synthesis of lecithin and triglycerides.
The hypothesis to consider is that triglycerides change the fate of methylated PP2A, by targeting it to the nucleus, there a methylesterase demethylates it; the phosphatase attacks new targets such as cell cycle proteins, inducing mitosis. Moreover, the phosphatase action on nuclear membrane proteins may render the nuclear membrane permeable to SAM the general methyl donor; promoters get methylated inducing epigenetic changes.
The relative decrease of methylated PP2A in the cytosol, not only cancels the brake over the signaling kinases, but also favors the inactivation of PK and PDH, which remain phosphorylated, contributing to the metabolic anomaly of tumor cells.
In order to prevent tumors, one should then favor the methylation route rather than the phosphorylation route for choline metabolism. This would decrease triglycerides, promote the methylation of PP2A and keep it in the cytosol, reestablishing the brake over signaling kinases.
Hypoxia is an essential issue to discuss
Many adequate “adult proteins” replace their fetal isoform: muscle proteins utrophine, switches to dystrophine; enzymes such as embryonic M2 PK [39] is replaced by M1. Hypoxic conditions seem to trigger back the expression of the fetal gene packet via HIF1-Von-Hippel signals. The mechanism would depend of a double switch since not all fetal genes become active after hypoxia. First, the histones have to be in an acetylated form, opening the way to transcription factors, this depends either of histone deacetylase (HDAC) inhibition or of histone acetyltransferase (HAT) activation, and represents the main switch. Second, a more specific switch must be open, indicating the adult/fetal gene couple concerned, or more generally the isoform of a given gene that is more adapted to the specific situation. When the adult gene mutates, an unbound ligand may indeed indicate, directly or indirectly, the particular fetal copy gene to reactivate [40]. In anoxia, lactate is more difficult to release against its external gradient, leading to a cytosolic increase of up-stream glycolytic products, 3P glycerate or others. These products may then be a second signal controlling the specific switch for triggering the expression of fetal genes, such as fetal hemoglobin or the embryonic M2 PK; this takes place if histones (main switch) are in an acetylated form.
Growth hormone-IGF actions, the control of asymmetrical mitosis
When IGF – Growth hormone operate, the fatty acid source of acetyl CoA takes over. Indeed, GH stimulates a triglyceride lipase in adipocytes, increasing the release of fatty acids and their β oxidation. In parallel, GH would close the glycolytic source of acetyl CoA, perhaps inhibiting the hexokinase interaction with the mitochondrial ANT site. This effect, which renders apoptosis possible, does not occur in tumor cells. GH mobilizes the fatty acid source of acetyl CoA from adipocytes, which should help the formation of ketone bodies, but since citrate synthase activity is elevated in tumors, ketone bodies do not form.
Compounds for correcting tumor metabolism
The figure figure1 indicates interrupted and enhanced metabolic pathways in tumor cells.
In table table1,1, the numbered pathways represent possible therapeutic targets; they cover several enzymes. When the activity of the pathway is increased, one may give inhibitors; when the activity of the pathway decreases, we propose possible activators

Table 1 Mol Cancer. 2011; 10 70. Published online Jun 7, 2011. doi 10.1186_1476-4598-10-70
The origin of Cancers by means of metabolic selection
The disruption of cells by internal or external compounds, releases substrates stimulating the tyrosine kinase signals for anabolism proliferation and stem cell repair, like for most oncogenes. If such signals are not limited, there is a parallel blockade of key metabolic enzymes by activated kinases or inhibited phosphatases. The result is a metabolism typical of tumor cells, which gives them a selective advantage; stabilized by epigenetic changes. A proliferation process, in which the two daughter cells divide, increases the tumor mass at the detriment of the body. Inevitable mutations follow.
What are lncRNAs?
It was traditionally thought that the transcriptome would be mostly comprised of mRNAs, however advances in high-throughput RNA sequencing technologies have revealed the complexity of our genome. Non-coding RNA is now known to make up the majority of transcribed RNAs and in addition to those that carry out well-known housekeeping functions (e.g. tRNA, rRNA etc), many different types of regulatory RNAs have been and continue to be discovered.
Long noncoding RNAs (lncRNAs) are a large and diverse class of transcribed RNA molecules with a length of more than 200 nucleotides that do not encode proteins. Their expression is developmentally regulated and lncRNAs can be tissue- and cell-type specific. A significant proportion of lncRNAs are located exclusively in the nucleus. They are comprised of many types of transcripts that can structurally resemble mRNAs, and are sometimes transcribed as whole or partial antisense transcripts to coding genes. LncRNAs are thought to carry out important regulatory functions, adding yet another layer of complexity to our understanding of genomic regulation.

The evolution of genome-scale models of cancer metabolism
The importance of metabolism in cancer is becoming increasingly apparent with the identification of metabolic enzyme mutations and the growing awareness of the influence of metabolism on signaling, epigenetic markers, and transcription. However, the complexity of these processes has challenged our ability to make sense of the metabolic changes in cancer. Fortunately, constraint-based modeling, a systems biology approach, now enables one to study the entirety of cancer metabolism and simulate basic phenotypes. With the newness of this field, there has been a rapid evolution of both the scope of these models and their applications. (NE Lewis and AM.Abdel-Haleem. frontiers physiol 2013;4(237): 1 http://dx.doi.org/10.3389/fphys.2013.00237)
Here we review the various constraint-based models built for cancer metabolism and how their predictions are shedding new light on basic cancer phenotypes, elucidating pathway differences between tumors, and discovering putative anti-cancer targets. As the field continues to evolve, the scope of these genome-scale cancer models must expand beyond central metabolism to address questions related to the diverse processes contributing to tumor development and metastasis.
“One of the goals of cancer research is to ascertain the mechanisms of cancer.”These words, penned by Dulbecco (1986), began a treatise on how a mechanistic understanding of cancer requires a sequenced human genome. Now with the abundance of sequence data, we are finding diverse genetic changes among different cancers (Vogelstein et al.,2013). While we are cataloging these mutations, the associated mechanisms leading to phenotypic changes are often unclear since mutations occur in the context of complex biological networks. For example, mutations to isocitrate dehydrogenase lead to oncometabolite synthesis, which alters DNA methylation and ultimately changes gene expression and the balance of normal cell processes (Sasakietal.,2012). Furthermore, many different combinations of mutations can lead to cancer. Since the genetic heterogeneity between tumors can be large, the biomolecular mechanisms underlying tumor physiology can vary substantially.
This is apparent in metabolism, where tumors can differ in serine metabolism dependence (Possematoetal., 2011) or TCA cycle function (Frezzaetal., 2011b). In addition, diverse mutations can alter NADPH synthesis by differentially regulat ing signaling pathways, such as the AMPK pathway (Cairnsetal., 2011; Jeonetal., 2012). The challenges regarding complexity and heterogeneity in cancer metabolism are beginning to be addressed with the COnstraint-Based Reconstruction and Analysis (COBRA) approach (Hernández Patiñoetal., 2012; Sharma and König, 2013), an emerging field in systems biology.Specifically, it accounts for the complexity of the perturbed biochemical processes by using genome-scale metabolic network reconstructions (Duarteetal., 2007; Maetal., 2007;Thieleetal., 2013).
In a reconstruction, the stoichiometric chemical reactions in a cell are carefully annotated and stitched together into a large network, often containing thousands of reactions. Genes and enzymes associated with each reaction are also delineated. The networks are converted into computational models and analyzed using many algorithms (Lewisetal., 2012). COBRA approaches are also beginning to address heterogeneity in cancer by integrating experimental data with the reconstructions (Blazier and Papin, 2012; Hydukeetal., 2013) to tailor the models to the unique gene expression profiles of general cancer tissue, and even individual cell lines and tumors. Here we describe the recent conceptual evolution that has occurred for constraint-based cancer modeling.
Targeting of gene expression
Tumor Suppressor Genes and its Implications in Human Cancer
Gain-of-function mutations in oncogenes and loss-of-function mutations in tumor suppressor genes (TSG) lead to cancer. In most human cancers, these mutations occur in somatic tissues. However, hereditary forms of cancer exist for which individuals are heterozygous for a germline mutation in a TSG locus at birth. The second allele is frequently inactivated by gene deletion, point mutation, or promoter methylation in classical TSGs that meet Knudson’s two-hit hypothesis. Conversely, the second allele remains as wild-type, even in tumors in which the gene is haplo-insufficient for tumor suppression. (K Inoue, EA Fry and Pj Taneja. Recent Progress in Mouse Models for Tumor Suppressor Genes and its Implications in Human Cancer. Clinical Medicine Insights: Oncology2013:7 103–122). This article highlights the importance of PTEN, APC, and other tumor suppressors for counteracting aberrant PI3K, β-catenin, and other oncogenic signaling pathways. We discuss the use of gene-engineered mouse models (GEMM) of human cancer focusing on Pten and Apc knockout mice that recapitulate key genetic events involved in initiation and progression of human neoplasia.
Targeting cancer metabolism – aiming at a tumour’s sweet-spot
Neil P. Jones and Almut Schulze
Drug Discovery Today January 2012
Targeting cancer metabolism has emerged as a hot topic for drug discovery. Most cancers have a high demand for metabolic inputs (i.e. glucose/glutamine), which aid proliferation and survival. Interest in targeting cancer metabolism has been renewed in recent years with the discovery that many cancer related (e.g. oncogenic and tumor suppressor) pathways have a profound effect on metabolism and that many tumors become dependent on specific metabolic processes. Considering the recent increase in our understanding of cancer metabolism and the increasing knowledge of the enzymes and pathways involved, the question arises: could metabolism be cancer’s Achilles heel?
During recent years, interest into the possible therapeutic benefit of targeting metabolic pathways in cancer has increased dramatically with academic and pharmaceutical groups actively pursuing this aspect of tumor physiology. Therefore, what has fuelled this revived interest in targeting cancer metabolism and what are the major advances and potential challenges faced in the race to develop new therapeutics in this area? This review will attempt to answer these questions and illustrate why we, and others, believe that targeting metabolism in cancer presents such a promising therapeutic rationale.
Oncogenes and cancer metabolism
Glycolysis TCA cycle Pentose phosphate pathway
FIGURE 1
Schematic representation of the regulation of cancer metabolism pathways. Metabolic enzymes are regulated by signaling pathways involving oncogenes and tumor suppressors. Complex regulatory mechanisms, key pathway interactions and enzymes are shown along with key metabolic endpoints (shown in purple) necessary for proliferation and survival (biosynthetic intermediates and NADPH). Key oncogenic pathways are shown in green and key tumor suppressor pathways are shown in red. Mutant IDH (mIDH) pathway is listed but is only functional in cancers containing mIDH.
FIGURE 2
Schematic representation of key components of the pentose phosphate pathway (PPP). Key enzymes are shown in blue boxes and key intermediates in purple text/box outline. DNA damage can activate ATM which in turn activates G6PDH to upregulate nucleotide synthesis for DNA repair and NAPDH to combat reactive oxygen species. PPP is also regulated by the tumour suppressor p53. The PPP can function as two separate branches (oxidative and non-oxidative) or be coupled into a recycling pathway – the pentose phosphate shunt – for maximum NADPH production.
Serine biosynthesis
Another branch diverting from glycolysis recently implicated in cancer is the serine biosynthesis pathway which converts the glycolytic intermediate 3-phosphoglycerate into serine (Fig. 3). Serine is an amino acid and an important neurotransmitter but can also provide fuel for the synthesis of other amino acids and nucleotides. The serine biosynthesis pathway also provides another key metabolic intermediate, a-KG, from glutamate breakdown via the action of phosphoserine aminotransferase (PSAT1). This pathway couples glycolysis (via 3-phosphoglycerate) with glutaminolysis (via glutamate), thereby linking two metabolic pathways known to be activated in many cancers.
FIGURE 3
Schematic representation of the serine biosynthesis pathway. Synthesis of serine involves integration of metabolites from glycolysis and glutaminolysis pathways and generates a-ketoglutarate, a key biosynthetic intermediate, and serine. Serine has many essential uses in the cell including amino acid, phospholipid and nucleotide synthesis.
Silencing of tumor suppressor genes by recruiting DNA methyltransferase 1 (DNMT1)
Ubiquitin-like containing PHD and Ring finger 1 (UHRF1) contributes to silencing of tumor suppressorgenes by recruiting DNA methyltransferase 1 (DNMT1) to their hemi-methylated promoters. Conversely,demethylation of these promoters has been ascribed to the natural anti-cancer drug, epigallocatechin-3-gallate (EGCG). The aim of the present study was to investigate whether the UHRF1/DNMT1 pair is an important target of EGCG action. (Mayada Achour, et al. Epigallocatechin-3-gallate up-regulates tumor suppressor gene expression via a reactive oxygen species-dependent down-regulation of UHRF1. Biochemical and Biophysical Research Communications 430 (2013) 208–212. http://dx.doi.org/10.1016/j.bbrc.2012.11.087)
Here, we show that EGCG down-regulates UHRF1 and DNMT1 expression in Jurkat cells, with subsequent up-regulation of p73 and p16INK4A genes. The down-regulation of UHRF1 is dependent upon the generation of reactive oxygen species by EGCG. Up-regulation of p16INK4A is strongly correlated with decreased promoter binding by UHRF1. UHRF1 over-expression counteracted EGCG-induced G1-arrested cells, apoptosis, and up-regulation of p16INK4A and p73. Mutants of the Set and Ring Associated (SRA) domain of UHRF1 were unable to down-regulate p16INK4A and p73, either in the presence or absence of EGCG. Our results show that down-regulation of UHRF1 is upstream to many cellular events, including G1 cell arrest, up-regulation of tumor suppressor genes and apoptosis.
Tumor Suppressor Activity of a Constitutively-Active ErbB4 Mutant
ErbB4 (HER4) is a member of the ErbB family of receptor tyrosine kinases, which includes the Epidermal Growth Factor Receptor (EGFR/ErbB1), ErbB2 (HER2/Neu), and ErbB3 (HER3). Mounting evidence indicates that ErbB4, unlike EGFR or ErbB2, functions as a tumor suppressor in many human malignancies. Previous analyses of the constitutively-dimerized and –active ErbB4 Q646C mutant indicate that ErbB4 kinase activity and phosphorylation of ErbB4 Tyr1056 are both required for the tumor suppressor activity of this mutant in human breast, prostate, and pancreatic cancer cell lines. However, the cytoplasmic region of ErbB4 possesses additional putative functional motifs, and the contributions of these functional motifs to ErbB4 tumor suppressor activity have been largely underexplored. (Citation: Richard M. Gallo, et al. (2013) Multiple Functional Motifs Are Required for the Tumor Suppressor Activity of a Constitutively-Active ErbB4 Mutant. J Cancer Res Therap Oncol 1: 1-10)
Here we demonstrate that ErbB4 BH3 and LXXLL motifs, which are thought to mediate interactions with Bcl family proteins and steroid hormone receptors, respectively, are required for the tumor suppressor activity of the ErbB4 Q646C mutant. Furthermore, abrogation of the site of ErbB4 cleavage by gamma-secretase also disrupts the tumor suppressor activity of the ErbB4 Q646C mutant. This last result suggests that ErbB4 cleavage and subcellular trafficking of the ErbB4 cytoplasmic domain may be required for the tumor suppressor activity of the ErbB4 Q646C mutant. Indeed, here we demonstrate that mutants that disrupt ErbB4 kinase activity, ErbB4 phosphorylation at Tyr1056, or ErbB4 cleavage by gamma-secretase also disrupt ErbB4 trafficking away from the plasma membrane and to the cytoplasm. This supports a model for ErbB4 function in which ErbB4 tumor suppressor activity is dependent on ErbB4 trafficking away from the plasma membrane and to the cytoplasm, mitochondria, and/or the nucleus.
EGF Receptor
Initiation of pancreatic ductal adenocarcinoma (PDA) is definitively linked to activating mutations in the KRAS oncogene. However, PDA mouse models show that mutant Kras expression early in development gives rise to a normal pancreas, with tumors forming only after a long latency or pancreatitis induction.
(CM Ardito,BM Gruner. ,EGF Receptor Is Required for KRAS-Induced Pancreatic Tumorigenesis. http://dx.doi.org/10.1016/j.ccr.2012.07.024)
Here, we show that oncogenic KRAS upregulates endogenous EGFR expression and activation, the latter being dependent on the EGFR ligand sheddase, ADAM17. Genetic ablation or pharmacological inhibition of EGFR or ADAM17 effectively eliminates KRAS-driven tumorigenesis in vivo. Without EGFR activity, active RAS levels are not sufficient to induce robust MEK/ERK activity, a requirement for epithelial transformation
The almost universal lethality of PDA has led to the intense study of genetic mutations responsible for its formation and progression. The most common oncogenic mutations associated with all PDA stages are found in the KRAS gene, suggesting it as the primary initiator of pancreatic neoplasia. However, mutant Kras expression throughout the mouse pancreatic parenchyma shows that the oncogene remains largely indolent until secondary events, such as pancreatitis, unlock its transforming potential. We find KRAS requires an inside-outside-in signaling axis that involves ligand-dependent EGFR activation to initiate the signal transduction and cell biological changes that link PDA and pancreatitis. (Cancer Cell (2012); 22: 304–317).
HER4 (EGFR/ErbB, HER2/Neu, HER3)
ErbB4 (HER4) is a member of the ErbB family of receptor tyrosine kinases, which includes the Epidermal Growth Factor Receptor (EGFR/ErbB1), ErbB2 (HER2/Neu), and ErbB3 (HER3). Mounting evidence indicates that ErbB4, unlike EGFR or ErbB2, functions as a tumor suppressor in many human malignancies. Previous analyses of the constitutively-dimerized and –active ErbB4 Q646C mutant indicate that ErbB4 kinase activity and phosphorylation of ErbB4 Tyr1056 are both required for the tumor suppressor activity of this mutant in human breast, prostate, and pancreatic cancer cell lines. However, the cytoplasmic region of ErbB4 possesses additional putative functional motifs, and the contributions of these functional motifs to ErbB4 tumor suppressor activity have been largely underexplored.
ErbB4 Possesses Multiple Functional Motifs and Mutations Have Been Engineered to Target These Motifs.
The organization of ErbB4 is as indicated in this schematic. The extracellular ligand-binding motifs reside in the amino-terminal region upstream of amino acid residue 651. The singlepass transmembrane domain consists of amino acid residues 652-675. The cytoplasmic tyrosine kinase domain consists of amino acid residues 713-989. The majority of cytoplasmic sites of tyrosine phosphorylation reside in amino acid residues 990-1308, most notably Tyr1056. Additional putative functional motifs include a TACE cleavage site, a gamma-secretase cleavage site, two LXXLL (steroid hormone receptor binding) motifs, a BH3 domain, three WW domain binding motifs, and a PDZ domain binding motif. Mutations that disrupt these motifs are noted. Finally, note the two locations of alternative transcriptional splicing, resulting in a total of four different splicing isoforms.
Here we demonstrate that ErbB4 BH3 and LXXLL motifs, which are thought to mediate interactions with Bcl family proteins and steroid hormone receptors, respectively, are required for the tumor suppressor activity of the ErbB4 Q646C mutant. Furthermore, abrogation of the site of ErbB4 cleavageby gamma-secretase also disrupts the tumor suppressor activity of the ErbB4 Q646C mutant. This last result suggests that ErbB4 cleavage and subcellular trafficking of the ErbB4 cytoplasmic domain may be required for the tumor suppressor activity of the ErbB4 Q646C mutant. Indeed, here we demonstrate that mutants that disrupt ErbB4 kinase activity, ErbB4 phosphorylation at Tyr1056, or ErbB4 cleavage by gamma-secretase also disrupt ErbB4 trafficking away from the plasma membrane and to the cytoplasm. This supports a model for ErbB4 function in which ErbB4 tumor suppressor activity is dependent on ErbB4 trafficking away from the plasma membrane and to the cytoplasm, mitochondria, and/or the nucleus.
(Richard M. Gallo, et al. (2013) Multiple Functional Motifs Are Required for the Tumor Suppressor Activity of a Constitutively-Active ErbB4 Mutant. J Cancer Res Therap Oncol 1: 1-10)
Resistance to Receptor Tyrosine Kinase Inhibition
Receptor tyrosine kinases (RTKs) are activated by somatic genetic alterations in a subset of cancers, and such cancers are often sensitive to specific inhibitors of the activated kinase. Two well-established examples of this paradigm include lung cancers with either EGFR mutations or ALK translocations. In these cancers, inhibition of the corresponding RTK leads to suppression of key downstream signaling pathways, such as the PI3K (phosphatidylinositol 3-kinase)/AKT and MEK (mitogen-activated protein kinase kinase)/ERK (extracellular signal–regulated kinase) pathways, resulting in cell growth arrest and death. Despite the initial clinical efficacy of ALK (anaplastic lymphoma kinase) and EGFR (epidermal growth factor receptor) inhibitors in these cancers, resistance invariably develops, typically within 1 to 2 years. (MJ Niederst and JA Engelman. Sci Signal, 24 Sep 2013; 6(294), p. re6 . http://dx.doi.org/10.1126/scisignal.2004652)
Over the past several years, multiple molecular mechanisms of resistance have been identified, and some common themes have emerged. One is the development of resistance mutations in the drug target that prevent the drug from effectively inhibiting the respective RTK. A second is activation of alternative RTKs that maintain the signaling of key downstream pathways despite sustained inhibition of the original drug target. Indeed, several different RTKs have been implicated in promoting resistance to EGFR and ALK inhibitors in both laboratory studies and patient samples. In this mini-review, we summarize the concepts underlying RTK-mediated resistance, the specific examples known to date, and the challenges of applying this knowledge to develop improved therapeutic strategies to prevent or overcome resistance.
The TGF-β Pathway
Aberrations in the enzymes that modify ubiquitin moieties have been observed to cause a myriad of diseases, including cancer. Therefore a better understanding of these enzymes and their substrates will lead to the identification of prospective druggable targets. Here we discuss the role of ubiquitin modifying enzymes in the canonical TGF-β pathway highlighting the ubiquitin regulating enzymes, which may potentially be targeted by small molecule inhibitors. (Pieter Eichhorn. (DE) -Ubiquitination in The TGF-β Pathway. J Cancer Res Therap Oncol 2013; 1: 1-6).
TGF-β is a multifunctional cytokine that plays a key role in embryogenesis and adult tissue homoeostasis. TGF-β is secreted by a myriad of cell types triggering a varied array of cellular functions including apoptosis, proliferation, migration, endothelial and mesenchymal transition, and extracellular matrix production. Downstream TGFβ responses can also be modulated by other signalling pathways (i.e. PI3K, ERK, WNT, etc.) resulting in a complex web of TGF-β pathway activation or repression depending on the nature of the signal and cellular context. Apart from TGF-β mediated cell autonomous effects TGF-β can further play an important function in regulating tumour microenvironments effecting the interaction between stromal fibroblasts and tumour cells.
Due to the central role of TGF-β in cellular processes it is therefore unsurprising that loss of TGF-β pathway integrity is frequently observed in a variety of human diseases, including cancer. However, the TGF-β pathway plays a complex dual role in cancer. In normal epithelial cells and premalignant cells TGF-β acts a potent tumor suppressor eliciting a cytostatic response inhibiting tumor progression. Supporting this notion, inactivating mutations in members of the TGF-βpathway have been observed in a variety of cancers including pancreatic, colorectal, and head and neck cancer.
In contrast, during tumor progression the TGF-β antiproliferative function is lost, and in certain advanced cancers TGF-β becomes an oncogenic factor inducing cellular proliferation, invasion, angiogenesis, and immune suppression. As a consequence, the TGFβ pathway is currently considered a therapeutic target in advanced cancers and several anti- TGF-β agents in clinical trials have shown promising results. However, due to the complex dichotomous role of TGF-β in oncogenesis a detailed understanding of TGF-β biology is required in order to design successful therapeutic strategies to identify patient populations that will benefit most from these compounds.
G protein receptor
G protein-coupled receptors (GPCRs) modulate a vast array of cellular processes. The current review gives an overview of the general characteristics of GPCRs and their role in physiological conditions. In addition, it describes the current knowledge of the physiological and pathophysiological functions of GPR55, an orphan GPCR, and how it can be exploited as a therapeutic target to combat various cancers.
(D Leyva-Illades, S DeMorrow . Orphan G protein receptor GPR55 as an emerging target in cancer therapy and management. Cancer Management and Research 2013:5 147–155)
Signal transduction is essential for maintaining cellular homeostasis and to coordinate the activity of cells in all organisms. Proteins localized in the cell membrane serve as the interface between the outside and inside of the cell. G protein-coupled receptors (GPCRs) are the largest and most diverse group of membrane receptors in eukaryotes and are encoded by at least 800 genes in the human genome. GPCRs are also known as seven-transmembrane domain receptors, 7TM receptors, heptahelical receptors, serpentine receptors, and G protein-linked receptors. GPCRs can detect an expansive array of extracellular signals or ligands that include photons, ions, odors, pheromones, hormones, and neurotransmitters. Nonsensory GPCRs (excluding light, odor, and taste receptors) have been classified into four families: class A rhodopsin-like, class B secretin-like, class C metabotropic glutamate/pheromone, and frizzled receptors. They have a peculiar structure that has been highly conserved over the course of evolution and are made up of an amino acid chain, the N-terminal of which is localized outside of the cellular membrane and the C-terminal in the cytoplasm. The amino acid chain spans the cellular membrane seven times and has three intracellular and three extracellular loops.
GPCRs are called that because they exert their actions by associating with a family of heterotrimeric proteins (made up of α, β, and γ subunits) that are capable of binding and hydrolyzing guanosine triphosphate (GTP).To date, 16 different α subunits, five β subunits, and 11 γ subunits have been described in mammalian tissues. When activated, these receptors undergo conformational changes that are mechanically transduced to the G proteins, which then initiate a cycle of activation and inactivationassociated with the binding and hydrolysis of GTP. Activated G proteins can then positively or negatively modulate ion channels (mainly potassium and calcium) or the second messenger generating enzymes (ie, adenylate cyclase and phospholipase C [PLC]) that allow the signal to be propagated to the interior of the cell to ultimately affect cell function.
Matrix Metalloproteinases
Degradation of extracellular matrix is crucial for malignant tumour growth, invasion, metastasis and angiogenesis. Matrix metalloproteinases (MMPs) are a family of zinc-dependent neutral endopeptidases collectively capable of degrading essentially all components of the ECM. Elevated levels of distinct MMPs can be detected in tumour tissue or serumof patients with advanced cancer and their role as prognostic indicators in cancer is studied. In addition, therapeutic intervention of tumour growth and invasion based on inhibition of MMP activity is under intensive investigation and several MMP inhibitors are in clinical trials in cancer. In this review, we discuss the current view on the feasibility of MMPs as prognostic markers and as targets for therapeutic intervention in cancer.
(MATRIX METALLOPROTEINASES IN CANCER: PROGNOSTIC MARKERS AND THERAPEUTIC TARGETS.
Pia Vihinen and Veli-Matti Kahari. Int. J. Cancer 2002;99: 157–166. http://dx.doi.org/10.1002/ijc.10329
Common properties of the MMPs include the requirement of zinc in their catalytic site for activity and their synthesis as inactive zymogens that generally need to be proteolytically cleaved to be active. Normally the MMPs are expressed only when and where needed for tissue remodeling accompanies various processes such as during embryonic development, wound healing, uterine and mammary involution, cartilage-to-bone transition during ossification, and trophoblast invasion into the endometrial stoma during placenta development. However, aberrant expression of various MMPs has been correlated with pathological conditions, such as periodontitis, rheumatoid arthritis, and tumor cell invasion and metastasis .
There are now over 20 members of the MMP family, and they can be subgrouped based on their structures. The minimal domain structure consists of a signal peptide, prodomain, and catalytic domain. The propeptide domain contains a conserved cysteine residue (the “cysteine switch”) that coordinates to the catalytic zinc to maintain inactivity. MMPs with only the minimal domain are referred to as matrilysins (MMP-7 and -26). The most common structures for secreted MMPs, including collagenases and stromelysins, have an additional hemopexin-like domain connected by a hinge region to the catalytic domain (MMP-1, -3, -8, -10, -12, -13, -19, and -20).
Terms: 1FN, fibronectin; 2M, 2-macroglobulin; 1PI, 1-proteinase inhibitor; COMP, cartilage oligomeric matrix protein; ND, not determined; TACE, TNF-converting enzyme; OP, osteopontin

FIGURE 1 – Structure of human matrix metalloproteinases. The signal peptide directs the proenzyme for secretion. The propeptide contains a conserved sequence (PRCGxPD), in which the cysteine forms a covalent bond (cysteine switch), with the catalytic zinc (Zn2_) to maintain the latency of proMMPs. Catalytic domain contains the highly conserved zinc binding site (HExGHxxGxxHS) in which Zn2_is coordinated by 3 histidines. The proline-rich hinge region links the catalytic domain to the hemopexin domain, which determines the substrate specificity of specific MMPs. The hemopexin domain is absent in matrilysin (MMP-7) and matrilysin-2 (endometase, MMP-26). Gelatinases A and B (MMP-2 and MMP-9, respectively) contain 3 repeats of the fibronectin-type II domain inserted in the catalytic domain. MT1-, MT2-, MT3- and MT5-MMP contain a transmembrane domain and MT4- and MT6-MMPs contain a glycosylphosphatidylinositol (GPI) anchor in the C-terminus of the molecule, which attach these MMPs to the cell surface. MT-MMPs, MMP-11, MMP-23 and MMP-28 contain a furin cleavage site (RxKR) between the propeptide and catalytic domain, making these proenzymes susceptible to activation by intracellular furin convertases. MMP-23 contains an N-terminal signal anchor, which anchors proMMP-23 to the Golgi complex and has a different C-terminal domain instead of hemopexin-like domain.
The physiologic expression of MMP-13 in vivo is limited to situations, such as fetal bone development and fetal wound repair, in which rapid remodeling of collagenous ECM is required. MMP-13 is expressed in pathologic conditions, such as arthritis, chronic dermal and intestinal ulcers, chronic periodontal inflammation and atherosclerotic plaques. The expression of MMP-13 is detected in vivo in invasive malignant tumours, breast carcinomas, squamous cell carcinomas (SCCs) of the head and neck and vulva, malignant melanomas, chondrosarcomas and urinary bladder carcinomas.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
MMP expression and activity are regulated at several levels. In most cases, MMPs are not synthesized until needed. Transcription can be induced by various signals including cytokines, growth factors, and mechanical stress. In certain cases, regulation of mRNA stability and translational efficiencyhave been reported. Because most MMPs are secreted as inactive zymogens, they need to be activated, usually by proteolytic cleavage of their NH2-terminal prodomains. Some MMPs are activated by other serine proteases such as plasmin and furin, whereas some of the MMPs can activate other members of their family. The most well characterized is the activation of pro-MMP-2 by MT1-MMP.
A number of MMPs have been strongly implicated in multiple stages of cancer progression including the acquisition of invasive and metastatic properties. Thus, efforts have been made for the past 20 years to develop MMPIs that can be used to halt the spread of cancer, which is what ultimately kills the person. However, initial clinical trials using first generation MMPIs proved to be disappointing . In the ensuing years, much has been learned about the roles of specific MMPs in the different processes of carcinogenesis and more specific MMPIs are being developed and brought to clinical trials.
However, the dosing and scheduling for optimal efficacy is not the same as required for conventional cytotoxic drugs because the MMPIs do not directly kill cancer cells, but instead target such processes as angiogenesis (the development of new blood vessels), invasion, and metastatic spread. (Matrix Metalloproteinases, Angiogenesis, and Cancer. Joyce E. Rundhaug. Commentary re: A. C. Lockhart et al., Reduction of Wound Angiogenesis in Patients Treated with BMS-275291, a Broad Spectrum Matrix Metalloproteinase Inhibitor. Clin. Cancer Res., 2003; 9551–554).
Role of p38 MAP Kinase Signal Transduction in Solid Tumors
HK Koul, M Pal, and S Koul. Genes & Cancer 2013 ; 4(9-10) 342–359. http://dx.doi.org/10.1177/ 1947601913507951
Mitogen-activated protein kinases (MAPKs) mediate a wide variety of cellular behaviors in response to extracellular stimuli. One of the main subgroups, the p38 MAP kinases, has been implicated in a wide range of complex biologic processes, such as cell proliferation, cell differentiation, cell death, cell migration, and invasion. Dysregulation of p38 MAPK levels in patients are associated with advanced stages and short survival in cancer patients (e.g., prostate, breast, bladder, liver, and lung cancer). p38 MAPK plays a dual role as a regulator of cell death, and it can either mediate cell survival or cell death depending not only on the type of stimulus but also in a cell type specific manner. In addition to modulating cell survival, an essential role of p38 MAPK in modulation of cell migration and invasion offers a distinct opportunity to target this pathway with respect to tumor metastasis. The specific function of p38 MAPK appears to depend not only on the cell type but also on the stimuli and/or the isoform that is activated.
Mitogen-activated protein kinase (MAPK) signal transduction pathways are evolutionarily conserved among eukaryotes and have been implicated to play key roles in a number of biological processes, including cell growth, differentiation, apoptosis, inflammation, and responses to environmental stresses.
They are typically organized in 3-tiered architecture consisting of a MAPK, a MAPK activator (MAPK kinase), and a MAPKK activator (MAPKK kinase). The MAPK pathways can be regulated at multiple levels as well as via multiple mechanisms, of which the regulation of mitogen-activated protein kinase kinase kinase (MAPKKK/MAP3K) has been proved to be the most challenging due to the great diversity and versatility between different modules at this level. The complex array of growth factors and other ligands that can initiate intracellular cell signaling requires a very high level of coordination among the different proteins involved.
GTP cyclohydrolase (GCH1)
GTP cyclohydrolase (GCH1) is the key-enzyme to produce the essential enzyme cofactor, tetrahydrobiopterin. The byproduct, neopterin is increased in advanced human cancer and used as cancer-biomarker, suggesting that pathologically increased GCH1 activity may promote tumor growth.
(G Picker, Hee-Young Lim, et al. Inhibition of GTP cyclohydrolase attenuates tumor growth by reducing angiogenesis and M2-like polarization of tumor associated macrophages. Int. J. Cancer 2003; 132: 591–604 (2013) http://dx.doi.org/10.1002/ijc.27706 )
We found that inhibition or silencing of GCH1 reduced tumor cell proliferation and survival and the tube formation of human umbilical vein endothelial cells, which upon hypoxia increased GCH1 and
endothelial NOS expression, the latter prevented by inhibition of GCH1. In nude mice xenografted with HT29-Luc colon cancer cells GCH1 inhibition reduced tumor growth and angiogenesis, determined by in vivo luciferase and near-infrared imaging of newly formed blood vessels. The treatment with the GCH1 inhibitor shifted the phenotype of tumor associated macrophages from the proangiogenic M2 towards M1, accompanied with a shift of plasma chemokine profiles towards tumor-attacking chemokines including CXCL10 and RANTES. GCH1 expression was increased in mouse AOM/DSS-induced colon tumors and in high grade human colon and skin cancer and oppositely, the growth of GCH1-deficient HT29-Luc tumor cells in mice was strongly reduced. The data suggest that GCH1 inhibition reduces tumor growth by (i) direct killing of tumor cells, (ii) by inhibiting angiogenesis, and (iii) by enhancing the antitumoral immune response.
The Role of Stroma in Tumour-Host Co-Existence
Molnár et al., The Role of Stroma in Tumour-Host Co-Existence: Some Perspectives in Stroma-Targeted Therapy of Cancer Biochem Pharmacol 2013, 2:1 http://dx.doi.org/10.4172/2167-0501.1000107
Cancer grows at the expense of the host as a parasite or superparasite following the second law of thermodynamics (conservation of energy). When the cancer cell progresses via replication to the special state called “spheroid”, a new phase begins with its intimate interaction and development of responses from the stroma which together assist in the formation of a full blown cancer. Among the processes involved are the development of blood vessels and lymphatic channels which are essential for maintenance and further growth of the cancer mass. In this way the condition of “parasitism” is completed with simultaneous suppression of the immune response of the host to the histo-incompatability of the tumor mass. Stroma/parenchyma promotes cancer invasion by feeding cancer cells and inducing immune tolerance. The dynamic changes in composition of stroma and biological consequences as feeder of cancer cells and immune tolerance can give a perspective for rational drug design in anti-stromal therapy. There are differences between normal and cancer cells at subcellular level such as compartmentalzation and structure of cytoskeleton and energy distribution (that is low generally, but locally high in normal cells). In cancer cannibalism of normal cells, the growing cancer mass is a factor for progression and invasion.
Cancer cells have been shown to kill normal cells and the products of cell death used for progression of growth of the cancer cell. Serum and growth factors produced by tumor stroma also provide the needed nutrients and conditions for further tumor growth. Cancer cannot feed off other cancer cells and therefore grow poorly. Probably, although not yet proven, the inability of cancer to “parasitise” other cancer cell types is probably due to some kind of competition or interference. The tumor is in charge of its own development due to its induction proteinases, lipid mobilization factors and angiogenetic factors as well as its ability to negate immune responses of the host response to what is in essence a foreign body.
In our review co-existence of normal and cancer cells in tumor with the growth promoting factors, and the immune tolerance mediating factors produced in the stromal and cancer cells/tissues will be discussed with perspective of stroma targeted therapy.
The clinical significance of cell cannibalism is well defined and described in a large number of publications. The direction of process of cancer development is defined as the tumor invades the normal tissue which never occurs in the reverse direction. This suggests that the cancer cell strives to achieve the lowest energy level possible. Therefore the first of the development of a full blown cancer can be considered as the 2nd Thermodynamic principle that explains, describes and drives the invading cancer into normal surrounding tissue.
From the normal living state, under particular conditions such as hypoxia, where ATP synthesis is decreased resulting in a switch to glycolytic pathways, cancer cells are selected from a fraction of the population [4]. Energetically, in the presence of electron transfer, by using high energy from respiration, the proliferating state is more stable than resting cells where a higher degree of protein stabilization occurs such as that needed for maintainance of the cytoskeleton of the cell. It was proposed that tumor-promotion might be controlled or modulated by small electronic currents originating from reactive oxygen species and transported through the cytoskeletal microfilament network of the cancer cell.
Aerobic glycolysis is the main energy producing process in cancer cells. Among many other aspects, recently the mitochondria have also been regarded as potential targets in the therapy of cancer. Several small molecules have been tested to restore their dysfunctional functions either by direct or indirect effects. Because of poorly functioning mitochondria, the electron transfer component of the respiration cycle is inefficient; therefore, cancer cells have smaller Gibbs energy than healthy cells. This means, that these cancer cells exists in a metastable state and are not able maintain normal cell structure.
Therefore, the cytoskeleton system is collapsed and dielectric bilayers are formed as a lower grade of cellular structure with decreased electron conductivity. Consequently, to halt cancer growth, one has to evaluate the process of cancer cell development in situ, where the primary tumor is growing as well as that of the metastatic cell that is invading surrounding or distal tissues. This affords one to suggest that the stroma is formed first during long term repeated oxidative stress, a process that is initially accompanied with inflammation due to an active immune response to the histoincompatability antigens present on the surface of the cancer cell. If the cancer cell evades the activity of killer T cells (Treg cells) by either secreting agents that reduce the response of the Treg cells or the immune system for whatever reason is ineffective (immunosuppressed states such as HIV/AIDS, pregnancy, transplantation therapy, etc.), the formed cancer cells have the opportunity to initiate tumor development. Because of the limited capacity of its electron transfer cycle, cancer cells are essentially starving cells that require glycolytically useful substrates. These substrates are obtained from the killing of normal cells by agents secreted by the cancer cell and the products yielded from dead normal cells “eaten” (phagocytosed) by the starving cancer cell which is digested by the cancer cells lysosomal system. This autophagic process of cannibalism keeps the cancer cell alive and thriving and is known as cytophagy, i.e., cannibalism of normal cells. This type of autophagocytosis results in a parasitic co-existence of tumor cells with normal cells and will determine the main pathway of interaction between the growing cancer tissue (tumor) and normal tissue where the cancer tissue gradually destroys normal tissues. This process obeys the second law of thermodynamics-conservation of energy within a defined system.
Treatments for Cancer
Bosutinib: a SRC–ABL tyrosine kinase inhibitor for treatment of chronic myeloid leukemia.
FE Rassi, HJ Khoury. Pharmacogenomics and Personalized Medicine 2013:6 57–62.
Bosutinib is one of five tyrosine kinase inhibitors commercially available in the United States for the treatment of chronic myeloid leukemia. This review of bosutinib summarizes the mode of action, pharmacokinetics, efficacy and safety data, as well as the patient-focused perspective through quality-of-life data. Bosutinib has shown considerable and sustained efficacy in chronic myeloid leukemia, especially in the chronic phase, with resistance or intolerance to prior tyrosine kinase inhibitors. Bosutinib has distinct but manageable adverse events. In the absence of T315I and V299L mutations, there are no absolute contraindications for the use of bosutinib in this patient population
Chronic myeloid leukemia (CML) is a clonal myeloproliferative stem cell disorder characterized by the presence of a signature hybrid oncogene, the BCR–ABL. The Philadelphia chromosome (Ph+) results from a reciprocal translocation between chromosome 9 and chromosome 22 that juxtaposes the two genes BCR and ABL and drives the leukemogenesis in CML. The ABL gene encodes for a nonreceptor tyrosine kinase that becomes deregulated and constitutively active after the juxtaposition of BCR. BCR–ABL is central in controlling downstream pathways involved in cell proliferation, regulation of cellular adhesion, and apoptosis.The understanding of the importance of this kinase activity in the pathophysiology of CML led to the development of tyrosine kinase inhibitors (TKI) that specifically target BCR–ABL. These agents became the mainstay of modern therapy in CML. CML has a triphasic clinical course, and the majority of patients (∼80%) are diagnosed during the early phase or the chronic phase (CP). However, and without effective treatment, CML invariably progresses to the advanced phases of the disease – the accelerated phase (AP) and the blast phase (BP). BP CML is a lethal refractory secondary leukemia with a short predicted survival.
Comprehensive molecular portraits of human breast tumors
The Cancer Genome Atlas Network
Nature. 2012 October 4; 490(7418): 61–70. http://dx.doi.org/10.1038/nature11412.
We analyzed primary breast cancers by genomic DNA copy number arrays, DNA methylation, exome sequencing, mRNA arrays, microRNA sequencing and reverse phase protein arrays. Our ability to integrate information across platforms provided key insights into previously-defined gene expression subtypes and demonstrated the existence of four main breast cancer classes when combining data from five platforms, each of which shows significant molecular heterogeneity.
Somatic mutations in only three genes (TP53, PIK3CA and GATA3) occurred at > 10% incidence across all breast cancers; however, there were numerous subtype-associated and novel gene mutations including the enrichment of specific mutations in GATA3, PIK3CA and MAP3K1 with the Luminal A subtype. We identified two novel protein expression-defined subgroups, possibly contributed by stromal/microenvironmental elements, and integrated analyses identified specific signaling pathways dominant in each molecular subtype including a HER2/p-HER2/HER1/p-HER1 signature within the HER2-Enriched expression subtype. Comparison of Basal-like breast tumors with high-grade Serous Ovarian tumors showed many molecular commonalities, suggesting a related etiology and similar therapeutic opportunities. The biologic finding of the four main breast cancer subtypes caused by different subsets of genetic and epigenetic abnormalities raises the hypothesis that much of the clinically observable plasticity and heterogeneity occurs within, and not across, these major biologic subtypes of breast cancer.
Most molecular studies of breast cancer have focused on just one or two high information content platforms, most frequently mRNA expression profiling or DNA copy number analysis, and more recently massively parallel sequencing. Supervised clustering of mRNA expression data has reproducibly established that breast cancers encompass several distinct disease entities, often referred to as the intrinsic subtypes of breast cancer. The recent development of additional high information content assays focused on abnormalities in DNA methylation, microRNA expression and protein expression, provide further opportunities to more completely characterize the molecular architecture of breast cancer.
Synbiology contribution and Nanotechnology
Synthetic RNAs Designed to Fight Cancer
Xiaowei Wang and his colleagues at Washington University School of Medicine in St. Louis have designed synthetic molecules that combine the advantages of two experimental RNA therapies against cancer. They have designed synthetic molecules that combine the advantages of two experimental RNA therapies against cancer. RNA plays an important role in how genes are turned on and off in the body. Both siRNAs and microRNAs are snippets of RNA known to modulate a gene’s signal or shut it down entirely. Separately, siRNA and microRNA treatment strategies are in early clinical trials against cancer, but few groups have attempted to marry the two.
“We are trying to merge two largely separate fields of RNA research and harness the advantages of both,” said Xiaowei Wang, assistant professor of radiation oncology and a research member of the Siteman Cancer Center. The study appears in the December issue of the journal RNA.
“We designed an artificial RNA that is a combination of siRNA and microRNA,” Wang said “our artificial RNA simultaneously inhibits both cell migration and proliferation.” For therapeutic purposes, “small interfering” RNAs, or siRNAs, are designed and assembled in a lab and can be made to shut down– or interfere with– a single specific gene that drives cancer. The siRNA molecules work extremely well at silencing a gene target because the siRNA sequence is made to perfectly complement the target sequence, thereby silencing a gene’s expression.
Though siRNAs are great at turning off the gene target, they also have potentially dangerous side effects: siRNAs inadvertently can shut down other genes that need to be expressed to carry out tasks that keep the body healthy. The siRNAs interfere with off-target genesthat closely complement their “seed region,” a section of the siRNA that governs binding to a gene target. “In the past, we tried to block the seed region in an attempt to reduce the side effects. Until now, we never tried to replace the seed region completely.”
Wang and his colleagues asked whether they could replace the siRNA’s seed region with the seed region from microRNA. Unlike siRNA, microRNA is a natural part of the body’s gene expression. And it can also shut down genes. As such, the microRNA seed region (with its natural targets) might reduce the toxic side effects caused by the artificial siRNA seed region. Plus, the microRNA seed region would add a new tool to shut down other genes that also may be driving cancer.
Wang’s group started with a bioinformatics approach, using a computer algorithm to design siRNA sequences against a common driver of cancer, a gene called AKT1 that encourages uncontrolled cell division. The program also selected siRNAs against AKT1 that had a seed region highly similar to the seed region of a microRNA known to inhibit a cell’s ability to move, thus potentially reducing the cancer’s ability to spread.
A Neutralizing RNA Aptamer
Nucleic acid aptamers have been developed as high-affinity ligands that may act as antagonists of disease-associated proteins. Aptamers are non immunogenic and characterised by high specificity and low toxicity thus representing a valid alternative to antibodies or soluble ligand receptor traps/decoys to target specific cancer cell surface proteins in clinical diagnosis and therapy. The epidermal growth factor receptor (EGFR) has been implicated in the development of a wide range of human cancers including breast, glioma and lung. The observation that its inhibition can interfere with the growth of such tumors has led to the design of new drugs including monoclonal antibodies and tyrosine kinase inhibitors currently used in clinic. However, some of these molecules can result in toxicity and acquired resistance, hence the need to develop novel kinds of EGFR-targeting drugs with high specificity and low toxicity.
(CL Esposito, D Passaro, et al. A Neutralizing RNA Aptamer against EGFR Causes Selective Apoptotic Cell Death. PLoS ONE 6(9): e24071. http://dx.doi.org/10.1371/journal.pone.0024071)
Here we generated, by a cell-Systematic Evolution of Ligands by EXponential enrichment (SELEX) approach, a nuclease resistant RNA-aptamer that specifically binds to EGFR with a binding constant of 10 nM. When applied to EGFR-expressing cancer cells the aptamer inhibits EGFR-mediated signal pathways causing selective cell death. Furthermore, at low doses it induces apoptosis even of cells that are resistant to the most frequently used EGFR-inhibitors, such as gefitinib and cetuximab, and inhibits tumor growth in a mouse xenograft model of human non-small-cell lung cancer (NSCLC). Interestingly, combined treatment with cetuximab and the aptamer shows clear synergy in inducing apoptosis in vitro and in vivo. In conclusion, we demonstrate that this neutralizing RNA aptamer is a promising bio-molecule that can be developed as a more effective alternative to the repertoire of already existing EGFR-inhibitors.
In-Silico Molecular Docking Analysis of Cancer Biomarkers
Currently, in the research scenario for cancer, the identification of anti-cancer drugs using immuno-modulatory proteins and other molecular agents to initiate apoptosis in cancer cells and to inhibit the signaling pathways of cancer biomarkers as a drug targeted therapy, for cancer cell proliferation assays by the researchers. In-Silico analysis is used to recognize anticancer compounds as a future prospective for In-Vitro and In-Vivo analysis. A large number of herbal remedies (e.g. garlic, mistletoe) are used by cancer patients for treating the cancer and/or reducing the toxicities of chemotherapeutic drugs. Some herbal medicines have shown potentially beneficial effects on cancer progression and may ameliorate chemotherapy-induced toxicities. (K. Gowri Shankar et al., In-Silico Molecular Docking Analysis of Cancer Biomarkers with Bioactive Compounds of Tribulus terrestris. Intl J NOVEL TRENDS PHARMAL SCI. 2013; 3(4).
Tribulus terrestris is mentioned in ancient Indian Ayurvedic medical texts dating back thousands of years. Tribulus terrestris has been widely used in the Ayurvedic system of medicine for the treatment of sexualdysfunction and various urinary disorders. The aim of the present study is to evaluate the interactions of some bioactive compounds of Tribulus terrestris for In-Silico anticancer analysis with cancer biomarkers as targets. The targeted biomarkers for analysis include NSE-Lung cancer, Follistatin-Prostrate cancer, GGT Hepatocellular carcinoma, Human Prostasin-Ovarian cancer.
GC-MS analysis of Tribulus terrestris whole plant methanol extract revealed the existence of the major compound like 3,7,11,15-tetramethylhexadec-2-en-1-ol, 1,2-Benzenedicarboxylic acid, disooctyl ester, 9,12,15-Octadecatrienoic acid, (z,z,z)-, 9,12-Octadecadienoic acid (z,z)-, Hexadecadienoic acid, ethyl ester, n-Hexadecadienoic acid, Octadecanoic acid, Phytol, α-Amyrin are chosen as ligands. Hence, by analyzing the minimum binding energy of the ligand binding complex with the receptors by dockinganalysis using AutoDock tools will show effective nature of inhibition of these receptors by the unique ligands. Based on the results low minimum binding energy ligands are identified and used as a future studies can be done for specific receptors docking.
Anti-Cancerous Effect of4,4′-Dihydroxychalcone ((2E,2′E)-3,3′-(1,4-Phenylene) Bis (1-(4-hydroxyphenyl) Prop-2-en-1-one)) on T47D Breast Cancer Cell Line
Narges Mahmoodi, T Besharati-Seidani, N Motamed, and NO Mahmoodi*
Annual Research & Review in Biology 2014; 4(12): 2045-2052
SCIENCEDOMAIN international www.sciencedomain.org
Aims: The majority of human breast tumors are estrogen receptor α (ERα) positive. However, not all of the ERα+ breast cancers respond to anti-estrogens drugs for those women who do respond, initial positive responses can be of short duration. Thus, more effective drugs are needed to enhance the efficacy of anti-estrogens drugs or to be used separately in a period of time. In view of potential cytotoxicity associated with silybin as polyhydroxy compounds a synthetic 4-hydroxychalcones (bis-phenol) was considered to explore its anti-carcinogenic effects in comparison to silybin on ERα+ breast cancer cell line.
Methodology: We have studied the inhibitory effect of 4,4′-dihydroxychalcone on the T47D breast cancer cell line by MTT test and the IC50s were estimated using Pharm PCS.
Results: The 4,4′-dihydroxychalcone showed significant dose- and time-dependent cell growth inhibitory effects on T47D breast cancer cells. The IC50 of 4,4′-dihydroxychalcone on T47D cells after 24 and 48 hours was 160.88+/–1 μM, 62.20+/–1 μM and for silybin was 373.42+/-1 μM,176.98+/–1 μM respectively.
Conclusion: Our results strongly suggests that this premade synthetic 4,4′-dihydroxychalcone can promote anti carcinogenic actions on T47D cell line. All 4,4′-dihydroxychalcone doses had a much larger inhibitory effect on cell viability than silybin doses in T47D cells. The ratio of the IC50 of 4,4′-dihydroxychalcone to silybin after 24 and 48 hours was 1: 2.3 and 1: 2.8 respectively.
Anticancer and multidrug resistance-reversal effects of solanidine analogs synthetized from pregnadienolone acetate.
István Zupkó, Judit Molnár, Borbála Réthy, Renáta Minorics, Eva Frank, et al.
Molecules (Impact Factor: 2.43). 01/2014; 19(2):2061-76. http://dx.doi.org/10.3390/molecules19022061
Source: PubMed
ABSTRACT A set of solanidine analogs with antiproliferative properties were recently synthetized from pregnadienolone acetate, which occurs in Nature. The aim of the present study was an in vitro characterization of their antiproliferative action and an investigation of their multidrug resistance-reversal activity on cancer cells. Six of the compounds elicited the accumulation of a hypodiploid population of HeLa cells, indicating their apoptosis-inducing character, and another one caused cell cycle arrest at the G2/M phase. The most effective agents inhibited the activity of topoisomerase I, as evidenced by plasmid supercoil relaxation assays. One of the most potent analogs down-regulated the expression of cell-cycle related genes at the mRNA level, including tumor necrosis factor alpha and S-phase kinase-associated protein 2, and induced growth arrest and DNA damage protein 45 alpha. Some of the investigated compounds inhibited the ABCB1 transporter and caused rhodamine-123 accumulation in murine lymphoma cells transfected by human MDR1 gene, expressing the efflux pump (L5178). One of the most active agents in this aspect potentiated the antiproliferative action of doxorubicin without substantial intrinsic cytostatic capacity. The current results indicate that the modified solanidine skeleton is a suitable substrate for the rational design and synthesis of further innovative drug candidates with anticancer activities.
Nutrition and Cancer
Ascorbic Acid and Selenium Interaction: Its Relevance in Carcinogenesis
Michael J. Gonzalez
Journal of Orthomolecular Medicine 1990; 5(2)
Ascorbic acid and selenium are two nutrients that seem to have a preventive potential in the process of carcinogenesis; because of a possible synergistic action that may produce an enhanced anticarcinogenic effect. Interaction between these nutrients have been reported. Results indicate that the protective effect of the inorganic form of selenium (Na Selenite) was nullified by ascorbic acid, whereas the chemopreventive action of the organic form (seleno-DL-methionine) was not affected.
A possibility exists that Selenite is reduced by ascorbic acid to elemental selenium and is therefore not available for tissue uptake. In experiments using Selenite; plasma and erythrocyte glutathione peroxidase enzyme activity was directly related to the level of ascorbic acid fed.
Complementary RNA and Protein Profiling Identifies Iron as a Key Regulator of Mitochondrial Biogenesis
J W. Rensvold, Shao-En On, A Jeevananthan, et al.
Cell Rep. 2013 January 31; 3(1): . http://dx.doi.org/10.1016/j.celrep.2012.11.029
Mitochondria are centers of metabolism and signaling whose content and function must adapt to
changing cellular environments. The biological signals that initiate mitochondrial restructuring
and the cellular processes that drive this adaptive response are largely obscure. To better define
these systems, we performed matched quantitative genomic and proteomic analyses of mouse
muscle cells as they performed mitochondrial biogenesis. We find that proteins involved in
cellular iron homeostasis are highly coordinated with this process and that depletion of cellular
iron results in a rapid, dose-dependent decrease of select mitochondrial protein levels and
oxidative capacity. We further show that this process is universal across a broad range of cell
types and fully reversed when iron is reintroduced. Collectively, our work reveals that cellular iron
is a key regulator of mitochondrial biogenesis, and provides quantitative data sets that can be
leveraged to explore posttranscriptional and posttranslational processes that are essential for
mitochondrial adaptation.
Avemar outshines new cancer ‘breakthrough’ drug
by Michael Traub
Townsend Letter / Oct, 2010
Many of us in the cancer research community were happy to hear about progress against metastatic melanoma reported this June at the annual meeting of the American Society of Clinical
Oncology (ASCO). since there has not been an improvement in overall survival from chemotherapy in over three decades.
Data from a phase III clinical trial of the experimental monoclonal antibody ipilimumab (pronounced “ep-eh-lim-uemab”) showed that patients with melanoma survived longer if they were taking ipilimumab than if they were not, regardless of whether they also were taking the other drug in the study, an experimental cancer vaccine. (1)
A Closer Look: How Big an Improvement, at What Cost to Patients?
Overall Survival: the ‘Gold Standard’ for Judging Cancer Therapies
Overall survival (OS) is the length of time that a patient actuallysurvives a cancer after treatment. It can also be measured as the percentage of patients surviving a specific time. It is the gold
standard by which the usefulness of a cancer treatment should be determined. Many things can help a patient, but the most important goal of doctors and patients is for the cancer patient to live longer, with a decent quality of life (QOL).
Among patients taking ipilimumab with or without the experimental vaccine, median overall survival was about 10 months. That is compared with 6.4 months’ overall survival among patients receiving the vaccine by itself. About 45.6% of patients taking ipilimumab survived one year, an improvement of some 7% over the 38% seen in some earlier studies. This very modest improvement in survival comes at quite a price.
Severe Side Effects in More Than One in Four Ipilimumab Patients Ipilimumab has some side effects that can be “both severe and long-lasting,” according to the study report. Among patients taking ipilimumab by itself (without the vaccine), 19.1% had side effects requiring hospitalization or invasive intervention, 3.8% died from the effects of the drug, and another 33.8% had life-threatening or disabling side effects. All totaled, 26.7% of the patients taking ipilimumab by itself– more than 1 in 4-had side effects that were severe, very severe, or fatal. Severe side effects included diarrhea, nausea, constipation, vomiting, abdominal pain, fatigue, cough, and headache. Vernon Sondak, MD, of the H. Lee Moffitt Cancer and Research Institute, said that “using the drug requires the medical team to be on guard to manage toxicity at all times.” But even with its severe side effects, the researchers said that the drug should be welcomed because it can increase median survival from 6.4 months to 10.1 months. That is because any lengthening of lives is welcome in a disease that hasn’t seen a new drug that can do that in many years.
Fermented Wheat Germ (Avemar) Improves Melanoma Survival Without Harsh Side Effects
But what if there already were such a treatment available-not a drug, but a safe, natural substance shown in clinical trials to have a remarkably similar ability to lengthen the lives of melanoma patients, without the severe side effects of the new drug?
What if the other substance had no significant side effects at all?
What if, instead of causing severe and sometimes fatal side effects, that other substance actually helped prevent and reduce serious side effects caused by chemotherapy and radiotherapy?
In fact, there is just such a treatment available. It is known as fermented wheat germ extract (FWGE) and by its trade name Avemar. It has been approved as a medical nutriment for cancer
patients in Europe for years and is available in the US as a dietary supplement. It has been compared to dacarbazine (DTIC), standard melanoma therapy, in a clinical trial with longer
follow-up than the ipilimumab trial. And with better results.
In 2008, data were published in the research journal Cancer Biotherapy and Radiopharmaceuticals from seven years’ follow-up on a trial at the N. N. Blokhin Cancer Center in Moscow,
Russia, involving 52 patients who had taken or not taken Avemar while taking dacarbazine for the year following surgical removal of their stage III melanoma tumors. (2) Patients who got only dacarbazine survived 44.7 months. Those who got Avemar along with their dacarbazine survived 66.2 months. This is an improvement in overall survival time of over 48%. In the Russian study,
just as it has in other studies, Avemar reduced side effects of the chemotherapy. Among those taking only dacarbazine, 11 % experienced severe (grade 3 or grade 4) side effects that required hospitalization or invasive intervention. None of the Avemar patients had grade 3 or 4 side effects. Since it is difficult to compare length of survival between the recent ipilimumab study and the Avemar melanoma study, because the ipilimumab study tested mostly stage 4 melanoma patients and the Avemar study tested mostly stage 3 melanoma patients, it is most instructive to look at
the percentage improvement in overall survival from adding either treatment to the regimen. Ipilimumab and Avemar both produced very similar improvements in OS (56% vs. 48%, respectively),
Avemar Ameliorates Conventional Treatment Side Effects
The improvement of survival and the amelioration of chemotherapy side effects by Avemar seen in the Russian melanoma study is typical of Avemar’s effects when used in treating other cancers, including in combination with chemotherapy or radiotherapy. Among 170 colorectal cancer patients in a 2003 study published in the British journal of Cancer, Avemar improved overall survival
and reduced metastasis and recurrences after surgery, chemotherapy, and radiotherapy. (3) Taking Avemar for six months during and after those conventional treatments resulted in a 61.8% reduction in the death rate among those patients, compared with those who received only the conventional treatment. Those taking Avemar experienced lower rates of recurrences and metastases
as well, even though most patients in the Avemar group came into the study with more advanced disease, had more radiation earlier, and had been diagnosed longer. Side effects of Avemar, as in
other Avemar trials., were rare, mild, and transient, with no serious adverse events occurring.
In a 2004 study published in the journal of Pediatric Hematology and Oncology, childhood cancer patients taking Avemar during and after conventional therapies had a 42.8% reduction in the
low white blood cell counts and high fever known as febrile neutropenia, which can be a life-threatening consequence of chemotherapy and radiation. (4) This and similar results with
Avemar in other cancers are consistent with animal studies showing that Avemar helps the immune system recover a full white blood cell count after chemotherapy and radiation faster
than would otherwise happen. This study also demonstrated the safety of Avemar for children.
Why Avemar Works in Many Different Kinds of Cancer
Extensive studies in cells and animals have shown how Avemar works. Perhaps its most important action is to restrict cancer cells’ use of glucose. (5) Cancer cells use up to 50 times more glucose
than normal cells, a phenomenon known as the Warburg effect. (6) They use those enormous amounts of glucose to make ribose, the backbone sugar of DNA, much faster than normal cells can. To
do this, they must use a different series of biochemical reactions (“pathway”) than normal cells. Avemar makes this very difficult for cancer cells to do, because it inhibits the activity of the key enzyme in that pathway, transketolase (TK). (7) With the TK pathway blocked, cancer cells cannot use large amounts of glucose to make DNA fast enough to support the proliferation that makes them so dangerous.(8-10)
In experiments in the US and abroad, scientists have learned that Avemar has these additional effects. It:
* lowers the levels of a DNA repair enzyme known as poly (ADPribose) polymerase (PARP).” With this effect, cancer cells are forced to self-destruct, preventing them from proliferating and
producing a synergistic cancer-cell killing effect when given with chemotherapy, which also works to damage cancer cells’ DNA;
* reduces the number of molecules on cancer cells that identify them as originating within the body (MHC-1 molecules). (12) With cancer cells stripped of that protection, the immune system,
which recognizes the cancer cells as abnormal, no longer gives them the pass given to cells originating in the body. The cancer cells are attacked by the immune system’s natural killer (NK)
cells and destroyed;
* increases levels of molecules called intercellular adhesion molecule-1 (ICAM-1) on the blood vessels of cancer tumors. (13). The increase helps immune system cells pass through the walls of the blood vessels supplying the tumor blood flow, moving directly into the tumor to attack its cancer cells; increases the activity of the primary anticancer cytokine, tumor necrosis factor alpha (TNF-a), and produces a synergistic effect in interaction with other anticancer cytokines. (14) Cytokines are substances produced by cells to act directly on other cells. TNF-a helps force cancer cells into the programmed death known as apoptosis and inhibits tumorigenesis, the process through which new tumors are formed;
* inhibits the activity of ribonucleotide reductase (RR), a key enzyme that cells must have to make new DNA so that each cancer cell can divide to make two more like it. (15) With DNA
production slowed, increases in cancer cell growth and replication are inhibited.
Antimetastatic and Immune-Boosting Effects Are Key to Survival
Because the biochemical changes listed above have consistently been shown in both animal and human studies to be directly linked to reducing cancer’s ability to metastasize and to
improving the immune system’s ability to fight cancer, scientists count them as among the most likely main causes of improved survival seen in cancer patients when Avemar is used alone or,
more often, as an adjuvant in addition to standard-of-care therapies such as chemotherapy, radiotherapy, or the combination of the two. (16-23)
Extending Life: How Long, Exactly, and At What Cost in Quality of Life?
Any improvement in advanced melanoma survival, no matter how small, is certainly an achievement. But ipilimumab had severe side effects requiring hospitalization or invasive intervention in
over one-quarter of patients treated with it. And it increased median survival only by 3-plus months. On the other hand, Avemar added to dacarbazine improved survival very markedly, with no severe side effects. If actually improving overall survival substantially without significant side effects means that a drug should be considered as the new standard of care for first-line therapy, then there is no need to wait for further results. Avemar has already demonstrated very significant improvement in survival over chemotherapy alone and has a safety profile unmatched by
conventional therapies.
Michael Traub, ND, FABNO, is in private practice and serves as a member of Oncology Association of Naturopathic Physicians board of examiners.
Notes
(1.) Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010 Jun 14.
(2.) Demidov LV. Manziuk LV, Kharkevitch GY, Pirogova NA, Artamonova EV. Adjuvant fermented wheat germ extract (Avemar) nutraceutical improves survival of high-risk skin
melanoma patients; a randomized, pilot, phase ll clinical study with a 7-year follow-up. Cancer Biother Radiopharm. 2008 Aug. 23(4):477-482. Erratum in: Cancer Biother Radiopharm. 2008
Oct;2315):669.
(3.) Jakab F, Shoenfeld Y, Balogh A. et al. A medical nutriment has supportive value in the treatment of colorectal cancer. Br J Cancer. 2001 Aug 4;89(3):465-9.
(4.) Garami M, Schuler D, Babosa M, et al. Fermented wheat germ extract reduces chemotherapy-induced febrile neutropenia in pediatric cancer patients, J Pediatr Hematol Oncol. 2004
Oct;26(10):631-635.
(5.) Boros I.G, Lapis K, Szende B, et al. Wheat germ extract decreases glucose uptake and RNA ribose formation but increases fatty acid synthesis in MIA pancreatic adenocarcinoma
cells. Pancreas. 2001 Aug:23(2):141-147.
(6.) Warburg, O. On the origin of cancer cells. Science. 1956 Feb 24; 123(31 91):309-314.
(7.) Boros LG, Lee VVN, Go VL., A metabolic hypothesis of cell growth and death in pancreatic cancer, Pancreas. 2002 Jan;
24:(1):26 33.
(8.) Boros LG, Lapis K, Szende B, et al. Op cit.
(9.) Comin-Anduix B, Boros LG, Marin S, et al. Fermented wheat germ extract inhibits glycolysis/pentose cycle enzymes and induces apoptosis through poly(ADP-ribose) polymerase
activation in Jurkat T-cell leukemia tumor cells. J Biol Chem. 2002 Nov 29;277 (48):46408-46414. Epub 2002 Sep 25.
(23.) Garami M, Schuler D, Babosa M, et al. Fermented wheat germ extract reduces chemotherapy-induced febrile neutropenia in pediatric cancer patients. J Pediatr Hematol Oncol. 2004 Oct;
26(10):631-635.
by Michael Traub, ND, FABNO
COPYRIGHT 2010 The Townsend Letter Group
COPYRIGHT 2010 Gale, Cengage Learning
Nanotechnology in Cancer Drug Delivery and Selective Targeting
Nanoparticles are rapidly being developed and trialed to overcome several limitations of traditional drug delivery systems and are coming up as a distinct therapeutics for cancer treatment. Conventional chemotherapeutics possess some serious side effects including damage of the immune system and other organs with rapidly proliferating cells due to nonspecific targeting, lack of solubility, and inability to enter the core of the tumors resulting in impaired treatment with reduced dose and with low survival rate.
Nanotechnology has provided the opportunity to get direct access of the cancerous cells selectively with increased drug localization and cellular uptake. Nanoparticles can be programmed for recognizing the cancerous cells and giving selective and accurate drug delivery avoiding interaction with the healthy cells. This review focuses on cell recognizing ability of nanoparticles by various strategies having unique identifying properties that distinguish them from previous anticancer therapies. It also discusses specific drug delivery by nanoparticles inside the cells illustrating many successful researches and how nanoparticles remove the side effects of conventional therapies with tailored cancer treatment.
(Kumar Bishwajit Sutradhar and Md. Lutful Amin. Hindawi Publ. Corp. 2014, Article ID 939378, 12 pages
http://dx.doi.org/10.1155/2014/939378)
Cancer, the uncontrolled proliferation of cells where apoptosis is greatly disappeared, requires very complex process of treatment. Because of complexity in genetic and phenotypic levels, it shows clinical diversity and therapeutic resistance. A variety of approaches are being practiced for the treatment of cancer each of which has some significant limitations and side effects. Cancer treatment includes surgical removal, chemotherapy, radiation, and hormone therapy. Chemotherapy, a very common treatment, delivers anticancer drugs systemically to patients for quenching the uncontrolled proliferation of cancerous cells. Unfortunately, due to nonspecific targeting by anticancer agents, many side effects occur and poor drug delivery of those agents cannot bring out the desired outcome in most of the cases. Cancer drug development involves a very complex procedure which is associated with advanced polymer chemistry and electronic engineering.
The main challenge of cancer therapeutics is to differentiate the cancerous cells and the normal body cells. That is why the main objective becomes engineering the drug in such a way as it can identify the cancer cells to diminish their growth and proliferation. Conventional chemotherapy fails to target the cancerous cells selectively without interacting with the normal body cells. Thus they cause serious side effects including organ damage resulting in impaired treatment with lower dose and ultimately low survival rates.
Nanotechnology is the science that usually deals with the size range from a few nanometers (nm) to several hundrednm, depending on their intended use. It has been the area of interest over the last decade for developing precise drug delivery systems as it offers numerous benefits to overcome the limitations of conventional formulations . It is very promising both in cancer diagnosis and treatment since it can enter the tissues at molecular level.
Cisplatin-incorporated nanoparticles of poly(acrylic acid-co-methyl methacrylate) copolymer
K Dong Lee, Young-Il Jeong, DH Kim, Gyun-Taek Lim, Ki-Choon Choi. Intl J Nanomedicine 2013:8 2835–2845.
Although cisplatin is extensively used in the clinical field, its intrinsic toxicity limits its clinical use. We investigated nanoparticle formations of poly(acrylic acid-co-methyl methacrylate) (PAA-MMA) incorporating cisplatin and their antitumor activity in vitro and in vivo.
Methods: Cisplatin-incorporated nanoparticles were prepared through the ion-complex formation between acrylic acid and cisplatin. The anticancer activity of cisplatin-incorporated nanoparticles was assessed with CT26 colorectal carcinoma cells.
Results: Cisplatin-incorporated nanoparticles have small particle sizes of less than 200 nm with spherical shapes. Drug content was increased according to the increase of the feeding amount of cisplatin and acrylic acid content in the copolymer. The higher acrylic acid content in the copolymer induced increase of particle size and decrease of zeta potential. Cisplatin-incorporated nanoparticles showed a similar growth-inhibitory effect against CT26 tumor cells in vitro. However, cisplatin-incorporated nanoparticles showed improved antitumor activity against an animal tumor xenograft model.
Conclusion: We suggest that PAA-MMA nanoparticles incorporating cisplatin are promising carriers for an antitumor drug-delivery system.
Researchers Say Molecule May Help Overcome Cancer Drug Resistance
By Estel Grace Masangkay
A group of researchers from the University of Delaware has discovered that a deubiquitinase (DUB) complex, USP1-UAF1, may present a key target in helping fight resistance to platinum-based anticancer drugs. The research team’s findings were published online in Nature Chemical Biology.
Zhihao Zhuang, associate professor in the Department of Chemistry and Biochemistry at UD, and his team studied a DNA damage tolerance mechanism called translesion synthesis (TLS). Enzymes known as TLS polymerases synthesize DNA over damaged nucleotide bases, followed by replication after lesion. The enzymes have been linked with building cancer cell resistance to certain cancer drugs including cisplatin. Cisplatin is used in treatment of ovarian, bladder, and testicular cancers which have spread.
“Cancer drugs like cisplatin work by damaging DNA and thereby preventing cancer cells from replicating the genomic DNA and dividing. However, cancer cells quickly develop resistance to cisplatin, and we and other researchers suspect that a polymerase known as Pol η is involved in overcoming cisplatin-induced lesions,” Professor Zhuang said.
The team found that USP1-UAF1 may play a crucial role in regulating DNA damage response. A new molecule ML323 can be used to inhibit processes such as translesion synthesis. Zhuang said, “Using ML323, we studied the cellular response to DNA damage and revealed new insights into the role of deubiquitination in both the TLS pathway and another one called the Fanconi anemia, or FA, pathway. We’re very encouraged by the fact that a single molecule is effective at inhibiting the USP1-UAF1 DUB complex and disrupting two essential DNA damage tolerance pathways.”
A novel small peptide as an epidermal growth factor receptor targeting ligand for nanodelivery in vitro
Cui-yan Han, Li-ling Yue, Ling-yu Tai, Li Zhou et al. Intl J Nanomedicine 2013:8 1541–1549
The discovery of suitable ligands that bind to cancer cells is important for drug delivery specifically targeted to tumors. Monoclonal antibodies and fragments that serve as ligands have specific targets. Natural ligands have strong mitogenic and neoangiogenic activities. Currently, small peptides are pursued as targeting moieties because of their small size, low immunogenicity, and their ability to be incorporated into certain delivery vectors.
The epidermal growth factor receptor (EGFR) serves an important function in the proliferation of tumors in humans and is an effective target for the treatment of cancer. The epidermal growth factor receptor (EGFR) is a transmembrane protein on the cell surface that is overexpressed in a wide variety of human cancers. EGFR is an effective tumor-specific target because of its significant functions in tumor cell growth, differentiation, and migration. EGFR-targeted small molecule peptides such as YHWYGYTPQNVI have been successfully identified using phage display library screening; by contrast, the peptide LARLLT has been generated using computer-assisted design (CAD).
These peptides can be conjugated to the surfaces of liposomes that are then delivered selectively to tumors by the specific and efficient binding of these peptides to cancer cells that express high levels of EGFR.
In this paper, we studied the targeting characteristics of small peptides (AEYLR, EYINQ, and PDYQQD) These small peptides were labeled with fluorescein isothiocyanate (FITC) and used the peptide LARLLT as a positive control, which bound to putative EGFR selected from a virtual peptide library by computer-aided design, and the independent peptide RALEL as a negative control.
Analyses with flow cytometry and an internalization assay using NCI-H1299 and K562 with high EGFR and no EGFR expression, respectively, indicated that FITC-AEYLR had high EGFR targeting activity. Biotin-AEYLR that was specifically bound to human EGFR proteins demonstrated a high affinity for human non-small-cell lung tumors.
We found that AEYLR peptide-conjugated, nanostructured lipid carriers enhanced specific cellular uptake in vitro during a process that was apparently mediated by tumor cells with high-expression EGFR. Analysis of the MTT assay indicated that the AEYLR peptide did not significantly stimulate or inhibit the growth activity of the cells. These findings suggest that, when mediated by EGFR, AEYLR may be a potentially safe and efficient delivery ligand for targeted chemotherapy, radiotherapy, and gene therapy.
Arginine-based cationic liposomes for efficient in vitro plasmid DNA delivery with low cytotoxicity
SR Sarker Y Aoshima, R Hokama T Inoue et al. Intl J Nanomedicine 2013:8 1361–1375.
Currently available gene delivery vehicles have many limitations such as low gene delivery efficiency and high cytotoxicity. To overcome these drawbacks, we designed and synthesized two cationic lipids comprised of n-tetradecyl alcohol as the hydrophobic moiety, 3-hydrocarbon chain as the spacer, and different counterions (eg, hydrogen chloride [HCl] salt or trifluoroacetic acid [TFA] salt) in the arginine head group.
Cationic lipids were hydrated in 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer to prepare cationic liposomes and characterized in terms of their size, zeta potential, phase transition temperature, and morphology. Lipoplexes were then prepared and characterized in terms of their size and zeta potential in the absence or presence of serum. The morphology of the lipoplexes was determined using transmission electron microscopy and atomic force microscopy. The gene delivery efficiency was evaluated in neuronal cells and HeLa cells and compared with that of lysine-based cationic assemblies and Lipofectamine™ 2000. The cytotoxicity level of the cationic lipids was investigated and compared with that of Lipofectamine™ 2000.
We synthesized arginine-based cationic lipids having different counterions (ie, HCl-salt or TFA-salt) that formed cationic liposomes of around 100 nm in size. In the absence of serum, lipoplexes prepared from the arginine-based cationic liposomes and plasmid (p) DNA formed large aggregates and attained a positive zeta potential. However, in the presence of serum, the lipoplexes were smaller in size and negative in zeta potential. The morphology of the lipoplexes was vesicular.
Arginine-based cationic liposomes with HCl-salt showed the highest transfection efficiency in PC-12 cells. However, arginine-based cationic liposomes with TFA salt showed the highest transfection efficiency in HeLa cells, regardless of the presence of serum, with very low associated cytotoxicity.
The gene delivery efficiency of amino acid-based cationic assemblies is influenced by the amino acids (ie, arginine or lysine) present as the hydrophilic head group and their associated counterions.
Molecularly targeted approaches herald a new era of non-small-cell lung cancer treatment
H Kaneda, T Yoshida, I Okamoto. Cancer Management and Research 2013:5 91–101.
The discovery of activating mutations in the epidermal growth-factor receptor (EGFR) gene in 2004 opened a new era of personalized treatment for non-small-cell lung cancer (NSCLC). EGFR mutations are associated with a high sensitivity to EGFR tyrosine kinase inhibitors, such as gefitinib and erlotinib. Treatment with these agents in EGFR-mutant NSCLC patients results in dramatically high response rates and prolonged progression-free survival compared with conventional standard chemotherapy. Subsequently, echinoderm microtubule-associated protein-like 4 (EML4)–anaplastic lymphoma kinase (ALK), a novel driver oncogene, has been found in 2007. Crizotinib, the first clinically available ALK tyrosine kinase inhibitor, appeared more effective compared with standard chemotherapy in NSCLC patients harboring EML4-ALK. The identification of EGFR mutations and ALK rearrangement in NSCLC has further accelerated the shift to personalized treatmentbased on the appropriate patient selection according to detailed molecular genetic characterization. This review summarizes these genetic biomarker-based approaches to NSCLC, which allow the instigation of individualized therapy to provide the desired clinical outcome.
Non-small-cell lung cancer (NSCLC) has a poor prognosis and remains the leading cause of death related to cancer worldwide. For most individuals with advanced, metastatic NSCLC, cytotoxic chemotherapy is the mainstay of treatment on the basis of the associated moderate improvement in survival and quality of life. However, the outcome of chemotherapy in such patients has reached a plateau in terms of overall response rate (25%–35%) and overall survival (OS; 8–10 months). This poor outcome, even for patients with advanced NSCLC who respond to such chemotherapy, has motivated a search for new therapeutic approaches.
Recent years have seen rapid progress in the development of new treatment strategies for advanced NSCLC, in particular the introduction of molecularly targeted therapiesand appropriate patient selection. First, the most important change has been customization of treatment according to patient selection based on the genetic profile of the tumor. Small-molecule tyrosine kinase inhibitors (TKIs) that target the epidermal growth-factor receptor (EGFR), such as gefitinib and erlotinib, are especially effective in the treatment of NSCLC patients who harbor activating EGFR mutations.
Surgical Nanorobotics using nanorobots made from advanced DNA origami and Synthetic Biology
Ido Bachelet’s moonshot to use nanorobotics for surgery has the potential to change lives globally. But who is the man behind the moonshot?
Ido graduated from the Hebrew University of Jerusalem with a PhD in pharmacology and experimental therapeutics. Afterwards he did two postdocs; one in engineering at MIT and one in synthetic biology in the lab of George Church at the Wyss Institute at Harvard.
Now, his group at Bar-Ilan University designs and studies diverse technologies inspired by nature.
They will deliver enzymes that break down cells via programmable nanoparticles.
Delivering insulin to tell cells to grow and regenerate tissue at the desired location.
Surgery would be performed by putting the programmable nanoparticles into saline and injecting them into the body to seek out remove bad cells and grow new cells and perform other medical work.


http://www.youtube.com/watch?feature=player_embedded&v=aA-H0L3eEo0

http://www.youtube.com/watch?feature=player_embedded&v=aA-H0L3eEo0#t=236
http://www.youtube.com/watch?feature=player_embedded&v=aA-H0L3eEo0#t=283
http://www.youtube.com/watch?feature=player_embedded&v=aA-H0L3eEo0#t=287
http://www.youtube.com/watch?feature=player_embedded&v=aA-H0L3eEo0#t=292
http://www.youtube.com/watch?feature=player_embedded&v=aA-H0L3eEo0#t=333
http://www.youtube.com/watch?feature=player_embedded&v=aA-H0L3eEo0#t=397


http://www.youtube.com/watch?feature=player_embedded&v=aA-H0L3eEo0#t=470
Nanoparticles with computational logic has already been done
http://www.youtube.com/watch?feature=player_embedded&v=aA-H0L3eEo0#t=501
http://www.youtube.com/watch?feature=player_embedded&v=aA-H0L3eEo0#t=521

Load an ensemble of drugs into many particles for programmed release based on situation that is found in the body
http://www.youtube.com/watch?feature=player_embedded&v=aA-H0L3eEo0#t=572
http://www.youtube.com/watch?feature=player_embedded&v=aA-H0L3eEo0#t=577


Chemoprevention in Model Experiments
Effects of Two Disiloxanes ALIS-409 and ALIS-421 on Chemoprevention in Model Experiments
H TOKUDA,…. L AMARAL and J MOLNAR.. ANTICANCER RESEARCH 33: 2021-2028 (2013).

Figure 1. Chemical structures of ALIS-409 and ALIS-421.
Morpholino-disiloxane (ALIS-409) and piperazinodisiloxane (ALIS-421) compounds were developed as inhibitors of multidrug resistance of various types of cancer cells. In the present study, the effects of ALIS-409 and ALIS-421 compounds were investigated on cancer promotion and on co-existence of
tumor and normal cells. The two compounds were evaluated for their inhibitory effects on Epstein-Barr virus immediate early antigen (EBV-EA) expression induced by tetradecanoylphorbolacetate (TPA) in Raji cell cultures. The method is known as a primary screening test for antitumor effect, below the (IC50) concentration. ALIS-409 was more effective in inhibiting EBV-EA (100 μg/ml) and tumor promotion, than
ALIS-421, in the concentration range up to 1000 μg/ml. However, neither of the compounds were able to reduce tumor promotion significantly, expressed as inhibition of TPA-induced tumor antigen activation. Based on the in vitro results, the two disiloxanes were investigated in vivo for their effects on mouse skin tumors in a two-stage mouse skin carcinogenesis study.




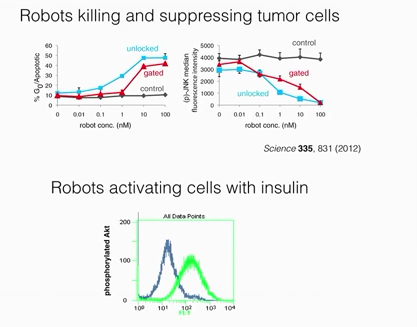.png){kind=link}
.png){kind=link}
.png){kind=link}
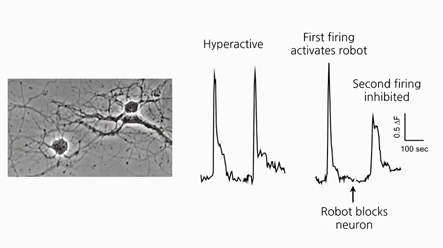.png){kind=link}
.png){kind=link}
.png){kind=link}
.png){kind=link}
{kind=link}