Lung Cancer (NSCLC), drug administration and nanotechnology
Author: Tilda Barliya PhD

Dr. Saxena has greatly introduced us to lung cancer , the associated drug treatments and their market share in the post titled ” NSCLC and where the future lie?”. Since lung cancer is the most leading cause of death in both man and women, and have gained lots of attention I am interested in elaborating on NSCLC and explore the potential use of nanotechnology in this matter.
As previously mentioned, there are 3 common types of lung cancer:
- Adenocarcinomas are often found in an outer area of the lung. (Most common)
- Squamous cell carcinomas are usually found in the center of the lung next to an air tube (bronchus).
- Large cell carcinomas can occur in any part of the lung. They tend to grow and spread faster than the other two types. (Least common).
Figure 1. The Signs and symptoms of lung cancer anatomy.

Since each type develops in different areas/part of the lung, it is hypothesized that they might need different routs of administration. The possible routes of administration are:
- IV (systemic)————->through the blood
- Inhaled aerosols (more localized)———–>through the airways
In order to understand what does “different routs of administration” refers to, we need to dig into the anatomy of the lung, i.e, airways and blood circulation as well as understand the lung-blood barriers components that may affect drug absorption.
The Blood Circulation
Two different circulatory systems, the bronchial and the pulmonary, supply the lungs with blood (Staub, 1991). The bronchial circulation is a part of the systemic circulation and is under high pressure. It receives about 1% of the cardiac output and supplies the airways (from the trachea to the terminal bronchioles), pulmonary blood vessels and lymph nodes with oxygenated blood and nutrients and conditions the inspired air (Staub, 1991). In addition, it may be important to the distribution of systemically administered drugs to the airways and to the absorption of inhaled drugs from the airways (Chediak et al., 1990). The pulmonary circulation comprise an extensive low pressure vascular bed, which receives the entire cardiac output. It perfuses the alveolar capillaries to secure efficient gas exchange and supplies nutrients to the alveolar walls. Anastomoses between bronchial and pulmonary arterial circulations have been found in the walls of medium-sized bronchi and bronchioles (Chediak et al., 1990; Kröll et al.,1987)

Advantages:
- Fast: 15–30 seconds to 1-2 hours
- suitable for drugs not absorbed by the digestive system
- IV can deliver continuous medication
Disadvantages:
- Patients are not typically able to self-administer
- It is the most dangerous route of administration because it bypasses most of the body’s natural defenses, exposing the user to health problems, known as chemo side affects.
- Finally dose at the organ site is much lower than the administrated dose
Most of the conventional chemotherapy are mainly administrated IV (Docetaxel, Paxlitaxel, Gemcitiabine, Avastin etc).
The Airways
The human respiratory system can be divided in two functional regions: the conducting airways and the respiratory region. The conducting airways, which are composed of the nasal cavity and associated sinuses, the pharynx, larynx, trachea, bronchi, and bronchioles, filter and condition the inspired air. From trachea to the periphery of the airway tree, the airways repeatedly branch dichotomously into two daughter branches with smaller diameters and shorter length than the parent branch (Weibel, 1991). For each new generation of airways, the number of branches is doubled and the crosssectional area is exponentially increased. The conducting region of the airways generally constitutes generation 0 (trachea) to 16 (terminal bronchioles). The respiratory region, where gas exchange takes place, generally constitutes generation 17-23 and is composed of respiratory bronchioles, the alveolar ducts, and the alveolar sacs.
The air-blood barrier of the gas exchange area is composed of the alveolar epithelial cells (surface area 140 m2) on one side and the capillary bed (surface area 130 m2) on the other side of a thin basement membrane (Simionescu, 1991; Stone et al., 1992). The extensive surface area of the air-blood barrier in combination with its extreme thinness (0.1-0.5 μm) permit rapid gas exchange by passive diffusion (Plopper, 1996).
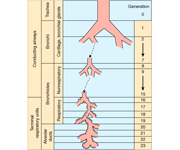
The lung is a very attractive target for drug delivery. It provides direct access to disease in the treatment of respiratory diseases, while providing an enormous surface area and a relatively low enzymatic, controlled environment for systemic absorption of medications. (http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1884307/)
Advantages:
- Can be self medicated
- Easy to use
- Reduced side effects associated with systemic delivery
Disadvantages:
- Slower route of action
- Potential problem of deposition to the deeper alveolar (higher generations, like G 8-10)
- Immuno-defense system
- Difficulty in measuring the exact dose inside the lung
- inhaled aerosol is entrapped in the mucus in the conducting airways
Need to be reminded that in addition, a drug’s efficacy may be affected by where in the respiratory tract it is deposited, its delivered dose and the disease it may be trying to treat.
Major components of the lung – barriers to drug absorption
As one of the primary interfaces between the organism and the environment, the respiratory system is constantly exposed to airborne particles, potential pathogens, and toxic gases in the inspired air (Plopper, 1996). As a result a sophisticated respiratory host defense system, present from the nostrils to the alveoli, has evolved to clear offending agents (Twigg, 1998).
The system comprises of:
- mechanical (i.e. air filtration,cough, sneezing, and mucociliary clearance),
- chemical (antioxidants, antiproteases and surfactant lipids),
- immunological defense mechanisms and is tightly regulated to minimize inflammatory reactions that could impair the vital gas-exchange
**Intratracheal inhalation is another administration option but will be left out of the discussion for now
From a drug delivery perspective, the components of the host defense system comprise barriers that must be overcome to ensure efficient drug deposition and absorption from the respiratory tract.
Generally, lung physiological investigations show that the airway and alveolar epithelia, not the interstitium and the endothelium, constitute the main barrier that restricts the movement of drugs and solutes from the airway lumen into the cells or the blood circulation.
Aerosols are defined as An aerosol is a suspensions of fine solid particles or liquid droplets in a gas.The major aspect affect the efficacy of aerosols as a drug delivery system is Drug Deposition.
Aerosol Drug deposition is affected by:
- particle properties (e.g. size, shape, density, and charge),
- respiratory tract morphology,
- the breathing pattern (e.g. airflow rate and tidal volume)
These parameters determine not only the quantity of particles that are deposited but also in what region of the respiratory tract the particles are deposited.
Particle properties
As the cross-sectional area of the airways increases, the airflow rate rapidly decreases, and consequently the residence time of the particles in the lung increases from the large conducting airways towards the lung periphery. The most important mechanisms of particle deposition in the respiratory tract are (1) inertial impaction, (2) sedimentation, and (3) diffusion.
- Inertial impaction – Inertial impaction occurs predominantly in the extrathoracic airways and in the tracheobronchial tree, where the airflow velocity is high and rapid changes in airflow direction occurs. Generally, particles with a diameter larger than 10 μm are most likely deposited in the extrathoracic region, whereas 2- to 10-μm particles are deposited in the tracheobronchial tree by inertial impaction. A long residence time of the inspired air favors particle deposition by sedimentation and diffusion.
- Sedimentation – Sedimentation is of greatest importance in the small airways and alveoli and is most pronounced for particles with a diameter of 0.5-2 μm, Ultrafine particles (<0.5 μm in diameter) are deposited mainly by diffusional transport in the small airways and lung parenchyma where there is a maximal residence time of the inspired air.
Most therapeutic aerosols are almost always heterodisperse, consisting of a wide range of particle sizes and described by the log-normal distribution with the log of the particle diameters plotted against particle number, surface area or volume (mass) on a linear or probability scale and expressed as absolute values or cumulative percentage (http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1884307/)
Optimal drug delivery to the lungs depends on an interaction between;
- the inhaler device,
- the drug formulation properties,
- the inhalation maneuver
The devices currently available for pulmonary drug administration of pharmaceutical aerosols in clinical therapy include nebulizers, pressurized metered dose inhalers (pMDIs), and dry powder inhalers (DPIs).
However, much effort is put into the development of new inhaler devices and formulations to optimize the pulmonary delivery system for local or systemic drug targeting.
One of the major problems in aerosol delivery is
One disadvantage of the aerosol inhalation is, however, that a substantial portion of the aerosolized drug is not delivered to the lungs (i.e. delivered to the nose, mouth, skin, exhaled). only 10–15% of the emitted dose in the lungs.
In general the aerosol exposure techniques have a low dosing effectiveness, which often requires longer exposure times to administer the target dose and renders investigations of rapid kinetic events difficult. In addition, aerosol exposure requires an advanced equipment for exposure and ml-quantities of test formulation to fill up the device.
Airway geometry and humidity
Progressive branching and narrowing of the airways encourage impaction of particles. The larger the particle size, the greater the velocity of incoming air, the greater the bend angle of bifurcations and the smaller the airway radius, the greater the probability of deposition by impaction. The lung has a relative humidity of approximately 99.5%. The addition and removal of water can significantly affect the particle size of a hygroscopic aerosol and thus deposition. Drug particles are known to be hygroscopic and grow or shrink in size in high humidity, such as in the lung. A hygroscopic aerosol that is delivered at relatively low temperature and humidity into one of high humidity and temperature would be expected to increase in size when inhaled into the lung. The rate of growth is a function of the initial diameter of the particle, with the potential for the diameter of fine particles <1 µm to increase five-fold compared with two-to-three-fold for particles >2 µm. he increase in particle size above the initial size should affect the amount of drug deposited and particularly, the distribution of the aerosolized drug within the lung,
Lung Clearance Mechanism
Once deposited in the lungs, inhaled drugs are either cleared from the lungs, absorbed into the systemic circulation or degraded via drug metabolism. Drug particles deposited in the conducting airways are primarily removed through mucociliary clearance and, to a lesser extent, are absorbed through the airway . epithelium into the blood or lymphatic system. a low-viscosity periciliary or sol layer covered by a high-viscosity gel layer. Insoluble particles are trapped in the gel layer and are moved toward the pharynx (and ultimately to the gastrointestinal tract) by the upward movement of mucus generated by the metachronous beating of cilia. In the normal lung, the rate of mucus movement varies with the airway region and is determined by the number of ciliated cells and their beat frequency. Movement is faster in the trachea than in the small airways and is affected by factors influencing ciliary functioning and the quantity and quality of mucus.
Drugs deposited in the alveolar region may be phagocytosed and cleared by alveolar macrophages or absorbed into the pulmonary circulation. Alveolar macrophages are the predominant phagocytic cell for the lung defence against inhaled microorganisms, particles and other toxic agents. There are approximately five to seven alveolar macrophages per alveolus in the lungs of healthy nonsmokers. Macrophages phagocytose insoluble particles that are deposited in the alveolar region and are either cleared by the lymphatic system or moved into the ciliated airways along currents in alveolar fluid and then cleared via the mucociliary escalator.
Very little is known about how the drug-metabolizing activities of the lung affect the concentration and therapeutic efficacy of inhaled drugs. All metabolizing enzymes found in the liver are found to a lesser extent in the lung. Therefore assuming, drug deposition could have been calculated it would be hard to impossible to evaluate it’s metabolism.
In summary:
As the end organ for the treatment of local diseases or as the route of administration for systemic therapies, the lung is a very attractive target for drug delivery. It provides direct access the site of disease for the treatment of respiratory diseases without the inefficiencies and unwanted effects of systemic drug delivery. It provides an enormous surface area and a relatively low enzymatic, controlled environment for systemic absorption of medications. But it is not without barriers. Airway geometry, humidity, clearance mechanisms and presence of lung disease influence the deposition of aerosols and therefore influence the therapeutic effectiveness of inhaled medications. A drug’s efficacy may be affected by the site of deposition in the respiratory tract and the delivered dose to that site. To provide an efficient and effective inhalant therapy, these factors must be considered. Aerosol particle size characteristics can play an important role in avoiding the physiological barriers of the lung, as well as targeting the drug to the appropriate lung region.
Drug formulations and chemo drug delivery will be further discussed in a another post.
Ref:
1. N R Labiris and M B Dolovich. “Pulmonary drug delivery. Part I: Physiological factors affecting therapeutic effectiveness of aerosolized medications”. Br J Clin Pharmacol. 2003 December; 56(6): 588–599. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1884307/.
2. Tronde A. “Pulmonary drug absorption”. Acta Universities Upsalninesis Uppsala 2002. uu.diva-portal.org/smash/get/diva2:161887/FULLTEXT01
3. Naushad Khan Ghilzai. Pulmonary drug delivery. http://www.drugdel.com/Pulm_review.pdf.






")