Notable Contributions to Regenerative Cardiology by Richard T. Lee – Part I
Author and Curator: Larry H Bernstein, MD, FCAP
and
Article Commissioner: Aviva Lev-Ari, PhD, RD
Introduction
This presentation is a two part discussion of selected articles of a large body of research from Dr. Richard T. Lee, at Harvard Medical School’s Lee Laboratory and Brigham & Womens Hospital. This work is innovative in the field of stem cell research and myocardial regeneration. It devolves the complex cellular processes that are involved in the management of a cell transforming from a progenitor to a functional cardiomyocyte. The cell engineering involves investigating interactions between a cell placed into the layer derived from the interstitial layer between viable cardiomyocytes. This is only possible from a through actionable knowledge of the mechanism involved in the transformation process, which has occupied the Lee Laboratory for many years. Part II will cover the cellular mechanisms underlying the conceptual approach to cardiac myocyte regeneration.
The Lee Laboratory uses emerging biotechnologies to discover and design new approaches to cardiovascular diseases. A central theme of the laboratory is that merging bioengineering and molecular biology approaches can yield novel approaches. Thus, the Lee Laboratory works at this interface using a broad variety of techniques in genomics, imaging, nanotechnology, physiology, cell biology, and molecular biology. The approach is to understand problems and design solutions in the laboratory and then demonstrate the effectiveness of these solutions in vivo. Ongoing projects in the laboratory include studies of cardiac regeneration, diabetic vascular disease, wound healing, heart failure, and cardiac hypertrophy.
Richard T. Lee is Professor of Medicine at Harvard Medical School and lecturer in Biological Engineering at the Massachusetts Institute of Technology. He is a 1979 graduate of Harvard College in Biochemical Sciences and received his M.D. from Cornell University Medical College in 1983. He went on to complete his residency in 1986 and cardiology fellowship in 1989, both at Brigham and Women’s Hospital in Boston, and he obtained post-doctoral training at MIT in Bioengineering.
Dr. Lee is certified by the American Board of Internal Medicine in cardiovascular disease and is a Fellow of the American College of Cardiology. He is Leader of the Cardiovascular Program of the Harvard Stem Cell Institute. He is a member of the Editorial Boards of the journals Circulation Research, Journal of Clinical Investigation, and Circulation, and has published over 180 peer-reviewed articles based on his research, which combines approaches in biotechnology and molecular biology to discover new avenues to manage and treat heart disease.
Regeneration of the heart
Matthew L. Steinhauser, Richard T. Lee
Division of Cardiovascular Medicine, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, and (2) Harvard Stem Cell Institute, Cambridge, MA
EMBO Mol Med 2011; 3: 701–712 http://dx.doi.org/10.1002/emmm.201100175
The death of cardiac myocytes diminishes the heart’s pump function and is a major cause of heart failure. With the exception of heart transplantation and implantation of mechanical ventricular assist devices, current therapies do not address the central problem of decreased pumping capacity owing to a depleted pool of cardiac myocytes. The field is evolving in two important directions. First, although endogenous mammalian cardiac regeneration clearly seems to decline rapidly after birth, it may still persist in adulthood. The careful elucidation of the cellular and molecular mechanisms of endogenous heart regeneration may therefore provide an opportunity for developing therapeutic interventions that amplify this process. Second, recent breakthroughs have enabled reprogramming of cells that were apparently terminally differentiated, either by dedifferentiation into pluripotent stem cells or by trans-differentiation into cardiac myocytes.
The longstanding paradigm held that the mammalian heart is a terminally differentiated organ, incapable of replenishing any myocyte attrition. During the past decade, however, studies revealed not only that mammalian cardiac myocytes retain some capacity for division (Beltrami et al, 2001), but also identified endogenous cardiac progenitor cells in the heart (Beltrami et al, 2003) or bone marrow (Orlic et al, 2001). These cells retain some potential for differentiation into the cellular components of the heart, including endothelial cells, smooth muscle cells and cardiac myocytes.
If progenitor cells residing in the adult are capable of producing new heart cells, the therapeutic delivery of such progenitors might facilitate the generation of de novo functional myocardium. In this context, cell-based therapies for the heart have been rapidly translated into the clinic to treat heart disease, but randomized clinical trials with bone marrow progenitors have shown at best modest improvements in ventricular function (Martin-Rendon et al, 2008). In short, the promise of complete cardiac regeneration has not yet been realized. Therefore, it is worth revisiting both the foundations of cardiac regeneration and highlight recent advances that may portend future directions in the field.
We will first define the problem, that is elucidating the scope of endogenous mammalian regeneration and, by extension, the scale of the regenerative deficit. We will then summarize current regenerative approaches, including both cell-based therapies and pharmacoregenerative strategies. In this context, we will summarize the many challenges that stand in the way of cardiac regeneration, both endogenous repair processes and exogenous regenerative therapies.
The regenerative deficit of the mammalian heart is obvious when compared with organisms such as zebrafish and newts, which demonstrate a remarkable survival capacity after removal of up to 20% of the heart by transection of the ventricular apex. Pre-existing cardiac myocytes adjacent to the site appear to undergo a process of dedifferentiation, characterized by dissolution of sarcomeric structures. This is followed by incorporation of deoxyribonucleic acid (DNA) synthesis markers (e.g. nucleotide analogues) consistent with proliferation. Ultimately, newly generated cardiac myocytes are functionally integrated with the preexisting myocardium. The heart is left with little residual evidence of the injury, thus providing a natural example of complete myocardial regeneration.
Evidence for heart regeneration in mammals
During embryonic development and the early post-natal period, mice also demonstrate a remarkable regenerative capacity. Embryos heterozygous for a cardiac myocyte-specific null mutation in the x-linked holocytochrome c synthase (Hccs) gene demonstrate cardiac myocyte replacement during foetal development (Drenckhahn et al, 2008): when one of two X-chromosomes is randomly inactivated in each female somatic cell, approximately 50% of the cardiac myocytes are rendered Hccs-null and hence dysfunctional. Proliferative functional Hccs-expressing cardiac myocytes compensate for dysfunctional Hccs-null myocytes, such that, at birth, 90% of the heart is derived from myocytes containing one functional Hccs allele. However, after the first week in post-natal mice, injured myocardium is largely replaced by fibrosis and scarring. Distinguishing whether the adult mammalian heart is incapable of cardiac myocyte replacement or whether it retains a low-level capacity for repair is therefore fundamentally important. This is the basis for an evolving view of a more plastic mammalian heart.
Arguments against the age old view of the terminally differentiated quiescent cardiac myocyte:
- evidence supporting cardiac myocyte plasticity relied on mathematical modelling of the myocyte population based on cytometric indices. (the measured average volume increase of cardiac myocytes was calculated to fall short of the increase predicted by the observed volumetric changes in the whole heart
- changes in heart volume could not be explained by hypertrophy alone, and that cardiac myocyte hyperplasia contributed to changes in heart mass, but the conclusions relied on a number of assumptions about myocyte size and DNA content.
- detecting cell cycle markers such as Ki67 or the incorporation of nucleotide analogues (e.g. iododeoxyuridine or 3H-thymidine) into newly synthesized DNA further support the notion that the mammalian heart may generate new myocytes
- human cardiac myocytes can reenter the cell cycle, but the described rates of this phenomenon differ by more than one order of magnitude
- experiments, made possible by nuclear arms testing in the middle of the 20th century, provide the most convincing evidence for post-natal human cardiac myocyte turnover.
- the period of nuclear testing serves as a historical DNA labelling pulse, and the period after the test ban treaty serves as a chase.
- the genomic DNA of cells generated during either the pulse or the chase reflect the earth’s atmospheric 14C concentration at that point in time, which allows investigators to date the age of cardiac myocytes by measuring the concentration of 14C in their nuclei
- Listed are a number of problems in detecting the generation of cardiac myocytes:
- small errors may magnify projections of absolute yearly or lifetime myocyte turnover
- mis-identification of cellular components by light microscopy
- autofluorescence of myocardium, which complicates any method that relies on the detection of a fluorescent signal
- confounders could also affect the 14C dating method, because it requires the isolation of cardiac myocyte nuclei by digestion and flow cytometric sorting
The heart also presents a unique challenge compared to other organs owing to the propensity of cardiac myocytes to synthesize DNA during S-phase without completing either mitosis and/or cytokinesis (Fig 1).
Figure 1. The majority of post-natal human DNA synthesis in the heart does not lead to new myocyte formation.
Cardiac myocytes can complete S-phase, followed by mitosis and cytokinesis (centre) resulting in myocyte doubling. Cardiac myocytes can also complete mitosis without cytokinesis (left), resulting in a binucleated cell. Cardiac myocytes can also undergo chromosomal replication without completing either mitosis or cytokinesis (right), resulting in polyploidy nuclei. By the completion of post-natal development, the majority of human myocyte nuclei contain ~4n chromosomal copies.
During early post-natal development, for example, the majority of rodent cardiac myocytes and an estimated 25–57% of human cardiac myocytes become binucleated. By adulthood, most cardiac myocyte nuclei have also become polyploid with at least one or two additional rounds of chromosomal replication.
The ploidy state of cardiac myocytes may increase with myocardial hypertrophy or injury, which could be mistaken for myocyte division. Conversely, hearts that have been unloaded by implantation of a ventricular assist device may have a lower percentage of polyploid myocytes, because more 2n cardiac myocytes are being generated. These aspects of cardiac myocyte biology inevitably represent potential confounders that must be considered in any quantification of cardiac myocyte formation.
Defining the cellular source of new cardiac myocytes
The majority of reports suggest some endogenous capacity for cardiac myocyte renewal, which has generated a broad focus on finding the cellular source of newly generated cardiac myocytes. Newly generated adult mammalian cardiac myocytes may arise from an endogenous pool of progenitor cells after injury. The Lee Laboratory developed a genetic lineage mapping approach to quantify progenitor-dependent cardiac myocyte turnover (Fig 2) (Hsieh et al, 2007). In the bitransgenic MerCreMer/ZEG inducible cardiac myocyte reporter mouse, mature cardiac myocytes undergo an irreversible genetic switch from constitutive 3-galactosidase expression to green fluorescent protein (GFP) expression upon tamoxifen pulse. During a chase period, we evaluated the effect of myocardial injury on the proportion of GFP+ or 3-gal+ cardiac myocytes. Pressure overload or myocardial infarction resulted in a significant reduction in the percentage of GFP+ cardiac myocytes and a corresponding increase in the percentage of B-gal+ cardiac myocytes, consistent with repletion of the myocyte pool by B-gal— expressing progenitors. This approach cannot directly identify the molecular identity or anatomic location of the progenitor pool.
One approach to characterizing the molecular phenotype of cardiac progenitors is to study cardiac embryologic develop-ment, guided by the assumption that developmental paradigms are recapitulated during post-natal repair. When examined through a developmental lens, an increasingly detailed picture emerges of the soluble and transcriptional signals that guide the cardiogenic programme from gastrulation (formation of distinct germ layers) through the ultimate maturation of cardiac myocytes. The induction of mesoderm posterior (MESP)-1 expression by brachyury-expressing primitive mesodermal cells is a proximal require¬ment for the ultimate production of differentiated heart cells. As the developing embryo grows beyond the germ layer phase, its developing heart receives cells from distinct anatomic progeni¬tor sources: the 1st and 2nd heart fields provide the majority of the myocardium, with some contribution from epicardial progenitors.
Also, Certain fields may be preferentially marked by specific transcription factors;
- the first heart field by T-box transcription factor 5 (Tbx5)
- the second heart field by
- Lim-homeodomain protein Islet1 (Isl1)
- and epicardial progenitors by Wilms tumour-1 (WT1) or
- T-box transcription factor 18 (Tbx18)
- identified by embryonic lineage tracing or analysis of gene silencing include
- homeobox protein nkx2.5
- myocyte enhancer factor 2C (Mef2c)
- GATA4
- there is no consensus yet about the molecular identity of post-natal mammalian cardiac progenitor cells or ‘adult cardiac stem cells’
Figure 2. Lineage-mapping in the adult heart.
Left: Theoretical progenitor lineage-mapping is depicted. Progenitors would be selectively marked by fluorescent protein expression. After injury, the appearance of fluorescently labelled cardiac myocytes would support the concept that these progenitors were contributing to new myocyte formation. Right: Differentiated cell (cardiac myocyte) lineage-mapping. Upon treatment of the MerCreMer-ZEG mouse with OH-tamoxifen, approximately 80% of the cardiac myocytes undergo a permanent switch from I3-galactosidase to GFP expression. The dilution of the GFPþ cardiac myocyte pool after injury is consistent with repletion by I3-galþ progenitors.
A number of laboratories have identified cell populations within the post-natal mouse, which fulfil some criteria of cardiac progenitors:
- expression of a developmentally important gene (isl-1(Laugwitz et al, 2005))
- specific cell surface receptor profile (c-kit (Beltrami et al, 2003)
- or sca-1 (Oh et al, 2003))
- capacity to actively exclude Hoechst dye (so-called side population cells (Martin et al, 2004)) or based on the outgrowth of typical spherical colonies in tissue culture
In general, the label of ‘cardiac stem cell’ results from the observation of self-propagation and cardiac myocyte transdifferentiation when exposed to cardiogenic conditions in vitro or when delivered in vivo after injury. However, the field will benefit from careful in vivo lineage tracing studies—without ex vivo culture steps—to study if and how a given cell type contributes to cardiac myocyte replenishment during either normal homeostasis or after injury (Fig 2). The lack of such publications to date owes in part to the lack of specificity of many stem cell markers (Fig 3).
Figure 3. Possible recapitulation of developmental paradigms by endogenous post-natal cardiac stem cells.
Between mesodermal development and the emergence of cardiac myocytes, cardiovascular progenitors express a number of markers that have also been detected in the various post-natal cardiac stem cell (CSC) preparations. Expression as measured by messenger RNA (mRNA) or protein expression is denoted with (þ). Absent expression is denoted by (-). Blank1/4 untested.
Moving towards a regenerative therapy
The therapeutic challenge is considerable: a typical large myocardial infarction that leads to heart failure will kill around 1 billion cardiac myocytes, roughly a quarter of the heart’s myocytes. A possible therapeutic approach would coax an endogenous stem cell population or an exogenously delivered cell-based therapy to replace lost cardiac myocytes in a coordinated fashion. Amongst the myriad of potential cell-based therapies, no clear winning strategy has so far emerged (Segers & Lee, 2008).
Bone marrow derived progenitors
Conflicting studies sparked excitement and also uncertainty about a possible adult cardiogenic progenitor originating outside of the heart. A post-mortem examination of male heart transplant patients who had received female donor hearts found that approximately 10% of -sarcomeric actin-positive cardiac myocytes had Y-chromosomes, and two cases in which a bone marrow cell population with a higher density of the cell surface receptor c-kit, showed repopulation of murine cardiac myocytes after experimental myocardial infarction. A number of studies that followed failed to demonstrate similar rates of chimerism in transplanted hearts or potency of bone marrow stem cell. However, some therapeutic effect was observed even in studies with no detectable transdifferentiation.
Figure 4. The challenge of regenerating the heart.
Both exogenously delivered cell therapies and progenitors in the endogenous niche encounter a similar hostile environment after myocardial injury, often including inadequate blood supply (ischemia), inflammation and fibrosis/scarring. Regenerative pathways may be activated by as yet unknown paracrine pathways, responsible for recruiting progenitors from the niche, stimulating proliferation and coaxing differentiation. Cell-based therapy using autologous bone marrow
progenitors was rapidly translated into the clinic to treat human ischemic heart disease. A number of randomized trials, using bone marrow mononuclear cells have been performed and most studies demonstrated modest cell therapy-mediated improvements in ventricular function.
Pluripotent stem cells
Embryonic stem (ES) cells represent the prototypical stem cells with the hallmarks of clonogenicity, self-renewal and pluripotency. The potency of these cells also represents a real safety concern, given their tendency to form teratomas. One approach to overcoming this prohibitive safety problem has been to generate pluripotent-derived progenitors that have already committed to a cardiogenic pathway. As a proof-of-principle example of such a strategy, cells with an expression profile of Oct4, stage-specific embryonic antigen 1 (SSEA-1) and MESP1 demonstrated some regenerative potency when delivered therapeutically in a primate infarct model, without detectable teratoma formation. One could envision a similar strategy using cardiogenic intermediates that express any of the previously mentioned transcription factors associated with cardiac progenitors or cell surface markers such as the receptor for vascular endothelial growth factor (Flk1/KDR). Yet, such a strategy should still demonstrate both substantial preclinical efficacy without tumorigenicity before human translation. If such criteria are met, ES-derived therapies have the potential of providing ‘off-the-shelf’ cardiac myocytes to treat acute myocardial infarctions or chronic heart failure.
A second approach, which may also obviate the risk of teratomas, is to generate a pure population of ES-derived cardiac myocytes for therapeutic delivery either as a cell suspension or after ex vivo tissue engineering. There has already been enormous progress during the past decade in defining the factors and transcription signals to differentiate cardiac myocytes from ES-cells. As discussed in greater detail, cardiac myocyte development is dictated by the time and dose-dependent exposure to a series of growth factors from the wingless-type MMTV integration site (Wnt), fibroblast growth factor (FGF), bone morphogenetic protein (BMP) and nodal families. Several laboratories have successfully generated ES-derived preparations with more than 50% of functional cardiac myocytes. The most realistic future for such technical advances may be as an unlimited source of cardiac myocytes for engineering myocardial grafts.
The generation of induced pluripotent stem (iPS) cells may overcome two important limitations of ES cells: ethical concerns about harvesting ES cells from embryos and graft rejection
- iPS cells can be custom-engineered from a patient’s stromal cells for autologous transplantation.
- immunogenecity in syngeneically transplanted iPS cells, suggests that the immune system cannot yet be discounted in the development of iPS-based therapies
The initial protocols for iPS cell generation involved retro-viral-mediated expression of four stem-cell genes.
But virally reprogrammed cells may harbour an associated risk of neoplastic conversion. Alternative reprogramming strategies, such as the use of small molecules (Shi et al, 2008) or non-viral gene modifying approaches (Warren et al, 2010) will probably be a necessary component of any future therapeutic strategies. However, the most important lesson from these landmark studies may be the remarkable plasticity of even the most terminally differentiated cells when exposed to the right combination of signals.
Tissue engineering
Historically, the greatest challenge in tissue engineering has been an adequate supply of oxygen and nutrients for metabolically active tissues such as the heart. One approach has been to engineer thin cardiac sheets, which can then be stacked for in vivo delivery. Although these layered sheets demonstrate some degree of electromechanical coordination and neovascularization in vivo, it is not clear yet if such an approach can be optimized to yield full-thickness myocardium with an adequate blood supply. The addition of non-myocyte cellular components, such as fibroblasts and endothelial cells, leads to the formation of primitive vascular structures within engineered grafts, but the electro-mechanical properties are not sufficient for normal functionality.
Circumventing cell-based therapy with pharmacoregeneration?
A short-term goal may be to exploit paracrine signalling to amplify the existent endogenous regenerative response. Recent cell transplantation experiments conducted in our laboratory, using an inducible cre-based genetic lineage mapping approach, tested the hypothesis that cell-based therapies might exert proregenerative effects via a paracrine mechanism (Loffredo et al, 2011) (Fig 5). Consistent with some prior studies, we found no evidence for transdifferentiation of exogenously delivered bone marrow cells into cardiac myocytes. However, we did find increased generation of cardiac myocytes from endogenous progenitors in mice, which were administered c-kit+ bone marrow cells but not mesenchymal stem cells. This finding suggests paracrine signalling between exogenously delivered cells and endogenous resident progenitors. It provides a rationale for therapeutic interventions aimed at activating progenitors or recruiting them from their niche to the injury site.
Figure 5. Proposed of action for cell-based therapies.
In theory, exogenously delivered cells may directly differentiate into endothelial cells, smooth muscle cells and cardiac myocytes. They may also release paracrine factors which may result in non-regenerative effects, such as immunomodulation, angiogenesis or cardioprotection. Recent work from our laboratory suggests that a dominant mechanism achieved with bone marrow progenitor therapy may be via the activation of endogenous progenitor recruitment (Loffredo et al, 2011).
Controlling the mitotic activity of mononucleated cardiac myocytes may provide an alternative approach to replenishing cardiac myocytes. A major concern with systemic growth factor therapy, however, is the potential for mitogenic effects that may impact other organs. Thus, the future of pharmacologic regeneration may lie in the local delivery of engineered proteins and small molecules that target
Future directions
In this review, we have described the current status of research on cardiac regeneration, highlighting important recent discoveries and ongoing controversies. The initial hope that a cell progenitor would emerge with the capacity to regenerate the injured mammalian heart in the same manner that bone marrow may be reconstituted has not been realized.
Cardiac myocyte regeneration may lie in the local delivery of engineered proteins and small molecules that target specific survival, growth and differentiation pathways.
Pending issues
Dissect the mechanistic differences between adult mammals with limited regenerative capacity and organisms, such as neonatal mice, zebrafish and newts, that demonstrate unambiguous cardiac myocyte regeneration. Understanding these differences may reveal new pathways that can be therapeutically targeted to achieve more robust regeneration.
Complete molecular and functional characterization of endogenous cardiac myocyte progenitors. Multiple laboratories have isolated progenitors from the heart with different molecular characteristics. What are the in vivo functional roles of these progenitors? Do the observed molecular differences between these isolated cells represent functionally distinct cell types?
Identify paracrine signalling pathways responsible for activation and recruitment of endogenous cardiac myocyte progenitors. This may facilitate a pharmacoregenerative therapy, in which treatment with a protein or small molecule would hold the promise of amplifying endogenous regeneration.
Refine reprogramming strategies to more efficiently produce mature cardiac myocytes, both in vitro and in vivo. The ultimate bioengineering goal is to produce a pure population of mature, fully functional cardiac myocytes for ex vivo tissue engineering (or) to control the proliferation and differentiation of endogenous cell populations or exogenously delivered cell therapies such that scar tissue is replaced by myocardium. These different strategies are unified by an underlying requirement to understand the fundamental pathways involved in cardiac myocyte differentiation and maturation.
There is reason for optimism for a regenerative medicine approach to heart failure, given the intense research efforts and the capacity of higher organisms, including the neonatal mouse, to regenerate myocardium. Perhaps the most important issue in this field is identifying which findings are consistently supported by multiple experimental approaches. Ultimately, the findings that are easily reproduced by most or all laboratories will most likely benefit patients.
Selected references
Hsieh et al, 2007. Hsieh PC, Segers VF, Davis ME, MacGillivray C, Gannon J, Molkentin JD, Robbins J, Lee RT (2007) Evidence from a genetic fate-mapping study that stem cells refresh adult mammalian cardiomyocytes after injury. Nat Med 13: 970¬974
Laugwitz et al, 2005. Laugwitz KL, Moretti A, Lam J, Gruber P, Chen Y, Woodard S, Lin LZ, Cai CL, Lu MM, Reth M et al (2005) Postnatal isl1þ cardioblasts enter fully differentiated cardiomyocyte lineages. Nature 433: 647-653
Beltrami et al, 2003. Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, Kasahara H, Rota M, Musso E, Urbanek K et al (2003) Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell 114: 763¬776
Oh et al, 2003. Oh H, Bradfute SB, Gallardo TD, Nakamura T, Gaussin V, Mishina Y, Pocius J, Michael LH, Behringer RR, Garry DJ et al (2003) Cardiac progenitor cells from adult myocardium: homing, differentiation, and fusion after infarction. Proc Natl Acad Sci USA 100: 12313-12318
Segers & Lee, 2008. Segers VF, Lee RT (2008) Stem-cell therapy for cardiac disease. Nature 451:937-942
Loffredo et al, 2011. Loffredo FS, Steinhauser ML, Gannon J, Lee RT (2011) Bone marrow-derived cell therapy stimulates endogenous cardiomyocyte progenitors and promotes cardiac repair. Cell Stem Cell 8: 389-398.
Shi et al, 2008. Shi Y, Desponts C, Do JT, Hahm HS, Scholer HR, Ding S (2008) Induction of pluripotent stem cells from mouse embryonic fibroblasts by Oct4 and Klf4 with small-molecule compounds. Cell Stem Cell 3: 568-574.
Warren et al, 2010. Warren L, Manos PD, Ahfeldt T, Loh YH, Li H, Lau F, Ebina W, Mandal PK, Smith ZD, Meissner A et al (2010) Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 7: 618-630.
Mammalian Heart Renewal by Preexisting Cardiomyocytes
SE Senyo, ML Steinhauser, CL Pizzimenti, VK. Yang, Lei Cai, Mei Wang, …,and Richard T. Lee
Cardiovascular and Genetics Divisions, Brigham and Women’s Hospital and Harvard Medical School,
INSERM, Orsay (Fr), Institut Curie, Laboratoire de Microscopie Ionique, Orsay (Fr), National Resource for Imaging Mass Spectrometry, Harvard Stem Cell Institute
Nature. 2013 January 17; 493(7432): 433–436. http://dx.doi.org/10.1038/nature11682
Although recent studies have revealed that heart cells are generated in adult mammals, the frequency and source of new heart cells is unclear. Some studies suggest a high rate of stem cell activity with differentiation of progenitors to cardiomyocytes. Other studies suggest that new cardiomyocytes are born at a very low rate, and that they may be derived from division of pre-existing cardiomyocytes. Thus, the origin of cardiomyocytes in adult mammals remains unknown. Here we combined two different pulse-chase approaches — genetic fate-mapping with stable isotope labeling and Multi-isotope Imaging Mass Spectrometry (MIMS). We show that genesis of cardiomyocytes occurs at a low rate by division of pre-existing cardiomyocytes during normal aging, a process that increases by four-fold adjacent to areas of myocardial injury. Cell cycle activity during normal aging and after injury led to polyploidy and multinucleation, but also to new diploid, mononucleated cardiomyocytes. These data reveal pre-existing cardiomyocytes as the dominant source of cardiomyocyte replacement in normal mammalian myocardial homeostasis as well as after myocardial injury.
Despite intensive research, fundamental aspects of the mammalian heart’s capacity for self-renewal are actively debated. Estimates of cardiomyocyte turnover range from less than 1% per year to more than 40% per year. Turnover has been reported to either decrease or increase with age, while the source of new cardiomyocytes has been attributed to both division of existing myocytes and to progenitors residing within the heart or in exogenous niches such as bone marrow. Controversy persists regarding the plasticity of the adult heart in part due to methodological challenges associated with studying slowly replenished tissues. Toxicity attributed to radiolabeled thymidine and halogenated nucleotide analogues limits the duration of labeling and may produce direct biological effects. The challenge of measuring cardiomyocyte turnover is further compounded by the faster rate of turnover of cardiac stromal cells relative to cardiomyocytes.
We used Multi-isotope Imaging Mass Spectrometry (MIMS) to study cardiomyocyte turnover and to determine whether new cardiomyocytes are derived from preexisting myocytes or from a progenitor pool (Fig 1a). MIMS uses ion microscopy and mass spectrometry to generate high resolution quantitative mass images and localize stable isotope reporters in domains smaller than one micron cubed15,16,17. MIMS generates 14N quantitative mass images by measuring the atomic composition of the sample surface with a lateral resolution of under 50nm and a depth resolution of a few atomic layers. Cardiomyocyte cell borders and intracellular organelles were easily resolved (Fig 1b). Regions of interest can be analyzed at higher resolution, demonstrating cardiomyocyte-specific subcellular ultrastructure, including sarcomeres (Fig 1c, Supplemental Fig 1a). In all subsequent analyses, cardiomyocyte nuclei were identified by their location within sarcomere-containing cells, distinguishing them from adjacent stromal cells.
An immense advantage of MIMS is the detection of nonradioactive stable isotope tracers. As an integral part of animate and inanimate matter, they do not alter biochemical reactions and are not harmful to the organism18. MIMS localizes stable isotope tracers by simultaneously quantifying multiple masses from each analyzed domain; this enables the generation of a quantitative ratio image of two stable isotopes of the same element15. The incorporation of a tracer tagged with the rare stable isotope of nitrogen (15N) is detectable with high precision by an increase in 15N:14N above the natural ratio (0.37%). Nuclear incorporation of 15N-thymidine is evident in cells having divided during a one-week labeling period, as observed in the small intestinal epithelium, which turns over completely in 3–5 days16 (Fig 1d); in contrast, 15N-thymidine labeled cells are rarely observed in the heart (Fig 1e) after 1 week of labeling. In subsequent studies, small intestine was used as a positive control to confirm label delivery.
To evaluate for an age-related change in cell cycle activity, we administered 15N-thymidine for 8 weeks to three age groups of C57BL6 mice starting at day-4 (neonate), at 10-weeks (young adult) and at 22-months (old adult) (Supplemental Fig 2). We then performed MIMS analysis (Fig 2a, b, Supplemental Fig 3). In the neonatal group, 56% (±3% s.e.m., n=3 mice) of cardiomyocytes demonstrated 15N nuclear labeling, consistent with the well-accepted occurrence of cardiomyocyte DNA synthesis during post-natal development19. We observed a marked decline in the frequency of 15N-labeled cardiomyocyte nuclei (15N+CM) in the young adult (neonate= 1.00%15N+CM/day ±0.05 s.e.m. vs young adult=0.015% 15N+CM/ day ±0.003 s.e.m., n=3 mice/group, p<0.001) (Fig 2a, c; Supplemental Fig 3). We found a further reduction in cardiomyocyte DNA synthesis in old mice (young adult=0.015%15N+CM/day ±0.003 s.e.m. vs. old adult=0.007 %15N+CM/day ±0.002 s.e.m., n=3/group, p<0.05) (Fig 2c). The observed pattern of nuclear 15N-labeling in cardiomyocytes is consistent with the known chromatin distribution pattern in cardiomyocytes20 (Supplemental Fig 1b) and was measured at levels that could not be explained by DNA repair (Supplemental Fig 4). Extrapolating DNA synthesis measured in cardiomyocytes over 8 weeks yields a yearly rate of 5.5% in the young adult and 2.6% in the old mice. Given that cardiomyocytes are known to undergo DNA replication without completing the cell cycle19,21,22, these calculations represent the upper limit of cardiomyocyte generation under normal homeostatic conditions, indicating a low rate of cardiogenesis.
To test whether cell cycle activity occurred in preexisting cardiomyocytes or was dependent on a progenitor pool, we performed 15N-thymidine labeling of double-transgenic MerCreMer/ZEG mice, previously developed for genetic lineage mapping (Fig 3a)23,24. MerCreMer/ZEG cardiomyocytes irreversibly express green fluorescent protein (GFP) after treatment with 4OH-tamoxifen, allowing pulse labeling of existing cardiomyocytes with a reproducible efficiency of approximately 80%. Although some have reported rare GFP expression by non-cardiomyocytes with this approach25, we did not detect GFP expression in interstitial cells isolated from MerCreMer/ZEG hearts nor did we detect GFP expression by Sca1 or ckit-expressing progenitors in histological sections (Supplemental Fig 5). Thus, during a chase period, cardiomyocytes generated from progenitors should be GFP−, whereas cardiomyocytes arising from preexisting cardiomyocytes should express GFP at a frequency similar to the background rate induced by 4OH-tamoxifen. We administered 4OH-tamoxifen for two weeks to 8 wk-old mice (n=4); during a subsequent 10-week chase, mice received 15N-thymidine via osmotic minipump.
We next used MIMS and genetic fate mapping to study myocardial injury. Cardiomyocyte GFP labeling was induced in MerCreMer/ZEG mice with 4OH-tamoxifen. Mice then underwent experimental myocardial infarction or sham surgery followed by continuous labeling with 15N-thymidine for 8wks. The frequency of 15N-labeled cardiomyocytes in sham-operated mice was similar to prior experiments in unoperated mice (yearly projected rates: sham=6.8%; unoperated=4.4%), but increased significantly adjacent to infarcted myocardium (total 15N+ cardiomyocyte nuclei: MI=23.0% vs sham=1.1%, Fig 4a–b, Supplemental Fig 8). We examined GFP expression, nucleation and ploidy status of 15N-labeled cardiomyocytes and surrounding unlabeled cardiomyocytes. We found a significant dilution of the GFP+ cardiomyocyte pool at the border region as previously shown23,24 (67% vs. 79%, p<0.05, Table 2, Supplemental Fig 9); however, 15N+ myocytes demonstrated a similar frequency of GFP expression compared to unlabeled myocytes (71% vs. 67%, Fisher’s exact=n.s.), suggesting that DNA synthesis was primarily occurring in pre-existing cardiomyocytes. Of 15N-labeled cardiomyocytes, approximately 14% were mononucleated and diploid consistent with division of pre-existing cardiomyocytes (Supplemental Fig 6, 7). We observed higher DNA content (>2N) in the remaining cardiomyocytes as expected with compensatory hypertrophy after injury. Thus, in the 8wks after myocardial infarction, approximately 3.2% of the cardiomyocytes adjacent to the infarct had unambiguously undergone division (total 15N+ × mononucleated diploid fraction = 23% × 0.14 = 3.2%). The low rate of cardiomyocyte cell cycle completion is further supported by the absence of detectable Aurora B Kinase, a transiently expressed cytokinesis marker, which was detected in rapidly proliferating small intestinal cells but not in cardiomyocytes (Supplemental Fig 10). We also considered the possibility that a subset of 15N+ myocytes that were multinucleated and/or polyploid resulted from division followed by additional rounds of DNA synthesis without division. However, quantitative analysis of the 15N+ population did not identify a subpopulation that had accumulated additional 15N-label as would be expected in such a scenario (Supplemental Fig 11). Together, these data suggest that adult cardiomyocytes retain some capacity to reenter the cell cycle, but that the majority of DNA synthesis after injury occurs in preexisting cardiomyocytes without completion of cell division.
If dilution of the GFP+ cardiomyocyte pool cannot be attributed to division and differentiation of endogenous progenitors, do these data exclude a role for progenitors in the adult mammalian heart? These data could be explained by preferential loss of GFP+ cardiomyocytes after injury, a process that we have previously considered but for which we have not found supporting evidence23. Such an explanation excludes a role for endogenous progenitors in cardiac repair and would be consistent with data emerging from lower vertebrates8,26 and the neonatal mouse27 in which preexisting cardiomyocytes are the cellular source for cardiomyocyte repletion. A second possibility to explain the dilution of the GFP+ cardiomyocyte pool is that injury stimulates progenitor differentiation without division; inevitably, this would lead to exhaustion of the progenitor pool, which if true could explain the limited regenerative potential of the adult mammalian heart.
In summary, this study demonstrates birth of cardiomyocytes from preexisting cardiomyocytes at a projected rate of approximately 0.76%/year (15N+ annual rate × mononucleated diploid fraction = 4.4% × 0.17) in the young adult mouse under normal homeostatic conditions, a rate that declines with age but increases by approximately four-fold after myocardial injury in the border region. This study shows that cardiac progenitors do not play a significant role in myocardial homeostasis in mammals and suggests that their role after injury is also limited.
Engineering insulin-like growth factor-1 for local delivery
T Tokunou, R Miller, P Patwari, ME Davis, VFM Segers, AJ Grodzinsky, and RT Lee
Cardiovascular Division, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA and Biological Engineering, MIT, Cambridge, MA
FASEB J. 2008 June ; 22(6): 1886–1893. http://dx.doi.org/10.1096/fj.07-100925
Insulin-like growth factor-1 (IGF-1) is a small protein that promotes cell survival and growth, often acting over long distances. Although for decades IGF-1 has been considered to have therapeutic potential, systemic side effects of IGF-1 are significant, and local delivery of IGF-1 for tissue repair has been a long-standing challenge. In this study, we designed and purified a novel protein, heparin-binding IGF-1 (Xp-HB-IGF-1), which is a fusion protein of native IGF-1 with the heparin-binding domain of heparin-binding epidermal growth factor-like growth factor. Xp-HB-IGF-1 bound selectively to heparin as well as the cell surfaces of 3T3 fibroblasts, neonatal cardiac myocytes and differentiating ES cells. Xp-HB-IGF-1 activated the IGF-1 receptor and Akt with identical kinetics and dose response, indicating no compromise of biological activity due to the heparin-binding domain. Because cartilage is a proteoglycan-rich environment and IGF-1 is a known stimulus for chondrocyte biosynthesis, we then studied the effectiveness of Xp-HB-IGF-1 in cartilage. Xp-HB-IGF-1 was selectively retained by cartilage explants and led to sustained chondrocyte proteoglycan biosynthesis compared to IGF-1. These data show that the strategy of engineering a “long-distance” growth factor like IGF-1 for local delivery may be useful for tissue repair and minimizing systemic effects.
INSULIN-LIKE GROWTH FACTOR-1 (IGF-1) is a growth factor well known as an important mediator of cell growth and differentiation. IGF-1 stimulates several signaling pathways through the tyrosine kinase IGF-1 receptor, including phosphatidylinositol (PI) 3-kinase and mitogen-activated protein kinases (MAPKs). PI3-kinase has many downstream targets, including the kinase Akt, and activation of Akt promotes survival, proliferation, and growth.
IGF-1 has been extensively studied for its therapeutic potential in tissue repair and regeneration. IGF-1 is a small and highly diffusible protein that can act over long distances. However, systemic administration of IGF-1 has significant side effects as well as the potential to promote diabetic retinopathy and cancer. Therefore, local delivery of IGF-1 has been a longstanding challenge. Here, we describe the design of a new protein, formed by fusion of IGF-1 with the heparin-binding (HB) domain of heparin-binding epidermal growth factor-like growth factor (HB-EGF). HB-EGF binds selectively to glycosaminoglycans through its highly positively charged heparin-binding domain.
Thus, we hypothesized that engineering IGF-1 to bind to glycosaminoglycans could provide selective delivery of IGF-1 to cell surfaces or to specific tissues. We demonstrate that this heparin-binding IGF-1 (Xp-HB-IGF-1) can bind to cell surfaces as well as the proteoglycan-rich tissue of cartilage; furthermore, Xp-HB-IGF-1 prolongs the stimulation of chondrocyte biosynthesis, demonstrating its potential for tissue specific repair.
Purification of Xp-HB-IGF-1
Figure 1A—C shows the constructs for Xp-HB-IGF-1 and the control Xp-IGF-1 fusion proteins.
IGF-1 has 3 disulfide bonds and includes 70 amino acids. The IGF-1 fusion proteins both contain polyhistidine tags for protein purification and Xpress tags for protein detection. The expected molecular masses of Xp-HB-IGF-1 and Xp-IGF-1 are 14,018 and 11,548 Da, respectively. Xp-HB-IGF-1 has the HB domain on the N terminus of IGF-1. The HB domain has 21 amino acids and includes 12 positively charged amino acids. Final purification of the new fusion proteins after refolding was performed with RP-HPLC (Fig. 1D, E). Identification of the correctly folded protein was performed as described previously and confirmed with bioactivity assays. These 3 IGF-1s (Xp-HB-IGF-1, Xp-IGF-1, and unmodified IGF-1) yielded similar intensities.
Xp-HB-IGF-1 binds to heparin and cell surfaces
1. Xp-HB-IGF-1 binds selectively to heparin compared with Xp-IGF-1 (Fig. 2A).
2. Xp-HB-IGF-1 bound to 3T3 fibroblast cells when treated with 10 and 100 nM concentrations.
3. Xp-HB-IGF-1 binds with neonatal cardiac myocytes, with clear selective binding of Xp-HB-IGF-1 (Fig 2C)
4. These results are consistent with binding of this HB domain to heparan sulfate in the submicromolar range
5. Xp-HB-IGF-1 was readily detected on the surfaces of ES cells in embryoid bodies — which contain multiple cell types.
6. There is more Xpress epitope tag in Xp-HB-IGF-1 group than the Xp-IGF-1 group, suggesting that Xp-HB-IGF-1 binds with heparan sulfate on the cell surface.
Xp-HB-IGF-1 bioactivity
Bioassays for IGF-1 receptor phosphorylation and Akt activation were performed. Control IGF-1, Xp-HB-IGF-1, and Xp-IGF-1 all activated the IGF-1 receptor of neonatal cardiac myocytes dose-dependently and induced Akt phosphorylation identically (Fig. 3A), and they activated Akt with a similar time course (Fig. 3B), indicating — addition of the heparin-binding domain does not interfere with the bioactivity of IGF-1.
- Xp-HB-IGF-1 transport in cartilage
- Cartilage is a proteoglycan-rich tissue, and chondrocytes respond to IGF-1 with increased extracellular matrix synthesis (19). Because prolonged local stimulation of IGF-1 signaling could thus be beneficial for cartilage repair, we studied the ability of Xp-HB-IGF-1 to bind to cartilage.
- Xp-HB-IGF-1 is selectively retained by cartilage, while Xp-IGF-1 is rapidly lost.
- Xp-HB-IGF-1 can bind to cartilage after chondroitin sulfate digestion
To explore the possibility of nonspecific binding of Xp-HB-IGF-1 to glycosaminoglycans other than heparan sulfate, we studied the binding of Xp-HB-IGF-1 after chondroitinase ABC digestion.
Xp-HB-IGF-1 retention is not mediated by the pool of chondroitin sulfated proteoglycans in the cartilage matrix.
- Xp-HB-IGF-1 increases chondrocyte biosynthesis
- Xp-HB-IGF-1, which is selectively retained in the cartilage, stimulates chondrocyte biosynthesis over a more sustained period.
DISCUSSION
In this study, we describe a novel IGF-1 protein, Xp-HB-IGF-1, which binds to proteoglycan-rich tissue and cell surfaces but has the same bioactivity as IGF-1. Our data indicate that Xp-HB-IGF-1 can activate the IGF-1 receptor and Akt and thus that the heparin-binding domain does not interfere with interactions of IGF-1 and its receptor. IGF-1 has four domains: B domain (aa 1–29), C domain (aa 30 – 41), A domain (aa 42–62) and D domain (aa 63–70), with the C domain playing the most important role in binding to the IGF-1 receptor. Replacement of the entire C domain causes a 30-fold decrease in affinity for the IGF-1 receptor. Thus, the addition of the heparin-binding domain to the N terminus of IGF-1 was not anticipated to interfere with interactions with the IGF-1 C domain.
Both extracellular matrix and cell surfaces are rich in proteoglycans and can serve as reservoirs for proteoglycan-binding growth factors. A classic example is the fibroblast growth factor-2 (FGF-2) system, where a low-affinity, high-capacity pool of proteoglycan receptors serves as a reservoir of FGF-2 for its high-affinity receptor. Our experiments suggest that Xp-HB-IGF-1 could function in some circumstances in a similar manner, since Xp-HB-IGF-1 is selectively retained on cell surfaces. Many growth factors are known to interact with heparan sulfate, including HB-EGF (10-12), FGF-2 (26), vascular endothelial growth factor-A (VEGF-A), transforming growth factor beta (TGF-β) (28), platelet-derived growth factors (PDGFs), and hepatocyte growth factor (HGF). However, other proteins such as nerve growth factor (NGF), which induces differentiation and reduces apoptosis of neurons, does not have the heparin-binding domain. Thus, the strategy of engineering growth factors for selective matrix or cell surface binding could be used for other growth factors.
IGF-1 can also bind with extracellular matrix via IGF binding proteins (IGFBPs); in the circulation, at least 99% of IGF-1 is bound to IGFBPs (IGFBP-1 to −6). Further experiments are necessary to determine whether addition of a heparin-binding domain to IGF-1 changes interactions with IGFBPs and whether this changes its biological activity.
IGF-1 can promote the synthesis of cartilage extracellular matrix and inhibit cartilage degradation (19); however, a practical mode of IGF-1 delivery to cartilage has yet to be developed (33). Heparan sulfate proteoglycans are prevalent in the pericellular matrix of cartilage, particularly as chains on perlecan and syndecan-2, and are known to bind other ligands such as FGF-2 (34). Our experiments suggest that Xp-HB-IGF-1 protein can bind with matrix and increase local, long-term bioavailability to chondrocytes and thus may improve cartilage repair.
Selected References
Hameed M, Orrell RW, Cobbold M, Goldspink G, Harridge SD. Expression of IGF-I splice variants in young and old human skeletal muscle after high resistance exercise. J. Physiol 2003;547:247–254. [PubMed: 12562960]
Shavlakadze T, Winn N, Rosenthal N, Grounds MD. Reconciling data from transgenic mice that overexpress IGF-I specifically in skeletal muscle. Growth Horm. IGF Res 2005;15:4–18. [PubMed: 15701567]
Milner SJ, Francis GL, Wallace JC, Magee BA, Ballard FJ. Mutations in the B-domain of insulin-like growth factor-I influence the oxidative folding to yield products with modified biological properties. Biochem. J 1995;308(Pt 3):865–871. [PubMed: 8948444]
Milner SJ, Carver JA, Ballard FJ, Francis GL. Probing the disulfide folding pathway of insulin-like growth factor-I. Biotechnol. Bioeng 1999;62:693–703. [PubMed: 9951525]
Bonassar LJ, Grodzinsky AJ, Srinivasan A, Davila SG, Trippel SB. Mechanical and physicochemical regulation of the action of insulin-like growth factor-I on articular cartilage. Arch. Biochem. Biophys 2000;379:57–63. [PubMed: 10864441]
Denley A, Cosgrove LJ, Booker GW, Wallace JC, Forbes BE. Molecular interactions of the IGF system. Cytokine Growth Factor Rev 2005;16:421–439. [PubMed: 15936977]
Musaro A, Dobrowolny G, Rosenthal N. The neuroprotective effects of a locally acting IGF-1 isoform. Exp. Gerontol 2007;42:76–80. [PubMed: 16782294]
Farndale RW, Buttle DJ, Barrett AJ. Improved quantitation and discrimination of sulphated glycosaminoglycans by use of dimethylmethylene blue. Biochim. Biophys. Acta 1986;883:173–177. [PubMed: 3091074]
Yayon A, Klagsbrun M, Esko JD, Leder P, Ornitz DM. Cell surface, heparin-like molecules are required for binding of basic fibroblast growth factor to its high affinity receptor. Cell 1991;64:841– 848. [PubMed: 1847668]
Martin P. Wound healing—aiming for perfect skin regeneration. Science 1997;276:75–81. [PubMed: 9082989]

Figure 1. Construction and purification of a new Xp-HB-IGF-1 fusion protein.
A) The heparin binding domain of HB-EGF was inserted N-terminal to IGF-1 to generate the fusion protein. The construct included the hexahistidine and Xpress tags from the pTrcHis vector for purification and detection. B) The resulting amino acid sequence of HB-IGF-1. C) Schematic for the structure of HB-IGF-1. Red circles: positively charged amino acids; blue circles: negatively charged amino acids; yellow circles: cysteines. The arrow shows the HB domain. In this figure the epitope tags are not shown. D, E) Representative reverse-phase high-performance liquid chromatography (RP-HPLC) elution profiles with single peaks containing correctly folded protein. Readings of optical density at 214 nm are in blue; readings at 280 nm are in red; elution is by acetonitrile (ACN) gradient. F) After RP-HPLC, Coomassie blue staining and Western blot analysis demonstrate isolation of single bands containing Xpress-tagged protein. The right panel shows that the Western blot analysis of IGF-1, and the two engineered IGF-1 proteins yield similar results using an anti-IGF-1 antibody.
Protein Therapeutics for Cardiac Regeneration after Myocardial Infarction
Vincent F.M. Segers and Richard T. Lee
Provasculon Inc, 14 Cambridge Center, and Harvard Stem Cell Institute and the Cardiovascular Division, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Cambridge, MA
J Cardiovasc Transl Res. 2010 October ; 3(5): 469–477. http://dx.doi./10.1007/s12265-010-9207-5.
Although most medicines have historically been small molecules, many newly approved drugs are derived from proteins. Protein therapies have been developed for treatment of diseases in almost every organ system, including the heart. Great excitement has now arisen in the field of regenerative medicine, particularly for cardiac regeneration after myocardial infarction. Every year, millions of people suffer from acute myocardial infarction, but the adult mammalian myocardium has limited regeneration potential. Regeneration of the heart after myocardium infarction is therefore an exciting target for protein therapeutics.
In this review, we discuss different classes of proteins that have therapeutic potential to regenerate the heart after myocardial infarction. Protein candidates have been described that induce angiogenesis, including fibroblast growth factors and vascular endothelial growth factors, although thus far clinical development has been disappointing. Chemotactic factors that attract stem cells, e.g. hepatocyte growth factor and stromal cell derived factor-1, may also be useful. Finally, neuregulins and periostin are proteins that induce cell cycle reentry of cardiomyocytes, and growth factors like IGF-1 can induce growth and differentiation of stem cells. As our knowledge of the biology of regenerative processes and the role of specific proteins in these processes increases, the use of proteins as regenerative drugs could develop as a cardiac therapy.
Keywords: protein therapeutics; myocardial infarction; regeneration; heart failure
The current standard of care for MI is early reperfusion of the occluded vessel with angioplasty or thrombolysis to reverse ischemia and increase the number of surviving myocytes. Efforts to decrease delays between onset of symptoms and reperfusion have resulted in decreased morbidity and mortality, but the maximal benefit of early reperfusion has reached a point close to practical limits. Besides early reperfusion therapy, ACE inhibitors and beta-blockers are used to prevent remodeling after MI and progression to heart failure. Both ACE inhibitors and beta-blockers improve long term survival but no therapies besides cardiac transplantation are currently available that restore cardiac function.
In the last decade, a large number of pre-clinical and clinical studies have been published on the potential use of stem cells for cardiac regeneration after MI. Different stem cell types have been shown to improve cardiac function in animal studies and can induce a small but potentially significant increase in ejection fraction in clinical studies. Stem cell therapy is a promising treatment option for heart failure, but numerous technical challenges and gaps in our understanding of stem cell behavior may limit translation to the clinic.
With the advent of biotechnology, protein and peptide drugs are becoming increasingly important in modern medicine. Drugs based on naturally-occurring proteins have the advantage of efficacy based on a mechanism of action refined by millions of years of biological evolution. Though promising as therapeutics, proteins might behave differently when used at pharmacological instead of physiological concentrations with an increase in adverse effects on other organs. Proteins used as therapeutics have been modified in different ways to limit immunogenicity and rapid degradation in plasma and tissues.
We discuss four different classes of proteins that could potentially benefit patients with MI (Figure 1); all of these proteins have been shown to improve cardiac function in animal models of MI or heart failure. They include angiogenic growth factors, proteins that increase recruitment of progenitor cells to the heart, proteins that induce mitosis of existing myocytes, and proteins that increase differentiation and growth of stem cells and myocytes. As more is learned about cardiac regeneration and why mammals lack sufficient myocardial regeneration, more proteins are likely to be added to this list of candidates.
A decade of extensive research on cardiac stem cell biology revealed 1 protein (G-CSF) that can be used to mobilize hematopoietic stem cells and just 2 proteins with chemotactic properties on stem cells: SDF-1 on endothelial progenitor cells and HGF on cardiac stem cells. Another protein that has been identified as a stem cell attractant is monocyte chemotactic protein-3 which attracts mesenchymal stem cells [42]. It is unknown if local administration of MCP-3 improves cardiac function. Identification of new stem cell chemotactic proteins is important because it could lead to the development of new and feasible therapeutics for treatment of MI and heart failure. At the same time, the true regenerative potential of most stem cells remains highly controversial; indicating that even if a chemotactic factor attracting stem cells to the heart is identified, formation of functional myocardial is still a challenging task.
Proteins like periostin and neuregulin which stimulate mitosis of surviving myocytes can partially restore the damage inflicted by MI. However, some requirements have to be met before this will result in a viable therapy. An inherent selectivity for myocytes would also allow for systemic delivery as opposed to the use of more complicated local delivery methods. An important factor to consider is the duration of the signal necessary to induce mitosis in a significant number of myocytes. A protein that induces cell cycle reentry in a significant fraction of myocytes with a single pulse has more therapeutical potential than a protein that needs sustained or repeated delivery. Ideally, pro-mitotic proteins will be not only specific for myocytes in general but might also be specific for myocytes in the border zone of the MI. This has drawbacks, among which is that formation of new myocytes, either by stem cell differentiation or by myocyte mitosis, carries an increased risk of ventricular arrhythmias.
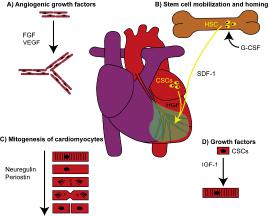
Figure 1. Regeneration of the heart by 4 different classes of proteins
See text for details. A) FGF and VEGF increase angiogenesis. B) G-CSF mobilizes bone marrow hematopoietic stem cells and SDF-1 attracts endothelial progenitor cells. HGF attracts cardiac stem cells. C) Neuregulin and periostin can induce division of adult cardiomyocytes. D) IGF-1 induces maturation and differentiation of cardiac stem cells.
Read Full Post »


