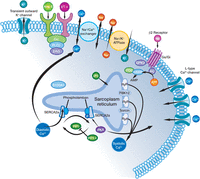
Renal Distal Tubular Ca2+ Exchange Mechanism in Health and Disease
Author and Curator: Larry H. Bernstein, MD, FCAP
Curator: Stephen J. Williams, PhD
and
Curator: Aviva Lev-Ari, PhD, RN

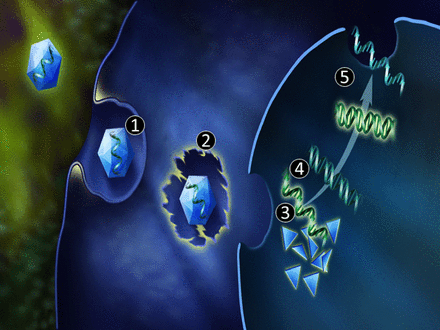
Image generated by Adina Hazan, 06/30/2021
This is Part III in a series of articles on the role of Calcium Release Mechanism in cell biology and physiology.
The Series consists of the following articles:
Part I: Identification of Biomarkers that are Related to the Actin Cytoskeleton
Larry H Bernstein, MD, FCAP
Part II: Role of Calcium, the Actin Skeleton, and Lipid Structures in Signaling and Cell Motility
Larry H. Bernstein, MD, FCAP, Stephen Williams, PhD and Aviva Lev-Ari, PhD, RN
Part III: Renal Distal Tubular Ca2+ Exchange Mechanism in Health and Disease
Larry H. Bernstein, MD, FCAP, Stephen J. Williams, PhD and Aviva Lev-Ari, PhD, RN
Part IV: The Centrality of Ca(2+) Signaling and Cytoskeleton Involving Calmodulin Kinases and Ryanodine Receptors in Cardiac Failure, Arterial Smooth Muscle, Post-ischemic Arrhythmia, Similarities and Differences, and Pharmaceutical Targets
Larry H Bernstein, MD, FCAP, Justin Pearlman, MD, PhD, FACC and Aviva Lev-Ari, PhD, RN
Part V: Ca2+-Stimulated Exocytosis: The Role of Calmodulin and Protein Kinase C in Ca2+ Regulation of Hormone and Neurotransmitter
Larry H Bernstein, MD, FCAP and Aviva Lev-Ari, PhD, RN
Part VI: Calcium Cycling (ATPase Pump) in Cardiac Gene Therapy: Inhalable Gene Therapy for Pulmonary Arterial Hypertension and Percutaneous Intra-coronary Artery Infusion for Heart Failure: Contributions by Roger J. Hajjar, MD
Aviva Lev-Ari, PhD, RN
Part VII: Cardiac Contractility & Myocardium Performance: Ventricular Arrhythmias and Non-ischemic Heart Failure – Therapeutic Implications for Cardiomyocyte Ryanopathy (Calcium Release-related Contractile Dysfunction) and Catecholamine Responses
Justin Pearlman, MD, PhD, FACC, Larry H Bernstein, MD, FCAP and Aviva Lev-Ari, PhD, RN
Part VIII: Disruption of Calcium Homeostasis: Cardiomyocytes and Vascular Smooth Muscle Cells: The Cardiac and Cardiovascular Calcium Signaling Mechanism
Justin Pearlman, MD, PhD, FACC, Larry H Bernstein, MD, FCAP and Aviva Lev-Ari, PhD, RN
Part IX: Calcium-Channel Blockers, Calcium Release-related Contractile Dysfunction (Ryanopathy) and Calcium as Neurotransmitter Sensor
Justin Pearlman, MD, PhD, FACC, Larry H Bernstein, MD, FCAP and Aviva Lev-Ari, PhD, RN
Part X: Synaptotagmin functions as a Calcium Sensor: How Calcium Ions Regulate the fusion of vesicles with cell membranes during Neurotransmission
Larry H Bernstein, MD, FCAP and Aviva Lev-Ari, PhD, RN
Part XI: Sensors and Signaling in Oxidative Stress
Larry H. Bernstein, MD, FCAP
http://pharmaceuticalintelligence.com/2013/11/01/sensors-and-signaling-in-oxidative-stress/
Part XII: Atherosclerosis Independence: Genetic Polymorphisms of Ion Channels Role in the Pathogenesis of Coronary Microvascular Dysfunction and Myocardial Ischemia (Coronary Artery Disease (CAD))
Larry H Bernstein, MD, FCAP and Aviva Lev-Ari, PhD, RN
Renal Distal Tubular Ca2+ Exchange Mechanism
This is the Third article of a multipart series covering Ca(2+) signaling and the cytoskeleton, and two on Ca2+ in cardiac contractility governed by the activations involving a ryanodine (RyR2) receptor and a specific calmodulin protein CaKIIδ with B and C splice variants. In all of these discussions, Ca(2+) has a crucial role in many cellular events, not all of which are detailed, and its importance to cardiac function and function disorders is critical. We shall next undertake the difficult examination of Ca(2+) movements in the kidney, which has a special relationship to vitamin D and bone mineral metabolism that is not of interest here. Nor will we go into any depth on the importance of the kidney to maintenance of plasma H+ and K+ balance and metabolic acidosis. Whereas the lung has a large role in pH maintenance by the respiratory rate (under sympathetic control), it maintains the balance through the expiration of CO2, with H+ tied up in water via the carbonic anhydrase reaction.
Key words, abbreviations:
calcium, magnesium, phosphate, renal calcium transport, calcium channels, diltiazem, mibefradil, ω-conotoxin. FGF23, Parathyroid hormone (PTH), Thick Ascending Loop (TAL), cTAL, proximal tubule, distal convoluted tubule (DCL), chronic kidney disease, Ca2+-ATPase, Ca2+-stimulated adenosine triphosphatase, Na++K-E-ATPase: (Na++K+)-stimulated adenosine triphosphatase, Na+-K+-2Cl cotransporter (NKCC@),TRPV6, calbindin- D9K, Ca2+ – ATPase, 4-(2-hydroxyethyl)-1-piperazine-ethanesulphonic acid, Non-hypertensive uremic, de novo cardiomyopathy, renal transplantation, karyotypes, isoform, Angiotensin converting enzyme (ACE), Basic fibroblast growth factor (BFGF), Extracellular signal regulated kinase (ERK), Friend leukemia integration-1 transcription factor (Fli-1), Growth hormone (GSH), oxidative stress, Nitric oxide (NO), Protein kinase C (PKC),angiotensin II, Renin-angiotensin system (RAS), Transforming growth factor-beta (TGF-b), Vascular endothelial growth factor (VEGF), 22-oxacalcitriol (OCT), Calcium-sensing receptor (CaSR), Claudin14, Claudin 16, bradykinin, bradykinin B2 receptor antagonists, inosine, marino-bufagenin (MBG), ramipril, nifedipine or moxonidine, calcitriol, Vitamin D receptor (VDR), Alpha-Kloth and FGf23
The first part in the Series, excludes calcium related heart failure and arrhythmias of calcium and includes the following:
(Part I) Identification of Biomarkers that are Related to the Actin Cytoskeleton Curator: Larry H Bernstein, MD, FCAP
(Part II) Role of Calcium, the Actin Skeleton, and Lipid Structures in Signaling and Cell Motility Larry H. Bernstein, MD, FCAP, Stephen Williams, PhD and Aviva Lev-Ari, PhD, RN
(Part III) Renal Distal Tubular Ca2+ Exchange Mechanism in Health and Disease Larry H. Bernstein, MD, FCAP, Stephen J. Williams, PhD and
Aviva Lev-Ari, PhD, RN
This article is a continuation to the following article series on tightly related topics:
Part I: Identification of Biomarkers that are Related to the Actin Cytoskeleton
Larry H Bernstein, MD, FCAP
(Part II) Role of Calcium, the Actin Skeleton, and Lipid Structures in Signaling and Cell Motility Larry H. Bernstein, MD, FCAP, Stephen Williams, PhD and Aviva Lev-Ari, PhD, RN
(Part III) Renal Distal Tubular Ca2+ Exchange Mechanism in Health and Disease
Larry H. Bernstein, MD, FCAP, Stephen J. Williams, PhD and Aviva Lev-Ari, PhD, RN
(Part IV) The Centrality of Ca(2+) Signaling and Cytoskeleton Involving Calmodulin Kinases and Ryanodine Receptors in Cardiac Failure, Arterial Smooth Muscle, Post-ischemic Arrhythmia, Similarities and Differences, and Pharmaceutical Targets
Larry H Bernstein, MD, FCAP, Justin Pearlman, MD, PhD, FACC and Aviva Lev-Ari, PhD, RN
Part V: Heart Failure and Arrhythmia: Potential for Targeted Intervention — The Effects of Ca 2+ -calmodulin (Ca-CaM) phosphorylation/dephosphorylation/hyperphosphorylation
Larry H Bernstein, MD, FCAP, Justin Pearlman, MD, PhD, FACC and Aviva Lev-Ari, PhD, RN
(VI) Calcium Cycling (ATPase Pump) in Cardiac Gene Therapy: Inhalable Gene Therapy for Pulmonary Arterial Hypertension and Percutaneous Intra-coronary Artery Infusion for Heart Failure: Contributions by Roger J. Hajjar, MD
Curator: Aviva Lev-Ari, PhD, RN
http://pharmaceuticalintelligence.com/2013/08/01/calcium-molecule-in-cardiac-gene-therapy-inhalable-gene-therapy-for-pulmonary-arterial-hypertension-and-percutaneous-intra-coronary-artery-infusion-for-heart-failure-contributions-by-roger-j-hajjar/
Cardiac Contractility & Myocardium Performance: Ventricular Arrhythmias and Non-ischemic Heart Failure – Therapeutic Implications for Cardiomyocyte Ryanopathy (Calcium Release-related Contractile Dysfunction) and Catecholamine Responses in the Human Heart
Justin Pearlman, MD, PhD, FACC, Larry H Bernstein, MD, FCAP and Aviva Lev-Ari, PhD, RN
http://pharmaceuticalintelligence.com/2013/08/28/cardiac-contractility-myocardium-performance-ventricular-arrhythmias-and-non-ischemic-heart-failure-therapeutic-implications-for-cardiomyocyte-ryanopathy-calcium-release-related-contractile/
Calcium Ion Transport across Plasma Membranes
Basal-lateral-plasma-membrane vesicles and brush-border-membrane vesicles were isolated from rat kidney cortex by differential centrifugation followed by free-flow electrophoresis. Ca2+ uptake into these vesicles was investigated by a rapid filtration method. Both membranes show a considerable binding of Ca2+ to the vesicle interior, making the analysis of passive fluxes in uptake experiments difficult. Only the basal-lateral-plasma-membrane vesicles exhibit an ATP-dependent pump activity which can be distinguished from the activity in mitochondrial and endoplasmic reticulum by virtue of the different distribution during free-flow electrophoresis and its lack of sensitivity to oligomycin. The basal-lateral plasma membranes contain in addition a Na+/Ca2+-exchange system which mediates a probably rheogenic counter-transport of Ca2+ and Na+ across the basal cell border. The latter system is probably involved in the secondary active Na+-dependent and ouabain-inhibitable Ca2+ reabsorption in the proximal tubule, the ATP-driven system is probably more important for the maintenance of a low concentration of intracellular Ca2+.
In recent micropuncture studies using simultaneously tubular and capillary perfusion it could be demonstrated that in the rat kidney proximal tubule Ca2+ reabsorption is dependent on the presence of Na+- ions and sensitive to ouabain (Ullrich et al., 1976). On the other hand cell-fractionation studies on the distribution of plasma-membrane-bound enzymes in rat proximal tubular epithelial cells revealed a contraluminal localization of a Ca2+-stimulated ATPase (Kinne-Saffran & Kinne, 1974). These results suggested that both Na+-driven and ATP-driven Ca2+ transport systems might be involved in proximal tubular transepithelial Ca2+ transport. Considering the low concentration of intracellular Ca2+ one could expect that these active steps in Ca2+ reabsorption are located at the basal cell pole.
To our knowledge there have been two attempts to study the role of ATP in the Ca2+ transport of renal membranes. In one study increase in Ca2+ uptake by rabbit kidney membranes was observed, but this increase was attributed to a phosphorylation of the membranes and a concomitant binding of Ca2+ to the negative charges newly generated at the membrane surface. Moore et al. (1974) observed an ATP-dependent Ca2+ uptake distinct from that of the mitochondria in a crude fraction of renal plasma membranes as well as in rat renal microsomes. The two uptake systems differed in their capacity, their sensitivity to Na+ and their apparent Km values for Mg2+-ATP.
Experiments are described on the Ca2+ transport into brush-border-membrane vesicles and basal-lateral plasma-membrane vesicles isolated from rat renal cortex. The results show that a primary active ATP-driven Ca2+ pump and an Na+/Ca2+-exchange system are present in the basal-lateral plasma membranes, but not in the brush-border membrane.
These findings indicate that trans-epithelial Ca2+ transport in rat proximal tubule can be
- primarily active via the ATP-driven system as well as
- secondarily active if the Na+/Ca2+ exchange system is involved.
It appears that the Na+/Ca2+ exchange system
- is responsible for the bulk flow of Ca2+ across the epithelium, whereas
- the ATP-driven system might be involved in the fine regulation of the concentration of intracellular Ca2+.
(Gmaj P, Murer H, and Kinne R. 1979)
The Renal Na+/Ca2+ Exchange System of the Nephron
The movement of Ca2+ across the basolateral plasma membrane was studied from rabbit proximal and distal convoluted tubules and ATP-dependent Ca2+ uptake was found in both. But the activity was higher distal. The distal tubular membranes had a very active Na+/Ca2+ exchange system, which was absent in the proximal segment. The ATP-dependent Ca2+ uptake in the distal tubular membrane preparations was gradually inhibited by Na+ outside the vesicles, and was a function of the imposed Na+ gradient. The results indicate that an active Na+/Ca2+ exchange system is absent in the proximal tubule. Ramachandram & Brunette, 1989). Parathyroid hormone (PTH) and calcitonin increase Ca2+ uptake by purified distal tubular luminal membranes (DTLM), and both hormone stimulate adenylate cyclase and phospholipase C. Therefore, distal tubules were incubated with dibutyryl cAMP (dbcAMP) and the result was that dbcAMP increased the Ca2+ transport by luminal membranes, but phorbol 12-myristate 13 acetate (PMA) had no such effect. But when PMA was added to low concentrations of dbcAMP the uptake significantly increased. Protein kinase C inhibitors prevented the effect. This indicated that in the distal tubule Ca2+ transport required both the combined effect of PK A and C involves both components of the transport kinetics. (Hila, Claveau, Laclerc, Brunette, 1997)
In the rabbit, calcitonin enhances Ca2+ reabsorption in the distal tubule. Tubules were incubated with or in the absence of calcitonin, and the luminal or basolateral membranes were purified and Ca2+ transport was measured through the vesicles. The results were compared with those obtained from proximal tubule membranes, and the results were no effect of calcitonin on Ca2+ uptake in the proximal tubules. In the distal tubules there was the expected uptake, but the presence of Na+ in the suspension decreased the Ca2+ uptake. The uptake was partially restored by preincubation with calcitonin. Recall the experiment demonstrating a requirement for PK A and C in Ca2+ uptake indicating a dual kinetics of Ca2+ uptake by the distal luminal membranes. Calcitonin enhanced Ca2+ transport by the low affinity component, increasing the Vmax and leaving the K(m) unchanged. Renal calcitonin receptors usually couple to both adenylate cyclase and phospholipase C. Calcitonin stimulates cAMP and IP3 release. Incubation of the distal tubules with 10(-7) M calcitonin significantly increased both messengers. In contrast, calcitonin did not influence the IP3 nor the cAMP content of proximal tubules. Incubation of distal tubule suspensions with dbcAMP significantly increased Ca2+ uptake by the luminal membranes. However, incubation of these tubules with various concentrations of PMA (10 nM, 100 nM and 1 microM) had no effect on this uptake. Calcitonin also influenced Ca2+ transport by the distal basolateral membrane. Incubation of distal tubule suspensions with 10(-7) M calcitonin activated the Na+/Ca2+ exchanger activity, almost doubling the Na+ dependent Ca2+ uptake. Here again this action was mimicked by cAMP. The researchers concluded that calcitonin increases Ca2+ transport by the distal tubule through two mechanisms:
- the opening of low affinity Ca2+ channels in the luminal membrane and
- the stimulation of the Na+/Ca2+ exchanger in the basolateral membrane, both actions depending on the activation of adenylate cyclase.
(Zuo Q, Claveau D, Hilal G, Leclerc M, Brunette MG. 1997)
Calcium (Ca2+) filtered in the glomerulus is reabsorbed by the luminal membrane of the proximal and distal nephron. Ca2+ enters cells across apical plasma membranes along a steep electrochemical gradient, through Ca2+ channels. Regulation by hormones requires
- binding of these hormones to the basolateral membrane,
- interaction with G proteins,
- liberation of messengers,
- activation of kinases
- opening of the channels at the opposite pole of the cells.
It follows that if the Ca2+ entry through the luminal membranes of proximal and distal tubules is a membrane-limited process, then G proteins have a regulatory role. Luminal membranes were purified from rabbit proximal and distal tubule suspensions, and their vesicles were loaded with GTPγs or the carrier. Then, the 45Ca2+ uptake by these membrane vesicles was measured in the presence and absence of 100 mM NaCl. In the absence of Na+, intravesicular GTPγs significantly enhanced 0.5 mM Ca2+ uptake by the proximal membrane vesicles (p < 0.05). In the presence of Na+, however, this effect disappeared. In the distal tubules, intravesicular GTPγs increased 0.5 mM Ca2+ uptake in the absence (p < 0.02) and in the presence (p < 0.02) of Na+. The action of GTPγs, when present, was dose dependent. The distal luminal membrane is the site of two Ca2+ channels with different kinetics parameters. GTPγs increased the Vmax value of the low-affinity component exclusively, in the presence as in the absence of Na+. Finally, Ca2+ uptake by the membranes of the two segments was differently influenced by toxins: cholera toxin slightly stimulated transport by the proximal membrane, but had no influence on the distal membrane, whereas pertussis toxin decreased the cation uptake by the distal tubule membrane exclusively. We conclude that the nature of Ca2+ channels differs in the proximal and distal luminal membranes: Ca2+ channels present in the proximal tubule and the low-affinity Ca2+ channels present in the distal tubule membranes are directly regulated by Gs and Gi proteins respectively, whereas the high-affinity Ca2+ channel in the distal tubule membrane is insensitive to any of them.
(Brunette MG, Hilal G, Mailloux J, Leclerc M. 2000)
We previously reported a dual kinetics of Ca2+ transport by the distal tubule luminal membrane of the kidney, suggesting the presence of several types of channels. We, therefore, examined the effects of specific inhibitors (i.e., diltiazem, an L-type channel; ω-conotoxin MVIIC, a P/Q-type channel; and mibefradil, a T-type channel antagonist) on Ca2+ uptake by rabbit nephron luminal membranes. None of these inhibitors influenced Ca2+ uptake by the proximal tubule membranes. In contrast, in the absence of sodium (Na+), the three channel antagonists decreased Ca2+ transport by the distal membranes, and their action depended on the substrate concentrations: (P < 0.05) without influencing 0.5 mM Ca2+ transport, whereas ω-conotoxin MVIIC decreased 0.5 mM Ca2+ (P < 0.02) and 1 µM mibefradil decreased it (P < 0.05); the latter two inhibitors [P/Q type, T-type] left 0.1 mM Ca2+ transport unchanged. Diltiazem [L-type] decreased the Vmax of the high-channels, whereas ω-conotoxin MVIIC and mibefradil influenced exclusively the Vmax of the low-affinity channels. These results not only confirm that the distal luminal membrane is the site of Ca2+ channels, but they suggest that these channels belong to the L, P/Q, and T types. (M G Brunette, M Leclerc, D Couchourel, J Mailloux, Y Bourgeois. 2000)
Calcium (Ca2+) transport by the distal tubule (DT) luminal membrane
Calcium (Ca2+) transport by the distal tubule (DT) luminal membrane is regulated by
- the parathyroid hormone (PTH) and calcitonin (CT) through the action of messengers,
- protein kinases, and
- ATP as the phosphate donor.
Could ATP itself, when directly applied to the cytosolic surface of the membrane influence the Ca2+ channels previously detected in this membrane. We purified the luminal membranes of rabbit proximal (PT) and DT separately and measured Ca2+ uptake by these vesicles loaded with ATP or the carrier. The presence of 100 μM ATP in the DT membrane vesicles significantly enhanced 0.5 mM Ca2+ uptake in the absence of Na+ (P < 0.01) and in the presence of 100 mM Na+ (P < 0.01). This effect was dose dependent with an EC50 value of approximately 40 μM. ATP action involved the high-affinity component of Ca2+ transport, decreasing the Km from 0.08 ± 0.01 to 0.04 ± 0.01 mM (P< 0.02). Replacement of the nucleotide by the nonhydrolyzable ATPγs abolished this action. Because ATP has been reported to be necessary for cytoskeleton integrity, they investigated the effect of intravesicular cytochalasin on Ca2+ transport. Cytochalasin B decreased 0.5 mM Ca2+ uptake (P< 0.01). However, when both ATP and cytochalasin were present in the vesicles, the uptake was not different from that observed with ATP alone. Neither ATP nor cytochalasin had any influence on Ca2+ uptake by the PT luminal membrane. They conclude from this that the high-affinity Ca2+ channel of the DT luminal membrane is regulated by ATP and that ATP plays a crucial role in the integrity of the cytoskeleton which is also involved in the control of Ca2+ channels within this membrane. (MG. Brunette*, J Mailloux, G Hilal. 1999)
Proximal tubular sodium-calcium exchanger
The functional expression of the renal sodium-calcium exchanger has been amply documented in studies on renal cortical basolateral membranes. In perfused renal tubules, other investigators have shown sodium-calcium exchange activity in the
- proximal convolution
- in the distal convolution,
- the connecting tubule, and
- the collecting tubule of the rabbit.
In rat proximal tubules, we found that the sodium-calcium exchanger is an important determinant of cytosolic calcium homeostasis, since
- inhibition of sodium-dependent calcium efflux mode caused a large accumulation of tubular calcium.
In membranes from rat proximal tubules sodium-calcium activity was high, and in intact proximal tubules,
- the tubular sodium-calcium exchanger exhibited a high affinity for cytosolic calcium
and had a substantial transport capacity, which may be absolute requirements for the maintenance of stable cytosolic calcium in proximal tubules. (Dominguez JH, Juhaszova M, Feister HA. 1992.)
Proximal tubule Na(+)-Ca2+ exchanger protein is same as the cardiac protein
The activity of the Na(+)-Ca2+ exchanger, a membrane transporter that mediates Ca2+ efflux, has been described in amphibian and mammalian renal proximal tubules. However, demonstration of cell-specific
- expression of the Na(+)-Ca2+ exchanger in proximal renal tubules has been restricted to functional assays.
In this work, Na(+)-Ca2+ exchanger gene expression in rat proximal tubules was characterized by three additional criteria:
- functional assay of transport activity in membrane vesicles derived from proximal tubules, expression of
- specific Na(+)-Ca2+ exchanger protein detected on Western blots, and
- determination of specific mRNA encoding Na(+)-Ca2+ exchanger protein on Northern blots.
A new transport activity assay showed that proximal tubule membranes
- contained the highest Na(+)-Ca2+ exchanger transport activity reported in renal tissues.
In dog renal proximal tubules and sarcolemma, a specific protein of approximately 70 kDa was detected, whereas in rat proximal tubules and sarcolemma, the specific protein approximated 65 kDa and was localized to the basolateral membrane. On Northern blots, a single 7-kb transcript isolated from rat
- proximal tubules,
- whole kidney, and
- heart
hybridized with rat heart cDNA.
These data indicate that Na(+)-Ca2+ exchanger protein expressed in rat proximal tubule is similar, if not identical, to the cardiac protein. We suggest that the tubular Na(+)-Ca2+ exchanger characterized herein represents the Na(+)-Ca2+ exchanger described in functional assays of renal proximal tubules. (Dominguez JH, Juhaszova M, Kleiboeker SB, Hale CC, Feister HA. 1992.)
Calcium reabsorption regulated by the distal tubules
Extracellular calcium homeostasis involves coordinated calcium absorption by
- the intestine,
- calcium resorption from bone, and
- calcium reabsorption by the kidney.
This review addresses the mechanism and regulation of renal calcium transport. Calcium reabsorption occurs throughout the nephron. However, distal tubules are the nephron site at which calcium reabsorption is regulated by
- parathyroid hormone,
- calcitonin, and
- 1 alpha,25-dihydroxyvitamin D3 and
where the magnitude of net reabsorption is largely determined. These and related observations underscore the view that distal tubules are highly specialized
- to permit fine regulation of calcium excretion in response to
- alterations in extracellular calcium levels.
Progress in understanding the mechanism and regulation of calcium transport has emerged from application of
- single cell fluorescence,
- patch clamp, and
- molecular biological approaches.
These techniques permit the examination of
- ion transport at the cellular level and
- its regulation at subcellular and molecular levels.
This editorial review focuses on recent and emerging observations and attempts to integrate them into models of cellular calcium transport. (Friedman PA , Gesek FA. 1993)
Calcium-Sensing Receptor (CSR)
Renal tubular calcium reabsorption is a critical determinant of extracellular fluid (ECF) calcium concentration; for the need of constancy of ECF calcium concentration,
- the renal tubular handling of calcium is tightly controlled
- in order to match renal calcium excretion to the net amount of calcium entering the ECF.
Both parathyroid hormone (PTH) and vitamin D metabolites are involved in
- the control of renal tubular calcium reabsorption and
- ECF calcium concentration [1].
Besides this hormonal control, it has been recognized recently that
- ECF calcium is able to regulate its own reabsorption by the mammalian tubule.
Indeed, a large body of evidence supports the view that ECF calcium exerts this action
- by activating the calcium/polyvalent cation-sensing receptor (CaSR)
- located in the plasma membrane of many tubular cell types.
First, increasing ECF calcium concentration
- elicits a marked increase in urinary calcium (and magnesium) excretion [2,3] and
- this occurs independently of any change in the calcium-regulating hormones [2,3].
Second, the inhibitory effect of ECF calcium on its own reabsorption is shared by other CaSR agonists, e.g. magnesium [4].
Third, the relationship between ECF calcium and urinary calcium excretion
is altered in patients bearing mutations of the CASR gene: renal tubular calcium reabsorption
- is enhanced in patients with inactivating mutations [5,6]
- and decreased in patients with activating mutations.
Therefore, there is abundant evidence that renal tubular CaSR plays a role
- in the control of divalent cations reabsorption under
- both normal and pathological conditions.
Localization of the extracellular CaSR
Transcripts of the CASR gene are expressed in many nephron segments of rat kidney, extending from glomeruli to the inner medullary collecting duct (IMCD) [7]. The CaSR protein is expressed in
- the proximal tubule,
- medullary and cortical thick ascending limb (TAL) segments,
- macula densa cells,
- distal convoluted tubule (DCT) and
- type-A intercalated cells in the distal tubule and cortical collecting duct [8]
- and in inner medullary collecting duct cells [9].
The polarity of expression varies from segment to segment, the protein being expressed in
- the apical membrane of proximal tubule and
- IMCD cells and
- in the basolateral membrane of TAL and DCT cells [8,9].
Interestingly, the highest density of protein expression has been observed in the cortical TAL (cTAL),
- known to reabsorb calcium and magnesium in a regulated manner.
CaSR under physiological conditions
Consistent with its polarized plasma membrane localization,
- CaSR has been shown to be involved in the control of thick ascending limb (TAL) calcium and magnesium reabsorption.
In the mouse and rat TAL,
- both calcium and magnesium are reabsorbed selectively in the cortical portion (cTAL) [10]
- and this reabsorption is passive along an electrical gradient
through the paracellular pathway [10,11]. The electrical gradient is related to
- transcellular NaCl reabsorption.
The first step is NaCl entry into the cell via
- the electroneutral apical Na- K-2Cl co-transporter BSC1 (NKCC2).
Subsequently, most of the potassium recycles back to the lumen, through an apical potassium channel,
- necessary to maintain NaCl absorption via BSC1 (NKCC2).
In the absence of recycling, NaCl absorption is inhibited because of
- the low availability of potassium in luminal fluid.
In addition, potassium recycling hyperpolarizes the apical membrane.
Chloride exits the cell
- across the basolateral membrane
- mainly via the CLC-Kb channel,
- which depolarizes the basolateral membrane.
The overall consequence is a lumen-positive transepithelial voltage that
- drives calcium, magnesium and also sodium through the paracellular pathway.
The pathway permeability for calcium and magnesium requires the presence of a specific protein,
- paracellin-1 (also known as claudin-16),
- co-expressed with occludin
- in the tight junctions of thick ascending limb (TAL) [12].
Inactivating mutations of the paracellin-1 gene cause a specific
- decrease in cTAL calcium and magnesium reabsorption and
- renal loss of both cations without renal sodium loss,
which is the landmark of an inherited disease referred to as hypercalciuric hypomagnesaemia with nephrocalcinosis [4].
Calcium and magnesium reabsorption in the cTAL is tightly regulated. Micropuncture studies have shown that peptide hormones, such as
- PTH,
- arginine vasopressin,
- calcitonin and
- glucagon,
stimulate NaCl as well as calcium and magnesium reabsorption in the loop of Henle and decrease their excretion in final urine. PTH, the most important peptide hormone for the stimulation of renal calcium transport, elicits an increase in calcium and magnesium reabsorption cTAL.
Wittner et al. [14] demonstrated that PTH stimulation of calcium and magnesium transport
- involves an increase in paracellular pathway permeability.
The activation of CaSR also affects a number of intracellular events in TAL cells and
- modulates transport processes along the cTAL epithelium.
Activating CaSR increases intracellular free calcium concentration in
- cTAL,
- DCT and
- cortical as well as
- outer medullary collecting duct.
This also decreases hormone-dependent cAMP accumulation in cTAL by
- inhibition of type-6 adenylyl cyclase [20],
- increases inositol phosphate formation [21] and
- elicits an increase in phospholipase A2 activity and
- in intracellular cellular production of 20-hydroxyeicosatetraenoic acid [22]. ….
In conclusion, a large body of evidence supports the view that CaSR is
- a major regulator of calcium and magnesium reabsorption in the cTAL and,
- of overall tubular divalent cation handling.
However, several issues remain unresolved. It is still unclear whether CaSR activation in the cTAL decreases NaCl reabsorption in this segment or not. The mechanism through which CaSR activation could alter the function of paracellin-1 and the paracellular pathway permeability also remains unsettled. Finally, the role of CaSR in the medullary part of TAL should be investigated: a CaSR-dependent inhibition of NaCl reabsorption could explain at least part of the polyuria that accompanies hypercalcaemic states. (P Houillier and M Paillar. 2003)
Alpha-Kloth and FGf23
Recent advances that have given rise to marked progress in clarifying actions of alpha(α)-Klotho (alpha-Kl) and FGf23 can be summarized as follows ;
(i) α-Kl binds to Na(+), K(+)-ATPase, and Na(+), K(+)-ATPase is recruited to the plasma membrane by a novel α-Kl dependent pathway in correlation with cleavage and secretion of α-Kl in response to extracellular Ca(2+) fluctuation.
(ii) The increased Na(+) gradient created by Na(+), K(+)-ATPase activity drives the transepithelial transport of Ca(2+) in the choroid plexus and the kidney, this is defective in α-kl(-/-) mice.
(iii) The regulated PTH secretion in the parathyroid glands is triggered via recruitment of Na(+), K(+)-ATPase to the cell surface in response to extracellular Ca(2+) concentrations.
(iv) α-Kl, in combination with FGF23, regulates the production of 1,25 (OH) (2)D in the kidney. In this pathway, α-Kl binds to FGF23, and α-Kl converts the canonical FGF receptor 1c to a specific receptor for FGF23, enabling the high affinity binding of FGF23 to the cell surface of the distal convoluted tubule where α-Kl is expressed.
(v) FGF23 signal down-regulates serum phosphate levels, due to decreased NaPi-IIa abundance in the apical membrane of the kidney proximal tubule cells.
(vi) α-Kl in urine increases TRPV5 channel abundance at the luminal cell surface by hydrolyzing the N-linked extracellular sugar residues of TRPV5, resulting in increased Ca(2+) influx from the lumen.
These findings revealed a comprehensive regulatory scheme of mineral homeostasis that is illustrated by the mutually regulated positive/negative feedback actions of α-Kl, FGF23, PTH and 1,25 (OH) (2)D. In this regard, α-Kl and FGF23 might play pivotal roles in mineral metabolism as regulators that integrate calcium and phosphate homeostasis, although this concept requires further verification in the light of related findings. Here, the unveiling of the molecular functions of α-Klotho and FGF23 has recently given new insight into the field of calcium and phosphate homeostasis. Unveiled molecular functions of α-Kl and FGF23 provided answers for several important questions regarding the mechanisms of calcium and phosphate homeostasis that remained to be solved, such as :
(i) what is the non-hormonal regulatory system that directly responds to the fluctuation of extracellular Ca(2+),
(ii) how is Na(+), K(+)-ATPase activity enhanced in response to low calcium stimuli in the parathyroid glands,
(iii) what is the exact role of FGF23 in calcium and phosphorus metabolism,
(iv) how is Ca(2+) influx through TRPV5 controlled in the DCT nephron, and finally
(v) how is calcium homeostasis regulated in cerebrospinal fluid. However, several critical questions still remain to be solved. So far reported,
- α-Kl binds to Na(+),
- K(+)-ATPase,
- FGF receptors and FGF23, and
- α-Kl hydrolyzes the sugar moieties of TRPV5.
The following questions are unresolved:
Does alpha-Kl recognize these proteins directly or indirectly?
Is there any common mechanism?
How can we reconcile such diverse functions of alpha-Kl?What is the Ca(2+) sensor machinery and how can we isolate it?
How do hypervitaminosis D and the subsequently altered mineral-ion balance lead to the multiple phenotypes?
What is the phosphate sensor machinery and how can we isolate it?
How does the Fgf23/α-Kl system regulate phosphorus homeostasis?
How are serum concentrations of Ca(2+) and phosphate mutually regulated?
(Nabeshima Y. 2008)
Cilium and Calcium Signal
We tested the hypothesis that the primary cilium of renal epithelia is mechanically sensitive and serves as a flow sensor in MDCK cells using differential interference contrast and fluorescence microscopy. Bending the cilium, either by suction with a micropipette or by increasing the flow rate of perfusate, causes intracellular calcium to substantially increase as indicated by the fluorescent indicator, Fluo-4. This calcium signal is initiated by Ca2+-influx through mechanically sensitive channels that probably reside in the cilium or its base. The influx is followed by calcium release from IP3-sensitive stores. The calcium signal then spreads as a wave from the perturbed cell to its neighbors by diffusion of a second messenger through gap junctions. This spreading of the calcium wave points to flow sensing as a coordinated event within the tissue, rather than an isolated phenomenon in a single cell. Measurement of the membrane potential difference by microelectrode during perfusate flow reveals a profound hyperpolarization during the period of elevated intracellular calcium. We conclude that the primary cilium in MDCK cells is mechanically sensitive and responds to flow by greatly increasing intracellular calcium. (Praetorius HA, Spring KR. 2001)
Fgf23 regulation in chronic renal disease
The mechanism of FGF23 action in calcium/phosphorus metabolism of patients with chronic kidney disease (CKD) was studied using a mathematical model and clinical data in a public domain. We have previously built a physiological model that describes interactions of PTH, calcitriol, and FGF23 in mineral metabolism encompassing organs such as bone, intestine, kidney, and parathyroid glands. Since an elevated FGF23 level in serum is a characteristic symptom of CKD patients, we evaluate herein potential metabolic alterations in response to administration of a neutralizing antibody against FGF23. Using the parameters identified from available clinical data, we observed that a transient decrease in the FGF23 level elevated the serum concentrations of PTH, calcitriol, and phosphorus. The model also predicted that the administration reduced a urinary output of phosphorous. This model-based prediction indicated that the therapeutic reduction of FGF23 by the neutralizing antibody did not reduce phosphorus burden of CKD patients and decreased the urinary phosphorous excretion. Thus, the high FGF23 level in CKD patients was predicted to be a failure of FGF23-mediated phosphorous excretion. The results herein indicate that it is necessary to understand the mechanism in CKD in which the level of FGF23 is elevated without effectively regulating phosphorus.
A traditional, physiological model with PTH and calcitriol needs to be rebuilt in accordance with the emerging role of FGF23 and its interacting molecules. To understand probable interactions among FGF23, PTH and calcitriol, we previously developed a minimum physiological model of calcium/phosphorus metabolism and investigated potential influences of FGF23 on the observable state variables such as the serum concentrations of PTH, calcitriol, calcium (Ca), and phosphorous (P), as well as the urinary excretion of Ca and P.3 In this study, we extended the model and evaluated the mechanism of FGF23-mediated regulation in chronic kidney diseases (CKD).
The FGF23 gene was identified by its mutations associated with autosomal dominant hypophosphatemic rickets (ADHR), which is an inherited phosphate wasting disorder.4 Thereafter, a variety of disorders resulting from gain or loss of FGF23 bioactivity have been reported.5 These disorders, which are caused by mutations in the genes that directly or indirectly interact with FGF23, include hyperphosphatemic familial tumoral calcinosis (HFTC), hereditary hypo-phosphatemic rickets with hypercalciuria (HHRH), autosomal recessive hypophosphatemic rickets (ARHR), and X-linked dominant hypophosphatemic rickets (XLH, HYP). CKD patients who need dialysis have very high levels of FGF23 in serum that are linked with increased rates of death.6
We examined the effect of reduction of FGF23 by neutralizing antibody would modulate phosphorus balance of CKD patients. We evaluated the levels of physiological variables such as the levels of PTH, calcitriol, FGF23, Ca, and P in serum as well as urinary outputs of Ca and P using clinical data. Since a glomerular filtration rate (GFR) is a good indicator of severity of CKD, data were processed as a function of GFR. We then employed the previously developed mathematical model for mineral metabolism, and conducted numerical simulations in response to the modulation of FGF23 by neutralizing antibody.
Estimation of the relationship of the FGF23 level to other physiological variables
The FGF23 concentrations, reported in literature, considerably varied among available datasets, presumably caused by differential baseline levels or sensitivity variations among individual assays. To predict a quantitative relationship among the FGF23 level and other physiological variables, the reported FGF23 level was linearly modified:
[FGF23]AB = {[FGF23]-A}/B (1)
in which [FGF23] = reported FGF23 level, [FGF23]AB = linearly modified FGF23 level, and A and B = two correction factors. Note that these correction factors are constant and they were chosen independently for each of the physiological variables such as the serum level of PTH and the urinary output of P. The “+” and “-” values of the factor B indicate positive and negative correlations to the FGF23 level, respectively. We applied the described modification in analyzing clinical data since the observed FGF23 variation was larger than others. Without this procedure, it was difficult to estimate a quantitative relationship of its concentrations to other variables. [With the significant variation around the linear fit, it might well have been warranted to use the log transform of the modified level, LHB].
Mathematical model and prediction of effects of FGF23 antibody
We previously developed a pair of metabolism models of calcium and phosphorus with and without including the predicted action of FGF23.3,20 In this study we considered an additional state variable, GFRf, as a multiplicative term pertaining to the calcium and phosphorus renal thresholds and the kidney production of calcitriol:
GFRf = (GFR/GFR0)k (2)
in which GFR0 and GFR = glomerular filtration rates in the control state and at any given degree of renal failure, respectively, and a factor k (>0) was chosen so as to fit the clinical data as described previously.7
To predict the effects of intravenous administration of a neutralizing antibody against FGF23, we numerically examined 5 different dosages for i.v. administration at 0.003, 0.01, 0.03, 0.1 and 0.3 mg/kg (dosage levels 1–5). These dosages corresponded to a clinical trial study being proposed for a dose-escalation study of KRN23 (Kyowa Hakko Kirin Pharma Inc.). A primary outcome measure of this Phase I clinical trial is a change in a serum phosphate level, and a single dose by intravenous or subcutaneous administration is planned. The initial target is X-linked hypophosphatemia but no clinical data regarding efficacy and side effects are available. To simulate a probable injection procedure, we assumed a form of a single, smoothed-out pulse. The rise in the antibody concentration was modeled using a Gaussian type diffusion profile with a period dependent on the distribution volume and cardiac output.
Glomerular filtration rate (GFR) as an indicator in cKD patients
We plotted physiological variables of CKD patients as a function of GFR in ml/min/1.73 m2. Figure 1 illustrated the levels of PTH (pg/ml), calcitriol (pg/ml), Ca (mg/dl), and P (mg/dl) in serum as well as urinary outputs of Ca and P expressed as a fraction of the glomerular loads. The numbers in the brackets in Figure 1 were the numbers of patients. The average and SEM values were obtained in each of the sampling bins. As GFR was normal above 90, the levels of PTH and P in serum as well as the fractions of urinary Ca and P outputs were lowered. On the contrary, the level of calcitriol in serum was higher as GFR increased.
Estimation of FGF23 levels in serum in cKD patients
The relationships of the linearly modified FGF23 concentration in serum, [FGF23]AB, to the selected physiological variables in CKD patients were illustrated in Figure 2. First, a strong correlation was observed between log.e(GFR) and a negative form of log.e[FGF23]AB, indicating that the FGF23 level was sharply elevated in CKD patients with reduction in GFR. Second, an increase in [FGF23]AB was correlated to the levels of PTH, calcitriol, P in serum, and the renal threshold for P. Note that a positive correlation (i.e. B > 0) was observed for the levels of PTH and P in serum, while a negative correlation (i.e. B < 0) for the serum level of calcitriol and the renal threshold for P. Note that a majority of data points had the PTH level above 50 pg/ml, indicating a poor balance of mineral metabolism in CKD patients.
Linkage of FGF23 and P levels in serum
In all groups, a positive correction was observed between the level of P and the modified level of FGF23 in serum. Note that CKD data in Figure 2D showed the elevated P level up to 6 mg/dl, while the higher bound of the P level was ∼2 mg/dl (Tumor Induced Osteomalacia), 3.5 ∼4 mg/dl (Fibrous Dysplasia and XLH), and 4.5 mg/dl (healthy populations).
Predicted effects of the antibody specific to FGF23
Although the observed increase of FGF23 in CKD is apparently a physiological response to hyperphos-phatemia, the use of FGF23 antibody is suggested for transplanted hypophosphatemic patients of CKD with a high level of FGF23.21 In response to intravenous administration of the antibody specific to FGF23, we evaluated the predicted changes in the serum levels of PTH, calcitriol, and P as well as a normalized urinary output of P. The results were positive.
(Yokota H, Pires A, Raposa JF, Ferreira HG. 2010.)
Overview of renal Ca2+ handling
About 50% of plasma calcium (ionized and complexed form; ultrafilterable fraction, excluding the protein bound form) is freely filtered through the renal glomerulus, and 99% of the filtered calcium is actually reabsorbed along renal tubules (Table 1- see Fig below on right)). The excreted calcium in the final urine is about 200 mg per day in an adult person with an average diet. Several factors are involved in the regulation of calcium in renal tubules. PTH and activated vitamin D enhance calcium reabsorption in the thick ascending limb (TAL), distal convoluted tubule (DCT) and/or connecting tubule (CNT).
Acidosis contributes to hypercalciuria by reducing calcium reabsorption in the proximal tubule (PT) and DCT, and alkalosis vice versa3). Diuretics like thiazide and furosemide also alter calcium absorption in the renal tubules; thiazide promotes calcium reabsorption and furosemide inhibits it. Plasma calcium itself also controls renal calcium absorption through altered PTH secretion as well as via binding to the calcium sensing receptor (CaSR) in the TAL.
To facilitate Ca2+ reabsorption along renal tubules;
(i) voltage difference between the lumen and blood compartment should be favorable for Ca2+ passage, i.e., a positive voltage in the lumen;
(ii) concentration difference should be favorable for Ca2+ passage with a higher Ca2+ concentration in the lumen;
(iii) an active transporter should exist if the voltage or concentration difference is not favorable for Ca2+ reabsorption. Each renal tubular segment has a different Ca2+ concentration difference or voltage environment for its unique mechanism for calcium re-absorption.
Calcium handling along the tubules
- In the collecting duct (CD), there is no evidence that Ca2+ reabsorption occurs even though calcium channel (TRPV6) was documented to be expressed in CD cells.
- Each renal tubule has a unique environment and plays a different role in Ca2+ reabsorption.
- The coordinated play of different renal tubules could maintain harmony of renal Ca2+ handling.
Transient receptor potential (TRP) channel is a super-family of ion channels permeable to monovalent and/or divalent cations with six-transmembrane domains. The mammalian TRP family consists of six subfamilies like TRPC (canonical), TRPV (vanilloid), TRPM (melastatin), TRPP (polycystin), TRPML (mucolipin), and TRPA (ankyrin). TRPV is one of them and consists of six members in mammalians; TRPV1 to TRPV6. TRPV5 (previously known as ECaC1) and TRPV6 (ECaC2), both cloned in 1999, have characteristics distinguished from other TRPV channels; (i) constitutively active at low intracellular Ca2+ concentration, and (ii) exclusively selective for Ca2+ (PCa/PNa >100)9). TRPV5 and TRPV6 have the highest sequence homology (~730 amino acids, amino-terminal ankyrin repeats, TM5 and TM6 each forming the pore-region composed with tetramer, on human chromosome 7q34-35) (Fig. 3a). TRPV5 is exclusively expressed in the DCT and CNT in the kidney10) (Fig. 3b). On the contrary, TRPV6 is more ubiquitously distributed, especially in the intestine, and also found from the DCT to the CD in the kidney11) (Fig. 3b). Both TRPV5 and TRPV6 are located in the apical plasma membrane of the tubular epithelium, and serve as the entrance of Ca2+ from the lumen into the cytoplasm. TRPV5 knockout mice exhibited severe hypercalciuria (more than 6 times of wild type mouse) and low bone densities, but without hypocalcemia due to the compensatory elevation of activated vitamin D, clearly demonstrating that TRPV5 plays a crucial role in renal calcium reabsorption12). TRPV6 knockout mice also showed significant hypercalciuria and bone disease13). Even though TRPV5 and TRPV6 knockout mice showed congenital hypercalciuria, the mutation of the proteins has not been found in the human. Until now, TRPV5 is known
as the main entry of Ca2+ in renal tubular epithelial cells in the DCT and CNT, and TRPV6 is also known to contribute to renal Ca2+ reabsorption in the distal nephron.
Several factors (PTH, 1,25(OH)2D3, calcitonin, estrogen, i[Ca2+], acid-base status, klotho, diuretics, and im-munosuppressive drugs, etc) are involved in the regulation of both TRPV5 and TRPV610) (Table 2). Alteration of TRPV5 and TRPV6 by these factors contributes in disturbance of calcium metabolism: dyscalcemia, hypo- and hypercalciuria. 1,25(OH)2D3-depleted rats showed decreased expression of TRPV5 and calbindin-D28K mRNA and protein, and repletion of the hormone restored the expression of them.
TRPV
- On the contrary, TRPV6 is more ubiquitously distributed, especially in the intestine, and also found from the DCT to the CD in the kidney
- Both TRPV5 and TRPV6 are located in the apical plasma membrane of the tubular epithelium, and serve as the entrance of Ca2+ from the lumen into the cytoplasm.
| Factors | TRPV5 TRPV6 Calbindin- Mechanisms | |||
|
D28K |
||||
| PTH | + | NC | + | transcription |
| Vit D | + | + | + | transcription |
| Estrogen | + | + | + | transcription |
| Low Ca2+ diet | + | + | NC | transcription |
| Acidosis | – | ND | – | transcription |
| Thiazide | C | ND | C | transcription |
| Furosemide | + | + | + | transcription |
| Tacrolimus | – | ND | – | transcription |
| [Ca2+] | – | – | – | Channel activity |
| Calbindin-D28K | + | NC | Channel activity | |
| Klotho | + | + | ND | trafficking |
FGF23
FGF23, a member of the FGF family (type I trans-membrane phosphotyrosine kinase receptors), is a 30 kDa secreted protein and inactivated by cleavage into two smaller fragments (N-terminal 18 kDa fragment and C-terminal 12 kDa fragment) by a pro-convertase enzyme, furin . It was first cloned as the candidate gene for autosomal dominant hypophosphatemic rickets (ADHR). FGF23 is primarily expressed in the osteoblasts and osteocytes. Because Fgf23 knockout mice showed very similar phenotype to Klotho knockout mice including severe hyperphophatemia and osteoporosis, and gain of function mutation of Fgf23 gene was observed in ADHR patients. The main studies about the role of FGF23 in the kidney have focused on phosphate metabolism rather than calcium metabolism.
It is unknown how the FGF23:klotho complex from the DCT acts in the PT because the main action site of FGF23 in the kidney is the PT, whereas the FGF23:klotho complex is most abundant in the DCT. Both overexpression and deficiency of FGF23 cause several clinical diseases including ADHR and HFTC (hyperphosphatemic familial tumorial calcino-sis). Recently, FGF23 was suggested as a potential bio-marker for management of phosphate balance in chronic kidney disease (CKD) patients because the circulating FGF23 level was higher in CKD patients than healthy controls and the increased FGF23 level was an independent risk factor for higher mortality among dialysis patients26). FGF23 also plays some roles in the parathyroid glands and other organs like the choroid plexus, pituitary gland, and bone. However, further studies are needed to clarify the roles and the mechanisms.
Conclusion
The kidney has been known as the central organ for calcium homeostasis through fine regulation of renal calcium excretion. For the past decade, there has been big progress in the understanding of the roles of the kidney in calcium homeostasis. The identification of calcium transport proteins and the molecular approach to the regulatory mechanisms achieved a major contribution to this progress. TRPV5, TRPV6, calbindin-D28K, NCX1, and PMCA1b have been identified as the main calcium transport proteins in the distal nephron. PTH, vitamin D, i[Ca2+], CaSR, and other various conditions control renal calcium excretion through the regulation of these transport proteins. Klotho and FGF23 emerged as new players in calcium metabolism in the kidney. Thus, the role of the klotho-FGF23 axis in the regulatory mechanisms of calcium transport needs to be addressed.
Disorders of Calcium, Phosphorus and Magnesium Metabolism
Infrequently patients might present in the outpatient settings with non-specific symptoms that might be due to abnormalities of divalent cation (magnesium, calcium) or phosphorous metabolism. Several inherited disorders have been identified that result in renal or intestinal wasting of these elements. Physicians need to have a thorough understanding of the mechanism of calcium, magnesium and phosphorous metabolism and diagnoses disorders due to excess or deficiency of these elements. Prompt identification and treatment of the underlying disorders result in prevention of serious morbidity and mortality.
Maintenance of serum calcium in the extra cellular fluid space (ECF) is tightly regulated. Most calcium (around 99%) is bound and complexed in the bones. Calcium in the ECF is found in three fractions, of which 45% is in biological ionized fraction, 45% is protein bound and not filterable in the kidney and 10% is complexed with anions such as bicarbonate, citrate, phosphate, and lactate (Fig. 1 ). Most of the protein bound calcium is complexed with albumin, and a smaller amount to globulin. Each 1 g/dL of albumin binds 0.8 mg/dL (0.2 mmol/L) calcium. Hence, for each 1g/ dl decrease in serum albumin below normal value of 4.0 g/dl, one needs to add 0.8 mg/ dl to the measured serum calcium. Levels of calcium are also influenced by acid-base status, with acidosis increasing serum calcium while alkalosis decreases serum calcium levels.
Maintenance of normal calcium in ECF is dependent on fluxes of calcium between the intestine, kidneys and bone. The regulation of calcium in serum is regulated by calcium itself, through a calcium sensing receptor (Ca RG) and hormones like parathormone (PTH) and 1, 25-dihydroxyvitamin D3.
Calcium transport across the intestine occurs in two directions, absorption and secretion. The factors that influence calcium absorption in the intestine include daily amount of calcium that is ingested and 1, 25-dihydroxyvitamin D3 that binds to and activates the Vitamin D receptor (VDR) and induces the expression of calcium channel TRPV6, calbindin- D9K, and Ca2+ – ATPase. Other hormones like PTH, estrogen, prolactin and growth hormone may play a minor role in calcium absorption. Conditions that result in decreased intestinal calcium transport include high vegetable fiber and fat content of food, corticosteroid deficiency, estrogen deficiency, advanced age, gastrectomy, intestinal malabsorption, diabetes mellitus, renal failure and low Ca2+ phosphate ratio in the food.
PTH and 1, 25- dihydroxyvitamin D3 stimulate osteoclasts in bones and promote release of calcium in ECF. PTH promotes hydroxylation of 25(OH) D3 to 1, 25(OH) D3 and distal tubular calcium reabsorption.
Hypocalcaemia occurs when the loss of calcium from the ECF via renal excretion is greater than influx of Ca 2+ from intestine or bones. One of the commonest cause of low calcium is hypoalbuminemia, though the level of ionized Ca2+ is normal. The causes of hypocalcaemia is summarized in Table 1 . Acute hypocalcaemia is often seen in acute respiratory alkalosis due to hyperventilation. Idiopathic or acquired (post surgery, radiotherapy) hypoparathyroid states are usually accompanied with elevated phosphate level. Pseudo hypoparathyroidism is characterized by short neck, round face and short metacarpal and results from end-organ resistance to PTH. Chronic kidney disease and massive phosphate administration can result in hypocalcaemia with high serum phosphate levels. Familial hypocalcaemia is linked with activating mutation of Ca RG. Hypocalcaemia with low phosphate levels occur in Vitamin D deficiency, resistance to calcitriol (Type 2 vitamin D- dependent rickets) acute pancreatitis and magnesium deficiency.
Table 1 : Causes of Hypocalcemia
Idiopathic Hypoparathyroidism
Post parathyroidectomy (Hungry bones syndrome)
Pseudo-hypoparathyroidism
Familial hypocalcemia
Rapid correction of severe acidosis with dialysis
Acute respiratory and metabolic alkalosis
Acute pancreatitis
Rhabdomyolysis
Hypomagnesemia
Septic shock
Ethylene glycol toxicity
Vitamin D deficiency
Chronic kidney disease
Massive transfusion- Citrate toxicity
Hypercalcemia occurs when in influx of calcium into the ECF exceeds the efflux of calcium from intestine and kidneys. The normal calcium level ranges from 8.9- 10.1 mg/ dL. The range of serum calcium levels in mild hypercalcemia is (10.1- 12.0 mg/dL), moderate hypercalcemia (12.0 – 14.0 mg/dl) and severe hypercalcemia > 14.0 mg/ dL respectively. The various causes of hypercalcemia is depicted in Table 2. Mutation of the gene for Ca RG results in hypercalcemia in few cases.
Table 2. : Causes of hypercalcemia
Parathormone Primary hyperparathyroidism
(PTH) mediated Lithium induced
Familial hypocalciuric hypercalcemia
Tertiary hyperparathyroidism
Cancer Multiple myeloma
PTHrp mediated-Breast, lung,
Exogenous Vitamin D
Dialysis patients (exogenous Vit D)
Other causes Vitamin A toxicity
Thyrotoxicosis
Paget’s disease
Adrenal insufficiency
Thiazide use
Deficiency of calcium, magnesium and phosphorous are common in general practice. A thorough understanding of pathophysiology of these elements, common dietary sources of these elements and pharmacological measures that might be necessary to correct these deficiencies could guide the physician to make an accurate diagnosis, initiate appropriate treatment and prevent future recurrences. (Ghosh AK*, Joshi SR. 2008.)
Renal Disease and the Cardiovascular System
Cardiovascular disease is a leading cause of death among patients with end stage renal failure. Animal models have played a crucial role in teasing apart the complex pathological processes involved. In addition to the anatomical and histological characteristics humans share with other species, human diseases can be reproduced in these species using pharmacological, surgical or genetic manipulation. Experimentation still provides the best evidence for disease causation, and only with this evidence can clinical science proceed to developing treatments. However, experimentation is often not possible or ethical in human subjects, and thus without these animal models the advancement in knowledge of the patho-physiology of disease would come to a standstill.
The way in which kidneys succumb to disease and the development of renal failure involves complex interactions between numerous different systems, mediated by a multitude of chemicals. Current understanding of renal disease is merely the tip of the metaphorical iceberg. The history of renal pathology is plagued by controversy, and nowhere is this more evident than in the development of cardiovascular disease in patients with chronic renal failure. Impairment of renal function increases the risk of cardiac disease to 15-20 times that of individuals with normal renal function. The result is that cardiac disease causes 40% of deaths in patients on dialysis.
This review discusses the principles of using animal models, the history of their use in the study of renal hypertension, the controversies arising from experimental models of non-hypertensive uraemic cardiomyopathy and the lessons learned from these models, and highlights important areas of future research in this field, including de novo cardiomyopathy secondary to renal transplantation.
Myocardial Interstitial Fibrosis, Cardiac Compliance and Vascular Architecture
Using subtotally nephrectomised Sprague-Dawley rats, Mall et al. showed that the increase in total heart weight demonstrated by Rambausek et al. after 21 days of uremia (as well as an increase in both right and left ventricular weight) was secondary to an increase in true interstitial volume, both cellular and non-cellular, with increased deposition of collagen. This was associated with activated interstitial cells, and a reduced capillary cross-sectional area. In 1992, this latter point was confirmed using stereological techniques to analyse perfusion-fixed hearts of subtotally nephrectomised Sprague-Dawley rats. Uremia resulted in increased blood pressure and reduced capillary length per unit myocardial volume, as well as reduced capillary luminal surface density and volume density, compared to control rats. The same group found a blood pressure-independent increase in the wall to lumen ratio of intramyocardial arteries, and in the aorta media thickness of subtotally nephrectomised rats. The intramyocardial arterial wall thickening has been found to be due to hypertrophy rather than hyperplasia, independent of blood pressure. These architectural changes were reported again in 1996. In that experiment, nephrectomised Sprague-Dawley rats were given ramipril, nifedipine or moxonidine to normalise blood pressure; these drugs had differential effects on the above architectural changes, and also acted to prevent these changes. The different changes in interstitial and capillary density in uremic cardiomyopathy have not yet been explained, but the role of growth factors such as basic fibroblast growth factor (BFGF) and vascular endothelial growth factor (VEGF) has been proposed.
Cardiac Function and Energetics in Uremia
The above experiments provided some insight into the structural changes seen in uraemic hearts. They were followed by a study using the subtotal (5/6) nephrectomy model on Wistar rats, in which the authors focused on the mechanical effects of these structural changes in vitro, thereby removing neurohormonal influences on cardiac contractility. Four weeks after surgery, isolated perfusing working heart preparations demonstrated reduced cardiac output. However, blood pressure was not controlled during the four weeks post-operatively, and could have contributed to the effects. An increased susceptibility to ischemic damage was also shown via decreased phosphocreatine content, and an increased release of inosine (a marker of ischaemic damage). These hearts failed in response to increases in calcium; the authors proposed that impaired cytosolic calcium control played a role in the relationship between renal failure and impaired cardiac function.
This in vitro experiment demonstrated the fact that impaired cardiac function was independent of circulating urea and creatinine, as the hearts were perfused with physiological saline, with no effect from the addition of urea and creatinine. The opposite has been shown in spontaneously beating mouse cardiac myocytes, in response to sera from patients on haemodialysis for chronic renal failure. Urea, creatinine, and combinations of the two reduced the cardiac inotropy and resulted in arrhythmias and asynchronies.
These experiments make a good case for uremic cardiomyopathy to be a distinct entity from hypertensive cardiac dysfunction and atherosclerotic cardiac disease secondary to the risk factors common to both heart and kidney disease. The cause of this phenomenon is still controversial, with parathyroid hormone (PTH), angiotensin II, marino-bufagenin (MBG), oxidative stress, and growth hormone.
The Role of Calcium in Uremic Cardiomyopathy
Calcium ions play a crucial role in cardiac physiology, particularly in myocardial excitation-contraction coupling. Therefore, PTH was one of the first culprits to be suspected of playing a role in the pathophysiology of uremic cardiomyopathy; this was as early as 1984. As reviewed by Rostand and Drüeke, there are numerous theories pertaining to the mechanisms whereby PTH could act as an intermediary between renal impairment and cardiomyopathy. These include
- direct trophic effects on myocytes
- interstitial fibroblasts,
- indirect effects via anaemia or large and small vessel changes.
Rostand and Drüeke suggest an increase in blood pressure via hypercalcemia, but the effects on the heart appear to be independent of blood pressure.
Rambausek et al. noted increased cardiac calcium content in experimental rats, and that an increase in heart weight still occurred after parathyroidectomy with calcium supplementation. This was followed in the 1990s by in vitro experiments that demonstrated; an increased cytosolic calcium concentration in isolated rat myocytes in response to PTH, a reduced expression of PTH-related peptide receptor mRNA in rat hearts secondary to hyperparathyroidism due to chronic renal failure, and increased force and frequency of contraction of isolated, beating rat cardiomyocytes.
Subsequent to “chance observations” in the laboratory, Amann et al. argued for the role of PTH in the wall thickening of intramyocardial arterioles and for fibroblast activation and subsequent cardiac fibrosis. Abolishing hyperparathyroidism prevented the cardiac fibrosis and capillary changes normally seen in nephrectomised rats, which was independent of blood pressure.
The Renin-Angiotensin System (RAS) and Endothelin
Many studies have highlighted the importance of the RAS in the development of uremic cardiomyopathy. Tornig et al. showed that in nephrectomised rats, ramipril, an ACE inhibitor, prevented the increased wall thickness of the intramyocardial arterioles, as well as the expansion of nonvascular cardiac interstitial volume and the aortic wall and lumen changes, but not the reduced capillary length density. The same group subsequently repeated these observations, and demonstrated that the beneficial effects of ramipril were prevented by the use of specific bradykinin B2 receptor antagonists, suggesting a role for increased bradykinin as a mediator for the effects of ramipril.
CONCLUSIONS
Experimental models have played a crucial role in the study of the complex interplay between the heart and the kidney in chronic renal disease. In view of the numerous differences in animal and human anatomy, physiology and pathology, the results of these experiments should be interpreted with caution, but in some areas, these studies have led directly to advances in therapeutics.
(RC Grossman. 2010.)
Deficiency of the Calcium-Sensing Receptor
Rare loss-of-function mutations in the calcium-sensing receptor (Casr) gene lead to decreased urinary calcium excretion in the context of parathyroid hormone (PTH)–dependent hypercalcemia, but the role of Casr in the kidney is unknown. Using animals expressing Cre recombinase driven by the Six2 promoter, we generated mice that appeared grossly normal but had undetectable levels of Casr mRNA and protein in the kidney. Baseline serum calcium, phosphorus, magnesium, and PTH levels were similar to control mice. When challenged with dietary calcium supplementation, however, these mice had significantly lower urinary calcium excretion than controls (urinary calcium to creatinine, 0.31±0.03 versus 0.63±0.14; P=0.001). Western blot analysis on whole-kidney lysates suggested an approximately four-fold increase in activated Na+-K+-2Cl cotransporter (NKCC2). In addition, experimental animals exhibited significant downregulation of Claudin14, a negative regulator of paracellular cation permeability in the thick ascending limb, and small but significant upregulation of Claudin16, a positive regulator of paracellular cation permeability. Taken together, these data suggest that renal Casr regulates calcium reabsorption in the thick ascending limb, independent of any change in PTH, by increasing the lumen-positive driving force for paracellular Ca2+ transport.
(Toka HR, Al-Romaih K, Koshy JM, DiBartolo, III S, et al. 2012)
References:
The renal sodium-calcium exchanger.
Dominguez JH, Juhaszova M, Feister HA.
J Lab Clin Med. 1992 Jun;119(6):640-9.
http://www.ncbi.nlm.nih.gov/pubmed/1593210
Na(+)-Ca2+ exchanger of rat proximal tubule: gene expression and subcellular localization.
Dominguez JH, Juhaszova M, Kleiboeker SB, Hale CC, Feister HA.
Am J Physiol. 1992 Nov;263(5 Pt 2):F945-50.
http://www.ncbi.nlm.nih.gov/pubmed/1443182
Calcium transport in renal epithelial cells
PA Friedman, FA Gesek
Am J Physiol – Renal Physiology 1993; 264(F181-F198)
http://ajprenal.physiology.org/content/264/2/F181
Effect of calcitonin on calcium transport by the luminal and basolateral membranes of the rabbit nephron.
Zuo Q, Claveau D, Hilal G, Leclerc M, Brunette MG.
Kidney Int. 1997; 51(6):1991-9.
http://www.ncbi.nlm.nih.gov/pubmed/9186893
Ca2+ transport by the luminal membrane of the distal nephron: action and interaction of protein kinases A and C.
Hilal G, Claveau D, Leclerc M, Brunette MG.
Biochem J. 1997 Dec 1;328 ( Pt 2):371-5
http://www.ncbi.nlm.nih.gov/pubmed/9371690
Calcium-sensing receptor and renal cation handling
Pascal Houillier and Michel Paillar
Nephrol. Dial. Transplant. (2003) 18 (12): 2467-2470. http://ndt.oxfordjournals.org/content/18/12/2467.full
http://dx.doi.org/10.1093/ndt/gfg420
Patch-clamp evidence for calcium channels in apical membranes of rabbit kidney connecting tubules.
S Tan and K Lau
J Clin Invest. 1993; 92(6): 2731–2736
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC288471/
http://dx.doi.org/10.1172/JCI116890
Bending the MDCK Cell Primary Cilium Increases Intracellular Calcium
H.A. Praetorius, K.R. Spring
J Membrane Biol 2001; 184(1), pp 71-7
http://link.springer.com/article/10.1007/s00232-001-0075-4
Branching points of renal resistance arteries are enriched in L-type calcium channels and initiate vasoconstriction.
M S Goligorsky, D Colflesh, D Gordienko, L C Moore
Am J Physiol 03/1995; 268(2 Pt 2):F251-7.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC9186893/
G proteins regulate calcium channels in the luminal membranes of the rabbit nephron
Brunette MG, Hilal G, Mailloux J, Leclerc M.
Nephron. 2000 Jul;85(3):238-47.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC10867539/
Characterization of three types of calcium channel in the luminal membrane of the distal nephron
MG Brunette, M Leclerc, D Couchourel, J Mailloux, Y Bourgeois
Can J Phy Pharma 2004; 82(1): 30-37 http://dx.doi.org/10.1139/y03-127
ATP directly enhances calcium channels in the luminal membrane of the distal nephron
MG Brunette, J Mailloux, G Hilal
J Cell Phys 1999; 181(3), pp 416–423
http://dx.doi.org/10.1002/(SICI)1097-4652(199912)181:3<416::AID-JCP5>3.0.CO;2-X
Discovery of alpha-Klotho and FGF23 unveiled new insight into calcium and phosphate homeostasis
Nabeshima Y.
Clin Calcium. 2008;18(7):923-34. http://dx.doi.org/CliCa0807923934
Deficiency of the calcium-sensing receptor in the kidney causes parathyroid hormone-independent hypocalciuria
Toka HR, Al-Romaih K, Koshy JM, DiBartolo S, III, Kos, CH, et al.
J Am Soc Nephrol 2012; 23: 1879-1890. http://dx.doi.org/10.1681/ASN.2012030323
The renal Na+/Ca2+ exchange system is located exclusively in the distal tubule
Ramachandram C, Brunette MG.
Biochem J 1989;257:259-264
Calcium ion transport across plasma membranes isolated from rat kidney cortex
Gmaj P, Murer H, Kinne R
Biochem J 1979; 178:549-557
Model-based analysis of Fgf23 regulation in chronic renal disease
Yokota H, Pires A, Raposo JF, Ferreira HG.
Gene Reg and Systems Biol 2010; 4: 53-60
Kidney and Calcium Homeostasis
Un Sil Jeon
Electrolyte & Blood Pressure 2008; 6:68-76
Related Articles including Pharma Intel
- Cardiac Contractility & Myocardium Performance: Ventricular Arrhythmias and Non-ischemic Heart Failure – Therapeutic Implications for Cardiomyocyte Ryanopathy (Calcium Release-related Contractile Dysfunction) and Catecholamine Responses in the Human Heart (pharmaceuticalintelligence.com)
- Ryanodine Receptor (RyR2 Subunits) in Heart Failure and Arrhythmia: Potential for Targeted Intervention – The Effects of Ca 2+ -calmodulin (Ca-CaM) phosphorylation/dephosphorylation/hyperphosphorylation (pharmaceuticalintelligence.com)
- Role of Calcium, the Actin Skeleton, and Lipid Structures in Signaling and Cell Motility (pharmaceuticalintelligence.com)
- Finding the Best Drug Combination for High Blood Pressure Control (everydayhealth.com)
- Calcium Molecule in Cardiac Gene Therapy: Inhalable Gene Therapy for Pulmonary Arterial Hypertension and Percutaneous Intra-coronary Artery Infusion for Heart Failure: Contributions by Roger J. Hajjar, MD (pharmaceuticalintelligence.com)
- Congestive Heart Failure-Pharmacological approach I (Cardiac Glycosides-Digoxin) (medicine1journal.wordpress.com)
The Amazing Structure and Adaptive Functioning of the Kidneys: Nitric Oxide – Part I
http://pharmaceuticalintelligence.com/2012/11/26/the-amazing-structure-and-adaptive-functioning-of-the-kidneys/
Nitric Oxide and iNOS have Key Roles in Kidney Diseases – Part II Larry H Bernstein, writer and curator
http://pharmaceuticalintelligence.com/2012/11/26/nitric-oxide-and-inos-have-key-roles-in-kidney-diseases/
The Molecular Biology of Renal Disorders: Nitric Oxide- Part III.
Curator and Author: Larry H. Bernstein, MD, FCAP
https://pharmaceuticalintelligence.com/wp-admin/post.php?post=5009&action=edit
New Insights on Nitric Oxide donors- Part IV.
Curator and Author: Larry H. Bernstein, MD, FCAP
https://pharmaceuticalintelligence.com/wp-admin/post.php?post=5043&action=edit
The Essential Role of Nitric Oxide and Therapeutic NO Donor Targets in Renal Pharmacotherapy- Part V.
Curator and Author: Larry H. Bernstein, MD, FCAP
https://pharmaceuticalintelligence.com/wp-admin/post.php?post=5060&action=edit
Prostacyclin and Nitric Oxide: Adventures in vascular biology – a tale of two mediators
Reporter: Aviva Lev-Ari, PhD, RN
https://pharmaceuticalintelligence.com/wp-admin/post.php?post=12604&action=edit
Nitric Oxide and it’s impact on Cardiothoracic Surgery
Author, curator: Tilda Barliya PhD
https://pharmaceuticalintelligence.com/wp-admin/post.php?post=5908&action=edit
Advanced Topics in Sepsis and the Cardiovascular System at its End Stage Larry H Bernstein, writer and curator
http://pharmaceuticalintelligence.com/2013/08/18/advanced-topics-in-sepsis-and-the-cardiovascular-system-at-its-end-stage/
The Cardio-Renal Syndrome (CRS) in Heart Failure (HF) Larry H Bernstein, writer and curator
http://pharmaceuticalintelligence.com/2013/06/30/the-cardiorenal-syndrome-in-heart-failure/
Hypertension and Vascular Compliance: 2013 Thought Frontier – An Arterial Elasticity Focus
Curator: Aviva Lev-Ari, PhD, RN
http://pharmaceuticalintelligence.com/2013/05/11/arterial-elasticity-in-quest-for-a-drug-stabilizer-isolated-systolic-hypertension-caused-by-arterial-stiffening-ineffectively-treated-by-vasodilatation-antihypertensives/
Inhaled Nitric Oxide in Adults: Clinical Trials and Meta Analysis Studies – Recent Findings
Curator: Aviva Lev-Ari, PhD, RN
https://pharmaceuticalintelligence.com/wp-admin/post.php?post=13840&action=edit
Low Bioavailability of Nitric Oxide due to Misbalance in Cell Free Hemoglobin in Sickle Cell Disease – A Computational Model
Author and Reporter: Anamika Sarkar, Ph.D.
https://pharmaceuticalintelligence.com/wp-admin/post.php?post=4655&action=edit
Differential Distribution of Nitric Oxide – A 3-D Mathematical Model
Author and Reporter: Anamika Sarkar, Ph.D.
https://pharmaceuticalintelligence.com/wp-admin/post.php?post=4268&action=edit