Quantum dots
Writer and Curator: Larry H. Bernstein, MD, FCAP
7.1 Quantum dots
7.1.1 Bioconjugated quantum dots for cancer research: present status, prospects and remaining issues.
7.1.2 Bioconjugated quantum dots for in vivo molecular and cellular imaging
7.1.3 In vivo molecular and cellular imaging with quantum dots
7.1.4 Luminescent quantum dots for multiplexed biological detection and imaging
7.1.5 Multifunctional quantum dots
7.1.6 Potentials and pitfalls of fluorescent quantum dots for biological imaging
7.1.1 Bioconjugated quantum dots for cancer research: present status, prospects and remaining issues.
Biju V, Mundayoor S, Omkumar RV, Anas A, Ishikawa M.
Biotechnol Adv. 2010 Mar-Apr;28(2):199-213
http://dx.doi.org:/10.1016/j.biotechadv.2009.11.007
Semiconductor quantum dots (QDs) are nanoparticles in which charge carriers are three dimensionally confined or quantum confined. The quantum confinement provides size-tunable absorption bands and emission color to QDs. Also, the photoluminescence (PL) of QDs is exceptionally bright and stable, making them potential candidates for biomedical imaging and therapeutic interventions. Although fluorescence imaging and photodynamic therapy (PDT) of cancer have many advantages over imaging using ionizing radiations and chemo and radiation therapies, advancement of PDT is limited due to the poor availability of photostable and NIR fluorophores and photosensitizing (PS) drugs. With the introduction of biocompatible and NIR QDs, fluorescence imaging and PDT of cancer have received new dimensions and drive. In this review, we summarize the prospects of QDs for imaging and PDT of cancer. Specifically, synthesis of visible and NIR QDs, targeting cancer cells with QDs, in vitro and in vivo cancer imaging, multimodality, preparation of QD-PS conjugates and their energy transfer, photosensitized production of reactive oxygen intermediates (ROI), and the prospects and remaining issues in the advancement of QD probes for imaging and PDT of cancer are summarized.
Fluorescence imaging and photodynamic therapy (PDT) are advancing clinical trials for efficient detection and curing of cancers. Fluorescence imaging of cancer is facilitated by targeting tumor milieus using fluorescent dyes conjugated with anticancer antibodies followed by exciting the dyes with visible or NIR light sources.In PDT, cancers are treated by applying a photosensitizing (PS) drug followed by light.The principle underlying PDT is that a photoactivated PS drug transfers energy or electron to oxygen or other molecules, and creates reactive oxygen intermediates (ROI), which immediately react with and damage vital biomolecules in cell organelles resulting in cell death. The main advantage of fluorescence imaging over other biomedical imaging techniques such as X-rays, CT and PET is that visible and NIR excitation in fluorescence imaging is non-ionizing and less hazardous. The main advantage of PDT over chemotherapy and radiation therapy is that site-specific photoactivation of targeted PS drugs using visible or NIR light offers selective therapy, leaving the immune system and normal cells intact. However, fluorescence imaging and PDT of cancer are challenging due to the limited availability of photostable and NIR dyes as PS drugs. The center of fluorescence imaging and PDT of cancer is the selective delivery of fluorescent dyes and PS drugs in tumor milieu.The basic principle underlying PDT is that photoactivation of a PS drug results in the formation of ROI such as singlet oxygen (1O2), hydroxyl radical (UOH), superoxide anion(−∙O2) and hydrogen peroxide (H2O2) through a series of energy and electron transfer reactions initiated between PS and dissolved oxygen (3O2) [(Ochsner, 1997) and (Oleinick and Evans, 1998)].
Fig. 1 shows various photophysical and photochemical processes involved in PDT. Briefly, photoactivation of a PS drug places it at the excited singlet (S1) and triplet (T1) states.The lifetime of the T1 states for most PS drugs ranges from several hundred nanoseconds to milliseconds, much longer than the S1 lifetime. A PS drug in the T1 state either relaxesto the groundstate (S0) by transferring excess energy to molecular oxygen or transfers an electron (also, at S1 state) to oxygen, water or a proximal molecule and enters into a series of photochemical reactions [(Ochsner, 1997) and (Oleinick and Evans, 1998)]. By the energy transfer from a PS to 3O2, an electron in the πx */πy * orbital in 3O2 changes its spin quantum number and forms 1O2, for which the energy required is only 94.3 kJ/ mol. 1O2 is an unstable species and it reacts with water, generating a sequence of ROI such as UOH, −∙O2 and H2O2. On the other hand, electron transfer from a PS drug directly produces ROI. However, electron transfer creates the cation radical of a PS, which irreversibly reactswithothermoleculeandresultsinthechemicaltransformation of PS (Lachheb et al., 2002). On the other hand, photosensitized production of ROI through energy transfer is a renewable process. Thus, energy transfer is preferred over electron transfer for the durability of PS drugs. In both the mechanisms, cell death is initiated by the photochemical reactions of ROI with biomolecules and cell organelles such as amino acids, endoplasmic reticulum, mitochondrion, lysosomes and Golgi apparatus. Examples of standard PS drugs for PDT are porphyrins, phthalocyanines, and chlorine derivatives. In the earlier days, a mixture of porphyrins, called the first generation PS drugs was used for PDT. For example, Dougherty etal. (1975) successfully cured breast cancer in a mouse model by applying hematoporphyrin derivatives as the PS drug. Later, with the introduction of purified PS drugs, also called the second generation PS drugs, such as porphyrins, phthalocyanines and chlorine derivatives, research on PDT has infiltrated into clinical trials. For example, superficial bladder cancer was treated by non-specific administration of photofrin as the PS drug followed by illuminating the bladder with red light (Nseyoetal.,1998). However,this approach suffered from severe side effects due to non-specific drug delivery and photoactivation. Recently,with the advancements such as synthesis of new generation PS drugs, targeted drug delivery, image-guided PDT, and introduction of tunable and fiber-optic laser light sources, imaging and PDT of cancer have become more popular methods for skin cancers, Barrett’s esophagus, bronchial cancers, head and neck cancer, lung cancer, prostate cancer, and bladder cancer. Recently, metal, semiconductor, polymer and ceramic nanoparticles have gained much attraction in the imaging and PDT of cancer (Brigger et al. 2002). Polymer and ceramic nanoparticles have been widely employed as drug carriers, whereas metal and semiconductor nanoparticles act as probes for imaging and therapy. Among various nanoparticles, semiconductor quantum dots (QDs) attracted much attention as probes for bioimaging [(Chan and Nie, 1998), (Bruchez et al.,1998),(Alivisatosetal.,2005),(Gaoetal.,2005),(Paraketal.,2005), (Medintz et al., 2005), (Michalet et al., 2005), (Klostranec and Chan, 2006), (Bijuetal.,2007a), (Hoshinoetal.,2007), (Jamiesonetal.,2007), (Hild et al., 2008), (Biju et al., 2008), (Smith et al., 2008), (Anas et al., 2009), (Delehanty et al., 2009), and (Walling et al., 2009)] and PDT [(Samiaetal.,2003), (Lovricetal.,2005), (Shietal.,2006), (Hsiehetal., 2006), (Tsayetal.,2007), (Bagalkotetal.,2007),(Anasetal.,2008), (Ma etal.,2008), (Juzenasetal.,2008a), (Wallingetal.,2009), and (Yaghini et al., 2009)]. QDs are nanoparticles in which electrons and holes are three dimensionally confined within the exciton Bohr radius of the material,providinguniqueopticalproperties,suchasbroadabsorption and sharp emission bands and size-tunable photoluminescence color [(Brus,1984),(Murrayetal.,1993),(Alivisatos,1996),(Dabbousietal., 1997) and (Biju et al., 2008)]. Also, bright emission, exceptional photostability, large-surface area, large two-photon absorption crosssection, availability in multicolor and with NIR photoluminescence are the most attractive properties of QDs for imaging and PDT of cancer.
Fig. 1. Photophysical and photochemical processes involved in PDT.
http://ars.els-cdn.com/content/image/1-s2.0-S073497500900192X-gr1.sml
Surface functionalization of quantum dots
High quality core-only and core/shell QDs with absorption and photoluminescence in the visible and NIR regions can be prepared by the methods described above. However, surface of such QDs is covered by hydrophobic molecules such as TOPO, TOP and TBP. On the other hand, QDs with hydrophilic surface-molecules and reactive functional groups are necessary for biological applications. Thus, conversion of hydrophobic-capped core and core/shell QDs from organic phase into an aqueous phase was extensively investigated. The conversion was carried out by coating or conjugating hydrophilic and amphiphilic molecules such as mercapto acids, hydrophilic dendrimers, silica-shells, amphiphilic polymers, proteins, and sugars on the surface of core and core/shell QDs. These methods are gracefully summarized by Medintz et al. (2005). For example, Chan and Nie (1998) successfully converted CdSe QDs from an organic to aqueous phase by exchanging hydrophobic molecules on the surface of QDs with mercaptoacetic acid. By a similar approach, Uyeda et al. (2005) tethered bidentate dihydrolipoic acid (DHLA) on the surface of CdSe/ZnS QDs and prepared water-soluble QDs. Now, surface modification of QDs using DHLA has become a popular method. The formation of disulfide bond with ZnS shell is the key in these preparations. Conjugation of biomolecules on the surface of QDs dispersed in water is another important requirement for biological applications. For this purpose, antibodies, nucleic acids, peptides, etc. can be attached either covalently or non-covalently on the surface of QDs. In particular, conjugation of anticancer antibodies, peptides and PS drugs on the surface of QDs is required for imaging and PDT of cancer. QDs bearing surface functional groups such as carboxylic acids, primary amine and thiol can be conjugated with antibodies and peptides by exploiting cross-linking chemistry of carbodiimide, maleimide and succinimide. Also, avidin–biotin cross-linking is one of the most popular methods for conjugating biomolecules on the surface of QDs. These methods are summarized in Fig. 2.
Fig. 2. Schematic presentation of steps involved in the bioconjugation of QDs.
http://ars.els-cdn.com/content/image/1-s2.0-S073497500900192X-gr2.sml
Absorption and photoluminescence properties of quantum dots
Broad absorption bands, sharp and symmetrical photoluminescence bands, large two-photon absorption cross-section, size-tunable absorption and photoluminescence spectra, and exceptional photostability are the optical properties of QDs attractive for biological applications. These properties, in particular, the size-tunable absorption and photoluminescence spectra of QDs originate from the large surface to volume ratios and the quantum confinement effect [(Brus, 1984)]. Due to the broad absorption band and the large two-photon absorption cross-section, QDs can be photoactivated using one- or multi-photon excitation. Also, the sharp and size-tunable photoluminescence of QDs is beneficial for multiplexed bioimaging. The absorption spectra of semiconductor QDs are broad due to a combined effect of a distribution of electronic transitions in the bulk semiconductoranddiscreteelectronictransitionssuchass–s,p–pand d–d transitionsdueto the quantumconfinement effect. However,the sharp photoluminescence bands of QDs are contributed by carrier recombination in the band-edge states. The band-edge states are quantum confined or size-dependent, and are 8-fold degenerate in CdSe QDs due to asymmetric and crystal-filed splitting, and mixing of carrier exchange perturbations with angular momentum of the charge carriers [(Norris and Bawendi, 1995), (Nirmal et al., 1995), (Efros et al.,1996) and (Nirmal and Brus,1999)]. Thus, for example, in the case of CdSe QDs the photoluminescence color shifts from near visible to NIR region with an increase in the size of QDs. In CdSeQDs, the highest occupied states are contributed by the 4p orbitals of selenium and the lowest unoccupied states are contributed by the 5s orbitals of cadmium. Similar to the size-dependent absorption and photoluminescence spectra for a given QD, the absorption and photoluminescence spectra can be tuned from UV to NIR regions by varying the core material.Forexample,2.5 nmdiameterCdS,CdSe,InP,CdTe,PbS,PbSe and PbTe QDs show near visible to NIR band-edge absorption and photoluminescence.Thus,QDs with suitable absorption spectrum and photoluminescence color for bioimaging and PDT can be easily selected based on either the core size or the core material. The merits of the broad absorption and sharp photoluminescence bands of QDs for cancer imaging and PDT are many. For example, QDs can be photoactivated at any wavelength below the band-edge absorption.
Fig. 3. (A) Schematic presentation of an immunoliposome internalized with doxorubicin and conjugated with QDs and anti-Her2 antibody. (B) Fluorescence images of human pancreatic cancer cells incubated with (a) InP QD-anti-Claudin-4 antibody conjugate and (b) InP QD without antibody. Reprinted with permission from (A) Weng et al. (2008) and (B) Yong et al. (2009). Copyright (2008, 2009) American Chemical Society.
http://ars.els-cdn.com/content/image/1-s2.0-S073497500900192X-gr3.sml
Targeted imaging of cancer cells using quantum dot-ligand conjugates
Anticancer antibodies are specific but expensive agents for targeting certain over-expressed receptors in cancer cells. Thus, alternative bioconjugates of QDs for targeted imaging of cancer cells were investigated by many researchers. For example, biomolecules such as arginine–glycine–aspartic acid (RGD peptide), folic acid, epidermal growth factor, transferrin and a few aptamers were investigated for targeting particular cancer cells. Like in the case of antibodies, these biomolecules target specific receptors over-expressed in cancer cells. For example, Cai et al. (2006) targeted MDA-MB-435 human breast cancer cells and U87MG human glioblastoma cells using QD conjugated with RGD peptide. The advantage of QD-RGD peptide conjugate is that the peptide selectively labels over-expressed αvβ3 integrin in the above cell lines. They also found that RGD peptide effectively distinguishes MCF-7 human breast cancer cells, in which αvβ3 integrin is not upregulated, from other cancer cells such as MDA-MB-435 and U87MG cells. Bharali et al. (2005) successfully labeled human nasopharyngeal epidermal carcinoma cells (KB cells) using InPQDs conjugated with folic acid. The advantages of InPQD-folic acid conjugate are twofold: InPQD is less toxic than QDs derived from heavy metals such as Cd, Pb, and Hg, and folic acid selectively recognizes over-expressed folate receptor in KB cells.Onthe other hand, human lung carcinoma cells (A549), in which folate receptor is not up-regulated, were not labeled by QD-folic acid conjugates.Bagalkotetal.(2007)foundthatQDslabeledwithaptamers were selectively delivered in prostate cancer cells. They labeled PSMA positive LNCaP prostate cells using QDs conjugated with an A10 RNA aptamer, but not PSMA-negative PC3 prostate adenocarcinoma cells. The QD-aptamer conjugate was found to be equally efficient as QDPSMA antibody conjugate for selectively labeling and imaging prostate cancer cells. Thus, the aptamer-based targeting is cost effective than antibody-based targeting. Like antibodies, ligands for membrane receptors are ideal candidates for targeting cancer cells. For example, Lidke et al. (2004) and Kawashima et al. (2010) found that CHO and A431 cells were efficiently labeled by QD-epidermal growth factor(EGF) conjugates due to the specific binding of EGF to EGFR. The advantage of QD-EGF conjugate is that it can be utilized for labeling various cancer cells because EGFR is over-expressed in many cancers. Although the QD-conjugates discussed above efficiently label over-expressed receptors in various cancer cells, the receptors are signaling proteins important for the regular growth and functioning of normal cells as well.
In vivo targeted imaging of cancer using quantum dots
In vivo targeted imaging of cancer cells using quantum dot-antibody conjugates
The basic principles underlying in vitro targeting of cancer cells can be applied in vivo. However, the main challenges for in vivo targeting and imaging of cancers using QDs are biodistribution of QD bioconjugates, penetration depths of excitation light and photoluminescence, tissue autofluorescence, toxicity and pharmacokinetics. Bioconjugated QDs were applied in vivo either systemically for deep cancers or subcutaneously for peripheral cancers. However,compared with local administration, systemic administration needs more attention owing to possible interactions of QD-conjugates with blood components and stimulation of immune response. Although it was found that QDs conjugated with various anticancer antibodies were selectively and uniformly distributed in tumor milieu, little evidence supports that QDs have the ability to extravasate to reach tumor cells in vivo. Indeed, biodistribution of QDs and non-specific uptake in the reticulo endothelial system that includes the liver, spleen and lymphatic system is an important issue remaining in the in vivo applications of QDs. InvivoapplicationofQDswas firsttestedbyAkermanetal.(2002). They injected CdSe/ZnS QDs coated with peptides into the tail vein in mouse, and found that the injected QDs preferentially distribute in endothelial cells in the lung blood vessels. Also, based on ex vivo fluorescence microscopic imaging of tissue sections, they found that the QD-peptide conjugates were preferentially bound to tumors. Subsequently, QDs conjugated with various cancer markers such as PSMA antibody (Gao et al., 2004), RGD peptide (Cai et al., 2006), alpha-fetoprotein (Yu et al., 2007) and anti-Her2 antibody (Weng et al.,2008) were tested in vivo in mouse models.Gao etal.(2004) were the first to apply QD-antibody conjugates in vivo and perform whole animal cancer imaging. They systemically administered QD-PSMA antibody conjugates in mouse bearing subcutaneous human prostate cancer. The QD-antibody conjugate was efficiently and uniformly distributed in prostate tumor due to the specific binding between PSMA antigen in prostate cancer cells and PSMA antibody on QDs (Fig. 4A). By using RGD peptide conjugated NIR QDs, Cai et al. (2006) investigated in vivo targeting and imaging of cancers. They targeted glioblastoma with NIR QD-RGD peptide conjugate and investigated the selective targeting by in vivo whole animal imaging and ex vivo tumor imaging. As described in the previous section, the key factor underlying in this targeting is the selective binding of RGD peptide to over-expressed αvβ3 integrin in U87MG glioblastoma cells and MDAMB-435 human breast cancer cells. Fig. 4B shows the signal to background ratio for NIR QD-RGD peptide conjugates in the cancer. More recently, Yu et al. (2007) found that QDs conjugated with an antibody to alpha-fetoprotein (anti-AFP) is an ideal candidate for in vivotargetedimagingofHCCLM6humanhepatacarcinomacells.They subcutaneously implanted HCCLM6 cancer cells in mice, and intravenously injected the QD-anti-AFP antibody conjugates. AFP, a main component in mammalian serum, is an important marker protein for liver cancer. Thus, the systemically administered QD-antiAFP conjugate was effectively accumulated in human hepatocarcinoma cells. Weng et al. (2008) developed multifunctional immunoliposomes for in vivo targeted imaging of cancers, drug delivery, and chemotherapy. As discussed in the previous section, they conjugated NIR QDs and anti-Her2 antibody on the surface of a liposome, and encompassed the liposome with doxorubicin, ananticancerdrug. The immunoliposome was applied to MCF-7/Her2 Xenografts implanted
in nude mouse. This multimodal approach of targeted imaging of cancersand drug deliveryhas great potentialsfor imaging and PDT of cancer.
4.2.2. Non-specific imaging of tumor vasculature and lymph nodes using quantum dots Withtheclassicalworkonmulti-photoninvivo fluorescenceimaging using QDs by Larson et al. (2003), targeted and two-photon imaging of tumor vasculature and lymph node using bioconjugated QDs attracted much attention in cancer research. Larson et al. (2003) systemically administeredwater-solubleCdSe/ZnSQDsinlivingmice,andvisualized capillaries in the adipose tissue and skin using NIR excited two-photon fluorescence.Thelargetwo-photonabsorptioncross-sectionofQDsisthe keyforNIRexcitationofvisibleQDs.Soonafterthisreport,non-specificin vivoimagingoftumorvasculature,lymphnodes,andlymphaticdrainage using bioconjugated QDs emerged into active research topics. For example, Stroh et al. (2005) targeted and imaged tumor vasculature associatedwithMCaIVisogenicmouseadenocarcinomatumorimplants in C3H mice using PEG-phosphatidylethanolamine-labeled core/shell CdS/ZnS and CdSe/ZnCdS QDs and two-photon excitation. Kim et al. (2004) applied QDs for in vivo lymph node mapping. They subcutaneously injected oligo-phosphine coated NIR CdTe/CdSe QDs in a mouse and a pig, and found that the QDs were drained within a few minutes after the injection into the sentinel lymph node (SLN) 1cm below the skin. The NIR photoluminescence of QDs enabled them not only to visualize the drainage of QDs towards SLN, but image-guided resection of samples as well. More recently, Ballou et al. (2007) successfully imaged lymph nodes in mice model using QDs without any specific surface functional group.
Fig. 4. (A) Fluorescence image of human prostate cancer implanted in a mouse. The tumor is targeted with anti-PSMA antigen conjugated CdSe/ZnS QDs. Reprinted by permission from Mcmillan publishers Ltd: [Nature Biotechnology], Ref. Gao et al. (2004).(B)Histogramof fluorescencesignalfromU87MGtumor-bearingmiceinjected with an NIR QD-RGD peptide conjugate. Reprinted with permission from Ref. Cai et al. (2006). Copyright (2006) American Chemical Society.
http://ars.els-cdn.com/content/image/1-s2.0-S073497500900192X-gr4.sml
Quantum dots for multimodal imaging
Magnetic resonance imaging (MRI), radiography, and fluorescence imaging are powerful biomedical imaging modalities. Each imaging modality has its merits and demerits and hence cannot achieve comprehensive imaging. Quality imaging requires high spatial and temporal resolutions, 3D tomography, excellent signal-to-noise ratio, and noninvasiveness. Individual modalities lack one or more of these qualities and therefore, multimodality has been sought as active imaging technology in basic research and biomedical applications. Independent implementation of imaging probes for different modalities cannot be an ideal solution to achieve multimodal imaging because different probes very often differ in their biodistribution and other pharmacodynamic properties. Thus, grouping the properties for different imaging modalities in the same chemical entity has been sought after. Multimodal imaging probes have components that function synergistically, complementing and enhancing the functionality of each other. Notably, QDs are promising multimodal probes as it is possible to combine multiple probe characteristics in QDs. For example, fluorescence imaging using QDs can be combined with MRI and radiography imaging if interfaced with molecules/materials having paramagnetism and radioactivity on the surface of QDs [(Cheon and Lee, 2008) and (Jennings and Long, 2009)]. Examples of bimodal imaging using QD probes are MRI-fluorescence imaging and scintigraphy-fluorescence imaging.The main advantage of QDs for multimodal imaging is the durability of the probe.On the other hand, fluorescence imaging using multimodal probes based on organic dyes such as FITC and rhodamine is less promising due to photobleaching. Typical example for MR-fluorescence bimodal imaging using QDs was investigated by Mulder et al. (2006) using multifunctional CdSe/ ZnSQDprobes.They coated QDs with pegylated phospholipid micelle, a Gd-diethylene triamine pentaacetic acid (DTPA) conjugate as MRI probe, and an RGD peptide for targeting cancer cells. By using this multifunctional probe, they successfully targeted endothelial cells and detected both by fluorescence and MRI imaging. This approach was extended to QD-based bimoda lprobes contained in a silica nanoparticle which is known to improve biocompatibility (Koole et al., 2008). AnotherexampleforQD-basedMR-fluorescencebimodalimagingisthe detection of apoptosis in a culture of Jurkat cells as well as in a murine carotid artery injury model by using QDs conjugated with annexin A5 andaGd-DTPAconjugate(Prinzenetal.,2007).Similarly,bycombining fluorescence and radioactivity in a single nanoprobe, Kobayashi et al. (2007)demonstrated dualmodalinvivolymphatic imaging in mice. In another report, Duconge et al. (2008) successfully demonstrated the utility of CdSe/ZnS QDs encapsulated in Fluorine-18 labeled phospholipids micelle as bimodal imaging probes for combined positron emission tomography (PET) and in vivo fibered confocal fluorescence imaging in mice. In short, as individual imaging technologies are now well-developed, biomedical imaging of cancer should receive a new dimension and momentum with the design and synthesis of suitable multimodal probes based on QDs. This appears achievable in the context of the rapid growth in the field of QDs and the wealth of information on the molecular mechanisms of cancer and other diseases.
Quantum dots for photodynamic therapy of cancer
The quality of a PS drug for PDT depends on its efficiency for energy and or electron transfer to molecular oxygen and the subsequent production of ROI. Compared with electron transfer, energy transfer is desirable for PDT because electron transfer products such as cation and anion radicals undergo irreversible chemical transformations, which prevent subsequent photoactivation of a PS drug and continuous generation of ROI. The concept “QDs for PDT” was proposed and investigated first by Samia et al. (2003). Exceptional photostability of QDs is the most promising property for PDT. Additionally, broad absorption band and large two-photon absorption cross-section of QDs are advantages for photoactivation using various visible and NIR light sources. Despite these advantages, photosensitized production of ROI at high efficiency is the primary requirement for a standard PS drug. Although targeted delivery of QDs in cancer cells and tumor milieu by using anticancer antibodies and other biomolecules have became possible recently, compared with conventional PS drugs such as porphyrins and phthalocyanines, the efficiency of QDs to produce ROI under direct photoactivation is low. Thus, preparation of conjugates between QDs and conventional PS drugs, investigation of energy transfer efficiencies from QDs to PS drugs and ROI production by the conjugates are being widely investigated.
Quantum dots vs conventional PS drugs for PDT
Samia et al. (2003) found that direct photoactivation of QDs produces 1O2 due to energy transfer from the dark exciton state of QDs to 3O2.
Fig. 5. Nude mouse bearing M21 melanoma, dorsal view 3 min after injection into the tumor using 655nmPEG5k-COOH quantum dots. Left, visible light; right, fluorescence at 655nm. Reprinted with permission from Ballouetal. (2007). Copyright (2007) AmericanChemical Society.
http://ars.els-cdn.com/content/image/1-s2.0-S073497500900192X-gr5.sml
Despite the low efficiency for 1O2, QDs offer prolonged photoactivation and persistent production of 1O2 and other ROI owing to the incredible photostability. Thus, in contrast to conventional PS drugs that are less photostable, QDs offer cumulative effects in PDT. For example,Anasetal.(2008)foundthatprolongedphotoactivationofa QD-plasmid DNA conjugate at 512 nm results in the breakage and damage of DNA. The breakage and damage of DNA were due to the photosensitized production of ROI, which was determined using nitroblue tetrazolium (NBT) chloride as the ROI scavenger. Also, the strand breakage of DNA was characterized by atomic force microscopy imaging and nucleobase damage was characterized by gel electrophoresis and base excision repair enzyme assays. ROI such as hydroxyl radical abstract hydrogen atoms from the bases or pyranose ring and create radical centers in DNA. Subsequent rearrangement of free radicals in DNA results in the strand breakage and nucleobase damage in DNA. Fig. 6 shows the photoactivation of a QD, various relaxation processes in a photoactivated QD, ROI production and subsequent breakage and damage of DNA. The photosensitized strand breakage and nucleobase damage of DNA suggest that QDs are promising PS drugs for nucleus targeted PDT if combined with intranuclear delivery of QDs in cancer cells. Also, Liang et al. (2007) reported that UV illumination of a mixture of calf thymus DNA and CdSe QDs results in DNA nicking, which was attributed to the reactions of DNA with ROI. Similarly, Clarke et al. (2006) reported that photoactivation of QD dopamine complex internalized in A9 cells results in DNA damage due to the production of 1O2. However, the production of 1O2 was due to electron transfer from QD to dopamine followed by the oxidation of dopamine. More recently, the potential of QDs as PS drugs for PDT was investigated by Juzenas et al. (2008b). They found that NIR photoactivation of QDs in cancer cells results in the production of ROI and reactive nitrogen species (RNS) such as superoxide and peroxynitrite. They employed dihydrorhodamine 123 as a sensor for the oxidation, and found that RONS generated by QDs results in the breakage of lysosomes. In contrast to the reports by Samia et al. (2003) and Anas et al. (2008), specific tests made by Juzenas et al. (2008a,b) using 9,10-dimethylanthracene, a 1O2 scavenger, and 1O2 sensorgreenindicatedthat 1O2 wasnotproducedbyQDsunderdirect photoactivation. The properties of QDs such as photostability, photosensitized production of ROI and RNS, and damage and breakage of DNA and lysosomes show the potentials of QDs for PDT. However, cytotoxicity of QDs due to photo-oxidation and chemical degradation should be resolved. For example, Derfus et al. (2004) found that CdSe QDs release toxic levels of cadmium ions inside cells and result in cell death. Similarly, Cho et al. (2007) found that human breast cancer cells MCF-7 treated with cysteine or mercaptoacetic acid capped CdTe QDs results in severe mitochondrial impairment and cell death due to both the release of cadmium ions through surface-etching and the production of superoxide through electron transfer.
Fig. 6. Schematic presentation of ROI production by a QD (center part) and reactions of a DNA molecule with hydroxyl radical and subsequent nucleobase damage and strand breakage (peripheral part). Reprinted with permission from Anas et al. (2008). Copyright (2008) American Chemical Society.
http://ars.els-cdn.com/content/image/1-s2.0-S073497500900192X-gr6.sml
Quantum dot-PS hybrids as drugs for PDT
There are several advantages and limitations for both conventional PS drugs and QDs when individually applied for PDT. For example, the properties of QDs such as NIR absorption, large two-photon absorption cross-section,broad absorption band and photostability are promising for PDT. In contrast to these unique optical properties of QDs, narrow absorption band, poor photostability, visible light absorption and small two-photon absorption cross-section of conventional PS drugs are less attractive for PDT. However, the efficiency (N75%) for ROI production by PS drugs is superior to that by QDs (∼5%). In other words, the advantages and limitations of QDs and PS drugs complement each other. Thus, in order to utilize the photostability of QDs and improve the production of 1O2, several conjugates/hybrids of QDs and conventional PS drugs were investigated as new generation drugs for PDT.In such hybrid QD-PS systems, the excited singlet (1PS*) and triplet (3PS*) states of PS drugs are indirectly generated by nonradiative energy transfer, also called Förster resonance energy transfer (FRET) from photoactivated QDs (QD*). Due to the indirect photoactivation, photobleaching of PS drugs was minimized. Also, due to the large surface area and biocompatibility of QDs multiple PS drug molecules, which are hydrophobic, can be conjugated to QDs. The indirectly excited PS drugs form collision-complexes (QD-3PS*-3O2) with oxygen, transfer energy to 3O2, and generate 1O2 and other ROI. Fig.7 shows steps involved in the photoactivation of a QD-PS conjugate and the production of ROI. The concept of FRET-based production of 1O2 by QD-PS hybrid systems was first envisaged and demonstrated by Samia et al. (2003) by preparing a non-covalent mixture composed of CdSe QDs and a silicon phthalocyanine (Pc4). They selected Pc4 due to its high 1O2 efficiency (43%) under direct photoactivation. In the QD-Pc4 hybrid system, QD acts as the energy donor to Pc4, and Pc4 acts as both an energy acceptor from QD and an energy donor to 3O2. Thus, high quantum efficiency for 1O2 and stability for the hybrid system were anticipated. However,according to the principle underlyingFRET,the energy transfer efficiencyinversely varies withthe sixth powerof the distance between a donor and an acceptor [(Lakowicz, 1986), (Medintz and Mattoussi, 2009), (Biju et al., 2006), and (Kanemoto et al., 2008)]. Thus, close conjugation, typically within 10 nm, of PS drugs to QDs is necessary for efficient energy transfer and ROI production. Simply, the construction of energy donor–acceptor QD-PS systems should follow the standards described by Medintz and Mattoussi (2009) and in the reference therein. Following the first investigation of QD-PS system by Samia et al. (2003), many researchers were attracted to the energy transfer properties of covalent and non-covalent QD-PS systems composed of CdSe, CdSe/ CdS/ZnS, CdSe/ZnS, and CdTe QDs as energy donors and various chromophores such as porphyrins, phthalocyanines, inorganic complexes and other organic dyes as energy acceptors. Depending on the energy acceptor, the QD-PS systems can be classified into QDphthalocyanines, QD-porphines, QD-organic dyes, and QD-inorganic dyes.
Quantum dot-phthalocyanine conjugates for FRET and single oxygen production
Phthalocynaine-conjugated QDs (QD-Pc) were widely investigated for energy transfer and 1O2 production due to the high triplet quantum efficiency and long-living triplet state for Pc. Burda and coworkers extended investigations of energy transfer and 1O2 production into a large number of QD-Pc conjugates as functions of donor–acceptor distance, relative numbers of QDs and Pcs, terminal functional group in Pc, bulkiness of spacers between donors and acceptors, the mode of binding between QD and Pc, and the size and surface states of QDs [(Dayal et al., 2006), (Samia et al., 2006), (Dayal and Burda, 2007), and (Dayal et al., 2008)]. For example, they employed fluorescence up-conversion and transient absorption measurements, which are valuable methods for characterizing the energy transfer kinetics from various exciton-states in photoactivated QDs, and investigated energy transfer from CdSe QDs to silicon Pc molecules bearing one or two axial functional groups such as thiol, hydroxyl, tertiary alkyl and tertiary amine [(Dayal et al., 2006), and (Dayal et al., 2008)]. Examples of Pc molecules that were used as energy acceptors in QD-Pc systems are shown in Fig. 8. For these molecules, the energy transfer efficiency decreased with increase in both the length and the bulkiness of spacers between QD and Pc [(Dayal et al. (2006), and (Dayal et al., 2008)]. Also, they found that functional groups such as amine and thiol in Pc played important roles on both QD to Pc bonding and quenching of the excited state of QDs.In particular, the energy transfer efficiency was found higher when Pc molecules were linked to QDs through two axial amine or thiol groups. Dayal et al. (2006) detected up to 70% efficiency for energy transferfrom QDsto aprimary amine-terminatedPc. Also,quenching of QD’s photoluminescence was effective for 1:1 and 1:2 conjugates between QD and Pc, but the energy transfer efficiency has decreased with increase in the number of Pc per QD due to self absorbance (Dayaletal.,2006),indicatingthatalargenumberofPSonthesurface of a QD will be less attractive for PDT. One of the reasons for different energy transfer efficiencies for QD-Pc systems linked through bulky or amine/thiol/alkyl functional groups was different electronic coupling between the donor and acceptor.
Fig. 7. Energy transfer processes in a photoactivated QD-PS system, and the production of ROI.
http://ars.els-cdn.com/content/image/1-s2.0-S073497500900192X-gr7.sml
Another important factor involved in the energy transfer efficiency is the surface states of QDs, which was identified by Dayal et al.(2008) from non-linear relationship between spectral overlap integral and energy transfer efficiency for QD-Pc systems. In short, Burda and coworkers have concluded that 1:1 or 1:2 complexes between QDs and PS molecules bearing two axial amine or thiol functional groups and non-bulky and short spacers would be ideal QD-PS donor–acceptor systems for efficient energy transfer and 1O2 production. Investigations such as preparation of QD-Pc systems, energy transfer from QD to Pc and the generation of 1O2 were further extended into complexes between CdTe QDs and tetrasulfonated aluminum Pc (AlTSPc) systems [(Idowu et al., 2008), (Moeno and Nyokong, 2008), and (Moeno and Nyokong, 2009)]. Here, Nyokong and coworkers prepared CdTe–AlTSPc mixtures by adding solutions of AlTSPc having varying concentrations to solutions of CdTe QDs tethered with mercaptocarboxylic acids such as thioglycolic acid (TDA), 3-mercaptopropionic acid (MPA) and L-lysine (Idowu et al., 2008). In this mixture, the excited state of QDs was quenched and resulted in an increase in the triplet yield for AlTSPc along with fluorescence emission from AlTSPc. Among the CdTe QDs with three different capping ligands stated above, MPA capped CdTe QDs provided long-living triplet state of AlTSPc, which was attributed to the strong binding between AlTSPc and MPA. Later, they found that the CdTe–AlTSPc complex produces 1O2 at 9.5–15% yield that was determined using phosphorescence decay of 1O2 in the presence and absence of sodium azide, a 1O2 scavenger (Moeno and Nyokong, 2008). Recently, they extended energy transfer investigations to various metallophthalocyanines (TSPc) linked to CdTe QDs through sulfonic acid, carboxylic acid, and pyridinium group (Moeno and Nyokong, 2009). By varying the metal ion and the functional groups in Pc, they obtained QD-Pc systems with exceptionally high triplet yields and energy transfer efficiencies (up to 80%). The most important properties of the CdTe-TSPc systems are their water solubility and photosensitized production of 1O2. However, the mode of binding between CdTe QDs and sulfonated Pcs, correlation between the quenching of QD’s excited state and the formation of both the triplet and singlet states of TSPcs, toxicity due to cadmium, and potentials of QD-Pc systems for in vitro and in vivo PDT need further attention.
Quantum dot-porphine conjugates for FRET and singlet oxygen production
Porphines are classical photosensitizers clinically applied for PDT of various cancers due to their high triplet yields and high efficiencies for ROI production. However, as with most phthalocyanines, poor water solubility, inadequate mechanism for selective delivery in tumor milieu and lack of NIR absorption are major drawbacks of porphines for PDT. Recently, Tsay et al. (2007) lifted most of these drawbacks by coating Chlorin e6 on the surface of CdSe/CdS/ZnS QDs either non-covalently using an alkylamine linker or covalently using a lysine-terminated peptide linker (Fig. 9). They found that the photoluminescence lifetime of QDs was decreased as a result of energy transfer from QDs to Chlorin e6. Also, in contrast to the previousreportbyDayaletal.2006),Tsayetal.(2007)foundthatthe energy transfer efficiency from QD to Chlorin e6 has increased with increase in the number of Chlorin e6 molecules attached to a single QD. The QD-Chlorin e6 conjugate provided 1O2 at 31% efficiency. Another example for water-soluble QD-porphine system for 1O2 production is CdTe QDs electrostatically coated by a meso-tetra(4sulfonatophenyl)porphine (TSPP), investigated by Shi et al. (2006). The CdTe-TSPP composite produced 1O2 at 43% efficiency when photoactivated at 355 nm. At this wavelength, both the donor and acceptor were directly photoactivated. Thus, the quantum efficiency for FRET-based 1O2 production was probably overrated. However, based on an assumption that QDs quench the directly-excited triplet stateofanacceptor,Tsayet al.(2007)ruledoutthe productionof 1O2 throughdirectphotoactivationofanacceptorintheproximityofaQD. Also,incontrasttotheproductionof1O2 andotherROIbyCdSeQDsas reportedbySamiaetal.(2003)andAnasetal.(2008), 1O2 production was not detected for CdTe QDs alone, indicating that QD-PS systems are ideal candidates for PDT compared with QDs alone. Despite the above two reports on QD-porphine systems for energy transfer and 1O2 production, systematic investigations of the relations among energy transfer, donor–acceptor distance, size of QDs, dielectric constant of the medium and the efficiency for 1O2 production remain.
Fig. 8. Examples of Pc molecules having different bridging units and terminal functional groups. With kind permission from Springer Science+Business Media; Dayal et al. (2006).
http://ars.els-cdn.com/content/image/1-s2.0-S073497500900192X-gr8.sml
Quantum dot-organic/inorganic dye systems for FRET and singlet oxygen production
Organic and inorganic dyes having high triplet quantum efficiencies are potential energy acceptors for the construction of QD-PS systems for 1O2 and other ROI production and PDT. Typical example for QD-dye conjugates was investigated by Tsay et al. (2007) by conjugating Rose Bengal on the surface of CdSe/CdS/ZnSQDs through alysine-terminated peptide linker (Fig. 9). As a result of the conjugation of Rose Bengal the photoluminescence lifetime of QD was considerably decreased, indicating efficient FRET from QD to Rose Bengal. Furthermore, they investigated the production of 1O2 by recording the steady-state absorption spectrum of anthracene dipropionic acid, a well-known 1O2 scavenger, and the phosphorescence spectrum of 1O2 at 1270nm. The 1O2 quantum efficiency for QD-Rose Bengal conjugate excited at 355nm was 17%. Here,the production of 1O2 through direct excitation of the acceptor was ruled as stated in the previous section. The low quantum efficiency for 1O2 production was attributed to inefficient energy transfer because of poor donor–acceptor spectral overlap integral.Interestingly,by selecting Chlorine6 as the energy acceptor, they achieved 31% quantum efficiency for 1O2 owing to better overlap between the photoluminescence spectrum of QDs and the absorption spectrum of Chlorin e6. Other examples of organic dyes for the preparation of QD-PS systems are Merocyanine 540 (MC540) and Toluidine Blue O (TBO) [(Narayanan et al., 2008), and (Narband et al., 2008)]. From steady-state and timeresolved fluorescence measurements, Narayanan et al. (2008) detected efficient FRET from CdSe/ZnS QDs to MC540, a chemotherapeutic drug. Here,FRET efficiency was determined from the quenching of the steady state and time-resolved photoluminescence of QDs. Narband et al.(2008) utilized the advantages of QD-PS systems for photodynamic killing of bacteria by applying a mixture of NIR QD and TBO. Photoactivation of QDs resulted in FRET from QD to TBO and the production of 1O2.Here,the high molar extinction coefficient of QDs in the short wavelength region and efficient overlap between the photoluminescence spectrum of QDs and the absorption spectrum of TBO were advantageous for the generation of various cytotoxic species including 1O2. Energy transfer, 1O2 production and bactericidal action for TBO:QD mixtures were discussed in terms of ionic interactions between QD andTBO.
Covalent conjugates and physical mixtures between QDs and inorganic dyes are another class of donor–acceptor systems with potentials for PDT. For example, Hsieh et al. (2006) conjugated iridium complexes with CdSe/ZnS QDs and prepared covalent donor– acceptor systems. Photoactivation of a de-oxygenated solution of the conjugate resulted in a weak phosphorescence emission with a 2.1 μs decay component from the Ir complex, which disappeared when the solution was aerated. Here, the excited state of the Ir complex was generated through FRET from QDs. The disappearance of the phosphorescence during aeration was due to the quenching of the excited state of Ir complex by 3O2 and the formation of 1O2. Although high quantum efficiency (97%) for 1O2 production was estimated for the QD-Ir complex system, the roles of non-radiative relaxations of QDs and the Ir complex, spectral overlap integral, anddonor–acceptor distance are yet to be addressed. Another example for QD-inorganic dye systems was investigated by Neuman et al.(2008) by preparing a physical mixture between CdSe/ZnS QDs and trans-Cr(cyclam)Cl2. Here, the excited state of QD was quenched by the Cr complex, evidenced from a non-linear Stern–Volmer quenching kinetics and a decrease in the photoluminescence lifetime of QDs with increase in the concentration of Cr complex. The spectral overlap integral for the QD-Cr complex was ideal for efficient FRET. Preparation of QD-PS systems such as non-covalent and covalent assemblies between QDs and organic chromophores as well as investigation of energy transfer and 1O2 production are emerging research topics with great potentials for environment and health management. The significance of QD-PS systems compared to conventional PS is that the exceptional photostability of QDs offers durability. Despite the reports discussed above, systematic investigationsofthedonor–acceptordistance,donor–acceptorspectraloverlap integral, donor–acceptor orientation, efficiency of 1O2 production, toxicity of the donor–acceptor systems and in vitro andi n vivo PDT are important issues remaining. Inparticular, QD-PS systems composed of QDs with small size and without heavy metals would bring radical changes to PDT of cancer.
Fig. 9. QD-Chlorin e6 (top) and QD-Rose Bengal (bottom) FRET pairs conjugated using peptide linkers. Reproduced with permission from Tsay et al. (2007). Copyright (2007) American Chemical Society
http://ars.els-cdn.com/content/image/1-s2.0-S073497500900192X-gr9.sml
7.1.2 Bioconjugated quantum dots for in vivo molecular and cellular imaging
Smith AM, Duan H, Mohs AM, Nie S.
Adv Drug Deliv Rev. 2008 Aug 17;60(11):1226-40
http://dx.doi.org:/10.1016/j.addr.2008.03.015
http://www.ncbi.nlm.nih.gov/pmc/articles/instance/2649798/bin/nihms62165f2.gif
http://www.ncbi.nlm.nih.gov/pmc/articles/instance/2649798/bin/nihms62165f3.gif
http://www.ncbi.nlm.nih.gov/pmc/articles/instance/2649798/bin/nihms62165f5.gif
Semiconductor quantum dots (QDs) are tiny light-emitting particles on the nanometer scale, and are emerging as a new class of fluorescent labels for biology and medicine. In comparison with organic dyes and fluorescent proteins, they have unique optical and electronic properties, with size-tunable light emission, superior signal brightness, resistance to photobleaching, and broad absorption spectra for simultaneous excitation of multiple fluorescence colors. QDs also provide a versatile nanoscale scaffold for designing multifunctional nanoparticles with both imaging and therapeutic functions. When linked with targeting ligands such as antibodies, peptides or small molecules, QDs can be used to target tumor biomarkers as well as tumor vasculatures with high affinity and specificity. Here we discuss the synthesis and development of state-of-the-art QD probes and their use for molecular and cellular imaging. We also examine key issues for in vivo imaging and therapy, such as nanoparticle biodistribution, pharmacokinetics, and toxicology.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2649798/ FREE PMC Article
The development of biocompatible nanoparticles for molecular imaging and targeted therapy is an area of considerable current interest [1–9]. The basic rationale is that nanometer-sized particles have functional and structural properties that are not available from either discrete molecules or bulk materials [1–3]. When conjugated with biomolecular affinity ligands, such as antibodies, peptides or small molecules, these nanoparticles can be used to target malignant tumors with high specificity [10–13]. Structurally, nanoparticles also have large surface areas for the attachment of multiple diagnostic (e.g., optical, radioisotopic, or magnetic) and therapeutic (e.g., anticancer) agents. Recent advances have led to the development of biodegradable nanostructures for drug delivery [14–18], iron oxide nanocrystals for magnetic resonance imaging (MRI) [19, 20], luminescent quantum dots (QDs) for multiplexed molecular diagnosis and in vivoimaging [21–25], as well as nanoscale carriers for siRNA delivery [26, 27].
Due to their novel optical and electronic properties, semiconductor QDs are being intensely studied as a new class of nanoparticle probe for molecular, cellular, and in vivo imaging [10–24]. Over the past decade, researchers have generated highly monodispersed QDs encapsulated in stable polymers with versatile surface chemistries. These nanocrystals are brightly fluorescent, enabling their use as imaging probes both in vitroand in vivo. In this article, we discuss recent developments in the synthesis and modification of QD nanocrystals, and their use as imaging probes for living cells and animals. We also discuss the use of QDs as a nanoscale carrier to develop multifunctional nanoparticles for integrated imaging and therapy. In addition, we describe QD biodistribution, pharmacokinetics, toxicology, as well as the challenges and opportunities in developing nanoparticle agents for in vivo imaging and therapy.
QD Chemistry and Probe Development
QDs are nearly spherical semiconductor particles with diameters on the order of 2–10 nanometers, containing roughly 200–10,000 atoms. The semiconducting nature and the size-dependent fluorescence of these nanocrystals have made them very attractive for use in optoelectronic devices, biological detection, and also as fundamental prototypes for the study of colloids and the size-dependent properties of nanomaterials [28]. Bulk semiconductors are characterized by a composition-dependent bandgap energy, which is the minimum energy required to excite an electron to an energy level above its ground state, commonly through the absorption of a photon of energy greater than the bandgap energy. Relaxation of the excited electron back to its ground state may be accompanied by the fluorescent emission of a photon. Small nanocrystals of semiconductors are characterized by a bandgap energy that is dependent on the particle size, allowing the optical characteristics of a QD to be tuned by adjusting its size. Figure 1 shows the optical properties of CdSe QDs at four different sizes (2.2 nm, 2.9 nm, 4.1 nm, and 7.3 nm). In comparison with organic dyes and fluorescent proteins, QDs are about 10–100 times brighter, mainly due to their large absorption cross sections, 100–1000 times more stable against photobleaching, and show narrower and more symmetric emission spectra. In addition, a single light source can be used to excite QDs with different emission wavelengths, which can be tuned from the ultraviolet [29], throughout the visible and near-infrared spectra [30–33], and even into the mid-infrared [34]. However QDs are macromolecules that are an order of magnitude larger than organic dyes, which may limit their use in applications in which the size of the fluorescent label must be minimized. Yet, this macromolecular structure allows the QD surface chemistry and biological functionality to be modified independently from its optical properties.

2.1. QD Synthesis
QD synthesis was first described in 1982 by Efros and Ekimov [35, 36], who grew nanocrystals and microcrystals of semiconductors in glass matrices. Since this work, a wide variety of synthetic methods have been devised for the preparation of QDs in different media, including aqueous solution, high-temperature organic solvents, and solid substrates [28, 37, 38]. Colloidal suspensions of QDs are commonly synthesized through the introduction of semiconductor precursors under conditions that thermodynamically favor crystal growth, in the presence of semiconductor-binding agents, which function to kinetically control crystal growth and maintain their size within the quantum-confinement size regime.
The size-dependent optical properties of QDs can only be harnessed if the nanoparticles are prepared with narrow size distributions. Major progress toward this goal was made in 1993 by Bawendi and coworkers [39], with the introduction of a synthetic method for monodisperse QDs made from cadmium sulfide (CdS), cadmium selenide (CdSe), or cadmium telluride (CdTe). Following this report, the synthetic chemistry of CdSe QDs quickly advanced, generating brightly fluorescent QDs that can span the visible spectrum. As a result, CdSe has become the most common chemical composition for QD synthesis, especially for biological applications. Many techniques have been implemented to post-synthetically modify QDs for various purposes, such as coating with a protective inorganic shell [40, 41], surface modification to render colloidal stability [42, 43], and direct linkage to biologically active molecules [44, 45]. QD production has now become an elaborate molecular engineering process, best exemplified in the synthesis of polymer-encapsulated (CdSe)ZnS (core)shell QDs. In this method, CdSe cores are prepared in a nonpolar solvent, and a shell of zinc sulfide (ZnS) is grown on their surfaces. The QDs are then transferred to aqueous solution through encapsulation with an amphiphilic polymer, which can then be cross-linked to biomolecules to yield targeted molecular imaging agents.
In the design of a QD imaging probe, the selection of a QD core composition is determined by the desired wavelength of emission. For example, CdSe QDs may be size-tuned to emit in the 450–650 nm range, whereas CdTe can emit in the 500–750 nm range. QDs of this composition are then grown to the appropriate wavelength-dependent size. In a typical synthesis of CdSe, a room-temperature selenium precursor (commonly trioctylphosphine-selenide or tributylphosphine-selenide) is swiftly injected into a hot (~300°C) solution containing both a cadmium precursor (dimethylcadmium or cadmium oleate) and a coordinating ligand (trioctylphosphine oxide or hexadecylamine) under inert conditions (nitrogen or argon atmosphere). The cadmium and selenium precursors react quickly at this high temperature, forming CdSe nanocrystal nuclei. The coordinating ligands bind to metal atoms on the surfaces of the growing nanocrystals, stabilizing them colloidally in solution, and controlling their rate of growth. This injection of a cool solution quickly reduces the temperature of the reaction mixture, causing nucleation to cease. The remaining cadmium and selenium precursors then can grow on the existing nuclei at a slower rate at lower temperature (240–270°C). Once the QDs have reached the desired size and emission wavelength, the reaction mixture may be cooled to room temperature to arrest growth. The resulting QDs are coated in aliphatic coordinating ligands and are highly hydrophobic, allowing them to be purified through liquid-liquid extractions or via precipitation from a polar solvent.
Because QDs have high surface area to volume ratios, a large fraction of the constituent atoms are exposed to the surface, and therefore have atomic or molecular orbitals that are not completely bonded. These “dangling” orbitals serve as defect sites that quench QD fluorescence. For this reason, it is advantageous to grow a shell of another semiconductor with a wider bandgap on the core surface after synthesis to provide electronic insulation. The growth of a shell of ZnS on the surface of CdSe cores has been found to dramatically enhance photoluminescence efficiency [40, 41]. ZnS is also less prone to oxidation than CdSe, increasing the chemical stability of the QDs, and greatly decreasing their rate of oxidative photobleaching [46]. As well, the Zn2+ atoms on the surface of the QD bind more strongly than Cd2+ to most basic ligands, such as alkyl phosphines and alkylamines, increasing the colloidal stability of the nanoparticles [47]. In a typical shell growth of ZnS on CdSe, the purified cores are again mixed with coordinating ligands, and heated to an elevated temperature (140–240°C). Molecular precursors of the shell, usually diethylzinc and hexamethyldisilathiane dissolved in TOP, are then slowly added [40]. The (CdSe)ZnS nanocrystals may then be purified just like the cores.
More recently, it has become possible to widely engineer the fluorescence of QDs by changing the material composition while maintaining the same size. The technological advances that made this possible were the development of alloyed QDs [29, 30] and type-II heterostructures [32]. For example, homogeneously alloying the semiconductors CdTe and CdSe in different ratios allows one to prepare QDs of 5 nm diameter with emission wavelengths of 620 nm for CdSe, 700 nm for CdTe, and 800 nm for the CdSe0.34Te0.67 alloy [30]. Alternatively, type-II QDs allow one to physically separate the charge carriers (the electron and its cationic counterpart, known as the hole) into different regions of a QD by growing an appropriately chosen material on the QD as a shell [32]. For example, both the valence and conduction band energy levels of CdSe are lower in energy than those of CdTe. This means that in a heterostructure composed of CdTe and CdSe domains, electrons will segregate to the CdSe region to the lowest energy of the conduction band, whereas the hole will segregate to the CdTe region, where the valence band is highest in energy. This will effectively decrease the bandgap due to the smaller energy separating the two charge carriers, and emission will occur at a longer wavelength. By using different sizes of the core and different shell thicknesses, one can engineer QDs with the same size but different wavelengths of emission.
Surface Modification
QDs produced in nonpolar solutions using aliphatic coordinating ligands are only soluble in nonpolar organic solvents, making phase transfer an essential and nontrivial step for the QDs to be useful as biological reporters. Alternatively, QD syntheses have been performed directly in aqueous solution, generating QDs ready to use in biological environments [48], but these protocols rarely achieve the level of monodispersity, crystallinity, stability, and fluorescent efficiency as the QDs produced in high-temperature coordinating solvents. Two general strategies have been developed to render hydrophobic QDs soluble in aqueous solution: ligand exchange, and encapsulation by an amphiphilic polymer. For ligand exchange, a suspension of TOPO-coated QDs are mixed with a solution containing an excess of a heterobifunctional ligand, which has one functional group that binds to the QD surface, and another functional group that is hydrophilic. Thereby, hydrophobic TOPO ligands are displaced from the QD through mass action, as the new bifunctional ligand adsorbs to render water solubility. Using this method, (CdSe)ZnS QDs have been coated with mercaptoacetic acid and (3-mercaptopropyl) trimethoxysilane, both of which contain basic thiol groups to bind to the QD surface atoms, yielding QDs displaying carboxylic acids or silane monomers, respectively [44, 45]. These methods generate QDs that are useful for biological assays, but ligand exchange is commonly associated with decreased fluorescence efficiency and a propensity to aggregate and precipitate in biological buffers. More recently it has been shown that these problems can be alleviated by retaining the native coordinating ligands on the surface, and covering the hydrophobic QDs with amphiphilic polymers [10, 23,49]. This encapsulation method yields QDs that can be dispersed in aqueous solution and remain stable for long periods of time due to a protective hydrophobic bilayer surrounding each QD through hydrophobic interactions. No matter what method is used to suspend the QDs in aqueous buffers, they should be purified from residual ligands and excess amphiphiles before use in biological assays, using ultracentrifugation, dialysis, or filtration. Also, when choosing a water solubilization method, it should be noted that many biological and physical properties of the QDs may be affected by the surface coating, and the overall physical dimensions of the QDs are dependent on the coating thickness. Typically the QDs are much larger when coated with amphiphiles, compared to those coated with a monolayer of ligand.
Bioconjugation
Water-soluble QDs may be cross-linked to biomolecules such antibodies, oligonucleotides, or small molecule ligands to render them specific to biological targets. This may be accomplished using standard bioconjugation protocols, such as the coupling of maleimide-activated QDs to the thiols of reduced antibodies [22]. The reactivities of many types of biomolecules have been found to remain after conjugation to nanoparticles surfaces, although possibly at a decreased binding strength. The optimization of surface immobilization of biomolecules is currently an active area of research [50, 51]. The surfaces of QDs may also be modified with bio-inert, hydrophilic molecules such as polyethylene glycol, to eliminate possible nonspecific binding, or to decrease the rate of clearance from the bloodstream following intravenous injection. QDs have also emerged as a new class of sensor, mediated by energy transfer to organic dyes (fluorescence resonance energy transfer, FRET) [52–54]. It has also recently been reported that QDs can emit fluorescence without an external source of excitation when conjugated to enzymes that catalyze bioluminescent reactions, due to bioluminescence resonance energy transfer (BRET) [55].
Figure 2 depicts the most commonly used and technologically advanced QD probes. Biologically nonfunctional QDs may be prepared by using a variety of methods. As shown from left to right (top), QDs coated with a monolayer of hydrophilic thiols (e.g. mercaptoacetic acid) are generally stabilized ionically in solution [45]; QDs coated with a cross-linked silica shell can be readily modified with a variety of organic functionalities using well developed silane chemistry [44]; QDs encapsulated in amphiphilic polymers form highly stable, micelle-like structures [23, 49]; and any of these QDs may be modified to contain polyethylene glycol (PEG) to decrease surface charge and increase colloidal stability [56]. Also, water-soluble QDs may be covalently or electrostatically bound to a wide range of biologically active molecules to render specificity to a biological target. As shown in Figure 2 (middle), QDs conjugated to streptavidin may be readily bound to many biotinylated molecules of interest with high affinity [23]; QDs conjugated to antibodies can yield specificity for a variety of antigens, and are often prepared through the reaction between reduced antibody fragments with maleimide-PEG-activated QDs [22, 57]; QDs cross-linked to small molecule ligands, inhibitors, peptides, or aptamers can bind with high specificity to many different cellular receptors and targets [58, 59]; and QDs conjugated to cationic peptides, such as the HIV Tat peptide, can quickly associate with cells and become internalized via endocytosis [60]. Further, QDs have been used to detect the presence of biomolecules using intricate probe designs incorporating energy donors or acceptors. For example, QDs can be adapted to sense the presence of the sugar maltose by conjugating the maltose binding protein to the nanocrystal surface (Figure 2, bottom) [53]. By initially incubating the QDs with an energy-accepting dye that is conjugated to a sugar recognized by the receptor, excitation of the QD (blue) yields little fluorescence, as the energy is nonradiatively transferred (grey) to the dye. Upon addition of maltose, the quencher-sugar conjugate is displaced, restoring fluorescence (green) in a concentration-dependent manner. QDs can also be sensors for specific DNA sequences [52]. By mixing the ssDNA to be detected with (a) an acceptor fluorophores conjugated to a DNA fragment complementary to one end of the target DNA and (b) a biotinylated DNA fragment complementary to the opposite end of the target DNA, these nucleotides hybridize to yield a biotin-DNA-fluorophore conjugate. Upon mixing this conjugate with QDs, QD fluorescence (green) is quenched via nonradiative energy transfer (grey) to the fluorophore conjugate. This dye acceptor then becomes fluorescent (red), specifically and quantitatively indicating the presence of the target DNA. Finally, QDs conjugated to the luciferase enzyme can nonradiatively accept energy from the enzymatic bioluminescent oxidation of luciferins on the QD surface, exciting the QDs without the need for external illumination [55].

nonfunctionalized and bioconjugated QD probes nihms62165f2
Researchers have achieved considerable success in using QDs for in vitro bioassays [61, 62], labeling fixed cells [23] and tissue specimens [63, 64], and for imaging membrane proteins on living cells [58, 65]. However, only limited progress has been made in developing QD probes for imaging inside living cells. A major problem is the lack of efficient methods for delivering monodispersed (that is, single) QDs into the cytoplasms of living cells. A common observation is that QDs tend to aggregate inside cells, and are often trapped in endocytotic vesicles such as endosomes and lysosomes.
Imaging and Tracking of Membrane Receptors
QD bioconjugates have been found to be powerful imaging agents for specific recognition and tracking of plasma membrane antigens on living cells. In 2002 Lidke et al. coupled red-light emitting (CdSe)ZnS QDs to epidermal growth factor, a small protein with a specific affinity for the erbB/HER membrane receptor [58]. After addition of these conjugates to cultured human cancer cells, receptor-bound QDs could be identified at the single-molecule level (single QDs may be distinguished from aggregates because the fluorescent intensity from discrete dots is intermittent, or “blinking”). The bright, stable fluorescence emitted from these QDs allowed the continuous observation of protein diffusion on the cellular membrane, and could even be visualized after the proteins were internalized. Dahan et al. similarly reported that QDs conjugated to an antibody fragment specific for glycine receptors on the membranes of living neurons allowed tracking of single receptors [65]. These conjugates showed superior photostability, lateral resolution, and sensitivity relative to organic dyes. These applications have inspired the use QDs for monitoring other plasma membrane proteins such as integrins [50, 66], tyrosine kinases [67, 68], G-protein coupled receptors [69], and membrane lipids associated with apoptosis [70, 71]. As well, detailed procedures for receptor labeling and visualization of receptor dynamics with QDs have recently been published [72, 73], and new techniques to label plasma membrane proteins using versatile molecular biology methods have been developed [74, 75].
Intracellular Delivery of QDs
A variety of techniques have been explored to label cells internally with QDs, using passive uptake, receptor-mediated internalization, chemical transfection, and mechanical delivery. QDs have been loaded passively into cells by exploiting the innate capacity of many cell types to uptake their extracellular space through endocytosis [76–78]. It has been found that the efficiency of this process may be dramatically enhanced by coupling the QDs to membrane receptors. This is likely due to the avidity-induced increase in local concentration of QDs at the surface of the cell, as well as an active enhancement caused by receptor-induced internalization [58, 77, 79]. However, these methods lead to sequestration of aggregated QDs in vesicles, showing strong colocalization with membrane dyes. Although these QDs cannot diffuse to specific intracellular targets, this is a simple way to label cells with QDs, and an easy method to fluorescently image the process of endocytosis. Nonspecific endocytosis was also utilized by Parak et al. to fluorescently monitor the motility of cells on a QD-coated substrate [78]. The path traversed by each cell became dark, and the cells increased in fluorescence as they took up more QDs. Chemically-mediated delivery enhances plasma membrane translocation with the use of cationic lipids or peptides, and was originally developed for the intracellular delivery of a wide variety of drugs and biomolecules [60, 80–83]. The efficacy of these carriers for the intracellular deliver of QDs is discussed below (Section 3.3 and Section 3.4). Mechanical delivery methods include microinjection of QDs into individual cells, and electroporation of cells in the presence of QDs. Microinjection has been reported to deliver QDs homogeneously into the cytoplasms of cells [49, 83], however this method is of low statistical value, as careful manipulation of single cells prevents the use of large sample sizes. Electroporation makes use of the increased permeability of cellular membranes under pulsed electric fields to deliver QDs, but this method was reported to result in aggregation of QDs in the cytoplasm [83], and generally results in widespread cell death.
Despite the current technical challenges, QDs are garnering interest as intracellular probes due to their intense, stable fluorescence, and recent reports have demonstrated that intracellular targeting is not far off. In 2004, Derfus et al. demonstrated that QDs conjugated to organelle-targeting peptides could specifically stain either cellular mitochondria or nuclei, following microinjection into fibroblast cytoplasms [83]. Similarly, Chen et al. targeted peptide-QD conjugates to cellular nuclei, using electroporation to overcome the plasma membrane barrier [60]. These schemes have resulted in organelle-level resolution of intracellular targets for living cells, yielding fluorescent contrast of vesicles, mitochondria, and nuclei, but not the ability to visualize single molecules. Recently Courty et al. demonstrated the capacity to image individual kinesin motors in HeLa cells using QDs delivered into the cytoplasm via osmotic lysis of pinocytotic vesicles [84]. By incubating the cells in a hypertonic solution containing QDs, water efflux resulted in membrane invagination and pinocytosis, trapping extracellular QDs in endosomal vesicles. Then a brief incubation in hypotonic medium induced intracellular water influx, rupturing the newly formed vesicles, and releasing single QDs into the cytosol. All of the QDs were observed to undergo random Brownian motion in the cytoplasm. However if these QDs were first conjugated to kinesin motor proteins, a significant population of the QDs exhibited directional motion. The velocity of the directed motion and its processivity (average time before cessation of directed motion) were remarkably close to those observed for the motion of these conjugates on purified microtubules in vitro. Although this work managed to overcome the plasma membrane diffusion barrier, it highlighted a different problem fundamental to intracellular imaging of living cells, which is the impossibility of removing probes that have not found their target. In this report, the behavior of the QDs was sufficient to distinguish bound QDs from those that were not bound, but this will not be the case for the majority of other protein targets. Without the ability to wash away unbound probes, which is a crucial step for intracellular labeling of fixed, permeabilized cells, the need for activateable probes that are ‘off’ until they reach their intended target is apparent. However QDs have already found a niche for quantitative monitoring of motor protein transport and for tracking the fate of internalized receptors, allowing the study of downstream signaling pathways in real time with high signal-to-noise and high temporal and spatial resolution [58, 67, 68, 85, 86].
Tat-QD Conjugates
Cell-penetrating peptides are a class of chemical transfectants that have garnered widespread interest due to the high transfection efficiency of their conjugated cargo, versatility of conjugation, and low toxicity. For this reason, cell-penetrating peptides such as polyarginine and Tat have been investigated for their capacity to deliver QDs into living cells [81, 85, 87], but the delivery mechanism and the behavior of intracellular QDs are still a matter of debate. Considerable effort has been devoted to understanding the delivery mechanism of these cationic carrier, especially the HIV-1-derived Tat peptide, which has emerged as a widely used cellular delivery vector [88–93]. The delivery process was initially thought to be independent of endocytosis because of its apparent temperature-independence [89–93]. However, later research showed that the earlier work failed to exclude the Tat peptide conjugated cargos bound to plasma membranes, and was largely an artifact caused by cellular fixation. More recent studies based on improved experimental methods indicate that Tat peptide-mediated delivery occurs via macropinocytosis [94], a fluid-phase endocytosis process that is initiated by the binding of Tat-QD to the cell surface [90]. These new results, however, did not shed any light on the downstream events or the intracellular behavior of the internalized cargo. This kind of detailed and mechanistic investigation would be possible with QDs, which are sufficiently bright and photostable for extended imaging and tracking of intracellular events. In addition, most previous studies on Tat peptide-mediated delivery are based on the use of small dye molecules and proteins as cargo [89–93], so it is not clear whether larger nanoparticles would undergo the same processes of cellular uptake and transport. This understanding is needed for the design and development of imaging and therapeutic nanoparticles for biology and medicine.
Ruan et al. have recently used Tat peptide-conjugated QDs (Tat-QDs) as a model system to examine the cellular uptake and intracellular transport of nanoparticles in live cells [95]. The authors used a spinning-disk confocal microscope for dynamic fluorescence imaging of quantum dots in living cells at 10 frames per second. The results indicate that the peptide-conjugated QDs are internalized by macropinocytosis, in agreement with the recent work of Dowdy and coworkers [90]. It is interesting, however, that the internalized Tat-QDs are tethered to the inner surface of vesicles, and are trapped in intracellular organelles. An important finding is that the QD-loaded vesicles are actively transported by molecular machines (such as dyneins) along microtubule tracks to an asymmetric perinuclear region called the microtubule organizing center (MTOC) [96]. Furthermore, it was found that Tat-QDs strongly bind to cellular membrane structures such as filopodia, and that large QD-containing vesicles are able to pinch off from the tips of filopodia. These results not only provide new insight into the mechanisms of Tat peptide-mediated delivery, but also are important for the development of nanoparticle probes for intracellular targeting and imaging.
QDs with Endosome-Disrupting Coatings
Duan and Nie [97] developed a new class of cell-penetrating quantum dots (QDs) based on the use of multivalent and endosome-disrupting (endosomolytic) surface coatings (Figure 3). Hyperbranched copolymer ligands such as PEG-grafted polyethylenimine (PEI-g-PEG) were found to encapsulate and stabilize luminescent quantum dots in aqueous solution through direct ligand binding to the QD surface. Due to the cationic charges and a “proton sponge effect” [98–100] associated with multivalent amine groups, these QDs could penetrate cell membranes and disrupt endosomal organelles in living cells. This mechanism arises from the presence of a large number of weak bases (with buffering capabilities at pH 5–6), which lead to proton absorption in acidic organelles, and an osmotic pressure buildup across the organelle membrane [100]. This osmotic pressure causes swelling and/or rupture of the acidic endosomes and a release of the trapped materials into the cytoplasm. PEI and other polycations are known to be cytotoxic, however the grafted PEG segment was found to significantly reduce the toxicity and improve the overall nanoparticle stability and biocompatibility. In comparison with previous QDs encapsulated with amphiphilic polymers, the cell-penetrating QDs were smaller in size and exceedingly stable in acidic environments [56]. Cellular uptake and imaging studies revealed that these dots were rapidly internalized by endocytosis, and the pathways of the QDs inside the cells showed dependence on the number of PEG grafts of the polymer ligands. While higher PEG content led to QD sequestration in vesicles, the QDs coated by PEI-g-PEG with fewer PEG grafts are able to escape from endosomes and release into the cytoplasm.

Encapsulation and solubilization of core-shell CdSe-CdS-ZnS quantum dots nihms62165f3
Lovric et al. [101] recently reported that very small QDs (2.2 nm) coated with small molecule ligands (cysteamine) spontaneously translocated to the nuclei of murine microglial cells following cellular uptake through passive endocytosis. In contrast, larger QDs (5.5 nm) and small QDs bound to albumin remained in the cytosol only. This is fascinating because these QDs could not only escape from endocytotic vesicles, but were also subjected to an unknown type of active machinery that attracted the QDs to the nucleus. Nabiev et al. [102] studied a similar trend of size-dependent QD segregation in human macrophages, and found that small QDs may target nuclear histones and nucleoli after active transport across the nuclear membrane. They found that the size cut-off for this effect was around 3.0 nm. Larger QDs eventually ended up in vesicles in the MTOC region, although some QDs were found to be free in the cytoplasm. This group proposed that the proton sponge effect was also responsible for endosomal escape, as small carboxyl-coated QDs could buffer in the pH 5–7 range. These insights are important for the design and development of nanoparticle agents for intracellular imaging and therapeutic applications.
Compared to the study of living cells in culture, different challenges arise with the increase in complexity to a multicellular organism, and with the accompanying increase in size. Unlike monolayers of cultured cells and thin tissue sections, tissue thickness becomes a major concern because biological tissue attenuates most signals used for imaging. Optical imaging, especially fluorescence imaging, has been used in living animal models, but it is still limited by the poor transmission of visible light through biological tissue. It has been suggested that there is a near-infrared optical window in most biological tissue that is the key to deep-tissue optical imaging [103]. The rationale is that Rayleigh scattering decreases with increasing wavelength, and that the major chromophores in mammals, hemoglobin and water, have local minima in absorption in this window. Few organic dyes are available that emit brightly in this spectral region, and they suffer from the same photobleaching problems as their visible counterparts, although this has not prevented their successful use as contrast agents for living organisms [104]. One of the greatest advantages of QDs for imaging in living tissue is that their emission wavelengths can be tuned throughout the near-infrared spectrum by adjusting their composition and size, resulting in photostable fluorophores that are stable in biological buffers [24].
Biodistribution of QDs
For most in vivo imaging applications using QDs and other nanoparticle contrast agents, systemic intravenous delivery into the bloodstream will be the main mode of administration. For this reason, the interaction of the nanoparticles with the components of plasma, the specific and nonspecific adsorption to blood cells and the vascular endothelium, and the eventual biodistribution in various organs are of great interest. Immediately upon exposure to blood, QDs may be quickly adsorbed by opsonins, in turn flagging them for phagocytosis. In addition, platelet coagulation may occur, the complement system may be activated, or the immune system can be stimulated or repressed (Figure 4). Although it is important for each of these potential biological effects to be addressed in detail, so far there are no studies that directly examine blood or immune system biocompatibility of QDs in vivo or ex vivo. However, a recent review article by Dobrovolskaia and McNeil addresses the immunological properties of polymeric, liposomal, carbon-based, and magnetic nanoparticles [105]. Considering the many factors that may affect systemically administered QDs, such as size, shape, charge, targeting ligands, etc., the two most important parameters that affect biodistribution are likely size and the propensity for serum protein adsorption.

QD interactions with blood immune cells and plasma proteins nihms62165f4
The number of papers published on quantum dot pharmacokinetics and biodistribution is limited, but several common trends can be identified. It has been consistently reported that QDs are taken up nonspecifically by the reticuloendothelial system (RES), including the liver and spleen, and the lymphatic system [106–108]. These findings are not necessarily intrinsic to QDs, but are strictly predicated upon the size of the QDs and their surface coatings. Ballou and coworkers reported that (CdSe)ZnS QDs were rapidly removed from the bloodstream into organs of the RES, and remained there for at least 4 months with detectable fluorescence [107]. TEM of these tissues revealed that these QDs retained their morphology, suggesting that given the proper coating, QDs are stable in vivo for very long periods of time without degradation into their potentially toxic elemental components. A complimentary work by Fischer, et al. showed that nearly 100% of albumin-coated QDs were removed from circulation and sequestered in the liver within hours after a tail vein injection, much faster than QDs that were not bound to albumin [108]. Within the liver, QDs conjugated to albumin were primarily associated with Kupffer cells (resident macrophages). From a clinical perspective, it may be possible to completely inhibit the accumulation of QDs and avoid potential toxic effects if they are within the size range of renal excretion. Recent publications have focused on this insight. Frangioni and coworkers demonstrated that the renal clearance of quantum dots is closely related to the hydrodynamic diameter of the nanoparticle and the renal filtration threshold (~5–6 nm) [109]. Of equal importance to the QD size, is that the surface does not promote protein adsorption, which could significantly increase QD size above that of the renal threshold, and promote phagocytosis. However, it is unlikely that even small QDs could be entirely eliminated from the kidneys, as it has also been found that small QDs (~9 nm) may directly extravasate out of blood vessels, into interstitial fluid [110].
For targeted imaging, specific modulation of the biodistribution of QD contrast agents is the main goal. One way to increase the probability of bioaffinity ligand-specific distribution is to increase the circulation time of the contrast agent in the bloodstream. QD structure and surface properties have been found to strongly impact the plasma half-life. It was demonstrated by Ballou et al. [107] that the lifetime of anionic, carboxylated QDs in the bloodstream of mice (4.6 minutes half-life) is significantly increased if the QDs are coated with PEG polymer chains (71 minutes half-life). This effect has also been documented for other types of nanoparticles and small molecules, in part due to decreased nonspecific adsorption of the nanoparticles, an increase in size, and decreased antigenicity [111]. In a more recent study using perfused porcine skin in vitro, Lee, et al.demonstrated that carboxylated QDs were extracted more rapidly from circulation, and had greater tissue deposition than PEG coated QDs [112]. It is important to note that a bioaffinity molecule may also be prone to RES uptake, despite a strong affinity for its intended target. For example, Jayagopal et al. reported that QD-antibody conjugates have a significantly longer circulation time if the Fc antibody regions (non-antigen binding domains) are immunologically shielded to reduce nonspecific interactions [113].
In Vivo Vascular Imaging
One of the most immediately successful applications of QDs in vivo has been their use as contrast agents for the two major circulatory systems of mammals, the cardiovascular system and the lymphatic system. In 2003, Larson et. al demonstrated that green-light emitting QDs remained fluorescent and detectable in capillaries of adipose tissue and skin of a living mouse following intravenous injection [114]. This work was aided by the use of near-infrared two-photon excitation for deeper penetration of excitation light, and by the extremely large two-photon cross-sections of QDs, 100–20,000 times that of organic dyes [115]. In other work, Lim et al. used near-infrared QDs to image the coronary vasculature of a rat heart [116], and Smith et al. imaged the blood vessels of chicken embryos with a variety of near-infrared and visible QDs [117]. The later report showed that QDs could be detected with higher sensitivity than traditionally used fluorescein-dextran conjugates, and resulted in a higher uniformity in image contrast across vessel lumena. Jayagopal et al. [113] recently demonstrated the potential for QDs to serve as molecular imaging agents for vascular imaging. Spectrally distinct QDs were conjugated to three different cell adhesion molecules (CAMs), and intravenously injected in a diabetic rat model. Fluorescence angiography of the retinal vasculature revealed CAM-specific increases in fluorescence, and allowed imaging of the inflammation-specific behavior of individual leukocytes, as they freely floated in the vessels, rolled along the endothelium, and underwent leukostasis. The unique spectral properties of QDs allowed the authors to simultaneously image up to four spectrally distinct QD tags.
For imaging of the lymphatic system, the overall size of the probe is an important parameter for determining biodistribution and clearance. For example, Kim et al. [24] intradermally injected ~16–19 nm near-infrared QDs in mice and pigs. QDs translocated to sentinel lymph nodes, likely due to a combination of passive flow in lymphatic vessels, and active migration of dendritic cells that engulfed the nanoparticles. Fluorescence contrast of these nodes could be observed up to 1 cm beneath the skin surface. It was found that if these QDs were formulated to have a smaller overall hydrodynamic size (~9 nm), they could migrate further into the lymphatic system, with up to 5 nodes showing fluorescence [110]. This technique could have great clinical impact due to the quick speed of lymphatic drainage and the ease of identification of lymph nodes, enabling surgeons to fluorescently identify and excise nodes draining from primary metastatic tumors for the staging of cancer. This technique has been used to identify lymph nodes downstream from the lungs [106, 118], esophagus [119], and from subcutaneous tumors [120]. Recently the multiplexing capabilities of QDs have been exploited for mapping lymphatic drainage networks. By injection of QDs of different color at different intradermal locations, these QDs could be fluorescently observed to drain to common nodes [121], or up to 5 different nodes in real time [122]. A current problem is that a major fraction of the QDs remain at the site of injection for an unknown length of time [123].
In Vivo Tracking of QD-Loaded Cells
Cells can also be loaded with QDs in vitro, and then administered to an organism, providing a means to identify the original cells and their progeny within the organism. This was first demonstrated on a small organism scale by microinjecting QDs into the cytoplasms of single frog embryos [49]. As the embryos grew, the cells divided, and each cell that descended from the original labeled cell retained a portion of the fluorescent cytoplasm, which could be fluorescently imaged in real time under continuous illumination. In reports by Hoshino et al. [124] and Voura et al. [82], cells loaded with QDs were injected intravenously into mice, and their distributions in the animals were later determined through tissue dissection, followed by fluorescence imaging. Also Gao et al. loaded human cancer cells with QDs, and injected these cells subcutaneously in an immune-compromised mouse [10]. The cancer cells divided to form a solid tumor, which could be visualized fluorescently through the skin of the mouse. Rosen et al. recently reported that human mesenchymal stem cells loaded with QDs could be implanted into an extracellular matrix patch for use as a regenerative implant for canine hearts with a surgically-induced defect [125]. Eight weeks following implantation, it was found that the QDs remained fluorescent within the cells, and could be used to track the locations and fates of these cells. This group also directly injected QD-labeled stem cells into the canine myocardium, and used the fluorescence signals in cardiac tissue sections to elaborately reconstruct the locations of these cells in the heart. With reports that cells may be labeled with QDs at a high degree of specificity [80, 81], it is foreseeable that multiple types of cells may be simultaneously monitored in living organisms, and also identified using their distinct optical codes.
In Vivo Tumor Imaging
Imaging of tumors presents a unique challenge not only because of the urgent need for sensitive and specific imaging agents of cancer, but also because of the unique biological attributes inherent to cancerous tissue. Blood vessels are abnormally formed during tumorinduced angiogenesis, having erratic architectures and wide endothelial pores. These pores are large enough to allow the extravasation of large macromolecules up to ~400 nm in size, which accumulate in the tumor microenvironment due to a lack of effective lymphatic drainage [126–129]. This “enhanced permeability and retention” effect (EPR effect) has inspired the development of a variety of nanotherapeutics and nanoparticulates for the treatment and imaging of cancer (Figure 5). Because cancerous cells are effectively exposed to the constituents of the bloodstream, their surface receptors may also be used as active targets of bioaffinity molecules. In the case of imaging probes, active targeting of cancer antigens (molecular imaging) has become an area of tremendous interest to the field of medicine because of the potential to detect early stage cancers and their metastases. QDs hold great promise for these applications mainly due to their intense fluorescent signals and multiplexing capabilities, which could allow a high degree of sensitivity and selectivity in cancer imaging with multiple antigens.

The first steps toward this goal were undertaken in 2002 by Akerman et al., who conjugated QDs to peptides with affinity for various tumor cells and their vasculatures [130]. After intravenous injection of these probes into tumor-bearing mice, microscopic fluorescence imaging of tissue sections demonstrated that the QDs specifically homed to the tumor vasculature. In 2004 Gao et al. demonstrated that tumor targeting with QDs could generate tumor contrast on the scale of whole-animal imaging [10]. QDs were conjugated to an antibody against the prostate-specific membrane antigen (PSMA), and intravenously injected into mice bearing subcutaneous human prostate cancers. Tumor fluorescence was significantly greater for the actively targeted conjugates compared to nonconjugated QDs, which also accumulated passively though the EPR effect. Using similar methods, Yu et al. were able to actively target and image mouse models of human liver cancer with QDs conjugated to an antibody against alpha-fetoprotein [131], and Cai et al. showed that labeling QDs with RGD peptide significantly increased their uptake in human glioblastoma tumors [132].
The development of clinically relevant QD contrast agents for in vivo imaging is certain to encounter many roadblocks in the near future (see Section 5), however QDs can currently be used as powerful imaging agents for the study of the complex anatomy and pathophysiology of cancer in animal models. Stroh et al.[133] demonstrated that QDs greatly enhance current intravital microscopy techniques for the imaging of tumor microenvironment. The authors used QDs as fluorescent contrast agents for blood vessels using two-photon excitation, and simultaneously captured images of extracellular matrix from autofluorescent collagen, and perivascular cell contrast from fluorescent protein expression. The use of QDs allowed stark contrast between the tumor constituents due to their intense brightness, tunable wavelengths, and reduced propensity to extravasate into the tumor, compared to organic dye conjugates. In this work, the authors also used QD-tagged beads with variable sizes to model the size-dependent distribution of various nanotherapeutics in tumors. As well, the authors demonstrated that bone marrow lineage-negative cells, which are thought to be progenitors for neovascular endothelium, were labeled ex vivo with QDs and imaged in vivo as they flowed and adhered to tumor blood vessels following intravenous administration. More recently, Tada et al. used QDs to study the biological processes involved in active targeting of nanoparticles. The authors used QDs labeled with an antibody against human epidermal growth factor receptor 2 (HER2) to target human breast cancer in a mouse model [134]. Through intravital fluorescence microscopy of the tumor following systemic QD administration, the authors could distinctly observe individual QDs as they circulated in the bloodstream, extravasated into the tumor, diffused in extracellular matrix, bound to their receptors on tumor cells, and then translocated into the perinuclear region of the cells. The combination of sensitive QD probes with powerful techniques like intravital microscopy and in vivo animal imaging could soon lead to major breakthroughs in the current understanding of tumor biology, improve early detection schemes, and guide new therapeutic designs.
Great concern has been raised over the use of quantum dots in living cells and animals due to their chemical composition of toxic heavy metal atoms (e.g. Cd, Hg, Pb, As, Pb). Presently the most commonly used QDs contain divalent cadmium, a nephrotoxin in its ionic form. Although this element is incorporated into a nanocrystalline core, surrounded by biologically inert zinc sulfide, and encapsulated within a stable polymer, it is still unclear if these toxic ions will impact the use of QDs as clinical contrast agents. It may be of greater concern that QDs, and many other types of nanoparticles, have been found to aggregate, bind nonspecifically to cellular membranes and intracellular proteins, and induce the formation of reactive oxygen species. As previously stated, QDs larger than the renal filtration threshold quickly accumulate in the reticuloendothelial system following intravenous administration. The eventual fate of these nanoparticles is of vital importance, but so far has yet to be elucidated.
Cadmium Toxicity
In the only long-term, quantitative study on QD biodistribution to date, Yang, et al. showed that after intravenous administration of cadmium-based QDs, the concentration of cadmium in the liver and kidneys gradually increased over the course of 28 days, as determined via ICP-MS [135]. The cadmium levels in the kidneys eventually reached nearly 10% of the injected dose, compared to 40% in the liver. Although it was not apparent if the cadmium was in the form of a free ion, or remained in the nanocrystalline form, fluorescence microscopy revealed the presence of intact QDs in both the liver and kidneys. However the redistribution of the cadmium over time may signify the degradation of QDs in vivo, since the natural accumulation sites of Cd2+ ions are the liver and kidneys [79, 136, 137]. In acute exposures, free cadmium also may be redistributed to the kidneys via hepatic production of metallothionein [138]. Whether or not this is the specific mechanism observed in this report should be the focus of detailed in vivo validation studies. Nevertheless, these findings stress that (a) QD size and nonspecific protein interaction should be minimized to allow renal filtration, or else QDs will inevitably accumulate in organs and tissues of the RES, lung, and kidney, and (b) the potential release of the elements of the QD and their distribution in specific organs, tissues, cell types, and subcellular locations must be well understood.
In general, most in vitro studies on the exposure of cells to QDs have attempted to relate cytotoxic events to the release of potentially toxic elements and/or to the size, shape, surface, and cellular uptake of QDs. Because the toxicity of Cd2+ ions is well documented, a significant body of work has focused on the intracellular release of free cadmium from the QDs. Cd2+ ions can be released through oxidative degradation of the QD, and may then bind to sulfhydryl groups on a variety of intracellular proteins, causing decreased functionality in many subcellular organelles [139]. Several groups have investigated methods to quantify the amount of free Cd2+ ions released from QDs, either intracellularly or into culture media, by ICP-MS or fluorometric assays, leading to the conclusion that Cd2+ release correlates with cytotoxic manifestations [79,140, 141]. Derfus, et al. facilitated oxidative release of cadmium ions from the surface of CdSe QDs by exposure to air or ultraviolet irradiation [79]. Under these conditions, CdSe QD cores coated with small thiolate ligands were toxic. Capping these QDs with ZnS shells or coating with BSA rendered the QD cores less susceptible to oxidative degradation and less toxic to primary rat hepatocytes, implicating the potential role of cadmium in QDs cytotoxicity. The decrease in QD cytotoxicity of CdSe QDs with the overgrowth of a ZnS shell has since been verified in several reports [139, 142]. If it is revealed in the future that Cd2+release is a major hindrance for the use of QDs in cells and in animals, several new types of QDs that have no heavy metals atoms may be useful for advancing this field [143, 144].
Toxicity Induced by Colloidal Instability
Presently it is nearly impossible to drawing firm conclusions about the toxicity of QDs in cultured cells due to (a) the immense variety of QDs and variations of surface coatings used by different labs and (b) a technical disparity in experimental conditions, such as the duration of the nanoparticle exposure, use of relevant cell lines, media choice (e.g. with or without serum), and even the units of concentration (e.g. mg/ml versus nM). Nonetheless, the cytotoxicity of QDs reported in the literature has strongly correlated with the stability and surface coatings of these nanoparticles, which can be separated into three categories. (1) Core CdTe QDs that are synthesized in aqueous solution and stabilized by small thiolate ligands (e.g. mercaptopropionic acid or mercaptoacetic acid). These QDs have been widely used due to their ease of synthesis, low cost, and immediate utility in biological buffers. However, because these QDs are protected only by a weakly bound ligand, they are highly prone to degradation and aggregation, and their cytotoxicity toward cells in culture has been widely reported [140, 145]. (2) Core/shell CdSe/ZnS QDs synthesized in nonpolar solvents and transferred to water using thiolate ligands. CdSe is less prone to oxidation than CdTe, and ZnS is even more inert, and therefore these QDs are much more chemically stable. With direct comparison to CdTe QDs, these nanocrystals are significantly less cytotoxic, although high concentrations have been found to illicit toxic responses from cells [140]. Because these QDs are coated with a ZnS shell, the origin of this cytotoxicity is still unclear, whether it is from degradation of the shell, leading to cadmium release, or if it is caused by other effects. When coated with small ligands, these QDs have similar surface chemistries compared to aqueous CdTe QDs, burdened by significant dissociation of ligands from the QDs, rendering the nanoparticles colloidally unstable [146]. This propensity for aggregation may contribute to their cytotoxicity, even if free cadmium is not released. Importantly for the comparison between CdSe/ZnS QDs and their cadmium-only counterparts (CdSe or CdTe core QDs), thiolate ligands bind more strongly to zinc than to cadmium, which may contribute colloidal stability. (3) Core/shell CdSe/ZnS QDs synthesized in nonpolar solvents and transferred to water via encapsulation in amphiphilic polymers or cross-linked silica. These QDs have been found to be significantly more stable colloidally, chemically, and optically when compared to their counterparts coated in small ligands [56]. For this reason, they have been found to be nearly biologically inert in both living cells and living animals [10, 24, 49, 60, 79, 107, 114, 147]. Only when exposed to extreme conditions or when directly injected into cells at immensely high concentrations have these QDs been found to elicit toxic or inflammatory responses [49, 142].
It is feasible that a significant amount of toxicological data obtained for QDs thus far has been considerably influenced by the colloidal nature of these nanoparticles. The tendency for nanoparticles to aggregate, precipitate on cells in culture, nonspecifically adsorb to biomolecules, and catalyze the formation of reactive oxygen species (ROS) may be just as important as heavy metal toxicity contributions to toxicity. For example, Kircher et al. found that CdSe/ZnS QDs coated with an amphiphilic polymer shell induced the detachment of human breast cancer cells from their cell culture substrate [139]. This effect was found to also occur for biologically inert gold nanoparticles coated with the same polymer, thus ruling out the possibility of heavy metal atom poisoning. Microscopic examination of the cells revealed that the nanoparticles precipitated on the cells, causing physical harm. Indeed, carbon nanotubes, which are entirely composed of harmless carbon, have been found to be capable of impaling cells and causing major problems in the lungs of mammals [148]. Nonspecific adsorption to intracellular proteins may also impair cellular function, especially for very small QDs (3 nm and below), which can invade the cellular nucleus [101], binding to histones and nucleosomes [102], and damage DNA in vitro [149, 150]. QDs are also known to catalyze the formation of ROS [145, 151], especially when exposed to ultraviolet radiation. In fact, Cho et al. exposed cells to CdTe QDs in cell culture and determined that their cytotoxicity could only be accounted for with the effects of ROS generation, as there was no dose-dependent relationship with intracellular Cd2+ release, as determined with a cadmium-reactive dye [140]. However, protection of the surface of QDs with a thick ZnS shell may greatly reduce ROS production [152, 153]. Despite a significant surge of interest in the cytotoxicity of nanoparticles, there is still much to learn about the cytological and physiological mediators of nanoparticle toxicology. If it is determined that heavy metal composition plays a negligible role in QD toxicity, QDs will have as good of a chance as any other nanoparticle at being used as clinical contrast agents.
In comparison with small organic fluorophores, QDs have large surfaces that can be modified through versatile chemistry. This makes QDs convenient scaffolds to accommodate multiple imaging (e.g., radionuclide-based or paramagnetic probes) and therapeutic agents (e.g. anticancer drugs), through chemical linkage or by simple physical immobilization. This may enable the development of a nearly limitless library of multifunctional nanostructures for multimodality imaging, as well as for integrated imaging and therapy.
Dual-Modality Imaging
The applications of QDs described above for in vivo imaging are limited by tissue penetration depth, quantification problems, and a lack of anatomic resolution and spatial information. To address these limitations, several research groups have led efforts to couple QD-based optical imaging with other imaging modalities that are not limited by penetration depth, such as MRI, positron emission tomography (PET) and single photon emission computed tomography (SPECT) [154–158]. For example, Mulder et al. [154] developed a dual-modality imaging probe for both optical imaging and MRI by chemically incorporating paramagnetic gadolinium complexes in the lipid coating layer of QDs [154, 155]. In vitro experiments showed that labeling of cultured cells with these QDs led to significant T1 contrast enhancement with a brightening effect in MRI, as well as an easily detectable fluorescence signal from QDs. However, the in vivoimaging potential of this specific dual-modality contrast agent is uncertain due to the unstable nature of the lipid coating that was used. More recently, Chen and coworkers used a similar approach to attach the PET-detectable radionuclide 64Cu to the polymeric coating of QDs through a covalently bound chelation compound [158]. The use of this probe for targeted in vivo imaging of a subcutaneous mouse tumor model was achieved by also attaching αvβ3 integrin-binding RGD peptides on the QD surface. The quantification ability and ultrahigh sensitivity of PET imaging enabled the quantitative analysis of the biodistribution and targeting efficacy of this dual-modality imaging probe. However, the full potential of in vivo dual-modality imaging was not realized in this study, as fluorescence was only used as an ex vivo imaging tool to validate the in vivo results of PET imaging, primarily due to the lower sensitivity of optical imaging in comparison with PET. This imbalance in sensitivity is fundamental to the differences in the physics of these imaging modalities, and points to an inherent difficulty in designing useful multimodal imaging probes. The majority of these probes are still at an early stage of development. The clinical relevance of these nanoplatforms still needs further improvement in sensitivity and better integration of different imaging modalities, as well as validation of their biocompatibility and safety.
It is also noteworthy that recent advances in the synthesis of QDs containing paramagnetic dopants, such as manganese, have led to a new class of QDs that are intrinsically fluorescent and magnetic [159, 160]. However the utility of these new probes for bioimaging application is unclear because they are currently limited to the ultraviolet and visible emission windows, and their stability (e.g., photochemical and colloidal) and biocompatibility have yet to be systematically investigated [144]. As well, inorganic heterodimers of QDs and magnetic nanoparticles have generated dual-functional nanoparticles [161, 162]. Although these new materials are of great interest, they are still in development and have only recently shown applicability in cell culture, but not yet in living animals [160, 163].
Integration of Imaging and Therapy
Drug-containing nanoparticles have shown great promise for treating tumors in animal models and even in clinical trials [157]. Both passive and active targeting of nanotherapeutics have been used to increase the local concentration of chemotherapeutics in the tumor. Due to the size and structural similarities between imaging and therapeutic nanoparticles, it is possible that their functions can be integrated to directly monitor therapeutic biodistribution, to improve treatment specificity, and to reduce side effects. This synergy has become the principle foundation for the development of multi-functional nanoparticles for integrated imaging and cancer treatment. Most studies are still at a proof-of-concept stage using cultured cancer cells, and are not immediately relevant to in vivo imaging and treatment of solid tumors. However, these studies will guide the future design and optimization of multifunctional nanoparticle agents for in vivo imaging and therapy [164–167].
In one example, Farokhzad et al. reported a ternary system composed of a QD, an aptamer, and the small molecular anticancer drug doxorubicin (Dox) for in vitro targeted imaging, therapy and sensing of drug release [165]. As illustrated in Figure 6, aptamers were conjugated to QDs to serve as targeting units, and Dox was attached to the stem region of the aptamers, taking advantage of the nucleic acid binding ability of doxorubicin. Two donor-quencher pairs of fluorescence resonance energy transfer occurred in this construct, as the QD fluorescence were quenched by Dox, and Dox was quenched by the double-stranded RNA aptamers. As a result, gradual release of Dox from the conjugate was found to “turn on” the fluorescence of both QDs and Dox, providing a means to sense the release of the drug. However it is clear that the current design of this conjugate will not be sufficient for in vivo use unless the drug loading capacity can be greatly increased (currently 7–8 Dox molecules per QD).

QD-Aptamer-Dox FRET system and its targeted delivery nihms62165f6
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2649798/bin/nihms62165f6.jpg
QDs also provide a versatile nanoscale scaffold to develop multifunctional nanoparticles for siRNA delivery and imaging. RNA interference (RNAi) is a powerful technology for sequence-specific suppression of genes, and has broad applications ranging from functional gene analysis to targeted therapy [168–172]. However, these applications are limited by the same delivery problems that hinder intracellular imaging with QDs (Section 3.2), namely intracellular delivery and endosomal escape, in addition to dissociation from the delivery vehicle (i.e. unpacking), and coupling with cellular machines (such as the RNA-induced silencing complex or RISC). For cellular and in vivo siRNA delivery, a number of approaches have been developed (see ref. [168] for a review), but these methods have various shortcomings and do not allow a balanced optimization of gene silencing efficacy and toxicity. For example, previous work has used QDs and iron oxide nanoparticles for siRNA delivery and imaging [27, 166, 167, 173], but the QD probes were either mixed with conventional siRNA delivery agents [166] or an exogenous compound, such as the antimalaria drug chloroquine, was needed for endosomal rupture and gene silencing activity [173].
Gao et al. have recently fine-tuned the colloidal and chemical properties of QDs for use as delivery vehicles for siRNA, resulting in highly effective and safe RNA interference, as well as fluorescence contrast [174]. The authors balanced the proton-absorbing capacity of the QD surface in order to induce endosomal release of the siRNA through the proton sponge effect (see Section 3.4). A major finding is that this effect can be precisely controlled by partially converting the carboxylic acid groups on a QD into tertiary amines. When both are linked to the surface of nanometer-sized particles, these two functional groups provide steric and electrostatic interactions that are highly responsive to the acidic organelles, and are also well suited for siRNA binding and cellular entry. As a result, these conjugates can improve gene silencing activity by 10–20 fold, and reduce cellular toxicity by 5–6 fold, compared with current siRNA delivery agents (lipofectamine, JetPEI, and TransIT). In addition, QDs are inherently dual-modality optical and electron microscopy probes, allowing real-time tracking and ultrastructural localization of QDs during transfection.
Quantum dots have been received as technological marvels with characteristics that could greatly improve biological imaging and detection. In the near future, there are a number of areas of research that are particularly promising but will require concerted effort for success:
(1) Design and development of nanoparticles with multiple functions
For cancer and other medical applications, important functions include imaging (single or dual-modality), therapy (single drug or combination of two or more drugs), and targeting (one or more ligands). With each added function, nanoparticles could be designed to have novel properties and applications. For example, binary nanoparticles with two functions could be developed for molecular imaging, targeted therapy, or for simultaneous imaging and therapy. Ternary nanoparticles with three functions could be designed for simultaneous imaging and therapy with targeting, targeted dual-modality imaging, or for targeted dual-drug therapy. Quaternary nanoparticles with four functions can be conceptualized in the future to have the capabilities of tumor targeting, dual-drug therapy and imaging.
(2) Use of multiplexed QD bioconjugates for analyzing a panel of biomarkers and for correlation with disease behavior, clinical outcome, and treatment response
This application should begin with retrospective studies of archived specimens in which the patient outcome is already known. A key hypothesis to be tested is that the analysis of a panel of tumor markers will allow more accurate correlations than single tumor markers. As well, the analysis of the relationship between gene expression from cancer cells and the host stroma may help to define important cancer subclasses, identify aggressive phenotypes of cancer, and determine the response of early stage disease to treatment (chemotherapy, radiation, or surgery).
(3) Design and development of biocompatible nanoparticles to overcome nonspecific organ uptake and RES scavenging
There is an urgent need to develop nanoparticles that are capable of escaping RES uptake, and able to target tumors by active binding mechanisms. This in vivo biodistribution barrier might be mitigated or overcome by systematically optimizing the size, shape, and surface chemistry of imaging and therapeutic nanoparticles.
(4) Penetration of imaging and therapeutic nanoparticles into solid tumors beyond the vascular endothelium
This task will likely require active pumping mechanisms such as caveolin transcytosis and receptor-mediated endocytosis, or cell-based strategies such as nanoparticle-loaded macrophages.
(5) Release of drug payloads inside targeted cells or organs
This task will likely require the development of biodegradable nanoparticle carriers that are responsive to pH, temperature, or enzymatic reactions.
(6) Nanotoxicology studies including nanoparticle distribution, excretion, metabolism, pharmacokinetics, and pharmacodynamics in animal models in vivo
These investigations will be vital for the development of nanoparticles beyond their current use as research tools, toward clinical applications in cancer imaging and therapy.
7.1.3 In vivo molecular and cellular imaging with quantum dots
Gao X, Yang L, Petros JA, Marshall FF, Simons JW, Nie S.
Curr Opin Biotechnol. 2005 Feb;16(1):63-72.
http://dx.doi.org:/10.1016/j.copbio.2004.11.003
http://ars.els-cdn.com/content/image/1-s2.0-S0958166904001648-gr1.sml
The structure of a multifunctional QD probe. Schematic illustration showing the capping ligand TOPO, an encapsulating copolymer layer, tumor-targeting ligands (such as peptides, antibodies or small-molecule inhibitors), and polyethylene glycol (PEG).
http://ars.els-cdn.com/content/image/1-s2.0-S0958166904001648-gr2.sml
Methods for conjugating QDs to biomolecules. (a) Traditional covalent cross-linking chemistry using EDAC (ethyl-3-dimethyl amino propyl carbodiimide) as a catalyst. (b) Conjugation of antibody fragments to QDs via reduced sulfhydryl-amine coupling. SMCC, succinimidyl-4-Nmaleimidomethyl-cyclohexane carboxylate. (c) Conjugation of antibodies to QDs via an adaptor protein. (d) Conjugation of histidine-tagged peptides and proteins to Ni-NTA-modified QDs, with potential control of the attachment site and QD:ligand molar ratios.
http://ars.els-cdn.com/content/image/1-s2.0-S0958166904001648-gr4.sml
Quantum dots (QDs), tiny light-emitting particles on the nanometer scale, are emerging as a new class of fluorescent probe for in vivo biomolecular and cellular imaging. In comparison with organic dyes and fluorescent proteins, QDs have unique optical and electronic properties: size-tunable light emission, improved signal brightness, resistance against photobleaching, and simultaneous excitation of multiple fluorescence colors. Recent advances have led to the development of multifunctional nanoparticle probes that are very bright and stable under complex in vivo conditions. A new structural design involves encapsulating luminescent QDs with amphiphilic block copolymers and linking the polymer coating to tumor-targeting ligands and drug delivery functionalities. Polymer-encapsulated QDs are essentially nontoxic to cells and animals, but their long-term in vivo toxicity and degradation need more careful study. Bioconjugated QDs have raised new possibilities for ultrasensitive and multiplexed imaging of molecular targets in living cells, animal models and possibly in humans.
7.1.4 Luminescent quantum dots for multiplexed biological detection and imaging
Chan WC1, Maxwell DJ, Gao X, Bailey RE, Han M, Nie S.
Curr Opin Biotechnol. 2002 Feb;13(1):40-6.
http://dx.doi.org/10.1016/S0958-1669(02)00282-3


http://ars.els-cdn.com/content/image/1-s2.0-S0958166902002823-gr2.sml

http://ars.els-cdn.com/content/image/1-s2.0-S0958166902002823-gr3.sml
http://ars.els-cdn.com/content/image/1-s2.0-S0958166902002823-gr5.sml
Recent advances in nanomaterials have produced a new class of fluorescent labels by conjugating semiconductor quantum dots with biorecognition molecules. These nanometer-sized conjugates are water-soluble and biocompatible, and provide important advantages over organic dyes and lanthanide probes. In particular, the emission wavelength of quantum-dot nanocrystals can be continuously tuned by changing the particle size, and a single light source can be used for simultaneous excitation of all different-sized dots. High-quality dots are also highly stable against photobleaching and have narrow, symmetric emission spectra. These novel optical properties render quantum dots ideal fluorophores for ultrasensitive, multicolor, and multiplexing applications in molecular biotechnology and bioengineering.
7.1.5 Multifunctional quantum dots for Personalized Medicine
Pavel Zrazhevskiy and Xiaohu Gao
Nano Today. 2009 Oct 5; 4(5): 414–428.
http://dx.doi.org:/10.1016/j.nantod.2009.07.004
Successes in biomedical research and state-of-the-art medicine have undoubtedly improved the quality of life. However, a number of diseases, such as cancer, immunodeficiencies, and neurological disorders, still evade conventional diagnostic and therapeutic approaches. A transformation towards personalized medicine may help to combat these diseases. For this, identification of disease molecular fingerprints and their association with prognosis and targeted therapy must become available. Quantum dots (QDs), semiconductor nanocrystals with unique photo-physical properties, represent a novel class of fluorescence probes to address many of the needs of personalized medicine. This review outlines the properties of QDs that make them a suitable platform for advancing personalized medicine, examines several proof-of-concept studies showing utility of QDs for clinically relevant applications, and discusses current challenges in introducing QDs into clinical practice.
State-of-the-art medicine is an indispensable part of the human society. Wealth of medical knowledge accumulated over centuries of observation and experimentation, advanced diagnostic techniques made possible by the technological revolution, and innovative biomedical research done on the cellular and molecular levels provide a formidable weapon against nearly any threat to human health. However, the most devastating diseases, such as cancer, immunodeficiencies and neurological disorders to name a few, are notorious for their ability to evade current diagnostic methods and resist therapy. It is not easy to pinpoint the main reasons for poor success in combating these diseases, as they might range from a lack of understanding of patho-physiology to the absence of appropriate diagnostic techniques capable of addressing the complexity of these diseases. One potential issue is that utilization of generalized diagnostic and treatment approaches based on identifying and targeting disease symptoms (often with limited information about the underlying cause) is inefficient in addressing the great genetic and phenotypic variability of cancer and immune system disorders. Significant heterogeneity on molecular level, complex interlinking of subcellular mechanisms along with integrated pathophysiological effects on organs and systems of the human body, and an often unclear origin and cause of the disease represent major challenges for current biomedical research and clinical practice.
Personalized medicine, a practice of addressing individual diseases in a pathology-specific and patient-specific manner spanning all levels from whole-body symptoms down to molecular signatures of the disease, is an emerging field of medicine promising to provide efficient tools against cancer and other challenging diseases. A personalized approach offers unique opportunities to accurate diagnosis (i.e. pinpoint exact changes that occurred within healthy cells and tissues), prognosis (i.e. predict progression of a disease based on these changes), and treatment (i.e. specifically reverse the changes or, if not possible, target and kill the diseased cells without affecting healthy ones). Such an approach relies on advances in basic research as well as integration of novel diagnostic and therapeutic techniques into clinical practice.
Currently, attempts of introducing personalized approach in medicine rely on screening for genetic alterations in diseased cells; yet diagnostic and predictive power of genetic screening alone is questionable due to insufficient knowledge of how certain alterations on the DNA level propagate along the DNA-RNA-protein chain [1, 2] and the requirement of performing analysis on a homogenized mixture of different cell types, including a variety of healthy cells [3]. Therefore, complementary analysis of phenotypic changes (i.e. changes in protein expression) as well as assessment of the effect of diseased cells on the healthy tissues (e.g. activation of angiogenesis in tumors) is necessary for comprehensive analysis of a pathological process. Compilation of a database of genetic and phenotypic signatures of individual diseases will provide an access to a more accurate prognosis and personalized treatment targeted directly against the biomarkers expressed. Realizing this, significant research effort is being focused at understanding the physiology of normal cellular processes as well as patho-physiology of diseases in order to determine specific disease-causing changes in individual cells, organs, and systems.
A key challenge is presented by the complexity of inter- and intracellular networks with multiple inputs, controllers, and feedback loops, which is hard to assess using conventional biomedical techniques (such as immunohistochemistry, Western blot, ELISA, etc) that suffer from a limitation in the number of biomarkers that can be analyzed simultaneously, lack real-time monitoring capacity for intracellular processes, provide limited single-cell information resulting from the need to analyze signals averaged over many cells, and utilize qualitative rather than quantitative analytical techniques [4–6]. Consequently, diagnosis and prognosis are limited by the lack of knowledge about the predictive biomarkers that would unambiguously discriminate between disease and normal function as well as distinguish different disease types and provide information about possible progression of the pathological process.
Advances in nanotechnology have enabled the design of nanoparticle-based tools for improved diagnosis and personalized treatment of many complex diseases. In particular, semiconductor QDs have emerged as a new platform for high-throughput quantitative characterization of multiple biomarkers in cells and clinical tissue specimens ex vivo, detection of diseased cells in vivo, and potentially targeted and traceable drug delivery [3,7, 8].
QDs are semiconductor nanoparticles with size ranging between 2 and 10 nm in diameter (hydrodynamic size often larger). Restricting the mobility of charge carriers (electrons and holes) within the nanoscale dimensions generates the quantum confinement effect responsible for unique size-dependent photo-physical properties of QDs [9–11]. Additionally, nanometer-scale size of QDs comparable with the size of large proteins enables integration of nanoparticles and biomolecules yielding biologically functional nanomaterials suitable for probing physiological processes on a molecular level [12–14]. While a relatively large size (compared to small drug molecules or organic fluorescent dyes) might be associated with slower diffusion, limited permeability, complex bio-distribution, and possible interference with intracellular processes [15], QDs possess a wide range of features essential for addressing the most urgent needs of personalized medicine. Among such features are size-tunable and spectrally narrow light emission, simultaneous excitation of multiple colors, improved brightness, resistance to photobleaching, and an extremely large Stokes shift.
The cornerstone of personalized medicine is the ability to uniquely identify the disease by its “molecular fingerprint” (i.e. pattern of biomarker expression), associate the fingerprint with possible progression of the disease, and assign a treatment which targets diseased cells with the identified fingerprint. Achieving this goal is not a trivial task – many diseased cells look very much like the healthy ones (especially in case of cancer), and screening for a large panel of biomarkers is required. It is quite possible that certain diseases have one or few biomarkers specific enough for unique identification, yet finding these biomarkers de novo using low-throughput conventional approaches is like looking for a needle in a haystack. QDs open access to a multi-parameter biomarker screening on intact specimens via multiplexed detection [16]. This feature is based on two properties of QDs: spectrally narrow size-tunable light emission [17–19] and effective light absorption throughout a wide spectrum [12] (Fig. 1). Excitation of multiple QD probes with a single light source (e.g. laser) significantly reduces the complexity and cost of imaging instrumentation and simplifies data analysis. Utilization of hyperspectral imaging, a technique that allows deconvolution of an image into spectral components, further improves the multiplexing capabilities of QD technology (Fig. 2) [20]. It is worth mentioning that highly multiplexed molecular analysis would be limited if hyperspectral imaging or QDs are used separately. Combination of these two complementary technologies enhances each other’s capability.


An indispensable part of disease molecular profiling is the ability to quantify biomarker expression in an accurate and consistent manner. So far, this requirement has been only partially fulfilled. The problem lies in the fact that colorimetric assays usually rely on amplification mechanisms, which are difficult to control, thus providing inconsistent and mostly qualitative information about the biomarker expression. Quantitative analysis with fluorescence imaging using organic fluorophores is often compromised by the quick photobleaching of the dyes and unstable signal intensity. Destructive techniques, while allowing protein quantification (e.g. Western blot, RT-PCR, protein chips), do not preserve tissue morphology and cannot properly address the heterogeneity of specimens. QD probes, on the other hand, are well-suited for addressing these issues. First of all, QDs are highly resistant to photobleaching and photodegradation: in one example QDs retained constant signal intensity for over 30 minutes of illumination, while organic dyes faded by more than 90% in less than one minute under identical experimental conditions [21]. Second, QDs do not rely on chemical amplification (in contrast to assays such as horse radish peroxidase mediated color development and Au catalyzed Ag-enhancement) and have a promise of providing imaging probes with a 1:1 stoichiometry. It is necessary to note, though, that the intensity of different color QDs under identical illumination conditions differ significantly, showing enhancement of red QD signal over green/blue QDs. Such discordance has been observed by Ghazani and coworkers in a three-color staining of lung carcinoma xenografts for epidermal growth factor receptor, E-cadherin, and cytokeratin using QDs emitting at 655, 605, and 565 nm [22]. While quantitative analysis of individual QD signals was readily achievable, comparison between different QD signals was not possible through this study. The discordance in fluorescence intensity of individual probes directly relates to light absorption properties and the quantum yield of QDs (i.e. red particles having larger cross-section absorb light more efficiently) and can be accounted for in signal analysis algorithm. For example, Yezhelyev et al used bulk fluorescence measurement of equal concentrations of QDs and determined that QD655 were 8 times as bright and QD605 4 times as bright as QD565 [23]. However, other effects associated with high QD concentration, such as steric hindrance between the probes, self-quenching, and fluorescence resonance energy transfer (FRET) from smaller to larger particles [22], might be possible in cases of high biomarker density and deserves further investigation for achieving accurate quantitative analysis.
A variety of nanomaterials have already shown utility in addressing tough questions posed by unmet clinical needs. In particular, QDs have proven to be well suited for sensitive quantitative molecular profiling of cells and tissues, holding tremendous promise for unraveling the complex gene expression profiles of diseases, accurate clinical diagnosis and personalized treatment of patients [3, 24]. Possessing advantageous photo-physical properties and being compatible with conventional biomedical assays, QDs have found use in most techniques where fluorescence or colorimetric imaging of target biomarker is utilized (e.g. cell and tissue staining, Western blot, ELISA, etc.) and have launched many novel applications (e.g. targeted in vivoimaging, single-molecule tracking, traceable drug delivery, etc.). The number of biomedical applications of QDs continues growing, ranging from ultrasensitive detection in vitro to targeted drug delivery and imagingin vivo.
Identification of molecular fingerprints of diseases
Molecular fingerprinting of diseases implies characterization of biomarker expression schemes in diseased cells in comparison to healthy ones. QD-based probes are uniquely suited for this task when employed by both multi-parameter flow-cytometry analysis of cell populations and quantitative multiplexed analysis of biomarker expression in intact tissue specimens. For example, Chattopadhyay et al, by utilizing a 17-parameter flow-cytometry (based on 8 QD probes and 9 organic fluorophores), revealed significant phenotypic differences between T-cells specific to distinct epitopes of the same pathogen (Fig. 3) [25]. Access to molecular profiles of individual cell populations not only improves our understanding of immune response, but also enables analysis of changes occurring during immune system disorders, sensitive detection of metastasizing cancer cells in a bloodstream, and accurate phenotyping of heterogeneous cell populations.

Moving towards introducing QD technology into clinical diagnostics, five-parameter characterization of breast cancer tissue specimens obtained from biopsies has been demonstrated [23]. Comparison of the three specimens revealed distinct molecular profiles, where one tumor over-expressed such biomarkers as ER and PR, another tumor primarily expressed EGFR, and third tumor showed abundance of ER and HER2 (Fig. 4). Besides diagnostic and prognostic value of such analysis, potential targets for anti-cancer treatment can also be identified, thus enabling a “personalized” approach in therapy.

Accuracy of molecular fingerprinting based on protein expression can be further improved by analysis of gene expression via quantification of mRNA using fluorescent in situ hybridization (FISH). Relying on binding of oligonucleotide probes to complimentary mRNA molecules in 1:1 probe-to-target ratio, this technique offers high level of specificity, yields direct quantitative correlation between gene amplification (i.e. number of mRNA molecules present) and signal intensity, and provides accurate information about mRNA localization within the cell. Similar to protein-based staining, quantitative potential and sensitivity of FISH might be significantly improved by utilization of QD probes [14]. In early proof-of-concept studies Xiao and Barker have used highly stable QD-Streptavidin bioconjugates for monochromatic visualization of biotinylated oligonucleotide probes in FISH analysis of amplification of clinically important erbB2 gene [26]. Using a slightly modified procedure, Tholouli et al have achieved multiplexed staining of 3 mRNA targets within one specimen [27]. In order to reduce the size of imaging probe and improve binding stoichiometry, Chan et al have developed a monovalent FISH probe by blocking extra streptavidin sites with biocytin (water-soluble biotin derivative) [28]. High-resolution multiplexed FISH has been demonstrated in simultaneous detection of four mRNA targets using two different QD probes and two different organic fluorophore probes within a single mouse midbrain neuron (Fig. 5). Notably, reduced size of FISH probes enabled staining in milder, protein-compatible specimen permeabilization conditions, which is essential for combined QD-based FISH and QD-based immunohistochemistry (IHC), thus offering the possibility of correlating gene expression at the mRNA level with the number of corresponding protein copies in diseased cells or tissue specimens [14].

While molecular fingerprinting of diseases holds tremendous diagnostic and therapeutic value, uncovering intracellular pathways leading to disorder is essential for understanding the patho-physiology of a disease, identification of an underlying cause of the pathologic changes, and design of therapies targeting dysfunctional pathways on a molecular level. Study of patho-physiology on sub-cellular level involves the characterization of intracellular distribution and relative expression of biomarkers (proteins, mRNA, etc.), analysis of phenotypic changes in cells upon certain stimulation, and real-time monitoring of changes in intracellular processes (e.g. phagocytosis, intracellular trafficking, and cell motility) in live cells.
One interesting study of intracellular morphology was demonstrated by Matsuno et al who combined QD-based FISH and IHC along with confocal laser scanning microscopy for three-dimensional imaging of the intracellular localization of growth hormone (GH), prolactin (PRL), and of their mRNAs within tissue specimens [29]. With further improvements in design of QD probes suitable for multiplexed FISH and IHC, this technology will allow three-dimensional mapping of the relative position of biomarkers and corresponding mRNAs inside cells and tissues with high resolution and sensitivity, thus providing access to studies of intricate signaling pathways and mechanisms of pathogenesis.
Further improvement in imaging resolution can be achieved by utilization of transmission electron microscopy (TEM). For example, relatively high electron density of QDs was successfully employed by Giepmans et al for high-resolution study of intracellular biomarker distribution [30]. In this study initial optimization of staining conditions was achieved using fluorescence imaging, while further examination with TEM revealed intracellular localization of QD probes (and corresponding biomarkers) with respect to sub-cellular structures. Due to direct correlation between fluorescence emission color and QD size, detection of three QD-labeled biomarkers distinguishable at both fluorescence (by color) and TEM (by size) levels was achieved [30]. Enhancement in multiplexing functionality of this technique can be obtained from discrimination of QDs based on their elemental composition. Nisman et al have proposed the use of electron spectroscopic imaging (ESI, a technique for generating elemental maps of materials with high resolution and detection sensitivity) for mapping the distribution of QDs in cells and tissues based on QD internal chemistry in addition to discriminating probes by size [31].
Monitoring of intracellular processes in live cells, although more difficult and less flexible in terms of multiplexing, provides information about dynamics of cellular functioning and real-time cellular response to applied stimuli. Design of biocompatible coatings and unprecedented photostability render QDs well-suited for this task, as long exposure to excitation source and constant signal intensity are often not achievable with conventional techniques. The relatively large size of QD probes creates a barrier for intracellular targeting, yet biomarkers expressed on the cell membrane are readily accessible. As a result the majority of reports on real-time tracking describe dynamics of membrane proteins rather than intracellular targets. For example, Lidke et al used QDs conjugated to epidermal growth factor (EGF) to study erbB/HER receptor-mediated cellular response to EGF in living human epidermoid carcinoma A431 cells, assigning the mechanism of EGF-induced signaling to heterodimerization of erbB1 and erbB2 monomers and uncovering retrograde transport of endocytosed QD probes (Fig. 6) [32]. Murcia et al utilized QD-lipid bioconjugates for high-speed tracking of single-probe movement on cell surface and accurate measurement of diffusion coefficient [33], while Roullier et al labeled two subunits of type I interferon receptor with QD probes and monitored diffusion and interaction of these subunits in real-time [34].

One highly informative method of intracellular tracking involves endocytosis of QD probes with consequent monitoring of endosome dynamics. Cui et al utilized pseudo-TIRF (total internal reflection fluorescence) microscopy for long-term real-time tracking of intracellular transport of QD-labeled nerve growth factor (NGF) along axons of rat dorsal root ganglion neurons and described the dynamics of axonal internalization and neuronal retrograde transport of QD-NGF [35]. In another example, Zhang et al induced single QD uptake into synaptic vesicles and monitored fluorescence of each QD probe to discriminate between complete vesicle fusion (full-collapse fusion) and transient fusion (so-called kiss-and-run behavior), thus characterizing dynamics of neuronal transmission with respect to time and frequency of impulse firing [36].
The challenge that is yet to be overcome is labeling of intracellular components in live cells. Integrity of cellular membrane and crowded intracellular environment have proven to be an obstacle for QD entry into live cells. While endosomal uptake of bare QDs is readily achievable, escape from endosomal compartments and labeling of specific components is challenging. Further, elimination of unbound probes from intracellular compartments to avoid false positive detection is often hampered, because, unlike fixed cells, unbound nanoparticles cannot be washed away. Recently a few reports on delivery of nanoparticles within live cells have been published. In mechano-chemical approach, Yum et al utilized gold-coated boron nitride nanotubes (with a diameter of 50 nm) to deliver QDs within the cytoplasm or nucleus of live HeLa cells with consequent 30-minute monitoring of QD diffusion within those compartments (Fig. 7A) [37]. Park et alengineered arrays of vertically aligned carbon nanosyringes for intracellular delivery of QDs and therapeutic agents (Fig. 7B) [38]. While efficiently delivering nanoparticles within cells, both techniques are quite labor-intensive and low-throughput. Design of nanoparticles capable of escaping endosomes or entering cells without inducing endocytosis remains the most promising approach for intracellular delivery [39–42]. For example, Kim et al encapsulated multiple QDs within the biodegradable polymer poly(D,L-lactide-co-glycolide) (PLGA) that induced cellular uptake, endosomal escape, and release of QD load within the cytoplasm (Fig. 7C), providing efficient high-throughput method for intracellular delivery of multicolor QDs and enabling multiplexed staining within live cells [40]. In a single-particle approach, Qi and Gao coated individual QDs with a pH-responsive amphyphilic polymer [42]. Besides achieving efficient cellular uptake and endosomal escape facilitated by a proton sponge effect, polymer-coated QDs allowed delivery of intact siRNA inside the cells and monitoring of siRNA release within the cytoplasm.

delivery-of-qd-probes-inside-cells-represents-a-challenge-for-labeling-intracellular-target-nihms137729f7
In vivo imaging of diseased cells and tissues provides many benefits for personalized medicine, including high-throughput screening and potential for diagnosis at early stages of disease, obtaining patient-specific information about the localization and size of the disease core, assessment of adverse effects on healthy tissues, and monitoring of disease progression and response to therapy. Therefore, non-invasive in vivoimaging represents one of the major goals of current biomedical research. Conventional medical imaging techniques, such as ultrasound imaging, magnetic resonance imaging (MRI), and positron emission tomography (PET), in most cases do not offer sensitivity and resolution simultaneously for early-stage diagnosis (e.g. MRI provides high resolution, yet poor sensitivity; while PET offers high sensitivity with low resolution) as well as specificity for conveying disease molecular information.
Fluorescence microscopy remains the most potent technique for molecular profiling of diseased cells. However, presence of tissue barriers between disease sites and imaging equipment complicates the utilization of fluorescence microscopy for in vivo imaging, as biological tissues efficiently absorb and scatter visible light along with producing intense autofluorescence over a broad spectrum. Unlike organic fluorophores, QDs possess high brightness and multiplexing capabilities along with large Stokes shifts, thus representing a promising tool for in vivo molecular imaging and profiling [16, 22, 29–31, 43–45]. In particular, the spectral gap between excitation and emission for QDs is significantly larger than that of organic fluorophores and can be as large as 300–400 nm, depending on the wavelength of the excitation light [46, 47], thus moving QD signal into a region with reduced tissue autofluorescence. For example, in early studies Akerman et aldemonstrated targeted imaging of tumor vasculature using QD-peptide bioconjugates [48]. However, utilization of green and red QDs limited deep-tissue imaging in live animals, and post-mortem histological examination of tissue specimens was used to evaluate QD biodistribution. Taking advantage of large stokes shift, Gao et al have demonstrated the utility of PEG-coated red QDs (emission peak around 640 nm) conjugated to antibodies against prostate-specific membrane antigen (PSMA) for in vivo tumor imaging in mice [49]. Further signal processing with spectral unmixing algorithm allowed clear separation of QD signal from the background fluorescence (Fig. 8).

Utilization of near-infrared imaging probes might further reduce interference from tissue autofluorescence and enable in vivo imaging with deeper penetration and better resolution. Modeling studies performed by Limet al have identified two spectral windows in far-red (700–900 nm) and infrared (1200–1600 nm) regions suitable for nearly background-free deep-tissue imaging [50]. Kim et al utilized model predictions on practice in mapping sentinel lymph nodes (SLN) with NIR QDs, providing accurate identification and image-guided resection of SLN – an indispensable tool in surgical treatment of metastatic cancers [51]. Targeted in vivoimaging of human glioblastoma vasculature in mouse model was demonstrated by Cai et al, who used NIR CdTe/ZnS QDs conjugated to targeting peptide against integrin αvβ3, which is significantly up-regulated in tumors [52]. Recently, Diagaradjane et al reported on in vivo imaging and quantitative analysis of EGFR with NIR QDs (emission peak at 800 nm), showing QD capability to distinguish EGFR over-expression in tumor site compared to normal expression levels in surrounding tissues [53]. Meanwhile Shi et al used NIR QDs to achieve a deep-tissue high-sensitivity detection of prostate cancer xenografts grown in mouse tibia [54].
An alternative NIR QD probe was developed by So et al, who conjugated luciferase to QD surface to yield self-illuminating fluorescent probes via bioluminescence resonance energy transfer process (Fig. 9) [55]. By making external excitation unnecessary, bioluminescent QDs completely eliminated the tissue autofluorescence and provided higher sensitivity of detection. Increased size of luciferase-QD bioconjugates and requirement for supplying the substrate coelenterazine put certain limitations on in vivo imaging applications. Therefore, development of compact self-illuminating QDs utilizing naturally occurring biomolecules as a substrate might further advance this technology and provide high-sensitivity in vivoimaging probes.

shallow-depth-of-in-vivo-imaging-with-qd-probes-imposes-significant-limits-nihms137729f9
Two-photon microscopy represents another promising alternative to standard in vivo fluorescence imaging. Despite some technical limitations, two-photon microscopy represents a powerful tool for multiplexed and highly sensitive in vivo imaging. This technique uses low-energy photons (in red and infrared regions) for excitation of QDs emitting in visible range, achieving dramatically reduced attenuation of excitation light by tissues along with reducing the autofluorescense, while allowing utilization of QDs emitting over a full visible spectrum. Moreover, the high two-photon cross-section of QDs enables deeper-tissue imaging with higher sensitivity. The first study of QD-based multiphoton fluorescence in vivo imaging was reported by Larson et al, when green CdSe/ZnS QDs were used for imaging of capillaries under the dermis layer of skin [56]. Levene et al have combined needle-like gradient index lens imaging setup together with multiphoton microscopy to obtain high-resolution microangiographs of deep brain blood vessels labeled with QDs [57]. In a recent in vivo study of tumor morphology Stroh et al utilized two-photon microscopy for simultaneous imaging of tumor vessels (stained with blue QDs) and perivascular cells (expressing GFP) [58]. Further incorporation of second harmonic generation signal emanating from collagen provided information about the distribution and morphology of extracellular matrix (Fig. 10).

Overall, NIR QDs have already proven to be a great tool for characterization of disease models in small animals and post-mortem evaluation of tissue specimens. Moving towards in vivo imaging in human subjects, mapping of lymph nodes and image-guided resection of tumors represent most promising clinical applications of QD probes. Additionally, recent reports on decorating QD probes with TAT peptide [59] and wheat germ agglutinin [60] for improving QD transport over a blood-brain barrier and targeting cells of the central nervous system might enable highly accurate and conservative image-guided surgeries on brain tissue.
Targeted and traceable drug delivery
Accurate identification of key molecular targets distinguishing diseased cells from healthy ones enables targeted drug delivery with minimal side-effects. Nanoparticle-based drug carriers show great potential for efficient targeted delivery applications, as they can provide sufficiently long blood circulation, protect the cargo from degradation, possess large drug loading capacity and controlled drug release profile, and integrate multiple targeting ligands on their surface. Additionally, QD probes provide unique functionality of traceable drug delivery, as biodistribution of carriers and intracellular uptake can be monitored via fluorescence.
Several drug delivery applications employing tracing functionality of QDs have been developed recently. For example, Chen et al co-transfected QDs and siRNA using Lipofectamine 2000 and monitored transfection efficiency via QD fluorescence [61]. Mixing QDs with transfection reagent in 1:1 mass ratio provided correlation between the QD signal intensity and the degree of gene silencing. Such an approach enabled the collection of uniformly silenced cell population by fluorescence-activated cell sorting, while introducing minimal modifications into standard siRNA transfection protocol and requiring no chemical modifications of siRNA. Interestingly, additional co-transfection of different siRNA molecules with different QD colors might allow multiplexed monitoring of gene silencing. Yet, indirect link between siRNA and QD transfection imposes certain limitations on this technology, as there is a possibility of interference between QD probes and siRNA transfection resulting in inaccurate correlation of fluorescence signal with the degree of gene silencing. More reliable quantitative information about the number of siRNA molecules delivered into cells can be achieved by using QD-doped chitosan nanobeads developed by Tan et al [62]. In such an approach siRNA molecules are deposited on the surface of nanobeads, and intracellular delivery can be directly monitored by the nanobead fluorescence. Further improvement can be gained from a direct labeling approach demonstrated by Jia et al, who used carbon nanotubes for intracellular delivery of antisense oligonucleotides tagged with QDs [63]. This technology might not only enable a reliable method of quantification of intracellular oligonucleotide concentration, but also provide spatial information about the localization of oligonucleotides within the cell. For example, direct labeling of plasmid DNA with QDs followed by Lipofectamine-mediated transfection enabled long-term study of intracellular and intranuclear localization and transport of plasmid DNA, while preserving the ability of expressing reporter protein encoded by the plasmid [64].
Initial success of highly efficient and traceable intracellular drug delivery utilizing supplementary transfection reagents inspired the design of compact single-QD based carriers with integrated functionalities. Utilization of single-QD drug delivery vehicles for in vivo applications is desirable, as intermediate size of such carriers (~10–20 nm in diameter) reduces the renal clearance as well as uptake by reticulo-endothelial system (RES), thus increasing the blood circulation time and improving the delivery efficiency. Further, QD core can serve as a structural scaffold for loading of various types of drug molecules. For example, small-molecule hydrophobic drugs can be embedded between the inorganic core and the amphiphilic polymer coating layer [65], while hydrophilic therapeutic agents (such as siRNA and antisense oligonucleotides) can be deposited onto the hydrophilic exterior of the polymeric shell (Fig. 11) [41]. Flexibility of the shell design enables engineering of drug carriers with different physical properties (e.g. size, charge, biodegradability, etc), thus providing a large platform for a variety of specific applications.

Integration of functionality for enhanced cellular uptake and endosomal escape within single-QD probes was demonstrated by Qi and Gao [42]. Encapsulation of QDs with zwitterionic amphipols enabled non-covalent deposition of up to 10 siRNA molecules on the surface of each particle via electrostatic interaction. Efficient endosomal uptake of such particles followed by decrease in pH and shift in particle surface charge resulted in endosomal escape and release of intact siRNA within the cells. While outperforming transfection efficiency of common reagents (such as PEI and Lipofectamine), QD carriers showed substantially lower toxicity in cell cultures. Further, real-time monitoring of particle uptake (via QD fluorescence) and release of siRNA (via labeling of individual siRNA molecules with FITC) was achieved. Targeted siRNA transfection to tumor cells was demonstrated by Defrus et al, who used PEG-coated QDs as a platform for deposition of siRNA and tumor-homing peptide [66]. Attachment of siRNA molecules via cleavable chemical bonds ensured efficient intracellular release of active siRNAs. However, deposition of targeting ligands and cargo molecules in a “parallel” manner introduced competition between the loading capacity and targeting capabilities of the delivery vehicles. In light of successful RNAi experiments with aptamer-siRNA chimeras performed by McNamara et al [67] it is reasonable to expect higher efficiency from vehicles with “serial” attachment of therapeutic molecule and targeting ligand. For small-molecule drug delivery, Bagalkot et al functionalized the QDs with targeting RNA aptamers and loaded anti-cancer drug Doxorubicin via intercalation within the aptamer [68]. Notably, bi-FRET from QD core to Doxorubicin and then to aptamer enabled monitoring of the vehicle disintegration and drug release within the cells via restoration of QD fluorescence.
In vivo drug delivery with QD carriers was demonstrated by Manabe et al [69]. Conjugation of an antihypertensive drug captopril to the QD surface provided the therapeutic effect similar to that of the free drug, while also enabling the monitoring of QD-drug biodistribution over a 96-hour period. With advancements in design of biocompatible QD surface coatings and identification of suitable molecular targets for therapy, QD-based drug delivery vehicles promise to provide an indispensable tool for modeling of pharmacokinetics and pharmacodynamics of nanoparticle-drug carriers.
Challenges of integrating QD technology into clinical practice
Nanotechnology represents a highly dynamic field of research developing novel platforms for a variety of applications. Unique properties of nanomaterials inspire enthusiasm for overcoming limitations of current technology and hold promise of advancing the field of personalized medicine. An increasing number of proof-of-concept studies along with more applied and clinically relevant QD-based tools appearing in a variety of fields ranging from ex vivo molecular fingerprinting of individual cells to in vivo diagnostics and image-guided therapy will undoubtedly make their way into clinical practice. However, there are still a number of challenges on the way of integrating QD technology into biomedical applications.
Unique behavior of nanomaterials compared with small molecules and lack of clinical experience of utilizing nanoparticle-based assays often raise concerns of result reproducibility, reliability, and comparability between each other and conventional techniques. However, increasing numbers of proof-of-concept studies are actively exploring a wide range of possible areas of QD applications. A forthcoming leap towards technologies working in clinical settings along with wide-scale “test-drives” of QD tools and training of technical personnel should encourage interest in QD-based tools, increase familiarity and hands-on experience working with QD probes, and establish confidence in this technology within scientific and medical communities. Among first steps towards this goal, standardization of QD-based assays will be beneficial for making data from different research centers comparable and enabling large-scale clinical studies.
Increasing efforts are focused on the study of the effect of QDs on human health and environment. Partially due to the novelty of nanotechnology, there is not much information about these effects available yet. Short-term and long-term toxicity and immunogenicity of nanoparticles as well as disposal of nanoparticle-containing waste remain a highly debatable area of research and deserve thorough investigation to ensure safety of QD technology in clinical practice [70–72]. While early studies of QD toxicity by Derfus et alindicated significant cytotoxicity of unprotected CdSe QDs due to nanoparticle photo-oxidation upon exposure to UV light and release of toxic Cd2+ ions [73, 74], capping of CdSe core with ZnS layer and deposition of a stable coating seemed to dramatically reduce QD toxicity in cell cultures. In a more adequate model based on 3D cell culture (liver tissue spheroids) Lee et al observed substantially decreased nanoparticle-induced toxicity compared to 2D cell cultures, emphasizing the impact of tissue morphology on toxicity [75]. Sometimes conflicting toxicity data might also result from significant over- or under-estimation of cell toxicity determined with standard cell viability assays [76]. In addition to in vitro assays, greater complexity of live organisms with plethora of mechanisms for QD accumulation, degradation, and excretion might require more thorough in vivo toxicity studies. For example, Mancini et al suggested that hypochlorous acid together with hydrogen peroxide produced by phagocytes can diffuse through an otherwise stable secondary coating, causing solubilization of the QD core and release of toxic ions [77]. Additionally, the QD surface coating and particle size play important role in the particle biodistribution and toxicity [71, 78, 79]. Pharmacokinetics studies performed on rat models by Fischer et al have shown that QDs coated with bovine serum albumin (BSA) are efficiently eliminated from the bloodstream by liver uptake, while QDs lacking BSA on their surface are cleared at slower rate [80]. As each QD probe appears to be unique, development of comprehensive assays for QD toxicity assessment should improve our understanding of potential risks of this technology, provide guidance for design of QD probes with minimized adverse effects, and increase the public confidence in QD-based diagnostics and therapeutics.
As promising benefits of QD technology might be hampered by potential adverse effects, design of biocompatible and non-toxic QD probes has become an active area of research. One way of resolving an issue of heavy metal toxicity involves utilization of QD probes made of benign materials. For example, Yonget al recently prepared Cd-free InP/ZnS QDs and utilized these probes for targeting of pancreatic cancer cell lines [81]; however, low quantum yield (~30%) and large size (~30 nm in diameter) might limit utility of such probes for in vivo imaging. Higher-quality probes with quantum yield of up to ~60% and hydrodynamic diameter of 17 nm were developed by Li et al on the basis of CuInS2/ZnS QDs [82]. Further, engineering of low-cost, non-toxic, and potentially biodegradable in vivo imaging probes might become available through utilization of recently developed technology for preparation of water-soluble QDs made of silicon [83, 84] – intert, biocompatible, and abundant material.
While being an attractive approach, Cd-free QDs still suffer from poor stability and inferior photo-physical properties compared to high-quality QDs made of toxic materials (such as CdSe). Therefore, improving biocompatibility of potentially toxic QD probes remains a sound and highly promising alternative, and elimination or reduction of cadmium interaction with live cells seem to be the cornerstone of such approach. There are several feasible strategies to achieving this goal. The toxicity associated with cadmium poisoning comes from a quick release of large amounts of this metal into a bloodstream, its preferential accumulation in kidneys, and consequent nephrotoxicity. However, up to 30 ug/day of dietary Cd (coming from fish, vegetables, and other sources) can be consumed by a healthy adult without adverse effects on kidney function [85]. Therefore, slow degradation of QD probes within a human body followed by urinary excretion might offer a way of safe and efficient elimination of QDs. Adapting technology developed for controlled drug release and coating nanoparticles with biodegradable polymers might provide one strategy for gaining control over QD degradation and Cd release in vivo.
Complete and quick elimination of intact QD probes from the body via renal excretion represents another approach to overcoming possible toxicity. This approach seems especially favorable in light of sparse information on in vivo QD degradation mechanisms and long-term effect of QD accumulation in organs. Systematic investigation of the renal clearance of QDs on rat and mice models done by Choi et al has defined the renal clearance threshold of 5.5 nm and emphasized the role of zwitterionic surface coatings in preventing protein absorption and retaining the original nanoparticles size [79]. Working along this direction, Law et alprepared ultrasmall (3–5 nm in diameter) cysteine-coated CdTe/ZnTe QDs and tested biodistribution of these probes in mice, finding no QDs in liver and spleen 2 weeks post-injection [86]. However, bio-functionalization of QDs, required for targeted imaging and therapy, increases the QD size, thus making renal clearance of functional QD probes difficult. Further, quick renal clearance is often undesirable, as prolonged QD circulation is required for specific targeting, high-sensitivity imaging, and therapeutic efficiency. Therefore, high molecular weight coatings are routinely applied to QD probes to increase their circulation time and improve bioavailability. Ballou et al emphasized the importance of coating with high molecular weight PEG to reduce accumulation of QDs in liver and bone marrow [87], and Prencipe et alachieved remarkably long blood circulation of nanomaterials encapsulated with branched PEG [88]. Utilization of biodegradable ligands that would detach from QD probes after prolonged circulation in blood or due to degradation in target cells, thus releasing single nanoparticles with original size below 5.5 nm, might render renal excretion of functional QD probes feasible.
In some cases complete elimination of QD probes from the body via renal excretion or other means might prove challenging or undesirable. Engineering of ultra-stable QDs encapsulated with inert biocompatible materials might provide an alternative strategy for addressing Cd toxicity issue. If QD integrity within a human body can be retained for many years, biological systems might never be exposed to heavy metal components of the QD core. For example, Ballou et al indicating that intact PEG-coated QDs remained in bone marrow and lymph nodes of mice for several months after injection [87]. While organic coatings, such as polymers and lipids, might still degrade due to exposure to biological environment, utilization of more stable inorganic materials should protect the cores of QD probes for extended periods of time.
Advancement of personalized medicine is essential for making progress towards combating such complex diseases as cancer and immune system disorders, and incorporation of novel QD-based tools will undoubtedly play a major role in this process. Design of compact, stable, and biocompatible coatings functionalized with targeting agents have already converted QDs into multifunctional nanodevices suitable for in vitro as well as in vivo applications. While certain challenges and concerns regarding QD incorporation into clinical practice remain, and cautiously enthusiastic attitude towards QD-based tools prevails in scientific community, the benefits of this technology will ensure the increasing interest in QDs as more practical and clinically relevant applications are demonstrated and comprehensive toxicity data is made available. With further advances in design and engineering of biocompatible QD probes such applications as image-guided surgery, molecular fingerprinting of diseases, and personalized diagnosis and therapy will become widely available.
7.1.6 Potentials and pitfalls of fluorescent quantum dots for biological imaging
Jaiswal JK, Simon SM.
Trends Cell Biol. 2004 Sep; 14(9):497-504.
http://dx.doi.org/10.1016/j.tcb.2004.07.012
Fluorescent semiconductor nanocrystals, known as quantum dots (QDs), have several unique optical and chemical features. These features make them desirable fluorescent tags for cell and developmental biological applications that require long-term, multi-target and highly sensitive imaging. The improved synthesis of water-stable QDs, the development of approaches to label cells efficiently with QDs, and improvements in conjugating QDs to specific biomolecules have triggered the recent explosion in their use in biological imaging. Although there have been many successes in using QDs for biological applications, limitations remain that must be overcome before these powerful tools can be used routinely by biologists.
Glossary
Fluorescence blinking: a property of a single fluorophore to transit between a fluorescent (on) and non-fluorescent (off) phase, which is caused by its transition between a singlet (fluorescent) and a triplet (non fluorescent) state. Blinking occurs in quantum dots because a specific process causes them to switch between their ionized and neutralized states.
Multiphoton microscopy: a process in which more than one photon, each with a fraction of the energy needed to excite fluorescent molecules, is simultaneously absorbed by the fluorophore, resulting in fluoresce emission. This process facilitates the use of infrared light (which, owing to its longer wavelength, penetrates deeper into the tissue) for animal imaging.
Quantum yield: the ratio of photons absorbed to photons emitted by a fluorescent molecule. The quantum yield quantifies the probability that a molecule in an excited state will relax by emitting fluorescence rather than by decaying non-radiatively.
Semiconductor: a material that is an insulator at very low temperature but has considerable electrical conductivity at room temperature. Stoke’s shift: the separation in energy (and thus wavelength) between the excitation and emission spectra.
Box 1. History of biocompatible quantum dots
Ekimov and Onuschenko [46] carried out the first controlled synthesis of semiconductor crystals of nanometer size by heating glass containers with supersaturated solutions of copper and chlorine compounds at high temperatures to cause the controlled precipitation of copper chloride (CuCl). They used additional heating to create, systematically, collections of small crystalline CuCl particles ranging from tens to hundreds of A ˚ ngstroms, which were initially called quantum droplets and later given other names including nanoparticles, nanocrystals, nanocrystallites and Q-dots. This approach provided particles that remained trapped in the glass and thus could not be easily manipulated after synthesis. In 1993, Bawendi’s group [47] developed an approach for quantum dot (QD) synthesis that facilitated the production of high-quality (see Ref. [2]) monodisperse nanoparticle QDs. Their approach allowed the synthesis of QDs that could be dispersed in various solvents and whose surface could be derivatized. These QDs still had poor fluorescence quantum yields (w10%). A subsequent approach led to the large-scale synthesis of more uniform and monodisperse QDs with higher quantum yields (O20%) [48]. It was, however, the approach of coating the QDs with a few layers of zinc sulfide (ZnS) that provided the greatest enhancement of quantum yield (Figure 1a) [3,49]. Because ZnS-coated QDs are hydrophobic, several methods have been used to stabilize them in aqueous solution and to facilitate their conjugation to biomolecules to make them useful for biological imaging. These include (i) embedding them in a silica or siloxane shell with a thickness of 1–5 nm and with amine, thiol or carboxyl functional groups on its surface [17,50]; (ii) derivatizing their surface with mercaptoacetic acid [18]; (iii) encapsulating them in phospholipid micelles [16]; (iv) derivatizing their surface with dihydroxylipoic acid [2]; and (v) coating them with an amine-modified polyacrylic acid [13].

http://ars.els-cdn.com/content/image/1-s2.0-S0962892404001916-gr1.sml
Figure 1. Properties of bioconjugatable quantum dots (QDs). (a) QDs are inorganic fluorophores and consist of a cadmium selenide (CdSe) core with several layers of a thick zinc sulfide (ZnS) shell to improve quantum yield and photostability. (b) The excitation spectrum (broken lines) of a QD (green) is very broad, whereas that of an organic dye (rhodamine, orange) is narrow. The emission spectrum (unbroken lines) is narrower for a QD (green) than for organic dyes (rhodamine, orange). Values indicate the full spectral width at half-maximum intensity (FWHM value). (c) The emission of the QDs can be tuned by controlling the size of the CdSe core: an increase in the size of the core shifts the emission to the red end of the spectrum. The combined size of the core and the shell of QDs emitting in the visible region of spectra are in the size range of commonly used fluorescent proteins such as green fluorescent protein (GFP) and DsRed. (d,e) To provide specificity of binding, QDs are conjugated with antibody molecules (blue) by using avidin (purple) or protein A (green) as linkers. Between 10 and 15 linker molecules can be attached covalently or electrostatically to a single QD, which facilitates the binding of many or a few (note the presence offree linker molecules) antibody molecules on each QD. Note that, although the QDs and molecules are drawn to size, their binding sites and relative topologies are shown hypothetically
http://ars.els-cdn.com/content/image/1-s2.0-S0962892404001916-gr2.sml
Figure 2. Specific labeling of live cells with quantum dots (QDs). (a) Positively charged avidin and maltose-binding protein containing a positively charged tail (MBPzb) selfassemble on the negatively charged surface of QDs capped with dihydrolipoic acid (DHLA) and can bind to biotinylated molecules such as antibodies specific for Pgp. (b) Transient transfection of HeLa cells with Pgp–GFP (green fluorescent protein) results in its expression in a subset of cells (not marked with arrows). The subsequent incubation of all cells with biotin-conjugated antibodies specific for Pgp, followed by avidin-conjugated QDs, leads to labeling of the cell membrane with the QD bioconjugates: only cells that express detectable levels of Pgp–GFP, and not those that do not express Pgp–GFP (marked with arrows), are labeled [48]. Yellow coloring in the fluorescence image indicates an overlap of green (Pgp–GFP) and red (QD bioconjugate) fluorescence emission. (See Ref. [2] for further details).
Box 2. Specific labeling of biomolecules in vitro
Quantum dots (QDs) have been used to tag molecules of interest both selectively and stably. One approach involves capping the surface of QDs with dihydroxylipoic acid (DHLA), which makes the QD surface negatively charged [2]; this enables QDs to bind to linker molecules, such as protein G engineered to carry a positively charged tail (PGzb) or avidin, which is innately positively charged. These linker molecules provide the specificity to bind the molecule of interest through interactions between either PGzb and antibody or avidin andbiotin (Figure2a). Such QDbioconjugateshave beenused to detect simultaneously as little as 10K9 g of single or multiple toxins and small molecules in vitro [6,20]. Specific biomolecules can be detected despite an excess of other nonspecific biomolecules; the specificity is limited only by the specificity of the antibody used [6]. Collectively, theseresults haveprovedthatQDscanbe conjugatedto biomolecules without compromising their biological activity. Because QDs are brighter than most conventional fluorophores, their use should increase the sensitivity of all fluorescence-based assays. In addition, QDs have been shown to be inert when conjugated via other approaches and when used to detect other molecules such as protein ligands [11,51]. For example, QDs have found a major application in the area of nucleic acid detection [52–54], where QD-tagged probes are being used for the simultaneous detection of multiple nucleic acids [52,53]. The ability to identify simultaneously (not sequentially) and specifically different molecules in a single solution significantly expedites high-throughput chemical screening and holds the potential to revolutionize microarray-based approaches for large-scale studies of the gene expression profiles of organisms.
Box 3. How to get quantum dots into cells
Owing to their size and chemical nature, quantum dots (QDs) cannot diffuse through the cell membrane. To use QDs for labeling and imaging cytoplasmic proteins, the QDs must be delivered by invasive approaches such as microinjection [16], cationic lipidbased reagents [7] or conjugation to membrane-permeable peptides [30]. However, these approaches can cause the intracellular QDs to aggregate in punctae or to end up in endosomes [26,55], instead of being dispersed in the cytosol. Crucial challenges to using QDs for intracellular imaging are (i) the development of non-invasive approaches for the efficient intracellular deliveryanddispersalofQDs;(ii)thedevelopmentofmethodstolabel intracellularproteinsthatarelocatedinanenvironmentvastlydifferent from the extracellular space; and (iii) the development of QDs that eitherareinerttothecytoplasmicenvironmentorrespondinadefined manner to selective changes of the cytoplasmic environment.
![Trageting the Wnt Pathway [7.11]](https://i0.wp.com/pharmaceuticalintelligence.com/wp-content/uploads/2020/08/trageting-the-wnt-pathway-7.11-1.png?resize=500%2C307&ssl=1)





















































































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
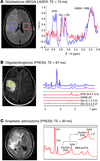{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Would be good to talk about how different Wnt isoforms activate either the noncanonical or canonical pathways in different cancer types. Different Wnts have different specificities for different tissues and activate different pathways. For reference see work by Rugang Zhang in ovarian.
In addition it would be good to find literature on why, after nearly a decade, drug development strategies against the Wnt pathway, have never come to fruition, much like the early days of the farnesylation inhibitors. I believe the early inhibitors were too toxic. In addition, in relation to the OMP trial, Wnt inhibitors target the cancer stem cell and results showing a few months benefit in survival.
good review on current status of Wnt inhibitor development
http://link.springer.com/chapter/10.1007/978-1-4419-8023-6_9
also this is a reference which should be linked to great article by Emma Hill
http://www.onclive.com/publications/oncology-live/2012/december-2012/wnt-signaling-inhibition-will-decades-of-effort-be-fruitful-at-last/2
shows all the trials and tribulations of a decade worth of effort.