Healthcare analytics, AI solutions for biological big data, providing an AI platform for the biotech, life sciences, medical and pharmaceutical industries, as well as for related technological approaches, i.e., curation and text analysis with machine learning and other activities related to AI applications to these industries.
Crowdsourcing Genetic Data Yields Discovery of DNA loci associated with Major Depressive Disorder (MDD) in European Descendants, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 1: Next Generation Sequencing (NGS)
Crowdsourcing Genetic Data Yields Discovery of DNA loci associated with Major Depressive Disorder (MDD) in European Descendants
Reporter: Kelly Perlman, Life Sciences Student and Research Assistant, McGill University
UPDATED on 11/24/2019
Can AI help diagnose depression? It’s a long shot
At the moment, machine intelligence is just as subjective as human intelligence
Researchers from Pfizer Global Research and Development, 23andMe, and the Massachusetts General Hospital have published a study in Nature Genetics, pinpointing 15 genetic loci associated with the risk of developing major depressive disorder (MDD) in individuals of European ancestry. Evidence from previous research suggests that MDD is heritable, but the details of the specific gene correlates are unclear. The identification of loci where single nucleotide polymorphisms (SNPs) related to MDD exist could provide better insight into the neurobiology of depression, and therefore better treatment options.
23andMe, a private biotechnology company situated in California, offers a DNA sequencing service in which consumers send in a saliva swab for testing, and later receive a report listing the findings of the analysis related to ancestry, physical and behavioral traits, along with risk of inheriting certain diseases. The participants of this study had agreed to provide the results of their genetic testing for scientific research.
The results of 75,607 participants with self-reported diagnoses of depression were compared to the results of 231,747 participants reporting having never experienced depression. This data was combined with the results of previously published MDD genome-wide association studies (GWAS). To test the whether these results could be replicated, another set of results from 23andMe was analyzed, in which there were 45,773 MDD subjects, and 106,354 controls.
After the joint analysis, 17 SNPs were identified at 15 different loci. Tissue and gene enrichment assays showed that the genes that were over-expressed in the CNS were related to functions including neurodevelopment, histone methylation, neurogenesis and synaptic modification.
The team then created a weighted genetic risk score (GRS) in which they compared the 17 SNPs with factors including medication use, comorbid diseases and behavioral phenotypes, all of which were correlated with the GRS. Of note, the GRS was very highly correlated with age of onset of MDD.
The crowdsourcing of genetic data proves to be an efficient and powerful tool for large-scale MDD studies. Pooling large subject databases together is essential in order to account for the heterogeneous nature of the disease. Despite not being able to precisely assess each subject’s disease phenotype, scientists can make more rapid headway by collaborating with biotechnology companies in the quest to better understand the biological mechanisms of depression. Ron Perlis, M.D., M.Sc., of the Massachusetts General Hospital and co-author of this paper explained that “finding genes associated with depression should help make clear that this is a brain disease, which we hope will decrease the stigma still associated with these kinds of illnesses”.
Hyde, C. L., Nagle, M. W., Tian, C., Chen, X., Paciga, S. A., Wendland, J. R., . . . Winslow, A. R. (2016). Identification of 15 genetic loci associated with risk of major depression in individuals of European descent. Nature Genetics Nat Genet. doi:10.1038/ng.3623
Using Online Mendelian Inheritance in Man (OMIM) database and the Human Genome Mutation Database (HGMD) Pro 2015.2 for Quantification of the growth in gene-disease and variant-disease associations, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 1: Next Generation Sequencing (NGS)
Using Online Mendelian Inheritance in Man (OMIM) database and the Human Genome Mutation Database (HGMD) Pro 2015.2 for Quantification of the growth in gene-disease and variant-disease associations
Reporter: Aviva Lev-Ari, PhD, RN
Reanalysis of Clinical Exome Data Over Time Could Yield New Diagnoses
NEW YORK (GenomeWeb) – Clinical exomes that are re-evaluated in a systematic way could yield new diagnoses and prove useful to clinicians, according to a study published yesterday in Genetics in Medicine.
A team of researchers from Stanford University set out to examine whether nondiagnostic clinical exomes could provide new information for patients if they were re-examined with current bioinformatics software and knowledge of disease-related variants as presented in the literature.
Clinical exome sequencing yields no diagnosis for about 75 percent of patients evaluated for possible Mendelian disorders, wrote senior author Gill Bejerano and his colleagues. But a reanalysis of exome and phenotypic data from 40 such individuals using current methods identified a definitive diagnosis for four of them — 10 percent — the team said.
In these cases, the causative variant was de novo and found in a relevant autosomal-dominant disease gene. At the time these exomes were first sequenced, the researchers wrote, the existing literature on these causative genes was either “weak, nonexistent, or not readily located.” When the exomes were re-examined by his team, Bejerano noted, the supporting literature was more robust.
In addition to re-analyzing exome data, the researchers have been working on establishing causality for novel candidate disease genes through patient matches. For this, the team has been using the GeneMatcher website, which allows them to find other clinicians and researchers around the world who have patients, or animal models, with mutations in the same genes as their own patients. Through an API developed by the Matchmaker Exchange project, GeneMatcher submitters can also query the PhenomeCentral and Decipher databases. As of March, more than 4,000 genes had been submitted to GeneMatcher from more than 1,300 submitters in 48 countries, and 1,900 matches had been made, Sobreira reported.
Her team has so far submitted data from 104 families, involving 280 genes, and has had 314 matches so far, involving 113 genes. Several cases have been successes, meaning the researchers could establish that a candidate gene is indeed disease causing, and several others are pending, both from Hopkins and from other groups. The total number of solved cases tracing their success to GeneMatcher is currently unknown, Sobreira said, but the organizers are planning to survey submitters about their success rate in the near future.
mRNA Data Survival Analysis, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 1: Next Generation Sequencing (NGS)
mRNA Data Survival Analysis
Curators: Larry H. Bernstein, MD, FCAP and Aviva Lev-Ari, PhD, RN
SURVIV for survival analysis of mRNA isoform variation
The rapid accumulation of clinical RNA-seq data sets has provided the opportunity to associate mRNA isoform variations to clinical outcomes. Here we report a statistical method SURVIV (Survival analysis of mRNA Isoform Variation), designed for identifying mRNA isoform variation associated with patient survival time. A unique feature and major strength of SURVIV is that it models the measurement uncertainty of mRNA isoform ratio in RNA-seq data. Simulation studies suggest that SURVIV outperforms the conventional Cox regression survival analysis, especially for data sets with modest sequencing depth. We applied SURVIV to TCGA RNA-seq data of invasive ductal carcinoma as well as five additional cancer types. Alternative splicing-based survival predictors consistently outperform gene expression-based survival predictors, and the integration of clinical, gene expression and alternative splicing profiles leads to the best survival prediction. We anticipate that SURVIV will have broad utilities for analysing diverse types of mRNA isoform variation in large-scale clinical RNA-seq projects.
Eukaryotic cells generate remarkable regulatory and functional complexity from a finite set of genes. Production of mRNA isoforms through alternative processing and modification of RNA is essential for generating this complexity. A prevalent mechanism for producing mRNA isoforms is the alternative splicing of precursor mRNA1. Over 95% of the multi-exon human genes undergo alternative splicing2, 3, resulting in an enormous level of plasticity in the regulation of gene function and protein diversity. In the last decade, extensive genomic and functional studies have firmly established the critical role of alternative splicing in cancer4, 5, 6. Alternative splicing is involved in a full spectrum of oncogenic processes including cell proliferation, apoptosis, hypoxia, angiogenesis, immune escape and metastasis7, 8. These cancer-associated alternative splicing patterns are not merely the consequences of disrupted gene regulation in cancer but in numerous instances actively contribute to cancer development and progression. For example, alternative splicing of genes encoding the Bcl-2 family of apoptosis regulators generates both anti-apoptotic and pro-apoptotic protein isoforms9. Alternative splicing of the pyruvate kinase M (PKM) gene has a significant impact on cancer cell metabolism and tumour growth10. A transcriptome-wide switch of the alternative splicing programme during the epithelial–mesenchymal transition plays an important role in cancer cell invasion and metastasis11, 12.
RNA sequencing (RNA-seq) has become a popular and cost-effective technology to study transcriptome regulation and mRNA isoform variation13, 14. As the cost of RNA-seq continues to decline, it has been widely adopted in large-scale clinical transcriptome projects, especially for profiling transcriptome changes in cancer. For example, as of April 2015 The Cancer Genome Atlas (TCGA) consortium had generated RNA-seq data on over 11,000 cancer patient specimens from 34 different cancer types. Within the TCGA data, breast invasive carcinoma (BRCA) has the largest sample size of RNA-seq data covering over 1,000 patients, and clinical information such as survival times, tumour stages and histological subtypes is available for the majority of the BRCA patients15. Moreover, the median follow-up time of BRCA patients is ~400 days, and 25% of the patients have more than 1,200 days of follow-up. Collectively, the large sample size and long follow-up time of the TCGA BRCA data set allow us to correlate genomic and transcriptomic profiles to clinical outcomes and patient survival times.
To date, systematic analyses have been performed to reveal the association between copy number variation, DNA methylation, gene expression and microRNA expression profiles with cancer patient survival16, 17. By contrast, despite the importance of mRNA isoform variation and alternative splicing, there have been limited efforts in transcriptome-wide survival analysis of alternative splicing in cancer patients. Most RNA-seq studies of alternative splicing in cancer transcriptomes focus on identifying ‘cancer-specific’ alternative splicing events by comparing cancer tissues with normal controls (see refs 18, 19, 20, 21, 22, 23 for examples). A recent analysis of TCGA RNA-seq data identified 163 recurrent differential alternative splicing events between cancer and normal tissues of three cancer types, among which five were found to have suggestive survival signals for breast cancer at a nominal P-value cutoff of 0.05 (ref. 21). Some other studies reported a significant survival difference between cancer patient subgroups after stratifying patients with overall mRNA isoform expression profiles24, 25. However, systematic cancer survival analyses of alternative splicing at the individual exon resolution have been lacking. Two main challenges exist for survival analyses of mRNA isoform variation and alternative splicing using RNA-seq data. The first challenge is to account for the estimation uncertainty of mRNA isoform ratios inferred from RNA-seq read counts. The statistical confidence of mRNA isoform ratio estimation depends on the RNA-seq read coverage for the events of interest, with larger read coverage leading to a more reliable estimation14. Modelling the estimation uncertainty of mRNA isoform ratio is an essential component of RNA-seq analyses of alternative splicing, as shown by various statistical algorithms developed for detecting differential alternative splicing from multi-group RNA-seq data14, 26, 27, 28,29. The second challenge, which is a general issue in survival analysis, is to properly model the association of mRNA isoform ratio with survival time, while accounting for missing data in survival time because of censoring, that is, patients still alive at the end of the survival study, whose precise survival time would be uncertain. To date, no algorithm has been developed for survival analyses of mRNA isoform variation that accounts for these sources of uncertainty simultaneously.
Here we introduce SURVIV (Survival analysis of mRNA Isoform Variation), a statistical model for identifying mRNA isoform ratios associated with patient survival times in large-scale cancer RNA-seq data sets. SURVIV models the estimation uncertainty of mRNA isoform ratios in RNA-seq data and tests the survival effects of isoform variation in both censored and uncensored survival data. In simulation studies, SURVIV consistently outperforms the conventional Cox regression survival analysis that ignores the measurement uncertainty of mRNA isoform ratio. We used SURVIV to identify alternatively spliced exons whose exon-inclusion levels significantly correlated with the survival times of invasive ductal carcinoma (IDC) patients from the TCGA breast cancer cohort. Survival-associated alternative splicing events are identified in gene pathways associated with apoptosis, oxidative stress and DNA damage repair. Importantly, we show that alternative splicing-based survival predictors outperform gene expression-based survival predictors in the TCGA IDC RNA-seq data set, as well as in TCGA data of five additional cancer types. Moreover, the integration of clinical information, gene expression and alternative splicing profiles leads to the best prediction of survival time.
SURVIV statistical model
The statistical model of SURVIV assesses the association between mRNA isoform ratio and patient survival time. While the model is generic for many types of alternative isoform variation, here we use the exon-skipping type of alternative splicing to illustrate the model (Fig. 1a). For each alternative exon involved in exon-skipping, we can use the RNA-seq reads mapping to its exon-inclusion or -skipping isoform to estimate its exon-inclusion level (denoted as ψ, or PSI that is Per cent Spliced In14). A key feature of SURVIV is that it models the RNA-seq estimation uncertainty of exon-inclusion level as influenced by the sequencing coverage for the alternative splicing event of interest. This is a critical issue in accurate quantitative analyses of mRNA isoform ratio in large-scale RNA-seq data sets14, 26, 27, 28, 29. Therefore, SURVIV contains two major components: the first to model the association of mRNA isoform ratio with patient survival time across all patients, and the second to model the estimation uncertainty of mRNA isoform ratio in each individual patient (Fig. 1a).
Figure 1: The statistical framework of the SURVIV model.
(a) For each patient k, the patient’s hazard rate λk(t) is associated with the baseline hazard rate λ0(t) and this patient’s exon-inclusion level ψk. The association of exon-inclusion level with patient survival is estimated by the survival coefficient β. The exon-inclusion level ψk is estimated from the read counts for the exon-inclusion isoform ICk and the exon-skipping isoform SCk. The proportion of the inclusion and skipping reads is adjusted by a normalization function f that considers the lengths of the exon-inclusion and -skipping isoforms (see details in Results and Supplementary Methods). (b) A hypothetical example to illustrate the association of exon-inclusion level with patient survival probability over time Sk(t), with the survival coefficient β=−1 and a constant baseline hazard rate λ0(t)=1. In this example, patients with higher exon-inclusion levels have lower hazard rates and higher survival probabilities. (c) The schematic diagram of an exon-skipping event. The exon-inclusion reads ICk are the reads from the upstream splice junction, the alternative exon itself and the downstream splice junction. The exon-skipping reads SCk are the reads from the skipping splice junction that directly connects the upstream exon to the downstream exon.
Briefly, for any individual exon-skipping event, the first component of SURVIV uses a proportional hazards model to establish the relationship between patient k’s exon-inclusion level ψk and hazard rate λk(t).
For each exon, the association between the exon-inclusion level and patient survival time is reflected by the survival coefficient β. A positive β means increased exon inclusion is associated with higher hazard rate and poorer survival, while a negative β means increased exon inclusion is associated with lower hazard rate and better survival. λ0(t) is the baseline hazard rate estimated from the survival data of all patients (see Supplementary Methods for the detailed estimation procedure). A particular patient’s survival probability over time Sk(t) can be calculated from the patient-specific hazard rate λk(t) as . Figure 1b illustrates a simple example with a negative β=−1 and a constant baseline hazard rate λ0(t)=1, where higher exon-inclusion levels are associated with lower hazard rates and higher survival probabilities.
The second component of SURVIV models the exon-inclusion level and its estimation uncertainty in individual patient samples. As illustrated in Fig. 1c, the exon-inclusion level ψk of a given exon in a particular sample can be estimated by the RNA-seq read count specific to the exon inclusion isoform (ICk) and the exon-skipping isoform (SCk). Other types of alternative splicing and mRNA isoform variation can be similarly modelled by this framework29. Given the effective lengths (that is, the number of unique isoform-specific read positions) of the exon-inclusion isoform (lI) and the exon-skipping isoform (lS), the exon-inclusion level ψk can be estimated as . Assuming that the exon-inclusion read count ICk follows a binomial distribution with the total read count nk=ICk+SCk, we have:
The binomial distribution models the estimation uncertainty of ψk as influenced by the total read count nk, in which the parameter pk represents the proportion of reads from the exon-inclusion isoform, given the exon-inclusion level ψk adjusted by a length normalization function f(ψk) based on the effective lengths of the isoforms. The definitions of effective lengths for all basic types of alternative splicing patterns are described in ref. 29.
Distinct from conventional survival analyses in which predictors do not have estimation uncertainty, the predictors in SURVIV are exon-inclusion levels ψk estimated from RNA-seq count data, and the confidence of ψk estimate for a given exon in a particular sample depends on the RNA-seq read coverage. We use the statistical framework of survival measurement error model30 to incorporate the estimation uncertainty of isoform ratio in the proportional hazards model. Using a likelihood ratio test, we test whether the exon-inclusion levels have a significant association with patient survival over the null hypothesis H0:β=0. The false discovery rate (FDR) is estimated using the Benjamini and Hochberg approach31. Details of the parameter estimation and likelihood ratio test in SURVIV are described in Supplementary Methods.
Figure 2: Simulation studies to assess the performance of SURVIV and the importance of modelling the estimation uncertainty of mRNA isoform ratio.
We compared our SURVIV model with Cox regression using point estimates of exon-inclusion levels, which does not consider the estimation uncertainty of the mRNA isoform ratio. (a) To study the effect of RNA-seq depth, we simulated the mean total splice junction read counts equal to 5, 10, 20, 50, 80 and 100 reads. We generated two sets of simulations with and without data-censoring. For each simulation, the true-positive rate (TPR) at 5% false-positive rate is plotted. The inset figure shows the empirical distribution of the mean total splice junction read counts in the TCGA IDC RNA-seq data (x axis in the log10 scale). (b) To faithfully represent the read count distribution in a real data set, we performed another simulation with read counts directly sampled from the TCGA IDC data. Sampled read counts were then multiplied by different factors ranging from 10 to 300% to simulate data sets with different RNA-seq read depth. Continuous and dashed lines represent the performance of SURVIV and Cox regression, respectively. Red lines represent the area under curve (AUC) of the ROC curve (TPR versus false-positive rate plot). Black lines represent the TPR at 5% false-positive rate.
Using these simulated data, we compared SURVIV with Cox regression in two settings, without or with censoring of the survival time. In the setting without censoring, the death and survival time of each individual is known. In the setting with censoring, certain individuals are still alive at the end of the survival study. Consequently, these patients have unknown death and survival time. Here, in the simulation with censoring, we assumed that 85% of the patients were still alive at the end of the study, similar to the censoring rate of the TCGA IDC data set. In both settings and with different depths of RNA-seq coverage, SURVIV consistently outperformed Cox regression in the true-positive rate at the same false-positive rate of 5% (Fig. 2a). As expected, we observed a more significant improvement in SURVIV over Cox regression when the RNA-seq read coverage was low (Fig. 2a).
To more faithfully recapitulate the read count distribution in a real cancer RNA-seq data set, we performed another simulation study with read counts directly sampled from the TCGA IDC data. To assess the influence of RNA-seq read depth on the performance of SURVIV and Cox regression, sampled read counts were then multiplied by different factors ranging from 10 to 300% to simulate data sets with different RNA-seq read depths (Fig. 2b). The TCGA IDC data set has an average RNA-seq depth of ~60 million paired-end reads per patient. Thus, the read depth of these simulated RNA-seq data sets ranged from ~6 million reads to 180 million reads per patient, representing low-coverage RNA-seq studies designed primarily for gene expression analysis32 up to high-coverage RNA-seq studies designed primarily for alternative isoform analysis29. At all levels of RNA-seq depth, SURVIV consistently outperformed Cox regression, as reflected by the area under curve of the receiver operating characteristic (ROC) curve as well as the true-positive rate at 5% false-positive rate (Fig. 2b). The improvement of SURVIV over Cox regression was particularly prominent when the read depth was low. For example, at 10% read depth, SURVIV had 7% improvement in area under curve (68% versus 61%) and 8% improvement in the true-positive rate at 5% false-positive rate (46% versus 38%). Collectively, these simulation results suggest that SURVIV achieves a higher accuracy by accounting for the estimation uncertainty of mRNA isoform ratio in RNA-seq data.
SURVIV analysis of TCGA IDC breast cancer data
To illustrate the practical utility of SURVIV, we used it to analyse the overall survival time of 682 IDC patients from the TCGA breast cancer (BRCA) RNA-seq data set (see Methods for details of the data source and processing pipeline). We chose to analyse IDC because it is the most frequent type of breast cancer33, comprising ~70% of patients in the TCGA breast cancer data set. To control for the effects of significant clinical parameters such as tumour stage and subtype and identify alternative splicing events associated with patient outcomes across multiple molecular and clinical subtypes, we followed the procedure of Croce and colleagues in analysing mRNA and microRNA prognostic signature of IDC33 and stratified the patients according to their clinical parameters. We then conducted SURVIV analysis in 26 clinical subgroups with at least 50 patients in each subgroup. We identified 229 exon-skipping events associated with patient survival in multiple clinical subgroups that met the criteria of SURVIV P-value≤0.01 in at least two subgroups of the same clinical parameter (cancer subtype, stage, lymph node, metastasis, tumour size, oestrogen receptor status, progesterone receptor status, HER2 status and age as shown in Fig. 3). DAVID (Database for Annotation, Visualization and Integrated Discovery) Gene Ontology analyses34 of the 229 alternative splicing events suggest an enrichment of genes in cancer-related functional categories such as intracellular signalling, apoptosis, oxidative stress and response to DNA damage (Supplementary Fig. 1). Table 1 shows a few selected examples of survival-associated alternative splicing events in cancer-related genes. Using two-means clustering of each individual exon’s inclusion levels, the 682 IDC patients can be segregated into two subgroups with significantly different survival times as illustrated by the Kaplan–Meier survival plot (Fig. 4). We also carried out hierarchical clustering of IDC patients using 176 survival-associated alternative exons (P≤0.01; SURVIV analysis of all IDC patients). Using the exon-inclusion levels of these 176 exons, we clustered IDC patients into three major subgroups, with 95, 194 and 389 patients, respectively. As illustrated by the Kaplan–Meier survival plots, the three subgroups had significantly different survival times (Supplementary Fig. 2).
Figure 3: SURVIV analysis of exon-skipping events in the TCGA IDC RNA-seq data set.
IDC patients are stratified into multiple clinical subgroups based on clinical parameters including cancer subtype, stage, lymph node status, metastasis, tumour size, oestrogen receptor status, progesterone receptor status, HER2 status and age. Only clinical subgroups with at least 50 patients are included in further analyses. Numbers of patients in the subgroups are indicated next to the names of the subgroups. Shown in the heatmap are the log10 SURVIV P-values of the 229 exons associated with patient survival (P≤0.01) in at least two subgroups of the same class of clinical parameters. Turquoise colour indicates positive correlation that higher exon-inclusion levels are associated with higher survival probabilities. Magenta colour indicates negative correlation that lower exon-inclusion levels are associated with higher survival probabilities.
Figure 4: Kaplan–Meier survival plots of IDC patients stratified by two-means clustering of the exon-inclusion levels of four survival-associated alternative splicing events.
Clustering was generated for each of the four exons separately. Black lines represent patients with high exon-inclusion levels. Red lines represent patients with low exon-inclusion levels. The P-values are from SURVIV analysis of the TCGA IDC RNA-seq data. (a) ATRIP. (b) BCL2L11. (c) CD74. (d) PCBP4.
Figure 5: Alternative splicing of STAT5A exon 5 is significantly associated with IDC patient survival.
(a) The gene structure of the STAT5A full-length isoform compared to the ΔEx5 isoform skipping the 5th exon. (b) Kaplan–Meier survival plot of IDC patients stratified by two-means clustering using exon-inclusion levels of STAT5A exon 5. The 420 patients in Group 1 (average exon 5 inclusion level=95%) have significantly higher survival probabilities than the 262 patients in Group 2 (average exon 5 inclusion level=85%) (SURVIV P=6.8e−4). (c) Exon 5 inclusion levels of IDC patients stratified by two-means clustering using exon 5 inclusion levels. Group 1 has 420 patients with average exon-inclusion level at 95%. Group 2 has 262 patients with average exon-inclusion level at 85%. (d) STAT5A exon 5 inclusion levels in normal breast tissues versus breast cancer tumour samples. Exon-inclusion levels are extracted from 86 TCGA breast cancer patients with matched normal and tumour samples. Normal breast tissues have average exon 5 inclusion level at 95%, compared to 91% average exon-inclusion level in tumour samples. Error bars represent 95% confidence interval of the mean.
Figure 6: Splicing factor regulatory network of survival-associated alternative splicing events in IDC.
(a–c) Kaplan–Meier survival plots of IDC patients stratified by the gene expression levels of three splicing factors: TRA2B (a, Cox regression P=1.8e−4), HNRNPH1 (b, P=3.4e−4) and SFRS3 (c, P=2.8e−3). Black lines represent patients with high gene expression levels. Red lines represent patients with low gene expression levels. (d) The exon-inclusion levels of a DHX30 alternative exon are negatively correlated with TRA2B gene expression levels (robust correlation coefficient r=−0.26, correlation P=1.2e−17). (e) The exon-inclusion levels of a MAP3K4 alternative exon are positively correlated withHNRNPH1 gene expression levels (robust correlation coefficient r=0.16, correlation P=2.6e−06). (f) A splicing co-expression network of the three splicing factors and their correlated survival-associated alternative exons. In total, 84 survival-associated alternative exons are significantly correlated with the three splicing factors. The positive/negative correlation between splicing factors and alternative exons is represented by blue/red lines, respectively. Exons whose inclusion levels are positively/negatively correlated with survival times are represented by blue/red dots, respectively. The size of the splicing factor circles is proportional to the number of correlated exons within the network.
Figure 7: Cross-validation of different classes of IDC survival predictors measured by the C-index
A C-index of 1 indicates perfect prediction accuracy and a C-index of 0.5 indicates random guess. The plots indicate the distribution of C-indexes from 100 rounds of cross-validation. The centre value of the box plot is the median C-index from 100 rounds of cross-validation. The notch represents the 95%confidence interval of the median. The box represents the 25 and 75% quantiles. The whiskers extended out from the box represent the 5 and 95% quantiles. Two-sided Wilcoxon test was used to compare different survival predictors. The different classes of predictors are: (a) clinical information (median C-index 0.67). (b) Gene expression (median C-index 0.68). (c) Alternative splicing (median C-index 0.71). (d) Clinical information+gene expression (median C-index 0.69). (e) Clinical information+alternative splicing (median C-index 0.73). (f) Clinical information+gene expression+alternative splicing (median C-index 0.74). Note that ‘Gene’ refers to ‘Gene-level expression’ in these plots.
Next, we carried out the SURVIV analysis in five additional cancer types in TCGA, including GBM (glioblastoma multiforme), KIRC (kidney renal clear cell carcinoma), LGG (lower grade glioma), LUSC (lung squamous cell carcinoma) and OV (ovarian serous cystadenocarcinoma). As expected, the number of significant events at different FDR or P-value significance cutoffs varied across cancer types, with LGG having the strongest survival-associated alternative splicing signals with 660 significant exon-skipping events at FDR≤5% (Supplementary Data 3 and 4). Strikingly, regardless of the number of significant events, alternative splicing-based survival predictors outperformed gene expression-based survival predictors across all cancer types (Supplementary Fig. 3), consistent with our initial observation on the IDC data set.
Alternative processing and modification of mRNA, such as alternative splicing, allow cells to generate a large number of mRNA and protein isoforms with diverse regulatory and functional properties. The plasticity of alternative splicing is often exploited by cancer cells to produce isoform switches that promote cancer cell survival, proliferation and metastasis7, 8. The widespread use of RNA-seq in cancer transcriptome studies15, 47, 48 has provided the opportunity to comprehensively elucidate the landscape of alternative splicing in cancer tissues. While existing studies of alternative splicing in large-scale cancer transcriptome data largely focused on the comparison of splicing patterns between cancer and normal tissues or between different subtypes of cancer18, 21, 49, additional computational tools are needed to characterize the clinical relevance of alternative splicing using massive RNA-seq data sets, including the association of alternative splicing with phenotypes and patient outcomes.
We have developed SURVIV, a novel statistical model for survival analysis of alternative isoform variation using cancer RNA-seq data. SURVIV uses a survival measurement error model to simultaneously model the estimation uncertainty of mRNA isoform ratio in individual patients and the association of mRNA isoform ratio with survival time across patients. Compared with the conventional Cox regression model that uses each patient’s mRNA isoform ratio as a point estimate, SURVIV achieves a higher accuracy as indicated by simulation studies under a variety of settings. Of note, we observed a particularly marked improvement of SURVIV over Cox regression for low- and moderate-depth RNA-seq data (Fig. 2b). This has important practical value because many clinical RNA-seq data sets have large sample size but relatively modest sequencing depth.
Using the TCGA IDC breast cancer RNA-seq data of 682 patients, SURVIV identified 229 alternative splicing events associated with patient survival time, which met the criteria of SURVIVP-values≤0.01 in multiple clinical subgroups. While the statistical threshold seemed loose, several lines of evidence suggest the functional and clinical relevance of these survival-associated alternative splicing events. These alternative splicing events were frequently identified and enriched in the gene functional groups important for cancer development and progression, including apoptosis, DNA damage response and oxidative stress. While some of these events may simply reflect correlation but not causal effect on cancer patient survival, other events may play an active role in regulating cancer cell phenotypes. For example, a survival-associated alternative splicing event involving exon 5 of STAT5A is known to regulate the activity of this transcription factor with important roles in epithelial cell growth and apoptosis37. Using a co-expression network analysis of splicing factor to exon correlation across all patients, we identified three splicing factors (TRA2B, HNRNPH1 and SFRS3) as potential hubs of the survival-associated alternative splicing network of IDC. The expression levels of all three splicing factors were negatively associated with patient survival times (Fig. 6a–c), and both TRA2B and HNRNPH1 were previously reported to have an impact on cancer-related molecular pathways40, 41, 42, 43, 44, 45. Finally, despite the limited power in detecting individual events, we show that the survival-associated alternative splicing events can be used to construct a predictor for patient survival, with an accuracy higher than predictors based on clinical parameters or gene expression profiles (Fig. 7). This further demonstrates the potential biological relevance and clinical utility of the identified alternative splicing events.
We performed cross-validation analyses to evaluate and compare the prognostic value of alternative splicing, gene expression and clinical information for predicting patient survival, either independently or in combination. As expected, the combined use of all three types of information led to the best prediction accuracy. Because we used penalized regression to build the prediction model, combining information from multiple layers of data did not necessarily increase the number of predictors in the model. The perhaps more surprising and intriguing result is that alternative splicing-based predictors appear to outperform gene expression-based predictors when used alone and when either type of data was combined with clinical information (Fig. 7). We observed the same trend in five additional cancer types (Supplementary Fig. 3). We note that this finding was consistent with a previous report that cancer subtype classification based on splicing isoform expression performed better than gene expression-based classification25. While this trend seems counterintuitive because accurate estimation of gene expression requires much lower RNA-seq depth than accurate estimation of alternative splicing29, one possible explanation may be the inherent characteristic of isoform ratio data. By definition, mRNA isoform ratio is estimated as the ratio of multiple mRNA isoforms from a single gene. Therefore, mRNA isoform ratio data have a ‘built-in’ internal control that could be more robust against certain artefacts and confounding issues that influence gene expression estimates across large clinical RNA-seq data sets, such as poor sample quality and RNA degradation12. Regardless of the reasons, our data call for further studies to fully explore the utility of mRNA isoform ratio data for various clinical research applications.
The SURVIV source code is available for download at https://github.com/Xinglab/SURVIV. SURVIV is a general statistical model for survival analysis of mRNA isoform ratio using RNA-seq data. The current statistical framework of SURVIV is applicable to RNA-seq based count data for all basic types of alternative splicing patterns involving two isoform choices from an alternatively spliced region, such as exon-skipping, alternative 5′ splice sites, alternative 3′ splice sites, mutually exclusive exons and retained introns, as well as other forms of alternative isoform variation such as RNA editing. With the rapid accumulation of clinical RNA-seq data sets, SURVIV will be a useful tool for elucidating the clinical relevance and potential functional significance of alternative isoform variation in cancer and other diseases.
Two pediatric siblings with recurrent multifocal glioblastoma multiforme (GBM) refractory to current standard therapies exhibited “remarkable and durable” responses to immune checkpoint inhibition with single-agent nivolumab (Opdivo), researchers said.
Following pre-clinical testing in 37 biallelic mismatch repair deficiency (bMMRD) cancers, a regimen of 3 mg/kg nivolumab every 2 weeks resulted in clinically significant responses and a profound radiologic response, Uri Tabori, MD, of The Hospital for Sick Children, Toronto, Ontario, Canada, and colleagues reported in the Journal of Clinical Oncology.
The 6-year-old white female patient and her 3.5-year-old brother resumed normal schooling and daily activities after 9 and 5 months of therapy, respectively, the researchers said.
“This observation is especially encouraging because these children are still clinically stable, whereas most relapsed pediatric GBMs will progress within 1 to 2 months despite salvage treatment, and survival is usually 3 to 6 months post-recurrence. It also highlights the utility of germline predisposition in guiding novel treatment options — in this case, immunotherapy — for cancer treatment.”
Findings from this lab study and small case series report may have implications for GBM as well as for other hypermutant cancers arising from primary (genetic predisposition) or secondary MMRD, the researchers said. “Given the increasing availability of commercial sequencing platforms, analysis of mutation burden and neoantigens can play a role in transforming treatment of these patients.”
Still, they added that these results, while encouraging, need to be validated in multinational prospective clinical trials of these “universally lethal” bMMRD-driven hypermutant cancers.
“Sometimes very small studies can yield meaningful results,” Robert Fenstermaker, MD, of Roswell Park Cancer Institute in Buffalo, N.Y., told MedPage Today via email. “Although anecdotal, the results of this study are quite encouraging because they tend to confirm current theory about immunotherapy for glioblastoma.”
Although these kinds of clinical responses to single-agent drug therapy in GBM are uncommon and the results may not be broadly applicable to all glioblastoma patients, this paper “is of much greater importance than just these few cases,” Fenstermaker emphasized. “The excellent responses in these particular cases suggest that an immune checkpoint inhibitor (nivolumab) may have enabled the immune system to respond fully.”
This “very small case series” report of a “compelling clinical experience” is a “fascinating and beautiful example of how mechanistic insight can be linked to rationally designed clinical applications — in turn, stimulating new downstream ideas,” Stephanie Weiss, MD, a radiation oncologist at Fox Chase Cancer Center in Philadelphia, commented in an email.
“This series also tests ‘proof of principle,’ that bMMRD tumors are hypermutated and associated with a high neoantigen load, and therefore may respond much like other immune checkpoint inhibitor-sensitive tumors. In this sense, the results reveal a tantalizing glimpse into the disease process of at least a subset of GBMs and can guide high-quality study of novel treatment for GBM.”
For the study, Tabori and colleagues performed exome sequencing and neoantigen prediction on 37 bMMRD-associated tumors, including 21 GBMs, and compared them with childhood and adult brain neoplasms.
The bMMRD GBMs were found to be hypermutant and to have an extremely strong neoantigen load — up to 16 times higher than the signature commonly seen in known immune checkpoint inhibitors (P<.001).
The female patient, diagnosed with a left parietal GBM, underwent near-total resection and focal irradiation over 6.5 weeks. After a clinical remission lasting 3 months, surveillance MRI revealed recurrence in the initial tumor bed and a second lesion in the left temporal lobe.
Six months earlier, the index patient’s brother had been diagnosed with a right frontoparietal GBM and treated with surgery, focal irradiation, and temozolomide (Temodal). Ten months after diagnosis, surveillance MRI revealed an asymptomatic diffuse multinodular GBM recurrence.
When given nivolumab as a last-resort therapeutic agent, both children initially experienced serious symptoms that on imaging mimicked tutor progression. After symptomatic management and observation, both stabilized, and follow-up imaging demonstrated significant improvement in tumor-related abnormalities.
Fenstermaker said that important next steps lie ahead, such as combining immune checkpoint inhibitors with specific cancer vaccines designed to immunize patients with glioblastomas other than this rare hypermutated type. “There are a number of prospective vaccines currently in the glioblastoma drug pipeline that would be candidates for this kind of approach,” he told MedPage Today. Examples include SurVaxM, NeoVax, HSPPC-96, and various dendritic cell vaccines.
In addition, newer genomic techniques are being developed that could make it possible to create a personalized profile of the mutant proteins in a given patient’s tumor, he noted. “One can imagine combining such a personalized vaccine against these mutant proteins together with an immune checkpoint inhibitor. Such a combination might result in many more responses like the ones seen in this small study.”
PD-1 Blockade in Tumors with Mismatch-Repair Deficiency
Somatic mutations have the potential to encode “non-self” immunogenic antigens. We hypothesized that tumors with a large number of somatic mutations due to mismatch-repair defects may be susceptible to immune checkpoint blockade.
We conducted a phase 2 study to evaluate the clinical activity of pembrolizumab, an anti–programmed death 1 immune checkpoint inhibitor, in 41 patients with progressive metastatic carcinoma with or without mismatch-repair deficiency. Pembrolizumab was administered intravenously at a dose of 10 mg per kilogram of body weight every 14 days in patients with mismatch repair–deficient colorectal cancers, patients with mismatch repair–proficient colorectal cancers, and patients with mismatch repair–deficient cancers that were not colorectal. The coprimary end points were the immune-related objective response rate and the 20-week immune-related progression-free survival rate.
The immune-related objective response rate and immune-related progression-free survival rate were 40% (4 of 10 patients) and 78% (7 of 9 patients), respectively, for mismatch repair–deficient colorectal cancers and 0% (0 of 18 patients) and 11% (2 of 18 patients) for mismatch repair–proficient colorectal cancers. The median progression-free survival and overall survival were not reached in the cohort with mismatch repair–deficient colorectal cancer but were 2.2 and 5.0 months, respectively, in the cohort with mismatch repair–proficient colorectal cancer (hazard ratio for disease progression or death, 0.10 [P<0.001], and hazard ratio for death, 0.22 [P=0.05]). Patients with mismatch repair–deficient noncolorectal cancer had responses similar to those of patients with mismatch repair–deficient colorectal cancer (immune-related objective response rate, 71% [5 of 7 patients]; immune-related progression-free survival rate, 67% [4 of 6 patients]). Whole-exome sequencing revealed a mean of 1782 somatic mutations per tumor in mismatch repair–deficient tumors, as compared with 73 in mismatch repair–proficient tumors (P=0.007), and high somatic mutation loads were associated with prolonged progression-free survival (P=0.02).
This study showed that mismatch-repair status predicted clinical benefit of immune checkpoint blockade with pembrolizumab. (Funded by Johns Hopkins University and others; ClinicalTrials.gov number, NCT01876511.)
Purpose Recurrent glioblastoma multiforme (GBM) is incurable with current therapies. Biallelic mismatch repair deficiency (bMMRD) is a highly penetrant childhood cancer syndrome often resulting in GBM characterized by a high mutational burden. Evidence suggests that high mutation and neoantigen loads are associated with response to immune checkpoint inhibition.
Patients and Methods We performed exome sequencing and neoantigen prediction on 37 bMMRD cancers and compared them with childhood and adult brain neoplasms. Neoantigen prediction bMMRD GBM was compared with responsive adult cancers from multiple tissues. Two siblings with recurrent multifocal bMMRD GBM were treated with the immune checkpoint inhibitor nivolumab.
Results All malignant tumors (n = 32) were hypermutant. Although bMMRD brain tumors had the highest mutational load because of secondary polymerase mutations (mean, 17,740 ± standard deviation, 7,703), all other high-grade tumors were hypermutant (mean, 1,589 ± standard deviation, 1,043), similar to other cancers that responded favorably to immune checkpoint inhibitors. bMMRD GBM had a significantly higher mutational load than sporadic pediatric and adult gliomas and all other brain tumors (P < .001). bMMRD GBM harbored mean neoantigen loads seven to 16 times higher than those in immunoresponsive melanomas, lung cancers, or microsatellite-unstable GI cancers (P < .001). On the basis of these preclinical data, we treated two bMMRD siblings with recurrent multifocal GBM with the anti–programmed death-1 inhibitor nivolumab, which resulted in clinically significant responses and a profound radiologic response.
Conclusion This report of initial and durable responses of recurrent GBM to immune checkpoint inhibition may have implications for GBM in general and other hypermutant cancers arising from primary (genetic predisposition) or secondary MMRD.
Glioblastoma multiforme (GBM) is a highly malignant brain tumor and the most common cause of death among children with CNS neoplasms.1 Despite primary management, which consists of surgical resection followed by radiation therapy and chemotherapy, most GBMs will recur, resulting in rapid death. Patients with recurrent disease have a particularly poor prognosis, with a median survival of fewer than 6 months; no effective therapies currently exist.
In contrast to adult CNS malignancies, a significant proportion of childhood brain tumors occur in the context of cancer predisposition syndromes.2 Pediatric GBMs are associated with germline mutations in TP53 (Li-Fraumeni syndrome)1 and the mismatch repair (MMR) genes (biallelic MMR deficiency syndrome [bMMRD]).3 Patients with bMMRD are unique in both the molecular events that lead to GBM formation and opportunities for innovative management of these tumors to possibly improve survival.
bMMRD is caused by homozygous germline mutations in one of the four MMR genes (PMS2, MLH1, MSH2, and MSH6) and is arguably the most penetrant cancer predisposition syndrome, with 100% of biallelic mutation carriers developing cancers in the first two decades of life. These are most commonly malignant gliomas, hematologic malignancies, and GI cancers.3,4 Understanding the relationship between the bMMRD somatic mutational landscape and tumor biology can lead to development of novel therapies and improved patient outcomes.
bMMRD GBMs harbor the highest mutation load among human cancers.5 Combined germline mutations in the MMR genes and somatic mutations in DNA polymerase result in complete ablation of proofreading during DNA replication and underpin this phenomenon. bMMRD GBMs, in contrast to other childhood cancers and adult MMR-proficient gliomas, exhibit a molecular signature characterized by single-nucleotide changes present in exponentially higher numbers. An important characteristic of non-bMMRD cancers exhibiting high mutation loads—subsets of malignant melanomas and lung, bladder, and microsatellite-unstable GI cancers—is responsiveness to immune checkpoint inhibitors.6–9
Checkpoint inhibitors target the immunomodulatory effect of CTLA-4 (cytotoxic T lymphocyte–associated protein 4) and programmed death-1 (PD-1)/programmed death-ligand 1, restoring effector T-cell function and antitumor activity. Recent reports have shown that patients whose tumors bear a high mutation load and/or definedtumor-associated antigen (neoantigen) signatures derive enhanced clinical benefit from checkpoint inhibitor therapy.10
Nivolumab is an anti–PD-1–directed immune checkpoint inhibitor approved for use in the treatment of non–small-cell lung cancer11and melanoma and under clinical investigation in multiple adult and pediatric tumors.12,13 However, this response is currently unknown in bMMRD-associated cancers and the uniformly lethal GBM.
Fig 1. Clinical and molecular features of the biallelic mismatch repair (MMR) deficiency (bMMRD) family. (A) Pedigree of the family with both bMMRD-affected children (solid square and circle). Both siblings presented with glioblastoma multiforme (GBM), whereas parents remained unaffected, as observed in other bMMRD families. (B) Immunohistochemistry staining of the index patient’s GBM for the four MMR genes: MSH2, MSH6,MLH1, and PMS2. A PMS2-negative stain in both tumor and normal cells prompted subsequent genetic testing that confirmed the diagnosis of bMMRD. NF1, neurofibromatosis type 1. http://jco.ascopubs.org/content/early/2016/03/17/JCO.2016.66.6552/F1.small.gif
To examine whether immune checkpoint inhibitors would be applicable for bMMRD cancers, we surveyed the extent of hypermutation across bMMRD tumors form various tissues. Exome sequencing of 37 cancers collected from the bMMRD consortium revealed that all malignant tumors (n = 32) were hypermutant. Although bMMRD brain tumors had the highest mutational load resulting from secondary polymerase mutations (mean, 17,740 ± standard deviation [SD], 7,703), all other high-grade tumors were hypermutant, harboring more than 100 exonic mutations (mean, 1,589 ± SD, 1,043; Fig 2A). Lower-grade bMMRD tumors (n = 5) did not exhibit hypermutation (mean, 40 ± SD, 18). Importantly, bMMRD GBMs had a significantly higher mutational load than sporadic pediatric and adult gliomas and all other brain tumors (P < .001; Fig 2A). To test the extent to which hypermutation translates to a strong neoantigen signature, a current predictor of response to immune checkpoint inhibition, we performed genome-wide somatic neoepitope analysis using similar algorithms previously used for melanoma, lung, and colon cancers.9,14,15 For each study, we compared our cohort of tumors with other tumors that were reported to respond to immune checkpoint inhibitors (Fig 2B). Strikingly, bMMRD GBMs had a significantly higher number of predicted neoantigens, whereas other tumors responded with a fraction of the neoantigens found in our patients (P < .001; Fig 2B). The mean neoantigen load was seven to 16 times higher than those of immunoresponsive melanomas, lung cancers, and microsatellite-unstable GI cancers.
Fig 2. Tumor mutation and neoantigen analysis. (A) Boxplot comparing the number of mutations per tumor exome in several biallelic mismatch repair deficiency (bMMRD) cancer types with pediatric and adult brain tumors. (B) Ratio of the number of neoantigens found in immunoresponsive tumors from melanoma (n = 27), lung cancer (n = 14), and colon cancer (n = 7) data sets compared with median number of neoantigens in bMMRD glioblastoma multiforme (GBM; n = 13). ATRT, atypical teratoid rhabdoid tumor; DIPG, diffuse intrinsic pontine glioma; L/L, leukemia/lymphoma; LGG, low-grade glioma; MB, medulloblastoma; PA, pilocytic astrocytoma; PNET, primitive neuroectodermal tumor.
We describe two pediatric patients with recurrent multifocal GBM refractory to current standard therapies who exhibited remarkable and durable responses to immune checkpoint inhibition with single-agent nivolumab. This observation is especially encouraging because these children are still clinically stable, whereas most relapsed pediatric GBMs will progress within 1 to 2 months19 despite salvage treatment, and survival is usually 3 to 6 months postrecurrence.20 Furthermore, bMMRD GBMs have outcomes similar to those of sporadic childhood GBMs,21 and data gathered from the consortium reveal a mean time from relapse to death of 2.6 months in bMMRD GBM. To our knowledge, this is the first report of such a response in childhood or adult GBM. It also highlights the utility of germline predisposition in guiding novel treatment options—in this case, immunotherapy—for cancer treatment. ….
sjwilliamspa
Not sure if the link between PD-L1 response and MMR status is causal in this ase. there are many tumors with MMR and especially all tumors had high degree of MMR. Perhaps they need to look at tumors that have a more stable genome like certain hepatocarcinomas.
CRISPR-Cas9 Screening by Horizon Discovery, Cambridge, UK – HDx™ Reference Standards, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 2: CRISPR for Gene Editing and DNA Repair
CRISPR-Cas9 Screening by Horizon Discovery, Cambridge, UK – HDx™ Reference Standards
Reporters: David Orchard-Webb, PhD and Aviva Lev-Ari, PhD, RN
They have leveraged our cutting-edge technologies to build a ground-breaking platform for Immuno-Oncology drug discovery.
Their collection of high throughput assays allows you to rapidly identify single agents or combinations that affect T cell and natural killer (NK) cell function.
Gene-Editing in Immune Cells
Gene-editing in immune cells for your Immuno-Oncology project is now even faster than before. Leveraging the latest genome engineering tools, including CRISPR-Cas9, we’re able to precisely edit over 10 pre-characterized T-cell, B-cell, and monocytes lines.
Power Your Immune-Oncology Projects with Custom Engineered Immune Cells
Gene-editing in immune cell lines can be challenging due to low targeting efficiency and difficulties in single cell derivation of suspension cells. Horizon has validated 10+ immune cell lines including THP-1, Jurkat and NALM-6 cells for gene-editing projects. CRISPR, rAAV and ZFN gene-editing technologies are available depending on project requirements.
Take advantage of the largest panel of pre-characterized immune cell lines available and benefit from Horizon’s exceptional knowhow and experience in completing over 2,000 gene-editing projects.
It was discovered some time ago that eukaryotic cells regularly secrete such structures as microvesicles, macromolecular complexes, and small molecules into their ambient environment. Exosomes are one of the types of natural nanoparticles (or nanovesicles) that have shown promise in many areas of research, diagnostics and therapy. They are small lipid membrane vesicles (30-120 nm) generated by fusion of cytoplasmic endosomal multivesicular bodies within the cell surface. Exosomes are found throughout the body in such fluids as blood, saliva, urine, and breast milk. Furthermore, all types of cells secrete them in in vitro culture. It is believed that they have many natural functions, including acting as transporters of nucleic acids (mostly RNA), cytosolic proteins and metabolites to many cells, tissues or organs throughout the body. Much remains to be understood regarding how they are formed, as well as of their targeting and ultimate physiological activity. But many don’t realize that some activities have been rather thoroughly demonstrated─ such as their function in some sort of either local or more systemic intercellular communication.
Exosomes as Tools
General interest in exosomes is now growing for many reasons. One is because of the observation of their natural activity with antigen-presenting cells and in immune responses in the body. Their potential as very powerful biomedical tools of both diagnostic and therapeutic value is now being more widely reported. Applications described include using them as immunotherapeutic reagents, vectors of engineered genetic constructs, and vaccine particles. They’ve also been described as tools in the diagnosis or prognosis of a wide variety of disorders, such as cancer and neurodegenerative diseases. Also, their potential in tissue-level microcommunication is driving interest in such therapeutic activities as cardiac repair following heart attacks. Their potential as biomarkers is being explored because their content has been described as a “fingerprint” of differentiation or signaling or regulation status of the cell generating them. For example, by monitoring the exosomes secreted by transplanted cells, one may be able to predict the status or potentially even the outcome of cell therapy procedures. Clinical trials are in progress for exosomes in many therapeutic functions, for many indications. One example is using dendritic cell-derived exosomes to initiate immune response to cancers.
Exosome Manufacturing
Exosome product manufacturing involves many distinct areas of study. First of all, we are interested in their efficient and robust generation at a sufficient scale. Also, because they are found in such raw materials as animal serum, avoiding process-related contaminants is a concern. Finally, a variety of means of separating them from other types of extracellular vesicles and cell debris is under study. As exosomes are being examined in so many applications, their production involves many distinct platforms and concerns. First of all, an appropriate and effective culture mode is required for any cell line that is specifically required by the application. Also, one must consider the quality systems and regulatory status of the materials and manufacturing environment for the particular product addressed. Finally, a robust process must be described for the scale and duration of production demanded. As things exist now, their production can be described as
1) the at-scale expansion and culture of the parent cell-line,
2) the collection or harvest of the culture media containing the secreted exosomes, and
3) the isolation or purification of the desired exosomes from not only other macrovesicles, macromolecular complexes, and small molecules, but from such other process contaminants as cellular debris and culture media components.
Genomics and epigenetics link to DNA structure, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 1: Next Generation Sequencing (NGS)
Genomics and epigenetics link to DNA structure
Larry H. Bernstein, MD, FCAP, Curator
LPBI
Sequence and Epigenetic Factors Determine Overall DNA Structure
Atomic-level simulations show electrostatic forces between each atom. [Alek Aksimentiev, University of Illinois at Urbana-Champaign]
The traditionally held hypothesis about the highly ordered organization of DNA describes the interaction of various proteins with DNA sequences to mediate the dynamic structure of the molecule. However, recent evidence has emerged that stretches of homologous DNA sequences can associate preferentially with one another, even in the absence of proteins.
Researchers at the University of Illinois Center for the Physics of Living Cells, Johns Hopkins University, and Ulsan National Institute of Science and Technology (UNIST) in South Korea found that DNA molecules interact directly with one another in ways that are dependent on the sequence of the DNA and epigenetic factors, such as methylation.
The researchers described evidence they found for sequence-dependent attractive interactions between double-stranded DNA molecules that neither involve intermolecular strand exchange nor are mediated by DNA-binding proteins.
“DNA molecules tend to repel each other in water, but in the presence of special types of cations, they can attract each other just like nuclei pulling each other by sharing electrons in between,” explained lead study author Hajin Kim, Ph.D., assistant professor of biophysics at UNIST. “Our study suggests that the attractive force strongly depends on the nucleic acid sequence and also the epigenetic modifications.”
The investigators used atomic-level supercomputer simulations to measure the forces between a pair of double-stranded DNA helices and proposed that the distribution of methyl groups on the DNA was the key to regulating this sequence-dependent attraction. To verify their findings experimentally, the scientists were able to observe a single pair of DNA molecules within nanoscale bubbles.
“Here we combine molecular dynamics simulations with single-molecule fluorescence resonance energy transfer experiments to examine the interactions between duplex DNA in the presence of spermine, a biological polycation,” the authors wrote. “We find that AT-rich DNA duplexes associate more strongly than GC-rich duplexes, regardless of the sequence homology. Methyl groups of thymine act as a steric block, relocating spermine from major grooves to interhelical regions, thereby increasing DNA–DNA attraction.”
The findings from this study were published recently in Nature Communications in an article entitled “Direct Evidence for Sequence-Dependent Attraction Between Double-Stranded DNA Controlled by Methylation.”
After conducting numerous further simulations, the research team concluded that direct DNA–DNA interactions could play a central role in how chromosomes are organized in the cell and which ones are expanded or folded up compactly, ultimately determining functions of different cell types or regulating the cell cycle.
“Biophysics is a fascinating subject that explores the fundamental principles behind a variety of biological processes and life phenomena,” Dr. Kim noted. “Our study requires cross-disciplinary efforts from physicists, biologists, chemists, and engineering scientists and we pursue the diversity of scientific disciplines within the group.”
Dr. Kim concluded by stating that “in our lab, we try to unravel the mysteries within human cells based on the principles of physics and the mechanisms of biology. In the long run, we are seeking for ways to prevent chronic illnesses and diseases associated with aging.”
Direct evidence for sequence-dependent attraction between double-stranded DNA controlled by methylation
Jejoong Yoo, Hajin Kim, Aleksei Aksimentiev, and Taekjip Ha Nature Communications 7 11045 (2016) DOI:10.1038/ncomms11045BibTex
Although proteins mediate highly ordered DNA organization in vivo, theoretical studies suggest that homologous DNA duplexes can preferentially associate with one another even in the absence of proteins. Here we combine molecular dynamics simulations with single-molecule fluorescence resonance energy transfer experiments to examine the interactions between duplex DNA in the presence of spermine, a biological polycation. We find that AT-rich DNA duplexes associate more strongly than GC-rich duplexes, regardless of the sequence homology. Methyl groups of thymine acts as a steric block, relocating spermine from major grooves to interhelical regions, thereby increasing DNA–DNA attraction. Indeed, methylation of cytosines makes attraction between GC-rich DNA as strong as that between AT-rich DNA. Recent genome-wide chromosome organization studies showed that remote contact frequencies are higher for AT-rich and methylated DNA, suggesting that direct DNA–DNA interactions that we report here may play a role in the chromosome organization and gene regulation.
Formation of a DNA double helix occurs through Watson–Crick pairing mediated by the complementary hydrogen bond patterns of the two DNA strands and base stacking. Interactions between double-stranded (ds)DNA molecules in typical experimental conditions containing mono- and divalent cations are repulsive1, but can turn attractive in the presence of high-valence cations2. Theoretical studies have identified the ion–ion correlation effect as a possible microscopic mechanism of the DNA condensation phenomena3, 4, 5. Theoretical investigations have also suggested that sequence-specific attractive forces might exist between two homologous fragments of dsDNA6, and this ‘homology recognition’ hypothesis was supported by in vitro atomic force microscopy7 and in vivo point mutation assays8. However, the systems used in these measurements were too complex to rule out other possible causes such as Watson–Crick strand exchange between partially melted DNA or protein-mediated association of DNA.
Here we present direct evidence for sequence-dependent attractive interactions between dsDNA molecules that neither involve intermolecular strand exchange nor are mediated by proteins. Further, we find that the sequence-dependent attraction is controlled not by homology—contradictory to the ‘homology recognition’ hypothesis6—but by a methylation pattern. Unlike the previous in vitro study that used monovalent (Na+) or divalent (Mg2+) cations7, we presumed that for the sequence-dependent attractive interactions to operate polyamines would have to be present. Polyamine is a biological polycation present at a millimolar concentration in most eukaryotic cells and essential for cell growth and proliferation9, 10. Polyamines are also known to condense DNA in a concentration-dependent manner2, 11. In this study, we use spermine4+(Sm4+) that contains four positively charged amine groups per molecule.
Sequence dependence of DNA–DNA forces
To characterize the molecular mechanisms of DNA–DNA attraction mediated by polyamines, we performed molecular dynamics (MD) simulations where two effectively infinite parallel dsDNA molecules, 20 base pairs (bp) each in a periodic unit cell, were restrained to maintain a prescribed inter-DNA distance; the DNA molecules were free to rotate about their axes. The two DNA molecules were submerged in 100mM aqueous solution of NaCl that also contained 20 Sm4+molecules; thus, the total charge of Sm4+, 80 e, was equal in magnitude to the total charge of DNA (2 × 2 × 20 e, two unit charges per base pair; Fig. 1a). Repeating such simulations at various inter-DNA distances and applying weighted histogram analysis12 yielded the change in the interaction free energy (ΔG) as a function of the DNA–DNA distance (Fig. 1b,c). In a broad agreement with previous experimental findings13, ΔG had a minimum, ΔGmin, at the inter-DNA distance of 25−30Å for all sequences examined, indeed showing that two duplex DNA molecules can attract each other. The free energy of inter-duplex attraction was at least an order of magnitude smaller than the Watson–Crick interaction free energy of the same length DNA duplex. A minimum of ΔG was not observed in the absence of polyamines, for example, when divalent or monovalent ions were used instead14, 15.
Figure 1: Polyamine-mediated DNA sequence recognition observed in MD simulations and smFRET experiments.
(a) Set-up of MD simulations. A pair of parallel 20-bp dsDNA duplexes is surrounded by aqueous solution (semi-transparent surface) containing 20 Sm4+ molecules (which compensates exactly the charge of DNA) and 100mM NaCl. Under periodic boundary conditions, the DNA molecules are effectively infinite. A harmonic potential (not shown) is applied to maintain the prescribed distance between the dsDNA molecules. (b,c) Interaction free energy of the two DNA helices as a function of the DNA–DNA distance for repeat-sequence DNA fragments (b) and DNA homopolymers (c). (d) Schematic of experimental design. A pair of 120-bp dsDNA labelled with a Cy3/Cy5 FRET pair was encapsulated in a ~200-nm diameter lipid vesicle; the vesicles were immobilized on a quartz slide through biotin–neutravidin binding. Sm4+ molecules added after immobilization penetrated into the porous vesicles. The fluorescence signals were measured using a total internal reflection microscope. (e) Typical fluorescence signals indicative of DNA–DNA binding. Brief jumps in the FRET signal indicate binding events. (f) The fraction of traces exhibiting binding events at different Sm4+ concentrations for AT-rich, GC-rich, AT nonhomologous and CpG-methylated DNA pairs. The sequence of the CpG-methylated DNA specifies the methylation sites (CG sequence, orange), restriction sites (BstUI, triangle) and primer region (underlined). The degree of attractive interaction for the AT nonhomologous and CpG-methylated DNA pairs was similar to that of the AT-rich pair. All measurements were done at [NaCl]=50mM and T=25°C. (g) Design of the hybrid DNA constructs: 40-bp AT-rich and 40-bp GC-rich regions were flanked by 20-bp common primers. The two labelling configurations permit distinguishing parallel from anti-parallel orientation of the DNA. (h) The fraction of traces exhibiting binding events as a function of NaCl concentration at fixed concentration of Sm4+ (1mM). The fraction is significantly higher for parallel orientation of the DNA fragments.
Unexpectedly, we found that DNA sequence has a profound impact on the strength of attractive interaction. The absolute value of ΔG at minimum relative to the value at maximum separation, |ΔGmin|, showed a clearly rank-ordered dependence on the DNA sequence: |ΔGmin| of (A)20>|ΔGmin| of (AT)10>|ΔGmin| of (GC)10>|ΔGmin| of (G)20. Two trends can be noted. First, AT-rich sequences attract each other more strongly than GC-rich sequences16. For example, |ΔGmin| of (AT)10 (1.5kcalmol−1 per turn) is about twice |ΔGmin| of (GC)10 (0.8kcalmol−1 per turn) (Fig. 1b). Second, duplexes having identical AT content but different partitioning of the nucleotides between the strands (that is, (A)20 versus (AT)10 or (G)20 versus (GC)10) exhibit statistically significant differences (~0.3kcalmol−1 per turn) in the value of |ΔGmin|.
To validate the findings of MD simulations, we performed single-molecule fluorescence resonance energy transfer (smFRET)17 experiments of vesicle-encapsulated DNA molecules. Equimolar mixture of donor- and acceptor-labelled 120-bp dsDNA molecules was encapsulated in sub-micron size, porous lipid vesicles18 so that we could observe and quantitate rare binding events between a pair of dsDNA molecules without triggering large-scale DNA condensation2. Our DNA constructs were long enough to ensure dsDNA–dsDNA binding that is stable on the timescale of an smFRET measurement, but shorter than the DNA’s persistence length (~150bp (ref. 19)) to avoid intramolecular condensation20. The vesicles were immobilized on a polymer-passivated surface, and fluorescence signals from individual vesicles containing one donor and one acceptor were selectively analysed (Fig. 1d). Binding of two dsDNA molecules brings their fluorescent labels in close proximity, increasing the FRET efficiency (Fig. 1e).
FRET signals from individual vesicles were diverse. Sporadic binding events were observed in some vesicles, while others exhibited stable binding; traces indicative of frequent conformational transitions were also observed (Supplementary Fig. 1A). Such diverse behaviours could be expected from non-specific interactions of two large biomolecules having structural degrees of freedom. No binding events were observed in the absence of Sm4+ (Supplementary Fig. 1B) or when no DNA molecules were present. To quantitatively assess the propensity of forming a bound state, we chose to use the fraction of single-molecule traces that showed any binding events within the observation time of 2min (Methods). This binding fraction for the pair of AT-rich dsDNAs (AT1, 100% AT in the middle 80-bp section of the 120-bp construct) reached a maximum at ~2mM Sm4+(Fig. 1f), which is consistent with the results of previous experimental studies2, 3. In accordance with the prediction of our MD simulations, GC-rich dsDNAs (GC1, 75% GC in the middle 80bp) showed much lower binding fraction at all Sm4+ concentrations (Fig. 1b,c). Regardless of the DNA sequence, the binding fraction reduced back to zero at high Sm4+ concentrations, likely due to the resolubilization of now positively charged DNA–Sm4+ complexes2, 3, 13.
Because the donor and acceptor fluorophores were attached to the same sequence of DNA, it remained possible that the sequence homology between the donor-labelled DNA and the acceptor-labelled DNA was necessary for their interaction6. To test this possibility, we designed another AT-rich DNA construct AT2 by scrambling the central 80-bp section of AT1 to remove the sequence homology (Supplementary Table 1). The fraction of binding traces for this nonhomologous pair of donor-labelled AT1 and acceptor-labelled AT2 was comparable to that for the homologous AT-rich pair (donor-labelled AT1 and acceptor-labelled AT1) at all Sm4+ concentrations tested (Fig. 1f). Furthermore, this data set rules out the possibility that the higher binding fraction observed experimentally for the AT-rich constructs was caused by inter-duplex Watson–Crick base pairing of the partially melted constructs.
Next, we designed a DNA construct named ATGC, containing, in its middle section, a 40-bp AT-rich segment followed by a 40-bp GC-rich segment (Fig. 1g). By attaching the acceptor to the end of either the AT-rich or GC-rich segments, we could compare the likelihood of observing the parallel binding mode that brings the two AT-rich segments together and the anti-parallel binding mode. Measurements at 1mM Sm4+ and 25 or 50mM NaCl indicated a preference for the parallel binding mode by ~30% (Fig. 1h). Therefore, AT content can modulate DNA–DNA interactions even in a complex sequence context. Note that increasing the concentration of NaCl while keeping the concentration of Sm4+ constant enhances competition between Na+ and Sm4+ counterions, which reduces the concentration of Sm4+ near DNA and hence the frequency of dsDNA–dsDNA binding events (Supplementary Fig. 2).
Methylation determines the strength of DNA–DNA attraction
Analysis of the MD simulations revealed the molecular mechanism of the polyamine-mediated sequence-dependent attraction (Fig. 2). In the case of the AT-rich fragments, the bulky methyl group of thymine base blocks Sm4+ binding to the N7 nitrogen atom of adenine, which is the cation-binding hotspot21, 22. As a result, Sm4+ is not found in the major grooves of the AT-rich duplexes and resides mostly near the DNA backbone (Fig. 2a,d). Such relocated Sm4+ molecules bridge the two DNA duplexes better, accounting for the stronger attraction16, 23, 24, 25. In contrast, significant amount of Sm4+ is adsorbed to the major groove of the GC-rich helices that lacks cation-blocking methyl group (Fig. 2b,e).
Figure 2: Molecular mechanism of polyamine-mediated DNA sequence recognition.
(a–c) Representative configurations of Sm4+ molecules at the DNA–DNA distance of 28Å for the (AT)10–(AT)10 (a), (GC)10–(GC)10 (b) and (GmC)10–(GmC)10 (c) DNA pairs. The backbone and bases of DNA are shown as ribbon and molecular bond, respectively; Sm4+ molecules are shown as molecular bonds. Spheres indicate the location of the N7 atoms and the methyl groups. (d–f) The average distributions of cations for the three sequence pairs featured in a–c. Top: density of Sm4+ nitrogen atoms (d=28Å) averaged over the corresponding MD trajectory and the z axis. White circles (20Å in diameter) indicate the location of the DNA helices. Bottom: the average density of Sm4+ nitrogen (blue), DNA phosphate (black) and sodium (red) atoms projected onto the DNA–DNA distance axis (x axis). The plot was obtained by averaging the corresponding heat map data over y=[−10, 10] Å. See Supplementary Figs 4 and 5 for the cation distributions at d=30, 32, 34 and 36Å.
If indeed the extra methyl group in thymine, which is not found in cytosine, is responsible for stronger DNA–DNA interactions, we can predict that cytosine methylation, which occurs naturally in many eukaryotic organisms and is an essential epigenetic regulation mechanism26, would also increase the strength of DNA–DNA attraction. MD simulations showed that the GC-rich helices containing methylated cytosines (mC) lose the adsorbed Sm4+ (Fig. 2c,f) and that |ΔGmin| of (GC)10 increases on methylation of cytosines to become similar to |ΔGmin| of (AT)10 (Fig. 1b).
To experimentally assess the effect of cytosine methylation, we designed another GC-rich construct GC2 that had the same GC content as GC1 but a higher density of CpG sites (Supplementary Table 1). The CpG sites were then fully methylated using M. SssI methyltransferase (Supplementary Fig. 3; Methods). As predicted from the MD simulations, methylation of the GC-rich constructs increased the binding fraction to the level of the AT-rich constructs (Fig. 1f).
The sequence dependence of |ΔGmin| and its relation to the Sm4+ adsorption patterns can be rationalized by examining the number of Sm4+ molecules shared by the dsDNA molecules (Fig. 3a). An Sm4+ cation adsorbed to the major groove of one dsDNA is separated from the other dsDNA by at least 10Å, contributing much less to the effective DNA–DNA attractive force than a cation positioned between the helices, that is, the ‘bridging’ Sm4+ (ref. 23). An adsorbed Sm4+ also repels other Sm4+ molecules due to like-charge repulsion, lowering the concentration of bridging Sm4+. To demonstrate that the concentration of bridging Sm4+ controls the strength of DNA–DNA attraction, we computed the number of bridging Sm4+ molecules, Nspm (Fig. 3b). Indeed, the number of bridging Sm4+ molecules ranks in the same order as |ΔGmin|: Nspm of (A)20>Nspm of (AT)10≈Nspm of (GmC)10>Nspm of (GC)10>Nspm of (G)20. Thus, the number density of nucleotides carrying a methyl group (T and mC) is the primary determinant of the strength of attractive interaction between two dsDNA molecules. At the same time, the spatial arrangement of the methyl group carrying nucleotides can affect the interaction strength as well (Fig. 3c). The number of methyl groups and their distribution in the (AT)10 and (GmC)10 duplex DNA are identical, and so are their interaction free energies, |ΔGmin| of (AT)10≈|ΔGmin| of (GmC)10. For AT-rich DNA sequences, clustering of the methyl groups repels Sm4+ from the major groove more efficiently than when the same number of methyl groups is distributed along the DNA (Fig. 3b). Hence, |ΔGmin| of (A)20>|ΔGmin| of (AT)10. For GC-rich DNA sequences, clustering of the cation-binding sites (N7 nitrogen) attracts more Sm4+ than when such sites are distributed along the DNA (Fig. 3b), hence |ΔGmin| is larger for (GC)10 than for (G)20.
Figure 3: Methylation modulates the interaction free energy of two dsDNA molecules by altering the number of bridging Sm4+.
(a) Typical spatial arrangement of Sm4+ molecules around a pair of DNA helices. The phosphates groups of DNA and the amine groups of Sm4+ are shown as red and blue spheres, respectively. ‘Bridging’ Sm4+molecules reside between the DNA helices. Orange rectangles illustrate the volume used for counting the number of bridging Sm4+ molecules. (b) The number of bridging amine groups as a function of the inter-DNA distance. The total number of Sm4+ nitrogen atoms was computed by averaging over the corresponding MD trajectory and the 10Å (x axis) by 20Å (y axis) rectangle prism volume (a) centred between the DNA molecules. (c) Schematic representation of the dependence of the interaction free energy of two DNA molecules on their nucleotide sequence. The number and spatial arrangement of nucleotides carrying a methyl group (T or mC) determine the interaction free energy of two dsDNA molecules.
Genome-wide investigations of chromosome conformations using the Hi–C technique revealed that AT-rich loci form tight clusters in human nucleus27, 28. Gene or chromosome inactivation is often accompanied by increased methylation of DNA29 and compaction of facultative heterochromatin regions30. The consistency between those phenomena and our findings suggest the possibility that the polyamine-mediated sequence-dependent DNA–DNA interaction might play a role in chromosome folding and epigenetic regulation of gene expression.
Rau, D. C., Lee, B. & Parsegian, V. A.Measurement of the repulsive force between polyelectrolyte molecules in ionic solution: hydration forces between parallel DNA double helices. Proc. Natl Acad. Sci. USA81, 2621–2625 (1984).
Raspaud, E., Olvera de la Cruz, M., Sikorav, J. L. & Livolant, F.Precipitation of DNA by polyamines: a polyelectrolyte behavior. Biophys. J.74, 381–393 (1998).
Grosberg, A. Y., Nguyen, T. T. & Shklovskii, B. I.The physics of charge inversion in chemical and biological systems. Rev. Mod. Phys.74, 329–345 (2002).
Thomas, T. & Thomas, T. J.Polyamines in cell growth and cell death: molecular mechanisms and therapeutic applications. Cell. Mol. Life Sci.58, 244–258 (2001).
Controversial Case of Sarepta’s eteplirsen, for DMD gained a Support Letter to FDA by Site Investigators and Advisers working on the drug
Reporter: Aviva Lev-Ari, PhD, RN
UPDATED on 7/21/2025
What does Elevidys’ future look like now?
Some backstory first: The FDA first approved Elevidys in June 2023, granting it accelerated clearance for Duchenne patients who were 4 or 5 years old and could still walk. One year later, the agency expanded the treatment’s OK to include ambulatory and non-ambulatory Duchenne patients who were 4 years of age or older.
Both decisions were controversial, as Sarepta’s trial data for Elevidys didn’t prove the therapy could significantly improve motor function. But the Duchenne community mostly embraced Elevidys, propelling it to the fastest market launch of any gene therapy approved in the U.S.
Safety concerns prompted by the recent deaths could now change that. But without Sarepta’s cooperation, the FDA’s options for stopping Elevidys sales altogether are limited. And those it does have at its disposal are likely to take time.
In refusing the FDA’s request to halt sales, Sarepta maintained there has been no new information indicating greater safety risk in younger, ambulatory patients. Both of the Elevidys patients who died were teenagers whose disease had eroded their ability to walk.
The distinction is clinically important as Elevidys is dosed by weight, so younger, lighter patients receive less of the drug. And as they’re earlier in their disease course, the treatment window to forestall further damage may be greater.
The distinction also has regulatory implications. In June 2024, when the FDA expanded Elevidys’ approval it did so by converting its accelerated approval to full for ambulatory patients. In non-ambulatory patients, Elevidys’ clearance remains “accelerated,” which indicates Sarepta is required to produce further evidence of its benefit in this group.
For the time being, the FDA has informed Sarepta that Elevidys’ indication should be limited to ambulatory patients only. The agency said in a statement that it is “committed to further investigating the safety of the product in ambulatory patients and will take additional steps to protect patients as needed.” — Ned Pagliarulo
Why did the FDA wait to step in?
The FDA claimed Friday it took “swift action” by suspending multiple Sarepta trials and requesting the company halt all Elevidys shipments. It took those steps following “new safety concerns” that the agency said showed patients may be exposed to “unreasonable and significant risk of illness or injury.”
“We believe in access to drugs for unmet medical needs but are not afraid to take immediate action when a serious safety signal emerges,” FDA Commissioner Martin Makary said in the statement.
In stunning twist, FDA approves Sarepta’s Duchenne drug it rejected
Vyondys 53, a type of nucleic acid therapy, is now conditionally cleared for the roughly 8% of patients with Duchenne who are “amenable” to exon 53 skipping, the mechanism by which the drug works. Sarepta said it would price Vyondys 53 “at parity” with Exondys 51, which controversially won FDA approval in 2016. Exondys 51 costs about $300,000 per year per child.
Exondys 51 was the first medicine specifically approved to treat Duchenne. Its approval sparked internal debate at the FDA, with agency officials at the Division of Neurology Products, the director of the Office of New Drugs and the acting chief scientist arguing in favor of rejection. They were overruled by Janet Woodcock, head of the FDA’s Center for Drug Evaluation and Research.
The FDA’s rejection of Vyondys 53 had stirred speculation the agency had done so for political reasons tied to the controversy over Exondys 51.
Company CEO Doug Ingram steadfastly refused to say much about the decision other than that it related to infection and kidney toxicity risks, and asked the patient community to not pressure the agency to change its mind.
Approval is conditional on Sarepta running a confirmatory study to prove Vyondys 53 delivers a clinical benefit. The data Sarepta used to support its application showed that treatment increased levels of dystrophin, a critical muscle protein missing in patients with DMD, from 0.1% of normal to 1.02% on average after 48 weeks.
Sarepta ($SRPT) enjoyed a rare spike in its share price today after several dozen experts in Duchenne muscular dystrophy, including a host of site investigators and advisers working on the drug, circulated a detailed letter explaining why the FDA should support the company’s application for eteplirsen–despite a withering internal assessment from agency insiders.
On a point-by-point basis these physicians took exception to the FDA review, noting that despite the extremely small number of patients in the key study–with data on only 12 boys–their experience observing patients in their practices suggest that the drug was clearly effective. As news of the letter spread, Sarepta’s share price surged 20%.
“The collective signatories note that the group of 12 eteplirsen treated boys, even accounting for daily deflazacort usage or twice-weekly prednisone, is clearly performing better than our collective clinical experience and the published literature would predict,” the lineup of physicians asserts. “Collectively, a portion of us represent a group of physicians who have observed over 5,000 DMD patients in our practices over an average of more than 15 years. Published external natural history data and our clinical experience strongly support that the 12 boys treated for over 4 years show a milder clinical progression, likely due to a positive treatment effect of eteplirsen.”
A quick check of Sarepta’s websites and online records also revealed that many of the as-advertised experts have close ties to Sarepta, often listed as the very investigators who have been helping Sarepta gather the controversial data together and analyze it for regulators. Among those who have worked on drug trials related to eteplirsen and signed the letter are UCLA’s Perry Shieh (a principal investigator for one study), Stanford’s John Day (who lists Confirmatory Study of Eteplirsen in DMD Patients on his resume; and Kathy Mathews at the University of Iowa (a hub site investigator and principal). Harvard Med School’s Louis Kunkel and the University of Washington’s Jeff Chamberlain, who signed on to the company’s advisory board, are also signers on the lobbying letter, along with many trial site leaders, including Anne Connolly, Susan Apkon, Nancy Kuntz and Basil Darras, all listed on Sarepta’s website.
Dr. M. Carrie Miceli and Dr. Stanley Nelson, co-director of the Center for Muscular Dystrophy at UCLA, took the lead on the letter, which was dated February 24 and addressed to Dr. Billy Dunn, director of the division of neurology products at the FDA. The wife/husband team launched a public campaign advocating for new drugs to be approved for Duchenne, which afflicts their teenage son. Miceli, Nelson and Chamberlain also sit on the advisory board of CureDuchenne, an advocacy group which has offered its full-throated support of eteplirsen’s approval, along with Sarepta CEO Ed Kaye.
In their view, which you can see here, the best approach would be to go ahead and approve eteplirsen and then go ahead and let upcoming data provide confirmatory results.
“The FDA Briefing Document also implies that the ongoing non-placebo controlled confirmatory eteplirsen trial (NCT02255552) and additional eteplirsen safety studies (NCT02420379 and NCT02286947) initiated in response to FDA guidance may not be considered sufficiently robust to allow for approval,” the letter reads. “Given the relative paucity of patients with amenable mutations, the flexibility afforded by FDASIA, and the fact that many of the boys between the ages of 4 and 21 years with relevant mutations are already receiving eteplirsen in the context of these trials, it would be difficult to conduct a large placebo controlled study in the near future. Thus, it would be dubiously ethical to veer from the currently recommended study path at this point. In keeping with the criteria imposed by FDASIA for accelerated approval for rare disease with unmet need, we conclude that the aggregate data, described in the briefing documents, are providing substantial evidence of efficacy and use in the greater population of boys amenable to exon 51 skipping is appropriate. We suggest that the most scientifically robust way forward and the most ethical choice for the Duchenne community is in the context of an accelerated approval followed by a confirmatory trial.”
Just how persuasive this group can be, with such close ties to Sarepta, won’t be clear until April 25, when the FDA’s advisory committee will finally meet for a review. The FDA’s internal assessment virtually dismissed Sarepta’s case. But the biotech has enjoyed intense support from parents and patients–as well as the professional community, which has played a big role in testing the drug.
Article ID #201: Correspondence on Leadership in Genomics and other Gene Curations: Dr. Williams with Dr. Lev-Ari. Published on 2/18/2016
WordCloud Image Produced by Adam Tubman
Correspondence on Leadership in Genomics and other Gene Curations: Dr. Williams with Dr. Lev-Ari, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 1: Next Generation Sequencing (NGS)
Correspondence on Leadership in Genomics and other Gene Curations: Dr. Williams with Dr. Lev-Ari
Authors: Stephen J Williams, PhD and Aviva Lev-Ari, PhD, RN
Subject: Re: In light of — >>>>>> Leadership in Genomics: VarElect Variants in Disease and UCSC Genome Technology Center | Leaders in Pharmaceutical Business Intelligence
Leadership in Genomics: VarElect Variants in Disease and UCSC Genome Technology Center
Subject: re: In light of — >>>>>> Leadership in Genomics: VarElect Variants in Disease and UCSC Genome Technology Center | Leaders in Pharmaceutical Business Intelligence
Hi Aviva,
I am not sure what is being proposed here. In the cancer area, there are at least 1,200 genes implicated somehow in this disease and new ones are reported every day. This is a colossal task!
Subject: In light of — >>>>>> Leadership in Genomics: VarElect Variants in Disease and UCSC Genome Technology Center | Leaders in Pharmaceutical Business Intelligence
Dear Dr. Williams,
HERE I am thinking LOUD
Is it possible to go to the dashboard, all posts and click on your Name, you will get the Universe of ~200 articles that you published.
HOW one could search or one needs to visually glance at the title of each — so as to pull a subset of posts that are dedicated to a GENE.
Create an Excel File, place each gene inside and go to Weizmann Institute’s genecards.org and pullout from them respective data on that gene
By so doing we will have LPBI’s Gene Inventory which we could reference in the Drug Discovery process, we do more and more, as we are aggregating all Biologics under the Joint Venture with SBH Sciences, Inc.
In light of :
Leadership in Genomics: VarElect Variants in Disease and UCSC Genome Technology Center
1. HOW could we take this “to be create Excel File” to be published a PAGE, Password Protected as your Curation, it needs to have a Parent or a Hierarchy of Nesting in the Website architecture
And subject that to your our search into New Medicine, Inc. NM/OK DB for data complementarity compilation?
2. What Foundation Medicine, Now Roche, does have vs. Weizmann Institute’s genecards.org
3. Will Weizmann Institute’s genecards.org be interested in New Medicine, Inc., NM/OK DB?
4. I have explored with Foundation Medicine, Now Roche regarding New Medicine, Inc., NM/OK DB and their reply was that they focus ONLY on Genomics data in Cancer, thus,, no interest in New Medicine, Inc. NM/OK DB, there
5. What is in Weizmann Institute’s genecards.org that is NOT in UC Santa Cruz DBs ?
8. Doudna started her professorship at Yale University in 1994. While the group was able to grow high-quality crystals, they struggled with thephase problem due to unspecific binding of the metal ions. One of her early graduate students and later her husband, Jamie Cate decided to soak the crystals in osmiumhexamine to imitate magnesium. Using this strategy, they were able to solve the structure, the second solved folded RNA structure since tRNA.[9][10] The magnesium ions would cluster at the center of the ribozyme and would serve as a core for RNA folding similar to that of a hydrophobic core of a protein.[5]
9. In 2015, Doudna gave a TED Talk about the bioethics of using CRISPR. [13]
Precision medicines have the ability to transform healthcare and treat a myriad of unmet medical needs. The Caribou technology platform has the ability to generate transformative medicines in multiple different market segments.
Our current therapeutic areas of exploration include anti-microbials, animal health, and therapeutic bioproduction.
Human therapeutics
In 2014, Caribou co-founded Intellia Therapeutics to develop curative medicines utilizing the Caribou CRISPR-Cas9 platform. Rachel Haurwitz, President and Chief Executive Officer of Caribou, is a member of Intellia’s Board of Directors.
Intellia is developing human gene and cell therapies for both ex vivo and in vivo applications using CRISPR-Cas9 gene editing technology. Near-term ex vivo applications include the treatment of blood disorders and cancer. In January 2015, Intellia announced a five-year research and development collaboration with Novartis to accelerate the ex vivo development of new CRISPR-Cas9-based therapies using chimeric antigen receptor T cells (CARTs) and hematopoetic stem cells (HSCs).
Subject: Re: Leadership in Genomics: VarElect Variants in Disease and UCSC Genome Technology Center | Leaders in Pharmaceutical Business Intelligence
Every post I do that contains a gene in the post is curated with a link to genecards database so later it not only can be searched but is an integrated knowledge-analysis base integrated with a knowledge and fully integrated Omics database as gene cards . org also contains protein, structure and functional databases.
This is where I always felt the power of LPBI was in the genomic space, integration of a deep analysis curated database
Subject: Fwd: Leadership in Genomics: VarElect – Variants in Disease and UCSC Genome Technology Center | Leaders in Pharmaceutical Business Intelligence
Subject: Re: The Science Coming in 2016 – OpenMind
I want you to go to http://www.genecards.org/ then pick a gene and scroll down. You will see a database there for CRISPR products available from different distributors including Qiagen, Promega, Fisher Scientific, Santa Cruz as well as others. This seems to be already underway. It is possible to copy what these companies are already doing but I don’t see the business advantage in that. Please remember that 3D printing involves layering a of first and second dimension to a third dimension product. So for instance the cell would be the “first dimension” even though it is three dimensional but the effect of layering MULTIPLE layers of cells is what gives their 3D effect. The biomaterial you put in each tube is, in essence, your first dimension you are going to layer into a multilayered “3D” structure.
DNA can be made by synthesizers, there is no need to bioprint it, especially short fragments and in fact you wouldn’t. They can handle even longer material. Possibly if you want to replace a whole nucleosome but the chemistry is not there. That is fine working with Jennifer Duodna making a library of small guide RNA’s to be used in CRISPR however it seems to be in process as I said before. This would need to be done with her system and optimized for her system. You would also need a huge operation to do validation as well. In addition the number of mutations, SNPs, variants are extremely large and many are not disease specific.
Again each would have to be validated. In addition, unless you are doing embryo manipulation, you will need to partner with a company that has a good gene delivery system. This will cost $, probably around 500 million.
This gene fragment in red color — I am suggesting to build with 3D BioPrinting,
at the Oligonucleotide level.
Create a library of fragments for the most common mismatch in transcriptions, as well as on demand for rare deletions.
Per University of California, Santa Cruz, Database of Variations, prepare an INVENTORY of GENE REPAIR PARTS, manage the inventory by Analytics, where each part was implanted and monthly interval monitoring of segment incorporation and new function of protein folding achieved.
Trace the genetic therapy achieved by Gene editing.
Regulatory DNA Engineered, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 2: CRISPR for Gene Editing and DNA Repair
Regulatory DNA engineered
Larry H. Bernstein, MD, FCAP, Curator
LPBI
New Type of CRISPR Screen Probes the Regulatory Genome
February 8, 2016 | When a geneticist stares down the 3 billion DNA base pairs of the human genome, searching for a clue to what’s gone awry in a single patient, it helps to narrow the field. One of the most popular places to look is the exome, the tiny fraction of our DNA―less than 2%―that actually codes for proteins. For patients with rare genetic diseases, which might be fully explained by one key mutation, many studies sequence the whole exome and leave all the noncoding DNA out. Similarly, personalized cancer tests, which can help bring to light unexpected treatment options, often sequence the tumor exome, or a smaller panel of protein-coding genes.
Unfortunately, we know that’s not the whole picture. “There are a substantial number of noncoding regions that are just as effective at turning off a gene as a mutation in the gene itself,” says Richard Sherwood, a geneticist at Brigham and Women’s Hospital in Boston. “Exome sequencing is not going to be a good proxy for what genes are working.”
Sherwood studies regulatory DNA, the vast segment of the genome that governs which genes are turned on or off in any cell at a given time. It’s a confounding area of genetics; we don’t even know how much of the genome is made up of these regulatory elements. While genes can be recognized by the presence of “start” and “stop” codons―sequences of three DNA letters that tell the cell’s molecular machinery which stretches of DNA to transcribe into RNA, and eventually into protein―there are no definite signs like this for regulatory DNA.
Instead, studies to discover new regulatory elements have been somewhat trial-and-error. If you suspect a gene’s activity might be regulated by a nearby DNA element, you can inhibit that element in a living cell, and see if your gene shuts down with it.
With these painstaking experiments, scientists can slowly work their way through potential regulatory regions―but they can’t sweep across the genome with the kind of high-throughput testing that other areas of genetics thrive on. “Previously, you couldn’t do these sorts of tests in a large form, like 4,000 of them at once,” says David Gifford, a computational biologist at MIT. “You would really need to have a more hypothesis-directed methodology.”
Recently, Gifford and Sherwood collaborated on a paper, published in Nature Biotechnology, which presents a new method for testing thousands of DNA loci for regulatory activity at once. Their assay, called MERA (multiplexed editing regulatory assay), is built on the recent technology boom in CRISPR-Cas9 gene editing, which lets scientists quickly and easily cut specific sequences of DNA out of the genome.
So far, their team, including lead author Nisha Rajagopal from Gifford’s lab, has used MERA to study the regulation of four genes involved in the development of embryonic stem cells. Already, the results have defied the accepted wisdom about regulatory DNA. Many areas of the genome flagged by MERA as important factors in gene expression do not fall into any known categories of regulatory elements, and would likely never have been tested with previous-generation methods.
“Our approach allows you to look away from the lampposts,” says Sherwood. “The more unbiased you can be, the more we’ll actually know.”
A New Kind of CRISPR Screen
In the past three years, CRISPR-Cas9 experiments have taken all areas of molecular biology by storm, and Sherwood and Gifford are far from the first to use the technology to run large numbers of tests in parallel. CRISPR screens are an excellent way to learn which genes are involved in a cellular process, like tumor growth or drug resistance. In these assays, scientists knock out entire genes, one by one, and see what happens to cells without them.
This kind of CRISPR screen, however, operates on too small a scale to study the regulatory genome. For each gene knocked out in a CRISPR screen, you have to engineer a strain of virus to deliver a “guide RNA” into the cellular genome, showing the vicelike Cas9 molecule which DNA region to cut. That works well if you know exactly where a gene lies and only need to cut it once—but in a high-throughput regulatory test, you would want to blanket vast stretches of DNA with cuts, not knowing which areas will turn out to contain regulatory elements. Creating a new virus for each of these cuts is hugely impractical.
The insight behind MERA is that, with the right preparation, most of the genetic engineering can be done in advance. Gifford and Sherwood’s team used a standard viral vector to put a “dummy” guide RNA sequence, one that wouldn’t tell Cas9 to cut anything, into an embryonic stem cell’s genome. Then they grew plenty of cells with this prebuilt CRISPR system inside, and attacked each one with a Cas9 molecule targeted to the dummy sequence, chopping out the fake guide.
Normally, the result would just be a gap in the CRISPR system where the guide once was. But along with Cas9, the researchers also exposed the cells to new, “real” guide RNA sequences. Through a DNA repair mechanism called homologous recombination, the cells dutifully patched over the gaps with new guides, whose sequences were very similar to the missing dummy code. At the end of the process, each cell had a unique guide sequence ready to make cuts at a specific DNA locus—just like in a standard CRISPR screen, but with much less hands-on engineering.
By using a large enough library of guide RNA molecules, a MERA screen can include thousands of cuts that completely tile a broad region of the genome, providing an agnostic look at anywhere regulatory elements might be hiding. “It’s a lot easier [than a typical CRISPR screen],” says Sherwood. “The day the library comes in, you just perform one PCR reaction, and the cells do the rest of the work.”
In the team’s first batch of MERA screens, they created almost 4,000 guide RNAs for each gene they studied, covering roughly 40,000 DNA bases of the “cis-regulatory region,” or the area surrounding the gene where most regulatory elements are thought to lie. It’s unclear just how large any gene’s cis-regulatory region is, but 40,000 bases is a big leap from the highly targeted assays that have come before.
“We’re now starting to do follow-up studies where we increase the number of guide RNAs,” Sherwood adds. “Eventually, what you’d like is to be able to tile an entire chromosome.”
Far From the Lampposts
Sherwood and Gifford tried to focus their assays on regions that would be rich in regulatory elements. To that end, they made sure their guide RNAs covered parts of the genome with well-known signs of regulatory activity, like histone markers and transcription factor binding sites. For many of these areas, Cas9 cuts did, in fact, shut down gene expression in the MERA screens.
But the study also targeted regions around each gene that were empty of any known regulatory features. “We tiled some other regions that we thought might serve as negative controls,” explains Gifford. “But they turned out not to be negative at all.”
The study’s most surprising finding was that several cuts to seemingly random areas of the genome caused genes to become nonfunctional. The authors named these DNA regions “unmarked regulatory elements,” or UREs. They were especially prevalent around the genes Tdgf1 and Zfp42, and in many cases, seemed to be every bit as necessary to gene activity as more predictable hits on the MERA screen.
These results caught the researchers so off guard that it was natural to wonder if MERA screens are prone to false positives. Yet follow-up experiments strongly supported the existence of UREs. Switching the guide RNAs from aTdgf1 MERA screen and a Zfp42 screen, for example, produced almost no positive results: the UREs’ regulatory effects were indeed specific to the genes near them.
In a more specific test, the researchers chose a particular URE connected to Tdgf1, and cut it out of a brand new population of cells for a closer look. “We showed that, if we deleted that region from the genome, the cells lost expression of the gene,” says Sherwood. “And then when we put it back in, the gene became expressed again. Which was good proof to us that the URE itself was responsible.”
From these results, it seems likely that follow-up MERA screens will find even more unknown stretches of regulatory DNA. Gifford and Sherwood’s experiments didn’t try to cover as much ground around their target genes as they might have, because the researchers assumed that MERA would mostly confirm what was already known. At best, they hoped MERA would rule out some suspected regulatory regions, and help show which regulatory elements have the biggest effect on gene expression.
“We tended to prioritize regions that had been known before,” Sherwood says. “Unfortunately, in the end, our datasets weren’t ideally suited to discovering these UREs.”
Getting to Basic Principles
MERA could open up huge swaths of the regulatory genome to investigation. Compared to an ordinary CRISPR screen, says Sherwood, “there’s only upside,” as MERA is cheaper, easier, and faster to run.
Still, interpreting the results is not trivial. Like other CRISPR screens, MERA makes cuts at precise points in the genome, but does not tell cells to repair those cuts in any particular way. As a result, a population of cells all carrying the same guide RNA can have a huge variety of different gaps and scars in their genomes, typically deletions in the range of 10 to 100 bases long. Gifford and Sherwood created up to 100 cells for each of their guides, and sometimes found that gene expression was affected in some but not all of them; only sequencing the genomes of their mutated cells could reveal exactly what changes had been made.
By repeating these experiments many times, and learning which mutations affect gene expression, it will eventually be possible to pin down the exact DNA bases that make up each regulatory element. Future studies might even be able to distinguish between regulatory elements with small and large effects on gene expression. In Gifford and Sherwood’s MERA screens, the target genes were altered to produce a green fluorescent protein, so the results were read in terms of whether cells gave off fluorescent light. But a more precise, though expensive, approach would be to perform RNA sequencing, to learn which cuts reduced the cell’s ability to transcribe a gene into RNA, and by how much.
A MERA screen offers a rich volume of data on the behavior of the regulatory genome. Yet, as with so much else in genetics, there are few robust principles to let scientists know where they should be focusing their efforts. Histone markers provide only a very rough sketch of regulatory elements, often proving to be red herrings on closer examination. And the existence of UREs, if confirmed by future experiments, shows that we don’t yet even know which areas of the genome to rule out in the hunt for regulatory regions.
“Every dataset we get comes closer and closer to computational principles that let us predict these regions,” says Sherwood. As more studies are conducted, patterns may emerge in the DNA sequences of regulatory elements that link UREs together, or reveal which histone markers truly point toward regulatory effects. There might also be functional clues hidden in these sequences, hinting at what is happening on a molecular level as regulatory elements turn genes on and off in the course of a cell’s development.
For now, however, the data is still rough and disorganized. For better and for worse, high-throughput tools like MERA are becoming the foundation for most discoveries in genetics—and that means there is a lot more work to do before the regulatory genome begins to come into focus.
CORRECTED 2/9/16: Originally, this story incorrectly stated that only certain cell types could be assayed with MERA for reasons related to homologous recombination. In fact, the authors see no reason MERA could not be applied to any in vitro cell line, and hope to perform screens in a wide range of cell types. The text has been edited to correct the error.




 .
.  . Assuming that the exon-inclusion read count ICk follows a binomial distribution with the total read count nk
. Assuming that the exon-inclusion read count ICk follows a binomial distribution with the total read count nk















{kind=link}
{kind=link}
{kind=link}
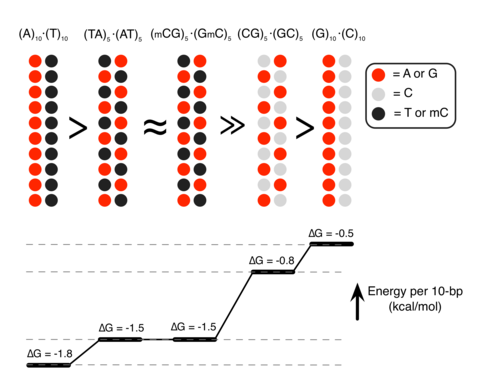{kind=link}