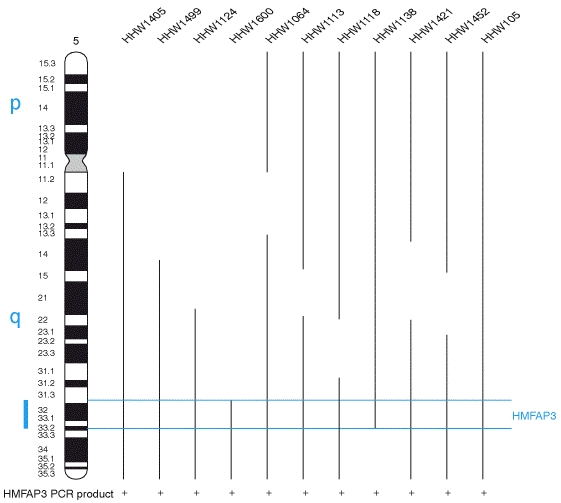
This diagram shows the chromosomes of Drosophila melanogaster approximately to scale. Chromosome sizes were based on basepair lengths given on the NCBI map viewer, and A. B. Carvalho, 2002. Curr. Op. Genet. & Devel. 12:664-668. Centimorgan distances were derived from selected loci listed in the NCBI website. (credit Wikipedia)
Introduction
Generally speaking sexually reproducing species are composed of individuals of two complementary mating types or sexes. An essential aspect of the developmental history of each individual is thus sex determination and differentiation. There exist two sex determination mechanisms, somatic and germline, that based on the chromosomal mechanism in the Drosophila melanogaster. In the somatic sex determination mechanism, each individual assesses the ratio of X-chromosomes to autosomal chromosome sets), the X:A ratio provides the primary sex-determining signal (reviewed by Cline and Meyer, 1996). When X:A=1, female differentiation ensues (Bridges, 1925), along with the male-mode of X-chromosome dosage compensation. The X:A ratio is calculated within each cell of the developing embryo, 2 hrs after fertilization. The X:A ratio determines the sex in Drosophila (Bridges, 1916, 1921, 1925) in a somatic-cell-autonomous manner that occurs early in embryonic development (Baker and Belote, 1983; Baker, 1989). Females possess two X-chromosomes, and males possess one X-chromosome and one Y-chromosome. The Y-chromosome is required only for spermatogenesis (Lindsley and Tokuyasu 1980; Bridges 1986), and will not be considered further. The number of X-chromosomes is counted through a mechanism involving positive-acting X-chromosome-encoded transcription factors, termed X-numerator elements (Cline, 1988), negative-acting autosome-encoded transcription factors or denominators, and signal transduction factors provided maternally. Among the X-numerators are sisterless-a, sisterless-b (sis-b), sisterless-c, and runt (Schurpbach, 1985; Cline, 1986, 1988; Steinmann-Zwicky et al., 1989; Parkhurst et al., 1990; Ericson and Cline, 1991, 1993; Estes, 1995; Hoshijima et al., 1995; reviewed by Cline, 1993).
The best candidate for a denominator gene is the deadpan (dpn) locus. Both daughterless (da) and extramacrochaete (emc) fulfill the role of maternally contributed transduction loci (Cline, 1976; Cronmiller et al., 1988). Both in vitro biochemical evidence and in vivo genetic evidence support the idea that transcription factors of the basic-helix-loop-helix (bHLH) family are able to form homo- and hetero-dimers; thus the X:A ratio counting mechanism seems to involve the relative affinities and chromosome-dependent stoiciometries of the bHLH proteins SIS-B, DA, EMC, and DPN. When X:A=1, sufficient SIS-B protein is synthesized so that it can effectively compete with the EMC and DPN proteins for binding to DA protein. DA:SIS:B heterodimers then bind to so-called establishment promoter (Pe) elements of the SXL gene and activates its transcription, resulting in an early burst of SXL protein that sets splicing and dosage compensation in to female-specific modes. When X:A=0.5, too little SIS-B is produced, and DA protein remains sequestered with EMC and DPN. The Sxl Pe remains inactive, and splicing and dosage compensation enters male-specific modes. In response to X:A ratio=1, an embryo specific promoter of the gene called Sex-lethal (Sxl) is activated (Keyes et al., 1932).
Sxl protein that acts as a master gene for the somatic germline sex determination, has three somatic functions. First, Sxl protein carries out autoregulation at the level of pre-mRNA splicing. Second, Sxl controls female-specific differentiation at the level of pre-RNA splicing and polyadenylation at least two genes that code for transcription factors that effect terminal differentiation. Third, Sxl protein negatively regulates X-chromosome dosage compensation. It does so in two ways, by alternative RNA splicing of a normally male-specific gene, and by translation-level regulation of many X-chromosomal transcripts during embryogenesis. In the male, with Sxl in the off state, male differentiation occurs because tra is in the off state and therefore the differentiation-effector transcription factors are produced in alternative male-specific modes. Dosage compensation is active, and the male X-chromosome is decorated by a minimum of four proteins and two RNA molecules that form a complex along the entire chromosome (reviewed by Cline and Meyer, 1996). Transcription of the male X-chromosome is elevated two-fold, and it produces the same amount of RNA per template as found in females.
Germline pathway for sex determination and dosage compensation is different than the somatic sex determination mechanism. (Figure 1) Figure 1: Sex determination of D. melanogaster (1998)The vast majority of somatic sex determination loci have no function in germline cells. For example, none of the X-chromosome numerators is required for proper oogenesis (Granadino et al., 1989, 1992; Steinmann-Zwicky 1991), despite the fact that proper oogenesis requires that X:A =1 in the germline (Schupbach, 1982, 1985) nor are tra, tra-2, and dsxF required for oogenesis. Sxl and snf have germline functions but the former is not a binary switch gene between oogenesis and spermatogenesis (Despande et al., 1996; Bopp et al., 1993, 1995; Hager et al., 1997). Systematic screens for female-sterile mutations have identified a large number of genes required for normal oogenesis (e.g. Gans et al., 1975; Mohler, 1977; Perrimon et al., 1986; Schupbach and Wieschaus, 19889, 1991). Female-sterility can arise in diverse ways, but one interesting class of mutations is germline-dependent and causes an “ovarian tumor” phenotype. “Ovarian tumor” mutations cause under-developed ovaries, in which egg chambers and ovarioles are filled with an excess of undifferentiated germ cells that have adopted male-like characteristics that include a prominent spherical nucleus, assembly of mitocondria around the nucleus, and mis-expression of male-specific marker genes (Oliver et al., 1988, 1990, 1993; Steinmann-Zwicky, 1988, 1992; Bopp et al., 1993; Pauli et al., Wei et al., 1994). Among the “ovarian tumor” class of genes are ovo, ovarian tumor (otu), fused, and two genes with somatic phenotypes, namely snf and Sxl. Strong mutations at the ovo and otu loci result in ovaries totally devoid of germ cells (King and Killey, 1982; Busson et al., 1983; Oliver et al., 1987; Mevel-Ninio et al., 1989; Rodesh et al., 1995), Weaker mutations at both loci result in viable germline cells that have abnormal male-like splicing at the Sxl gene (Oliver et al, 1993). The overall conclusion is that oogenesis requires a chromosomally female germline is wild type for ovo, otu, Sxl, and snf. If one of these genes is defective, either the germline will die or male-like differentiation and tumor formation ensure.
However, there are soma-germline interactions for a normal sex determination. (Figure 2) Figure 2: Somatic-Germline Interactions. (1998)Unlike the somatic regulatory hierarchy, which genetic mosaic experiments clearly showed functions in cell-autonomous fashion, sexual differentiation of the germline requires inductive signaling from somatic cells. This was shown by use of pole cell transplantation, the method of making mosaics in which germline cells surgically transferred from donor embryos (Schubach. 1985; Steinmann-Zwicky et al., 1989). These experiments show that proper germline differentiation requires a combination of germline-autonomous chromosomal cues and proper signaling from the soma. Evidence with tra and dsx mutant somatic hosts indicates these soma-germline interactions have detectable effects by larval stages (Steinmann-Zwicky., 1996).
The ovo gene is genetically complex. At least three transcripts are produced from the ovo region (Mevel-Ninio et al, 1991, 1995, 1996; Garfinkel et al., 1992, 1994). Two of these are germline-specific and correspond to the ovo function, while the third corresponds to the somatic-epidermal, non-sex-specific shavenbaby (svb) function. (For a schematic of the gene map please refer to Figure3)
The ovo function is transcribed from two closely spaced germline-specific promoters, ovo a and ovob, give rise to 5-kb mRNAs (Mevel-Ninio et al., 1991, 1995; Garfinkel et al., 1992, 1994). First identified promoter was ovob Garfinkel et al., (1994) and the leader exon it forms is called Exon 1b, 1028-codon-long open reading frame that contains four Cys2-His2 fingers at the carboxy terminus; protein MW of 110.6 kD. A second germline promoter, ovoa, was identified by Mevel-Ninio et al (1995), 1400 codons long, and predicts a 150.8-kD protein. This Exon 1a contains an in-frame AUG upstream of the translation start in Exon 2 utilized by the OvoB open reading frame. The OvoB mRNA isoforms is predominant during adult life, with the OvoA isoforms only appearing during Stage 14 of oogenesis (Mevel-Ninio et al., 1991, 1996; Garfinkel., 1994). The ovo zinc finger domain binds to its own germline promoter regions, to the otu promoter region (Garfinkel et al., 1997; Lee, 1998; Lee and Garfinkel 1998). This is consistent with ovo playing an important role in a sex determination hierarchy operating in germline cells that involves these other genes. The svb function is transcribed from an incompletely characterized somatic promoter that forms a 7.1 kb poly(A)+ mRNA (Garfinkel et al., 1994). This transcript accumulates 9-12-hr post-fertilization, in the somatic tissues that later in embryogenesis form the cuticular structures affected by svb mutations. Wieschaus et al. (1984) observed that ventral denticle belts and dorsal hairs are defective in svb mutations; hence the name, and svb mutations are polyphasic larval lethals. Exons and exon segments that are found in all mRNA forms coded by the region correspond to genomic DNA where so-called svb-ovo- mutations map (Mevel-Ninio et al., 1989; Garfinkel 1992). Finally, somatic-specific exons, exon segments, and transcriptional regions correspond to region mutable to the svb- ovo- phenotype. Since al known mRNA forms utilize the same splice junctions to join Exon3 to Exon4, all protein forms coded by the locus are believed to contain the same four zinc fingers at the carboxy terminus. A wide variety of evidence points to ovo playing a critical role in germline sex determination. High-level of ovo transcription in germline cells, as detected with Xgal staining of ovo promoter-lacZ constructs requires that they have a female karyotype (Oliver et al., 1994). Chromosomally male germline cells have low levels of ovo transcription even if the soma is transformed towards female through the use of hs-traF cDNA minigenes. Likewise, chromosomally female germline cells have high levels of ovo transcription even if the soma is anatomically male through the action of tra loss-of-function mutations. This argues that high-level of ovo transcription is a germline X: A ratio-autonomous property, and stands in contrast to related experiments with otu. In the case of otu, there is evidence that chromosomally male germline cells, which normally have no need of otu+ function at all, require otu- for proliferation when they are in a female host (Nagoshi et al., 1995). The D. melanogaster ovo gene is required for cell viability and differentiation of female germ cells, apparently playing a role in germline sex determination. While female X: A ratio in germline cells is required for high levels of ovo germline promoters. Therefore we undertook to identify trans-acting regulatory regions of the X-chromosome, with a particular interest in identifying candidate germline X-chromosome numerator elements. In this study, I screened X-chromosome using 45 deficiency strains, I found that these trans-regulating regions were grouped into 12 loci based on overlapping cytology. Five regions were trans-regulating activators, and seven were trans-regulating repressors; extrapolating to the entire genome, this result predicts nearly 85 loci. A subset of the dozen X-chromosomal regions correlated with previously identified E(ovoD) and Su(ovoD) loci (Pauli et al., 1995).
Materials and Methods
Fly Strains and Growth Flies were maintained on standard yeast/cornmeal medium and kept at 25oC and 18oC unless otherwise indicated. Mutants are described in Lindsley and Zimm (1992). The ovo3U21 and ovo4B8 were obtained from Brian Oliver of NIH; OvoD1rS1 FM3 is from the Garfinkel lab collection. The remaining stocks were obtained from the Bloomington Stock Center (see Table 2.1 for the list of stocks that had been used and Figure 2.1 for their location on the X Chromosome).
Outcrosses Outcrosses were designed to create transgenic flies so that screening of the X chromosome for trans-regulators of ovo in the germline can be done. Virgin female flies were collected 14 hour long windows at 18oC or 8 hour long windows at 25oC, during which newly emerged males remained immature. Collected females were kept 3-5 days to make sure they are virgin before outcrossing them. Heterozygous virgin females (5-7), carrying deficiency X-chromosomes balanced over first chromosome balancers were mated with males homozygous for either of two P-element transformation constructs of a lacZ reporter gene fused to the ovo promoter. Both events were inserted on third chromosome. They were grown at 25oC unless otherwise noted. The control class of F1 progeny has a complete X-chromosome pair, whereas the experimental class has one complete and one deficient X chromosome in its genome. The [ovo::lacZ constructs] were designed by Oliver et al., (1994). In this study two of their strains, ovo4B8 (pCOW+1.9) and ovo3U21 (pCOW-2.1) respectively, were used to determine the ovo promoter activity.
Outcrosses to Remove Duplications Several X-chromosome deficiencies in the Bloomington collection are carried in males, with compensatory duplications of X material on an autosome. These had to be crossed to eliminate the duplications (Fig 2.4). This was done as follows: FM3/FM7a virgin flies were mated to Df/Y; Dp males. Among the F1 progeny, half of the Df/(FM3 or FM7a) daughters will carry the unwanted duplication, and half will be free of the duplication. In some cases, presence of the duplication could be determined from the females’ phenotypes. In other cases, up to twenty individuals virgin Df(FM3 or FM7) F1 progeny were backcrossed to FM7a/Y males to establish stocks. In the F2, absence of the duplication could be established by examining sons; in all cases, the Df is male-lethal unless “rescued” by the duplication. Also FM3 is itself male lethal. Thus, single-female stocks that produce only FM7a sons had the desired genotypes and were kept for experiments.
X-Gal Staining In this assay ovaries from two-day-old adults were dissected in Drosophila Ringer’s solution (182 mM KCl, 46 mM NaCl, 3 mM CaCl2, 10mM TrisHCl, pH 6.8). Then, these tissues were transferred to a microtiter plate and fixed in 1% gluteraldehyde, 50mM Na-cacodylyte acid solution for 15 minutes. After rinsing the tissues, three times for 5 minutes each staining buffer (7.2 mM Na2HPO4, 2.8 mM NaH2PO4, 1.0 mM MgCl2, 0.15 mM NaCl), they were transferred to incubation buffer (staining buffer, 5 mM Fe2 (CN)3, 5 mM Fe3 (CN)2, 0.2% X-Gal) for an hour at 37oC. Next, tissues were washed three times 5 minutes each in washing buffer, which is a 1 mM EDTA, added PBS (130 mM NaCl, 7 mM Na2HPO4*2H2O, 3 mM NaH2PO4*2H2O, pH 7.0) solution. Finally, the tissues were dehydrated in ethanol solutions of increasing concentrations (50%, 75%, 95%) and mounted on a slide in Permount. Preparate concentrations were examined under a compound microscope to make correlations between staining and gene activity. Although it was easy to determine positive and negative controls, but this assay wasn’t sensitive enough to see subtle differences due to effects of deleted regions on ovo promoters driving LacZ.
Histochemical Assay of LacZ Activity This method allowed us to make quantitative measurements of lacZ activity due to ovo promoter function in animals heterozygous for X-chromosome deletions. Emerging F1 flies were collected and aged for two days before dissecting ovaries under a dissecting microscope. For each soluble assay, 10 flies were dissected. This is repeated at least seven assays (N, sample number) completed per stock for each construct. Ovaries from ten dissected outcrossed flies were out into eppendorf tubes containing 100ml of Assay Buffer (50 mM K-phosphate, 1 mM MgCl2 at pH 7.8) and homogenized about 20 strokes. For each dissected pair of ovaries 100 ml of assay buffer was used and the volume was completed to appropriate amount. After centrifuging for one minute, 20 ml of the supernatant was transferred into 980 ml of assay buffer (Simon and Lis, 1987; Ashburner, 1989) to make 2mM chlorophenol red-beta-D-galactopyranoside (CPRG). Absorbance at 574 nm was measured at half hour time intervals starting from zero to two hours hydrolysis of CPRG by chlorophenol (red CPRG). CPR has a molar extinction coefficient of 75,000 M-1 cm-1 (Boehringer-Manheim data sheet) and this is a very easily detected product of b-galactoside enzyme activity. Range finding experiments showed that 2mM of CPRG gives linear data for 2-3 hours often, color changes could be seen with the unaided eye. Two controls are shown in Figure 2.8 that validates CPRG for this work. Ovaries from a non-transformed strain (y w RD) were used to prepare soluble extracts. A near zero-absorbance at 574 nm was observed that did not appreciably change over several hours. In contrast, ovarian extracts from the ovo promoter-lacZ transformant strain ovo3U21 and ovo4B8 (Oliver et al, 1994) showed a steep linear increase in A 574 during the same period. The slopes of these lines were proportional to the amount of ovo3U21 and ovo4B8 extract added.
Bradford (1976) Assay For Protein This protein determination method is based on the binding of Coomasie Brilliant Blue G-250 to the protein. Preparation of protein reagent was done according to Bradford (1976). After 100 mg of Coomasie Brilliant Blue G-250 was dissolved in 50 ml 95% ethanol, and then 100 ml 85% (w/v) phosphoric acid was added. The resulting solution was diluted to a final volume of 1 liter [final concentrations in the reagent were 0.01% (w/v) Coomasie Brilliant Blue G-250, 4.7% (w/v) ethanol, and 8.5% (w/v) phosphoric acid]. 20ml of prepared soluble extract from the dissected tissues were used. This volume is diluted to 0.1ml with ddH2O, then 5ml of protein reagent was added to the test tube and contents were mixed. The absorbance at 595nm was measured after 2 min and before 1 hr in 3 ml cuvettes against a reagent blank prepared from 0.1 ml of the appropriate buffer and 5 ml of protein reagent. A standard curve using known quantities of bovine serum albumin (BSA) was constructed. Soluble extract absorbances were plotted on the standard curve and protein amount interpolated.
Statistical Analysis Average specific activity is calculated as nanomoles of substrate used per hour per nanogram protein expressed (nmole CPRG liberated /ng / hr). Sample number (N) always exceeded seven. Mean specific activity and standard error of the mean (SEM) were calculated for each experimental and control class. The F test was used to determine whether variances were equal, and therefore,, which type of student’s t-test calculation was appropriate. A significant difference between experimental and control values was identified by a P < 0.05 for the t-test score.
RESULTS
The results are given in three sections: X chromosome deficiency screening, negative autoregulation of ovo exhibited by deficiencies removing ovo, and gene dose analysis using P element transformants carrying extra copies of ovo.
X Chromosome Screening The presence of polytene chromosomes in the salivary glands, which have distinctive, banding patterns allows the map positions of genes to be correlated with physical features of the chromosomes. Breakpoint locations rearrangements, and the locations of cloned sequences can be easily established. Each of the major chromosome arms is divided into 20 numbered segments, except chromosome 4, which is divided into 4 regions. Each numbered region is then divided into six consecutive lettered regions, and each lettered region into numbered bands, for example 4E1. The precise relationship between physical length and the numbering scheme depends on local topography (Lefevre, 1976). In the summary tables, each deficiency listed according to cytological positions. The map of the X chromosome, including the deficiencies used in this study is given in Materials and Methods (Fig 1). Figure 1: Sex determination of D. melanogaster (1998) In Drosophila melanogaster germ cells, ovo has a primary role in female sex specific cell viability, proliferation and differentiation. Ovo responds to the number of X-chromosomes as assessed by high level expression (Oliver et al., 1994). Thus, the ovo promoter may be dependent upon X germline numerator elements. To identify possible trans-regulators of the ovo germline promoter (and, I hope, to identify germline numerators) I undertook deficiency screen for quantitative effects on ovo::lacZ reporter constructs. Determination of trans-regulation effect by any of the deletion mutant, was based on two general rules. If the excised part of the X chromosomes has any genes with the positive regulatory effects on ovo gene activity, then the levels of LacZ reporter gene function will be reduced in experimentals compared to control siblings. If the experimental class results in the elevation of the LacZ activity by producing high levels of enzyme compared to controls, the elevated region having removed a repression locus. Significant effects were determined by statistical analysis, which using a student’s t-test P value is less than or equal to 0.05. X-chromosome screening results are presented in Table 3.1 and 3.2. The entire X-chromosome deficiency set was tested twice: once with a 3.3kb ovo promoter fragment driving LacZ (strain ovo3u21), and separately with a 3.1kb ovo promoter (ovo4B8). Of 45 deficiencies that represent about 70% of the X-chromosome 17 deficiencies had significant effects in both ovo3U21 and ovo4B8 reporter activity, 1 deficiency had significant effects on only ovo3U21 and only 1 deficiency effect on ovo4B8. Some of these deficiencies partly overlap, allowing the identification of 11 regions that apparently contain trans-acting modifiers of ovo promoter activity six are positive regulators and five are negative.
Region 1-4. This region covers the eight overlapping deficiency lines, Df(1) BA1, Df(1)sc14, Df(1)64c18, Df(1)JC19, Df(1)dm75e19, Df(1)N8, Df(1)A113, DF(1)JC70. For three of them, Df(1)A113, Df(1)JC70, and Df(1)BA1, the student’s t-test probabilities show a significant difference between control and experimental siblings. The remaining strain has no significant trans-regulation effect on ovo gene activity. Df(1)BA1 enhanced the ovo gene expression activity about 20% when either ovo3U21 or ovo4B8 is used. It was suggested that a suppressor of ovoD (1F-2B+ locus) maps within 1E3-4 to 2B3-4 because of the dramatic gene dose effect of this region on the development of ovoD2/+ ovaries (Pauli et al, 1995). In contrast, it was found that Df(1)A113 and Df(1)JC70 have repressing effects on ovo expression. Df(1)A113 (3D6-E1; 4F5) removes several genes beside ovo, showed a very significant repression effect in outcrosses, about 82% and 47% (e/C), in ovo3U21 and ovo4B8 respectively. That data obtained in Df/+ females has a particular quantitative significance, which implies that the missing loci have the complementary effect. It was shown that this region is contains a gene or genes resulting in genetic unbalance (Cline et al., 1987). Also, Oliver et al., (1988) show that in deficiency lines, which they have used, strains removing both ovo and snf together are reducing viability of the progeny, that is, there is a synergistic interaction between ovo and snf.
Region 5-8. Twelve overlapping deletions have been tested in this region. Two deletions Df(1)N73 (5C3-5;5E-8) and Df(1)Lz90b24 (8B-D) caused very significant repressing effects, implying the presence of trans-activating loci, one deletion Df(1)RA2 (7D10;8A4-5) resulted in heterozygous experimentals with significant elevation in LacZ compared to siblings, implying a trans-repressor locus. It has been reposted that Df(1)RA2 strongly enhances ovoD phenotypes due to the function of otu+ in germline sex determination (Pauli et al., 1993). However, since out protein is cytoplasmic, it is unlikely that the Df(1)RA2 effect on ovo::lacZ promoter activity is due to changing dosage of otu. It is also suggested that there is a synergistic interaction between ovo and lozenge, eye phenotype, which is deleted by Df(1)Lz90b24, and here the data showed a trans-activating effect due to this deletion. The other deletions do not cause any significant effect on gene activity.
Region 9-10. In this cytological position nine deficiency lines had been tested. Since this region was very dense for putative trans-regulation repressors, it was group in a small region. Among nine of the deficiencies were used six of them showed a repressor effect. These effective regions were: Df91)vL15, Df(1)N110, Df(1)HC133, Df(1)vL11, Df(1)KA7, and Df(1)N71. This region seems to have a very important effect on ovo, since in the 9Bto 10F interval there are various levels of repressor effect. Two common overlapping regions were found; one was from 9C4 to 9D1-2, and the other was from 10A to10F6. Other repressor effects from strongest to weakest was Df(1)vL11 (9C4;10A1-2), Df(1)HC133 (9B9-10;9E-F), Df(1)N110 (9B3-4;9D1-2), and Df(1)v-L15 (9B1-2;10A1-2), Df(1)KA7 (10A9;10F6-7) breakpoint was outside the first loci in the examined region. Df(1)Ka7 and Df(1)vL15 show about 20% increase in the heterozygous siblings, the longest and the shortest breakpoints, respectively. Three out of five repressing effect intervals, Df(1)v-L11 (9C4; 10A1-2), Df (1)HC133 (9B9-10; 9E-F), Df(1) N110 (9C4; 10A1-2) is the strongest of all in Df/+ and bearing the common region among the five strains, which is 9C4; 10A1-2.
Region 11-13. Eight deficiency lines were in this region, Df(1)JA26, Df(1)HF368, Df(1)N12, Df(1)C246, Df(1)g, Df(1) RK2, Df(1)RK4, and Df(1) sd 72b . It has been found that this region involves five overlapping deletions that gave rise to repressing effect on ovo gene expression. According to common regions of the cytological positions, these overlapping deletions were grouped into three loci. These three common regions, which are responsible from trans-regulation activity of ovo, reside on 11D0F; 12B-D, and 13F-B regions of the X-chromosome. Df(1)N12 (11D12;11F1-2) and Df(1)C246 (11D-E; 12A1-2) were in the 11D-F loci, Df(1)g (12B;12E8) and Df(1)RK2 (12D2-E1; 13A2-5) were in the 12B0D region, and Df(1)sd72B (13F1-14B1) in the 13B-14B loci, all of which in this examined region showed a repressor activity. The strongest effect among the X-chromosome screening was located in 11D1-11F1-2 excised region of X-chromosome, this deletion corresponds to Df(1)N12 strain, which shows a significant effect as well as high gene activity repression, Around 140% to 240% E/C in Df/+ flies for both ovo::LacZ constructs. In addition, it has been reported that reduced dose of the 11D-F region results in synergistic mutant phenotypes with a number of somatic sex determination genes (Belote et., 1985). Furthermore, Flybase reports that this region seems to include locus involved in early sex determination examined by Scott and baker (1986). However, ambiguities in deficiency breakpoint assignments complicate interpretation. For example, first loci, which includes Df(1)N12 and Df(1)C246 due to uncertainty at the distal end breakpoints of Df(1)C246 (12D-e; 12A1-2); the trans-acting repressor of ovo maybe located in 11E-F rather than 11D-F. Similarly, for the second loci in this region ambiguity at the distal breakpoint of Df(1)RK2 also cause a dilemma about the location of the trans-acting repressor, since the question was the common region between Df(1)g and Df(1)RK2 was whether in the 12D-E or in the 2E1-2E8 of X-chromosome. On the other hand, the last loci were determined by the only one deficiency strain. In this case, the problem was whether determination of the loci was accurate enough, or whether another locus is involved in repressing of ovo reporter activity which Df(1)sd72b (13F1–14B1) may have a common region with. This deficiency removes several lethal mutations, Myb, sd (scalloped), shi (shibiri), and exd (extradenticle). Two genes previously cloned in the 13F cytological region are the Drosophila c-myb oncogene homolog (Katzen et al, 1985) and a G protein b-subunit (Yarfitz et al 1988). It has been suggested that the sd+ gene might be associated with more than one product (perhaps a differential processing) or it might reflect differential tissue and/or temporal regulation (Campbell et al., 1991).
Region 14-20. In this region eight deficiency strains, Df(1)4b18, Df(1)rD1, Df(1)B, Df(1)N19, Df(1)JA27, Df(1)HF396, DF(1)DCB1, and Df(1) A-209, were tested. According to measured specific activities Df(1)4b18 (14B8; 14C1) and DF(1) B (15F9=16A6-7) showed significant activating effect on ovo promoter, activity of the former was weaker than that of latter. Since there is no common region between these two putative trans-acting activators, interpretations of the results gave rise to two loci, 14B8-14C1 and 15F-16A1; 16A6-9. In addition, the Flybase report for Df(1) shows that 70 deletion that breaks within the second exon of the non A (no on or transient A) gene from Stanewsky et al (1993). As a result of X-chromosome screening, 45 deficiency strains were tested and found 17 regions were trans-regulating ovo promoter. These regions were classified into 12 loci according to their overlapping common regions. Among these, six, of which were showing trans-acting activator effect, and seven, of which were responsible for trans-acting repressor effect on ovo promoter. Furthermore, one deficiency strain, Df(1)sc14, showed a significant trans-acting repressor effect in only ovo4B8 strain but not in ovo3U21 strain. This maybe explained by position effect of P[ovo::LacZ] construct due to landing on P element transposase onto insertion site or by difference between the size of the ovo::LacZ constructs, e.g. ovo3U21 carries 200 bp longer than ovo4B8 at the N-terminal end that may cause a better translation product. Consequently, among the X-chromosome screening data, it was found that two of the deficiency lines. Df(1)A113 and Df(1)JC70, which are removing ovo and snf along with the several genes due to deletions, and correspond to one loci acting as an repressor, were taking into more detailed investigations. These results suggested a negative autoregulation mechanism in the ovo promoter. Therefore, negative autoregulation of ovo was examined with three approaches: ovo point mutations, more defined deficiency strain, and downstream genes.
DISCUSSION
The sex determination involves complex set of mechanisms. The fly is chosen to be studied since Drosophila is inexpensive to rear, generates large numbers of progeny, and has nearly a century of accumulated data upon which to design experiments. Mutational analysis of cell biological and developmental process is relatively simple, even if the resulting mutations are organism-lethal when homozygous. This is decided advantage over mammalian genetics, in which lethal mutations often die in utero, which complicates the ability to examine and interpret mutant phenotypes. The Drosophila genome is one-twentieth the size of the mammalian genome, making insertional mutagenesis and positional cloning much less difficult. Additionally, mammalian genetics lacks genetic tools such as balancers that make the maintenance of sterile and lethal-mutations nearly trouble free in Drosophila. Nematodes have many of the same conveniences as Drosophila, with the added advantage of a highly stereotyped pattern of embryonic (and post-hatching) cell lineages. The more-regulative character of Drosophila development induces complications lacking from worm genetics, with respect to cellular level analysis of mutant phenotypes. Perhaps, the most compelling reason to take advantage of the specialized properties of Drosophila, is the extent to which prior studies have shown that genes, proteins, and developmental pathways and processes are conserved among metazoan groups. We can, with high confidence, study sex determination in Drosophila with a reasonable confidence that what we learn can be extrapolated to other species, including man and his clinical diseases.
The deletion mapping technique was used to identify the locations of genes that are required for ovo trans-regulation. Each deficiency line removes several to many genes from the genome. A sufficiently complete set of overlapping deletions can allow, potentially, every individual trans-acting gene to be localized. Seventeen deficiencies that have effects on the ovo germline promoters are shown in Table 4.1. Twelve deficiencies showed repressor effects, and five deficiencies showed activator effects. Deleted regions may affect any of several processes, such as numerator elements, cell viability and differentiation, dosage compensation, and response to inductive signals from soma. Determination of which gene within a specific region is responsible for the effect on ovo requires more defined deletions or having null alleles for each gene. Estimation of the Number of Trans-Regulators. Among the seventeen deficiencies in Table 4.1, overlapping common regions identify seven that function as trans-acting repressor loci, and five that function as trans-acting activator loci. Thus, the entire euchromatic X-chromosome may have as many as ≈10 repressor genes and ≈7 activator genes for the ovo germline promoters. If these results were extrapolated to the entire fly genome, ≈50 repressors and ≈35 activators of ovo transcription are predicted. These are underestimates from the data, since any given deleted common region need not remove exactly one relevant gene. Is it reasonable for nearly 85 genes to be involved in regulating the ovo germline promoters? Precedents from other developmental control systems suggest this is not an implausibly high number.
Regulation of the master sex determination gene Sxl is complex. To establish somatic sex determination in the early embryo, nine genes are required to activate the Sxl early promoter. These are sis-a, sis-b, sis-c, run, da, emc, gro, dpn, and her. In biochemical terms, most are DNA-binding proteins. In genetic terms, some are positive and are others are negative regulators. Maintenance of Sxl expression involves positive autoregulation at the level of pre-mRNA alternative splicing. At least five genes are known to play specific roles in this process: Sxl itself, snf, vir, her, and fl(2)d. Function of Sxl in the germline is regulated in several ways. Germline-specific transcriptional control of Sxl is still conjectural, but it is clear that the somatic functioning numerator elements play no role in the germline. It is possible that ovo may play an important role in germline transcriptional control of Sxl (e.g., Lee. 1998); certainly it has an indirect role (e.g., Oliver et al., 1993). Splicing-level autoregulation of Sxl is active in the female germline, and it involves the same genes that function in this process in somatic cells. Once Sxl protein is produced in female germline cells, the otu protein plays an important role in this relocalization into the nucleus. Thus, a minimum of sixteen genes is required for proper regulation of Sxl.
Establishment of the body plan in Drosophila is also under complex transcriptional control. Maternally localized RNA and protein molecules establish the gross body axes: anterior-posterior and dorsal-ventral. Hierarchically organized sets of zygotically activated genes are transcribed, and their protein products serve to refine the body axes into progressively finer-grained structures. The metameric anterior-posterior body axis is specified by so-called gap genes, pair rule genes, and segment polarity genes, which create the segment-sized repeating units of the body. Homeotic genes encoded by the Antennapedia Complex (ANT-C) and bithorax Complex (BX-C) then confer position-specific identities upon each segment. During the cellular blastoderm stage, gap genes and maternal coordinate genes regulated the activation of primary pair rule genes such as even-skipped (eve). These are expressed in seven one-segment-wide stripes that alternate with on-segment-wide regions of non-expressing cells. For example, the second stripe of eve expression is positively regulated by hunchback and bicoid, and negatively regulated by giant and Kruppel. All four proteins directly bind to a 500-bp-long “eve-stripe 2 enhancer.” Binding have giant and Kruppel is competitive with binding of hunchback and bicoid, and vice versa. Thus, spatially controlled concentrations of giant, Kruppel, bicoid, and hunchback proteins result in spatially restricted activation or repression of the eve stripe 2 enhancer. The remaining six stripes of eve expression are similarly controlled by other DNA-binding proteins, which are acting another discrete stripe-specific enhancers. Ectopic expression of homeotic genes can have disastrous effects on development. Thus, a special heterochromatin-like mechanism functions to ensure that ANT-C and BX-C genes are inactive in cells and tissues that do not require their expression. Stable repression is mediated by the Polycomb class of proteins, which number over forty. Each of these examples illustrates that developmental control of individual gene transcription is mediated by both positive and negative effectors, and that sometimes the number of such upstream regulators numbers between one and several dozen. Thus, our estimate of 85 regulators of the ovo germline promoters is not out of line with other developmentally regulated systems.
Evaluation of Candidate Loci Within Common Regions. Based overlapping cytology, seventeen deficiencies that affected the ovo germline promoter fell into twelve common regions. Each of these will be discussed in turn below. Of particular interest was the relationship each of our trans-acting may have with Su(ovoD) and E(ovoD) loci identified in a generic screen by Pauli et al. (1995). In general, it is not straightforward to suggest identities between Su(ovoD) or E(ovoD) loci and our trans-acting repressor or activator loci because of the dissimilar means of assaying these gene-dose-sensitive interactions. We use quantitative measures of LacZ reporter activity as a proxy for ovo transcription, while Pauli et al. (1995) use semi-quantitative measures of vitellogenesis.
Region 1 (polytene bands 1A1; 2A1-4): The distal region of the X-chromosome showed a trans-regulating activator effect on the ovo promoters. This region includes the acheate-scute complex (AS-C), home of the X-chromosome numerator element sis-b (Cline, 1988; Parkhurst and Ish-Horowicz, 1990), also known as scute-T4. This numerator has no function in the female germline (Granadino et al., 1989). Pauli et al., (1995), using other deficiency strains affecting this section of the X-chromosome, identified a strong Su(ovoD) locus in the polytene region 1E3-4; 2B3-4 that may correspond with our trans-activator. Flybase indicates that this region contains over 100 genes, among them 23 unassigned open reading frames, 33 genes defined by apparent visible mutations, 53 lethal genes,, and two female sterile loci.
Region 2 (polytene bands 4C15-16; 4F15): This region includes the ovo and snf loci, and was identified by Pauli et al., (1995) as a strong E(ovoD) due to the effects of these loci. Further discussion is deferred to mechanism of ovo autoregulation, which deal with ovo negative regulation. Region 3 (polytene bands 5C3-5; 5E8): This region has a trans-regulatory activation effect on the ovo germline promoters. Deficiency for this region showed no interaction with ovoD in the vitellogenesis assay (Pauli et al., 1995). Examination of Flybase records for this region reveals over twenty genes, and no strong candidates that may account for the interaction with the ovo promoters.
Region 4 (polytene bands 7D10; 8A4-5): Results showed that this region contains a transacting-repressor of ovo germline promoter activity. This region reported by Pauli et al. (1995) to contain a strong E(ovoD) locus, which was identified as the ovarian tumor gene (Pauli et al., 1993, 1995). It is virtually certain that the repressor-of-ovo is distinct from otu. First, the otu protein is cytoplasmic and plays a role in egg chamber cytoskeletal function (Nagoshi et al., 1997). Second, the ovo protein binds to the otu promoter in vitro (Garfinkel et al., 1997; Lee, 1998, Lee and Garfinkel 1998; Lu et al., 1998). Third, under certain conditions, in vivo activity of the otu promoter is dependent upon ovo protein production (Hager and Cline, 1997; Lu et al., 1998). Examination of Flybase reveals that this region contains fifty genes mutable to lethal, visible, or female-sterile phenotypes, but none appear to be a strong candidate for the repressor-of-ovo locus.
Region 5 (polytene bands 8B5-8; 8DE): This region also has an apparent repressor of ovo germline promoter activity. Deficiency for this region showed no interaction with ovoD mutations in the Pauli et al. (1995) vitellogenesis assay. Examination of Flybase reveals that this region contains thirty genes mutable to lethal, visible, or female sterile phenotypes. One gene stands out as a candidate for the repressor, namely, lozenge. This is a complex locus that is mutable to female sterility (Green and Green, 1949, 1956), and it is named for a reduced-eye, smoothened-eye, mutant phenotypes. Interestingly, certain ovo-mutant alleles are called “lozenge-like” in recognition of a similar eye defect (Oliver et al., 1987; Mevel-Ninio et al., 1989; Garfinkel et al., 1992). The lz gene codes for a transcription factor (Dag et al., 1996). Region 6 (polytene bands 9C4; 9D1-2): The cytological assignment of this region is based on the overlap of three deficiencies: Df(1)N110, Df(1)H133, and Df(1)v L11. Together, they mark a trans-acting repressor of ovo promoter activity. According to Pauli et al. (1995), only two of these three deficiencies behaved as if they exposed an E(ovoD) locus, while the third had no effect. In combination with positive results from other deficiencies, Pauli et al. positioned the E(ovoD) locus at cytological region 9E-F. Thus, it is again possible that the repressor-of-ovo we identified is distinct from a nearby E(ovoD) locus, and is among the half-dozen loci identified by Flybase as mapping into this interval.
Region 7 (polytene bands 10A6; 10F6-7): This region contains a trans-acting repressor of ovo promoter activity. According to Pauli et al. (1995), the defining deficiency had no significant interaction with ovoD alleles. Examination of Flybase reveals that this region includes the somatic X-chromosome numerator element sis-a, which also has no function in germline development (Granadino et al., 1989, 1990, 1997). Given the extent of this region, it is not surprising that Flybase identifies 65 genes with diverse phenotypes and biochemical roles; however no strong candidate locus that may count for the repressor-of-ovo locus is apparent.
Region 8 (polytene bands11D1-2; 11F1-2): This region contains perhaps the strongest trans-acting repressor of ovo promoter activity in the survey: deficiency heterozygous experimentals had 2-2.5 fold more lacZ specific activity in their ovaries that the balancer carrying controls. According to Pauli et al (1995), one of the two deficiencies defining this common region showed a statistically weak enhancement of ovoDalleles, while the other had a significant Su(ovoD) phenotype. Likewise, Belote et al. (1985) and Scott and Baker (1986) reported that the same deficiency later shown to have Su(ovoD) activity also interacted with loci in the somatic sex determination pathway. It is an open question how these three results relate to one another. Among sixteen genes that map into this region are two signal transduction loci: the Mek3 gene, a serine-threonine-specific protein kinase in the MAP kinase pathway, and a beta subunit of the heterotrimeric GTP-binding protein. A solitary female-sterile, fs(1) K4, also maps roughly into this region; it is germline-dependent, and yields fragile eggs, a phenotype occasionally seen in the eggs laid by ovoD3/+ females.
Region 9 (polytene bands 12D2-12E1; 12E8): This region contains a trans-acting repressor of ovo promoter activity. According to Pauli et al. (1995), neither deficiency defining this common region interacted with ovoDalleles. This region contains the yolkless gene (DiMario et al., 1987), which has been cloned and codes for a member of 35 known genes, including a cluster of tRNA genes, the male-germline-specific Stellate genes, and several lethal and female-sterile genes.
Region 10 (polytene bands 13F1; 14B1): This region contains a trans-acting repressor of ovo promoter activity. Again, no significant interaction with ovoD allel4es was observed by Pauli et al. (1995). Podry, Katzen and others have extensively mutagenized this region due to its containing shibiri (the Drosophila homolog of dynamin), c-myb, another Gb subunit, and the homeodomain protein extradenticle. Their work revealed a total of twenty lethal genes, ten apparent visibles, and over a half-dozen unassigned open reading frames.
Region 11 (polytene bands 14B8; 14C1): This region contains a trans-acting activator of ovo promoter activity. According to Pauli et al., (1995), the defining deficiency had no significant interaction with ovoD alleles. This region is surprisingly dense genetically, as it apparently contains over forty genes. Several behavioral genes coding for neuronal functions map here, including nonA, paralytic, and easily shocked. The nonA gene codes for an RNA-binding protein, and is mutable to a variety of phenotypes including recessive lethality, male-courtship-strong abnormalities, and defective vision. The location of para (a sodium channel) is particularly intriguing since parats mutations fail to complement certain napts alleles, and nap genetically overlaps the dosage compensation function maleless. Mutations in maleless are unique among the known dosage compensation loci in having a mutant phenotype in germline clones, and they are said to suppress the female-germline-lethality of ovo null mutations. The easily shocked locus codes for ethanolmine kinase, and mutations at this locus also interact with mle.
Region 12 (polytene bands 15F9-16A1; 16A7): This region contains a trans-acting activator of ovo promoter activity. According to Pauli et al. (1995), the defining deficiency had no significant interaction with ovoDalleles. Examination of Flybase reveals that this region contains at least a dozen female-sterile loci, a dozen lethal loci (including the Bar homeodomain protein gene). There is an ambiguity in compared mean of activities. According to the negative autoregulation mechanism, there suppose to be a linear decrease pattern correlated to increase in copy of ovo. However, the pattern of the gene dose was reaching plato, when three copies of ovo were present in the genome. Yet, this also shows that there is a protection mechanism that counts the number of ovo versus number of X chromosome exists. Therefore, the sex determination mechanism turns off the extra ovo in the system immediately.
Consequently, the system prohibits more wrong information to be processed according to its default setting where if the X:A ratio equals to one the outcome is going to be prepared as female, if not turn off the mechanism towards male-like, sterile mode, or death at the embryonic stage. This discontinuity in the linear correlation may be due to position effect of P[w+ ovo+]. Future Directions and Concluding Remarks The results of this study suggest that the ovo germline promoters are regulated by a large set of upstream factors. Nearly a dozen of these maps to the X-chromosome, some to region that are well characterized genetically. Further deficiency mapping experiments, and assessment of the phenotypes of single-P insertion lines with female-sterile or perhaps lethal phenotypes, would be required to identify the relevant genes. Some regions contain candidate loci that have been cloned (e.g. lozenge); in this example, either in vitro DNA-binding experiments using Lz protein and the ovo promoter region, or computational assessment of the likelihood that the ovo promoter contains binding sites for Lz can be done. Another potential upstream factor not assessed in these experiments is the ecdysone regulatory hierarchy. The steroid ecdysone is the endocrine hormone that controls molting and metamorphosis in arthropods. It is an allosteric effector for a heterodimeric receptor of the steroid-receptor superfamily. The ovaries of adult females manufacture their own ecdysone, and the gene for the rate-limiting steroidogenic enzyme transcribed beginning in Stage 7-8 egg chambers. This stage immediately precedes the onset of the highest level of ovo transcription (Mevel-Ninio et al., 1991; Garfinkel et al., 1994). Mutations in the E74 and E75 genes, when made homozygous in germline clones, cause arrest of oogenesis at Stage 7-8, as if egg chambers are unable to respond to endogenous ecdysone and continue differentiation. Both E74 and E75 code for transcription factors that are induced as immediate-early primary responses to added ecdysone both in-vivo and in tissue culture assays. Thus, it is reasonable to suggest that one or both of these proteins will bind to the ovo germline promoter in an in vivo effect on expression of the ovo::lacZ reporter using the methods established in this dissertation.
Acknowledgement: This work had been comppleted in the laboratory of Dr. Mark Garfinkel at Illinois Institute of Technology. Dr. Demet Sag initiated the project with her own ideas, was fully supported by Turkish National Merit Fellowship, and earned NATO Advanced Science institute Grant on Genome Structure and Functional Genomics, Elba Island, Italy, accepted to work with Dr. Mevel Ninio, based on the proposal submitted by Demet Sag on Molecular Mechanism of ovo, through EMBO long term scholarship in France.
BIBLIOGRAPHY
- Ashburner, M., Drosophila Laboratory Manual, Cold Spring Harbor Laboratory Press, pp. 317-318, 1989
- Baker, B.S., “Sex in flies: the splice of life,” Nature 340, pp.521-524, 1989.
- Baker, B.S. and Belote, J.M., “Sex determination an dosage compensation in Drosophila melanogaster” Ann. Rev of Genetics 17, pp345-393, 1983.
- Baker B.S. and Ridge, K.A., “Sex and the single cell I. On the action of major loci affecting sex determination in Drosophila melanogaster,” Genetics 94, pp.383-423, 1980.
- Bell, L.R. Horabin, J.I., Schedl, P., and Cline, T.W., “Sex-lethal, a Drosophila sex determination switch gene, exhibits sex specific RNA spilicing and sequence similarity to RNA binding proteins,” Cell 55, pp.1037-1046, 1988.
- Bell, L.R. Horabin, J.I., Schedl, P., and Cline, T.W., Positive autoregulation of Sex-lethal by alternative spilicing maintains the female determined state in Drosophila, Cell Vol 65, pp.229-239, 1991.
- Belote, J.M., Handler, A.M., Wolfner, M.F., Livak, K.J., and Baker, B.S., Sex-specific regulation of yolk protein expression in Drosophila, Cell 40, pp.339-348, 1985.
- Bohringer- Manheim Product Support, Chloroform Red-beta-D_Galactopyranoside sodium salt (CPRG) provided by ID#0223p, June 1986 dated information, fax received on 2/26/1996.
- Bopp, D., Bell, L.R., Cline, T.W., Schedl, P. Developmental distribution of female specific Sex-Lethal proteins in Drosophila melanogaster, Genes and Development 5, pp.403-415,, 1991.
- Bopp, D., Sex-specific control of Sex-lethal is a conserved mechanism for sex determination in the genus Drosophila, Development 122, pp.971-982, 1996.
- Bopp, D., Horabin, J.I., Lersch, R.A., Cline, T.W., Schedl, P., Expression of the Sex-lethal gene is controlled at multiple levels during Drosophila oogenesis, Development 118, pp.797-812, 1993.
- Bradford, M. M., A Rapid and sensitive method for the Qantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding, Analytical Biochemistry 72, pp.248-254, 1976.
- Bridges, C.B., Sex in relation to chromosomes, Am. Nat.59, pp.127-137, 1925.
- Bridges, C.B., Triploid intersexes in Drosophila melanogaster, Science 54, pp.252-254, 1921.
- Bridges, C.B., Non-disjunction as proof of the chromosome theory of heredity, Genetics 1, pp.1-52, 1916.
- Burtis, K.C. and Baker, B.S., Drosophila doublesex gene controls somatic sexual differentiation by producing alternatively spliced mRNAs encoded related sex-specific yolk protein gene enhancer, The EMBO J., Vol. 10, No. 9, pp.2557-2582, 1991.
- Busson, D., Gans, M., Komitopoulou, and Masson, M., Genetic Analysis of three dominant female-sterile mutations located on the X-chromosome of Drosophila melanogaster, Genetics 105, pp.309-325, 1983.
- Campbell, S.D., Duttoroy, A., Katzen, A.L., and Chovnick, A., Cloning and characterization of the scalloped region of Drosophila melanogaster, Genetics 127, pp.367-380, 1991.
- Cline, T.W., The Drosophila sex determination signals: how do flies count two?, Ann. Rev. Genet 30, pp637-702, 1996.
- Cline, T.W., Evidence that sisterless-a and sisterless-b are two of several discrete numerator elements of the X/A ratio sex-determination signal in Drosophila that switch Sxl between two alternative stable expression states, Genetics 119, pp.829-862, 1988.
- Cline, T.W., Re-evaluation of the functional relationship in Drosophila between a small region on the X-chromosome (3E8-4F11) and the sex determination gene, Sex-lethal, Genetics 116, s:12, 1987.
- Cline, T.W., A female specific lethal lesion in an X-linked positive regulator of the Drosophila sex determination gene, Sex-lethal, Genetics 113, pp.641-663, 1986.
- Cline, T.W., Autoregulatory functioning of a Drosophila gene product that establishes and maintains the sexuality determined state, Genetics 107, pp.231-277, 1984.
- Cline, T.W., Functioning of the gene daughterless and Sex-lethal in Drosophila germ cells, Genetics 107, s16-17, 1983.
- Cline, T.W., A sex specific temperature-sensitive maternal effect of the daughterless mutation of Drosophila melanogaster, Genetics 84, pp.723-742, 1976.
- Cronmiller, C., Schedl, P., Cline, T.W., Molecular characterization of daughterless, a Drosophila sex determination gene with multiple roles in development, Genes Dev. 2: 155-167, 1988.
- Despande, G., Samuels., M.E., and Schedl, P.D. “Sex-lethal interacts with splicing in vitro and in vivo, Molecular and Cellular Biology, Vol. 16, No 8, pp. 5036-5047, 1996.
- Despande, Stukey, J., and Schedl, P., scute (sis-b) function in Drosophila sex determination, Moll. Cell Biology 15, pp. 4430-4440, 1995.
- DiMario, P.J., and Mahowald, A.P., Female sterile (1) yolkless: A recessive female sterile mutation in Drosophila melanogaster with depressed numbers of coated pits and coated vesicles within the developing oocytes, J. Cell Biology 105: 199-206, 1987.
- Erickson, J.W. and Cline, T.W., A bZIP protein, sisterless-a, collaborates with bHLH transcription factors early in Drosophila development to determine sex, Genes and Dev. 7: 1688-1702, 1993.
- Erickson, J.W. and Cline, T.W., Molecular nature of the Drosophila sex determination signal and its link to neuorogenesis, Science 251, pp. 1071-1074, 1991.
- Estes, P.A., Keyes, L.N., and Schedl, P., Multiple response elements in the sex-lethal early promoter ensure its female-specific expression pattern, Mol Cell Biol 15, pp. 904-917, 1995.
- Flickinger, T.W. and Salz, H.K., The Drosophila sex determination gene snf encodes a nuclear protein with sequence and functional similarity to the mammalian U1A snRNP, Gene and Development 8, pp. 914-925, 1994.
- Flybase, The Drosophila genetic database, http://www.flybase.bio.indiana.edu, 1998.
- Gans, M.C., Audit and Massson, M., Isolation and characterization of sex-linked female-sterile mutations in Drosophila melanogaster, Genetics 81, pp. 683-704, 1975.
- Garfinkel, M.D., Lee, S., and Sigar, I., DNA-binding targets of the Drosophila melanogaster OVO protein, 38th Annual Drosophila Research Conference, Chicago, IL, 1997.
- Garfinkel, M.D., Wang, J., Liang, Y., and Mahowald, A. P., Multiple products from the shavenbaby-ovo gene region of Drosophila melanogaster: relationship to genetic complexity, Molecular and Cell Biology, Vol. 14, No., 10, pp. 6809-6818, 1994.
- Garfinkel, M.D., Lohe, A.H., and Mahowald, A.P., Molecular genetics of the Drosophila melanogaster ovo locus, a gene required for sex determination of germline cells, Genetics 130, pp. 791-803, 1992.
- Gollin, S. M. and King, R. C., Studies on fs(1)1621, a mutation producing ovarian tumors in Drosophila melanogaster, Developmental Genetics 2, pp. 203-218, 1981.
- Granadino, B., Compuzano, S., and Sanchez, L., The Drosophila melanogaster fl(2)d, a gene needed for Sex-lethal expression in Drosophila melanogaster, Genetics 130, pp. 597-612, 1990.
- Granadino, B., Santamaria, P., and Sanchez, L., Sex-determination in the germ line of Drosophila melanogaster: activation of the gene Sex-lethal, Development 118, pp. 813-816, 1993.
- Granadino, B., Juan, A. B. S. B, Santamaria, P., Sanchez, L., Distinct mechanisms of splicing regulation in vivo by the Drosoophila protein Sex-lethal, PNAS USA, 94, pp. 7343-7348, 1997.
- Hager, J.H. and Cline, T.W., Induction of female Sex-lethal RNA splicing in male germ cells: implications for Drosophila germline sex-determination, Development 124, pp. 5033-5048, 1997.
- Hilfiker, A., Amrein, H., H., Dobendorfer, A., Schneiter, R, and Nuthiger, R., The gene virilizer is required for female-specific splicing controlled by Sxl, master gene for development in Drosophila, Development 121, pp. 4017-4026, 1995.
- Horabin, J.I., Bopp, D., Waterburry, J., and Schedl, P., Selection and maintenance of sexual identity in the Drosophila melanogaster, Genetics 141, pp. 1521-1565, 1995.
- Horabin, J. I. And Schedl, P., Regulated spilicing of the Drosophila Sex-lethal male exon involves a blockage mechanism, Moll. Cell. Biol. 13, pp. 1408-1414, 1993.
- Horabin, J.L., and Schedl, P., Sex-lethal autoregulation requires multiple cis-acting elements upstream and downstream of the male exon and appears to depend largely on controlling the use of the male exon 5’ splice site, Moll. Cell. Biol. 13: pp. 7734-7746, 1993.
- Hoshijima, K., Kohyama, A., Watakabe, I., Inonue, K., Sakamato, H., and Shimura, Y., Transcriptiuonal regulation of the Sex-lethal gene by helix-loop-helix proteins, Nucleic Acids Res. 23, pp. 3441-3448, 1995.
- Inonue, K., Hojhijima, K., Sakamato,, H., and Shimura, Y., Binding of the Drosophila Sex-lethal gene product to the alternative splice site of transformer primary transcript, Nature 344, pp. 461-463, 1990.
- Keyes, L.N., Cline, T.W., and Schedl, P., The primary sex-determination signal of Drosophila acts at the level of transcription, Cell, Vol. 68, pp. 933-943, 1992.
- Komitopoulou, K., Gans, M., Margaritis, L.H., Kafatos, F.C., and Masson, M., Isolation and characterization of sex-linked female-sterile mutants in Drosophila melanogaster with special attention to eggshell mutants, Genetics 105: 897-921, 1983.
- Lee, S., DNA binding targets of the Drosophila melanogaster OVO protein, PhD Dissertation, Illinois Institute of Technology, Chicago, IL, USA, 1998.
- Lee, S. and Garfinkel, M.D., DNA-binding targets of the Drosophila melanogaster OVO protein, Nucleic Acid. Res.
- Linsley, D.L., and Zimm, G., The genome of the Drosophila melanogaster, Academic Press, San Diego, New York, 1980.
- Lu, J., Andrews, J., Pauli, D., and Oliver, B., Drosophila OVO zinc finger protein regulates ovo and ovarian tumor target promoters, Dev. Genes. Evol., pp. 1-10, 1998.
- Luccesi, J.C. and Manning, E., Gene dosage compaensation in Drosophila melanogaster, Adv. Genetics 24, pp. 371-429, 1987.
- Madl, J.E., and Herman, R.K., Polyploids and sex determination in Caenornabtidis elegans, Genetics 93, pp. 393-402, 1979.
- Mevel-Ninio, M., Mariol, M.C. and Gans, M., Mobilization of the gypsy and copia retrotransposans in Drosophila melanogaster induces reversion of the ovoD dominant female-sterile-mutations: molecular analysis of revertant alleles, EMBO J. 8, pp. 1549-1558, 1989.
- Mevel-Ninio, M., Terracol, R., and Kafatos, F.C., The ovo gene of Drosophila encodes a zinc finger protein required for female germ line development, EMBO J. 10, pp.2259-2266, 1991.
- Mevel-Ninio, M., Guenal, I., and Limburg-Bouchen, B., Production of dominant female sterility in Drosophila melanogaster by insertion of the ovoD1 allele autosomes: use of transformed starins to generate germline mosaic, Mechanism of development 45, pp. 155-162, 1994.
- Mevel-Ninio, M., Terracol, R., Salles, C., Vincent, A., and Payre, F., ovo, a Drosophila gene required for ovarian development, is specially expressed in the germline and shares most of its coding sequences with shavenbaby, a gene involved in embryo patterning, Mecahnism of Development 49, pp. 83-95, 1995.
- Mevel-Ninio, M., Fouilloux, E., Genal, I. and Vincent, A., The three point dominant female-staerile mutations of Drosophila ovo gene are point mutations that create new translation-initiatorAUG codons, Development 122, pp. 4131-4138, 1996.
- Mohler, J.D., Developmental genetics of the Drosophila egg. I.: Identification of 50 sex-linked cistrons with maternal effects on embryonic development, genetics 85, pp. 259-272, 1977.
- Nagai, K., Oubridge, C., Jessen, T. H., Li, J., and Evans, P.R., Crystal structure of the RNA-binding domain of the U1 small nuclear ribonucleoprotein A, Nature 348, pp. 515-520, 1990.
- Nagoshi, R.N., McKeown, M., Burtis, K.C., Belote, J.M., and Baker, B., The control of alternative spilicing at genes regulating differentiation in D. melanogaster, Cell, Vol. 53, pp.229-236, 1988.
- Nagoshi, R.N., and Baker, B., Regulation of sex-specific RNA splicing at the Drosophila doublesex gene: cis-acting mutations in exon sequences alter sex specific RNA splicing patterns, Genes and Development 4, pp. 89-97, 1990.
- Nagoshi, R.N., Patton, J.S., Bae, E., and Geyer, P., The somatic sex determines the requirement for ovarian tumor gene activity in the proliferation of the Drosophila germline, development 121, pp.579-587, 1995.
- Nothiger, R., and Steinmann-Zwicky, M., Meier-Gerschwiller, P., and Weber, T., Sex determination in the germline of Drosophila depends on genetic signals and inductive somatic factors, development 107, pp.505-518, 1989.
- Oliver, B., Singer, J., Laget, V., Pennetta, G. and Pauli, D., Function of Drosophila melanogaster ovo– in germ line sex determination depend on X-Chromosome number, Development 120, pp.1-11, 1994.
- Oliver, B., Kim, Y. and Baker, B., Sex-lethal, master and slave: a hierarchy of germ line sex determination in Drosophila, Development 119, pp. 897-908, 1993.
- Oliver, B., Pauli, D., and Mahowald, A.P., Genetic evidence that the ovo locus is involved in Drosophila germ line sex determination, Genetics 125, pp. 535-550, 1990.
- Oliver, B., Perrimon, N, an Mahowald, A.P., The ovo locus is required for sex-specific germ line maintenance in Drosophila, Genes and Development 1, pp. 913-923, 1987.
- Oubridge, C., Ito, N., Evans, P.R., Teo, C.H., and Nagai, K., Crystal structure at the 1.92A resolution of the RNA binding domain of the U1A splicesomal protein completed with an RNA hairpin, Nature 372, pp.432-438, 1994.
- Pauli, D., Oliver, B., and Mahowald, A.P., Identifications of regions interacting with ovo D mutations: potential new genes involved in germline sex determination in Drosophila melanogaster, Genetics 139, pp.713-732, 1995.
- Pauli, D., Oliver, B., and Mahowal, A.P., The role of the ovarian tumor locus in Drosophila melanogaster germ line sex determination, Development 119, pp.123-134, 1993.
- Pauli, D. and Mahowald, A.P., Germline sex determination in Drosophila melanogaster, Trends in Genetics, Vol. 6, No. 8, pp.259-264, 1990.
- Parkhurst, S.M., Bopp, D., and Ish-Horowicz, X:A ratio, the primary sex determining signal in Drosophila, is transduced by helix-loop-helix proteins, Cell, Vol. 63, pp.1179-1191, 1990.
- Perrimon, N., Mohler, D., Engsttrom, L., and Mahowald, A.P., X-linked female-sterile loci in Drosophila melanogaster, Genetics 113, pp.695-712, 1986.
- Perrimon, N., Engstrom, L., and Mahowald, A.P., The effects of zygotic lethal mutations on female-germ-line functions in Drosophila, Developmental Biology 105, pp. 404-414, 1984.
- Perrimon, N., Clonal analysis of dominant female-sterile, germline-dependent mutations in Drosophila melanogaster, Genetics 108, pp.927-939, 1984.
- Perrimon, N. and Gans, M., Clonalo analysis of the tissue specificity of recessive female-sterile mutations of Drosophila melanogaster using a dominant female sterile mutation Fs(1)K1237, Developmental Biology 100, pp. 365-373, 1983.
- Rodesch, C., Geyer, P.K., Patton, J.S., Bae, E., and Nagoshi, R.N., Developmental analysis of the ovarian tumor gene during Drosophila oogenesis, Genetics 141, pp.191-202, 1995.
- Sag-Ozkol, D., Tekin, S., Garfinkel, M.D., Gene-dose sensitive trans-acting regulators of the Drosophila melanogaster germline promoter, 38th Annual Drosophila Research Conference, Chicago, IL, USA, 1997.
- Sag-Ozkol, D., and Garfinkel, M.D., Negative autoregulation of Drosophila melanogaster female germline specific gene, ovo (in preparation).
- Sag-Ozkol, D., and Garfinkel, M.D., X-chromosome screening of Drosophila melanogaster to find numerator elements of germline sex determination (in preparation).
- Salz, H.K. and Flickinger, T.W., Both loss of function and gain-of-function mutations in snf define a role for snRNP proteins in regulating Sex-lethal pre-mRNA splicing in Drosophila development, Genetics 144, pp.95-108, 1996.
- Salz, H.K., Maine, E.M., Keyes, L.N., Samuels, M.E., Cline, T.W., and Schedl, P., The Drosophila female-specific sex-determination gene, Sex-lethal has stage-, tissue-, and sex-specific RNAs suggesting multiple models of regulation, Genes and Development 3, pp.708-709, 1989.
- Salz, H.K., Cline, T.W., and Schedl, P., Functional changes associated with structural alterations induced by mobilization of a p element inserted in the Sex-lethal gene of Drosophila, Genetics 117, pp.221-231, 1987.
- Sanchez, L., Granadino, B., and Torres, M., Sex determination in Drosophila melanogaster, X-linked genes involved in the initial step of Sex-lethal activation, Developmental Genetics 15: 251-264, 1994.
- Sass, G., Mohler, J.D., Walsh, R.C., Kalfayan, L.J. and Searles, L.L., Structure an the expression of hybrid dysgenesis-induced alleles of the ovarian-tumor (otu) gene in Drosophila melanogaster, Genetics 133, pp.253-263, 1993.
- Sass, G., Comer, A.R. and Searles, L.L., The ovarian tumor protein isoforms of Drosophila melanogaster exhibit differences in function, expression, and localization, developmental Biology 167, pp.201-212, 1995.
- Schedl, A, Ross, A., Lee, M., Engelkamp, D., Rashbass, van Heyningen, V., and Hastie, N., Influence of PAX6 gene dosage on development: over-expression causes sever eye abnormalities, Cell 86, pp.71-82, 1992.
- Schupbach, T., and Wieschhaus, E., Female sterile mutations on the second chromosome of Drosophila melanogaster II mutations blocking oogenesis an altering egg morphology, Genetics 129, pp.1119-1136, 1991.
- Shupbach, T., an Wieschaus, E., Female sterile mutations on the second chromosome of Drosophila melanogaster I. Maternal effect mutations, Genetics 121, pp.101-17, 1989.
- Schupbach, T., Normal female germ cell differentiation requires the female X-chromosome to autosome ratio and expression of Sex-lethal in Drosophila melanogaster, Genetics 109, pp.529-548, 1985.
- Simon, J.A. and Lis, J.T., A germline transformation analysis reveals flexibility in the organization of the heat-shock consensus elements, Nucleic Acids Research, Vol 15, No.7, 1987.
- Staab, H., Heller, A., Steinmann-Zwicky, M., Somatic sex determining signals act on XX germ cells in Drosophila embryos, Development 122, pp.4065-4071, 1996.
- Staab, H., and Steinmann-Zwicky, M., Female germ cells of Drosophila require zygotic ovo and out product for survival in larvae and pupae, Mech. Dev. 54, pp.205-210, 1995.
- Stanewsky, R., Rendahl, K.G., Dill, M., and Saumweber, H., Genetic and molecular analysis of the X-chromosomal region 14B17-14C4 in Drosophila melanogaster: Loss of function in NONA, a nuclear protein common to many cell types, results in specific physiological and behavioral defects, Genetics 135, pp.419-442, 1993.
- Steinman-Zwicky, M., Sex determination of the Drosophila germ line: tra and dsx control somatic inductive signals, Development 120, pp. 707-716, 1994.
- Steinman-Zwicky, M., Sxl in the germline of Drosophila: A target for somatic late induction, Developmental Genetics 15, pp.265-274, 1994.
- Steinman-Zwicky, M., Sex determination in Drosophila: sis-b, a major numerator element of the X:A ratio in the soma, does not contribute to the X:A ratio in germ line, Development 117, pp. 763-767, 1993.
- Steinman-Zwicky, M., How do the germ cells choose their sex? Drosophila as a paradigm, Bioassays 14 (8), pp.513-518, 1992.
- Steinman-Zwicky, M., Anrein, H. and Nothiger, R., Genetic control of sex determination in Drosophila, Advanced Genetics 27, pp.189-237, 1990.
- Steinman-Zwicky, M., Schmid, H. and Nothiger, R., Cell-autonomous an inductive signals can determine the sex of the germ line of Drosophila by regulating the gene Sxl, Cell, Vol. 57, pp.157-166, 1989.
- Steinman-Zwicky, M., Sex determination in Drosophila. The X-chromosomal gene liz is required for Sxl activity, The EMBO Journal 7, pp.3889-3898, 1988.
- Steinman-Zwicky, M. and Nothiger, R., The small region on the X chromosome of Drosophila regulates a key gene that controls sex determination and dosage compensation, Cell, Vol. 42, pp.877-887, 1985.
- Sosnowski, B. A., Belote, J. M. and McKeown, M., Sex specific alternative spilicing of RNA gene results from sequence-dependent splice site blockage, Cell, Vol. 3, pp.449-459, 1989.
- Yarfitz, S., Provost, N. M., and Hurley, J. B., Cloning of Drosophila melanogaster guanine nucleotide regulatory protein subunit gene and characterization of its expression during development, PNAS USA 85, pp.7134-7138, 1988.
- Wieschaus, E., Audit, C., and Masson, M., A clonal analysis of the rules of somatic cells and germline during oogenesis in Drosophila, Developmental Biology 88, pp.92-103, 1981.
- Wieschaus, E., Nusslein-Volhard, C., an Jurgen, G., Mutations affecting the pattern of the larval cuticle in Drosophila melanogaster. Part III. Zygotic loci on the X-chromosome and fourth chromosome, Roux. Arch. Dev. Biol., 193, pp.296-307, 1984.
FIGURES and TABLES:
Figure 1: Sex determination of D. melanogaster (1998)
Figure 2: Somatic-Germline Interactions. (1998)
Figure 3: Molecular Structure of the ovo locus
Figure 4: In vivo Biochemical_genetic Assay for Regulators
Figure 5: ovo-LacZ Reporter Construction. (1998)
Figure 6 : Establishing Stocks From Duplication Carrying Lines.
Figure 7: Control Assay for b-galactosidase Assay. (1998).
Table 1: List of Stocks for X-chromosome Screening (1998)
Table 2: Stocks Made in This Study for X-Chromosome Screening
Table 3: LacZ Specific Activities Obtained by Screening X-Chromosome with ovo3U21
Table 4: LacZ Specific Activities Obtained by Screening X-Chromosome with ovo4B8 (Results)
Table 5: Deficiency Lines Affecting the ovo Gene Activity (X-chromosome screening result)
Previously Posted:






… and targeted therapies for personalized medicine based on signal transduction, primary developmental pathways, biomarkers and functional …

Read Full Post »