CRACKING THE CODE OF HUMAN LIFE: The Birth of BioInformatics & Computational Genomics – Part IIB
Curator: Larry H Bernstein, MD, FCAP
Part I: The Initiation and Growth of Molecular Biology and Genomics – Part I From Molecular Biology to Translational Medicine: How Far Have We Come, and Where Does It Lead Us?
http://pharmaceuticalintelligence.com/wp-admin/post.php?post=8634&action=edit&message=1
Part II: CRACKING THE CODE OF HUMAN LIFE is divided into a three part series.
Part IIA. “CRACKING THE CODE OF HUMAN LIFE: Milestones along the Way” reviews the Human Genome Project and the decade beyond.
Part IIB. “CRACKING THE CODE OF HUMAN LIFE: The Birth of BioInformatics & Computational Genomics” lays the manifold multivariate systems analytical tools that has moved the science forward to a groung that ensures clinical application.
Part IIC. “CRACKING THE CODE OF HUMAN LIFE: Recent Advances in Genomic Analysis and Disease “ will extend the discussion to advances in the management of patients as well as providing a roadmap for pharmaceutical drug targeting.
To be followed by:
Part III will conclude with Ubiquitin, it’s role in Signaling and Regulatory Control.
Part IIB. “CRACKING THE CODE OF HUMAN LIFE: The Birth of BioInformatics & Computational Genomics” is a continuation of a previous discussion on the role of genomics in discovery of therapeutic targets titled, Directions for Genomics in Personalized Medicine, which focused on:
- key drivers of cellular proliferation,
- stepwise mutational changes coinciding with cancer progression, and
- potential therapeutic targets for reversal of the process.
It is a direct extension of The Initiation and Growth of Molecular Biology and Genomics – Part I
- chemical structure,
- the basic building blocks of DNA and proteins, of
- nucleotide and protein-protein interactions,
- protein folding,
- allostericity,
- genomic structure,
- DNA replication,
- nuclear polyribosome interaction, and
- metabolic control.

In addition, the emergence of methods for
- copying,
- removal
- insertion, and
- improvements in structural analysis
- developments in applied mathematics have transformed the research framework.
This last point,
- developments in applied mathematics have transformed the research framework, is been developed in this very article
CRACKING THE CODE OF HUMAN LIFE: The Birth of BioInformatics & Computational Genomics – Part IIB
Computational Genomics
1. Three-Dimensional Folding and Functional Organization Principles of The Drosophila Genome
Sexton T, Yaffe E, Kenigeberg E, Bantignies F,…Cavalli G. Institute de Genetique Humaine, Montpelliere GenomiX, and Weissman Institute, France and Israel. Cell 2012; 148(3): 458-472.
http://dx.doi.org/10.1016/j.cell.2012.01.010/
http://www.cell.com/retrieve/pii/S0092867412000165
http://www.ncbi.nlm.nih.gov/pubmed/22265598
Chromosomes are the physical realization of genetic information and thus form the basis for its readout and propagation.


Here we present a high-resolution chromosomal contact map derived from
- a modified genome-wide chromosome conformation capture approach applied to Drosophila embryonic nuclei.
- the entire genome is linearly partitioned into well-demarcated physical domains that overlap extensively with active and repressive epigenetic marks.
- Chromosomal contacts are hierarchically organized between domains.
- Global modeling of contact density and clustering of domains show that inactive
- domains are condensed and confined to their chromosomal territories, whereas
- active domains reach out of the territory to form remote intra- and interchromosomal contacts.
Moreover, we systematically identify
- specific long-range intrachromosomal contacts between Polycomb-repressed domains.
Together, these observations
- allow for quantitative prediction of the Drosophila chromosomal contact map,
- laying the foundation for detailed studies of chromosome structure and function in a genetically tractable system.

2A. Architecture Reveals Genome’s Secrets
Three-dimensional genome maps – Human chromosome
Genome sequencing projects have provided rich troves of information about
- stretches of DNA that regulate gene expression, as well as
- how different genetic sequences contribute to health and disease.
But these studies miss a key element of the genome—its spatial organization—which has long been recognized as an important regulator of gene expression.
- Regulatory elements often lie thousands of base pairs away from their target genes, and recent technological advances are allowing scientists to begin examining
- how distant chromosome locations interact inside a nucleus.
- The creation and function of 3-D genome organization, some say, is the next frontier of genetics.
Mapping and sequencing may be completely separate processes. For example, it’s possible to determine the location of a gene—to “map” the gene—without sequencing it. Thus, a map may tell you nothing about the sequence of the genome, and a sequence may tell you nothing about the map. But the landmarks on a map are DNA sequences, and mapping is the cousin of sequencing. A map of a sequence might look like this:
On this map, GCC is one landmark; CCCC is another. Here we find, the sequence is a landmark on a map. In general, particularly for humans and other species with large genomes,
- creating a reasonably comprehensive genome map is quicker and cheaper than sequencing the entire genome.
- mapping involves less information to collect and organize than sequencing does.
Completed in 2003, the Human Genome Project (HGP) was a 13-year project. The goals were:
- identify all the approximately 20,000-25,000 genes in human DNA,
- determine the sequences of the 3 billion chemical base pairs that make up human DNA,
- store this information in databases,
- improve tools for data analysis,
- transfer related technologies to the private sector, and
- address the ethical, legal, and social issues (ELSI) that may arise from the project.
Though the HGP is finished, analyses of the data will continue for many years. By licensing technologies to private companies and awarding grants for innovative research, the project catalyzed the multibillion-dollar U.S. biotechnology industry and fostered the development of new medical applications. When genes are expressed, their sequences are first converted into messenger RNA transcripts, which can be isolated in the form of complementary DNAs (cDNAs). A small portion of each cDNA sequence is all that is needed to develop unique gene markers, known as sequence tagged sites or STSs, which can be detected using the polymerase chain reaction (PCR). To construct a transcript map, cDNA sequences from a master catalog of human genes were distributed to mapping laboratories in North America, Europe, and Japan. These cDNAs were converted to STSs and their physical locations on chromosomes determined on one of two radiation hybrid (RH) panels or a yeast artificial chromosome (YAC) library containing human genomic DNA. This mapping data was integrated relative to the human genetic map and then cross-referenced to cytogenetic band maps of the chromosomes. (Further details are available in the accompanying article in the 25 October issue of SCIENCE).
Tremendous progress has been made in the mapping of human genes, a major milestone in the Human Genome Project. Apart from its utility in advancing our understanding of the genetic basis of disease, it provides a framework and focus for accelerated sequencing efforts by highlighting key landmarks (gene-rich regions) of the chromosomes. The construction of this map has been possible through the cooperative efforts of an international consortium of scientists who provide equal, full and unrestricted access to the data for the advancement of biology and human health.

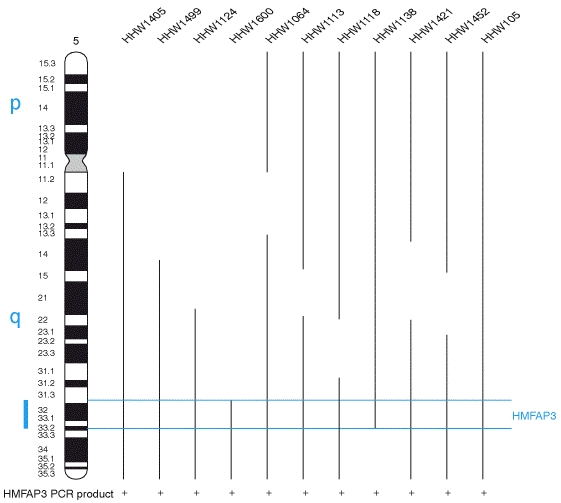
2B. Genome-nuclear lamina interactions and gene regulation.
- large genomic domains that are in contact with the lamina.
- the lamina can actively contribute to this repression.
- individual transcription units and multigene regions and
- affects many genes that determine cellular identity.
- genes that move away from the lamina are concomitantly activated;
- others, remain inactive yet become unlocked for activation in a next differentiation step.
lamina-genome interactions are widely involved in the control of gene expression programs during lineage commitment and terminal differentiation.
- Various cell types share a core architecture of genome-nuclear lamina interactions
- During differentiation, hundreds of genes change their lamina interactions
- Changes in lamina interactions reflect cell identity
- Release from the lamina may unlock some genes for activation
Fractal “globule”
About 10 years ago—just as the human genome project was completing its first draft sequence—Dekker pioneered a new technique, called chromosome conformation capture (C3) that allowed researchers to get a glimpse of how chromosomes are arranged relative to each other in the nucleus. The technique relies on the physical cross-linking of chromosomal regions that lie in close proximity to one another. The regions are then sequenced to identify which regions have been cross-linked. In 2009, using a high throughput version of this basic method, called Hi-C, Dekker and his collaborators discovered that the human genome appears to adopt a “fractal globule” conformation—
- a manner of crumpling without knotting.

In the last 3 years, Jobe Dekker and others have advanced technology even further, allowing them to paint a more refined picture of how the genome folds—and how this influences gene expression and disease states. Dekker’s 2009 findings were a breakthrough in modeling genome folding, but the resolution—about 1 million base pairs— was too crude to allow scientists to really understand how genes interacted with specific regulatory elements. The researchers report two striking findings.
First, the human genome is organized into two separate compartments, keeping
- active genes separate and accessible
- while sequestering unused DNA in a denser storage compartment.
- Chromosomes snake in and out of the two compartments repeatedly
- as their DNA alternates between active, gene-rich and inactive, gene-poor stretches.
Second, at a finer scale, the genome adopts an unusual organization known in mathematics as a “fractal.” The specific architecture the scientists found, called
- a “fractal globule,” enables the cell to pack DNA incredibly tightly —
the information density in the nucleus is trillions of times higher than on a computer chip — while avoiding the knots and tangles that might interfere with the cell’s ability to read its own genome. Moreover, the DNA can easily Unfold and Refold during
- gene activation,
- gene repression, and
- cell replication.
Dekker and his colleagues discovered, for example, that chromosomes can be divided into folding domains—megabase-long segments within which
- genes and regulatory elements associate more often with one another than with other chromosome sections.
The DNA forms loops within the domains that bring a gene into close proximity with a specific regulatory element at a distant location along the chromosome. Another group, that of molecular biologist Bing Ren at the University of California, San Diego, published a similar finding in the same issue of Nature. Dekker thinks the discovery of [folding] domains will be one of the most fundamental [genetics] discoveries of the last 10 years. The big questions now are
- how these domains are formed, and
- what determines which elements are looped into proximity.
“By breaking the genome into millions of pieces, we created a spatial map showing how close different parts are to one another,” says co-first author Nynke van Berkum, a postdoctoral researcher at UMass Medical School in Dekker‘s laboratory. “We made a fantastic three-dimensional jigsaw puzzle and then, with a computer, solved the puzzle.”
Lieberman-Aiden, van Berkum, Lander, and Dekker’s co-authors are Bryan R. Lajoie of UMMS; Louise Williams, Ido Amit, and Andreas Gnirke of the Broad Institute; Maxim Imakaev and Leonid A. Mirny of MIT; Tobias Ragoczy, Agnes Telling, and Mark Groudine of the Fred Hutchison, Cancer Research Center and the University of Washington; Peter J. Sabo, Michael O. Dorschner, Richard Sandstrom, M.A. Bender, and John Stamatoyannopoulos of the University of Washington; and Bradley Bernstein of the Broad Institute and Harvard Medical School.
2C. three-dimensional structure of the human genome
Lieberman-Aiden et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science, 2009; DOI: 10.1126/science.1181369.
Harvard University (2009, October 11). 3-D Structure Of Human Genome: Fractal Globule Architecture Packs Two Meters Of DNA Into Each Cell. ScienceDaily. Retrieved February 2, 2013, from http://www.sciencedaily.com/releases/2009/10/091008142957
Using a new technology called Hi-C and applying it to answer the thorny question of how each of our cells stows some three billion base pairs of DNA while maintaining access to functionally crucial segments. The paper comes from a team led by scientists at Harvard University, the Broad Institute of Harvard and MIT, University of Massachusetts Medical School, and the Massachusetts Institute of Technology. “We’ve long known that on a small scale, DNA is a double helix,” says co-first author Erez Lieberman-Aiden, a graduate student in the Harvard-MIT Division of Health Science and Technology and a researcher at Harvard’s School of Engineering and Applied Sciences and in the laboratory of Eric Lander at the Broad Institute. “But if the double helix didn’t fold further, the genome in each cell would be two meters long. Scientists have not really understood how the double helix folds to fit into the nucleus of a human cell, which is only about a hundredth of a millimeter in diameter. This new approach enabled us to probe exactly that question.”
The mapping technique that Aiden and his colleagues have come up with bridges a crucial gap in knowledge—between what goes on at the smallest levels of genetics (the double helix of DNA and the base pairs) and the largest levels (the way DNA is gathered up into the 23 chromosomes that contain much of the human genome). The intermediate level, on the order of thousands or millions of base pairs, has remained murky. As the genome is so closely wound, base pairs in one end can be close to others at another end in ways that are not obvious merely by knowing the sequence of base pairs. Borrowing from work that was started in the 1990s, Aiden and others have been able to figure out which base pairs have wound up next to one another. From there, they can begin to reconstruct the genome—in three dimensions.

Even as the multi-dimensional mapping techniques remain in their early stages, their importance in basic biological research is becoming ever more apparent. “The three-dimensional genome is a powerful thing to know,” Aiden says. “A central mystery of biology is the question of how different cells perform different functions—despite the fact that they share the same genome.” How does a liver cell, for example, “know” to perform its liver duties when it contains the same genome as a cell in the eye? As Aiden and others reconstruct the trail of letters into a three-dimensional entity, they have begun to see that “the way the genome is folded determines which genes were
2D. “Mr. President; The Genome is Fractal !”
Eric Lander (Science Adviser to the President and Director of Broad Institute) et al. delivered the message on Science Magazine cover (Oct. 9, 2009) and generated interest in this by the International HoloGenomics Society at a Sept meeting.
First, it may seem to be trivial to rectify the statement in “About cover” of Science Magazine by AAAS.
- The statement “the Hilbert curve is a one-dimensional fractal trajectory” needs mathematical clarification.
The mathematical concept of a Hilbert space, named after David Hilbert, generalizes the notion of Euclidean space. It extends the methods of vector algebra and calculus from the two-dimensional Euclidean plane and three-dimensional space to spaces with any finite or infinite number of dimensions. A Hilbert space is an abstract vector space possessing the structure of an inner product that allows length and angle to be measured. Furthermore, Hilbert spaces must be complete, a property that stipulates the existence of enough limits in the space to allow the techniques of calculus to be used. A Hilbert curve (also known as a Hilbert space-filling curve) is a continuous fractal space-filling curve first described by the German mathematician David Hilbert in 1891,[1] as a variant of the space-filling curves discovered by Giuseppe Peano in 1890.[2] For multidimensional databases, Hilbert order has been proposed to be used instead of Z order because it has better locality-preserving behavior.
Representation as Lindenmayer system
The Hilbert Curve can be expressed by a rewrite system (L-system).
Alphabet : A, B
Constants : F + – 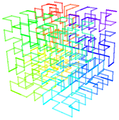

Axiom : A
Production rules:
A → – B F + A F A + F B –
B → + A F – B F B – F A +
Here, F means “draw forward”, – means “turn left 90°”, and + means “turn right 90°” (see turtle graphics).

While the paper itself does not make this statement, the new Editorship of the AAAS Magazine might be even more advanced if the previous Editorship did not reject (without review) a Manuscript by 20+ Founders of (formerly) International PostGenetics Society in December, 2006.
Second, it may not be sufficiently clear for the reader that the reasonable requirement for the DNA polymerase to crawl along a “knot-free” (or “low knot”) structure does not need fractals. A “knot-free” structure could be spooled by an ordinary “knitting globule” (such that the DNA polymerase does not bump into a “knot” when duplicating the strand; just like someone knitting can go through the entire thread without encountering an annoying knot): Just to be “knot-free” you don’t need fractals. Note, however, that
- the “strand” can be accessed only at its beginning – it is impossible to e.g. to pluck a segment from deep inside the “globulus”.
This is where certain fractals provide a major advantage – that could be the “Eureka” moment for many readers. For instance,
- the mentioned Hilbert-curve is not only “knot free” –
- but provides an easy access to “linearly remote” segments of the strand.
If the Hilbert curve starts from the lower right corner and ends at the lower left corner, for instance
- the path shows the very easy access of what would be the mid-point
- if the Hilbert-curve is measured by the Euclidean distance along the zig-zagged path.
Likewise, even the path from the beginning of the Hilbert-curve is about equally easy to access – easier than to reach from the origin a point that is about 2/3 down the path. The Hilbert-curve provides an easy access between two points within the “spooled thread”; from a point that is about 1/5 of the overall length to about 3/5 is also in a “close neighborhood”.
This may be the “Eureka-moment” for some readers, to realize that
- the strand of “the Double Helix” requires quite a finess to fold into the densest possible globuli (the chromosomes) in a clever way
- that various segments can be easily accessed. Moreover, in a way that distances between various segments are minimized.
This marvellous fractal structure is illustrated by the 3D rendering of the Hilbert-curve. Once you observe such fractal structure, you’ll never again think of a chromosome as a “brillo mess”, would you? It will dawn on you that the genome is orders of magnitudes more finessed than we ever thought so.
Those embarking at a somewhat complex review of some historical aspects of the power of fractals may wish to consult the ouvre of Mandelbrot (also, to celebrate his 85th birthday). For the more sophisticated readers, even the fairly simple Hilbert-curve (a representative of the Peano-class) becomes even more stunningly brilliant than just some “see through density”. Those who are familiar with the classic “Traveling Salesman Problem” know that “the shortest path along which every given n locations can be visited once, and only once” requires fairly sophisticated algorithms (and tremendous amount of computation if n>10 (or much more). Some readers will be amazed, therefore, that for n=9 the underlying Hilbert-curve helps to provide an empirical solution.
refer to pellionisz@junkdna.com
Briefly, the significance of the above realization, that the (recursive) Fractal Hilbert Curve is intimately connected to the (recursive) solution of TravelingSalesman Problem, a core-concept of Artificial Neural Networks can be summarized as below.
Accomplished physicist John Hopfield (already a member of the National Academy of Science) aroused great excitement in 1982 with his (recursive) design of artificial neural networks and learning algorithms which were able to find reasonable solutions to combinatorial problems such as the Traveling SalesmanProblem. (Book review Clark Jeffries, 1991, see also 2. J. Anderson, R. Rosenfeld, and A. Pellionisz (eds.), Neurocomputing 2: Directions for research, MIT Press, Cambridge, MA, 1990):
“Perceptions were modeled chiefly with neural connections in a “forward” direction: A -> B -* C — D. The analysis of networks with strong backward coupling proved intractable. All our interesting results arise as consequences of the strong back-coupling” (Hopfield, 1982).
The Principle of Recursive Genome Function surpassed obsolete axioms that blocked, for half a Century, entry of recursive algorithms to interpretation of the structure-and function of (Holo)Genome. This breakthrough, by uniting the two largely separate fields of Neural Networks and Genome Informatics, is particularly important for
- those who focused on Biological (actually occurring) Neural Networks (rather than abstract algorithms that may not, or because of their core-axioms, simply could not
- represent neural networks under the governance of DNA information).

3A. The FractoGene Decade
from Inception in 2002 to Proofs of Concept and Impending Clinical Applications by 2012
- Junk DNA Revisited (SF Gate, 2002)
- The Future of Life, 50th Anniversary of DNA (Monterey, 2003)
- Mandelbrot and Pellionisz (Stanford, 2004)
- Morphogenesis, Physiology and Biophysics (Simons, Pellionisz 2005)
- PostGenetics; Genetics beyond Genes (Budapest, 2006)
- ENCODE-conclusion (Collins, 2007)
The Principle of Recursive Genome Function (paper, YouTube, 2008)
- Cold Spring Harbor presentation of FractoGene (Cold Spring Harbor, 2009)
- Mr. President, the Genome is Fractal! (2009)
- HolGenTech, Inc. Founded (2010)
- Pellionisz on the Board of Advisers in the USA and India (2011)
- ENCODE – final admission (2012)
- Recursive Genome Function is Clogged by Fractal Defects in Hilbert-Curve (2012)
- Geometric Unification of Neuroscience and Genomics (2012)
- US Patent Office issues FractoGene 8,280,641 to Pellionisz (2012)
http://www.junkdna.com/the_fractogene_decade.pdf
http://www.scribd.com/doc/116159052/The-Decade-of-FractoGene-From-Discovery-to-Utility-Proofs-of-Concept-Open-Genome-Based-Clinical-Applications
http://fractogene.com/full_genome/morphogenesis.html
When the human genome was first sequenced in June 2000, there were two pretty big surprises. The first was thathumans have only about 30,000-40,000 identifiable genes, not the 100,000 or more many researchers were expecting. The lower –and more humbling — number
- means humans have just one-third more genes than a common species of worm.
The second stunner was
- how much human genetic material — more than 90 percent — is made up of what scientists were calling “junk DNA.”
The term was coined to describe similar but not completely identical repetitive sequences of amino acids (the same substances that make genes), which appeared to have no function or purpose. The main theory at the time was that these apparently non-working sections of DNA were just evolutionary leftovers, much like our earlobes.
If biophysicist Andras Pellionisz is correct, genetic science may be on the verge of yielding its third — and by far biggest — surprise.
With a doctorate in physics, Pellionisz is the holder of Ph.D.’s in computer sciences and experimental biology from the prestigious Budapest Technical University and the Hungarian National Academy of Sciences. A biophysicist by training, the 59-year-old is a former research associate professor of physiology and biophysics at New York University, author of numerous papers in respected scientific journals and textbooks, a past winner of the prestigious Humboldt Prize for scientific research, a former consultant to NASA and holder of a patent on the world’s first artificial cerebellum, a technology that has already been integrated into research on advanced avionics systems. Because of his background, the Hungarian-born brain researcher might also become one of the first people to successfully launch a new company by using the Internet to gather momentum for a novel scientific idea.
The genes we know about today, Pellionisz says, can be thought of as something similar to machines that make bricks (proteins, in the case of genes), with certain junk-DNA sections providing a blueprint for the different ways those proteins are assembled. The notion that at least certain parts of junk DNA might have a purpose for example, many researchers now refer to with a far less derogatory term: introns.
In a provisional patent application filed July 31, Pellionisz claims to have unlocked a key to the hidden role junk DNA plays in growth — and in life itself. His patent application covers all attempts to count, measure and compare the fractal properties of introns for diagnostic and therapeutic purposes.
3B. The Hidden Fractal Language of Intron DNA
To fully understand Pellionisz’ idea, one must first know what a fractal is.
Fractals are a way that nature organizes matter. Fractal patterns can be found in anything that has a nonsmooth surface (unlike a billiard ball), such as coastal seashores, the branches of a tree or the contours of a neuron (a nerve cell in the brain). Some, but not all, fractals are self-similar and stop repeating their patterns at some stage; the branches of a tree, for example, can get only so small. Because they are geometric, meaning they have a shape, fractals can be described in mathematical terms. It’s similar to the way a circle can be described by using a number to represent its radius (the distance from its center to its outer edge). When that number is known, it’s possible to draw the circle it represents without ever having seen it before.
Although the math is much more complicated, the same is true of fractals. If one has the formula for a given fractal, it’s possible to use that formula
- to construct, or reconstruct,
- an image of whatever structure it represents,
- no matter how complicated.
The mysteriously repetitive but not identical strands of genetic material are in reality building instructions organized in a special type
- of pattern known as a fractal. It’s this pattern of fractal instructions, he says, that
- tells genes what they must do in order to form living tissue,
- everything from the wings of a fly to the entire body of a full-grown human.
In a move sure to alienate some scientists, Pellionisz has chosen the unorthodox route of making his initial disclosures online on his own Web site. He picked that strategy, he says, because it is the fastest way he can document his claims and find scientific collaborators and investors. Most mainstream scientists usually blanch at such approaches, preferring more traditionally credible methods, such as publishing articles in peer-reviewed journals.
Basically, Pellionisz’ idea is that a fractal set of building instructions in the DNA plays a similar role in organizing life itself. Decode the way that language works, he says, and in theory it could be reverse engineered. Just as knowing the radius of a circle lets one create that circle, the more complicated fractal-based formula would allow us to understand how nature creates a heart or simpler structures, such as disease-fighting antibodies. At a minimum, we’d get a far better understanding of how nature gets that job done.
The complicated quality of the idea is helping encourage new collaborations across the boundaries that sometimes separate the increasingly intertwined disciplines of biology, mathematics and computer sciences.
Hal Plotkin, Special to SF Gate. Thursday, November 21, 2002. http://www.junkdna.com/Special to SF Gate/plotkin.htm (1 of 10)2012.12.13. 12:11:58/

3C. multifractal analysis
The human genome: a multifractal analysis. Moreno PA, Vélez PE, Martínez E, et al.
BMC Genomics 2011, 12:506. http://www.biomedcentral.com/1471-2164/12/506
Background: Several studies have shown that genomes can be studied via a multifractal formalism. Recently, we used a multifractal approach to study the genetic information content of the Caenorhabditis elegans genome. Here we investigate the possibility that the human genome shows a similar behavior to that observed in the nematode.
Results: We report here multifractality in the human genome sequence. This behavior correlates strongly on the
- presence of Alu elements and
- to a lesser extent on CpG islands and (G+C) content.
In contrast, no or low relationship was found for LINE, MIR, MER, LTRs elements and DNA regions poor in genetic information.
- Gene function,
- cluster of orthologous genes,
- metabolic pathways, and
- exons tended to increase their frequencies with ranges of multifractality and
- large gene families were located in genomic regions with varied multifractality.
Additionally, a multifractal map and classification for human chromosomes are proposed.
Conclusions
we propose a descriptive non-linear model for the structure of the human genome,
This model reveals
- a multifractal regionalization where many regions coexist that are far from equilibrium and
- this non-linear organization has significant molecular and medical genetic implications for understanding the role of
- Alu elements in genome stability and structure of the human genome.
Given the role of Alu sequences in
- gene regulation,
- genetic diseases,
- human genetic diversity,
- adaptation
- and phylogenetic analyses,
these quantifications are especially useful.
MiIP: The Monomer Identification and Isolation Program
Bun C, Ziccardi W, Doering J and Putonti C.Evolutionary Bioinformatics 2012:8 293-300. http://dx.goi.org/10.4137/EBO.S9248
Repetitive elements within genomic DNA are both functionally and evolutionarilly informative. Discovering these sequences ab initio is
- computationally challenging, compounded by the fact that
- sequence identity between repetitive elements can vary significantly.
Here we present a new application, the Monomer Identification and Isolation Program (MiIP), which provides functionality to both
- search for a particular repeat as well as
- discover repetitive elements within a larger genomic sequence.
To compare MiIP’s performance with other repeat detection tools, analysis was conducted for
- synthetic sequences as well as
- several a21-II clones and
- HC21 BAC sequences.
The primary benefit of MiIP is the fact that it is a single tool capable of searching for both
- known monomeric sequences as well as
- discovering the occurrence of repeats ab initio, per the user’s required sensitivity of the search.
Methods for Examining Genomic and Proteomic Interactions
1. An Integrated Statistical Approach to Compare Transcriptomics Data Across Experiments: A Case Study on the Identification of Candidate Target Genes of the Transcription Factor PPARα
Ullah MO, Müller M and Hooiveld GJEJ. Bioinformatics and Biology Insights 2012:6 145–154. http://dx.doi.org/10.4137/BBI.S9529
http://www.la- press.com/
http://bionformaticsandBiologyInsights.com/An_Integrated_Statistical_Approach_to_Compare_ transcriptomic_Data_Across_Experiments-A-Case_Study_on_the_Identification_ of_Candidate_Target_Genes_of_the Transcription_Factor_PPARα/
Corresponding author email: guido.hooiveld@wur.nl
An effective strategy to elucidate the signal transduction cascades activated by a transcription factor is to compare the transcriptional profiles of wild type and transcription factor knockout models. Many statistical tests have been proposed for analyzing gene expression data, but most
- tests are based on pair-wise comparisons. Since the analysis of microarrays involves the testing of multiple hypotheses within one study, it is
- generally accepted that one should control for false positives by the false discovery rate (FDR). However, it has been reported that
- this may be an inappropriate metric for comparing data across different experiments.
Here we propose an approach that addresses the above mentioned problem by the simultaneous testing and integration of the three hypotheses (contrasts) using the cell means ANOVA model.
These three contrasts test for the effect of
- a treatment in wild type,
- gene knockout, and
- globally over all experimental groups.
We illustrate our approach on microarray experiments that focused on the identification of candidate target genes and biological processes governed by the fatty acid sensing transcription factor PPARα in liver. Compared to the often applied FDR based across experiment comparison, our approach identified a conservative but less noisy set of candidate genes with same sensitivity and specificity. However, our method had the advantage of
- properly adjusting for multiple testing while
- integrating data from two experiments, and
- was driven by biological inference.
We present a simple, yet efficient strategy to compare
- differential expression of genes across experiments
- while controlling for multiple hypothesis testing.
Vincent VanBuren & Hailin Chen. Scientific Reports 2, Article number: 1011 Received 02 October 2012 Accepted 04 December 2012 Published 20 December 2012
http://dx.doi.org/10.1038/srep01011
The complexity of biomolecular interactions and influences is a major obstacle to their comprehension and elucidation. Visualizing knowledge of biomolecular interactions increases comprehension and facilitates the development of new hypotheses. The rapidly changing landscape of high-content experimental results also presents a challenge for the maintenance of comprehensive knowledgebases. Distributing the responsibility for maintenance of a knowledgebase to a community of subject matter experts is an effective strategy for large, complex and rapidly changing knowledgebases.
Cognoscente serves these needs by
- building visualizations for queries of biomolecular interactions on demand,
- by managing the complexity of those visualizations, and
- by crowdsourcing to promote the incorporation of current knowledge from the literature.
Imputing functional associations between biomolecules and imputing directionality of regulation for those predictions each
- require a corpus of existing knowledge as a framework to build upon. Comprehension of the complexity of this corpus of knowledge
- will be facilitated by effective visualizations of the corresponding biomolecular interaction networks.
Cognoscente
http://vanburenlab.medicine.tamhsc.edu/cognoscente.html
was designed and implemented to serve these roles as
- a knowledgebase and
- as an effective visualization tool for systems biology research and education.
Cognoscente currently contains over 413,000 documented interactions, with coverage across multiple species. Perl, HTML, GraphViz1, and a MySQL database were used in the development of Cognoscente. Cognoscente was motivated by the need to
- update the knowledgebase of biomolecular interactions at the user level, and
- flexibly visualize multi-molecule query results for heterogeneous interaction types across different orthologs.
Satisfying these needs provides a strong foundation for developing new hypotheses about regulatory and metabolic pathway topologies. Several existing tools provide functions that are similar to Cognoscente, so we selected several popular alternatives to
- assess how their feature sets compare with Cognoscente ( Table 1 ). All databases assessed had
- easily traceable documentation for each interaction, and
- included protein-protein interactions in the database.
Most databases, with the exception of BIND,
- provide an open-access database that can be downloaded as a whole.
Most databases, with the exceptions of EcoCyc and HPRD, provide
- support for multiple organisms.
Most databases support web services for interacting with the database contents programatically, whereas this is a planned feature for Cognoscente.
- INT, STRING, IntAct, EcoCyc, DIP and Cognoscente provide built-in visualizations of query results,
- which we consider among the most important features for facilitating comprehension of query results.
- BIND supports visualizations via Cytoscape. Cognoscente is among a few other tools that support multiple organisms in the same query,
- protein->DNA interactions, and
- multi-molecule queries.
Cognoscente has planned support for small molecule interactants (i.e. pharmacological agents). MINT, STRING, and IntAct provide a prediction (i.e. score) of functional associations, whereas
Cognoscente does not currently support this. Cognoscente provides support for multiple edge encodings to visualize different types of interactions in the same display,
- a crowdsourcing web portal that allows users to submit interactions
- that are then automatically incorporated in the knowledgebase, and displays orthologs as compound nodes to provide clues about potential
- orthologous interactions.
The main strengths of Cognoscente are that
- it provides a combined feature set that is superior to any existing database,
- it provides a unique visualization feature for orthologous molecules, and relatively unique support for
- multiple edge encodings,
- crowdsourcing, and
- connectivity parameterization.
The current weaknesses of Cognoscente relative to these other tools are
- that it does not fully support web service interactions with the database,
- it does not fully support small molecule interactants, and
- it does not score interactions to predict functional associations.
Web services and support for small molecule interactants are currently under development.
Other related articles on thie Open Access Online Sceintific Journal, include the following:
Big Data in Genomic Medicine lhb http://pharmaceuticalintelligence.com/2012/12/17/big-data-in-genomic-medicine/
BRCA1 a tumour suppressor in breast and ovarian cancer – functions in transcription, ubiquitination and DNA repair S Saha http://pharmaceuticalintelligence.com/2012/12/04/brca1-a-tumour-suppressor-in-breast-and-ovarian-cancer-functions-in-transcription-ubiquitination-and-dna-repair/
Computational Genomics Center: New Unification of Computational Technologies at Stanford A Lev-Ari http://pharmaceuticalintelligence.com/2012/12/03/computational-genomics-center-new-unification-of-computational-technologies-at-stanford/
Paradigm Shift in Human Genomics – Predictive Biomarkers and Personalized Medicine – Part 1 (pharmaceuticalintelligence.com) A Lev-Ari http://pharmaceuticalintelligence.com/2013/01/13/paradigm-shift-in-human-genomics-predictive-biomarkers-and-personalized-medicine-part-1/
LEADERS in Genome Sequencing of Genetic Mutations for Therapeutic Drug Selection in Cancer Personalized Treatment: Part 2 A Lev-Ari
http://pharmaceuticalintelligence.com/2013/01/13/leaders-in-genome-sequencing-of-genetic-mutations-for-therapeutic-drug-selection-in-cancer-personalized-treatment-part-2/
Personalized Medicine: An Institute Profile – Coriell Institute for Medical Research: Part 3 A Lev-Ari http://pharmaceuticalintelligence.com/2013/01/13/personalized-medicine-an-institute-profile-coriell-institute-for-medical-research-part-3/
GSK for Personalized Medicine using Cancer Drugs needs Alacris systems biology model to determine the in silico effect of the inhibitor in its “virtual clinical trial” A Lev-Ari http://pharmaceuticalintelligence.com/2012/11/14/gsk-for-personalized-medicine-using-cancer-drugs-needs-alacris-systems-biology-model-to-determine-the-in-silico-effect-of-the-inhibitor-in-its-virtual-clinical-trial/
Recurrent somatic mutations in chromatin-remodeling and ubiquitin ligase complex genes in serous endometrial tumors S Saha
http://pharmaceuticalintelligence.com/2012/11/19/recurrent-somatic-mutations-in-chromatin-remodeling-and-ubiquitin-ligase-complex-genes-in-serous-endometrial-tumors/
Human Variome Project: encyclopedic catalog of sequence variants indexed to the human genome sequence A Lev-Ari
Prostate Cancer Cells: Histone Deacetylase Inhibitors Induce Epithelial-to-Mesenchymal Transition sjwilliams
http://pharmaceuticalintelligence.com/2012/11/30/histone-deacetylase-inhibitors-induce-epithelial-to-mesenchymal-transition-in-prostate-cancer-cells/
Directions for genomics in personalized medicine lhb http://pharmaceuticalintelligence.com/2013/01/27/directions-for-genomics-in-personalized-medicine/
How mobile elements in “Junk” DNA promote cancer. Part 1: Transposon-mediated tumorigenesis. Sjwilliams
http://pharmaceuticalintelligence.com/2012/10/31/how-mobile-elements-in-junk-dna-prote-cancer-part1-transposon-mediated-tumorigenesis/
Mitochondrial fission and fusion: potential therapeutic targets? Ritu saxena http://pharmaceuticalintelligence.com/2012/10/31/mitochondrial-fission-and-fusion-potential-therapeutic-target/
Mitochondrial mutation analysis might be “1-step” away ritu saxena http://pharmaceuticalintelligence.com/2012/08/14/mitochondrial-mutation-analysis-might-be-1-step-away/
mRNA interference with cancer expression lhb http://pharmaceuticalintelligence.com/2012/10/26/mrna-interference-with-cancer-expression/
Expanding the Genetic Alphabet and linking the genome to the metabolome http://pharmaceuticalintelligence.com/2012/09/24/expanding-the-genetic-alphabet-and-linking-the-genome-to-the-metabolome/
Breast Cancer: Genomic profiling to predict Survival: Combination of Histopathology and Gene Expression Analysis A Lev-Ari
Ubiquinin-Proteosome pathway, autophagy, the mitochondrion, proteolysis and cell apoptosis lhb http://pharmaceuticalintelligence.com/2012/10/30/ubiquinin-proteosome-pathway-autophagy-the-mitochondrion-proteolysis-and-cell-apoptosis/
Genomic Analysis: FLUIDIGM Technology in the Life Science and Agricultural Biotechnology A Lev-Ari http://pharmaceuticalintelligence.com/2012/08/22/genomic-analysis-fluidigm-technology-in-the-life-science-and-agricultural-biotechnology/
2013 Genomics: The Era Beyond the Sequencing Human Genome: Francis Collins, Craig Venter, Eric Lander, et al. http://pharmaceuticalintelligence.com/2013_Genomics
Paradigm Shift in Human Genomics – Predictive Biomarkers and Personalized Medicine – Part 1 http://pharmaceuticalintelligence.com/Paradigm Shift in Human Genomics_/

English: DNA replication or DNA synthesis is the process of copying a double-stranded DNA molecule. This process is paramount to all life as we know it. (Photo credit: Wikipedia)

Français : Deletion chromosomique (Photo credit: Wikipedia)

A slight mutation in the matched nucleotides can lead to chromosomal aberrations and unintentional genetic rearrangement. (Photo credit: Wikipedia)
Related articles
- Ryogen Receives Nine Human Gene Patents in 2012 for Total of 23-Patent Portfolio (prweb.com)
- Unraveling the Human Genome: 6 Molecular Milestones (livescience.com)
- Life Stands on the shoulders of Giants (Viruses) (bytesizebio.net)
- New insights into the human genome by ENCODE project (slideshare.net)
- Melanoma Genes Found In “Junk” DNA (medicalnewstoday.com)
- Learning the alphabet of gene control (esciencenews.com)
- BEST OF THE WEB: On viral ‘junk’ DNA, a DNA-enhancing Ketogenic diet, and cometary kicks (sott.net)


