developments in medical spectroscopy
Larry H. Bernstein, MD, FCAP, Curator
LPBI
Using QCLs for MIR-Based Spectral Imaging — Applications in Tissue Pathology
A quantum cascade laser (QCL) microscope allows for fast data acquisition, real-time chemical imaging and the ability to collect only spectral frequencies of interest. Due to their high-quality, highly tunable illumination characteristics and excellent signal-to-noise performance, QCLs are paving the way for the next generation of mid-infrared (MIR) imaging methodologies.
http://www.photonics.com/images/Web/Articles/2015/9/8/Imaging_Prostate.png
Efficient Spectroscopic Imaging Demonstrated In Vivo
Although optical spectroscopy is routinely used study molecules in cell samples, it is currently not practical to perform in vivo. Now, a converted Raman spectroscopy system has been used to reveal the chemical composition of living tissues in seconds.

http://www.photonics.com/images2/EmailBlasts%5CSpectroscopy/2015/11/Efficient_Spectroscopic_Imaging_Demonstrated_In_Vivo.jpg
Broadband Laser Aimed at Cancer Detection
Covering a wide swath of the mid-infrared region, a new laser system offers greater spectral sensitivity.

http://www.photonics.com/images/Web/Articles/2015/9/25/REAS_molecular_1.jpg
Using QCLs for MIR-Based Spectral Imaging — Applications in Tissue Pathology
A quantum cascade laser (QCL) microscope allows for fast data acquisition, real-time chemical imaging and the ability to collect only spectral frequencies of interest. Due to their high-quality, highly tunable illumination characteristics and excellent signal-to-noise performance, QCLs are paving the way for the next generation of mid-infrared (MIR) imaging methodologies.
MICHAEL WALSH, UNIVERSITY OF ILLINOIS AT CHICAGO; MATTHEW BARRE & BENJAMIN BIRD, DAYLIGHT SOLUTIONS
H. Sreedhar*1, V. Varma*2, A. Graham3, Z. Richards1, F. Gambacorata4, A. Bhatt1,
P. Nguyen1, K. Meinke1, L. Nonn1, G. Guzman1, E. Fotheringham5, M. Weida5,
D. Arnone5, B. Mohar5, J. Rowlette5
1 Department of Bioengineering, University of Illinois at Chicago
2 Department of Pathology, University of Illinois at Chicago
3 Department of Bioengineering, University of Illinois at Urbana-Champaign
4 Department of Chemical Engineering, University of Illinois at Chicago
5 Daylight Solutions, San Diego
*Contributed Equally
Real-time, MIR chemical imaging microscopes could soon become powerful frontline screening tools for practicing pathologists. The ability to see differences in the biochemical makeup across a tissue sample greatly enhances a practioner’s ability to detect early stages of disease or disease variants. Today, this is accomplished much as it was 100 years ago — through the use of specially formulated stains and dyes in combination with white light microscopy. A new MIR, QCL-based microscope from Daylight Solutions enables real-time, nondestructive biochemical imaging of tissues without the need to perturb the sample with chemical or heat treatments, thus preserving the sample for follow-on fluorescence tagging, histochemical staining or other “omics” testing within the workflow.
MIR chemical imaging is a well-established absorbance spectroscopy technique; it senses the relative amount of light that molecules absorb due to their unique vibrational resonances falling within the MIR portion of the electromagnetic spectrum (i.e., wavelengths from approximately 2 to 15 µm). This absorption can be detected with a variety of MIR detector types and can provide detailed information about the sample’s chemical composition.
The most common instrument for this type of measurement is known as a Fourier transform infrared (FTIR) spectrometer. FTIR systems use a broadband MIR light source, known as a globar, to illuminate a sample; the absorption spectrum is generated by the use of interferometry. Throughout the past decade, FTIR systems have incorporated linear arrays and 2D focal plane arrays (FPAs) in a microscope configuration to enable a technique known as chemical imaging.
With this approach, the illumination beam is expanded across a sample area, and the data produced is transformed into a hyperspectral data cube — a 2D image of the sample with an absorption profile associated with every pixel. This is a very versatile technique that allows the detailed spatial distribution of chemical content to be analyzed across a sample. Recently, this technique has proved to be very useful within the biomedical imaging sector for label-free, biochemical analyses of cells, tissue and biofluids.
While FTIR microscopy now is established as a powerful technique for a wide variety of applications, the instruments used for this methodology are fundamentally limited by the brightness of the globar source. Users looking to maximize the signal-to-noise ratios, and the associated resolutions of the images produced are forced to use synchrotron facilities, which replace the globar light source with a MIR beam generated by a particle accelerator. This approach can yield excellent results but clearly is not practical for benchtop applications; it is particularly unfit for biomedical imaging applications within clinical settings.
The recent advent of QCLs has provided an ideal light source for next-generation MIR microscopy. They are compact, semiconductor-based lasers that produce high-brightness light in the MIR region. The devices can be manufactured in an external cavity configuration to provide broadly tunable output with a narrow spectral bandwidth at each frequency. In this configuration, a QCL can be tuned across the MIR spectrum to sequentially capture an absorption profile for chemical identification.
Daylight Solutions’ IR microscope incorporates a broadly tunable and high-brightness QCL light source (it is an order of magnitude brighter than a synchrotron), a set of high numerical aperture (NA) diffraction-limited objectives, and an uncooled microbolometer FPA into a compact, benchtop instrument, as shown in Figure 1. The instrument provides rapid, high-resolution chemical images across very large fields of view and also provides a real-time chemical imaging mode. By overcoming the physical size, camera cooling and data collection time requirements of FTIR-based instruments, the microscope is positioned to bring MIR microscopy beyond research settings and into clinical use.

Figure 1. Schematic of a quantum cascade laser (QCL) microscope. Courtesy of Daylight Solutions.
Dr. Michael Walsh of the University of Illinois at Chicago (UIC) conducts research within the pathology department’s Spectral Pathology Lab, which has been using the IR microscope for the past several months. Walsh has been focused on developing chemical imaging techniques, with the ultimate goal of improving diagnoses within the field of tissue pathology.
Currently, the state-of-the-art method-ology used for the diagnosis of most solid-organ diseases is to extract a tissue sample via a biopsy. Tissue inherently has very little contrast and needs to be stained with dyes or probes to visualize and identify cell types and tissue structures. The field of pathology is based on examining the stained tissues, typically using white light, to determine if the tissue morphology deviates from a normal pattern. If the tissue looks abnormal, the disease state may be further subclassified by grade or by predicted outcome. However, the field of pathology is limited by the information that can be derived from the stained tissues and the subjective interpretation of the tissue by a highly trained pathologist.

Spero microscope. Courtesy of Daylight Solutions.
UIC’s Spectral Pathology Lab is focused on identifying areas in pathology where current techniques fail, or where there is a need for additional diagnostic or prognostic information that can help improve patient care. Potentially, MIR imaging is a very valuable adjunct to the current practice of pathology. Rather than using only stains, MIR imaging can interrogate the entire biochemistry of the tissue and render a diagnosis in an objective fashion. Traditionally, MIR imaging with an FTIR system has been limited by slow data acquisition speeds and the need to collect the entire spectral data cube. QCL imaging with the Spero microscope has the potential to speed up the data acquisition of images obtained from a tissue sample and to collect only the spectral frequencies of interest. The device also provides real-time imaging of samples at 30 fps, which could allow pathologists to very rapidly identify areas of interest on a tissue biopsy in a manner that is similar to their current clinical workflows. Some examples of the comparison of FTIR-derived and QCL-derived images from multiple organ tissues of interest are presented.

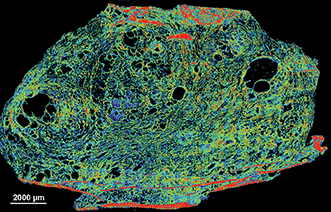
Figure 2. (a) H&E-stained image of a mouse brain section on IR reflective slide, with selected regions labeled: hypothalamus, thalamus, and dentate gyrus. (b) Transflectance QCL IR image of same region, prior to staining, at 1652 cm−1, in which the thalamus is clearly distinguished from surrounding regions. (c) Same region at 1548 cm−1. (d) Same region at 1500 cm−1. Courtesy of University of Illinois at Chicago (UIC)/Spectral Pathology Lab.
A tissue section from a mouse brain was scanned using the Spero microscope’s high-magnification objective (12.5×; 0.7 NA; 1.4 × 1.4-µm pixels) at various MIR frequencies in transflection mode, as shown in Figure 2. The tissue then was stained using hematoxylin and eosin (H&E), the most common stain in histopathology, and is displayed in Figure 2a. Using the H&E stain, regions were identified in the brain (thalamus, dentate gyrus and hypothalamus) that correlated with structures in the IR image. By illuminating the tissue at various wavelengths, discrete tissue features exhibit contrast due to the difference in absorption, as highlighted in the IR images taken at 1652, 1548 and 1500 cm−1 in Figure 2b-d, respectively. The microscope also makes it possible to visualize tissue at these individual wavelengths in real time. The identification of cell types and their biochemical changes is of particular interest in neuropathology.

Figure 3. (a) Transmission FTIR image of a 4-µm thick section from a human liver tissue microarray on barium fluoride at 1650 cm-1. The image was taken with 64 coadditions of successive scans. (b) Transmission image from the Spero microscope of the same tissue at 1652 cm-1, both baseline corrected between 1796 cm-1 and 904 cm-1. In both images, the bright white stripe dividing the tissue core roughly in half is a region of fibrosis (red arrow), while the rest of the tissue on either side is composed primarily of hepatocytes (blue arrow). Courtesy of UIC/Spectral Pathology Lab.
A single biopsy core obtained from human liver tissue was scanned in transmission mode on a barium fluoride substrate by an Agilent Cary 600 Series FTIR microscope (Figure 3a). The FTIR image was acquired using a 36× Cassegrain collecting objective and a 15× Cassegrain condenser for a pixel size of 2.2 × 2.2 µm. Figure 3b shows the same liver core acquired using the Spero microscope with the high-magnification collecting objective (12.5×, 0.7 NA) and condenser objective for a pixel size of 1.4 × 1.4 µm. High-definition IR imaging enables clear contrast and identification of the band of fibrosis in the center of the core and the surrounding regions of liver cells, known as hepatocytes, and is indicated within Figure 3a-b. Acquisition of IR imaging data at the diffraction limit enables chemical information to be recorded from tissue structures at the single-cell level, allowing accurate characterization of individual tissue components, different cell types, varied disease states or other aspects of a tissue section.

Figure 4. (a) Averaged spectra for regions of interest corresponding to the hepatocytes and the fibrotic area on the FTIR image in Figure 3a. Spectra have been truncated from 1800 to 900 cm-1, normalized to 1650 cm-1, and baseline corrected between 1796 and 904 cm-1. (b) Averaged spectra for regions of interest corresponding to the hepatocytes and the fibrotic area on the Spero microscope image in Figure 3b. Spectra have been normalized to 1652 cm-1 and baseline corrected between 1796 and 904 cm-1.
Figure 4 displays average spectra calculated from homogenous tissue regions that describe hepatocytes and fibrosis within the liver tissue core shown in Figure 3. The spectra acquired from both FTIR and QCL systems are very similar. Walsh is focused on developing spectral classifiers that can aid pathologists in making very difficult diagnoses in the precancerous stages of liver cancer.

Figure 5. H&E-stained section of human colon tissue, and FTIR (with 16 coadditions) and Spero microscope transmission images of a 4-µm thick serial section of the same sample on barium fluoride. FTIR image shown at 1650 cm-1, Spero microscope image shown at 1652 cm-1. The red circle indicates mucin, the green circle indicates malignant colon carcinoma epithelium, and the blue circle indicates fibroblastic stroma. The raw spectra (taken from single pixels in approximately the same location for each of the three tissue features) are shown below their respective IR images. The FTIR spectra were truncated to match the Spero microscope’s spectral range of 1800 to 900 cm-1. Courtesy of UIC/Spectral Pathology Lab.
Point spectra from individual pixels were obtained and compared from a human colon sample on barium fluoride scanned in transmission on the same FTIR and QCL systems, which is shown in Figure 5. A serial section was obtained and stained with H&E to identify the different tissue structures. Using the H&E image as a reference, spectra from mucin (red), malignant colon carcinoma epithelium (green) and fibroblastic stroma (blue) were collected from a single pixel at approximately the same location. The unprocessed QCL and FTIR spectra are shown directly beneath their respective images. The FTIR system has an FPA size of 128 × 128 detector elements, while the Spero system has a microbolometer of 480 × 480 detector elements. Therefore, the FTIR image was collected as a mosaic and then stitched together.

Figure 6. (a) FTIR and Spero microscope spectra from a single pixel of mucin, from the tissue shown in Figure 5. (b) FTIR and Spero microscope spectra from a single pixel of malignant colon carcinoma epithelium, from the same tissue. (c) FTIR and Spero microscope spectra from a single pixel of fibroblastic stroma. All spectra have been normalized (FTIR to 1650 cm-1, Spero to 1652 cm-1) and baseline corrected between 1796 and 904 cm-1, with the FTIR spectra truncated to match the Spero microscope’s spectral range of 1800 to 900 cm-1. Note that pixels for each tissue feature were located in approximately the same region, and that the two images have different pixel sizes (2.2 × 2.2 µm for FTIR, 1.4 × 1.4 µm for Spero microscope). Courtesy of UIC/Spectral Pathology Lab.
The spectra obtained from the regions of interest depicted in Figure 5 were preprocessed, as shown in Figure 6. The data was peak height normalized to the Amide I band. The FTIR data and QCL data were processed using a simple, two-point linear baseline correction between 1796 and 904 cm−1. Figure 6a-c shows the processed data from single pixels looking at the biochemistry of mucin, malignant colon carcinoma epithelium and fibroblastic stroma, respectively. The spectra from the QCL and FTIR systems are very similar on an individual-pixel level.
Finally, Figure 7 shows the scan of a frozen prostate tissue section captured with the microscope. Once thawed, the system can quickly image these sections at a single frequency of interest. The real-time capabilities of the system combined with the capacity for scanning frozen samples could someday allow for the analysis of samples in a time-critical intraoperative setting.

Figure 7. Transflectance scan of a 5-µm frozen human prostate tissue section on Kevley low-emissivity substrate captured with the Spero microscope. Visualized with a false color map at 1640 cm-1. Data was baseline corrected between 1796 and 904 cm-1. Courtesy of UIC/Spectral Pathology Lab. University of Illinois at Chicago — Spectral Pathology Lab members, from left to right: David Martinez, Francesca Gambacorta, Vishal Varma, Andrew Graham and Michael Walsh. Courtesy of Daylight Solutions.
While there has been significant interest in MIR imaging for pathology applications for a number of years1-5, the technology has lacked the maturity to be ready for clinical implementation due to slow scanning speeds, low spatial resolutions and by a lack of computational power to fully handle large multispectral datasets. The Spero microscope, coupled with modern computing power, overcomes these limitations. The information detailed above demonstrates that the quality of the images and spectra obtained from the instrument are similar to those offered by FTIR imaging methods but with the additional benefits associated with the use of a QCL-based system. Recent advances in large multielement FPAs6-8) and high-resolution imaging approaches9-11 for tissue pathology have made this a much more attractive approach for fast and detailed image acquisition. QCLs represent the next step toward clinical implementation — they have demonstrated fast data acquisition, live-imaging capabilities and the ability to collect only spectral frequencies of diagnostic value.
Meet the authors
Michael Walsh holds a PhD in biological sciences and is an assistant professor at the University of Illinois at Chicago in Chicago; email: walshm@uic.edu. Matthew Barre is the business development manager at Daylight Solutions in San Diego; email: mbarre@daylightsolutions.com. Benjamin Bird is an applications scientist at Daylight Solutions in San Diego; email: bbird@daylightsolutions.com.
References
1. D.C. Fernandez et al. (2005). Infrared spectroscopic imaging for histopathologic recognition. Nat Biotechnol, Vol. 23, Issue 4, pp. 469-474.
2. C. Matthaus et al. (2008). Chapter 10: Infrared and Raman microscopy in cell biology. Methods Cell Biol, Vol. 89, pp. 275-308.
3. C. Kendall et al. (2009). Vibrational spectroscopy: a clinical tool for cancer diagnostics. Analyst, Vol. 134, Issue 6, pp. 1029-1045.
4. C. Krafft et al. (2009). Disease recognition by infrared and Raman spectroscopy. J Biophotonics, Vol. 2, Issue 1-2, pp. 13-28.
5. F.L. Martin et al. (2010). Distinguishing cell types or populations based on the computational analysis of their infrared spectra. Nat Protoc, Vol. 5, Issue 11, pp. 1748-1760.
Broadband Laser Aimed at Cancer Detection
Covering a wide swath of the mid-infrared, a new system offers greater spectral sensitivity
BY JAMES F. LOWE, WEB MANAGING EDITOR, JAMES.LOWE@PHOTONICS.COM
MUNICH, Sept. 25, 2015 — Mid-infrared (MIR) light is rich with molecular “fingerprint” information that can be used to detect substances from atmospheric pollutants to cancer cells.
While some lasers already operate in this region, enabling a variety of spectroscopy applications, their linewidth is relatively narrow, which limits the types of substances they can detect at any given moment.
Now a team of researchers from Germany and Spain has developed a laser system with phase-coherent emission from 6.8 to 16.4 μm and output power of 0.1 W. That is broad and powerful enough, they said, to detect subtle signs of cancer early in its development.
Molecules absorb portions of the MIR spectrum in ways that are unique to their atomic structures, and their absorption patterns provide a means of identifying the molecules with great specificity, even in low concentrations.

The emission spectrum of the laser and corresponding molecular fingerprint regions. Courtesy of the Institute of Photonic Sciences (ICFO).
“Cancer causes subtle modification in protein structure and content within a cell,” said professor Dr. Jens Biegert, a group leader at the Institute of Photonic Sciences (ICFO) in Barcelona. “Looking at only a few nanometer range, the probability of detection is extremely low. But comparing many of such intervals, one can have an extremely high confidence level.”
The new laser system generates MIR pulses via difference-frequency generation driven by the nonlinearly compressed pulses of a Kerr-lens mode-locked Yb:YAG thin-disc oscillator. It features a repetition rate of 100 MHz and pulse durations of 66 fs — so short that the electric field oscillates only twice per pulse.

Staff scientist Dr. Ioachim Pupeza (left) and postdoctoral researcher Oleg Pronin helped develop a laser system that emits ultrashort pulses of mid-infrared light. These pulses can be used to detect trace molecules in gaseous and liquid media. Courtesy of Thorsten Naeser/Ludwig Maximilian University.
“Since we now possess a compact source of high-intensity and coherent infrared light, we have a tool that can serve as an extremely sensitive sensor for the detection of molecules, and is suitable for serial production,” said project leader Dr. Ioachim Pupeza, a staff scientist at Ludwig Maximilian University of Munich (LMU).
The LMU and ICFO researchers aim to use their MIR laser to identify and quantify disease markers in exhaled air. Many diseases, including some types of cancer, are thought to produce specific molecules that end up in the air expelled from the lungs.
“We assume that exhaled breath contains well over 1000 different molecular species,” said Dr. Alexander Apolonskiy, an LMU group leader.
However, the amount of molecular biomarkers present in exhaled breath is extraordinarily low, meaning a diagnostic tool would need to be capable of detecting concentrations of at least one part per billion. The next step will be to couple the new laser system with a novel amplifier that would increase its brightness and boost sensitivity one part per trillion.
Detecting MIR signatures
The laser’s output spans more than one octave. Until now, the researchers said, such broadband emission has only been available from large-scale synchrotron sources.
Other more compact MIR sources, such as quantum cascade lasers (QCLs), have narrower linewidths. Tuning them to different sensing bands is time consuming, and combining multiple QCLs emitting in different parts of the MIR would be cost-prohibitive, Biegert said.
Meanwhile, the laser system’s 100-MHz pulse train is hundreds to thousands of times more powerful than state-of-the-art frequency combs that emit in the same range, the researchers said.
Detecting broadband MIR signals presents its own problems, however. Detectors for this region have poor signal-to-noise ratios unless cooled with liquid nitrogen, the researchers said.
In this case, electro-optical sampling proved to be a better option. Well-established for the terahertz range, the technique is less common in the fingerprint region.
“In the MIR range, there are not many groups who have implemented this already, because you need a broadband, phase-stable MIR pulse and an ultrashort sample pulse at the same time, which is quite challenging,” Pupeza said.
Having solved that problem with their broadband laser, the team now could use electro-optical sampling to extract the data they wanted.
In a nutshell, the process works like this: The electric field of an MIR pulse alters the birefringence of a crystal. This change can be measured by observing how the polarization of slightly shorter near-infrared (NIR) pulse is changed while propagating through the same crystal at the same time. In the end, only the NIR pulse is measured directly.
“Therefore, one big advantage is low-noise detection in the NIR, even though one obtains information on spectral components in the MIR,” said Ioachim Pupeza. “You only need to perform a Fourier transform numerically to get the spectrum of the pulse once you have its electric field.”
http://www.photonics.com/Article.aspx?AID=57757
The research was published in Nature Photonics (doi: 10.1038/nphoton.2015.179).
Read Full Post »





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}