The Colors of Life Function
Writer and Curator: Larry H. Bernstein, MD, FCAP
2.5.1 Type 1 Copper Proteins
The Cu(II) state of this category has an intense blue color due to a thiolate ligand
to Cu(II) charge transfer, and unusual EPR properties arising from the asymmetrical
Cu site (distorted trigonal-pyramidal). The proteins all have a low molecular
mass and have, so far, rather arbitrarily been divided into sub-groups, such as
azurins, plastocyanins, pseudoazurins, amicyanins and various other blue
proteins. Of these the azurins, amicyanins, pseudo-azurins and plastocyanins
apparently have similar copper coordination by two histidine, one cysteine and
one methionine residue. Where the function of Type I copper proteins is known,
it is invariably electron transfer. As yet the names for these proteins are all trivial
and are often derived from source, function or color. The different classes are
usually discerned on the basis of their primary and tertiary structure.
The first bacterial blue proteins to be described were called azurins. Rusticyanin is
another example of a bacterial protein. It has unusual properties with a reduction
potential of 680 mV, and is functional at pH 2. The azurins have well-defined electron
-transfer functions.
The so-called pseudo-azurins differ from the azurins in the N-terminal amino acid
sequence and the optical spectra, which resemble those of plastocyanins.
The blue proteins known as plastocyanins occur in plants, blue-green and green
algae. Their electron transfer role is well defined, i.e. from the bc1 complex
(EC 1.10.2.2) to the photooxidized P-700.
Amicyanins are electron carriers between methylamine dehydrogenase and
cytochrome c, with a characteristic amino acid sequence.
Of the remaining blue proteins stellacyanin is a well- known example. Umecyanin,
plantacyanin and mavicyanin are also considered to belong to this group.
Although these proteins undergo redox reactions in vitro, their true biological
function remains unknown. Most of these proteins exhibit an unusual EPR signal
in which the copper hyperfine splitting pattern is poorly resolved. There is good
evidence that at least for stellacyanin, methionine does not function as a ligand
for copper.
2.5.2 Type 2 Copper Proteins
The copper centres in these proteins are spectroscopically consistent with square
planar or pyramidal coordination, containing oxygen and/or nitrogen ligation.
The Cu(II) is EPR active, with a ‘normal’ signal. There is no intense blue color.
This group includes the copper/zinc superoxide dismutase (EC 1.15.1.1),
dopamine b-monooxygenase (EC 1.14.17.1), galactose oxidase (EC 1.1.3.9)
and the various copper-containing amine oxidases. Some members of this last
group may also contain an organic prosthetic group, such as PQQ
(see section 10), or a modified amino-acid residue.
2.5.3 Type 3 Copper Proteins
In this group a pair of copper atoms comprise a dinuclear centre, with no EPR
activity as for single Cu’s. The best known example of an enzyme containing a
single Type 3 centre is tyrosinase (catechol oxidase, EC 1.10.3.1). This protein
contains a metal center which is a structural analogue of the dinuclear copper
center in hemocyanin (ref 31).
2.5.4 Multi-Copper Oxidases
In addition to the above, there are several proteins with catalytic activity that
contain Types 1, 2 and 3 centres in various stoichiometric ratios. These
include L-ascorbate oxidase (EC 1.10.3.3), laccase (EC 1.10.3.2) and
ceruloplasmin (ferro-oxidase, EC 1.16.3.1), the latter two having aromatic diamine
and diphenol oxidase activity. There is growing evidence that in these proteins
the Type 2 and Type 3 copper centres are juxtaposed. Recently it has been
shown that in L-ascorbate oxidase, a trinuclear copper site is present, consisting
of a type 3 copper site, very close (3.9 Å) and possibly bridged to a type 2 copper
site (ref 32). There is a view that ceruloplasmin functions as a ferro-oxidase
and the Fe(III) produced in this reaction can then oxidize the same substrates
as laccase.
2.5.5 Copper Centres in Cytochrome Oxidase
There are two copper centres that appear to be unique. Both are present in
cytochrome-c oxidase (EC 1.9.3.1). The first appears to be an isolated metal ion
and has been referred to as Cud and CuA. The second appears to be part
of a dinuclear centre with cytochrome a3. It has been referred to as Cuu,
Cua3 and CuB. At the moment the ascriptions CuA and CuB are most frequently
used; however, the recent discovery (ref 33) of a cytochrome oxidase in which
cytochrome a has been replaced by cytochrome b, leads to the recommendation
that CuB shall be referred to as Cua3.
There is a striking similarity between two of the Cu centres of N2O reductase
and CuA (ref 34, 35).
2.5.6 Molybdenum enzymes (general)
Molybdenum enzymes contain molybdenum at the catalytic center responsible
for reaction with substrate. They may be divided into those that contain
the iron-molybdenum cofactor and those that contain the pterin-molybdenum
cofactor.
2.5.7 Additional centers
If a molybdenum enzyme contains flavin, it may be called either a molybdenum
flavoprotein or a flavomolybdenum protein, as indicated above. Other centers
should be treated similarly, e.g. an iron-sulfur molybdenum protein.
2.5.8 Molybdenum enzymes containing the iron-molybdenum cofactor
The only enzymes at present known to belong to this group are the nitrogenases
(EC 1.18.6.1; and EC 1.19.6.1): see pp 89-116 in (ref 36) and pp 91-100 in (ref 37).
2.5.9 Molybdenum enzymes containing the pterin-molybdenum cofactor
These enzymes [see pp 411-415 in (ref 36) and (ref 38)] may be divided
into those in which the molybdenum bears a cyanide-labile sulfido (or thio
– see Note 1) ligand i.e. containing the S2- ligand as Mo=S) and those
lacking this ligand. The former group includes xanthine oxidase (EC 1.1.3.22),
xanthine dehydrogenase (EC 1.1.1.204), aldehyde oxidase (EC 1.2.3.1) and
purine hydroxylase (EC: see Note 2 and 3). These may be called ‘molybdenum-
containing hydroxylase’ as is widely done. Molybdenum enzymes lacking the
sulfide (thio) ligand include sulfite oxidase (EC 1.8.3.1), NAD(P)+-independent
aldehyde dehydrogenase and nitrate reductases (assimilatory and dissimilatory)
(EC 1.6.6.1-3).
2.5.10 Molybdenum enzymes containing the pterin-molybdenum cofactor
These enzymes [see pp 411-415 in (ref 36) and (ref 38)] may be divided into those
in which the molybdenum bears a cyanide-labile sulfido (or thio – see Note 1)
ligand i.e. containing the S2- ligand as Mo=S) and those lacking this ligand. The
former group includes xanthine oxidase (EC 1.1.3.22), xanthine dehydrogenase
(EC 1.1.1.204), aldehyde oxidase (EC 1.2.3.1) and purine hydroxylase. These
may be called ‘molybdenum-containing hydroxylase’ as is widely done.
Molybdenum enzymes lacking the sulfide (thio) ligand include sulfite oxidase
(EC 1.8.3.1), NAD(P)+-independent aldehyde dehydrogenase and nitrate
reductases (assimilatory and dissimilatory) (EC 1.6.6.1-3).
2.5.11 Metal-Substituted Metalloproteins
Scientists from several areas, dealing with spectroscopy and electron-transfer
mechanisms, often use metalloproteins in which a metal at the active site has
been substituted by another metal ion, like Co, Zn, Hg, Cd. Examples are zinc-
substituted cytochromes and cobalt-substituted ferredoxins.
The names for such modified proteins are easily given by using indications
like: ‘zinc-substituted ….’. In case of multi-metal proteins, where ambiguity might
arise about which metal has been substituted, one could easily add in parentheses
the name of the metal that has been replaced, such as: cobalt- substituted [Fe]
nitrogenase.
In formulae fragments or short names one could use the following notation:
[3Fe1Co-4S]2+, cytochrome c'[Fe[arrow right]CoFe], plastocyanin[Cu
[arrow right]Hg].
Ambler, R.P. (1980) in From Cyclotrons to Cytochromes (Kaplan, N.O. &
Robinson, A., eds) Academic Press, New York
Moore, G. & Pettigrew, F.(1987) Cytochromes c, Springer-Verlag, Berlin
Bartsch, R.G. (1963) in Bacterial Photosynthesis (Gest, H., San Pietro, A. &
Vernon, L.P., ed.) p. 315, Antioch Press, Yellow Springs, Ohio.
Stiefel, E.I. & Cramer, S.P. (1985) in Molybdenum Enzymes (Spiro, T.G., ed.),
Wiley-Interscience, New York, 89-116.
Smith B.E. et al. (1988), in Nitrogen Fixation Hundred Years After (Bothe,
H., de Bruijn, F.J. & Newton, W.E., ed.), Gustav Fischer, Stuttgart, New York,
91-100
Type-2 copper-containing enzymes.
MacPherson IS1, Murphy ME.
Cell Mol Life Sci. 2007 Nov;64(22):2887-99.
Type-2 Cu sites are found in all the major branches of life and are often
involved in the catalysis of oxygen species. Four type-2 Cu protein
families are selected as model systems for review: amine oxidases,
Cu monooxygenases, nitrite reductase/multicopper oxidase, and
CuZn superoxide dismutase. For each model protein, the availability
of multiple crystal structures and detailed enzymological studies provides
a detailed molecular view of the type-2 Cu site and delineation of the
mechanistic role of the Cu in biological function. Comparison of these
model proteins leads to the identification of common properties of the
Cu sites and insight into the evolution of the trinuclear active site found
in multicopper oxidases.
Copper proteins and copper enzymes.
Cass AE, Hill HA.
Ciba Found Symp. 1980;79:71-91.
http://www.chm.bris.ac.uk/motm/caeruloplasmin/copper_proteins/t1.htm
The copper proteins that function in homeostasis, electron transport, dioxygen
transport and oxidation are discussed. Particular emphasis is placed on the
role of the ligands, their type and disposition which, in conjunction with other
residues in the active site, determine the role of the copper ion. It is proposed that
copper proteins can be considered in four groups. Those in Group I contain a
single copper ion in an approximately tetrahedral environment with nitrogen and
sulphur-containing ligands. Group II proteins have a single copper ion in a square-
planar-like arrangement. Group III proteins have two copper ions in close
proximity. Group IV consists of multi-opper proteins, composed of sites
representative of the other three groups.
Such centers owe their name to the intense blue coloration of the corresponding
Cu(II) proteins. The color is particularly distinctive since the metal centers are
so optically diluted in these metalloenzymes that only intense absorption in the
visible region, resulting from symmetry allowed electronic transitions, can give
rise to conspicuous colors. In contrast, the comparatively pale blue color of normal
Cu(II)) is the result of forbidden electronic transitions between d-orbitals of
different symmetry; in Cu2+(aq) this gives a molar extinction coefficient of
10 M-1cm-1 from a broad absorption between 10,000 cm-1 and 15,000 cm-1
compared to about 3000 M-1cm-1 observed for blue Cu(II) centers. For the
T1 centers the intense absorption is attributed to a ligand-to-metal charge
transfer between the Cu2+ and a bonded cysteinate ligand. Typically, as in
azurin or plastocyanin this occurs around 16,000 cm-1. Ceruloplasmin has
three T1 centers, and the blue absorption is at 16,400 cm-1 (610nm).
Plastocyanine geometry
around the copper Crystal structures show a very irregular ‘tetrahedral’ coordination
with two sulphurs from methionine and cysteinate, and two histidine nitrogens.
However a comparison of azurin with plastocyanin shows that the geometry
is in some ways closer to a trigonal bipyramid, with and without one extra apical
ligand, so that azurin has a weakly bound glutamine oxygen, and plastocyanine
does not. The T1 coppers in caruloplasmin are in plastocyanine-type domains.
Each of these are coordinated to two histidines and a cysteine, in two of the T1
domains there is also a methionine residue, the third T1 domain has a leucine
residue which may only have a van der Waals type contact with the copper.
T1 copper centers are functional in the reversible electron transfer:
Cu2+ + e- = Cu+
The strongly distorted geometry represents a compromise (entactic-state
situation) between d10 Cu(I), with its preferred tetrahedral or trigonal
coordination through soft sulfur ligands, and d9 Cu(II) with preferential
square planar or square pyramidal geometry and nitrogen ligand
coordination. This irregular, high energy arrangement at the metal
center resembles the transition-state geometry between the tetrahedral
and square planar equilibrium configurations of the two oxidation states
involved and permits enhanced rates of electron transfer. The potential
range for proteins with T1 copper centers runs from 180 mV in
stellacyanin to 680 mV in rusticyanin.
Zinc proteins: enzymes, storage proteins, transcription factors, and replication
proteins.
Coleman JE.
Annu Rev Biochem. 1992;61:897-946.
In the past five years there has been a great expansion in our knowledge of
the role of zinc in the structure and function of proteins. Not only is zinc
required for essential catalytic functions in enzymes (more than 300 are known
at present), but also it stabilizes and even induces the folding of protein
subdomains. The latter functions have been most dramatically illustrated
by the discovery of the essential role of zinc in the folding of the DNA-binding
domains of eukaryotic transcription factors, including the zinc
finger transcription factors, the large family of hormone receptor proteins,
and the zinc cluster transcription factors from yeasts. Similar functions are
highly probable for the zinc found in the RNA polymerases and the zinc-
containing accessory proteins involved in nucleic acid replication. The rapid
increase in the number and nature of the proteins in which zinc functions
is not unexpected since zinc is the second most abundant trace metal found in
eukaryotic organisms, second only to iron. If one subtracts the amount of iron
found in hemoglobin, zinc becomes the most abundant trace metal found
in the human body.
Zinc Coordination Spheres in Protein Structures
ACS ChemWorx
Mikko Laitaoja , Jarkko Valjakka , and Janne Jänis
Inorg. Chem., 2013, 52 (19), pp 10983–10991
http://dx.doi.org:/10.1021/ic401072d
Sept 23, 2013
Synopsis
A statistical analysis in terms of zinc coordinating amino acids, metal-to-ligand
bond lengths, coordination number, and structural classification was performed,
revealing coordination spheres from classical tetrahedral cysteine/histidine binding
sites to more complex binuclear sites with carboxylated lysine residues. According
to the results, coordination spheres of hundreds of crystal structures in the PDB
could be misinterpreted due to symmetry-related molecules or missing electron
densities for ligands.
Protein-folding location can regulate manganese-binding versus copper- or
zinc-binding.
Tottey S, Waldron KJ, Firbank SJ, Reale B, Bessant C, Sato K, Cheek TR, et al.
Nature. 2008 Oct 23;455(7216):1138-42. http://dx.doi.org:/10.1038/nature07340
Metals are needed by at least one-quarter of all proteins. Although metallo-
chaperones insert the correct metal into some proteins, they have not been
found for the vast majority, and the view is that most metalloproteins acquire
their metals directly from cellular pools. However, some metals form more
stable complexes with proteins than do others. For instance, as described
in the Irving-Williams series, Cu(2+) and Zn(2+) typically form more stable
complexes than Mn(2+). Thus it is unclear what cellular mechanisms manage
metal acquisition by most nascent proteins. To investigate this question, we
identified the most abundant Cu(2+)-protein, CucA (Cu(2+)-cupin A), and the
most abundant Mn(2+)-protein, MncA (Mn(2+)-cupin A), in the periplasm of
the cyanobacterium Synechocystis PCC 6803. Each of these newly identified
proteins binds its respective metal via identical ligands within a cupin fold.
Consistent with the Irving-Williams series, MncA only binds Mn(2+) after
folding in solutions containing at least a 10(4) times molar excess of Mn(2+)
over Cu(2+) or Zn(2+). However once MncA has bound Mn(2+), the metal
does not exchange with Cu(2+). MncA and CucA have signal peptides for
different export pathways into the periplasm, Tat and Sec respectively. Export
by the Tat pathway allows MncA to fold in the cytoplasm, which contains only
tightly bound copper or Zn(2+) (refs 10-12) but micromolar Mn(2+) (ref. 13). In
contrast, CucA folds in the periplasm to acquire Cu(2+). These results reveal
a mechanism whereby the compartment in which a protein folds overrides its
binding preference to control its metal content. They explain why the cytoplasm
must contain only tightly bound and buffered copper and Zn(2+).
Predicting copper-, iron-, and zinc-binding proteins in pathogenic species of the
Paracoccidioides genus
GB Tristão, L do Prado Assunção, LPA dos Santos, CL Borges, MG Silva-Bailão,
CM de Almeida Soares, G Cavallaro and AM Bailão*
Front. Microbiol., 9 Jan 2015 http://dx.doi.org:/10.3389/fmicb.2014.00761
Approximately one-third of all proteins have been estimated to contain at least
one metal cofactor, and these proteins are referred to as metalloproteins. These
represent one of the most diverse classes of proteins, containing metal ions that
bind to specific sites to perform catalytic, regulatory and structural functions.
Bioinformatic tools have been developed to predict metalloproteins encoded by
an organism based only on its genome sequence. Its function and the type of
metal binder can also be predicted via a bioinformatics approach. Paracoccidioides
complex includes termodimorphic pathogenic fungi that are found as saprobic
mycelia in the environment and as yeast, the parasitic form, in host tissues. They
are the etiologic agents of Paracoccidioidomycosis, a prevalent systemic mycosis
in Latin America. Many metalloproteins are important for the virulence of several
pathogenic microorganisms. Accordingly, the present work aimed to predict the
copper, iron and zinc proteins encoded by the genomes of three phylogenetic species
of Paracoccidioides (Pb01, Pb03, andPb18). The metalloproteins were identified
using bioinformatics approaches based on structure, annotation and domains. Cu-,
Fe-, and Zn-binding proteins represent 7% of the total proteins encoded by
Paracoccidioides spp. genomes. Zinc proteins were the most abundant metallo-
proteins, representing 5.7% of the fungus proteome, whereas copper and iron
proteins represent 0.3 and 1.2%, respectively. Functional classification revealed that
metalloproteins are related to many cellular processes. Furthermore, it was observed
that many of these metalloproteins serve as virulence factors in the biology of the
fungus. Thus, it is concluded that the Cu, Fe, and Zn metalloproteomes of the
Paracoccidioides spp. are of the utmost importance for the biology and virulence
of these particular human pathogens.
Zinc finger proteins: new insights into structural and functional diversity
John H Laity, Brian M Lee, Peter E Wright
Current Opinion in Structural Biology Feb 2001; 11(1): 39–46
http://epigenie.com/key-epigenetic-players/chromatin-modifying-and-dna-
binding-proteins/zinc-finger-proteins/
Zinc finger proteins are among the most abundant proteins in eukaryotic genomes.
Their functions are extraordinarily diverse and include DNA recognition, RNA
packaging, transcriptional activation, regulation of apoptosis, protein folding
and assembly, and lipid binding. Zinc finger structures are as diverse as their
functions. Structures have recently been reported for many new zinc finger
domains with novel topologies, providing important insights into structure/function
relationships. In addition, new structural studies of proteins containing the
classical Cys2His2 zinc finger motif have led to novel insights into mechanisms
of DNA binding and to a better understanding of their broader functions in
transcriptional regulation.
Zinc Finger Proteins
Zinc finger (ZnF) proteins are a massive, diverse family of proteins that serve a
wide variety of biological functions. Due to their diversity, it is difficult to come up
with a simple definition of what unites all ZnF proteins; however, the most common
approach is to define them as all small, functional domains that require coordination
by at least one zinc ion (Laity et al., 2001). The zinc ion serves to stabilize the
integration of the protein itself, and is generally not involved in binding targets.
The “finger” refers to the secondary structures (α-helix and β-sheet) that are
held together by the Zn ion. Zinc finger containing domains typically serve
as interactors, binding DNA, RNA, proteins or small molecules (Laity et al., 2001).
ZnF Protein Families
Cys2His2 was the first domain discovered (also known as Krüppel-type). It was
initially discovered as a repeating domain in the IIIA transcription factor in
Xenopus laevis (Brown et al., 1985; Miller et al., 1985). IIIA has nine repeats
of the 30 amino acids that make up the Cys2His2 domain. Each domain forms
a left-handed ββα secondary structure, and coordinates a Zn ion between
two cysteines on the β-sheet hairpin and two histidines in the α-helix, hence
the name Cys2His2 (Lee et al., 1989). These resides are highly conserved,
as well as a general hydrophobic core that allows the helix to form. The other
residues can show great sequence diversity (Michael et al., 1992). Cys2His2
zinc fingers that bind DNA tend to have 2-4 tandem domains as part of a
larger protein. The residues of the alpha helices form specific contacts with a
specific DNA sequence motif by “reading” the nucleotides in major groove
of DNA (Elrod-Erickson et al., 1996; Pavletich and Pabo, 1991). Cys2His2
proteins are the biggest group of transcription factors in most species. Non-
DNA binding proteins can have much more flexible tertiary structure.
Examples of Cys2His2 proteins include the Inhibitor of Apoptosis (IAP) family
of proteins and the CTFC transcription factor.
Treble clef fingers are a very diverse group of ZnF protiens both in terms of
structure and function. What makes them a family is a shared fold at their core
that looks a little like a musical treble clef, especially if you squint (Grishin,
2001). Most treble clef finger motifs have a β hairpin, a variable loop region,
a β hairpin, and an α helix. The “knuckle” of the β hairpin and the α helix contain
the Cys-x-x-Cys sequence necessary to coordinate the Zn ion. Treble clef
fingers often form the core of protein structures, for example the L24E and
S14 ribosomal proteins and the RING finger family.
Zinc ribbons are a little less structurally complex than the other two major groups.
Zinc ribbons contain two zinc knuckles, often β hairpins, coordinating a zinc ion via
a two Cys residures separated by 2-4 other residues on one knuckle, and a Cys-x-x-
Cys on the other (Hahn and Roberts, 2000). Examples of zinc ribbon-containing
proteins include the basal transcription factors TFIIS and TFIIB that for a complex
with RNAPII to bind DNA, and the Npl4 nuclear core protein that uses a zinc ribbon
to bind ubiquitin (Alam et al., 2004). Cys2His2, treble clef fingers, and zinc ribbons
form the majority of zinc fingers, but there are several other smaller groups that
don’t fit neatly into these three. Green fluorescent protein as a marker for gene
expression.
Metallothionein proteins expression, copper and zinc concentrations, and lipid
peroxidation level in a rodent model for amyotrophic lateral sclerosis
E Tokuda, Shin-Ichi Ono, K Ishige, A Naganuma, Y Ito, T Suzuki
Toxicology Jan 2007; 229(1–2): 33–41
It has been hypothesized that copper-mediated oxidative stress contributes to the
pathogenesis of familial amyotrophic lateral sclerosis (ALS), a fatal motor neuron
disease in humans. To verify this hypothesis, we examined the copper and zinc
concentrations and the amounts of lipid peroxides, together with that of the
expression of metallothionein (MT) isoforms in a mouse model [superoxide
dismutase1 transgenic (SOD1 Tg) mouse] of ALS. The expression of MT-I and
MT-II (MT-I/II) isoforms were measured together with Western blotting, copper
level, and lipid peroxides amounts increased in an age-dependent manner in the
spinal cord, the region responsible for motor paralysis. A significant increase was
already seen as early as 8-week-old SOD1 Tg mice, at which time the mice had not
yet exhibited motor paralysis, and showed a further increase at 16 weeks of age,
when paralysis was evident. Inversely, the spinal zinc level had significantly
decreased at both 8 and 16 weeks of age. The third isoform, the MT-III level,
remained at the same level as an 8-week-old wild-type mouse, finally increasing
to a significant level at 16 weeks of age. It has been believed that a mutant SOD1
protein, encoded by a mutant SOD1, gains a novel cytotoxic function while
maintaining its original enzymatic activity, and causes motor neuron death
(gain-of-toxic function). Copper-mediated oxidative stress seems to be a probable
underlying pathogenesis of gain-of-toxic function. Taking the above current
concepts and the classic functions of MT into account, MTs could have a disease
modifying property: the MT-I/II isoform for attenuating the gain-of-toxic function
at the early stage of the disease, and the MT-III isoform at an advanced stage.
Prion protein expression level alters regional copper, iron and zinc content in
the mouse brain
MJ Pushie, IJ Pickering, GR Martin, S Tsutsui, FR Jirik and GN George
Metallomics, 2011,3, 206-214 http://dx.doi.org:/10.1039/C0MT00037J
The central role of the prion protein (PrP) in a family of fatal neurodegenerate
diseases has garnered considerable research interest over the past two decades.
Moreover, the role of PrP in neuronal development, as well as its apparent role
in metal homeostasis, is increasingly of interest. The host-encoded form of the
prion protein (PrPC) binds multiple copper atoms via its N-terminal domain
and can influence brain copper and iron levels. The importance of PrPC to the
regulation of brain metal homeostasis and metal distribution, however, is not
fully understood. We therefore employed synchrotron-based X-ray fluorescence
imaging to map the level and distributions of several key metals in the brains of
mice that express different levels of PrPC. Brain sections from wild-type, prion
gene knockout (Prnp−/−) and PrPC over-expressing mice revealed striking
variation in the levels of iron, copper, and even zinc in specific brain regions as
a function of PrPC expression. Our results indicate that one important function
of PrPC may be to regulate the amount and distribution of specific metals within
the central nervous system. This raises the possibility that PrPC levels, or its
activity, might regulate the progression of diseases in which altered metal
homeostasis is thought to play a pathogenic role such as Alzheimer’s,
Parkinson’s and Wilson’s diseases and disorders such as hemochromatosis.
The status of zinc and copper levels may have profound implications for
many people. Much has been written about the significance of these two
trace elements for many, many years. Many health conditions may be
directly caused by abnormal zinc and copper levels.
With all of the recent attention given to methylation status, gene mutations,
MTHFR, and the associated neurological and mental/behavioral disorders
that may ensue, zinc and copper status remains a pivotal ratio in these regards.
While zinc toxicity and copper deficiency are possible, the subject of this
article is on the more common imbalance: copper toxicity and zinc deficiency.
The Physiological Roles Of Zinc & Copper
Zinc and copper are antagonists. The balance between these two trace
elements is an example of the effects of biological dualism. While zinc
toxicity is possible, far more common is zinc deficiency and copper toxicity.
Both zinc and copper play essential roles in the body, and there can be a
number of causes for why imbalances ensue.
It may be easier to identify the roles that zinc doesn’t play in the body,
than the roles it does play. Zinc is an essential trace element that activates
several hundred enzymatic reactions. These reactions are fundamental
to life and biological activity. Some of the activities that zinc are involved in:
- DNA & RNA synthesis
- Gene expression
- Nervous system function
- Immune function & immune signaling such as cell
apoptosis
- Neuronal transmission
- Brain function
- Zinc possesses powerful anabolic activities in the cells
- Formation of zinc proteins known as “zinc fingers”
- Zinc is essential for blood clotting and platelet formation
- Zinc is involved in Vitamin A synthesis
- Folate is made available through zinc enzyme reactions
- Along with copper, Zinc makes up the antioxidant
enzyme
system, ZnCu superoxide dismutase
- Steroidal hormone synthesis
- Growth & development of children
- Testosterone and semen formation
- The highest concentration of zinc is found in the
male prostate gland
Copper is an essential trace element serving many important functions
as well. However, copper is well documented to induce several toxic effects
in the body, when elevated. Because copper is a pro-oxidant when free and
unbound, it can quickly generate free radicals.
The major sources for copper toxicity are: exposure to industrial forms
of copper such as copper pipes, copper cookware, birth control, exposure
to copper-based fungicides. Diets high in copper and low in zinc may play
a role in copper toxicity. Pyrrole disorder, which causes depletion of zinc,
may result in elevated levels of copper.
Some of the essential roles copper plays in the body:
- Connective tissue formation
- ATP synthesis
- Iron metabolism
- Brain health via neurotransmitter synthesis
- Gene transcription
- Synthesis of the antioxidant superoxide dismutase
- Skin pigmentation
- Nerve tissue: myelin sheath formation
- Copper tends to rise when estrogen is dominant
Perhaps one of the first reports that zinc and copper imbalances play
a role in human health and disease was their detection in mental
disorders made by Carl Pfeiffer, MD, PhD. Dr. Pfeiffer identified a
condition known as pyrrole disorder, sometimes referred to as
pyrroluria or “mauve factor”.
As it turns out, pyrrole disorder is a major biochemical imbalance
in many people with chronic illnesses such as chronic Lyme disease,
autism, schizophrenia, depression, bi-polar, and chronic fatigue
syndrome. Pyrroles are a byproduct of hemoglobin synthesis.
Apparently, some individuals are more predisposed towards producing
higher amounts of pyrroles. When pyrroles are excessive, they irreversibly
bind to zinc and vitamin B6, causing their excretion. Consequently,
it is common that once zinc levels become depleted, copper levels tend to rise.
Copper Toxicity
Problems associated with copper toxicity include: pyrrole disorder,
estrogen dominance, schizophrenia, depression, anxiety disorder,
chronic fatigue, migraines, liver toxicity, thyroid conditions, chronic
candida yeast infections, PMS, to name a few. Some research has
even implicated copper toxicity with Alzheimer’s Disease and with
cardiovascular disease. Perhaps one of the primary mechanisms
through which copper toxicity can damage tissues is through its
initiation of oxidative stress and free radical formation. Free copper
ions that are not bound to copper proteins such as ceruloplasmin,
are pro-oxidants, and are highly reactive.
Empirical research from clinicians, indicates that there are different
types of copper imbalances. For example, if there is a lot of free,
unbound copper present, this may cause a situation of nutritive
copper deficiency. Another copper imbalance is when high pyrroles
depress zinc levels, and copper levels concomintantly rise. If high
pyrroles are present, B6 will also be lost in high amounts. In a general
but very real sense, all forms of copper excess will affect zinc status,
due to the dualistic nature of zinc and copper.
Copper & Estrogen
It has been known for many years that copper can cause a rise in
estrogen, and conversely estrogen may raise copper. Estrogen
dominance has been extensively studied in its role in breast
cancer development. One possible, critical role that can cause
estrogen to become carcinogenic, is through its oxidation induced by
copper. Once oxidized, estrogen forms volatile hydroxyl radicals and
the associated DNA damage and “mutagenesis”.
Zinc Deficiency
As mentioned previously, pyrrole disorder will directly depress
zinc status, causing high levels of its excretion. When zinc is
lost, copper rises. Because of their essential roles in neuro-
transmitter synthesis, low zinc and high copper levels can
directly effect cognition, behavior and thought processes.
Zinc has been studied in biochemical reactions involving
calcium-driven, synaptic neurotransmission, as well as in
glutamate/GABA balance and with limbic brain function.
Zinc & Reproduction
Zinc is essential for steroidal hormone synthesis, and is a
well known catalyst for testosterone synthesis, as well as
leutinizing hormone. Zinc has demonstrated its ability to
prevent miscarriage and toxicity during pregnancy. The male
prostate gland reportedly contains the highest concentration
of zinc in the body.
Zinc & Brain Function
Much attention has been given to excitotoxicity, such as the
effects induced by MSG (monosodium glutamtate). Excess
stimulation of the excitatory neurotransmitter glutamate,
may cause severe physical and psychological reactions in
certain individuals. Zinc has been studied for its ability to
enhance GABA (glutamate’s antagonistic neurotransmitter)
activity and to suppress excess glutamate.
Studies on mice demonstrated that when depleted of zinc
for two weeks, the mice developed seizures, most likely due
to GABA deficiencies and glutamate excess.
There is an emerging body of evidence that demonstrates
that Alzheimer’s disease may involve copper toxicity and
zinc deficiency. Not only can excess copper cause zinc
depletion, but so can excess lead.
The hippocampus, a major part of the limbic brain, records
memories and is responsible for processing meaningful
experiences. Numerous studies site that if hippocampal
cells are deprived of zinc, the hippocampal cells die. In
addition to hippocampus cell death induced by zinc
deprivation, the amygdala, the other major limbic gland
experiences cell death as well, when deprived of zinc.
Green Fluorescent Protein
Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC.
Science. 1994 Feb 11;263(5148):802-5.
http://www.ncbi.nlm.nih.gov/pubmed/8303295
A complementary DNA for the Aequorea victoria green fluorescent protein (GFP)
produces a fluorescent product when expressed in prokaryotic (Escherichia coli)
or eukaryotic (Caenorhabditis elegans) cells. Because exogenous substrates and
cofactors are not required for this fluorescence, GFP expression can be used
to monitor gene expression and protein localization in living organisms.
http://en.wikipedia.org/wiki/Green_fluorescent_protein
The green fluorescent protein (GFP) is a protein composed of 238 amino acid
residues (26.9 kDa) that exhibits bright green fluorescence when exposed
to light in the blue to ultraviolet range. Although many other marine organisms
have similar green fluorescent proteins, GFP traditionally refers to the protein
first isolated from the jellyfish Aequorea victoria. The GFP from A. victoria
has a major excitation peak at a wavelength of 395 nm and a minor one at
475 nm. Its emission peak is at 509 nm, which is in the lower green portion
of the visible spectrum. The fluorescence quantum yield (QY) of GFP is 0.79.
The GFP from the sea pansy (Renilla reniformis) has a single major excitation
peak at 498 nm.
In cell and molecular biology, the GFP gene is frequently used as a reporter of
expression. In modified forms it has been used to make biosensors, and many
animals have been created that express GFP as a proof-of-concept that a gene
can be expressed throughout a given organism. The GFP gene can be introduced
into organisms and maintained in their genome through breeding, injection with a
viral vector, or cell transformation. To date, the GFP gene has been introduced
and expressed in many Bacteria, Yeast and other Fungi, fish (such as zebrafish),
plant, fly, and mammalian cells, including human. Martin Chalfie, Osamu Shimomura,
and Roger Y. Tsien were awarded the 2008 Nobel Prize in Chemistry on 10 October
2008 for their discovery and development of the green fluorescent protein.
http://www.conncoll.edu/ccacad/zimmer/GFP-ww/GFP-1.htm
In Aequorea victoria a protein called aequorin releases blue light upon binding
with calcium. This blue light is then totally absorbed by the GFP, which in turn
gives off the green light as in the animation below.
In 1994 GFP was cloned. Now GFP is found in laboratories all over the world where
it is used in every conceivable plant and animal. Flatworms, algae, E. coli and pigs
have all been made to fluoresce with GFP.
The importance of GFP was recognized in 2008 when the Nobel Committee awarded
Osamu Shimomura, Marty Chalfie and Roger Tsien the Chemistry Nobel Prize ”
for the discovery and development of the green fluorescent protein, GFP.”
Why is it so popular? Well, I like to think of GFP as the microscope of the twenty-
first century. Using GFP we can see when proteins are made, and where they can go.
This is done by joining the GFP gene to the gene of the protein of interest so that
when the protein is made it will have GFP hanging off it. Since GFP fluoresces, one
can shine light at the cell and wait for the distinctive green fluorescence associated
with GFP to appear.
A variant of yellow fluorescent protein with fast and efficient maturation for
cell-biological applications
T Nagai, K Ibata, E Sun Park, M Kubota, K Mikoshiba & A Miyawaki
Nature Biotechnology 20, 87 – 90 (2002) http://dx.doi.org:/10.1038/nbt0102-87
The green fluorescent protein (GFP) from the jellyfish Aequorea victoria
has provided a myriad of applications for biological systems. Over the last
several years, mutagenesis studies have improved folding properties of GFP.
However, slow maturation is still a big obstacle to the use of GFP variants for
visualization. These problems are exacerbated when GFP variants are expressed
at 37°C and/or targeted to certain organelles. Thus, obtaining GFP variants that
mature more efficiently is crucial for the development of expanded research
applications. Among Aequorea GFP variants, yellow fluorescent proteins (YFPs)
are relatively acid-sensitive,and uniquely quenched by chloride ion (Cl−)3. For
YFP to be fully and stably fluorescent, mutations that decrease the sensitivity
to both pH and Cl− are desired. Here we describe the development of an
improved version of YFP named “Venus”. Venus contains a novel mutation,
F46L, which at 37°C greatly accelerates oxidation of the chromophore, the rate-
limiting step of maturation. As a result of other mutations, F64L/M153T/
V163A/S175G, Venus folds well and is relatively tolerant of exposure
to acidosis and Cl−. We succeeded in efficiently targeting a neuropeptide
Y-Venus fusion protein to the dense-core granules of PC12 cells. Its secretion
was readily monitored by measuring release of fluorescence into the medium.
The use of Venus as an acceptor allowed early detection of reliable signals of
fluorescence resonance energy transfer (FRET) for Ca2+ measurements in brain
slices. With the improved speed and efficiency of maturation and the increased
resistance to environment, Venus will enable fluorescent labelings that were not
possible before.
Rhodopsin-like Protein from the Purple Membrane of Halobacterium halobium
DIETER OESTERHELT & WALTHER STOECKENIUS
Nature New Biology 29 Sep 1971; 233, 149-152 | http://dx.doi.org:/10.1038/
newbio233149a0
HALOPHILIC bacteria require high concentrations of sodium chloride and lower
concentrations of KCl and MgCl2 for growth. The cell membrane dissociates into
fragments of varying size when the salt is removed1. One characteristic fragment—
termed the “purple membrane” because of its characteristic deep purple colour—
has been isolated in relatively pure form from Halobacterium halobium. We can
now show that the purple colour is due to retinal bound to an opsin-like protein,
the only protein present in this membrane fragment.
References
Stoeckenius, W. , and Rowen, R. , J. Cell Biol., 34, 365 (1967).
Stoeckenius, W. , and Kunau, W. H. , J. Cell Biol., 38, 337 (1968).
Blaurock, A. E. , and Stoeckenius, W. , Nature New Biology, 233, 152 (1971).
Sehgal, S. N. , and Gibbons, N. E. , Canad. J. Microbiol., 6, 165 (1960).
Kelly, M. , Norgård, S. , and Liaach-Jensen, S. , Acta Chem. Scand., 2A, 2169 (1970).
Shapiro, A. L. , Vinnela, E. , and Maizel, jun., J. V. , Biochem. Biophys. Res.
Commun., 28, 815 (1967).
The monomerization of the Purple protein, a member of the GFP-family
Corning, Brooke
Green fluorescent protein (GFP) has been used extensively since its discovery
in the 1960s to report and visualize gene expression. For years it has been the only
known naturally occurring fluorescent pigment that is encoded by a single gene,
making it extremely useful in various fields of biology, because the expression of
this gene directly leads to the appearance of the fluorescent green color. Recently,
however, many more proteins with similar properties to GFP, and available in a
variety of colors, have been isolated from the class of marine organisms called
Anthozoa, which includes the corals. This increase in the availability of colored
proteins in GFP family in turn has expanded the number of available biotech-
nology applications. However, some of these newly discovered GFP-like
proteins do not have wild-type forms that readily allow for the creation of
fusion proteins, particularly because of oligomerization. It is widely accepted
that almost all members of the GFP-family form dimers or tetramers in their
functional forms.
This study investigates a GFP-ike protein, Purple, isolated from two species,
Galaxea fascicularis and Montipora efflorescens. Purple protein forms oligomers
when expressed, which would then interfere with the normal expression of a protein
to be tagged in gene fusion experiments. We selectively mutated 3 amino acids,
which we believed were responsible for oligomerization in Purple. These 3
residues were chosen based on sequence similarities to a very similar protein,
a mutant form of the Rtms5 chromoprotein from Montipora efflorescens. While
we had hoped that the resulting triple-mutant Purple protein would form
monomers in vivo while retaining its purple coloration, this turned out to
be incorrect. The resulting mutants had lost their ability to turn purple. However,
we also determined that we had successfully changed the oligomerization
state of Purple by examining the relative molecular mass of one our
mutant proteins, which turned out to be half the size of the original
purple protein. It is possible that by adding additional mutations in
the future, the original spectral properties could be recovered. If
successful, this would further expand the utility of the GFP family.
Rhodopsin, also known as visual purple, from Ancient Greek ῥόδον
(rhódon, “rose”), due to its pinkish color, and ὄψις (ópsis, “sight”), is
a light-sensitive receptor protein. It is a biological pigment in photo-
receptor cells of the retina. Rhodopsin is the primary pigment found
in rod photoreceptors. Rhodopsins belong to the G-protein-coupled
receptor (GPCR) family. They are extremely sensitive to light, enabling
vision in low-light conditions. Exposed to light, the pigment
immediately photobleaches, and it takes about 45 minutes to regenerate
fully in humans. Its discovery was reported by German physiologist
Franz Christian Boll in 1876.
Read Full Post »


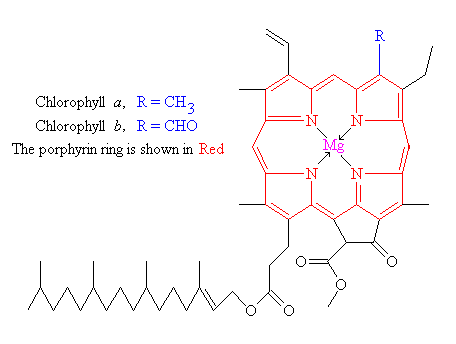
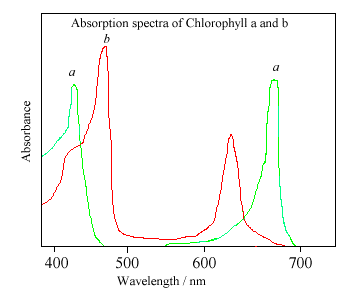






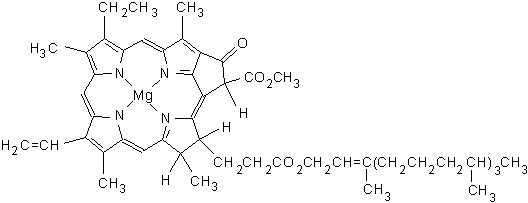














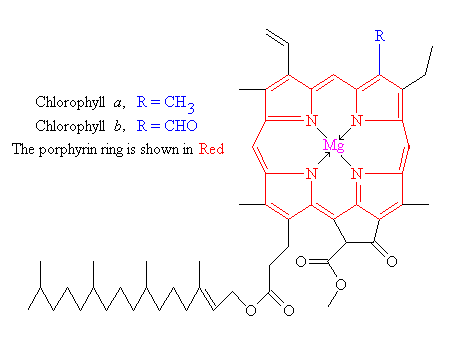
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
.PNG/220px-Spectra_Chlorophyll_ab_oenin_(1).PNG){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}