Healthcare analytics, AI solutions for biological big data, providing an AI platform for the biotech, life sciences, medical and pharmaceutical industries, as well as for related technological approaches, i.e., curation and text analysis with machine learning and other activities related to AI applications to these industries.
2.2.8 RNA-Based Drugs Turn CRISPR/Cas9 On and Off, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 2: CRISPR for Gene Editing and DNA Repair
This image depicts a conventional CRISPR-Cas9 system. The Cas9 enzyme acts like a wrench, and specific RNA guides act as different socket heads. Conventional CRISPR-Cas9 systems act continuously, raising the risk of off-target effects. But CRISPR-Cas9 systems that incorporate specially engineered RNAs could act transiently, potentially reducing unwanted changes. [Ernesto del Aguila III, NHGRI]
By removing parts of the CRISPR/Cas9 gene-editing system, and replacing them with specially engineered molecules, researchers at the University of California, San Diego (UCSD) and Isis Pharmaceutical hope to limit the CRISPR/Cas9 system’s propensity for off-target effects. The researchers say that CRISPR/Cas9 needn’t remain continuously active. Instead, it could be transiently activated and deactivated. Such on/off control could prevent residual gene-editing activity that might go awry. Also, such control could be exploited for therapeutic purposes.
The key, report the scientists, is the introduction of RNA-based drugs that can replace the guide RNA that usually serves to guide the Cas9 enzyme to a particular DNA sequence. When Cas9 is guided by a synthetic RNA-based drug, its cutting action can be suspended whenever the RNA-based drug is cleared. The Cas9’s cutting action can be stopped even more quickly if a second, chemically modified RNA drug is added, provided that it is engineered to direct inactivation of the gene encoding the Cas9 enzyme.
Details about temporarily activated CRISPR/Cas9 systems appeared November 16 in the Proceedings of the National Academy of Sciences, in a paper entitled, “Synthetic CRISPR RNA-Cas9–guided genome editing in human cells.” The paper’s senior author, the USCD’s Don Cleveland, Ph.D., noted that the RNA-based drugs described in the study “provide many advantages over the current CRISPR/Cas9 system,” such as increased editing efficiency and potential selectivity.
“Here we develop a chemically modified, 29-nucleotide synthetic CRISPR RNA (scrRNA), which in combination with unmodified transactivating crRNA (tracrRNA) is shown to functionally replace the natural guide RNA in the CRISPR-Cas9 nuclease system and to mediate efficient genome editing in human cells,” wrote the authors of the PNAS paper. “Incorporation of rational chemical modifications known to protect against nuclease digestion and stabilize RNA–RNA interactions in the tracrRNA hybridization region of CRISPR RNA (crRNA) yields a scrRNA with enhanced activity compared with the unmodified crRNA and comparable gene disruption activity to the previously published single guide RNA.”
Not only did the synthetic RNA functionally replace the natural crRNA, it produced enhanced cleavage activity at a target DNA site with apparently reduced off-target cleavage. These findings, Dr. Cleveland explained, could provide a platform for multiple therapeutic applications, especially for nervous system diseases, using successive application of cell-permeable, synthetic CRISPR RNAs to activate and then silence Cas9 activity. “In addition,” he said, “[these designer RNAs] can be synthesized efficiently, on an industrial scale and in a commercially feasible manner today.”
Alternative CRISPR discovered @MIT, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 2: CRISPR for Gene Editing and DNA Repair
Alternative CRISPR Discovered @MIT
Reporter & Curator: Larry H. Bernstein, MD, FCAP
New breakthrough! – A better alternative CRISPR system just identified
CRISPR-Cas9 system has revolutionized the field of genome editing since its first application in human cells was reported in 2012. A recent publication in Cell reported the identification of a different CRISPR system with the potential for even simpler and more precise genome editing. The newly identified CRISPR-Cpf1 system mediates robust DNA interference with features different from Cas9. Cpf1 possesses several advantages over the currently used Cas9 system.
The Cpf1 system is simpler than Cas9 system as it requires only a single RNA for its DNA-cutting enzymatic activity.
Cpf1 cut has shot overhangs on the exposed ends, allowing more efficient and precise genome engineering; while Cas9 cut produces blunt ends that often undergo mutations when rejoined.
Cpf1 is smaller than Cas9, thus easier to deliver into the cells or tissues.
Cpf1 cut is far away from the recognition site, leaving space for further editing if mutation occurred at the cutting site.
The Cpf1 complex recognize very different PAM sequences than those of Cas9, adding more flexibility in choosing target sites.
These properties of Cpf1 and its potential with more precise gene editing expanded the application scope of CRISPR, from gene knock-out and knock-ins, genomic deletions, to even gene therapy.
•Cpf1 is a CRISPR-associated two-component RNA-programmable DNA nuclease
•Targeted DNA is cleaved as a 5-nt staggered cut distal to a 5′ T-rich PAM
•Two Cpf1 orthologs exhibit robust nuclease activity in human cells
Summary
The microbial adaptive immune system CRISPR mediates defense against foreign genetic elements through two classes of RNA-guided nuclease effectors. Class 1 effectors utilize multi-protein complexes, whereas class 2 effectors rely on single-component effector proteins such as the well-characterized Cas9. Here, we report characterization of Cpf1, a putative class 2 CRISPR effector. We demonstrate that Cpf1 mediates robust DNA interference with features distinct from Cas9. Cpf1 is a single RNA-guided endonuclease lacking tracrRNA, and it utilizes a T-rich protospacer-adjacent motif. Moreover, Cpf1 cleaves DNA via a staggered DNA double-stranded break. Out of 16 Cpf1-family proteins, we identified two candidate enzymes from Acidaminococcus and Lachnospiraceae, with efficient genome-editing activity in human cells. Identifying this mechanism of interference broadens our understanding of CRISPR-Cas systems and advances their genome editing applications.
Almost all archaea and many bacteria achieve adaptive immunity through a diverse set of CRISPR-Cas (clustered regularly interspaced short palindromicrepeats and CRISPR-associated proteins) systems, each of which consists of a combination of Cas effector proteins and CRISPR RNAs (crRNAs) (Makarova et al., 2011, Makarova et al., 2015). The defense activity of the CRISPR-Cas systems includes three stages: (1) adaptation, when a complex of Cas proteins excises a segment of the target DNA (known as a protospacer) and inserts it into the CRISPR array (where this sequence becomes a spacer); (2) expression and processing of the precursor CRISPR (pre-cr) RNA resulting in the formation of mature crRNAs; and (3) interference, when the effector module—either another Cas protein complex or a single large protein—is guided by a crRNA to recognize and cleave target DNA (or in some cases, RNA) (Horvath and Barrangou, 2010,Sorek et al., 2013, Barrangou and Marraffini, 2014). The adaptation stage is mediated by the complex of the Cas1 and Cas2 proteins, which are shared by all known CRISPR-Cas systems, and sometimes involves additional Cas proteins. Diversity is observed at the level of processing of the pre-crRNA to mature crRNA guides, proceeding via either a Cas6-related ribonuclease or a housekeeping RNaseIII that specifically cleaves double-stranded RNA hybrids of pre-crRNA and tracrRNA. Moreover, the effector modules differ substantially among the CRISPR-Cas systems (Makarova et al., 2011, Makarova et al., 2015,Charpentier et al., 2015). In the latest classification, the diverse CRISPR-Cas systems are divided into two classes according to the configuration of their effector modules: class 1 CRISPR systems utilize several Cas proteins and the crRNA to form an effector complex, whereas class 2 CRISPR systems employ a large single-component Cas protein in conjunction with crRNAs to mediate interference (Makarova et al., 2015).
Multiple class 1 CRISPR-Cas systems, which include the type I and type III systems, have been identified and functionally characterized in detail, revealing the complex architecture and dynamics of the effector complexes (Brouns et al., 2008, Marraffini and Sontheimer, 2008, Hale et al., 2009, Sinkunas et al., 2013,Jackson et al., 2014, Mulepati et al., 2014). Several class 2 CRISPR-Cas systems have also been identified and experimentally characterized, but they are all type II and employ homologous RNA-guided endonucleases of the Cas9 family as effectors (Barrangou et al., 2007, Garneau et al., 2010, Deltcheva et al., 2011, Sapranauskas et al., 2011, Jinek et al., 2012, Gasiunas et al., 2012). A second, putative class 2 CRISPR system, tentatively assigned to type V, has been recently identified in several bacterial genomes (http://www.jcvi.org/cgi-bin/tigrfams/HmmReportPage.cgi?acc=TIGR04330) (Schunder et al., 2013, Vestergaard et al., 2014, Makarova et al., 2015). The putative type V CRISPR-Cas systems contain a large, ∼1,300 amino acid protein called Cpf1 (CRISPR from Prevotella and Francisella 1). It remains unknown, however, whether Cpf1-containing CRISPR loci indeed represent functional CRISPR systems. Given the broad applications of Cas9 as a genome-engineering tool (Hsu et al., 2014, Jiang and Marraffini, 2015), we sought to explore the function of Cpf1-based putative CRISPR systems.
Here, we show that Cpf1-containing CRISPR-Cas loci of Francisella novicida U112 encode functional defense systems capable of mediating plasmid interference in bacterial cells guided by the CRISPR spacers. Unlike Cas9 systems, Cpf1-containing CRISPR systems have three features. First, Cpf1-associated CRISPR arrays are processed into mature crRNAs without the requirement of an additional trans-activating crRNA (tracrRNA) (Deltcheva et al., 2011, Chylinski et al., 2013). Second, Cpf1-crRNA complexes efficiently cleave target DNA proceeded by a short T-rich protospacer-adjacent motif (PAM), in contrast to the G-rich PAM following the target DNA for Cas9 systems. Third, Cpf1 introduces a staggered DNA double-stranded break with a 4 or 5-nt 5′ overhang.
To explore the suitability of Cpf1 for genome-editing applications, we characterized the RNA-guided DNA-targeting requirements for 16 Cpf1-family proteins from diverse bacteria, and we identified two Cpf1 enzymes fromAcidaminococcus sp. BV3L6 and Lachnospiraceae bacterium ND2006 that are capable of mediating robust genome editing in human cells. Collectively, these results establish Cpf1 as a class 2 CRISPR-Cas system that includes an effective single RNA-guided endonuclease with distinct properties that has the potential to substantially advance our ability to manipulate eukaryotic genomes.
Results
Figure 1
The Francisella novicida U112 Cpf1 CRISPR Locus Provides Immunity against Transformation of Plasmids Containing Protospacers Flanked by a 5′-TTN PAM
(A) Organization of two CRISPR loci found in Francisella novicida U112 (NC_008601). The domain architectures of FnCas9 and FnCpf1 are compared.
(B) Schematic illustrating the plasmid depletion assay for discovering the PAM position and identity. Competent E. coliharboring either the heterologous FnCpf1 locus plasmid (pFnCpf1) or the empty vector control were transformed with a library of plasmids containing the matching protospacer flanked by randomized 5′ or 3′ PAM sequences and selected with antibiotic to deplete plasmids carrying successfully targeted PAM. Plasmids from surviving colonies were extracted and sequenced to determine depleted PAM sequences.
(C and D) Sequence logo for the FnCpf1 PAM as determined by the plasmid depletion assay. Letter height at each position is measured by information content (C) or frequency (D); error bars show 95% Bayesian confidence interval.
(E) E. coli harboring pFnCpf1 provides robust interference against plasmids carrying 5′-TTN PAMs (n = 3; error bars represent mean ± SEM).
Cpf1-Containing CRISPR Loci Are Active Bacterial Immune Systems
The Cpf1-Associated CRISPR Array Is Processed Independent of TracrRNA
Cpf1 Is a Single crRNA-Guided Endonuclease
The RuvC-like Domain of Cpf1 Mediates RNA-Guided DNA Cleavage
Sequence and Structural Requirements for the Cpf1 crRNA
Cpf1-Family Proteins from Diverse Bacteria Share Common crRNA Structures and PAMs
Cpf1 Can Be Harnessed to Facilitate Genome Editing in Human Cells
In this work, we characterize Cpf1-containing class 2 CRISPR systems, classified as type V, and show that its effector protein, Cpf1, is a single RNA-guided endonuclease. Cpf1 substantially differs from Cas9—to date, the only other experimentally characterized class 2 effector—in terms of structure and function and might provide important advantages for genome-editing applications. Specifically, Cpf1 contains a single identified nuclease domain, in contrast to the two nuclease domains present in Cas9. The results presented here show that, in FnCpf1, inactivation of RuvC-like domain abolishes cleavage of both DNA strands. Conceivably, FnCpf1 forms a homodimer (Figure S2B), with the RuvC-like domains of each of the two subunits cleaving one DNA strand. However, we cannot rule out that FnCpf1 contains a second yet-to-be-identified nuclease domain. Structural characterization of Cpf1-RNA-DNA complexes will allow testing of these hypotheses and elucidation of the cleavage mechanism.
February 8, 2016 | When a geneticist stares down the 3 billion DNA base pairs of the human genome, searching for a clue to what’s gone awry in a single patient, it helps to narrow the field. One of the most popular places to look is the exome, the tiny fraction of our DNA―less than 2%―that actually codes for proteins. For patients with rare genetic diseases, which might be fully explained by one key mutation, many studies sequence the whole exome and leave all the noncoding DNA out. Similarly, personalized cancer tests, which can help bring to light unexpected treatment options, often sequence the tumor exome, or a smaller panel of protein-coding genes.
Unfortunately, we know that’s not the whole picture. “There are a substantial number of noncoding regions that are just as effective at turning off a gene as a mutation in the gene itself,” says Richard Sherwood, a geneticist at Brigham and Women’s Hospital in Boston. “Exome sequencing is not going to be a good proxy for what genes are working.”
Sherwood studies regulatory DNA, the vast segment of the genome that governs which genes are turned on or off in any cell at a given time. It’s a confounding area of genetics; we don’t even know how much of the genome is made up of these regulatory elements. While genes can be recognized by the presence of “start” and “stop” codons―sequences of three DNA letters that tell the cell’s molecular machinery which stretches of DNA to transcribe into RNA, and eventually into protein―there are no definite signs like this for regulatory DNA.
Instead, studies to discover new regulatory elements have been somewhat trial-and-error. If you suspect a gene’s activity might be regulated by a nearby DNA element, you can inhibit that element in a living cell, and see if your gene shuts down with it.
With these painstaking experiments, scientists can slowly work their way through potential regulatory regions―but they can’t sweep across the genome with the kind of high-throughput testing that other areas of genetics thrive on. “Previously, you couldn’t do these sorts of tests in a large form, like 4,000 of them at once,” says David Gifford, a computational biologist at MIT. “You would really need to have a more hypothesis-directed methodology.”
Recently, Gifford and Sherwood collaborated on a paper, published in Nature Biotechnology, which presents a new method for testing thousands of DNA loci for regulatory activity at once. Their assay, called MERA (multiplexed editing regulatory assay), is built on the recent technology boom in CRISPR-Cas9 gene editing, which lets scientists quickly and easily cut specific sequences of DNA out of the genome.
So far, their team, including lead author Nisha Rajagopal from Gifford’s lab, has used MERA to study the regulation of four genes involved in the development of embryonic stem cells. Already, the results have defied the accepted wisdom about regulatory DNA. Many areas of the genome flagged by MERA as important factors in gene expression do not fall into any known categories of regulatory elements, and would likely never have been tested with previous-generation methods.
“Our approach allows you to look away from the lampposts,” says Sherwood. “The more unbiased you can be, the more we’ll actually know.”
A New Kind of CRISPR Screen
In the past three years, CRISPR-Cas9 experiments have taken all areas of molecular biology by storm, and Sherwood and Gifford are far from the first to use the technology to run large numbers of tests in parallel. CRISPR screens are an excellent way to learn which genes are involved in a cellular process, like tumor growth or drug resistance. In these assays, scientists knock out entire genes, one by one, and see what happens to cells without them.
This kind of CRISPR screen, however, operates on too small a scale to study the regulatory genome. For each gene knocked out in a CRISPR screen, you have to engineer a strain of virus to deliver a “guide RNA” into the cellular genome, showing the vicelike Cas9 molecule which DNA region to cut. That works well if you know exactly where a gene lies and only need to cut it once—but in a high-throughput regulatory test, you would want to blanket vast stretches of DNA with cuts, not knowing which areas will turn out to contain regulatory elements. Creating a new virus for each of these cuts is hugely impractical.
The insight behind MERA is that, with the right preparation, most of the genetic engineering can be done in advance. Gifford and Sherwood’s team used a standard viral vector to put a “dummy” guide RNA sequence, one that wouldn’t tell Cas9 to cut anything, into an embryonic stem cell’s genome. Then they grew plenty of cells with this prebuilt CRISPR system inside, and attacked each one with a Cas9 molecule targeted to the dummy sequence, chopping out the fake guide.
Normally, the result would just be a gap in the CRISPR system where the guide once was. But along with Cas9, the researchers also exposed the cells to new, “real” guide RNA sequences. Through a DNA repair mechanism called homologous recombination, the cells dutifully patched over the gaps with new guides, whose sequences were very similar to the missing dummy code. At the end of the process, each cell had a unique guide sequence ready to make cuts at a specific DNA locus—just like in a standard CRISPR screen, but with much less hands-on engineering.
By using a large enough library of guide RNA molecules, a MERA screen can include thousands of cuts that completely tile a broad region of the genome, providing an agnostic look at anywhere regulatory elements might be hiding. “It’s a lot easier [than a typical CRISPR screen],” says Sherwood. “The day the library comes in, you just perform one PCR reaction, and the cells do the rest of the work.”
In the team’s first batch of MERA screens, they created almost 4,000 guide RNAs for each gene they studied, covering roughly 40,000 DNA bases of the “cis-regulatory region,” or the area surrounding the gene where most regulatory elements are thought to lie. It’s unclear just how large any gene’s cis-regulatory region is, but 40,000 bases is a big leap from the highly targeted assays that have come before.
“We’re now starting to do follow-up studies where we increase the number of guide RNAs,” Sherwood adds. “Eventually, what you’d like is to be able to tile an entire chromosome.”
Far From the Lampposts
Sherwood and Gifford tried to focus their assays on regions that would be rich in regulatory elements. To that end, they made sure their guide RNAs covered parts of the genome with well-known signs of regulatory activity, like histone markers and transcription factor binding sites. For many of these areas, Cas9 cuts did, in fact, shut down gene expression in the MERA screens.
But the study also targeted regions around each gene that were empty of any known regulatory features. “We tiled some other regions that we thought might serve as negative controls,” explains Gifford. “But they turned out not to be negative at all.”
The study’s most surprising finding was that several cuts to seemingly random areas of the genome caused genes to become nonfunctional. The authors named these DNA regions “unmarked regulatory elements,” or UREs. They were especially prevalent around the genes Tdgf1 and Zfp42, and in many cases, seemed to be every bit as necessary to gene activity as more predictable hits on the MERA screen.
These results caught the researchers so off guard that it was natural to wonder if MERA screens are prone to false positives. Yet follow-up experiments strongly supported the existence of UREs. Switching the guide RNAs from aTdgf1 MERA screen and aZfp42 screen, for example, produced almost no positive results: the UREs’ regulatory effects were indeed specific to the genes near them.
In a more specific test, the researchers chose a particular URE connected to Tdgf1, and cut it out of a brand new population of cells for a closer look. “We showed that, if we deleted that region from the genome, the cells lost expression of the gene,” says Sherwood. “And then when we put it back in, the gene became expressed again. Which was good proof to us that the URE itself was responsible.”
From these results, it seems likely that follow-up MERA screens will find even more unknown stretches of regulatory DNA. Gifford and Sherwood’s experiments didn’t try to cover as much ground around their target genes as they might have, because the researchers assumed that MERA would mostly confirm what was already known. At best, they hoped MERA would rule out some suspected regulatory regions, and help show which regulatory elements have the biggest effect on gene expression.
“We tended to prioritize regions that had been known before,” Sherwood says. “Unfortunately, in the end, our datasets weren’t ideally suited to discovering these UREs.”
Getting to Basic Principles
MERA could open up huge swaths of the regulatory genome to investigation. Compared to an ordinary CRISPR screen, says Sherwood, “there’s only upside,” as MERA is cheaper, easier, and faster to run.
Still, interpreting the results is not trivial. Like other CRISPR screens, MERA makes cuts at precise points in the genome, but does not tell cells to repair those cuts in any particular way. As a result, a population of cells all carrying the same guide RNA can have a huge variety of different gaps and scars in their genomes, typically deletions in the range of 10 to 100 bases long. Gifford and Sherwood created up to 100 cells for each of their guides, and sometimes found that gene expression was affected in some but not all of them; only sequencing the genomes of their mutated cells could reveal exactly what changes had been made.
By repeating these experiments many times, and learning which mutations affect gene expression, it will eventually be possible to pin down the exact DNA bases that make up each regulatory element. Future studies might even be able to distinguish between regulatory elements with small and large effects on gene expression. In Gifford and Sherwood’s MERA screens, the target genes were altered to produce a green fluorescent protein, so the results were read in terms of whether cells gave off fluorescent light. But a more precise, though expensive, approach would be to perform RNA sequencing, to learn which cuts reduced the cell’s ability to transcribe a gene into RNA, and by how much.
A MERA screen offers a rich volume of data on the behavior of the regulatory genome. Yet, as with so much else in genetics, there are few robust principles to let scientists know where they should be focusing their efforts. Histone markers provide only a very rough sketch of regulatory elements, often proving to be red herrings on closer examination. And the existence of UREs, if confirmed by future experiments, shows that we don’t yet even know which areas of the genome to rule out in the hunt for regulatory regions.
“Every dataset we get comes closer and closer to computational principles that let us predict these regions,” says Sherwood. As more studies are conducted, patterns may emerge in the DNA sequences of regulatory elements that link UREs together, or reveal which histone markers truly point toward regulatory effects. There might also be functional clues hidden in these sequences, hinting at what is happening on a molecular level as regulatory elements turn genes on and off in the course of a cell’s development.
For now, however, the data is still rough and disorganized. For better and for worse, high-throughput tools like MERA are becoming the foundation for most discoveries in genetics—and that means there is a lot more work to do before the regulatory genome begins to come into focus.
CORRECTED 2/9/16: Originally, this story incorrectly stated that only certain cell types could be assayed with MERA for reasons related to homologous recombination. In fact, the authors see no reason MERA could not be applied to any in vitro cell line, and hope to perform screens in a wide range of cell types. The text has been edited to correct the error.
Gene Editing for Exon 51: Why CRISPR Snipping might be better than Exon Skipping for DMD
Why CRISPR might be better than exon skipping for DMD: Snipping vs. skipping for DMD
By Lauren Martz, Senior Writer
Published on Thursday, January 21, 2016
As if to preempt the regulatory setbacks in Duchenne muscular dystrophy (DMD) that last week disappointed the field, a trio of preclinical studies emerged two weeks earlier showing that cutting out DMD mutations with gene editing might offer a viable alternative to the exon-skipping strategies that have dominated the pipeline. Now, the question is whether there’s reason to believe the mouse studies will translate any better to the clinic.
The studies, published Dec. 31 in Science, provide in vivo proof of concept for the first time that CRISPR-Cas9 used postnatally can have a disease-modifying effect. Despite the hype around its therapeutic promise, the technology has so far proved itself primarily in research applications, for example, in modifying cells for in vitro screening or creating animal models of disease.
RNA interference (RNAi) silences, or knocks down, the translation of a gene by inducing degradation of a gene target’s transcript. To advance RNAi applications, Thermo Fisher Scientific has developed two types of small RNA molecules: short interfering RNAs and microRNAs. The company also offers products for RNAi analysis in vitro and in vivo, including libraries for high-throughput applications.
Genes can be knocked down with RNA interference (RNAi) or knocked out with CRISPR-Cas9. RNAi, the screening workhorse, knocks down the translation of genes by inducing rapid degradation of a gene target’s transcript.
CRISPR-Cas9, the new but already celebrated genome-editing technology, cleaves specific DNA sequences to render genes inoperative. Although mechanistically different, the two techniques complement one another, and when used together facilitate discovery and validation of scientific findings.
RNAi technologies along with other developments in functional genomics screening were discussed by industry leaders at the recent Discovery on Target conference. The conference, which emphasized the identification and validation of novel drug targets and the exploration of unknown cellular pathways, included a symposium on the development of CRISPR-based therapies.
RNAi screening can be performed in either pooled or arrayed formats. Pooled screening provides an affordable benchtop option, but requires back-end deconvolution and deep sequencing to identify the shRNA causing the specific phenotype. Targets are much easier to identify using the arrayed format since each shRNA clone is in an individual well.
“CRISPR complements RNAi screens,” commented Ryan Raver, Ph.D., global product manager of functional genomics at Sigma-Aldrich. “You can do a whole genome screen with either small interfering RNA (siRNA) or small hairpin RNA (shRNA), then validate with individual CRISPRs to ensure it is a true result.”
A powerful and useful validation method for knockdown or knockout studies is to use lentiviral open reading frames (ORFs) for gene re-expression, for rescue experiments, or to detect whether the wild-type phenotype is restored. However, the ORF randomly integrates into the genome. Also, with this validation technique, gene expression is not acting under the endogenous promoter. Accordingly, physiologically relevant levels of the gene may not be expressed unless controlled for via an inducible system.
In the future, CRISPR activators may provide more efficient ways to express not only wild-type but also mutant forms of genes under the endogenous promoter.
Choice of screening technique depends on the researcher and the research question. Whole gene knockout may be necessary to observe a phenotype, while partial knockdown might be required to investigate functions of essential or lethal genes. Use of both techniques is recommended to identify all potential targets.
For example, recently, a whole genome siRNA screen on a human glioblastoma cell line identified a gene, known as FAT1, as a negative regulator of apoptosis. A CRISPR-mediated knockout of the gene also conferred sensitivity to death receptor–induced apoptosis with an even stronger phenotype, thereby validating FAT1’s new role and link to extrinsic apoptosis, a new model system.
Dr. Raver indicated that next-generation RNAi libraries that are microRNA-adapted might have a more robust knockdown of gene expression, up to 90–95% in some cases. Ultracomplex shRNA libraries help to minimize both false-negative and false-positive rates by targeting each gene with ~25 independent shRNAs and by including thousands of negative-control shRNAs.
Recently, a relevant paper emerged from the laboratory of Jonathan Weissman, Ph.D., a professor of cellular and molecular pharmacology at the University of California, San Francisco. The paper described how a new ultracomplex pooled shRNA library was optimized by means of a microRNA-adapted system. This system, which was able to achieve high specificity in the detection of hit genes, produced robust results. In fact, they were comparable to results obtained with a CRISPR pooled screen. Members of the Weissman group systematically optimized the promoter and microRNA contexts for shRNA expression along with a selection of guide strands.
Using a sublibrary of proteostasis genes (targeting 2,933 genes), the investigators compared CRISPR and RNAi pooled screens. Data showed 48 hits unique to RNAi, 40 unique to CRISPR, and an overlap of 21 hits (with a 5% false discovery rate cut-off). Together, the technologies provided a more complete research story.
Arrayed CRISPR Screens
Click Image To Enlarge +
Synthetic crRNA:tracrRNA reagents can be used in a similar manner to siRNA reagents for assessment of phenotypes in a cell population. Top row: A reporter cell line stably expressing Cas9 nuclease was transfected with GE Dharmacon’s Edit-R synthetic crRNA:tracrRNA system, which was used to target three positive control genes (PSMD7, PSMD14, and VCP) and a negative control gene (PPIB). Green cells indicate EGFP signaling occuring as a result of proteasome pathway disruption. Bottom row: A siGENOME siRNA pool targeting the same genes was used in the same reporter cell line.
“RNA screens are well accepted and will continue to be used, but it is important biologically that researchers step away from the RNA mechanism to further study and validate their hits to eliminate potential bias,” explained Louise Baskin, senior product manager, Dharmacon, part of GE Healthcare. “The natural progression is to adopt CRISPR in the later stages.”
RNAi uses the cell’s endogenous mechanism. All of the components required for gene knockdown are already within the cell, and the delivery of the siRNA starts the process. With the CRISPR gene-editing system, which is derived from a bacterial immune defense system, delivery of both the guide RNA and the Cas9 nuclease, often the rate limiter in terms of knockout efficiency, are required.
In pooled approaches, the cell has to either drop out or overexpress so that it is sortable, limiting the types of addressable biological questions. A CRISPR-arrayed approach opens up the door for use of other analytical tools, such as high-content imaging, to identify hits of interest.
To facilitate use of CRISPR, GE recently introduced Dharmacon Edit-R synthetic CRISPR RNA (crRNA) libraries that can be used to carry out high-throughput arrayed analysis of multiple genes. Rather than a vector- or plasmid-based gRNA to guide the targeting of the Cas9 cleavage, a synthetic crRNA and tracrRNA are used. These algorithm-designed crRNA reagents can be delivered into the cells very much like siRNA, opening up the capability to screen multiple target regions for many different genes simultaneously.
GE showed a very strong overlap between CRISPR and RNAi using this arrayed approach to validate RNAi screen hits with synthetic crRNA. The data concluded that CRISPR can be used for medium- or high-throughput validation of knockdown studies.
“We will continue to see a lot of cooperation between RNAi and gene editing,” declared Baskin. “Using the CRISPR mechanism to knockin could introduce mutations to help understand gene function at a much deeper level, including a more thorough functional analysis of noncoding genes.
“These regulatory RNAs often act in the nucleus to control translation and transcription, so to knockdown these genes with RNAi would require export to the cytoplasm. Precision gene editing, which takes place in the nucleus, will help us understand the noncoding transcriptome and dive deeper into how those genes regulate disease progression, cellular development and other aspects of human health and biology.”
Tool Selection
Click Image To Enlarge +
Schematic of a pooled shRNA screening workflow developed by Transomic Technologies. Cells are transduced, and positive or negative selection screens are performed. PCR amplification and sequencing of the shRNA integrated into the target cell genome allows the determination of shRNA representation in the population.
The functional genomics tool should fit the specific biology; the biology should not be forced to fit the tool. Points to consider include the regulation of expression, the cell line or model system, as well as assay scale and design. For example, there may be a need for regulatable expression. There may be limitations around the cell line or model system. And assay scale and design may include delivery conditions and timing to optimally complete perturbation and reporting.
“Both RNAi- and CRISPR-based gene modulation strategies have pros and cons that should be considered based on the biology of the genes being studied,” commented Gwen Fewell, Ph.D., chief commercial officer, Transomic Technologies.
RNAi reagents, which can produce hypomorphic or transient gene-suppression states, are well known for their use in probing drug targets. In addition, these reagents are enriching studies of gene function. CRISPR-Cas9 reagents, which produce clean heterozygous and null mutations, are important for studying tumor suppressors and other genes where complete loss of function is desired.
Timing to readout the effects of gene perturbation must be considered to ensure that the biological assay is feasible. RNAi gene knockdown effects can be seen in as little as 24–72 hours, and inducible and reversible gene knockdown can be realized. CRISPR-based gene knockout effects may become complete and permanent only after 10 days.
Both RNAi and CRISPR reagents work well for pooled positive selection screens; however, for negative selection screens, RNAi is the more mature tool. Current versions of CRISPR pooled reagents can produce mixed populations containing a fraction of non-null mutations, which can reduce the overall accuracy of the readout.
To meet the needs of varied and complex biological questions, Transomic Technologies has developed both RNAi and CRISPR tools with options for optimal expression, selection, and assay scale. For example, the company’s shERWOOD-UltramiR shRNA reagents incorporate advances in design and small RNA processing to produce increased potency and specificity of knockdown, particularly important for pooled screens.
Sequence-verified pooled shRNA screening libraries provide flexibility in promoter choice, in vitro formats, in vivo formats, and a choice of viral vectors for optimal delivery and expression in biologically relevant cell lines, primary cells or in vivo.
The company’s line of lentiviral-based CRISPR-Cas9 reagents has variable selectable markers such that guide RNA- and Cas9-expressing vectors, including inducible Cas9, can be co-delivered and selected for in the same cell to increase editing efficiency. Promoter options are available to ensure expression across a range of cell types.
“Researchers are using RNAi and CRISPR reagents individually and in combination as cross-validation tools, or to engineer CRISPR-based models to perform RNAi-based assays,” informs Dr. Fewell. “Most exciting are parallel CRISPR and RNAi screens that have tremendous potential to uncover novel biology.”
Converging Technologies
The convergence of RNAi technology with genome-editing tools, such as CRISPR-Cas9 and transcription activator-like effector nucleases, combined with next-generation sequencing will allow researchers to dissect biological systems in a way not previously possible.
“From a purely technical standpoint, the challenges for traditional RNAi screens come down to efficient delivery of the RNAi reagents and having those reagents provide significant, consistent, and lasting knockdown of the target mRNAs,” states Ross Whittaker, Ph.D., a product manager for genome editing products at Thermo Fisher Scientific. “We have approached these challenges with a series of reagents and siRNA libraries designed to increase the success of RNAi screens.”
Thermo Fisher’ provides lipid-transfection RNAiMax reagents, which effectively deliver siRNA. In addition, the company’s Silencer and Silencer Select siRNA libraries provide consistent and significant knockdown of the target mRNAs. These siRNA libraries utilize highly stringent bioinformatic designs that ensure accurate and potent targeting for gene-silencing studies. The Silencer Select technology adds a higher level of efficacy and specificity due to chemical modifications with locked nucleic acid (LNA) chemistry.
The libraries alleviate concerns for false-positive or false-negative data. The high potency allows less reagent use; thus, more screens or validations can be conducted per library.
Dr. Whittaker believes that researchers will migrate regularly between RNAi and CRISPR-Cas9 technology in the future. CRISPR-Cas9 will be used to create engineered cell lines not only to validate RNAi hits but also to follow up on the underlying mechanisms. Cell lines engineered with CRISPR-Cas9 will be utilized in RNAi screens. In the long term, CRISPR-Cas9 screening will likely replace RNAi screening in many cases, especially with the introduction of arrayed CRISPR libraries.
Validating Antibodies with RNAi
Unreliable antibody specificity is a widespread problem for researchers, but RNAi is assuaging scientists’ concerns as a validation method.
The procedure introduces short hairpin RNAs (shRNAs) to reduce expression levels of a targeted protein. The associated antibody follows. With its protein knocked down, a truly specific antibody shows dramatically reduced or no signal on a Western blot. Short of knockout animal models, RNAi is arguably the most effective method of validating research antibodies.
The method is not common among antibody suppliers—time and cost being the chief barriers to its adoption, although some companies are beginning to embrace RNAi validation.
“In the interest of fostering better science, Proteintech felt it was necessary to implement this practice,” said Jason Li, Ph.D., founder and CEO of Proteintech Group, which made RNAi standard protocol in February 2015. “When researchers can depend on reproducibility, they execute more thorough experiments and advance the treatment of human diseases and conditions.”
Junk DNA Kept in Good Repair by Nuclear Membrane
Heterochromatin has the dubious distinction of being called the “dark matter” of DNA, and it has even suffered the indignity of being dismissed as “junk DNA.” But it seems to get more respectful treatment inside the nucleus, where it has the benefit of a special repair mechanism. This mechanism, discovered by scientists based at the University of Southern California (USC), transports broken heterochromatin sequences from the hurly-burly of the heterochromatin domain so that they can be repaired in the relative peace and quiet of the nuclear periphery.
This finding suggests that the nuclear membrane is more versatile than is generally appreciated. Yes, it serves as a protective container for nuclear material, and it uses its pores to manage the transport of molecules in and out of the nucleus. But it may also play a special role in maintaining the integrity of heterochromatin, which tends to be overlooked because it consists largely of noncoding DNA, including repetitive stretches of no apparent function.
“Scientists are now starting to pay a lot of attention to this mysterious component of the genome,” said Irene E. Chiolo, Ph.D., an assistant professor at USC. “Heterochromatin is not only essential for chromosome maintenance during cell division; it also poses specific threats to genome stability. Heterochromatin is potentially one of the most powerful driving forces for cancer formation, but it is the ‘dark matter’ of the genome. We are just beginning to unravel how repair works here.”
Dr. Chilo led an effort to understand how heterochromatin stays in good repair, even though it is particularly vulnerable to a kind of repair error called ectopic recombination. This kind of error is apt to occur when flaws in repeated sequences undergo homologous recombination (HR) by means of double-strand break (DSB) repair. Specifically, repeated sequences tend to recombine with each other during DNA repair.
Working with the fruit fly Drosophila melanogaster, Dr. Chilo’s team observed that breaks in heterochromatin are repaired after damaged sequences move away from the rest of the chromosome to the inner wall of the nuclear membrane. There, a trio of proteins mends the break in a safe environment, where it cannot accidentally get tangled up with incorrect chromosomes.
The details appeared October 26 in Nature Cell Biology, in an article entitled, “Heterochromatic breaks move to the nuclear periphery to continue recombinational repair.”
“[Heterochromatic] DSBs move to the nuclear periphery to continue HR repair,” the authors wrote. “Relocalization depends on nuclear pores and inner nuclear membrane proteins (INMPs) that anchor repair sites to the nuclear periphery through the Smc5/6-interacting proteins STUbL/RENi. Both the initial block to HR progression inside the heterochromatin domain, and the targeting of repair sites to the nuclear periphery, rely on SUMO and SUMO E3 ligases.”
“We knew that nuclear membrane dysfunctions are common in cancer cells,” Dr. Chiolo said. “Our studies now suggest how these dysfunctions can affect heterochromatin repair and have a causative role in cancer progression.”
This study may help reveal how and why organisms become more predisposed to cancer as they age—the nuclear membrane progressively deteriorates as an organism ages, removing this bulwark against genome instability.
Next, Dr. Chiolo and her team will explore how the movement of broken sequences is accomplished and regulated, and what happens in cells and organisms when this membrane-based repair mechanism fails. Their ultimate goal is to understand how this mechanism functions in human cells and identify new strategies to prevent their catastrophic failure and cancer formation.
Gene Found that Regulates Stem Cell Number Production
The gene Prkci promotes the generation of differentiated cells (red). However if Prkci activity is reduced or absent, neural stem cells (green) are promoted. [In Kyoung Mah]
A scientific team from the University of Southern California (USC) and the University of California, San Diego have described an important gene that maintains a critical balance between producing too many and too few stem cells. Called Prkci, the gene influences whether stem cells self-renew to produce more stem cells, or differentiate into more specialized cell types, such as blood or nerves.
When it comes to stem cells, too much of a good thing isn’t necessarily a benefit: producing too many new stem cells may lead to cancer; making too few inhibits the repair and maintenance of the body.
In their experiments, the researchers grew mouse embryonic stem cells, which lacked Prkci, into embryo-like structures in the laboratory. Without Prkci, the stem cells favored self-renewal, generating large numbers of stem cells and, subsequently, an abundance of secondary structures.
Upon closer inspection, the stem cells lacking Prkci had many activated genes typical of stem cells, and some activated genes typical of neural, cardiac, and blood-forming cells. Therefore, the loss of Prkci can also encourage stem cells to differentiate into the progenitor cells that form neurons, heart muscle, and blood.
Prkci achieves these effects by activating or deactivating a well-known group of interacting genes that are part of the Notch signaling pathway. In the absence of Prkci, the Notch pathway produces a protein that signals to stem cells to make more stem cells. In the presence of Prkci, the Notch pathway remains silent, and stem cells differentiate into specific cell types.
These findings have implications for developing patient therapies. Even though Prkci can be active in certain skin cancers, inhibiting it might lead to unintended consequences, such as tumor overgrowth. However, for patients with certain injuries or diseases, it could be therapeutic to use small molecule inhibitors to block the activity of Prkci, thus boosting stem cell production.
“We expect that our findings will be applicable in diverse contexts and make it possible to easily generate stem cells that have typically been difficult to generate,” said Francesca Mariani, Ph.D., principal investigator at the Eli and Edythe Broad Center for Regenerative Medicine and Stem Cell Research at USC.
Their study (“Atypical PKC-iota Controls Stem Cell Expansion via Regulation of the Notch Pathway”) was published in a Stem Cell Reports.
Atypical PKC-iota Controls Stem Cell Expansion via Regulation of the Notch Pathway
In Kyoung Mah,1 Rachel Soloff,2,3 Stephen M. Hedrick,2 and Francesca V. Mariani1, *
The number of stem/progenitor cells available can profoundly impact tissue homeostasis and the response to injury or disease. Here, we propose that an atypical PKC, Prkci, is a key player in regulating the switch from an expansion to a differentiation/maintenance phase via regulation of Notch, thus linking the polarity pathway with the control of stem cell self-renewal. Prkci is known to influence symmetric cell division in invertebrates; however a definitive role in mammals has not yet emerged. Using a genetic approach, we find that loss of Prkci results in a marked increase in the number of various stem/progenitor cells. The mechanism used likely involves inactivation and symmetric localization of NUMB, leading to the activation of NOTCH1 and its downstream effectors. Inhibition of atypical PKCs may be useful for boosting the production of pluripotent stem cells, multipotent stem cells, or possibly even primordial germ cells by promoting the stem cell/progenitor fate.
The control of asymmetric versus symmetric cell division in stem and progenitor cells balances self-renewal and differentiation to mediate tissue homeostasis and repair and involves key proteins that control cell polarity. In the case of excess symmetric division, too many stem-cell-like daughter cells are generated that can lead to tumor initiation and growth. Conversely, excess asymmetric cell division can severely limit the number of cells available for homeostasis and repair (Go´mez-Lo´pez et al., 2014; Inaba and Yamashita, 2012). The Notch pathway has been implicated in controlling stem cell self-renewal in a number of different contexts (Hori et al., 2013). However, how cell polarity, asymmetric cell division, and the activation of determinants ultimately impinges upon the control of stem cell expansion and maintenance is not fully understood. In this study, we examine the role of an atypical protein kinase C (aPKC), PRKCi, in stem cell self-renewal and, in particular, determine whether PRKCi acts via the Notch pathway. PKCs are serine-threonine kinases that control many basic cellular processes and are typically classified into three subgroups—conventional, novel, and the aPKCs iota and zeta, which, in contrast to the others, are not activated by diacylglyceride or calcium. The aPKC proteins are best known for being central components of an evolutionarily conserved Par3-Par6-aPKC trimeric complex that controls cell polarity in C. elegans, Drosophila, Xenopus, zebrafish, and mammalian cells (Suzuki and Ohno, 2006).
Before Notch influences stem cell self-renewal, the regulation of cell polarity, asymmetric versus symmetric cell division, and the segregation of cell fate determinants such as NUMB may first be required (Knoblich, 2008). For example, mutational analysis in Drosophila has demonstrated that the aPKC-containing trimeric complex is required for maintaining polarity and for mediating asymmetric cell division during neurogenesis via activation and segregation of NUMB (Wirtz-Peitz et al., 2008). NUMB then functions as a cell fate determinant by inhibiting Notch signaling and preventing self-renewal (Wang et al., 2006). In mammals, the PAR3-PAR6-aPKC complex also can bind and phosphorylate NUMB in epithelial cells and can regulate the unequal distribution of Numb during asymmetric cell division (Smith et al., 2007). During mammalian neurogenesis, asymmetric division is also thought to involve the PAR3-PAR6-aPKC complex, NUMB segregation, and NOTCH activation (Bultje et al., 2009).
Mice deficient in Prkcz are grossly normal, with mild defects in secondary lymphoid organs (Leitges et al., 2001). In contrast, deficiency of the Prkci isozyme results in early embryonic lethality at embryonic day (E)9.5 (Seidl et al., 2013; Soloff et al., 2004). A few studies have investigated the conditional inactivation of Prkci; however, no dramatic changes in progenitor generation were detected in hematopoietic stem cells (HSCs) or the brain (Imai et al., 2006; Sengupta et al., 2011), although one study found evidence of a role for Prkci in controlling asymmetric cell division in the skin (Niessen et al., 2013). Analysis may be complicated by functional redundancy between the iota and zeta isoforms and/or because further studies perturbing aPKCs in specific cell lineages and/or at specific developmental stages are needed.
Here, we investigate the requirement of Prkci in mouse cells using an in vitro system that bypasses early embryonic lethality. Embryonic stem (ES) cells are used to make embryoid bodies (EBs) that develop like the early post-implantation embryo in terms of lineage specification and morphology and can also be maintained in culture long enough to observe advanced stages of cellular differentiation (Desbaillets et al., 2000). Using this approach, we provide genetic evidence that inactivation of Prkci signaling leads to enhanced generation of pluripotent cells and some types of multipotent stem cells, including cells with primordial germ cell (PGC) characteristics. In addition, we provide evidence that aPKCs ultimately regulate stem cell fate via the Notch pathway.
Figure 1. Prkci/ EBs Contain Cells with Pluripotency Characteristics (A and A0 ) Day (d) 12 heterozygous EBs have few OCT4/E-CAD+ cells, while null EBs contain many in clusters at the EB periphery. Inset: OCT4 (nucleus)/E-CAD (cytoplasm) double-positive cells. (B and B0 ) Adjacent sections in a null EB show that OCT4+ cells are likely also SSEA1+. (C) Dissociated day-12 Prkci/ EBs contain five to six times more OCT4+ and approximately three times more SSEA1+ cells than heterozygous EBs (three independent experiments). (D and D0 ) After 2 days in ES cell culture, no colonies are visible in null SSEA1 cultures while present in null SSEA1+ cultures (red arrows). (E–E00) SSEA1+ sorted cells can be maintained for many passages, 27+. (E) Prkci+/ sorted cells make colonies with differentiated cells at the outer edges (n = 27/35). (E0 ) Null cells form colonies with distinct edges (n = 39/45). (E00) The percentage of undifferentiated colonies is shown. ***p < 0.001. (F) Sorted null cells express stem cell and differentiation markers at similar levels to normal ES cells (versus heterozygous EBs) (three independent experiments). (G) EBs made from null SSEA1+ sorted cells express germ layer marker genes at the indicated days. Error bars indicate mean ± SEM, three independent experiments. Scale bars, 100 mm in (A, D, and E); 25 mm in (B). See also Figure S1.
RESULTS
Prkci/ Cultures Have More Pluripotent Cells Even under Differentiation Conditions First, we compared Prkci null EB development to that of Prkci/ embryos. Consistent with another null allele (Seidl et al., 2013), both null embryos and EBs fail to properly cavitate (Figures S1A and S1B). The failure to cavitate is unlikely to be due to the inability to form one of the three germ layers, as null EBs express germ-layer-specific genes (Figure S1E). A failure of cavitation could alternatively be caused by an accumulation of pluripotent cells. For example, EBs generated from Timeless knockdown cells do not cavitate and contain large numbers of OCT4-expressing cells (O’Reilly et al., 2011). In addition, EBs generated with Prkcz isoform knockdown cells contain OCT4+ cells under differentiation conditions (Dutta et al., 2011; Rajendran et al., 2013). Thus, we first evaluated ES colony differentiation by alkaline phosphatase (AP) staining. After 4 days without leukemia inhibitory factor (LIF), Prkci/ ES cell colonies retained crisp boundaries and strong AP staining. In contrast, Prkci+/ colonies had uneven colony boundaries with diffuse AP staining (Figures S1F–S1F00). To definitively detect pluripotent cells, day-12 EBs were assayed for OCT4 and E-CADHERIN (E-CAD) protein expression. Prkci+/ EBs had very few OCT4/E-CAD double-positive cells (Figure 1A); however, null EBs contained large clusters of OCT4/E-CAD double-positive cells, concentrated in a peripheral zone (Figure 1A0 ). By examining adjacent sections, we found that OCT4+ cells could also be positive for stage-specific embryonic antigen 1 (SSEA1) (Figures 1B and 1B0 ). Quantification by fluorescence-activated cell sorting (FACS) analysis showed that day-12 Prkci/ EBs had more OCT4+ and SSEA1+ cells than Prkci+/ EBs (Figure 1C). We did not find any difference between heterozygous and wild-type cells with respect to the number of OCT4+ or SSEA1+ cells or in their levels of expression for Oct4, Nanog, and Sox2 (Figures S1I, S1I0 and S1J). However, we did find that Oct4, Nanog, and Sox2 were highly upregulated in OCT4+ null cells (Figure S1G). Thus, together, these data indicate that Prkci/ EBs contain large numbers of pluripotent stem cells, despite being cultured under differentiation conditions.
Functional Pluripotency Tests If primary EBs have a pluripotent population with the capacity to undergo self-renewal, they can easily form secondary EBs (O’Reilly et al., 2011). Using this assay, we found that more secondary EBs could be generated from Prkci/ versus Prkci+/ EBs, especially at days 6, 10, and 16; even when plated at a low density to control for aggregation (Figure S1H). To test whether SSEA1+ cells could maintain pluripotency long term, FACS-sorted Prkci/ SSEA1+ and SSEA1 cells were plated at a low density and maintained under ES cell culture conditions. SSEA1 cells were never able to form identifiable colonies and could not be maintained in culture (Figure 1D). SSEA1+ cells, however, formed many distinct colonies after 2 days of culture, and these cells could be maintained for over 27 passages (Figures 1D0 , 1E0 , and 1E00). Prkci+/ SSEA1+ cells formed colonies that easily differentiated at the outer edge, even in the presence of LIF (Figure 1E). In contrast Prkci/ SSEA1+ cells maintained distinct round colonies (Figure 1E0 ). Next, we determined whether null SSEA1+ cells expressed pluripotency and differentiation markers similarly to normal ES cells. Indeed, we found that Oct4, Nanog, and Sox2 were upregulated in both null SSEA1+ EB cells and heterozygous ES cells. In addition, differentiated markers (Fgf5, T, Wnt3, and Afp) and tissue stem/progenitor cell markers (neural: Nestin, Sox1, and NeuroD; cardiac: Nkx2-5 and Isl1; and hematopoietic: Gata1 and Hba-x) were downregulated in both SSEA1+ cells and heterozygous ES cells (Figure 1F). SSEA1+ cells likely have a wide range of potential, since EBs generated from these cells expressed markers for all three germ layers (Figure 1G).
Figure 2. Prkci and Pluripotency Pathways (A) ERK1/2 phosphorylation (Y202/Y204) is reduced in null ES cells and early day (d)-6 null EBs compared to heterozygous EBs and strongly increased at later stages. The first lane shows ES cells activated (A) by serum treatment 1 day after serum depletion. (B) Quantification of pERK1/2 normalized to non-phosphorylated ERK1/2 (three independent experiments; mean ± SEM; **p < 0.01). (C) pERK1/2 Y202/Y204 is strongly expressed in the columnar epithelium of heterozygous EBs that have just cavitated. Null EBs have lower expression. OCT4 and pERK1/2 expression do not co-localize. Scale bar, 100 mm. (D) pERK1/2Y202/Y204 levels are lower in null SSEA1+ sorted cells than in heterozygous or in null day-12 EBs that have undergone further differentiation. pSTAT3 and STAT levels are unchanged. See also Figure S2.
ERK1/2 Signaling during EB Development Stem cell self-renewal has been shown to require the activation of the JAK/STAT3 and PI3K/AKT pathways and the inhibition of ERK1/2 and GSK3 pathways (Kunath et al., 2007; Niwa et al., 1998; Sato et al., 2004; Watanabe et al., 2006). We found that both STAT3 and phosphorylated STAT3 levels were not grossly altered and that the p-STAT3/STAT3 ratio was similar between heterozygous and null ES cells and EBs (Figures S2A and S2B). In addition we did not see any difference in AKT, pAKT, or b-CATENIN levels when comparing heterozygous to null ES cells or EBs (Figures S2A and S2C). Thus, the effects observed by the loss of Prkci are unlikely to be due to a significant alteration in the JAK/STAT3, PI3K/AKT, or GSK3 pathways.
Next, we investigated ERK1/2 expression and activation. Consistent with other studies showing ERK1/2 activation to be downstream of Prkci in some mammalian cell types (Boeckeler et al., 2010; Litherland et al., 2010), pERK1/2 was markedly inactivated in Prkci null versus heterozygous ES cells. In addition, during differentiation, null EBs displayed strong pERK1/2 inhibition early (until day 6). Later, pERK1/2 was activated strongly, as the EB began differentiating (Figures 2A and 2B). By immunofluorescence, pERK1/2 was strongly enriched in the columnar epithelium of control EBs, while overall levels were much lower in Prkci/ EBs (Figure 2C). In addition, high OCT4 expression correlated with a marked inactivation of pERK1/2 (Figure 2C). Next, we examined Prkci/ SSEA1+ cells by western blot. We found that SSEA1+ cells isolated from day-12 null EBs had pSTAT3 expression levels similar to whole EBs, while pERK1/2 levels were low (Figure 2D). Thus, these experiments indicate that the higher numbers of pluripotent cells in null EBs correlate with a strong inactivation of ERK1/2.
Neural Stem Cell Fate Is Favored in Prkci/ EBs It is well known that ERK/MEK inhibition is not sufficient for pluripotent stem cell maintenance (Ying et al., 2008); thus, other pathways are likely involved. Therefore, we used a TaqMan Mouse Stem Cell Pluripotency Panel (#4385363) on an OpenArray platform to investigate the mechanism of Prkci action. Day 13 and day 20 Prkci/ EBs expressed high levels of pluripotency and stemness markers versus heterozygous EBs, including Oct4, Utf1, Nodal, Xist, Fgf4, Gal, Lefty1, and Lefty2. However, interestingly, EBs also expressed markers for differentiated cell types and tissue stem cells, including Sst, Syp, and Sycp3 (neural-related genes), Isl1 (cardiac progenitor marker), Hba-x, and Cd34 (hematopoietic markers). Based on this first-pass test, we sought to determine whether loss of Prkci might favor the generation of neural, cardiac, and hematopoietic cell types and/or their progenitors.
Figure 3. Neural Stem Cell Populations Are Increased in Null EBs (A–C0 ) Prkci/ EBs (B) have more NESTINpositive cells than Prkci+/ EBs (A). (C and C0 ) MAP2 and TUJ1 are expressed in null EBs, similarly to heterozygous EBs (data not shown). (D) EBs were assessed for PAX6 expression, and the images were used for quantification (Figures S3A and S3B). The pixel count ratio of PAX6+ cells in null EBs (green) is substantially higher than that found in heterozygous EBs (black) (three independent experiments; mean ± SEM; *p < 0.05). (E–F000) Day 4 after RA treatment, Prkci/ EBs have more NESTIN- than TUJ1-positive neurons (E and F). However, null cells can still terminally differentiate into NEUROD-, NEUN-, and MAP2-positive cells (F0 –F000). Scale bars, 25 mm in (A and C) and 50 mm in (E). See also Figure S3. Ste
The Generation of Cardiomyocyte and Erythrocyte Progenitors Is Also Favored Next, we examined ISL1 expression (a cardiac stem cell marker) by immunofluorescence and found that Prkci/ EBs contained larger ISL1 clusters compared with Prkci+/ EBs; this was confirmed using an image quantification assay (Figures 4A, 4A0 , and 4C). Differentiated cardiac cells and ventral spinal neurons can also express ISL1 (Ericson et al., 1992); therefore, we also examined Nkx2-5 expression, a better stem cell marker and regulator of cardiac progenitor determination (Brown et al., 2004), by RT-PCR and immunofluorescence. In null EBs, Nkx2-5 was upregulated (Figure 4D). In addition, in response to RA, which can promote cardiac fates in vitro (Niebruegge et al., 2008), cells expressing NKX2-5 were more prevalent in null versus heterozygous EBs (Figures 4B and 4B0 ).The abundant cardiac progenitors found in null EBs were still capable of undergoing differentiation (Figures 4E–4F0 ).
Figure 4. Cardiomyocyte and Erythrocyte Progenitors Are Increased in Prkci/ EBs (A–F0 ) In (A, A0 , E, and E0 ), Prkci/ EBs cultured without LIF have more ISL1 (cardiac progenitor marker) and a-ACTININ-positive cells compared to heterozygous EBs. (C) At day (d) 9, the pixel count ratio for ISL1 expression indicates that null EBs (green) have larger ISL1 populations than heterozygous EBs (black) (three independent experiments, n = 20 heterozygous EBs, 21 null EBs total; mean ± SEM; *p < 0.05). In (B, B0 , D, F, and F0 ), RA treatment induces more NKX2-5 (both nuclear and cytoplasmic) and a-ACTININ expression in null EBs. Arrows point to fibers in (F0 ). (G) Null EBs (green) generate more beating EBs with RA treatment compared to heterozygous EBs (black) (four independent experiments; mean ± SEM; *p < 0.05, ***p < 0.001). (H) Dissociated null EBs of different stages (green) generate more erythrocytes in a colony-forming assay (CFU-E) (four independent experiments; mean ± SEM; **p < 0.01). (I) Examples of red colonies. (J) Gene expression for primitive HSC markers is upregulated in null EBs (relative to heterozygous EBs) (three independent experiments; mean ± SEM). Scale bars, 50 mm in (A, B, and E); 100 mm in (F), and 25 mm in (I). See also Figure S4. 6
Hba-x expression is restricted to yolk sac blood islands and primitive erythrocyte populations (Lux et al., 2008; Trimborn et al., 1999). Cd34 is also a primitive HSC marker (Sutherland et al., 1992). Next, we determined whether the elevated expression of these markers observed with OpenArray might represent higher numbers of primitive hematopoietic progenitors. Using a colony-forming assay (Baum et al., 1992), we found that red colonies (indicative of erythrocyte differentiation; examples in Figure 4I) were produced significantly earlier and more readily from cells isolated from null versus heterozygous EBs (Figure 4H). By quantitative real-time PCR, upregulation of Hba-x and Cd34 genes confirmed the OpenArray results (Figure 4J). In addition, we found Gata1, an erythropoiesis-specific factor, and Epor, an erythropoietin receptor that mediates erythroid cell proliferation and differentiation (Chiba et al., 1991), to be highly upregulated in null versus heterozygous EBs (Figure 4J). These data suggest that the loss of Prkci promotes the generation of primitive erythroid progenitors that can differentiate into erythrocytes.
To determine whether the aforementioned tissue stem cells identified were represented in the OCT4+ population that we described earlier, we examined the expression of PAX6, ISL1, and OCT4 in adjacent EB sections. We found that cells expressing OCT4 appeared to represent a distinct population from those expressing PAX6 and ISL1 (although some cells were PAX6 and ISL1 double-positive) (Figures S4A–S4C).
Prkci/ Cells Are More Likely to Inherit NUMB/aNOTCH1 Symmetrically The enhanced production of both pluripotent and tissue stem cells suggests that the mechanism underlying the action of Prkci in these different contexts is fundamentally similar. Because the Notch pathway controls stem cell self-renewal in many contexts (Hori et al., 2013), and because previous studies implicated a connection between PRKCi function and the Notch pathway (Bultje et al., 2009; Smith et al., 2007), we examined the localization and activation of a key player in the Notch pathway, NUMB, (Inaba and Yamashita, 2012). Differences in NUMB expression were first evident in whole EBs, where polarized expression was evident in the ectodermal and endodermal epithelia of heterozygous EBs, while Prkci/ EBs exhibited a more even distribution (Figures 5A–5B0 ). To more definitively determine the inheritance of NUMB during cell division, doublets undergoing telophase or cytokinesis were scored for symmetric (evenly distributed in both cells) or asymmetric (unequally distributed) NUMB localization (examples: Figures 5C and 5C0 ).
Because NUMB can be directly phosphorylated by aPKCs (both PRKCi and PRKCz) (Smith et al., 2007; Zhou et al., 2011), loss of Prkci might be expected to lead to decreased NUMB phosphorylation. Three NUMB phosphorylation sites—Ser7, Ser276, and Ser295—could be aPKC mediated (Smith et al., 2007). By immunofluorescence, we found that one of the most well-characterized sites (Ser276), was strongly inactivated in null versus heterozygous EBs, especially in the core (Figures 5F and 5G). Western analysis also confirmed that the levels of pNUMB (Ser276) were decreased in null versus heterozygous EBs (Figure S5F). Thus, genetic inactivation of Prkci leads to a marked decrease in the phosphorylation status of NUMB.
Notch pathway inhibition by NUMB has been observed in flies and mammals (Berdnik et al., 2002; French et al., 2002). Therefore, we investigated whether reduced Numb activity in Prkci/ EBs might lead to enhanced NOTCH1 activity and the upregulation of the downstream transcriptional readouts (Meier-Stiegen et al., 2010). An overall increase in NOTCH1 activation was supported by western blot analysis showing that the level of activated NOTCH1 (aNOTCH1) was strongly increased in day 6 and day 10 null versus heterozygous EBs (Figure S5G). This was supported by immunofluorescence in EBs, where widespread strong expression of aNOTCH1 was seen in most null cells (Figures 5I and 5I0 ), while in heterozygous EBs, this pattern was observed only in the OCT4+ cells (Figures 5H and 5H0 ).
Figure 5. Prkci/ Cells Preferentially Inherit Symmetric Localization of NUMB and aNOTCH1 and Notch Signaling Is Required for Stem Cell Self-Renewal in Null Cells (A–B0 ) In (A and B), day (d)-7 heterozygous EBs have polarized NUMB localization within epithelia and strong expression in the endoderm, while null EBs have a more even distribution. (A0 and B0 ) Enlarged views. (C and C0 ) Asymmetric and symmetric NUMB expression examples. (D) Doublets from day-10 null EBs have more symmetric inheritance when compared to day-10 heterozygous doublets (three independent experiments; mean ± SEM; **p < 0.01). A red line indicates a ratio of 1 (equal percent symmetric and asymmetric). (E) CD24high null doublets exhibited more symmetric NUMB inheritance than CD24high heterozygous doublets (three independent experiments; mean ± SEM; *p < 0.05). A red line indicates where the ratio is 1. (F and G) Decreased pNUMB (Ser276) is evident in the core of null versus heterozygous EBs (n = 10 of each genotype). (H–I0 ) In (H and I), aNOTCH1 is strongly expressed in heterozygous EBs, including both OCT4+ and OCT4 cells, while strong aNOTCH1 expression is predominant in OCT4+ cells of null EBs (n = 10 of each genotype)). (H0 and I0 ) Enlarged views of boxed regions. OCT4+ cells are demarcated with dotted lines. (J and J0 ) OCT4+ cells express HES5 strongly in the nucleus (three independent experiments). (K) Null doublets from dissociated EBs have more symmetric aNOTCH1 inheritance compared to heterozygous doublets (three independent experiments; mean ± SEM; **p < 0.01). A red line indicates where the ratio is 1. (L) CD24high Prkci/ doublets exhibit more symmetric aNOTCH1 than CD24high heterozygous doublets (three independent experiments; mean ± SEM; *p < 0.05). A red line indicates where the ratio is 1. (M and M0 ) Examples of asymmetric and symmetric aNOTCH1 localization. (N and O) Day-3 DMSO-treated null ES colonies show strong AP staining all the way to the colony edge in (N). Treatment with 3 mM DAPT led to more differentiation in (O). (P–R) OCT4 is strongly expressed in day-4 DMSO-treated null ES cultures (P). With DAPT (Q,R), OCT4 expression is decreased. (S) Working model: In daughter cells that undergo differentiation, PRKCi can associate with PAR3 and PAR6. NUMB is recruited and directly phosphorylated. The activation of NUMB then leads to an inhibition in NOTCH1 activation and stimulation of a differentiation/maintenance program. In the absence of Prkci, the PAR3/PAR6 complex cannot assemble (although it may do so minimally with Prkcz). NUMB asymmetric localization and phosphorylation is reduced. Low levels of pNUMB are not sufficient to block NOTCH1 activation, and activated NOTCH1 preserves the stem cell self-renewal program. We suggest that PRKCi functions to drive differentiation by pushing the switch from an expansion phase that is symmetric to a differentiation and/or maintenance phase that is predominantly asymmetric. In situations of low or absent PRKCi, we propose that the expansion phase is prolonged. Scale bars, 50 mm in (A, B, F, G, H, I, J, J0 , P–R); 200 mm in (A0 and B0 ); 25 mm in (C, C0 , M, and M0 ); and 100 mm in (H0 , I0 , N, and O). See also Figure S5.
Figure 6. Additional Inhibition of PRKCz Results in an Even Higher Percentage of OCT4-, SSEA1-, and STELLA-Positive Cells (A and A0 ) After day 4 without LIF, heterozygous ES cells undergo differentiation in the presence of Go¨6983, while null ES cells stay as distinct colonies in (A0 ). (B and B0 ) Go¨6983 stimulates an increase in OCT4+ populations in heterozygous EBs and an even larger OCT4+ population in null EBs in (B0 , insets: green and red channels separately). (C–D0 ) An even higher percentage of cells are OCT4+ (C and C0 ) and SSEA1+ (D and D0 ) with Go¨6983 treatment (day 12, three independent experiments). (E and F) More STELLA+ clusters containing a larger number of cells are present in drugtreated heterozygous EBs. (G and H) Null EBs also have more STELLA+ clusters and cells. Drug-treated null EBs exhibit a dramatic increase in the number of STELLA+ cells. (I–K) Some cells are double positive for STELLA and VASA in drug-treated null EBs (yellow arrows). There are also VASAonly (green arrows) and STELLA-only cells (red arrows) (three independent experiments). (L–P) Treatment with ZIP results in an increase in OCT4+ and STELLA+ cells. ZIP treatment also results in more cells that are VASA+ (three independent experiments); n = 11 for Prkci+/, and n = 13 for Prkci+/ + ZIP; n = 14 for Prkci/, and n = 20 for Prkci/ + ZIP; eight EBs assayed for both STELLA and VASA expression). Scale bars, 100 mm in (A and A0 ); 50 mm in (B and B0 ); and 25 mm in (E, I, and L).
DISCUSSION In this report, we suggest that Prkci controls the balance between stem cell expansion and differentiation/maintenance by regulating the activation of NUMB, NOTCH1, and Hes /Hey downstream effector genes. In the absence of Prkci, the pluripotent cell fate is favored, even without LIF, yet cells still retain a broad capacity to differentiate. In addition, loss of Prkci results in enhanced generation of tissue progenitors such as neural stem cells and cardiomyocyte and erythrocyte progenitors. In contrast to recent findings on Prkcz (Dutta et al., 2011), loss of Prkci does not appear to influence STAT3, AKT, or GSK3 signaling but results in decreased ERK1/2 activation. We hypothesize that, in the absence of Prkci, although ERK1/2 inhibition may be involved, it is the decreased NUMB phosphorylation and increased NOTCH1 activation that promotes stem and progenitor cell fate. Thus, we conclude that PRKCi, a protein known to be required for cell polarity, also plays an essential role in controlling stem cell fate and generation via regulating NOTCH1 activation.
Notch Activation Drives the Decision to Self-Renew versus Differentiate Notch plays an important role in balancing stem cell selfrenewal and differentiation in a variety of stem cell types and may be one of the key downstream effectors of Prkci signaling. Sustained Notch1 activity in embryonic neural progenitors has been shown to maintain their undifferentiated state (Jadhav et al., 2006). Similarly, sustained constitutive activation of NOTCH1 stimulates the proliferation of immature cardiomyocytes in the rat myocardium (Collesi et al., 2008). In HSCs, overexpression of constitutively active NOTCH1 in hematopoietic progenitors and stem cells supports both primitive and definitive HSC selfrenewal (Stier et al., 2002). Together, these studies suggest that activation and/or sustained Notch signaling can lead to an increase in certain tissue stem cell populations. Thus, a working model for how tissue stem cell populations are favored in the absence of Prkci involves a sequence of events that ultimately leads to Notch activation. Recent studies have shown that aPKCs can be found in a complex with NUMB in both Drosophila and mammalian cells (Smith et al., 2007; Zhou et al., 2011); hence, in our working model (Figure 5S), we propose that the localization and phosphorylation of NUMB is highly dependent on the activity of PRKCi. When Prkci is downregulated or absent (as shown here), cell polarity is not promoted, leading to diffuse distribution and decreased phosphorylation of NUMB. Without active NUMB, NOTCH1 activation is enhanced, Hes/Hey genes are upregulated, and stem/progenitor fate generation is favored. To initiate differentiation, polarization could be stochastically determined but could also be dependent on external cues such as the presentation of certain ligands or extracellular matrix (ECM) proteins (Habib et al., 2013). When PRKCi is active and the cell becomes polarized, a trimeric complex is formed with PRKCi, PAR3, and PAR6. Numb is then recruited and phosphorylated, leading to Notch inactivation, the repression of downstream Hes/Hey genes, and differentiation is favored (see Figure 5S). Support for this working model comes from studies in Drosophila showing that the aPKC complex is essential for Numb activation and asymmetric localization (Knoblich, 2008; Smith et al., 2007; Wang et al., 2006). Additional studies on mouse neural progenitors show that regulating Numb localization and Notch activation is critical for maintaining the proper number of stem/progenitor cells in balance with differentiation (Bultje et al., 2009). Thus, an important function for PRKCi may be to regulate the switch between symmetric expansion of stem/progenitor cells to an asymmetric differentiation/maintenance phase. In situations of low or absent PRKCi, we propose that the expansion phase is favored. Thus, temporarily blocking either, or both, of the aPKC isozymes may be a powerful approach for expanding specific stem/progenitor populations for use in basic research or for therapeutic applications.
Although we do not see changes in the activation status of the STAT3, AKT, or GSK3 pathway, loss of Prkci results in an inhibition of ERK1/2 (Figures 2A and 2B). This result is consistent with the findings that ERK1/2 inhibition is both correlated with and directly increases ES cell selfrenewal (Burdon et al., 1999). Modulation of ERK1/2 activity by Prkci has been observed in cancer cells and chondrocytes (Litherland et al., 2010; Murray et al., 2011). Although it is not clear whether a direct interaction exists between Prkci and ERK1/2, Prkcz directly interacts with ERK1/2 in the mouse liver and in hypoxia-exposed cells (Das et al., 2008; Peng et al., 2008). The Prkcz isozyme is still expressed in Prkci null cells but evidently cannot suf- ficiently compensate and activate the pathway normally. Furthermore, knocking down Prkcz function in ES cells does not result in ERK1/2 inhibition, suggesting that this isozyme does not impact ERK1/2 signaling in ES cells (Dutta et al., 2011). Therefore, although PRKCi may interact with ERK1/2 and be directly required for its activation, ERK1/2 inhibition could also be a readout for cells that are more stem-like. Further studies will be needed to address this question.
Utility of Inhibiting aPKC Function Loss of Prkci resulted in EBs that contained slightly more STELLA+ cells than EBs made from +/ cells. Furthermore, inhibition of both aPKC isozymes by treating Prkci null cells with the PKC inhibitor Go¨6983 or the more specific inhibitor, ZIP, strongly promoted the generation of large clusters of STELLA+ and VASA+ cells, suggesting that inhibition of both isozymes is important for PGC progenitor expansion (Figure 6). It is unclear what the mechanism for this might be; however, one possibility is that blocking both aPKCs is necessary to promote NOTCH1 activation in PGCs or in PGC progenitor cells that may ordinarily have strong inhibitions to expansion (Feng et al., 2014). Regardless of mechanism, the ability to generate PGC-like cells in culture is notoriously challenging, and our results provide a method for future studies on PGC specification and differentiation. Expansion of stem/progenitor pools may not be desirable in the context of cancer. Prkci has been characterized as a human oncogene, a useful prognostic cancer marker, and a therapeutic target for cancer treatment. Overexpression of Prkci is found in epithelial cancers (Fields and Regala, 2007), and Prkci inhibitors are being evaluated as candidate cancer therapies (Atwood et al., 2013; Mansfield et al., 2013). However, because our results show that Prkci inhibition leads to enhanced stem cell production in vitro, Prkci inhibitor treatment as a cancer therapy might lead to unintended consequences (tumor overgrowth), depending on the context and treatment regimen. Thus, extending our findings to human stem and cancer stem cells is needed.
In summary, here, we demonstrate that loss of Prkci leads to the generation of abundant pluripotent cells, even under differentiation conditions. In addition, we show that tissue stem cells such as neural stem cells, primitive erythrocytes, and cardiomyocyte progenitors can also be abundantly produced in the absence of Prkci. These increases in stem cell production correlate with decreased NUMB activation and symmetric NUMB localization and require Notch signaling. Further inhibition of Prkcz may have an additive effect and can enhance the production of PGC-like cells. Thus, Prkci (along with Prkcz) may play key roles in stem cell self-renewal and differentiation by regulating the Notch pathway. Furthermore, inhibition of Prkci and or Prkcz activity with specific small-molecule inhibitors might be a powerful method to boost stem cell production in the context of injury or disease.
Use of CRISPR/CAS9 to Edit Genome of Pigs: Recominetics announces $10M Funding Round, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 2: CRISPR for Gene Editing and DNA Repair
Gene-editing startup raising $10M to expand staff
Nov 25, 2015
Katharine Grayson
Staff reporter
Minneapolis / St. Paul Business Journal
Recombinetics Inc. is seeking $10 million in funding as it ramps up sales of its genetically tweaked animals.
The St. Paul-based biotech company’s recent round has already brought in about about $2.8 million from friends and family, said Chief Operating Officer Kyle Dawley. Company officials hope to close out the round within the next two months and add about 10 employees to its staff of 25.
Recombinetics edits pigs’ genes for biomedical research purposes. Photo source: Simone Van Den Berg
Recombinetics uses gene-editing technology to tweak animals for the agribusiness and biomedical markets. It’s biomedical business centers around pigs, which the company modifies for research purposes. That side of the company’s business already generates revenue, Dawley said, though he declined to reveal sales figures.
The company focuses on pigs, touting them as better research subjects than mice when it comes to testing medical devices and drugs for use in humans.
“Pigs are — size-wise and genetically — a lot more like humans than rats and mice,” Dawley said.
One of Recombinetics’ long-term goals is grow human organs inside pigs.
The company aims to modify livestock for food consumption as well. One of its projects calls for creating hornless cattle by taking a gene from one breed and putting into another.
Disease Disablers, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 2: CRISPR for Gene Editing and DNA Repair
The Gene Hackers
A powerful new technology enables us to manipulate our DNA more easily than ever before.
At thirty-four, Feng Zhang is the youngest member of the core faculty at the Broad Institute of Harvard and M.I.T. He is also among the most accomplished. In 1999, while still a high-school student, in Des Moines, Zhang found a structural protein capable of preventing retroviruses like H.I.V. from infecting human cells. The project earned him third place in the Intel Science Talent Search, and he applied the fifty thousand dollars in prize money toward tuition at Harvard, where he studied chemistry and physics. By the time he received his doctorate, from Stanford, in 2009, he had shifted gears, helping to create optogenetics, a powerful new discipline that enables scientists to use light to study the behavior of individual neurons.
Zhang decided to become a biological engineer, forging tools to repair the broken genes that are responsible for many of humanity’s most intractable afflictions. The following year, he returned to Harvard, as a member of the Society of Fellows, and became the first scientist to use a modular set of proteins, called TALEs, to control the genes of a mammal. “Imagine being able to manipulate a specific region of DNA . . . almost as easily as correcting a typo,” one molecular biologist wrote, referring to TALEs, which stands for transcription activator-like effectors. He concluded that although such an advance “will probably never happen,” the new technology was as close as scientists might get.
Having already helped assemble two critical constituents of the genetic toolbox used in thousands of labs throughout the world, Zhang was invited, at the age of twenty-nine, to create his own research team at the Broad. One day soon after his arrival, he attended a meeting during which one of his colleagues mentioned that he had encountered a curious region of DNA in some bacteria he had been studying. He referred to it as a CRISPR sequence.
“I had never heard that word,” Zhang told me recently as we sat in his office, which looks out across the Charles River and Beacon Hill. Zhang has a perfectly round face, its shape accentuated by rectangular wire-rimmed glasses and a bowl cut. “So I went to Google just to see what was there,” he said. Zhang read every paper he could; five years later, he still seemed surprised by what he found. CRISPR, he learned, was a strange cluster of DNA sequences that could recognize invading viruses, deploy a special enzyme to chop them into pieces, and use the viral shards that remained to form a rudimentary immune system. The sequences, identical strings of nucleotides that could be read the same way backward and forward, looked like Morse code, a series of dashes punctuated by an occasional dot. The system had an awkward name—clustered regularly interspaced short palindromic repeats—but a memorable acronym.
CRISPR has two components. The first is essentially a cellular scalpel that cuts DNA. The other consists of RNA, the molecule most often used to transmit biological information throughout the genome. It serves as a guide, leading the scalpel on a search past thousands of genes until it finds and fixes itself to the precise string of nucleotides it needs to cut. It has been clear at least since Louis Pasteur did some of his earliest experiments into the germ theory of disease, in the nineteenth century, that the immune systems of humans and other vertebrates are capable of adapting to new threats. But few scientists had considered the possibility that single bacterial cells could defend themselves in the same way. The day after Zhang heard about CRISPR, he flew to Florida for a genetics conference. Rather than attend the meetings, however, he stayed in his hotel room and kept Googling. “I just sat there reading every paper on CRISPR I could find,” he said. “The more I read, the harder it was to contain my excitement.”
It didn’t take Zhang or other scientists long to realize that, if nature could turn these molecules into the genetic equivalent of a global positioning system, so could we. Researchers soon learned how to create synthetic versions of the RNA guides and program them to deliver their cargo to virtually any cell. Once the enzyme locks onto the matching DNA sequence, it can cut and paste nucleotides with the precision we have come to expect from the search-and-replace function of a word processor. “This was a finding of mind-boggling importance,” Zhang told me. “And it set off a cascade of experiments that have transformed genetic research.”
With CRISPR, scientists can change, delete, and replace genes in any animal, including us. Working mostly with mice, researchers have already deployed the tool to correct the genetic errors responsible for sickle-cell anemia, muscular dystrophy, and the fundamental defect associated with cystic fibrosis. One group has replaced a mutation that causes cataracts; another has destroyed receptors that H.I.V. uses to infiltrate our immune system.
The potential impact of CRISPR on the biosphere is equally profound. Last year, by deleting all three copies of a single wheat gene, a team led by the Chinese geneticist Gao Caixia created a strain that is fully resistant to powdery mildew, one of the world’s most pervasive blights. In September, Japanese scientists used the technique to prolong the life of tomatoes by turning off genes that control how quickly they ripen. Agricultural researchers hope that such an approach to enhancing crops will prove far less controversial than using genetically modified organisms, a process that requires technicians to introduce foreign DNA into the genes of many of the foods we eat.
The technology has also made it possible to study complicated illnesses in an entirely new way. A few well-known disorders, such as Huntington’s disease and sickle-cell anemia, are caused by defects in a single gene. But most devastating illnesses, among them diabetes, autism, Alzheimer’s, and cancer, are almost always the result of a constantly shifting dynamic that can include hundreds of genes. The best way to understand those connections has been to test them in animal models, a process of trial and error that can take years. CRISPR promises to make that process easier, more accurate, and exponentially faster.
Inevitably, the technology will also permit scientists to correct genetic flaws in human embryos. Any such change, though, would infiltrate the entire genome and eventually be passed down to children, grandchildren, great-grandchildren, and every subsequent generation. That raises the possibility, more realistically than ever before, that scientists will be able to rewrite the fundamental code of life, with consequences for future generations that we may never be able to anticipate. Vague fears of a dystopian world, full of manufactured humans, long ago became a standard part of any debate about scientific progress. Yet not since J. Robert Oppenheimer realized that the atomic bomb he built to protect the world might actually destroy it have the scientists responsible for a discovery been so leery of using it.
For much of the past century, biology has been consumed with three essential questions: What does each gene do? How do we find the genetic mutations that make us sick? And how can we overcome them? With CRISPR, the answers have become attainable, and we are closing in on a sort of grand unified theory of genetics. “I am not sure what a Golden Age looks like,” Winston Yan, a member of Zhang’s research team, told me one day when I was with him in the lab, “but I think we are in one.”
At least since 1953, when James Watson and Francis Crick characterized the helical structure of DNA, the central project of biology has been the effort to understand how the shifting arrangement of four compounds—adenine, guanine, cytosine, and thymine—determines the ways in which humans differ from each other and from everything else alive. CRISPR is not the first system to help scientists pursue that goal, but it is the first that anyone with basic skills and a few hundred dollars’ worth of equipment can use.
“CRISPR is the Model T of genetics,” Hank Greely told me when I visited him recently, at Stanford Law School, where he is a professor and the director of the Center for Law and the Biosciences. “The Model T wasn’t the first car, but it changed the way we drive, work, and live. CRISPR has made a difficult process cheap and reliable. It’s incredibly precise. But an important part of the history of molecular biology is the history of editing genes.”
Scientists took the first serious step toward controlling our genes in the early nineteen-seventies, when they learned to cut chains of DNA by using proteins called restriction enzymes. Suddenly, genes from organisms that would never have been able to mate in nature could be combined in the laboratory. But those initial tools were more hatchet than scalpel, and, because they could recognize only short stretches within the vast universe of the human genome, the editing was rarely precise. (Imagine searching through all of Shakespeare for Hamlet’s soliloquy on suicide, relying solely on the phrase “to be.” You’d find the passage, but only after landing on several hundred unrelated citations.)
When the first draft of the Human Genome Project was published, in 2001, the results were expected to transform our understanding of life. In fundamental ways, they have; the map has helped researchers locate thousands of genes associated with particular illnesses, including hundreds that cause specific types of cancer. To understand the role that those genes play in the evolution of a disease, however, and repair them, scientists need to turn genes on and off systematically and in many combinations. Until recently, though, altering even a single gene took months or years of work.
That began to change with the growing use of zinc fingers, a set of molecular tools that, like CRISPR clusters, were discovered by accident. In 1985, scientists studying the genetic code of the African clawed frog noticed a finger-shaped protein wrapped around its DNA. They soon figured out how to combine that tenacious grip with an enzyme that could cut the DNA like a knife. Two decades later, geneticists began using TALEs, which are made up of proteins secreted by bacteria. But both engineering methods are expensive and cumbersome. Even Zhang, who published the first report on using TALEs to alter the genes of mammals, realized that the system was little more than an interim measure. “It is difficult to use,” he told me. “I had to assign a graduate student just to make the proteins and test them before I could begin to use them in an experiment. The procedure was not easy.”
Zhang’s obsession with science began in middle school, when his mother prodded him to attend a Saturday-morning class in molecular biology. “I was thirteen and had no idea what molecular biology was,” he said one evening as we walked across the M.I.T. campus on the way to the fiftieth-anniversary celebration of the Department of Brain and Cognitive Sciences, where Zhang is also a faculty member. “It really opened my imagination.” His parents, both engineers, moved the family to Iowa when he was eleven. They stayed largely because they thought he would get a better education in the United States than in China.
In 1997, when Zhang was fifteen, he was offered an internship in a biosafety facility at the Des Moines Human Gene Therapy Research Institute—but he was told that federal law prohibited him from working in a secure lab until he was sixteen. “So I had to wait,” he said. On his birthday, Zhang went to the lab and met the scientists. “I was assigned to a man who had a Ph.D. in chemistry but trained as a molecular biologist,” he continued. “He had a lot of passion for science, and he had a very big impact on me and my research.” On his first day, Zhang spent five hours in the lab, and nearly as much time every day after school until he graduated.
Zhang is unusually reserved, and he speaks in low, almost sleepy tones. I asked him if he considered himself to be mellow, a characteristic rarely associated with prize-winning molecular biologists. “You came to the lab meeting, right?” he replied. Earlier that morning, I had caught the tail end of a weekly meeting that Zhang holds for his group. I watched as he gently but relentlessly demolished a presentation given by one of the people on his team. When I mentioned it to one of the scientists who was at the meeting, he responded, “That was nothing. You should have been there from the start.”
At his Saturday-morning classes, Zhang learned how to extract DNA from cells and determine the length of each sequence. But that isn’t what he remembers best. “They showed us ‘Jurassic Park,’ ” he said, his voice moving up a register. “And it was amazing to me. The teacher explained the different scientific concepts in the movie, and they all seemed completely feasible.”
We had reached the cocktail party, a tepid affair crowded with men in khakis and women wearing sensible shoes. Zhang left after barely twenty minutes and headed back to the lab. He retains his position on the cognitive-sciences faculty, because he hopes that his research will help neuroscientists study the brain in greater detail. He told me that when he was young he had a friend who suffered from serious depression, and he had been surprised to find that there was almost no treatment available. It spurred a lasting interest in psychiatry. “People think you are weak if you are depressed,” he said. “It is still a common prejudice. But many people suffer from problems we cannot begin to address. The brain is still the place in the universe with the most unanswered questions.”
The Broad Institute was founded, in 2003, by the entrepreneur Eli Broad and his wife, Edythe, to foster research into the molecular components of life and their connections to disease. One afternoon in Zhang’s laboratory, Winston Yan offered to walk me through the mechanics of using CRISPR to edit a gene. “We need to be able to break DNA in a very precise place in the genome,” he said as I watched him at work. He swivelled in his chair and pointed to a row of vials that contained DNA samples to be analyzed and edited. Yan, a thin, bespectacled man, wore black laboratory gloves and a white Apple Watch; he clapped his hands and shrugged, as if to suggest that the work was simple.
Ordering the genetic parts required to tailor DNA isn’t as easy as buying a pair of shoes from Zappos, but it seems to be headed in that direction. Yan turned on the computer at his lab station and navigated to an order form for a company called Integrated DNA Technologies, which synthesizes biological parts. “It takes orders online, so if I want a particular sequence I can have it here in a day or two,” he said. That is not unusual. Researchers can now order online almost any biological component, including DNA, RNA, and the chemicals necessary to use them. One can buy the parts required to assemble a working version of the polio virus (it’s been done) or genes that, when put together properly, can make feces smell like wintergreen. In Cambridge, I.D.T. often makes same-day deliveries. Another organization, Addgene, was established, more than a decade ago, as a nonprofit repository that houses tens of thousands of ready-made sequences, including nearly every guide used to edit genes with CRISPR. When researchers at the Broad, and at many other institutions, create a new guide, they typically donate a copy to Addgene.
The RNA that CRISPR relies upon to guide the molecular scalpel to its target is made of twenty base pairs. Humans have twenty thousand genes, and twenty base pairs occupy roughly the same percentage of space in a single gene as would one person standing in a circle that contained the entire population of the United States. CRISPR is better at locating specific genes than any other system, but it isn’t perfect, and sometimes it cuts the wrong target. Yan would order a ready-made probe from Addgene. When it arrives, he pairs it with a cutting enzyme and sends it to the designated gene.
Yan joined Zhang’s lab just before what he described as “the CRISPR craze” began. But, he added, the technology has already transformed the field. “For many years, there was a reductionist approach to genetics,” he said. “A kind of wishful thinking: ‘We will find the gene that causes cancer or the gene that makes you prone to heart disease.’ It is almost never that simple.”
The next morning, I walked over to the Broad’s new Stanley Building and rode the elevator to the top floor, where I emptied my pockets, put on a mask and gown, and slipped booties over my shoes. Then I passed through an air chamber that was sealed with special gaskets and had a fan blowing continuously to keep out foreign microbes. I entered the vivarium, a long, clean floor that looked like a combination of research unit and hospital ward. The vivarium, which opened last year, provides thousands of mice with some of the world’s most carefully monitored accommodations.
Despite our growing knowledge of the way that cancer develops in human cells, mutations can’t be studied effectively in a petri dish, and, since the late nineteen-eighties, genetically modified mice have served as the standard proxy. What cures (or kills) a mouse won’t necessarily have the same effect on a human, but the mouse genome is surprisingly similar to our own, and the animals are cheap and easy to maintain. Like humans, and many other mammals, mice develop complex diseases that affect the immune system and the brain. They get cancer, atherosclerosis, hypertension, and diabetes, among other chronic illnesses. Mice also reproduce every three weeks, which allows researchers to follow several generations at once. Typically, technicians would remove a stem cell from the mouse, then edit it in a lab to produce a particular gene or to prevent the gene from working properly. After putting the stem cell back into the developing embryo of the mouse, and waiting for it to multiply, they can study the gene’s effect on the animal’s development. The process works well, but it generally allows for the study of only one characteristic in one gene at a time.
The vivarium at the Broad houses an entirely different kind of mouse, one that carries the protein Cas9 (which stands for CRISPR-associated nuclease) in every cell. Cas9, the part of the CRISPR system that acts like a genetic scalpel, is an enzyme. When scientists originally began editing DNA with CRISPR, they had to inject both the Cas9 enzyme and the probe required to guide it. A year ago, Randall Platt, another member of Zhang’s team, realized that it would be possible to cut the CRISPR system in two. He implanted the surgical enzyme into a mouse embryo, which made it a part of the animal’s permanent genome. Every time a cell divided, the Cas9 enzyme would go with it. In other words, he and his colleagues created a mouse that was easy to edit. Last year, they published a study explaining their methodology, and since then Platt has shared the technique with more than a thousand laboratories around the world.
The “Cas9 mouse” has become the first essential tool in the emerging CRISPR arsenal. With the enzyme that acts as molecular scissors already present in every cell, scientists no longer have to fit it onto an RNA guide. They can dispatch many probes at once and simply make mutations in the genes they want to study.
To demonstrate a potential application for cancer research, the team used the Cas9 mouse to model lung adenocarcinoma, the most common form of lung cancer. Previously, scientists working with animal models had to modify one gene at a time or cross-breed animals to produce a colony with the needed genetic modifications. Both processes were challenging and time-consuming. “Now we can activate CRISPR directly in the cells we’re interested in studying, and modify the genome in whatever way we want,” Platt said, as he showed me around the vivarium. We entered a small exam room with a commanding view of Cambridge. I watched as a technician placed a Cas9 mouse in a harness inside a biological safety cabinet. Then, peering through a Leica microscope, she used a fine capillary needle to inject a single cell into the mouse’s tail.
“And now we have our model,” Platt said, explaining that the mouse had just received an injection that carried three probes, each of which was programmed to carry a mutation that scientists believe is associated with lung cancer. “The cells will carry as many mutations as we want to study. That really is a revolutionary development.”
“In the past, this would have taken the field a decade, and would have required a consortium,” Platt said. “With CRISPR, it took me four months to do it by myself.” In September, Zhang published a report, in the journal Cell, describing yet another CRISPR protein, called Cpf1, that is smaller and easier to program than Cas9.
The lab employs a similar approach to studying autism. Recent experiments suggest that certain psychiatric conditions can be caused by just a few malfunctioning neurons out of the trillions in every brain. Studying the way neurons function within the brain is difficult. But by re-creating, in the lab, genetic mutations that others have linked to autism and schizophrenia Zhang’s team has been able to investigate faulty neurons that may play a role in those conditions.
As the price of sequencing plunges, cancer clinics throughout the United States have begun to study their patients’ tumors in greater detail. Tumors are almost never uniform; one may have five mutations or fifty, which means, essentially, that every cancer is a specific, personal disease. Until CRISPR became available, the wide genetic variations in cancer cells often made it hard to develop effective treatments.
“What I love most about the CRISPR process is that you can take any cancer-cell line, knock out every gene, and identify every one of the cell’s Achilles’ heels,” Eric Lander, the fifty-eight-year-old director of the Broad, told me recently. Lander, who was among the leaders of the Human Genome Project, said that he had never encountered a more promising research tool. “You can also use CRISPR to systematically study the ways that a cancer cell can escape from a treatment,” he said. “That should make it possible to build a comprehensive road map for cancer.”
Lander went on to say that each vulnerability of a tumor might be attacked by a single drug. But cancer cells elude drugs in many ways, and, to succeed, a therapy may need to block them all. That strategy has proved effective for infectious diseases like AIDS. “Remember the pessimism about H.I.V.,” he said, referring to the early years of the AIDS epidemic, when a diagnosis was essentially a death sentence. Eventually, virologists developed a series of drugs that interfere with the virus’s ability to replicate. The therapy became truly successful, however, only when those drugs, working together, could block the virus completely.
The same approach has proved successful in treating tuberculosis. Lander is convinced that it will also work for many cancers: “With triple-drug therapy,” for H.I.V., “we reached an inflection point: we were losing badly, and one day suddenly we were winning.”
He stood up and walked across the office toward his desk, then pointed at the wall and described his vision for the future of cancer treatment. “There will be an enormous chart,” he said. “Well, it will be electronic, and it will contain the therapeutic road map of every trick that cancer cells have—how they form, all the ways you can defeat them, and all the ways they can escape and defeat a treatment. And when we have that we win. Because every cancer cell starts naïve. It doesn’t know what we have waiting in the freezer for it. Infectious diseases are a different story; they share their knowledge as they spread. They learn from us as they move from person to person. But every person’s cancer starts naïve. And this is why we will beat it.”
Developing any technology as complex and widely used as CRISPR invariably involves contributions from many scientists. Patent fights over claims of discovery and licensing rights are common. Zhang, the Broad Institute, and M.I.T. are now embroiled in such a dispute with Jennifer Doudna and the University of California; she is a professor of chemistry and of molecular biology at Berkeley. By 2012, Doudna, along with Emmanuelle Charpentier, a medical microbiologist who studies pathogens at the Helmholtz Centre for Infection Research, in Germany, and their lab teams, demonstrated, for the first time, that CRISPR could edit purified DNA. Their paper was published that June. In January of 2013, though, Zhang and George Church, a professor of genetics at both Harvard Medical School and M.I.T., published the first studies demonstrating that CRISPR could be used to edit human cells. Today, patents are generally awarded to the first people to file—in this case, Doudna and Charpentier. But Zhang and the Broad argued that the earlier success with CRISPR had no bearing on whether the technique would work in the complex organisms that matter most to scientists looking for ways to treat and prevent diseases.
Zhang was awarded the patent, but the University of California has requested an official reassessment, and a ruling has not yet been issued. Both he and Doudna described the suit to me as “a distraction” that they wished would go away. Both pledged to release all intellectual property to researchers without charge (and they have). But both are also involved in new companies that intend to develop CRISPR technology as therapies, as do many pharmaceutical firms and other profit-seeking enterprises.
CRISPR research is becoming big business: venture-capital firms are competing with one another to invest millions, and any patent holder would have the right to impose licensing fees. Whoever wins stands to make a fortune. Other achievements are also at stake, possibly including a Nobel Prize. (Doudna’s supporters have described her as America’s next female Nobel Prize winner, and at times the publicity war seems a bit like the battles waged by movie studios during Academy Award season.) Last year, the National Science Foundation presented Zhang with its most prestigious award, saying that his fundamental research “moves us in the direction” of eliminating schizophrenia, autism, and other brain disorders. A few months later, Doudna and Charpentier received three million dollars each for the Breakthrough Prize, awarded each year for scientific achievement. The prize was established, in 2012, by several Silicon Valley billionaires who are seeking to make science a more attractive career path. The two women also appeared on Time’s annual list of the world’s hundred most influential people.
In fact, neither group was involved in the earliest identification of CRISPR or in the first studies to demonstrate how it works. In December, 1987, biologists at the Research Institute of Microbial Diseases, in Osaka, Japan, published the DNA sequence of a gene taken from the common intestinal bacterium E. coli. Those were early days in the genomic era, and thousands of labs around the world had embarked on similar attempts to map the genes of species ranging from fruit flies to humans. In an effort to better understand how this particular gene functioned, the Japanese scientists also sequenced some of the DNA that surrounded it. When they examined the data, they were surprised to see cellular structures that none of them recognized: they had no idea what to make of the strange phenomenon, but they took note of it, writing in the final sentence of their report, published in the Journal of Bacteriology, that the “biological significance of these sequences is not known.”
The mystery remained until 2005, when Francisco Mojica, a microbiologist at the University of Alicante, who had long sought to understand CRISPR, decided to compare its DNA with the DNA of tens of thousands of similar organisms. What he saw amazed him: every unknown sequence turned out to be a fragment of DNA from an invading virus.
The pace of research quickened. In 2007, Rodolphe Barrangou and Philippe Horvath, microbiologists then working for Danisco, the Danish food company, had noticed that some of its yogurt cultures were routinely destroyed by viruses and others were not. They decided to find out why. The scientists infected the microbe Streptococcus thermophilus, which is widely used to make yogurt, with two viruses. Most of the bacteria died, but those which survived had one property in common: they all contained CRISPR molecules to defend them.
“No single person discovers things anymore,” George Church told me when we met in his office at Harvard Medical School. “The whole patent battle is silly. There has been much research. And if anybody should be making a fuss about this I should be making a fuss. But I am not doing that, because I don’t think it matters. They are all nice people. They are all doing important work. It’s a tempest in a teapot.”
From the moment that manipulating genes became possible, many people, including some of those involved in the experiments, were horrified by the idea of scientists in lab coats rearranging the basic elements of life. In 1974, David Baltimore, the pioneering molecular biologist, who was then at M.I.T., and Paul Berg, of Stanford, both of whom went on to win a Nobel Prize for their research into the fundamentals of viral genetics, called for a moratorium on gene-editing research until scientists could develop safety principles for handling organisms that contained recombinant DNA. That meeting, which took place in 1975, at a conference center in Asilomar, California, has come to be regarded as biotechnology’s Constitutional Convention.
Roughly a hundred and fifty participants, most of them scientists, gathered to discuss ways to limit the risks of accidentally releasing genetically modified organisms. At the time, the possibility of creating “designer babies”—a prospect that, no matter how unlikely, is attached to almost everything written or said about CRISPR—was too remote to consider. Nevertheless, the technology seemed frightening. In Cambridge, home to both M.I.T. and Harvard, the city council nearly banned such research altogether. The work went on, but decoding sequences of DNA wasn’t easy. “In 1974, thirty base pairs”—thirty rungs on the helical ladder of the six billion nucleotides that make up our DNA—“was a good year’s work,” George Church told me. Now the same work would take seconds.
At least for the foreseeable future, CRISPR’s greatest impact will lie in its ability to help scientists rapidly rewrite the genomes of animal and plant species. In laboratories, agricultural companies have already begun to use CRISPR to edit soybeans, rice, and potatoes in an effort to make them more nutritious and more resistant to drought. Scientists might even be able to edit allergens out of foods like peanuts.
Normally, it takes years for genetic changes to spread through a population. That is because, during sexual reproduction, each of the two versions of any gene has only a fifty per cent chance of being inherited. But a “gene drive”—which is named for its ability to propel genes through populations over many generations—manages to override the traditional rules of genetics. A mutation made by CRISPR on one chromosome can copy itself in every generation, so that nearly all descendants would inherit the change. A mutation engineered into a mosquito that would block the parasite responsible for malaria, for instance, could be driven through a large population of mosquitoes within a year or two. If the mutation reduced the number of eggs produced by that mosquito, the population could be wiped out, along with any malaria parasites it carried.
Kevin Esvelt, an evolutionary biologist at Harvard, was the first to demonstrate how gene drives and CRISPR could combine to alter the traits of wild populations. Recently, he has begun to study the possibility of using the technology to eliminate Lyme disease by rewriting the genes of mice in the wild. Lyme disease is caused by a bacterium and transmitted by ticks, and more than eighty-five per cent of the time they become infected after biting a mouse. Once exposed, however, some mice naturally acquire resistance or immunity. “My idea is to take the existing genes that confer resistance to Lyme and make sure that all mice have the most effective version,’’ Esvelt said. To do that, scientists could encode the most protective genes next to the CRISPR system and force them to be passed on together. Esvelt stressed that such an approach would become possible only after much more research and a lengthy series of public discussions on the risks and benefits of the process.
The promise of CRISPR research becomes more evident almost every month. Recently, Church reported that he had edited sixty-two genes simultaneously in a pig cell. The technique, if it proves accurate and easy to repeat, could help alleviate the constant shortage of organ donors in the U.S. For years, scientists have tried to find a way to use pig organs for transplants, but a pig’s DNA is filled with retroviruses that have been shown in labs to infect human cells. Church and his colleagues discovered that those viruses share a common genetic sequence. He deployed CRISPR to their exact locations and snipped them out of the genome. In the most successful of the experiments, the CRISPR system deleted all sixty-two of the retroviruses embedded in the pig’s DNA. Church then mixed those edited cells with human cells in the laboratory, and none became infected.
While CRISPR will clearly make it possible to alter our DNA, serious risks remain. Jennifer Doudna has been among the most vocal of those calling for caution on what she sees as the inevitable march toward editing human genes. “It’s going to happen,” she told me the first time we met, in her office at Berkeley. “As a research tool, CRISPR could hardly be more valuable—but we are far from the day when it should be used in a clinical setting.” Doudna was a principal author of a letter published in Science this spring calling for a temporary research moratorium. She and others have organized a conference to discuss the ethics of editing DNA, a sort of Asilomar redux. The conference, to be attended by more than two hundred scientists—from the U.S., England, and China, among other countries—will take place during the first week of December at the National Academy of Sciences, in Washington.
Until April, the ethical debate over the uses of CRISPR technology in humans was largely theoretical. Then a group at Sun Yat-sen University, in southern China, attempted to repair, in eighty-six human embryos, the gene responsible for betathalassemia, a rare but often fatal blood disorder. If those disease genes, and genes that cause conditions like cystic fibrosis, could be modified successfully in a fertilized egg, the alteration could not only protect a single individual but eventually eliminate the malady from that person’s hereditary lineage. Given enough time, the changes would affect all of humanity. The response to the experiment was largely one of fear and outrage. The Times carried the story under the headline “CHINESE SCIENTISTS EDIT GENES OF HUMAN EMBRYOS, RAISING CONCERNS.”
Critics called the experiment irresponsible and suggested that the scientists had violated an established code of conduct. “This paper demonstrates the enormous safety risks that any such attempt would entail, and underlines the urgency of working to forestall other such efforts,” Marcy Darnovsky, of the Center for Genetics and Society, told National Public Radio when the report was published. “The social dangers of creating genetically modified human beings cannot be overstated.”
There seems to be little disagreement about that. But the Chinese researchers were not trying to create genetically modified humans. They were testing the process, and every CRISPR researcher I spoke to considered the experiment to have been well planned and carried out with extraordinary care. The scientists also agreed that the results were illuminating. “That was an ethical paper, and a highly responsible project,’’ Lander told me. “What did they do? They took triploid zygotes’’—a relatively common genetic aberration—“from I.V.F. clinics. They deliberately chose those because they knew no human could ever develop from them. And what did the paper say? ‘Boy, we see problems everywhere.’ That was good science, and it was cautionary.”
Fewer than half the embryos were edited successfully, and, of those, most retained none of the new DNA that was inserted into the genes. The experiment, which was published in the Beijing-based journal Protein & Cell, demonstrated clearly that the day when scientists could safely edit humans is far off. The CRISPR system also made unintended cuts and substitutions, the potential effects of which are unknown. In other cases, it made the right changes in some cells of the embryo but not in all of them, which could cause other problems. “These authors did a very good job, pointing out the challenges,” Dieter Egli, a stem-cell researcher at Columbia University, said when the study was published. “They say themselves that this type of technology is not ready for any kind of application.”
Doudna agreed that the Chinese experiment yielded valuable results. She is fifty-one, and has been at Berkeley since 2002, when she and her husband, the biochemist Jamie Cate, were offered joint appointments to the departments of chemistry and molecular and cell biology. Their offices are next to each other, with the same commanding view of San Francisco Bay and the Golden Gate Bridge. Doudna’s work, unlike that of the scientists at the Broad, has been focussed on molecules, not mammalian genetics. For years, she has been leading investigations into the shape, structure, and capabilities of RNA, and in 2011 Charpentier asked for her help in exploring the mechanism of CRISPR. Doudna is tall, with graying blond hair and piercing blue eyes. She grew up in Hawaii, where her parents were academics; when it was time for college, she decided to leave the island and study in California, at Pomona. She earned her doctorate at Harvard and then moved on to Yale. “I have always been a bit of a restless soul,” she said. “I may spend too much time wondering what comes next.”
Doudna is a highly regarded biochemist, but she told me that not long ago she considered attending medical school or perhaps going into business. She said that she wanted to have an effect on the world and had begun to fear that the impact of her laboratory research might be limited. The promise of her work on CRISPR, however, has persuaded her to remain in the lab. She told me that she was constantly amazed by its potential, but when I asked if she had ever wondered whether the powerful new tool might do more harm than good she looked uncomfortable. “I lie in bed almost every night and ask myself that question,” she said. “When I’m ninety, will I look back and be glad about what we have accomplished with this technology? Or will I wish I’d never discovered how it works?”
Her eyes narrowed, and she lowered her voice almost to a whisper. “I have never said this in public, but it will show you where my psyche is,” she said. “I had a dream recently, and in my dream”—she mentioned the name of a leading scientific researcher—“had come to see me and said, ‘I have somebody very powerful with me who I want you to meet, and I want you to explain to him how this technology functions.’ So I said, Sure, who is it? It was Adolf Hitler. I was really horrified, but I went into a room and there was Hitler. He had a pig face and I could only see him from behind and he was taking notes and he said, ‘I want to understand the uses and implications of this amazing technology.’ I woke up in a cold sweat. And that dream has haunted me from that day. Because suppose somebody like Hitler had access to this—we can only imagine the kind of horrible uses he could put it to.”
Nobody is going to employ CRISPR technology to design a baby, let alone transform the genetic profile of humanity, anytime soon. Even if scientists become capable of editing human embryos, it would take years for the genetically modified baby to grow old enough to reproduce—and then many generations for the alteration to disseminate throughout the population.
But there are long-term consequences to consider. Modern medicine already shapes our genome, by preserving genes that might otherwise have been edited out of our genome by natural selection. Today, millions of people suffer from myopia, and many of them are legally blind. Were it not for the invention of glasses, which have turned poor eyesight largely into a nuisance rather than an existential threat, the genes responsible for myopia might be less prevalent than they are today. The same could be said about many infectious diseases, and even chronic conditions like diabetes.
Humans also carry genes that protect us from one disease but increase our susceptibility to others, and it’s impossible to predict the impact of changing all or even most of them. The AIDS virus often enters our blood cells through a protein called CCR5. One particular genetic variant of that protein, called the Delta32 mutation, prevents H.I.V. from locking onto the cell. If every person carried that mutation, nobody would get AIDS. So why not introduce that mutation into the human genome? Several research teams are working to develop drugs that do that in people who have already been infected.
Yet it’s important to note that, while such a procedure would prevent H.I.V. infection, it would also elevate our susceptibility to West Nile virus. Today, that trade-off may seem worth the risk, but there’s no way of knowing whether it would be true seven or ten generations from now. For example, sickle cells, which cause anemia, evolved as a protection against malaria; the shape of the cell blocks the spread of the parasite. If CRISPR technology had been available two hundred thousand years ago, it might have seemed sensible to edit sickle cells into the entire human population. But the results would have been devastating.
“This is a little bit like geoengineering,” Zhang told me, referring to attempts to deliberately alter the climate to offset damages associated with global warming. “Once you go down that path, it may not be so reversible.”
George Church disagrees. “It strikes me as a fake argument to say that something is irreversible,” he told me. “There are tons of technologies that are irreversible. But genetics is not one of them. In my lab, we make mutations all the time and then we change them back. Eleven generations from now, if we alter something and it doesn’t work properly we will simply fix it.”
In 1997, Scottish scientists shocked the world by announcing that they had cloned a lamb, which they named Dolly. Scores of journalists (including me) descended on Edinburgh, and wrote that the achievement, while wondrous, also carried the ominous implication that scientists had finally pried open Pandora’s box. Many articles about cloning and the value of human life were published. Evil people and dictators would clone themselves, their children, their pets. A new class of humans would arise.
Eighteen years later, the closest we have come to cloning a person was a failed attempt at a monkey, in 2007. Nobody spends much time worrying about it today. In Cambridge this summer, one of the researchers at the Broad told me that he and Louise Brown, the first success of in-vitro fertilization, were both born in 1978. “Did that set off an uproar?” he asked. It did. Even seven years earlier, James Watson had written, in The Atlantic, that the coming era of designer babies might overwhelm us all. Today, though, with more than five million children on earth born through in-vitro fertilization, that particular furor, too, seems to have passed.
CRISPR technology offers a new outlet for the inchoate fear of tinkering with the fundamentals of life. There are many valid reasons to worry. But it is essential to assess both the risks and the benefits of any new technology. Most people would consider it dangerous to fundamentally alter the human gene pool to treat a disease like AIDS if we could cure it with medicine or a vaccine. But risks always depend on the potential result. If CRISPR helps unravel the mysteries of autism, contributes to a cure for a form of cancer, or makes it easier for farmers to grow more nutritious food while reducing environmental damage, the fears, like the many others before them, will almost certainly disappear.
2.1.3.10 Can CRISPR/Cas9 Target Multiple Targets? Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 2: CRISPR for Gene Editing and DNA Repair
CRISPR/Cas9 has advanced genome editing and revolutionized molecular biology perhaps even more than the restriction enzyme. But can it edit multiple targets efficiently?
For CRISPR/Cas9 editing, single guide RNAs (sgRNAs) direct the bacterial Cas9 endonuclease to specific loci, allowing targeting of almost any gene. But is it possible to efficiently target multiple genes? “You can express one, two, or even three sgRNAs pretty easily, but if you want to do four, five, or more, it becomes difficult,” Yinong Yang at Pennsylvania State University said.
Yang’s team addressed this question in a Proceedings of the National Academy of Science paper by turning to the cell’s own tRNA processing systems. The group created polycistronic tRNA-gRNA (PTG) constructs that consisted of an sgRNA flanked by a pre-tRNA gene; the cell’s endogenous RNases can then cleave one or multiple transcribed gRNAs from the cistron to direct Cas9 to target genes.
Schematic depiction of the synthetic tRNA-gRNA gene. Credit: Yinong Yang.
“The beauty of this approach is that the 77 bp pre-tRNA gene contains internal promoter elements (box A and B) to recruit the RNA Pol III complex, so maybe you don’t even need a promoter. The Pol III promoter [which is currently used to drive expression of the sgRNA] isn’t very strong, so the tRNA will give you enhanced expression of multiple RNAs.”
The group first tested the PTG in rice protoplasts and soon realized that existing CRISPR/Cas9 vectors can be used to express PTGs. They also observed that the PTGs were more effective at introducing insertions or deletions than sgRNAs, perhaps owing to their higher expression levels from the endogenous tRNA enhancers.
Yang and his colleagues next asked if it was possible to introduce deletions in multiple genes by targeting the MAP kinase components MPK1, MPK2, MPK5, and MPK6 individually and in combinations of two or four. The PTG system introduced deletions for up to four genes, although there was a two-fold reduction in editing efficiency, which the authors attribute to competition for Cas9 among the multiple gRNAs. They then usedAgrobacterium-mediated transformation to transform mature rice plants with sgRNAs or PTGs for MPK genes and observed a higher mutational frequency of bi-allelic mutations and deletions in the plants transformed with the PTGs. Finally, they were able to target the phytoene desaturase (PDS) gene to generate a photo-bleached phenotype in the resulting plants. While they only obtained a single line carrying the fragment deletion of PDS, the mutational efficiency for PTGs was 100 percent.
Reference:
Xie, K, Minkenberg B, and Yang, Y. Boosting CRISPR/Cas9 multiplex editing capability with the endogenous tRNA-processing system. Proc Natl Acad Sci U S A. 2015 Mar 17;112(11):3570-5. doi: 10.1073/pnas.1420294112.
Boosting CRISPR/Cas9 multiplex editing capability with the endogenous tRNA-processing system
Kabin Xie, Bastian Minkenberg, and Yinong Yang1
Department of Plant Pathology and Environmental Microbiology and the Huck Institutes of the Life Sciences,
Pennsylvania State University, University Park, PA 16802
Edited by Jennifer A. Doudna, University of California, Berkeley, CA, and approved February 3, 2015
The clustered regularly interspaced short palindromic repeat (CRISPR)/ CRISPR-associated protein 9 nuclease (Cas9) system is being harnessed as a powerful tool for genome engineering in basic research, molecular therapy, and crop improvement. This system uses a small guide RNA (gRNA) to direct Cas9 endonuclease to a specific DNA site; thus, its targeting capability is largely constrained by the gRNA-expressing device. In this study, we developed a general strategy to produce numerous gRNAs from a single polycistronic gene. The endogenous tRNA-processing system, which precisely cleaves both ends of the tRNA precursor, was engineered as a simple and robust platform to boost the targeting and multiplex editing capability of the CRISPR/ Cas9 system. We demonstrated that synthetic genes with tandemly arrayed tRNA–gRNA architecture were efficiently and precisely processed into gRNAs with desired 5′ targeting sequences in vivo, which directed Cas9 to edit multiple chromosomal targets. Using this strategy, multiplex genome editing and chromosomal-fragment deletion were readily achieved in stable transgenic rice plants with a high efficiency (up to 100%). Because tRNA and its processing system are virtually conserved in all living organisms, this method could be broadly used to boost the targeting capability and editing efficiency of CRISPR/Cas9 toolkits.
Significance The clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated protein 9 nuclease (Cas9) system has recently emerged as an efficient and versatile tool for genome editing in various organisms. However, its targeting capability and multiplex editing efficiency are often limited by the guide RNA (gRNA)-expressing device. This study demonstrates a general strategy and platform for precise processing and efficient production of numerous gRNAs in vivo from a synthetic polycistronic gene via the endogenous tRNA-processing system. This strategy is shown to significantly increase CRISPR/Cas9 multiplex editing capability and efficiency in plants and is expected to have broad applications for small RNA expression and genome engineering.
Fig. 1. Engineering the endogenous tRNA system for multiplex genome editing with CRISPR/Cas9. (A) The eukaryotic pre-tRNA with 5′ leader and 3′ trailer is cleaved by RNase P and RNase Z at specific sites. (B) Transcription of tRNA gene with RNA polymerase III (Pol III). The box A and box B elements in the tRNA gene function as internal transcriptional elements and are bound by transcription factor IIIC (TFIII C), which recruits TFIIIB and Pol III to start transcription. (C) Schematic depiction of the PTG/Cas9 method for simultaneously targeting multiple sites. The synthetic PTG consists of tandemly arrayed tRNA-gRNA units, with each gRNA containing a target-specific spacer (labeled as a diamond with different color) and conserved gRNA scaffold (rectangle). The tRNA containing box A and B elements is shown as round rectangles. The primary transcript of PTG is cleaved by endogenous RNase P and RNase Z (labeled as scissors) to release mature gRNAs and tRNA (red lines of cloverleaf structure). The excised mature gRNAs direct Cas9 to multiple targets.
Strategy to Engineer a tRNA-processing System for Producing Numerous gRNAs
Precise Processing of PTG to Produce Functional gRNAs with Desired Targeting Sequences
Fig. 2. Precise excision of functional gRNAs in vivo from synthetic PTG genes. (A) The architecture of two sgRNA genes and four PTGs to produce one gRNA. (B) Sequence and predicted secondary structure of tRNA–gRNA–tRNA fusion of PTG gene. The bases of the tRNA region are indicated with red color and the tRNA 5′ leader is shown in lowercase. The gRNA is indicated in black, and the gRNA spacer sequence is shown as N. (C–F) Examination of mature gRNAs produced from sgRNA or PTGs with cRT-PCR. Total RNAs from the protoplasts expressing empty vector were used as control (CK). Arrows indicate mature gRNAs amplified by cRT-PCR, and asterisks indicate the nonspecifically amplified rRNA. (G) Summary of excision sites in PTG according to mapped gRNA ends from cRT-PCR (SI Appendix, Figs. S3–S5). Arrows indicate the cleavage sites in PTG to release gRNA. The mature gRNA 5′ ends were excised from PTG exactly at the tRNA–gRNA fusion site in all cRT-PCR results whereas its 3′ ends shifted 1–4 nt within the tRNA 5′ leader (lowercase). (H) gRNA produced from U3p:sgRNA. All detected U3p:sgRNA-produced gRNA started with ribonucleotide A and terminated with multiple Us. (I) Introduction of indels at the desired sites by PTG1:Cas9 or PTG2:Cas9 in rice protoplasts as shown by PCR/RE. Arrows indicate mutated fragments resistant to RE digestion. The indel frequency is indicated at the bottom. (J) Relative expression of sgRNA1/2 and PTG1/2 in rice protoplasts. Data represent mean ± SD. ND, not detected. CK, empty vector control.
Efficient Multiplex Genome Editing in Rice Protoplasts via PTG/Cas9.
Fig. 3. Simultaneous editing of multiple genomic sites in rice protoplasts expressing PTG:Cas9. (A) Architecture, gRNA components, and targets of PTGs for multiplex genome editing. (B) PCR detection of chromosomal fragment deletion at targeted loci in rice protoplasts expressing respective PTGs with Cas9. Successful deletion is shown as truncated PCR product (indicated with arrows). The chromosomal fragment deletion frequency (del %) is indicated at the bottom of each lane. The protoplast samples expressing an empty vector were used as control (CK). (C) Representative sequences of chromosomal fragment deletion aligned with that of WT. The gRNA paired region is labeled with green color, and the PAM region is shown in red color letters. The number at the end indicates deleted (−) or inserted (+) bases between two Cas9 cuts. The total length between two Cas9 cut sites (labeled with scissor) is indicated on the top. Short lines in the aligned sequences indicate deletions.
Improving Multiplex Genome Editing in Stable Transgenic Plants with PTG/Cas9
Table 1. Targeted mutation efficiency in PTG:Cas9 vs. sgRNA:Cas9 plants
Fig. 4. Highly efficient targeted mutagenesis in transgenic rice expressing PTG:Cas9. (A and B) Chromosomal fragment deletion in PTG7:Cas9 plant at T0 generation. Of note, only mpk1 with 358-bp deletion (Δ358) was detected in genomic DNA. Sequence analysis of the PCR products (the number in parentheses) reveals an identical deletion pattern in the transgenic plant. (C) Albino seedlings were regenerated from calli transformed with PTG10:Cas9. Most T0 seedlings (87%, n = 15) exhibited a similar photo-bleach phenotype, suggesting a very high efficiency of knocking out PDS with PTG10:Cas9. Vec, control plants transformed with empty vector. (Scale bar: 5 cm.)
We developed a general strategy and platform to produce multiple gRNAs from a single synthetic PTG gene by hijacking the endogenous tRNA-processing system (Fig. 1). We also provided a framework to design, synthesize, and use PTG for multiplex genome editing with Cas9. These PTGs were expressed with Pol III promoters (e.g., U3p) in the same manner as sgRNA genes but were not obligated to start with a specific nucleotide (Fig. 2). As a result, current CRISPR/Cas9 vectors for expressing sgRNA could be directly used to express PTG for multiplex genome editing as we demonstrated in this study.
By producing multiple gRNAs from a single polycistronic gene, the PTG technology could be used to improve simultaneous mutagenesis of multiple genomic loci or deletion of short chromosomal fragments (Figs. 3 and 4). Such a genome engineering approach may lead to simultaneous knock-out of multiple protein coding genes or deletion of noncoding RNA regions and other genetic elements. In addition to targeted mutagenesis/ deletion, the PTG approach could facilitate other Cas9-based applications in which multiple gRNAs are required. For example, PTG could be used with Cas9 nickase to improve targeting fidelity (13, 33, 34), or with deactivated Cas9 transcriptionalactivator or -repressor to manipulate multiple gene expression (35, 36). Given the high processing accuracy and capability of RNase P and RNase Z that we observed (Fig. 2), the tRNAprocessing system also could be used as a general platform to produce other regulatory RNAs (e.g., short hairpin RNA or artificial microRNA) from a single synthetic gene. These different classes of regulatory RNAs, like gRNA and short hairpin RNA, also could be compacted into a single polycistronic gene to develop more sophisticated devices for genetic engineering.
Recently, polycistronic transcripts that fused gRNA with a 28-nt RNA (referred to as gRNA-28nt) were successfully used to guide Cas9 to target up to four targets in human cells (12, 13). The synthetic gene with a gRNA-28nt architecture produced mature gRNAs with a 28-nt extra 3′ sequence and also required coexpressing the endonuclease Csy4 from Pseudomonas aeruginosa to cleave the transcript. In comparison with the gRNA-28nt strategy, our approach uses a robust endogenous tRNA-processing system that enables precise production of gRNAs with only a 1- to 4-nt extra sequence at the gRNA 3′ end (Figs. 1 and 2) and carries no additional risk of endonuclease Csy4 toxicity to recipients. Given the extremely large number of tRNA genes with variable sequences and the fact that RNase P and RNase Z precisely recognize RNA substrates with tRNA-like structures (18, 37), there are many choices of tRNA sequences to be embedded in PTG. Furthermore, the tRNA-processing system is universal in all living organisms; thus, the PTG technology could be directly adapted to other organisms for Cas9-mediated genome engineering.
When multiple double-strand breaks (DSBs) in genomic DNA were generated by PTG/Cas9 in rice plants, indels resulting from error-prone NHEJ repairing occurred more frequently than fragment deletions generated by directly joining two DSBs (SI Appendix, Figs. S10 and S11). To date, the molecular mechanism by which two DSBs directly link together to generate chromosomal translocation or fragment deletion in vivo is largely unclear. We speculate that the process leading to such a chromosomal disorder may require two DSBs at the same time interval and is likely determined by the highly dynamic interaction between gRNA/Cas9 cutting and endogenous DNA repairing and also by the distance between DSBs. Due to the differences in the delivery, expression, and activity of gRNAs and Cas9, it is not surprising to see some discrepancies in fragment-deletion frequency between protoplasts (Fig. 3B) and stable transgenic plants and among different PTG transgenic lines (Fig. 4A and SI Appendix, Figs. S9–S11). Because the PTG technology enables the generation of many DSBs in genomic DNAs, it may provide an efficient tool to help dissect the molecular process of chromosomal deletion. More importantly, the PTG technology significantly improves multiplex editing capability and efficiency and is expected to facilitate more sophisticated Cas9 applications, such as targeted mutagenesis and deletion of redundant genes or Fig. 4. Highly efficient targeted mutagenesis in transgenic rice expressing PTG:Cas9. (A and B) Chromosomal fragment deletion in PTG7:Cas9 plant at T0 generation. Of note, only mpk1 with 358-bp deletion (Δ358) was detected in genomic DNA. Sequence analysis of the PCR products (the number in parentheses) reveals an identical deletion pattern in the transgenic plant. (C) Albino seedlings were regenerated from calli transformed with PTG10:Cas9. Most T0 seedlings (87%, n = 15) exhibited a similar photo-bleach phenotype, suggesting a very high efficiency of knocking out PDS with PTG10:Cas9. Vec, control plants transformed with empty vector. (Scale bar: 5 cm.) genetic elements, transcriptional modulation of multiple genes and pathways, modification and labeling of numerous genomic sites, site-specific integration, and gene replacement.
3570-3575 | www.pnas.org/cgi/doi/10.1073/pnas.1420294112 Xie et al. genetic elements, transcriptional modulation of multiple genes and pathways, modification and labeling of numerous genomic sites, site-specific integration, and gene replacement
Validating “predicted” regulatory elements through CRISPR editing of the non-coding genome
CRISPR/Cas9-mediated genome editing is not only an efficient way to create gene KO & KI, but is a uniquely powerful tool to functionally characterize the >98% of the genome that does not encode protein. A new study demonstrates how CRISPR can be used to systematically validate putative regulatory elements described by the ENCODE and EPIGENOME projects: even in a repeat-rich genomic region, a genomic insulator upstream of mouse tyrosinase was efficiently deleted or inverted, with no significant off-target effects and high efficiency in vivo, demonstrating a functional role for this noncoding region in regulating tyrosinase gene expression and mouse coat pigmentation.
How can CRISPR/Cas9 bring us closer to finding a cure for HIV?
GenScript makes it easy for your lab to start using CRISPR to create transgenic animals or isogenic cell lines. Our license with the Broad Institute and our extensive in-house experience making CRISPR-edited mammalian cell lines enable us to make your transition to CRISPR seamless:
Complementary custom design of gRNA for any target in any species – request it here.
Choose from vectors developed in Feng Zhang’s laboratory, including all-in-one and improved GeCKO lentiviral vectors – FREE when you order gRNA constructs.
Newly developed genome-editing tools, such as the clustered regularly interspaced short palindromic repeat (CRISPR)–Cas9 system, allow simple and rapid genetic modification in most model organisms and human cell lines. Here, we report the production and analysis of mice carrying the inactivation via deletion of a genomic insulator, a key non-coding regulatory DNA element found 5′ upstream of the mouse tyrosinase (Tyr) gene. Targeting sequences flanking this boundary in mouse fertilized eggs resulted in the efficient deletion or inversion of large intervening DNA fragments delineated by the RNA guides. The resulting genome-edited mice showed a dramatic decrease in Tyr gene expression as inferred from the evident decrease of coat pigmentation, thus supporting the functionality of this boundary sequence in vivo, at the endogenous locus. Several potential off-targets bearing sequence similarity with each of the two RNA guides used were analyzed and found to be largely intact. This study reports how non-coding DNA elements, even if located in repeat-rich genomic sequences, can be efficiently and functionally evaluated in vivo and, furthermore, it illustrates how the regulatory elements described by the ENCODE and EPIGENOME projects, in the mouse and human genomes, can be systematically validated.
Non-coding DNA regulatory elements are composed of arrays of DNA–protein binding sites extending over tens to hundreds of base pairs that are occupied by multiple groups of transcription factors. DNA methylation, covalent modification of histone proteins and DNase I hypersensitivity profiles allow unbiased identification of such elements as regions of active chromatin that might be relevant in the regulation of different genes in a particular tissue or condition. Systematic ChIP-Sequencing (chromatin immunoprecipitation coupled with massive parallel sequencing) using antibodies specific for a variety of nuclear factors, applied to several human cell lines (1) and mouse tissues (2), served to identify cell type-specific regulatory elements accounting for almost 80% of the non-coding fraction of the genome. These studies, globally known as the ENCODE project (Encyclopaedia of DNA Elements; (3)) underline the rich proportion of functional elements existing within the non-coding areas of mammalian genomes. The recent publication of the human EPIGENOME project has provided additional evidence for the relevance of DNA regulatory elements in controlling gene expression (4). However, many functional experiments are required to unequivocally demonstrate the links between the observed biochemical chromatin features and the predicted biological function (5).
In the past years, the relevance of non-coding regions has been typically addressed, in vivo, using genomic-type transgenes (mostly bacterial and yeast artificial chromosomes, BACs and YACs; reviewed in (6)) carrying the inactivation of putative regulatory elements surrounded by tens to hundreds of kilo bases of genomic sequences of a suitable endogenous gene or coupled to a reporter gene (7–11). In this manner, large genomic fragments have been easily manipulated using homologous recombination in bacteria (12) and yeast (13) and then introduced into the mouse germline by standard procedures (14–15). However, variability is often observed between transgenic lines generated with BAC- or YAC-type transgenes, suggesting that position effects can influence transgene expression, even on large constructs (15–21). In addition, not all loci fit in such artificial chromosome-type transgenes, for example, large multi-gene syntenic blocks or gene clusters, whose transcriptional regulation programs during development are coordinated (22).
Here, we propose a simple strategy to functionally validate the relevance of non-coding regulatory elements in the mouse genome, in vivo. We have applied CRISPR–Cas9-mediated mutagenesis tools to inactivate, via deletion, a key regulatory sequence identified in the mouse Tyr gene (48).
We previously reported a DNAse hypersensitive (HS) site, located at ∼12 kb 5′-upstream of the mouse Tyr transcription start site (TSS), associated with a melanocyte-specific enhancer that was required for the correct expression of the Tyr gene (39). The deletion or inactivation of this element, in the context of YAC transgenesis, produced mice displaying variegation with severely reduced coat color pigmentation, supporting the notion that this key element was acting as a Locus Control Region (LCR) (7)). Homologous sequences to this mouse Tyr 5′ element were also found within the 5′ end of the human TYR locus, suggesting that mutations in this element could also impair the function of the human TYR gene (54). Traditional molecular diagnosis efforts for OCA1 patients regularly fail to detect all TYR mutations, beyond coding, promoter and limited intronic DNA sequences routinely explored. Consequently, it has been repeatedly suggested that mutations in non-coding regions could be responsible for some of these unknown non-functional TYR alleles (38,55,56). Interestingly, the recent human epigenome data released for many cellular types, including skin melanocytes, describes a regulatory element (a DNAse HS) located at ∼10 kb 5′ upstream of the human TYR gene promoter ((4); Supplementary Figure S8) at the same genomic location as was previously predicted (54). Until now, the direct relevance of TYR or Tyrregulatory elements could not be adequately studied at the endogenous loci. Instead, their role had to be inferred from results obtained using diverse standard and chromosome-type transgenes in mice (17,35).
Further studies revealed that the Tyr LCR had properties typical of genomic boundaries or insulators (57), including the capacity of establishing barriers that prevent spreading of heterochromatin and epigenetic silencing (29), and enhancer-blocking activity (40). The function of insulators is rather complex and strictly dependent on the interactions with other proximal and distal sequences in the genomic locus (43,58–60). The context-dependent activity of insulators should be therefore characterized in their native chromosomal context by gene targeting. However, the presence of repetitive sequences surrounding theTyr 5′ boundary element (29) invalidated the application of standard gene targeting approaches. As an alternative, we decided to use CRISPR–Cas9-mediated mutagenesis to overcome the limitations of classical gene targeting strategies.
Similar approaches have been recently reported to address the role of a distal Sox2 enhancer in mouse ES cells (5). Endonuclease-mediated deletions, using Transcription Activator-Like Effector Nucleases (TALENs) and Zinc-Finger Nucleases (ZFN), have been described in zebrafish (61). CRISPR–Cas9 was also used to characterize mutations found at the distal enhancer of the TAL1 oncogene in human tumor cell lines (62). Additionally, mouse models were generated using CRISPR–Cas9 in mouse ES cells to reproduce structural variants, including deletions and inversions, found in human disease (63).
In this work, we report that defined deletions and inversions in non-coding regions can be efficiently generated in vivo by CRISPR–Cas9 approaches using sgRNAs directed to adjacent genomic target sites. CRISPR–Cas9 RNA species are injected into fertilized eggs where they generate mutations at the target sequences. These mutations are then efficiently transmitted through the germ line. Using this strategy, mouse embryos are exposed to a limited amount of Cas9 nuclease for a short time, thus minimizing the risk of off-target mutations. Indeed, in our screen, no undesired mutations were detected at the six genomic loci highly similar to the targeted sequences under investigation. In contrast to this, approaches based on the delivery of CRISPR–Cas9 plasmids to somatic or ES cells may increase the associated risk of off-target mutations since exposure to the Cas9 nuclease is massive and prolonged (31).
Inactivation of the Tyr 5′ boundary element in genomic-type transgenes resulted in a severe reduction in coat color pigmentation, pointing to a relevant role for this non-coding sequence (7). However, these results were based on ectopic chromosomal locations, where variables such as transgene integrity, copy number and integration site could affect the overall gene expression program (15–21). Because of this, our vision was to target this 5′ boundary element directly at the Tyr endogenous locus, where we could unequivocally link this element to the observed phenotype without further uncontrolled variables. In actual fact, a comparative analysis of Tyr expression patterns in YAC Tyr transgenic mouse lines and TYRINS5 edited lines reveals fundamental differences in both melanocytes and RPE cells (Figures 4A, C, D, 5A, B and C). Deleting the Tyr 5′ boundary appears to have a milder effect in skin and choroidal melanocytes and a more limited impact in RPE cells, suggesting that additional regulatory elements may be responsible for controlling Tyr gene expression in RPE cells. Indeed, the presence of RPE-specific regulatory elements further upstream had been previously proposed and investigated in mice using BAC Tyr transgenes engineered with a lacZ reporter gene and variable combinations of Tyr 5′ genomic sequences (64).
CRISPR genome editing in human cells: improved targeting with the H1 promoter
A recent paper in Nature Communications reports success with a clever technique to make CRISPR-mediated genome editing easier in human cells. Compared to the commonly-used U6 promoter, driving guide RNA expression from the H1 promoter more than doubles the number of targetable sites within the genomes of humans and other eukaryotes.
Why is H1 more versatile than U6? The U6 promoter initiates transcription from a guanosine (G) nucleotide, while the H1 promoter can initiate transcription from A or G. In designing a gRNA sequence, the requirement for the protospacer adjacent motif (PAM) sequence “NGG” at the end of a 20-mer means that U6-driven gRNA must fit the pattern GN19NGG. But H1-driven gRNAs can also target sequences of the form AN19NGG, which occur 15% more frequently than GN19NGG within the human genome.
To support your genome editing efforts, GenScript offers:
The repurposed CRISPR–Cas9 system has recently emerged as a revolutionary genome-editing tool. Here we report a modification in the expression of the guide RNA (gRNA) required for targeting that greatly expands the targetable genome. gRNA expression through the commonly used U6 promoter requires a guanosine nucleotide to initiate transcription, thus constraining genomic-targeting sites to GN19NGG. We demonstrate the ability to modify endogenous genes using H1 promoter-expressed gRNAs, which can be used to target both AN19NGG and GN19NGG genomic sites. AN19NGG sites occur ~15% more frequently than GN19NGG sites in the human genome and the increase in targeting space is also enriched at human genes and disease loci. Together, our results enhance the versatility of the CRISPR technology by more than doubling the number of targetable sites within the human genome and other eukaryotic species.
Figure 1: Evaluating the ability to direct CRISPR targeting via gRNA synthesis from the H1 promoter.
(a) Schematic illustration depicting the gRNA expression constructs. Above, the U6 promoter only expresses gRNAs with a +1 guanosine nucleotide; below, the H1 promoter can drive expression of gRNAs initiating at either purine (adenosine…
Figure 2: Bioinformatics analysis of GN19NGG and AN19NGG sites in the genome.
(a) Circos plot depicting the frequency of CRISPR sites in the human genome. The outside circle depicts the human chromosome ideograms. Moving inwards, GN19NGG (orange), AN19NGG (blue) and RN19NGG (purple) CRISPR sites frequency is indi…
Could CRISPR technology be used to cure AIDS and other devastating viral diseases?
Why are viral diseases like AIDS still incurable? Although antiretroviral drugs can effectively control viral load in many patients, the permanent integration of viral DNA into a host genome means that patients remain vulnerable to re-activation of a latent virus. Exciting new research now shows that CRISPR technology can remove HIV DNA that has integrated into the host genome in human cells, re-igniting our hopes for developing a true cure for AIDS.
CRISPR-mediated genome editing is revolutionizing biomedical research due to its precise targeting, high efficiency, and ease of use in any cell type or experimental system. CRISPR has been used to create new transgenic animal models for basic and translational research, and it holds promise for use in gene therapy and other medical applications.
Our gene synthesis services have been cited in landmark publications in Nature Methods, Genetics, and Development by researchers who’ve pioneered CRISPR/Cas9 technology and applied it to new species: see references
For more than three decades since the discovery of HIV-1, AIDS remains a major public health problem affecting greater than 35.3 million people worldwide. Current antiretroviral therapy has failed to eradicate HIV-1, partly due to the persistence of viral reservoirs. RNA-guided HIV-1 genome cleavage by the Cas9 technology has shown promising efficacy in disrupting the HIV-1 genome in latently infected cells, suppressing viral gene expression and replication, and immunizing uninfected cells against HIV-1 infection. These properties may provide a viable path toward a permanent cure for AIDS, and provide a means to vaccinate against other pathogenic viruses. Given the ease and rapidity of Cas9/guide RNA development, personalized therapies for individual patients with HIV-1 variants can be developed instantly.
AIDS remains incurable due to the permanent integration of HIV-1 into the host genome, imparting risk of viral reactivation even after antiretroviral therapy. New strategies are needed to ablate the viral genome from latently infected cells, because current methods are too inefficient and prone to adverse off-target effects. To eliminate the integrated HIV-1 genome, we used the Cas9/guide RNA (gRNA) system, in single and multiplex configurations. We identified highly specific targets within the HIV-1 LTR U3 region that were efficiently edited by Cas9/gRNA, inactivating viral gene expression and replication in latently infected microglial, promonocytic, and T cells. Cas9/gRNAs caused neither genotoxicity nor off-target editing to the host cells, and completely excised a 9,709-bp fragment of integrated proviral DNA that spanned from its 5′ to 3′ LTRs. Furthermore, the presence of multiplex gRNAs within Cas9-expressing cells prevented HIV-1 infection. Our results suggest that Cas9/gRNA can be engineered to provide a specific, efficacious prophylactic and therapeutic approach against AIDS.
Infection with HIV-1 is a major public health problem affecting more than 35 million people worldwide (1). Current therapy for controlling HIV-1 infection and impeding AIDS development (highly active antiretroviral therapy; HAART) includes a mixture of compounds that suppress various steps of the viral life cycle (2). HAART profoundly reduces viral replication in cells that support HIV-1 infection and reduces plasma viremia to a minimal level but neither suppresses low-level viral genome expression and replication in tissues nor targets the latently infected cells that serve as a reservoir for HIV-1, including brain macrophages, microglia, and astrocytes, gut-associated lymphoid cells, and others (3, 4). HIV-1 persists in ∼106 cells per patient during HAART, and is linked to comorbidities including heart and renal diseases, osteopenia, and neurological disorders (5). Because current therapies are unable to suppress viral gene transcription from integrated proviral DNA or eliminate the transcriptionally silent proviral genomes, low-level viral protein production by latently infected cells may contribute to multiple illnesses in the aging HIV-1–infected patient population. Supporting this notion, pathogenic viral proteins including transactivator of transcription (Tat) are present in the cerebrospinal fluid of HIV-1–positive patients receiving HAART (6). To prevent viral protein expression and viral reactivation in latently infected host cells, new strategies are thus needed to permanently disable the HIV-1 genome by eradicating large segments of integrated proviral DNA.
Advances in the engineered nucleases including zinc finger nuclease (ZFN), transcription activator-like effector nuclease (TALEN), and clustered regularly interspaced short palindromic repeats (CRISPR) associated 9 (Cas9) that can disrupt target genes have raised prospects of selectively deleting HIV-1 proviral DNA integrated into the host genome (7⇓⇓–10). These approaches have been used to disrupt HIV-1 entry coreceptors C-C chemokine receptor 5 (CCR5) or C-C-C chemokine receptor 4 (CXCR4) and proviral DNA-encoding viral proteins (8, 9). CCR5 gene-targeting ZFNs are in phase II clinical trials for HIV-1/AIDS treatment (11). Also, various gene editing technologies have recently been shown to remove the proviral HIV-1 DNA from the host cell genome by targeting its highly conserved 5′ and 3′ long terminal repeats (LTRs) (12, 13). However, introduction of nucleases into cells via these nuclease-based genomic editing approaches remains inefficient and partially selective to remove the entire HIV-1 genome. Thus, the key barrier to their clinical translation is insufficient gene specificity to prevent potential off-target effects (toxicities). To achieve highly specific HIV-1 genome editing, we combined approaches to identify HIV-1 targets while circumventing host off-target effects. The resulting highly specific Cas9-based method proved capable of eradicating integrated HIV-1 DNA with high efficiency from latently infected human “reservoir” cell types, and prevented their infection by HIV-1.
Here, we found that LTR-directed gRNA/Cas9 eradicates the HIV-1 genome and effectively immunizes target cells against HIV-1 reactivation and infection with high specificity and efficiency. These properties may provide a viable path toward a permanent or “sterile” HIV-1 cure, and perhaps provide a means to eradicate and vaccinate against other pathogenic viruses. In the current study, we have mainly focused our efforts on myeloid lineage cells (microglia/macrophage), which are the primary cell types that harbor HIV-1 in the brain. However, this proof of concept is certainly applicable to any other cell type, including T-lymphoid cells (Fig. S6) (12, 13), astrocytes, and neural stem cells.
Our combined approaches minimized off-target effects while achieving high efficiency and complete ablation of the genomically integrated HIV-1 provirus. In addition to an extremely low homology between the foreign viral genome and host cellular genome including endogenous retroviral DNA, the key design attributes in our study included: bioinformatic screening using the strictest 12-bp+NGG target selection criteria to exclude off-target human transcriptome or (even rarely) untranslated genomic sites; avoiding transcription factor binding sites within the HIV-1 LTR promoter (potentially conserved in the host genome); selection of LTR-A- and -B-directed, 30-bp protospacer and also precrRNA system reflecting the original bacterial immune mechanism to enhance specificity/efficiency vs. 20-bp protospacer-based, chimeric crRNA-tracRNA system (16, 30); and WGS, Sanger sequencing, and SURVEYOR assay, to identify and exclude potential off-target effects. Indeed, the use of newly developed Cas9 double-nicking (23) and RNA-guided FokI nuclease (31, 32) may further assist identification of new targets within the various conserved regions of HIV-1 with reduced off-target effects.
More recently, a clinical trial using the ZFN gene editing strategy was launched to disrupt the gene encoding the HIV-1 coreceptor, CCR5 (8, 9, 11). Functional knockout of CCR5 in autologous CD4 T cells of a small cohort of patients revealed that in one out of four enrolled subjects, the viral load remained undetectable at the time of treatment (33). Similarly, TALEN and Cas9 have been tested experimentally for efficient disruption of CCR5 and CXCR4 (9, 28, 34⇓⇓–37); therefore, taking them into consideration for clinical trials is anticipated. Whether or not the strategies targeting HIV-1 entry can reach the “sterile” cure of AIDS remains to be seen. Our results show that the HIV-1 Cas9/gRNA system has the ability to target more than one copy of the LTR, which are positioned on different chromosomes, suggesting that this genome-editing system can alter the DNA sequence of HIV-1 in latently infected patient’s cells harboring multiple proviral DNAs. To further ensure high editing efficacy and consistency of our technology, one may consider the most stable region of HIV-1 genome as a target to eradicate HIV-1 in patient samples, which may not harbor only one strain of HIV-1. Alternatively, one may develop personalized treatment modalities based on the data from deep sequencing of the patient-derived viral genome before engineering therapeutic Cas9/gRNA molecules.
Our results also demonstrate, for the first time to our knowledge, that Cas9/gRNA genome editing can be used to immunize cells against HIV-1 infection. The preventative vaccination is independent of HIV-1 strain’s diversity because the system targets genomic sequences regardless of how the viruses enter the infected cells. Interestingly, the preexistence of the Cas9/gRNA system in cells leads to a rapid elimination of the new HIV-1 before it integrates into the host genome, just like the way by which the bacteria defense system evolved to combat phage infection (38). Similarly, a gene-editing-based vaccine strategy may be effective in eradicating postintegrated HIV-1 genome and newly packaged proviruses in cells. Therefore, investigation of such HIV-1 vaccination in various latent reservoir cells and animal models with stable expression of Cas9/LTR-gRNAs presents an important next step to assess the ability of Cas9 to eradicate viral reservoirs in vivo. Moreover, in light of recent data illustrating efficient in vitro genome editing using a mixture of Cas9/gRNA and DNA (39⇓⇓–42), one may explore various systems for delivery of Cas9/LTR-gRNA via various routes for immunizing high-risk subjects. Once advanced, one may use gene therapies (viral vector and nanoparticle) and transplantation of autologous Cas9/gRNA-modified bone marrow stem/progenitor cells (43, 44) or inducible pluripotent stem cells for eradicating HIV-1 infection.
Here, we demonstrated the high specificity of Cas9/gRNAs in editing HIV-1 target genome. Results from subclone data revealed the strict dependence of genome editing on the presence of both Cas9 and gRNA. Moreover, only one nucleotide mismatch in the designed gRNA target will disable the editing potency. In addition, all four of our designed LTR gRNAs worked well with different cell lines, indicating that the editing is more efficient in the HIV-1 genome than the host cellular genome, wherein not all designed gRNAs are functional, which may be due to different epigenetic regulation, variable genome accessibility, or other reasons. Given the ease and rapidity of Cas9/gRNA development, even if HIV-1 mutations confer resistance to one Cas9/gRNA-based therapy, as described above, HIV-1 variants can be genotyped to enable another personalized therapy for individual patients (10).
CRISPR-Cas9 Gene Editing: Check Three Times, Cut Once
Two new studies from UC Berkeley should give scientists who use CRISPR-Cas9 for genome engineering greater confidence that they won’t inadvertently edit the wrong DNA.
The gene editing technique, created by UC Berkeley biochemist Jennifer Doudna and her colleague, Emmanuelle Charpentier, director of the Max Planck Institute of Infection Biology in Berlin, has taken the research and clinical communities by storm as an easy and cheap way to make precise changes in DNA in order to disable genes, correct genetic disorders or insert mutated genes into animals to create models of human disease.
The two new reports from Doudna’s lab and that of UC Berkeley colleague Robert Tjian show in much greater detail how the Cas9 protein searches through billions of base pairs in a cell to find the right DNA sequence, and how Cas9 determines whether to bind, or bind and cut, thereby initiating gene editing. Based on these experiments, Cas9 appears to have at least three ways of checking to make sure it finds the right target DNA before it takes the irrevocable step of making a cut.
“CRISPR-Cas9 has evolved for accurate DNA targeting, and we now understand the molecular basis for its seek-and-cleave activity, which helps limit off-target DNA editing,” said Doudna, a Howard Hughes Medical Institute investigator at UC Berkeley and professor of molecular and cell biology and of chemistry. Tjian is president of the Howard Hughes Medical Institute and a UC Berkeley professor of molecular and cell biology.
The studies also illustrate how well CRISPR/Cas9 works in human and animal cells – eukaryotes – even though “the technique was invented by bacteria to protect themselves from getting the flu,” Doudna said.
CRISPR-Cas9 is a hybrid of protein and RNA – the cousin to DNA – that functions as an efficient search-and-snip system in bacteria. It arose as a way to recognize and kill viruses, but Doudna and Charpentier realized that it could also work well in other cells, including humans, to facilitate genome editing. The Cas9 protein, obtained from the bacteria Streptococcus pyogenes, functions together with a “guide” RNA that targets a complementary 20-nucleotide stretch of DNA. Once the RNA identifies a sequence matching these nucleotides, Cas9 cuts the double-stranded DNA helix.
One study tracked Cas9-RNA molecules though the nucleus of mammalian cells as they rapidly searched through the entire genome to find and bind just the region targeted and no other.
“It’s crazy that the Cas9 complex manages to scan the vast space of eukaryotic genomes,” said graduate student Spencer Knight, first author of the paper.
Previous studies had suggested that there are many similar-looking DNA regions that Cas9 could bind and cut, which could limit its usefulness if precision were important. These off-target regions might share as few as four or five nucleotides with the 20-nucleotide primer, just enough for Cas9 to recognize.
“There is a lot of off-target binding by Cas9, but we found that these interactions are very brief – from milliseconds to seconds – before Cas9 moves on,” he said.
Because these exploratory bindings – perhaps as many as 300,000 of them – are often very short-lived, a few thousand CRISPR-Cas9 complexes can scour the entire genome to find one targeted stretch of DNA. Cas9 must also recognize a short three-base-pair DNA sequence immediately following the primer sequence, dubbed PAM, which occurs about 300 million times within the human genome.
“If Cas9 bound for tens of seconds or minutes at each off-target site, it would never, ever be able to find a target and cut in a timely manner,” Knight said.
Cas9’s final checkpoint
The other study, published online Oct. 28 in Nature, showed that once Cas9 binds to a region of DNA, it performs another check before two distant sections of the Cas9 protein complex come together, like the blades of a scissors, to precisely align the active sites that cut double-stranded DNA.
“We found that RNA-guided Cas9 can bind some off-target DNA sequences, which differ from the correct target by just a few mutations, very tightly. Surprisingly, though, the region of Cas9 that does the cutting is inhibited because of the imperfect match. But when the correctly matching DNA is located, Cas9 undergoes a large structural change that releases this inhibition and triggers DNA cutting,” said first author Samuel Sternberg, who recently received his Ph.D. at UC Berkeley. He was able to observe these changes using a fluorescently labeled version of the Cas9 complex.
“We think that this structural change is the last checkpoint, or proofreading stage, of the DNA targeting reaction,” he said. “First, Cas9 recognizes a short DNA segment next to the target – the PAM – then the target DNA is matched up with the guide RNA via Watson-Crick base-pairing. Finally, when a perfect match is identified, the last part of the protein swings into place to enable cutting and initiate genome editing.”
A smaller Cas9 protein from a different species of bacteria, Staphylococcus aureus, likely exploits the same strategy to improve the precision of DNA targeting, suggesting that “this important feature has been preserved throughout evolutionary time,” he added.
“This is good news, in that it suggests that you have more than one checkpoint to ensure correct Cas9 binding,” Knight said. “There’s not just sequence regulation, there is also temporal regulation: it has to engage with the DNA and park long enough that it can actually rearrange and cut.”
The discoveries from Doudna, Tjian and their teams shed light on the molecular basis of off-target effects during genome editing applications, and may guide the future design of more accurate Cas9 variants.
2.1.4.6 Unchecked Spread of Engineered Genes, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 2: CRISPR for Gene Editing and DNA Repair
“Gene drive” is a phenomenon that causes a gene to be inherited at a rate faster than Mendelian principles would dictate. It relies on genes that can copy themselves onto a corresponding location in a paired chromosome, thereby overriding typical allele inheritance patterns. In conjunction with CRISPR/Cas9, gene drives can be created with almost any DNA sequence, raising questions about the risk of engineered genes spreading quickly through a population. But a team of researchers from Harvard University published a study this week (November 16) in Nature Biology that offers some safety constraints on the system.
description of the first CRISPR/Cas9 gene drive system was published in March by a team at the University of California, San Diego, and showed rapid spreading of a normally recessive phenotype inDrosophila. Other labs are researching the system’s potential to wipe out insect-borne diseases such as malaria by spreading mutated genes throughout a mosquito population. But the strategy carries the risk of accidental contamination of wild populations.
“We have a responsibility to keep our experiments confined to the laboratory,” Kevin Esvelt, an evolutionary engineer and coauthor on the paper, told Nature. “The basic lesson is: if you don’t have to build a gene drive that can spread through a wild population, then don’t.”
Esvelt’s team developed safety protocols—ways to prevent or reverse a released gene drive—using the yeast Saccharomyces cerevisia. One technique genetically separates the components necessary to create a gene drive, putting one half directly in the yeast genome and the other half on an external strand of DNA. The researchers also developed a method that uses one gene drive to overwrite the effects of another.
While these techniques are intended to stem the potential for gene drives in light of safety concerns, Esvelt told Nature that he hopes the scientific community will thoughtfully evaluate gene drives rather than dismiss them. “Should we use gene drive to eliminate malaria? Should we use it to replace broadly toxic insecticides? These questions all have to be considered separately,” he told Nature. “This paper is really about making sure we don’t blow it in the meantime and obviate the chance to talk about all of this.”
RNA-guided gene drives capable of spreading genomic alterations made in laboratory organisms through wild populations could be used to address environmental and public health problems. However, the possibility of unintended genome editing occurring through the escape of strains from laboratories, coupled with the prospect of unanticipated ecological change, demands caution. We report the efficacy of CRISPR-Cas9 gene drive systems in wild and laboratory strains of the yeastSaccharomyces cerevisiae. Furthermore, we address concerns surrounding accidental genome editing by developing and validating methods of molecular confinement that minimize the risk of unwanted genome editing. We also present a drive system capable of overwriting the changes introduced by an earlier gene drive. These molecular safeguards should enable the development of safe CRISPR gene drives for diverse organisms.
Figure 1: Mechanism and population-level effect of endonuclease gene drives.close
(a) Homing endonucleases cut competing alleles, inducing the cell to repair the damage by copying the endonuclease gene. (b) By converting heterozygous germline cells into homozygotes containing two copies (teal), gene drives increase
Figure 3: Gene drives and cargo genes remain intact upon copying and can spread by targeting both nonessential and essential genes.close
(a) The ADE2-targeting gene drive was modified to carry URA3 as a cargo gene. (b) Diploids produced by mating wild-typeURA3− haploid yeast with haploids encoding the gene drive carrying URA3 were allowed to sporulate and tetrads dissec…
CRISPR Chain Reaction
A powerful new CRISPR/Cas9 tool can be used to produce homozygous mutations within a generation, but scientists call for caution.
A new genetic-editing technique based on integratingCRISPR/Cas9 technology into a Drosophila melanogaster genome can make homozygous mutants in half the time it would take using traditional crosses, according to a paper published today (March 19) in Science.
“The study is well done and also very elegant,” said Ji-Long Liu of the University of Oxford who was not involved in the research, but helped to develop CRISPR/Cas9 in Drosophila. Liu called the method “a really clever way to . . . make the magic happen.”
A rare mosaic female fly, with a lighter left half mutated by MCR and a wild-type darker right half. UCSD, VALENTINO GANTZ AND ETHAN BIER
Safety upgrade found for gene-editing technique
Tweak reduces chance of a mutation escaping into the wild, and can help to undo a mutation after it has spread.
A method that can spread genetic changes rapidly through populations could aid the fight against the malaria parasite, shown here infecting red blood cells.
A genome-editing method that could allow researchers to rapidly engineer entire populations has had an important upgrade. A US team has added safeguards to reduce the chances that such ‘gene drives’ will escape the laboratory, and found a way to erase the genetic mutations after they have spread.
Gene drives hold the potential to wipe out insect-borne diseases and can speed up some genetic studies in the laboratory. But if released into the wild — whether intentionally or not — gene drives could irrevocably scar entire ecosystems.
The safeguards, published today in Nature Biotechnology1, may calm some fears about the technology. One of the techniques provides a way of genetically separating the components that fuel a gene drive, so that the engineered mutation will not spread as rapidly through a population. Another is a molecular ‘undo’ button: sending a second gene drive out to undo the effects of the first.
“We have a responsibility to keep our experiments confined to the laboratory,” says Kevin Esvelt, an evolutionary engineer at the Wyss Institute for Biologically Inspired Engineering at Harvard University in Boston, Massachusetts, and an author of the paper. “The basic lesson is: if you don’t have to build a gene drive that can spread through a wild population, then don’t.”
New life
The concept of a gene drive is an old one that was given new life by the advent of a genome-editing technique called CRISPR–Cas9. It allows researchers to make targeted changes to a genome with unprecedented ease and versatility.
Esvelt and others quickly realized that this technique could be used to engineer a gene drive by incorporating the genes encoding the Cas9 enzyme, which cuts DNA, and the guide RNAs, which direct Cas9 to a specific site, into the genome. Once present on one chromosome, the system can copy itself and the desired genome modification to the other chromosome, thus racing more rapidly through a population than a mutation would normally spread.
The first demonstration of this was published in March2 by developmental biologists Valentino Gantz and Ethan Bier at the University of California, San Diego. The team used gene drives to speed up genetic studies in certain species of fruit flies. But the publication kicked off concerns that the gene drive might escape from the lab into the wild, and the US National Academies of Sciences, Engineering and Medicine tasked a committee with evaluating the benefits and risks of the technology.
Even so, some researchers have embraced the approach, particularly as a means to prevent the transmission of insect-borne diseases such as malaria, says Esvelt. George Church, a bioengineer also at the Wyss Institute and a co-author on the latest report, predicts that gene drives to wipe out malaria and the tick-borne Lyme disease will be developed within the next two years. Esvelt is also collaborating with tropical-disease specialist Paul Brindley of George Washington University in Washington DC to study the application of gene drive to wiping out schistosomiasis, a disease caused by parasitic trematode worms.
Safety measures
But Esvelt worries that an accident could undermine the technique before it has a chance to prove its worth. “If anyone messes up and a gene drive gets out into the wild, there will be a huge media circus,” he says. “The message will be that scientists cannot be trusted to deal with this technology, and we will be set back by years.”
So he and his colleagues decided to develop safety measures using the yeast Saccharomyces cerevisiae. The organism is easy to work with in the laboratory and unlikely to spread a gene drive into wild populations because of its infrequent sexual reproduction.
Cambridge Healthtech Institute’s Second Annual New Frontiers in Gene Editing
Reporter: Stephen J. Williams, PhD
Gene editing is rapidly progressing from being a research/screening tool to one that promises important applications downstream in drug development, cell therapy and bioprocessing. Cambridge Healthtech Institute’s second annual symposium on New Frontiers in Gene Editing will bring together experts from all aspects of basic science and clinical research to talk about the progress being made in gene editing and how it’s being applied. Knowing the strengths and limitations of the different tools, how does one decide when to use the CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)/Cas system, as opposed to Transcription Activator-Like Effector Nucleases (TALENs), zinc finger nucleases (ZFNs) and other systems? What is being done to overcome some of the inherent challenges with design, delivery and off-target effects, associated with each of these techniques? Experts from pharma/biotech, academic and government labs will share their experiences leveraging the utility of gene editing for diverse applications.
Striving for better design, targeted delivery and performance
CRISPR Screening for drug target identification
Gene editing in stem cells
Gene editing for cell therapy and regenerative medicine
Understanding the pitfalls of gene editing
Dealing with off-target effects
Interested in Gene Editing? You may also want to attend our focused short course:
Cambridge Healthtech Institute 250 First Avenue, Suite 300 | Needham, MA 02494 | 781-972-5400 | www.healthtech.com
2.1.5.26 Cambridge Healthtech Institute’s Second Annual New Frontiers in Gene Editing, SF, 3/10-3/11, 2016, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 2: CRISPR for Gene Editing and DNA Repair
CRISPR Gene Editing Trial, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 2: CRISPR for Gene Editing and DNA Repair
CRISPR Gene Editing Trial
Larry H Bernstein, MD, FCAP, Curator
LPBI
CRISPR Gene Editing to Be Tested on People by 2017
The biotechnology startup Editas Medicine intends to begin tests of a powerful new form of gene repair in humans within two years.
Speaking this week at the EmTech conference in Cambridge, Massachusetts, Editas CEO Katrine Bosley said the company hopes to start a clinical trial in 2017 to treat a rare form of blindness using CRISPR, a groundbreaking gene-editing technology.
If Editas’s plans move forward, the study would likely be the first to use CRISPR to edit the DNA of a person.
CRISPR technology was invented just three years ago but is so precise and cheap to use it has quickly spread through biology laboratories. Already, scientists have used it to generate genetically engineered monkeys, and the technique has stirred debate over whether modified humans are next (see “Engineering the Perfect Baby”).
Editas is one of several startups, including Intellia Therapeutics and CRISPR Therapeutics, that have plans to use the technique to correct DNA disorders that affect children and adults. Bosley said that because CRISPR can “repair broken genes” it holds promise for treating several thousand inherited disorders caused by gene mistakes, most of which, like Huntington’s disease and cystic fibrosis, have no cure.
Editas, which had not previously given a timeline for an initial human test of CRISPR, will try to treat one form of a rare eye disease called Leber congenital amaurosis, which affects the light-receiving cells of the retina.
The condition Editas is targeting affects only about 600 people in the U.S., says Jean Bennet, director of advanced retinal and ocular therapeutics at the University of Pennsylvania’s medical school. “The target that they have selected is fantastic; it has all the right characteristics in terms of making a correction easily,” says Bennett, who isn’t involved in the Editas study.
Children with LCA are born seeing only large, bright shapes, and infants are diagnosed when they don’t look into their mother’s eyes or react to colorful balloons. Their poor vision can progress to “stone cold blindness where everything is black,” says Bennett.
Editas picked the disease in part because it is relatively easy to address with CRISPR, Bosley said. The exact gene error is known, and the eye is easy to reach with genetic treatments. “It feels fast, but we are going at the pace science allows,” she said. There are still questions about how well gene-editing will work in the retina and whether side effects could be caused by unintentional changes to DNA.
Editas plans to deliver the CRISPR technology as a gene therapy. The treatment will involve injecting into the retina a soup of viruses loaded with the DNA instructions needed to manufacture the components of CRISPR, including a protein that can cut a gene at a precise location. Bosley said in order to treat LCA, the company intends to delete about 1,000 DNA letters from a gene called CEP290 in a patient’s photoreceptor cells.
After the edit, preliminary lab experiments show, the gene should function correctly again. Bosley said Editas still needs to test the approach further in the lab and in animals before a human study starts.
Editas was created by venture capital funds including Third Rock Ventures in 2013 and was cofounded by scientists including Feng Zhang of the MIT/Harvard Broad Institute. It has raised more than $160 million in capital, allowing it to pursue ideas for several treatments simultaneously, Bosley said.
Although the Editas study could be the first for CRISPR in humans, it wouldn’t be the first clinical study of gene editing. An older method called zinc fingers is already in testing to treat HIV infection by the biotechnology company Sangamo Biosciences. But the versatility and ease with which CRISPR can change DNA means it may outpace earlier approaches.
Theoretically, gene editing could be used to repair broken genes in any part of the body. But in practice it is difficult to make DNA repairs in most cell types, such as brain cells. The eye is an exception because doctors can inject treatment directly under the retina.
There is already a gene-therapy treatment for one form of LCA in late-stage clinical testing by Philadelphia biotech Spark Therapeutics, says Bennett, who helped develop that treatment. In that case, an entire, healthy version of a gene is being added to eye cells. Typically, gene therapy can only add genes, not edit them.
LCA has several different genetic causes, and standard gene therapy won’t work for the form of the disease Editas is looking at. That is because the required gene, CEP290, is too big to fit inside a virus, says Bennett, and so there is no easy way to add it.
By targeting an exceptionally rare illness, Editas may have an easier time getting a treatment tested and approved. However, the eventual cost of such a treatment could be extraordinarily high, given the small number of people who would need it. Bennett says only around 3,000 Americans have LCA, and about 20 percent of those have the form being targeted by Editas.
Gene Silencing, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 2: CRISPR for Gene Editing and DNA Repair
The scientists fused Cas9 to zinc finger proteins to reduce off-target activity, essentially replacing the other nucleases that had been used with zinc fingers.
A study in Molecular Cellshows CRISPR-Cas9-like genome editing in human cells and suggests one protein could be an RNA-guided, RNA-cleaving endonuclease.
The work could potentially make lab-based blood production cost-effective, as well as improve the creation of specific cell populations from stem cells.
EDITING Researchers are learning how to use synthetic RNA sequences to control the cutting of any piece of DNA they choose. The cell will repair the cut, but an imperfect repair may disable the gene. Or a snippet of different DNA can be inserted to fill the gap, effectively editing the DNA sequence.
By:
Wallace Ravven
2.2.20 Principles of Gene Editing, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 2: CRISPR for Gene Editing and DNA Repair
The New York Times calls it “a scientific frenzy.” Science magazine dubbed it “red hot” — “The CRISPR Craze.”
It’s been less than two years since Berkeley biochemist Jennifer Doudna reported in Science a startlingly versatile strategy to precisely target and snip out DNA at multiple sites in the cells of microbes, plants and animals.
But since her landmark paper, more than 100 labs have already taken up the new genomic engineering technique to delete, add or suppress genes in fruit flies, mice, zebrafish and other animals widely used to model genetic function in human disease.
Jennifer Doudna in her lab. Photo: Roy Kaltschmidt
Last year, Doudna and her colleagues showed that this “molecular scissors” approach, known as CRISPR/Cas9, can be used with great precision to selectively disable or add several genes at once in human cells, offering a potent new tool to understand and treat complex genetic diseases.
Journal articles now appear almost weekly as researchers around the word apply the technique in basic and clinical research. Patents have been filed and licensed, and companies founded last year in Cambridge, London and Berkeley have begun zeroing in on agricultural, industrial and biomedical applications.
“I’ve never experienced anything like the pace of discovery before in my life,” Doudna says of the flurry of experimentation flowing from her 2012 paper co-authored with Emmanuelle Charpentier, now at the Helmholtz Centre for Infection Research in Germany.
HOW can we create a SNIPPET using 3D Printer? The SNIPPET with Transcription Error will be removed REPAIR the gene by a new Snippet wihtout the Transcription error
Zinc-finger proteins of the Cys2His2 type bind DNA-RNA hybrids with affinities comparable to those for DNA duplexes. Such zinc-finger proteins were converted into site-specific cleaving enzymes by fusing them to the FokI cleavage domain. The fusion proteins are active and under optimal conditions cleave DNA duplexes in a sequence-specific manner. These fusions also exhibit site-specific cleavage of the DNA strand within DNA-RNA hybrids albeit at a lower efficiency (approximately 50-fold) compared to the cleavage of the DNA duplexes. These engineered endonucleases represent the first of their kind in terms of their DNA-RNA cleavage properties, and they may have important biological applications.
Construction of vectors producing ZF–FN.
Chembiochem. 2009 May 25;10(8):1279-88. doi: 10.1002/cbic.200900040.
Artificial restriction DNA cutters as new tools for gene manipulation.
The final cut. Two types of artificial tools (artificial restriction DNA cutter and zinc finger nuclease) that cut double-stranded DNA through hydrolysis of target phosphodiester linkages, have been recently developed. The chemical structures, preparation, properties, and typical applications of these two man-made tools are reviewed.Two types of artificial tools that cut double-stranded DNA through hydrolysis of target phosphodiester linkages have been recently developed. One is the chemistry-based artificial restriction DNA cutter (ARCUT) that is composed of a Ce(IV)-EDTA complex, which catalyses DNA hydrolysis, and a pair of pseudo-complementary peptide nucleic acid fragments for sequence recognition. Another type of DNA cutter, zinc finger nuclease (ZFN), is composed of the nuclease domain of naturally occurring FokI restriction endonuclease and a designed zinc finger DNA-binding domain. For both of these artificial tools, the scission site and specificity can be freely chosen according to our needs, so that even huge genomic DNA sequences can be selectively cut at the target site. In this article, the chemical structures, preparation, properties, and typical applications of these two man-made tools are described.
Figure 1: DNA Binding Overview (original image) (crystal image rendered from PDB: 4UN3 Anders et al. 2014.)
CRISPR/Cas9 systems use a guide RNA with a region complementary to the target DNA to specifically bind their target sequences. However, there is an immediate and inherent issue with this. In order to achieve specificity, longer guide RNAs are beneficial, as each nucleotide in the RNA guide increases the specificity of the nuclease about 4-fold. However, in order for the DNA to melt and accommodate base-pairing to the guide RNA, the longer the RNA guide, the less efficient the nuclease. How can CRISPR/Cas9 systems have such dramatically increased specificity over other nucleases such as TALENS and ZFNS and still maintain roughly the same, if not better, efficiency? (Mali et al. 2013)
The answer is that the CRISPR/Cas9 system uses the Protospacer Adjacent Motif (PAM) binding as a preliminary step in locating the target sequence. As was determined by single molecule fluorescence microscopy, the initial binding of Cas9 to PAM (N-G-G) sequences allows the enzyme to quickly screen for potential target sequences. The enzyme will rapidly detach from DNA that does not have the proper PAM sequence. If the protein finds a potential target with the appropriate PAM, it will to melt the remaining DNA on the target to test whether the remaining target sequence is complementary to its guide sequence. The PAM binding step allows the protein to quickly screen potential targets and avoid melting many non-target sequences in its search for fully complementary sequences to cut. (Sternberg et al. 2014)
In July of 2014, Anders et al. published a crystal structure that led to a model for PAM-dependent target DNA binding, unwinding, and recognition by the Cas9 nuclease. The following images are created based off of figure 4 of the paper, or are images rendered inPymol (distributed by Schrödinger) using the crystal structure from that paper (obtained from the Protein Data Bank).
Proposed model for PAM-dependent target DNA binding, melting, and recognition by Cas9:
1. PAM Binding:
The Protospacer Adjacent Motif (PAM) NGG bases of the target DNA strand are shown in yellow. Arginine residues 1333 and 1335 of the PAM Interacting (PI) domain bind to the major groove of the guanine bases in the PAM. A lysine residue in the Phosphate Lock Loop, also in the PI domain, binds the minor groove.
2. Phosphate Lock Loop:
This positions the PAM and target DNA such that serine 1109 in the phosphate lock loop, and two nitrogens of the phosphate lock loop’s backbone, can form hydrogen bonds to the phosphate at position +1 of the PAM. This stabilizes the target DNA such that the first bases of the target sequence (or the protospacer) can melt and rotate upwards towards the guide RNA.
3. Guide RNA:
If the target DNA is complementary to the guide RNA strand, the two strands will base pair. This will allow the target DNA to unzip, as the bases flip up and bind the guide RNA. Without the initial PAM binding and stabilization of the +1 phosphate, the guide RNA would very rarely be able to bind the target DNA, and Cas9 would be very inefficient. This illustrates a mechanism that explains why Cas9 is able to have both high efficiency and high specificity, thus making it a powerful genome editing tool.
4. Cleavage:
Finally, complete annealing of the guide RNA to the target DNA allows the HNH and RuvC nucleases to cleave their respective strands. These nucleases cleave very specifically between the 3rd and 4th nucleotides from the PAM. Again, this specificity of cleavage, as well as the fact that the individual nucleases may be mutated independently and without affecting the ability of Cas9 to bind specific sequences, make the CRISPR/Cas9 system a simultaneously powerful and flexible genome editing tool.
Seminal studies showed that CRISPR-Cas systems provide adaptive immunity in prokaryotes and promising gene-editing tools from bacteria to humans. Yet, reports diverged on whether some CRISPR systems naturally target DNA or RNA. Here, Samai and colleagues unify the studies, showing that a single type III CRISPR-Cas system cleaves both DNA and RNA targets, independently.
More on Cleavage
Supplementary Figure 11: Base-skipping CRISPR mutants mediated efficient cleavage with Cas9 and D10A Cas9.
HEK293T cells were transfected with the indicated plasmids and the genomic DNA harvested 48 h later was assessed using the Surveyor assay. The mutant name is as described in Fig. 2. wt: wild type; UD, undetectable.
September 18, 2015 | After Chinese scientists announced in April that they had edited the genes in human embryos, many researchers said it shouldn’t be done. Scientists in London say they want to do it for research only. NPR.org
October 29, 2015 | BGI ― formerly the Beijing Genomics Institute, China’s contribution to the Human Genome Project, and now a hybrid state agency and private corporation ― is one of the world’s largest scientific research and industrial powers. From its headquarters in Shenzhen and outposts across Asia, Europe and the United States, BGI performs population-scale genomics studies, runs the world’s largest on-demand DNA sequencing service, and sells a small but growing suite of commercial products. Last week, BGIrevealed the first sequencing instrument to be developed and produced in China, the BGISEQ-500, launched exclusively to Chinese markets.
Like other recent Chinese accomplishments in high-tech fields, the sequencer is as much a point of national pride as it is a commercial venture. “Shenzhen has transformed itself from labor-intensive industry to high tech,” says He Jiankui, a specialist in genomics and biochemistry who teaches at the city’s South University of Science and Technology of China. “The government has ambitions. They’re trying to switch from ‘Made in China’ to ‘Invented in China.’”
October 1, 2015 | This Wednesday, in a surprise announcement, Pacific Biosciences of Menlo Park, Calif., confirmed rumors that it has been working on a smaller, more price-effective version of its RS II gene sequencer. But rather than push out a scaled-down benchtop instrument for simple use cases, as many had anticipated, the company unveiled a machine that improves on the RS II in every particular: less than half the cost, a third the size, and most importantly, almost seven times as powerful.
New and Unusual DNA Repair Activity Identified
Click Image To Enlarge +
The new type of DNA repair enzyme, AlkD on the left, can identify and remove a damaged DNA base without forcing it to physically “flip” to the outside of the DNA backbone, which is how all the other DNA repair enzymes in its family work, as illustrated by the human AAG enzyme on the right. The enzymes are shown in grey, the DNA backbone is orange, normal DNA base pairs are yellow, the damaged base is blue and its pair base is green. [Brandt Eichman, Vanderbilt University]
Hot on the heels of the recent announcement of the Nobel Prize in Chemistry being awarded for seminal discoveries in the area of DNA repair, researchers at Vanderbilt University have published data describing new enzymatic activity for a DNA glycosylase discovered previously in the bacteria Bacillus cereus.
When Watson and Crick first published their now famous double-helix structure of DNA, many scientists imagined the molecule to be extremely chemically stable—acting as the template for passing along inheritable genetic traits. However, over the years investigators have since discovered DNA’s susceptibility to damage and its dynamic nature to repair itself, to maintain genomic stability.
“It’s a double-edged sword,” remarked senior author and project leader Brandt Eichman, Ph.D., associate professor of biological sciences and biochemistry at Vanderbilt. “If DNA were too reactive then it wouldn’t be capable of storing genetic information. But, if it were too stable, then it wouldn’t allow organisms to evolve.”
There are many ways that DNA can become damaged, but they can be classified into two basic groups: environmental sources including ultraviolet light, toxic chemicals, and ionizing radiation and internal sources, which include, reactive oxygen species, a number of chemicals the cell produces during normal metabolism, and even water.
“More than 10,000 DNA damage events occur each day within every cell of the human body, which must be repaired for DNA to function properly,” explained lead author Elwood Mullins, Ph.D., a postdoctoral research associate in Dr. Eichman’s laboratory.
The Vanderbilt team discovered the new repair activity while studying the DNA glycosylase AlkD. Glycosylases are part of a family of enzymes discovered by Tomas Lindahl, Ph.D., who received this year’s Nobel prize for recognizing that these enzymes removed damaged DNA bases through a process called base-excision repair (BER).
Briefly, during BER, a specific glycosylase molecule binds to DNA at the location of a lesion and bends the double-helix in a way that causes the damaged base to flip from the inside of the helix to the outside. The enzyme fits around the flipped out base and holds it in a position that exposes its link to the DNA’s sugar backbone, allowing the enzyme to detach it. After the damaged base has been removed, additional DNA-repair proteins move in to replace it with a new, undamaged base.
Dr. Eichman and his team found that AlkD from B. cereus works in a totally different fashion—as it does not require base flipping to recognize damaged DNA or repair it. Using crystallography techniques, the researchers were able to determine that AlkD forms a series of interactions with the DNA backbone at and around the lesion while the lesion is still stacked in the double helix. Several of these interactions are contributed by three amino acids in the enzyme that catalyze excision of the damaged base.
The findings from this study were published recently in Nature through an article entitled “The DNA glycosylase AlkD uses a non-base-flipping mechanism to excise bulky lesions.”
Additionally, the investigators found that AlkD identifies lesions by interacting with the DNA backbone without contacting the damaged base itself and can repair many different types of lesions as long as they are positively charged. Since the enzyme doesn’t have the same type of binding pocket, it isn’t restricted in the same way as other glycosylases. Lastly, AlkD can excise much bulkier lesions than other glycosylases. Base excision repair is limited to relatively small lesions. A different pathway called nucleotide excision repair typically handles larger lesions like those caused by UV radiation damage. However, Dr. Eichman’s team discovered that AlkD could excise lesions that would normally default to other DNA repair pathways.
“Our discovery shows that we still have a lot to learn about DNA repair and that there may be alternative repair pathways yet to be discovered. It certainly shows us that a much broader range of DNA damage can be removed in ways that we didn’t think were possible,” Dr. Eichman stated. “Bacteria are using this to their advantage to protect themselves against the antibacterial agents they produce. Humans may even have DNA-repair enzymes that operate in similar fashion to remove complex types of DNA damage. This could have clinical relevance because these enzymes if they exist, could be reducing the effectiveness of drugs designed to kill cancer cells by shutting down their ability to replicate.”

 This image depicts a conventional CRISPR-Cas9 system. The Cas9 enzyme acts like a wrench, and specific RNA guides act as different socket heads. Conventional CRISPR-Cas9 systems act continuously, raising the risk of off-target effects. But CRISPR-Cas9 systems that incorporate specially engineered RNAs could act transiently, potentially reducing unwanted changes. [Ernesto del Aguila III, NHGRI]
This image depicts a conventional CRISPR-Cas9 system. The Cas9 enzyme acts like a wrench, and specific RNA guides act as different socket heads. Conventional CRISPR-Cas9 systems act continuously, raising the risk of off-target effects. But CRISPR-Cas9 systems that incorporate specially engineered RNAs could act transiently, potentially reducing unwanted changes. [Ernesto del Aguila III, NHGRI]
















 2.1.4.6 Unchecked Spread of Engineered Genes, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 2: CRISPR for Gene Editing and DNA Repair
2.1.4.6 Unchecked Spread of Engineered Genes, Volume 2 (Volume Two: Latest in Genomics Methodologies for Therapeutics: Gene Editing, NGS and BioInformatics, Simulations and the Genome Ontology), Part 2: CRISPR for Gene Editing and DNA Repair





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
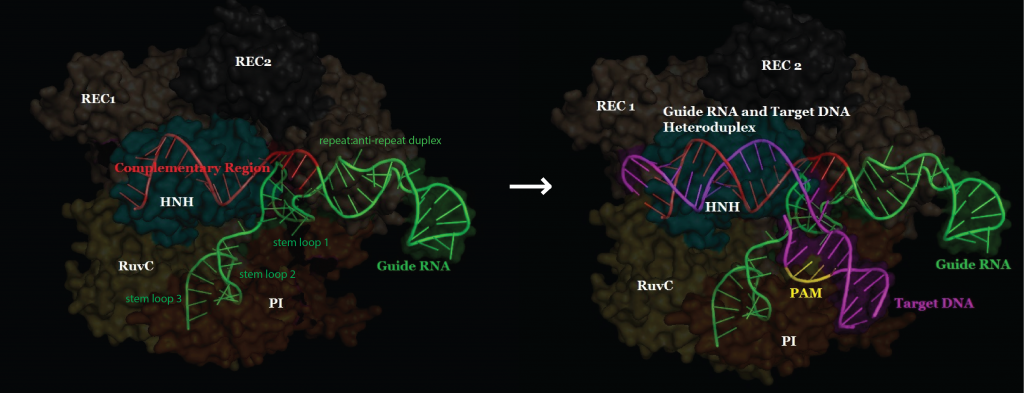{kind=link}